RESEARCH ARTICLE Functional adaptation of microbial communities from jet fuel- contaminated soil under bioremediation treatment: simulation of pollutant rebound Olesya Korotkevych 1 , Jirina Josefiova 1 , Martina Praveckova 1 , Tomas Cajthaml 1 , Monika Stavelova 2 & Maria V. Brennerova 1 1 Department of Cell Molecular Microbiology, Institute of Microbiology, Prague, Czech Republic; and 2 AECOM CZ Ltd, Prague, Czech Republic Correspondence: Maria V. Brennerova, Department of Cell Molecular Microbiology, Institute of Microbiology v. v. i., Videnska 1083, 142 20 Prague, Czech Republic. Tel.: 1420 241 062 781; fax: 1420 241 722 257; e-mail: [email protected] Received 5 December 2010; revised 21 June 2011; accepted 27 June 2011. Final version published online 1 August 2011. DOI:10.1111/j.1574-6941.2011.01169.x Editor: Michael Schloter Keywords mesocosms; biodegradation; phytoremediation; qPCR of catabolic genes; DGGE. Abstract To investigate the link between the functionality and the diversity of microbial communities under strong selective pressure from pollutants, two types of meso- cosms that simulate natural attenuation and phytoremediation were generated using soil from a site highly contaminated with jet fuel and under air-sparging treatment. An increase in the petroleum hydrocarbon concentration from 4900 to 18 500 mg kg 1 dw soil simulated a pollutant rebound (postremediation pollutant reversal due to residual contamination). Analysis of soil bacterial communities by denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA gene fragments showed stronger changes and selection for a phylogenetically diverse microbial population in the mesocosms with pollutant-tolerant willow trees. Enumeration of the main subfamilies of catabolic genes characteristic to the site detected a rapid increase in the degradation potential of both systems. A marked increase in the abundance of genes encoding extradiol dioxygenases with a high affinity towards various catecholic substrates was found in the planted mesocosms. The observed adaptive response to the simulated pollutant rebound, characterized by increased catabolic gene abundance, but with different changes in the microbial structure, can be explained by functional redundancy in biodegrading microbial communities. Introduction Subsurface spills of petroleum compounds that have disas- trous consequences for the biotic and abiotic components of ecosystems are the most frequently cited cause of groundwater contamination (Okoh, 2006; Glick, 2010; Das & Chandran, 2011). The large family of several hundred hydrocarbon compounds that originally come from crude oil is described by the term total petroleum hydrocarbons (TPH), which is defined as the measurable amount of hydrocarbons (Thompson & Nathanail, 2003). In addition to aliphatic hydrocarbons, petroleum hydrocarbons contain benzene, toluene, ethylbenzene and xylenes (BTEX). These hazardous substances are major components of gasoline and jet fuels, and they are regulated by many nations. The US Environmental Protection Agency and the Agency for Toxic Substances and Disease Registry have compiled a list of the most frequently observed toxic compounds, wherein TPH components such as n-hexane, monoaromatic and polyaro- matic hydrocarbons, fuel oils, gasoline and hydraulic fluids are cataloged (Glick, 2010). Biodegradation is a process whereby microorganisms play a major role in the biological conversion of hazardous pollutants to innocuous products and has been reported to be one of the primary mechanisms by which petroleum and other hydrocarbon pollutants can be removed from the environment (Das & Chandran, 2011). Biodegradation and the use of plants to remediate polluted soils (phytoremedia- tion) are believed to be noninvasive, effective and inexpen- sive technologies (Glick, 2010; Tang et al., 2010). Over the past three decades, investigations of the bio- chemical pathways, genes and proteins responsible for the enzymatic degradation of mono- and polyaromatic pollu- tants have focused on cultivation techniques and the isola- tion of bacterial strains useful for biodegradation (Yeates et al., 2000; Witzig et al., 2006). Aerobic bacteria have been FEMS Microbiol Ecol 78 (2011) 137–149 c 2011 Federation of European Microbiological Societies Published by Blackwell Publishing Ltd. All rights reserved MICROBIOLOGY ECOLOGY

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

R E S E A R C H A R T I C L E
Functional adaptationofmicrobial communities from jet fuel-contaminated soil under bioremediation treatment: simulationofpollutant reboundOlesya Korotkevych1, Jirina Josefiova1, Martina Praveckova1, Tomas Cajthaml1, Monika Stavelova2 &Maria V. Brennerova1
1Department of Cell Molecular Microbiology, Institute of Microbiology, Prague, Czech Republic; and 2AECOM CZ Ltd, Prague, Czech Republic
Correspondence: Maria V. Brennerova,
Department of Cell Molecular Microbiology,
Institute of Microbiology v. v. i., Videnska
1083, 142 20 Prague, Czech Republic. Tel.:
1420 241 062 781; fax: 1420 241 722 257;
e-mail: [email protected]
Received 5 December 2010; revised 21 June
2011; accepted 27 June 2011.
Final version published online 1 August 2011.
DOI:10.1111/j.1574-6941.2011.01169.x
Editor: Michael Schloter
Keywords
mesocosms; biodegradation;
phytoremediation; qPCR of catabolic genes;
DGGE.
Abstract
To investigate the link between the functionality and the diversity of microbial
communities under strong selective pressure from pollutants, two types of meso-
cosms that simulate natural attenuation and phytoremediation were generated using
soil from a site highly contaminated with jet fuel and under air-sparging treatment.
An increase in the petroleum hydrocarbon concentration from 4900 to
18 500 mg kg�1 dw soil simulated a pollutant rebound (postremediation pollutant
reversal due to residual contamination). Analysis of soil bacterial communities by
denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA gene fragments
showed stronger changes and selection for a phylogenetically diverse microbial
population in the mesocosms with pollutant-tolerant willow trees. Enumeration of
the main subfamilies of catabolic genes characteristic to the site detected a rapid
increase in the degradation potential of both systems. A marked increase in the
abundance of genes encoding extradiol dioxygenases with a high affinity towards
various catecholic substrates was found in the planted mesocosms. The observed
adaptive response to the simulated pollutant rebound, characterized by increased
catabolic gene abundance, but with different changes in the microbial structure, can
be explained by functional redundancy in biodegrading microbial communities.
Introduction
Subsurface spills of petroleum compounds that have disas-
trous consequences for the biotic and abiotic components
of ecosystems are the most frequently cited cause of
groundwater contamination (Okoh, 2006; Glick, 2010; Das
& Chandran, 2011). The large family of several hundred
hydrocarbon compounds that originally come from crude
oil is described by the term total petroleum hydrocarbons
(TPH), which is defined as the measurable amount of
hydrocarbons (Thompson & Nathanail, 2003). In addition
to aliphatic hydrocarbons, petroleum hydrocarbons contain
benzene, toluene, ethylbenzene and xylenes (BTEX). These
hazardous substances are major components of gasoline and
jet fuels, and they are regulated by many nations. The US
Environmental Protection Agency and the Agency for Toxic
Substances and Disease Registry have compiled a list of the
most frequently observed toxic compounds, wherein TPH
components such as n-hexane, monoaromatic and polyaro-
matic hydrocarbons, fuel oils, gasoline and hydraulic fluids
are cataloged (Glick, 2010).
Biodegradation is a process whereby microorganisms play
a major role in the biological conversion of hazardous
pollutants to innocuous products and has been reported to
be one of the primary mechanisms by which petroleum and
other hydrocarbon pollutants can be removed from the
environment (Das & Chandran, 2011). Biodegradation and
the use of plants to remediate polluted soils (phytoremedia-
tion) are believed to be noninvasive, effective and inexpen-
sive technologies (Glick, 2010; Tang et al., 2010).
Over the past three decades, investigations of the bio-
chemical pathways, genes and proteins responsible for the
enzymatic degradation of mono- and polyaromatic pollu-
tants have focused on cultivation techniques and the isola-
tion of bacterial strains useful for biodegradation (Yeates
et al., 2000; Witzig et al., 2006). Aerobic bacteria have been
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
MIC
ROBI
OLO
GY
EC
OLO
GY

extensively studied with regard to their ability to utilize
aromatic compounds as the sole carbon and energy sources
and their environmental plasticity (Harwood & Parales,
1996; Pieper, 2005). The organization and regulation of
catabolic genes has been characterized, and catabolic operon
prototypes have been described (Harayama et al., 1987; Eltis
& Bolin, 1996; Jimenez et al., 2002; Parales & Harwood,
2002; McLeod & Eltis, 2008). The metagenomic approach
has further enabled the direct and detailed evaluation of
microbial community networks without cultivation (Rie-
senfeld et al., 2004) and the identification of enzymes
involved in the metabolism of aromatic compounds (Sue-
naga et al., 2007; Brennerova et al., 2009). These achieve-
ments have led to new horizons for monitoring natural and
anthropogenically endangered environments for new genes
and attenuation processes.
Hradcany is a former military airbase (5013709.95500N,
14143054.53100E) that occupies a region that is the most
intensively polluted with petroleum hydrocarbons in North-
ern Bohemia, the Czech Republic. More than 28 ha were
contaminated by military activities from 1940 to 1991. Jet
fuel comprised 70% of the contamination. TPH concentra-
tions varied from 5000 to 55 000 mg kg�1 dry soil and BTEX
was detected at 1000 mg kg�1 (Machackova et al., 2008).
Based on risk assessments, the Czech Ministry of Environ-
ment dictated the TPH clean-up target limit for soil to be
5000 mg kg�1 dry soil. A remediation project was initiated in
1997 that involved in situ soil vacuum extraction, injection
of contaminant-free air into the subsurface saturated zone,
known as air sparging, and additional stimulation of the soil
microorganisms by nutrient amendment. At the end of
2008, the decontamination measures were terminated. Re-
vitalization and utilization of the site with the introduction
of plants as facilitators of residual contaminant removal was
planned by 2012. Many crop plants and grasses (maize, rice,
legumes, sorghum, ryegrass, Bermuda grass and beggar
ticks) are effective in degrading TPH (Shirdam et al.,
2008; Glick, 2010; Tang et al., 2010). Willows and poplar
trees are alternative phytoremediation species. Their reme-
diation potential is proportional to survival, fast growth,
height, deep root system and volume production, which are
measures of potential benefit for long-term petroleum
degradation by the trees and their rhizosphere-associated
microorganisms (Trapp et al., 2001; Zalesny et al., 2005).
Willows (Salix euxina, also known as Salix fragilis) are part
of the native vegetation of Hradcany. Their use for revitali-
zation of the site requires the collection of missing experi-
mental evidence under conditions that are similar to the
field environment (Glick, 2010).
Pollutants in the soil can frequently return to levels
detected before remediation initiation (Switzer & Kosson,
2007). The contamination reversal, called pollutant re-
bound, is a post-treatment phenomenon that follows the
interruption of soil vacuum extraction and air sparging, and
is caused by alterations in the pollution equilibrium between
groundwater, soil and soil gas. This postremediation pollu-
tant concentration increase may impose environmental
stress on the microbial communities present in the soil and
rhizosphere. On the other hand, the in situ stimulation of
the natural microbial communities at the Hradcany site has
enabled strong selection for meta-cleavage degradation
pathways (Brennerova et al., 2009); thereby, the soil’s
microbial populations evolving after a bioremediation treat-
ment are ideal model systems to study the pollutant
rebound related to changes in microbial biodegradation
potential and adaptability.
The main objectives of this study were (1) to define the
level of tolerance of willow trees to the contaminant, (2) to
compare changes in the soil microbial populations in two
different mesocosm systems modeling natural attenuation
and phytoremediation, (3) to study the ability of microbial
populations to adapt their catabolic machinery to the
biodegradation process following a sudden increase in
pollutant concentration and (4) to investigate the possible
dependence of catabolic potential on shifts in the structure
of the microbial community. This work furthers the devel-
opment of phytoremediation techniques by undertaking a
methodical assessment of phytoadaptation to petroleum
hydrocarbons and provides new insight into the relationship
between the dynamics of microbial populations and their
degradation potential.
Materials and methods
Soil analysis
The contaminated soil used in the study was excavated at a
depth of 1.5–2.5 m from an area in Hradcany (field G) that
had been intensively cleaned for 7.5 years. The analysis of
grain sizes and petroleum hydrocarbons in the soil was
carried out by the accredited laboratory ALS Czech Republic
Ltd. Fractions of soil particles from 4 2 to 0.063 mm were
assessed using the sieving method, and other fractions were
identified from the o 0.063-mm fraction by a laser particle
size analyzer using the liquid dispersion mode (test method
CZ_SOP_D06_07_N11). The TPH concentration in the soil
was estimated as the average of nine samples measured using
Fourier transform infrared spectroscopy (Czech standards
CSN 757505 and CSN 757506). In light non-aqueous-phase
liquid (LNAPL), the analysis of hydrocarbon fractions with
carbon numbers C10 through C40 was performed by GC
(CSN EN ISO 9377-2). Additional BTEX analyses, in both
soil and LNAPL, were conducted according to methods for
the chromatographic/mass spectrometric (GC–MS) detec-
tion of volatile organic compounds (VOC) in EPA 624 and
EPA 8260. PAHs were analyzed as semi-VOC by GC–MS
FEMS Microbiol Ecol 78 (2011) 137–149c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
138 O. Korotkevych et al.

according to methods EPA 8270, EPA 8131, EPA 8091 and
Czech normative CSN EN ISO 6468.
Soil was suspended in water at a 1 : 1 ratio by shaking in the
dark for 1 h, and the pH was measured using a PH114 (Snail
Instruments, Czech Republic). The water content was deter-
mined by drying the soil at 105 1C for 10 h and was expressed
as a percentage of the sample weight (CSN ISO 11465).
The numbers of cultivable heterotrophic bacteria were
determined to monitor the influence of pollutant rebound
on the soil microbiota. Soil samples were homogenized in
0.9% NaCl and agitated for 1 h at 4 1C. Appropriately
diluted aliquots were spread on R2A agar (BD DifcoTM)
and incubated at 24 1C in closed glass jars in the presence of
LNAPL vapors. Three independent determinations of the
CFU g�1 dry soil were performed in triplicate.
Mesocosm experiments
Mesocosm cultivation systems were constructed outdoors
using six 45-cm� 45-cm� 50-cm cultivation boxes built
from white polypropylene (Fig. 1). The boxes were filled
with a 5-cm layer of keramzite gravel and 60 kg of
Fig. 1. Mesocosm setup: (a) graphic representation of mesocosm systems that model natural bioremediation (pot N) and phytoremediation (pot P).
(b) Picture of the mesocosms on day 30 of the experiment.
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
139Functional redundancy in biodegrading microbial communities

contaminated soil mixed with LNAPL. Three mesocosm
boxes were assigned as models for natural attenuation (N).
Another three mesocosms were used for a comparative
analysis of rhizosphere-enhanced phytoremediation (P).
Pilot experiments were carried out to determine the thresh-
old concentration of pollutants tolerated by the young trees.
Three-month-old hydroponically rooted willow cuttings
were compared with cuttings rooted in a garden substrate.
Direct transfer into soil containing TPH at 13 000 mg kg�1 dw
was lethal for the hydroponically pregrown willows and had
detrimental effects on the young trees when their rhizospheres
had developed in soil. In parallel mesocosms with a twofold
lower soil TPH concentration, the organic phase had a toxic
impact on the hydroponic plants, but was tolerated by the
willow cuttings with soil-grown rhizospheres. Therefore, slow
adaptation of the trees was chosen for the final mesocosm
experiment. Crack willow (S. euxina, also known as S. fragilis)
cuttings that were 30–35 cm in length and approximately 1 cm
in diameter and had 3-month-old root systems developed in
1.5 L of garden substrate were gradually adapted to the
contaminant by transfer to pots containing 3 L of soil from
field G. They were grown for an additional month under
conditions of a gradually increasing TPH concentration by
weekly admixing of LNAPL aliquots into the substrate to a
final concentration of 13 000 mg kg�1 dry soil. Four adapted
young trees were transferred into each P container, and the
water content of the soil was maintained between 8.3% and
14.7%. The sandy soil, which quickly drained and dried out,
was mixed at the sides and the center of the pots once every 2
weeks, thus providing additional air access. Soil sampling was
carried out on days 1, 15, 32, 62, 90 and 126. Bulk soil was
collected at five sampling points in each pot by augering (100 in
diameter) to a depth of 20–25 cm and, in the case of P pots,
10–13 cm from the plants. The control sample, termed ‘day 0’,
was taken before mixing the soil with the LNAPL. An
additional control, termed ‘rhizosphere’, was collected from
the rhizosphere-associated soil at the termination of meso-
cosm P cultivation. All soil samples were homogenized, stored
in airtight sterile glass bottles, placed on ice and then used for
DNA isolation, enumeration of cultivable soil microorganisms
and analyses of TPH concentration, pH and water content.
Changes in the microbial community structure and in cata-
bolic gene concentrations in both the N and the P systems
were followed over a period of 4 months during the growing
season (June 15, 2006–November 2, 2006).
Soil DNA extraction
High-molecular-weight DNA was recovered from 10 g of soil
as described previously (Brennerova et al., 2009). The DNA
was purified further by humic acid removal using the
PowerClean DNA Clean-Up Kit (MoBio Laboratories,
Carlsbad, CA). The concentration of DNA was determined
using a spectrophotometer (BioMate 5, Thermo Spectronic)
and a NanoDrop ND-1000 UV-VIS (NanoDrop Technolo-
gies). DNA was stored at 4 1C during the course of the study.
Denaturing gradient gel electrophoresis (DGGE)analysis of partial 16S rRNA genes
The V3 region of the 16S rRNA gene was amplified by a PCR
using the 338f forward primer, which is specific for a region
conserved among members of the domain Bacteria (Ovreas
et al., 1997), and the 518r universal primer (Muyzer et al., 1993)
to amplify a product with an average length of 196 bp. A 40-bp
GC clamp (50-CGC CCG CCG CGC CCC GCG CCC GGC
CCG CCG CCG CCG CCG C-30) was attached to the 50 end of
the forward primer (Myers et al., 1987) for DGGE system
adaptation. For sequencing, 16S rRNA gene sequences from the
V3-V4 hypervariable regions were retrieved by PCR with the
primers GC-338f (Ovreas et al., 1997) and 802r (Tamura &
Hatano, 2001), which span positions 802-785 of the Escherichia
coli 16S rRNA gene. Amplification was performed using a
Mastercycler ep (Eppendorf AG, Hamburg, Germany). The 50-
mL reaction mixture contained 50 ng of soil DNA, 10 pmol each
primer, 200mM each dNTP, 2.5 mM MgCl2, 5mL 10�PCR
buffer and 1.25 U Thermo-Starts DNA Polymerase (ABgene,
UK). The enzyme was activated at 95 1C for 15 min. To increase
the specificity of amplification and reduce the formation of
spurious byproducts, a ‘touchdown’ hotstart PCR was per-
formed. The initial annealing temperature (61 1C) was de-
creased by 1 1C during every cycle until an annealing
temperature of 55 1C was reached, and 28 additional cycles
were carried out at 94 1C (denaturing), 55 1C (annealing) and
72 1C (elongation) for 45 s at each temperature. The final
primer extension was performed at 72 1C for 10 min. The PCR
products, together with pBR322 DNA/AluI Marker (Fermentas,
Lithuania), were separated on 2% agarose gels, visualized by
ethidium bromide staining and quantified using KODAK mole-
cular imaging software v4.5 (Rochester). PCR amplicons were
then separated by DGGE using a BioRad D-Code System
(BioRad, Hercules). Sample volumes containing 500–600 ng of
PCR product were applied to an 8% w/v polyacrylamide–bisa-
crylamide (37.5 : 1) gel in 1�TAE. The PCR amplicons
obtained with the primers GC-338f and 802r were separated
in a 7% gel. Optimal separation was achieved at 60 1C with a
parallel 45–60% urea–formamide denaturing gradient increas-
ing in the direction of electrophoresis. Denaturant at 100%
corresponded to 7 M urea and 40% v/v formamide. The gels
were electrophoresed for 15 min at 20 V, then for 24 h at 55 V
and stained with SYBRs Green I (1 : 10 000; Molecular Probes,
Eugene) in 25 mM Tris-HCl (pH 8) for 20 min and photo-
graphed using a Kodak EDAS 290 camera. PCR runs for DGGE
and DGGE fingerprinting were performed in triplicate, and the
resulting fingerprints were analyzed using GELCOMPAR II software
v5.1 (Applied Maths, Belgium).
FEMS Microbiol Ecol 78 (2011) 137–149c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
140 O. Korotkevych et al.

Sequencing of DNA from DGGE bands andphylogenetic analysis
DGGE separation of amplicons with approximate lengths of
456 bp was achieved under the conditions described above.
Selected resolved bands were excised from the polyacryl-
amide gels under UV illumination and incubated in 20 mL
nanopure water at 4 1C for 20 h. Aliquots (5 mL) served as
templates for PCR reamplification with the same primers
(without GC clamp), followed by purification using the
Wizards SV Gel and PCR Clean-Up System (Promega,
Madison, WI) and cloning into the pGEM-T easy vector
system (Promega). Transformant colonies were suspended
in water and incubated at 95 1C for 10 min, and plasmid
inserts were amplified by PCR with vector-specific M13
forward and reverse primers. Restriction fragment length
polymorphism (RFLP) analysis of inserts from 20 transfor-
mants was carried out by restriction with BsuRI, FspBI and
Tru1I purchased from Fermentas, in 10-mL reaction mix-
tures, with subsequent separation on 3.0% SeaKems LE
agarose gels (Cambrex Bio Science Rockland). Distinct
RFLP types were selected for sequencing of both strands
using M13 primers by BigDyes Terminator v3.1 chemistry
according to the manufacturer’s instructions (Applied Bio-
systems, Foster City, CA) on an ABI PRISM 3100 Genetic
Analyzer (Applied Biosystems).
DNA similarity searches of GenBank databases were per-
formed using the BLASTN program from the National Center for
Biotechnology Information (NCBI) website. Vector sequences
were removed using the KODON program v3.6 (Applied Maths),
and the sequences were oriented 50–30 relative to known 16S
rRNA genes using ORIENTATIONCHECKER (Bioinformatics Toolkit,
Cardiff School of Biosciences, UK). The sequences were
analyzed for potential chimeric sequences using the services
available at the Ribosomal Database Project II (Cole et al.,
2009). Additional potential chimeras were assessed using the
program BELLEROPHON (Huber et al., 2004). Alignments were
generated using the CLUSTALX 1.83 Windows interface of the
CLUSTALW program running default values. A block of sequence
alignments was selected using the GENEDOC multiple sequence
alignment editor software (Nicholas et al., 1997). Phylogenetic
trees were obtained by MEGA 4.1 software (Tamura et al., 2007)
using the neighbor-joining algorithm (Saitou & Nei, 1987)
with a p-distance model and pairwise deletion of gaps and
missing data. A consensus tree was inferred from a total of
1000 bootstrap trees generated for each data set. The sequences
of the 16S rRNA gene PCR fragments obtained in this study
are available under the EMBL/GenBank/DBBJ accession num-
bers GU560728–GU560730 and GU568253–GU568336.
Quantitative real-time PCR (qPCR) analysis ofcatabolic genes in soil samples
A real-time PCR method for the absolute quantification of the
main catabolic genes characteristic of the Hradcany site
(Brennerova et al., 2009) was developed. The primers intro-
duced were specific for four groups of dioxygenase genes
(Table 1). The forward and reverse primers bphAf668-3 and
bphAr1153-2 (Witzig et al., 2006) target conserved regions of
genes encoding the catalytic a-subunits of the Rieske non-
heme iron oxygenases, which are members of the toluene/
biphenyl oxygenase subfamily of aromatic ring-hydroxylating
dioxygenases (RHDO). The primer set EXDO-Dbt-F/EXDO-
Dbt-R allowed the amplification of metagenomic gene frag-
ments that encode peptides showing 55–64% sequence iden-
tity to extradiol dioxygenases involved in the degradation of
naphthalene, specifically to DbtC (Brennerova et al., 2009).
The group of targeted genes was named EXDO-Dbt after their
phylogenetic affiliation with dbtC (AF404408) of Burkholderia
sp. DBT1 (Di Gregorio et al., 2004). The primer set EXDO-
K2-F/EXDO-K2-R targets a third group of genes (EXDO-K2
genes) encoding proteins with 78–81% identity to the I.3.A
extradiol dioxygenases (Eltis & Bolin, 1996), which is reported
to be involved in the degradation of isopropylbenzene and
ethylbenzene. EXDO-K2 genes include ipbC (U53507) in
Pseudomonas sp. JR1 (degrading isopropylbenzene) (Pflugma-
cher et al., 1996) and ebdC (AJ293587) in P. putida 01G3
(degrading ethylbenzene) (Chablain et al., 2001). In our
previous study, the highly degenerate primer pair EXDO-D-
F/EXDO-D-R was used to amplify phylogenetically diverse
genes from the EXDO-D group, which corresponds to the
I.2.C subfamily of catechol 2,3-dioxygenases (Brennerova
Table 1. Oligonucleotide primers used in this study
Primers Sequence (50–30)
Amplicon
size (bp)
Annealing
temperature ( 1C)
Elongation
time (s) References
bphAF668-3 GTTCCGTGTAACTGGAARTWYGC 535 58 55 Witzig et al. (2006)
bphAR1153-2 CCAGTTCTCGCCRTCRTCYTGHTC
EXDO-K2-F GAAAAAGTGGGTTTGATGGAGG 810 63 70 Brennerova et al. (2009)
EXDO-K2-R CGCTTATGCCKCGTCATCACCC
EXDO-Dbt-F TCCGCATGGATTACAACC 423 58 45 Brennerova et al. (2009)
EXDO-Dbt-R GATCTGTGGAACGGGCAA
EXDO-D2-F GCTGGATCATTGCCTGTTG 314 64 35 This study
EXDO-D2-R GCGTGGGGGTGACATCM
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
141Functional redundancy in biodegrading microbial communities

et al., 2009). The formation of primer-dimers during qPCR
was avoided by designing the more specific primer set EXDO-
D2-F/EXDO-D2-R. OLIGO EXPLORER and OLIGO ANALYZER soft-
ware (Teemu Kuulasmaa, Finland) were used for primer
construction and for confirming both the optimal DG values
and the lack of hairpin structures formation. In contrast to the
originally used highly degenerate EXDO-D primer set that was
used for screening of metagenomic libraries (Brennerova et al.,
2009), the new EXDO-D2-F primer was not degenerate and
the EXDO-D2-R primer had only one degenerate nucleotide
at the 30-end position (Supporting Information, Fig. S4). The
new primers allowed the amplification of an abundant cluster
of genes (EXDO-D2 genes) showing similarity to btxH
(DQ834383) in Ralstonia sp. strain PHS1 (which is able to
utilize toluene, ethylbenzene, o-xylene and cresols) and to tbuE
(U20258) in Ralstonia pickettii strain PK01 (which degrades
toluene, benzene, phenol and cresols) (Kukor & Olsen, 1991).
An absolute calibration model was developed from a
series of DNA standards that utilizes recombinant fosmids
of known copy number (ABI, 2006; Sivaganesan et al., 2010)
and their associated quantification cycle (Cq). For the
purpose of generating standard curves, DNA was isolated
from the metagenomic fosmid clones s115, s37, s79 and s76,
which harbor the RHDO, EXDO-D2, EXDO-K2 and
EXDO-Dbt genes, respectively (Brennerova et al., 2009).
Fosmid DNA was isolated under conditions that induce
high-copy amplification and was purified using the Qiagen
Plasmid Mini Kit system (Qiagen, Hilden, Germany). The
plasmids were linearized by NotI digestion and their size was
determined by pulse-field gel electrophoresis. The standards
were divided into small aliquots, stored at � 20 1C and
thawed only once before use. The standard curve was
generated from Cq measurements of 10-fold serial dilutions
of the standard (103–108 gene copies). Soil DNA with an
unknown concentration of the target genes was analyzed in
the same instrument run. Real-time PCR was conducted on
a Mastercycler ep Realplex Thermocycler (Eppendorf AG).
Amplifications were performed in 25-mL reaction volumes
containing 1� Express SYBR GreenER qPCR SuperMix
(Invitrogen, Carlsbad, CA), 0.2 mM each primer and 2mL
soil DNA (10–20 ngmL�1) or 5 mL of plasmid DNA used as
the qPCR standard. A mixture of all PCR reagents without
any DNA was used as a no-template control (NTC).
Temperature gradient qPCR with subsequent melting curve
DNA analysis and agarose gel electrophoresis was performed
to determine the optimal conditions (annealing temperature
and extension time; Table 1) required for the amplification
of a specific product with each primer set. The final qPCR
runs were carried out under the following conditions: step
one, 50 1C for 1 min (UDG incubation); step two, 95 1C for
2 min; and step three, 40 cycles of 95 1C for 15 s, for 20 s at
the primer-specific annealing temperature and elongation
time at 72 1C (Table 1). SYBR Green I fluorescence signal
intensity was measured at 0.5 1C increments every 15 s, from
60 to 95 1C. The amplified products were analyzed in
ethidium bromide-stained agarose gels (Fig. S5) and by
real-time fluorescence incorporation. The phylogenetic af-
filiation with the expected dioxygenases was confirmed by
sequencing of the PCR products as described previously
(Brennerova et al., 2009). The postrun data analyses were
performed using EPPENDORF REALPLEX software v1.5 (Eppen-
dorf AG). Triplicate Cq measurements were collected to
generate a standard curve. Instrument runs with NTCs Cq
values Z35 were further evaluated. Potential PCR inhibition
due to the coextraction of humic substances with the soil
DNA (Van Doorn et al., 2009) was assessed by comparing
gene copy quantification in serial dilutions of the template.
The final number of target genes, reported as gene co-
pies ng�1 DNA, was an average of triplicate measurements
from two independent DNA extractions made from each
soil sample. For both standard and target DNA, the ampli-
fication efficiency (E) was estimated at 95% for qPCR with
the primer set bphAf668-3/bphAr1153-2, 88% for EXDO-
K2-, 90% for EXDO-Dbt- and 87% for EXDO-D2-. The
correlation coefficient values (r2) were 0.99 for all runs.
Results
Mesocosm simulation of pollutant rebound
Soil microbial communities and their degradation potential
were examined in natural remediation (N) and phytoreme-
diation (P) systems under conditions of pollutant rebound.
The test was performed outdoors to simulate real growing
conditions (Fig. 1). The soil consisted of 78% medium- and
coarse-grained sand particles, 9% silt and 2% clay. Its TPH
concentration was estimated at 4900 mg kg�1 dry soil. After
mixing with LNAPL, its TPH concentration was measured at
18 500 mg kg�1 dry soil. Thus, at the start of the degradation
experiment, the TPH content of the soil was elevated 3.8-
fold. Analyses of the LNAPL revealed that C6–C20 hydro-
carbons consisting of 59.9% C10–C12 and 38.2% C12–C16
hydrocarbons (characteristic of kerosene-type jet fuel) were
predominant among the C10–C40 analytical fractions
(69 800mg L�1) (Fig. S1). In addition, BTEX (8270mg L�1),
190.08 mg L�1 PAHs, such as naphthalene (184mg L�1), ace-
naphthalene (0.789mg L�1), acenaphthene (2.08mg L�1),
fluorene (1.89mg L�1), phenanthrene (0.736mg L�1), fluor-
anthene (0.136mg L�1), pyrene (0.141mg L�1), benzanthra-
cene (0.021mg L�1), chrysene (0.023mg L�1) and
benzo(b)fluoranthene (0.036mg L�1) were present. Willows
that had been gradually adapted to high TPH concentra-
tions (13 000 mg kg�1 dw) tolerated subsequent transfer to
the P mesocosms, where the pollutant content was brought
up to 18 500 mg kg�1 dw soil. Ten of the 12 plants survived
the first 10 days of cultivation. The average pH value of the
FEMS Microbiol Ecol 78 (2011) 137–149c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
142 O. Korotkevych et al.

soil in all the mesocosms was 5.8 during the first 62 days and
decreased to 5.0 by the end of cultivation.
DGGE assay of changes in the bacterialcommunity structure
Control 16S rRNA gene fingerprints of the control DNA
(day 0) were compared with the community profiles of bulk
soil samples taken at days 15, 32, 62, 90 and 126 of
mesocosm development without (pot N) or with (pot P)
young willows adapted to LNAPL. GELCOMPAR II software
analysis of the bacterial fingerprints is shown in Fig. 2. The
3.8-fold increase in the pollutant concentration to
18 500 mg kg�1 dry soil did not cause significant changes in
the microbial community profiles during the first month of
mesocosm cultivation. The DGGE band patterns on days 15
and 32 of the P mesocosms showed 50% similarity and those
of the N mesocosms showed 60% similarity. In the later
phases of mesocosm development, more pronounced mi-
crobial shifts were detected in the phytoremediation systems
(P). Between days 62 and 90, the microbial fingerprints of P
soil samples showed 30.8% similarity. Further changes were
observed in the microbial fingerprint of bulk soil from the P
mesocosms on day 126. Thus, at the end of the experiment,
the bacterial population in the P pots retained 20.2%
similarity to the other DGGE patterns. On the other hand,
the phylogenetic profile of the control ‘rhizosphere’ (soil
adhering to roots of the willows) shared only 7.7% of the
bands of the bulk soil community patterns of the N and P
mesocosms. In parallel with the described phylogenetic
changes, the growth of cultivable bacteria was also observed
in both systems using the CFU-detection method, which
indicated that the sudden increase in the TPH concentration
did not have an adverse effect on population density.
CFU g�1 dw soil increased 100-fold by the third month of
cultivation (as shown in Fig. S2). At the end of the experi-
ment, the average values for cultivable soil bacteria remained
20–50-fold higher than the initial values. At this point, the
final average TPH concentration in the N (4700 mg kg�1 dry
soil) and P (5000 mg kg�1 dry soil) mesocosms was compar-
able to the TPH content of the soil before simulation of the
rebound (4900 mg kg�1 dw soil).
Phylogenetic affiliations of the dominant 16SrRNA gene sequences
Pollutant rebound-driven changes in soil microbial popula-
tions were assessed by cloning the bands from the DGGE
fingerprints that displayed the most intense shifts on days 90
and 126 (Fig. S3). DNA was extracted from 20 bands from the
N mesocosms and 22 bands from the P mesocosms. RFLP
screening of reamplified inserts distinguished 71 N- and 61 P-
derived clones that contained sequences with unique restric-
tion patterns. After removing chimeras, the sequences were
grouped into 46 phylotypes (defined as having a minimum of
91% similarity) from the N mesocosms and 40 phylotypes
from the P mesocosms. Notably, up to seven different 16S
rRNA gene sequences were retrieved from a single DGGE
band (e.g. band P51 in Figs 3 and S3), each having different
annotations in the NCBI database. In addition, different bands
containing sequences (clones N23a, N24f, N27a and N28e)
assigned to the same bacterial genus were observed, which can
be attributed to the divergence and redundancy of 16S rRNA
gene sequences in genomes with multiple rrn operons (Acinas
et al., 2004; Lee et al., 2009).
Figure 3 presents a comparative phylogenetic analysis of
the predominant bacterial representatives of the N and P soil
microbial communities. In both types of mesocosm, the
priority proteobacterial classes of Alpha-, Beta- and Gamma-
proteobacteria comprised 75–76% of all phylotypes. The
major group of sequences retrieved from the N mesocosm
was affiliated with Betaproteobacteria (34.8%), followed by
Fig. 2. Dynamic changes in microbial
community structure at different stages of
mesocosm cultivation. DGGE analysis-generated
profiles of 16S rRNA gene products amplified
from DNA extracted from control soil and from
bulk soil sampled from the N and P mesocosms.
The DGGE patterns were compared using
GELCOMPAR II v5.1 software. Percent similarities
were calculated using the band-based Dice
coefficient. The numbers at the nodes represent
the degree of similarity.
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
143Functional redundancy in biodegrading microbial communities
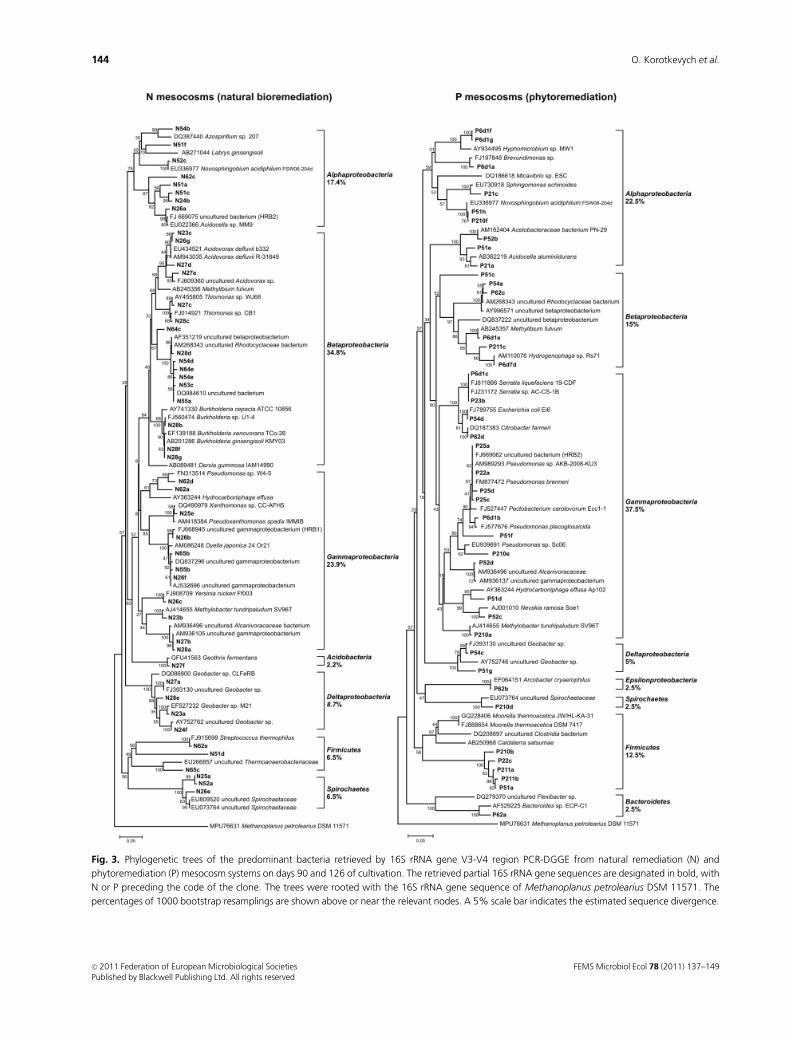
Fig. 3. Phylogenetic trees of the predominant bacteria retrieved by 16S rRNA gene V3-V4 region PCR-DGGE from natural remediation (N) and
phytoremediation (P) mesocosm systems on days 90 and 126 of cultivation. The retrieved partial 16S rRNA gene sequences are designated in bold, with
N or P preceding the code of the clone. The trees were rooted with the 16S rRNA gene sequence of Methanoplanus petrolearius DSM 11571. The
percentages of 1000 bootstrap resamplings are shown above or near the relevant nodes. A 5% scale bar indicates the estimated sequence divergence.
FEMS Microbiol Ecol 78 (2011) 137–149c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
144 O. Korotkevych et al.
Gammaproteobacteria (23.9%) and Alphaproteobacteria
(17.4%). In the P mesocosm, Gammaproteobacteria were the
most abundant, accounting for 37.5% of all phylotypes, and
Betaproteobacteria were the third most abundant bacterial
group (15%).
Degradation potential dynamics duringmesocosm development
Soil DNA was extracted at different phases of mesocosm
cultivation and examined by an absolute qPCR assay using
primers that target four groups of catabolic genes and the
optimized quantification conditions shown in Table 1.
Changes in gene copy number per ng soil DNA were used
to assess shifts in the degradation potential in situ (Fig. 4).
The sudden increase in the TPH concentration from 4900 to
18 500 mg kg�1 dry soil failed to negatively influence the
catabolic potential for the aerobic biodegradation of aro-
matic hydrocarbons in either mesocosm system. Moreover,
all four assessed groups of dioxygenase genes increased in
number and reached maximum levels after the second
month of mesocosm cultivation (Fig. 4). The quantification
of RHDO genes and EXDO-K2 genes showed common
dynamic patterns that can be ascribed to their colocalization
(Brennerova et al., 2009). In N mesocosm soil, the EXDO-K2
and RHDO genes reached maximum copy numbers of
5.57� 104 and 6.76� 104 gene copies ng�1 total DNA, respec-
tively, on day 90. In P mesocosm soil, the highest numbers,
4.19� 104 for EXDO-K2 genes and 8.61� 104 for RHDO
genes, were detected on day 126. At the end of cultivation, both
mesocosm systems showed a greater than threefold increase in
biodegradation potential relative to the untreated soil values
(1.46� 104 for EXDO-K2 and 2.11� 104 for RHDO).
The strongest shifts were observed for EXDO-D2 genes,
which encode proteins with an exceptionally high affinity
for various catecholic substrates (Brennerova et al., 2009).
The newly designed qPCR primers identified up to
8.15� 103 gene copies ng�1 DNA in the N soil on day 90
and 1.83� 104 gene copies ng�1 DNA in the P soil on day 62.
Hence, in a simulation of the rebound effect, biodegradation
potential dependent on EXDO-D2 genes increased 6.5- and
14.6-fold in the N and P microbial populations, respectively.
A similar tendency for the gene copy number to increase
was observed for the least abundant catabolic gene EXDO-
Dbt. The encoded enzyme exhibits broad specificity for
different substrates, including 1,2-dihydroxynaphthalene,
dihydroxybiphenyl, 3-methylcatechol and catechol (Bren-
nerova et al., 2009). On day 62, soil DNA from the P
mesocosms produced a maximum amplification signal of
1.02� 104 copies, which is three times higher than the peak
signal detected on day 32 in the N mesocosms and 8.4 times
higher than the gene concentration in the control soil before
mixing with the LNAPL fraction.
Discussion
In the current study, we adopted a metagenomic approach
to monitoring changes in the degradation potential of soil
microbial communities under conditions that simulate
pollutant rebound. Little is known about (1) the ability of
soil microbiota to resist sudden increases in petroleum
hydrocarbon concentrations, (2) the relationship between
microbial diversity and the functionality of microbial eco-
systems and (3) the influence of plants on the behavior and
biodegradation potential of bulk soil bacteria that are not in
close association with the rhizosphere. Understanding these
relationships is necessary for the design and implementation
of effective and economical remediation and revitalization
measures on sites with a long history of environmental
contamination.
We initiated this investigation by searching for conditions
under which TPH concentrations are several times higher
than the remediation limit (5000 mg kg�1 dw soil), but are
still not lethal to willow trees. To our knowledge, there is a
lack of evidence regarding the pollutant tolerance of plant
species that can be used for in situ remediation. A field study
Fig. 4. The abundance of four groups of catabolic genes during the
development of the N and P mesocosms. Gene copy numbers were
determined by qPCR of soil DNA extracted before the addition of LNAPL
(control) and on days 15, 32, 62, 90 and 126 of mesocosm cultivation.
Each bar represents an average of triplicate PCR runs performed on two
independent DNA extractions (n = 6). Error bars indicate the SD.
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
145Functional redundancy in biodegrading microbial communities
Maria
Highlight
Maria
Highlight
Maria
Highlight

using hydroponically rooted willow and poplar trees found
that weathered gasoline and diesel at a concentration of
4000 mg kg�1 reduced transpiration by 50%, and it was
concluded that at high TPH concentrations, for example,
4 5000 mg kg�1 dw soil, willows cannot be used for phytor-
emediation due to toxic effects (Trapp et al., 2001). Our
pilot mesocosm experiments confirmed the aforementioned
results. Although 6500 mg kg�1 dw soil had a lethal effect on
young willows prerooted in water, the control cuttings
developed larger root systems in a garden substrate; their
vitality was not affected by transfer to contaminated soil
with subsequent increases in TPH up to 18 500 mg kg�1 dw
soil. Thus, their successful adaptation to increasing pollu-
tant concentrations and their development of large roots
that reach over 6 m in depth make willow trees suitable for
the remediation and revitalization of soil contaminated with
petroleum hydrocarbons.
We were specifically interested in comparing microbial
populations and their catabolic properties in the bulk soil of
two systems. In the N mesocosm, the main factor that
defined specific changes in the bacterial community was the
rapid increase in hydrocarbon contamination from 4900 to
18 500 mg kg�1 dw soil. An additional determinant of phy-
logenetic changes in the P mesocosm was the rhizosphere of
pollutant-tolerant willows. Comparative 16S rRNA gene
DGGE analysis of the bacterial communities revealed a clear
change in their structure, reflecting a net adaptation to the
new conditions in the soil (Fig. 2). Distinct microbial
populations developed in the N and P mesocosms during
the 4 months of active vegetative growth under outdoor
conditions. Changes in the microbial community structure
in the N pots were the most intense during the first month
of mesocosm development. The DGGE fingerprints of the
15- and 32-day-old P mesocosms clustered together with
those of the 15- and 32-day-old N mesocosms. In the later
phases of cultivation, stronger shifts were detected for the P
mesocosms, indicating phylogenetic diversification that can
be ascribed to the influence of the willows.
The soil used in our study originated in field G of
Hradcany, which is directly adjacent to the HRB-1 site, well
characterized by biomolecular tools. Phospholipid fatty acid
profiling and 16S rRNA gene library analysis revealed a
highly diverse soil microbial population (Kabelitz et al.,
2009). The latter study, together with a metagenomic
characterization of the diversity and abundance of active
meta-cleavage pathways (Brennerova et al., 2009), demon-
strate that the majority of biodegradation genes are encoded
by Betaproteobacteria species, which represent the largest
phylogenetic group in the soil microbial community.
Although PCR-DGGE does not allow the full complexity of
the system to be determined, we were able to monitor
dynamic changes in the soil microbial communities and
compare differences between the two types of mesocosms.
Bacterial population shifts in the plant-free N mesocosms
were less intense, and the predominant phylotypes were
affiliated mainly with Betaproteobacteria at the final stages of
cultivation. More dynamic changes in the DGGE finger-
prints of the mesocosms with pollutant-tolerant willow trees
indicated selection for phylogenetically diverse bacterial
populations. Sequence analysis confirmed that Gamma-
proteobacteria species comprised the major and predomi-
nant phylotype groups in the P mesocosms. However, the N
and P systems shared eight common phylotypes showing
at least 91% similarity to isolates and uncultured
bacteria previously reported in hydrocarbon-contaminated
environments (Palleroni et al., 2004). We assume that the
representatives affiliated with the Sphingomonadaceae
(Novosphingobium acidiphilum), Acetobacteraceae (Acidocel-
la sp.), Rhodocyclaceae, Alcanivoracaceae, Sinobacteraceae
(Hydrocarboniphaga effusa), Spirochaetaceae and Geobacter-
aceae (Geobacter sp.) families are phylogenetic signatures of
the biodegrading community. A characteristic of the N
mesocosms was 16S rRNA gene sequences showing the
presence of Acidovorax species-related patterns in the soil
microbial community, sequences with similarity to the
gammaproteobacterium Pseudoxanthomonas spadix isolated
from oil-contaminated soil and to the gram-positive Ther-
moanaerobacteriaceae clone found in tar oil-impacted aqui-
fers where BTEX degradation depends mainly on sulfate
reduction (Winderl et al., 2008). Bacteria characteristic of
the soil community in the P mesocosms were phylogeneti-
cally affiliated to (1) Brevundimonas sp. 39, which was
isolated from contaminated river sediment, (2) aerobic
benzene-degrading Hydrogenophaga sp. Rs71, (3) Citrobac-
ter farmeri from offshore oil fields and (4) alicyclic hydro-
carbon-degrading Bacteroides sp. ECP-C1, which was
isolated in sulfate-reducing conditions from a gas conden-
sate-contaminated aquifer (Rios-Hernandez et al., 2003).
Direct evidence that TPH removal is a primary effect of
bioremediation processes occurring in the soil can be
obtained by specifically targeting genes responsible for the
functionality of the autochthonous microbial community.
The majority of culture-independent surveys of catabolic
gene diversity in contaminated environments have used
conserved nucleotide sequences to design primers to detect
the presence, abundance and diversity of catabolic genes that
encode a defined group of enzymes thought to be critical in
the target environment. Our previous study improved upon
this by adopting a metagenomic approach in combination
with functional selection and PCR screening with newly
designed primers to identify key catabolic gene groups in
soil (Brennerova et al., 2009). Detailed enumeration of the
degradation genes characteristic to the locality revealed that
simulated pollutant rebound failed to impair the overall
degradative capacity of the soil microbiota in either the N or
the P mesocosm. Moreover, an increase in gene copy per ng
FEMS Microbiol Ecol 78 (2011) 137–149c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
146 O. Korotkevych et al.
Maria
Underline
Maria
Underline
Maria
Underline
Maria
Underline

soil DNA was demonstrated for all EXDO and related
RHDO genes in the microbial community at the contami-
nated site (Fig. 4). It may be that during cultivation of the P
mesocosms, the evolution of taxonomically distinct popula-
tions was concomitant with the remedial response to the
hydrocarbon contamination, resulting in an increase of the
catabolic potential, understood as an increase of gene copies
per genome equivalence at the microbial community. In
addition, the abundance of EXDO-D2 and EXDO-Dbt
genes in the P soil was over two and three times higher than
that in the natural attenuation system. The latter two groups
of EXDO genes encode enzymes that have an exceptionally
high affinity for various catecholic substrates (Brennerova
et al., 2009) and could promote the environmental fitness of
microbial community members. Thus, the relative increases
in EXDO-D2 and EXDO-Dbt gene copy numbers in the P
community would confer the bacterial hosts with enhanced
biodegradative capabilities.
According to contemporary ecological theory, the stabi-
lity of soil-based biotic components is of fundamental
importance to the functionality of ecosystems (Wardle &
Giller, 1996). The ability of ecosystems to resist changes in
environmental conditions is positively correlated with spe-
cies diversity and functional redundancy (Gitay & Wilson,
1996; Griffiths et al., 2000). It was found that the relative
abundances of genes with similar functions are more highly
conserved than are the relative abundances of phylotypes
(Turnbaugh et al., 2009). Turnbaugh and colleagues sug-
gested that the microbial communities exist at the level of
shared genes and different species assemblages converge on
shared core functions provided by distinctive components.
The microbial community of the site under study includes
members of at least 14 bacterial orders (Kabelitz et al., 2009).
Under the selective pressure of a chronic and massive
pollutant spill, clusters of ecologically equivalent specialized
microbial species play overlapping roles in the maintenance
and regulation of the ecosystem. The rapid increase in the
degradation potential of both mesocosm systems in response
to pollutant rebound can be explained by the functional
redundancy of biodegrading microbial communities. Thus,
the increased concentration of organic matter served as a
primary environmental cue that triggered the expansion of
microbial density and catabolic gene abundance. The more
intense microbial shifts that took place in the mesocosms
containing plants could be attributable to the presence of the
rhizosphere as an additional factor supporting the activity of
the biodegrading microbial community.
The current work demonstrates that the taxonomical
affiliations of the microbial communities are of little
descriptive power in terms of catabolic degradation per-
formance in complex in situ conditions. Our results support,
in a culture-independent manner and directly in environ-
mental samples, the observations made in well-studied
cultured bacterial pollutant degraders, where there is no
clear association between the extradiol dioxygenases gene
phylogeny and the taxonomy of the host (Vilchez-Vargas
et al., 2010), indicating once more the importance of
detecting catabolic genes in studies defining the influence
of pollutants in the environment.
Acknowledgements
We wish to thank V. Reimannova for excellent technical
assistance; Jirina Machackova, Stanislava Proksova and J. Jurak
from AECOM CZ for providing soil samples, analytical data
and helpful discussions; and M. Havelkova for graphic work.
We would like to thank Howard Junca and Miroslav Patek for
their suggestions and advice. We also thank the anonymous
reviewers for their constructive comments and criticism. This
work was supported by the Czech Ministry of Education
research programs 2B06156 and 1M06011 and by the Institu-
tional Research Concept # AVOZ50200510.
Statement
GenBank accession numbers: GU560728–GU560730,
GU568253–GU568336.
Authors’contribution
O.K. and J.J. contributed equally to this work.
References
ABI (2006) Creating Standard Curves with Genomic DNA or
Plasmid DNA Templates for Use in Quantitative PCR. Applied
Biosystems, Carlsbad, California.
Acinas SG, Marcelino LA, Klepac-Ceraj V & Polz MF (2004)
Divergence and redundancy of 16S rRNA sequences in
genomes with multiple rrn operons. J Bacteriol 186:
2629–2635.
Brennerova MV, Josefiova J, Brenner V, Pieper DH & Junca H
(2009) Metagenomics reveals diversity and abundance of
meta-cleavage pathways in microbial communities from soil
highly contaminated with jet fuel under air-sparging
bioremediation. Environ Microbiol 11: 2216–2227.
Chablain PA, Zgoda AL, Sarde CO & Truffaut N (2001) Genetic
and molecular organization of the alkylbenzene catabolism
operon in the psychrotrophic strain Pseudomonas putida
01G3. Appl Environ Microb 67: 453–458.
Cole JR, Wang Q, Cardenas E et al. (2009) The Ribosomal
Database Project: improved alignments and new tools for
rRNA analysis. Nucleic Acids Res 37: D141–D145.
Das N & Chandran P (2011) Microbial degradation of petroleum
hydrocarbon contaminants: an overview. Biotechnol Res Int
2011, doi:10.4061/2011/941810.
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
147Functional redundancy in biodegrading microbial communities
Maria
Underline

Di Gregorio S, Zocca C, Sidler S, Toffanin A, Lizzari D & Vallini G
(2004) Identification of two new sets of genes for
dibenzothiophene transformation in Burkholderia sp. DBT1.
Biodegradation 15: 111–123.
Eltis LD & Bolin JT (1996) Evolutionary relationships among
extradiol dioxygenases. J Bacteriol 178: 5930–5937.
Gitay H & Wilson JBB (1996) Species redundancy: a redundant
concept? J Ecol 84: 121–124.
Glick BR (2010) Using soil bacteria to facilitate
phytoremediation. Biotechnol Adv 28: 367–374.
Griffiths BS, Ritz K, Bardgett RD et al. (2000) Ecosystem response
of pasture soil communities to fumigation-induced microbial
diversity reductions: an examination of the biodiversity–
ecosystem function relationship. Oikos 90: 279–294.
Harayama S, Mermod N, Rekik M, Lehrbach PR & Timmis KN
(1987) Roles of the divergent branches of the meta-cleavage
pathway in the degradation of benzoate and substituted
benzoates. J Bacteriol 169: 558–564.
Harwood CS & Parales RE (1996) The beta-ketoadipate pathway
and the biology of self-identity. Annu Rev Microbiol 50:
553–590.
Huber T, Faulkner G & Hugenholtz P (2004) Bellerophon: a
program to detect chimeric sequences in multiple sequence
alignments. Bioinformatics 20: 2317–2319.
Jimenez JI, Minambres B, Garcıa JL & Dıaz E (2002) Genomic
analysis of the aromatic catabolic pathways from Pseudomonas
putida KT2440. Environ Microbiol 4: 824–841.
Kabelitz N, Machackova J, Imfeld G, Brennerova M, Pieper DH,
Heipieper HJ & Junca H (2009) Enhancement of the microbial
community biomass and diversity during air sparging
bioremediation of a Northern Bohemia soil highly
contaminated with kerosene and BTEX. Appl Microbiol Biot
82: 565–577.
Kukor JJ & Olsen RH (1991) Genetic organization and regulation
of a meta cleavage pathway for catechols produced from
catabolism of toluene, benzene, phenol, and cresols by
Pseudomonas pickettii PKO1. J Bacteriol 173: 4587–4594.
Lee ZM-P, Bussema C III & Schmidt TM (2009) rrnDB:
documenting the number of rRNA and tRNA genes in bacteria
and archaea. Nucleic Acids Res 37: D489–D493.
Machackova J, Wittlingerova Z, Vlk K, Zima J & Linka A (2008)
Comparison of two methods for assessment of in situ jet-fuel
remediation efficiency. Water Air Soil Poll 187: 181–194.
McLeod MP & Eltis LD (2008) Genomic insights into the aerobic
pathways for degradation of organic pollutants. Microbial
Biodegradation. ISBN: 978-1-904455-17-2 (Dıaz E, ed), pp.
1–23. Caister Academic Press, Norfolk, UK.
Muyzer G, de Waal EC & Uitterlinden AG (1993) Profiling of
complex microbial populations by denaturing gradient gel
electrophoresis analysis of polymerase chain reaction-
amplified genes coding for 16S rRNA. Appl Environ Microb 59:
695–700.
Myers RM, Maniatis T, Lerman LS & Ray W (1987) Detection and
localization of single base changes by denaturing gradient gel
electrophoresis. Method Enzymol 155: 501–527.
Nicholas KB, Nicholas HB Jr & Deerfield DW II (1997) GeneDoc:
analysis and visualization of genetic variation.
EMBNET.NEWS 4: 1–4.
Okoh AI (2006) Biodegradation alternative in the cleanup of
petroleum hydrocarbon pollutants. Biotechnol Mol Biol Rev 1:
38–50.
Ovreas L, Forney L, Daae FL & Torsvik V (1997) Distribution of
bacterioplankton in meromictic Lake Saelenvannet, as
determined by denaturing gradient gel electrophoresis of PCR-
amplified gene fragments coding for 16S rRNA. Appl Environ
Microb 63: 3367–3373.
Palleroni NJ, Port AM, Chang H-K & Zylstra GJ (2004)
Hydrocarboniphaga effusa gen. nov., sp. nov., a novel member
of the gamma-proteobacteria active in alkane and aromatic
hydrocarbon degradation. Int J Syst Evol Micr 54: 1203–1207.
Parales RE & Harwood CS (2002) Bacterial chemotaxis to
pollutants and plant-derived aromatic molecules. Curr Opin
Microbiol 5: 266–273.
Pflugmacher U, Averhoff B & Gottschalk G (1996) Cloning,
sequencing, and expression of isopropylbenzene degradation
genes from Pseudomonas sp. strain JR1: identification of
isopropylbenzene dioxygenase that mediates trichloroethene
oxidation. Appl Environ Microb 62: 3967–3977.
Pieper DH (2005) Aerobic degradation of polychlorinated
biphenyls. Appl Microbiol Biot 67: 170–191.
Riesenfeld CS, Goodman RM & Handelsman J (2004)
Uncultured soil bacteria are a reservoir of new antibiotic
resistance genes. Environ Microbiol 6: 981–989.
Rios-Hernandez LA, Gieg LM & Suflita JM (2003)
Biodegradation of an alicyclic hydrocarbon by a sulfate-
reducing enrichment from a gas condensate-contaminated
aquifer. Appl Environ Microb 69: 434–443.
Saitou N & Nei M (1987) The neighbor-joining method: a new
method for reconstructing phylogenetic trees. Mol Biol Evol 4:
406–425.
Shirdam R, Zand AD, Bidhendi GN & Mehrdadi N (2008)
Phytoremediation of hydrocarbon-contaminated soils with
emphasis on the effect of petroleum hydrocarbons on the
growth of plant species. Phytoprotection 89: 21–29.
Sivaganesan M, Haugland RA, Chern EC & Shanks OC (2010)
Improved strategies and optimization of calibration models
for real-time PCR absolute quantification. Water Res 44:
4726–4735.
Suenaga H, Ohnuki T & Miyazaki K (2007) Functional screening
of a metagenomic library for genes involved in microbial
degradation of aromatic compounds. Environ Microbiol 9:
2289–2297.
Switzer C & Kosson DS (2007) Soil vapor extraction performance
in layered vadose zone materials. Vadose Zone J 6: 397–405.
Tamura K, Dudley J, Nei M & Kumar S (2007) MEGA4:
Molecular Evolutionary Genetics Analysis (MEGA) software
version 4.0. Mol Biol Evol 24: 1596–1599.
Tamura T & Hatano K (2001) Phylogenetic analysis of the genus
Actinoplanes and transfer of Actinoplanes minutisporangius
Ruan et al. 1986 and ‘Actinoplanes aurantiacus’ to
FEMS Microbiol Ecol 78 (2011) 137–149c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
148 O. Korotkevych et al.

Cryptosporangium minutisporangium comb. nov. and
Cryptosporangium aurantiacum sp. nov. Int J Syst Evol Micr 51:
2119–2125.
Tang JC, Wang RG, Niu XW, Wang M, Chu HR & Zhou QX
(2010) Characterisation of the rhizoremediation of
petroleum-contaminated soil: effect of different influencing
factors. Biogeosciences 7: 3961–3969.
Thompson C & Nathanail P (2003) Chemical Analysis of
Contaminated Land. ISBN: 1-84127-334-1. Blackwell
Publishing, Oxford, UK, 305 pp..
Trapp S, Kohler A, Larsen LC, Zambrano KC & Karlson U (2001)
Phytotoxicity of fresh and weathered diesel and gasoline to
willow and poplar trees. J Soils Sediments 1: 71–76.
Turnbaugh PJ, Hamady M, Yatsunenko T et al. (2009) A core gut
microbiome in obese and lean twins. Nature 457: 480–484.
Van Doorn R, Klerks MM, van Gent-Pelzer MPE, Speksnijder
AGCL, Kowalchuk GA & Schoen CD (2009) Accurate
quantification of microorganisms in PCR-inhibiting
environmental DNA extracts by a novel internal amplification
control approach using Biotrove OpenArrays. Appl Environ
Microb 75: 7253–7260.
Vilchez-Vargas R, Junca H & Pieper DH (2010) Metabolic
networks, microbial ecology and ‘omics’ technologies: towards
understanding in situ biodegradation processes. Environ
Microbiol 12: 3089–3104.
Wardle DA & Giller KE (1996) The quest for a contemporary
ecological dimension to soil biology. Soil Biol Biochem 28:
1549–1554.
Winderl C, Anneser B, Griebler C, Meckenstock RU & Lueders T
(2008) Depth-resolved quantification of anaerobic toluene
degraders and aquifer microbial community patterns in
distinct redox zones of a tar oil contaminant plume. Appl
Environ Microb 74: 792–801.
Witzig R, Junca H, Hecht H-J & Pieper DH (2006) Assessment of
toluene/biphenyl dioxygenase gene diversity in benzene-
polluted soils: links between benzene biodegradation and
genes similar to those encoding isopropylbenzene
dioxygenases. Appl Environ Microb 72: 3504–3514.
Yeates C, Holmes AJ & Gillings MR (2000) Novel forms of ring-
hydroxylating dioxygenases are widespread in pristine and
contaminated soils. Environ Microbiol 2: 644–653.
Zalesny RS, Bauer EO, Hall RB, Zalesny JA, Kunzman J, Rog CJ &
Riemenschneider DE (2005) Clonal variation in survival and
growth of hybrid poplar and willow in an in situ trial on soils
heavily contaminated with petroleum hydrocarbons. Int J
Phytoremediat 7: 177–197.
Supporting Information
Additional Supporting Information may be found in the
online version of this article:
Fig. S1. GC chromatograms of LNAPL and kerosene.
Fig. S2. Changes in the concentrations of cultivable bacteria
and TPH in bulk soil from mesocosms N (natural biode-
gradation) and P (phytoremediation model).
Fig. S3. Negative image of DGGE gel used for band excision
and 16S rRNA gene sequencing analysis.
Fig. S4. The fosmid clones used to design the primers
EXDO-D2-F (a) and EXDO-D2-R (b).
Fig. S5. Agarose-gel electrophoresis for verification the
specificity of qPCR products after using the primers target-
ing (a) RHDO, (b) EXDO-K2, (c) EXDO-D2 and (d)
EXDO-Dbt genes (Table 1).
Please note: Wiley-Blackwell is not responsible for the
content or functionality of any supporting materials sup-
plied by the authors. Any queries (other than missing
material) should be directed to the corresponding author
for the article.
FEMS Microbiol Ecol 78 (2011) 137–149 c� 2011 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. All rights reserved
149Functional redundancy in biodegrading microbial communities
Related Documents