Gallic acid based steroidal phenstatin analogues for selective targeting of breast cancer cells through inhibiting tubulin polymerization Swati Parihar a , Atul Gupta a , Amit K. Chaturvedi a , Jyoti Agarwal a , Suaib Luqman a , Bendangla Changkija b , Murli Manohar b , Debabrata Chanda a , C.S. Chanotiya a , Karuna Shanker a , Anila Dwivedi b , Rituraj Konwar b , Arvind S. Negi a,⇑ a Central Institute of Medicinal and Aromatic Plants (CSIR-CIMAP), Kukrail Picnic Spot Road, Lucknow 226015, UP, India b Central Drug Research Institute (CSIR-CDRI), Chattar Manzil Palace, Lucknow 226001, UP, India article info Article history: Received 28 February 2012 Received in revised form 23 March 2012 Accepted 26 March 2012 Available online 5 April 2012 Keywords: Gallic acid Phenstatin Anticancer Tubulin polymerization inhibition Acute oral toxicity abstract Phenstatin analogues were synthesized on steroidal framework, for selective targeting of breast cancer cells. These analogues were evaluated for anticancer efficacy against breast cancer cell lines. Analogues 12 and 19 exhibited significant anticancer activity against MCF-7, hormone dependent breast cancer cell line. While analogues 10–14 exhibited significant anticancer activity against MDA-MB-231, hormone independent breast cancer cell line. Compound 10 showed significant oestrogen antagonistic activities with low agonistic activity in in vivo rat model. These analogues also retain tubulin polymerization inhi- bition activity. The most active analogue 10 was found to be non-toxic in Swiss albino mice up to 300 mg/ kg dose. Gallic acid based phenstatin analogues may further be optimized as selective anti-breast cancer agents. Ó 2012 Elsevier Inc. All rights reserved. 1. Introduction Hormone-dependent breast cancer over-expresses oestrogen receptor alpha (ERa) in comparison to normal breast tissue [1] and serves as prognostic indicator of the disease [2]. ERa is a well validated therapeutic target for breast cancer with gold standard example of tamoxifen, a Selective Estrogen Receptor Modulator (SERM) [3]. However, several reports suggest tamoxifen to be potentially associated with risk of endometrial cancers [4]. Tamox- ifen also exhibits drug resistance in breast cancer [5]. These critical factors impel to develop newer alternatives of targeted breast can- cer with improved profile. Plant molecules for a long have made immense contribution in enriching repertoire of chemotherapeutic agents. Taxol, vincristine, vinblastine, podophyllotoxin, camptothecin, indole 3-carbinol and combretastatin A4 (CA-4) are some of the notable anticancer leads [6,7]. Combretastatin A4 is an antimitotic agent originally isolated from the bark of the South African Willow tree Combretum caffrum. Combretastatin binds to the b-subunit of tubulin at the colchicine site, thereby strongly inhibiting polymerization of tubulin, causing cellular disorganization and toxicity. Combretastatin A 4 (1) is an attractive lead for developing anticancer drugs and several analogues with potent activity has been developed [8,9]. To enhance the bioavailability of these lead molecules (CA-1 and CA- 4), water soluble prodrugs CA-4P (3) and CA-1P (4) have been pre- pared as phosphates, which are in the final stage of clinical trials [10]. Further, structure activity relationship (SAR) based synthesis of other analogues yielded phenstatin (5) as one of the most active compounds of this series (Fig. 1) [11,12]. However, like other che- motherapeutic agents combretastatin often fails to selectively tar- get cancer cells and also undesired toxicity being a major concern. Therefore, we thought to target ERa with tubulin poisoning properties of combretastatin as a new strategy for specifically tar- geting ERa over-expressing hormone dependent breast cancer. In the present study, we designed several phenstatin analogues on steroidal framework synthesized through modification of gallic acid, an abundantly available plant phenolic acid. In our approach to induce cytotoxicity in the desired molecule, phenstatin (5) was set as a model molecule, where 3,4,5-trimethoxybenzoyl part was introduced from gallic acid and the ring A of estradiol unit was used as second aryl ring to have a phenstatin type arrangement. The structure activity relationship (SAR) of phenstatin and combre- tastatin revealed that a diaryl system separated by a linker group having restricted rotation was essentially required for inducing anticancer activity of these molecules and one of the rings should essentially possess 3,4,5-trimethoxyphenyl type arrangement [13]. A series of compounds has been synthesized based on above prototype. All the compounds were evaluated against breast cancer 0039-128X/$ - see front matter Ó 2012 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.steroids.2012.03.012 ⇑ Corresponding author. Tel.: +91 522 2342676; fax: +91 522 2342666. E-mail address: [email protected] (A.S. Negi). Steroids 77 (2012) 878–886 Contents lists available at SciVerse ScienceDirect Steroids journal homepage: www.elsevier.com/locate/steroids

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Steroids 77 (2012) 878–886
Contents lists available at SciVerse ScienceDirect
Steroids
journal homepage: www.elsevier .com/locate /s teroids
Gallic acid based steroidal phenstatin analogues for selective targetingof breast cancer cells through inhibiting tubulin polymerization
Swati Parihar a, Atul Gupta a, Amit K. Chaturvedi a, Jyoti Agarwal a, Suaib Luqman a, Bendangla Changkija b,Murli Manohar b, Debabrata Chanda a, C.S. Chanotiya a, Karuna Shanker a, Anila Dwivedi b,Rituraj Konwar b, Arvind S. Negi a,⇑a Central Institute of Medicinal and Aromatic Plants (CSIR-CIMAP), Kukrail Picnic Spot Road, Lucknow 226015, UP, Indiab Central Drug Research Institute (CSIR-CDRI), Chattar Manzil Palace, Lucknow 226001, UP, India
a r t i c l e i n f o
Article history:Received 28 February 2012Received in revised form 23 March 2012Accepted 26 March 2012Available online 5 April 2012
Keywords:Gallic acidPhenstatinAnticancerTubulin polymerization inhibitionAcute oral toxicity
0039-128X/$ - see front matter � 2012 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.steroids.2012.03.012
⇑ Corresponding author. Tel.: +91 522 2342676; faxE-mail address: [email protected] (A.S.
a b s t r a c t
Phenstatin analogues were synthesized on steroidal framework, for selective targeting of breast cancercells. These analogues were evaluated for anticancer efficacy against breast cancer cell lines. Analogues12 and 19 exhibited significant anticancer activity against MCF-7, hormone dependent breast cancer cellline. While analogues 10–14 exhibited significant anticancer activity against MDA-MB-231, hormoneindependent breast cancer cell line. Compound 10 showed significant oestrogen antagonistic activitieswith low agonistic activity in in vivo rat model. These analogues also retain tubulin polymerization inhi-bition activity. The most active analogue 10 was found to be non-toxic in Swiss albino mice up to 300 mg/kg dose. Gallic acid based phenstatin analogues may further be optimized as selective anti-breast canceragents.
� 2012 Elsevier Inc. All rights reserved.
1. Introduction
Hormone-dependent breast cancer over-expresses oestrogenreceptor alpha (ERa) in comparison to normal breast tissue [1]and serves as prognostic indicator of the disease [2]. ERa is a wellvalidated therapeutic target for breast cancer with gold standardexample of tamoxifen, a Selective Estrogen Receptor Modulator(SERM) [3]. However, several reports suggest tamoxifen to bepotentially associated with risk of endometrial cancers [4]. Tamox-ifen also exhibits drug resistance in breast cancer [5]. These criticalfactors impel to develop newer alternatives of targeted breast can-cer with improved profile.
Plant molecules for a long have made immense contribution inenriching repertoire of chemotherapeutic agents. Taxol, vincristine,vinblastine, podophyllotoxin, camptothecin, indole 3-carbinol andcombretastatin A4 (CA-4) are some of the notable anticancer leads[6,7]. Combretastatin A4 is an antimitotic agent originally isolatedfrom the bark of the South African Willow tree Combretum caffrum.Combretastatin binds to the b-subunit of tubulin at the colchicinesite, thereby strongly inhibiting polymerization of tubulin, causingcellular disorganization and toxicity. Combretastatin A 4 (1) is anattractive lead for developing anticancer drugs and several
ll rights reserved.
: +91 522 2342666.Negi).
analogues with potent activity has been developed [8,9]. Toenhance the bioavailability of these lead molecules (CA-1 and CA-4), water soluble prodrugs CA-4P (3) and CA-1P (4) have been pre-pared as phosphates, which are in the final stage of clinical trials[10]. Further, structure activity relationship (SAR) based synthesisof other analogues yielded phenstatin (5) as one of the most activecompounds of this series (Fig. 1) [11,12]. However, like other che-motherapeutic agents combretastatin often fails to selectively tar-get cancer cells and also undesired toxicity being a major concern.
Therefore, we thought to target ERa with tubulin poisoningproperties of combretastatin as a new strategy for specifically tar-geting ERa over-expressing hormone dependent breast cancer. Inthe present study, we designed several phenstatin analogues onsteroidal framework synthesized through modification of gallicacid, an abundantly available plant phenolic acid. In our approachto induce cytotoxicity in the desired molecule, phenstatin (5) wasset as a model molecule, where 3,4,5-trimethoxybenzoyl part wasintroduced from gallic acid and the ring A of estradiol unit wasused as second aryl ring to have a phenstatin type arrangement.The structure activity relationship (SAR) of phenstatin and combre-tastatin revealed that a diaryl system separated by a linker grouphaving restricted rotation was essentially required for inducinganticancer activity of these molecules and one of the rings shouldessentially possess 3,4,5-trimethoxyphenyl type arrangement [13].
A series of compounds has been synthesized based on aboveprototype. All the compounds were evaluated against breast cancer

ON
OCH3
H3CO
H3CO
OCH3
R2
R1
Tamoxifen1: R1=H, R2=OH; Combretastatin A4;2: R1=R2=OH; Combretastatin A1;3: R1=H, R2=OP(O)(OH)2 ; Combretastatin A4 phosphate;4: R1=R2=OP(O)(OH)2; Combretastatin A1 diphosphate;
O
OCH3
H3CO
H3CO
OCH3
OH
O
H3CO
H3CO
OR2
R1OOCH3
Prototype5: Phenstatin
Fig. 1. Tamoxifen, combretastatins, phenstatin and proposed prototype.
S. Parihar et al. / Steroids 77 (2012) 878–886 879
cell lines, ERa expressing (MCF-7) and ER-a negative (MDA-MB-231) for in vitro cytotoxicity and mode of action studies as tubulinpolymerization inhibition. To estimate bioactivity due to steroidalframework, in vivo oestrogenicity and anti-oestrogenicity infemale Sprague–Dawley rats were carried out. The most activeanalogue 10, was further evaluated for in vivo acute oral toxicityin Swiss albino mice.
2. Experimental protocols
2.1. General procedures
Melting points were determined on E-Z Melt automated melt-ing point apparatus, Stanford Research System, USA and wereuncorrected. Gallic acid was procured from S.d. Fine Chemicals, In-dia and estrone was procured for Sigma, USA. All the reactionswere monitored on Merck aluminium thin layer chromatography(TLC, UV254 nm) plates. TLC visualization was accomplished byspraying with a solution of 2% ceric sulphate in 10% aqueous sul-phuric acid and charring at 100–110 �C. Column chromatographywas carried out on silica gel (60–120 mesh, Thomas Baker). NMRspectra were obtained on Bruker Avance-300 MHz instrumentwith tetramethylsilane (TMS, chemical shifts in d ppm) as an inter-nal standard. EI mass spectra were recorded on Perkin–Elmer Tur-boMass GC–MS system after dissolving the compounds inmethanol, while ESI mass spectra were recorded on ShimadzuLC–MS after dissolving the compounds in methanol. FT-IR spectrawere recorded on Perkin–Elmer SpectrumBX. Nomenclature of ste-roid derivatives has been given as per the recommendations pub-lished by the Joint Commission on the Biochemical Nomenclature(JCBN) of IUPAC [14]. Paclitaxel (Taxol), podophyllotoxin andtamoxifen were procured from Sigma–Aldrich, USA and centchro-man was taken from Central Drug Research Institute, India. Allthese were used as standard cytotoxic, tubulin polymerizationinhibitor and anti-oestrogenic drugs, respectively.
2.2. Chemical synthesis
2.2.1. General procedure for the synthesis of steroidal benzophenones(10–16)2.2.1.1. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(3,4,5-trimethoxyphenyl)-methanone (10). 3,4,5-Trimethoxy-benzoic acid (250 mg, 1.18 mmol) was taken in thionyl chloride(2 mL). The reaction mixture was heated at 80 �C for 2 h. On com-pletion, the excess of thionyl chloride was evaporated in vacuo andthe residue thus obtained was as such processed for next step. Theresidue was dissolved in dry dichloromethane (15 mL) and anhy-drous aluminium chloride (200 mg, 1.5 mmol) was added to itand stirred at room temperature for 15 min. To the stirred solution,estradiol 3-methyl ether, 17-acetate (6, 256 mg, 0.78 mmol) wasadded and further stirred for 18–20 h. On completion, it was acid-ified with dil. HCl (10%, 5 mL) and extracted with chloroform(3 � 30 mL). Organic layer was washed with water and dried over
anhydrous sodium sulphate. On evaporation, a residue was ob-tained which on purification over column silica gel yielded purebenzophenone derivative 10 as light yellow solid (253 mg).
Yield 62%; mp 177–178 �C; 1H NMR (CDCl3, 300 MHz): d 0.84 (s,3H, 18-CH3), 1.24–2.26 (m, 13H, rest of the 5XCH2 and 3XCH of ste-roidal ring), 2.06 (s, 3H, 17-OCOCH3), 2.92 (bs, 2H, 6-CH2), 3.72 (s,3H, 3-OCH3), 3.88 (s, 6H, 30,50-OCH3), 3.93 (s, 3H, 40-OCH3), 4.69 (t,1H, 17-CH, J = 8.2 Hz), 6.69 (s, 1H, 4-CH), 7.11 (s, 2H, 20 and 60-CH),7.27 (s, 1H, 1-CH). 13C NMR (CDCl3, 75 MHz): d 12.38, 21.23, 23.60,26.56, 27.45, 27.98, 30.31, 37.27, 39.01, 43.34, 44.09, 50.37, 56.19,56.83, 56.83, 61.10, 82.94, 108.66, 108.66, 112.49, 127.01, 127.19,133.03, 133.77, 141.27, 143.48, 153.29, 153.29, 155.79, 171.13,195.34. ESI mass (MeOH): 523 [M+H]+, 545 [M+Na]+, 561 [M+K]+.IR (KBr, cm�1): 2938, 1736, 1656, 1582, 1501, 1246, 1127.
2.2.1.2. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-methoxyphenyl)-methanone (11). Yield 53%; mp 137–138 �C; 1H NMR (CDCl3, 300 MHz): d 0.83 (s, 3H, 18-CH3), 1.30–2.24 (m, 13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.05(s, 3H, 17-OCOCH3), 2.92 (bs, 2H, 6-CH2), 3.68 (s, 3H, 3-OCH3),3.86 (s, 3H, 40-OCH3), 4.68 (t, 1H, 17-CH), 6.67 (s, 1H, 4-CH), 6.89(d, 2H, 30 and 40-CH), 7.24 (s, 1H, 1-CH), 7.81 (d, 2H, 20 and 60-CH). 13C NMR (CDCl3, 75 MHz): d 12.43, 21.39, 23.64, 26.50,27.53, 27.99, 30.36, 37.23, 38.96, 43.32, 44.09, 50.28, 55.76,56.12, 83.00, 112.28, 113.76, 113.76, 127.01, 127.41, 131.65,132.54, 132.54, 132.92, 140.89, 155.54, 163.77, 171.32, 195.47.ESI mass (MeOH): 463 [M+H]+, 485 [M+Na]+, Negative mode: 461[M�H]�. IR (KBr, cm�1): 2931, 1735, 1655, 1601, 1509, 1251.
2.2.1.3. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(3,4-dimethoxyphenyl)-methanone (12). Yield 52%; mp 86–87 �C; 1H NMR (CDCl3, 300 MHz): d 0.83 (s, 3H, 18-CH3), 1.30–2.24 (m, 13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.05(s, 3H, 17-OCOCH3), 2.92 (bs, 2H, 6-CH2), 3.68 (s, 3H, 3-OCH3),3.86 (s, 6H, 30 and 40-OCH3), 4.68 (t, 1H, 17-CH), 6.67 (s, 1H, 4-CH), 6.89 (d, 2H, 30 and 40-CH), 7.24 (s, 1H, 1-CH), 7.81 (d, 2H, 20
and 60-CH). 13C NMR (CDCl3, 75 MHz): d 12.37, 21.23, 23.61,26.52, 27.50, 27.99, 30.29, 37.27, 39.02, 43.35, 44.11, 50.38,56.21, 56.36, 56.45, 82.96, 110.62, 112.47, 112.47, 125.78, 126.92,127.50, 131.92, 132.54, 132.97, 140.80, 153.82, 155.65, 171.11,195.32. ESI mass (MeOH): 493 [M+H]+, 515 [M+Na]+. IR (KBr,cm�1): 2934, 1736, 1655, 1510, 1270.
2.2.1.4. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(3,4-methylenedioxyphenyl)-methanone (13). Yield 69%; mp123–124 �C; 1H NMR (CDCl3, 300 MHz): d 0.78 (s, 3H, 18-CH3),1.23–2.21 (m, 13H, rest of the 5XCH2 and 3XCH of steroidal ring),1.98 (s, 3H, 17-OCOCH3), 2.84 (bs, 2H, 6-CH2), 3.62 (s, 3H, 3-OCH3), 4.61 (t, 1H, 17-CH, J = 8.4 Hz), 5.96 (s, 2H, –O–CH2–O),6.59 (s, 1H, 4-CH), 6.72 (d, 1H, 50-CH), 7.15 (s, 1H, 1-CH), 7.28 (d,1H, 60-CH), 7.30 (s, 1H, 20-CH). 13C NMR (CDCl3, 75 MHz): d 12.39,21.27, 23.62, 26.51, 27.51, 28.00, 30.32, 37.43, 39.01, 43.35,44.11, 50.37, 56.18, 82.99, 102.01, 107.90, 109.70, 112.41, 126.87,

880 S. Parihar et al. / Steroids 77 (2012) 878–886
126.96, 127.45, 133.04, 133.62, 140.92, 148.27, 151.93, 155.57,171.20, 194.99. ESI mass (MeOH): 477 [M+H]+, 499 [M+Na]+, 515[M+K]+. IR (KBr, cm�1): 2934, 1734, 1651, 1605, 1501, 1251.
2.2.1.5. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-methylphenyl)-methanone (14). Yield 58%; mp 81–82 �C;1H NMR (CDCl3, 300 MHz): d 0.84 (s, 3H, 18-CH3), 0.86–2.26 (m,13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.06 (s, 3H,17-OCOCH3), 2.42 (s, 3H, CH3), 2.93 (bs, 2H, 6-CH2), 3.69 (s, 3H,3-OCH3), 4.69 (t, 1H, 17-CH, J = 8.4 Hz), 6.68 (s, 1H, 4-CH), 7.21(d, 2H, 30 and 40-CH, J = 4.1 Hz), 7.27 (s, 1H, 1-CH), 7.72 (d, 2H, 20
and 60-CH). 13C NMR (CDCl3, 75 MHz): d 14.31,21.29, 21.88,23.63, 26.51, 27.52, 28.01, 30.34, 37.27, 39.01, 43.35, 44.12,50.36, 56.12, 82.99, 112.43, 127.16, 127.43, 129.12, 129.34,129.91, 130.30, 133.01, 136.33, 141.12, 143.55, 155.82, 171.18,196.36. ESI mass (MeOH): 447 [M+H]+, 469 [M+Na]+, 485 [M+K]+,Negative mode: 445 [M�H]�. IR (KBr, cm�1): 2932, 1734, 1654,1607, 1496, 1249.
2.2.1.6. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-bromophenyl)-methanone (15). Yield 49%; mp 108–109 �C; 1H NMR (CDCl3, 300 MHz): d 0.83 (s, 3H, 18-CH3),1.25–2.27 (m, 13H, rest of the 5XCH2 and 3XCH of steroidalring), 2.05 (s, 3H, 17-OCOCH3), 2.93 (bs, 2H, 6-CH2), 3.66 (s,3H, 3-OCH3), 4.68 (t, 1H, 17-CH, J = 8.4 Hz), 6.67 (s, 1H, 4-CH),7.56 (d, 2H, 20 and 60-CH), 7.30 (s, 1H, 1-CH), 7.65 (d, 2H, 30
and 50-CH). ESI mass (MeOH): 511 [M+H]+, 533 [M+Na]+, 549[M+K]+, Negative mode: 509 [M�H]�. IR (KBr, cm�1): 2931,1736, 1654, 1607, 1586, 1248.
2.2.1.7. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17-one-2yl]-(3,4,5-trimethoxyphenyl)-methanone (16). Yield 46%; mp 151–152 �C; 1H NMR (CDCl3, 300 MHz): d 0.88 (s, 3H, 18-CH3), 1.24–2.51 (m, 13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.99(bs, 2H, 6-CH2), 3.73 (s, 3H, 3-OCH3), 3.84 (s, 6H, 30 and 50-OCH3),3.95 (s, 3H, 40-OCH3), 6.71 (s, 1H, 4-CH), 7.11 (s, 2H, 20 and 60-CH), 7.28 (s, 1H, 1-CH). 13C NMR (CDCl3, 75 MHz): d 14.23, 21.94,26.41, 26.77, 30.32, 32.01, 36.06, 38.83, 44.31, 48.23, 51.00,56.27, 56.86, 56.86, 61.12, 108.71, 108.71, 112.53, 127.15, 128.83,132.59, 133.72, 141.06, 153.32, 153.32, 153.68, 155.92, 195.28,221.11. ESI mass (MeOH): 479 [M+H]+, 501 [M+Na]+, 517 [M+K]+.IR (KBr, cm�1): 2936, 1737, 1657, 1580, 1500, 1227, 1126.
2.2.2. General procedure for the synthesis of demethylated products(17–20)2.2.2.1. Synthesis of [3-hydroxylestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-hydroxy,3-methoxyphenyl)-methanone (19). Compound 12(100 mg, 0.20 mmol) was taken in dry dichloromethane (15 mL).To this stirred solution anhydrous aluminium chloride (200 mg,1.50 mmol) was added and further stirred at room temperaturefor 3 h. On completion, dil. HCl (5% v/v, 2 mL) was added to itand extracted with chloroform (3 � 25 mL). Organic layer waswashed with water, dried over anhydrous sodium sulphate andevaporated to dryness in vacuo. The residue thus obtained wasrecrystallised with chloroform-hexane (1:3) to get a creamishwhite solid (82 mg).
Yield 87%; mp 105–106 �C; 1H NMR (CDCl3, 300 MHz): d 0.83 (s,3H, 18-CH3), 1.25–2.23 (m, 13H, rest of the 5XCH2 and 3XCH of ste-roidal ring), 2.91 (bs, 2H, 6-CH2), 3.95 (s, 3H, 3-OCH3), 4.76 (t, 1H,17-CH, J = 8.7 Hz), 6.76 (s, 1H, 4-CH), 7.26 (d, 2H, 50 and 60-CH), 7.31(s, 1H, 20-CH), 7.54 (s, 1H, 1-CH), 11.71 (s, 1H, exchangeable, Phe-nolic OH), 11.78 (s, 1H, exchangeable, Phenolic OH). ESI mass(MeOH): 465 [M+H]+, 487 [M+Na]+. IR (KBr, cm�1): 3449, 2931,1719, 1704, 1636, 1579, 1271.
2.2.2.2. Synthesis of [3-hydroxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-hydroxy,3,5-dimethoxyphenyl)-methanone (17). Yield 76%; mp133–134 �C. 1H NMR (CDCl3, 300 MHz): d 0.84 (s, 3H, 18-CH3),1.27–2.26 (m, 13H, rest of the 5XCH2 and 3XCH of steroidal ring),2.94 (bs, 2H, 6-CH2), 3.95 (s, 6H, 3 and 5-OCH3), 4.70 (dt, 1H, 17-CH), 6.79 (s, 1H, 4-CH), 7.06 (s, 2H, 20 and 60-CH), 7.61 (s, 1H, 1-CH), 11.42 (s, 1H, exchangeable, Phenolic OH), 11.72 (s, 1H,exchangeable, Phenolic OH). 13C NMR (CDCl3, 75 MHz): 12.34,21.11, 23.55, 26.77, 27.16, 27.94, 30.17, 37.18, 38.92, 43.28,43.87, 50.37, 56.95, 56.95, 82.87, 108.14, 108.14, 117.76, 118.00,129.64, 130.25, 131.35, 139.68, 146.83, 147.27, 147.27, 161.15,171.14, 199.52. ESI mass (MeOH): 495 [M+H]+, 517 [M+Na]+, neg-ative ion mode: 493 [M�H]�. IR (KBr, cm�1): 3495, 3423, 2937,1737, 1704, 1587, 1510, 1214.
2.2.2.3. Synthesis of [3-hydroxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-methoxyphenyl)-methanone (18). Yield 83%; mp 97–98 �C; 1HNMR (CDCl3, 300 MHz): d 0.83 (s, 3H, 18-CH3), 1.23–2.23 (m,13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.92 (bs, 2H,6-CH2), 3.90 (s, 3H, 3-OCH3), 4.68 (t, 1H, 17-CH, J = 8.4 Hz), 6.77(s, 1H, 4-CH), 6.97 (d, 2H, 30 and 50-CH), 7.52 (s, 1H, 1-CH), 7.68(d, 2H, 20 and 60-CH), 11.92 (s, 1H, exchangeable, Phenolic OH).ESI mass (MeOH): 449 [M+H]+, 471 [M+Na]+. IR (KBr, cm�1):3448, 3424, 2930, 1718, 1705, 1618, 1510, 1265.
2.2.2.4. Synthesis of [3-hydroxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-methylphenyl)-methanone (20). Yield 92%; mp 115–116 �C; 1HNMR (CDCl3, 300 MHz): d 0.84 (s, 3H, 18-CH3), 1.25–2.24 (m,13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.09 (s, 3H, –OCOCH3), 2.43 (s, 3H, 40-CH3), 2. 94 (bs, 2H, 6-CH2), 4.69 (t, 1H,17-CH, J = 8.4 Hz), 6.79 (s, 1H, 4-CH), 7.31 (d, 2H, 30 and 50-CH),7.52 (s, 1H, 1-CH), 7.59 (d, 2H, 20 and 60-CH), 11.88 (s, 1H,exchangeable, Phenolic OH). ESI mass (MeOH): 433 [M+H]+, 455[M+Na]+, negative ion mode: 431 [M�H]�. IR (KBr, cm�1): 3448,2926, 1735, 1704, 1602, 1262.
2.2.3. General procedure for synthesizing 2-benzyl analogues (21–23)2.2.3.1. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(3,4,5-trimethoxyphenyl)-methane (21). Steroidal benzophe-none 10 (100 mg, 0.19 mmol) was taken in trifluoroacetic acid(TFA, 3 mL). The reaction mixture was cooled to 0–10 �C and so-dium borohydride (80 mg, 2.1 mmol) was added to it in portionsand stirred for 1 h with cooling and then at room temperaturefor 2 h. On completion, the reaction mixture was acidified withdil. HCl (5 mL, 5% v/v) and extracted with chloroform(3 � 20 mL). The organic layer was washed with water and evapo-rated in vacuo and the residue thus obtained was recrystallisedwith CHCl3:hexane (1:5) to get compound 21 as creamish whitesolid (77 mg).
Yield 79%; mp 139–140 �C; 1H NMR (CDCl3, 300 MHz): d 0.81 (s,3H, 18-CH3), 1.26–2.20 (m, 13H, rest of the 5XCH2 and 3XCH of ste-roidal ring), 2.05 (s, 3H, –OCOCH3), 2. 85 (bs, 2H, 6-CH2), 3.81 (s,12H, 30,40,50-OCH3 and 3-OCH3), 3.86 (s, 2H, –CH2–Ar), 4.67 (t,1H, 17-CH), 6.45 (s, 2H, 20 and 60-CH), 6.59 (s, 1H, 4-CH), 7.00 (s,1H, 1-CH). EI mass (MeOH): 508 [M+]. IR (KBr, cm�1): 2935,1736, 1686, 1592, 1506, 1217, 1166.
2.2.3.2. Synthesis of [3-hydroxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-hydroxy,3,5-dimethoxyphenyl)-methane (22). Yield 62%; mp 98–99 �C; 1H NMR (CDCl3, 300 MHz): d 0.83 (s, 3H, 18-CH3), 1.26–2.24(m, 13H, rest of the 5XCH2 and 3XCH of steroidal ring), 2.06 (s, 3H,–OCOCH3), 2.77 (bs, 2H, 6-CH2), 3.78 (s, 6H, 30 and 50-OCH3), 3.93(s, 2H, –CH2–Ar), 4.68 (t, 1H, 17-CH), 6.47 (s, 2H, 20 and 60-CH),6.52 (s, 1H, 4-CH), 7.01 (s, 1H, 1-CH), 11.7 (s, 1H, exchangeable,Phenolic OH). ESI mass (MeOH): 503 [M+Na]+, 519 [M+K]+,

S. Parihar et al. / Steroids 77 (2012) 878–886 881
negative ion mode: 479 [M�H]�. IR (KBr, cm�1): 3424, 2930, 1718,1705, 1618, 1510, 1265, 1216.
2.2.3.3. Synthesis of [3-methoxyestra-1,3,5(10)-trien,17b-acetate-2yl]-(4-methoxyphenyl)-methane (23). Yield 73%; oil; 1H NMR(CDCl3, 300 MHz): d 0.83 (s, 3H, 18-CH3), 1.25–2.19 (m, 13H, restof the 5XCH2 and 3XCH of steroidal ring), 2.07 (s, 3H, –OCOCH3),2.84 (bs, 2H, 6-CH2), 3.84 (s, 6H, 30 and 40-OCH3), 3.88 (bs, 2H, –CH2–Ar), 4.70 (t, 1H, 17-CH), 6.57 (s, 1H, 4-CH), 6.81 (s, 2H, 30
and 50-CH), 7.14 (s, 2H, 20 and 60-CH), 7.25 (s, 1H, 1-CH). ESI mass(MeOH): 517 [M+K]+. IR (neat, cm�1): 2933, 1735, 1509, 1246,1212.
2.3. Pharmacology
2.3.1. Cell cultureHuman breast cancer cell lines MCF-7 and MDA-MB-231 were
American type of cell culture collection (ATCC), USA. Cells weregrown in tissue culture flasks in complete growth medium, DMEMwith 2 mM glutamine, pH 7.4 supplemented with 10% fetal bovineserum, 100 lg/mL streptomycin and 100 units/mL penicillin in aCO2 incubator (37 �C, 5% CO2, 90% RH). The cells at subconfluentstage were harvested from the flask by treatment with trypsin(0.05% in PBS, pH 7.4, containing 0.02% EDTA) and seeded in tissueculture plates for assay.
2.3.2. In vitro cytotoxicity evaluationThe cytotoxic activities of the compounds were determined
using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazoliumbromide) reduction assay [15]. 1 � 104 cells/well were seeded in200 lL DMEM, supplemented with 10% FBS in each well of 96-wellculture plates and incubated for 24 h at 37 �C in a CO2 incubator.Compounds, diluted to the desired concentrations in culture med-ium, were added to the wells with respective vehicle control. After18 h of incubation, 10 lL MTT (5 mg/mL) was added to each welland the plates were further incubated for 4 h. Then the supernatantfrom each well was carefully removed, formazon crystals were dis-solved in 100 lL of DMSO and absorbance at 540 nm wavelengthwas recorded. IC50 were determined from sigmoidal dose-responseanalysis of drug concentration versus percent cell inhibition usingGraphpad Prism software version 3.0. Mean and standard error ofmean (SEM) were calculated using Microsoft Excel and data are ex-pressed as mean ± SEM obtained from three independentexperiments.
2.3.3. Tubulin polymerization inhibition assayTubulin polymerization experiment was done as per reported
method [16,17] using ‘assay kit’ from Cytoskeleton, USA. In brief,tubulin protein (3 mg/mL) in tubulin polymerization buffer(80 mM PIPES, pH 6.9, 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTPand 15% glycerol) was placed in pre-warmed 96-well microtiterplates at 37 �C in the presence of test compounds with variableconcentrations. All samples were mixed well and polymerizationwas monitored kinetically at 340 nm every min for 1 h using Spec-tramax plate reader. Podophyllotoxin was used as standard inhib-itor of tubulin polymerase and DMSO as negative control. The IC50
value was determined from dose-dependent analysis and is de-fined as the concentration that inhibits the rate of polymerizationby 50%.
2.3.4. In vivo oestrogenicityTwenty-one-days-old immature female Sprague–Dawley rats
were bilaterally ovariectomized under light ether anaesthesiaand after post-operative rest for 7 days were randomized into dif-ferent treatment groups. Each group comprised of four or five ani-mals. Each rat received the compound once daily for three
consecutive days by oral route. A separate group of animals re-ceived only the vehicle for similar duration served as control. Atautopsy 24 h after the last treatment on day 31 of age, vaginalsmear of each rat was taken and uterus was carefully excised,gently blotted, and weighed. Premature opening of vagina, cornifi-cation of vaginal epithelium, and increase in uterine fresh weightwere taken as parameters for evaluation of oestrogen agonisticactivity in comparison to rats of vehicle control group. The objec-tive was to evaluate oestrogen agonistic effect of the compoundson the uterus and vagina [18].
2.3.5. In vivo anti-oestrogenicityTwenty-one days old immature female Sprague–Dawley rats
were bilaterally ovariectomized under light ether anaesthesiaand after post-operative rest for 7 days were randomized into dif-ferent treatment groups. Each group comprised of four or five ani-mals. Each rat received the compound of the invention and0.02 mg/kg dose of 17a-ethynylestradiol in 10% ethanol–distilledwater once daily for three consecutive days by oral route. A sepa-rate group of animals receiving only 17a-ethynylestradiol(0.02 mg/kg) in 10% ethanol–distilled water for similar durationwas used for comparison. At autopsy on day 31 of age, vaginalsmear of each rat was taken and uterus was carefully excised,gently blotted and weighed. Inhibition in ethynylestradiol inducedcornification of vaginal epithelium and increase in uterine freshweight were taken as parameters for evaluation of oestrogenantagonistic effect of the compounds [18].
2.3.6. Oestrogen receptor binding affinityThe relative binding affinity of these compounds for oestrogen
receptor was determined by competitive assay radiometrically asdescribed earlier [15]. Radiolabeled estradiol 3H[E2] was used asreference compound.
2.3.7. In vivo acute oral toxicityIn view of anti-cancer activity of compound 10 in in vitro mod-
el, acute oral toxicity of the same was carried out in Swiss albinomice. Experiment was conducted in accordance with the Organiza-tion for Economic Co-operation and Development (OECD) testGuideline No. 423.
For the study, 16 mice (eight male and eight female) were takenand divided into four groups comprising two male and two femalemice in each group weighing between 20 and 25 g. The animalswere maintained at 22 ± 5 �C with humidity control and also onan automatic dark and light cycle of 12 h. The animals were fedwith the standard rat feed and provided ad libitum drinking water.Mice of group 1 were kept as control and animals of groups 2–4were kept as experimental. The animals were acclimatized for7 days in the experimental environment prior to the actual exper-imentation. The test compound was suspended in distilled waterusing traces of ethanol as a co-solvent and was given at 5, 50and 300 mg/kg body weight to animals of groups 2–4, respectively.Control animals received only vehicle.
2.3.7.1. Observational, haematological, biochemical and gross patho-logical study. The animals were checked for mortality and any signsof ill health at hourly interval on the day of administration of drugand there after a daily general case side clinical examination wascarried out including changes in skin, mucous membrane, eyes,occurrence of secretion and excretion and also responses like lach-rymation, pilo-erection respiratory patterns etc. Also changes ingait, posture and response to handling were also recorded. Inaddition to observational study, body weights were recorded andblood and serum samples were collected from all the animals on7th day after experiment and were analysed for total RBC, WBC, dif-ferential leucocyte count, haemoglobin percentage and biochemical

882 S. Parihar et al. / Steroids 77 (2012) 878–886
parameters like total cholesterol, triglycerides, creatinine, SGPT andSGOT activity. The animals were then sacrificed and were necrop-sed for any gross pathological changes. Weights of vital organs likeliver, heart, kidney etc. were recorded.
3. Results and discussion
3.1. Chemistry
The synthetic strategy adopted was as depicted in Scheme 1. Es-trone and gallic acid were modified suitably to get the compoundsof desired prototype. Compound 6 was synthesized from estrone asreported earlier [19]. Gallic acid was methylated with dimethylsulphate in 20% aqueous alkali to produce 3,4,5-trimethoxybenzoicacid (8) [20]. Compound 8 was transformed to its acid chloride i.e.,3,4,5-trimethoxybenzoyl chloride (9) by heating it with thionylchloride [21]. To get benzophenone 10, Friedel-Craft’s acylationreaction was done on 6 and 9 in presence of anhydrous aluminiumchloride in dry dichloromethane. Similarly, various other acid chlo-rides were used to get other analogues 11–15.
These benzophenone moieties on treatment with excess of alu-minium chloride in DCM yielded o and p-demethylated analogues17–20. Initially, these demethylated products were synthesized inthe separate steps but when the molar ratio of aluminium chloridewas enhanced by several folds in the Friedel–Craft’s reaction step,we got both the methylated and demethylated products in thesame reaction. Some of the benzophenone analogues were treatedwith sodium borohydride in TFA to yield corresponding benzylderivatives (21–23).
3.2. Biological results and discussion
In MTT assay [15] compounds 10, 11, 12, 13, 14 and 19 exhib-ited significant cytotoxicity against human breast cancer cell lines
H3CO
OAc
6
+C Cl
O
vi
B
R1
R1
1112131415
HO
OAc
O
R1
17: R1=4-hydroxy, 3,5-dimethoxy-18: R1=4-methoxy-;19: R1=3-methoxy, 4-hydroxy-;20: R1=4-methyl-;
vii
A
H3CO
6: R1=H, R2=OAc;7: R1-R2=O;
+
C Cl
O
OCH3
H3CO
H3CO
9
R2R1
Scheme 1. Reagents and conditions: (i) Me2SO4, K2CO3, dry acetone-DCM (1:1), reflux, 83 h, 94%; (iv) Me2SO4, 20% aqueous KOH, 0–10 �C for 30 min, then reflux, 2 h, 68%; (v) SO(vii) DCM, anhydrous AlCl3, RT, 2–3 h, 76–92%; (viii) NaBH4, TFA, 0 �C, 1 h then RT, 3–4
MCF-7 and MDA-MB-231 (Table 1), while rest of the compoundswere either poorly active or inactive (IC50 > 15 lM) Out of these,compounds 12 and 19 showed significantly higher activity inER-positive MCF-7 cells indicating that their action may involveoestrogen receptor (ER). Whereas compounds 10, 11, 13 and 14showed higher activity in ER-negative MDA-MB-231 cells possiblyindicating lack of role of ER involvement. Most selectively activeanalogues in ER-bearing cell MCF-7 i.e., 12 and 19 possess a 3,4-dimethoxybenzoyl or 3-methoxy,4-hydroxybenzoyl unit attachedto estradiol unit.
Analogue 10 with highest activity in MDA-MB-231 possessed3,4,5-trimethoxybenzoyl arrangement. On removing methoxygroups from the benzoyl ring cytotoxicity was reduced. p-Methylsubstitution at benzoyl group (compound 14) exhibited good activ-ity and comparable to 3,4,5-trimethoxy group. p-Bromo substitu-tion showed very poor cytotoxicity. o and p-Demethylatedcompounds (compounds 17–20) showed comparable cytotoxicityto methylated products but on reducing keto group of benzophe-none to methylene group, the cytotoxicity was reduced drastically.It might be due to loss of restricted rotation of the phenyl rings,which was pre-requisite for inducing cytotoxicity. We also evalu-ated non-specific cytotoxicity of the molecules. For this purpose,we evaluated their cytotoxicity against human cell lines of normaltissue origin, HEK-293 (human embryonic kidney cells). Only com-pound 12 and 17 showed moderate toxicity against normal humancell line, whereas other compounds are significantly safe with IC50
higher than 50 lM. This further supports that our hybrid analoguesare acting on cancer cells in selective fashion.
The Table 2 shows the tubulin polymerization inhibition activ-ity of these analogues [16,17]. Analogue compound 10 possessedantitubulin activity comparable to podophyllotoxin, a standardantitubulin drug. Rest of the compounds were considered inactivedue to low inhibition (% inhibition < 30% at 20 lg/mL) concentration.Both the compounds 10 and 21 possessed a 3,4,5-trimethoxyphenyl
H3CO
OAc
O
R1
OAc
R2
: R1=4-methoxy-;: R1=3,4-dimethoxy-;: R1=3,4-methylenedioxy-;: R1=4-methyl-;: R1=4-bromo-;
21: R1=OCH3, R2=3,4.5-trimethoxy-;22: R1=OH, R2=4-hydroxy, 3,5-dimethoxy-;23: R1=OCH3, R2=4-OCH3
viii
viii
H3CO
O
viH3CO
H3CO
H3CO
10: R1= H, R2=OAc;16: R1-R2=O
R1 R2
7%; (ii) NaBH4, MeOH:DCM (2:1), RT, 2 h, 91%; (iii) Ac2O, dry pyridine, dry DCM, RT,Cl2, 80 �C, 1 h; (vi) Crude mass of (v), dry DCM, anhydrous AlCl3, RT, 3–4 h, 46–69%;h, 62–79%.
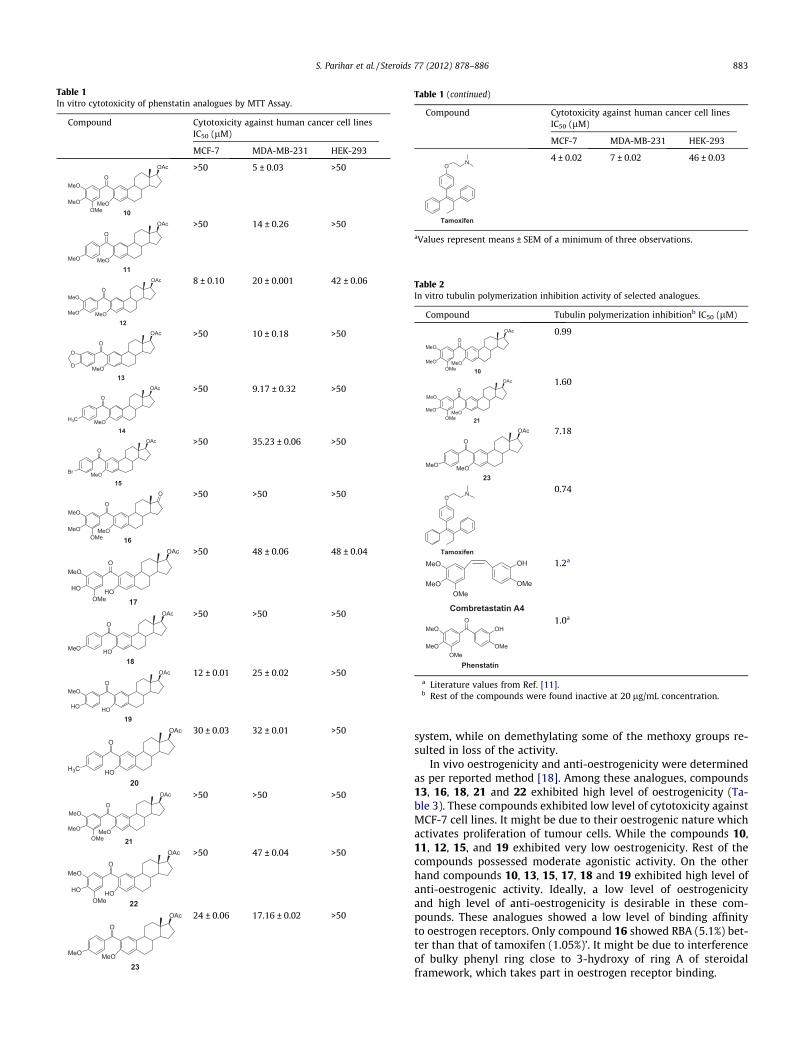
Table 1In vitro cytotoxicity of phenstatin analogues by MTT Assay.
Compound Cytotoxicity against human cancer cell linesIC50 (lM)
MCF-7 MDA-MB-231 HEK-293
MeO
OAc
O
OMeMeO
MeO
10
>50 5 ± 0.03 >50
MeO
OAc
O
MeO
11
>50 14 ± 0.26 >50
MeO
OAc
O
MeO
12
MeO
8 ± 0.10 20 ± 0.001 42 ± 0.06
MeO
OAc
O
13
O
O
>50 10 ± 0.18 >50
MeO
OAc
O
14
H3C
>50 9.17 ± 0.32 >50
MeO
OAc
O
15
Br
>50 35.23 ± 0.06 >50
MeO
O
OMeMeO
MeO
16
O >50 >50 >50
HO
OAc
O
17
HOOMe
MeO
>50 48 ± 0.06 48 ± 0.04
HO
OAc
O
18
MeO
>50 >50 >50
HO
OAc
O
19
HO
MeO
12 ± 0.01 25 ± 0.02 >50
HO
OAc
O
20
H3C
30 ± 0.03 32 ± 0.01 >50
MeO
OAc
O
21
MeOOMe
MeO
>50 >50 >50
HO
OAc
O
22
HOOMe
MeO
>50 47 ± 0.04 >50
MeO
OAc
O
23
MeO
24 ± 0.06 17.16 ± 0.02 >50
Table 1 (continued)
Compound Cytotoxicity against human cancer cell linesIC50 (lM)
MCF-7 MDA-MB-231 HEK-293
ON
Tamoxifen
4 ± 0.02 7 ± 0.02 46 ± 0.03
aValues represent means ± SEM of a minimum of three observations.
Table 2In vitro tubulin polymerization inhibition activity of selected analogues.
Compound Tubulin polymerization inhibitionb IC50 (lM)
MeO
OAc
O
OMeMeO
MeO
10
0.99
MeO
OAc
O
21
MeOOMe
MeO
1.60
MeO
OAc
O
23
MeO
7.18
ON
Tamoxifen
0.74
Combretastatin A4
OH
OMeOMe
MeO
MeO 1.2a
O
OMeMeO
MeO
OMe
OH
Phenstatin
1.0a
a Literature values from Ref. [11].b Rest of the compounds were found inactive at 20 lg/mL concentration.
S. Parihar et al. / Steroids 77 (2012) 878–886 883
system, while on demethylating some of the methoxy groups re-sulted in loss of the activity.
In vivo oestrogenicity and anti-oestrogenicity were determinedas per reported method [18]. Among these analogues, compounds13, 16, 18, 21 and 22 exhibited high level of oestrogenicity (Ta-ble 3). These compounds exhibited low level of cytotoxicity againstMCF-7 cell lines. It might be due to their oestrogenic nature whichactivates proliferation of tumour cells. While the compounds 10,11, 12, 15, and 19 exhibited very low oestrogenicity. Rest of thecompounds possessed moderate agonistic activity. On the otherhand compounds 10, 13, 15, 17, 18 and 19 exhibited high level ofanti-oestrogenic activity. Ideally, a low level of oestrogenicityand high level of anti-oestrogenicity is desirable in these com-pounds. These analogues showed a low level of binding affinityto oestrogen receptors. Only compound 16 showed RBA (5.1%) bet-ter than that of tamoxifen (1.05%)’. It might be due to interferenceof bulky phenyl ring close to 3-hydroxy of ring A of steroidalframework, which takes part in oestrogen receptor binding.
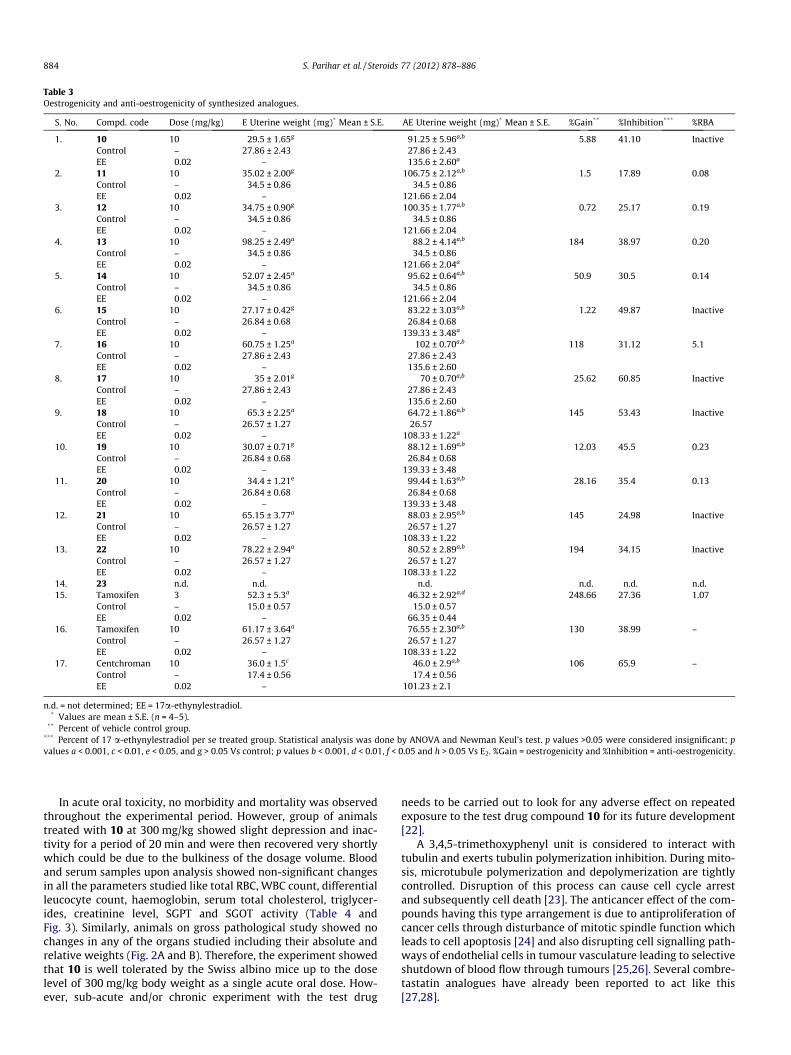
Table 3Oestrogenicity and anti-oestrogenicity of synthesized analogues.
S. No. Compd. code Dose (mg/kg) E Uterine weight (mg)* Mean ± S.E. AE Uterine weight (mg)* Mean ± S.E. %Gain** %Inhibition*** %RBA
1. 10 10 29.5 ± 1.65g 91.25 ± 5.96a,b 5.88 41.10 InactiveControl – 27.86 ± 2.43 27.86 ± 2.43EE 0.02 – 135.6 ± 2.60a
2. 11 10 35.02 ± 2.00g 106.75 ± 2.12a,b 1.5 17.89 0.08Control – 34.5 ± 0.86 34.5 ± 0.86EE 0.02 – 121.66 ± 2.04
3. 12 10 34.75 ± 0.90g 100.35 ± 1.77a,b 0.72 25.17 0.19Control – 34.5 ± 0.86 34.5 ± 0.86EE 0.02 – 121.66 ± 2.04
4. 13 10 98.25 ± 2.49a 88.2 ± 4.14a,b 184 38.97 0.20Control – 34.5 ± 0.86 34.5 ± 0.86EE 0.02 – 121.66 ± 2.04a
5. 14 10 52.07 ± 2.45a 95.62 ± 0.64a,b 50.9 30.5 0.14Control – 34.5 ± 0.86 34.5 ± 0.86EE 0.02 – 121.66 ± 2.04
6. 15 10 27.17 ± 0.42g 83.22 ± 3.03a,b 1.22 49.87 InactiveControl – 26.84 ± 0.68 26.84 ± 0.68EE 0.02 – 139.33 ± 3.48a
7. 16 10 60.75 ± 1.25a 102 ± 0.70a,b 118 31.12 5.1Control – 27.86 ± 2.43 27.86 ± 2.43EE 0.02 – 135.6 ± 2.60
8. 17 10 35 ± 2.01g 70 ± 0.70a,b 25.62 60.85 InactiveControl – 27.86 ± 2.43 27.86 ± 2.43EE 0.02 – 135.6 ± 2.60
9. 18 10 65.3 ± 2.25a 64.72 ± 1.86a,b 145 53.43 InactiveControl – 26.57 ± 1.27 26.57EE 0.02 – 108.33 ± 1.22a
10. 19 10 30.07 ± 0.71g 88.12 ± 1.69a,b 12.03 45.5 0.23Control – 26.84 ± 0.68 26.84 ± 0.68EE 0.02 – 139.33 ± 3.48
11. 20 10 34.4 ± 1.21e 99.44 ± 1.63a,b 28.16 35.4 0.13Control – 26.84 ± 0.68 26.84 ± 0.68EE 0.02 – 139.33 ± 3.48
12. 21 10 65.15 ± 3.77a 88.03 ± 2.95a,b 145 24.98 InactiveControl – 26.57 ± 1.27 26.57 ± 1.27EE 0.02 – 108.33 ± 1.22
13. 22 10 78.22 ± 2.94a 80.52 ± 2.89a,b 194 34.15 InactiveControl – 26.57 ± 1.27 26.57 ± 1.27EE 0.02 – 108.33 ± 1.22
14. 23 n.d. n.d. n.d. n.d. n.d. n.d.15. Tamoxifen 3 52.3 ± 5.3a 46.32 ± 2.92a,d 248.66 27.36 1.07
Control – 15.0 ± 0.57 15.0 ± 0.57EE 0.02 – 66.35 ± 0.44
16. Tamoxifen 10 61.17 ± 3.64a 76.55 ± 2.30a,b 130 38.99 –Control – 26.57 ± 1.27 26.57 ± 1.27EE 0.02 – 108.33 ± 1.22
17. Centchroman 10 36.0 ± 1.5c 46.0 ± 2.9a,b 106 65.9 –Control – 17.4 ± 0.56 17.4 ± 0.56EE 0.02 – 101.23 ± 2.1
n.d. = not determined; EE = 17a-ethynylestradiol.* Values are mean ± S.E. (n = 4–5).
** Percent of vehicle control group.*** Percent of 17 a-ethynylestradiol per se treated group. Statistical analysis was done by ANOVA and Newman Keul’s test. p values >0.05 were considered insignificant; pvalues a < 0.001, c < 0.01, e < 0.05, and g > 0.05 Vs control; p values b < 0.001, d < 0.01, f < 0.05 and h > 0.05 Vs E2. %Gain = oestrogenicity and %Inhibition = anti-oestrogenicity.
884 S. Parihar et al. / Steroids 77 (2012) 878–886
In acute oral toxicity, no morbidity and mortality was observedthroughout the experimental period. However, group of animalstreated with 10 at 300 mg/kg showed slight depression and inac-tivity for a period of 20 min and were then recovered very shortlywhich could be due to the bulkiness of the dosage volume. Bloodand serum samples upon analysis showed non-significant changesin all the parameters studied like total RBC, WBC count, differentialleucocyte count, haemoglobin, serum total cholesterol, triglycer-ides, creatinine level, SGPT and SGOT activity (Table 4 andFig. 3). Similarly, animals on gross pathological study showed nochanges in any of the organs studied including their absolute andrelative weights (Fig. 2A and B). Therefore, the experiment showedthat 10 is well tolerated by the Swiss albino mice up to the doselevel of 300 mg/kg body weight as a single acute oral dose. How-ever, sub-acute and/or chronic experiment with the test drug
needs to be carried out to look for any adverse effect on repeatedexposure to the test drug compound 10 for its future development[22].
A 3,4,5-trimethoxyphenyl unit is considered to interact withtubulin and exerts tubulin polymerization inhibition. During mito-sis, microtubule polymerization and depolymerization are tightlycontrolled. Disruption of this process can cause cell cycle arrestand subsequently cell death [23]. The anticancer effect of the com-pounds having this type arrangement is due to antiproliferation ofcancer cells through disturbance of mitotic spindle function whichleads to cell apoptosis [24] and also disrupting cell signalling path-ways of endothelial cells in tumour vasculature leading to selectiveshutdown of blood flow through tumours [25,26]. Several combre-tastatin analogues have already been reported to act like this[27,28].

Table 4Effect of compound 10 on body weight, haemogram and serum biochemical parameters in Swiss albino mice (n = 4).
Parameters Dose of compound 10 at mg/kg body weight
Control 5 mg/kg 50 mg/kg 300 mg/kg
Body weight (g) 21.94 ± 0.85 23.84 ± 0.59 22.01 ± 1.16 22.67 ± 1.06Total RBC count (millions/mm3) 7.85 ± 0.45 8.62 ± 0.71 7.82 ± 0.86 10.24±.60Total WBC count (thousands/mm3) 5.05 ± 0.88 5.08 ± 0.75 5.03 ± 0.20 7.80 ± 0.67Haemoglobin (g/dL) 16.23 ± 1.07 14.51 ± 0.33 18.16 ± 1.81 17.05 ± 0.66Serum total cholesterol (mg/dL) 148.39 ± 20.63 165.38 ± 11.02 163.56 ± 20.04 146.63 ± 6.87Serum triglycerides (mg/dL) 137.95 ± 9.05 157.91 ± 16.31 146.91 ± 5.42 155.38 ± 17.35Serum creatinine (mg/dL) 0.55 ± 0.06 0.32 ± 0.06 0.43 ± 0.03 0.40 ± 0.09SGPT (U/L) 7.32 ± 0.88 12.63 ± 0.67 9.09 ± 0.90 12.20 ± 2.52SGOT (U/L) 26.61 ± 1.68 25.02 ± 1.38 26.79 ± 2.22 32.53 ± 2.30
Fig. 3. Effect of compound 10 on differential leucocytes counts in Swiss albino mice(n = 4).
Fig. 2. (A and B): Effect of compound 10 on absolute and relative organ weights in Swiss albino mice (n = 4).
S. Parihar et al. / Steroids 77 (2012) 878–886 885
4. Conclusion
Gallic acid based phenstatin analogues exhibited moderate tolow level of cytotoxicity. The most active analogue 10 exhibitedhigh level of inhibition of tubulin polymerization and was foundto be non-toxic in the Swiss albino mice up to 300 mg/kg dose.This compound also possessed low oestrogenicity and higheranti-oestrogenicity. Such an anticancer agent may have a dualaction as selective oestrogen receptor modulators along withtubulin interaction. However, these compounds may further bestudied in specific gene expression of oestrogen receptors a orb to establish their overall effect.
Acknowledgements
The authors thank to Director CIMAP for constant support andencouragement. This work was supported by a Grant from Depart-ment of Science and Technology (DST), Govt. of India.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.steroids.2012.03.012.
References
[1] (a) Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Cancer Biology2001;11:339–52;(b) Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA.Estrogen receptor b ligands: recent advances and biomedical applications.Med Res Rev 2011;31:364–442;(c) Sherwin BB. Estrogen and cognitive functioning in women. Endocr Rev2003;24:133–51.
[2] Godolphin W, Elwood JM, Spinelli JJ. Estrogen receptor quantitation andstaging as complementary prognostic indicators in breast cancer: a study of583 patients. Int J Cancer 1981;28:677–83.
[3] Jordan VC, O’Malley BW. Selective estrogen-receptor modulators andantihormonal resistance in breast cancer. J Clin Oncol 2007;25:5815–24.
[4] Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM.Endometrial cancer in tamoxifen-treated breast cancer patients: findings fromthe National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J NatCancer Inst 1994;86:527–37.
[5] Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacologyof antiestrogen action and resistance. Pharmacol Rev 2001;53:25–71.
[6] Srivastava V, Negi AS, Kumar JK, Gupta MM, Khanuja SPS. Plant basedanticancer molecules: a chemical and biological profile of some importantmolecules. Bioorg Med Chem 2005;13:5892–908.
[7] Brandi G, Paiardini M, Cervasi B, Fiorucci C, Filippone P, De Marco C, ZaffaroniN, MaGnani M. The chemistry and pharmacology of indole-3-carbinol (indole-3-methanol) and 3-(methoxymethyl)indole. Cancer Res 2003;63:4028–36.

886 S. Parihar et al. / Steroids 77 (2012) 878–886
[8] Ohsumi K, Nakagawa R, Fukuda Y, Hatanaka T, Morinaga Y, Nihei Y, Ohishi K,Suga Y, Akiyama Y, Tsuji T. Novel combretastatin analogues effective againstmurine solid tumors: design and structure-activity relationships. J Med Chem1998;41:3022–32.
[9] O’Boyle NM, Carr M, Greene LM, Bergin O, Nathwani SM, McCabe T, Lloyd DG,Zisterer DM, Meegan MJ. Synthesis and evaluation of azetidinone analogues ofcombretastatin A-4 as tubulin targeting agents. J Med Chem 2010;53:8569–84.
[10] Siemann DW, Chaplin DJ. A review and update of the current status of thevasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert OpinInvestig Drugs 2009;18:189–97.
[11] Pettit GR, Toki B, Herald DL, Verdier-Pinard P, Boyd MR, Hamel H, Pettit RK.Antineoplastic agents. 379. synthesis of phenstatin phosphate1a. J Med Chem1998;41:1688–95.
[12] Pettit GR, Grealish MP, Herald DL, Boyd MR, Hamel E, Pettit RK.Antineoplastic agents. 443. Synthesis of the cancer cell growth inhibitorhydroxyphenstatin and its sodium diphosphate prodrug. J Med Chem2000;43:2731–7.
[13] Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinalchemistry of combretastatin A4: present and future directions. J Med Chem2006;49:3033–44.
[14] IUPAC. Joint commission on biochemical nomenclature (JCBN). Nomenclatureof steroids. Pure Appl Chem 1989;61:1783–1822.
[15] Kumar S, Deshpande S, Chandra V, Kitchlu S, Dwivedi A, Nayak VL, Konwar R,Prabhakar YS, Sahu DP. Synthesis and biological evaluation of 2,3,4-triarylbenzopyran derivatives as SERM and therapeutic agent for breastcancer. Bioorg Med Chem 2009;17:6832–40.
[16] Shelanski ML, Gaskin F, Cantor CR. Microtubule assembly in the absence ofadded nucleotides. Proc Nat Acad Sci 1973;70:765–8.
[17] Lee JC, Timassheff SN. In vitro reconstitution of calf brain microtubules: effectsof solution variable. Biochemistry 1977;16:1754–62.
[18] Ghosh R, Kamboj VP, Singh MM. Interaction with anti-implantation andestrogen antagonistic activities of dl-ormeloxifene, a selective estrogenreceptor modulator, by tetracycline in female Sprague–Dawley rats.Contraception 2001;64:261–9.
[19] Saxena HO, Faridi U, Kumar JK, Luqman S, Darokar MP, Shanker K, ChanotiyaCS, Gupta MM, Negi AS. Synthesis of chalcone derivatives on steroidalframework and their anticancer activities. Steroids 2007;72:892–900.
[20] Gilman H, Blatt AH, Syntheses Organic. Collective, 2nd ed., vol. 1. NewYork: John Wiley & Sons Inc.; 1941. p 537–38.
[21] Li YW, Liu J, Liu N, Shi D, Zhou XT, Lv JG, Zheng CH, Zhou YJ. Imidazolone-amide bridges and their effects on tubulin polymerisation in cis-lockedvinylogous combretastatin-A4 analogues: synthesis and biological evaluation.Bioorg Med Chem 2011;19:3579–84.
[22] Ghosh MN. Fundamentals of Experimental Pharmacology. 1sted. Kolkata: Scientific Book Agency; 1984. p. 156.
[23] Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agentson signal transduction pathways of apoptosis: a review. Cancer ChemotherPharmacol 1999;44:355–61.
[24] Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat RevCancer 2004;4:253–65.
[25] Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat RevCancer 2005;5:423–35.
[26] Griggs J, Metcalfe JC, Hesketh R. Targeting tumour vasculature: thedevelopment of combretastatin A4. The Lancet Oncol 2001;2:82–7.
[27] Duan JX, Cai X, Meng F, Lan L, Matteucci M. Potent antitubulin tumor cellcytotoxins based on 3-aroyl indazoles. J Med Chem 2007;50:1001–6.
[28] Romagnoli R, Baraldi PG, Sarkar T, Carrion MD, Cara CL, Cruz-Lopez O, Preti D,Tabrizi A, Tolomeo M, Grimaudo S, Cristina AD, Zonta N, Balzarini J, Brancale A,Hsieh HP, Hamel E. Synthesis and biological evaluation of 1-Methyl-2-(30 ,40 ,50-trimethoxybenzoyl)-3-aminoindoles as a new class of antimitotic agents andtubulin inhibitors. J Med Chem 2008;51:1464–8.
Related Documents











