FTY720 Induces Apoptosis in Hepatocellular Carcinoma Cells through Activation of Protein Kinase C D Signaling Jui-Hsiang Hung, 1 Yen-Shen Lu, 3 Yu-Chieh Wang, 1 Yi-Hui Ma, 1 Da-Sheng Wang, 1 Samuel K. Kulp, 1 Natarajan Muthusamy, 2 John C. Byrd, 2 Ann-Lii Cheng, 3 and Ching-Shih Chen 1 1 Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy and 2 Division of Hematology and Oncology, Department of Internal Medicine, The Ohio State University, Columbus, Ohio and 3 Department of Oncology, National Taiwan University Hospital, Taipei, Taiwan Abstract This study was aimed at elucidating the mechanism by which FTY720, a synthetic sphingosine immunosuppressant, medi- ated antitumor effects in hepatocellular carcinoma (HCC) cells. The three HCC cell lines examined, Hep3B, Huh7, and PLC5, exhibited differential susceptibility to FTY720-mediated suppression of cell viability, with IC 50 values of 4.5, 6.3, and 11 Mmol/L, respectively. Although FTY720 altered the phospho- rylation state of protein kinase B and p38, our data refuted the role of these two signaling kinases in FTY720-mediated apoptosis. Evidence indicates that the antitumor effect of FTY720 was attributable to its ability to stimulate reactive oxygen species (ROS) production, which culminated in pro- tein kinase C (PKC)D activation and subsequent caspase- 3–dependent apoptosis. We showed that FTY720 activated PKCD through two distinct mechanisms: phosphorylation and caspase-3–dependent cleavage. Cotreatment with the caspase- 3 inhibitor Z-VAD-FMK abrogated the effect of FTY720 on facilitating PKCD proteolysis. Equally important, pharmaco- logic inhibition or shRNA-mediated knockdown of PKCD protected FTY720-treated Huh7 cells from caspase-3 activa- tion. Moreover, FTY720 induced ROS production to different extents among the three cell lines, in the order of Hep3B > Huh7 >> PLC5, which inversely correlated with the respective glutathione S-transferase P expression levels. The low level of ROS generation might underlie the resistant phenotype of PLC5 cells to the apoptotic effects of FTY720. Blockade of ROS production by an NADPH oxidase inhibitor protected Huh7 cells from FTY720-induced PKCD activation and caspase-3– dependent apoptosis. Together, this study provides a rationale to use FTY720 as a scaffold to develop potent PKCD-activating agents for HCC therapy. [Cancer Res 2008;68(4):1204–12] Introduction FTY720 is a synthetic sphingosine immunosuppressant, which is currently undergoing clinical trials for the prevention of kidney graft rejection (1) and the treatment of relapsing multiple sclerosis (2). Previous studies indicate that the effect of FTY720 on prolonging the survival of allografts is attributable to the ability of its phosphorylated metabolite to inhibit T-lymphocyte infiltra- tion by targeting several of the sphingosine-1-phosphate (S1P) receptors (3, 4). In addition to immunosuppression, FTY720 has also been shown to induce apoptosis in several human cancer cell lines, including Jurkat T cells (5), multiple myeloma cells (6), and those of liver (7, 8), prostate (9–12), breast (13), kidney (14), and bladder (15). This antitumor effect is noteworthy because of the involvement of S1P receptor–independent mechanisms (16). To date, a number of signaling pathways have been proposed to account for the ability of FTY720 to facilitate apoptosis in different cancer cell lines, including those mediated by protein kinase B (Akt) (5, 6, 8), mitogen-activated protein (MAP) kinases (6, 10), focal adhesion kinase (10), Rho-GTPase (12), signal transducers and activators of transcription 3, InBa, nuclear factor-nB, and Bcl-xL (6). Mechanistically, the targeting of this wide spectrum of signaling elements underscores the effectiveness of FTY720 in suppressing cell growth in a broad range of cancer cells that exhibit distinct mechanisms in governing cell cycle progression and apoptosis. Hepatocellular cancer occurs both sporadically and is also related to chronic viral infection, environmental exposure, and alternative causes of hepatic cirrhosis. The US Surveillance, Epidemiology, and End Results database estimates that 19,160 men and women will be diagnosed with and 16,780 men and women will die of cancer of the liver and intrahepatic bile duct in 2007 within the United States (17). The incidence of hepatocellular carcinoma is even higher in the Asian cultures due to the higher frequency of chronic active viral hepatitis. Until only recently, effective treatment of hepatocellular cancer has been essentially absent (18). Recently, the RAF inhibitor sornafinib has been shown to be beneficial for the treatment of metastatic hepatocellular carcinoma and was approved for marketing by the Food and Drug Administration in this indication (19). However, this therapy only works in a subset of patients and is not curative. This emphasizes both the potential for multitargeted kinase inhibitors in hepato- cellular carcinoma and the need to identify new therapies. In this study, we describe a novel mechanism by which FTY720 induces apoptosis in hepatocellular carcinoma cells (HCC). We obtain evidence that FTY720 facilitates reactive oxygen species (ROS)-dependent activation of protein kinase C y (PKCy), resulting in caspase-3–mediated apoptotic death in HCC cells. PKCy,a member of the novel PKC subfamily, has been shown to play a pivotal role in mediating apoptosis induced by oxidative stress (20, 21), Fas ligands (22), and various genotoxic agents including DNA damaging agents (23) and paclitaxel (24), thereby representing an important target for cancer therapy. Consequently, this mecha- nistic finding provides a molecular basis to pharmacologically exploit FTY720 to develop potent PKCy-targeted antitumor agents. Materials and Methods Reagents. FTY720 was synthesized according to a published procedure (25). The identity and purity were verified by nuclear magnetic resonance, Requests for reprints: Ching-Shih Chen, Division of Medicinal Chemistry, College of Pharmacy, Parks Halls, The Ohio State University, 500 West 12th Avenue, Columbus, OH 43210. Phone: 614-688-4008; Fax: 614-688-8556; E-mail: [email protected]. I2008 American Association for Cancer Research. doi:10.1158/0008-5472.CAN-07-2621 Cancer Res 2008; 68: (4). February 15, 2008 1204 www.aacrjournals.org Research Article

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

FTY720 Induces Apoptosis in Hepatocellular Carcinoma Cells
through Activation of Protein Kinase C D Signaling
Jui-Hsiang Hung,1Yen-Shen Lu,
3Yu-Chieh Wang,
1Yi-Hui Ma,
1Da-Sheng Wang,
1Samuel K. Kulp,
1
Natarajan Muthusamy,2John C. Byrd,
2Ann-Lii Cheng,
3and Ching-Shih Chen
1
1Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy and 2Division of Hematology and Oncology,Department of Internal Medicine, The Ohio State University, Columbus, Ohio and 3Department of Oncology,National Taiwan University Hospital, Taipei, Taiwan
Abstract
This study was aimed at elucidating the mechanism by whichFTY720, a synthetic sphingosine immunosuppressant, medi-ated antitumor effects in hepatocellular carcinoma (HCC)cells. The three HCC cell lines examined, Hep3B, Huh7, andPLC5, exhibited differential susceptibility to FTY720-mediatedsuppression of cell viability, with IC50 values of 4.5, 6.3, and 11Mmol/L, respectively. Although FTY720 altered the phospho-rylation state of protein kinase B and p38, our data refutedthe role of these two signaling kinases in FTY720-mediatedapoptosis. Evidence indicates that the antitumor effect ofFTY720 was attributable to its ability to stimulate reactiveoxygen species (ROS) production, which culminated in pro-tein kinase C (PKC)D activation and subsequent caspase-3–dependent apoptosis. We showed that FTY720 activatedPKCD through two distinct mechanisms: phosphorylation andcaspase-3–dependent cleavage. Cotreatment with the caspase-3 inhibitor Z-VAD-FMK abrogated the effect of FTY720 onfacilitating PKCD proteolysis. Equally important, pharmaco-logic inhibition or shRNA-mediated knockdown of PKCDprotected FTY720-treated Huh7 cells from caspase-3 activa-tion. Moreover, FTY720 induced ROS production to differentextents among the three cell lines, in the order of Hep3B >Huh7 >> PLC5, which inversely correlated with the respectiveglutathione S-transferase P expression levels. The low level ofROS generation might underlie the resistant phenotype ofPLC5 cells to the apoptotic effects of FTY720. Blockade of ROSproduction by an NADPH oxidase inhibitor protected Huh7cells from FTY720-induced PKCD activation and caspase-3–dependent apoptosis. Together, this study provides a rationaleto use FTY720 as a scaffold to develop potent PKCD-activatingagents for HCC therapy. [Cancer Res 2008;68(4):1204–12]
Introduction
FTY720 is a synthetic sphingosine immunosuppressant, which iscurrently undergoing clinical trials for the prevention of kidneygraft rejection (1) and the treatment of relapsing multiple sclerosis(2). Previous studies indicate that the effect of FTY720 onprolonging the survival of allografts is attributable to the abilityof its phosphorylated metabolite to inhibit T-lymphocyte infiltra-tion by targeting several of the sphingosine-1-phosphate (S1P)receptors (3, 4). In addition to immunosuppression, FTY720 has
also been shown to induce apoptosis in several human cancer celllines, including Jurkat T cells (5), multiple myeloma cells (6), andthose of liver (7, 8), prostate (9–12), breast (13), kidney (14), andbladder (15). This antitumor effect is noteworthy because of theinvolvement of S1P receptor–independent mechanisms (16). Todate, a number of signaling pathways have been proposed toaccount for the ability of FTY720 to facilitate apoptosis in differentcancer cell lines, including those mediated by protein kinase B(Akt) (5, 6, 8), mitogen-activated protein (MAP) kinases (6, 10),focal adhesion kinase (10), Rho-GTPase (12), signal transducers andactivators of transcription 3, InBa, nuclear factor-nB, and Bcl-xL(6). Mechanistically, the targeting of this wide spectrum of signalingelements underscores the effectiveness of FTY720 in suppressingcell growth in a broad range of cancer cells that exhibit distinctmechanisms in governing cell cycle progression and apoptosis.Hepatocellular cancer occurs both sporadically and is also
related to chronic viral infection, environmental exposure, andalternative causes of hepatic cirrhosis. The US Surveillance,Epidemiology, and End Results database estimates that 19,160men and women will be diagnosed with and 16,780 men andwomen will die of cancer of the liver and intrahepatic bile duct in2007 within the United States (17). The incidence of hepatocellularcarcinoma is even higher in the Asian cultures due to the higherfrequency of chronic active viral hepatitis. Until only recently,effective treatment of hepatocellular cancer has been essentiallyabsent (18). Recently, the RAF inhibitor sornafinib has been shownto be beneficial for the treatment of metastatic hepatocellularcarcinoma and was approved for marketing by the Food and DrugAdministration in this indication (19). However, this therapy onlyworks in a subset of patients and is not curative. This emphasizesboth the potential for multitargeted kinase inhibitors in hepato-cellular carcinoma and the need to identify new therapies.In this study, we describe a novel mechanism by which FTY720
induces apoptosis in hepatocellular carcinoma cells (HCC). Weobtain evidence that FTY720 facilitates reactive oxygen species(ROS)-dependent activation of protein kinase C y (PKCy), resultingin caspase-3–mediated apoptotic death in HCC cells. PKCy, amember of the novel PKC subfamily, has been shown to play apivotal role in mediating apoptosis induced by oxidative stress(20, 21), Fas ligands (22), and various genotoxic agents includingDNA damaging agents (23) and paclitaxel (24), thereby representingan important target for cancer therapy. Consequently, this mecha-nistic finding provides a molecular basis to pharmacologicallyexploit FTY720 to develop potent PKCy-targeted antitumor agents.
Materials and Methods
Reagents. FTY720 was synthesized according to a published procedure
(25). The identity and purity were verified by nuclear magnetic resonance,
Requests for reprints: Ching-Shih Chen, Division of Medicinal Chemistry, Collegeof Pharmacy, Parks Halls, The Ohio State University, 500 West 12th Avenue, Columbus,OH 43210. Phone: 614-688-4008; Fax: 614-688-8556; E-mail: [email protected].
I2008 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-07-2621
Cancer Res 2008; 68: (4). February 15, 2008 1204 www.aacrjournals.org
Research Article

high-solution mass spectrometry, and elemental analysis. The followingpharmacologic agents were purchased from the respective vendors: okadaic
acid, PD98059, SB203580, Ro6976, rottlerin, and U3122 (Calbiochem);
diphenyleneiodonium chloride (DPI; Cayman Chemical); 2,7-dichlorofluor-
escein diacetate (DCFDA; Invitrogen); (Ac-DMQD)2-Rh110 (Anaspec);3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H -tetrazolium bromide (MTT;
TCI America). Cell death detection ELISA kit and the NE-PER Nuclear
and Cytoplasmic Extraction Reagents kit were obtained from Roche
Diagnostic and Pierce, respectively. Antibodies against various proteinswere obtained from the following sources: PKCa, PKCq, PKCf, PKCy,p-Tyr155-PKCy, p-Thr507-PKCy, and p-Thr308-Akt (Santa Cruz Biotechno-
logy); p-Tyr311-PKCy, p-Ser664-PKCy (Abcam); Akt and p-Ser473-Akt (Cell
Signaling); glutathione S-transferase (GST)-k (DAKO). The pKD-PKCy-v2plasmid encoding shRNA against PKCy, goat anti-rabbit IgG-horseradish
peroxidase (HRP) conjugates, and rabbit anti-mouse IgG-HRP conjugates
was purchased from Upstate.Cell culture. Huh7, PLC5, and Hep3B human HCC cell lines were
purchased from the American Type Culture Collection. Cells were cultured
in DMEM containing 10% fetal bovine serum (FBS) and 10 Ag/mL
gentamicin at 37jC in a humidified incubator containing 5% CO2. Culturemedium was replaced every 2 days.
MTT assay. Cell viability was assessed by using the MTT assay in six
replicates. HCC cells were seeded at 3 � 105 per well in 24-well flat-
bottomed plates and incubated in 10% FBS–supplemented DMEM for 24 h.Cells were treated with FTY720 at various concentrations in the same
medium. Controls received DMSO vehicle at a concentration equal to that
in drug-treated cells. After 24 h, the drug-containing medium was replacedwith 200 AL of 10% FBS–supplemented DMEM containing 0.5 mg/mL MTT,
and cells were incubated in the CO2 incubator at 37jC for 4 h. Medium was
removed, the reduced MTT was solubilized in 500 AL per well of DMSO, and
100 AL aliquots from each well was transfer to 96-well plates to measureabsorbance at 570 nm.
Annexin V/propidium iodide assay. For assessment of apoptosis, both
floating and adherent cells were collected and analyzed. Briefly, f7 � 105
cells per dish were plated onto 10-cm dishes and incubated at 37jC for 16 h.The cells were treated with DMSO or 10 Amol/L FTY720 for the time
indicated. The cells were washed twice with PBS and collected by
trypsinization. After centrifugation at 400 � g for 5 min at roomtemperature, the cells were stained with Annexin V and propidium iodide
(1 Ag/mL). The cell apoptosis distributions were determined on a FACScort
flow cytometer and analyzed by ModFitLT V3.0 software program.
Western blotting. Cells, seeded in 10-cm dishes (7 � 105 cells per dish),
were incubated for 16 h, subjected to different drug treatments, and
harvested by scrapping. Cell lysates were prepared by exposing cells to 2 �SDS lysis buffer [0.1 mol/L Tris, 0.4% SDS, and 20% glycerol (pH 6.8)].
Protein concentrations of cell lysates were measured using a Micro BCA
protein assay reagent kit (Pierce). To the cell lysate, the same volume of
SDS-PAGE sample loading buffer [100 mmol/L Tris-HCl, 4% SDS, 5%
h-mercaptoethanol, 20% glycerol, and 0.1% bromphenol blue (pH 6.8)] was
added, and the cells were boiled for 10 min. Equal amounts of proteins were
resolved in SDS-polyacrylamide gels. After electrophoresis, gel was
transferred to nitrocellulose membranes using a semidry transfer cell. The
transblotted membrane was washed twice with TBS containing 0.1% Tween
20 (TBST). After blocking with TBST containing 5% nonfat milk for 1 h, the
membrane was incubated with the appropriate primary antibody in 1%
TBST nonfat milk at 4jC overnight. The membrane was washed thrice with
TBST for a total of 15 min. The secondary anti-mouse IgG-HRP conjugates
or anti-rabbit IgG-HRP conjugates (1:2,000 dilution) was subsequently
incubated with the membrane for 1 h at room temperature and was washed
extensively for 50 min with TBST. The blots were visualized with the
enhanced chemiluminescence (Amersham) Western blot detection system
according to the manufacturer’s instructions.
Cell death detection ELISA assay. Induction of apoptosis was assessedwith a Cell Death Detection ELISA kit, which is based on the quantitative
determination of cytoplasmic histone–associated DNA fragments in the
form of mononucleosomes and oligonucleosomes after induced apoptotic
death. In brief, Huh7 cells (5 � 104) were seeded and incubated in 96-well
plates containing DMEM supplemented with 10% FBS for 16 h and werethen subjected to various drug treatments for 24 h. After incubation, the
plates were centrifuged at 200 � g for 10 min and incubated with 200 AL of
lysis buffer at room temperature for 30 min. Twenty microliters of
supernatant from each sample were used in the ELISA by following themanufacturer’s instruction.
Detection of ROS. DCFDA is an indicator for ROS that is nonfluorescent
until its acetate groups are removed by intracellular esterases and oxidation
occurs in the cell. The ROS production in Huh7 cells after FTY720 treatmentwas detected using 5 Amol/L DCFDA. Huh7 cells (7 � 105) in 10 cm-dishes
were treated with FTY720 for 1 h, washed with PBS twice, and then exposed
to 5 Amol/L DCFDA for 30 min at 37jC. Cells were collected and analyzed
by flow cytometry.Detection of hydrogen peroxide. Quantitative determination of
hydrogen peroxide in the culture medium was performed by using a
colorimetric hydrogen peroxide kit (Assay Designs, Inc.) according to the
vendor’s instruction. In brief, Huh7 cells (7� 105) were seeded and incubated
in 10-cm plates containing 10% FBS–supplemented DMEM for 16 h. Because
the culture mediumwould interfere with the detection of hydrogen peroxide,
it was replaced with an equal volume of Hanks’ balanced Salt Solution
(HBSS) containing 10 Amol/L FTY720. After 3-h of exposure, 50 AL of the
cultured HBSS solution was incubated with 100 AL of the colorimetric
solution provided in the kit in 96-well plates, and incubated at room
temperature for 30 min. Absorbance at 570 nm was measured.
ShRNA-mediated knockdown of PKCA, PKCE, and PKCD. Huh7 cells
(1 � 106) were cotransfected with 1.8 Ag of individual shRNA plasmids
(pKD-PKCa-v4, pKD-PKCy-v2, and pKD-PKCq-v5) and 0.2 Ag of pCDNA 3.1(+) plasmids using Invitrogen Lipofectamine 2000 reagent according to the
manufacturer’s protocol. Cells were then selected by G418 for 2 weeks. The
stable clones were established and confirmed by Western blotting usingindividual antibodies against PKCa, PKCq, and PKCy.
Ectopic expression of constitutively active Akt. The pcDNA 3.1
(+)/CA-Akt-HA plasmid that encodes AktT308D/S473D, a constitutively active
form of Akt, was provided by Dr. Matthew D. Ringel (The Ohio StateUniversity, Columbus, OH). Huh7 cells were transfected via nucleofection by
using program T-022 of the AMAXA Nucleofector system according to the
manufacturer’s instructions. Expression of constitutively active Akt was
confirmed by Western blotting using antibodies against Akt andhemagglutinin tag.
Subcellular fractionation. Subcellular fractionation was performed
using NE-PER Nuclear and Cytoplasmic Extraction Reagents kit accordingto the manufacturer’s instruction. In brief, Huh7 (7 � 105) cells in 10-cm
dishes were exposed to 10 Amol/L FTY720 in 10% FBS–containing DMEM
for different time intervals. After treatment, the cells were washed with
cold PBS, scraped, and harvested by centrifugation. The cell pellets weresuspended in 200 AL of Cytoplasmic Extraction Reagent I solution and
incubated on ice for 10 min, followed by adding 11 AL of Cytoplasmic
Extraction Reagent II solution and incubation on ice for 1 min. The
cell suspensions were centrifuged at 16,000 � g for 5 min to collectsupernatant as the cytoplasmic fraction. The pellets were resuspended with
100 AL of Nuclear Extraction Reagent on ice for 40 min. The cell suspension
was centrifuged at 16,000 � g for 10 min at 4jC to collect supernatant as
the nuclear fraction.Analysis of caspase-3 activity. Caspase-3 activity was determined using
(Ac-DMQD)2-Rh110 as the fluorogenic substrate for active caspase-3.
Briefly, Huh7 (7 � 105) cells in 10-cm dishes were subjected to different drugtreatments for 24 h and were resuspended in 100 AL of PBS containing
10 Amol/L 2-Rh110 for 15 min at room temperature. Each sample was then
added with 400 AL PBS, and fluorescence signals generated by caspase-3–
cleaved substrate were analyzed by flow cytometry.Cytochrome c release. Drug-treated Huh7 cells were collected and
triturated with 100 AL of chilled hypotonic lysis solution [220 mmol/L
mannitol, 68 mmol/L sucrose, 50 mmol/L KCl, 5 mmol/L EDTA, 2 mmol/L
MgCl2, and 1 mmol/L DTT in 50 mmol/L PIPES-KOH (pH 7.4)] for 45 min.The solution was centrifuged at 600 � g for 10 min to collect the
supernatant. The supernatant was further centrifuged at 14,000 rpm for
30 min, and equal amounts of proteins (50 Ag) from the supernatant were
FTY720 Mediates Apoptosis through PKCd Activation
www.aacrjournals.org 1205 Cancer Res 2008; 68: (4). February 15, 2008

resolved in 15% SDS-polyacrylamide gel. Proteins were transferred tonitrocellulose membranes and analyzed by immunoblotting with anti-
cytochrome c antibodies.
Statistical analysis. The JMP5.0.1 software package was used to perform
all analyses. Data were analyzed by the Student’s t test. Differences wereconsidered significant at a P value of <0.05.
Results
Differential susceptibility of HCC cell lines to FTY720-induced apoptotic death. The in vitro antitumor efficacy ofFTY720 was evaluated in three human HCC cell lines, Huh7,Hep3B, and PLC5, which are resistant to cytotoxic drugs due to lossof p53 function and/or overexpression of Bcl-xL (26). As these threecell lines harbor different cellular and genetic abnormalities, theyshowed differential susceptibility to the antiproliferative effect ofFTY720. The IC50 values for Hep3B, Huh7, and PLC5 cells were 4.5,6.3, and 11 Amol/L, respectively (Fig. 1A). The antiproliferativeactivity of FTY720 was, at least in part, attributable to apoptosis, asevidenced by poly(ADP)ribose polymerase (PARP) cleavage in eachof these three cell lines and Annexin V/propodium iodide stainingin Huh7 cells in a dose- and/or time-dependent manner (Fig. 1Band C).Involvement of PKCD in FTY720-mediated apoptotic death.
To date, various signaling pathways have been reported to accountfor FTY720-induced apoptosis in different cancer cell systems,which might be reconciled by differences in the molecular
abnormalities associated with the oncogenesis of individual cancertypes. Of these proposed mechanisms, the ability of FTY720 toaffect the activation status of p38 MAP kinase and Akt in prostatecancer cells (10) and HCC cells (8), respectively, was especiallynoteworthy.To discern the role of Akt and p38 in FTY720-mediated apoptosis
in HCC cells, we investigated the effect of FTY720 on thephosphorylation of these two signaling kinases in Huh7, Hep3B,and PLC5 cells. As these HCC cell lines contained functional PTEN,they exhibited low levels of Akt phosphorylation at Ser473
compared with the PTEN-defective PC-3 cancer cells (Fig. 2A). Incontrast, this PTEN functional status had no direct correlation withthe Thr308 phosphorylation, as all these three HCC cell linesexhibited high levels of p-Thr308-Akt relative to PC-3 (Fig. 2B). Thissite-specific phenomenon is consistent with the finding that levelsof PTEN modulate Akt phosphorylation on Ser473, but not onThr308, in rhabdomyosarcomas cells (27).As shown in Fig. 2B , exposure to FTY720 for 24 h led to a dose-
dependent decrease in p-Thr308-Akt levels in these three HCC celllines (Fig. 2B), which is consistent with that previously reported (8).Moreover, the relative potency in Akt dephosphorylation correlatedwith the respective susceptibility to FTY720-induced cell death. Incontrast, the effect of FTY720 on modulating p38 phosphorylationwas cell line specific among the three cell lines examined, i.e., onlyPLC5 cells exhibited a dose-dependent increase in p-p38 levels afterdrug treatment.
Figure 1. Huh7, Hep3B, and PLC5cells exhibit differential susceptibilityto FTY720-induced apoptotic death.A, dose-dependent effect of FTY720 onsuppressing the cell viability of thethree cell lines. Cells were exposed toFTY720 at the indicated doses in 10%FBS–supplemented DMEM for 24 h, andthe cell viability was assessed by MTTassays. Points, mean; bars, SD (n = 6).B, dose- and time-dependent effect ofFTY720 on PARP cleavage in the three celllines by Western blot analysis. C, flowcytometric analysis of apoptotic death inHuh7 cells with DMSO vehicle or 10 Amol/LFTY720 in 10% FBS–containing DMEM for12 or 24 h. Huh7 cells were cultured withFTY720 for the time indicated. The cellswere analyzed by flow cytometry afterstaining with fluorescein-conjugatedAnnexin V and propidium iodide (PI ). Thepercentages in the graphs represent thepercent of cell numbers in the respectivequadrants. Columns, mean; bars, SD.
Cancer Research
Cancer Res 2008; 68: (4). February 15, 2008 1206 www.aacrjournals.org

To further examine whether Akt inhibition represented a majorunderlying antitumor mechanism for FTY720, we assessed theeffect of the ectopic expression of a constitutively active form ofAkt (AktT308D/S473D) on FTY720-induced cell death by transientlytransfecting Huh7 cells with HA-CA-Akt plasmids (Fig. 2C). Theconstitutively activated status of Akt was manifested by themultifold increase in the phosphorylation level of GSK3h. However,this ectopic AktT308D/S473D expression did not provide a significantprotection against the suppression of cell viability by 10 Amol/LFTY720. Together, these findings argued against the involvement ofAkt and p38 in FTY720-induced apoptosis at least in these twosensitive cell lines.To delineate the underlying mechanism, we further assessed the
effect of a panel of pharmacologic inhibitors, including those ofprotein phosphatase 2A (PP2A; okadaic acid), MAP kinase kinase(PD98059), p38 kinase (SB203580), PKCa/h (Ro6970), PKCy
(rottlerin), and phospholipase C (U3122) on rescuing Huh7 cellfrom FTY720-induced cell death. Some of these signaling enzymesmight potentially be targeted by FTY720 to trigger apoptosissignaling. For example, a recent report suggests the involvement ofPP2A activation in FTY720-mediated Akt dephosphorylation in aT-cell–derived leukemia cell line (5). However, of these inhibitors,only the PKCy inhibitor rottlerin exhibited the ability to counteractthe cell killing activity of 5 Amol/L FTY720, whereas others eitherexacerbated or had no appreciable effects on FTY720-mediatedsuppression of cell viability (Fig. 2D, left). In addition, rottlerin,even at 1 Amol/L, was effective in protecting Huh7 cells from theantitumor activity of FTY720. As PKCy mediates mitochondria-dependent apoptosis, cotreatment of Huh7 cells with rottlerincould block the effect of FTY720 on cytochrome c release into thecytoplasm and PARP cleavage (Fig. 2E). Consequently, thesefindings suggested that PKCy activation played a pivotal role in
Figure 2. Role of Akt inhibition and PKCyactivation in FTY720-mediated apoptosis.A, endogenous expression levels of PTEN,p-Ser473-Akt, and p-Thr308-Akt, in Huh7,PLC5, and Hep3B cells. PC-3 prostatecancer cells, which are PTEN null,were used as a negative control.B, dose-dependent effect of FTY720 on thephosphorylation states of Thr308-Akt andp38 in Huh7, Hep3B, and PLC5 cells byWestern blotting. C, ectopic expression ofAktT308D/S473D, a constitutively active formof Akt, does not protect Huh7 cellsfrom FTY720-induced cell death. Left,expression of AktT308D/S473D (CA-Akt-HA)in Huh7 transient transfection. Westernblot analysis used antibodies againsthemagglutinin (HA ) tag, Akt, p-GSK3h,and GSK3h. Right, viability of Huh7cells overexpressing AktT308D/S473D
vis-a-vis cells transfected with emptypCMV vector in the presence of DMSOvehicle or 10 Amol/L FTY720 in 10%FBS–supplemented DMEM for 24 h.Columns, mean; bars , FSD (n = 3).D, effect of the pharmacologic inhibitorsof various signaling enzymes onFTY720-mediated cell death in Huh7 cells(left). Huh7 cells were exposed to 5 Amol/LFTY720 in the presence of one of thefollowing inhibitors, okadaic acid(100 nmol/L), PD98059 (10 Amol/L),SB203580 (10 Amol/L), Ro6976(10 Amol/L), rottlerin (10 Amol/L), andU3122 (10 Amol/L), for 24 h in 10%FBS–supplemented DMEM for 24 h. Cellviability was assessed by MTT assays.Right, dose-dependent effect of rottlerinon protecting Huh7 cells againstFTY720-induced cell death. Huh7 cellswere exposed to DMSO vehicle or theindicated dose of rottlerin in the absence orpresence of 10 Amol/L FTY720 for 24 h,and cell viability was analyzed by MTTassay. Columns, mean; bars, FSD (n = 6).E , rottlerin (10 Amol/L) inhibits the effect ofFTY720 (10 Amol/L) on cytochrome c(left) and PARP cleavage (right ) in Huh7cells. Huh7 cells were exposed to10 Amol/L FTY720 in the presence of10 Amol/L rottlerin or DMSO vehicle in 10%FBS–containing DMEM for the indicatedtime intervals. Mitochondria-free lysatesand total lysates were prepared asdescribed in Materials and Methods for theWestern blot analysis of cytochrome crelease and PARP cleavage, respectively.
FTY720 Mediates Apoptosis through PKCd Activation
www.aacrjournals.org 1207 Cancer Res 2008; 68: (4). February 15, 2008
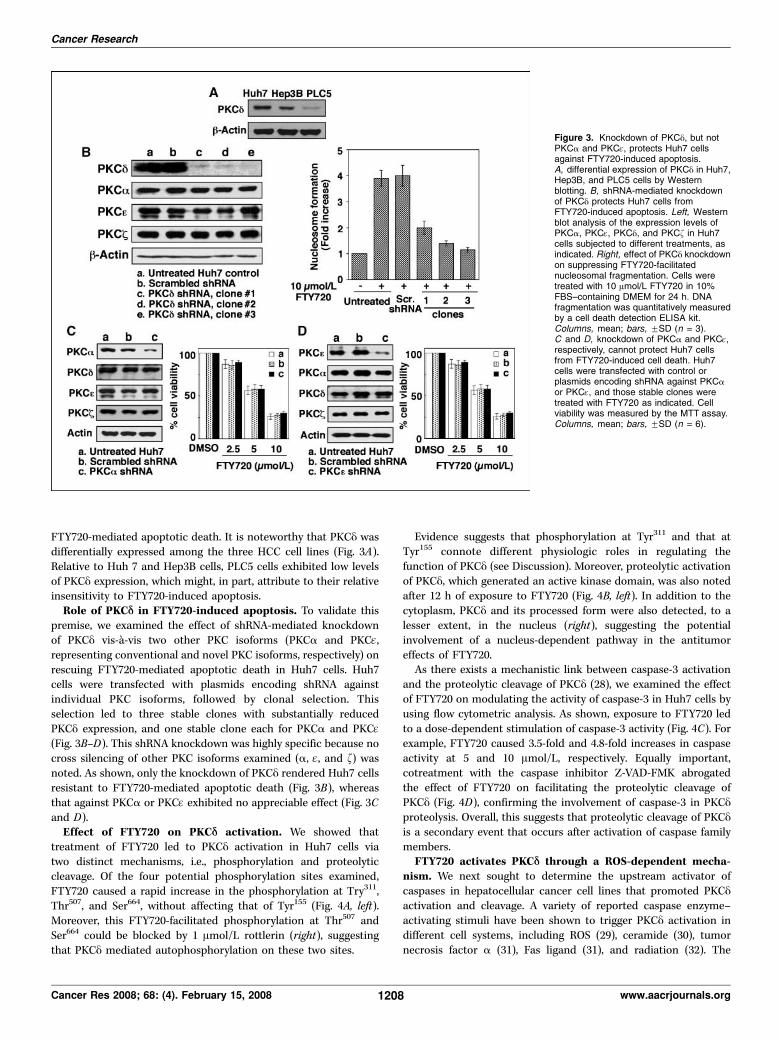
FTY720-mediated apoptotic death. It is noteworthy that PKCy wasdifferentially expressed among the three HCC cell lines (Fig. 3A).Relative to Huh 7 and Hep3B cells, PLC5 cells exhibited low levelsof PKCy expression, which might, in part, attribute to their relativeinsensitivity to FTY720-induced apoptosis.Role of PKCD in FTY720-induced apoptosis. To validate this
premise, we examined the effect of shRNA-mediated knockdownof PKCy vis-a-vis two other PKC isoforms (PKCa and PKCq,representing conventional and novel PKC isoforms, respectively) onrescuing FTY720-mediated apoptotic death in Huh7 cells. Huh7cells were transfected with plasmids encoding shRNA againstindividual PKC isoforms, followed by clonal selection. Thisselection led to three stable clones with substantially reducedPKCy expression, and one stable clone each for PKCa and PKCq(Fig. 3B–D). This shRNA knockdown was highly specific because nocross silencing of other PKC isoforms examined (a, q, and f) wasnoted. As shown, only the knockdown of PKCy rendered Huh7 cellsresistant to FTY720-mediated apoptotic death (Fig. 3B), whereasthat against PKCa or PKCq exhibited no appreciable effect (Fig. 3Cand D).Effect of FTY720 on PKCD activation. We showed that
treatment of FTY720 led to PKCy activation in Huh7 cells viatwo distinct mechanisms, i.e., phosphorylation and proteolyticcleavage. Of the four potential phosphorylation sites examined,FTY720 caused a rapid increase in the phosphorylation at Try311,Thr507, and Ser664, without affecting that of Tyr155 (Fig. 4A, left).Moreover, this FTY720-facilitated phosphorylation at Thr507 andSer664 could be blocked by 1 Amol/L rottlerin (right), suggestingthat PKCy mediated autophosphorylation on these two sites.
Evidence suggests that phosphorylation at Tyr311 and that atTyr155 connote different physiologic roles in regulating thefunction of PKCy (see Discussion). Moreover, proteolytic activationof PKCy, which generated an active kinase domain, was also notedafter 12 h of exposure to FTY720 (Fig. 4B, left). In addition to thecytoplasm, PKCy and its processed form were also detected, to alesser extent, in the nucleus (right), suggesting the potentialinvolvement of a nucleus-dependent pathway in the antitumoreffects of FTY720.As there exists a mechanistic link between caspase-3 activation
and the proteolytic cleavage of PKCy (28), we examined the effectof FTY720 on modulating the activity of caspase-3 in Huh7 cells byusing flow cytometric analysis. As shown, exposure to FTY720 ledto a dose-dependent stimulation of caspase-3 activity (Fig. 4C). Forexample, FTY720 caused 3.5-fold and 4.8-fold increases in caspaseactivity at 5 and 10 Amol/L, respectively. Equally important,cotreatment with the caspase inhibitor Z-VAD-FMK abrogatedthe effect of FTY720 on facilitating the proteolytic cleavage ofPKCy (Fig. 4D), confirming the involvement of caspase-3 in PKCyproteolysis. Overall, this suggests that proteolytic cleavage of PKCyis a secondary event that occurs after activation of caspase familymembers.FTY720 activates PKCD through a ROS-dependent mecha-
nism. We next sought to determine the upstream activator ofcaspases in hepatocellular cancer cell lines that promoted PKCyactivation and cleavage. A variety of reported caspase enzyme–activating stimuli have been shown to trigger PKCy activation indifferent cell systems, including ROS (29), ceramide (30), tumornecrosis factor a (31), Fas ligand (31), and radiation (32). The
Figure 3. Knockdown of PKCy, but notPKCa and PKCq, protects Huh7 cellsagainst FTY720-induced apoptosis.A, differential expression of PKCy in Huh7,Hep3B, and PLC5 cells by Westernblotting. B, shRNA-mediated knockdownof PKCy protects Huh7 cells fromFTY720-induced apoptosis. Left, Westernblot analysis of the expression levels ofPKCa, PKCq, PKCy, and PKCf in Huh7cells subjected to different treatments, asindicated. Right, effect of PKCy knockdownon suppressing FTY720-facilitatednucleosomal fragmentation. Cells weretreated with 10 Amol/L FTY720 in 10%FBS–containing DMEM for 24 h. DNAfragmentation was quantitatively measuredby a cell death detection ELISA kit.Columns, mean; bars, FSD (n = 3).C and D, knockdown of PKCa and PKCq,respectively, cannot protect Huh7 cellsfrom FTY720-induced cell death. Huh7cells were transfected with control orplasmids encoding shRNA against PKCaor PKCq, and those stable clones weretreated with FTY720 as indicated. Cellviability was measured by the MTT assay.Columns, mean; bars, FSD (n = 6).
Cancer Research
Cancer Res 2008; 68: (4). February 15, 2008 1208 www.aacrjournals.org

present study shows that the effect of FTY720 on PKCy activationwas attributable to its ability to stimulate ROS generation in HCCcells.Flow cytometric analysis using the ROS-sensitive probe DCFDA
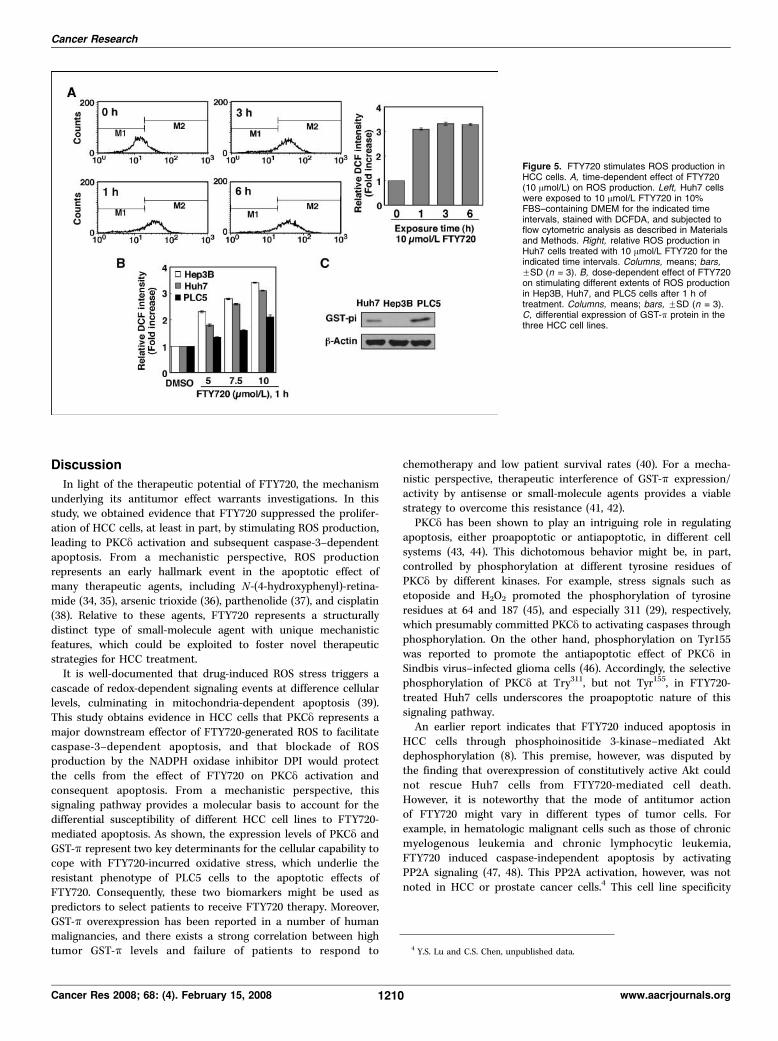
indicates that FTY720 at 10 Amol/L stimulated an immediaterobust increase in ROS production in Huh7 cells, and that the levelof intracellular ROS levels remained high even at 6 h after thetreatment (Fig. 5A). The time course of this ROS productionparalleled that of PKCy phosphorylation. Moreover, FTY720induced different levels of ROS production among the three HCCcell lines, in the order of Hep3B > Huh7 >> PLC5. It is noteworthythat this FTY720-stimulated ROS generation inversely correlatedwith the expression of GST-k (Fig. 5C), a phase II detoxificationenzyme, which has been implicated in protection against apoptosisand drug resistance in cancer cells (33). However, the major ROSproduced in response to FTY720 remained unclear because nosignificant increase in H2O2 generation was detected in FTY720-treated Huh7 cells (data not shown).
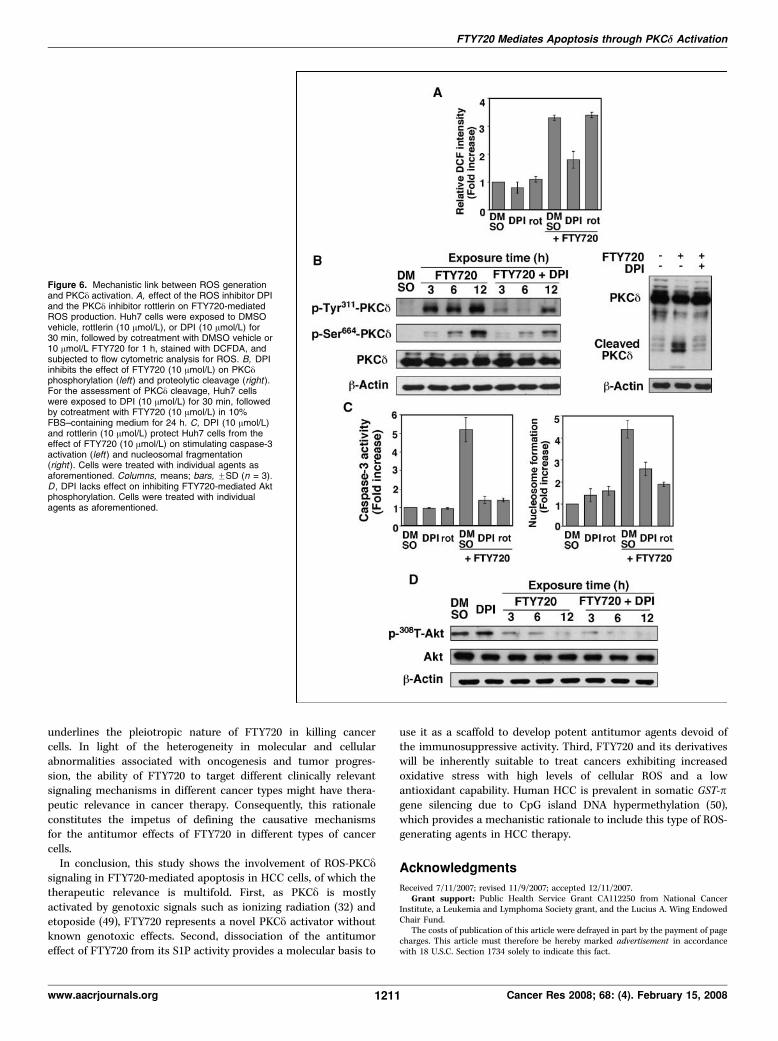
The effect of FTY720 on increasing intracellular ROS levels wasindependent of PKCy activity. As shown in Fig. 6A , although theNADPH oxidase inhibitor DPI could significantly inhibit FTY720-stimulated production of ROS, rottlerin exhibited no appreciableeffect on ROS levels. Moreover, DPI was able to suppress the effectof FTY720 on stimulating the phosphorylation and proteolyticcleavage of PKCy (Fig. 6B), indicating the causative role of ROS inPKCy activation. As a consequence, blockade of ROS generation byDPI protected Huh7 cells from FTY720-induced caspase-3 activa-tion and apoptotic death, with potency similar to that of rottlerin(Fig. 6C). In contrast, DPI lacked appreciable effect on rescuingFTY720-facilitated Akt dephosphorylation, suggesting that Aktdeactivation was not mechanistically linked to ROS-PKCy signalingand did not play a crucial role in FTY720-mediated cell death(Fig. 6D). Overall, this suggests that FTY720 production of ROS andsecondary activation of PKCy contributes to the cytotoxicityobserved in hepatocellular carcinoma cell lines treated with thisagent.
Figure 4. FTY720 facilitates phosphorylation andcaspase-3–dependent cleavage of PKCy in Huh7 cells.A, time-dependent effect of FTY720 (10 Amol/L) on thephosphorylation of PKCy at different sites (left ). Right,effect of 1 Amol/L rottlerin on FTY720-facilitated PKCyphosphorylation at Thr507 and Ser664. B, time-dependenteffect of FTY720 (10 Amol/L) on the proteolytic cleavage ofPKCy in whole cell lysates (left) and in the cytosolic versusnuclear fractions (right ). C, flow cytometric analysis ofdose-dependent effect of FTY720 on increasing caspase-3activity in Huh7 cells. Left, increased caspase-3 activitywas observed in a dose-dependent manner after24-h exposure to FTY720. Right, relative caspase-3activities, normalized to DMSO control, at the indicatedconcentrations of FTY720. Columns, means; bars, FSD(n = 3). D, the caspase inhibitor Z-VAD-FMK blocksthe effect of FTY720 (10 Amol/L) on proteolytic cleavageof PKCy. Cells were treated with DMSO vehicle or10 Amol/L FTY720 in the presence of increasing doses ofZ-VAD-FMK for 24 h, and cell lysates were subjectedto immunoblotting with antibodies against anti-PKCyand h-actin.
FTY720 Mediates Apoptosis through PKCd Activation
www.aacrjournals.org 1209 Cancer Res 2008; 68: (4). February 15, 2008
Discussion
In light of the therapeutic potential of FTY720, the mechanismunderlying its antitumor effect warrants investigations. In thisstudy, we obtained evidence that FTY720 suppressed the prolifer-ation of HCC cells, at least in part, by stimulating ROS production,leading to PKCy activation and subsequent caspase-3–dependentapoptosis. From a mechanistic perspective, ROS productionrepresents an early hallmark event in the apoptotic effect ofmany therapeutic agents, including N-(4-hydroxyphenyl)-retina-mide (34, 35), arsenic trioxide (36), parthenolide (37), and cisplatin(38). Relative to these agents, FTY720 represents a structurallydistinct type of small-molecule agent with unique mechanisticfeatures, which could be exploited to foster novel therapeuticstrategies for HCC treatment.It is well-documented that drug-induced ROS stress triggers a
cascade of redox-dependent signaling events at difference cellularlevels, culminating in mitochondria-dependent apoptosis (39).This study obtains evidence in HCC cells that PKCy represents amajor downstream effector of FTY720-generated ROS to facilitatecaspase-3–dependent apoptosis, and that blockade of ROSproduction by the NADPH oxidase inhibitor DPI would protectthe cells from the effect of FTY720 on PKCy activation andconsequent apoptosis. From a mechanistic perspective, thissignaling pathway provides a molecular basis to account for thedifferential susceptibility of different HCC cell lines to FTY720-mediated apoptosis. As shown, the expression levels of PKCy andGST-k represent two key determinants for the cellular capability tocope with FTY720-incurred oxidative stress, which underlie theresistant phenotype of PLC5 cells to the apoptotic effects ofFTY720. Consequently, these two biomarkers might be used aspredictors to select patients to receive FTY720 therapy. Moreover,GST-k overexpression has been reported in a number of humanmalignancies, and there exists a strong correlation between hightumor GST-k levels and failure of patients to respond to
chemotherapy and low patient survival rates (40). For a mecha-nistic perspective, therapeutic interference of GST-k expression/activity by antisense or small-molecule agents provides a viablestrategy to overcome this resistance (41, 42).PKCy has been shown to play an intriguing role in regulating
apoptosis, either proapoptotic or antiapoptotic, in different cellsystems (43, 44). This dichotomous behavior might be, in part,controlled by phosphorylation at different tyrosine residues ofPKCy by different kinases. For example, stress signals such asetoposide and H2O2 promoted the phosphorylation of tyrosineresidues at 64 and 187 (45), and especially 311 (29), respectively,which presumably committed PKCy to activating caspases throughphosphorylation. On the other hand, phosphorylation on Tyr155was reported to promote the antiapoptotic effect of PKCy inSindbis virus–infected glioma cells (46). Accordingly, the selectivephosphorylation of PKCy at Try311, but not Tyr155, in FTY720-treated Huh7 cells underscores the proapoptotic nature of thissignaling pathway.An earlier report indicates that FTY720 induced apoptosis in
HCC cells through phosphoinositide 3-kinase–mediated Aktdephosphorylation (8). This premise, however, was disputed bythe finding that overexpression of constitutively active Akt couldnot rescue Huh7 cells from FTY720-mediated cell death.However, it is noteworthy that the mode of antitumor actionof FTY720 might vary in different types of tumor cells. Forexample, in hematologic malignant cells such as those of chronicmyelogenous leukemia and chronic lymphocytic leukemia,FTY720 induced caspase-independent apoptosis by activatingPP2A signaling (47, 48). This PP2A activation, however, was notnoted in HCC or prostate cancer cells.4 This cell line specificity
Figure 5. FTY720 stimulates ROS production inHCC cells. A, time-dependent effect of FTY720(10 Amol/L) on ROS production. Left, Huh7 cellswere exposed to 10 Amol/L FTY720 in 10%FBS–containing DMEM for the indicated timeintervals, stained with DCFDA, and subjected toflow cytometric analysis as described in Materialsand Methods. Right, relative ROS production inHuh7 cells treated with 10 Amol/L FTY720 for theindicated time intervals. Columns, means; bars,FSD (n = 3). B, dose-dependent effect of FTY720on stimulating different extents of ROS productionin Hep3B, Huh7, and PLC5 cells after 1 h oftreatment. Columns, means; bars, FSD (n = 3).C, differential expression of GST-k protein in thethree HCC cell lines.
4 Y.S. Lu and C.S. Chen, unpublished data.
Cancer Research
Cancer Res 2008; 68: (4). February 15, 2008 1210 www.aacrjournals.org
underlines the pleiotropic nature of FTY720 in killing cancercells. In light of the heterogeneity in molecular and cellularabnormalities associated with oncogenesis and tumor progres-sion, the ability of FTY720 to target different clinically relevantsignaling mechanisms in different cancer types might have thera-peutic relevance in cancer therapy. Consequently, this rationaleconstitutes the impetus of defining the causative mechanismsfor the antitumor effects of FTY720 in different types of cancercells.In conclusion, this study shows the involvement of ROS-PKCy
signaling in FTY720-mediated apoptosis in HCC cells, of which thetherapeutic relevance is multifold. First, as PKCy is mostlyactivated by genotoxic signals such as ionizing radiation (32) andetoposide (49), FTY720 represents a novel PKCy activator withoutknown genotoxic effects. Second, dissociation of the antitumoreffect of FTY720 from its S1P activity provides a molecular basis to
use it as a scaffold to develop potent antitumor agents devoid ofthe immunosuppressive activity. Third, FTY720 and its derivativeswill be inherently suitable to treat cancers exhibiting increasedoxidative stress with high levels of cellular ROS and a lowantioxidant capability. Human HCC is prevalent in somatic GST-pgene silencing due to CpG island DNA hypermethylation (50),which provides a mechanistic rationale to include this type of ROS-generating agents in HCC therapy.
Acknowledgments
Received 7/11/2007; revised 11/9/2007; accepted 12/11/2007.Grant support: Public Health Service Grant CA112250 from National Cancer
Institute, a Leukemia and Lymphoma Society grant, and the Lucius A. Wing EndowedChair Fund.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
Figure 6. Mechanistic link between ROS generationand PKCy activation. A, effect of the ROS inhibitor DPIand the PKCy inhibitor rottlerin on FTY720-mediatedROS production. Huh7 cells were exposed to DMSOvehicle, rottlerin (10 Amol/L), or DPI (10 Amol/L) for30 min, followed by cotreatment with DMSO vehicle or10 Amol/L FTY720 for 1 h, stained with DCFDA, andsubjected to flow cytometric analysis for ROS. B, DPIinhibits the effect of FTY720 (10 Amol/L) on PKCyphosphorylation (left) and proteolytic cleavage (right ).For the assessment of PKCy cleavage, Huh7 cellswere exposed to DPI (10 Amol/L) for 30 min, followedby cotreatment with FTY720 (10 Amol/L) in 10%FBS–containing medium for 24 h. C, DPI (10 Amol/L)and rottlerin (10 Amol/L) protect Huh7 cells from theeffect of FTY720 (10 Amol/L) on stimulating caspase-3activation (left ) and nucleosomal fragmentation(right ). Cells were treated with individual agents asaforementioned. Columns, means; bars, FSD (n = 3).D , DPI lacks effect on inhibiting FTY720-mediated Aktphosphorylation. Cells were treated with individualagents as aforementioned.
FTY720 Mediates Apoptosis through PKCd Activation
www.aacrjournals.org 1211 Cancer Res 2008; 68: (4). February 15, 2008

Cancer Research
Cancer Res 2008; 68: (4). February 15, 2008 1212 www.aacrjournals.org
References1. Tedesco-Silva H, Mourad G, Kahan BD, et al. FTY720, anovel immunomodulator: efficacy and safety resultsfrom the first phase 2A study in de novo renaltransplantation. Transplantation 2004;77:1826–33.
2. Kappos L, Antel J, Comi G, et al. Oral fingolimod(FTY720) for relapsing multiple sclerosis. N Engl J Med2006;355:1124–40.
3. Brinkmann V, Davis MD, Heise CE, et al. The immunemodulator FTY720 targets sphingosine 1-phosphatereceptors. J Biol Chem 2002;277:21453–7.
4. Graler MH, Goetzl EJ. The immunosuppressant FTY720down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J 2004;18:551–3.
5. Matsuoka Y, Nagahara Y, Ikekita M, et al. A novelimmunosuppressive agent FTY720 induced Akt dephos-phorylation in leukemia cells. Br J Pharmacol 2003;138:1303–12.
6. Yasui H, Hideshima T, Raje N, et al. FTY720 inducesapoptosis in multiple myeloma cells and overcomesdrug resistance. Cancer Res 2005;65:7478–84.
7. Ho JW, Man K, Sun CK, et al. Effects of a novelimmunomodulating agent, FTY720, on tumor growthand angiogenesis in hepatocellular carcinoma. MolCancer Ther 2005;4:1430–8.
8. Lee TK, Man K, Ho JW, et al. FTY720 inducesapoptosis of human hepatoma cell lines through PI3-K-mediated Akt dephosphorylation. Carcinogenesis2004;25:2397–405.
9. Chua CW, Lee DT, Ling MT, et al. FTY720, a fungusmetabolite, inhibits in vivo growth of androgen-independent prostate cancer. Int J Cancer 2005;117:1039–48.
10. Permpongkosol S, Wang JD, Takahara S, et al.Anticarcinogenic effect of FTY720 in human prostatecarcinoma DU145 cells: modulation of mitogenicsignaling, FAK, cell-cycle entry and apoptosis. Int JCancer 2002;98:167–72.
11. Wang JD, Takahara S, Nonomura N, et al. Earlyinduction of apoptosis in androgen-independent pros-tate cancer cell line by FTY720 requires caspase-3activation. Prostate 1999;40:50–5.
12. Zhou C, Ling MT, Kin-Wah Lee T, et al. FTY720,a fungus metabolite, inhibits invasion ability ofandrogen-independent prostate cancer cells throughinactivation of RhoA-GTPase. Cancer Lett 2006;233:36–47.
13. Azuma H, Horie S, Muto S, et al. Selective cancer cellapoptosis induced by FTY720; evidence for a Bcl-dependent pathway and impairment in ERK activity.Anticancer Res 2003;23:3183–93.
14. Ubai T, Azuma H, Kotake Y, et al. FTY720 inducedBcl-associated and Fas-independent apoptosis in hu-man renal cancer cells in vitro and significantly reducedin vivo tumor growth in mouse xenograft. AnticancerRes 2007;27:75–88.
15. Tanaka T, Takahara S, Hatori M, et al. A novelimmunosuppressive drug, FTY720, prevents the cancerprogression induced by cyclosporine. Cancer Lett 2002;181:165–71.
16. Brinkmann V, Wilt C, Kristofic C, et al. FTY720:dissection of membrane receptor-operated, stereospe-cific effects on cell migration from receptor-indepen-
dent antiproliferative and apoptotic effects. TransplantProc 2001;33:3078–80.
17. Ries LAG, Melbert D, Krapcho M, et al. SEER CancerStatistics Review, 1975-2004, National Cancer Institute.Bethesda (MD): 2007.
18. Rougier P, Mitry E, Barbare JC, et al. Hepatocellularcarcinoma (HCC): an update. Semin Oncol 2007;34:S12–20.
19. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase IIstudy of sorafenib in patients with advanced hepatocel-lular carcinoma. J Clin Oncol 2006;24:4293–300.
20. Anantharam V, Kitazawa M, Wagner J, et al. Caspase-3-dependent proteolytic cleavage of protein kinase Cy isessential for oxidative stress-mediated dopaminergiccell death after exposure to methylcyclopentadienylmanganese tricarbonyl. J Neurosci 2002;22:1738–51.
21. Kitazawa M, Anantharam V, Kanthasamy AG. Diel-drin induces apoptosis by promoting caspase-3-depen-dent proteolytic cleavage of protein kinase Cy indopaminergic cells: relevance to oxidative stress anddopaminergic degeneration. Neuroscience 2003;119:945–64.
22. Frasch SC, Henson PM, Kailey JM, et al. Regulation ofphospholipid scramblase activity during apoptosis andcell activation by protein kinase Cy. J Biol Chem 2000;275:23065–73.
23. Emoto Y, Kisaki H, Manome Y, et al. Activation ofprotein kinase Cy in human myeloid leukemia cellstreated with 1-h-D-arabinofuranosylcytosine. Blood1996;87:1990–6.
24. Matassa AA, Carpenter L, Biden TJ, et al. PKCy isrequired for mitochondrial-dependent apoptosis insalivary epithelial cells. J Biol Chem 2001;276:29719–28.
25. Kiuchi M, Adachi K, Kohara T, et al. Synthesis andimmunosuppressive activity of 2-substituted 2-amino-propane-1,3-diols and 2-aminoethanols. J Med Chem2000;43:2946–61.
26. Takehara T, Liu X, Fujimoto J, et al. Expression androle of Bcl-xL in human hepatocellular carcinomas.Hepatology 2001;34:55–61.
27. Wan X, Helman LJ. Levels of PTEN protein modulateAkt phosphorylation on serine 473, but not on threonine308, in IGF-II-overexpressing rhabdomyosarcomas cells.Oncogene 2003;22:8205–11.
28. Denning MF, Wang Y, Tibudan S, et al. Caspaseactivation and disruption of mitochondrial membranepotential during UV radiation-induced apoptosis ofhuman keratinocytes requires activation of proteinkinase C. Cell Death Differ 2002;9:40–52.
29. Konishi H, Yamauchi E, Taniguchi H, et al. Phos-phorylation sites of protein kinase C y in H2O2-treatedcells and its activation by tyrosine kinase in vitro . ProcNatl Acad Sci U S A 2001;98:6587–92.
30. Kajimoto T, Ohmori S, Shirai Y, et al. Subtype-specifictranslocation of the y subtype of protein kinase C andits activation by tyrosine phosphorylation induced byceramide in HeLa cells. Mol Cell Biol 2001;21:1769–83.
31. Emoto Y, Manome Y, Meinhardt G, et al. Proteolyticactivation of protein kinase C y by an ICE-like proteasein apoptotic cells. EMBO J 1995;14:6148–56.
32. Denning MF, Wang Y, Nickoloff BJ, et al. Proteinkinase Cy is activated by caspase-dependent proteolysisduring ultraviolet radiation-induced apoptosis of humankeratinocytes. J Biol Chem 1998;273:29995–30002.
33. Aliya S, Reddanna P, Thyagaraju K. Does glutathioneS-transferase k (GST-k) a marker protein for cancer?Mol Cell Biochem 2003;253:319–27.
34. Simeone AM, Ekmekcioglu S, Broemeling LD, et al. Anovel mechanism by which N-(4-hydroxyphenyl)retina-mide inhibits breast cancer cell growth: the productionof nitric oxide. Mol Cancer Ther 2002;1:1009–17.
35. Wu JM, DiPietrantonio AM, Hsieh TC. Mechanism offenretinide (4-HPR)-induced cell death. Apoptosis 2001;6:377–88.
36. Jing Y, Dai J, Chalmers-Redman RM, et al. Arsenictrioxide selectively induces acute promyelocytic leuke-mia cell apoptosis via a hydrogen peroxide-dependentpathway. Blood 1999;94:2102–11.
37. Wen J, You KR, Lee SY, et al. Oxidative stress-mediated apoptosis. The anticancer effect of thesesquiterpene lactone parthenolide. J Biol Chem 2002;277:38954–64.
38. Siddik ZH. Cisplatin: mode of cytotoxic action andmolecular basis of resistance. Oncogene 2003;22:7265–79.
39. Pelicano H, Carney D, Huang P. ROS stress in cancercells and therapeutic implications. Drug Resist Updat2004;7:97–110.
40. McIlwain CC, Townsend DM, Tew KD. GlutathioneS-transferase polymorphisms: cancer incidence andtherapy. Oncogene 2006;25:1639–48.
41. Ban N, Takahashi Y, Takayama T, et al. Transfectionof glutathione S-transferase (GST)- k antisense comple-mentary DNA increases the sensitivity of a colon cancercell line to adriamycin, cisplatin, melphalan, andetoposide. Cancer Res 1996;56:3577–82.
42. Turcotte S, Averill-Bates DA. Sensitization to thecytotoxicity of melphalan by ethacrynic acid andhyperthermia in drug-sensitive and multidrug-resistantChinese hamster ovary cells. Radiat Res 2001;156:272–82.
43. Brodie C, Blumberg PM. Regulation of cell apoptosisby protein kinase c y. Apoptosis 2003;8:19–27.
44. Jackson DN, Foster DA. The enigmatic protein kinaseCy: complex roles in cell proliferation and survival.FASEB J 2004;18:627–36.
45. Blass M, Kronfeld I, Kazimirsky G, et al. Tyrosinephosphorylation of protein kinase Cy is essential for itsapoptotic effect in response to etoposide. Mol Cell Biol2002;22:182–95.
46. Zrachia A, Dobroslav M, Blass M, et al. Infection ofglioma cells with Sindbis virus induces selectiveactivation and tyrosine phosphorylation of proteinkinase C y. Implications for Sindbis virus-inducedapoptosis. J Biol Chem 2002;277:23693–701.
47. Liu Q, Zhao X, Frissora F, et al. FTY720 demonstratespromising pre-clinical activity for chronic lymphocyticleukemia and lymphoblastic leukemia/lymphoma.Blood 2007;111:275–84.
48. Neviani P, Santhanam R, Oaks JJ, et al. FTY720, a newalternative for treating blast crisis chronic myelogenousleukemia and Philadelphia chromosome-positive acutelymphocytic leukemia. J Clin Invest 2007;117:2408–21.
49. Reyland ME, Anderson SM, Matassa AA, et al. Proteinkinase C y is essential for etoposide-induced apoptosisin salivary gland acinar cells. J Biol Chem 1999;274:19115–23.
50. Tchou JC, Lin X, Freije D, et al. GSTP1 CpG islandDNA hypermethylation in hepatocellular carcinomas.Int J Oncol 2000;16:663–76.
Related Documents











