Gene predictions for eukaryotes attgccagtacgtagctagctacacgtatgctattacggatctgtagcttagcgtatctgtatgct gttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgtta gcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgta gtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtag tctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttc taggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctg tacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagctta gtcgtgtagtcttgatctacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcg tagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatgg ctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagggga gcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtac gtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgt acgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgta tgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtc gttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggct agtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagc ttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatggtcgtagtcgttagcatct gtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctag

Gene predictions for eukaryotes attgccagtacgtagctagctacacgtatgctattacggatctgtagcttagcgtatct gtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgt.
Dec 19, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Gene predictions for eukaryotes
attgccagtacgtagctagctacacgtatgctattacggatctgtagcttagcgtatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcttagtcgtgtagtcttgatctacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatggtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctag

Gene predictions for eukaryotes
attgccagtacgtagctagctacacgtatgctattacggatctgtagcttagcgtatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcttagtcgtgtagtcttgatctacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctagagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctagtcgtagtcgtagtcgttagcatctgtatggtcgtagtcgttagcatctgtatgctgttagctgtacgtacgtatttttctaggggagcttcgtagtctatggctag
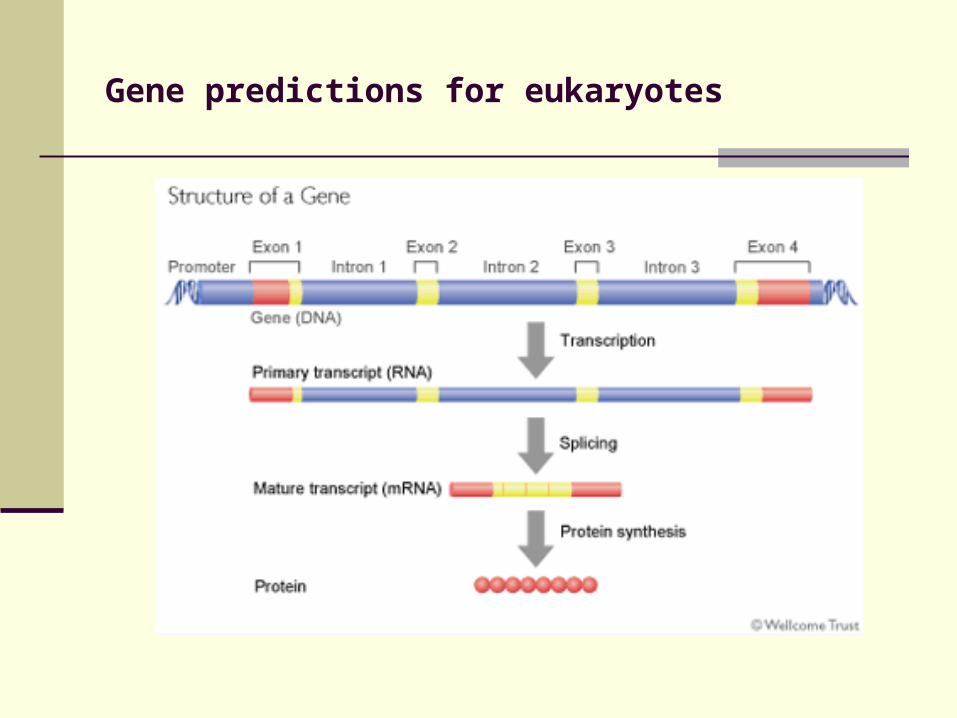
Gene predictions for eukaryotes

Gene predictions for eukaryotes
Three different approaches to computational gene-finding:
Intrinsic: use statistical information about known genes (Hidden Markov Models)
Extrinsic: compare genomic sequence with known proteins / genes
Cross-species sequence comparison: search for similarities among genomes
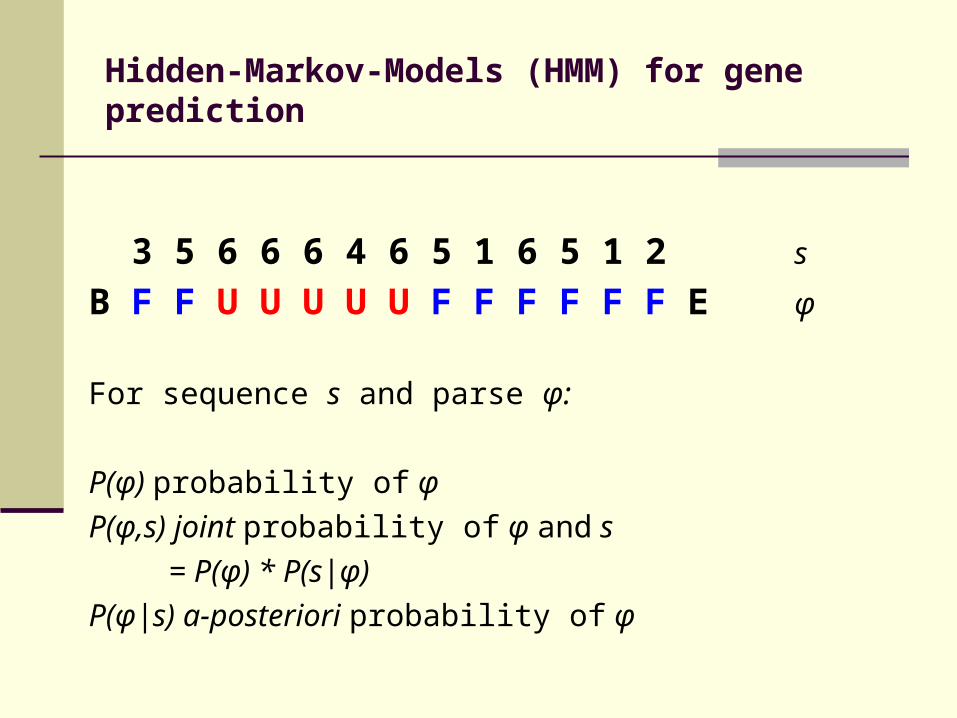
Hidden-Markov-Models (HMM) for gene prediction
3 5 6 6 6 4 6 5 1 6 5 1 2 s
B F F U U U U U F F F F F F E φ
For sequence s and parse φ:
P(φ) probability of φ P(φ,s) joint probability of φ and s = P(φ) * P(s|φ) P(φ|s) a-posteriori probability of φ
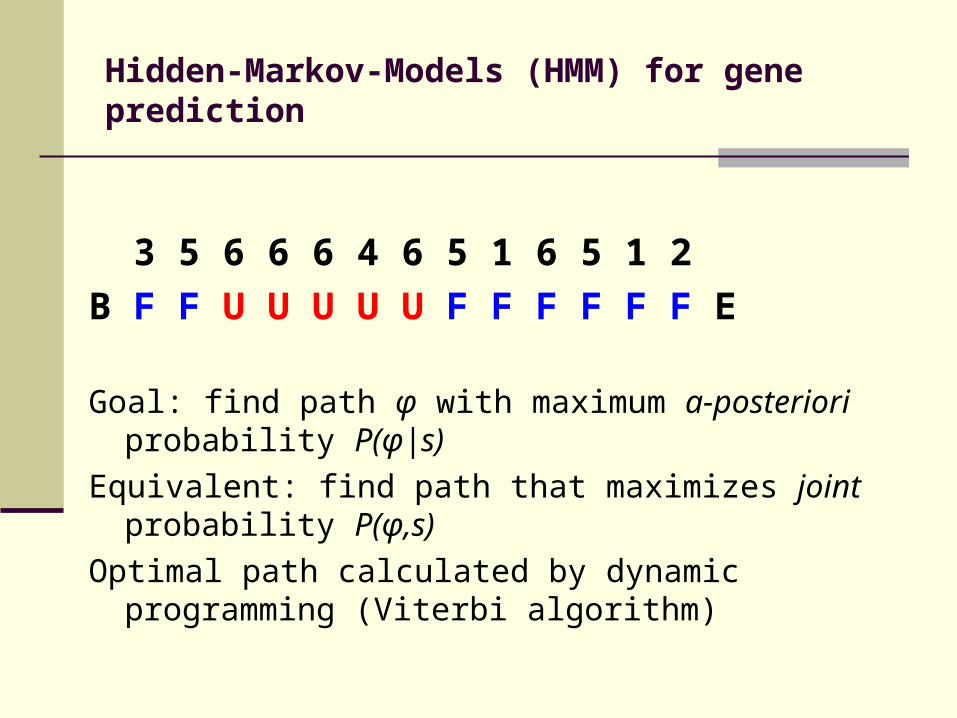
Hidden-Markov-Models (HMM) for gene prediction
3 5 6 6 6 4 6 5 1 6 5 1 2
B F F U U U U U F F F F F F E
Goal: find path φ with maximum a-posteriori probability P(φ|s)
Equivalent: find path that maximizes joint probability P(φ,s)
Optimal path calculated by dynamic programming (Viterbi algorithm)
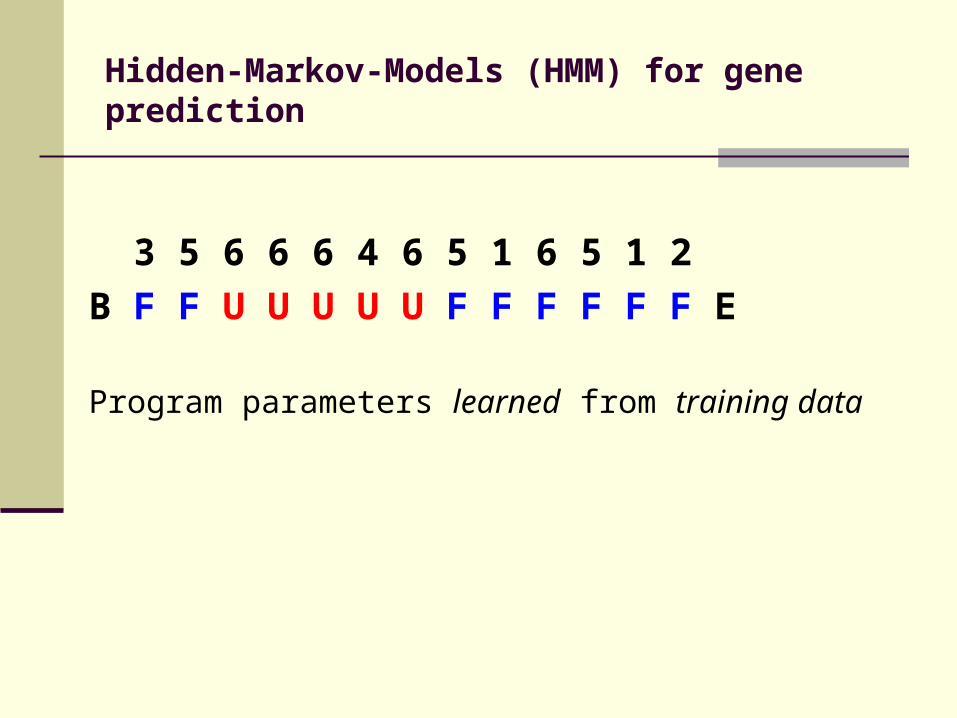
Hidden-Markov-Models (HMM) for gene prediction
3 5 6 6 6 4 6 5 1 6 5 1 2
B F F U U U U U F F F F F F E
Program parameters learned from training data
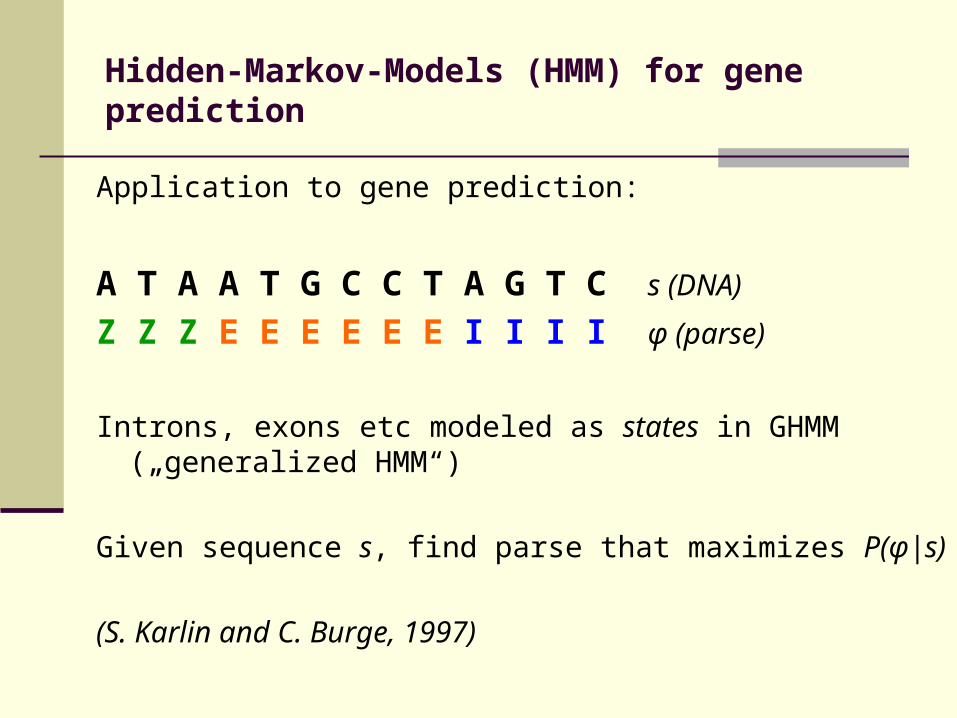
Hidden-Markov-Models (HMM) for gene prediction
Application to gene prediction:
A T A A T G C C T A G T C s (DNA) Z Z Z E E E E E E I I I I φ (parse)
Introns, exons etc modeled as states in GHMM („generalized HMM“)
Given sequence s, find parse that maximizes P(φ|s)
(S. Karlin and C. Burge, 1997)
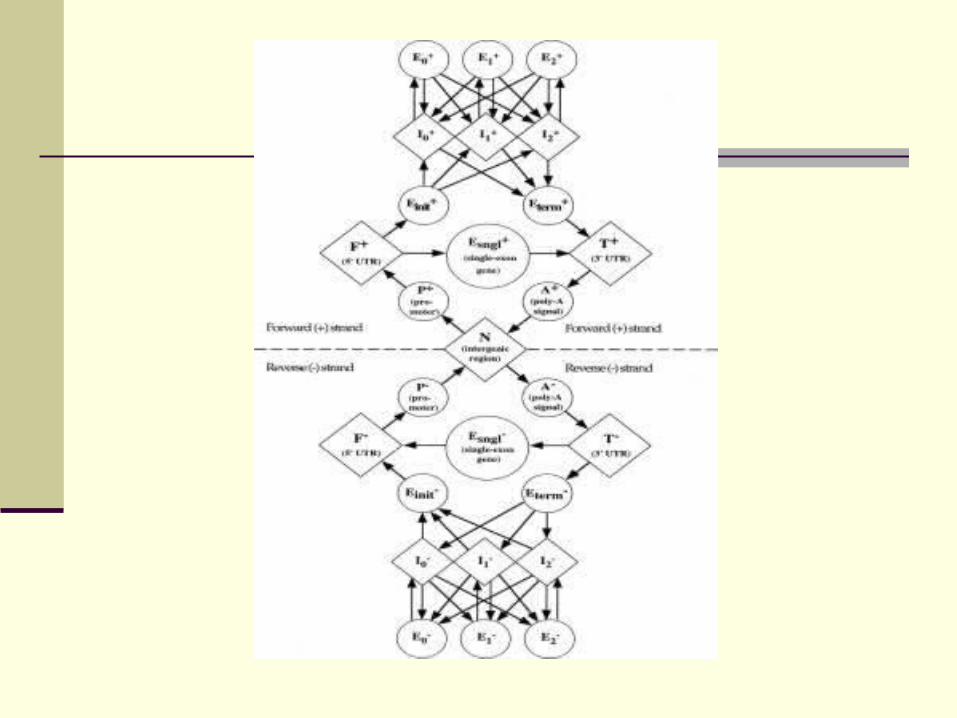

AUGUSTUS
Basic model for GHMM-based intrinsic gene finding comparable to GenScan (M. Stanke)
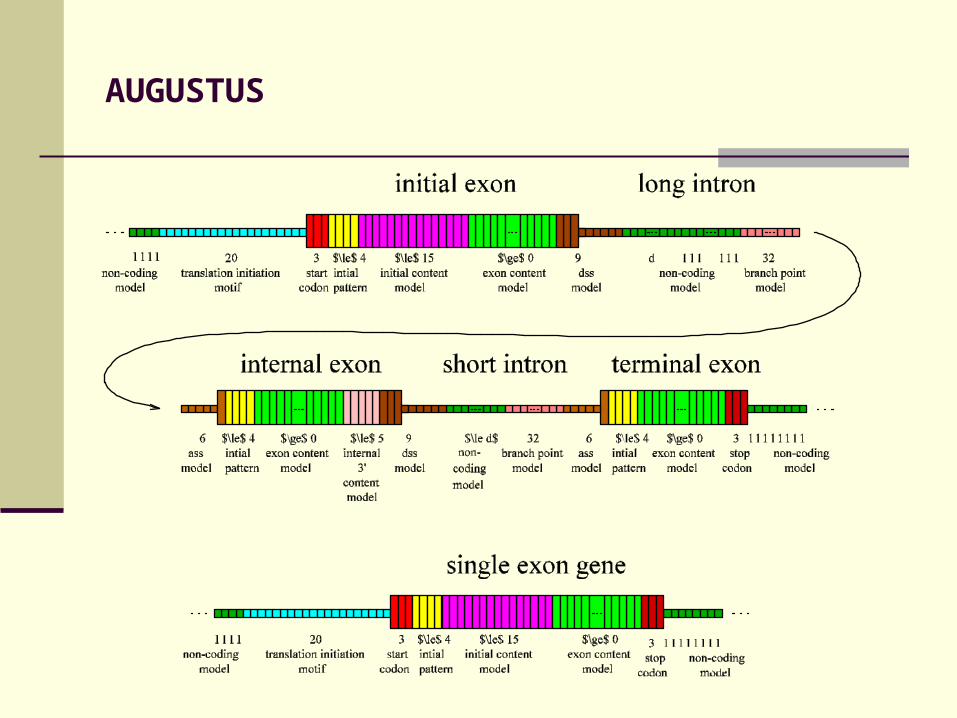
AUGUSTUS
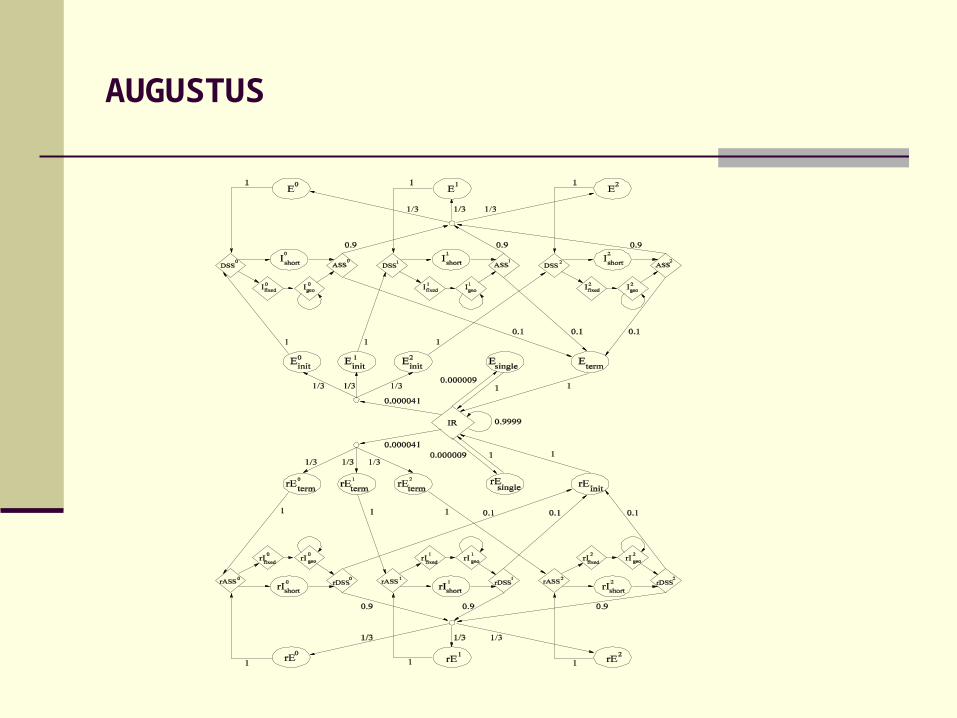
AUGUSTUS

AUGUSTUS
Features of AUGUSTUS:
Intron length model Initial pattern for exons Similarity-based weighting for splice sites Interpolated HMM Internal 3’ content model
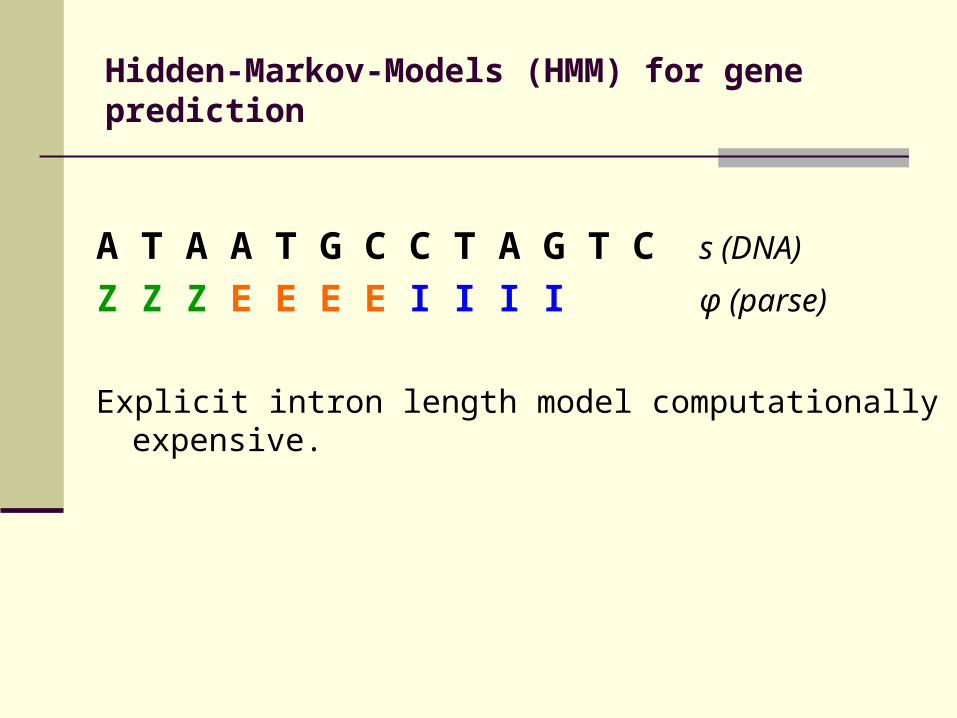
Hidden-Markov-Models (HMM) for gene prediction
A T A A T G C C T A G T C s (DNA) Z Z Z E E E E I I I I φ (parse)
Explicit intron length model computationally expensive.
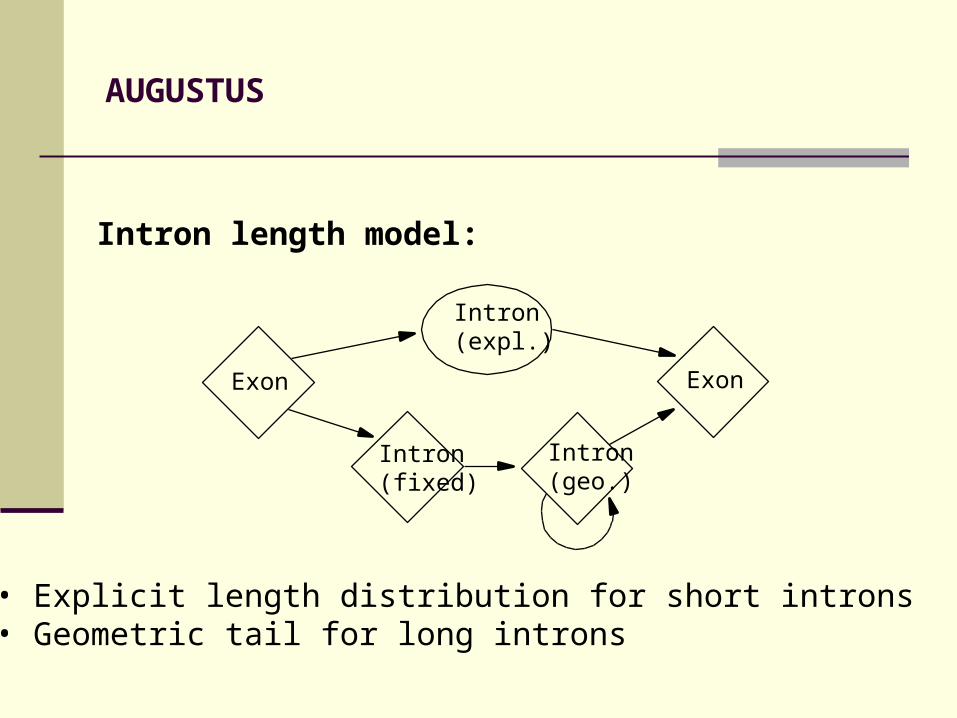
AUGUSTUS
Intron length model:
• Explicit length distribution for short introns• Geometric tail for long introns
Intron (fixed)
Exon
Intron (expl.)
Exon
Intron (geo.)
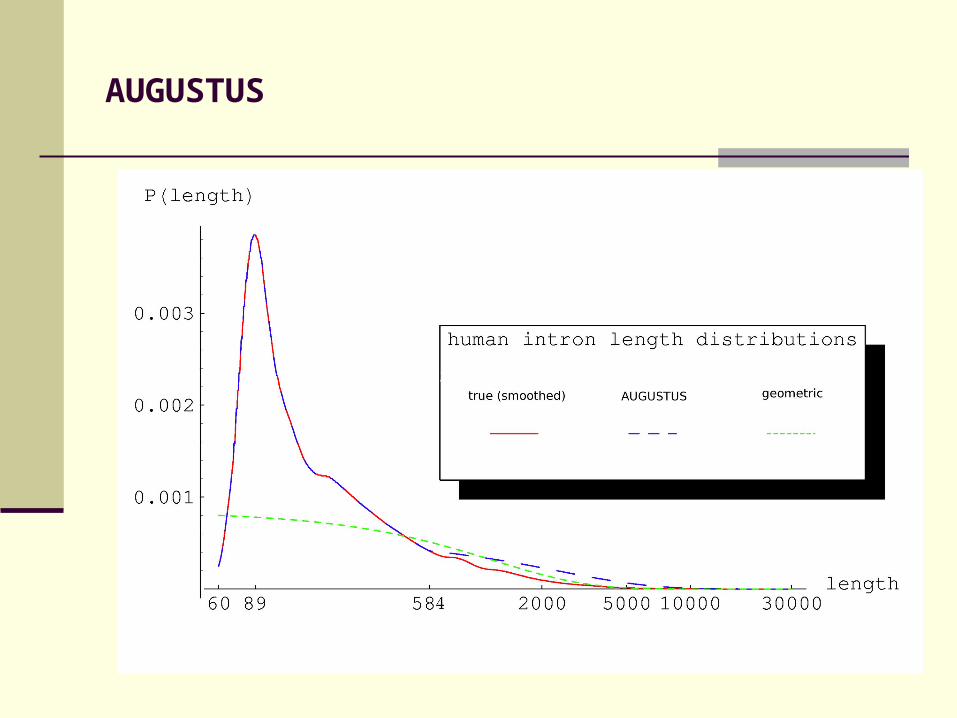
AUGUSTUS

AUGUSTUS+
Extension of AUGUSTUS using include extrinsic information:
Protein sequences EST sequences Syntenic genomic sequences User-defined constraints
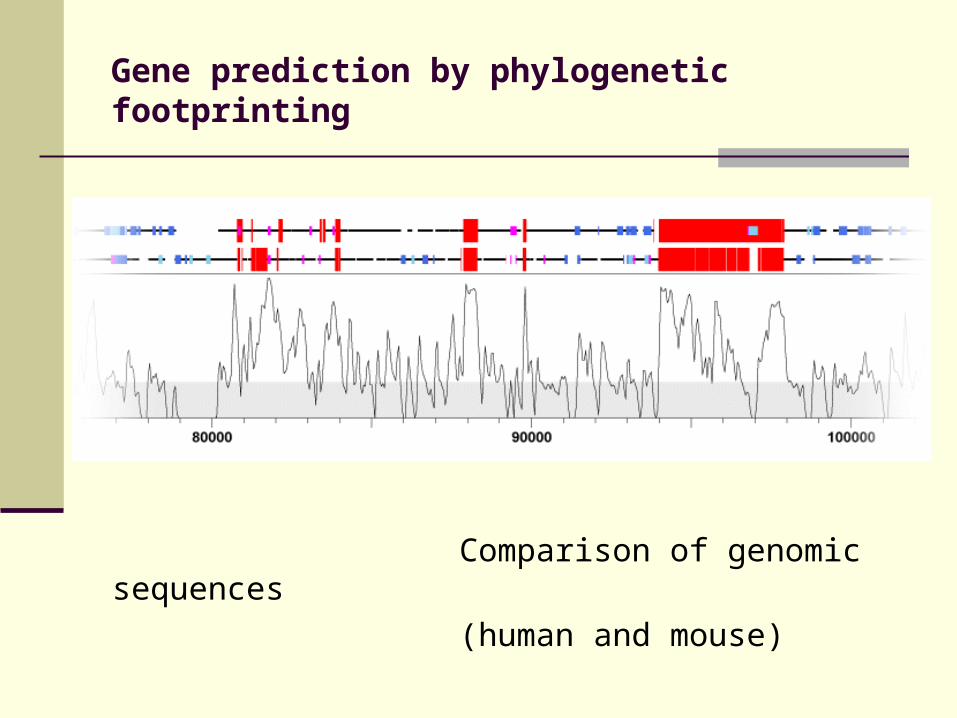
Gene prediction by phylogenetic footprinting
Comparison of genomic sequences
(human and mouse)
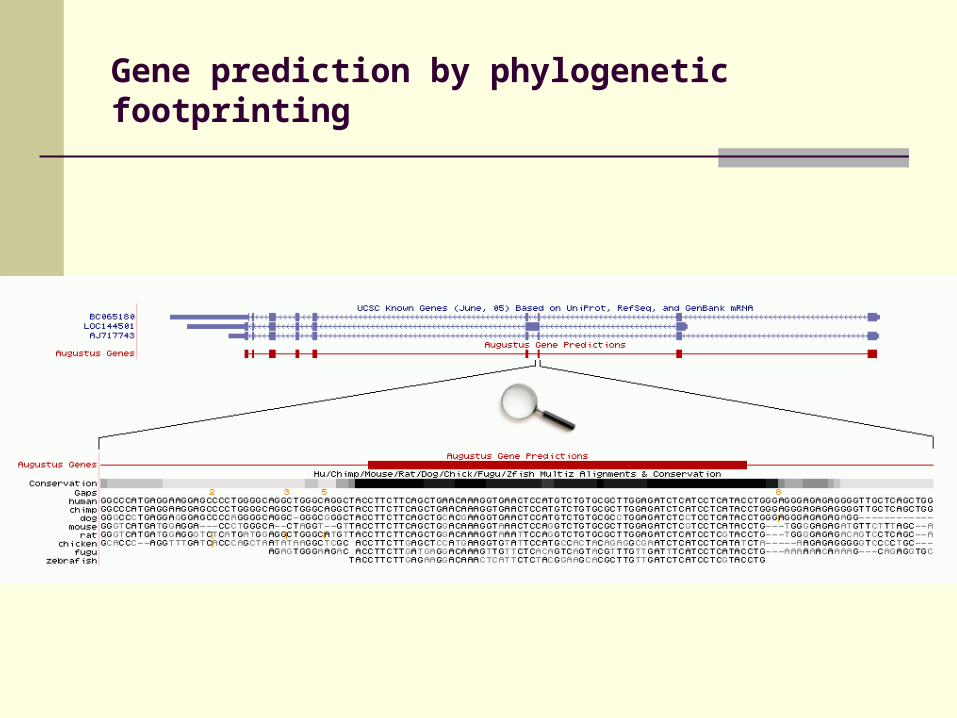
Gene prediction by phylogenetic footprinting

AUGUSTUS+
Extended GHMM using extrinsic information
Additional input data: collection h of `hints’ about possible gene structure φ for sequence s
Consider s, φ and h result of random process. Define probability P(s,h,φ)
Find parse φ that maximizes P(φ|s,h) for given s and h.

AUGUSTUS+
Hints created using
Alignments to EST sequences Alignments to protein sequences Combined EST and protein alignment (EST
alignments supported by protein alignments) Alignments of genomic sequences User-defined hints
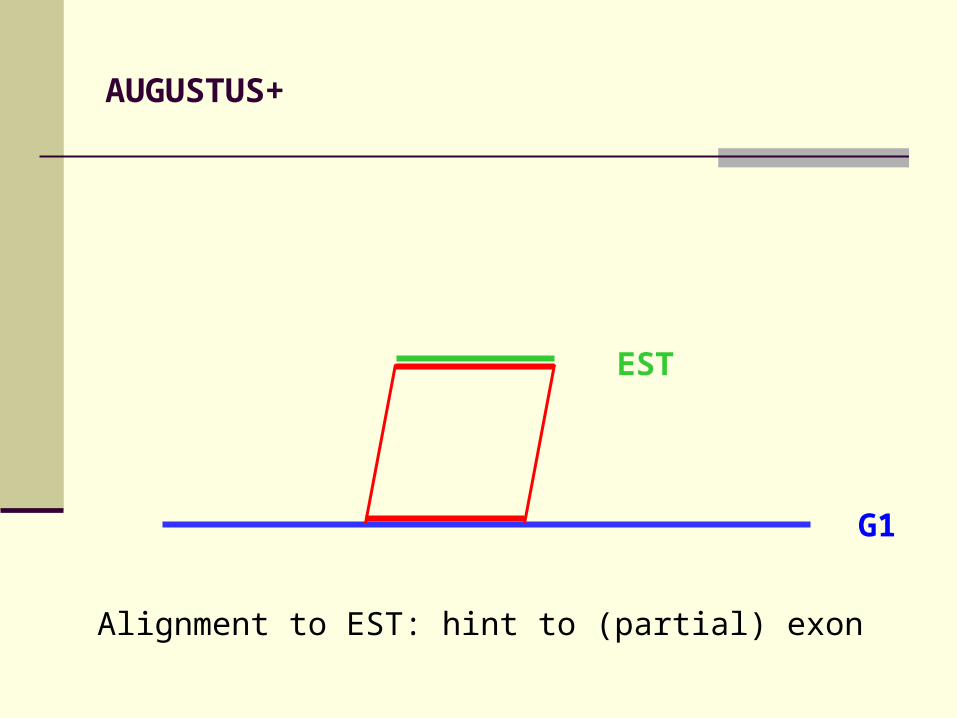
AUGUSTUS+
Alignment to EST: hint to (partial) exon
EST
G1
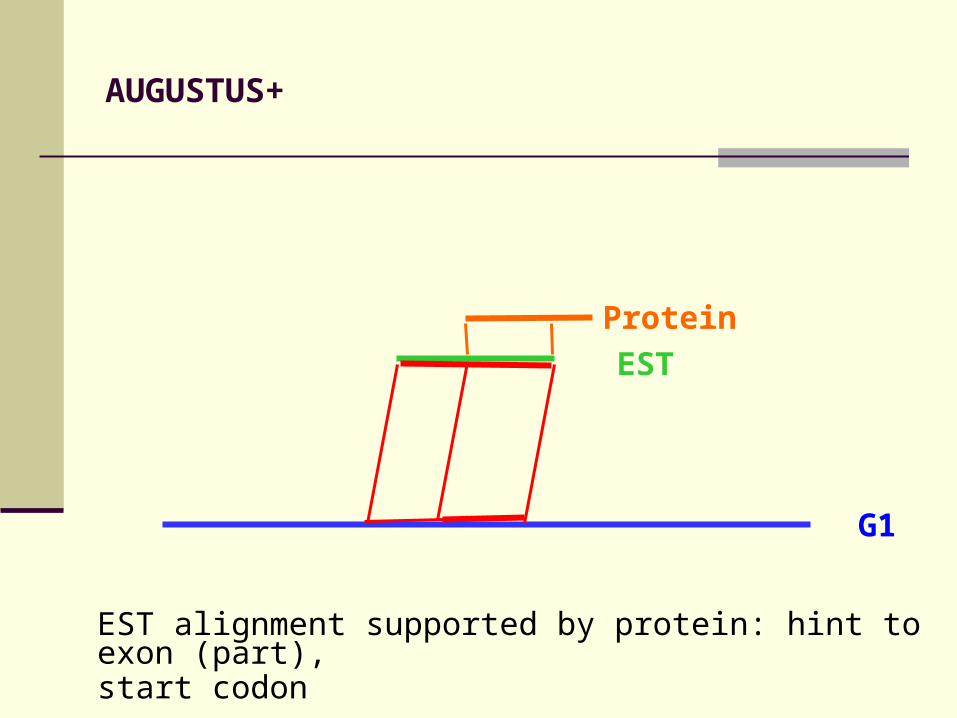
AUGUSTUS+
EST alignment supported by protein: hint to exon (part), start codon
EST
G1
Protein
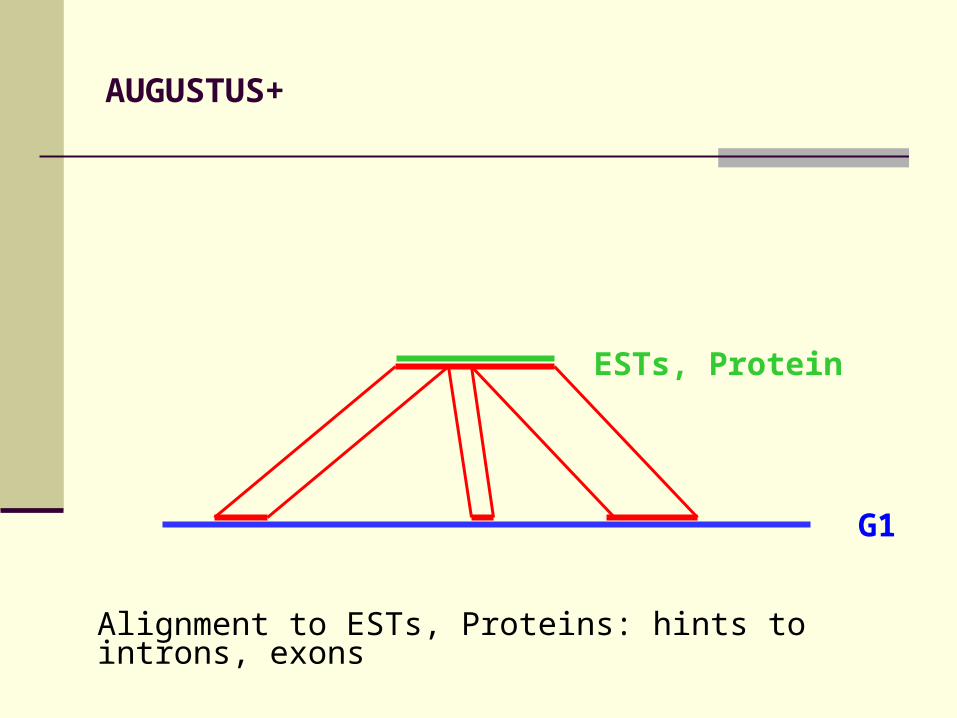
AUGUSTUS+
Alignment to ESTs, Proteins: hints to introns, exons
ESTs, Protein
G1
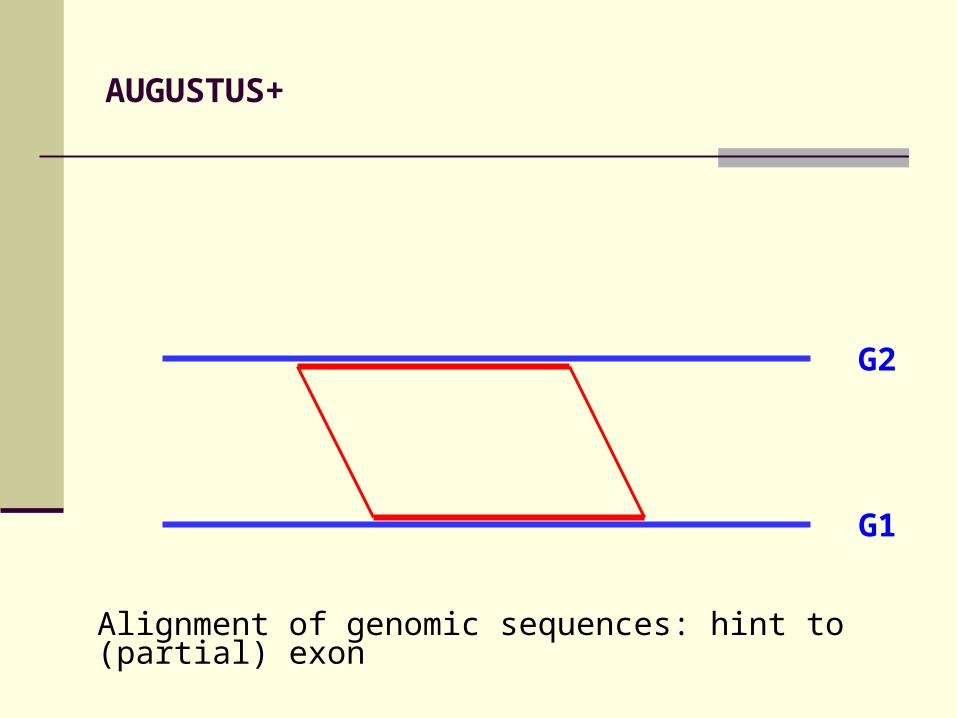
AUGUSTUS+
Alignment of genomic sequences: hint to (partial) exon
G2
G1

AUGUSTUS+
Consider different types of hints:
type of hints: start, stop, dss, ass, exonpart, exon, introns
Hint associated with position i in s (exons etc. associated with right end position) max. one hint of each type allowed per position in s Each hint associated with a grade g that indicates its source.

AUGUSTUS+
hi,t = information about hint of type t at position i
hi,t = [grade, strand, (length, reading frame)] if hint available
(hints created by protein alignments contain information about reading frame)
hi,t = $ if no hint of type t available at i

AUGUSTUS+
Standard program version, without hints
A T A A T G C C T A G T C s (sequence) Z Z Z E E E E E E I I I I φ (parse)
Find parse that maximizes P(φ|s)

AUGUSTUS+
AUGUSTUS+ using hints
A T A A T G C C T A G T C s (sequence) $ $ $ $ $ $ $ X $ $ $ $ $ h (type 1) $ $ $ $ $ $ $ $ $ $ $ $ $ h (type 2) $ $ $ $ X $ $ $ $ $ $ $ $ h (type 3) . . . .
Z Z Z E E E E E E I I I I φ (parse)
Find parse that maximizes P(φ|s,h)

AUGUSTUS+
As in standard HMM theory: maximize joint probability P(φ,s,h)
How to calculate P(φ,s,h) ?

AUGUSTUS+
Simplifying assumption: Hints of different types t and at different positions i independent of each other (for redundant hints: ignore „weaker“ types).

AUGUSTUS+
Simplifying assumption: Hints of different types t and at different positions i independent of each other (for redundant hints: ignore „weaker“ types).
),|(),(),,( shPsPhsP

AUGUSTUS+
Simplifying assumption: Hints of different types t and at different positions i independent of each other (for redundant hints: ignore „weaker“ types).
),|(),(),,( shPsPhsP
ti
ti shPsP,
, ),|(),(

AUGUSTUS+
Results:
Gene (sub-)structures supported by hints receive bonus compared to non-supported structures
Gene (sub-)structures not supported by hints receive malus
(M. Stanke et al. 2006, BMC Bioinformatics)

AUGUSTUS+

AUGUSTUS+
Using hints from DIALIGN alignments:
1. Obtain large human/mouse sequence pairs (up to 50kb) from UCSC
2. Run CHAOS to find anchor points3. Run DIALIGN using CHAOS anchor points4. Create hints h from DIALIGN fragments5. Run AUGUSTUS with hints

AUGUSTUS+
Hints from DIALIGN fragments:
Consider fragments with score ≥ 20
Distinguish high scores (≥ 45) from low scores Consider reading frame given by DIALIGN Consider strand given by DIALIGN
=> 2*2*2 = 8 grades

AUGUSTUS+
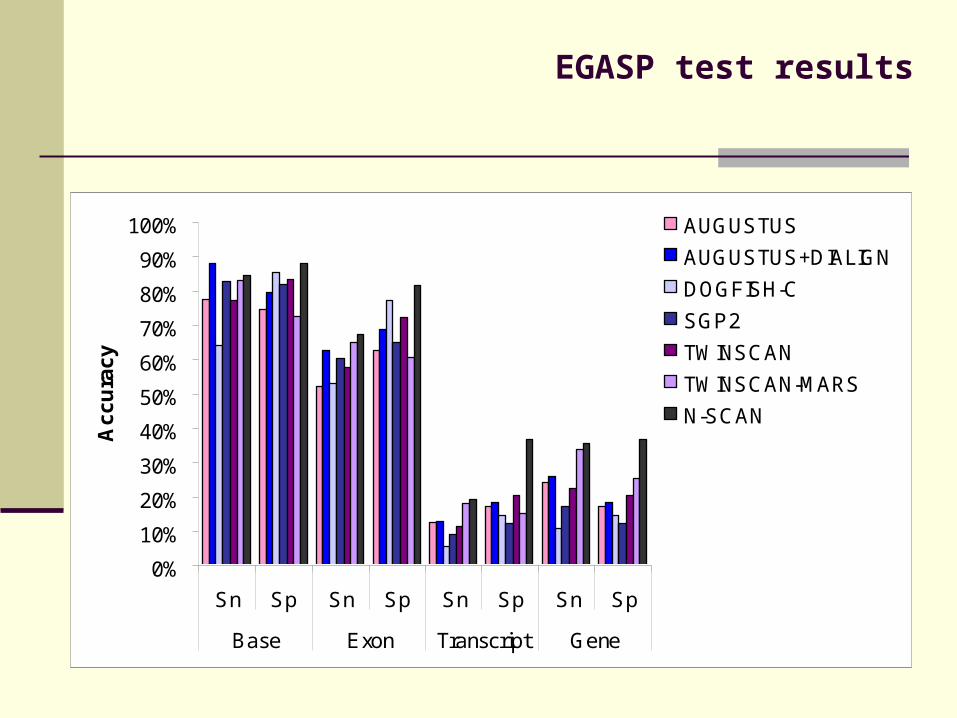
EGASP competition to evaulate and compare gene-prediction methods (Sanger Center, 2005)
AUGUSTUS best ab-initio method at EGASP

EGASP test results
AUGUSTUS
GENSCAN
geneid GeneMark.hmm
Genezilla
0
10
20
30
40
50
60
70
80
90
100 Nukleotid Level
Sensitivität
Spezifität

EGASP test results
AUGUSTUS
GENSCAN
geneid GeneMark.hmm
Genezilla
0
10
20
30
40
50
60
70
80
90
100 Exon Level
Sensitivität
Spezifität
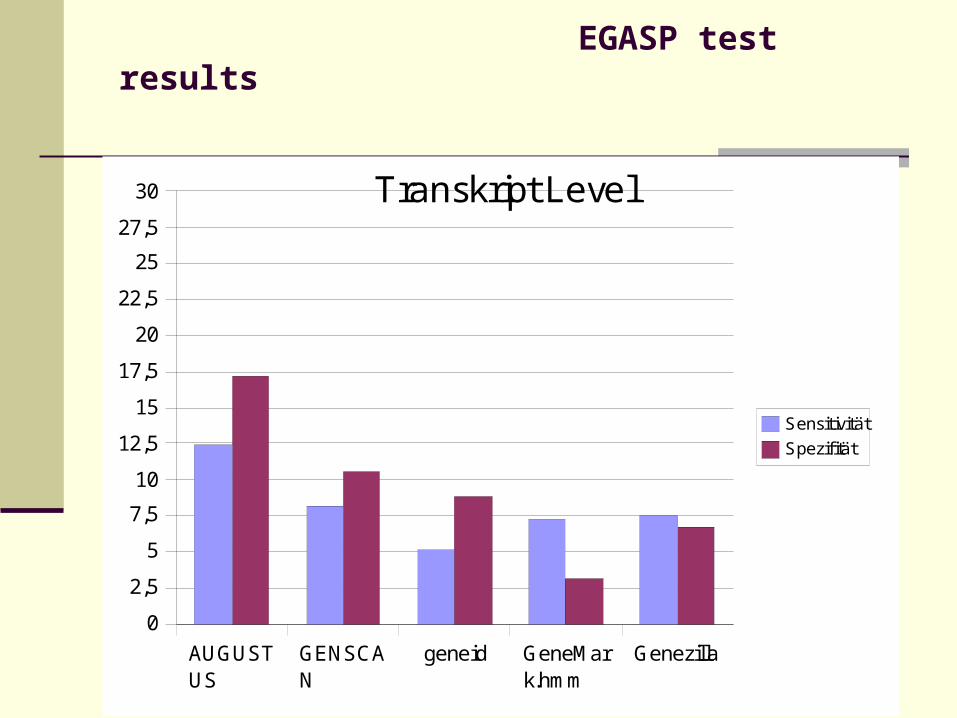
EGASP test results
AUGUSTUS
GENSCAN
geneid GeneMark.hmm
Genezilla
0
2,5
5
7,5
10
12,5
15
17,5
20
22,5
25
27,5
30 Transkript Level
Sensitivität
Spezifität
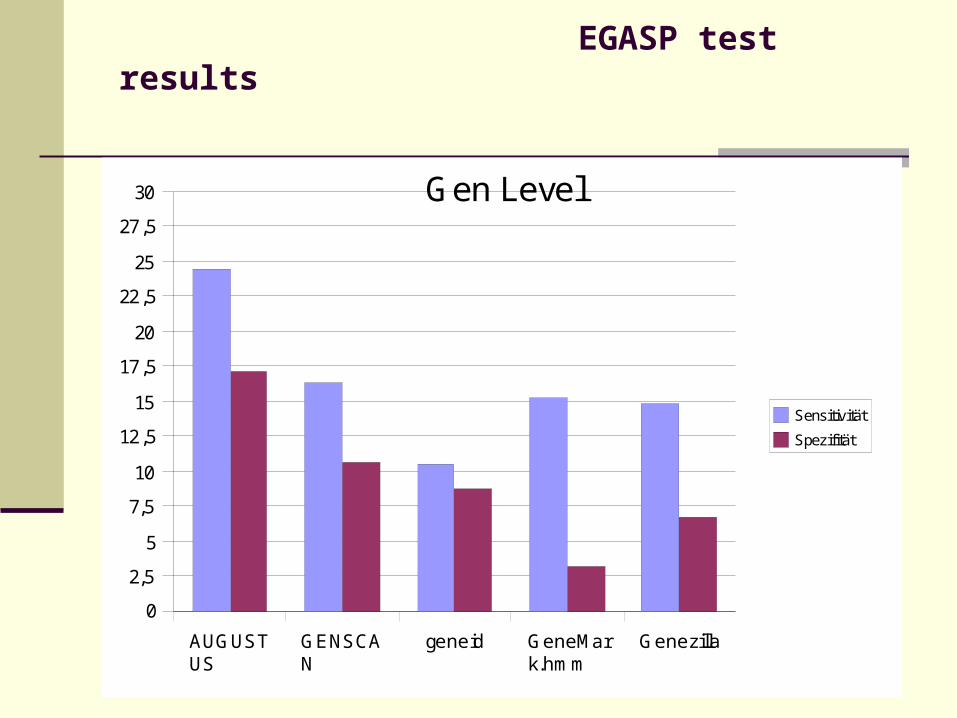
EGASP test results
AUGUSTUS
GENSCAN
geneid GeneMark.hmm
Genezilla
0
2,5
5
7,5
10
12,5
15
17,5
20
22,5
25
27,5
30 Gen Level
Sensitivität
Spezifität
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Sn Sp Sn Sp Sn Sp Sn Sp
Base Exon Transcript Gene
Ac
cu
rac
y
AUGUSTUS
AUGUSTUS+DIALIGN
DOGFISH-C
SGP2
TWINSCAN
TWINSCAN-MARS
N-SCAN
EGASP test results

Application of AUGUSTUS in genome projects
Brugia malayi (TIGR)
Aedes aegypti (TIGR)
Schistosoma mansoni (TIGR)
Tetrahymena thermophilia (TIGR)
Galdieria Sulphuraria (Michigan State Univ.)
Coprinus cinereus (Univ. Göttingen)
Tribolium castaneum (Univ. Göttingen)
Related Documents











