Notch ligand Delta-like 4 blockade attenuates atherosclerosis and metabolic disorders Daiju Fukuda a , Elena Aikawa a , Filip K. Swirski b , Tatiana I. Novobrantseva c , Victor Kotelianski c , Cem Z. Gorgun d , Aleksey Chudnovskiy b , Hiroyuki Yamazaki a , Kevin Croce a , Ralph Weissleder b , Jon C. Aster e , Gökhan S. Hotamisligil d , Hideo Yagita f , and Masanori Aikawa a,1 Departments of a Medicine and e Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; b Center for Systems Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; c Department of Research, Alnylam Pharmaceuticals, Cambridge, MA 02142; d Department of Genetics and Complex Diseases, Harvard School of Public Health, Boston, MA 02115; and f Department of Immunology, Juntendo University School of Medicine, Tokyo 113, Japan Edited by Monty Krieger, Massachusetts Institute of Technology, Cambridge, MA, and approved May 11, 2012 (received for review October 18, 2011) Atherosclerosis and insulin resistance are major components of the cardiometabolic syndrome, a global health threat associated with a systemic inflammatory state. Notch signaling regulates tissue de- velopment and participates in innate and adaptive immunity in adults. The role of Notch signaling in cardiometabolic inflamma- tion, however, remains obscure. We noted that a high-fat, high- cholesterol diet increased expression of the Notch ligand Delta-like 4 (Dll4) in atheromata and fat tissue in LDL-receptor–deficient mice. Blockade of Dll4-Notch signaling using neutralizing anti-Dll4 anti- body attenuated the development of atherosclerosis, diminished plaque calcification, improved insulin resistance, and decreased fat accumulation. These changes were accompanied by decreased macrophage accumulation, diminished expression of monocyte chemoattractant protein-1 (MCP-1), and lower levels of nuclear fac- tor-κB (NF-κB) activation. In vitro cell culture experiments revealed that Dll4-mediated Notch signaling increases MCP-1 expression via NF-κB, providing a possible mechanism for in vivo effects. Further- more, Dll4 skewed macrophages toward a proinflammatory phe- notype (“M1”). These results suggest that Dll4-Notch signaling plays a central role in the shared mechanism for the pathogenesis of cardiometabolic disorders. biotherapy | cardiovascular diseases | collagen | diabetes mellitus | obesity A therosclerosis and insulin resistance are cardinal features of the so-called cardiometabolic syndrome, a global health threat (1). Although chronic inflammation strongly associates with major components of this syndrome (2), the underlying proinflammatory mechanisms are incompletely understood. The Notch pathway regulates embryonic development (3) and con- tributes to physiological homeostasis and pathological processes in adult tissues (4–7). Notch receptors (Notch1–4) undergo proteolytic cleavage when bound by Delta-like (Dll1, Dll3, Dll4) or Serrate (Jagged1, Jagged2) ligands expressed on adjacent cells, allowing nuclear translocation of the Notch intracellular domain. Notch pathway components are expressed in a cell- type–specific fashion and have diverse context-dependent func- tions. Among previous studies on diverse roles of Notch signaling in physiology and pathology, recent reports have suggested that Notch signaling has metabolic functions and that Notch in- hibition is beneficial in the treatment for insulin resistance (8, 9). Dll4, originally found as an endothelial cell-specific ligand, par- ticipates in angiogenesis and may be a therapeutic target for solid tumors (10–12). However, the role of Dll4 in cardiometabolic inflammation remains unknown. We showed previously that Dll4-triggered Notch signaling promotes inflammatory responses in macrophages in vitro (13). Macrophages play pivotal roles in the development of chronic inflammatory diseases. We therefore tested the hypothesis in vivo that Dll4 contributes to the path- ogenesis of the cardiometabolic syndrome. In this study, we blocked Dll4-mediated Notch signaling using a previously described neutralizing anti-mouse Dll4 antibody (Ab) (14–20) in LDL-receptor–deficient (Ldlr -/- ) mice fed a high-fat, high-cholesterol diet, a model that produces atherosclerosis and metabolic disturbances resembling those seen in the car- diometabolic syndrome (21). Generation of anti-mouse Dll4 Ab was described in previous studies (14, 17). Previous studies thor- oughly characterized the same Ab (14–20). The selective binding of the Ab (HMD4-2) to mouse Dll4 was verified by flow cytometry, using mouse Dll4-expressing cells (14). The Ab blocks Notch1-Fc and Notch4-Fc binding to mouse Dll4-expressing cells in a dose- dependent manner and does not bind to cells expressing other mouse Notch ligands, such as Dll1, Jagged1, or Jagged2 (14, 17). Other reports demonstrated that the Ab blocked Dll4-dependent Notch signaling in vivo, using cancer transplant models (17, 20). Use of anti-Dll4 Ab enabled us to circumvent the embryonic le- thality of Dll4 deficiency (22), and to provide clinically translatable evidence for the proinflammatory role of Dll4. Multiple cellular and molecular pathways associate with the pathogenesis of car- diometabolic disorders. Although our approach did not aim to examine the relative contribution of metabolic cell types, in vivo and in vitro experiments in this study addressed the function of Dll4 in each cell type, proposing the shared mechanisms by which Dll4-mediated Notch signaling promotes cardiometabolic dis- eases. This study demonstrates unique in vivo evidence that Dll4- mediated Notch signaling contributes to the pathogenesis of car- diometabolic disorders and provides proof of concept that the Notch pathway is a new target for much-needed therapies against the cardiometabolic syndrome. Results Characterization of Dll4 Expression and the Effects of Neutralizing Anti-Dll4 Ab. Atherosclerotic lesions and epididymal adipose tis- sue of 32-wk-old Ldlr -/- mice fed a high-fat, high-cholesterol diet contained immunoreactive Dll4, as did human white adipose tissue (Fig. 1A) and atheromata, as described previously (13). Dll4 protein and RNA levels increased in the aorta and adipose tissue, compared with levels in wild-type mice (Fig. 1 B and C). Furthermore, high-fat feeding promoted Dll4 expression in these organs in a time-dependent manner (Fig. 1 D and E). These results suggest that an inflammatory environment triggered by overnutrition promotes Dll4 expression in vivo. Bolus injection of Dll4 Ab (250 μg) into Ldlr -/- mice significantly reduced the expression of the prototypical Notch target gene Hes1 in peri- toneal macrophages and of Hes1 and Hey1 in fat (SI Appendix, Fig. S1A), indicating that the Notch pathway is activated by Dll4 in important cardiometabolic organs. In addition, Dll4 Ab Author contributions: D.F., F.K.S., and M.A. designed research; D.F., E.A., F.K.S., T.I.N., V.K., C.Z.G., A.C., H. Yamazaki, K.C., G.S.H., and M.A. performed research; R.W. and H. Yagita contributed new reagents/analytic tools; D.F., E.A., F.K.S., T.I.N., V.K., C.Z.G., A.C., K.C., J.C.A., G.S.H., and M.A. analyzed data; and D.F., E.A., K.C., J.C.A., G.S.H., and M.A. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 To whom correspondence should be addressed. E-mail: [email protected]. See Author Summary on page 10763 (volume 109, number 27). This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1116889109/-/DCSupplemental. E1868–E1877 | PNAS | Published online June 13, 2012 www.pnas.org/cgi/doi/10.1073/pnas.1116889109

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Notch ligand Delta-like 4 blockade attenuatesatherosclerosis and metabolic disordersDaiju Fukudaa, Elena Aikawaa, Filip K. Swirskib, Tatiana I. Novobrantsevac, Victor Kotelianskic, Cem Z. Gorgund,Aleksey Chudnovskiyb, Hiroyuki Yamazakia, Kevin Crocea, Ralph Weisslederb, Jon C. Astere, Gökhan S. Hotamisligild,Hideo Yagitaf, and Masanori Aikawaa,1
Departments of aMedicine and ePathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; bCenter for Systems Biology,Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; cDepartment of Research, Alnylam Pharmaceuticals, Cambridge, MA 02142;dDepartment of Genetics and Complex Diseases, Harvard School of Public Health, Boston, MA 02115; and fDepartment of Immunology, Juntendo UniversitySchool of Medicine, Tokyo 113, Japan
Edited by Monty Krieger, Massachusetts Institute of Technology, Cambridge, MA, and approved May 11, 2012 (received for review October 18, 2011)
Atherosclerosis and insulin resistance are major components of the
cardiometabolic syndrome, a global health threat associated with
a systemic inflammatory state. Notch signaling regulates tissue de-
velopment and participates in innate and adaptive immunity in
adults. The role of Notch signaling in cardiometabolic inflamma-
tion, however, remains obscure. We noted that a high-fat, high-
cholesterol diet increased expression of the Notch ligand Delta-like
4 (Dll4) in atheromata and fat tissue in LDL-receptor–deficient mice.
Blockade of Dll4-Notch signaling using neutralizing anti-Dll4 anti-
body attenuated the development of atherosclerosis, diminished
plaque calcification, improved insulin resistance, and decreased
fat accumulation. These changes were accompanied by decreased
macrophage accumulation, diminished expression of monocyte
chemoattractant protein-1 (MCP-1), and lower levels of nuclear fac-
tor-κB (NF-κB) activation. In vitro cell culture experiments revealed
that Dll4-mediated Notch signaling increases MCP-1 expression via
NF-κB, providing a possible mechanism for in vivo effects. Further-
more, Dll4 skewed macrophages toward a proinflammatory phe-
notype (“M1”). These results suggest that Dll4-Notch signaling
plays a central role in the shared mechanism for the pathogenesis
of cardiometabolic disorders.
biotherapy | cardiovascular diseases | collagen | diabetes mellitus | obesity
Atherosclerosis and insulin resistance are cardinal features ofthe so-called cardiometabolic syndrome, a global health
threat (1). Although chronic inflammation strongly associateswith major components of this syndrome (2), the underlyingproinflammatory mechanisms are incompletely understood. TheNotch pathway regulates embryonic development (3) and con-tributes to physiological homeostasis and pathological processesin adult tissues (4–7). Notch receptors (Notch1–4) undergoproteolytic cleavage when bound by Delta-like (Dll1, Dll3, Dll4)or Serrate (Jagged1, Jagged2) ligands expressed on adjacentcells, allowing nuclear translocation of the Notch intracellulardomain. Notch pathway components are expressed in a cell-type–specific fashion and have diverse context-dependent func-tions. Among previous studies on diverse roles of Notch signalingin physiology and pathology, recent reports have suggested thatNotch signaling has metabolic functions and that Notch in-hibition is beneficial in the treatment for insulin resistance (8, 9).Dll4, originally found as an endothelial cell-specific ligand, par-ticipates in angiogenesis and may be a therapeutic target for solidtumors (10–12). However, the role of Dll4 in cardiometabolicinflammation remains unknown. We showed previously thatDll4-triggered Notch signaling promotes inflammatory responsesin macrophages in vitro (13). Macrophages play pivotal roles inthe development of chronic inflammatory diseases. We thereforetested the hypothesis in vivo that Dll4 contributes to the path-ogenesis of the cardiometabolic syndrome.In this study, we blocked Dll4-mediated Notch signaling using
a previously described neutralizing anti-mouse Dll4 antibody (Ab)(14–20) in LDL-receptor–deficient (Ldlr−/−) mice fed a high-fat,high-cholesterol diet, a model that produces atherosclerosis
and metabolic disturbances resembling those seen in the car-diometabolic syndrome (21). Generation of anti-mouse Dll4 Abwas described in previous studies (14, 17). Previous studies thor-oughly characterized the same Ab (14–20). The selective bindingof theAb (HMD4-2) tomouseDll4 was verified by flow cytometry,using mouse Dll4-expressing cells (14). The Ab blocks Notch1-Fcand Notch4-Fc binding to mouse Dll4-expressing cells in a dose-dependent manner and does not bind to cells expressing othermouse Notch ligands, such as Dll1, Jagged1, or Jagged2 (14, 17).Other reports demonstrated that the Ab blocked Dll4-dependentNotch signaling in vivo, using cancer transplant models (17, 20).Use of anti-Dll4 Ab enabled us to circumvent the embryonic le-thality of Dll4 deficiency (22), and to provide clinically translatableevidence for the proinflammatory role of Dll4. Multiple cellularand molecular pathways associate with the pathogenesis of car-diometabolic disorders. Although our approach did not aim toexamine the relative contribution of metabolic cell types, in vivoand in vitro experiments in this study addressed the function ofDll4 in each cell type, proposing the shared mechanisms by whichDll4-mediated Notch signaling promotes cardiometabolic dis-eases. This study demonstrates unique in vivo evidence that Dll4-mediated Notch signaling contributes to the pathogenesis of car-diometabolic disorders and provides proof of concept that theNotch pathway is a new target for much-needed therapies againstthe cardiometabolic syndrome.
Results
Characterization of Dll4 Expression and the Effects of Neutralizing
Anti-Dll4 Ab. Atherosclerotic lesions and epididymal adipose tis-sue of 32-wk-old Ldlr−/−mice fed a high-fat, high-cholesterol dietcontained immunoreactive Dll4, as did human white adiposetissue (Fig. 1A) and atheromata, as described previously (13).Dll4 protein and RNA levels increased in the aorta and adiposetissue, compared with levels in wild-type mice (Fig. 1 B and C).Furthermore, high-fat feeding promoted Dll4 expression in theseorgans in a time-dependent manner (Fig. 1 D and E). Theseresults suggest that an inflammatory environment triggered byovernutrition promotes Dll4 expression in vivo. Bolus injectionof Dll4 Ab (250 μg) into Ldlr−/− mice significantly reduced theexpression of the prototypical Notch target gene Hes1 in peri-toneal macrophages and of Hes1 and Hey1 in fat (SI Appendix,Fig. S1A), indicating that the Notch pathway is activated byDll4 in important cardiometabolic organs. In addition, Dll4 Ab
Author contributions: D.F., F.K.S., and M.A. designed research; D.F., E.A., F.K.S., T.I.N., V.K.,
C.Z.G., A.C., H. Yamazaki, K.C., G.S.H., and M.A. performed research; R.W. and H. Yagita
contributed new reagents/analytic tools; D.F., E.A., F.K.S., T.I.N., V.K., C.Z.G., A.C., K.C., J.C.A.,
G.S.H., and M.A. analyzed data; and D.F., E.A., K.C., J.C.A., G.S.H., and M.A. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
1To whom correspondence should be addressed. E-mail: [email protected].
See Author Summary on page 10763 (volume 109, number 27).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.
1073/pnas.1116889109/-/DCSupplemental.
E1868–E1877 | PNAS | Published online June 13, 2012 www.pnas.org/cgi/doi/10.1073/pnas.1116889109

inhibited Notch signaling in Notch reporter transgenic mice (23)(SI Appendix, Fig. S1B). As mentioned above, previous studiesverified the specificity of the same Dll4 blocking Ab (14–20).To investigate the role of Dll4-Notch signaling in the initiation
and progression of cardiometabolic disorders, we intraperi-toneally administered Dll4 Ab or isotype control IgG (250 μg,twice a week) for 12 wk to fat-fed Ldlr−/− mice beginning at 8 wkof age (early-phase treatment) or at 20 wk of age (late-phasetreatment), respectively (SI Appendix, Fig. S1C). Unlike pan-Notch inhibitors (e.g., γ-secretase inhibitors), which cause severeand potentially fatal gut toxicity and thymus atrophy (24), ourDll4 Ab did not exert any obvious adverse effects and was welltolerated (SI Appendix, Fig. S1 D and E).
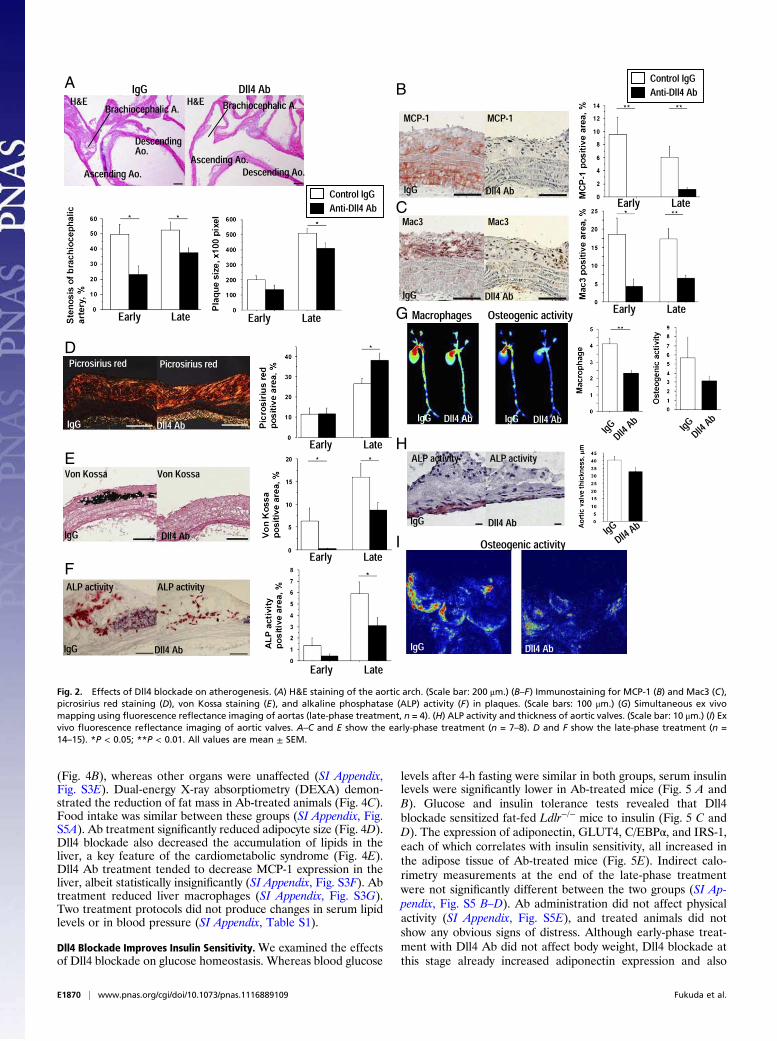
Dll4 Blockade Reduces Atherogenesis. Dll4 blockade lessened theseverity of atherosclerotic lesions in two vascular beds whereatherosclerotic changes often develop in hyperlipidemic mice—the aorta (Fig. 2) and brachiocephalic artery (SI Appendix, Fig.S2). Both early-phase and late-phase Dll4 Ab-treated miceshowed milder stenosis of brachiocephalic arteries, whereas late-phase treatment reduced the size of atherosclerotic lesions in theaortic arch (Fig. 2A). Dll4 Ab treatment markedly decreased
monocyte chemoattractant protein-1 (MCP-1) expression (Fig. 2Band SI Appendix, Fig. S2A) and macrophage accumulation (Fig.2C and SI Appendix, Fig. S2B) in atherosclerotic lesions in theaorta and brachiocephalic artery. MCP-1 immunoreactivity wasalso lower in the tunica media of the Ab-treated mice, indicatingthat Dll4 blockade may suppress MCP-1 expression in smoothmuscle cells.We and others have established the role of macrophage-de-
rived proteinases, including matrix metalloproteinases (MMP),in collagen degradation—which can lead to atheromatous plaque“instability” and acute vascular events (25, 26). Dll4 Ab-treatedmice demonstrated increased fibrillar collagen in atheroscleroticplaques (Fig. 2D and SI Appendix, Fig. S3C). Several studies,including ours, have shown that macrophages promote arterialcalcification (27–29), and other evidence indirectly suggests thatNotch signaling also regulates calcification (30). Ab treatmentsignificantly decreased advanced calcification (von Kossa) andosteogenic activity (alkaline phosphatase activity, ALP) in ath-erosclerotic lesions (Fig. 2 E and F and SI Appendix, Fig. S3 Dand E). Molecular imaging using fluorescence reflectance dem-onstrated that macrophage-targeted signal correlated positivelywith osteogenic activity, and that both of these features decreasedwith Dll4 Ab treatment in parallel (Fig. 2G). A recent study byFeig et al. reported that changes in total plaque area do notfollow the monotonic decline in macrophages and that the po-tential underlying mechanisms may involve increased plaquecollagen, which was observed in our Ab-treated animals (31).Our present results are consistent with this report. Furthermore,clinical evidence suggests that inflammation, rather than athe-romata size, participates critically in plaque instability and acutecomplications, and that antiinflammatory therapies such as lipidlowering attenuate macrophage burden and collagen loss andimprove clinical outcomes, even though they may not sub-stantially shrink lesions (26). We therefore believe that ourresults provide clinically relevant information on the effects ofDll4 Ab administration on an inflammatory plaque phenotype.Calcific aortic valve disease, an inflammatory disorder, causes
aortic stenosis and heart failure and is a major burden in clinicalpractice (32). Dll4 blockade trended toward a decrease in thethickness of aortic valve leaflets (40.0 ± 2.7 vs. 32.5 ± 2.7 μm, P=0.06) and in osteogenic activity detected by ALP activity andmolecular imaging (Fig. 2 H and I). Whereas Dll4 blockadesuppressed ectopic cardiovascular calcification, it did not de-crease bone mineral density (SI Appendix, Fig. S3A).Consistent with reduced calcification, Dll4 Ab treatment tended
to decrease aortic expression of osteogenic regulators and bonemorphogenetic proteins (BMPs) (Fig. 3 A and B). Dll4 Ab treat-ment also reduced the expression of MMP-9 and MMP-13,enzymes responsible for collagen degradation in plaques (33) (Fig.3C). Furthermore, the expression of proinflammatory mediators,including IL-1β, iNOS, and IL-6, was generally lower in the aortasof Ab-treated mice than in those of control mice (Fig. 3D). Dll4Ab also decreased aortic expression of the Notch target geneHey2, consistent with effective blockade of Notch signaling in thistissue (SI Appendix, Fig. S4A). In addition, Dll4 Ab decreased theexpression of BMP-2 andMMP-9 in peritoneal macrophages (Fig.3 E and F). To evaluate further whether blockade of Dll4 signalingin macrophages mediated effects onMMP expression in the aorta,in vitro experiments used the murine macrophage-like cell lineRAW264.7. siRNA against Dll4 reduced MMP-9 expression,whereas overexpression of Dll4 or exogenous immobilizedrecombinant Dll4 (34) tended to increaseMMP-9 expression (P=0.07 and 0.09, respectively) (SI Appendix, Fig. S3 B–D).
Dll4 Blockade Retards Excessive Fat Accumulation. Body weight gainin Ldlr−/− mice was similar in Dll4 Ab and IgG groups duringearly-phase treatment (8–20 wk of age), which was equivalent tothat of the nontreatment period in the late-phase treatmentprotocol (from 8 to 20 wk of age). Late-phase Dll4 Ab treatment(from 20 to 32 wk of age), however, retarded excessive bodyweight gain (Fig. 4A). Dll4 Ab reduced fat and liver weight
A
B D
C
E
Fig. 1. Characterization of Dll4 expression and effects of Dll4 blockade. (A)
Immunostaining of Dll4 in atheroma (Upper Left) and epididymal fat (Upper
Center) obtained from 32-wk-old Ldlr−/−mice on a high-fat and high-cholesterol
(HC) diet and in human white adipose tissue (Upper Right). (Lower) Staining
with nonimmune IgG (NC). (Scale bar: 100 μm.) (B andC) ImmunoblottingofDll4
in aortas (B) andwhite adipose tissue (C). (D and E) Effect ofHC diet onDll4 RNA
expression in aortas (D) and adipose tissue (E) of Ldlr−/−mice.Micewere fedaHC
diet from 8wk of age for 12 or 24 wk. HC diet promotes Dll4 expression. Longer
consumption of HC diet enhances the effect. NC, normal chow. A and B, n = 4.
D and E, n = 4. *P < 0.05; **P < 0.01. All values are mean ± SEM.
Fukuda et al. PNAS | Published online June 13, 2012 | E1869
MEDICALSCIENCES
PNASPLU
S
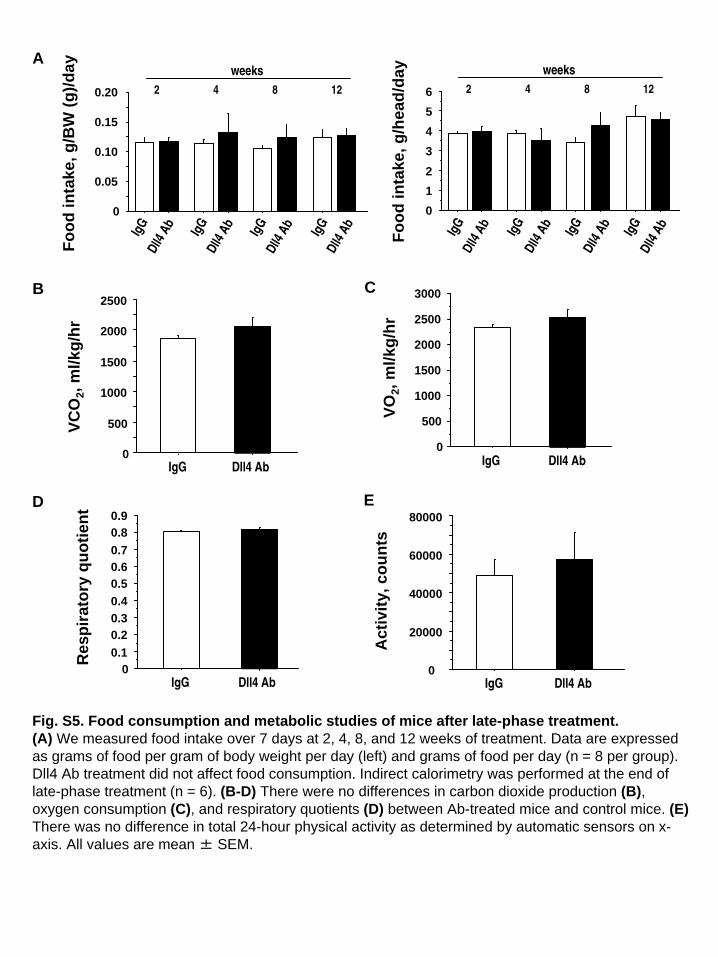
(Fig. 4B), whereas other organs were unaffected (SI Appendix,Fig. S3E). Dual-energy X-ray absorptiometry (DEXA) demon-strated the reduction of fat mass in Ab-treated animals (Fig. 4C).Food intake was similar between these groups (SI Appendix, Fig.S5A). Ab treatment significantly reduced adipocyte size (Fig. 4D).Dll4 blockade also decreased the accumulation of lipids in theliver, a key feature of the cardiometabolic syndrome (Fig. 4E).Dll4 Ab treatment tended to decrease MCP-1 expression in theliver, albeit statistically insignificantly (SI Appendix, Fig. S3F). Abtreatment reduced liver macrophages (SI Appendix, Fig. S3G).Two treatment protocols did not produce changes in serum lipidlevels or in blood pressure (SI Appendix, Table S1).
Dll4 Blockade Improves Insulin Sensitivity.We examined the effectsof Dll4 blockade on glucose homeostasis. Whereas blood glucose
levels after 4-h fasting were similar in both groups, serum insulinlevels were significantly lower in Ab-treated mice (Fig. 5 A andB). Glucose and insulin tolerance tests revealed that Dll4blockade sensitized fat-fed Ldlr−/− mice to insulin (Fig. 5 C andD). The expression of adiponectin, GLUT4, C/EBPα, and IRS-1,each of which correlates with insulin sensitivity, all increased inthe adipose tissue of Ab-treated mice (Fig. 5E). Indirect calo-rimetry measurements at the end of the late-phase treatmentwere not significantly different between the two groups (SI Ap-pendix, Fig. S5 B–D). Ab administration did not affect physicalactivity (SI Appendix, Fig. S5E), and treated animals did notshow any obvious signs of distress. Although early-phase treat-ment with Dll4 Ab did not affect body weight, Dll4 blockade atthis stage already increased adiponectin expression and also
AB
C
G
H
I
D
E
F
Fig. 2. Effects of Dll4 blockade on atherogenesis. (A) H&E staining of the aortic arch. (Scale bar: 200 μm.) (B–F) Immunostaining for MCP-1 (B) and Mac3 (C),
picrosirius red staining (D), von Kossa staining (E), and alkaline phosphatase (ALP) activity (F) in plaques. (Scale bars: 100 μm.) (G) Simultaneous ex vivo
mapping using fluorescence reflectance imaging of aortas (late-phase treatment, n = 4). (H) ALP activity and thickness of aortic valves. (Scale bar: 10 μm.) (I) Ex
vivo fluorescence reflectance imaging of aortic valves. A–C and E show the early-phase treatment (n = 7–8). D and F show the late-phase treatment (n =
14–15). *P < 0.05; **P < 0.01. All values are mean ± SEM.
E1870 | www.pnas.org/cgi/doi/10.1073/pnas.1116889109 Fukuda et al.

tended to increase GLUT4, C/EBPα, and IRS-1 expression inadipose tissues (SI Appendix, Fig. S6 A and B).
Dll4 Blockade Decreases Macrophage Accumulation in Fat. Insulinresistance associates with increased macrophage accumulation inadipose tissue (2). Late-phase Dll4 Ab administration decreasedthe numbers of macrophages found in adipose tissue (Fig. 5 Fand G). Although early-phase Dll4 blockade did not reduce ex-cessive weight gain or fat mass, it tended to decrease fat mac-rophage accumulation (SI Appendix, Fig. S6C). Adipose tissue inAb-treated animals also expressed lower levels of proin-flammatory molecules, including MCP-1 (Fig. 5H).To bolster in vivo evidence for the role of Dll4 in macrophage
accumulation, we also studied leptin-deficient (Lepob/Lepob)mice, another commonly used model of metabolic disorders.Dll4 Ab treatment reduced MCP-1 expression and macrophageaccumulation in fat tissues (SI Appendix, Fig. S7 A–C). AlthoughAb treatment did not affect total body weight gain, fat weightwas significantly lower in the Ab-treated group (SI Appendix, Fig.S7 D and E), and, as in Ldlr−/− mice, Dll4 blockade reducedserum insulin levels (SI Appendix, Fig. S7F).
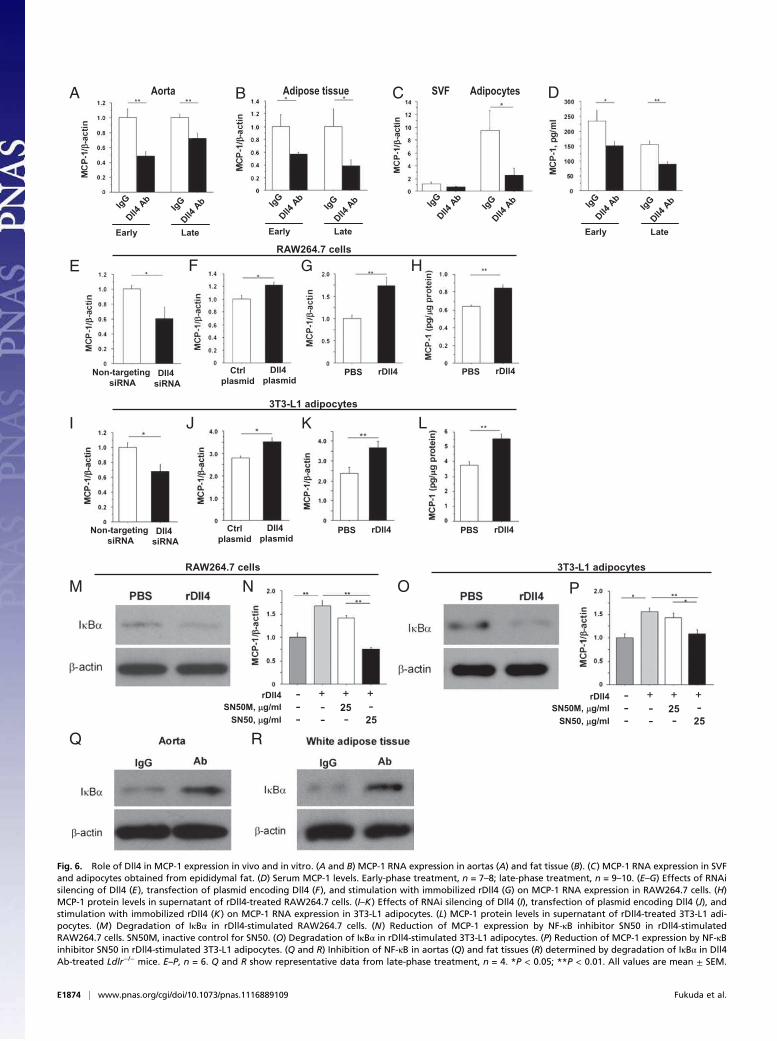
Dll4 Blockade Reduces MCP-1 Expression. Early-phase and late-phase Dll4 Ab administration decreased expression of MCP-1 inatheromata (Figs. 2B and 6A and SI Appendix, Fig S2A) and in
adipose tissue (Figs. 5H and 6B). Furthermore, Ab treatmentreduced MCP-1 expression in adipocytes and the stromal vas-cular fraction (SVF) isolated from epididymal fat (adipocytes,P = 0.04; SVF, P = 0.06) (Fig. 6C). Dll4 blockade decreasedexpression of the prototypical Notch target gene Hey1 in adi-pocytes (SI Appendix, Fig. S4B). MCP-1 levels in the peripheralblood of Ab-treated mice were also lower than in control animalsfrom both treatment protocols (Fig. 6D).We then asked whether Dll4-Notch signaling regulates MCP-1
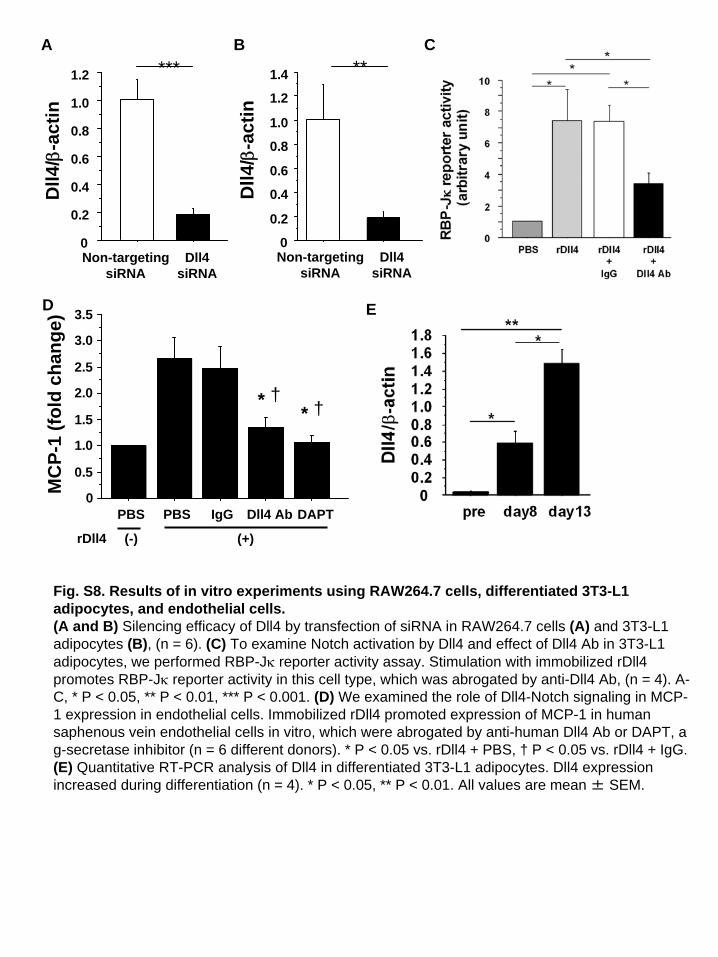
expression in RAW264.7 cells and differentiated 3T3-L1 adi-pocytes. RNAi silencing of Dll4 reduced MCP-1 RNA expres-sion in both cell types (Fig. 6 E and I and SI Appendix, Fig. S8 Aand B). In contrast, Dll4 overexpression enhanced MCP-1 ex-pression (Fig. 6 F and J). We also cultured these cells on dishescoated with mouse rDll4 (immobilized rDll4) (34), which in-creased MCP-1 expression at the RNA and protein levels (Fig. 6G, H, K, and L). We previously reported that Dll4 ligationactivates Notch signaling in human macrophages (13). Here, weshow that rDll4 triggers activation of Notch signaling in differ-entiated 3T3-L1 adipocytes as gauged via Notch reporter geneactivity, which was suppressed by Dll4 Ab (SI Appendix, Fig.S8C). This result also revealed that our anti-Dll4 Ab inhibits Dll4binding. Furthermore, rDll4 promoted expression of MCP-1 inhuman saphenous vein endothelial cells, an effect that was ab-
A
C D
E F
B
Fig. 3. Role of Dll4 in the expres-
sion of osteogenic regulators, MMPs,
and inflammatory molecules. (A–C)
Quantitative RT-PCR analysis of os-
teogenic regulators (A), BMPs (B),
and MMPs (C) in aortas. (D) Quanti-
tative RT-PCR analyses of the ex-
pression of inflammatory molecules
in aortas. (E and F) Quantitative RT-
PCR analyses of BMPs (E) and MMPs
(F) in peritoneal macrophages. A–F
show early-phase treatment (n = 7–
8). *P < 0.05; **P < 0.01. All values
are mean ± SEM.
Fukuda et al. PNAS | Published online June 13, 2012 | E1871
MEDICALSCIENCES
PNASPLU
S

rogated by anti-human Dll4 Ab or DAPT, a γ-secretase inhibitor(SI Appendix, Fig. S8D).Nuclear factor-κB (NF-κB) activates MCP-1 transcription (35,
36). Treatment of RAW264.7 cells and 3T3L-1 adipocytes withrDll4 decreased the levels of the NF-κB inhibitor IκBα, in-dicative of NF-κB activation (Fig. 6 M and O). SN50, a cell-permeable peptide inhibitor of NF-κB, abrogated Dll4-triggeredMCP-1 induction in both cell types (Fig. 6 N and P). Theseexperiments suggest that stimulation of the NF-κB pathway andMCP-1 expression in macrophages and adipocytes involve Dll4-mediated signaling, in part. Furthermore, our in vivo experi-ments revealed that Dll4 Ab treatment increased IκBα levels inthe aorta and epididymal fat, consistent with inhibition of NF-κB(Fig. 6 Q and R).
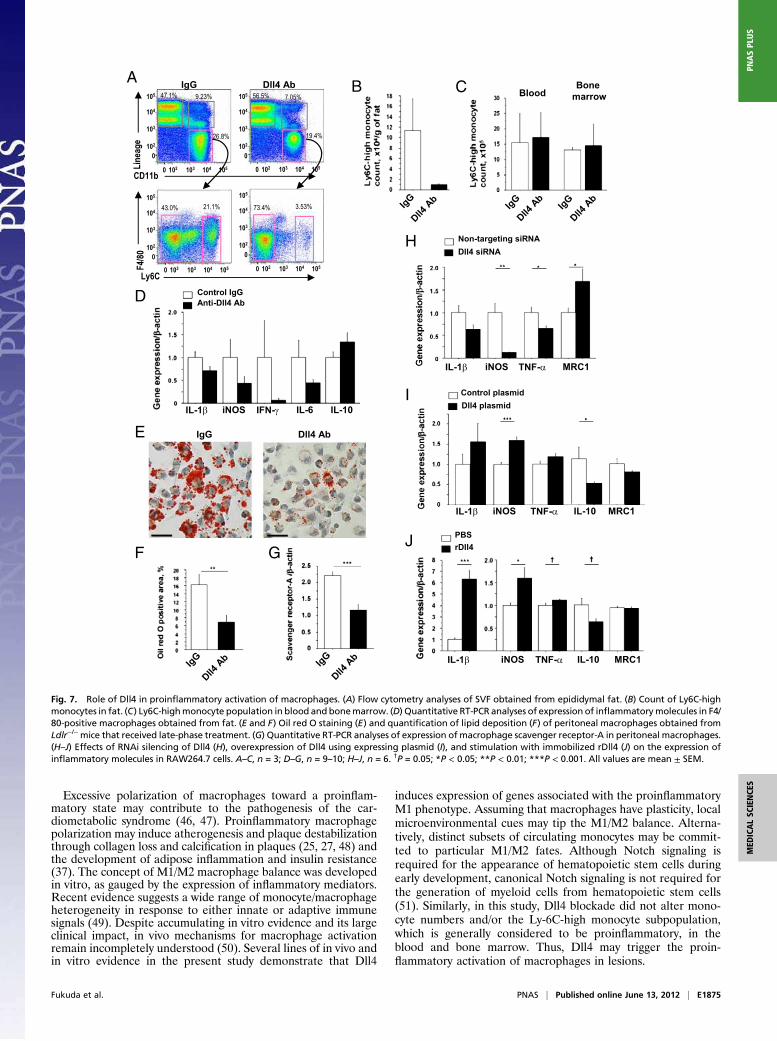
Dll4 Blockade Attenuates the Proinflammatory Phenotype of Macro-
phages. The concept is emerging that monocytes/macrophagescan be polarized to favor inflammation (Ly6C-high monocytesor “M1” macrophages) or to suppress inflammation (LyC6-lowmonocytes or “M2” macrophages). In addition to increasing innumber, macrophages that accumulate in tissues in the car-diometabolic syndrome are phenotypically skewed by unknownfactors toward the proinflammatory M1 phenotype (37–40). Toexplore whether Dll4 blockade affects macrophage polarization,we studied the effects of Dll4 blockade on monocytes/macro-
phages by flow cytometry, using SVF obtained from epididymalfat. Ab administration tended to decrease the Ly6C-high pop-ulation (Fig. 7 A and B), but had no effect on this population inthe blood and bone marrow (Fig. 7C). Flow cytometry analysesalso showed no significant differences in the percentage of pe-ripheral blood monocytes in Ldlr−/− mice treated with Dll4 Ab orIgG (IgG group vs. Ab group: 29.3 ± 11.6% vs. 27.4 ± 6.8%),suggesting that Dll4 blockade does not deplete circulatingmonocytes. Furthermore, F4/80-positive macrophages collectedfrom SVF of Ab-treated animals tended to express lower levels ofproinflammatory mediators, including iNOS, and slightly higherlevels of antiinflammatory mediators such as IL-10 (Fig. 7D).Macrophages take up lipids, become foam cells in vasculature,
and secrete various inflammatory cytokines, accelerating thedevelopment of atherosclerosis (41, 42). In this study, Dll4 Abtreatment markedly decreased lipid accumulation in peritonealmacrophages, as determined by Oil red O staining (Fig. 7 E andF). Furthermore, expression of scavenger receptor-A RNA waslower in peritoneal macrophages obtained from Ab-treated micethan in those from control mice (Fig. 7G).We used RAW264.7 cells to study further the mechanisms
underlying the observed proinflammatory role of Dll4. RNAi si-lencing with Dll4 siRNA decreased expression of typical proin-flammatory M1 mediators (e.g., iNOS and TNF-α) and increasedexpression of mannose receptor 1 (MRC1), an M2 macrophage
Dll4 AbIgG
Fat Lean
Control IgG
Anti-Dll4 Ab
8-20 8-20 20-32Treatment (+) (-) (+)
IgG
Dll4Ab
IgG
Dll4Ab
IgG
Dll4Ab
Epididymal Sub-
cutaneous
LiverBA
D
E
C
Weeks
Fat
IgG
Dll4Ab
IgG
Dll4Ab
IgG
Dll4Ab
Dll4 AbIgG
IgG
Dll4Ab
Fig. 4. Effects of Dll4 blockade on fat
accumulation. (A) Differences in body
weight gain during the study periods
(early-phase treatment, n = 7–8; late-
phase treatment, n = 19–20). (B)
Weight of fat and liver (late-phase
treatment, n = 19–20). (C) Results of
dual-energy X-ray absorptiometry
(DEXA), n = 5–6. (D) H&E staining and
quantification of adipocyte size in ep-
ididymal fat (n = 7). (Scale bar: 200 μm.)
(E) Oil red O staining of the liver and
quantification of lipid deposition (late-
phase treatment, n = 9–10). (Insets)
H&E staining. (Scale bar: 100 μm.) *P <
0.05; **P < 0.01; ***P < 0.001. All val-
ues are mean ± SEM.
E1872 | www.pnas.org/cgi/doi/10.1073/pnas.1116889109 Fukuda et al.

marker (Fig. 7H). In contrast, enforced Dll4 expression increasediNOS expression and suppressed IL-10 (Fig. 7I). Furthermore,stimulation with immobilized rDll4 increased proinflammatoryIL-1β and iNOS and decreased IL-10 (Fig. 7J). Collectively, theseresults suggest that Dll4-mediated Notch signaling shifts macro-phages toward a proinflammatory phenotype.
Discussion
Accumulating evidence supports the premise that chronic in-flammation is central to the pathobiology of atherosclerotic vas-cular disease and metabolic disorders (2, 25). Regulation of thecirculatory, metabolic, and immune systems is highly integrated.Dissecting the multiple intertwined mechanisms for cardiomet-abolic disorders and developing new therapies for their commonpathway thus require well-defined, relevant models (25, 43). Thepresent study demonstrates that inhibition of Dll4, one of theNotch ligands, reduces vascular and adipose inflammation, pos-sibly through the effects on NF-κB/MCP-1–mediated responses—implicating Dll4 as an important instigator of the cardiometabolicsyndrome. Our in vivo and in vitro results indicate the expressionof Dll4 in multiple cell types and its function related to the de-velopment of the cardiometabolic syndrome. Whereas geneticmanipulation—such as cell-type–specific loss-of-function and/orgain-of-function mouse strains—may provide insight into the
relative contribution of each cell type to Dll4-mediated cardio-metabolic inflammation, systemic administration of well-charac-terized anti-Dll4 Ab provides clinically translatable proof ofconcept for the role of Dll4 in the shared mechanisms for car-diometabolic syndrome and facilitates the development of newbiotherapies for this disease. Our study demonstrates that Dll4blockade using our Ab improves the cardiometabolic syndromeand represents clinically relevant evidence that Dll4 can be atherapeutic target.This study suggests that Dll4 induces MCP-1 expression in
arteries and adipose tissues. MCP-1 plays a key role in monocyte/macrophage recruitment and in macrophage-dependent inflam-matory responses that lead to the development of atherosclerosisand insulin resistance (44, 45). Our in vitro experiments dem-onstrate that Dll4 can induce MCP-1 expression in several celltypes responsible for the development of cardiometabolic dis-orders. In the present study, Dll4 blockade reduced MCP-1levels in the aorta, fat tissues, and peripheral blood. Enhancedexpression of MCP-1 by Dll4 in atheromata and adipose tissuescan promote accumulation of macrophages, another majorsource of MCP-1. Dll4-Notch signaling may thus amplify mac-rophage accumulation through the induction of MCP-1 expres-sion, an effect that Dll4 inhibition appears to reverse.
A D
FE
B
G
bA4llDGgI
3caM3caM
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4 Ab
IgG
Dll4 Ab
GTT ITT
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
H
C
Fig. 5. Effects of Dll4 blockade on insulin sensitivity and macrophage accumulation in fat tissue. (A and B) Blood glucose levels (A) and serum insulin levels (B)
after 4-h fasting (n = 9). (C and D) Glucose tolerance test (GTT) after 16-h fasting and insulin tolerance test (ITT) after 4-h fasting (n = 7). (E) Quantitative RT-
PCR analyses of genes related to insulin sensitivity in epididymal fat (n = 9–10). (F) Mac3 staining and population of Mac3-positive cells in epididymal fat (n =
7). (Scale bar: 100 μm.) (G) Quantitative RT-PCR analysis of F4/80 in epididymal fat (n = 9–10). (H) Quantitative RT-PCR analyses of expression of chemokines
and cytokines in epididymal fat (n = 9–10). All data are from late-phase treatment. *P < 0.05; **P < 0.01; ***P < 0.001. All values are mean ± SEM.
Fukuda et al. PNAS | Published online June 13, 2012 | E1873
MEDICALSCIENCES
PNASPLU
S
A DCB
HE F
I LJ
3T3-L1 adipocytes
RAW264.7 cells
NM
RAW264.7 cells
PO
3T3-L1 adipocytes
RQ
IgG
Dll4
Ab
IgG
Dll4
Ab
Early Late
IgG
Dll4
Ab
IgG
Dll4
Ab
Early Late
IgG
Dll4
Ab
IgG
Dll4
Ab
SVF Adipocytes
IgG
Dll4
Ab
IgG
Dll4
Ab
Early Late
Non-targeting
siRNADll4
siRNA
Non-targeting
siRNADll4
siRNA
Ctrl
plasmid
Dll4
plasmid
Ctrl
plasmid
Dll4
plasmid
PBS rDll4 PBS rDll4
PBS rDll4 PBS rDll4
rDll4
SN50M, g/ml
SN50, g/ml
-
-
-
+
-
-
-
25
25
-
+ + rDll4
SN50M, g/ml
SN50, g/ml
-
-
-
+
-
-
-
25
25
-
+ +
Adipose tissueAorta
G
K
Fig. 6. Role of Dll4 in MCP-1 expression in vivo and in vitro. (A and B) MCP-1 RNA expression in aortas (A) and fat tissue (B). (C) MCP-1 RNA expression in SVF
and adipocytes obtained from epididymal fat. (D) Serum MCP-1 levels. Early-phase treatment, n = 7–8; late-phase treatment, n = 9–10. (E–G) Effects of RNAi
silencing of Dll4 (E), transfection of plasmid encoding Dll4 (F), and stimulation with immobilized rDll4 (G) on MCP-1 RNA expression in RAW264.7 cells. (H)
MCP-1 protein levels in supernatant of rDll4-treated RAW264.7 cells. (I–K) Effects of RNAi silencing of Dll4 (I), transfection of plasmid encoding Dll4 (J), and
stimulation with immobilized rDll4 (K) on MCP-1 RNA expression in 3T3-L1 adipocytes. (L) MCP-1 protein levels in supernatant of rDll4-treated 3T3-L1 adi-
pocytes. (M) Degradation of IκBα in rDll4-stimulated RAW264.7 cells. (N) Reduction of MCP-1 expression by NF-κB inhibitor SN50 in rDll4-stimulated
RAW264.7 cells. SN50M, inactive control for SN50. (O) Degradation of IκBα in rDll4-stimulated 3T3-L1 adipocytes. (P) Reduction of MCP-1 expression by NF-κB
inhibitor SN50 in rDll4-stimulated 3T3-L1 adipocytes. (Q and R) Inhibition of NF-κB in aortas (Q) and fat tissues (R) determined by degradation of IκBα in Dll4
Ab-treated Ldlr−/− mice. E–P, n = 6. Q and R show representative data from late-phase treatment, n = 4. *P < 0.05; **P < 0.01. All values are mean ± SEM.
E1874 | www.pnas.org/cgi/doi/10.1073/pnas.1116889109 Fukuda et al.
Excessive polarization of macrophages toward a proinflam-matory state may contribute to the pathogenesis of the car-diometabolic syndrome (46, 47). Proinflammatory macrophagepolarization may induce atherogenesis and plaque destabilizationthrough collagen loss and calcification in plaques (25, 27, 48) andthe development of adipose inflammation and insulin resistance(37). The concept of M1/M2 macrophage balance was developedin vitro, as gauged by the expression of inflammatory mediators.Recent evidence suggests a wide range of monocyte/macrophageheterogeneity in response to either innate or adaptive immunesignals (49). Despite accumulating in vitro evidence and its largeclinical impact, in vivo mechanisms for macrophage activationremain incompletely understood (50). Several lines of in vivo andin vitro evidence in the present study demonstrate that Dll4
induces expression of genes associated with the proinflammatoryM1 phenotype. Assuming that macrophages have plasticity, localmicroenvironmental cues may tip the M1/M2 balance. Alterna-tively, distinct subsets of circulating monocytes may be commit-ted to particular M1/M2 fates. Although Notch signaling isrequired for the appearance of hematopoietic stem cells duringearly development, canonical Notch signaling is not required forthe generation of myeloid cells from hematopoietic stem cells(51). Similarly, in this study, Dll4 blockade did not alter mono-cyte numbers and/or the Ly-6C-high monocyte subpopulation,which is generally considered to be proinflammatory, in theblood and bone marrow. Thus, Dll4 may trigger the proin-flammatory activation of macrophages in lesions.
A
IgG
Dll4
Ab
IgG
Dll4
Ab iNOS IL-10 MRC1IL-1β TNF-α
TNF-α MRC1IL-10iNOSIL-1β
TNF-α MRC1iNOSIL-1β
iNOS IL-6IL-1β IFN-γ IL-10
Dll4 AbIgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
BloodBone
marrowB C
E
D
H
I
JF G
Anti-Dll4 Ab
Control IgG
PBS
rDll4
Control plasmid
Dll4 plasmid
Non-targeting siRNA
Dll4 siRNA
0
0
102
102
104
103
105
103 104 105
0
0
102
102
104
103
105
103 104 105
0
0
102
102
104
103
105
103 104 105
0
0
102
102
104
103
105
103 104 105
Dll4 AbIgG
CD11b
Ly6C
F4/
80L
inea
ge
47.1%
26.8%
9.23%
43.0% 21.1% 73.4% 3.53%
56.5% 7.05%
19.4%
Fig. 7. Role of Dll4 in proinflammatory activation of macrophages. (A) Flow cytometry analyses of SVF obtained from epididymal fat. (B) Count of Ly6C-high
monocytes in fat. (C) Ly6C-highmonocyte population in blood and bonemarrow. (D) Quantitative RT-PCR analyses of expression of inflammatorymolecules in F4/
80-positive macrophages obtained from fat. (E and F) Oil red O staining (E) and quantification of lipid deposition (F) of peritoneal macrophages obtained from
Ldlr−/−mice that received late-phase treatment. (G) Quantitative RT-PCR analyses of expression of macrophage scavenger receptor-A in peritoneal macrophages.
(H–J) Effects of RNAi silencing of Dll4 (H), overexpression of Dll4 using expressing plasmid (I), and stimulation with immobilized rDll4 (J) on the expression of
inflammatory molecules in RAW264.7 cells. A–C, n = 3; D–G, n = 9–10; H–J, n = 6. †P = 0.05; *P < 0.05; **P < 0.01; ***P < 0.001. All values are mean ± SEM.
Fukuda et al. PNAS | Published online June 13, 2012 | E1875
MEDICALSCIENCES
PNASPLU
S

Our results suggest that Dll4-Notch signaling augments NF-κBactivation. How Notch signaling leads to NF-κB activation ispoorly understood (13, 52–55). We and others have suggestedthe intertwined crosstalk between NF-κB and Notch pathways.Our previous study reported that proinflammatory stimuli (e.g.,IL-1β, minimally modified LDL, and LPS) induce Dll4 expres-sion in cultured human macrophages in a manner dependent onthe Toll-like receptor/IL-1 receptor superfamily and NF-κB andthat Dll4 promotes NF-κB activation, leading to the expressionof NF-κB–regulated genes, such as iNOS and Dll4 itself (13).The present study indicates in vivo and in vitro that Dll4-Notchmediates MCP-1 induction, in part via NF-κB activation, whichleads to increases of many proinflammatory genes, includingMCP-1, iNOS, and MMP-9, all of which were induced by Dll4 inthis study. Interaction of Notch intracellular domains and NF-κBpathway components is complex, appears to be context and cell-type dependent, and generally remains unclear. Because activa-tion of NF-κB represents the proinflammatory M1 environmentand contributes to atherosclerosis, insulin resistance, and obesity(38, 56, 57), its interplay with Dll4-Notch signaling deservesfurther investigation. Furthermore, our results suggest that theDll4-Notch axis promotes sustained macrophage activation viaa positive feedback loop—a vicious cycle that represents mech-anisms for uncontrolled macrophage activation typical of chronicinflammatory diseases. Overall, our results are consistent withthe intriguing possibility that the Dll4-Notch axis serves as a keyregulator of macrophage activation in vivo.Here we report that Dll4 blockade lessens the accumulation of
macrophages, decreases fat mass, and abrogates insulin resistancein two models of metabolic disorders. Our in vivo and in vitroexperiments fall short of providing definitive causal relationshipsbetween these three complex parameters. Reduced adipose ac-cumulation in these animals might help improve insulin resistanceand reduce macrophage accumulation, but data from the early-phase and late-phase treatments suggest that Dll4 blockademodulates the expression of proinflammatory factors in fat beforefat weight reduction. Furthermore, several studies reported thatmacrophage accumulation in fat precedes the development of in-sulin resistance inmice (58–60). Our finding that Dll4Ab treatmentreduced expression of M1 gene products and improved insulin re-sistance agrees with the recent evidence that accumulation ofproinflammatory macrophages leads to the development of insulinresistance (61, 62). MCP-1 also directly affects adipocyte functionand insulin sensitivity (44, 63) and participates in the developmentof fatty liver (64). We therefore speculate that reduction of MCP-1byDll4 blockade, observed in multiple cell types, also contributes tothe improvement of insulin sensitivity. Dll4 blockade disruptsangiogenesis and inhibits cancer growth by induction of hypoxia(10–12), and evidence links angiogenesis with adipose tissuedevelopment (65), fat accumulation, and insulin resistance (66).Therefore, the antiangiogenic effects of Dll4 blockade also mayhave contributed to the results reported here. Further studies areneeded to tease out the mechanisms underlying the effects of Dll4blockade on adipose tissue inflammation and insulin resistance.Whereas global Notch inhibition (e.g., γ-secretase inhibitors)
has acutely toxic effects (24), selective Dll4 inhibition produced nosigns of distress or toxicity in our mice. Furthermore, we did notobserve significant differences in body weight gain in mice thatreceived 12 wk of the early-phase administration of Dll4 Ab or IgGgroup (8–20 wk of age), whereas late-phase Dll4 blockade (20–32wk of age) retarded excessive fat accumulation. The effects duringthe late-phase treatment may account for increased Dll4 expres-sion in adipose tissue in fat-fed Ldlr−/− mice over time. Dll4 ex-pression in 3T3-L1 adipocytes also increased during differenti-ation in vitro (SI Appendix, Fig. S8E). Although a recent studysuggested that long-term (12 wk) Dll4 blockade can induce ad-verse effects in the liver in mice (67), our Dll4 Ab treatment for12 wk attenuated fatty liver. The same study also reported theformation of vascular neoplasm by Dll4 Ab administration (67). Inour study, two authors independently and thoroughly examinedorgans such as the aorta, adipose tissue, liver, and small intestine.
We did not find any signs of vascular neoplasm-like changes in ourAb-treatedmice. Differences in several factors (e.g., mouse strains,antibodies, and routes of administration) might have caused thesedifferences. Nevertheless, the feasibility of long-term Dll4 block-ade needs to be established by further evaluations.In summary, our study demonstrates that Notch signaling drives
proinflammatory programs of gene expression associated withcardiometabolic syndrome. Dll4 appears to function in homotypicand heterotypic crosstalk between pathways that control centralelements of inflammatory and metabolic responses in macro-phages and adipocytes, and thus constitutes a unique therapeutictarget in cardiometabolic disorders.
Materials and MethodsMice and Anti-Dll4 Ab Treatment. Ldlr−/− mice were fed a high-fat, high-
cholesterol diet (D12108; Research Diets) from 8 wk of age through the
completion of the study. Lepob/Lepob mice and Notch reporter transgenic
mice were fed normal chow. Animal care and experimentation were ap-
proved by the Harvard Medical School Institutional Animal Care and Use
Committee. Mice were treated with well-characterized hamster-derived
anti-mouse Dll4 antibody (14–20). Ldlr−/− mice were injected with 250 μg of
anti-mouse Dll4 antibody or isotype control IgG (BioXcell) intraperitoneally
twice a week from 8 wk of age (early phase) or from 20 wk of age (late
phase) for 12 wk. For Lepob/Lepob mice, the amount of Dll4 Ab and IgG was
adjusted according to body weight (10 μg/g) and administered for 10 wk.
Mice were weighed twice a week. For bolus injection, we administered
250 μg of Dll4 Ab or IgG to fat-fed 10-wk-old Ldlr−/− mice or Notch reporter
transgenic mice and harvested them at 6 h after injection.
Ex Vivo Fluorescence Reflectance Imaging of the Aorta and Aortic Valve. Ldlr−/−
mice that received late-phase treatment were used for fluorescence re-
flectance imaging. Two spectrally distinct near-infrared fluorescent agents
were administered to each mouse 24 h before imaging—cross-linked iron
oxide fluorescent iron nanoparticles for detection of macrophage accumu-
lation (CLIO750, 750 nm), and OsteoSense680 for detection of osteogenic
activity (680 nm; VisEn). The detailed method was described previously (68).
Analysis of Metabolic Parameters. Glucose and insulin tolerance tests were
performed after 16-h and 4-h fasting, respectively. Glucose and insulin sol-
utionswere injected into theperitoneal cavity at doses of 1.0 g/kg and0.5 unit/
kg, respectively. Indirect calorimetry andphysical activitymeasurements using
Ldlr−/−micewere performed at the end of 12wk of late-phase treatment. The
details of indirect experiments were described previously (69).
Flow Cytometry Analysis. To investigate effects of Dll4 blockade on mono-
cytes/macrophages and circulating leukocytes, we performed flow cytometry
analysis using SVF, bone marrow, and peripheral blood leukocytes. Anti-
bodies used in this study are described in SI Appendix.
Cell Culture Experiments. To activate Dll4-mediated Notch signaling, we
seeded RAW264.7 cells, differentiated 3T3-L1 adipocytes, and human sa-
phenous vein endothelial cells to plates coated with recombinant mouse or
human Dll4 (immobilized rDll4) (R&D Systems) (34). Day 10 3T3-L1 adipocytes
were used as differentiated 3T3-L1 adipocytes. We transfected siRNA against
mouse Dll4 (Dharmacon) and plasmid encoding mouse Dll4 (GeneCopoeia)
to RAW264.7 cells and 3T3-L1 adipocytes and RBP-Jκ reporter (SABiosciences)
to 3T3-L1 adipocytes by electroporation (Nucleofector system; Amaxa),
according to the manufacturer’s instructions.
Statistics. Data are expressed as mean ± SEM for continuous variables.
Comparisons between two groups were performed by unpaired Student’s
t test. Comparisons of multiple groups were made by one-way ANOVA, fol-
lowed by the Student–Newman–Keuls multiple-comparison test. P values
<0.05 were considered statistically significant.
For further details, please refer to SI Appendix.
ACKNOWLEDGMENTS. We thank Jose-Luiz Figueiredo, Eugenia Shvartz,Yevgenia Tesmenitsky, Jacob Aaron, and Mohammad Zafari for technicalassistance, and Sara Karwacki for her editorial expertise. This study wassupported in part by National Institutes of Health Grant R01HL107550 (toM.A.) and American Heart Association Grants 0655878T (to M.A.) and0835460N (to E.A.), Postdoctoral Fellowship Awards for Research Abroadfrom the Japanese Heart Foundation, the Japan Society for the Promotion ofScience, and the Uehara Memorial Foundation (D.F.).
E1876 | www.pnas.org/cgi/doi/10.1073/pnas.1116889109 Fukuda et al.

1. Eckel RH, Grundy SM, Zimmet PZ (2005) The metabolic syndrome. Lancet 365:
1415–1428.
2. Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867.
3. Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: Cell fate control and
signal integration in development. Science 284:770–776.
4. Aster JC, Pear WS, Blacklow SC (2008) Notch signaling in leukemia. Annu Rev Pathol 3:
587–613.
5. Radtke F, Fasnacht N, Macdonald HR (2010) Notch signaling in the immune system.
Immunity 32:14–27.
6. Hao L, et al. (2010) Notch-1 activates estrogen receptor-alpha-dependent
transcription via IKKalpha in breast cancer cells. Oncogene 29:201–213.
7. Ranganathan P, Weaver KL, Capobianco AJ (2011) Notch signalling in solid tumours: A
little bit of everything but not all the time. Nat Rev Cancer 11:338–351.
8. Pajvani UB, et al. (2011) Inhibition of Notch signaling ameliorates insulin resistance in
a FoxO1-dependent manner. Nat Med 17:961–967.
9. Rubio-Aliaga I, et al. (2009) Dll1 haploinsufficiency in adult mice leads to a complex
phenotype affecting metabolic and immunological processes. PLoS ONE 4:e6054.
10. Noguera-Troise I, et al. (2006) Blockade of Dll4 inhibits tumour growth by promoting
non-productive angiogenesis. Nature 444:1032–1037.
11. Ridgway J, et al. (2006) Inhibition of Dll4 signalling inhibits tumour growth by
deregulating angiogenesis. Nature 444:1083–1087.
12. Thurston G, Noguera-Troise I, Yancopoulos GD (2007) The Delta paradox: DLL4
blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer 7:
327–331.
13. Fung E, et al. (2007) Delta-like 4 induces notch signaling in macrophages: Implications
for inflammation. Circulation 115:2948–2956.
14. Moriyama Y, et al. (2008) Delta-like 1 is essential for the maintenance of marginal
zone B cells in normal mice but not in autoimmune mice. Int Immunol 20:763–773.
15. Fukushima A, et al. (2008) Notch ligand Delta-like4 inhibits the development of
murine experimental allergic conjunctivitis. Immunol Lett 121:140–147.
16. Sekine C, et al. (2009) Differential regulation of splenic CD8- dendritic cells and
marginal zone B cells by Notch ligands. Int Immunol 21:295–301.
17. Yamanda S, et al. (2009) Role of ephrinB2 in nonproductive angiogenesis induced by
Delta-like 4 blockade. Blood 113:3631–3639.
18. Kassner N, et al. (2010) Cutting edge: Plasmacytoid dendritic cells induce IL-10
production in T cells via the Delta-like-4/Notch axis. J Immunol 184:550–554.
19. Koyanagi A, Sekine C, Yagita H (2012) Expression of Notch receptors and ligands on
immature and mature T cells. Biochem Biophys Res Commun 418:799–805.
20. Oishi H, et al. (2010) Blockade of delta-like ligand 4 signaling inhibits both growth
and angiogenesis of pancreatic cancer. Pancreas 39:897–903.
21. Subramanian S, et al. (2008) Dietary cholesterol worsens adipose tissue macrophage
accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler
Thromb Vasc Biol 28:685–691.
22. Gale NW, et al. (2004) Haploinsufficiency of delta-like 4 ligand results in embryonic
lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci
USA 101:15949–15954.
23. Duncan AW, et al. (2005) Integration of Notch and Wnt signaling in hematopoietic
stem cell maintenance. Nat Immunol 6:314–322.
24. Fre S, et al. (2005) Notch signals control the fate of immature progenitor cells in the
intestine. Nature 435:964–968.
25. Aikawa M, Libby P (2004) The vulnerable atherosclerotic plaque: Pathogenesis and
therapeutic approach. Cardiovasc Pathol 13:125–138.
26. Libby P, Aikawa M (2002) Stabilization of atherosclerotic plaques: New mechanisms
and clinical targets. Nat Med 8:1257–1262.
27. Aikawa E, et al. (2009) Arterial and aortic valve calcification abolished by elastolytic
cathepsin S deficiency in chronic renal disease. Circulation 119:1785–1794.
28. Shao JS, Cheng SL, Sadhu J, Towler DA (2010) Inflammation and the osteogenic
regulation of vascular calcification: A review and perspective. Hypertension 55:
579–592.
29. New SE, Aikawa E (2011) Molecular imaging insights into early inflammatory stages
of arterial and aortic valve calcification. Circ Res 108:1381–1391.
30. Shimizu T, et al. (2009) Notch signaling induces osteogenic differentiation and
mineralization of vascular smooth muscle cells: Role of Msx2 gene induction via
Notch-RBP-Jk signaling. Arterioscler Thromb Vasc Biol 29:1104–1111.
31. Feig JE, et al. (2011) Reversal of hyperlipidemia with a genetic switch favorably affects
the content and inflammatory state of macrophages in atherosclerotic plaques.
Circulation 123:989–998.
32. Towler DA (2008) Oxidation, inflammation, and aortic valve calcification peroxide
paves an osteogenic path. J Am Coll Cardiol 52:851–854.
33. Deguchi JO, et al. (2005) Matrix metalloproteinase-13/collagenase-3 deletion
promotes collagen accumulation and organization in mouse atherosclerotic plaques.
Circulation 112:2708–2715.
34. Williams CK, Li JL, Murga M, Harris AL, Tosato G (2006) Up-regulation of the Notch
ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 107:
931–939.
35. Jiao P, et al. (2009) Obesity-related upregulation of monocyte chemotactic factors in
adipocytes: Involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase
pathways. Diabetes 58:104–115.
36. Shin WS, Szuba A, Rockson SG (2002) The role of chemokines in human cardiovascular
pathology: Enhanced biological insights. Atherosclerosis 160:91–102.
37. Chawla A (2010) Control of macrophage activation and function by PPARs. Circ Res
106:1559–1569.
38. Mantovani A, Garlanda C, Locati M (2009) Macrophage diversity and polarization in
atherosclerosis: A question of balance. Arterioscler Thromb Vasc Biol 29:1419–1423.
39. Gordon S (2007) Macrophage heterogeneity and tissue lipids. J Clin Invest 117:89–93.
40. Ley K, Miller YI, Hedrick CC (2011) Monocyte and macrophage dynamics during
atherogenesis. Arterioscler Thromb Vasc Biol 31:1506–1516.
41. Suzuki H, et al. (1997) A role for macrophage scavenger receptors in atherosclerosis
and susceptibility to infection. Nature 386:292–296.
42. Parthasarathy S, Steinberg D, Witztum JL (1992) The role of oxidized low-density
lipoproteins in the pathogenesis of atherosclerosis. Annu Rev Med 43:219–225.
43. Williams KJ, Feig JE, Fisher EA (2008) Rapid regression of atherosclerosis: Insights from
the clinical and experimental literature. Nat Clin Pract Cardiovasc Med 5:91–102.
44. Kanda H, et al. (2006) MCP-1 contributes to macrophage infiltration into adipose
tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116:1494–1505.
45. Liang CP, Han S, Senokuchi T, Tall AR (2007) The macrophage at the crossroads of
insulin resistance and atherosclerosis. Circ Res 100:1546–1555.
46. Swirski FK, et al. (2007) Ly-6Chi monocytes dominate hypercholesterolemia-associated
monocytosis and give rise to macrophages in atheromata. J Clin Invest 117:195–205.
47. Tacke F, et al. (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1
to accumulate within atherosclerotic plaques. J Clin Invest 117:185–194.
48. Shanahan CM (2007) Inflammation ushers in calcification: A cycle of damage and
protection? Circulation 116:2782–2785.
49. Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation.
Nat Rev Immunol 8:958–969.
50. Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of
metabolic disease. Cell 140:900–917.
51. Yuan JS, Kousis PC, Suliman S, Visan I, Guidos CJ (2010) Functions of notch signaling in
the immune system: Consensus and controversies. Annu Rev Immunol 28:343–365.
52. Osipo C, Golde TE, Osborne BA, Miele LA (2008) Off the beaten pathway: The
complex cross talk between Notch and NF-kappaB. Lab Invest 88:11–17.
53. Vacca A, et al. (2006) Notch3 and pre-TCR interaction unveils distinct NF-kappaB
pathways in T-cell development and leukemia. EMBO J 25:1000–1008.
54. Monsalve E, et al. (2009) Notch1 upregulates LPS-induced macrophage activation by
increasing NF-kappaB activity. Eur J Immunol 39:2556–2570.
55. Palaga T, et al. (2008) Notch signaling is activated by TLR stimulation and regulates
macrophage functions. Eur J Immunol 38:174–183.
56. Gao Z, et al. (2002) Serine phosphorylation of insulin receptor substrate 1 by inhibitor
kappa B kinase complex. J Biol Chem 277:48115–48121.
57. Hess K, Ushmorov A, Fiedler J, Brenner RE, Wirth T (2009) TNFalpha promotes
osteogenic differentiation of human mesenchymal stem cells by triggering the NF-
kappaB signaling pathway. Bone 45:367–376.
58. Suganami T, Ogawa Y (2010) Adipose tissue macrophages: Their role in adipose tissue
remodeling. J Leukoc Biol 88:33–39.
59. Weisberg SP, et al. (2003) Obesity is associated with macrophage accumulation in
adipose tissue. J Clin Invest 112:1796–1808.
60. Xu H, et al. (2003) Chronic inflammation in fat plays a crucial role in the development
of obesity-related insulin resistance. J Clin Invest 112:1821–1830.
61. Kang K, et al. (2008) Adipocyte-derived Th2 cytokines and myeloid PPARdelta
regulate macrophage polarization and insulin sensitivity. Cell Metab 7:485–495.
62. Odegaard JI, et al. (2007) Macrophage-specific PPARgamma controls alternative
activation and improves insulin resistance. Nature 447:1116–1120.
63. Sartipy P, Loskutoff DJ (2003) Monocyte chemoattractant protein 1 in obesity and
insulin resistance. Proc Natl Acad Sci USA 100:7265–7270.
64. Rull A, et al. (2009) Hepatic monocyte chemoattractant protein-1 is upregulated by
dietary cholesterol and contributes to liver steatosis. Cytokine 48:273–279.
65. Cao Y (2007) Angiogenesis modulates adipogenesis and obesity. J Clin Invest 117:
2362–2368.
66. Bråkenhielm E, et al. (2004) Angiogenesis inhibitor, TNP-470, prevents diet-induced
and genetic obesity in mice. Circ Res 94:1579–1588.
67. Yan M, et al. (2010) Chronic DLL4 blockade induces vascular neoplasms. Nature 463:
E6–E7.
68. Aikawa E, et al. (2007) Multimodality molecular imaging identifies proteolytic and
osteogenic activities in early aortic valve disease. Circulation 115:377–386.
69. Maeda K, et al. (2005) Adipocyte/macrophage fatty acid binding proteins control
integrated metabolic responses in obesity and diabetes. Cell Metab 1:107–119.
Fukuda et al. PNAS | Published online June 13, 2012 | E1877
MEDICALSCIENCES
PNASPLU
S

A B
C
D E
Fig. S1. Effects of anti-Dll4 Ab and study protocol.(A) Bolus injection of Dll4 Ab decreased expression of Notch target genes in peritoneal macrophages and adipose tissues (n =4, per group). (B) Bolus injection of Dll4 Ab reduced GFP signal in small intestines in Notch reporter transgenic mice determined by Western blotting (representative figures, n = 3). (C) Study protocols (in vivo). We administered 250 g of Dll4 Ab or isotype control hamster IgG by intraperitoneal injection twice a week to Ldlr-/- mice. We initiated Ab or IgG administration at 8 weeks (early phase) or 20 weeks (late phase) of age, to examine the effects on the initiation and progression of the cardiometabolic syndrome, respectively. All mice consumed a high-fat, high-cholesterol diet from 8 weeks of age to the harvest. (D) PAS staining of small intestine. Dll4 Ab treatment did not increase goblet cells (red). Scale bar, 100 m. (E) H&E staining of thymus. Dll4 Ab treatment did not cause atrophy of thymus. Scale bar, 200 m. D and E, Ldlr-/- mice received Dll4 Ab treatment for 12 weeks (n = 5). * P<0.05, ** P<0.01. Values are mean ± SEM.
A
B
C
D
E
Picrosirius red Picrosirius red
Von Kossa Von Kossa
ALP activity ALP activity
MCP-1 MCP-1
Mac3 Mac3
IgG Dll4 Ab
IgG Dll4 Ab
IgG Dll4 Ab
IgG Dll4 Ab
IgG Dll4 Ab
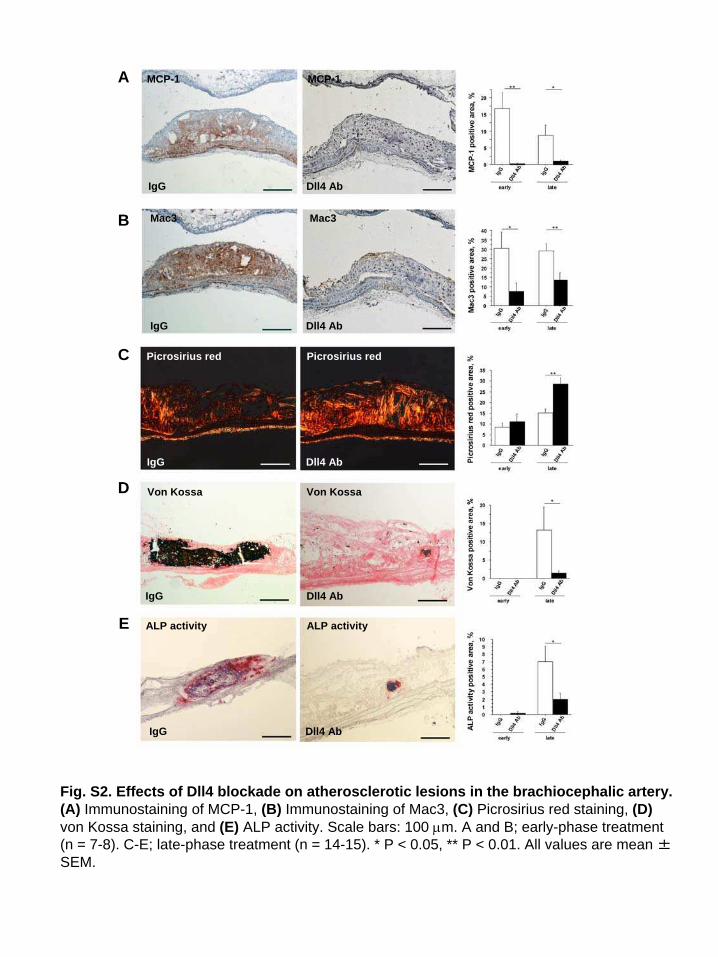
Fig. S2. Effects of Dll4 blockade on atherosclerotic lesions in the brachiocephalic artery.(A) Immunostaining of MCP-1, (B) Immunostaining of Mac3, (C) Picrosirius red staining, (D) von Kossa staining, and (E) ALP activity. Scale bars: 100 m. A and B; early-phase treatment (n = 7-8). C-E; late-phase treatment (n = 14-15). * P < 0.05, ** P < 0.01. All values are mean ±SEM.

A B C D
E F G
Fig. S3. Effects of Dll4 blockade on bone mineral density and liver, and the role of Dll4 in MMP9 expression in RAW264.7 cells.(A) Dll4 blockade by 12 weeks of Dll4 Ab administration did not decrease bone mineral density in Ab-treated animals (n = 6). (B) Effect of RNAi silencing of Dll4 on MMP9 expression in RAW264.7 cells. (C and D) Effect of transfection of the plasmid encoding mouse Dll4 (C) and stimulation with immobilized rDll4 (D) on MMP9 expression in RAW264.7 cells. G-I, (n = 6). (E) There were no differences in weight of muscle, spleen, and kidney after 12 weeks of Dll4 Ab administration (n = 19–20). (F and G) Quantitative RT-PCR analysis of expression of MCP-1 (F) and F4/80 (G) in the liver, (n = 7). * P < 0.05. Values are mean ± SEM.

A
B
Fig. S4. Expression of Notch target genes.Quantitative RT-PCR analysis of Notch target genes. Dll4 blockade using neutralizing Dll4 Ab decreased expression of Notch target genes in aortas (n = 7-8) (A) and SVF and adipocytes (n = 9-10) (B). ** P < 0.01. All values are mean ± SEM.
0
500
1000
1500
2000
2500
3000
Dll4 AbIgG
VO
2, m
l/kg
/hr
00.10.20.30.40.50.60.70.80.9
Dll4 AbIgG
Res
pir
ato
ry q
uo
tien
t
0
500
1000
1500
2000
2500
Dll4 AbIgG
VC
O2,
ml/k
g/h
r
0
20000
40000
60000
80000
Dll4 AbIgG
Act
ivit
y, c
ou
nts
0
0.05
0.10
0.15
0.20
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
Fo
od
inta
ke, g
/BW
(g
)/d
ay
0
1
2
3
4
5
6
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
Fo
od
inta
ke, g
/hea
d/d
ay
A
B C
D E
2
weeks
84 12 2
weeks
84 12
Fig. S5. Food consumption and metabolic studies of mice after late-phase treatment.(A) We measured food intake over 7 days at 2, 4, 8, and 12 weeks of treatment. Data are expressed as grams of food per gram of body weight per day (left) and grams of food per day (n = 8 per group). Dll4 Ab treatment did not affect food consumption. Indirect calorimetry was performed at the end of late-phase treatment (n = 6). (B-D) There were no differences in carbon dioxide production (B), oxygen consumption (C), and respiratory quotients (D) between Ab-treated mice and control mice. (E) There was no difference in total 24-hour physical activity as determined by automatic sensors on x- axis. All values are mean ± SEM.

0
0.2
0.4
0.6
0.8
1.0
1.2
IgG
Dll4 A
b
F4/
80/-a
ctin
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8Ig
GDll4
Ab
IgG
Dll4 A
b
Fat
wei
gh
t, g
Epididymal SubcutaneousA
B C
0
0.5
1.0
1.5
2.0
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Dll4
Ab
IgG
Adiponectin Glut4 IRS-1
Gen
e ex
pre
ssio
n/-a
ctin
C/EBP*
Fig. S6. Analyses of epididymal fat after early-phase treatment.(A) Dll4 blockade did not affect fat weight after early-phase treatment (n = 7-8). (B) Results of
quantitative RT-PCR analyses of genes related to insulin sensitivity showed that Dll4 Ab
administration significantly increased adiponectin and tended to increase GLUT4, C/EBP (both
P = 0.06) and IRS-1 (P = 0.09) expression in epididymal fat, regardless of the similarity in fat
weight (n = 6). (C) Dll4 blockade also tended to decrease macrophage accumulation in fat (P =
0.09) (n = 6). * P < 0.05. All values are mean ± SEM.

A
IgG Dll4 Ab
Mac3 Mac3Ig
G
Dll4Ab
IgG
Dll4Ab
IgG
Dll4Ab
IgG
Dll4Ab
IgG
Dll4Ab
Dll4Ab
Dll4Ab
IgG IgG
B
FDC
Fat
wei
gh
t, g
MC
P-1
/-act
in
E
Mac
3 p
osi
tive
cel
ls, %
F4/
80/-a
ctin
Bo
dy-
wei
gh
t g
ain
du
rin
g t
reat
men
t, g
Ser
um
insu
lin, n
g/m
l
Perigonadal Subcutaneous
Fig. S7. Effects of Dll4 blockade on white adipose tissue in Lepob/Lepob mice(A) Mac3 staining and population of Mac3-positive cells in perigonadal fat obtained from Lepob/Lepob mice after 10-week treatment. Scale bar: 100 m. (B and C) Quantitative RT-PCR analysis of F4/80 (B) and MCP-1 (C) expression in fat. (D and E) Effects of Dll4 Ab treatment on fat weight (D) and body weight gain (E). (F) Serum insulin levels. (n = 9). * P < 0.05, **P < 0.001, *** P < 0.05. All values are mean ± SEM.
0
0.2
0.4
0.6
0.8
1.0
1.2
Dll4siRNA
Non-targetingsiRNA
***D
ll4/-a
ctin
0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Dll4siRNA
Non-targetingsiRNA
**
Dll4
/-act
in
0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
rDll4
Dll4 Ab DAPTIgGPBS PBS
MC
P-1
(fo
ld c
han
ge)
(-) (+)
* †* †
A B C
D E
Fig. S8. Results of in vitro experiments using RAW264.7 cells, differentiated 3T3-L1 adipocytes, and endothelial cells.(A and B) Silencing efficacy of Dll4 by transfection of siRNA in RAW264.7 cells (A) and 3T3-L1 adipocytes (B), (n = 6). (C) To examine Notch activation by Dll4 and effect of Dll4 Ab in 3T3-L1 adipocytes, we performed RBP-J reporter activity assay. Stimulation with immobilized rDll4 promotes RBP-J reporter activity in this cell type, which was abrogated by anti-Dll4 Ab, (n = 4). A- C, * P < 0.05, ** P < 0.01, *** P < 0.001. (D) We examined the role of Dll4-Notch signaling in MCP- 1 expression in endothelial cells. Immobilized rDll4 promoted expression of MCP-1 in human saphenous vein endothelial cells in vitro, which were abrogated by anti-human Dll4 Ab or DAPT, a g-secretase inhibitor (n = 6 different donors). * P < 0.05 vs. rDll4 + PBS, † P < 0.05 vs. rDll4 + IgG. (E) Quantitative RT-PCR analysis of Dll4 in differentiated 3T3-L1 adipocytes. Dll4 expression increased during differentiation (n = 4). * P < 0.05, ** P < 0.01. All values are mean ± SEM.

NS161.9±15.4176.4±13.2Triglyceride, mg/dl
NS989.9±50.51051.4±74.7Total cholesterol, mg/dl
NS70.1±5.875.5±2.8Diastolic blood pressure, mmHg
NS91.5±4.998.4±2.4Systolic blood pressure, mmHg
Late-phase Treatment
NS190.6±7.7170.8±9.2Blood glucose, mg,dl
NS218.6±19.2249.9±28.1Triglyceride, mg/dl
NS716.1±57.3782.7±57.9Total cholesterol, mg/dl
NS88.0±6.882.5±3.5Diastolic blood pressure, mmHg
NS110.1±10.2102.3±5.3Systolic blood pressure, mmHg
Early-phase Treatment
P-valueDll4 AbIgG
Supporting Table. Blood pressure and serum lipid levels
All values are mean ± SEM. NS, not significant different.
Related Documents











