Volume 4, Number 6, December 2018 Neurology.org/NG A peer-reviewed clinical and translational neurology open access journal ARTICLE Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy e281 ARTICLE Development of a rapid functional assay that predicts GLUT1 disease severity e297 ARTICLE Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation e289 ARTICLE Amyloid- and tau-PET Imaging in a familial prion kindred e290

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Volume 4, Number 6, December 2018Neurology.org/NG
A peer-reviewed clinical and translational neurology open access journal
ARTICLE
Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy e281
ARTICLE
Development of a rapid functional assay that predicts GLUT1 disease severity e297
ARTICLE
Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation e289
ARTICLE
Amyloid- and tau-PET Imaging in a familial prion kindred e290

Academy OfficersRalph L. Sacco, MD, MS, FAAN, PresidentJames C. Stevens, MD, FAAN, President ElectAnn H. Tilton, MD, FAAN, Vice PresidentCarlayne E. Jackson, MD, FAAN, SecretaryJanis M. Miyasaki, MD, MEd, FRCPC, FAAN, TreasurerTerrence L. Cascino, MD, FAAN, Past President
Executive Office, American Academy of NeurologyCatherine M. Rydell, CAE, Executive Director/CEO20l Chicago AveMinneapolis, MN 55415Tel: 612-928-6000
Editorial OfficePatricia K. Baskin, MS, Executive EditorKathleen M. Pieper, Senior Managing Editor, NeurologyLee Ann Kleffman, Managing Editor, Neurology: GeneticsSharon L. Quimby, Managing Editor, Neurology® Clinical PracticeMorgan S. Sorenson, Managing Editor,Neurology® Neuroimmunology & NeuroinflammationCynthia S. Abair, MA, Senior Graphics EditorAndrea R. Rahkola, Production Editor, NeurologyRobert J. Witherow, Senior Editorial AssociateKaren Skaja, Senior Editorial AssociateKaitlyn Aman Ramm, Editorial AssistantKristen Swendsrud, Editorial AssistantAndrea Willgohs, Editorial Assistant
PublisherWolters KluwerBaltimore, MD
Publishing StaffKim Jansen, Executive PublisherJessica Heise, Production Team Leader, Neurology JournalsMegen Miller, Production EditorSteve Rose, Editorial AssistantStacy Drossner, Production Associate
Neurology® is a registered trademark of the American Academy of Neurology(registration valid in the United States).
Neurology® Genetics (eISSN 2376-7839) is an open access journal publishedonline for the American Academy of Neurology, 201 Chicago Avenue,Minneapolis, MN 55415, by Wolters Kluwer Health, Inc. at 14700 CiticorpDrive, Bldg. 3, Hagerstown, MD 21742. Business offices are located at TwoCommerce Square, 2001 Market Street, Philadelphia, PA 19103. Productionoffices are located at 351 West Camden Street, Baltimore, MD 21201-2436.© 2018 American Academy of Neurology.
Neurology® Genetics is an official journal of the American Academy ofNeurology. Journal website: Neurology.org/ng, AAN website: AAN.com
Copyright and Permission Information: Please go to the journal website(www.neurology.org/ng) and click the Permissions tab for the relevantarticle. Alternatively, send an email to [email protected] information about permissions can be found here: https://shop.lww.com/journal-permission.
Disclaimer: Opinions expressed by the authors and advertisers are notnecessarily those of the American Academy of Neurology, its affiliates, or ofthe Publisher. The American Academy of Neurology, its affiliates, and thePublisher disclaim any liability to any party for the accuracy, completeness,efficacy, or availability of the material contained in this publication(including drug dosages) or for any damages arising out of the useor non-use of any of the material contained in this publication.
Advertising Sales Representatives: Wolters Kluwer, 333 Seventh Avenue,New York, NY 10001. Contacts: Eileen Henry, tel: 732-778-2261, fax: 973-215-2485, [email protected] and in Europe: Craig Silver, tel: +447855 062 550 or e-mail: [email protected].
Careers & Events:Monique McLaughlin, Wolters Kluwer, Two CommerceSquare, 2001Market Street, Philadelphia, PA 19103, tel: 215-521-8468, fax: 215-521-8801; [email protected].
Reprints:Meredith Edelman, Commercial Reprint Sales, Wolters Kluwer, TwoCommerce Square, 2001Market Street, Philadelphia, PA 19103, tel: 215-356-2721;[email protected]; [email protected].
Special projects: US & Canada: Alan Moore, Wolters Kluwer, TwoCommerce Square, 2001 Market Street, Philadelphia, PA 19103, tel:215-521-8638, [email protected]. International: AndrewWible, Senior Manager, Rights, Licensing, and Partnerships, Wolters Kluwer;[email protected].

A peer-reviewed clinical and translational neurology open access journal Neurology.org/NG
Neurology® Genetics
EditorStefan M. Pulst, MD, Dr med, FAAN
Deputy EditorMassimo Pandolfo, MD, FAAN
Associate EditorsAlexandra Durr, MD, PhDMargherita Milone, MD, PhDRaymond P. Roos, MD, FAANJeffery M. Vance, MD, PhD
Editorial BoardHilaryCoon, PhDGiovanniCoppola,MDChantalDepondt,MD,PhDBrentL. Fogel,MD,PhD,FAANAnthony J.Griswold, PhDOrhunH.Kantarci,MDJulieR.Korenberg, PhD,MDMargheritaMilone,MD,PhDDavidePareyson,MDShojiTsuji,MD,PhDDinekeS. Verbeek, PhDDavidViskochil,MD,PhDJulianeWinkelmann,MDJuan I. Young, PhD
Neurology® Journals
Editor-in-ChiefRobert A. Gross, MD, PhD, FAAN
Deputy EditorBradford B. Worrall, MD, MSc, FAAN
Section Editors
BiostatisticsRichard J. Kryscio, PhDChristopher A. Beck, PhDSue Leurgans, PhD
Classification of Evidence EvaluationsGary S. Gronseth, MD, FAAN
PodcastsStacey L. Clardy, MD, PhDJeffrey B. Ratliff, MD, Deputy Podcast Editor
OmbudsmanDavid S. Knopman, MD, FAAN
Scientific Integrity AdvisorRobert B. Daroff, MD, FAAN
Classification of EvidenceReview Team
Melissa J. Armstrong,MDRichardL.Barbano,MD,PhD,FAANRichardM.Dubinsky,MD,MPH,FAANJeffrey J. Fletcher,MD,MScGaryM.Franklin,MD,MPH,FAANDavid S.Gloss II,MD,MPH&TMJohn J.Halperin,MD,FAANJasonLazarou,MSc,MDStevenR.Messe, MD, FAANPushpaNarayanaswami,MBBS,DM,
FAANAlexRae-Grant,MD
Vision Neurology®: Genetics will be the premier peer-reviewed journal in the field of neurogenetics.
Mission Neurology: Genetics will provide neurologistswith outstanding original contributions thatelucidate the role of genetic and epigeneticvariation in diseases and biological traits ofthe central and peripheral nervous systems.
EditorialInquiries
Tel: 612-928-6400Toll-free: 800-957-3182 (US)Fax: [email protected]
StayConnected
facebook.com/NeurologyGenetics
twitter.com/greenjournal
youtube.com/user/NeurologyJournal
TABLE OF CONTENTS Volume 4, Number 6, December 2018 Neurology.org/NG
Editorial
e299 The complex structure of ATXN2 genetic variationS.M. Pulst
Open Access Companion article, e283
Articles
e278 Anti-inflammatory effects of dietary vitamin D3 inpatients with multiple sclerosisR. Hashemi, M. Morshedi, M. Asghari Jafarabadi, D. Altafi,S. Saeed Hosseini-Asl, and S. Rafie-Arefhosseini
Open Access
e279 Novel genotype-phenotype and MRI correlations ina large cohort of patients with SPG7 mutationsC.A. Hewamadduma, N. Hoggard, R. O’Malley, M.K. Robinson,N.J. Beauchamp, R. Segamogaite, J. Martindale, T. Rodgers, G. Rao,P. Sarrigiannis, P. Shanmugarajah, P. Zis, B. Sharrack,C.J. McDermott, P.J. Shaw, and M. Hadjivassiliou
Open Access
e280 Molecular pathogenesis of human CD59 deficiencyN. Karbian, Y. Eshed-Eisenbach, A. Tabib, H. Hoizman, B.P. Morgan,O. Schueler-Furman, E. Peles, and D. Mevorach
Open Access
e281 Delineating FOXG1 syndrome: From congenitalmicrocephaly to hyperkinetic encephalopathyN. Vegas, M. Cavallin, C. Maillard, N. Boddaert, J. Toulouse,E. Schaefer, T. Lerman-Sagie, D. Lev, B. Magalie, S. Moutton,E. Haan, B. Isidor, D. Heron, M. Milh, S. Rondeau, C. Michot,S. Valence, S. Wagner, M. Hully, C. Mignot, A. Masurel, A. Datta,S. Odent, M. Nizon, L. Lazaro, M. Vincent, B. Cogne,A.M. Guerrot, S. Arpin, J.M. Pedespan, I. Caubel, B. Pontier,B. Troude, F. Rivier, C. Philippe, T. Bienvenu, M.-A. Spitz,A. Bery, and N. Bahi-Buisson
Open Access
e282 Identification of a new SYT2 variant validates anunusual distal motor neuropathy phenotypeN.I. Montes-Chinea, Z. Guan, M. Coutts, C. Vidal, S. Courel,A.P. Rebelo, L. Abreu, S. Zuchner, J.T. Littleton, andM.A. Saporta
Open Access
e285 TPP2 mutation associated with sterile braininflammation mimicking MSE.M. Reinthaler, E. Graf, T. Zrzavy, T. Wieland, C. Hotzy, C. Kopecky,S. Pferschy, C. Schmied, F. Leutmezer,M. Keilani, C.M. Lill, S. Hoffjan,J.T. Epplen, U.K. Zettl, M. Hecker, A. Deutschlander, S.G. Meuth,M. Ahram, B. Mustafa, M. El-Khateeb, C. Vilariño-Guell,A.D. Sadovnick, F. Zimprich, B. Tomkinson, T. Strom,W. Kristoferitsch, H. Lassmann, and A. Zimprich
Open Access
e286 Rare genetic variation implicated in non-Hispanicwhite families with Alzheimer diseaseG.W. Beecham, B. Vardarajan, E. Blue,W. Bush, J. Jaworski, S. Barral,A. DeStefano, K. Hamilton-Nelson, B. Kunkle, E.R. Martin, A. Naj,F. Rajabli, C. Reitz, T. Thornton, C. van Duijn, A. Goate, S. Seshadri,L.A. Farrer, E. Boerwinkle, G. Schellenberg, J.L. Haines, E. Wijsman,R. Mayeux, and M.A. Pericak-Vance, for The Alzheimer’s DiseaseSequencing Project
Open Access
e289 Mutation in POLR3K causes hypomyelinatingleukodystrophy and abnormal ribosomal RNAregulationI. Dorboz, H. Dumay-Odelot, K. Boussaid, Y. Bouyacoub, P. Barreau,S. Samaan, H. Jmel, E. Eymard-Pierre, C. Cances, C. Bar, A.-L. Poulat,C. Rousselle, F. Renaldo, M. Elmaleh- Berges, M. Teichmann, andO. Boespflug-Tanguy
Open Access
e290 Amyloid- and tau-PET imaging in a familial prionkindredD.T. Jones, R.A. Townley, J. Graff-Radford, H. Botha, D.S. Knopman,R.C. Petersen, C.R. Jack, Jr., V.J. Lowe, and B.F. Boeve
Open Access

e291 Copy number loss in SFMBT1 is commonamong Finnish and Norwegian patients withiNPHV.E. Korhonen, S. Helisalmi, A. Jokinen, I. Jokinen, J.-M. Lehtola,M. Oinas, K. Lonnrot, C. Avellan, A. Kotkansalo, J. Frantzen, J. Rinne,A. Ronkainen, M. Kauppinen, A. Junkkari, M. Hiltunen, H. Soininen,M. Kurki, J.E. Jaaskelainen, A.M. Koivisto, H. Sato, T. Kato,A.M. Remes, P.K. Eide, and V. Leinonen
Open Access
e292 Duplication and deletion upstream of LMNB1 inautosomal dominant adult-onset leukodystrophyN. Mezaki, T. Miura, K. Ogaki, M. Eriguchi, Y. Mizuno, K. Komatsu,H. Yamazaki, N. Suetsugu, S. Kawajiri, R. Yamasaki, T. Ishiguro,T. Konno, H. Nozaki, K. Kasuga, Y. Okuma, J.-I. Kira, H. Hara,O. Onodera, and T. Ikeuchi
Open Access
e293 Atrial fibrillation genetic risk differentiatescardioembolic stroke from other stroke subtypesS.L. Pulit, L.-C. Weng, P.F. McArdle, L. Trinquart, S.H. Choi,B.D. Mitchell, J. Rosand, P.I.W. de Bakker, E.J. Benjamin, P.T. Ellinor,S.J. Kittner, S.A. Lubitz, and C.D. Anderson, on behalf of the AtrialFibrillation Genetics Consortium and the International StrokeGenetics Consortium
Open Access
e294 Brain somatic mutations in SLC35A2 causeintractable epilepsy with aberrant N-glycosylationN.S. Sim, Y. Seo, J.S. Lim, W.K. Kim, H. Son, H.D. Kim, S. Kim, H.J. An,H.-C. Kang, S.H. Kim, D.-S. Kim, and J.H. Lee
Open Access
e295 Ataxia-telangiectasia-like disorder in a familydeficient for MRE11A, caused by a MRE11variantM. Sedghi, M. Salari, A.-R. Moslemi, A. Kariminejad, M. Davis,H. Goullee, B. Olsson, N. Laing, and H. Tajsharghi
Open Access Video
e296 Screening of novel restless legs syndrome–associatedgenes in French-Canadian familiesF. Akçimen, D. Spiegelman, A. Dionne-Laporte, Z. Gan-Or,P.A. Dion, and G.A. Rouleau
Open Access
e297 Development of a rapid functional assay thatpredicts GLUT1 disease severityS.M. Zaman, S.A. Mullen, S. Petrovski, S. Maljevic, E.V. Gazina,A.M. Phillips, G.D. Jones, M.S. Hildebrand, J. Damiano, S. Auvin,H. Lerche, Y.G. Weber, S.F. Berkovic, I.E. Scheffer, C.A. Reid, andS. Petrou
Open Access
e298 Leigh syndrome followed by parkinsonism in an adultwith homozygous c.626C>T mutation in MTFMTD.M. Hemelsoet, A.V. Vanlander, J. Smet, E. Vantroys, M. Acou,I. Goethals, T. Sante, S. Seneca, B. Menten, and R. Van Coster
Open Access
Clinical/Scientific Notes
e283 Homozygous 31 trinucleotide repeats in the SCA2allele are pathogenic for cerebellar ataxiaM. Tojima, G. Murakami, R. Hikawa, H. Yamakado, H. Yamashita,R. Takahashi, and M. Matsui
Open Access Editorial, e299
e284 Variable penetrance of Andersen-Tawil syndromein a family with a rare missense KCNJ2 mutationR. Deeb, A. Veerapandiyan, R. Tawil, and S. Treidler
Open Access
e287 A tropomyosin-receptor kinase-fused gene mutationassociates with vacuolar myopathyN.N.Madigan, J.A. Tracy,W.J. Litchy, Z.Niu,C.Chen,K. Ling, andM.Milone
Open Access
e288 Lysosomal dysfunction in TMEM106Bhypomyelinating leukodystrophyY. Ito, T. Hartley, S. Baird, S. Venkateswaran, C. Simons, N.I. Wolf,K.M. Boycott, D.A. Dyment, and K.D. Kernohan
Open Access
Correction
e300 Novel genotype-phenotype and MRI correlations ina large cohort of patients with SPG7 mutations
Cover imageDrosophila larval neuromuscular junctions stained for synaptotagmin(green) and neuronal membranes (blue). Mutations in humansynaptotagmin 2 disrupt synaptic transmission at neuromuscularjunctions.See e282
TABLE OF CONTENTS Volume 4, Number 6, December 2018 Neurology.org/NG

EDITORIAL OPEN ACCESS
The complex structure of ATXN2 geneticvariationStefan M. Pulst, MD, FAAN
Neurol Genet 2018;4:e299. doi:10.1212/NXG.0000000000000299
Correspondence
Dr. Pulst
In this issue, Tojima et al.1 describe the occurrence of a progressive cerebellar ataxia of 1-yearduration in an 81-year-old Japanese woman that was associated with the presence of 31 DNACAG repeats in the ATXN2 gene. The pathologic threshold for disease causing spinocerebellarataxia type 2 (SCA2) is usually considered to be 33 repeats and above, whereas 31 repeatswould not be considered to be causative for cerebellar neurodegeneration. The twist in this casereport is the fact that the patient carried 2 alleles with 31 repeats, suggesting that the 31-CAGrepeat allele acted in a recessive fashion.
The gene causing SCA2 was independently identified by 3 groups using different ethnic groupsin 1996.2–4 The mutation is an expansion of a CAG DNA repeat in the coding region of theATXN2 gene, encoding a polyglutamine. Although the lower threshold for dominant patho-logic alleles was originally thought to be ≥35 repeats, subsequent studies identified SCA2patients with ≥33 repeats.5,6 Consistent with dominant inheritance in human pedigrees, theCAG repeat expansion acts as a gain-of-function mutation. This is also supported by cerebellarneurodegeneration seen on transgenic overexpression of mutant ATXN27–9 and by absence ofa neurodegenerative phenotype in mice lacking functional Atxn2 alleles.10–12 Gain-of-functionof expanded ATXN2 is also supported by therapeutic responses to antisense oligonucleotidesthat lower ATXN2 expression in SCA2 mouse models.13
In most normal individuals, the repeat is once or twice interrupted by a CAA codon, which alsocodes for glutamine. In all populations, the 22-repeat allele is the most common, followed bythe 23-repeat allele. The frequency of the 27-repeat allele can be highly variable.
The ATXN2 gene is a good example for the complexities associated with genetic variation ina given gene and the associated risk for a number of diseases. At least 4 categories of variation canbe distinguished: dominant deterministic alleles leading to SCA2, a multisystem neurologicdisease affecting primarily or initially the cerebellum, repeat alleles that are unstable and althoughnot disease-causing in the carrier can expand to give rise to disease in the offspring, risk alleles forother neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) frontotemporaldementia (FTD) and ALS/FTD, dominant acting repeat alleles giving rise to noncerebellarphenotypes, and now also recessively acting alleles causing very late-onset cerebellar disease.
Other phenotypes associated with deterministicdominant allelesSCA2 patient phenotypes are dominated by cerebellar Purkinje cell and deep cerebellar nucleipathology. Careful clinical and pathologic examination also revealed the involvement of otherneurologic systems.14–17 Several years were needed, however, to appreciate that some of these“noncerebellar” phenotypes could occur in patients without cerebellar ataxia and that theycould even segregate in families. For example, parkinsonian signs and symptoms and L-dopa
From the Department of Neurology, University of Utah, Salt Lake City, UT.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
This work was supported by grants R01NS097903, RC4NS073009, and R56NS33123 from the National Institutes of Neurological Disorders and Stroke and the Noorda foundation.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
RELATED CLINICAL/
SCIENTIFIC NOTE
Homozygous 31trinucleotide repeats in theSCA2 allele are pathogenicfor cerebellar ataxia
Page e283
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

responsiveness are seen in many SCA2 patients in the pres-ence of cerebellar signs. In some patients, however, L-dopa–responsive Parkinson disease without overt cerebellar findingshas been described and this “restricted” phenotype can evensegregate in families.18–20
The importance of motor neuron degeneration in SCA2was highlighted initially by molecular studies that identifiedATXN2 as a protein interacting with TDP-43, a proteinmutated or aggregating inmost patients with ALS and in somewith FTD. These molecular insights prompted Elden et al.21
to examine ATXN2 alleles in patients with ALS. They showedthat alleles with ≥27 repeats were a risk factor for ALS. Sub-sequent meta-analyses in 2 nonoverlapping data sets led tomore precise assessments of risk associated with long normalalleles indicating that alleles with 27–29 repeats do not in-crease ALS risk22 and that the 27-repeat allele may actuallybe protective.23 For alleles with 30–34 repeats, ALS riskincreases in a length-dependent fashion. Pedigrees segre-gating an ataxia and an ALS phenotype in separate individ-uals also exist.24 Of note, the sister of the patient described inthe study by Tojima et al.1 developed ALS, although hergenotype is not known.
Meiotic and mitotic stabilityAs in other DNA repeat diseases, the ATXN2 CAG repeat ismeiotically and mitotically unstable. Meiotic instability leadsto the phenotypic phenomenon of anticipation. In one studyin Cuban SCA2 pedigrees, the repeat on average increased by;5 units, when inherited from the father, but only by ;1.5units when inherited from the mother.25 One-fifth of largeexpansions occurred in relatively short mutant alleles with 36repeats. The risk of expansion in normal alleles is unknown,although it seems likely that the risk increases with increasinglength of the normal allele and with the lack of interruptionsby CAA repeats. The presence of CAA interruptions may alsoinfluence phenotypic expression of ATXN2 repeat mutationsin that interrupted repeats are more stable in a lineage-dependent fashion during neurogenesis or during DNA repairin postmitotic cells.
ATXN2 variation in common diseaseIn addition to CAG repeat expansion, other genetic variationwithin or near the ATXN2 gene exists. This genetic variationhas largely been explored through genome-wide associationstudies. Common variants in ATXN2 have been associatedwith a number of disease traits such as obesity, insulin re-sistance, or glaucoma (reviewed in references 26 and 27). TheATXN2 locus is also thought to influence human longevity.28
The recessive mode of alleles with 31 repeats is not totallysurprising as an effect of normal alleles on age at onset ofSCA2 had been reported. These results, however, were largelyfocused on the more common alleles of 23–27 repeats and
showed that CAG repeat length in the normal allele was in-versely related to age at onset in SCA2.29
The results of the study by Tojima et al.1 deserve confirma-tion. Despite the most diligent efforts, phenocopies andpresence of other genetic variants or environmental effectscan never be completely excluded. Although a true causalrelationship between the 31/31 genotype and very late-onsetataxia is difficult to prove, the rarity of the CAG31 allele andespecially the 31/31 genotype would strengthen a causal re-lationship. A fertile population to examine the presence ofrecessive alleles and the importance of repeat interruptionsexists in the Holguin province, Cuba.25,30
In summary, genetic counseling for individuals with longnormal ATXN2 repeat alleles will require a very nuancedapproach, correct determination of repeat length, andknowledge of the precise repeat configuration. The instabilityof the repeat when transmitted to offspring needs to be dis-cussed as well as the increased relative risk for ALS. Fortu-nately, long normal ATXN2 repeat alleles are rare in thegeneral population.
AcknowledgmentThe author thank Daniel Scoles, PhD for critical reading ofthe manuscript and suggestions.
Study fundingSupported by NIH grants R37 NS033123, UO1 NS103883,and R21 NS103009.
DisclosureS.M. Pulst serves on the editorial boards of the Journal of Cere-bellum, NeuroMolecular Medicine, Experimental Neurology, Neu-rogenetics, Nature Clinical Practice, and Neurology: Genetics;receives research support from the NIH and the National AtaxiaFoundation; has served on the speakers’ bureau of AthenaDiagnostics; receives publishing royalties from Churchill Liv-ingston, AAN Press, Academic Press, and Oxford UniversityPress; has received license fee payments from Cedars-SinaiMedical Center; holds multiple patents; and receives an hono-rarium from the AAN as the Editor of Neurology: Genetics. Fulldisclosure form information provided by the authors is availablewith the full text of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics November 2, 2018. Accepted in finalform November 2, 2018.
References1. Tojima M, Murakami G, Hikawa R, et al. Homozygous 31 trinucleotide repeats in the
SCA2 allele are pathogenic for cerebellar ataxia. Neurol Genet 2018;4:e283.2. Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally
biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996;14:269–276.
3. Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2gene using a direct identification of repeat expansion and cloning technique, DI-RECT. Nat Genet 1996;14:277–284.
4. Imbert G, Saudou F, Yvert G, et al. Cloning of the gene for spinocerebellar ataxia 2reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet1996;14:285–291.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

5. Fernandez M,McClain ME, Martinez RA, et al. Late-onset SCA2: 33 CAG repeats aresufficient to cause disease. Neurology 2000;55:569–572.
6. Hoche F, Baliko L, den Dunnen W, et al. Spinocerebellar ataxia type 2 (SCA2):identification of early brain degeneration in one monozygous twin in the initial diseasestage. Cerebellum 2011;10:245–253.
7. Huynh DP, Figueroa K, Hoang N, Pulst SM. Nuclear localization or inclusion bodyformation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse of human.Nat Genet 2000;1:44–50.
8. Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and geneexpression precede behavioral pathology in mouse model of SCA2. Hum Mol Gene2013;22:271–273.
9. Dansithong W, Paul S, Figueroa KP, et al. Ataxin-2 regulates RGS8 translation ina new BAC-SCA2 transgenic mouse model. PLoS Genet 2015:11:e1005182
10. Kiehl TR, Nechiporuk A, Figueroa KP, Keating MT, Huynh DP, Pulst SM. Gener-ation and characterization of SCA2 (ataxian-2) knockout mice. Biochem Biophys ResCommun 2006;339:17–24.
11. Huynh DP, Maalouf M, Silva AJ, Schweizer FE, Pulst SM Dissociated fear and spatiallearning in mice with deficiency of ataxin-2. PLoS ONE 2009;4:e6235
12. Lasters-Becker I, Brodesser S, Lutjohann D, et al. Insulin receptor and lipid metab-olism pathology in ataxin-2 knock-out mice. Hum Mol Genet 2008;17:1465–1481.
13. Scoles DR, Meera P, Schneider MD, et al. Antisense oligonucleotide therapy forspinocerebellar ataxia type 2. Nature 2017;554:362–366.
14. Cancel G, Durr A, Didierjean O, et al. Molecular and clinical correlations in spino-cerebellar ataxia 2: a study of 32 families. Hum Mol Genet 1997;6:709–715.
15. Geschwind DH, Perlman S, Figueroa CP, Treiman LJ, Pulst SM. The prevalence andwide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat inpatients with autosomal dominant cerebellar ataxia. Am J Hum Genet 1997;60:842–850.
16. Schmitz-Hubsch T, Coudert M, Bauer P, et al. Spinocerebellar ataxia types 1, 2, 3, and6: disease severity and nonataxia symptoms. Neurology 2008;70:982–900.
17. Luo L, Wang J, Lo RY, et al. The initial symptom and motor progression in spino-cerebellar ataxias. Cerebellum 2017;16:615–622.
18. Gwinn-Hardy K, Chen JY, Liu HC, et al. Spinocerebellar ataxia type 2 with parkin-sonism in ethnic Chinese. Neurology 2000;55:800–805.
19. Furtado S, Payami H, Lockhart PJ, et al. Profile of families with parkinsonism: pre-dominant spinocerebellar ataxia type 2 (SCA2). Mov Disord 2004;19:622–629.
20. Simon-Sanchez J, Hanson M, Singleton A, et al. Analysis of SCA-2 and SCA-3 repeatsin parkinsonism: evidence of SCA-2 expansion in a family with autosomal dominantParkinson’s disease. Neurosci Lett 2005;382:191–194.
21. Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamineexpansions are associated with increased risk for ALS. Nature 2010;466:1069–1075.
22. Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM. Amyotrophic lateral sclerosisrisk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta–analysis.JAMA Neurol 2014;71:1529–1534.
23. SprovieroW, Shatunov A, Stahl D, et al. ATXN2 trinucleotide repeat length correlateswith risk of ALS. Neurobiol Aging 2017;51:178.e1–178.e9.
24. Tazen S, Figueroa K, Kwan JY, et al. Amyotrophic lateral sclerosis and spinocerebellarataxia type 2 in a family with full CAG repeat expansions of AXTN2. Neurol 2013;70:1302–1304.
25. Figueroa KP, Coon H, Santos N, Velazquez L, Mederos LA, Pulst SM. Genetic analysisof age at onset variation in spinocerebellar ataxia type 2. Neurol Genet 2017;3:e155
26. Meierhofer D, Halbach M, Sen NE, Gispert S, Auburger G. Ataxin-2 (atxn2)-knock–out mice show branched chain amino acids and fatty acids pathway alterations.Mol Cel Proteomics 2016;15:1728–1739.
27. Wiggs JL, Pasquale LR. Genetic of glaucoma. Hum Mol Genet 2017;26:R21–R27.28. Pilling LC, Kuo CL, Sicinski K, et al. Human longevity: 25 genetic loci associated in
389,166 UK biobank participants. Aging (Albany NY) 2017;9:2504–2520.29. van de Warrenburg BP, Sinke RJ, Verschuuren-Bemelmans CC, et al. Spinocerebellar
ataxias in The Netherlands: prevalence and age at onset variance analysis. Neurology2002;58:702–708.
30. Lafitta-Mesa JM, Velazquez-Perez LC, Santos Falcon N, et al. Unexpanded and in-termediate CAG polymorphism at the SCA2 locus (ATX2) in the Cuban population:evidence about the origin of expanded SCA2 alleles. Eur J Hum Genet 2012;20:41–49.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

ARTICLE OPEN ACCESS
Anti-inflammatory effects of dietary vitaminD3 inpatients with multiple sclerosisReza Hashemi, MSc, Mohammad Morshedi, MSc,* Mohammad Asghari Jafarabadi, PhD,* Davar Altafi, MD,*
Seyed Saeed Hosseini-Asl, PhD, and Seyed Rafie-Arefhosseini, PhD
Neurol Genet 2018;4:e278. doi:10.1212/NXG.0000000000000278
Correspondence
Dr. Rafie-Arefhosseini
AbstractObjectiveTo assess the effects of dietary vitamin D3 on proinflammatory (interleukin-17A [IL-17A] andIL-6) and anti-inflammatory (IL-10) cytokines.
MethodsOur study was conducted on 75 participants who were divided into 3 groups: multiple sclerosisparticipants (MSPs, n = 25), first-degree relative participants (FDRPs, n = 25), and healthyparticipants (HPs, n = 25). All groups received 50,000 IU vitamin D3/wk for 8 weeks. Serum25-(OH) vitamin D3 levels and messenger RNA (mRNA) expression levels of ILs were de-termined using electrochemiluminescence assay and real-time PCR, respectively.
ResultsVitamin D3 affected the levels of IL-17A, IL-10, and IL-6 among the 3 groups (p < 0.001 for all).Levels of IL-17A (MSPs: fold change [FC] = 5.9, p = 0.014; FDRPs: FC = 5.2, p = 0.006; HPs:FC = 4.2, p = 0.012) and IL-6 (MSPs: FC = 5.6, p = 0.003; FDRPs: FC = 5.5, p = 0.002; HPs:FC = 5.1, p < 0.001) were downregulated after vitamin D3 treatment. In addition, levels of IL-10(MSPs: FC = 6.2, p = 0.005; FDRPs: FC = 4.6, p < 0.001; HPs: FC = 5.2, p < 0.001) wereupregulated after 8 weeks.
ConclusionsAlthough supplementation with vitamin D3 reduced themRNA expression levels of IL-17A andIL-6, it increased the mRNA expression level of IL-10 in all groups. However, these effects weremore considerable in the MSP group than in the other groups. Of interest, in a deficiency stateof serum vitaminD3, IL-17A expression had a positive feedback effect on the expression of IL-6.Conversely, in the sufficient state, IL-10 expression had a negative feedback effect on theexpression of IL-17A and IL-6.
*These authors contributed equally to the manuscript.
From the School of Nutrition and Food Sciences (R.H., M.M), Tabriz University of Medical Sciences; Road Traffic Injury Research Center (M.A.-J), Tabriz University of Medical Sciences;Ardabil Province (D.A.); Department of Genetics (S.S.H.-A.), School of Medicine, Ardabil University of Medical Sciences; Department of Biochemistry and Diet Therapy (S.R.-A.), Schoolof Nutrition and Food Sciences, Tabriz University of Medical Sciences, Iran.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was waived at the discretion of the Editor.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

There are 2 theories that attempt to elucidate the process ofinflammation in patients with multiple sclerosis (MS). First, it isproposed that the disease may rely on the dysregulation of anti-inflammatory cytokines, such as interleukin-10 (IL-10) andinterleukin-4 (IL-4), as well as proinflammatory cytokines suchas tumor necrosis factor-α (TNFα), interleukin-2 (IL-2), in-terferon-γ (IFN-γ), and interleukin-1β (IL-1β). These pro- andanti-inflammatory cytokines are produced by T-helper 1 (Th-1)and Th-2 cells, respectively.1 Proinflammatory cytokines aug-ment the permeability of the blood-brain barrier (BBB), allowingfor the demyelization and neurodegeneration of the CNS,whereas anti-inflammatory cytokines quell the production ofproinflammatory cytokines.2,3 Second, besides the incompletenotion of Th-1/Th-2 disruption, other studies have proposedthat Th-17 cells and IL-17 family members, such as IL-17A andIL-17F, are involved in the disease process. Specially, IL-17Aplays a significant role in the stimulation and secretion ofproinflammatory cytokines and in chronic CNS inflammation.4,5
The development of MS may begin in individuals who aregenetically susceptible.6 Some studies have revealed that first-degree relatives of patients with MS are a 10–256 or 20–407
times more likely to develop MS than the general population.
Hence, in the present study, we have proposed a schematicmodel for the fluctuation of pro- and anti-inflammatorycytokines by vitamin D3, referred to as the See-Saw model(figure e-1, links.lww.com/NXG/A115). In fact, we assessedthe response of these interleukins to supplementation withvitamin D3, as well as the stabilization and balance of thisscheme in all 3 groups. Ultimately, our major aim was to findan appropriate way to ameliorate the intensity of MS inafflicted patients and perhaps prevent the disease in 2 othergroups via nutrigenomics, especially in first-degree relatives.
MethodsStandard protocol approvals, registrations,and patient consentsThis study was approved by the Ethical Committee of TabrizUniversity of Medical Sciences, Iran (ethical code: IR.TBZ-MED.REC.1395.780) and Iranian Registry of Clinical Trials(IRCT201703033655N3). Informed consent forms wereobtained from all participants, and they could withdraw fromthe study by their own decision.
Study design and interventionThe study started on February 19, 2017, and ended onJune 10, 2017. All randomized participants completed the
trial. Twenty-five participants were randomized to eachgroup through simple random sampling. Allocation to eachgroup was through lottery, and random paper was con-cealed in draw balls (figure 1), including multiple sclerosisparticipants (MSPs) as the first group (n = 25), first-degreerelative participants (FDRPs) of patients with MS, such asson, daughter, sister, or brother as the second group (n =25), and healthy participants (HPs) as the third group (n =25). All groups received 50,000 IU of vitamin D3 orally(Zahravi Pharmaceutical Co, Tabriz, Iran) every Fridayand between lunch meals for 8 weeks. Serum 25-(OH)vitamin D3 and mRNA expression levels of interleukinswere measured before and after supplementation in allgroups.
Participants and eligibility criteriaThe sample size was estimated based on a previous study inIran,8 with an odds ratio (OR) of 6, confidence level of 95%,and power of 80%. It was predicted that 25 persons in eachgroup would be sufficient for the detection of changes inserum parameters and gene expression, using G-power. MSPsand FDRPs were selected from the Ardabil MS society. MSPswere diagnosed by a neurologist, according to the McDonaldcriteria. HPs were chosen from Ardabil University of MedicalSciences. To be included in the study, HPs were at the age of30 ± 10 years, able to give blood samples, and willing to takepart in the study. Malabsorption, taking medicines that in-teract with vitamin D3, calcium, and vitamin D3 supplemen-tation in the last 30 days, gestation, and lactation wereconsidered exclusion criteria. Demographic and diseasecharacteristics are shown in table 1.
Serum 25-(OH) vitamin D3 assayWhole-blood samples (10 mL) were obtained from the par-ticipants before and after the trial. To separate the sera, 5 mLof the blood samples was centrifuged at 1,233g for 10 minutesat 4°C. Serum levels of 25-(OH) vitamin D3 were measuredusing electrochemiluminescence assay.
RNA extraction and cDNA synthesisFive milliliters of blood samples was collected in anticoagulantEDTA tubes. Total RNA was extracted using the MN kit(MACHEREY-NAGEL, Germany), according to the manu-facturer’s instructions. Concentration, integration, and purityof RNA samples were determined by spectrometry, Nano-Drop (Thermo Scientific, Waltham, MA), and gel electro-phoresis. Five micrograms of total RNA was used for cDNAsynthesis with a random hexamer primer through Hyper-Script Reverse Transcriptase (GeneAll, South Korea) in 20 μLtotal reaction mixture.
GlossaryAPC = antigen-presenting cell; BBB = blood-brain barrier; FC = fold change; FDRP = first-degree relative participant; HP =healthy participant; IFN-γ = interferon-γ; IL = interleukin;MS =multiple sclerosis;MSP =multiple sclerosis participant;TGF-β = transforming growth factor-β; Th = T helper; VDR = vitamin D receptor; VDRE = vitamin D response element.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Real-time PCR analysisReal-time PCR was performed on a Roche Light cycler 96(version: 1.1.0.1320, Germany), using primers specific for IL-6, IL-10, and IL-17A, with β-actin as a housekeeping control.Primer and probe sequences are presented in table 2.9–12
Real-time PCR reactions were performed in a total volume of25 μL containing 4 μL of synthesized cDNA solution, 12.5 μLof RealQ Plus 2x Master Mix for probe (Ampliqon, Den-mark), 500 nM of each forward and reverse primers, and 250nM of the TaqMan probe. The amplification program in-cluded a prewarming step (10 minutes at 94°C), denaturationstep (94°C for 15 seconds), and an annealing/extension step(60°C for 60 seconds).
Outcome measuresThe primary outcome of the study was to identify any changesin the serum levels of 25-(OH) vitamin D3 or in the levels ofIL-17A, IL-10, and IL-6 mRNA expression in all 3 groups after8 weeks.
Adverse eventsParticipants were continuously monitored for any side effectsof vitamin D3 consumption from baseline of the study.However, no particular events were found.
Statistical analysisStatistical analysis was performed using SPSS version 23.0software (SPSS Inc, Chicago, IL). Data are presented usingmean ± SD for numeric normal variables and frequency(percent) for categorical variables. The one-way analysis ofvariance (ANOVA), followed by the post hoc Tukey test, wasused for group comparisons. The Paired t test was applied forwithin-group comparisons. Relative mRNA expression nor-malized to β-actin was calculated by theDDCTmethod,13 andthe fold change (FC) expression of each gene was calculatedby the ratio formula (ratio = 2−DDCT). In all analysis, p < 0.05were considered statistically significant.
Table 1 Demographic and disease characteristics
Variables MSP (n = 25) FDRP (n = 25) HP (n = 25)
Sexa
Female 21 (84%) 17 (68%) 20 (80%)
Male 4 (16%) 8 (32%) 5 (20%)
Age (y)b 32.6 ± 6 27.4 ± 6 31.7 ± 4.3
Duration of disease (y)b 8.1 ± 5.8 — —
MS historya
Yes 5 (20%) 25 (100%) —
No 20 (80%) — —
MS familya
Brother 1 (4%) 5 (20%) —
Sister 3 (12%) 12 (48%) —
Daughter 1 (4%) 5 (20%) —
Son — 3 (12%) —
Abbreviations: MSP = multiple sclerosis participant; FDRP = first-degreerelative participant; HP = healthy participant.a Data are presented as frequency (percent) for categorical variables.b Data are presented as mean ± SD for numeric normal variables.
Figure 1 Enrollment and selection of participants allocated to groups by simple random sampling
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

Data availabilityThe datasets applied and analyzed during the current studyare available from the corresponding and first authors onreasonable request from any qualified investigator.
ResultsThe flow diagram of the study is shown in figure e-2 (links.lww.com/NXG/A116). Of 186 participants, 75 participants(25 per each group) were eligible to be assigned to the studyintervention (figure 1).
Association between vitamin D3 treatmentand mRNA expression levels of IL-6, IL-17A,and IL-10
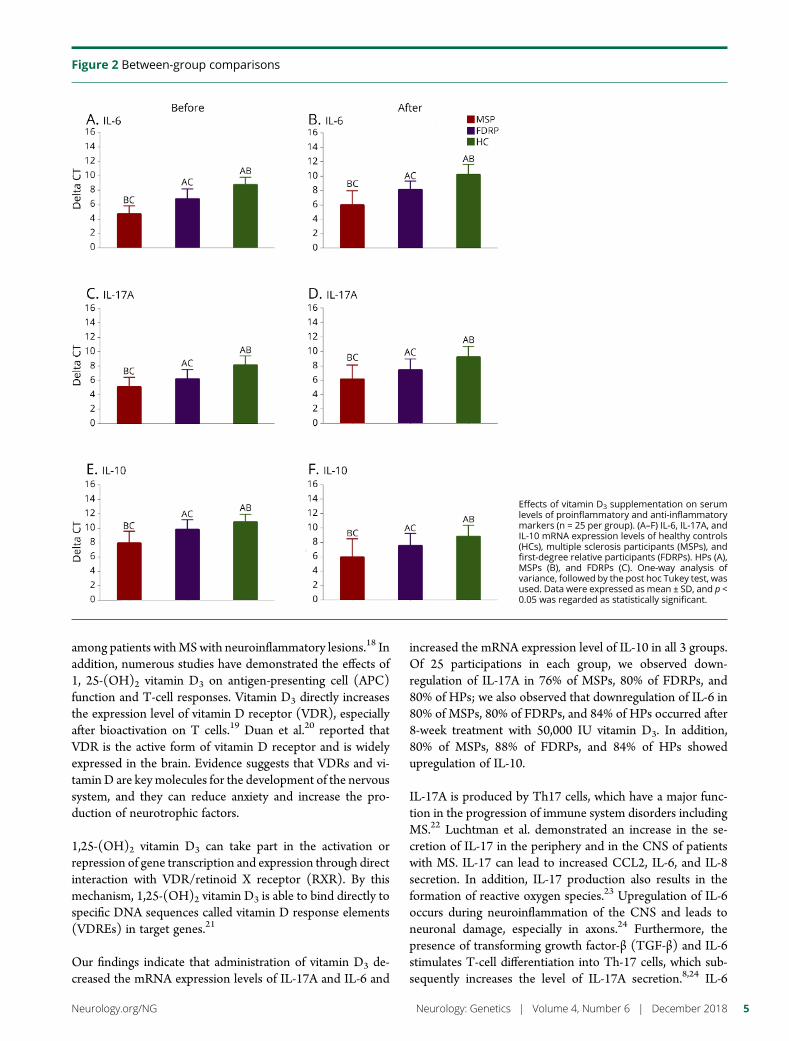
Between-group comparisonsBefore supplementationAt baseline, the results of one-way ANOVA showed that therewere differences in the mRNA expression levels of IL-6, IL-17A,and IL-10 (p < 0.001 for all) among the groups, but levels ofserumvitaminD3 (p= 0.063)were almost the same in all groups.The results of the pairwise comparison using the Tukey test alsorevealed differences between each 2 groups in IL-6 (MSPs andFDRPs: p < 0.001, FDRPs and HPs: p < 0.001, MSPs and HPs:p < 0.001), IL-17A (MSPs and FDRPs: p = 0.009, FDRPs andHPs: p = 0.025, MSPs and HPs: p < 0.001), and IL-10 (MSPsand FDRPs: p < 0.001, FDRPs and HPs: p < 0.001, MSPs andHPs: p < 0.001) (figures 2, A, C, and E and table 3).
After supplementationAfter 8 weeks of supplementation with vitamin D3, there weredifferences among groups in mRNA expression levels of IL-6(p < 0.001), IL-17A (p < 0.001), and IL-10 (p < 0.001), as wellas in the levels of serum vitamin D3 (p = 0.004). Pairwisecomparisons indicated that there were differences betweeneach 2 groups in IL-6 (MSPs and FDRPs: p < 0.001, FDRPsand HPs: p < 0.001, MSPs and HPs: p < 0.001), IL-17A(MSPs and FDRPs: p = 0.022, FDRPs and HPs: p = 0.001,MSPs and HPs: p < 0.001), IL-10 (MSPs and FDRPs: p =0.005, FDRPs and HPs: p < 0.001, MSPs and HPs: p < 0.001),and serum vitamin D3 (MSPs and FDRPs: p = 0.022, FDRPsand HPs: p = 0.887, MSPs and HPs: p = 0.006). However, nosignificant differences were found in the levels of serum vi-tamin D3 between the FDRP and HP groups (figures 2, B, D,and F and table 3).
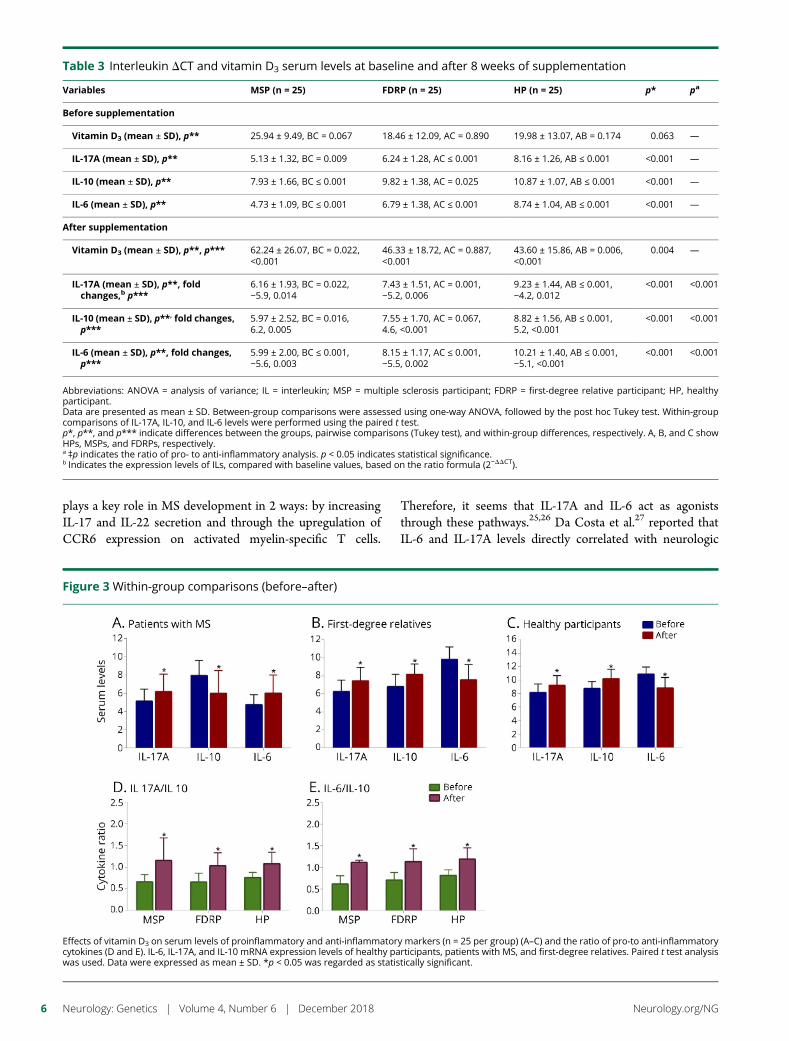
Within-group comparisons (before–after)The results of the paired t test analyses revealed that theproduction of proinflammatory cytokines (i.e., IL-17A and IL-6) decreased, whereas secretion of the anti-inflammatory cy-tokine, IL-10, increased in each group after intervention. Theobserved FCs in MSPs were −5.9 for IL-17A, (p = 0.014),−5.6 for IL-6 (p = 0.003), and 6.2 for IL-10 (p = 0.005). TheFCs in FDRPs were −5.2 for IL-17A (p = 0.006), −5.5 for IL-6(p = 0.002), and 4.6 for IL-10 (p < 0.001). In HPs, the FCswere −4.2 for IL-17A (p = 0.012), −5.1 for IL-6 (p < 0.001),and 5.2 for IL-10 (p < 0.001) (figures 3, A–C and table 3). Inaddition, the ratio of pro- to anti-inflammatory cytokines,including the ratio of IL-17A to IL-10 and IL-6 to IL-10, ineach group showed that there were differences within thegroups (MSPs, p < 0.001; FDRPs, p < 0.001; HPs, p < 0.001,figure 3, D and E).
DiscussionWith respect to the role of vitamin D3 in the immune systemand gene expression, we proposed a schematic balance forfluctuations of pro- (IL-17A and IL-6) and anti-inflammatory(IL-10) cytokines by vitamin D3 that we refer to as the See-Saw model.
In the present trial, 88% of MSPs, 84% of FDRPs, and 80% ofHPs showed a positive response after 8 weeks supplementa-tion with 50,000 IU vitamin D3. There were no differences inserum vitamin D3 levels among the groups at baseline. Thisfinding is similar to the results of other studies.14,15 However,our finding does not conform with that of other studies, whichindicated that serum levels of vitamin D3 in MSPs were lowerthan those in HPs.16,17 This contradiction may be justified byseveral reasons, including economic conditions, clothing, in-appropriate food habits, and above all, weather conditions andgeographical location (the mountainous weather of North-west of Iran with higher latitude and lower temperature).These may all cause lower exposure to sunlight and conse-quent insufficiency of serum vitamin D3. Several studies in-dicate that vitamin D3 deficiency is a common phenomenon
Table 2 Primers and probe sequences used for real-timePCR
Target Primer
IL-17A9
Forward AATCTCCACCGCAATGAGGA
Reverse ACGTTCCCATCAGCGTTGA
Probe FAM-CGGCACTTTGCCTCCCAGATCACA
IL-1010
Forward TGAGAACAGCTGCA CCCACTT
Reverse GCTGAAGGCATCTCGGAGAT
Probe FAM- CAGGCAACCTGCCTAACATGCTTCGA
IL-611
Forward GGTACATCCTCGACGGCATCT
Reverse GTGCCTCTTTGCTTTCAC
Probe FAM-TGTTACTCTTGTTACATGTCTCCTTTCTCAGGGCT
Beta-actin12
Forward TCACCCACACTGTGCCCATCTACGA
Reverse CAGCGGAACCGCTCATTGCCAATGG
Probe FAM-ATGCCCTCCCCCATGCCATC
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
among patients withMSwith neuroinflammatory lesions.18 Inaddition, numerous studies have demonstrated the effects of1, 25-(OH)2 vitamin D3 on antigen-presenting cell (APC)function and T-cell responses. Vitamin D3 directly increasesthe expression level of vitamin D receptor (VDR), especiallyafter bioactivation on T cells.19 Duan et al.20 reported thatVDR is the active form of vitamin D receptor and is widelyexpressed in the brain. Evidence suggests that VDRs and vi-taminD are keymolecules for the development of the nervoussystem, and they can reduce anxiety and increase the pro-duction of neurotrophic factors.
1,25-(OH)2 vitamin D3 can take part in the activation orrepression of gene transcription and expression through directinteraction with VDR/retinoid X receptor (RXR). By thismechanism, 1,25-(OH)2 vitamin D3 is able to bind directly tospecific DNA sequences called vitamin D response elements(VDREs) in target genes.21
Our findings indicate that administration of vitamin D3 de-creased the mRNA expression levels of IL-17A and IL-6 and
increased the mRNA expression level of IL-10 in all 3 groups.Of 25 participations in each group, we observed down-regulation of IL-17A in 76% of MSPs, 80% of FDRPs, and80% of HPs; we also observed that downregulation of IL-6 in80% of MSPs, 80% of FDRPs, and 84% of HPs occurred after8-week treatment with 50,000 IU vitamin D3. In addition,80% of MSPs, 88% of FDRPs, and 84% of HPs showedupregulation of IL-10.
IL-17A is produced by Th17 cells, which have a major func-tion in the progression of immune system disorders includingMS.22 Luchtman et al. demonstrated an increase in the se-cretion of IL-17 in the periphery and in the CNS of patientswith MS. IL-17 can lead to increased CCL2, IL-6, and IL-8secretion. In addition, IL-17 production also results in theformation of reactive oxygen species.23 Upregulation of IL-6occurs during neuroinflammation of the CNS and leads toneuronal damage, especially in axons.24 Furthermore, thepresence of transforming growth factor-β (TGF-β) and IL-6stimulates T-cell differentiation into Th-17 cells, which sub-sequently increases the level of IL-17A secretion.8,24 IL-6
Figure 2 Between-group comparisons
Effects of vitamin D3 supplementation on serumlevels of proinflammatory and anti-inflammatorymarkers (n = 25 per group). (A–F) IL-6, IL-17A, andIL-10 mRNA expression levels of healthy controls(HCs), multiple sclerosis participants (MSPs), andfirst-degree relative participants (FDRPs). HPs (A),MSPs (B), and FDRPs (C). One-way analysis ofvariance, followed by the post hoc Tukey test, wasused. Data were expressed as mean ± SD, and p <0.05 was regarded as statistically significant.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
plays a key role in MS development in 2 ways: by increasingIL-17 and IL-22 secretion and through the upregulation ofCCR6 expression on activated myelin-specific T cells.
Therefore, it seems that IL-17A and IL-6 act as agoniststhrough these pathways.25,26 Da Costa et al.27 reported thatIL-6 and IL-17A levels directly correlated with neurologic
Table 3 Interleukin DCT and vitamin D3 serum levels at baseline and after 8 weeks of supplementation
Variables MSP (n = 25) FDRP (n = 25) HP (n = 25) p* pa
Before supplementation
Vitamin D3 (mean ± SD), p** 25.94 ± 9.49, BC = 0.067 18.46 ± 12.09, AC = 0.890 19.98 ± 13.07, AB = 0.174 0.063 —
IL-17A (mean ± SD), p** 5.13 ± 1.32, BC = 0.009 6.24 ± 1.28, AC ≤ 0.001 8.16 ± 1.26, AB ≤ 0.001 <0.001 —
IL-10 (mean ± SD), p** 7.93 ± 1.66, BC ≤ 0.001 9.82 ± 1.38, AC = 0.025 10.87 ± 1.07, AB ≤ 0.001 <0.001 —
IL-6 (mean ± SD), p** 4.73 ± 1.09, BC ≤ 0.001 6.79 ± 1.38, AC ≤ 0.001 8.74 ± 1.04, AB ≤ 0.001 <0.001 —
After supplementation
Vitamin D3 (mean ± SD), p**, p*** 62.24 ± 26.07, BC = 0.022,<0.001
46.33 ± 18.72, AC = 0.887,<0.001
43.60 ± 15.86, AB = 0.006,<0.001
0.004 —
IL-17A (mean ± SD), p**, foldchanges,b p***
6.16 ± 1.93, BC = 0.022,−5.9, 0.014
7.43 ± 1.51, AC = 0.001,−5.2, 0.006
9.23 ± 1.44, AB ≤ 0.001,−4.2, 0.012
<0.001 <0.001
IL-10 (mean ± SD), p**, fold changes,p***
5.97 ± 2.52, BC = 0.016,6.2, 0.005
7.55 ± 1.70, AC = 0.067,4.6, <0.001
8.82 ± 1.56, AB ≤ 0.001,5.2, <0.001
<0.001 <0.001
IL-6 (mean ± SD), p**, fold changes,p***
5.99 ± 2.00, BC ≤ 0.001,−5.6, 0.003
8.15 ± 1.17, AC ≤ 0.001,−5.5, 0.002
10.21 ± 1.40, AB ≤ 0.001,−5.1, <0.001
<0.001 <0.001
Abbreviations: ANOVA = analysis of variance; IL = interleukin; MSP = multiple sclerosis participant; FDRP = first-degree relative participant; HP, healthyparticipant.Data are presented as mean ± SD. Between-group comparisons were assessed using one-way ANOVA, followed by the post hoc Tukey test. Within-groupcomparisons of IL-17A, IL-10, and IL-6 levels were performed using the paired t test.p*, p**, and p*** indicate differences between the groups, pairwise comparisons (Tukey test), and within-group differences, respectively. A, B, and C showHPs, MSPs, and FDRPs, respectively.a ‡p indicates the ratio of pro- to anti-inflammatory analysis. p < 0.05 indicates statistical significance.b Indicates the expression levels of ILs, compared with baseline values, based on the ratio formula (2−DDCT).
Figure 3 Within-group comparisons (before–after)
Effects of vitamin D3 on serum levels of proinflammatory and anti-inflammatory markers (n = 25 per group) (A–C) and the ratio of pro-to anti-inflammatorycytokines (D and E). IL-6, IL-17A, and IL-10 mRNA expression levels of healthy participants, patients with MS, and first-degree relatives. Paired t test analysiswas used. Data were expressed as mean ± SD. *p < 0.05 was regarded as statistically significant.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

disabilities in patients with MS. Moreover, 1,25-(OH)2 vita-min D3 decreases the secretion of proinflammatory cytokinesin vitro. Naghavi Gargari et al.8 indicated a positive associationbetween the mRNA expression of IL-6 and IL-17A. Supple-mentation of 1,25-(OH)2 vitamin D3 inhibits IL-17A pro-duction in T cells of patients with MS.28 In fact, vitamin D3
administration can ameliorate MS disease through to down-regulation of IL-17A and IL-6. On the other hand, a pivotalrole of anti-inflammatory cytokines, especially IL-10, stimu-lated by vitamin D3, has been established in the suppression ofT-cell activation through macrophages. Therefore, the arrestof IL-10 production may exacerbate inflammation.29,30
Thereby, vitamin D3 supplementation can increase IL-10 byT cells and reduce the production of IL-6 and IL-17 in bothpatient and control groups.31
Nevertheless, in several studies, contradictory results have alsobeen reported. For instance, Fujita et al. and Niino et al. haveboth shown that 1,25-(OH)2 vitamin D3 can downregulate theproduction of IL-10.32,33. In addition, studies by Naghavi Gar-gari et al. and Smolders et al. have demonstrated the upregu-lation of IL-6 and IL-17A by vitamin D.8,34 Meanwhile, Yaoet al.35 have shown that IL-6 has anti- and pro-inflammatoryeffects and that it can be secreted from various cell types,including lymphocytes, macrophages, and monocytes. Multiplefactors, including the variety of cell types studied, duration anddosage of 1,25(OH)2 vitamin D3 treatment, type of samples,genetic inheritance of patients, and polymorphisms in VDRgenes, have all been proposed to account for these contradictoryfindings as reported in several studies.8,14,29,36
In this study, we also compared the ratio of pro- to anti-inflammatory cytokines (IL-17A:IL-10 and IL-6:IL-10). In allgroups, both ratios were improved, but the most significantchanges occurred in MSPs. In addition, the changes in theratio of IL-6:IL-10 were found to be more important than thechanges in the ratio of IL-17A:IL-10. Therefore, these resultsmay confirm 2 points: First, vitamin D3 reduces the geneexpression levels of IL-6 and IL-17A. The expression level ofIL-6 decreases subsequent to the reduction in IL-17A ex-pression, which is why the ratio of in IL-6:IL10 was moretangible. Second, increased expression of IL-10 had a negativefeedback effect on the expression levels of IL-17A and IL-6.
To prove the accuracy of the proposed schematic (See-Sawmodel), the sample size should be increased in each group. Inaddition, a blank control group (out of the restricted criteria)should be more defined. In this state, a placebo would beadministered to determine the actual effects of vitamin D3 onthe expression levels of genes. Overall, the results suggest thatlong-term supplementation with a lower daily dosage may bebeneficial. Furthermore, participants should be assessed at themidpoint of the study to determine the temporal effect ofvitamin D3 supplementation. A reliable questionnaire shouldalso be devised and used for the effect of vitamin D3 sup-plements on the behavioral and mental performance of theparticipants before and after the intervention. The present
study and many other similar ones only examined the mRNAexpression levels of these cytokines; however, an analysis ofcytokine protein expression would be useful to confirm theeffect of vitamin D3. In addition, epigenetic studies may beuseful to address some of the contradictions presented byother studies. Epigenetics refers to the modification of geneexpression and changes in chromatin structure without al-teration of the DNA sequence.37 Recent studies suggest thatinteractions between environmental factors and epigeneticparameters are an underlying cause of MS.
Finally, we determined that upregulation of IL-10 anddownregulation of IL-17A and IL-6 occur at sufficient serumlevels of vitamin D3. These features were more noticeable inMSPs. As mentioned earlier, IL-17A and IL-6 augment theproduction of each other. Sufficient serum levels of vitaminD3 result in the production of IL-10 and have a negativefeedback effect on the expression levels of IL-17A and IL-6.Stabilization and proper balance of the schematic See-Sawmodel by sufficient levels of vitaminD3 seem to offer a feasiblemethod for protection from MS by dietary modifications.
Author contributionsS. Rafie-Arefhosseini and R. Hashemi proposed the See-Sawschematic model and wrote the study protocol and design.Davar Altafi, a neurologist, diagnosed patients with multiplesclerosis (MS). Reza Hashemi performed RNA extraction,cDNA synthesis, and real-time PCR under the supervision ofSeyed Saeed Hosseini-Asl and also performed analysis andinterpretation of the results. Reza Hashemi and MohammadMorshedi performed statistical analysis and interpretationunder the supervision of Mohammad Asghari-Jafarabadi.Mohammad Morshedi drew figures using Graph Prism soft-ware. Seyed Rafie-Arefhosseini, Reza Hashemi, and Moham-mad Morshedi involved in drafting the manuscript andrevising it critically for content. All authors have given theirfinal approval of the version to be published.
AcknowledgmentThe authors specially thank patients with MS for theirparticipation in the study and the Nutrition Research Centerof Tabriz University of Medical Sciences, Tabriz, Iran, andArdabil University of Medical Sciences and Imam KhomeiniHospital (Genetic Laboratory) and MS Society, Ardabil, Iran,for their financial support.
Study fundingThis study was financially supported by the Nutrition Re-search Center, Tabriz University of Medical Sciences, Tabriz,Iran, and Ardabil University of Medical Sciences, Ardabil,Iran. The results of this article were extracted from the MSc.Thesis of Reza Hashemi (Grant no.: 5/D/960060) was reg-istered at Tabriz University of Medical Sciences, Tabriz, Iran.
DisclosureR. Hashemi, M.Morshedi, M. Asghari Jafarabadi, and D. Altafireport no disclosures. S. Hosseini-Asl has received research
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

support from Ardabil University of Medical Sciences. S. Rafie-Arefhosseini is an employee of and has received researchsupport from Tabriz University of Medical Sciences. Fulldisclosure form information provided by the authors isavailable with the full text of this article at Neurology.org/NG.
Received January 9, 2018. Accepted in final form July 9, 2018.
References1. Kjølhede T, Dalgas U, Gade AB, et al. Acute and chronic cytokine responses to
resistance exercise and training in people with multiple sclerosis. Scand J Med SciSports 2016;26:824–834.
2. Martins TB, Rose JW, Jaskowski TD, et al. Analysis of proinflammatory and anti-inflammatory cytokine serum concentrations in patients with multiple sclerosis byusing a multiplexed immunoassay. Am J Clin Pathol 2011;136:696–704.
3. Siffrin V, Vogt J, Radbruch H, Nitsch R, Zipp F. Multiple sclerosis – candidatemechanisms underlying CNS atrophy. Trends Neurosci 2010;33:202–210.
4. Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood-brainbarrier disruption and central nervous system inflammation. 2007;13:1173–1175.
5. Camperio C, Muscolini M, Volpe E, et al. CD28 ligation in the absence of TCRstimulation up-regulates IL-17A and proinflammatory cytokines in relapsing-remitting multiple sclerosis T lymphocytes. Immunol Lett 2014;158:134–142.
6. Ramagopalan SV, Dobson R, Meier UC, Giovannoni G. Multiple sclerosis: risk fac-tors, prodromes, and potential causal pathways. Lancet Neurol 2010;9:727–739.
7. Xia Z, White CC, Owen EK, et al. Genes and environment in multiple sclerosis project:a platform to investigate multiple sclerosis risk. Ann Neurol 2016;79:178–189.
8. Naghavi Gargari B, Behmanesh M, Shirvani Farsani Z, Pahlevan Kakhki M, Azimi AR.Vitamin D supplementation up-regulates IL-6 and IL-17A gene expression in multiplesclerosis patients. Int Immunopharmacol 2015;28:414–419.
9. Furusawa H, Suzuki Y, Miyazaki Y, Inasea N, Eishib Y. Th1andTh17immuneres-ponsestoviable Propionibacteriumacnes in patientswithsarcoidosis. Respir Invest2012;50:104–109.
10. Wang Z, Zheng Y, Hou C, et al. DNA methylation impairs TLR9 induced Foxp3expression by attenuating IRF-7 binding activity in fulminant type 1 diabetes. Auto-immunity 2013;41:50–59.
11. Liang H, Hussey SE, Sanchez-Avila A, Tantiwong P, Musi N. Effect of lipopolysac-charide on inflammation and insulin action in human muscle. PLoS One 2013;8:e63983.
12. Seung-Hun S, Jae-Kwan L, Oye-Sun S, Ho-Suk S. The relationship etween cytokinesand HPV-16, HPV-16E6, E7 and high-risk HPV viral load in the uterin cervix.Gynecol Oncol 2007;104:732–738.
13. Yuan JS, Reed A, Chen F, Stewart CN. Statistical analysis of real-time PCR data. BMCBioinformatic 2006;7:85.
14. Eskandari G, Ghajarzadeh M, Yekaninejad MS, et al. Comparison of serum vitamin Dlevel in multiple sclerosis patients, their siblings, and healthy controls. Iran J Neurol2015;14:81.
15. Yildiz M, Tettenborn B, Putzki N. Vitamin D levels in Swiss multiple sclerosispatients. Swiss Med Wkly 2011;141:w13192.
16. Nikanfar M, Taheri-Aghdam AA, Yazdani M, et al. Serum 25(OH) vitamin D levels isnot associated with disability in multiple sclerosis patients: a case-control study. Iran JNeurol 2015;14:17–21.
17. Runia TF, Hop WC, de Rijke YB, Buljevac D, Hintzen RQ. Lower serum vitamin Dlevels are associated with a higher relapse risk in multiple sclerosis. Neurology 2012;79:261–266.
18. Delvin E, Souberbielle JC, Viard JP, Salle B. Role of vitamin D in acquired immuneand autoimmune diseases. Crit Rev Clin Lab Sci 2014;51:232–247.
19. Peelen E, Knippenberg S, Muris AH, et al. Effects of vitamin D on the peripheraladaptive immune system: a review. Autoimmun Rev 2011;10:733–743.
20. Duan Sh, Lv ZH, Fan X, et al. Vitamin D status and the risk of multiple sclerosis:a systematicreview and meta-analysis. Neurosci Lett 2014;570:108–113.
21. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metab-olism, molecular mechanism of action, and pleiotropic effects. Am Physiol Soc 2016;96:365–408.
22. Bolaños-Jimenez R, Arizmendi-Vargas J, Carrillo-Ruiz J, et al. Multiple sclerosis: anoverview of the disease and current concepts of its pathophysiology. J Neurosci BehavHealth 2010;3:44–50.
23. LuchtmanWD, Ellwardt E, Larochelle C, Zipp F. IL-17 and related cytokines involvedin the pathology and immunotherapy of multiple sclerosis. Curr Future Dev 2014;25:403–413.
24. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervoussystem. Int J Biol Sci 2012;8:1254.
25. Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol 2009;66:390–402.
26. Ferreira TB, Hygino J, Barros PO, et al. Endogenous interleukin-6 amplifiesinterleukin-17 production and corticoid-resistance in peripheral T cells from patientswith multiple sclerosis. Immunology 2014;143:560–568.
27. da Costa DS, Hygino J, Ferreira B, et al. Vitamin D modulates different IL-17-secreting T cell subsets in multiple sclerosis patients. J Neuroimmunol 2016;299:8–18.
28. Joshi S, Pantalena LC, Liu XK, et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17autoimmunity via transcriptional modulation of interleukin-17A. Mol Cel Biol 2011;31:3653–3669.
29. Korf H, Wenes M, Stijlemans B, et al. 1,25-Dihydroxyvitamin D3 curtails the in-flammatory and T cell stimulatory capacity of macrophages through an IL-10-dependent mechanism. Immunobiology 2012;217:1292–1300.
30. Farsani ZS, Behmanesh M, Sahraian MA. Interleukin-10 but not transforming growthfactor-a1 gene expression is up-regulated by vitamin D treatment in multiple sclerosispatients. J Neurol Sci 2015;350:18–23.
31. Correale J, Ysrraelit MC, Gaitan MI. Immunomodulatory effects of vitamin D inmultiple sclerosis. Brain 2009;132:1146–1160.
32. Fujita H, Asahina A, KomineM, Tamaki K. The direct action of 1α,25(OH)2- vitaminD3 on purified mouse Langerhans cells. Cell Immunol 2007;245:70–79.
33. Niino M, Fukazawa T, Miyazaki Y, et al. Suppression of IL-10 production by calcitriolin patients with multiple sclerosis. J Neuroimmunol 2014;270:86–94.
34. Smolders J, Hupperts R, Barkhof F, et al. Efficacy of vitamin D3 as add-on therapy inpatients with relapsing-remitting multiple sclerosis receiving subcutaneous interferonbeta-1a: a phase II, multicenter, double-blind, randomized, placebo-controlled trial.J Neurol Sci 2011;311:44–49.
35. Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmunediseases and cancers. Pharmacol Ther 2014;141:125–139.
36. Matilainen JM, Husso T, Toropainen S, et al. Primary effect of 1α,25(OH) 2D3 on IL-10 expression in monocytes is short-term down-regulation. Biochim Biophys Acta2010;1803:1276–1286.
37. Wise IA, Charchar FJ. Epigenetic modifications in essential hypertension. Int JMol Sci2016;17:451–465.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

ARTICLE OPEN ACCESS
Novel genotype-phenotype and MRIcorrelations in a large cohort of patientswith SPG7 mutationsChanna A. Hewamadduma, MBBS, FRCP, PhD, Nigel Hoggard, MBBS, MD, FRCP,
Ronan O’Malley, MBBS, MRCP, Megan K. Robinson, MBChB, MRCP, Nick J. Beauchamp, PhD,
Ruta Segamogaite, MSc, Jo Martindale, PhD, Tobias Rodgers, Ganesh Rao, MBBS, MD, FRCP,
Ptolemaios Sarrigiannis, MBBS, MD, FRCP, Priya Shanmugarajah, MBChB, MD, MRCP,
Panagiotis Zis, MBBS, MD, Basil Sharrack, MBBS, FRCP, PhD, Christopher J. McDermott, MBBS, FRCP, PhD,
Pamela J. Shaw, MBBS, MD, FRCP, FMedSci, and Marios Hadjivassiliou, MBBS, MD, FRCP
Neurol Genet 2018;4:e279. doi:10.1212/NXG.0000000000000279
Correspondence
Dr. Hewamadduma
Prof. Hadjivassiliou
AbstractObjectiveTo clinically, genetically, and radiologically characterize a large cohort of SPG7 patients.
MethodsWe used data from next-generation sequencing panels for ataxias and hereditary spasticparaplegia to identify a characteristic phenotype that helped direct genetic testing for variationsin SPG7. We analyzed MRI. We reviewed all published SPG7 mutations for correlations.
ResultsWe identified 42 cases with biallelic SPG7mutations, including 7 novelmutations, including a largemulti-exon deletion, representing one of the largest cohorts so far described. We identifieda characteristic phenotype comprising cerebellar ataxia with prominent cerebellar dysarthria, mildlower limb spasticity, and a waddling gait, predominantly from a cohort of idiopathic ataxia. Wereport a rare brain MRI finding of dentate nucleus hyperintensity on T2 sequences with SPG7mutations. We confirm that the c.1529C>T allele is frequently present in patients with long-standing British ancestry. Based on the findings of the present study and existing literature, weconfirm that patients with homozygous mutations involving the M41 peptidase domain of SPG7have a younger age at onset compared to individuals with mutations elsewhere in the gene (14years difference, p < 0.034), whereas c.1529C>T compound heterozygous mutations are associ-ated with a younger age at onset compared to homozygous cases (5.4 years difference, p < 0.022).
ConclusionsMutant SPG7 is common in sporadic ataxia. In patients with British ancestry, c.1529C>T allelerepresents the most frequent mutation. SPG7 mutations can be clinically predicted by thecharacteristic hybrid spastic-ataxic phenotype described above, along with T2 hyperintensity ofthe dentate nucleus on MRI.
From the Academic Directorate of Neurosciences (C.A.A.H., R.O’.M., M.K.R., S.P., Z.P., S.B., C.J.M., P.J.S., M.H.), Sheffield Teaching Hospitals NHS Foundation Trust, Royal HallamshireHospital; Sheffield Institute for Translational Neuroscience (SITraN) (C.A.A.H., R.S., T.R., C.J.M., P.J.S., M.H.), University of Sheffield; Sheffield Diagnostic Genetics Service (N.J.B., J.M.),Sheffield Children’s NHS Foundation Trust; Department of Clinical Neurophysiology (G.R., P.S.), Sheffield Teaching Hospitals NHS Foundation Trust, Royal Hallamshire Hospital;Academic Unit of Radiology (N.H.), University of Sheffield, Royal Hallamshire Hospital; and Sheffield NIHR Biomedical Research Centre for Translational Neuroscience (C.A.A.H., N.H.,R.S., P.S., S.B., C.J.M., P.J.S., M.H.), United Kingdom.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by NIHR BRC grant for SITRAN.
All patients consented to genetic testing and reporting of the findings. Study was conducted according to the departmental regulations. REC reference 09/H1310/79, IRAS 26259.
This is an open access article distributed under the terms of the Creative Commons Attribution License 4.0 (CC BY), which permits unrestricted use, distribution, and reproduction in anymedium, provided the original work is properly cited.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Hereditary spastic paraplegia (HSP) and hereditary cerebellarataxias (HCA) are heterogeneous groups of progressiveneurodegenerative conditions with considerable overlap.1,2
HSP is complicated when features such as ataxia, neuropathy,optic atrophy, and weakness are present.3 HCA can also beassociated with spastic paraplegia. There are over 80 differentgenetic loci associated with HSP and similar number associ-ated with cerebellar ataxias.4–6 This extensive genetic het-erogeneity together with the overlapping features of HCA andcomplicated HSP often causes difficulties in disease classifi-cation and clinical approach to genetic diagnosis.
Next-generation sequencing (NGS) gene panels are availablefor both HSP andHCA patients. However, such panel tests areexpensive and not always readily available. Our objective was todescribe clinical, genetic, and radiologic features of a Britishcohort of SPG7 cases where the phenotype may be helpful inproviding guidance to targeted genetic testing.We highlight theimportance of such clinical characterization through our ex-perience of diagnosing a large cohort of patients withmutationsin the SPG7 gene, implicated in both HSP and HCA.7 Inaddition, by reviewing all published SPG7 mutation data, wemake important new genotype-phenotype correlations.
MethodsPatient cohortsWe studied all cases positive for SPG7 mutation in our HSPand ataxia cohorts, which mainly include patients from theNorth of England (cohort study) and analyzed all clinical,genetic, and neuroimaging data.
Standard protocol approvals, registrationsPatient consent was obtained for genetic testing in accordancewith the departmental regulations. Healthy control cases forMRI were recruited as per ethics committee approval (RECreference 09/H1310/79, IRAS 26259). STROBE checklistfor cohort study adhered in reporting the data.
Genetic testingLibraries of sheared genomic DNA corresponding to panels ofeither HCA or HSP genes captured using a SureSelect XTcustom designed probe set (Agilent, Cheadle, UK), and pair-end sequenced using a HiSeq 2500 instrument (Illumina) wasused. Raw data were analyzed using the Genome AnalysisToolKit,8 (Broad Institute, Cambridge, MA) according toguidelines.9,10 After initial identification of 11 patients withSPG7 mutations using the ataxia and HSP gene panels, weevaluated the phenotype to identify a triad of spastic paraplegia(usually mild), cerebellar ataxia (with prominent cerebellardysarthria), and waddling gait indicative of proximal muscle
weakness. Thereafter, the majority of patients who presentedwho had the above triad were analyzed by bidirectional Sangersequencing and dosage analysis (multiplex ligation-dependentprobe amplification kit P213-B1 and B2, MRC-Holland) of all17 exons of the SPG7 gene. The remainder of the cohort wereidentified using eitherHSP orHCAgene panel testing as before.
Chromatographs were analyzed using Mutation surveyorv4.0.8 (softgenetics.com). Annotation of mutations was car-ried out in accordance withHumanGenome Variation societynomenclature (hgvs.org/mutnomen), with nomenclature basedon the reference sequence NM_003119.3. Novel variants in theSPG7 gene were assessed for pathogenicity using Alamut Visualversion 2.9.0 (Interactive Biosoftware, Rouen, France) andprediction software (Provean, MutPred, SNPS & GO andPolyPhen2). Allele frequencies for novel variants in normalcontrol populations were obtained from the Genome Ag-gregation Database (gnomAD).11
NeuroimagingMRIs, available for all patients who underwent MRI, wereanalyzed for cerebellar atrophy. Further subanalysis of thedentate nucleus was undertaken for all patients who un-derwent brain imaging on the same 3-TMR scanner (Ingenia,Philips Medical Systems, Eindhoven, The Netherlands) usingthe same T2-weighted sequence (avoid machine-related var-iability) (cases: n = 21 and controls: n = 16). This was com-pared with age- and sex-matched controls imaged with thissequence. The axial T2-weighted parameters were as follows:repetition time 3,000 ms, time to echo 80, echo train length 15,number of averages was 1 and 4 mm thick, 512 × 512 matrix.Matching criteria for healthy controls were age within 3 yearsand sex. Relative signal intensity of the dentate nucleus wascompared to normal-appearing pontine white matter and rednucleus. A region of interest (area 20 mm2) in these structureswas placed in the region of the dentate nucleus with the lowestsignal. The dentate nucleus signal was then dichotomizedby whether the ratio of the signals was less than or morethan 1 (i.e., hypointense or hyperintense compared to normal-appearing white matter in the pons).
Literature reviewTwo clinicians independently reviewed clinical and geneticdetails of all SPG7 cases thus far reported in the literature(until September 30, 2017). We searched the following termsin PubMed, MEDLINE, Web of Science, and Embase: SPG7,paraplegin, hereditary spastic paraparesis, HSP (mutations),spastic ataxia, and ataxia, and selected all the articles reportingSPG7 and/or paraplegin mutations and reviewed the pheno-type and genotype data published. We excluded publicationsthat were not in English or where English translation was not
GlossaryDN = dentate nuclei; HCA = hereditary cerebellar ataxias; HSP = hereditary spastic paraplegia; NGS = Next-generationsequencing; PEO = Progressive external ophthalmoplegia; RN = red nuclei; SARA = Scale for the assessment and rating of ataxia.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

available and articles that did not describe clinical features. Allmutations described by us and previously reported are depictedin a schematic diagram in relation to functionally importantdomains (figure 1) (e-table 1, links.lww.com/NXG/A89).
Statistical analysesStatistical analysis was performed using Prism GraphPadV7.0b and SPSS (2015) statistical software programs. One-way analysis of variance was used for multiple group com-parisons, and independent samples t test and χ2 test were usedto compare 2 groups.
Data availabilityAll anonymized data can be shared on a collaborative basis.
ResultsCharacterization of the phenotypeWe identified a total of 42 cases positive for pathogenic muta-tions in both alleles of the SPG7 gene (table 1). Initially, 11cases were identified using ataxia or HSP NGS gene panels (4patients using ataxia panel and 7 patients using the HSP panel).On reviewing the phenotype of these 11 cases, we noted that 9individuals had cerebellar ataxia with prominent slurring ofspeech, mild spasticity, and proximal muscle weakness resultingin a waddling gait.
Direct genetic screening based onthe phenotypeFollowing the clinical characterization of the initial 11 patients,we undertook direct testing for mutations in the SPG7 gene in
patients who demonstrated the above phenotype in a cohort ofpatients attending the Sheffield Ataxia Centre and HSP clinics.We identified a further 27 cases with pathogenic mutations(table 1). Four other cases were already diagnosed when re-ferred to us (panel tests).
The clinical characteristics of the 42 probands are summarizedin table 2. There was no history of consanguinity. Eighty-threepercent were male. The average age of symptom onset was41.7 years (SD: ± 11, median age 44 years). Female patientsdeveloped symptoms on average 4 years earlier than malepatients (38.5 vs 42.5 years) (table 2). The mean duration ofdisease at the time of diagnosis was 9.6 years (SD: ±5.2, mode5). Thirty-eight of 42 patients (90%) were of long-standingBritish ancestry. Four were UK citizens of Indian, Iranian,German, and Bulgarian descent.
Ninety-eight percent of cases presented with gait unsteadinessfollowed by dysarthric speech (76%). Thirty-two patients(76%) complained of mild spasticity. Two patients presentedwith the typical spastic gait characteristic of HSP (5%) and 7patients (16%) presented with moderate-to-severe spasticity.Seventy-six percent (32 of 42) had mildly increased lowerlimb tone and 93% had brisk reflexes, while the Babinski signwas positive in 51%.
At baseline, 38 patients (90%) were found to have at leastsome evidence of cerebellar ataxia and 33 (79%) were foundto have both mild spasticity and cerebellar ataxia. Despite thecerebellar features, only 2 cases were nonambulant, with a totalsymptomatic disease duration of 399 patient-years. None of the
Figure 1 Schematic diagram of the SPG7 protein with important functional domains and positioning of mutations in theSheffield cohort and all the published pathogenic mutations in the SPG7 gene
Mutations described in our cohort of patients are annotated above the SPG7 protein structure, while previously published mutations are below. Allelicfrequency is noted within parenthesis. New mutations detected in our cohort are highlighted in red font. Variations denoted in blue are matching com-plementary DNA sequence of the reported mutations. Some large exon deletions reported are indicated in the text box. Parentheses from mutationsremoved to create space. AAA = ATPases associated with diverse cellular activities; Coil1 and Coil2 = coiled domain; FtsH = filamentation temperature-sensitive mutant in Escherichia coli domain; TM1 and TM2 = transmembrane domain 1 and 2. Reference sequence: NM_003119.3.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
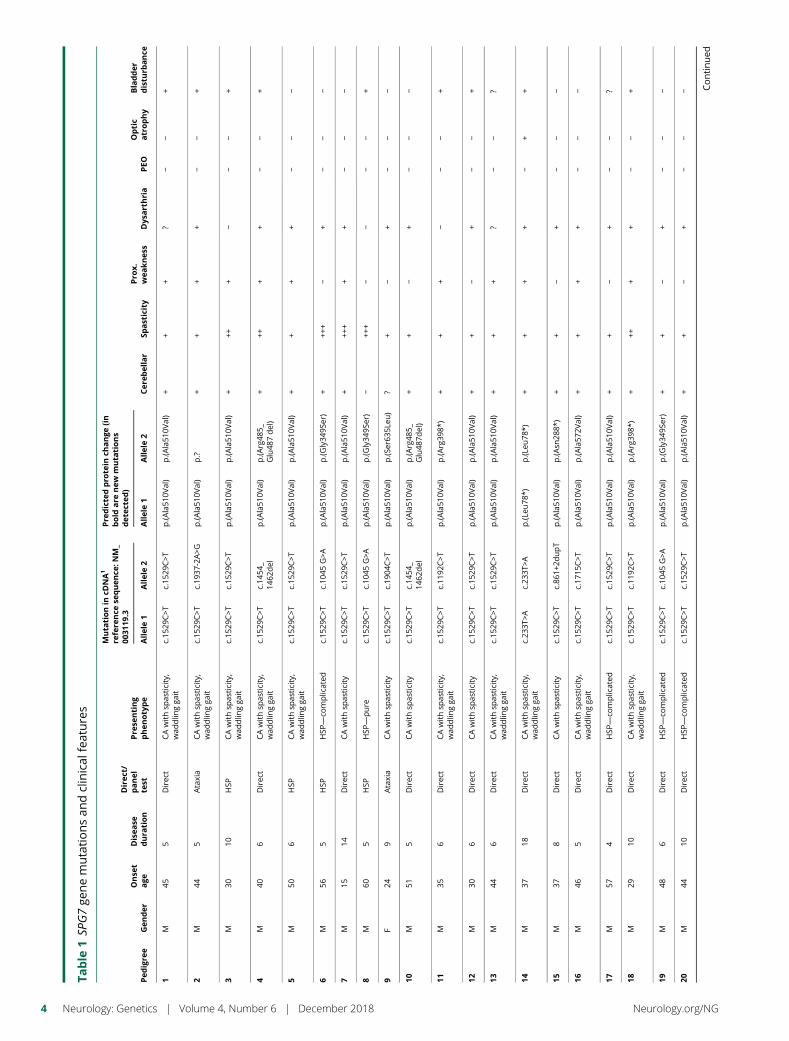
Table
1SP
G7ge
nemutationsan
dclinical
features
Pedigre
eGen
der
Onse
tage
Disea
sedura
tion
Direc
t/panel
test
Pre
senting
phenotype
Muta
tionin
cDNA1
refere
nce
sequen
ce:N
M_
0031
19.3
Pre
dicte
dpro
tein
change
(in
bold
are
new
muta
tions
det
ecte
d)
Cer
ebellar
Spasticity
Pro
x.wea
knes
sDys
arthria
PEO
Optic
atrophy
Bladder
distu
rbance
Allele1
Allele2
Allele1
Allele2
1M
455
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
+?
−−
+
2M
445
Ataxia
CAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.19
37-2A>G
p.(A
la51
0Val)
p.?
++
++
−−
+
3M
3010
HSP
CAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
+++
+−
−−
+
4M
406
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.14
54_
1462
del
p.(A
la51
0Val)
p.(A
rg48
5_Glu48
7del)
+++
++
−−
+
5M
506
HSP
CAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
++
−−
−
6M
565
HSP
HSP
—co
mplicated
c.15
29C>T
c.10
45G>A
p.(A
la51
0Val)
p.(G
ly34
9Ser)
+++
+−
+−
−−
7M
1514
Direc
tCAwithsp
asticity
c.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
+++
++
+−
−−
8M
605
HSP
HSP
—pure
c.15
29C>T
c.10
45G>A
p.(A
la51
0Val)
p.(G
ly34
9Ser)
−++
+−
−−
−+
9F
249
Ataxia
CAwithsp
asticity
c.15
29C>T
c.19
04C>T
p.(A
la51
0Val)
p.(S
er63
5Leu
)?
+−
+−
−−
10M
515
Direc
tCAwithsp
asticity
c.15
29C>T
c.14
54_
1462
del
p.(A
la51
0Val)
p.(A
rg48
5_Glu48
7del)
++
−+
−−
−
11M
356
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.11
92C>T
p.(A
la51
0Val)
p.(A
rg39
8*)
++
+−
−−
+
12M
306
Direc
tCAwithsp
asticity
c.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
−+
−−
+
13M
446
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
+?
−−
?
14M
3718
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.23
3T>A
c.23
3T>A
p.(L
eu78
*)p.(L
eu78
*)+
++
+−
++
15M
378
Direc
tCAwithsp
asticity
c.15
29C>T
c.86
1+2d
upT
p.(A
la51
0Val)
p.(A
sn28
8*)
++
−+
−−
−
16M
465
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.17
15C>T
p.(A
la51
0Val)
p.(A
la57
2Val)
++
++
−−
−
17M
574
Direc
tHSP
—co
mplicated
c.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
−+
−−
?
18M
2910
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.11
92C>T
p.(A
la51
0Val)
p.(A
rg39
8*)
+++
++
−−
+
19M
486
Direc
tHSP
—co
mplicated
c.15
29C>T
c.10
45G>A
p.(A
la51
0Val)
p.(G
ly34
9Ser)
++
−+
−−
−
20M
4410
Direc
tHSP
—co
mplicated
c.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
−+
−−
− Continued
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
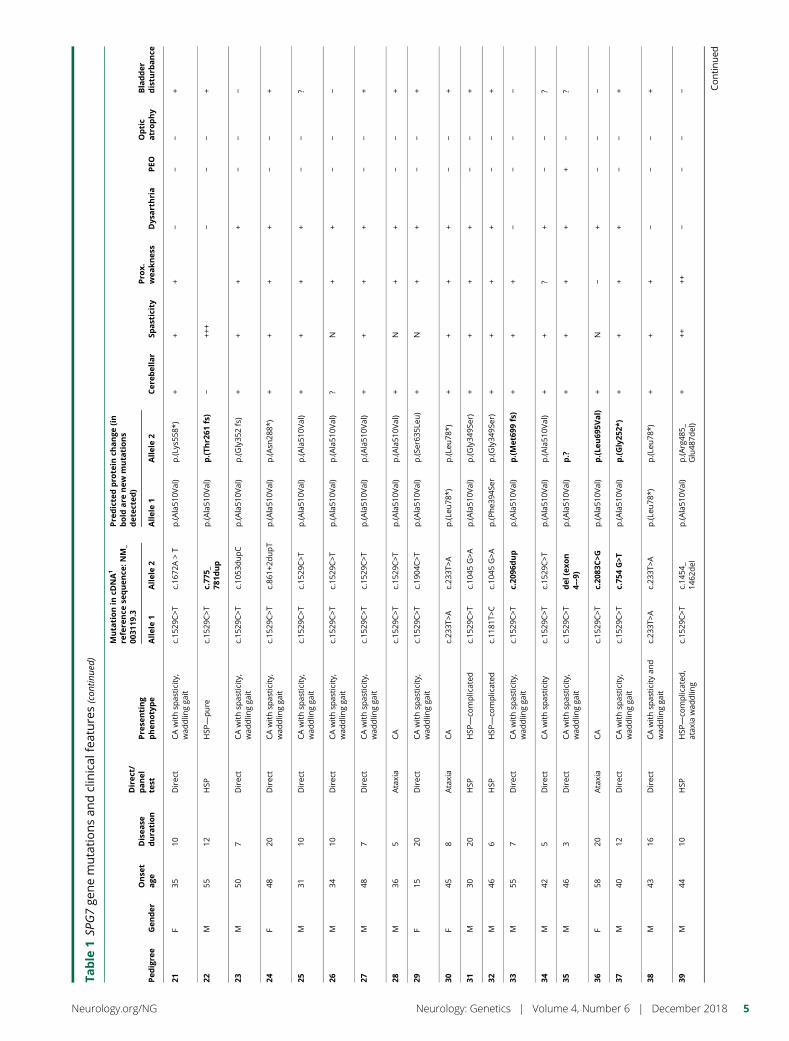
Table
1SP
G7ge
nemutationsan
dclinical
features(con
tinue
d)
Pedigre
eGen
der
Onse
tage
Disea
sedura
tion
Direc
t/panel
test
Pre
senting
phenotype
Muta
tionin
cDNA1
refere
nce
sequen
ce:N
M_
0031
19.3
Pre
dicte
dpro
tein
change
(in
bold
are
new
muta
tions
det
ecte
d)
Cer
ebellar
Spasticity
Pro
x.wea
knes
sDys
arthria
PEO
Optic
atrophy
Bladder
distu
rbance
Allele1
Allele2
Allele1
Allele2
21F
3510
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.16
72A>T
p.(A
la51
0Val)
p.(L
ys55
8*)
++
+−
−−
+
22M
5512
HSP
HSP
—pure
c.15
29C>T
c.77
5_78
1dup
p.(A
la51
0Val)
p.(Th
r261
fs)
−++
+−
−−
+
23M
507
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.10
53dupC
p.(A
la51
0Val)
p.(G
ly35
2fs)
++
++
−−
−
24F
4820
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.86
1+2d
upT
p.(A
la51
0Val)
p.(A
sn28
8*)
++
++
−−
+
25M
3110
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
++
−−
?
26M
3410
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
?N
++
−−
−
27M
487
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
++
−−
+
28M
365
Ataxia
CA
c.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
+N
++
−−
+
29F
1520
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.19
04C>T
p.(A
la51
0Val)
p.(S
er63
5Leu
)+
N+
+−
−+
30F
458
Ataxia
CA
c.23
3T>A
c.23
3T>A
p.(L
eu78
*)p.(L
eu78
*)+
++
+−
−+
31M
3020
HSP
HSP
—co
mplicated
c.15
29C>T
c.10
45G>A
p.(A
la51
0Val)
p.(G
ly34
9Ser)
++
++
−−
+
32M
466
HSP
HSP
—co
mplicated
c.11
81T>
Cc.10
45G>A
p.(P
he3
94Se
rp.(G
ly34
9Ser)
++
++
−−
+
33M
557
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.20
96dup
p.(A
la51
0Val)
p.(Met
699fs)
++
+−
−−
−
34M
425
Direc
tCAwithsp
asticity
c.15
29C>T
c.15
29C>T
p.(A
la51
0Val)
p.(A
la51
0Val)
++
?+
−−
?
35M
463
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
del
(exo
n4–
-9)
p.(A
la51
0Val)
p.?
++
++
+−
?
36F
5820
Ataxia
CA
c.15
29C>T
c.20
83C>G
p.(A
la51
0Val)
p.(Le
u69
5Val)
+N
−+
−−
−
37M
4012
Direc
tCAwithsp
asticity,
wad
dlin
gga
itc.15
29C>T
c.75
4G>T
p.(A
la51
0Val)
p.(Gly25
2*)
++
++
−−
+
38M
4316
Direc
tCAwithsp
asticity
and
wad
dlin
gga
itc.23
3T>A
c.23
3T>A
p.(L
eu78
*)p.(L
eu78
*)+
++
−−
−+
39M
4410
HSP
HSP
—co
mplicated
,atax
iawad
dlin
gc.15
29C>T
c.14
54_
1462
del
p.(A
la51
0Val)
p.(A
rg48
5_Glu48
7del)
+++
++−
−−
− Continued
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

patients could run, and 78% of the cases were using walking aid.The severity of the ataxia was less (median scale for the as-sessment and rating of ataxia [SARA] score 8, range: 3.5–13.5)when compared to spino cerebellar ataxia 6 (median score15.0) for the same duration of symptoms.
Sixty-four percent of cases demonstrated the triad of cere-bellar ataxia with dysarthria, spasticity, and waddling gait atpresentation, and 9 others developed the full clinical pictureduring follow-up (totaling 87%). Progressive external oph-thalmoplegia (PEO) was observed only in 1 patient. Anotherpatient had vertical gaze palsy. Nystagmus was present in 38%of patients. Optic atrophy was seen in 1 patient. Waddling gaitwas seen in 87% of our cases.
Although 4 patients were found to have reduced vibrationsense and 3 had reduced pinprick sensation on clinical ex-amination, none of the 19 patients who underwent neuro-physiologic assessment had evidence of large fiber peripheralneuropathy or myopathy.
Mutation analysisFifteen cases (36%) were homozygous for mutations in theSPG7 gene, while 27 cases (64%) were compound hetero-zygous. Twelve of the 15 homozygous cases had the commonmissense mutation in exon 11, c.1529C>T, p.(Ala510Val),while the other 3 cases were homozygous for the c.233T>A,p.(Leu78*) nonsense mutation. Ninety percent of our casesthat carried the common mutation p.(Ala510Val) in at leastone allele were of British ancestry. The 3 patients homozy-gous for the p. (Leu78*) nonsense mutation were second-generation British citizens of Indian, Iranian, or Bulgariandescent. The fourth case, of German descent, was compoundheterozygous for the c.1181T>C, p.(Phe394Ser) and c.1045G>A, p.(Gly349Ser) mutations.
The frequency of the c.1529C>T, p.(Ala510Val) mutation inour cohort was 60% (50 of 84 alleles assessed). The secondmost commonmutant allele, c.233T>A, p.(Leu78*), was seenin 3 patients in the homozygous state, while c.1045 G>A,p.(Gly349Ser) was seen in 5 cases in a compound hetero-zygous state. p.(Ala510Val) and p.Arg485_Glu487del muta-tions were observed in two-thirds of the disease alleles (50of 84). In addition, to the single case with a large deletion,several small insertions, duplications, deletions, and splicesite mutations were detected on 7 alleles, 5 of which havebeen previously described. Most of the pathogenic alleleswere missense mutations (63 of 84) while 21 were nonsensemutations (table 1).
Novel mutations in SPG7We discovered 7 novel likely pathogenic mutations in theSPG7 gene (table 1), of which 5 were null mutations, with 2frame-shift mutations c.775_781dup p.(Thr261 fs) andc.2096dup p.(Met699 fs), 2 nonsense mutations c.754G>T,p.(Gly252*) and c.300T>A, p.(Tyr100*), and a large de-letion encompassing at least exons 4 to 9 (c.377−?_1324+?Ta
ble
1SP
G7ge
nemutationsan
dclinical
features(con
tinue
d)
Pedigre
eGen
der
Onse
tage
Disea
sedura
tion
Direc
t/panel
test
Pre
senting
phenotype
Muta
tionin
cDNA1
refere
nce
sequen
ce:N
M_
0031
19.3
Pre
dicte
dpro
tein
change
(in
bold
are
new
muta
tions
det
ecte
d)
Cer
ebellar
Spasticity
Pro
x.wea
knes
sDys
arthria
PEO
Optic
atrophy
Bladder
distu
rbance
Allele1
Allele2
Allele1
Allele2
40F
4512
Ataxia
CAan
dmild
spas
ticity
butnowad
dlin
gc.15
29C>T
c.30
0T>
Ap.(A
la51
0Val)
p.(Ty
r100
*)++
+−
++−
−+
41M
4810
Direc
tHSP
—co
mplicated
,atax
iawad
dlin
gc.15
29C>T
c.10
49_
1077
del
p.(A
la51
0Val)
p.(P
ro35
0fs)
+++
++
−−
−
42M
4020
HSP
HSP
—co
mplicated
,atax
iawad
dlin
gc.15
29C>T
c.13
73C>T
p.(A
la51
0Val)
p.(Ala45
8Val)
+++
++
−−
+
Abbreviations:
CA=ce
rebellaratax
ia;H
SP=hered
itarysp
asticparap
legia.
+=mild
(orfeature
prese
nt);+
+=moderatese
verity,+
++=se
vere;?
=unkn
own;N
=norm
al;−
=reduce
d(orfeature
notprese
nt).
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
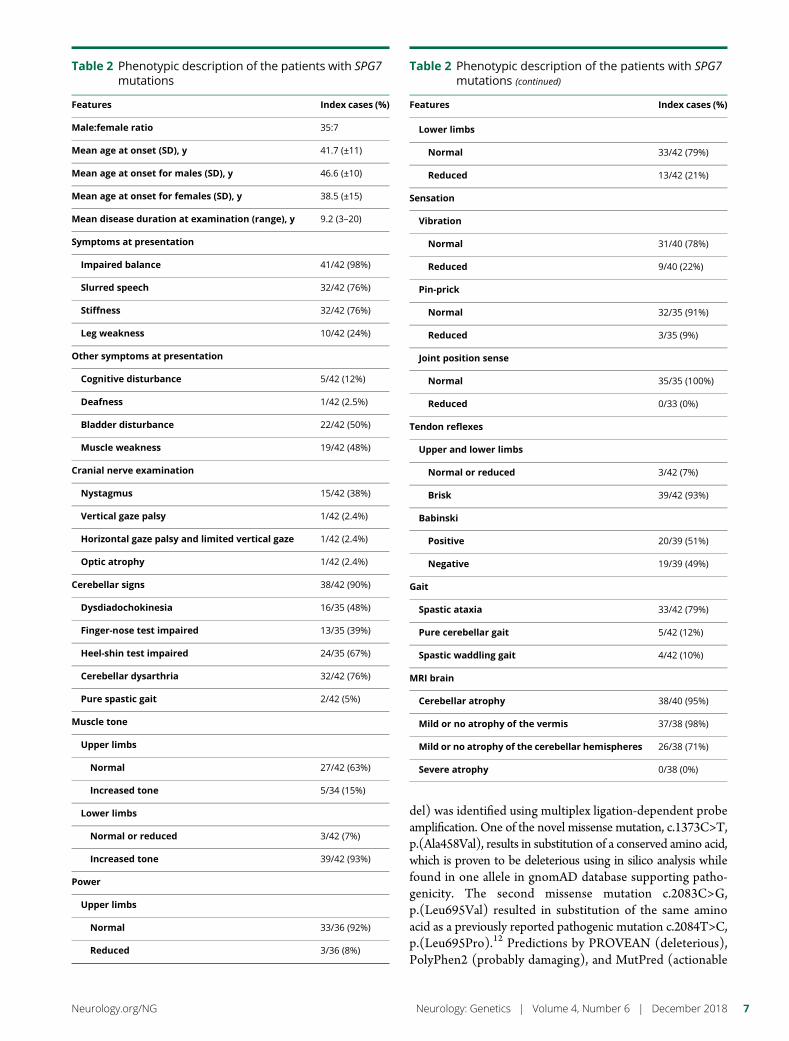
del) was identified using multiplex ligation-dependent probeamplification. One of the novel missense mutation, c.1373C>T,p.(Ala458Val), results in substitution of a conserved amino acid,which is proven to be deleterious using in silico analysis whilefound in one allele in gnomAD database supporting patho-genicity. The second missense mutation c.2083C>G,p.(Leu695Val) resulted in substitution of the same aminoacid as a previously reported pathogenic mutation c.2084T>C,p.(Leu695Pro).12 Predictions by PROVEAN (deleterious),PolyPhen2 (probably damaging), and MutPred (actionable
Table 2 Phenotypic description of the patients with SPG7mutations
Features Index cases (%)
Male:female ratio 35:7
Mean age at onset (SD), y 41.7 (±11)
Mean age at onset for males (SD), y 46.6 (±10)
Mean age at onset for females (SD), y 38.5 (±15)
Mean disease duration at examination (range), y 9.2 (3–20)
Symptoms at presentation
Impaired balance 41/42 (98%)
Slurred speech 32/42 (76%)
Stiffness 32/42 (76%)
Leg weakness 10/42 (24%)
Other symptoms at presentation
Cognitive disturbance 5/42 (12%)
Deafness 1/42 (2.5%)
Bladder disturbance 22/42 (50%)
Muscle weakness 19/42 (48%)
Cranial nerve examination
Nystagmus 15/42 (38%)
Vertical gaze palsy 1/42 (2.4%)
Horizontal gaze palsy and limited vertical gaze 1/42 (2.4%)
Optic atrophy 1/42 (2.4%)
Cerebellar signs 38/42 (90%)
Dysdiadochokinesia 16/35 (48%)
Finger-nose test impaired 13/35 (39%)
Heel-shin test impaired 24/35 (67%)
Cerebellar dysarthria 32/42 (76%)
Pure spastic gait 2/42 (5%)
Muscle tone
Upper limbs
Normal 27/42 (63%)
Increased tone 5/34 (15%)
Lower limbs
Normal or reduced 3/42 (7%)
Increased tone 39/42 (93%)
Power
Upper limbs
Normal 33/36 (92%)
Reduced 3/36 (8%)
Table 2 Phenotypic description of the patients with SPG7mutations (continued)
Features Index cases (%)
Lower limbs
Normal 33/42 (79%)
Reduced 13/42 (21%)
Sensation
Vibration
Normal 31/40 (78%)
Reduced 9/40 (22%)
Pin-prick
Normal 32/35 (91%)
Reduced 3/35 (9%)
Joint position sense
Normal 35/35 (100%)
Reduced 0/33 (0%)
Tendon reflexes
Upper and lower limbs
Normal or reduced 3/42 (7%)
Brisk 39/42 (93%)
Babinski
Positive 20/39 (51%)
Negative 19/39 (49%)
Gait
Spastic ataxia 33/42 (79%)
Pure cerebellar gait 5/42 (12%)
Spastic waddling gait 4/42 (10%)
MRI brain
Cerebellar atrophy 38/40 (95%)
Mild or no atrophy of the vermis 37/38 (98%)
Mild or no atrophy of the cerebellar hemispheres 26/38 (71%)
Severe atrophy 0/38 (0%)
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

hypothesis) suggested likely pathogenicity, but this was notsupported by SNPS & GO (neutral). This allele is present inthe East Asian gnomAD normal control population at a fre-quency of 0.4626%.
MRI brainMRI brain imaging was available in 40 cases. Cerebellar atrophywas noted in 95%, mostly mild atrophy of the vermis (table 2;figure 2, A and B). T1 sequences of both dentate nuclei (DN)and the red nuclei (RN) were not distinguishable betweencontrols and SPG7 cases (figure 2, A and C). The same T2sequence on 3-T imaging was available in 21 patients and thesewere matched with 17 healthy controls. In the 16 healthy con-trols, the DN were hypointense compared to normal-appearingwhite matter (figure 2C), and 1 healthy control had DN iso-intense relative to normal-appearing whitematter. TheDNwereisointense or hyperintense compared with normal-appearingwhite matter (T2 imaging) in 18 of the 21 SPG7-positive cases(figure 2D). Both controls and patients showed no difference inthe appearance of the RN, which were hypointense compared tonormal-appearing white matter in the pons (figure 2E). Theincrease in DN T2 hyperintensity on MRI in SPG7 cases wassignificant compared to the controls (χ2 test value 25.76, at p <0.001) (figure 2F).
Genotype-phenotype correlationfrom current and other studiesWe analyzed mutations in the different functionally importantdomains of SPG7 shown in figure 1 for any impact on age atonset of symptoms. Patients who had homozygous mutation inthe M41 peptidase domain had an earlier onset of diseasesymptoms (by 12 years) compared to patients with mutationsin a nonfunctionally assigned domain (p < 0.022) (figure 3A).Having homozygous, compound heterozygous mutationsor the presence of null alleles did not have an impact on ageat onset. However, we also noted that patients with thec.1529C>T mutation when in a compound-heterozygote statedeveloped symptoms 8 years earlier compared to c.1529C>Thomozygous cases (p < 0.019, unpaired t test) (figure 3B).
DiscussionWe describe a large British cohort of 42 unrelated and pre-viously unreported cases with mutations in the SPG7 gene.The largest other single-center cohort so far reported isa Dutch cohort of 46 unrelated families.13 We propose thatthe phenotype of cerebellar ataxia (with marked dysarthria),mild lower limb spasticity, and waddling gait is clinicallydistinct and should alert clinicians to direct genetic testingfor SPG7. Such an approach identified 64% of our cohort.While SPG7 biallelic mutations have historically been asso-ciated with HSP, it is now clear that ataxia is the majorclinical presentation, as only 26% of our cohort presentedwith an HSP-like phenotype.7,14,15 In another UK-basedstudy, SPG7 accounted for 18.6% of 70 patients with un-explained ataxia and pyramidal signs.7 SPG7 is the fourthcommonest cause of any genetic ataxia in the United
Kingdom and the second commonest recessive ataxia.16 Insupport of this finding, 90% of our SPG7 cohort demonstratedgait ataxia with cerebellar dysarthria (table 2).
Only 2 patients were wheelchair-dependent, indicating thatambulatory loss appears to be rare in SPG7 cases,15 with anaverage SARA score of 8 (range: 3.5–14). This favorableprognostic factor will be useful when counseling SPG7patients and their families.
A considerable proportion of our cohort was male (83%). Fe-male patients tended to develop symptoms about 4 years earlier.The median age at onset of symptoms was 44 years, indicatingthat SPG7-related disease is a late-onset disease in keeping withprevious reports.13,17 The age at onset, however, did range be-tween 15 and 60 years; therefore, SPG7 can rarely present withan early-onset ataxia. The recessive inheritance accounts for thelack of a positive family history. Absence of a family historyshould therefore not deter clinicians from SPG7 testing.
PEO was only seen in one of our patients but has beenreported in 11% of SPG7 cases worldwide. A previous reportfound that 13% of the patients with PEO have SPG7. PEOwasalso reported in 1 of 5 cases in a UK cohort of complex HSP.18
Overall, PEO in SPG7was also rare in other cohorts, includinga French (2/23),19 a Dutch (2/46),13 and a French Canadiancohort (none of the 22 individuals).15 A longitudinal study ofSPG7 patients from the United Kingdom reported a medianfollow-up duration of 23 years from presentation to detectingPEO,7 as did a Norwegian group (median follow-up of 24years).20 Clinicians should be aware that PEO-like features inSPG7 are rare but can develop late in the disease process.
Optic neuropathy was reported in 9.5% of the worldwideSPG7 cases, compared to one patient in our cohort. Ina French cohort of SPG7 patients, 44% had evidence of opticneuropathy based on optical coherence tomography, yet 40%of the patients with optic neuropathy had normal-appearingoptic discs on funduscopy. It is therefore likely that opticatrophy is common in SPG7-mutated patients but the clinicalsignificance of this remains unclear.
SPG7 cases have mild cerebellar atrophy and none had severeatrophy (table 2). The increased T2 signal from the dentatenucleus in SPG7 cases compared to controls has not beenpreviously reported. The dentate nucleus is a site of ironaccumulation in normal aging, and this is usually associatedwith reduced T2 signal. The high signal noted in SPG7 casesdoes not appear to be due to a globally reduced brain ironaccumulation. In support of theMRI findings are postmortemdata from an SPG7 case, which showed neuronal loss in thedentate nucleus.21 While the above imaging finding is notspecific for SPG7 mutations, yet it is an important charac-teristic and merits further consideration.22 We propose thatdentate nucleus hyperintensity on MRI T2 sequences, with-out severe overlying cerebellar atrophy and in the context ofa typical phenotype, aid the diagnosis of mutant SPG7.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
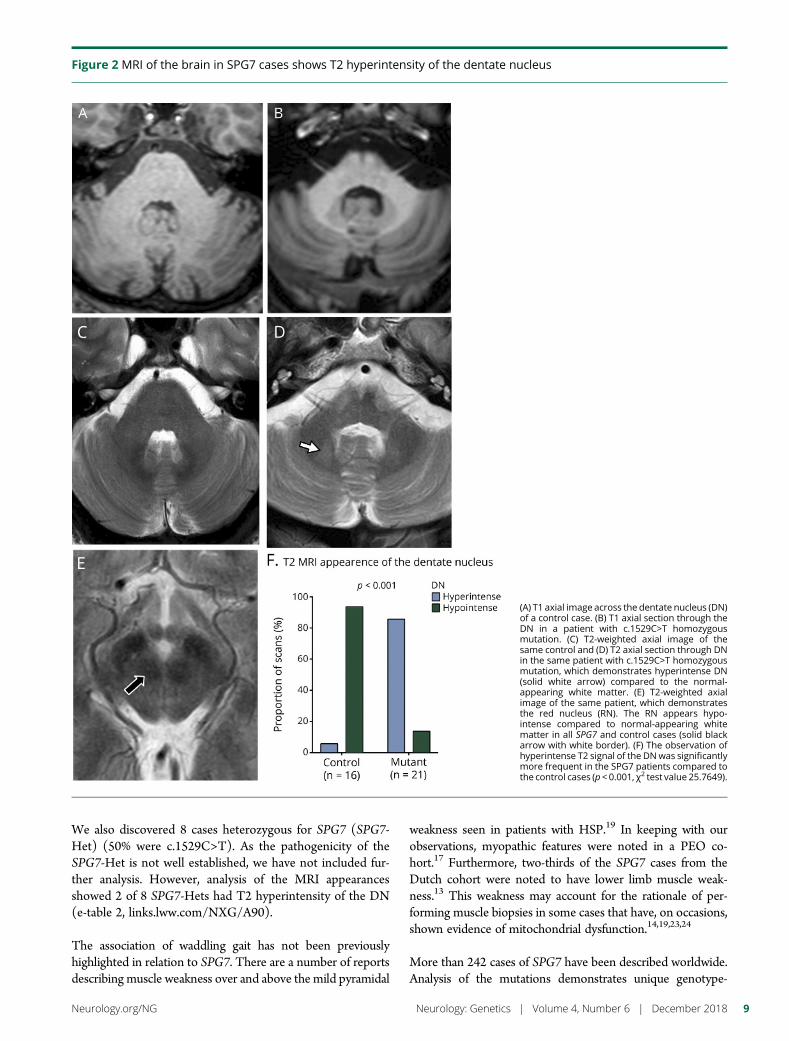
We also discovered 8 cases heterozygous for SPG7 (SPG7-Het) (50% were c.1529C>T). As the pathogenicity of theSPG7-Het is not well established, we have not included fur-ther analysis. However, analysis of the MRI appearancesshowed 2 of 8 SPG7-Hets had T2 hyperintensity of the DN(e-table 2, links.lww.com/NXG/A90).
The association of waddling gait has not been previouslyhighlighted in relation to SPG7. There are a number of reportsdescribing muscle weakness over and above the mild pyramidal
weakness seen in patients with HSP.19 In keeping with ourobservations, myopathic features were noted in a PEO co-hort.17 Furthermore, two-thirds of the SPG7 cases from theDutch cohort were noted to have lower limb muscle weak-ness.13 This weakness may account for the rationale of per-forming muscle biopsies in some cases that have, on occasions,shown evidence of mitochondrial dysfunction.14,19,23,24
More than 242 cases of SPG7 have been described worldwide.Analysis of the mutations demonstrates unique genotype-
Figure 2 MRI of the brain in SPG7 cases shows T2 hyperintensity of the dentate nucleus
(A) T1 axial image across the dentate nucleus (DN)of a control case. (B) T1 axial section through theDN in a patient with c.1529C>T homozygousmutation. (C) T2-weighted axial image of thesame control and (D) T2 axial section through DNin the same patient with c.1529C>T homozygousmutation, which demonstrates hyperintense DN(solid white arrow) compared to the normal-appearing white matter. (E) T2-weighted axialimage of the same patient, which demonstratesthe red nucleus (RN). The RN appears hypo-intense compared to normal-appearing whitematter in all SPG7 and control cases (solid blackarrow with white border). (F) The observation ofhyperintense T2 signal of the DNwas significantlymore frequent in the SPG7 patients compared tothe control cases (p < 0.001, χ2 test value 25.7649).
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9
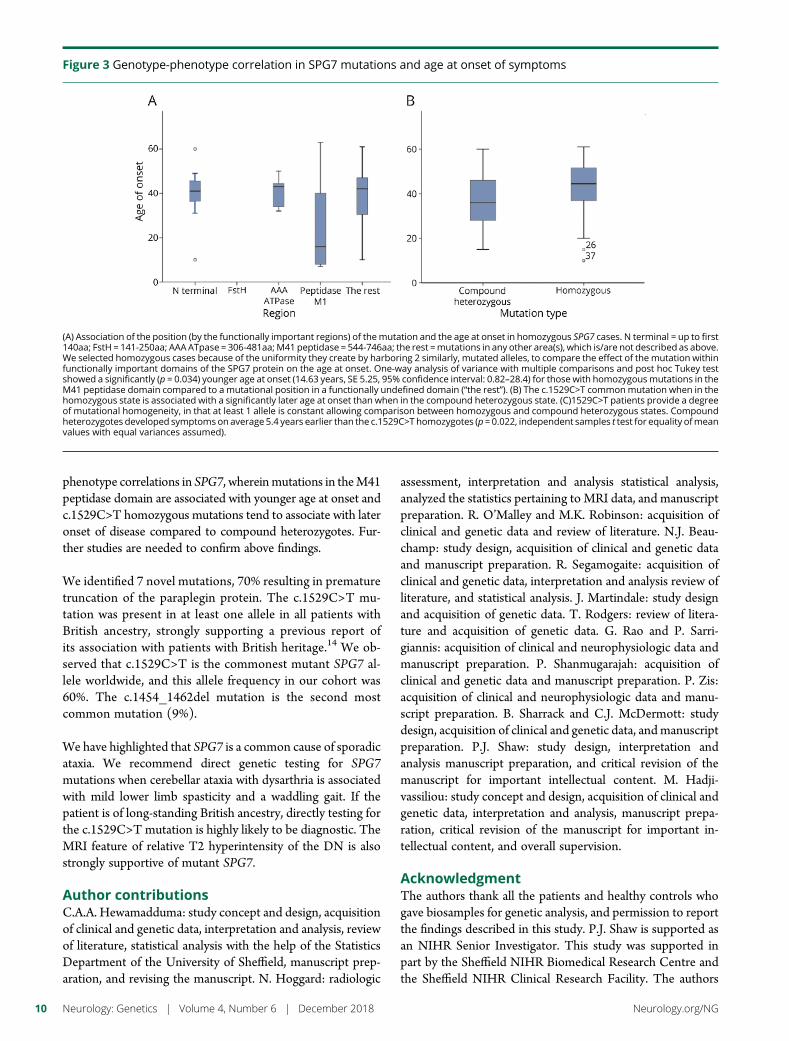
phenotype correlations in SPG7, whereinmutations in theM41peptidase domain are associated with younger age at onset andc.1529C>T homozygous mutations tend to associate with lateronset of disease compared to compound heterozygotes. Fur-ther studies are needed to confirm above findings.
We identified 7 novel mutations, 70% resulting in prematuretruncation of the paraplegin protein. The c.1529C>T mu-tation was present in at least one allele in all patients withBritish ancestry, strongly supporting a previous report ofits association with patients with British heritage.14 We ob-served that c.1529C>T is the commonest mutant SPG7 al-lele worldwide, and this allele frequency in our cohort was60%. The c.1454_1462del mutation is the second mostcommon mutation (9%).
We have highlighted that SPG7 is a common cause of sporadicataxia. We recommend direct genetic testing for SPG7mutations when cerebellar ataxia with dysarthria is associatedwith mild lower limb spasticity and a waddling gait. If thepatient is of long-standing British ancestry, directly testing forthe c.1529C>T mutation is highly likely to be diagnostic. TheMRI feature of relative T2 hyperintensity of the DN is alsostrongly supportive of mutant SPG7.
Author contributionsC.A.A. Hewamadduma: study concept and design, acquisitionof clinical and genetic data, interpretation and analysis, reviewof literature, statistical analysis with the help of the StatisticsDepartment of the University of Sheffield, manuscript prep-aration, and revising the manuscript. N. Hoggard: radiologic
assessment, interpretation and analysis statistical analysis,analyzed the statistics pertaining to MRI data, and manuscriptpreparation. R. O’Malley and M.K. Robinson: acquisition ofclinical and genetic data and review of literature. N.J. Beau-champ: study design, acquisition of clinical and genetic dataand manuscript preparation. R. Segamogaite: acquisition ofclinical and genetic data, interpretation and analysis review ofliterature, and statistical analysis. J. Martindale: study designand acquisition of genetic data. T. Rodgers: review of litera-ture and acquisition of genetic data. G. Rao and P. Sarri-giannis: acquisition of clinical and neurophysiologic data andmanuscript preparation. P. Shanmugarajah: acquisition ofclinical and genetic data and manuscript preparation. P. Zis:acquisition of clinical and neurophysiologic data and manu-script preparation. B. Sharrack and C.J. McDermott: studydesign, acquisition of clinical and genetic data, andmanuscriptpreparation. P.J. Shaw: study design, interpretation andanalysis manuscript preparation, and critical revision of themanuscript for important intellectual content. M. Hadji-vassiliou: study concept and design, acquisition of clinical andgenetic data, interpretation and analysis, manuscript prepa-ration, critical revision of the manuscript for important in-tellectual content, and overall supervision.
AcknowledgmentThe authors thank all the patients and healthy controls whogave biosamples for genetic analysis, and permission to reportthe findings described in this study. P.J. Shaw is supported asan NIHR Senior Investigator. This study was supported inpart by the Sheffield NIHR Biomedical Research Centre andthe Sheffield NIHR Clinical Research Facility. The authors
Figure 3 Genotype-phenotype correlation in SPG7 mutations and age at onset of symptoms
(A) Association of the position (by the functionally important regions) of themutation and the age at onset in homozygous SPG7 cases. N terminal = up to first140aa; FstH = 141-250aa; AAAATpase = 306-481aa; M41 peptidase = 544-746aa; the rest =mutations in any other area(s), which is/are not described as above.We selected homozygous cases because of the uniformity they create by harboring 2 similarly, mutated alleles, to compare the effect of the mutation withinfunctionally important domains of the SPG7 protein on the age at onset. One-way analysis of variance with multiple comparisons and post hoc Tukey testshowed a significantly (p = 0.034) younger age at onset (14.63 years, SE 5.25, 95% confidence interval: 0.82–28.4) for those with homozygousmutations in theM41 peptidase domain compared to a mutational position in a functionally undefined domain (“the rest”). (B) The c.1529C>T commonmutation when in thehomozygous state is associated with a significantly later age at onset than when in the compound heterozygous state. (C)1529C>T patients provide a degreeof mutational homogeneity, in that at least 1 allele is constant allowing comparison between homozygous and compound heterozygous states. Compoundheterozygotes developed symptomson average 5.4 years earlier than the c.1529C>T homozygotes (p = 0.022, independent samples t test for equality ofmeanvalues with equal variances assumed).
10 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

also thank the Statistics Department of the University ofSheffield for the assistance with statistical analysis.
Study fundingThis study did not receive sponsorship or corporate funding.
DisclosureC.A.A. Hewamadduma has received travel funding from Grif-fols Ltd.; is employed with the National Health Service (NHS)(professional affiliation) and holds anHonorary Senior ClinicalLectureship with the University of Sheffield (academic affilia-tion). N. Hoggard is a consultant Neuroradiologist employedby the NHS, professionally, and Professor of Neuroradiologyat the University of Sheffield. R. O’Malley, M.K. Robinson,N.J. Beauchamp, R. Segamogaite, J. Martindale, T. Rodgers,G. Rao, P Sarrigiannis, and P. Shanmugarajah report no dis-closures. R. Segamogaite was an MSc student supervised byDr. Hewamadduma at the University of Sheffield and con-ducted the data analysis with Dr. Hewamadduma. P. Zis serveson the editorial board of Pain and Therapy. B. Sharrack reportsno disclosures. C. McDermott received honoraria from con-sultancy work for Orion Pharma; holds a UK patent for cervicalorthosis; and has received research support from SynapseBiomedical, NIHR, theMarie Curie Foundation, and theMNDAssociation. P.J. Shaw has received research support fromNIHR, the MND Association, the UK Medical ResearchCouncil, the EU JPND and Horizon 2020 programs, Pfizer,Orion Pharma, Biogen, Heptares, the Saudi Arabian govern-ment, the Medical Research Council, EU Horizon, Mirocals,the Motor Neurone Disease Association, Yorkshire andHumber Clinical Research Network, and Wellcome Trust; hasperformed consultant and clinical trials work for Biogen,Treeway, Eclipse, Orion Pharma, Sanofi-Aventis, Ono Pharma,Vertex Pharmaceuticals, Eclipse, and Cytokinetics; serves onthe editorial boards of Amyotrophic Lateral Sclerosis and Fron-totemporal Lobar Degeneration; holds patents for therapeuticsfor neurologic disorders and for orphan drug designation forS-apomorphine in the treatment of amyotrophic lateral scle-rosis; and receives publishing royalties from Henry StewartTalks and the Oxford University Press. M.Hadjivassiliou serveson the editorial board of Cerebellum and Ataxias; has beena consultant for Celimune; and serves on the scientific advisoryboards of Ataxia UK and Coeliac UK. Full disclosure forminformation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Received April 24, 2018. Accepted in final form August 16, 2018.
References1. Refsum S, Skre H. Neurological approaches to the inherited ataxias. Adv Neurol 1978;
21:1–13.2. Polo JM, Calleja J, Combarros O, Berciano J. Hereditary ataxias and paraplegias in
Cantabria, Spain. An epidemiological and clinical study. Brain 1991;114(pt 2):855–866.
3. McDermott CJ, Shaw PJ. Chapter 17 Hereditary spastic paraparesis. Handb ClinNeurol 2007;82:327–352.
4. Matilla-Duenas A. The ever expanding spinocerebellar ataxias. Editorial. Cerebellum2012;11:821–827.
5. Campanella G, Filla A, De Michele G. Classifications of hereditary ataxias: a criticaloverview. Acta Neurol (Napoli) 1992;14:408–419.
6. Jayadev S, Bird TD. Hereditary ataxias: overview. Genet Med 2013;15:673–683.7. Pfeffer G, Pyle A, GriffinH, et al. SPG7mutations are a common cause of undiagnosed
ataxia. Neurology 2015;84:1174–1176.8. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce
framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303.
9. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery andgenotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
10. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidencevariant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bio-informatics 2013;43:11.10.1–11.10.33.
11. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variationin 60,706 humans. Nature 2016;536:285–291.
12. Yang Y, Zhang L, Lynch DR, et al. Compound heterozygote mutations in SPG7 ina family with adult-onset primary lateral sclerosis. Neurol Genet 2016;2:e60.
13. van Gassen KL, van der Heijden CD, de Bot ST, et al. Genotype-phenotype corre-lations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain 2012;135:2994–3004.
14. Roxburgh RH, Marquis-Nicholson R, Ashton F, et al. The p.Ala510Val mutation inthe SPG7 (paraplegin) gene is the most common mutation causing adult onsetneurogenetic disease in patients of British ancestry. J Neurol 2013;260:1286–1294.
15. Choquet K, Tetreault M, Yang S, et al. SPG7 mutations explain a significant pro-portion of French Canadian spastic ataxia cases. Eur J Hum Genet 2016;24:1016–1021.
16. Hadjivassiliou M, Martindale J, Shanmugarajah P, et al. Causes of progressive cere-bellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry2017;88:301–309.
17. Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronicprogressive external ophthalmoplegia through disordered mitochondrial DNAmaintenance. Brain 2014;137:1323–1336.
18. Kara E, Tucci A, Manzoni C, et al. Genetic and phenotypic characterization ofcomplex hereditary spastic paraplegia. Brain 2016;139:1904–1918.
19. Klebe S, Depienne C, Gerber S, et al. Spastic paraplegia gene 7 in patients withspasticity and/or optic neuropathy. Brain 2012;135:2980–2993.
20. Rydning SL, Wedding IM, Koht J, et al. A founder mutation p.H701P identified asa major cause of SPG7 in Norway. Eur J Neurol 2016;23:763–771.
21. Thal DR, Zuchner S, Gierer S, et al. Abnormal paraplegin expression in swollenneurites, tau- and alpha-synuclein pathology in a case of hereditary spastic paraplegiaSPG7 with an Ala510Val mutation. Int J Mol Sci 2015;16:25050–25066.
22. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentateupdate: imaging features of entities that affect the dentate nucleus. AJNR Am JNeuroradiol 2017;38:1467–1474.
23. Arnoldi A, Tonelli A, Crippa F, et al. A clinical, genetic, and biochemical character-ization of SPG7 mutations in a large cohort of patients with hereditary spastic para-plegia. Hum Mutat 2008;29:522–531.
24. McDermott CJ, Dayaratne RK, Tomkins J, et al. Paraplegin gene analysis in hereditaryspastic paraparesis (HSP) pedigrees in northeast England. Neurology 2001;56:467–471.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 11

ARTICLE OPEN ACCESS
Molecular pathogenesis of human CD59deficiencyNetanel Karbian, PhD, Yael Eshed-Eisenbach, PhD, Adi Tabib, MSc, Hila Hoizman, BSc, B. Paul Morgan, PhD,
Ora Schueler-Furman, PhD, Elior Peles, PhD, and Dror Mevorach, MD
Neurol Genet 2018;4:e280. doi:10.1212/NXG.0000000000000280
Correspondence
Dr. Mevorach
AbstractObjectiveTo characterize all 4 mutations described for CD59 congenital deficiency.
MethodsThe 4mutations, p.Cys64Tyr, p.Asp24Val, p.Asp24Valfs*, and p.Ala16Alafs*, were described in13 individuals with CD59 malfunction. All 13 presented with recurrent Guillain-Barre syn-drome or chronic inflammatory demyelinating polyneuropathy, recurrent strokes, and chronichemolysis. Here, we track the molecular consequences of the 4 mutations and their effects onCD59 expression, localization, glycosylation, degradation, secretion, and function. Mutantswere cloned and inserted into plasmids to analyze their expression, localization, andfunctionality.
ResultsImmunolabeling of myc-tagged wild-type (WT) and mutant CD59 proteins revealed cellsurface expression of p.Cys64Tyr and p.Asp24Val detected with the myc antibody, but nolabeling by anti-CD59 antibodies. In contrast, frameshift mutants p.Asp24Valfs* andp.Ala16Alafs* were detected only intracellularly and did not reach the cell surface. Western blotanalysis showed normal glycosylation but mutant-specific secretion patterns. All mutants sig-nificantly increased MAC-dependent cell lysis compared with WT. In contrast to CD59knockout mice previously used to characterize phenotypic effects of CD59 perturbation, all 4hCD59 mutations generate CD59 proteins that are expressed and may function intracellularly(4) or on the cell membrane (2). None of the 4 CD59 mutants are detected by known anti-CD59 antibodies, including the 2 variants present on the cell membrane. None of the 4 inhibitsmembrane attack complex (MAC) formation.
ConclusionsAll 4 mutants generate nonfunctional CD59, 2 are expressed as cell surface proteins that mayfunction in non–MAC-related interactions and 2 are expressed only intracellularly. Distinctsecretion of soluble CD59 may have also a role in disease pathogenesis.
From the Rheumatology Research Center (N.K., A.T., H.H., D.M.), Center of Rare Diseases, and Department of Medicine, Hadassah-Hebrew University Medical Center, Jerusalem; TheWeizmann Institute (Y.E.-E., E.P.), Rehovot, Israel; Systems Immunity Research Institute (B.P.M.), Cardiff University, Cardiff, Wales, UK; and Hebrew University (O.S.-F., D.M.),Jerusalem, Israel.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the Israel Science Foundation.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Complement activation triggers membrane attack complex(MAC) assembly to formpores in cell membrane lipid bilayers ofsusceptible bacteria.1 However, unregulated MAC formationmay cause host tissue damage.2 The glycosyl phosphatidylinositol(GPI)-anchored cell surface membrane glycoprotein CD59inhibits the final step of MAC formation to protect host cellsfrom MAC-mediated injury.3
Several mutations in the CD59-coding sequence are knownin human patients (figure 1A). We and others have pre-viously reported of 13 patients4–11 aged 1–4.5 years whosuffered from chronic hemolysis and recurrent episodes ofGuillain-Barre syndrome (GBS)-like disease from earlyinfancy, suggesting chronic inflammatory demyelinatingpolyradiculoneuropathy (CIDP), and recurrent strokes(table e-1, links.lww.com/NXG/A87). The mutations in-cluded p.Cys64Tyr, p.Asp24Val, p.Asp24Valfs*, andp.Ala16Alafs*, all leading to CD59 loss of function. In allmutations, no surface protein was detected by anti-CD59antibody staining. We were interested, in the current study,to verify whether indeed no proteins were produced,whether any proteins that were produced reached themembrane, and whether proteins or truncated proteinsexist intracellularly or secreted outside the cells. Thesepotential differences may have functional implications andclinical manifestations.
MethodsGeneration of myc-tagged wild-type (WT) andmutant hCD59 expression plasmidsTo verify the membrane and intracellular localization of themutated protein, we isolated, myc-tagged, and expressed WTCD59 and each mutant in mammalian cell lines. Total RNAwas extracted from human white blood cell samples usingTRI Reagent (Sigma, St Louis, MO). cDNAs generated bySuperScript II Reverse Transcriptase (Life Technologies,Carlsbad, CA) from RNAs isolated from both a Cys64Tyrhomozygous patient and a healthy individual were used astemplates for PCR reactions (details appear in a supplemen-tary section of Methods links.lww.com/NXG/A88).
AntibodiesTo test staining of WT and mutant CD59 by anti-CD59antibodies, human CD59 antibodies were used in im-munofluorescence labeling assays of overexpressingCOS7 cells and detected by flow cytometry and fluo-rescent microscopy. Details appear in supplementarysection.
Sodium dodecyl sulfate–polyacrylamide gelelectrophoresis (SDS-PAGE) and Western blotting andimmunoprecipitationSDS-PAGE, Western blot, and immunoprecipitation were usedforCD59 detection (details appear in the supplementary section).
Bortezomib (proteasome inhibitor) treatmentTo test the effect of ubiquitination on expression, we inhibitedubiquitination by bortezomib. Forty-eight hours after trans-fection, culture medium was supplemented with 10 nM bor-tezomib (Velcade, Cell Signaling) or vehicle-only control. Amouse antibody against the myc-tag peptide was used forWestern blot analysis.
In vitro cell lysis assay assessing MAC attack in WT andmutantsTo evaluate deposition of MAC and cell lysis, we used in vitrocell lysis assays. Transfections of Chinese hamster ovary(CHO) cells were performed using Lipofectamine 2000 re-agent. Details appear in the supplementary section. Cells wereharvested 24 hours after transfection, replated on 24-well plates(3 × 105 cells in 1 mL per well), and left until the plates wereconfluent. Cells were then washed twice with serum-free me-dium and incubated in 0.25 mL complete medium (7.5% FCSheat inactivated) containing a 1/750 dilution of calcein AM(Molecular Probes, Eugene, OR, 1 mg/mL stock in dimethylsulfoxide) for 1 hour at 37°C. After 1 wash with phosphatebuffered saline (PBS), duplicate wells were incubated with 1/5dilution of normal human serum and 40 μL of rabbit anti-CHOimmunoglobulin G (IgG). After incubation for 1 hour at 37°C,all fluid was removed from the cells and transferred to 96-wellplates for calcein measurement.
The remaining cells were lysed with 0.25 mL PBS containing0.1% Triton X-100 during a 15-minute incubation period atroom temperature, and the lysate was removed to 96-wellplates for calcein release measurement. Calcein fluorescenceof supernatants was read in a Cytation 3 Cell Imaging Multi-Mode Reader (BioTek, Winoosky, VT) with excitation andemission filters set at 485 and 530 nm, respectively. Percentlysis for each well was calculated as calcein release/total cal-cein loading. Mean values and SDs were determined fromduplicate samples. Analysis of variance (ANOVA) and Stu-dent t test were used for statistical analysis.
Structural biologyPymol (Schrodinger LLC, New York, NY) was used for in-spection of structures and figure generation. Models of theframeshift peptide sequences were generated using theI-TASSER server.12–14
GlossaryCIDP = chronic inflammatory demyelinating polyradiculoneuropathy; ER = endoplasmic reticulum; GBS = Guillain-Barresyndrome; MAC = membrane attack complex; PNH = paroxysmal nocturnal hemoglobinuria; SDS-PAGE = sodium dodecylsulfate–polyacrylamide gel electrophoresis; WT = wild type.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Standard protocol approvals, registrations, andpatient consentsHuman DNA and tissue samples were obtained in protocolsapproved by our institutional review board. All patients ortheir families provided informed consent.
Data availabilityStudy data for the primary analyses presented in this reportare available upon reasonable request from the correspondingand senior author.
ResultsPredicted effect of CD59 mutations on its 3Dprotein structureFigure 1 shows the sequences (figure 1A) and structures (figure1, B and C) of CD59 with residues mutated in the 2 describedpoint mutations (figure 1B). The similarities between the 2frameshift mutants are highlighted (figure 1C). The maturemembrane surface–expressed CD59 sequence consists of 77
residues after removal of a 25-residue N-terminal signal se-quence and a C-terminal GPI-anchoring signal.15
Inspection of solved structures of CD59,16 pdb IDs 2j8b,2uwr, 2ux2,17 4bik,18 and 2ofs19 shows that in the p.Cys64Tyrmutant, a local disulfide bridge near the carboxy terminus ofthe mature protein is interrupted (Cys64-Cys69) (figure 1B).This would probably merely loosen the loop that is followedby the C-terminal helix, which adopts different orientationsdepending on the solved structure (e.g., 2j8b vs 2uwr), in-dicating that it is not strongly stabilized in the WT structure.As for p.Asp24Val, Asp24 was previously reported to play animportant role in CD59 stability and activity: p.Asp24Alastrongly destabilizes CD59,20 whereas p.Asp24Arg permitsprotein folding and expression, but abolishes CD59 activity.21
Indeed, Asp24 is part of an exposed loop (Ser20-Asp24),located between the second and third β-strands (figure 1B),that is involved in modulating CD59 activity. Single mutationsto alanine in that loop increased, whereas multiple alanine
Figure 1 Sequence and structure of CD59 WT and mutants
(A) The sequences of WT CD59, pointmutations Cys64Tyr and Asp24Val, andframeshift mutations Asp24Valfs* andAla16Alafs*: The mature membranesurface CD59 primary sequence con-sists of 77 residues after removal ofa 25-residue N-terminal signal se-quence and a C-terminal GPI-anchor-ing signal (not shown). Colored arrowsand boxes represent β-strands andα-helices, respectively, as observed inWT CD59 (see B). Point mutations aremarked with an asterisk. The sequenceof the 2 frameshift mutants revealssignificantly shortened proteins thatlack most of the CD59 sequence, inparticular the GPI-anchoring signal. (B)The structure of CD59 highlights thevicinity of the point mutations toknown sites of activity and interaction.The mutated Cys64 and Asp24 posi-tions are highlighted as spheres andlabeled. The 3 characterized interfacesof CD59 are marked by numberedcrescents: (1) The classic site charac-terized originally described by Bodianet al.21 with the central Trp40 residueshown in sticks, (2) a loop spanningresidues 20–24 that modulates CD59activity20 (colored in white), and (3) theedge β-strand that interacts with ILY18
(to the left of the crescent). Disulfidebridges that stabilize CD59 are shownin sticks. Two solved structures (2j8band 2uwr) are shown, highlighting therelative flexibility of the C-terminal he-lix (colored in magenta). (C) Theframeshift mutants contain onlya small part of the original CD59 se-quence: The structure of CD59 isshown, with the regionwith a sequencein common with the Ala16Alafs* andAsp24Valfs* mutants colored in yellowand yellow-white, respectively. The I-TASSER model for the Asp24Valfs* se-quence aligns 2 additional β-strands(colored in green, ending with thesphere at CD59 position Trp40), beforeit diverges, and the remaining part (inblue) is unique to CD59. GPI = glyco-syl phosphatidylinositol; WT = wild type.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
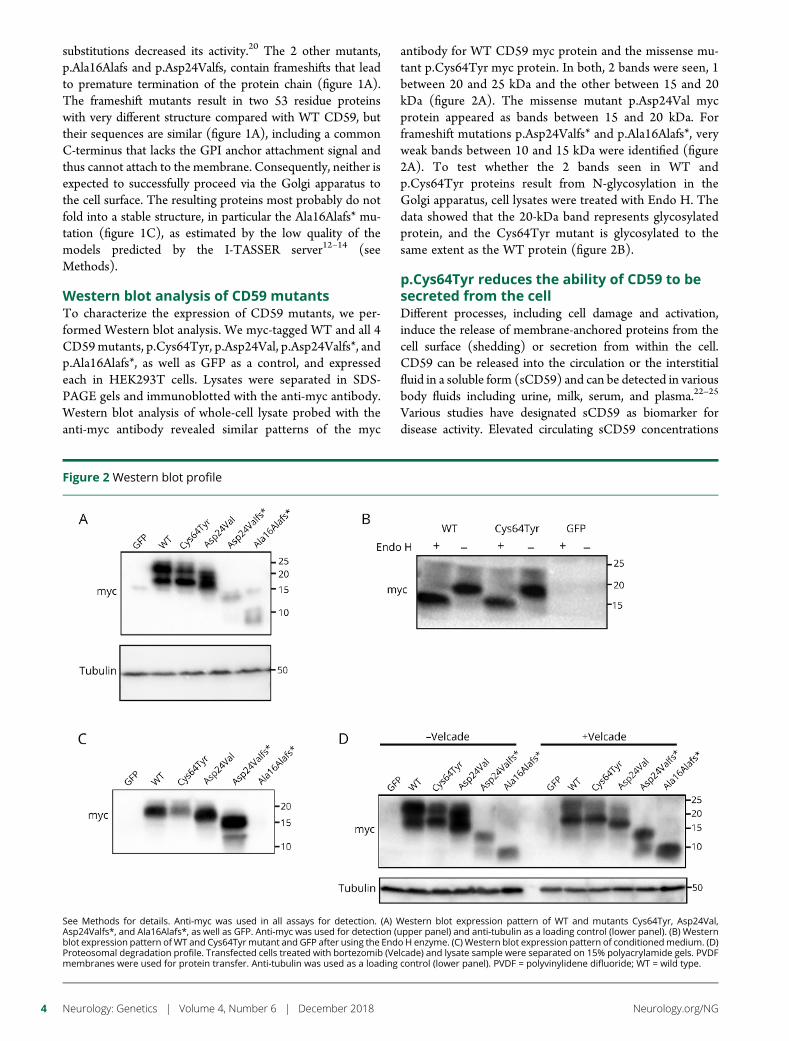
substitutions decreased its activity.20 The 2 other mutants,p.Ala16Alafs and p.Asp24Valfs, contain frameshifts that leadto premature termination of the protein chain (figure 1A).The frameshift mutants result in two 53 residue proteinswith very different structure compared with WT CD59, buttheir sequences are similar (figure 1A), including a commonC-terminus that lacks the GPI anchor attachment signal andthus cannot attach to the membrane. Consequently, neither isexpected to successfully proceed via the Golgi apparatus tothe cell surface. The resulting proteins most probably do notfold into a stable structure, in particular the Ala16Alafs* mu-tation (figure 1C), as estimated by the low quality of themodels predicted by the I-TASSER server12–14 (seeMethods).
Western blot analysis of CD59 mutantsTo characterize the expression of CD59 mutants, we per-formed Western blot analysis. We myc-tagged WT and all 4CD59mutants, p.Cys64Tyr, p.Asp24Val, p.Asp24Valfs*, andp.Ala16Alafs*, as well as GFP as a control, and expressedeach in HEK293T cells. Lysates were separated in SDS-PAGE gels and immunoblotted with the anti-myc antibody.Western blot analysis of whole-cell lysate probed with theanti-myc antibody revealed similar patterns of the myc
antibody for WT CD59 myc protein and the missense mu-tant p.Cys64Tyr myc protein. In both, 2 bands were seen, 1between 20 and 25 kDa and the other between 15 and 20kDa (figure 2A). The missense mutant p.Asp24Val mycprotein appeared as bands between 15 and 20 kDa. Forframeshift mutations p.Asp24Valfs* and p.Ala16Alafs*, veryweak bands between 10 and 15 kDa were identified (figure2A). To test whether the 2 bands seen in WT andp.Cys64Tyr proteins result from N-glycosylation in theGolgi apparatus, cell lysates were treated with Endo H. Thedata showed that the 20-kDa band represents glycosylatedprotein, and the Cys64Tyr mutant is glycosylated to thesame extent as the WT protein (figure 2B).
p.Cys64Tyr reduces the ability of CD59 to besecreted from the cellDifferent processes, including cell damage and activation,induce the release of membrane-anchored proteins from thecell surface (shedding) or secretion from within the cell.CD59 can be released into the circulation or the interstitialfluid in a soluble form (sCD59) and can be detected in variousbody fluids including urine, milk, serum, and plasma.22–25
Various studies have designated sCD59 as biomarker fordisease activity. Elevated circulating sCD59 concentrations
Figure 2 Western blot profile
See Methods for details. Anti-myc was used in all assays for detection. (A) Western blot expression pattern of WT and mutants Cys64Tyr, Asp24Val,Asp24Valfs*, and Ala16Alafs*, as well as GFP. Anti-myc was used for detection (upper panel) and anti-tubulin as a loading control (lower panel). (B) Westernblot expression pattern ofWT and Cys64Tyrmutant and GFP after using the Endo H enzyme. (C) Western blot expression pattern of conditionedmedium. (D)Proteosomal degradation profile. Transfected cells treated with bortezomib (Velcade) and lysate sample were separated on 15% polyacrylamide gels. PVDFmembranes were used for protein transfer. Anti-tubulin was used as a loading control (lower panel). PVDF = polyvinylidene difluoride; WT = wild type.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
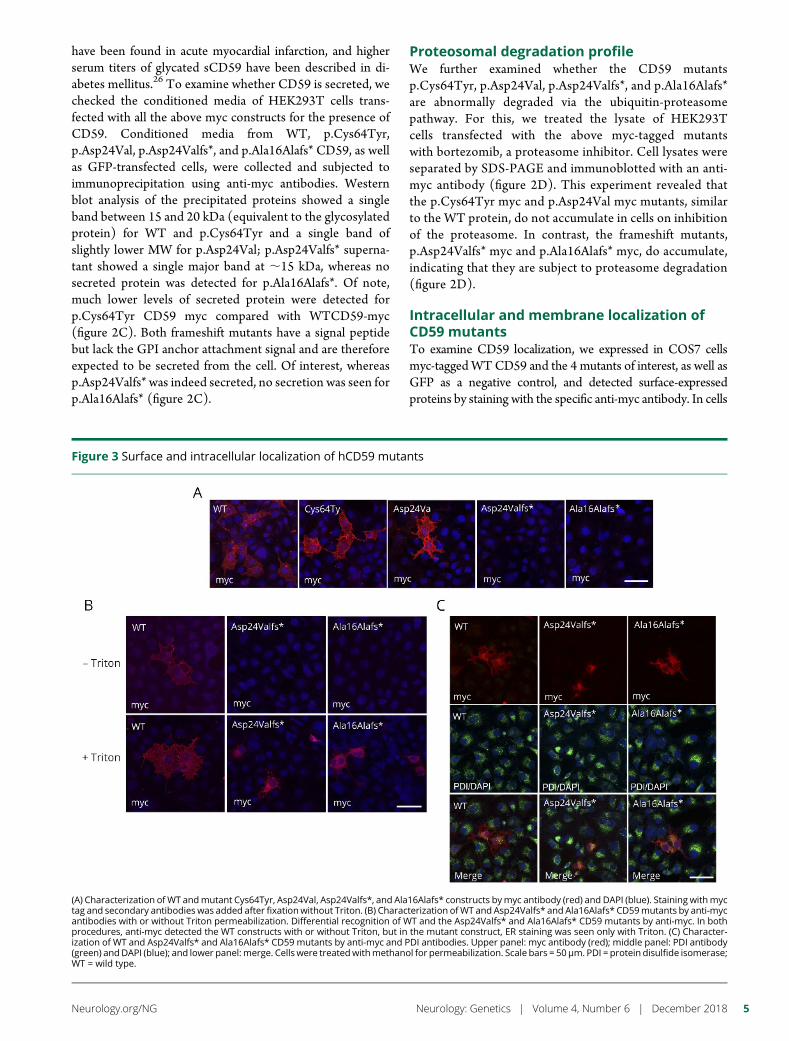
have been found in acute myocardial infarction, and higherserum titers of glycated sCD59 have been described in di-abetes mellitus.26 To examine whether CD59 is secreted, wechecked the conditioned media of HEK293T cells trans-fected with all the above myc constructs for the presence ofCD59. Conditioned media from WT, p.Cys64Tyr,p.Asp24Val, p.Asp24Valfs*, and p.Ala16Alafs* CD59, as wellas GFP-transfected cells, were collected and subjected toimmunoprecipitation using anti-myc antibodies. Westernblot analysis of the precipitated proteins showed a singleband between 15 and 20 kDa (equivalent to the glycosylatedprotein) for WT and p.Cys64Tyr and a single band ofslightly lower MW for p.Asp24Val; p.Asp24Valfs* superna-tant showed a single major band at ;15 kDa, whereas nosecreted protein was detected for p.Ala16Alafs*. Of note,much lower levels of secreted protein were detected forp.Cys64Tyr CD59 myc compared with WTCD59-myc(figure 2C). Both frameshift mutants have a signal peptidebut lack the GPI anchor attachment signal and are thereforeexpected to be secreted from the cell. Of interest, whereasp.Asp24Valfs* was indeed secreted, no secretion was seen forp.Ala16Alafs* (figure 2C).
Proteosomal degradation profileWe further examined whether the CD59 mutantsp.Cys64Tyr, p.Asp24Val, p.Asp24Valfs*, and p.Ala16Alafs*are abnormally degraded via the ubiquitin-proteasomepathway. For this, we treated the lysate of HEK293Tcells transfected with the above myc-tagged mutantswith bortezomib, a proteasome inhibitor. Cell lysates wereseparated by SDS-PAGE and immunoblotted with an anti-myc antibody (figure 2D). This experiment revealed thatthe p.Cys64Tyr myc and p.Asp24Val myc mutants, similarto the WT protein, do not accumulate in cells on inhibitionof the proteasome. In contrast, the frameshift mutants,p.Asp24Valfs* myc and p.Ala16Alafs* myc, do accumulate,indicating that they are subject to proteasome degradation(figure 2D).
Intracellular and membrane localization ofCD59 mutantsTo examine CD59 localization, we expressed in COS7 cellsmyc-taggedWTCD59 and the 4 mutants of interest, as well asGFP as a negative control, and detected surface-expressedproteins by staining with the specific anti-myc antibody. In cells
Figure 3 Surface and intracellular localization of hCD59 mutants
(A) Characterization ofWT andmutant Cys64Tyr, Asp24Val, Asp24Valfs*, and Ala16Alafs* constructs bymyc antibody (red) and DAPI (blue). Staining withmyctag and secondary antibodies was added after fixationwithout Triton. (B) Characterization ofWT and Asp24Valfs* and Ala16Alafs* CD59mutants by anti-mycantibodies with or without Triton permeabilization. Differential recognition of WT and the Asp24Valfs* and Ala16Alafs* CD59 mutants by anti-myc. In bothprocedures, anti-myc detected the WT constructs with or without Triton, but in the mutant construct, ER staining was seen only with Triton. (C) Character-ization of WT and Asp24Valfs* and Ala16Alafs* CD59 mutants by anti-myc and PDI antibodies. Upper panel: myc antibody (red); middle panel: PDI antibody(green) andDAPI (blue); and lower panel:merge. Cells were treatedwithmethanol for permeabilization. Scale bars = 50μm. PDI = protein disulfide isomerase;WT = wild type.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

expressing WT, p.Cys64Tyr, and p.Asp24Val constructs, cellsurface localization was detected by themyc antibody, whererasno surface staining was seen in cells expressing p.Asp24Valfs*and p.Ala16Alafs* constructs (figure 3A). Because we could notdetect any surface staining in the 2-frameshift mutants, we re-peated the immunolabeling procedures on fixed cells, with orwithout membrane permeabilization by Triton-X100. Whenimmunolabeling procedures were performed without Triton,themyc antibody clearly detectedWTCD59on the cell surface,but not the frameshift mutants p.Asp24Valfs* and p.Ala16Alafs*(figure 3B upper panel). After Triton permeabilization, how-ever, the myc antibody stained the 2 frameshift mutant CD59proteins intracellularly, presumably in the endoplasmic re-ticulum (ER) (figure 3B lower panel). To verify the ER stainingof the frameshift mutations, we double labeled the transfectedCOS7 cells with anti-myc and an anti-PDI antibody, an ERmarker. Immunolabeling revealed clear colocalization demon-strating that both frameshift mutants localized to the ER (figure3C). To summarize, although both missense mutantsp.Cys64Tyr and p.Asp24Val are expressed on the cell surface,the frameshift mutants p.Asp24Valfs* and p.Ala16Alafs* fail toreach the cell surface and remain in the ER.
These results are in agreement with Western blot expressionresults (figure 2, C and D), indicating that the missensemutants are expressed and stable at the cell surface, whereasthe frameshift mutants are retained in the ER, fromwhich theyare secreted from the cell (in the case of p.Asp24Valfs*) orsent to proteosomal degradation (both p.Asp24Valfs* andp.Ala16Alafs*).
CD59 mutants are not detected by anti-CD59 antibodiesTo investigate whether any of the available anti-CD59antibodies detect the surface-expressed mutants, patient-derived Cys64Tyr-expressing and healthy control lympho-blasts were exposed to 5 different hCD59 monoclonalantibodies representing mapped epitopes MEM43, HC1,BRIC229, YTH53.1, and A35,21 1 monoclonal antibody withan unknown epitope, 1.39, and a rabbit polyclonal antiserumraised against human CD59. By flow cytometry, detection ofCD59 by each of these 7 antibodies occurred only in thecontrol lymphoblasts and not in lymphoblasts expressingCD59 mutants (figure 4A). We further verified these resultsusing cells transfected with WT mutant, both missensemutants p.Cys64Tyr and p.Asp24Val and the 2 frameshiftmutants p.Asp24Valfs* and p.Ala16Alafs* by immunofluo-rescence. Transfected cells were immunolabeled with eachof the above human CD59 antibodies and, in parallel, withthe anti-myc antibody. Immunolabeling with the mono-clonal antibodies, MEM43, BRIC229 (figure 4B), HC1, 1.39(figure e-2) and rabbit polyclonal antibodies (data notshown) revealed clear cell surface detection of the WT butnot the mutant constructs (figure 4B). For technical reasons,A35 and YTH53.1 were not included in the staining. Resultsof the immunofluorescence labeling, including the myc an-tibody, are summarized in table e-2 (links.lww.com/NXG/
A87). Taken together, known anti-CD59 antibodies did notdetect surface expression of CD59 protein in either of the 2missense mutations. This may suggest that the missensemutations result in substantial structural changes that abol-ish the protein’s interaction with various antibodies and maysimilarly abolish protein-protein interactions necessary forits different functions.
CD59 p.Cys64Tyr and p.Asp24Val mutants arenonfunctional in protecting from MAC attackAll 7 patients described with the p.Cys64Tyr mutation, and all3 patients described with the Asp24Val mutation exhibitedparoxysmal nocturnal hemoglobinuria (PNH)-like RBC he-molysis and a hypercoagulability state6,9 due to increasedMAC formation. We expressed WT and mutant hCD59constructs in the CHO cells and examined MAC-dependentlysis of the transfected cells. As shown in figure 5, WT CD59transfection protected CHO cells from lysis (p < 0.05,ANOVA and Student t test); however, all mutants, includingp.Cys64Tyr and p.Asp24Val, showed significantly increasedMAC-dependent cell lysis (p < 0.05, ANOVA and Student ttest for frameshift mutants and p.Asp24Val) compared withthe level of MAC-dependent cell lysis in the presence of WThCD59. For the p.Cys64Tyr mutant, this measured lysis (p <0.002, ANOVA and Student t test) even surpassed the lysis ofCHO cells without any additional expression, indicatingperhaps an agonist effect on MAC-dependent lysis that needsfurther investigation.
Taken together, we conclude that although both p.Cys64Tyrand p.Asp24Val mutant CD59 are surface expressed asdemonstrated by myc staining, they are not detected by any ofa panel of anti-CD59 antibodies using flow cytometry orimmunofluorescence labeling and do not protect againstMAC killing. The data indicate that these mutant proteins aremisfolded and, similar to the frameshift mutations, show noCD59 function in relation to protection against MAC.
DiscussionSecondaryCD59 deficiency is a common finding in patients withPNH. This condition is characterized by clonal expansion ofhematopoietic stem cells that have acquired a somatic mutationin the PIGA gene (phosphatidylinositol N-acetylglucosaminyl-transferase subunit A). PIGA encodes a GPI biosynthesisprotein, phosphatidylinositol N-acetylglucosaminyltransferasesubunit A.27,28 The CD59 protein inhibits the final and mostimportant step of MAC formation. Erythrocytes that are de-ficient in GPI-anchored membrane proteins, including CD59,undergo complement-mediated hemolysis.29,30
The relationship of CD59 deficiency to the erythrocytephenotypes of PNH has been established.31–34 Indeed, ina murine model, targeted deletion of the CD59 gene resultedin spontaneous intravascular hemolysis and hemoglobinuria,findings characteristic of PNH.35
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
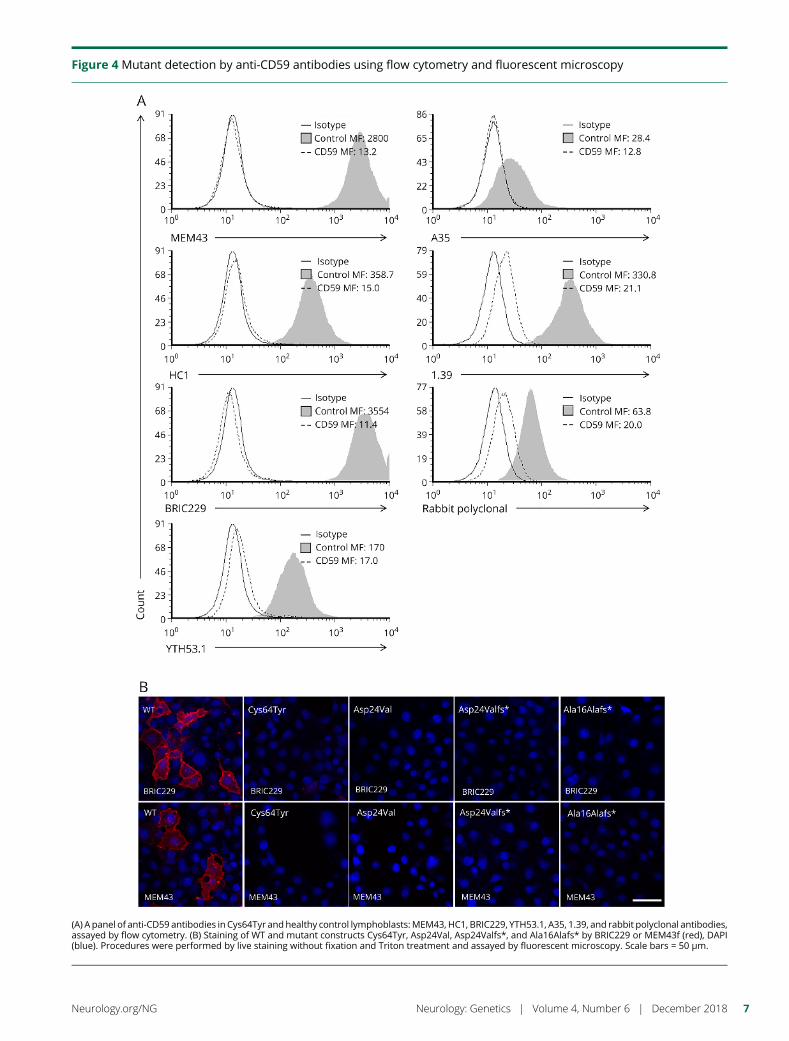
Figure 4 Mutant detection by anti-CD59 antibodies using flow cytometry and fluorescent microscopy
(A) A panel of anti-CD59 antibodies in Cys64Tyr andhealthy control lymphoblasts:MEM43, HC1, BRIC229, YTH53.1, A35, 1.39, and rabbit polyclonal antibodies,assayed by flow cytometry. (B) Staining of WT and mutant constructs Cys64Tyr, Asp24Val, Asp24Valfs*, and Ala16Alafs* by BRIC229 or MEM43f (red), DAPI(blue). Procedures were performed by live staining without fixation and Triton treatment and assayed by fluorescent microscopy. Scale bars = 50 μm.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7
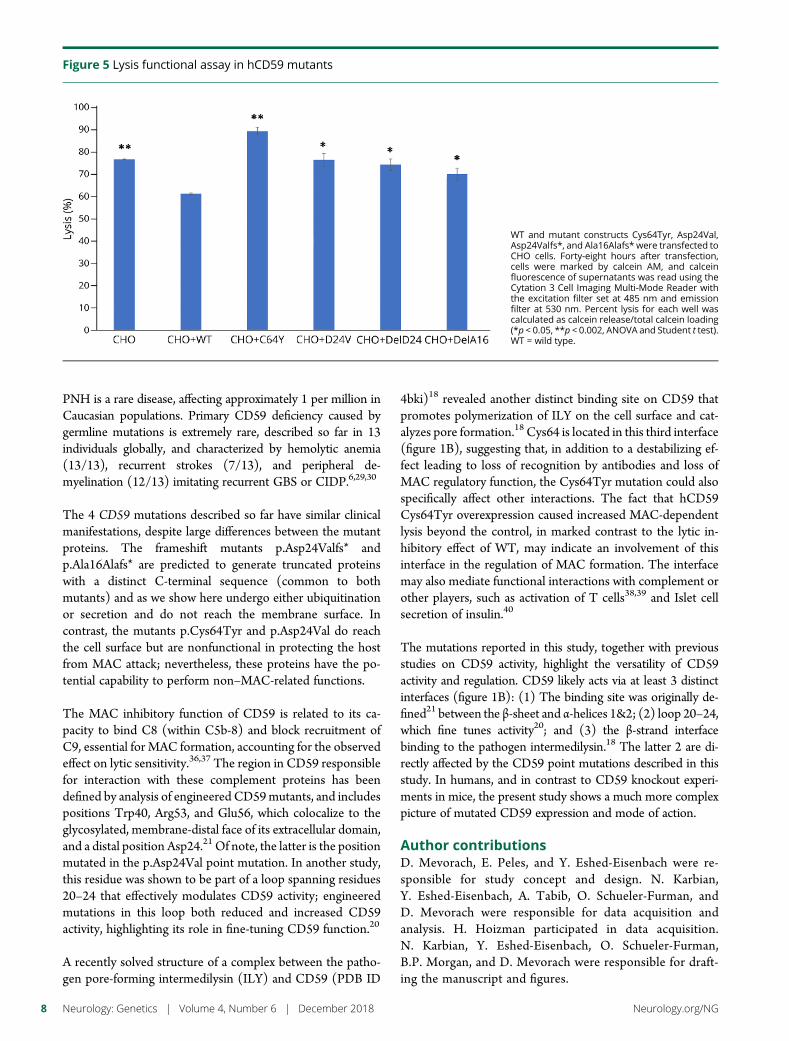
PNH is a rare disease, affecting approximately 1 per million inCaucasian populations. Primary CD59 deficiency caused bygermline mutations is extremely rare, described so far in 13individuals globally, and characterized by hemolytic anemia(13/13), recurrent strokes (7/13), and peripheral de-myelination (12/13) imitating recurrent GBS or CIDP.6,29,30
The 4 CD59 mutations described so far have similar clinicalmanifestations, despite large differences between the mutantproteins. The frameshift mutants p.Asp24Valfs* andp.Ala16Alafs* are predicted to generate truncated proteinswith a distinct C-terminal sequence (common to bothmutants) and as we show here undergo either ubiquitinationor secretion and do not reach the membrane surface. Incontrast, the mutants p.Cys64Tyr and p.Asp24Val do reachthe cell surface but are nonfunctional in protecting the hostfrom MAC attack; nevertheless, these proteins have the po-tential capability to perform non–MAC-related functions.
The MAC inhibitory function of CD59 is related to its ca-pacity to bind C8 (within C5b-8) and block recruitment ofC9, essential for MAC formation, accounting for the observedeffect on lytic sensitivity.36,37 The region in CD59 responsiblefor interaction with these complement proteins has beendefined by analysis of engineered CD59mutants, and includespositions Trp40, Arg53, and Glu56, which colocalize to theglycosylated, membrane-distal face of its extracellular domain,and a distal position Asp24.21 Of note, the latter is the positionmutated in the p.Asp24Val point mutation. In another study,this residue was shown to be part of a loop spanning residues20–24 that effectively modulates CD59 activity; engineeredmutations in this loop both reduced and increased CD59activity, highlighting its role in fine-tuning CD59 function.20
A recently solved structure of a complex between the patho-gen pore-forming intermedilysin (ILY) and CD59 (PDB ID
4bki)18 revealed another distinct binding site on CD59 thatpromotes polymerization of ILY on the cell surface and cat-alyzes pore formation.18 Cys64 is located in this third interface(figure 1B), suggesting that, in addition to a destabilizing ef-fect leading to loss of recognition by antibodies and loss ofMAC regulatory function, the Cys64Tyr mutation could alsospecifically affect other interactions. The fact that hCD59Cys64Tyr overexpression caused increased MAC-dependentlysis beyond the control, in marked contrast to the lytic in-hibitory effect of WT, may indicate an involvement of thisinterface in the regulation of MAC formation. The interfacemay also mediate functional interactions with complement orother players, such as activation of T cells38,39 and Islet cellsecretion of insulin.40
The mutations reported in this study, together with previousstudies on CD59 activity, highlight the versatility of CD59activity and regulation. CD59 likely acts via at least 3 distinctinterfaces (figure 1B): (1) The binding site was originally de-fined21 between the β-sheet and α-helices 1&2; (2) loop 20–24,which fine tunes activity20; and (3) the β-strand interfacebinding to the pathogen intermedilysin.18 The latter 2 are di-rectly affected by the CD59 point mutations described in thisstudy. In humans, and in contrast to CD59 knockout experi-ments in mice, the present study shows a much more complexpicture of mutated CD59 expression and mode of action.
Author contributionsD. Mevorach, E. Peles, and Y. Eshed-Eisenbach were re-sponsible for study concept and design. N. Karbian,Y. Eshed-Eisenbach, A. Tabib, O. Schueler-Furman, andD. Mevorach were responsible for data acquisition andanalysis. H. Hoizman participated in data acquisition.N. Karbian, Y. Eshed-Eisenbach, O. Schueler-Furman,B.P. Morgan, and D. Mevorach were responsible for draft-ing the manuscript and figures.
Figure 5 Lysis functional assay in hCD59 mutants
WT and mutant constructs Cys64Tyr, Asp24Val,Asp24Valfs*, and Ala16Alafs* were transfected toCHO cells. Forty-eight hours after transfection,cells were marked by calcein AM, and calceinfluorescence of supernatants was read using theCytation 3 Cell Imaging Multi-Mode Reader withthe excitation filter set at 485 nm and emissionfilter at 530 nm. Percent lysis for each well wascalculated as calcein release/total calcein loading(*p < 0.05, **p < 0.002, ANOVA and Student t test).WT = wild type.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

AcknowledgmentThis research was supported by the Legacy HeritageBioMedical Program of the Israel Science Foundation (grantno. 1070/15 to D.M.). The authors wish to thank ShifraFraifeld, Hadassah-Hebrew University Medical Center, forher editorial assistance.
Study fundingThis research was supported by the Legacy Heritage Bio-medical Program of the Israel Science Foundation (grant no.1070/15 to D.M.). No author has a financial relationship withany company in relation with this study.
DisclosureN. Karbian, Y. Eshed-Eisenbach, A. Tabib, and H. Hoizmanreport no disclosures. P. Morgan has served on the scientificadvisory boards of Roche, Alexion, and Achillion; has beena consultant for GSK (payments to university), Roche,Alexion, and Achillion; and has received research supportfrom GSK, the Medical Research Council, and WellcomeTrust. O. Schueler-Furman reports no disclosures. E. Peleshas served on the editorial boards of the Journal of CellBiology, ASN NEURO, Molecular and Cellular Neuroscience,Neuronal and Glial Biology, Faculty of 1,000, and F1000 Re-search and has received research support from the NIH andthe Israel Science Foundation. D. Mevorach has receivedconsultation fees from Enlivex; has received research sup-port from the Israel Science Foundation; and holds stock/stock options in Enlivex. Full disclosure form informationprovided by the authors is available with the full text of thisarticle at Neurology.org/NG.
Received March 21, 2018. Accepted in final form August 7, 2018.
References1. Podack ER. Molecular composition of the tubular structure of the membrane attack
complex of complement. J Biol Chem 1984;259:8641–8647.2. Morgan BP. Regulation of the complement membrane attack pathway. Crit Rev
Immunol 1999;19:173–198.3. Meri S, Morgan BP, Davies A, et al. Human protectin (CD59), an 18,000-20,000 MW
complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipidbilayers. Immunology 1990;71:1–9.
4. Nevo Y, Ben-Zeev B, Tabib A, et al. CD59 deficiency is associated with chronichemolysis and childhood relapsing immune-mediated polyneuropathy. Blood 2013;121:129–135.
5. Ben-Zeev B, Tabib A, Nissenkorn A, et al. Devastating recurrent brain ischemicinfarctions and retinal disease in pediatric patients with CD59 deficiency. Eur JPaediatr Neurol 2015;19:688–693.
6. Mevorach D, Reiner I, Grau A, et al. Therapy with eculizumab for patients with CD59p.Cys89Tyr mutation. Ann Neurol 2016;80:708–717.
7. Ardicli D, Taskiran EZ, Kosukcu C, et al. Neonatal-onset recurrent Guillain-Barresyndrome-like disease: clues for inherited CD59 deficiency. Neuropediatrics 2017;48:477–481.
8. Hochsmann B, Dohna-Schwake C, Kyrieleis HA, Pannicke U, Schrezenmeier H.Targeted therapy with eculizumab for inherited CD59 deficiency. N Engl J Med 2014;370:90–92.
9. Haliloglu G, Maluenda J, Sayinbatur B, et al. Early-onset chronic axonal neuropathy,strokes, and hemolysis: inherited CD59 deficiency. Neurology 2015;84:1220–1224.
10. Yamashina M, Ueda E, Kinoshita T, et al. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnalhemoglobinuria. N Engl J Med 1990;323:1184–1189.
11. Motoyama N, Okada N, Yamashina M, Okada H. Paroxysmal nocturnal hemoglo-binuria due to hereditary nucleotide deletion in the HRF20 (CD59) gene. Eur JImmunol 1992;22:2669–2673.
12. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated proteinstructure and function prediction. Nat Protoc 2010;5:725–738.
13. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: proteinstructure and function prediction. Nat Methods 2015;12:7–8.
14. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics2008;9:40.
15. Rudd PM, Morgan BP, Wormald MR, et al. The glycosylation of the complementregulatory protein, human erythrocyte CD59. J Biol Chem 1997;272:7229–7244.
16. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res2000;28:235–242.
17. Leath KJ, Johnson S, Roversi P, et al. High-resolution structures of bacteriallyexpressed soluble human CD59. Acta Crystallogr Sect F Struct Biol Cryst Commun2007;63:648–652.
18. Johnson S, Brooks NJ, Smith RA, Lea SM, Bubeck D. Structural basis for recognitionof the pore-forming toxin intermedilysin by human complement receptor CD59. CellRep 2013;3:1369–1377.
19. Huang Y, Fedarovich A, Tomlinson S, Davies C. Crystal structure of CD59: impli-cations for molecular recognition of the complement proteins C8 and C9 in themembrane-attack complex. Acta Crystallogr D Biol Crystallogr 2007;63:714–721.
20. Huang Y, Smith CA, Song H, Morgan BP, Abagyan R, Tomlinson S. Insights into thehuman CD59 complement binding interface toward engineering new therapeutics.J Biol Chem 2005;280:34073–34079.
21. Bodian DL, Davis SJ, Morgan BP, Rushmere NK. Mutational analysis of the active siteand antibody epitopes of the complement-inhibitory glycoprotein, CD59. J Exp Med1997;185:507–516.
22. Budding K, van de Graaf EA, Kardol-Hoefnagel T, et al. Soluble CD59 is a novelbiomarker for the prediction of obstructive chronic lung allograft dysfunction afterlung transplantation. Sci Rep 2016;6:26274.
23. Hakulinen J, Meri S. Shedding and enrichment of the glycolipid-anchored comple-ment lysis inhibitor protectin (CD59) into milk fat globules. Immunology 1995;85:495–501.
24. Vakeva A, Lehto T, Takala A, Meri S. Detection of a soluble form of the complementmembrane attack complex inhibitor CD59 in plasma after acute myocardial infarction.Scand J Immunol 2000;52:411–414.
25. Meri S, Lehto T, Sutton CW, Tyynela J, Baumann M. Structural composition andfunctional characterization of soluble CD59: heterogeneity of the oligosaccharide andglycophosphoinositol (GPI) anchor revealed by laser-desorption mass spectrometricanalysis. Biochem J 1996;316(pt 3):923–935.
26. Ghosh P, Sahoo R, Vaidya A, et al. A specific and sensitive assay for blood levels ofglycated CD59: a novel biomarker for diabetes. Am J Hematol 2013;88:670–676.
27. Miyata T, Takeda J, Iida Y, et al. The cloning of PIG-A, a component in the early stepof GPI-anchor biosynthesis. Science 1993;259:1318–1320.
28. Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused bya somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell1993;73:703–711.
29. Mevorach D. Paroxysmal nocturnal hemoglobinuria (PNH) and primary p.Cys89Tyrmutation in CD59: differences and similarities. Mol Immunol 2015;67:51–55.
30. Tabib A, Karbian N, Mevorach D. Demyelination, strokes, and eculizumab: lessonsfrom the congenital CD59 gene mutations. Mol Immunol 2017;89:69–72.
31. Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and char-acterization of a membrane protein from normal human erythrocytes that inhibitsreactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J ClinInvest 1989;84:7–17.
32. Okada N, Harada R, Fujita T, Okada H. A novel membrane glycoprotein capable ofinhibiting membrane attack by homologous complement. Int Immunol 1989;1:205–208.
33. Sugita Y, Nakano Y, Tomita M. Isolation from human erythrocytes of a new mem-brane protein which inhibits the formation of complement transmembrane channels.J Biochem 1988;104:633–637.
34. Holguin MH, Wilcox LA, Bernshaw NJ, Rosse WF, Parker CJ. Relationship betweenthe membrane inhibitor of reactive lysis and the erythrocyte phenotypes of parox-ysmal nocturnal hemoglobinuria. J Clin Invest 1989;84:1387–1394.
35. Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. Targeteddeletion of the CD59 gene causes spontaneous intravascular hemolysis and hemo-globinuria. Blood 2001;98:442–449.
36. Rollins SA, Sims PJ. The complement-inhibitory activity of CD59 resides in its ca-pacity to block incorporation of C9 into membrane C5b-9. J Immunol 1990;144:3478–3483.
37. Ninomiya H, Sims PJ. The human complement regulatory protein CD59 binds to thealpha-chain of C8 and to the “b”domain of C9. J Biol Chem 1992;267:13675–13680.
38. Deckert M, Ticchioni M, Mari B, Mary D, Bernard A. The glycosylphosphatidylinositol-anchored CD59 protein stimulates both T cell receptor zeta/ZAP-70-dependent and-independent signaling pathways in T cells. Eur J Immunol 1995;25:1815–1822.
39. Longhi MP, Harris CL, Morgan BP, Gallimore A. Holding T cells in check–a new rolefor complement regulators? Trends Immunol 2006;27:102–108.
40. Krus U, King BC, Nagaraj V, et al. The complement inhibitor CD59 regulates insulinsecretion by modulating exocytotic events. Cell Metab 2014;19:883–890.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

ARTICLE OPEN ACCESS
Delineating FOXG1 syndromeFrom congenital microcephaly to hyperkinetic encephalopathy
Nancy Vegas, MD, Mara Cavallin, MD, Camille Maillard, MD, Nathalie Boddaert, MD, PhD,
Joseph Toulouse, MD, Elise Schaefer, MD, Tally Lerman-Sagie, MD, Dorit Lev, MD, Barth Magalie, MD,
Sebastien Moutton, MD, Eric Haan, MD, Bertrand Isidor, MD, Delphine Heron, MD, Mathieu Milh, MD, PhD,
Stephane Rondeau, MD, Caroline Michot, MD, PhD, Stephanie Valence, MD, Sabrina Wagner, MD,
Marie Hully, MD, Cyril Mignot, MD, Alice Masurel, MD, Alexandre Datta, MD, Sylvie Odent, MD, PhD,
Mathilde Nizon, MD, Leila Lazaro, MD, Marie Vincent, MD, Benjamin Cogne, MD, Anne Marie Guerrot, MD,
Stephanie Arpin, MD, Jean Michel Pedespan, MD, Isabelle Caubel, MD, Benedicte Pontier, MD, PhD,
Baptiste Troude, MD, Francois Rivier, MD, PhD, Christophe Philippe, MD, PhD, Thierry Bienvenu, MD, PhD,
Marie-Aude Spitz, MD, Amandine Bery, PhD, and Nadia Bahi-Buisson, MD, PhD
Neurol Genet 2018;4:e281. doi:10.1212/NXG.0000000000000281
Correspondence
Pr. Bahi-Buisson
AbstractObjectiveTo provide new insights into the FOXG1-related clinical and imaging phenotypes and refine thephenotype-genotype correlation in FOXG1 syndrome.
MethodsWe analyzed the clinical and imaging phenotypes of a cohort of 45 patients with a pathogenic orlikely pathogenic FOXG1 variant and performed phenotype-genotype correlations.
ResultsA total of 37 FOXG1 different heterozygous mutations were identified, of which 18 are novel.We described a broad spectrum of neurodevelopmental phenotypes, characterized by severepostnatal microcephaly and developmental delay accompanied by a hyperkinetic movementdisorder, stereotypes and sleep disorders, and epileptic seizures. Our data highlighted 3 patternsof gyration, including frontal pachygyria in younger patients (26.7%), moderate simplifiedgyration (24.4%) and mildly simplified or normal gyration (48.9%), corpus callosum hypo-genesis mostly in its frontal part, combined with moderate-to-severe myelination delay thatimproved and normalized with age. Frameshift and nonsense mutations in the N-terminus ofFOXG1, which are the most common mutation types, show the most severe clinical featuresand MRI anomalies. However, patients with recurrent frameshift mutations c.460dupG andc.256dupC had variable clinical and imaging presentations.
ConclusionsThese findings have implications for genetic counseling, providing evidence that N-terminalmutations and large deletions lead to more severe FOXG1 syndrome, although genotype-phenotype correlations are not necessarily straightforward in recurrent mutations. Together,these analyses support the view that FOXG1 syndrome is a specific disorder characterized byfrontal pachygyria and delayed myelination in its most severe form and hypogenetic corpuscallosum in its milder form.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Mutations in the FOXG1 gene have been shown to causea rare neurodevelopmental disorder. Initially described asa “congenital variant of Rett syndrome,”1,2 subsequent reportsallowed delineation of the FOXG1 syndrome, which is nowconsidered a distinct clinical entity.3–7
To date, more than 90 individuals with FOXG1 mutationshave been described, mostly within small case series.5,7 Thedisorder comprises a complex constellation of clinical fea-tures, including severe postnatal microcephaly, deficient socialreciprocity, combined stereotypies and dyskinesias, epilepsy,poor sleep patterns, and unexplained episodes of crying.3 Inparallel to these clinical criteria, the importance of brain MRIfeatures has been emphasized.1,3,8 However, the spectrum ofMRI features in FOXG1 syndrome is yet to be fully defined.
FOXG1 encodes a transcription factor containing a highlyconserved domain spanning from the forkhead binding do-main (FBD) to the C-terminus and a variable N-terminus.9
FOXG1 mutations include frameshifts, deletions, and pointmutations.7,10 A recent study suggests that more severe phe-notypes are associated with truncating FOXG1 variants in theN-terminus and the FBD and milder phenotypes with mis-sense variants in the FBD. The most significant differenceswere related to motor and speech development, while onlyborderline differences were found concerning corpus cal-losum anomalies, delayed myelination, and microcephaly.7
In light of these recent findings, the aim of this study was toprovide a comprehensive overview of FOXG1-related clinicaland imaging phenotypes by thorough analysis of a cohort of 45clinically well-characterized patients with FOXG1 mutation andrefine the phenotype-genotype correlation in FOXG1 syndrome.
MethodsWe recruited patients with pathogenic or likely pathogenicFOXG1 mutations from different cohorts through a large na-tional and international network. Genetic testing was performedby array comparative genomic hybridization (CGH) (5/45),Sanger sequencing (31/45), targeted panel high-throughputsequencing (4/45), and whole-exome sequencing (4/45).
Standard protocol approvals, registrations,and patient consentsThe study was approved by the ethics committee of the Uni-versity Hospital of Necker Enfants Malades, Paris, France andthe relevant local institutional review boards. Parental writteninformed consent was obtained for all affected patients.
All patients were personally known to at least 1 of the co-authors and were reexamined for the purpose of the study.
Five patients had been reported previously and were reas-sessed for the study.8,11,12 Standardized clinical informationwas recorded. Movement disorders were characterized inperson by investigators and classified according to establishedcriteria.13 Epileptic seizures were classified according to therecommendations of the Commission on Classification andTerminology of the International League Against Epilepsy.
In addition, for patients filmed, we obtained additional autho-rization for disclosure of any recognizable persons in videos.
The genetic testings were performed in accordance with therespective national ethics guidelines and approved by the localauthorities in the participating study centers.
MRI studiesAs the MRI studies were performed over a period of 10 yearsat many different imaging centers and on many different typesof MR scanners, the imaging techniques that were useddiffered substantially, although a majority had at least axialand sagittal T1-weighted and axial T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences. Imagingassessment was based on agreement between 2 investigators(N.B. and N.B.-B.) who reviewed the images. Each made initialevaluations independently, and any disagreements regardingthe final conclusion were resolved by consensus.
Statistical analysisAll statistical analyses were performed in GraphPad Prismversion 6.00. Data are described as mean ± SEM. Differenceswere evaluated using the 2-way analysis of variance withmultiple comparison tests.
The study was approved by the ethics committee of the Uni-versity Hospital of Necker Enfants Malades, Paris, France andthe relevant local institutional review boards. Parental writteninformed consent was obtained for all affected patients.
ResultsOur cohort totaled 45 patients with FOXG1 mutations, 22males and 23 females ranging in age from 19 months to 42years (median: 5.73 years) at the time of evaluation (table e-1links.lww.com/NXG/A97).
A total of 37 FOXG1 different heterozygous mutations wereidentified, of which 18 are novel. They comprised 32 smallintragenic mutations and 5 large deletions of the whole FOXG1locus. All mutations were de novo, except 1 reported previouslyas a germinal mosaic.12 Point mutations were mostly frameshifts(14/32; 43.75%) and missense mutations (12/32; 37.5%), witha small number of nonsense (4/32; 12.5%) and in-frame
GlossaryFBD = forkhead binding domain.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

mutations (2/32; 6.25%) (figure 1, A and B). Three recurrentmutations, c.460dupG, c.256dupC, and c.256delC, wereidentified.
Clinical presentation in patients withFOXG1 mutationsPatients first came to medical attention at a median age of 3months (birth to 20 months) because of developmental delayand microcephaly (15/45; 33.3%) or with lack of eye contact,or strabismus (16/45; 35.6%). Epileptic seizures or move-ment disorder were less common (4/45; <10%). In 5 cases(11.1%), brain anomalies were diagnosed prenatally.
At birth, a majority of patients had normal body measure-ments and low normal birth head size (38/43; 88.4%). Severepostnatal microcephaly (−4 to −6 SD) became apparent afterthe age of 1 month.
At the age of the last evaluation (median: 5 years; 19months to42 years), all patients had profound developmental delay, withpermanent esotropia (38/42; 90.7%) (video 1 links.lww.com/NXG/A99). Hand usewas severely limited to involuntary grossmanipulation (13/44; 29.5%) (video 2 links.lww.com/NXG/A100). On examination, a complexmovement disorder was themost prominent feature characterized by generalized hyperki-netic and dyskinetic movements that was present at rest and
worsened with attempts to movement (videos 3 and 4 links.lww.com/NXG/A101 and links.lww.com/NXG/A102), withorolingual dyskinesias (12/33; 36.4%) (video 5 links.lww.com/NXG/A103); 34 of 43 patients (79.1%) also showed handstereotypies, consisting of hand pressing/wringing or handmouthing (videos 6 and 7 links.lww.com/NXG/A104, links.lww.com/NXG/A105), which are unusual in the context ofdyskinetic movement disorders. Thirty-two of 44 patients (72.7%) had feeding difficulties associated with gastroesophagealreflux (videos 8 and 9 links.lww.com/NXG/A106 and links.lww.com/NXG/A107). Sleep problems were frequent (27/42;64.3%) and included multiple nocturnal awakenings or diffi-culties in falling asleep with irritability and inconsolable cryingor inappropriate laughing (25/40; 62.5%). Seizures weredocumented in 77.8% (35/45) of patients and occurred ata mean age of 2.5 years (range: 2 days to 12 years). Generalizedtonic or tonic-clonic seizures were the most frequent seizuretype (21/35; 60%). Of the 35 patients, 17 (48.6%) developedrefractory epilepsy with multiple seizure types and 5 (14.3%)experienced at least 1 episode of status epilepticus (table 1).
Because, FOXG1mutations had been previously associated withcongenital Rett variant, we examined the prevalence of con-genital Rett-supportive manifestations. Overall, 2 of 21 females(9.5%) and 1 of 21males (4.76%) fulfilled the diagnostic criteriafor Rett syndrome14 (table e-2 links.lww.com/NXG/A98).
Figure 1 Schematic representation of FOXG1 gene, protein domain structure, and positions of FOXG1 mutations
(A) Schematic representation of FOXG1 gene and (B) FOXG1 protein domain structure and positions of the variations identified: N-terminal domain; FBDdomain (forkhead DNA binding domain, amino acids 181–275), GBD domain (Groucho binding domain, amino acids 307–317), JBD domain (JARID1B bindingdomain, amino acids 383–406), and C-terminal domain are indicated. Mutations are located all along the FOXG1 gene, within different protein domains.Missensemutations are predominantly located in the FBD (91.7%), whereas frameshiftmutations aremore prominent in theN-terminal domain (57.1%). Thenovel variants described in this article are highlighted in bold and the recurrent variants are underlined with the corresponding number of recurrencesindicated in brackets. FBD = forkhead binding domain; GBD = Groucho binding domain; JBD = JARID1B binding domain.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
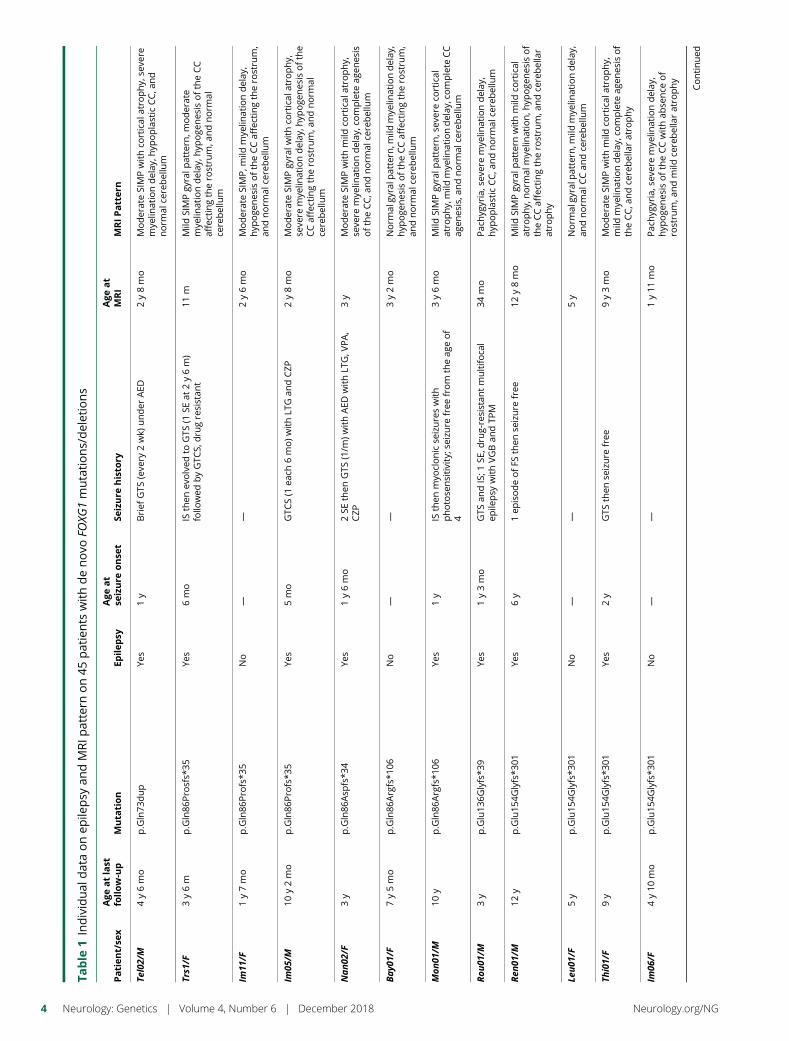
Table
1Individual
dataonep
ilepsy
andMRIp
attern
on45
patients
withdenovo
FOXG
1mutations/deletions
Patien
t/se
xAge
atlast
follow-up
Muta
tion
Epilep
syAge
at
seizure
onse
tSe
izure
histo
ryAge
at
MRI
MRIPattern
Tel02/M
4y6mo
p.Gln73
dup
Yes
1y
Brief
GTS
(eve
ry2wk)
under
AED
2y8mo
ModerateSIMPwithco
rtical
atrophy,
seve
remye
linationdelay
,hyp
oplastic
CC,a
nd
norm
alce
rebellum
Trs1
/F3y6m
p.Gln86
Prosfs*
35Ye
s6mo
ISthen
evolved
toGTS
(1SE
at2y6m)
follo
wed
byGTC
S,dru
gresistan
t11
mMild
SIMPgy
ralp
attern
,moderate
mye
linationdelay
,hyp
oge
nes
isoftheCC
affectingtherostru
m,a
ndnorm
alce
rebellum
Im11
/F1y7mo
p.Gln86
Profs*3
5No
——
2y6mo
ModerateSIMP,m
ildmye
linationdelay
,hyp
oge
nes
isoftheCCaffectingtherostru
m,
andnorm
alce
rebellum
Im05
/M10
y2mo
p.Gln86
Profs*3
5Ye
s5mo
GTC
S(1
each
6mo)w
ithLT
Gan
dCZP
2y8mo
ModerateSIMPgy
ralw
ithco
rtical
atrophy,
seve
remye
linationdelay
,hyp
oge
nes
isofthe
CCaffectingtherostru
m,a
ndnorm
alce
rebellum
Nan02
/F3y
p.Gln86
Asp
fs*3
4Ye
s1y6mo
2SE
then
GTS
(1/m
)withAED
withLT
G,V
PA,
CZP
3y
ModerateSIMPwithmild
cortical
atrophy,
seve
remye
linationdelay
,complete
agen
esis
oftheCC,a
ndnorm
alce
rebellum
Bay0
1/F
7y5mo
p.Gln86
Argfs*1
06No
——
3y2mo
Norm
algy
ralp
attern
,mild
mye
linationdelay
,hyp
oge
nes
isoftheCCaffectingtherostru
m,
andnorm
alce
rebellum
Mon
01/M
10y
p.Gln86
Argfs*1
06Ye
s1y
ISthen
myo
clonicse
izureswith
photose
nsitivity;seizu
refree
from
theag
eof
4
3y6mo
Mild
SIMPgy
ralp
attern
,sev
ereco
rtical
atrophy,mild
mye
linationdelay
,complete
CC
agen
esis,a
ndnorm
alce
rebellum
Rou
01/M
3y
p.Glu13
6Glyfs*3
9Ye
s1y3mo
GTS
andIS;1
SE,d
rug-resistan
tmultifo
cal
epile
psy
withVGBan
dTP
M34
mo
Pac
hyg
yria,sev
eremye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Ren
01/M
12y
p.Glu15
4Glyfs*3
01Ye
s6y
1ep
isodeofFS
then
seizure
free
12y8mo
Mild
SIMPgy
ralp
attern
withmild
cortical
atrophy,
norm
almye
lination,h
ypoge
nes
isof
theCCaffectingtherostru
m,a
ndce
rebellar
atrophy
Leu01
/F5y
p.Glu15
4Glyfs*3
01No
——
5y
Norm
algy
ralp
attern
,mild
mye
linationdelay
,an
dnorm
alCCan
dce
rebellum
Thi01/F
9y
p.Glu15
4Glyfs*3
01Ye
s2y
GTS
then
seizure
free
9y3mo
ModerateSIMPwithmild
cortical
atrophy,
mild
mye
linationdelay
,complete
agen
esisof
theCC,a
ndce
rebellaratrophy
Im06
/F4y10
mo
p.Glu15
4Glyfs*3
01No
——
1y11
mo
Pac
hyg
yria,sev
eremye
linationdelay
,hyp
oge
nes
isoftheCCwithab
sence
of
rostru
m,a
ndmild
cerebellaratrophy
Continued
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
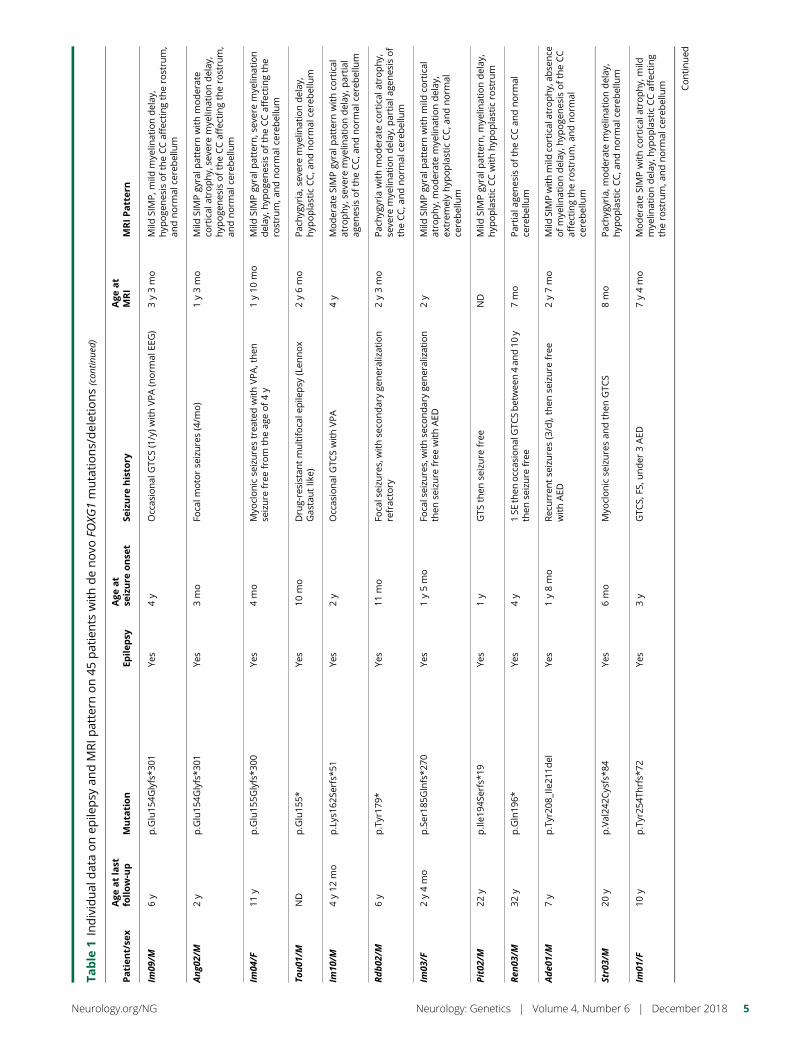
Table
1Individual
dataonep
ilepsy
andMRIp
attern
on45
patients
withdenovo
FOXG
1mutations/deletions(con
tinue
d)
Patien
t/se
xAge
atlast
follow-up
Muta
tion
Epilep
syAge
at
seizure
onse
tSe
izure
histo
ryAge
at
MRI
MRIPattern
Im09
/M6y
p.Glu15
4Glyfs*3
01Ye
s4y
Occas
ional
GTC
S(1/y)w
ithVPA(norm
alEE
G)
3y3mo
Mild
SIMP,m
ildmye
linationdelay
,hyp
oge
nes
isoftheCCaffectingtherostru
m,
andnorm
alce
rebellum
Ang0
2/M
2y
p.Glu15
4Glyfs*3
01Ye
s3mo
Foca
lmotorse
izures(4/m
o)
1y3mo
Mild
SIMPgy
ralp
attern
withmoderate
cortical
atrophy,
seve
remye
linationdelay
,hyp
oge
nes
isoftheCCaffectingtherostru
m,
andnorm
alce
rebellum
Im04
/F11
yp.Glu15
5Glyfs*3
00Ye
s4mo
Myo
clonicse
izurestrea
tedwithVPA,then
seizure
free
from
theag
eof4y
1y10
mo
Mild
SIMPgy
ralp
attern
,sev
eremye
lination
delay
,hyp
oge
nes
isoftheCCaffectingthe
rostru
m,a
ndnorm
alce
rebellum
Tou01
/MND
p.Glu15
5*Ye
s10
mo
Dru
g-resistan
tmultifo
cale
pile
psy
(Len
nox
Gas
tautlik
e)2y6mo
Pac
hyg
yria,sev
eremye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Im10
/M4y12
mo
p.Lys16
2Serfs*5
1Ye
s2y
Occas
ional
GTC
SwithVPA
4y
ModerateSIMPgy
ralp
attern
withco
rtical
atrophy,
seve
remye
linationdelay
,partial
agen
esisoftheCC,a
ndnorm
alce
rebellum
Rdb02
/M6y
p.Tyr17
9*Ye
s11
mo
Foca
lseizu
res,withse
condaryge
neralization
refrac
tory
2y3mo
Pac
hyg
yria
withmoderateco
rtical
atrophy,
seve
remye
linationdelay
,partial
agen
esisof
theCC,a
ndnorm
alce
rebellum
Im03
/F2y4mo
p.Ser18
5Glnfs*2
70Ye
s1y5mo
Foca
lseizu
res,withse
condaryge
neralization
then
seizure
free
withAED
2y
Mild
SIMPgy
ralp
attern
withmild
cortical
atrophy,
moderatemye
linationdelay
,ex
trem
elyhyp
oplastic
CC,a
ndnorm
alce
rebellum
Pit02/M
22y
p.Ile1
94Se
rfs*
19Ye
s1y
GTS
then
seizure
free
ND
Mild
SIMPgy
ralp
attern
,mye
linationdelay
,hyp
oplastic
CCwithhyp
oplasticrostru
m
Ren
03/M
32y
p.Gln19
6*Ye
s4y
1SE
then
occas
ionalGTC
Sbetwee
n4an
d10
ythen
seizure
free
7mo
Partial
agen
esisoftheCCan
dnorm
alce
rebellum
Ade0
1/M
7y
p.Tyr20
8_Ile
211d
elYe
s1y8mo
Rec
urren
tse
izures(3/d),then
seizure
free
withAED
2y7mo
Mild
SIMPwithmild
cortical
atrophy,ab
sence
ofmye
linationdelay
,hyp
oge
nes
isoftheCC
affectingtherostru
m,a
ndnorm
alce
rebellum
Str03/M
20y
p.Val24
2Cysfs*8
4Ye
s6mo
Myo
clonicse
izuresan
dthen
GTC
S8mo
Pac
hyg
yria,m
oderatemye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Im01
/F10
yp.Tyr25
4Thrfs*
72Ye
s3y
GTC
S,FS
,under
3AED
7y4mo
ModerateSIMPwithco
rtical
atrophy,
mild
mye
linationdelay
,hyp
oplastic
CCaffecting
therostru
m,a
ndnorm
alce
rebellum Continued
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
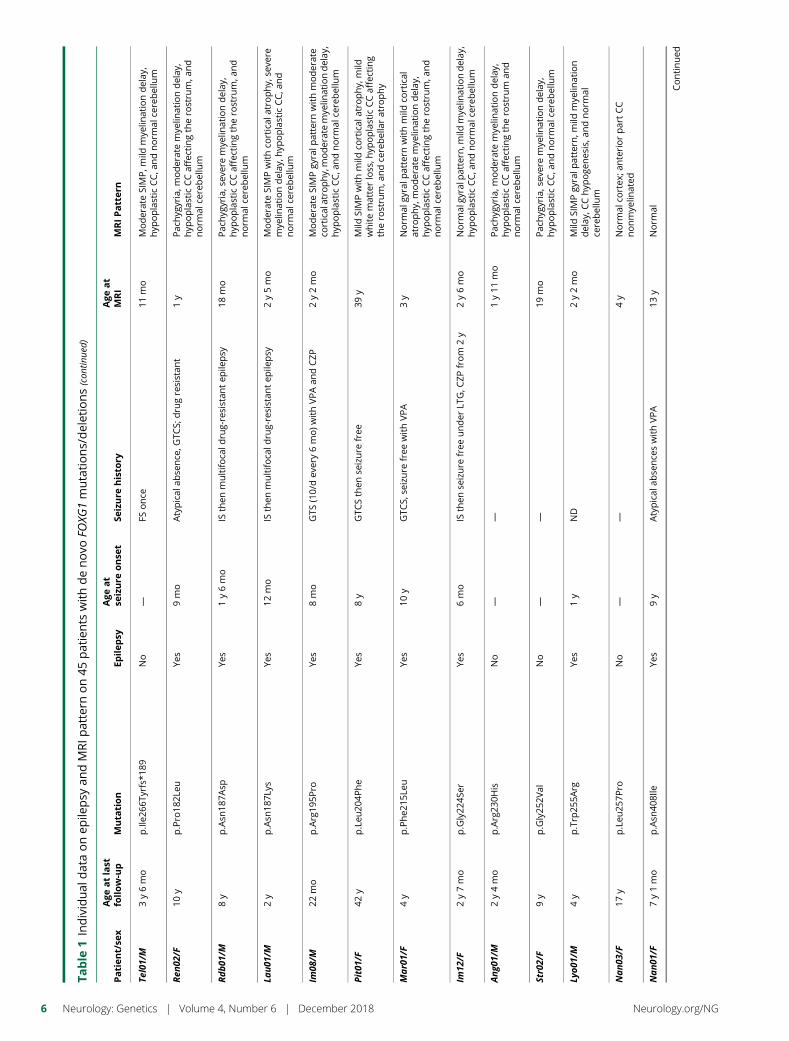
Table
1Individual
dataonep
ilepsy
andMRIp
attern
on45
patients
withdenovo
FOXG
1mutations/deletions(con
tinue
d)
Patien
t/se
xAge
atlast
follow-up
Muta
tion
Epilep
syAge
at
seizure
onse
tSe
izure
histo
ryAge
at
MRI
MRIPattern
Tel01/M
3y6mo
p.Ile2
66Ty
rfs*
189
No
—FS
once
11mo
ModerateSIMP,m
ildmye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Ren
02/F
10y
p.Pro18
2Leu
Yes
9mo
Atypical
abse
nce
,GTC
S;dru
gresistan
t1y
Pac
hyg
yria,m
oderatemye
linationdelay
,hyp
oplastic
CCaffectingtherostru
m,a
nd
norm
alce
rebellum
Rdb01
/M8y
p.Asn
187A
spYe
s1y6mo
ISthen
multifo
cald
rug-resistan
tep
ilepsy
18mo
Pac
hyg
yria,sev
eremye
linationdelay
,hyp
oplastic
CCaffectingtherostru
m,a
nd
norm
alce
rebellum
Lau01
/M2y
p.Asn
187L
ysYe
s12
mo
ISthen
multifo
cald
rug-resistan
tep
ilepsy
2y5mo
ModerateSIMPwithco
rtical
atrophy,
seve
remye
linationdelay
,hyp
oplastic
CC,a
nd
norm
alce
rebellum
Im08
/M22
mo
p.Arg19
5Pro
Yes
8mo
GTS
(10/dev
ery6mo)w
ithVPAan
dCZP
2y2mo
ModerateSIMPgy
ralp
attern
withmoderate
corticalatrophy,moderatemye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Pit01/F
42y
p.Leu
204P
he
Yes
8y
GTC
Sthen
seizure
free
39y
Mild
SIMPwithmild
cortical
atrophy,
mild
whitematterloss,h
ypoplastic
CCaffecting
therostru
m,a
ndce
rebellaratrophy
Mar01/F
4y
p.Phe2
15Le
uYe
s10
yGTC
S,se
izure
free
withVPA
3y
Norm
algy
ralp
attern
withmild
cortical
atrophy,
moderatemye
linationdelay
,hyp
oplastic
CCaffectingtherostru
m,a
nd
norm
alce
rebellum
Im12
/F2y7mo
p.Gly22
4Ser
Yes
6mo
ISthen
seizure
free
under
LTG,C
ZPfrom
2y
2y6mo
Norm
algy
ralp
attern
,mild
mye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Ang0
1/M
2y4mo
p.Arg23
0His
No
——
1y11
mo
Pac
hyg
yria,m
oderatemye
linationdelay
,hyp
oplastic
CCaffectingtherostru
man
dnorm
alce
rebellum
Str02/F
9y
p.Gly25
2Val
No
——
19mo
Pac
hyg
yria,sev
eremye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Lyo0
1/M
4y
p.Trp
255A
rgYe
s1y
ND
2y2mo
Mild
SIMPgy
ralp
attern
,mild
mye
lination
delay
,CChyp
oge
nes
is,a
ndnorm
alce
rebellum
Nan03
/F17
yp.Leu
257P
roNo
——
4y
Norm
alco
rtex
;anteriorpartCC
nonmye
linated
Nan01
/F7y1mo
p.Asn
408Ile
Yes
9y
Atypical
abse
nce
swithVPA
13y
Norm
al
Continued
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
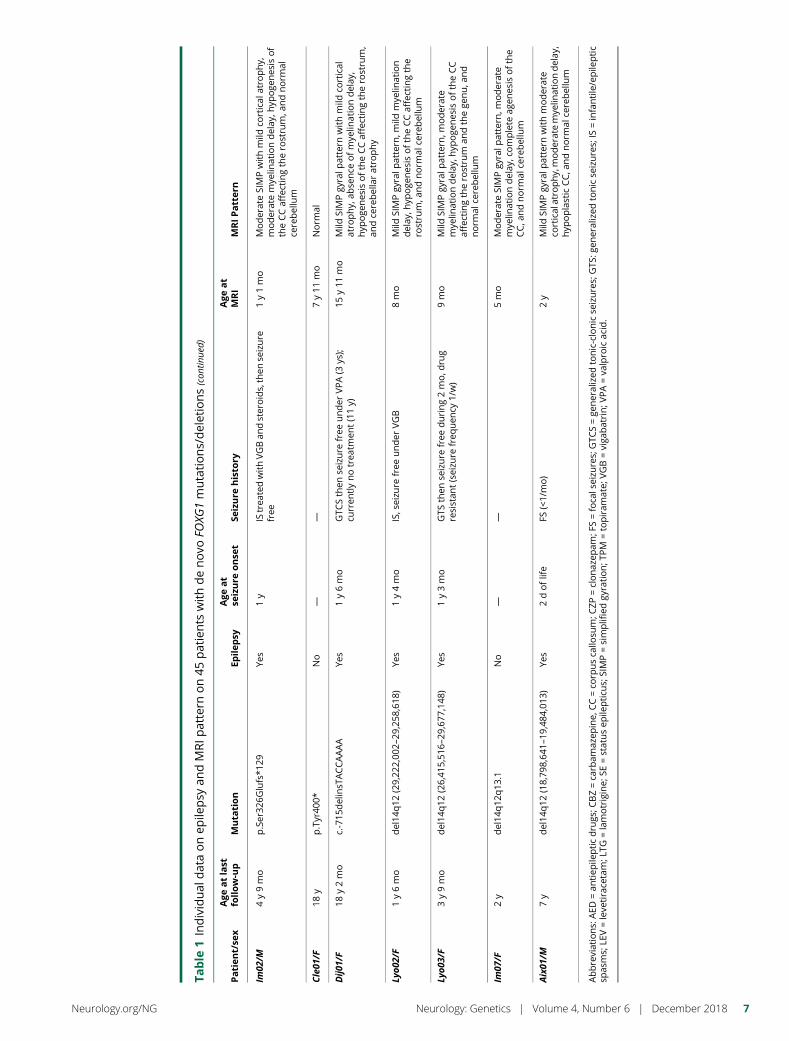
Table
1Individual
dataonep
ilepsy
andMRIp
attern
on45
patients
withdenovo
FOXG
1mutations/deletions(con
tinue
d)
Patien
t/se
xAge
atlast
follow-up
Muta
tion
Epilep
syAge
at
seizure
onse
tSe
izure
histo
ryAge
at
MRI
MRIPattern
Im02
/M4y9mo
p.Ser32
6Glufs*1
29Ye
s1y
IStrea
tedwithVGBan
dsteroids,then
seizure
free
1y1mo
ModerateSIMPwithmild
cortical
atrophy,
moderatemye
linationdelay
,hyp
oge
nes
isof
theCCaffectingtherostru
m,a
ndnorm
alce
rebellum
Cle01
/F18
yp.Tyr40
0*No
——
7y11
mo
Norm
al
Dij0
1/F
18y2mo
c.-715
delinsT
ACCAAAA
Yes
1y6mo
GTC
Sthen
seizure
free
under
VPA(3
ys);
curren
tlynotrea
tmen
t(11y)
15y11
mo
Mild
SIMPgy
ralp
attern
withmild
cortical
atrophy,
abse
nce
ofmye
linationdelay
,hyp
oge
nes
isoftheCCaffectingtherostru
m,
andce
rebellaratrophy
Lyo0
2/F
1y6mo
del14
q12
(29,22
2,00
2–29
,258
,618
)Ye
s1y4mo
IS,seizu
refree
under
VGB
8mo
Mild
SIMPgy
ralp
attern
,mild
mye
lination
delay
,hyp
oge
nes
isoftheCCaffectingthe
rostru
m,a
ndnorm
alce
rebellum
Lyo0
3/F
3y9mo
del14
q12
(26,41
5,51
6–29
,677
,148
)Ye
s1y3mo
GTS
then
seizure
free
during2mo,d
rug
resistan
t(seizu
refreq
uen
cy1/w)
9mo
Mild
SIMPgy
ralp
attern
,moderate
mye
linationdelay
,hyp
oge
nes
isoftheCC
affectingtherostru
man
dthege
nu,a
nd
norm
alce
rebellum
Im07
/F2y
del14
q12
q13
.1No
——
5mo
ModerateSIMPgy
ralp
attern
,moderate
mye
linationdelay
,complete
agen
esisofthe
CC,a
ndnorm
alce
rebellum
Aix01
/M7y
del14
q12
(18,79
8,64
1–19
,484
,013
)Ye
s2doflife
FS(<1/mo)
2y
Mild
SIMPgy
ralp
attern
withmoderate
corticalatrophy,moderatemye
linationdelay
,hyp
oplastic
CC,a
ndnorm
alce
rebellum
Abbreviations:AED
=an
tiep
ilepticdru
gs;C
BZ=ca
rbam
azep
ine,
CC=co
rpusca
llosu
m;C
ZP=clonaz
epam
;FS=foca
lseizu
res;GTC
S=ge
neralized
tonic-clonicse
izures;GTS
:gen
eralized
tonicse
izures;IS
=infantile/epile
ptic
spas
ms;
LEV=leve
tirace
tam;L
TG=lamotrigine;
SE=statusep
ilepticu
s;SIMP=simplifiedgy
ration;T
PM
=topiram
ate;
VGB=viga
batrin;V
PA=va
lproicac
id.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7
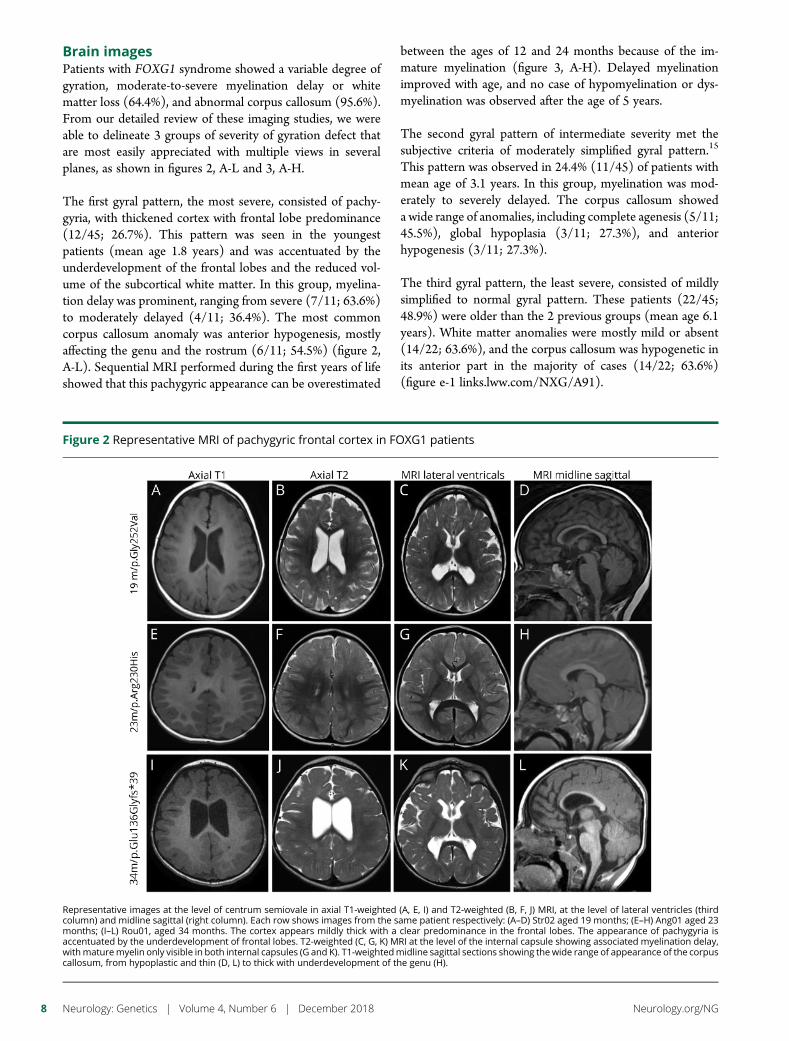
Brain imagesPatients with FOXG1 syndrome showed a variable degree ofgyration, moderate-to-severe myelination delay or whitematter loss (64.4%), and abnormal corpus callosum (95.6%).From our detailed review of these imaging studies, we wereable to delineate 3 groups of severity of gyration defect thatare most easily appreciated with multiple views in severalplanes, as shown in figures 2, A-L and 3, A-H.
The first gyral pattern, the most severe, consisted of pachy-gyria, with thickened cortex with frontal lobe predominance(12/45; 26.7%). This pattern was seen in the youngestpatients (mean age 1.8 years) and was accentuated by theunderdevelopment of the frontal lobes and the reduced vol-ume of the subcortical white matter. In this group, myelina-tion delay was prominent, ranging from severe (7/11; 63.6%)to moderately delayed (4/11; 36.4%). The most commoncorpus callosum anomaly was anterior hypogenesis, mostlyaffecting the genu and the rostrum (6/11; 54.5%) (figure 2,A-L). Sequential MRI performed during the first years of lifeshowed that this pachygyric appearance can be overestimated
between the ages of 12 and 24 months because of the im-mature myelination (figure 3, A-H). Delayed myelinationimproved with age, and no case of hypomyelination or dys-myelination was observed after the age of 5 years.
The second gyral pattern of intermediate severity met thesubjective criteria of moderately simplified gyral pattern.15
This pattern was observed in 24.4% (11/45) of patients withmean age of 3.1 years. In this group, myelination was mod-erately to severely delayed. The corpus callosum showeda wide range of anomalies, including complete agenesis (5/11;45.5%), global hypoplasia (3/11; 27.3%), and anteriorhypogenesis (3/11; 27.3%).
The third gyral pattern, the least severe, consisted of mildlysimplified to normal gyral pattern. These patients (22/45;48.9%) were older than the 2 previous groups (mean age 6.1years). White matter anomalies were mostly mild or absent(14/22; 63.6%), and the corpus callosum was hypogenetic inits anterior part in the majority of cases (14/22; 63.6%)(figure e-1 links.lww.com/NXG/A91).
Figure 2 Representative MRI of pachygyric frontal cortex in FOXG1 patients
Representative images at the level of centrum semiovale in axial T1-weighted (A, E, I) and T2-weighted (B, F, J) MRI, at the level of lateral ventricles (thirdcolumn) and midline sagittal (right column). Each row shows images from the same patient respectively: (A–D) Str02 aged 19 months; (E–H) Ang01 aged 23months; (I–L) Rou01, aged 34 months. The cortex appears mildly thick with a clear predominance in the frontal lobes. The appearance of pachygyria isaccentuated by the underdevelopment of frontal lobes. T2-weighted (C, G, K) MRI at the level of the internal capsule showing associated myelination delay,withmaturemyelin only visible in both internal capsules (G and K). T1-weightedmidline sagittal sections showing the wide range of appearance of the corpuscallosum, from hypoplastic and thin (D, L) to thick with underdevelopment of the genu (H).
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
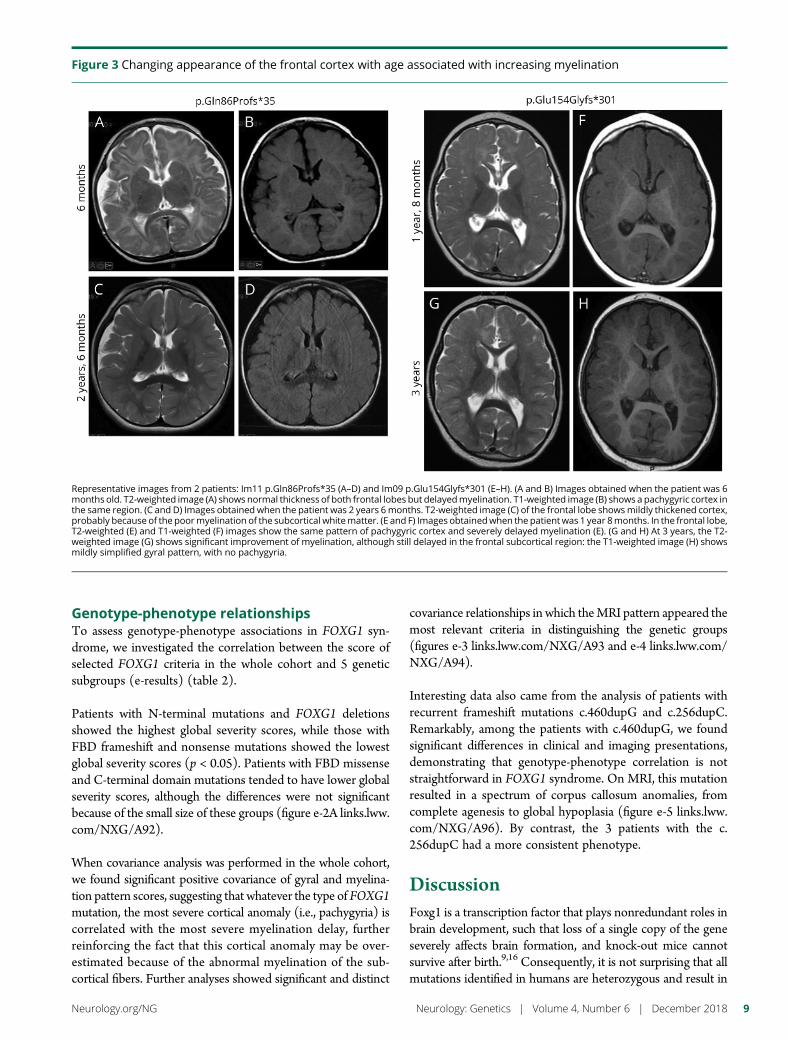
Genotype-phenotype relationshipsTo assess genotype-phenotype associations in FOXG1 syn-drome, we investigated the correlation between the score ofselected FOXG1 criteria in the whole cohort and 5 geneticsubgroups (e-results) (table 2).
Patients with N-terminal mutations and FOXG1 deletionsshowed the highest global severity scores, while those withFBD frameshift and nonsense mutations showed the lowestglobal severity scores (p < 0.05). Patients with FBD missenseand C-terminal domain mutations tended to have lower globalseverity scores, although the differences were not significantbecause of the small size of these groups (figure e-2A links.lww.com/NXG/A92).
When covariance analysis was performed in the whole cohort,we found significant positive covariance of gyral and myelina-tion pattern scores, suggesting that whatever the type of FOXG1mutation, the most severe cortical anomaly (i.e., pachygyria) iscorrelated with the most severe myelination delay, furtherreinforcing the fact that this cortical anomaly may be over-estimated because of the abnormal myelination of the sub-cortical fibers. Further analyses showed significant and distinct
covariance relationships in which theMRI pattern appeared themost relevant criteria in distinguishing the genetic groups(figures e-3 links.lww.com/NXG/A93 and e-4 links.lww.com/NXG/A94).
Interesting data also came from the analysis of patients withrecurrent frameshift mutations c.460dupG and c.256dupC.Remarkably, among the patients with c.460dupG, we foundsignificant differences in clinical and imaging presentations,demonstrating that genotype-phenotype correlation is notstraightforward in FOXG1 syndrome. On MRI, this mutationresulted in a spectrum of corpus callosum anomalies, fromcomplete agenesis to global hypoplasia (figure e-5 links.lww.com/NXG/A96). By contrast, the 3 patients with the c.256dupC had a more consistent phenotype.
DiscussionFoxg1 is a transcription factor that plays nonredundant roles inbrain development, such that loss of a single copy of the geneseverely affects brain formation, and knock-out mice cannotsurvive after birth.9,16 Consequently, it is not surprising that allmutations identified in humans are heterozygous and result in
Figure 3 Changing appearance of the frontal cortex with age associated with increasing myelination
Representative images from 2 patients: Im11 p.Gln86Profs*35 (A–D) and Im09 p.Glu154Glyfs*301 (E–H). (A and B) Images obtained when the patient was 6months old. T2-weighted image (A) shows normal thickness of both frontal lobes but delayedmyelination. T1-weighted image (B) shows a pachygyric cortex inthe same region. (C and D) Images obtained when the patient was 2 years 6months. T2-weighted image (C) of the frontal lobe showsmildly thickened cortex,probably because of the poormyelination of the subcortical whitematter. (E and F) Images obtainedwhen the patient was 1 year 8months. In the frontal lobe,T2-weighted (E) and T1-weighted (F) images show the same pattern of pachygyric cortex and severely delayed myelination (E). (G and H) At 3 years, the T2-weighted image (G) shows significant improvement of myelination, although still delayed in the frontal subcortical region: the T1-weighted image (H) showsmildly simplified gyral pattern, with no pachygyria.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9
Table
2Clin
ical
andneu
roim
agingfeaturesrelatedto
theFO
XG1ge
notypegroups
N-ter
minal
domain
variants
Fork
hea
ddomain
frames
hift
andnonse
nse
variants
Fork
hea
ddomain
misse
nse
variants
C-term
inaldomain
variants
Larg
edeletions
Case
snumber
,n(%
)19
39.5%
613
.3%
1225
.60%
37%
511
.6%
Sex,
M/F
10/9
52.6%/47.4%
4/2
66.7%/33.3%
6/6
50%/50%
1/2
33.3%/66.7%
1/4
20%/80%
Med
ianage
atlast
follow-up(y)
4.8
155.5
7.1
3.8
Pre
gnancy
andneo
nata
lper
iod
Pro
blemsin
pre
gnancy
/sca
ns
0/17
0%2/4
50%
4/11
36.4%
0/3
0%2/5
40%
Mea
nge
stationatdeliver
y(G
W)/n
39.7
1838
.26
39.1
1139
.23
38.9
5
Neo
nata
lissu
es4/17
23.7%
1/4
25%
2/11
18.20%
1/3
33.3%
4/5
80%
Feed
ingdifficu
ltiesatbirth
3/17
17.6%
1/4
25%
1/11
9.1%
1/3
33.3%
2/5
40%
Bodymea
sure
men
tsatbirth
Length<22S
D0/16
0%0/5
0%1/10
10%
0/3
0%0/5
0%
Weigh
t<22S
D0/17
0%0/5
0%0/11
0%0/3
0%0/5
0%
HC<22S
D1/18
5.6%
0/5
0%3/12
25%
0/3
0%0/5
0%
Bodymea
sure
men
tsatlast
evaluation
Med
ianage
(y)/n
4.7
186.6
57.5
127.1
33.8
5
Heigh
t<22S
D1/14
7.1%
1/3
33.3%
3/9
33.3%
0/2
0%2/5
40%
Weigh
t<22S
D4/17
23.5%
1/3
33.3%
4/11
36.4%
1/3
33.3%
3/5
60%
HC<22S
D18
/19
94.7%
5/5
100%
8/10
80%
3/3
100%
5/5
100%
HC<24S
D14
/19
73.7%
3/5
60%
4/10
40%
1/3
33.3%
4/5
80%
Micro
cephaly
score
0=Norm
ala
tbirth
andatlast
evaluation
1/18
5.6%
1/5
20%
2/11
18.2%
0/3
0%0/5
0%
1=Postnata
lmicro
cephaly
3/18
16.7%
1/5
20%
3/11
27.3%
2/3
66.7%
1/5
20%
2=Se
vere
postnata
lmicro
cephaly,2
4to
26SD
13/18
72.2%
3/5
60%
3/11
27.3%
1/3
33.3%
4/5
80%
3=Conge
nitala
ndpostnata
lmicro
cephaly
1/18
5.7%
0/5
0%3/11
27.3%
0/3
0%0/5
0% Continued
10 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Table
2Clin
ical
andneu
roim
agingfeaturesrelatedto
theFO
XG1ge
notypegroups(con
tinue
d)
N-ter
minal
domain
variants
Fork
hea
ddomain
frames
hift
andnonse
nse
variants
Fork
hea
ddomain
misse
nse
variants
C-term
inaldomain
variants
Larg
edeletions
Moto
randsp
eech
dev
elopmen
t
Socialinte
ractions(eye
conta
ctandsm
ilinginte
ntionally)
10/15
66.7%
3/3
100%
8/11
72.7%
3/3
100%
3/5
60%
Sitwithsu
pport
6/19
31.6%
2/6
40%
7/12
58.3%
2/3
66.7%
1/5
20%
Walked
indep
enden
tly
0/19
0%0/6
0%1/12
8.3%
2/3
66.7%
0/5
0%
Handuse
10/19
52.6%
3/5
60%
4/12
33.3%
1/3
33.3%
1/5
20%
Spee
ch(atleast
bisyllabisms)
1/17
5.9%
0/6
0%1/12
8.3%
1/3
33.3%
0/5
0%
Slee
pandbeh
aviordistu
rbance
s
Inappro
priate
laugh
ing/crying/scre
amingsp
ells
9/15
60%
3/5
60%
7/12
58.3%
3/3
100%
2/5
40%
Impaired
slee
ppatter
n11
/18
61.1%
4/5
80%
6/11
54.5%
2/3
66.7%
4/5
80%
Feed
ingdifficu
lties
13/19
68.4%
3/6
50%
9/12
75%
2/3
66.7%
5/5
100%
Firstco
nce
rnanddisea
seco
urse
Med
ianage
atfirstco
nce
rns(m
o)/n
3.7
183.5
63
116
30
5
Whatwer
eth
efirstco
nce
rns?
Micro
cephaly
7/19
36.8%
1/6
50%
4/12
33.3%
1/3
33.3%
2/5
40%
Stra
bismus/poorey
eco
nta
ct/abnorm
alo
cularpursuit
6/19
31.6%
2/6
33.3%
6/12
50%
0/3
0%2/5
40%
Dev
elopmen
talDelay
8/19
42.1%
4/6
66.7%
4/12
33.3%
2/3
66.7%
1/5
20%
Corp
usca
llosu
mabnorm
alities
0/18
0%0/6
0%1/12
8.3%
0/3
0%2/5
40%
Seizure
s2/19
10.5%
0/6
0%2/12
16.7%
0/3
0%0/5
0%
Move
men
tdisord
ers
2/19
10.5%
0/6
0%2/12
16.7%
1/3
33.3%
1/5
20%
Aper
iodofre
gres
sion
3/19
15.8%
2/5
40%
2/12
16.7%
2/3
66.7%
1/5
20%
Clinicale
xamination
Dys
morp
hic
featu
res
8/18
44.4%
2/5
40%
3/11
27.3%
1/3
33.3%
1/5
20%
Axialh
ypoto
nia
18/19
94.7%
5/6
83.3%
11/11
100%
3/3
100%
5/5
100%
Hyp
erto
nia/spasticity
13/19
68.4%
5/6
83.3%
8/12
66.7%
1/3
33.3%
3/5
60%
Continued
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 11
Table
2Clin
ical
andneu
roim
agingfeaturesrelatedto
theFO
XG1ge
notypegroups(con
tinue
d)
N-ter
minal
domain
variants
Fork
hea
ddomain
frames
hift
andnonse
nse
variants
Fork
hea
ddomain
misse
nse
variants
C-term
inaldomain
variants
Larg
edeletions
Move
men
tdisord
ers
19/19
100%
6/6
100%
12/12
100%
3/3
100%
5/5
100%
Ster
eotypic
move
men
ts15
/19
78.9%
3/4
75%
8/12
63.6%
3/3
100%
5/5
100%
Stra
bismus
16/18
88.9%
4/4
100%
10/12
81.8%
3/3
100%
5/5
100%
Scoliosis
5/19
26.3%
1/5
20%
1/11
9.1%
1/3
33.3%
1/5
20%
Epilep
sy
Seizure
occ
urren
ce15
/19
78.9%
5/6
80%
9/12
75%
2/3
66.7%
4/5
80%
Med
ianage
atse
izure
onse
t(y)
11.4
15
1.3
Seve
rity
ofep
ilep
sy
0=Nose
izure
s4/19
21%
1/6
16.7%
3/12
25%
1/3
33.3%
1/5
20%
1=Se
izure
onse
t>2
yandse
izure
free
after
withdra
walAE
2/15
13.3%
1/5
16.7%
1/9
11.1%
0/2
0%0/4
0%
2=Se
izure
onse
t>2
yandse
izure
free
withAE
2/15
13.3%
2/5
40%
2/9
22.2%
0/2
0%1/4
25%
3=Se
izure
onse
t<2
yandco
ntinuingse
izure
swithAE
6/15
40%
2/5
40%
3/9
33.3%
1/2
50%
1/4
25%
4=Se
vere
infantile
spasm
sorse
izure
onse
t<6
mo
5/15
33.3%
0/5
0%3/9
33.3%
1/2
50%
2/4
50%
MRIpatter
n
Med
ianage
atex
amination(y)
3.4
2.3
5.6
7.9
3.95
Corticala
nomalies
Norm
alo
rmildSIMPgy
ralp
atter
n8/19
41.7%
2/5
40%
5/11
45.5%
2/3
66.7%
4/5
80%
Moder
ate
SIMPgy
ralp
atter
n5/19
26.3%
2/5
40%
2/11
18.2%
1/3
33.3%
1/5
20%
Seve
reandpse
udopach
ygyr
icco
rtex
6/19
31.6%
1/5
20%
4/11
36.4%
0/3
0%0/5
0%
Corticala
trophy
12/19
63.2%
3/4
75%
7/11
63.6%
1/3
33.3%
2/5
20%
Mye
linationdelay
Abse
ntto
mildmye
linationdelay
6/19
31.6%
2/4
50%
3/10
30%
2/3
66.7%
2/5
40%
Moder
ate
mye
linationdelay
2/19
10.5%
1/4
25%
5/10
50%
1/3
33.3%
3/5
60%
Seve
remye
linationdelayorwhitematter
loss
11/19
57.9%
1/4
25%
2/10
20%
0/3
0%0/5
0% Continued
12 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

noticeable changes in brain size and mental development earlyin childhood. To date, FOXG1 has been linked to a wide rangeof human congenital brain disorders.1–7,17–19 In this study, wedescribe detailed clinical and neuroradiological data on 45patients with pathogenic single nucleotide variants and copynumber variations affecting FOXG1. This is one of the largestcohort of patients with FOXG1 syndrome and focuses onFOXG1 point mutations, which affect both sexes equally. Theaim of this study was to refine the phenotypic spectrum ofFOXG1 syndrome and its natural history and to further in-vestigate genotype-phenotype correlations. In keeping withpreviously published FOXG1-associated clinical features, wefound FOXG1 syndrome to be associated with severe postnatalmicrocephaly (−4 to −6 SD), dyskinetic-hyperkinetic movementdisorders, visual impairment, epilepsy, stereotypies, abnormalsleep patterns, and unexplained episodes of crying.1,3,5–8,18,20–23
Our data clearly confirm that head circumference is usuallynormal to borderline small at birth and evolves during infancyto severe microcephaly below −3 SD, with normal somaticgrowth. Although no longitudinal data on head circumferenceare available from our series, it is interesting to note thatmicrocephaly was the first concern in one-third of the cohortat the mean age of 3.47 months, suggesting that the slowdownin head growth occurs earlier than previously described. Ofnote, FOXG1-related postnatal microcephaly is characterizedby underdevelopment of the frontal lobes, a unique patternthat does not occur in other causes of progressive micro-cephaly.24 This underdevelopment of frontal lobes can beassociated with a mildly to moderately simplified gyral patternand reduced white matter or in the youngest patients witha pachygyric appearance. We observed this pattern on T2-weighted images in infants who showed mild gyral simplifi-cation later in childhood. The clue to the cause of the 2patterns came from studying serial MRI of patients Im09 andIm11. Frontal pachygyria, which was observed at 6 months ofage, changed into mild gyral simplification at 2.5 years of age.This finding suggested that the 2 cortical patterns did notrepresent differences of morphology but instead, differencesin the maturity of the subcortical white matter. It is note-worthy that this changing appearance has been observedpreviously in polymicrogyria.25,26
Another imaging hallmark of FOXG1 disorder is the delayedmyelination. While delayed myelination has a similar ap-pearance to hypomyelination on a single MRI, especially ifdone at an early age, sequential studies can distinguish be-tween them by demonstrating increasing myelin content indelayed myelination.27 This evolution of delayed myelinationtoward normalization in childhood is not specific to FOXG1syndrome, as it has been observed in other developmentaldisorders, such as MCT8 deficiency and Xq28 duplicationinvolving MECP2 or SPTAN1 encephalopathy.27–29
Taken together with the published literature, we suggest thatFOXG1 syndrome is a disorder in which hypogenetic corpuscallosum is the most frequent finding. More specifically, corpusTa
ble
2Clin
ical
andneu
roim
agingfeaturesrelatedto
theFO
XG1ge
notypegroups(con
tinue
d)
N-ter
minal
domain
variants
Fork
hea
ddomain
frames
hift
andnonse
nse
variants
Fork
hea
ddomain
misse
nse
variants
C-term
inaldomain
variants
Larg
edeletions
Corp
usca
llosu
masp
ect
Norm
al
1/19
5.3%
0/6
0%0/11
0%2/3
66.7%
0/5
0%
Hyp
oplasicbutco
mplete
3/19
15.8%
2/6
33.3%
3/11
27.30%
1/3
33.3%
3/5
60%
Hyp
oge
net
icwithabse
ntro
stru
m10
/19
52.6%
2/6
33.3%
6/11
54.5%
0/3
0%1/5
20%
Partialo
rco
mplete
age
nes
is5/19
26.3%
2/6
33.3%
2/11
18.2%
0/3
0%1/5
20%
Cer
ebellaratrophy
3/19
15.8%
0/5
0%1/11
9.1%
0/3
0%1/5
20%
Abbreviations:
AE=an
tiep
ilepticdru
g;CC=co
rpusca
llosu
m;S
E=statusep
ilepticu
s;SIMP=simplifiedgy
ration.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 13

callosum malformations in FOXG1 syndrome are frontal pre-dominant, similar to the gyral abnormality, suggesting that thesame pathogenic mechanism operates for both the frontalcortical abnormalities and the callosal abnormalities. Completeagenesis occurs occasionally and is likely to represent the mostsevere end of the spectrum of pathogenic mechanisms un-derlying hypoplasia. This also illustrates that hypoplasia andagenesis are related to a similar mechanism and that geneticmodifiers influence the severity of the callosal phenotype.30 Ofinterest, the severity of corpus callosum anomaly does notcorrelate with the degree of microcephaly, the degree of mye-lination abnormality, or the degree of gyral abnormalities. Thiscontrasts with data from congenital microcephaly that showeda correlation between the degree of microcephaly and the se-verity of the associated callosal anomaly.31
Hyperkinetic movement disorders have been recognized to bea key feature in FOXG1 syndrome since its originaldescription.6,18 Our data show that movement disorder israrely the presenting feature of FOXG1 syndrome; this hasnot been stressed previously. It is important that the combi-nation of hand stereotypes, mostly hand to mouth, withgeneralized dyskinesia is one of the key characteristics ofFOXG1 syndrome that distinguishes it from other monogenichyperkinetic movement disorders or neurodegenerativediseases.6,32 The hyperkinetic movement disorder, althoughaffecting quality of life, was stable over time, never evolvedinto status dystonicus, and did not lead to any of the com-plications of severe dystonia that can observed in other de-velopmental or degenerative neurologic disorders.6,32
A previous report suggested that FOXG1 syndrome could beclassified as an epileptic-dyskinetic encephalopathy18 likeARX- and STXBP1-related encephalopathies. Our data showthat epilepsy is not a consistent feature, unlike dyskinetic-hyperkinetic movements. Although epilepsy affected 79% ofpatients reported here, which is within the range of previousreports (from 57%7 to 86%5), it did not show a particularseizure pattern that could help the clinician to define a specificepilepsy syndrome.
Since the first report that FOXG1 mutations can be re-sponsible for congenital Rett variant, a number of publicationshave emphasized the differences between these disorders.33
Here, by applying the congenital Rett variant criteria,14 weconfirm that the majority of patients with the FOXG1 syn-drome do not meet the criteria for congenital Rett variant. Atall ages, FOXG1 syndrome is more severe with respect toambulation, reciprocity, and receptive language and has moredisordered sleep, compared with Rett syndrome, as well aslacking the regression observed in Rett syndrome. Thesefindings further reinforce that FOXG1 disorder is clinicallyseparable from Rett syndrome, with distinct clinical pre-sentation and natural history. It is important that patients withFOXG1 disorder receive appropriate counseling about med-ical comorbidities and natural history related to their disorder,avoiding the confusion with Rett syndrome.
The number of reported FOXG1 mutations is now largeenough to search for genotype-phenotype correlations inFOXG1 syndrome. We observed that patients carrying muta-tions in the N-terminal domain and large deletion of FOXG1,which are the most common mutation types, show the mostsevere presentation and MRI anomalies, while those carryingmutations in the FBD or C-terminal domain were less severelyaffected. In previous series, a milder phenotype was observed inpatients with missense variants in the FBD conserved site.7
However, the differences were found in items related to sitting,walking, and functional hand use, which are commonly severelyimpaired in all FOXG1mutation patients.7 Using covarianceand cluster analyses, we highlighted relationships betweengyral and myelination patterns in patients with FOXG1disorder. However, identical hotspot mutations c.256dupCand c.406dupG can be associated with highly variable fea-tures, such as variable epilepsy severity or degree of corpuscallosum anomalies, underlining the importance of being cau-tious about predicting phenotype on the basis of genotype in thecontext of genetic counseling. This suggests that factors beyondthe primary mutation can influence disease severity, includinggenetic modifiers and epigenetic and environmental factors.
The complexity and the poor reproducibility of genotype-phenotype relationships in FOXG1 syndrome probably reflectsthe pleiotropic and nonredundant roles of Foxg1 in vertebratebrain development.
This study, one of the largest to date, provides evidence thatFOXG1 mutations are responsible for a specific and recogniz-able neurodevelopmental disorder with a high degree of vari-ability.We have expanded the phenotypic spectrumby defining3 key brain imaging features of FOXG1 syndrome, noting thatthe degree of cortical abnormality is not correlated with theseverity of the corpus callosum malformation. Moreover, ourdata confirm that mutations leading to the loss of the FBDdomain, lead to themost severe clinical presentation of FOXG1syndrome. The pathophysiology of such complex genotype-phenotype relationships reflects the pleiotropic and non-redundant roles of Foxg1 during development.
AffiliationFrom the Imagine Institute (N.V., M.C., C. Maillard, A.B.,N.B.-B.), INSERM UMR 1163, Paris Descartes University,Necker Enfants Malades Hospital, Paris, France; PediatricNeurology APHP—Necker Enfants Malades Hospital (M.C.,M.H., N.B.-B.), Paris, France; Pediatric Radiology (N.B.),APHP—Necker Enfants Malades Hospital, Paris, France;Image—Imagine Institute (N.B.), INSERMUMR 1163, ParisDescartes University, Necker EnfantsMalades Hospital, Paris,France; Department of Paediatric Clinical Epileptology (J.T.),Sleep Disorders and Functional Neurology, University Hos-pitals of Lyon (HCL), France; Service de Genetique medicale(E.S.), Hopitaux Universitaires de Strasbourg, IGMA, France;Pediatric Neurology (T.L.-S.), Wolfson Medical Center, TelAviv, Israel; Wolfson Molecular Genetics Laboratory (D.L.),Wolfson Medical Center, Tel Aviv, Israel; Neurometabolism
14 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Department (B.M.), Angers Hospital and University, France;Centre de Genetique et Centre de Reference Maladies RaresAnomalies du Developpement (S.M., A.M.), CHU Dijon,France; South Australian Clinical Genetics Service (E.H.),SA Pathology (at Royal Adelaide Hospital), and School ofMedicine, University of Adelaide, Australia; Service deGenetique Medicale (B.I., M.N., M.V., B.C.), CHU Nantes,France; Departement de Genetique et Centre de ReferenceDeficiences Intellectuelles de Causes Rares (D.H., C. Mignot),Hopital de la Pitie-Salpetriere, APHP, Paris, France; GMGF(M.M.), INSERM UMR_S910, Aix-Marseille University, Pe-diatric Neurology Unit, Timone Children Hospital, Marseille,France; Department of Neonatal Medicine (S.R.), RouenUniversity Hospital, Haute-Normandie, France; DepartmentofMedical Genetics (C.Michot), Reference Center for SkeletalDysplasia, INSERM UMR 1163, Laboratory of Molecular andPhysiopathological Bases of Osteochondrodysplasia, ParisDescartes-Sorbonne Paris Cite University, AP-HP, InstitutImagine, and Hopital Universitaire Necker-Enfants Malades,Paris, France; APHP (S.V.), GHUEP, Hopital Trousseau,Neurologie Pediatrique, Paris, France; GRC ConCer-LD(S.V.), Sorbonne Universites, UPMCUniv 06, Paris, France;Hopital Nord Franche Comte (S.W.), CH HNFC—Site deBelfort, France; Pediatrics (A.D.), University of Basel Child-rens’ Hospital, Switzerland; CHU Rennes (S.O.), Service deGenetique Clinique, CNRS UMR6290, Universite Rennes1,France; Service de Pediatrie (L.L.), Centre Hospitalier dela Cote Basque, Bayonne, France; Department of Genetics(G.A.M.), Rouen University Hospital, France; Service deGenetique (A.S.), Hopital Bretonneau, Tours, France; Servicede Neurologie Pediatrique (J.M.P.), Hopital Pellegrin-Enfants,CHU de Bordeaux, France; Pediatrie generale (I.C.), Hopitalde Lorient, France; Genetique Medicale—CHU EstaingCLERMONT-FERRAND (B.P., B.T.), France; Service deNeurologie Pediatrique (F.R.), Hopital Gui de Chauliac,CHRU de Montpellier, France; Equipe Genetique desAnomalies du Developpement (C.P.), INSERM UMR1231,Universite de Bourgogne-Franche Comte, Dijon, France;Laboratoire de Genetique chromosomique moleculaire(C.P.), Plateau technique de Biologie, CHU, Dijon, France;Laboratory of Biochemistry and Molecular Genetics (T.B.),HUPC Paris Centre, Cochin Hospital, Paris, France; NationalRare Disease Center—Centre de Reference “deficiences intel-lectuelles de causes rares” (M.-A.S.), Strasbourg UniversityHospital, France; and National Rare Disease Center—Centrede Reference “deficiences intellectuelles de causes rares”(N.B.-B.), AP-HP, Necker Enfants Malades, Paris, France.
Author contributionsN. Vegas, M. Cavallin, C. Maillard: study concept and design,analysis and acquisition of clinical and molecular data. N.Boddaert: analysis and acquisition of MRI data. J. Toulouse, E.Schaefer, T. Lerman-Sagie, D. Lev, B. Magalie, S. Moutton,E. Haan, B. Isidor, D. Heron, M. Milh, S. Rondeau, C. Michot,S. Valence, S. Wagner, M. Hully, C. Mignot, A. Masurel, A.Datta, S. Odent, M. Nizon, L. Lazaro, M. Vincent, B. Cogne,
G.A. Marie, A. Stephanie, J.M. Pedespan, I. Caubel, B. Pontier,B. Troude, F. Rivier, M.-A. Spitz: acquisition of data andfollow-up of the patients. C. Philippe and T. Bienvenu:analysis molecular data. A. Bery and N. Bahi-Buisson: studysupervision, concept and critical revision of manuscript forintellectual content.
AcknowledgmentThe authors would like to thank the affected individuals andtheir families for participation in this study, as well as theclinicians in charge of these patients who may not be cited.The authors would like to sincerely thank Prof AlessandraPierani for her critical reading of the manuscript and helpfulcomments on our findings.
Study fundingResearch reported in this publication was supported by theAgence Nationale de la Recherche (ANR-16-CE16-0011 MC,AB, NBB), the Fondation Maladies Rares, and DESIRE (grantagreement 602531). The project was also supported by theEuropean Network on Brain Malformations (COST ActionCA16118). The authors have no conflict of interest to declare.
DisclosureN. Vegas, M. Cavallin, C. Maillard, N. Boddaert, J. Toulouse,E. Schaefer report no disclosures. T. Lerman-Sagie has servedon the editorial boards of the Journal of Child Neurology,Harefuah, and the European Journal of Paediatric Neurology. D.Lev has received research support from the Sackler School ofMedicine (Tel Aviv University). M. Barth and S. Mouttonreport no disclosures. E. Haan has received research supportfrom the Lipedema Foundation (USA). B. Isidor and D.Heron report no disclosures. M. Milh has received speakerhonoraria from Shire and Cyberonics. S. Rondeau, C. Michot,S. Valence, S. Wagner, M. Hully, C. Mignot, A. Masurel, A.Datta, S. Odent, M. Nizon, L. Lazaro, M. Vincent, B. Cogne,A.M. Guerrot, S. Arpin, J.M. Pedespan, I. Caubel, B. Pontier,B. Troude, F. Rivier, C. Philippe, T. Bienvenu,M. Spitz, and A.Bery report no disclosures. N. Bahi-Buisson has received re-search support from Agence Nationale de la recherche,Fondation pour la Recherche Medicale, Fondation NRJ—Institut de France, and the EU-FP7 project GENECODYS.Full disclosure form information provided by the authors isavailable with the full text of this article at Neurology.org/NG.
Received March 24, 2018. Accepted in final form July 12, 2018.
References1. Ariani F, Hayek G, Rondinella D, et al. FOXG1 is responsible for the congenital
variant of Rett syndrome. Am J Hum Genet 2008;83:89–93.2. Mencarelli MA, Spanhol-Rosseto A, Artuso R, et al. Novel FOXG1 mutations asso-
ciated with the congenital variant of Rett syndrome. J Med Genet 2010;47:49–53.3. Kortum F, Das S, Flindt M, et al. The core FOXG1 syndrome phenotype consists of
postnatal microcephaly, severe mental retardation, absent language, dyskinesia, andcorpus callosum hypogenesis. J Med Genet 2011;48:396–406.
4. Ellaway CJ, Ho G, Bettella E, et al. 14q12 Microdeletions excluding FOXG1 give riseto a congenital variant Rett syndrome-like phenotype. Eur J Hum Genet 2013;21:522–527.
5. Seltzer LE, MaM, Ahmed S, et al. Epilepsy and outcome in FOXG1-related disorders.Epilepsia 2014;55:1292–1300.
6. Papandreou A, Schneider RB, Augustine EF, et al. Delineation of the movementdisorders associated with FOXG1 mutations. Neurology 2016;86:1794–1800.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 15

7. Mitter D, Pringsheim M, Kaulisch M, et al. FOXG1 syndrome: genotype-phenotypeassociation in 83 patients with FOXG1 variants. Genet Med 2018;20:98–108.
8. Bahi-Buisson N, Nectoux J, Girard B, et al. Revisiting the phenotype associated withFOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics 2010;11:241–249.
9. Kumamoto T, Hanashima C. Evolutionary conservation and conversion of Foxg1function in brain development. Dev Growth Differ 2017;59:258–269.
10. Krishnaraj R, Ho G, Christodoulou J. RettBASE: Rett syndrome database update.Hum Mutat 2017;38:922–931.
11. Le Guen T, Bahi-Buisson N, Nectoux J, et al. A FOXG1 mutation in a boy withcongenital variant of Rett syndrome. Neurogenetics 2011;12:1–8.
12. Diebold B, Delepine C, Nectoux J, Bahi-Buisson N, Parent P, Bienvenu T. Somaticmosaicism for a FOXG1mutation: diagnostic implication. ClinGenet 2014;85:589–591.
13. Sanger TD, Chen D, Fehlings DL, et al. Definition and classification of hyperkineticmovements in childhood. Mov Disord 2010;25:1538–1549.
14. Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteriaand nomenclature. Ann Neurol 2010;68:944–950.
15. Adachi Y, Poduri A, Kawaguch A, et al. Congenital microcephaly with a simplifiedgyral pattern: associated findings and their significance. AJNR Am J Neuroradiol2011;32:1123–1129.
16. Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcriptionfactor BF-1 is essential for the development of the cerebral hemispheres. Neuron1995;14:1141–1152.
17. Striano P, Paravidino R, Sicca F, et al. West syndrome associated with 14q12 dupli-cations harboring FOXG1. Neurology 2011;76:1600–1602.
18. Cellini E, Vignoli A, Pisano T, et al. The hyperkinetic movement disorder of FOXG1-related epileptic-dyskinetic encephalopathy. Dev Med Child Neurol 2016;58:93–97.
19. Mariani J, Coppola G, Zhang P, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015;162:375–390.
20. De Bruyn C, Vanderhasselt T, Tanyalcin I, et al. Thin genu of the corpus callosumpoints to mutation in FOXG1 in a child with acquired microcephaly, trigonocephaly,and intellectual developmental disorder: a case report and review of literature. Eur JPaediatr Neurol 2014;18:420–426.
21. De Filippis R, Pancrazi L, Bjorgo K, et al. Expanding the phenotype associated withFOXG1mutations and in vivo FoxG1 chromatin-binding dynamics. Clin Genet 2012;82:395–403.
22. Florian C, Bahi-Buisson N, Bienvenu T. FOXG1-Related disorders: from clinicaldescription to molecular genetics. Mol Syndromol 2012;2:153–163.
23. Van der Aa N, Van den Bergh M, Ponomarenko N, Verstraete L, Ceulemans B, StormK. Analysis of FOXG1 is highly recommended in male and female patients with Rettsyndrome. Mol Syndromol 2011;1:290–293.
24. Seltzer LE, Paciorkowski AR. Genetic disorders associated with postnatal micro-cephaly. Am J Med Genet C Semin Med Genet 2014;166C:140–155.
25. Takanashi J, Barkovich AJ. The changing MR imaging appearance of polymicrogyria:a consequence of myelination. AJNR Am J Neuroradiol 2003;24:788–793.
26. Bahi-Buisson N, Poirier K, Boddaert N, et al. GPR56-related bilateral frontoparietalpolymicrogyria: further evidence for an overlap with the cobblestone complex. Brain2010;133:3194–3209.
27. van der Knaap MS, Wolf NI. Hypomyelination versus delayed myelination. AnnNeurol 2010;68:115.
28. El Chehadeh S, Faivre L, Mosca-Boidron AL, et al. Large national series of patientswith Xq28 duplication involving MECP2: delineation of brain MRI abnormalities in30 affected patients. Am J Med Genet A 2016;170A:116–129.
29. Syrbe S, Harms FL, Parrini E, et al. Delineating SPTAN1 associated phenotypes: fromisolated epilepsy to encephalopathy with progressive brain atrophy. Brain 2017;140:2322–2336.
30. Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imagingfindings identify new causes for corpus callosum development syndromes. Brain 2014;137:1579–1613.
31. Barkovich AJ, Kjos BO. Normal postnatal development of the corpus callosum asdemonstrated by MR imaging. AJNR Am J Neuroradiol 1988;9:487–491.
32. Carecchio M, Mencacci NE. Emerging monogenic complex hyperkinetic disorders.Curr Neurol Neurosci Rep 2017;17:97.
33. Ma M, Adams HR, Seltzer LE, Dobyns WB, Paciorkowski AR. Phenotype differen-tiation of FOXG1 and MECP2 disorders: a new method for characterization ofdevelopmental encephalopathies. J Pediatr 2016;178:233–240 e210.
16 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

ARTICLE OPEN ACCESS
Identification of a new SYT2 variant validatesan unusual distal motor neuropathy phenotypeNataly I. Montes-Chinea, MD, Zhuo Guan, PhD, Marcella Coutts, MD, Cecilia Vidal, MD, Steve Courel, BS,
Adriana P. Rebelo, PhD, Lisa Abreu, MPH, Stephan Zuchner, MD, PhD, J. Troy Littleton, PhD,
and Mario A. Saporta, MD, PhD
Neurol Genet 2018;4:e282. doi:10.1212/NXG.0000000000000282
Correspondence
Dr. Saporta
AbstractObjectiveTo report a new SYT2 missense mutation causing distal hereditary motor neuropathy andpresynaptic neuromuscular junction (NMJ) transmission dysfunction.
MethodsWe report a multigenerational family with a new missense mutation, c. 1112T>A (p. Ile371-Lys), in the C2B domain of SYT2, describe the clinical and electrophysiologic phenotypeassociated with this variant, and validate its pathogenicity in a Drosophila model.
ResultsBoth proband and her mother present a similar clinical phenotype characterized by a slowlyprogressive, predominantly motor neuropathy and clear evidence of presynaptic NMJ dys-function on nerve conduction studies. Validation of this new variant was accomplished bycharacterization of the mutation homologous to the human c. 1112T>A variant in Drosophila,confirming its dominant-negative effect on neurotransmitter release.
ConclusionsThis report provides further confirmation of the role of SYT2 in human disease and corrob-orates the resultant unique clinical phenotype consistent with heriditary distal motor neu-ropathy. SYT2-related motor neuropathy is a rare disease but should be suspected in patientspresenting with a combination of presynaptic NMJ dysfunction (resembling Lambert-Eatonmyasthenic syndrome) and a predominantly motor neuropathy, especially in the context ofa positive family history.
From the Department of Neurology (N.I.M.-C., M.C., C.V., M.A.S.), University of Miami Miller School of Medicine FL; Department of Biology (Z.G., J.T.L.) and Department of Brain andCognitive Sciences (Z.G., J.T.L.), The Picower Institute for Learning &Memory,Massachusetts Institute of Technology, Cambridge; andDepartment of HumanGenetics (S.C., A.P.R., L.A.,S.Z., M.A.S.), Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, FL.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Hereditary distal motor neuropathies (dHMNs) are a groupof rare diseases that share the common feature of a length-dependent predominantly motor neuropathy.1,2 A particulartype of dHMN associated with presynaptic neuromuscularjunction (NMJ) dysfunction has been recently reported in 2families harboring autosomal dominant pathogenic mutationsin synaptotagmin 2 (SYT2).3,4 Here, we describe a new variantin SYT2 causing this same phenotype in a multigenerationalfamily and present functional validation of a dominant-negativeeffect of the mutation on synaptic transmission in a Drosophilamodel.
MethodsStandard protocol approvals, registrations,and patient consentsResearch-related activities were only performed after in-formed consent was obtained, as part of a University of MiamiInstitutional Review Board-approved project.
Clinical evaluationThe proband was evaluated at the Charcot-Marie-Tooth clinicat theUniversity ofMiami. Her initial assessment included a fullhistory and physical examination. Disease severity was assessedusing the Charcot-Marie-Tooth Neuropathy Score (CMTNS)version 2.5 Subsequent evaluation included electrophysiologicstudies performed on a Cadwell Sierra Wave EMG machine,following standard protocols. Exercise facilitationwas examinedby performing a 10-second sustained contraction that was ap-plied to the right abductor pollicis brevis (APB), abductor digitiminimi (ADM), and abductor hallucis (AH) muscles followedby stimulation over their respective motor nerves. Repetitivenerve stimulation (RNS) was performed on the right ulnarnerve recording at the ADMmuscle. Ten supramaximal stimuliwere applied at 3 Hz, with percentage increment or decrementcalculated between the first and fourth response.
Amplification of SYT2 exons andSanger sequencingSYT2 sequencewas obtained fromUniversity of California SantaCruz genome browser to design amplification primers. Primersflanking each SYT2 exon were designed using Primer3 software.SYT2 exons were amplified from the proband’s and hermother’sgenomic DNA by PCR with Platinum Taq polymerase (Ther-moFisher). Amplifications were carried out in a thermal cycler(Applied Biosystem). PCR products were purified using QiagenPCR purification kit. Each purified PCR product, correspondingto an exon, was submitted with corresponding sequencing pri-mers to Eurofins for Sanger sequencing. Sequence traces wereanalyzed using Sequencher (Gene Codes, Ann Arbor, MI).
Following are the primers used for PCR and Sanger se-quencing: SYT2-Ex1F: CTTGGTCTCCTCCCCTCACT;SYT2-Ex1R: CCAACCCTACTCACCTCTCG; SYT2-Ex2F:GGCTGACTGTGTACTAATTGGATG; SYT2-Ex2R: CCCAGCCTGAAATCTAAGCA; SYT2-Ex3F: CTCACCCATTTTTCCCAATG; SYT2-Ex3R: TTAAGGAGGGGAGCAGGTTT; SYT2-Ex4F: GTTCCCACCACACACAGCTC; SYT2-Ex4R: GAGCTATAGGCCCTGCAGTTT;SYT2-Ex5F: CATTTCCCTGCCCCAACT; SYT2-Ex5R:GCCATTGTTCCAGGCTGAG; SYT2-Ex6F: TTTGTCTGTCTCGGCACACT; SYT2-Ex6R: AGGTCGTCTGCCTCCAAAG; SYT2-Ex7F: ACCTTCTCGGCCATCACATA; SYT2-Ex7R: GGCAGCAAAGTGTTCCTCTT;SYT2-Ex8F: TGGTCTCAGCGGAGTGAAG; SYT2-Ex8R:ACCCAGGCACCATTAGACCT; SYT2-Ex9F: TGGAGCA-GAGATGAAACCAA; SYT2-Ex9R: CAGAGCCAGGCTTCTCTTTC.
Genetic screen and Drosophila stocksDrosophila melanogaster was cultured on standard medium at22°C. Transgenic strains were generated using standard mi-croinjection intowhite (w−/−) embryos performed by BestGeneInc. UAS-syt1 transgenes were expressed using a GAL4 driverunder the control of the pan-neuronal elav promoter, as pre-viously described.6 DNA for rescue with individual pointmutants was generated using the QuikChange multisite-directed mutagenesis kit (Stratagene, Santa Clare, CA) withthe following primer sets: I426K-5’oligo: GGCACCTCCGAACCCAaaGGCCGCTGCATACTTG and I426K-3’oligo:CAAGTATGCAGCGGCCttTGGGTTCGGAGGTGCC.
Wild-type and mutant complementary DNAs were subclonedinto a modified pValum construct with an N-terminal myc tagto allow tracking of protein localization in overexpressedanimals containing endogenous synaptotagmin 1 (SYT1).These constructs were injected into a yv; attP third chro-mosome docking strain by BestGene Inc. (Chino Hills, CA).All constructs allowed use of the Gal4/UAS expression sys-tem to express the transgenic proteins. UAS-Syt1 transgeneswere expressed using a GAL4 driver under the control of thepan-neuronal C155 elav promoter in either control white orsyt1 null (syt1−/−) backgrounds. Null mutants lacking en-dogenous SYT1 were generated by crossing Syt1N13, an in-tragenic Syt1 deficiency,7 with Syt1AD4, which truncatesSYT1 before the transmembrane domain.8
Western blot analysisand immunocytochemistryWestern blotting of whole adult head lysates (1 head/lane)was performed using standard laboratory procedures with
GlossaryADM = abductor digiti minimi; AH = abductor hallucis; ANOVA = analysis of variance; APB = abductor pollicis brevis; CMT =Charcot-Marie-Tooth; CMTNS = Charcot-Marie-Tooth Neuropathy Score; dHMN = Hereditary distal motor neuropathy;eEJC = excitatory evoked junctional current;NMJ = neuromuscular junction;RNS = repetitive nerve stimulation;WT = wildtype.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

anti-SYT1 (1:1,000, kindly provided byNoreen Reist) or anti-syntaxin (1:1,000, Developmental Studies Hybridoma Bank,Iowa City, IA). Visualization and quantification were doneusing a LI-COR Odyssey Imaging System (LI-COR Bio-sciences, Lincoln, MA). Immunostaining was performed on 3rdinstar larvae at wandering stage larvae as described previously.9
Rabbit myc antibody (1:1,000; Genetex) and anti-horseradishperoxidase (1:1,000; Jackson ImmunoResearch, West Grove,PA)were used for immunostaining. Confocal stacks ofmuscle 6and 7NMJs containing immunoreactive proteins were capturedon a Zeiss Pascal Confocal with PASCAL software (Carl ZeissMicroImaging, Inc.) using a 63 × numerical aperture 1.3 PlanNeofluar oil immersion lens (Carl Zeiss, Inc) and fluorescentsecondary antibodies (Molecular Probes, Carlsbad, CA).
ElectrophysiologyPostsynaptic currents from third instar male larvae at thewandering stage from the indicated genotypes were recordedat muscle fiber 6 of segment A3 using 2-electrode voltageclamp with a −80 mV holding potential in hemolymph-like(HL) 3.1 saline solution as previously described.6,10 Finalcalcium concentration was adjusted to 2 mM. For evoked andmini analysis, n refers to the number of NMJs analyzed, withno more than 2 NMJs analyzed per animal, and with animalsderived from at least 3 independent experiments. Data ac-quisition and analysis was performed using Axoscope 9.0 andClampfit 9.0 software (Molecular Devices, Sunnyvale, CA).Motor nerves innervating the musculature were severed andplaced into a suction electrode, so action potential stimulationcould be applied at the indicated frequencies using a pro-grammable stimulator (Master8; AMPI, Jerusalem, Israel).
Data analysis and statisticsElectrophysiology analysis was performed using Clampfit 10software (Axon Instruments, Foster City, CA), as previouslydescribed.6 Statistical analysis and graphs were performed usingOrigin Software (OriginLab Corporation, Northampton, MA).Statistical significance was determined using 1-way analysis ofvariance (ANOVA) (nonparametric) with post hoc Sidakmultiple comparisons test. The p values associated with 1-wayANOVA tests were adjusted p values obtained from a post hocSidak multiple comparisons test. Appropriate sample size wasdetermined using GraphPad Statmate. In all figures, the dataare presented as mean ± SEM. Statistical comparisons are withcontrol, unless noted. The results were all shown: N.S. = nosignificant change (p > 0.05), *p < 0.05, **p < 0.005, ***p <0.001, and ****p < 0.0001. All error bars are SEM.
Data availabilityData, methods, and materials used to conduct this researchare documented in detail in the Methods section.
ResultsClinical evaluationThe Proband is a 50-year-old woman who was referred to theUniversity of Miami Comprehensive CMT clinic for evaluation
of suspected CMT because of gradually progressive weaknessof her extremities. She had normal developmental milestonesbut was found to have bilateral high arched feet and ham-mertoes and occasional falls around the age of 8 years. Shedeveloped progressive leg weakness, worsening bilateral handcramping, weak handgrip, and only mild paresthesia on distalextremities. The proband’s family history is remarkable forsimilar symptoms reported by her mother, maternal grandfa-ther, 2 maternal uncles, 1 maternal aunt, a younger sister, anda nephew (figure 1A). Initial physical examination revealedbilateral pes cavus and hammer toes (figure 1B), inability towalk on heels or toes, severe, nonfatigable, distal lower ex-tremity weakness, limited range of motion on ankles bi-laterally because of ankle fusions, nonspecific sensory changesin lower extremities, and diffuse hyporreflexia with postexer-cise normalization. Her initial CMTNS was 9. Her mother,a 68-year-old woman, was also evaluated. She has had a historyof high-arched feet and hammertoes since childhood. Exami-nation showed bilateral pes cavus and hammer toes (figure 1B).Strength testing revealed intrinsic hand muscle and plantar-flexion weakness. Deep tendon reflexes were absent initially,with postexercise facilitation noted. Sensory examination wasessentially normal for touch, pinprick, vibration, and pro-prioception. Her CMTNS was 4.
Clinical neurophysiology testingThe proband underwent electrodiagnostic testing to furtherevaluate for a hereditary peripheral neuropathy. Sensory nerveconduction studies were normal. However, compound mus-cle action potential amplitudes were significantly reducedthroughout all tested motor nerves. Examination of eachmotor nerve incidentally showed decreasing amplitudes withsuccessive stimulations. For this reason, a 10-second sustainedcontraction was applied at the right abductor digiti minimi(ADM), APB, tibialis anterior, and AH, and their respectivemotor nerves were stimulated afterward. This resulted insignificant incremental responses (figure 1C). RNS was sub-sequently performed on the right ulnar nerve recording at theright ADM and revealed a 42% decremental response anda 125% postexercise facilitation (figure 1, C and D). NeedleEMG examination revealed evidence of ongoing denervationin the form of positive waves and fibrillation potentials and ofchronic reinnervation in the form of reduced recruitment inthe medial gastrocnemius muscles. The proband’s motheralso underwent a limited nerve conduction studies and RNSexamination with similar findings, including normal suralsensory response, 17.6% decremental response and an 86%postexercise facilitation on repetitive stimulation study (figure1C). To rule out paraneoplastic Lambert-Eaton syndrome,the proband was investigated further and antibodies againstvoltage-gated calcium channel were negative, and a chest CTdid not show evidence of lung carcinoma.
Genetic analysisSanger sequencing of the SYT2 gene revealed a heterozygous1112T>A missense mutation in both the proband and hermother (figure 1E). This mutation causes an isoleucine (I) to
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
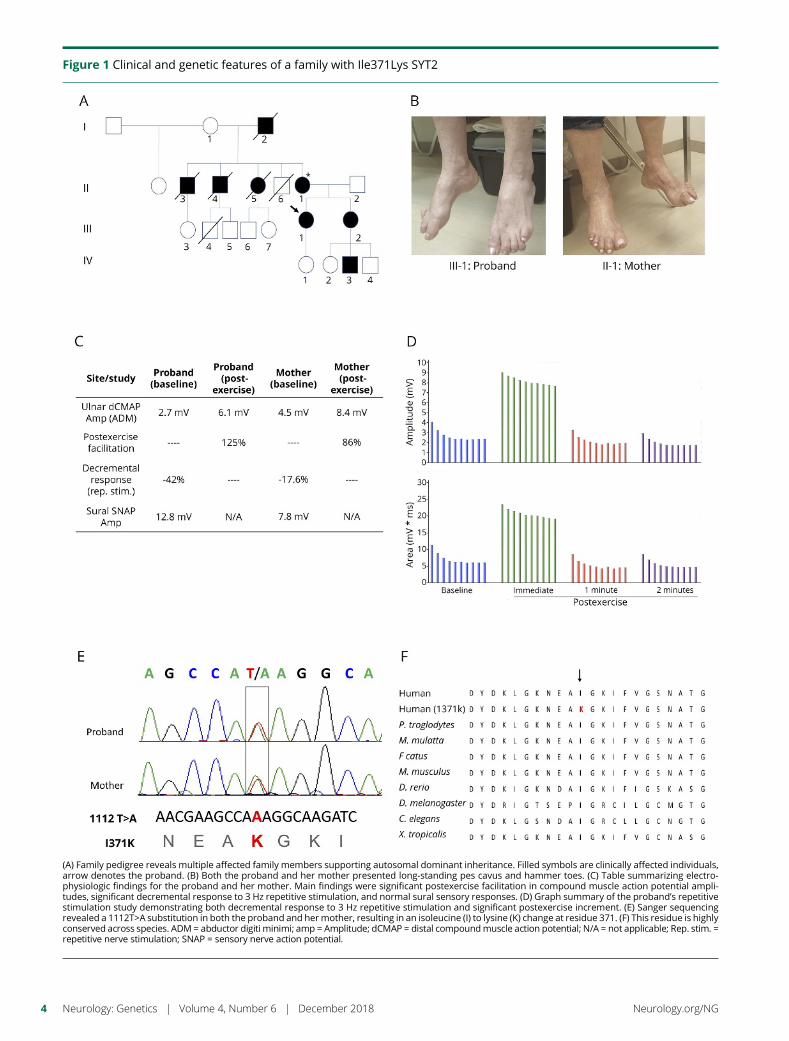
Figure 1 Clinical and genetic features of a family with Ile371Lys SYT2
(A) Family pedigree reveals multiple affected family members supporting autosomal dominant inheritance. Filled symbols are clinically affected individuals,arrow denotes the proband. (B) Both the proband and her mother presented long-standing pes cavus and hammer toes. (C) Table summarizing electro-physiologic findings for the proband and her mother. Main findings were significant postexercise facilitation in compound muscle action potential ampli-tudes, significant decremental response to 3 Hz repetitive stimulation, and normal sural sensory responses. (D) Graph summary of the proband’s repetitivestimulation study demonstrating both decremental response to 3 Hz repetitive stimulation and significant postexercise increment. (E) Sanger sequencingrevealed a 1112T>A substitution in both the proband and hermother, resulting in an isoleucine (I) to lysine (K) change at residue 371. (F) This residue is highlyconserved across species. ADM = abductor digiti minimi; amp = Amplitude; dCMAP = distal compoundmuscle action potential; N/A = not applicable; Rep. stim. =repetitive nerve stimulation; SNAP = sensory nerve action potential.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

lysine (K) change at residue 371 (I371K). The I371K residueis located within the C2B domain of SYT2, which spans res-idues 271 to 406. This variant was not found in the GenomeAggregation Database (gnomAD). Further variant analysisusing the PROVEAN (−6.243), SIFT (0.0), PolyPhen-2(1.000), and Align-GVGD (Grantham variation [GV]: 0,Grantham deviation [GD]: 101.61, class: C65) scores pre-dicted this variant to be disease causing. PhyloP (+4.83) andPhastCons (0.99) scores showed this variant to be highlyconserved throughout different species (figure 1F).
Functional validation of the new I371K SYT2variant in DrosophilaTo examine how the I371K mutation affects synaptic trans-mission directly, we generated transgenicDrosophila expressingthe mutant gene using the GAL4-UAS expression system.Drosophila has a single homolog (DSYT1) of the mammaliansynaptic vesicle SYT subfamily that includes SYT1, SYT2, andSYT9. DSYT1 and human SYT2 share strong homology withconservation of the key residues that form the C2B calciumbinding pocket, including I371 (corresponding to I426 inDrosophila) and 2 previous C2B residues that were foundmutated in patients (D307 and P308, figure 2A). We generatedwildtype (WT) and I371K UAS-DSyt1 transgenic lines andexpressed the transgenes in syt1−/− null mutants with the pan-neuronal GAL4 driver elavC155.Drosophila Syt1−/− null mutantsshow a reduction in viability during larval development (50%survival to the 3rd instar stage) because of defective synaptictransmission. In contrast to the ability of WT SYT1 to rescuelethality (100% survival), SYT1 I426K not only failed to rescuebut also caused a dramatic reduction in viability (2.9%) com-pared to null mutants alone (50%) (figure 2B). Western blotanalysis indicated that the I426K SYT1 protein was expressed atsimilar levels to WT (figure 2C). Immunocytochemistrydemonstrated that the protein also localized normally at NMJsynapses (figure 2D), suggesting the dominant effects on via-bility are secondary to aberrant function of SYT1 I426K versusdegradation or abnormal localization. To analyze neurotrans-mitter release in Syt1 nulls that were rescued with eitherWT orI426K SYT1, we performed current recordings of postsynapticresponses in voltage-clamp at 3rd instar NMJs. As observed inthe behavioral viability assays, I426K rescued animals displayedsynaptic transmission defects that were worse than the Syt1 nullmutant. I426K synapses displayed severely defective synchro-nous neurotransmitter release (figure 2, E–G), a large increasein failure rate following stimulation (figure 2H) and enhance-ment of the slower asynchronous phase of release (figure 2I).I426K also failed to rescue the elevated rate of spontaneousfusion observed in Syt1 null mutants. These results indicate thatthe SYT1 I426K mutation eliminates the ability of the proteinto drive calcium-triggered neurotransmitter release and actsdominantly to reduce the residual release that is normally ob-served in the absence of SYT1.
To model the dominant phenotype observed in patients thathave one endogenous copy of WT SYT2, we characterized theeffects of overexpressing I426K on synaptic transmission in
the presence of endogenous DSYT1. Two independenttransgenic insertions (I426K#1 and 1426K#2) were overex-pressed using the elavC155 driver. Compared to over-expression of WT SYT1, both I426K lines resulted in a strongdominant-negative effect on action potential evoked release,decreasing the excitatory evoked junctional current (eEJC) by68.5% and 64.8%, respectively (figure 3, A and B). In addition,both I426K transgenic overexpression lines showed facilita-tion of the eEJC during 10 Hz stimulation compared to thesynaptic depression observed in controls (figure 3, C and D),consistent with a reduction in the initial release probabilitycaused by SYT1 I426K. These findings indicate that SYTI426K lacks normal function for promoting synaptic vesiclefusion and exerts a strong dominant-negative effect on syn-aptic transmission even in the presence of WT SYT1.
DiscussionGain-of-function mutations in SYT2 have been recently associ-ated with an autosomal dominant presynaptic congenital my-asthenic syndrome in 2 families. SYT2 is an integral membraneprotein of synaptic vesicles and serves as a calcium sensor forneurotransmitter release, with calcium binding to its C2B do-main activating vesicle fusion.11–13 Of interest, the clinical pre-sentation varied between families and family members, rangingfrom a presynaptic myasthenic disorder resembling Lambert-Eaton syndrome and a distal motor neuropathy, or evena combination of both. Here, we studied a multigenerationalfamily with a new SYT2mutation presenting with clear evidenceof both presynaptic neuromuscular transmission impairment aswell as a distal motor neuropathy, both clinically and electro-physiologically. This report therefore reinforces the link be-tween SYT2mutations and distal hereditary motor neuropathy.
Synaptotagmins 1 and 2 have been shown to have several majorroles in regulating synaptic vesicle fusion. Their major functionis as calcium sensors for driving fast synchronous fusion ofsynaptic vesicles at synapses. However, they also function tosuppress the slower asynchronous fusion pathway, ensuringthat release is tightly linked to the action potential. As such, inthe absence of synaptotagmin 1, increased asynchronous re-lease is observed because of loss of this suppression function.The I426K mutant disrupts the normal function of synapto-tagmin 1, including its role in driving fast synchronous fusion,as well as its role in suppressing asynchronous release.
Synaptotagmin 1 null mutants die throughout development,with many dying during the larval stage. However, whenplaced directly on food where little movement is required, theanimals can occasionally survive to adulthood, although theyshow severe motor defects. Similar to other species, includingmammals, synaptotagmin 1 mutants still have residual slowneurotransmitter release. Although the identity of the residual“asynchronous” calcium sensor is still being debated, recentstudies suggest that another member of the synaptotagminfamily—synaptotagmin 7—may play this role.14,15 Thisexplains the 50% survival observed in SYT null mutants.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
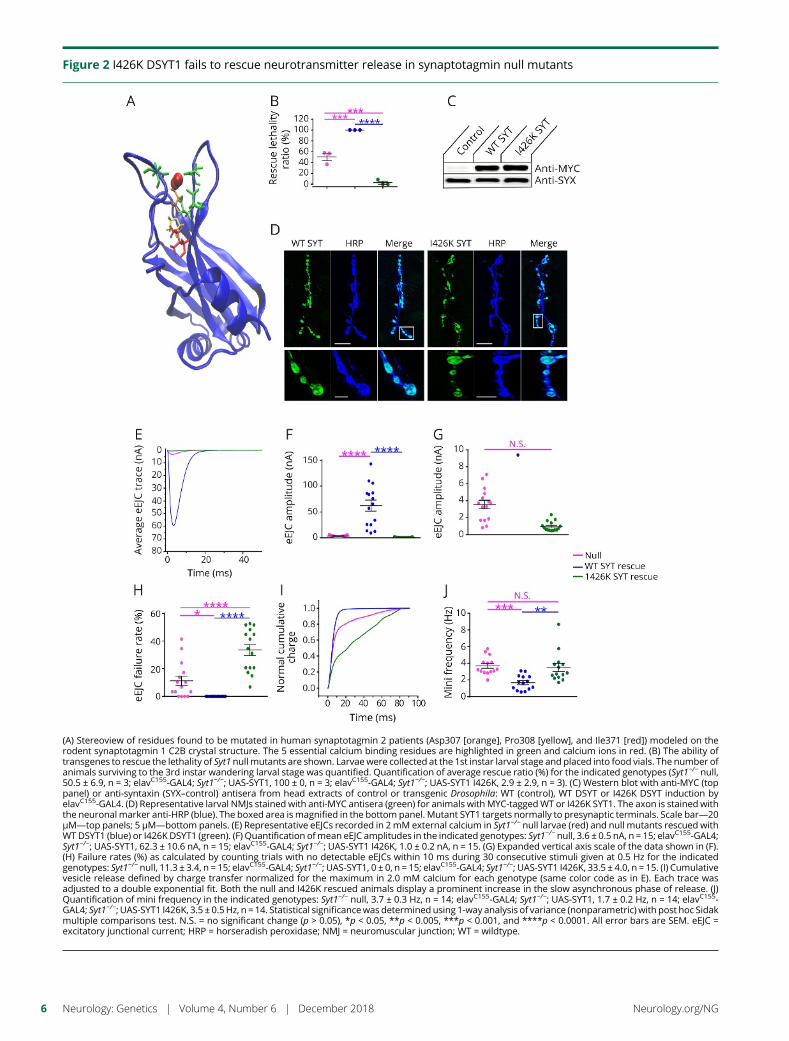
Figure 2 I426K DSYT1 fails to rescue neurotransmitter release in synaptotagmin null mutants
(A) Stereoview of residues found to be mutated in human synaptotagmin 2 patients (Asp307 [orange], Pro308 [yellow], and Ile371 [red]) modeled on therodent synaptotagmin 1 C2B crystal structure. The 5 essential calcium binding residues are highlighted in green and calcium ions in red. (B) The ability oftransgenes to rescue the lethality of Syt1 null mutants are shown. Larvae were collected at the 1st instar larval stage and placed into food vials. The number ofanimals surviving to the 3rd instar wandering larval stage was quantified. Quantification of average rescue ratio (%) for the indicated genotypes (Syt1−/− null,50.5 ± 6.9, n = 3; elavC155-GAL4; Syt1−/−; UAS-SYT1, 100 ± 0, n = 3; elavC155-GAL4; Syt1−/−; UAS-SYT1 I426K, 2.9 ± 2.9, n = 3). (C) Western blot with anti-MYC (toppanel) or anti-syntaxin (SYX–control) antisera from head extracts of control or transgenic Drosophila: WT (control), WT DSYT or I426K DSYT induction byelavC155-GAL4. (D) Representative larval NMJs stained with anti-MYC antisera (green) for animals with MYC-taggedWT or I426K SYT1. The axon is stained withthe neuronal marker anti-HRP (blue). The boxed area ismagnified in the bottom panel. Mutant SYT1 targets normally to presynaptic terminals. Scale bar—20μM—top panels; 5 μM—bottom panels. (E) Representative eEJCs recorded in 2mM external calcium in Syt1−/− null larvae (red) and null mutants rescued withWTDSYT1 (blue) or I426K DSYT1 (green). (F) Quantification ofmean eEJC amplitudes in the indicated genotypes: Syt1−/− null, 3.6 ± 0.5 nA, n = 15; elavC155-GAL4;Syt1−/−; UAS-SYT1, 62.3 ± 10.6 nA, n = 15; elavC155-GAL4; Syt1−/−; UAS-SYT1 I426K, 1.0 ± 0.2 nA, n = 15. (G) Expanded vertical axis scale of the data shown in (F).(H) Failure rates (%) as calculated by counting trials with no detectable eEJCs within 10 ms during 30 consecutive stimuli given at 0.5 Hz for the indicatedgenotypes: Syt1−/− null, 11.3 ± 3.4, n = 15; elavC155-GAL4; Syt1−/−; UAS-SYT1, 0 ± 0, n = 15; elavC155-GAL4; Syt1−/−; UAS-SYT1 I426K, 33.5 ± 4.0, n = 15. (I) Cumulativevesicle release defined by charge transfer normalized for the maximum in 2.0 mM calcium for each genotype (same color code as in E). Each trace wasadjusted to a double exponential fit. Both the null and I426K rescued animals display a prominent increase in the slow asynchronous phase of release. (J)Quantification of mini frequency in the indicated genotypes: Syt1−/− null, 3.7 ± 0.3 Hz, n = 14; elavC155-GAL4; Syt1−/−; UAS-SYT1, 1.7 ± 0.2 Hz, n = 14; elavC155-GAL4; Syt1−/−; UAS-SYT1 I426K, 3.5 ± 0.5Hz, n = 14. Statistical significancewas determined using 1-way analysis of variance (nonparametric) with post hoc Sidakmultiple comparisons test. N.S. = no significant change (p > 0.05), *p < 0.05, **p < 0.005, ***p < 0.001, and ****p < 0.0001. All error bars are SEM. eEJC =excitatory junctional current; HRP = horseradish peroxidase; NMJ = neuromuscular junction; WT = wildtype.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
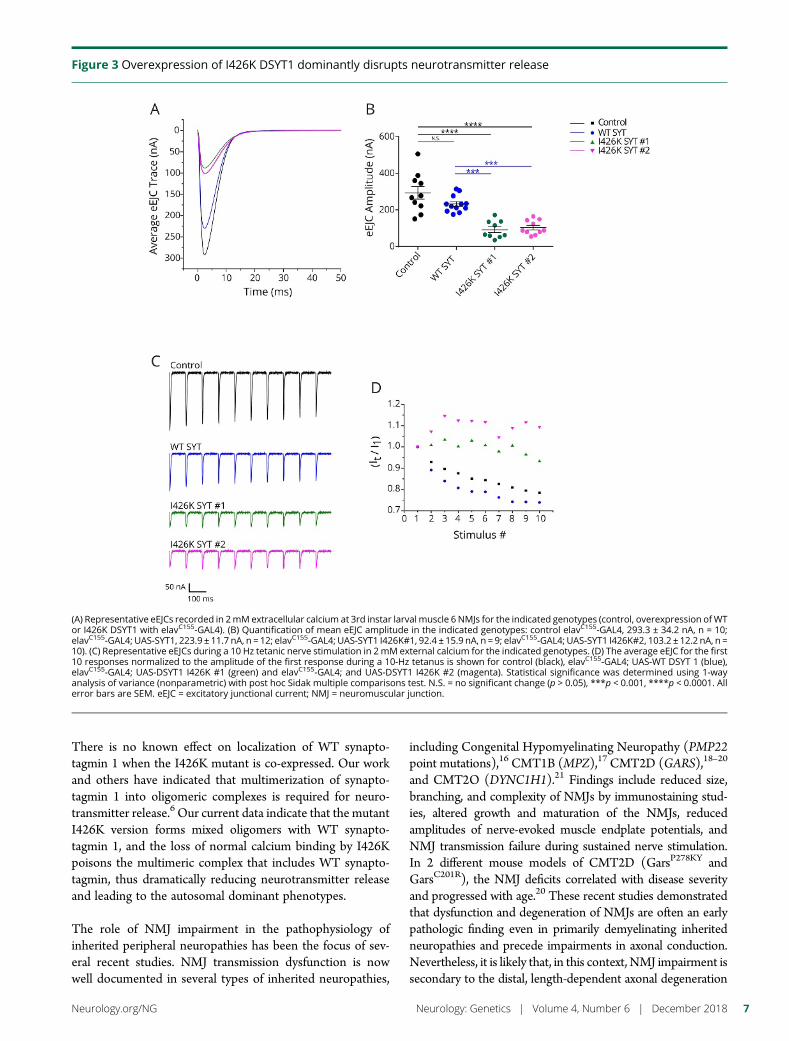
There is no known effect on localization of WT synapto-tagmin 1 when the I426K mutant is co-expressed. Our workand others have indicated that multimerization of synapto-tagmin 1 into oligomeric complexes is required for neuro-transmitter release.6 Our current data indicate that the mutantI426K version forms mixed oligomers with WT synapto-tagmin 1, and the loss of normal calcium binding by I426Kpoisons the multimeric complex that includes WT synapto-tagmin, thus dramatically reducing neurotransmitter releaseand leading to the autosomal dominant phenotypes.
The role of NMJ impairment in the pathophysiology ofinherited peripheral neuropathies has been the focus of sev-eral recent studies. NMJ transmission dysfunction is nowwell documented in several types of inherited neuropathies,
including Congenital Hypomyelinating Neuropathy (PMP22point mutations),16 CMT1B (MPZ),17 CMT2D (GARS),18–20
and CMT2O (DYNC1H1).21 Findings include reduced size,branching, and complexity of NMJs by immunostaining stud-ies, altered growth and maturation of the NMJs, reducedamplitudes of nerve-evoked muscle endplate potentials, andNMJ transmission failure during sustained nerve stimulation.In 2 different mouse models of CMT2D (GarsP278KY andGarsC201R), the NMJ deficits correlated with disease severityand progressed with age.20 These recent studies demonstratedthat dysfunction and degeneration of NMJs are often an earlypathologic finding even in primarily demyelinating inheritedneuropathies and precede impairments in axonal conduction.Nevertheless, it is likely that, in this context, NMJ impairment issecondary to the distal, length-dependent axonal degeneration
Figure 3 Overexpression of I426K DSYT1 dominantly disrupts neurotransmitter release
(A) Representative eEJCs recorded in 2mMextracellular calcium at 3rd instar larval muscle 6 NMJs for the indicated genotypes (control, overexpression ofWTor I426K DSYT1 with elavC155-GAL4). (B) Quantification of mean eEJC amplitude in the indicated genotypes: control elavC155-GAL4, 293.3 ± 34.2 nA, n = 10;elavC155-GAL4; UAS-SYT1, 223.9 ± 11.7 nA, n = 12; elavC155-GAL4; UAS-SYT1 I426K#1, 92.4 ± 15.9 nA, n = 9; elavC155-GAL4; UAS-SYT1 I426K#2, 103.2 ± 12.2 nA, n =10). (C) Representative eEJCs during a 10 Hz tetanic nerve stimulation in 2mM external calcium for the indicated genotypes. (D) The average eEJC for the first10 responses normalized to the amplitude of the first response during a 10-Hz tetanus is shown for control (black), elavC155-GAL4; UAS-WT DSYT 1 (blue),elavC155-GAL4; UAS-DSYT1 I426K #1 (green) and elavC155-GAL4; and UAS-DSYT1 I426K #2 (magenta). Statistical significance was determined using 1-wayanalysis of variance (nonparametric) with post hoc Sidak multiple comparisons test. N.S. = no significant change (p > 0.05), ***p < 0.001, ****p < 0.0001. Allerror bars are SEM. eEJC = excitatory junctional current; NMJ = neuromuscular junction.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

process characteristic of most types of inherited neuropathiesand not directly linked to specific presynaptic transmissiondysfunction. Of note, no electrophysiologic evidence of pre-synaptic NMJ transmission has been previously reported inpatients with CMT.
This concept was challenged recently by the identification ofmutations in the presynaptic choline transporter SLC5A7 asa cause of distal hereditary motor neuropathy type VII(dHMN VII).22 SLC5A7 is a Na+/Cl−-dependent high-affinity transporter that mediates uptake of choline for ace-tylcholine synthesis and therefore is a critical determinant ofsynaptic acetylcholine synthesis and release at the NMJ. Ofinterest, as is the case for SYT2 mutations, patients withdHMN VII do not present the hallmark clinical features ofa congenital myasthenic syndrome, namely, ophthalmopa-resis, ptosis, bulbar weakness, and respiratory fatigableweakness. Nonetheless, the role of these 2 presynaptic NMJproteins in the etiology of hereditary motor neuropathiesprovides a new framework to investigate the connectionsbetween NMJ transmission and axonal biology and function.
The clinical findings in our patients are in keeping with thephenotype described in 2 families with SYT2 mutations,3 in-cluding foot deformities since childhood, distal weakness,minimal sensory findings, and reduced deep tendon reflexeswith evidence of postexercise facilitation. The electrodiagnosticfeatures were also in agreement with the previous report,demonstrating clear presynaptic neuromuscular transmissiondysfunction characterized by decremental response to RNS andsignificant postexercise facilitation and normal sensory respon-ses. We identified a new SYT2 mutation causing autosomaldominant distal hereditary motor neuropathy, confirming theinteresting connection between presynaptic neuromusculartransmission dysfunction and motor axonopathy.
Study fundingNo targeted funding reported.
DisclosureN.I. Montes-Chinea, Z. Guan, M. Coutts, C. Vidal, S. Courel,A.P. Rebelo, L. Abreu, and S. Zuchner report no disclosures. J.T. Littleton has received research support from NIH. M.A.Saporta has served on scientific advisory boards for theCharcot-Marie-Tooth Association and Acceleron; serves onthe editorial board of the Journal of the Peripheral NervousSystem; has been a consultant for Alnylam, Strongbridge,Biogen, and Serepta; and has received research support from
the Charcot-Marie-Tooth Association. Full disclosure forminformation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Received June 11, 2018. Accepted in final form August 21, 2018.
References1. Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neu-
ropathies. J Neurol Neurosurg Psychiatry 2012;83:6–14.2. Bansagi B, Griffin H, Whittaker RG, et al. Genetic heterogeneity of motor neuropa-
thies. Neurology 2017;88:1226–1234.3. Herrmann DN, Horvath R, Sowden JE, et al. Synaptotagmin 2 mutations cause an
autosomal-dominant form of lambert-eaton myasthenic syndrome and non-progressive motor neuropathy. Am J Hum Genet 2014;95:332–339.
4. Whittaker RG, Herrmann DN, Bansagi B, et al. Electrophysiologic features of SYT2mutations causing a treatable neuromuscular syndrome. Neurology 2015;85:1964–1971.
5. Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neu-ropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst2011;16:191–198.
6. Guan Z, Bykhovskaia M, Jorquera RA, Sutton RB, Akbergenova Y, Littleton JT. Asynaptotagmin suppressor screen indicates SNARE binding controls the timing andCa2+ cooperativity of vesicle fusion. Elife 2017;6:e28409.
7. Littleton JT, Stern M, Perin M, Bellen HJ. Calcium dependence of neurotransmitterrelease and rate of spontaneous vesicle fusions are altered in Drosophila synapto-tagmin mutants. Proc Natl Acad Sci U S A 1994;91:10888–10892.
8. DiAntonio A, Schwarz TL. The effect on synaptic physiology of synaptotagminmutations in Drosophila. Neuron 1994;12:909–920.
9. Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neuro-transmitter release and synaptic growth. Nat Neurosci 2007;10:1235–1237.
10. Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. Com-plexin controls spontaneous and evoked neurotransmitter release by regulating thetiming and properties of synaptotagmin activity. J Neurosci 2012;32:18234–18245.
11. Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ. Mutational analysis of Dro-sophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neuro-transmitter release. Cell 1993;74:1125–1134.
12. Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)BCa(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo.Nature 2002;418:340–344.
13. Pang ZP, Melicoff E, Padgett D, et al. Synaptotagmin-2 is essential for survival andcontributes to Ca2+ triggering of neurotransmitter release in central and neuro-muscular synapses. J Neurosci 2006;26:13493–13504.
14. Luo F, Sudhof TC. Synaptotagmin-7-Mediated asynchronous release Boosts high-fidelity synchronous transmission at a central synapse. Neuron 2017;94:826–839.e3.
15. Turecek J, Regehr WG. Synaptotagmin 7 mediates both facilitation and asynchronousrelease at granule cell synapses. J Neurosci 2018;38:3240–3251.
16. Scurry AN, Heredia DJ, Feng CY, Gephart GB, Hennig GW, Gould TW. Structuraland functional abnormalities of the neuromuscular junction in the trembler-J ho-mozygote mouse model of congenital hypomyelinating neuropathy. J NeuropatholExp Neurol 2016;75:334–346.
17. Patzko A, Bai Y, Saporta MA, et al. Curcumin derivatives promote Schwann celldifferentiation and improve neuropathy in R98C CMT1B mice. Brain 2012;135:3551–3566.
18. Sleigh JN, Grice SJ, Burgess RW, Talbot K, Cader MZ. Neuromuscular junctionmaturation defects precede impaired lower motor neuron connectivity in Charcot-Marie-Tooth type 2D mice. Hum Mol Genet 2014;23:2639–2650.
19. Grice SJ, Sleigh JN, Motley WW, et al. Dominant, toxic gain-of-function mutations ingars lead to non-cell autonomous neuropathology. Hum Mol Genet 2015;24:4397–4406.
20. Spaulding EL, Sleigh JN, Morelli KH, Pinter MJ, Burgess RW, Seburn KL. Synapticdeficits at neuromuscular junctions in two mouse models of Charcot-Marie-Toothtype 2d. J Neurosci 2016;36:3254–3267.
21. Sabblah TT, Nandini S, Ledray AP, et al. A novel mouse model carrying a humancytoplasmic dynein mutation shows motor behavior deficits consistent with Charcot-Marie-Tooth type 2O disease. Sci Rep 2018;8:1739.
22. Barwick KES, Wright J, Al-Turki S, et al. Defective presynaptic choline transportunderlies hereditary motor neuropathy. Am J Hum Genet 2012;91:1103–1107.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
Appendix 1 Author contributions
Name Location Role Contribution
Nataly I. Montes-Chinea, MD University of Miami, Miami Author Major role in the acquisition of data design andconceptualized study; analyzed the data; draftedthe manuscript for intellectual content
Zhuo Guan, PhD Massachusetts Institute ofTechnology
Author Major role in the acquisition of data
Marcella Coutts, MD University of Miami, Miami Author Major role in the acquisition of data
Cecilia Vidal, MD University of Miami, Miami Author Major role in the acquisition of data
Steve Courel University of Miami, Miami Author Major role in the acquisition of data; analyzedthe data
Adriana P. Rebelo, PhD University of Miami, Miami Author Major role in the acquisition of data; analyzedthe data; drafted the manuscript forintellectual content
Lisa Abreu University of Miami, Miami Author Major role in the acquisition of data
Stephan Zuchner University of Miami, Miami Author Drafted the manuscript for intellectual content
J.Troy Littleton Massachusetts Institute ofTechnology
Author Design and conceptualized study; analyzed thedata; drafted the manuscript for intellectualcontent
Mario A. Saporta, MD, PhD,FAAN
University of Miami, Miami Correspondingauthor
Major role in the acquisition of data, design andconceptualized study; analyzed the data; draftedthe manuscript for intellectual content
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

ARTICLE OPEN ACCESS
TPP2 mutation associated with sterile braininflammation mimicking MSEva M. Reinthaler, PhD, Elisabeth Graf, PhD, Tobias Zrzavy, MD, Thomas Wieland, PhD, Christoph Hotzy, BSc,
Chantal Kopecky, PhD, Sandra Pferschy, Christiane Schmied, MD, Fritz Leutmezer, MD,
Mohammad Keilani, MD, Christina M. Lill, PhD, Sabine Hoffjan, MD, Jorg T. Epplen, MD, Uwe K. Zettl, MD,
Michael Hecker, Angela Deutschlander, MD, Sven G. Meuth, PhD, Mamoun Ahram, MD, Baha Mustafa, PhD,
Mohammed El-Khateeb, PhD, Carles Vilariño-Guell, PhD, A. Dessa Sadovnick, MD, Fritz Zimprich, MD, PhD,
Birgitta Tomkinson, PhD, Tim Strom, PhD, Wolfgang Kristoferitsch, PhD, Hans Lassmann, PhD,
and Alexander Zimprich, PhD
Neurol Genet 2018;4:e285. doi:10.1212/NXG.0000000000000285
Correspondence
Dr. A. Zimprich
alexander.zimprich@
meduniwien.ac.at
AbstractObjectiveTo ascertain the genetic cause of a consanguineous family from Syria suffering from a sterilebrain inflammation mimicking a mild nonprogressive form of MS.
MethodsWe used homozygosity mapping and next-generation sequencing to detect the disease-causinggene in the affected siblings. In addition, we performed RNA and protein expression studies,enzymatic activity assays, immunohistochemistry, and targeted sequencing of further MS casesfrom Austria, Germany, Canada and Jordan.
ResultsIn this study, we describe the identification of a homozygous missense mutation (c.82T>G,p.Cys28Gly) in the tripeptidyl peptidase II (TPP2) gene in all 3 affected siblings of the family.Sequencing of all TPP2-coding exons in 826 MS cases identified one further homozygousmissense variant (c.2027C>T, p.Thr676Ile) in a Jordanian MS patient. TPP2 protein expres-sion in whole blood was reduced in the affected siblings. In contrast, TPP2 protein expression inpostmortem brain tissue from MS patients without TPP2 mutations was highly upregulated.
ConclusionsThe homozygous TPP2 mutation (p.Cys28Gly) is likely responsible for the inflammation pheno-type in this family.TPP2 is an ubiquitously expressed serine peptidase that removes tripeptides fromtheN-terminal end of longer peptides. TPP2 is involved in various biological processes including thedestruction of major histocompatibility complex Class I epitopes. Recessive loss-of-functionmutations in TPP2 were described in patients with Evans syndrome, a rare autoimmune diseaseaffecting the hematopoietic system. Based on the gene expression results in our MS autopsy brainsamples, we further suggest that TPP2may play a broader role in the inflammatory process in MS.
From the Department of Neurology (E.M.R., S.P., C.S., F.L., F.Z., A.Z.), Medical University of Vienna, Austria; Institut fur Humangenetik (E.G., T.W., T.S.), Helmholtz ZentrumMunchen,Germany; Center for Brain Research (T.Z., H.L.), Medical University of Vienna; Division of Nephrology and Dialysis (C.K.), Department of Internal Medicine III, Medical University ofVienna; Department of Physical Medicine (M.K.), Rehabilitation and Occupational Medicine, Medical University of Vienna, Austria; Lubeck Interdisciplinary Platform for GenomeAnalytics (C.M.L.), Institutes of Neurogenetics and for Cardiogenetics, University of Lubeck; Department of Neurology and Neuroimaging Center (NIC) (C.M.L.), Focus ProgramTranslational Neuroscience (FTN), University Medical Center of the Johannes Gutenberg University Mainz; Department of Human Genetics (S.H., J.T.E.), Ruhr-University Bochum;Herdecke (J.T.E.), ZBAF, Faculty of Health, University Witten; Department of Neurology (U.K.Z., M.H.), Neuroimmunological Section, University of Rostock; Department of Neurology(A.D.), Department of Clinical Genomics (A.D.), Department of Neuroscience (A.D.), Jeweils Mayo Clinic, Jacksonville, FL; Department of Neurology (S.G.M.), University of Muenster,Germany; Department of Physiology and Biochemistry (M.A., B.M.), School of Medicine, the University of Jordan; The National Center (Institute) for Diabetes (M.E.-K.), EndocrinologyandGenetics (NCDEG), Amman, Jordan; Department of Medical Genetics (C.V.-G., A.D.S.), University of British Columbia, Vancouver, Canada; Department of Medical Biochemistry andMicrobiology (B.T.), Uppsala University, Sweden; Karl Landsteiner Institute for Neuroimmunological and Neurodegenerative Disorders (W.K.), SMZ-Ost-Donauspital, Vienna, Austria;and Institute for Neuroimmunological and Neurodegenerative Disorders (W.K.), SMZ-Ost-Donauspital, Vienna, Austria.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Evans syndrome (ES) is a rare autoimmune disorder, whichis defined by a combination of direct Coombs test positivehemolytic anemia and immune thrombocytopenia.1 Al-though most cases have no obvious underlying etiology,rare monogenic forms have been identified in the last years.2
ES can affect different organs, manifesting with hepatomegaly,splenomegaly,3–5 and lymphocytic infiltration of nonlymphoidorgans, including the brain.6,7 A case report from 2013 describeda patient who had ES in addition to a sterile brain in-flammation, which, from a clinical perspective, was in-distinguishable from MS.8
Recessive loss-of-function (LoF) mutations in the tripeptidylpeptidase II (TPP2) gene were found to cause a specific formof ES in patients, manifesting with neurodevelopmentaldelay and impaired glycolysis.9,10 TPP2 is a cytosolic protease;its main activity is the removal of tripeptides from theN-terminus of longer peptides, to generate free aminoacids for protein synthesis and energy production.11 ThisTPP2-linked phenotype was proposed to be designated as“TPP2-related immunodeficiency, autoimmunity and neuro-developmental delay with impaired glycolysis and lysosomalexpansion” (TRIANGLE) disease.9 Here, we report ona family with 3 affected siblings initially diagnosed with a mildand nonprogressive form of MS. We identified a homozygousmissense mutation in TPP2 as a likely cause for the disease inthis family.
MethodsStudy participantsWe clinically evaluated a consanguineous family from Syriawith 3 siblings diagnosed with MS and a suggestive pattern ofautosomal recessive inheritance. In brief, the disease courseobserved in the siblings is consistent with a benign relapsing-remitting MS even 27 (II.1) and 24 (II.2) years after the onsetof clinical symptoms. The female sibling (II.3) had a singleneurologic episode of demyelination and no further clinicalrelapses over the following 14 years. The Expanded DisabilityStatus Scale was not more than 1.5 in any of the siblings. All ofthem reported to have had frequent infections of the upperrespiratory tract during their childhood and adulthood. All 3siblings showed low but normal lymphocyte count between 1and 1.3 × 103/μL (normal range is between 1.0 and 4 × 103/μL) as tested several times between 2008 and 2017. However,they have never experienced other clinical signs typical for ES,such as thrombocytopenia or hemolytic anemia. A detailedclinical description in form of a timetable and magnetic
resonance tomography images are provided in figure e-1(links.lww.com/NXG/A108), figure e-2 (links.lww.com/NXG/A109; legends links.lww.com/NXG/A130), and in the supple-mentary information (links.lww.com/NXG/A110).
For the targeted sequence analysis, we ascertained 382 MSpatients from Europe (Austria and Germany) and 183 MScases from Jordan. In addition, we surveyed exome data from261 MS patients from Canada for mutations in the TPP2gene, which were collected through the longitudinal CanadianCollaborative Project on the Genetic Susceptibility to MS12
(table 1). All patients were diagnosed with MS according topublished criteria.13–15
Standard protocol approvals, registrations,and patient consentsWritten informed consent was obtained from all study par-ticipants; the study was approved by the local ethics com-mittee (EK.535/2004/20179 for the neuropathologic partand EK Nr:2195/2016 for MS patients).
Homozygositymapping and exome sequencingHomozygous regions shared between all 3 siblings weremapped using Affymetrix GenomeWideSNP 6 data from theaffected siblings and their healthy parents with the onlineHomozygosity Mapper tool (homozygositymapper.org).
Whole exome data were generated from individuals II.1 andII.2. Exomes were enriched with SureSelect Human All Exon50 Mb kit (AgilentTechnologies, Santa Clara, CA). Sequencingof postenrichment libraries was carried out on the IlluminaHiSeq 2000 sequencing instrument (Illumina, San Diego, CA)as 2 × 100 bp paired-end runs. Variants were filtered for ho-mozygosity and aminor allele frequency (MAF) smaller than 2%in our in-house data set of approximately 10,000 control exomesfrom patients with other unrelated diseases and exomes and inpublic available databases (exome aggregation consoritum[ExAC] database and 1000 Genomes).
Exome sequencing of the Canadian patients was done on anIon Proton sequencer (Life Technologies, Carlsbad, CA).Exome data were analyzed as previously described.16
Targeted capture sequencing, single-nucleotide polymorphism genotyping, and insilico predictionIllumina TruSeq Custom Amplicon Kit was used to target allexonic and flanking intronic regions of TPP2 and excisionrepair cross-complementation group 5 (ERCC5) in 382
GlossaryERCC5 = excision repair cross-complementation group 5; ExAC = exome aggregation consoritum; LoF = loss of function;MAF =minor allele frequency;MHC =major histocompatibility complex;NAWM = normal-appearing white matter; PBMC =peripheral blood mononuclear cell; TPP2 = tripeptidyl peptidase II; TRIANGLE = TPP2-related immunodeficiency,autoimmunity and neurodevelopmental delay with impaired glycolysis and lysosomal expansion.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

European and 183 Jordanian MS patients. Fast-QC fileswere subsequently analyzed with an in-house pipeline. Allidentified variants were subsequently validated with Sangersequencing. Homozygous variants in TPP2 (p.Cys28Gly,p.Thr676Ile) and ERCC5 (p.S1078A) were genotyped withcustom TaqMan single-nucleotide polymorphism geno-typing assays.
TPP2 protein expression in MS and controlbrain tissueExpression of TPP2 was assessed in formaldehyde-fixed andparaffin-embedded autopsy tissues from 13 MS patients and16 controls archived at the Center for Brain Research of theMedical University of Vienna (detailed clinical characteristicsin supplementary information, links.lww.com/NXG/A110).Expression of TPP2 was analyzed in the normal-appearingwhite matter (NAWM); initial, early active, late active, andinactive lesion sites in the white matter of MS patients; and inthe normal white matter of controls. Lesion stages were de-fined as described in detail before.17
TPP2 RNA Western blot analysisUsing a TaqMan gene expression assay, TPP2 whole bloodmRNA expression was quantitatively assessed for all individualsof the index family, 7 randomly chosen individuals affected withMS and 3 healthy controls (GAPDH, Hs03929097_g1). Im-munoblotting was performed with peripheral blood mono-nuclear cell (PBMC) lysates from the index patient (II.1), hisaffected brother (II.2), and 3 control individuals. Primaryantibodies for TPP2 (1:500, 14981S; Cell Signaling) and en-dogenous control beta-actin were used.
TPP2 enzymatic activityBlood samples from index patient II.1 and 2 healthy controlswere prepared according to the partial lysis method (wch.sa.gov.au/services/az/divisions/labs/geneticmed/nrl_methods.html).Enzymatic activity was measured as described previously.18
ResultsIdentificationof ahomozygousmutation in theTPP2 gene as a likely cause for a sterile braininflammation in a consanguineousSyrian familyTo identify the responsible gene of a benign brain in-flammation, mimicking a nonprogressive form of MS, ina consanguineous family with 3 affected siblings, we per-formed homozygosity mapping and whole exome sequencing.After filtering variants for allele frequency (MAF < 2%) andhomozygosity, only 2 novel autosomal homozygous missensevariants were left to be shared by all siblings. We identifieda c.82T>G, p.Cys28Gly variation in the TPP2 gene, a serinepeptidase that removes tripeptides from the N-terminus oflonger peptides and a c.3232T>G, p.Ser1078Ala variation inthe ERCC5 gene, known to be involved in DNA repair. These2 variants were confirmed in heterozygous state in the healthyparents. None of the 2 variants is present in any publiclyavailable databases. The 2 genes are in close vicinity, locatedon chromosome 13q33.1 and map to the only homozygousregion shared by all 3 affected siblings (figure 1, A–C).
Identification of a second rare homozygousvariant in the TPP2 gene in a JordanianMS patientAs the phenotype of the family was almost indistinguishablefrom a benign form of MS, we wondered whether otherpatients diagnosed with MS might carry biallelic variants ineither of the 2 genes. Sequencing of both genes in additional382 unrelated MS cases from Europe and 183 MS patientsfrom Jordan identified one further homozygous variation inTPP2 (c.2027C>G, p.Thr676Ile, rs760347832), in a Jorda-nian patient (table 2). This variant was also present in het-erozygous state in 2 additional MS patients from Jordan(figure 1D, clinical details in supplementary information,links.lww.com/NXG/A110). We did not find any rare
Table 1 Additional MS cases and controls for targeted sequence and genotyping analysis
Site Cases Male Female AaoPositive family history/recessivea
Targeted sequence analysis (TruSeq) of the TPP2 and ERCC5 gene in MS cases
Austria 352 150 202 29 56/9
Germany 30 10 20 30 20/20
Jordan 183 63 120 29 14/8
Canada 261 78 183 32 173/18
Targetedmutation screening of TPP2, Cys28Gly; and TPP2, Thr676Ile in Jordaniancases and controls
Jordan MS cases 233 82 151 29 10/3
Jordan controls 452 289 163 40
Abbreviations: Aao = age at onset; ERCC5 = excision repair cross-complementation group 5; TPP2 = tripeptidyl peptidase II.a Recessive: the number of cases who have a recessive mode of inheritance, which is here defined by at least one other affected sibling with both parentsbeing healthy or with no second-degree relative affected.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
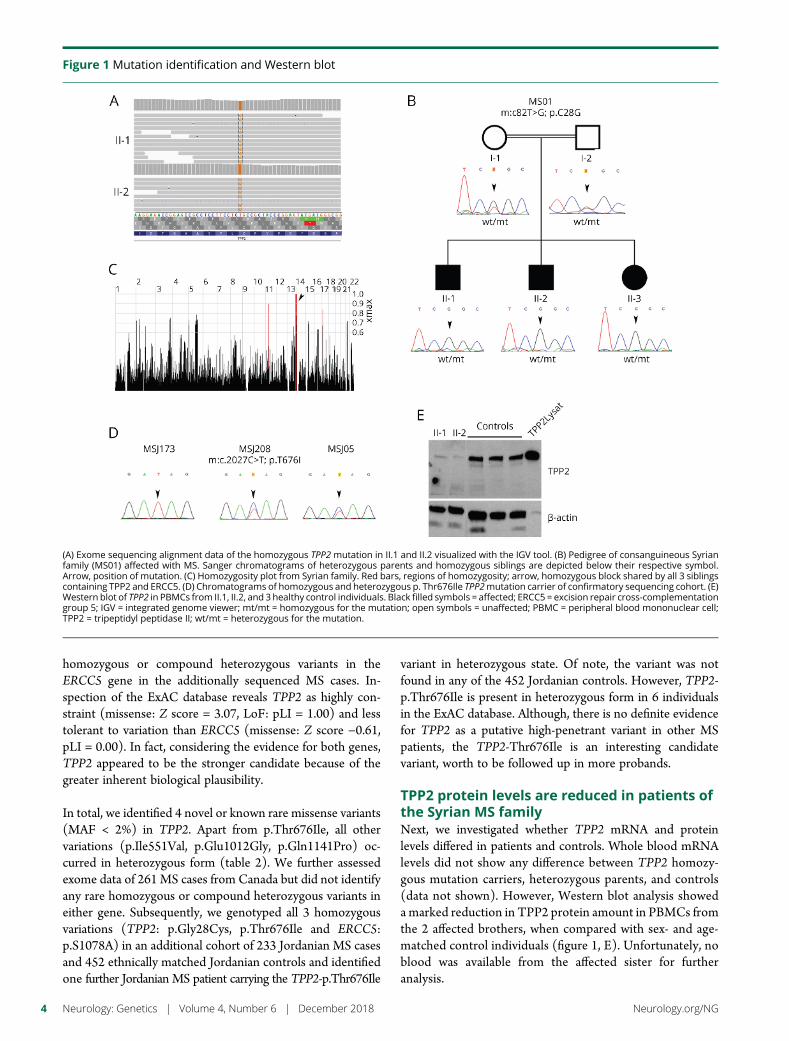
homozygous or compound heterozygous variants in theERCC5 gene in the additionally sequenced MS cases. In-spection of the ExAC database reveals TPP2 as highly con-straint (missense: Z score = 3.07, LoF: pLI = 1.00) and lesstolerant to variation than ERCC5 (missense: Z score −0.61,pLI = 0.00). In fact, considering the evidence for both genes,TPP2 appeared to be the stronger candidate because of thegreater inherent biological plausibility.
In total, we identified 4 novel or known rare missense variants(MAF < 2%) in TPP2. Apart from p.Thr676Ile, all othervariations (p.Ile551Val, p.Glu1012Gly, p.Gln1141Pro) oc-curred in heterozygous form (table 2). We further assessedexome data of 261 MS cases from Canada but did not identifyany rare homozygous or compound heterozygous variants ineither gene. Subsequently, we genotyped all 3 homozygousvariations (TPP2: p.Gly28Cys, p.Thr676Ile and ERCC5:p.S1078A) in an additional cohort of 233 Jordanian MS casesand 452 ethnically matched Jordanian controls and identifiedone further Jordanian MS patient carrying the TPP2-p.Thr676Ile
variant in heterozygous state. Of note, the variant was notfound in any of the 452 Jordanian controls. However, TPP2-p.Thr676Ile is present in heterozygous form in 6 individualsin the ExAC database. Although, there is no definite evidencefor TPP2 as a putative high-penetrant variant in other MSpatients, the TPP2-Thr676Ile is an interesting candidatevariant, worth to be followed up in more probands.
TPP2 protein levels are reduced in patients ofthe Syrian MS familyNext, we investigated whether TPP2 mRNA and proteinlevels differed in patients and controls. Whole blood mRNAlevels did not show any difference between TPP2 homozy-gous mutation carriers, heterozygous parents, and controls(data not shown). However, Western blot analysis showeda marked reduction in TPP2 protein amount in PBMCs fromthe 2 affected brothers, when compared with sex- and age-matched control individuals (figure 1, E). Unfortunately, noblood was available from the affected sister for furtheranalysis.
Figure 1 Mutation identification and Western blot
(A) Exome sequencing alignment data of the homozygous TPP2 mutation in II.1 and II.2 visualized with the IGV tool. (B) Pedigree of consanguineous Syrianfamily (MS01) affected with MS. Sanger chromatograms of heterozygous parents and homozygous siblings are depicted below their respective symbol.Arrow, position of mutation. (C) Homozygosity plot from Syrian family. Red bars, regions of homozygosity; arrow, homozygous block shared by all 3 siblingscontaining TPP2 and ERCC5. (D) Chromatograms of homozygous and heterozygous p. Thr676Ile TPP2mutation carrier of confirmatory sequencing cohort. (E)Western blot of TPP2 in PBMCs from II.1, II.2, and 3 healthy control individuals. Black filled symbols = affected; ERCC5 = excision repair cross-complementationgroup 5; IGV = integrated genome viewer; mt/mt = homozygous for the mutation; open symbols = unaffected; PBMC = peripheral blood mononuclear cell;TPP2 = tripeptidyl peptidase II; wt/mt = heterozygous for the mutation.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
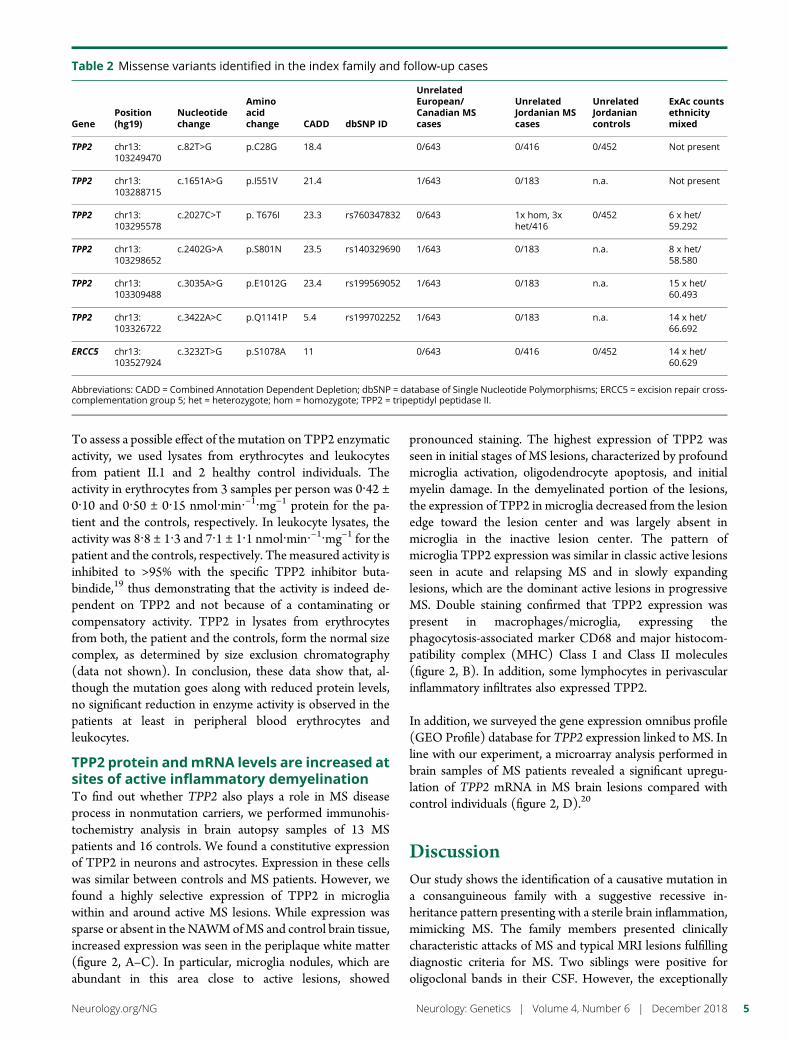
To assess a possible effect of the mutation on TPP2 enzymaticactivity, we used lysates from erythrocytes and leukocytesfrom patient II.1 and 2 healthy control individuals. Theactivity in erythrocytes from 3 samples per person was 0·42 ±0·10 and 0·50 ± 0·15 nmol·min·−1·mg−1 protein for the pa-tient and the controls, respectively. In leukocyte lysates, theactivity was 8·8 ± 1·3 and 7·1 ± 1·1 nmol·min·−1·mg−1 for thepatient and the controls, respectively. Themeasured activity isinhibited to >95% with the specific TPP2 inhibitor buta-bindide,19 thus demonstrating that the activity is indeed de-pendent on TPP2 and not because of a contaminating orcompensatory activity. TPP2 in lysates from erythrocytesfrom both, the patient and the controls, form the normal sizecomplex, as determined by size exclusion chromatography(data not shown). In conclusion, these data show that, al-though the mutation goes along with reduced protein levels,no significant reduction in enzyme activity is observed in thepatients at least in peripheral blood erythrocytes andleukocytes.
TPP2 protein andmRNA levels are increased atsites of active inflammatory demyelinationTo find out whether TPP2 also plays a role in MS diseaseprocess in nonmutation carriers, we performed immunohis-tochemistry analysis in brain autopsy samples of 13 MSpatients and 16 controls. We found a constitutive expressionof TPP2 in neurons and astrocytes. Expression in these cellswas similar between controls and MS patients. However, wefound a highly selective expression of TPP2 in microgliawithin and around active MS lesions. While expression wassparse or absent in the NAWMofMS and control brain tissue,increased expression was seen in the periplaque white matter(figure 2, A–C). In particular, microglia nodules, which areabundant in this area close to active lesions, showed
pronounced staining. The highest expression of TPP2 wasseen in initial stages of MS lesions, characterized by profoundmicroglia activation, oligodendrocyte apoptosis, and initialmyelin damage. In the demyelinated portion of the lesions,the expression of TPP2 in microglia decreased from the lesionedge toward the lesion center and was largely absent inmicroglia in the inactive lesion center. The pattern ofmicroglia TPP2 expression was similar in classic active lesionsseen in acute and relapsing MS and in slowly expandinglesions, which are the dominant active lesions in progressiveMS. Double staining confirmed that TPP2 expression waspresent in macrophages/microglia, expressing thephagocytosis-associated marker CD68 and major histocom-patibility complex (MHC) Class I and Class II molecules(figure 2, B). In addition, some lymphocytes in perivascularinflammatory infiltrates also expressed TPP2.
In addition, we surveyed the gene expression omnibus profile(GEO Profile) database for TPP2 expression linked to MS. Inline with our experiment, a microarray analysis performed inbrain samples of MS patients revealed a significant upregu-lation of TPP2 mRNA in MS brain lesions compared withcontrol individuals (figure 2, D).20
DiscussionOur study shows the identification of a causative mutation ina consanguineous family with a suggestive recessive in-heritance pattern presenting with a sterile brain inflammation,mimicking MS. The family members presented clinicallycharacteristic attacks of MS and typical MRI lesions fulfillingdiagnostic criteria for MS. Two siblings were positive foroligoclonal bands in their CSF. However, the exceptionally
Table 2 Missense variants identified in the index family and follow-up cases
GenePosition(hg19)
Nucleotidechange
Aminoacidchange CADD dbSNP ID
UnrelatedEuropean/Canadian MScases
UnrelatedJordanian MScases
UnrelatedJordaniancontrols
ExAc countsethnicitymixed
TPP2 chr13:103249470
c.82T>G p.C28G 18.4 0/643 0/416 0/452 Not present
TPP2 chr13:103288715
c.1651A>G p.I551V 21.4 1/643 0/183 n.a. Not present
TPP2 chr13:103295578
c.2027C>T p. T676I 23.3 rs760347832 0/643 1x hom, 3xhet/416
0/452 6 x het/59.292
TPP2 chr13:103298652
c.2402G>A p.S801N 23.5 rs140329690 1/643 0/183 n.a. 8 x het/58.580
TPP2 chr13:103309488
c.3035A>G p.E1012G 23.4 rs199569052 1/643 0/183 n.a. 15 x het/60.493
TPP2 chr13:103326722
c.3422A>C p.Q1141P 5.4 rs199702252 1/643 0/183 n.a. 14 x het/66.692
ERCC5 chr13:103527924
c.3232T>G p.S1078A 11 0/643 0/416 0/452 14 x het/60.629
Abbreviations: CADD = Combined Annotation Dependent Depletion; dbSNP = database of Single Nucleotide Polymorphisms; ERCC5 = excision repair cross-complementation group 5; het = heterozygote; hom = homozygote; TPP2 = tripeptidyl peptidase II.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
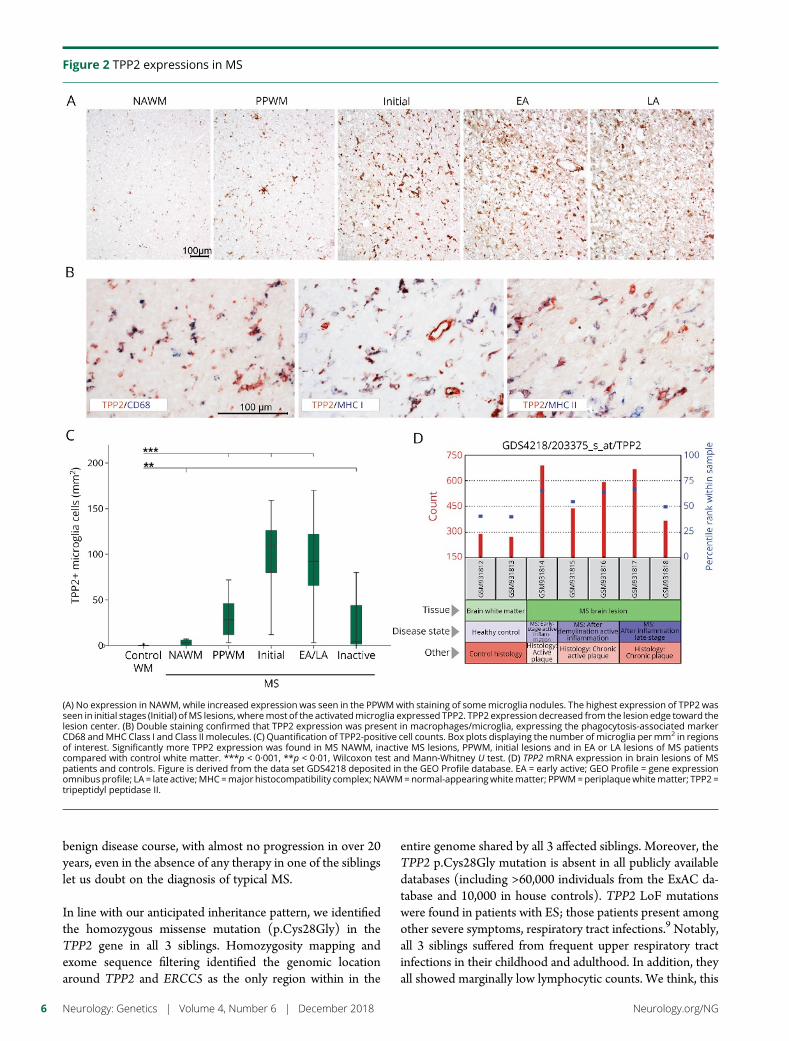
benign disease course, with almost no progression in over 20years, even in the absence of any therapy in one of the siblingslet us doubt on the diagnosis of typical MS.
In line with our anticipated inheritance pattern, we identifiedthe homozygous missense mutation (p.Cys28Gly) in theTPP2 gene in all 3 siblings. Homozygosity mapping andexome sequence filtering identified the genomic locationaround TPP2 and ERCC5 as the only region within in the
entire genome shared by all 3 affected siblings. Moreover, theTPP2 p.Cys28Gly mutation is absent in all publicly availabledatabases (including >60,000 individuals from the ExAC da-tabase and 10,000 in house controls). TPP2 LoF mutationswere found in patients with ES; those patients present amongother severe symptoms, respiratory tract infections.9 Notably,all 3 siblings suffered from frequent upper respiratory tractinfections in their childhood and adulthood. In addition, theyall showed marginally low lymphocytic counts. We think, this
Figure 2 TPP2 expressions in MS
(A) No expression in NAWM, while increased expression was seen in the PPWMwith staining of somemicroglia nodules. The highest expression of TPP2 wasseen in initial stages (Initial) ofMS lesions, wheremost of the activatedmicroglia expressed TPP2. TPP2 expression decreased from the lesion edge toward thelesion center. (B) Double staining confirmed that TPP2 expression was present in macrophages/microglia, expressing the phagocytosis-associated markerCD68 andMHC Class I and Class II molecules. (C) Quantification of TPP2-positive cell counts. Box plots displaying the number of microglia permm2 in regionsof interest. Significantly more TPP2 expression was found in MS NAWM, inactive MS lesions, PPWM, initial lesions and in EA or LA lesions of MS patientscompared with control white matter. ***p < 0·001, **p < 0·01, Wilcoxon test and Mann-Whitney U test. (D) TPP2 mRNA expression in brain lesions of MSpatients and controls. Figure is derived from the data set GDS4218 deposited in the GEO Profile database. EA = early active; GEO Profile = gene expressionomnibus profile; LA = late active;MHC=major histocompatibility complex; NAWM=normal-appearingwhitematter; PPWM=periplaquewhitematter; TPP2 =tripeptidyl peptidase II.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

possibly reflects subthreshold signs for ES and supports thatthe TPP2 variant is phenotypically effective.
As we assume recessively inherited TPP2 variations to belinked with the disease, we were interested in how frequentlycontrol individuals carry biallelic rare variants. We surveyedour in-house exome database, including more than 10,000control individuals for TPP2 variants. Notably, no single in-dividual was found to carry 2 rare TPP2 variants (MAF < 2%).These data indicate that rare variants in the TPP2 gene, par-ticularly when occurring on both alleles, are not well toleratedin healthy individuals.
Because of the close phenotypic resemblance of our family toMS, we wondered whether recessive TPP2 mutations mightalso be found in typical MS cases. We sequenced a large MScohort fromAustria, Germany, Canada, and Jordan and identified1 MS case from Jordan, with a homozygous TPP2 variant,p.Thr676Ile. However, as this variant also occurs in heterozygousform in other MS cases and in 6 ExAC control individuals, itremains questionable whether this variant is pathogenic.
It was found that TPP2 has a major role in maintaining thecellular homeostasis of amino acids. An increase in lysosomalfunction and, as a consequence, an inability of immune cells tomobilize aerobic glycolysis was seen in patients with TPP2LoF mutation.9 Like other cytosolic peptidases, TPP2 influ-ences the MHC Class I metabolism, usually through the de-struction of MHC Class I epitopes.21,22 Furthermore,increased MHC Class I and Class II expression has beenshown in TPP2-deficient mice.23,24 The familial mutation,p.Cys28Gly, results in reduced protein expression in thepatients, as seen in the Western blot experiments. However, itdoes not seem to drastically affect the activity of TPP2 in thepatients at least in peripheral blood leukocytes. One can onlyspeculate on possible reasons. TPP2 is a low abundant proteininvolved in immunologic processes; any circumstances af-fecting the immune status of a personmight also affect proteinlevels. It might also be that the sensitivity of the enzymatic testis not sufficient to capture small differences. The reducedprotein expression in the patients, however, might reflect in-stability in the complex formed by active TPP2.18 For ex-ample, dissociated TPP2 is more sensitive to proteolyticdegradation.25
Given the previously reported defect in aerobic glycolysisbecause of impairment of lysosmal function in TPP2-deficientpatients, one could anticipate a similar pathogenic mechanismin our TPP2-mutant patients.9 The aerobic glycolysis pathwayis used in situations of high-energy demand. Specifically, tu-mor cells and immune cells when activated are known toswitch their metabolism towards aerobic glycolysis.26 Partic-ularly, induction and suppressive function of regulatoryT cells have been shown to be critically dependent on thispathway.27–30 Intriguingly, it was shown that regulatoryT cells from relapsing-remittingMS patients have an impairedcapacity to induce aerobic glycolysis.29
Our work shows a marked upregulation of TPP2 at sites ofactive inflammatory demyelination in MS autopsy brainsamples without TPP2 mutations and a reduced TPP2 bloodexpression in mutation carriers. Why TPP2 is upregulated inMS brains and downregulated in TPP2mutation carriers withan MS phenotype cannot be answered here. Is TPP2 upre-gulation in brain secondary to MS onset and follows as a kindof compensatory mechanism to “defend” the inflammationprocess? Such a mechanism seems not unreasonable, re-garding its role in aerobic glycolysis for immune cell function.This speculation attributes TPP2 a rather protective role inthe MS process, which in TPP2mutation carriers is no longerfulfilled but instead leads to long-lasting impairment of aer-obic glycolysis and consequently to MS.
The pronounced and highly selective expression of TPP2 incells with high MHC Class I and Class II expression (figure 2,B) suggests an alternative disease mechanism. As mentionedbefore, TPP2 appears to play a role in peptide trimming and inthe destruction of MHC Class I epitopes. Notably, the fewknown TPP2 processed epitopes include the Epstein-Barrvirus–derived antigen latent membrane protein 1.31 In case ofa reduced efficiency or changed specificity of this processbecause of the TPP2 mutation, one can expect the escape ofself-peptides, which may then be recognized by autoreactiveT cells in a process that amplifies the inflammatory process.
We argue that the homozygous p.Cys28Gly mutation in theTPP2 gene is likely responsible for the MS-like phenotype inthe present family broadening the phenotypic spectrum ofTRIANGLE disease. We therefore consider it as an intriguingpossibility that other cases of TPP2-linked brain inflamma-tions might be covered under a diagnosis of MS. The strongincrease of TPP2 expression in MS brain lesions in non-mutation carriers suggests a broader role in MS physiopa-thology. The mechanisms by which TPP2 mutationscontribute to disease pathogenesis cannot be answered in ourwork. Previous studies claim aerobic glycolysis, lymphocyticimmunosenescence, or MHC peptide trimming as candidatemechanisms in disease development. Further functionalstudies on MS patients with TPP2 mutations in these par-ticular directions will help to elucidate this question.
Author contributionsE.M. Reinthaler and A. Zimprich designed the study, analyzedexome data, performed “Truseq” resequencing experiments,and Affymetrix GenomeWideSNP 6 array experiments forhomozygosity mapping as shown in figure 1, A and B. T.Zrzavy and H. Lassmann designed, executed, and analyzedexperiments shown in figures 2, A–C. C. Kopecky and S.Pferschy performed Western blot experiments as shown infigure 1, E. C. Hotzy helped and participated in Western blotand array experiments and performed Taqman analyses. B.Tomkinson executed and analyzed TPP2 enzymatic activityassays. W. Kristoferitsch recruited and clinically investigatedpatients from family MS01 and helped to generate data forsupplementary figure e-1 (links.lww.com/NXG/A108) and
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

provided and interpreted data for supplementary figure e-2(links.lww.com/NXG/A109). E. Graf, T. Wieland, and T.Strom performed Illlumina exome sequencing and providedthe bioinformatical pipeline for exome data analysis andsupported with the Truseq resequencing experiments. C.Schmied, F. Leutmezer, M. Keilani, C.M. Lill, S. Hoffjan, J.T.Epplen, U.K. Zettl, M. Hecker, A. Deutschlander, S.G. Meuth,M. Ahram, B. Mustafa, and M. El-Khateeb recruited andclinically investigated patients and control individuals forreplication analyses as shown in tables 1 and 2. C. Vilariño-Guell and A.D. Sadovnick recruited and clinically investigatedpatients, performed and analyzed exome data from Canadianpatients as shown in table 1. E.M. Reinthaler, F. Zimprich, W.Kristoferitsch, H. Lassmann, and A. Zimprich had importantintellectual content and wrote the paper.
AcknowledgmentThe authors thank all patients and family members for theirparticipation in this study.
Study fundingThe study was partly funded by the Austrian Science Foun-dation (FWF; Project I 2114 Meltra-BBB), Canada ResearchChair program (950-228408), Michael Smith Foundation forHealth Research (16827), Canadian Institute of Health Re-search (MOP-137051), Vancouver Coastal Health ResearchInstitute, the Milan & Maureen Ilich Foundation (11-32095000), and the Vancouver Foundation (ADV14-1597).
DisclosureE.M. Reinthaler, E. Graf, and T. Zrzavy report no disclosures.T. Wieland is an employee of Foundation Medicine Inc. C.Hotzy and C. Kopecky report no disclosures. S. Pferschy is anemployee of IQVIA. C. Schmied has received travel funding/speaker honoraria from Roche Austria and Sanofi-AventisOsterreich. F. Leutmetzer and M. Keilani report no dis-closures. C.M. Lill serves on the editorial boards of Gene andMetaGene. S. Hoffjan serves on the editorial board of thejournalMCP. J.T. Epplen serves on the editorial board of thejournal MCP and is an employee of Amedes Genetics(Hannover, Germany). U.K. Zettl has received speaker hon-oraria and travel funding from Bayer Pharma, Aventis Pharma,TEVA Pharma, Merck-Serono Pharma, and Biogen-IdecPharma. M. Hecker has received speaker honoraria and travelfunding from Bayer Health Care, Biogen, Novartis, and Teva.A. Deutschlander is supported by a gift from Carl EdwardBolch Jr and Susan Bass Bolch, and by the Sol GoldmanCharitable Trust; and has received research support fromAllergan. S.G. Meuth receives speaker honoraria and travelfunding from Almirall, Amicus Therapeutics Germany, BayerHealth Care, Biogen, Celgene, Diamed, Genzyme, MedDayPharmaceuticals, Merck Serono, Novartis, Novo Nordisk,ONO Pharma, Roche, Sanofi-Aventis, Chugai Pharma,QuintilesIMS, and Teva; receives research support from theGerman Ministry for Education and Research (BMBF),Deutsche Forschungsgesellschaft (DFG), Else Kroner Fre-senius Foundation, German Academic Exchange Service,
Hertie Foundation, Interdisciplinary Center for ClinicalStudies (IZKF) Muenster, German Foundation Neurologyand Almirall, Amicus Therapeutics Germany, Biogen,Diamed, Fresenius Medical Care, Genzyme, Merck Serono,Novartis, ONO Pharma, Roche, and Teva; serves on theeditorial board of PLoS One; and holds patents for effectivityof specific FXII/FXIIa inhibitors (particularly rHA-Infestin 4used to treat neuro-inflammatoral diseases) (WO 2013/113774 A1 and EP 2 263 110 A1), and for diagnosis of a novelautoimmune disease (European patent; 15001186.4—1402).M. Ahram has received research support from the Ministry ofHigher Education and Scientific Research, the University ofJordan, the King Abdullah II Fund for Development (KAFD),the Abdul Hameed Shoman Fund for Supporting ScientificResearch, and the Hashemite University. B. Mustafa, M. El-Khateeb, and C. Vilarino-Guell report no disclosures. A.D.Sadovnick has received travel funding from Biogen; has servedon the speakers’ bureau of the Consortium of Multiple Scle-rosis Centers (CMSC); and has received research supportfrom Biogen MA Inc, Novartis Pharmaceuticals Canada Inc,Genzyme Canada Inc, and Biogen Idec Inc (Canada). F.Zimprich serves on an editorial board (unspecified). B.Tomkinson reports no disclosures. T. Strom has receivedresearch support from the European Union; and receiveslicense fee payments from FGF23, Kirin Brewery. W. Kris-toferitsch has served on scientific advisory boards for BiogenAustria and Novartis; and serves on the editorial board ofJournal fur Neurologie, Neurochirurgie und Psychiatrie. H.Lassmann has received speaker and travel honoraria fromBiogen Idec, Novartis, Roche, and Teva; serves on the edi-torial boards of several journals in the fields of neurology andneuroscience (unspecified); has served as a consultant forMedday and Roche; and has received research support fromthe Austrian Science Fund and the European Union. A.Zimprich reports no disclosures. Full disclosure form in-formation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Received March 28, 2018. Accepted in final form September 26, 2018.
References1. Evans RS, Takahashi K, Duane RT, Payne R, Liu C. Primary thrombocytopenic
purpura and acquired hemolytic anemia; evidence for a common etiology. AMA ArchIntern Med 1951;87:48–65.
2. Besnard C, Levy E, Aladjidi N, et al. Pediatric-onset Evans syndrome: heterogeneouspresentation and high frequency of monogenic disorders including LRBA and CTLA4mutations. Clin Immunol 2018;188:52–57.
3. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults:new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–3172.
4. Wang W, Herrod H, Pui CH, Presbury G, Wilimas J. Immunoregulatory abnormal-ities in Evans syndrome. Am J Hematol 1983;15:381–390.
5. Aladjidi N, Fernandes H, Leblanc T, et al. Evans syndrome in children: long-term outcomein a prospective French national observational cohort. Front Pediatr 2015;3:79.
6. Schubert D, Bode C, Kenefeck R, et al. Autosomal dominant immune dysregulationsyndrome in humans with CTLA4 mutations. Nat Med 2014;20:1410–1416.
7. Kuehn HS, Ouyang W, Lo B, et al. Immune dysregulation in human subjects withheterozygous germline mutations in CTLA4. Science 2014;345:1623–1627.
8. Simon OJ, Kuhlmann T, Bittner S, et al. Evans syndrome associated with sterileinflammation of the central nervous system: a case report. J Med case Rep 2013;7:262.
9. LuW, Zhang Y,McDonald DO, et al. Dual proteolytic pathways govern glycolysis andimmune competence. Cell 2014;159:1578–1590.
10. Stepensky P, Rensing-Ehl A, Gather R, et al. Early-onset Evans syndrome, immu-nodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiency. Blood 2015;125:753–761.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

11. Tomkinson B, Lindas AC. Tripeptidyl-peptidase II: a multi-purpose peptidase. Int JBiochem Cell Biol 2005;37:1933–1937.
12. Sadovnick AD, Risch NJ, Ebers GC. Canadian collaborative project on genetic sus-ceptibility to MS, phase 2: rationale and method. Canadian Collaborative StudyGroup. Can J Neurol Sci 1998;25:216–221.
13. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis:guidelines for research protocols. Ann Neurol 1983;13:227–231.
14. McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria formultiple sclerosis: guidelines from the International Panel on the diagnosis of multiplesclerosis. Ann Neurol 2001;50:121–127.
15. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis:2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302.
16. Wang Z, Sadovnick AD, Traboulsee AL, et al. Nuclear receptor NR1H3 in familialmultiple sclerosis. Neuron 2016;90:948–954.
17. Lassmann H. Review: the architecture of inflammatory demyelinating lesions: implicationsfor studies on pathogenesis. Neuropathol Appl Neurobiol 2011;37:698–710.
18. Tomkinson B. Association and dissociation of the tripeptidyl-peptidase II complex asa way of regulating the enzyme activity. Arch Biochem Biophys 2000;376:275–280.
19. Rose C, Vargas F, Facchinetti P, et al. Characterization and inhibition of a cholecys-tokinin-inactivating serine peptidase. Nature 1996;380:403–409.
20. Han MH, Lundgren DH, Jaiswal S, et al. Janus-like opposing roles of CD47 in auto-immune brain inflammation in humans and mice. J Exp Med 2012;209:1325–1334.
21. Endert P. Role of tripeptidyl peptidase II in MHC class I antigen processing—the endof controversies? Eur J Immunol 2008;38:609–613.
22. Kawahara M, York IA, Hearn A, Farfan D, Rock KL. Analysis of the role of tripeptidylpeptidase II inMHCclass I antigen presentation in vivo. J Immunol 2009;183:6069–6077.
23. Huai J, Firat E, Nil A, et al. Activation of cellular death programs associated withimmunosenescence-like phenotype in TPPII knockout mice. Proc Natl Acad Sci U SA 2008;105:5177–5182.
24. Firat E, Huai J, Saveanu L, et al. Analysis of direct and cross-presentation of antigens inTPPII knockout mice. J Immunol 2007;179:8137–8145.
25. Tomkinson B, Zetterqvist O. Immunological cross-reactivity between human tri-peptidyl peptidase II and fibronectin. Biochem J 1990;267:149–154.
26. Palsson-McDermott EM, O’Neill LA. The Warburg effect then and now: from cancerto inflammatory diseases. Bioessays 2013;35:965–973.
27. Carbone F, De Rosa V, Carrieri PB, et al. Regulatory T cell proliferative potential isimpaired in human autoimmune disease. Nat Med 2014;20:69–74.
28. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppressionby CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med2004;199:971–979.
29. De Rosa V, Galgani M, Porcellini A, et al. Glycolysis controls the induction of humanregulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants.Nat Immunol 2015;16:1174–1184.
30. Chang CH, Curtis JD, Maggi LB Jr, et al. Posttranscriptional control of T cell effectorfunction by aerobic glycolysis. Cell 2013;153:1239–1251.
31. Diekmann J, Adamopoulou E, Beck O, et al. Processing of two latent membraneprotein 1 MHC class I epitopes requires tripeptidyl peptidase II involvement.J Immunol 2009;183:1587–1597.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

ARTICLE OPEN ACCESS
Rare genetic variation implicated innon-Hispanic white families withAlzheimer diseaseGary W. Beecham, PhD, Badri Vardarajan, PhD, Elizabeth Blue, PhD, William Bush, PhD, James Jaworski, MPH,
Sandra Barral, PhD, Anita DeStefano, PhD, Kara Hamilton-Nelson, MPH, Brian Kunkle, PhD,
Eden R. Martin, PhD, Adam Naj, PhD, Farid Rajabli, PhD, Christiane Reitz, MD, PhD, Timothy Thornton, PhD,
Cornelia vanDuijn, PhD, AllisonGoate, PhD, Sudha Seshadri, MD, Lindsay A. Farrer, PhD, Eric Boerwinkle, PhD,
Gerard Schellenberg, PhD, Jonathan L. Haines, PhD, Ellen Wijsman, PhD, Richard Mayeux, MD,
and Margaret A. Pericak-Vance, PhD, for The Alzheimer’s Disease Sequencing Project
Neurol Genet 2018;4:e286. doi:10.1212/NXG.0000000000000286
Correspondence
Dr. Beecham
AbstractObjectiveTo identify genetic variation influencing late-onset Alzheimer disease (LOAD), we used a largedata set of non-Hispanic white (NHW) extended families multiply-affected by LOAD byperforming whole genome sequencing (WGS).
MethodsAs part of the Alzheimer Disease Sequencing Project, WGS data were generated for 197 NHWparticipants from 42 families (affected individuals and unaffected, elderly relatives). A two-pronged approach was taken. First, variants were prioritized using heterogeneity logarithm ofthe odds (HLOD) and family-specific LOD scores as well as annotations based on function,frequency, and segregation with disease. Second, known Alzheimer disease (AD) candidategenes were assessed for rare variation using a family-based association test.
ResultsWe identified 41 rare, predicted-damaging variants that segregated with disease in the familiesthat contributed to the HLOD or family-specific LOD regions. These included a variant innitric oxide synthase 1 adaptor protein that segregates with disease in a family with 7 individualswith AD, as well as variants in RP11-433J8, ABCA1, and FISP2. Rare-variant associationidentified 2 LOAD candidate genes associated with disease in these families: FERMT2 (p-values= 0.001) and SLC24A4 (p-value = 0.009). These genes still showed association while con-trolling for common index variants, indicating the rare-variant signal is distinct from commonvariation that initially identified the genes as candidates.
ConclusionsWe identified multiple genes with putative damaging rare variants that segregate with disease inmultiplex AD families and showed that rare variation may influence AD risk at AD candidategenes. These results identify novel AD candidate genes and show a role for rare variation inLOAD etiology, even at genes previously identified by common variation.
From the John P. Hussman Institute for Human Genomics (G.W.B., J.J., K.H.-N., B.K., E.R.M., F.R., M.A.P.-V.), University of Miami, Miller School of Medicine; Dr. John T. Macdonald FoundationDepartment of Human Genetics (G.W.B., E.R.M., M.A.P.-V.), University of Miami, Miller School of Medicine, FL; The Taub Institute for Research on Alzheimer’s Disease and the Aging Brain(B.V., S.B., C.R., R.M.), Columbia University; TheGertrudeH. Segievsky Center (B.V., S.B., C.R., R.M.), ColumbiaUniversity, New York Presbyterian Hospital; Division ofMedical Genetics (E. Blue,E.W.), DepartmentofMedicine, University ofWashington, Seattle; Institute forComputational Biology (W.B., J.L.H.), CaseWestern ReserveUniversity, Cleveland,OH;Department ofNeurology(A.D., S.S., L.A.F.), Boston University School of Medicine; Department of Biostatistics (A.D., S.S., L.A.F.), Boston University School of Medicine, MA; School of Medicine (A.N., G.S.), University ofPennsylvania, Philadelphia;DepartmentofBiostatistics (T.T., E.W.),University ofWashington, Seattle; ErasmusMedicalUniversity (C.D.), Rotterdam,TheNetherlands; IcahnSchool ofMedicineat Mount Sinai (A.G.), New York, NY; Department of Medicine (L.A.F.), Boston University School of Medicine, MA; and University of Texas (E. Boerwinkle), Houston.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
Co-contributors are listed at links.lww.com/NXG/A132.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Late-onset Alzheimer disease (LOAD) is a neurodegenerativedisease, characterized by progressive dementia, and pathologicchanges include neuronal loss, neurofibrillary tangles, andamyloid-beta deposits.1 LOAD is highly heritable (60%–80%),but most of this heritability remains unexplained, despite theidentification of genetic factors that influence LOAD.2 Thesefactors include the APOE gene, as well as other genes identifiedthrough genome-wide association studies (GWAS) and a lim-ited number of studies of rare genetic variation.3–9 While thesefactors have replicable association with LOAD, few of the un-derlying causal variants have been definitively identified.
To identify additional genes influencing LOAD and to betterunderstand known LOAD genes, the Alzheimer Disease Se-quencing Project (ADSP) was established.10 A key componentof the ADSP is inclusion of whole genome sequencing (WGS) inlarge, multiply-affected LOAD families of non-Hispanic white(NHW) and Caribbean Hispanic (CH) ancestry. This family-based design enriches the study for risk variation, making it idealto identify novel risk variants.11 Family structure facilitates theprioritization of risk variation through linkage and segregation-based approaches. In this study, we report on analyses of theNHW families. Two primary approaches were taken: examina-tion of linkage regions segregating with disease in these familiesto identify novel genes and a gene-based association analysis torare variation at knownAlzheimer disease (AD) candidate genes.Results indicate that rare variants play a role in LOADmultiplexfamilies, both at novel genes identified through linkage andthrough rare variation at AD candidate genes.
MethodsThe Alzheimer Disease Sequencing ProjectFamilies were assembled as part of the ADSP. The ADSP isa collaboration of the LOAD genetics research community, theNational Institutes on Aging, and the National Human GenomeResearch Institute (NHGRI). The full design is described else-where.11 The ADSP includes contributors from the AlzheimerDisease Genetics Consortium, the neurology working group ofthe Cohorts for Heart and Aging Research in Genomic Epide-miology (CHARGE), as well as 3 NHGRI sequencing centers atBaylor University, the Broad Institute, and Washington Uni-versity. Data are available through dbGaP (phs000572).
Family selection and designApproximately 1,400 multiplex LOAD families were reviewedfor inclusion. Families were derived from the National
Institute on Aging Late Onset of Alzheimer’s Disease familystudy, the National Cell Repository for Alzheimer’s Disease,and families contributed by investigators from ColumbiaUniversity, University of Miami, University of Washington,University of Pennsylvania, Case Western Reserve University,and Erasmus University. Families analyzed here were ofNHW (CH descent families analyzed elsewhere)12 and wererequired to have multiple members with LOAD, availablegenomic DNA, and available APOE genotypes. We excludedfamilies known to carry mendelian AD mutations or werepathologically confirmed non-Alzheimer dementia.
Families meeting initial criteria were prioritized and chosenbased on the number of affected individuals, number ofgenerations affected, age at onset of clinical symptoms, andpresence of APOE e4 risk alleles (table 1). Details of criteriaand selection process are described elsewhere.10 No e4/e4individuals were included. Cognitively intact participants wereselected if available and informative for phasing.
All cases met National Institute of Neurological Diseases-Alzheimer’s NINCDS-ADRDA criteria for possible, probableor definite AD.13 Unaffected individuals were free of clinicalAD at themost recent cognitive assessment. In total, 42 NHWfamilies were included. These families included 208 affectedindividuals and 185 unaffected individuals with array dataavailable, of which 164 affected individuals and 33 unaffectedindividuals were included in the sequencing experiment.
Standard protocol approvals, registrations,and patient consentsAll individuals (or caregivers) provided written informedconsent for genomic studies, including broad data sharing,and were assessed with the approval of the relevant in-stitutional review boards.
NGS sequencingWGS was performed at the NHGRI sequencing centers at theBroad Institute (Boston, MA), Baylor College of Medicine(Houston, TX), and Washington University (St. Louis, MS).Samples were sequenced using Illumina instruments toa minimum average 30× depth. Details of the sequencingexperiments are described elsewhere.14
NGS calling and quality controlRaw NGS data were aligned to hg19 using BWA.15 Genotypecalling was performed using Atlas (v2).16 Extensive variant-level quality control (QC) was performed (appendix e-1,
GlossaryADSP = Alzheimer Disease Sequencing Project; CH = Caribbean Hispanic; HLOD = heterogeneity logarithm of the odds;LOAD = late-onset Alzheimer disease; LOD = logarithm of the odds; LRP1 = LDL-receptor-related protein 1;MAF = Minorallele frequency;NHGRI =National Human Genome Research Institute;NHW = non-Hispanic white;NOS1AP = nitric oxidesynthase 1 adaptor protein; QC = quality control; SNP = single nucleotide polymorphism; WES = whole exome sequencing;WGS = whole genome sequencing.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
links.lww.com/NXG/A117). Principal components analysiswas used to assess population substructure, using theEIGENSTRAT.17 Array data were compared with WGS datato assess and confirm the pedigree structure for all individuals.Additional details of QC are reported elsewhere.14
Linkage analysesMERLIN v1.1.2 software18 was used to perform parametricand nonparametric multipoint linkage analyses on the arraydata available for the entire family. Nonparametric analysesare described in detail elsewhere.19 For parametric multipoint
Table 1 Priority variants from consensus linkage regions
Chrm Position RS ID Family Gene Alleles Consequence CADD MAF (1kGP)
1 162,167,769 LD0254F NOS1AP C/T Intron variant 1.2 NA
1 162,207,390 LD0254F NOS1AP A/T Intron variant 0.2 NA
1 162,223,640 LD0254F NOS1AP A/G Intron variant 13.6 NA
1 162,479,200 UM0464F UHMK1 T/G Intron variant 0.4 0
1 162,564,187 LD1223F UAP1 A/G Intron variant 8.7 0.008
1 162,564,187 LD1223F UAP1 A/G Intron variant 8.7 0.008
1 162,700,025 UM0464F DDR2 A/C Intron variant 0.5 0.009
1 162,735,057 UM0464F DDR2 G/A Intron variant 0.7 0.009
1 162,739,064 UM0464F DDR2 G/A Intron variant 5.4 0.009
1 162,742,651 LD0254F DDR2 G/A Intron variant 0.1 0.001
1 162,751,967 LD0254F DDR2 T/A 3ʹ UTR variant 6.6 0.009
1 162,757,273 LD1223F DDR2 T/C Upstream gene variant 2.2 0.009
1 162,928,238 UM0464F G/A Intergenic variant 0.9 0.002
1 163,032,461 LD0254F T/A Intergenic variant 1.9 0.002
1 163,202,875 UM0464F RGS5 G/A Intron variant 8.3 0.002
1 163,578,427 UM0464F C/A Intergenic variant 4.7 0.003
1 163,749,571 LD0949F A/G Intergenic variant 7.2 0.002
1 163,841,625 UM0464F C/T Intergenic variant 7.1 0.010
1 164,034,469 rs187504850 UM0464F A/G Intergenic variant 19.6 NA
1 164,448,463 UM0464F G/A Intergenic variant 3.2 0.003
1 164,622,647 LD0949F PBX1 G/A Intron variant 1.2 0.006
1 164,887,661 UM0464F C/T Downstream gene variant 8.2 0.005
1 165,253,949 LD0254F LMX1A C/T Intron variant 14.7 0.002
1 165,342,593 UM0464F G/A Intergenic variant 5.9 0.010
1 165,532,785 LD1223F LRRC52 G/A Synonymous variant 18.3 0.004
14 95,913,507 rs191535004 LD0949F SYNE3 G/A Intron variant 0.7 0.008
14 95,923,822 UM0464F SYNE3 C/T Intron variant 3.5 0.008
14 96,449,175 UM0464F C/A Intergenic variant 0.8 NA
14 96,568,984 UM0464F C/A Regulatory region variant 1.5 NA
14 96,923,339 LD0254F AK7 C/T Noncoding transcript exon variant 0.1 0.008
14 97,029,358 UM0464F PAPOLA C/G Intron variant 10.4 NA
14 97,228,875 LD0949F RP11-433J8.2 A/G Intron variant 12.3 0.003
Abbreviations: CADD = Combined Annotation Dependent Depletion score; Chrm = chromosome; MAF (1kGP) = Minor Allele Frequency among the Europeansamples in the 1,000 Genomes Project data; UTR = untranslated region.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
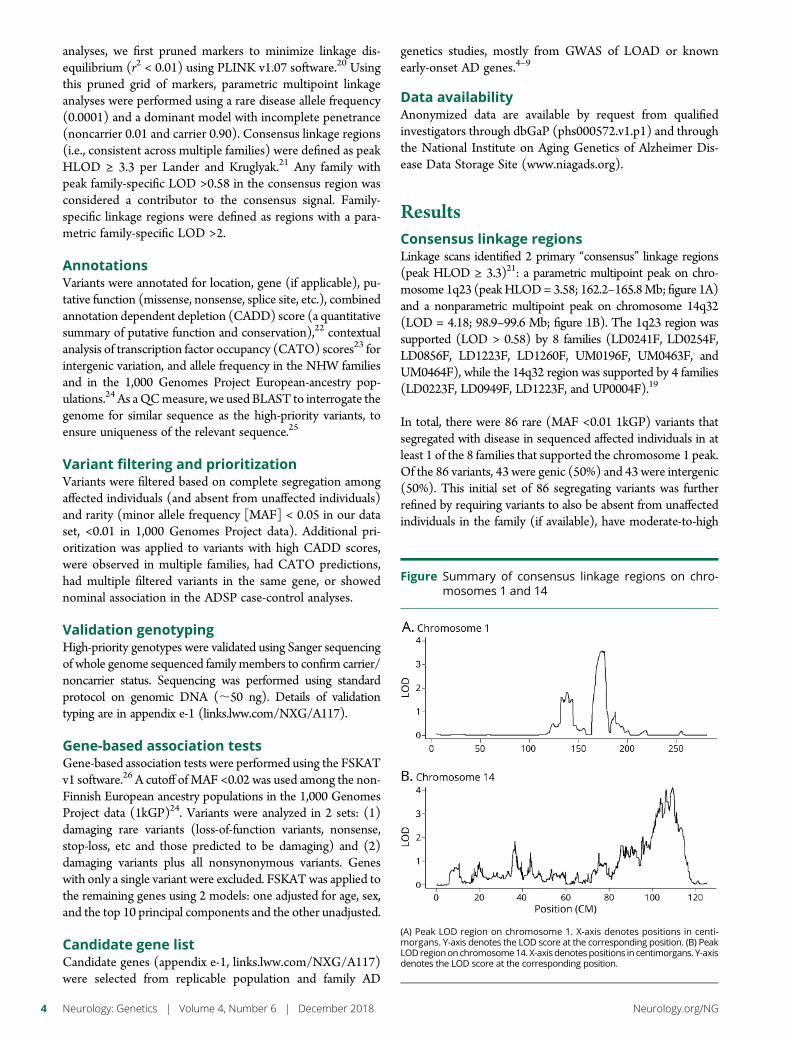
analyses, we first pruned markers to minimize linkage dis-equilibrium (r2 < 0.01) using PLINK v1.07 software.20 Usingthis pruned grid of markers, parametric multipoint linkageanalyses were performed using a rare disease allele frequency(0.0001) and a dominant model with incomplete penetrance(noncarrier 0.01 and carrier 0.90). Consensus linkage regions(i.e., consistent across multiple families) were defined as peakHLOD ≥ 3.3 per Lander and Kruglyak.21 Any family withpeak family-specific LOD >0.58 in the consensus region wasconsidered a contributor to the consensus signal. Family-specific linkage regions were defined as regions with a para-metric family-specific LOD >2.
AnnotationsVariants were annotated for location, gene (if applicable), pu-tative function (missense, nonsense, splice site, etc.), combinedannotation dependent depletion (CADD) score (a quantitativesummary of putative function and conservation),22 contextualanalysis of transcription factor occupancy (CATO) scores23 forintergenic variation, and allele frequency in the NHW familiesand in the 1,000 Genomes Project European-ancestry pop-ulations.24 As aQCmeasure, we used BLAST to interrogate thegenome for similar sequence as the high-priority variants, toensure uniqueness of the relevant sequence.25
Variant filtering and prioritizationVariants were filtered based on complete segregation amongaffected individuals (and absent from unaffected individuals)and rarity (minor allele frequency [MAF] < 0.05 in our dataset, <0.01 in 1,000 Genomes Project data). Additional pri-oritization was applied to variants with high CADD scores,were observed in multiple families, had CATO predictions,had multiple filtered variants in the same gene, or showednominal association in the ADSP case-control analyses.
Validation genotypingHigh-priority genotypes were validated using Sanger sequencingof whole genome sequenced family members to confirm carrier/noncarrier status. Sequencing was performed using standardprotocol on genomic DNA (;50 ng). Details of validationtyping are in appendix e-1 (links.lww.com/NXG/A117).
Gene-based association testsGene-based association tests were performed using the FSKATv1 software.26 A cutoff of MAF <0.02 was used among the non-Finnish European ancestry populations in the 1,000 GenomesProject data (1kGP)24. Variants were analyzed in 2 sets: (1)damaging rare variants (loss-of-function variants, nonsense,stop-loss, etc and those predicted to be damaging) and (2)damaging variants plus all nonsynonymous variants. Geneswith only a single variant were excluded. FSKATwas applied tothe remaining genes using 2 models: one adjusted for age, sex,and the top 10 principal components and the other unadjusted.
Candidate gene listCandidate genes (appendix e-1, links.lww.com/NXG/A117)were selected from replicable population and family AD
genetics studies, mostly from GWAS of LOAD or knownearly-onset AD genes.4–9
Data availabilityAnonymized data are available by request from qualifiedinvestigators through dbGaP (phs000572.v1.p1) and throughthe National Institute on Aging Genetics of Alzheimer Dis-ease Data Storage Site (www.niagads.org).
ResultsConsensus linkage regionsLinkage scans identified 2 primary “consensus” linkage regions(peak HLOD ≥ 3.3)21: a parametric multipoint peak on chro-mosome 1q23 (peakHLOD= 3.58; 162.2–165.8Mb; figure 1A)and a nonparametric multipoint peak on chromosome 14q32(LOD = 4.18; 98.9–99.6 Mb; figure 1B). The 1q23 region wassupported (LOD > 0.58) by 8 families (LD0241F, LD0254F,LD0856F, LD1223F, LD1260F, UM0196F, UM0463F, andUM0464F), while the 14q32 region was supported by 4 families(LD0223F, LD0949F, LD1223F, and UP0004F).19
In total, there were 86 rare (MAF <0.01 1kGP) variants thatsegregated with disease in sequenced affected individuals in atleast 1 of the 8 families that supported the chromosome 1 peak.Of the 86 variants, 43 were genic (50%) and 43 were intergenic(50%). This initial set of 86 segregating variants was furtherrefined by requiring variants to also be absent from unaffectedindividuals in the family (if available), have moderate-to-high
Figure Summary of consensus linkage regions on chro-mosomes 1 and 14
(A) Peak LOD region on chromosome 1. X-axis denotes positions in centi-morgans. Y-axis denotes the LOD score at the corresponding position. (B) PeakLODregiononchromosome14.X-axisdenotespositions in centimorgans. Y-axisdenotes the LOD score at the corresponding position.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
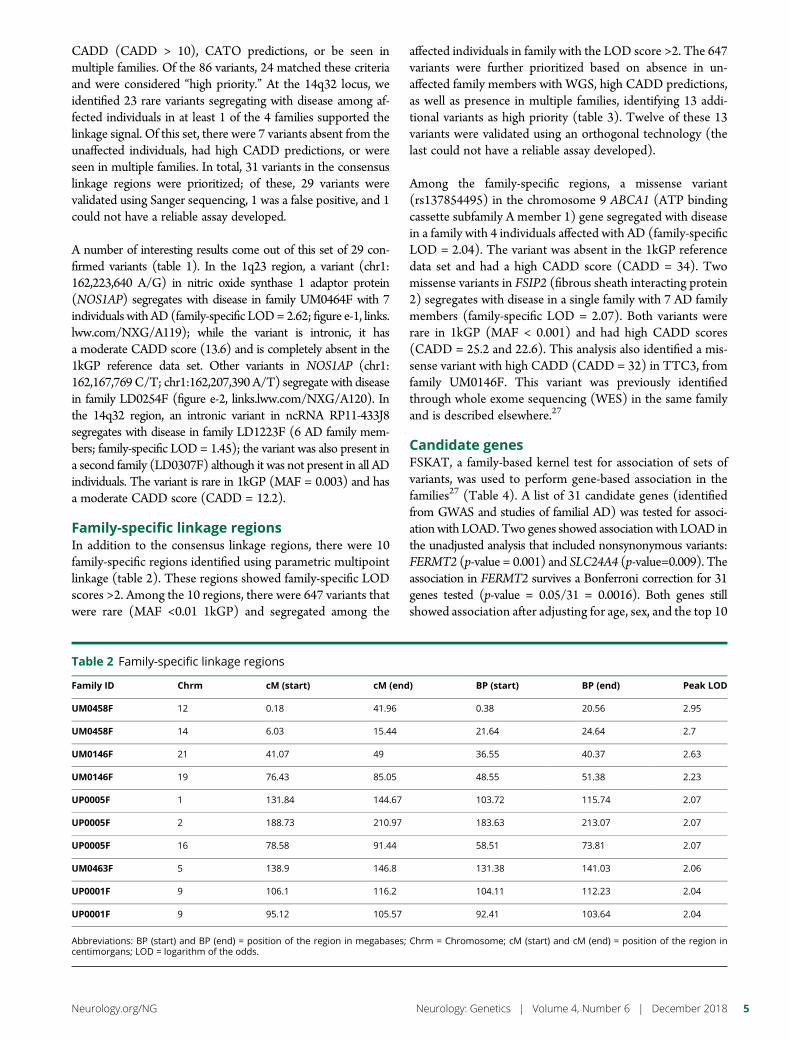
CADD (CADD > 10), CATO predictions, or be seen inmultiple families. Of the 86 variants, 24 matched these criteriaand were considered “high priority.” At the 14q32 locus, weidentified 23 rare variants segregating with disease among af-fected individuals in at least 1 of the 4 families supported thelinkage signal. Of this set, there were 7 variants absent from theunaffected individuals, had high CADD predictions, or wereseen in multiple families. In total, 31 variants in the consensuslinkage regions were prioritized; of these, 29 variants werevalidated using Sanger sequencing, 1 was a false positive, and 1could not have a reliable assay developed.
A number of interesting results come out of this set of 29 con-firmed variants (table 1). In the 1q23 region, a variant (chr1:162,223,640 A/G) in nitric oxide synthase 1 adaptor protein(NOS1AP) segregates with disease in family UM0464F with 7individuals with AD (family-specific LOD= 2.62; figure e-1, links.lww.com/NXG/A119); while the variant is intronic, it hasa moderate CADD score (13.6) and is completely absent in the1kGP reference data set. Other variants in NOS1AP (chr1:162,167,769C/T; chr1:162,207,390 A/T) segregate with diseasein family LD0254F (figure e-2, links.lww.com/NXG/A120). Inthe 14q32 region, an intronic variant in ncRNA RP11-433J8segregates with disease in family LD1223F (6 AD family mem-bers; family-specific LOD = 1.45); the variant was also present ina second family (LD0307F) although it was not present in all ADindividuals. The variant is rare in 1kGP (MAF = 0.003) and hasa moderate CADD score (CADD = 12.2).
Family-specific linkage regionsIn addition to the consensus linkage regions, there were 10family-specific regions identified using parametric multipointlinkage (table 2). These regions showed family-specific LODscores >2. Among the 10 regions, there were 647 variants thatwere rare (MAF <0.01 1kGP) and segregated among the
affected individuals in family with the LOD score >2. The 647variants were further prioritized based on absence in un-affected family members with WGS, high CADD predictions,as well as presence in multiple families, identifying 13 addi-tional variants as high priority (table 3). Twelve of these 13variants were validated using an orthogonal technology (thelast could not have a reliable assay developed).
Among the family-specific regions, a missense variant(rs137854495) in the chromosome 9 ABCA1 (ATP bindingcassette subfamily A member 1) gene segregated with diseasein a family with 4 individuals affected with AD (family-specificLOD = 2.04). The variant was absent in the 1kGP referencedata set and had a high CADD score (CADD = 34). Twomissense variants in FSIP2 (fibrous sheath interacting protein2) segregates with disease in a single family with 7 AD familymembers (family-specific LOD = 2.07). Both variants wererare in 1kGP (MAF < 0.001) and had high CADD scores(CADD = 25.2 and 22.6). This analysis also identified a mis-sense variant with high CADD (CADD = 32) in TTC3, fromfamily UM0146F. This variant was previously identifiedthrough whole exome sequencing (WES) in the same familyand is described elsewhere.27
Candidate genesFSKAT, a family-based kernel test for association of sets ofvariants, was used to perform gene-based association in thefamilies27 (Table 4). A list of 31 candidate genes (identifiedfrom GWAS and studies of familial AD) was tested for associ-ationwith LOAD. Two genes showed associationwith LOAD inthe unadjusted analysis that included nonsynonymous variants:FERMT2 (p-value = 0.001) and SLC24A4 (p-value=0.009). Theassociation in FERMT2 survives a Bonferroni correction for 31genes tested (p-value = 0.05/31 = 0.0016). Both genes stillshowed association after adjusting for age, sex, and the top 10
Table 2 Family-specific linkage regions
Family ID Chrm cM (start) cM (end) BP (start) BP (end) Peak LOD
UM0458F 12 0.18 41.96 0.38 20.56 2.95
UM0458F 14 6.03 15.44 21.64 24.64 2.7
UM0146F 21 41.07 49 36.55 40.37 2.63
UM0146F 19 76.43 85.05 48.55 51.38 2.23
UP0005F 1 131.84 144.67 103.72 115.74 2.07
UP0005F 2 188.73 210.97 183.63 213.07 2.07
UP0005F 16 78.58 91.44 58.51 73.81 2.07
UM0463F 5 138.9 146.8 131.38 141.03 2.06
UP0001F 9 106.1 116.2 104.11 112.23 2.04
UP0001F 9 95.12 105.57 92.41 103.64 2.04
Abbreviations: BP (start) and BP (end) = position of the region in megabases; Chrm = Chromosome; cM (start) and cM (end) = position of the region incentimorgans; LOD = logarithm of the odds.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
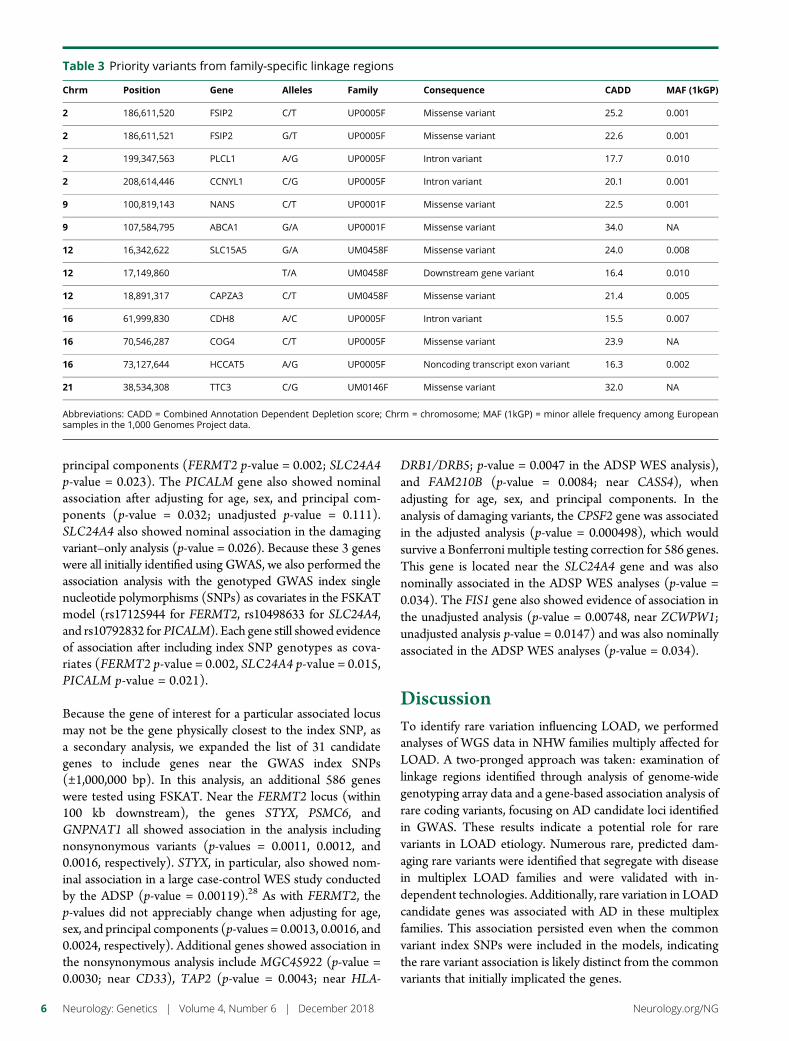
principal components (FERMT2 p-value = 0.002; SLC24A4p-value = 0.023). The PICALM gene also showed nominalassociation after adjusting for age, sex, and principal com-ponents (p-value = 0.032; unadjusted p-value = 0.111).SLC24A4 also showed nominal association in the damagingvariant–only analysis (p-value = 0.026). Because these 3 geneswere all initially identified using GWAS, we also performed theassociation analysis with the genotyped GWAS index singlenucleotide polymorphisms (SNPs) as covariates in the FSKATmodel (rs17125944 for FERMT2, rs10498633 for SLC24A4,and rs10792832 for PICALM). Each gene still showed evidenceof association after including index SNP genotypes as cova-riates (FERMT2 p-value = 0.002, SLC24A4 p-value = 0.015,PICALM p-value = 0.021).
Because the gene of interest for a particular associated locusmay not be the gene physically closest to the index SNP, asa secondary analysis, we expanded the list of 31 candidategenes to include genes near the GWAS index SNPs(±1,000,000 bp). In this analysis, an additional 586 geneswere tested using FSKAT. Near the FERMT2 locus (within100 kb downstream), the genes STYX, PSMC6, andGNPNAT1 all showed association in the analysis includingnonsynonymous variants (p-values = 0.0011, 0.0012, and0.0016, respectively). STYX, in particular, also showed nom-inal association in a large case-control WES study conductedby the ADSP (p-value = 0.00119).28 As with FERMT2, thep-values did not appreciably change when adjusting for age,sex, and principal components (p-values = 0.0013, 0.0016, and0.0024, respectively). Additional genes showed association inthe nonsynonymous analysis include MGC45922 (p-value =0.0030; near CD33), TAP2 (p-value = 0.0043; near HLA-
DRB1/DRB5; p-value = 0.0047 in the ADSP WES analysis),and FAM210B (p-value = 0.0084; near CASS4), whenadjusting for age, sex, and principal components. In theanalysis of damaging variants, the CPSF2 gene was associatedin the adjusted analysis (p-value = 0.000498), which wouldsurvive a Bonferroni multiple testing correction for 586 genes.This gene is located near the SLC24A4 gene and was alsonominally associated in the ADSP WES analyses (p-value =0.034). The FIS1 gene also showed evidence of association inthe unadjusted analysis (p-value = 0.00748, near ZCWPW1;unadjusted analysis p-value = 0.0147) and was also nominallyassociated in the ADSP WES analyses (p-value = 0.034).
DiscussionTo identify rare variation influencing LOAD, we performedanalyses of WGS data in NHW families multiply affected forLOAD. A two-pronged approach was taken: examination oflinkage regions identified through analysis of genome-widegenotyping array data and a gene-based association analysis ofrare coding variants, focusing on AD candidate loci identifiedin GWAS. These results indicate a potential role for rarevariants in LOAD etiology. Numerous rare, predicted dam-aging rare variants were identified that segregate with diseasein multiplex LOAD families and were validated with in-dependent technologies. Additionally, rare variation in LOADcandidate genes was associated with AD in these multiplexfamilies. This association persisted even when the commonvariant index SNPs were included in the models, indicatingthe rare variant association is likely distinct from the commonvariants that initially implicated the genes.
Table 3 Priority variants from family-specific linkage regions
Chrm Position Gene Alleles Family Consequence CADD MAF (1kGP)
2 186,611,520 FSIP2 C/T UP0005F Missense variant 25.2 0.001
2 186,611,521 FSIP2 G/T UP0005F Missense variant 22.6 0.001
2 199,347,563 PLCL1 A/G UP0005F Intron variant 17.7 0.010
2 208,614,446 CCNYL1 C/G UP0005F Intron variant 20.1 0.001
9 100,819,143 NANS C/T UP0001F Missense variant 22.5 0.001
9 107,584,795 ABCA1 G/A UP0001F Missense variant 34.0 NA
12 16,342,622 SLC15A5 G/A UM0458F Missense variant 24.0 0.008
12 17,149,860 T/A UM0458F Downstream gene variant 16.4 0.010
12 18,891,317 CAPZA3 C/T UM0458F Missense variant 21.4 0.005
16 61,999,830 CDH8 A/C UP0005F Intron variant 15.5 0.007
16 70,546,287 COG4 C/T UP0005F Missense variant 23.9 NA
16 73,127,644 HCCAT5 A/G UP0005F Noncoding transcript exon variant 16.3 0.002
21 38,534,308 TTC3 C/G UM0146F Missense variant 32.0 NA
Abbreviations: CADD = Combined Annotation Dependent Depletion score; Chrm = chromosome; MAF (1kGP) = minor allele frequency among Europeansamples in the 1,000 Genomes Project data.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
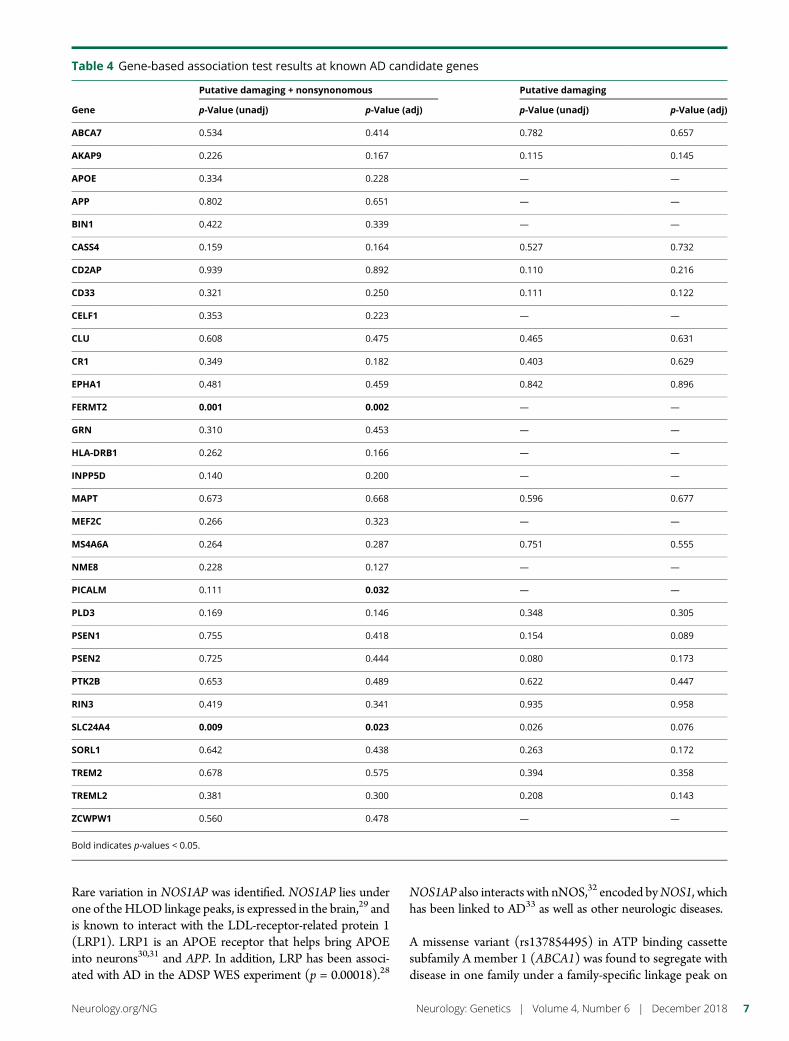
Rare variation in NOS1AP was identified. NOS1AP lies underone of theHLOD linkage peaks, is expressed in the brain,29 andis known to interact with the LDL-receptor-related protein 1(LRP1). LRP1 is an APOE receptor that helps bring APOEinto neurons30,31 and APP. In addition, LRP has been associ-ated with AD in the ADSP WES experiment (p = 0.00018).28
NOS1AP also interacts with nNOS,32 encoded byNOS1, whichhas been linked to AD33 as well as other neurologic diseases.
A missense variant (rs137854495) in ATP binding cassettesubfamily A member 1 (ABCA1) was found to segregate withdisease in one family under a family-specific linkage peak on
Table 4 Gene-based association test results at known AD candidate genes
Gene
Putative damaging + nonsynonomous Putative damaging
p-Value (unadj) p-Value (adj) p-Value (unadj) p-Value (adj)
ABCA7 0.534 0.414 0.782 0.657
AKAP9 0.226 0.167 0.115 0.145
APOE 0.334 0.228 — —
APP 0.802 0.651 — —
BIN1 0.422 0.339 — —
CASS4 0.159 0.164 0.527 0.732
CD2AP 0.939 0.892 0.110 0.216
CD33 0.321 0.250 0.111 0.122
CELF1 0.353 0.223 — —
CLU 0.608 0.475 0.465 0.631
CR1 0.349 0.182 0.403 0.629
EPHA1 0.481 0.459 0.842 0.896
FERMT2 0.001 0.002 — —
GRN 0.310 0.453 — —
HLA-DRB1 0.262 0.166 — —
INPP5D 0.140 0.200 — —
MAPT 0.673 0.668 0.596 0.677
MEF2C 0.266 0.323 — —
MS4A6A 0.264 0.287 0.751 0.555
NME8 0.228 0.127 — —
PICALM 0.111 0.032 — —
PLD3 0.169 0.146 0.348 0.305
PSEN1 0.755 0.418 0.154 0.089
PSEN2 0.725 0.444 0.080 0.173
PTK2B 0.653 0.489 0.622 0.447
RIN3 0.419 0.341 0.935 0.958
SLC24A4 0.009 0.023 0.026 0.076
SORL1 0.642 0.438 0.263 0.172
TREM2 0.678 0.575 0.394 0.358
TREML2 0.381 0.300 0.208 0.143
ZCWPW1 0.560 0.478 — —
Bold indicates p-values < 0.05.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

chromosome 9. The variant was rare, with a very high CADDscore (CADD= 34).ABCA1 is expressed in brain (though notexclusively; ABCA7 is expressed in many tissues) and is in-volved in lipid removal pathways.29 Variants in ABCA1 havebeen associated with HDL deficiency, familial hypercholes-terolemia, and APOA deficiency.34–37 The rs137854495 var-iant, in particular, has been noted in a family with Tangierdisease as part of a compound heterozygote.38 Dyslipidemiasand lipid pathways have long been linked to LOAD,39 startingwith the APOE gene,3 and more recently CLU, ABCA7,etc,4,22 although exact mechanisms remain unclear. Tangierdisease, in particular, has also been proposed as having links toAD, primarily through amyloid-β pathways, although evi-dence supporting this is mixed.40–44 The ADSP WES projectidentified nominal association with 2 additional apolipopro-teins (APOA2, p = 0.000636; APOA5, p = 0.0413).28
Gene-based association tests implicated fermitin familymember 2 (FERMT2) and surrounding genes STYX, PSMC6,and GNPNAT, all with similar levels of significance (p =0.0010–0.0016). FERMT2 is involved in cell adhesion, isexpressed in brain, and is near an SNP with strong associationto AD.4 STYX is likely involved in phosphatase activity andhas been associated with diabetes mellitus type 1.45 PSMC6 islikely involved in hydrolase activity; GNPNAT is involved insugar metabolism. SLC24A4 has been associated with ADthrough a genome-widemeta-analysis,4 and brainmethylationin SLC24A4 region has been associated with AD risk.46 Al-though FERMT2 and SLC24A4 were initially identified usingcommon variant approaches, the association observed at these2 genes was not greatly affected by including the GWAS indexSNPs as covariates in the model. If common variants weresolely responsible for the association, then we would expect tofail to reject the null hypothesis at the rare variants. Thisimplies the rare variation associated with disease in thesefamilies is distinct from the common variant index SNPsinitially used to identify the genes.
There are limitations to this study. The sample size wasmodest relative to GWAS approaches. This of course limitspower, particularly for the association-based approaches.However, the use of familial data and linkage and segregation-based approaches mitigates some of these power concerns.Increasing sample sizes and number of multiplex families is anongoing effort for future studies. Additional limitations in-clude the use of in silico predictions of function (e.g., CADD).While useful as a first pass, these predictions should be seen asa putative,47 and function will need to be validated by func-tional genetic approaches.
These results imply a role for rare variation in familial LOAD.The linkage analysis identified 41 high-priority variants, includingvariants inNOS1AP andABCA1, both with plausible roles in ADand AD-related pathways. The analysis of LOAD candidategenes identified several genes with rare variation associated withAD. The tests were still significant while controlling for thecommon index SNPs, implying a role for rare variation even at
GWAS-identified loci. Future directions include a thoroughanalysis on noncoding variation, particularly the role ofenhancers and other regulatory elements in the etiology of AD.
Author contributionsAll authors contributed to the work presented in this article.Critical revision: Primary manuscript was prepared by G.W.Beecham, with significant contributions from B. Vardarajan,E. Blue, E. Wijsman, and M.A. Pericak-Vance. All authorsparticipated in the revision and editing of the manuscript.Concept and design: There were significant contributions toconcept and design from G.W. Beecham, B. Vardarajan,E. Blue, W. Bush, A. DeStefano, E.R. Martin, A. Naj, C. Reitz,C. van Duijn, A. Goate, S. Seshadri, L.A. Farrer, E. Boerwinkle,G. Schellenberg, J.L. Haines, E. Wijsman, R. Mayeux, andM.A. Pericak-Vance. Analysis and interpretation: Review offamily data was performed by M. Pericak-Vance, R. Mayeux,E. Boerwinkle, S. Seshadri, and C. van Duijn. Primary statis-tical analyses were performed by G.W. Beecham, J. Jaworski,E.R. Martin, and K. Hamilton-Nelson, with additions fromB. Vardarajan, W. Bush, and E. Blue. All authors participatedin the interpretation and discussion of results. Acquisition ofdata: Sample data were contributed by C. van Duijn,A. DeStefano, L.A. Farrer, A. Goate, J.L. Haines, M.A. Pericak-Vance, E. Boerwinkle, R. Mayeux, S. Seshadri, and G. Schel-lenberg. Statistical analyses: Statistical analyses were primarilyconducted by G.W. Beecham; additional analyses conductedby J.C.B., A.C.N., E.R. Martin, S.H.C., A. DeStefano, and S.Seshadri (affiliations noted above, all academic). Study su-pervision and coordination: Primary study supervision andcoordination was by R. Mayeux, M.A. Pericak-Vance, and E.Wijsman. Funding: Primary funding was by G. Schellenberg,R. Mayeux, E. Boerwinkle, M.A. Pericak-Vance, J.L. Haines, S.Seshadri, A. Goate, L.A. Farrer, and E. Wijsman. A detailed listof funding is noted in the acknowledgements.
AcknowledgmentThe Alzheimer’s Disease Sequencing Project (ADSP)comprises 2 Alzheimer disease (AD) genetics consortia and3 National Human Genome Research Institute–funded LargeScale Sequencing and Analysis Centers (LSAC). The 2 ADgenetics consortia are the Alzheimer’s Disease GeneticsConsortium (ADGC) funded by the National Institute onAging (NIA; U01 AG032984), and the Cohorts for Heart andAging Research in Genomic Epidemiology (CHARGE)funded by NIA (R01 AG033193), the National Heart, Lung,and Blood Institute (NHLBI), other NIH institutes and otherforeign governmental and nongovernmental organizations.The Discovery Phase analysis of sequence data is supportedthrough UF1AG047133 (to Drs. Farrer, Haines, Mayeux,Pericak-Vance, and Schellenberg); U01AG049505 to Dr.Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507to Dr. Wijsman; and U01AG049508 to Dr. Goate; and theDiscovery Extension Phase analysis is supported throughU01AG052411 to Dr. Goate, U01AG052410 to Dr. Pericak-Vance, and U01 AG052409 to Drs. Seshadri and Fornage.Data generation and harmonization in the Follow-up Phases
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

is supported by U54AG052427 to Drs. Schellenberg andWang. The ADGC cohorts include: Adult Changes inThought (ACT supported by NIA grant U01AG006781 toDrs. Larson and Crane), the Alzheimer’s Disease Centers(ADC), the Chicago Health and Aging Project (CHAP), theMemory and Aging Project (MAP), Mayo Clinic (MAYO),Mayo Parkinson’s Disease controls, University of Miami, theMulti-Institutional Research in Alzheimer’s Genetic Epide-miology Study (MIRAGE), the National Cell Repository forAlzheimer’s Disease (NCRAD), theNational Institute on AgingLateOnset Alzheimer’s Disease Family Study (NIA-LOAD), theReligious Orders Study (ROS), the Texas Alzheimer’s Researchand Care Consortium (TARC), Vanderbilt University/CaseWestern Reserve University (VAN/CWRU), the WashingtonHeights-Inwood Columbia Aging Project (WHICAP supportedby NIA grant RF1AG054023 to Dr. Mayeux) and theWashington University Sequencing Project (WUSP), theColumbia University Hispanic-Estudio Familiar de InfluenciaGenetica de Alzheimer (EFIGA supported by NIA grantRF1AG015473 to Dr. Mayeux), the University of Toronto(UT), and Genetic Differences (GD). Analysis of ADGCcohorts was supported by NIA grants R01AG048927 andRF1AG057519 to Dr. Farrer. Efforts of ADGC investigatorswere also supported by grants from the NIA (R03AG054936)and National Library of Medicine (R01LM012535). TheCHARGE cohorts are supported in part by NHLBI in-frastructure grant HL105756 (Psaty) and RC2HL102419(Boerwinkle), and the neurology working group is supportedby the NIA R01 grant AG033193. The CHARGE cohortsparticipating in the ADSP include the following: AustrianStroke Prevention Study (ASPS), ASPS-Family study, and theProspective Dementia Registry-Austria (ASPS/PRODEM-Aus),the Atherosclerosis Risk in Communities (ARIC) Study, theCardiovascular Health Study (CHS), the Erasmus RucphenFamily Study (ERF), the Framingham Heart Study(FHS), and the Rotterdam Study (RS). ASPS is fundedby the Austrian Science Fond (FWF) grant numberP20545-P05 and P13180 and the Medical University ofGraz. The ASPS-Fam is funded by the Austrian ScienceFund (FWF) project I904, the EU Joint Programme—Neurodegenerative Disease Research (JPND) in frame ofthe BRIDGET project (Austria, Ministry of Science) andthe Medical University of Graz and the SteiermarkischeKrankenanstalten Gesellschaft. PRODEM-Austria is supportedby the Austrian Research Promotion agency (FFG) (ProjectNo. 827462) and by the Austrian National Bank (AnniversaryFund, project 15435). ARIC research is carried out asa collaborative study supported by NHLBI contracts(HHSN268201100005C, HHSN268201100006C,HHSN268201100007C, HHSN268201100008C,HHSN268201100009C, HHSN268201100010C,HHSN268201100011C, and HHSN268201100012C).Neurocognitive data in ARIC is collected by U012U01HL096812, 2U01HL096814, 2U01HL096899,2U01HL096902, 2U01HL096917 from the NIH (NHLBI,NINDS, NIA, and NIDCD) and with previous brain MRIexaminations funded by R01-HL70825 from the NHLBI. CHS
research was supported by contracts HHSN268201200036C,HHSN268200800007C, N01HC55222, N01HC85079,N01HC85080, N01HC85081, N01HC85082, N01HC85083,N01HC85086, and grants U01HL080295 and U01HL130114from the NHLBI with additional contribution from theNINDS. Additional support was provided by R01AG023629,R01AG15928, and R01AG20098 from the NIA. FHS researchis supported by NHLBI contracts N01-HC-25195 andHHSN268201500001I. This study was also supported byadditional grants from the NIA (R01s AG054076, AG049607,and AG033040) and NINDS (R01 NS017950). The ERFstudy as a part of EUROSPAN (European Special PopulationsResearch Network) was supported by European CommissionFP6 STRP grant number 018947 (LSHG-CT-2006-01947)and also received funding from the European Community’sSeventh Framework Programme (FP7/2007-2013)/grantagreement HEALTH-F4-2007-201413 by the EuropeanCommission under the programme “Quality of Life andManagement of the Living Resources” of 5th FrameworkProgramme (no. QLG2-CT-2002-01254). High-throughputanalysis of the ERF data was supported by a joint grant from theNetherlands Organization for Scientific Research and theRussian Foundation for Basic Research (NWO-RFBR047.017.043). The Rotterdam Study is funded by ErasmusMedical Center and Erasmus University, Rotterdam, theNetherlands Organization for Health Research and Develop-ment (ZonMw), the Research Institute for Diseases in theElderly (RIDE), the Ministry of Education, Culture andScience, the Ministry for Health, Welfare and Sports, theEuropean Commission (DG XII), and the municipality ofRotterdam. Genetic data sets are also supported by theNetherlands Organization of Scientific Research NWO Invest-ments (175.010.2005.011, 911-03-012), the Genetic Labora-tory of the Department of Internal Medicine, ErasmusMC, theResearch Institute for Diseases in the Elderly (014-93-015;RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO),Netherlands Consortium for Healthy Aging (NCHA), project050-060-810. All studies are grateful to their participants,faculty, and staff. The content of these manuscripts is solely theresponsibility of the authors and does not necessarily representthe official views of the NIH or the U.S. Department of Healthand Human Services. The ADES-FR study was funded bygrants from the Clinical Research Hospital Program from theFrench Ministry of Health (GMAJ, PHRC, 2008/067), theCNR-MAJ, the JPND PERADES, the GENMED labex(LABEX GENMED ANR-10-LABX-0013), and the FP7AgedBrainSysBio. Whole exome sequencing in the 3C-Dijonstudy was funded by the Fondation Leducq. This work wassupported by the France Genomique National infrastructure,funded as part of the Investissements d’Avenir programmanaged by the Agence Nationale pour la Recherche (ANR-10-INBS-09), the Centre National de Recherche enGenomiqueHumaine, theNational Foundation forAlzheimer’sdisease and related disorders, the Institut Pasteur de Lille,Inserm, the Lille Metropole Communaute Urbaine council,and the French government’s LABEX (laboratory of excellence
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

program investment for the future) DISTALZ grant (De-velopment of Innovative Strategies for a Transdisciplinaryapproach to Alzheimer’s disease). The 3C Study supports arelisted on the Study Website (three-city-study.com). TheFinnAD Study at the University of Tampere was supportedby The Academy of Finland: grants 286284 (T.L.), Compet-itive State Research Financing of the Expert Responsibility areaof Tampere University Hospitals (grant X51001); Juho VainioFoundation; Paavo Nurmi Foundation; Finnish Foundationfor Cardiovascular Research; Finnish Cultural Foundation;Tampere Tuberculosis Foundation; Yrjo Jahnsson Founda-tion; Signe and Ane Gyllenberg Foundation; and DiabetesResearch Foundation of Finnish Diabetes Association. Thethree LSACs are the Human Genome Sequencing Center atthe Baylor College of Medicine (U54 HG003273), the BroadInstitute Genome Center (U54HG003067), and the Wash-ington University Genome Institute (U54HG003079).
Study fundingSupported by the NIH, primarily the National Institute onAging (NIA), the National Heart, Lung, and Blood Institute,and the National Human Genome Research Institute. Pri-mary support includes the Alzheimer’s Disease GeneticsConsortium funded by NIA (U01 AG032984), and theCohorts for Heart and Aging Research in Genomic Epide-miology (CHARGE) funded by NIA (R01 AG033193), theHuman Genome Sequencing Center at the Baylor Collegeof Medicine (U54 HG003273), the Broad Institute Ge-nome Center (U54HG003067), and the WashingtonUniversity Genome Institute (U54HG003079). Additionalfunding of contributing sites is noted below in theacknowledgements.
DisclosureG. Beecham has received research support from the NIH andthe Department of Defense. B. Vardarajan reports no dis-closures. E. Blue has received research support from the NIHand the Cystic Fibrosis Foundation and has been a grant re-viewer for the NIH. W. Bush has served on the editorial boardfor BMC Biodata Mining and PLoS One and has receivedresearch support from the USDA, the Foundation for Foodand Agriculture, NIDDK, and the National Institute on Aging(NIA). J. Jaworski, S. Barral, and A. DeStefano report nodisclosures. K. Hamilton-Nelson has received research sup-port from the NIH. B. Kunkle reports no disclosures. E.Martin serves on the editorial board of Frontiers in StatisticalGenetics and Methodology; and holds a patent for Test forLinkage and Association in General Pedigrees: The PedigreeDisequilibrium Test. A. Naj, F. Rajabli, and C. Reitz report nodisclosures. T. Thornton has received research support fromthe NIH. C. van Duijn reports no disclosures. A. Goate hasserved on the scientific advisory boards of Denali Therapeu-tics, Pfizer, and DZNE; has received travel funding/speakerhonoraria from the Rainwater Foundation, the Indiana Uni-versity ADRC advisory board, andWellcome Trust; serves onthe editorial board of eLife; holds patents for PSENmutationsin AD, Tau mutations in FTD, and TDP43 mutations in
ALS\FTD; has been a consultant for Cognition Therapeutics,AbbVie, and Biogen; has received research support from FPrime, NIA, Rainwater Charitable Foundation, and JPBFoundation; and receives royalty payments from TaconicIndustries for tau mutation patent, and from Athena Diag-nostics for TDP43 mutation testing. S. Seshadri serves on theeditorial boards of the Journal of Alzheimer’s Disease, Stroke,and Neurology and has received research support from NIAand NINDS. L. Farrer serves on the editorial boards of theAmerican Journal of Alzheimer’s Disease & Other Dementias,Clinical Genetics, and the Journal of Clinical Medicine; holdsa patent (pending) for Use of PLXNA4 as a drug target andbiomarker for Alzheimer disease; has been a consultant forNovartis Pharmaceuticals, Gerson Lerman, and GuidepointGlobal; has received research support from the NIH, theFidelity Foundation, and the Thome Memorial Foundation;and was a consultant for legal proceedings involving Finnegan& Associates, LLP. E. Boerwinkle has received travel fundingand speaker honoraria from the Harvard School of PublicHealth and the Metabolomics Forum in Cambridge, UnitedKingdom; serves on the editorial board of Annals of Epide-miology; is a scientific officer at Codified Genomics, LLC; andhas received research support from the NIH. G. Schellenberghas served on scientific advisory boards for the Alzheimer’sAssociation, the Society of Progressive Supranuclear Palsy,the United Kingdom Parkinson Disease Center, UniversityCollege London, the Alzheimer’s Disease Sequence Project(co-chair), Structural Variant Work Group, the Alzheimer’sDisease Sequence Project, Mayo Clinic Rochester UdallCenter, University of Miami Udall Center, Discovery As-sessment Panel, and the Oxford Parkinson’s Disease Centre;has received travel funding/speaker honoraria from Alz-heimer’s Disease Center, CurePSP, the University of Cal-ifornia San Diego, Keystone Symposia, Southern CaliforniaAlzheimer’s Disease Research Conference, University ofCalifornia Institute for Memory Impairment and Neurologi-cal Disorders, NIH, Novartis, McKnight Brain Institute,University of Florida, Keep Memory Alive Event Center,Cleveland Clinic, Accelerated Medicines Program, PSP/LewyBody Disease Think-Tank, Alzheimer’s Disease Center,American Association of Neuropathologists, Fusion Confer-ences, Center for Public Health Genomics, Columbia Uni-versity, PSP Genetics Consortium, Tetra Institute,Rockefeller University, Blechman Foundation, Niigata Uni-versity, Xuan Wu Hospital (Capital Medical University),Genetics of Dementia Summit (United Kingdom), IcahnSchool of Medicine Mount Sinai, and Biogen; has served onthe editorial boards of the Journal of Neural Transmission,American Journal of Alzheimer’s Disease and Other Dementias,Alzheimer’s Research, Neurodegenerative Diseases, Current Alz-heimer Research, and Pathology and Laboratory Medicine In-ternational; is employed by the University of Pennsylvania;has been a consultant for Biogen; and has received researchsupport from the NIH, CurePSP, and CBD Solutions. J.Haines has served on the editorial boards of Neurogenetics,Current Protocols in Human Genetics, and Human MolecularGenetics; receives publishing royalties from John Wiley &
10 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
Sons; and has received research support from the NIH. E.Wijsman has served on the editorial boards of BMC Pro-ceedings and Faculty of 1000 and has received research supportfrom the NIH and the Metropolitan Life Foundation.
R. Mayeux has received research support from the NIH. R.Pericak-Vance reports no disclosures. Full disclosure forminformation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
APPENDIX 1 Co-investigators
Affiliation Full Name Contributions
Baylor College of Medicine Adam English Baylor College of Medicine site contributed expertise to the study design, sequencing of samples,bioinformatics analyses, quality control, data management, structural variation working group, aswell as input into both case-control and family study working groups
Divya Kalra
Donna Muzny
Evette Skinner
HarshaDoddapeneni
Huyen Dinh
Jianhong Hu
Jireh Santibanez
Joy Jayaseelan
Kim Worley
Michelle Bellair
Richard A. Gibbs
Sandra Lee
Shannon Dugan-Perez
Simon White
ViktoriyaKorchina
Waleed Nasser
William Salerno
Xiuping Liu
Yi Han
Yiming Zhu
Yue Liu
Ziad Khan
Boston University AdrienneCupples
The Boston University site contributed expertise to the study design, bioinformatics analyses,quality control, data management, structural variation working group, case-control working group,family study working group, as well as significant sample contributions
Alexa Beiser
Anita DeStefano*
Ching Ti Liu
Chloe Sarnowski
Claudia Satizabal
Dan Lancour
Devanshi Patel
Continued
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 11
APPENDIX 1 Co-investigators (continued)
Affiliation Full Name Contributions
Fangui Jenny Sun
Honghuang Lin
Jaeyoon Chung
John Farrell
Josee Dupuis
Kathy Lunetta
Lindsay Farrer*
Sudha Seshadri*
Xiaoling Zhang
Yiyi Ma
Yuning Chen
Broad Institute Eric Banks The Broad Institute site contributed expertise to the study design, sequencing of samples,bioinformatics analyses, quality control, and data management working group
Namrata Gupta
Seung Hoan Choi
Stacey Gabriel
Case Western ReserveUniversity
JonathanHaines*
The CaseWestern Reserve University site contributed expertise to the study design, bioinformaticsanalyses, quality control, data management, structural variation working group, case-controlworking group, family study working group, annotation working group, as well as samplecontributions
MariuszButkiewicz
Sandra Smieszek
Will Bush*
Yeunjoo Song
Columbia University BadriVardarajan*
The Columbia University site contributed expertise to the study design, bioinformatics analyses,quality control, datamanagement, structural variation working group, case-control working group,family study working group, annotation working group, as well as sample contributions
Christiane Reitz*
Dolly Reyes
Giuseppe Tosto
Phillip L De Jager
Richard Mayeux*
Sandra Barral*
Erasmus Medical University/Rotterdam
AshleyVanderspek
The Erasmus Medical University site contributed expertise to the study design, the family studyworking group, as well as sample contributions
Cornelia vanDuijn*
M Afran Ikram
Najaf Amin
Shahzad Amad
Sven van der Lee
Continued
12 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
APPENDIX 1 Co-investigators (continued)
Affiliation Full Name Contributions
Indiana University Kelley Faber The Indiana University site contributed expertise to the study design, data and sample collectionand management, participated in the family and case-control working groups
Tatiana Foroud
Medical University Graz,Austria
Helena Schmidt Medical University Grace contributed data and samples to the study
ReinholdSchmidt
Mount Sinai School ofMedicine
Alan Renton TheMount Sinai site contributed samples as well as expertise to the design of the study, the familyand case-control working groups, and the protective variant working group
Alison Goate*
EdoardoMarcora
Manav Kapoor
National Center forBiotechnology Information
Adam Stine National Center for Biotechnology Information site contributed expertise to data management
Michael Feolo
National Institutes of Aging Lenore J. Launer National Institute on Aging site contributed expertise to data and study management
Rush University David A Bennett Rush University site contributed samples and data to the study
Stanford University Li Charlie Xia The Stanford University site contributed expertise to the structural variation working group
University of Miami Brian Kunkle* The University of Miami site contributed expertise to the study design, bioinformatics analyses,quality control, data management, structural variation working group, case-control working group,family study working group, as well as significant sample contributions
Eden Martin*
Farid Rajabli*
Gary Beecham*
James Jaworski*
Kara Hamilton-Nelson*
MargaretPericak-Vance*
Michael Schmidt
University of Mississippi Thomas H.Mosley
The University of Mississippi site contributed samples and data to the study
University of Pennsylvania Amanda Kuzma The University of Pennsylvania site contributed expertise to the study design, bioinformaticsanalyses, quality control, data management, structural variation working group, case-controlworking group, and family study working groups
Han-Jen Lin
Liming Qu
Li-San Wang
Micah Childress
Otto Valladares
PrabhakaranGangadharan
Rebecca Cweibel
Yi Zhao
Yi-Fan Chou
Continued
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 13
APPENDIX 1 Co-investigators (continued)
Affiliation Full Name Contributions
Adam Naj*
ElisabethMlynarski
GerardSchellenberg*
John Malamon
Laura Cantwell
Nancy Zhang
Weixin Wang
Yuk Yee Leung
University of Texas Houston Eric Boerwinkle* The University of Texas Houston site contributed expertise to the study design, sequencing ofsamples, bioinformatics analyses, quality control, data management, structural variation workinggroup, as well as both case-control and family study working groups
Jan Bressler
Jennifer E. Below
Myriam Fornage
Xiaoming Liu
Xueqiu Jian
University of Washington Alejandro Q NatoJr.
The University of Washington site contributed expertise to the bioinformatics analyses, qualitycontrol, data management, structural variation working group, case-control working group, andfamily study working group
Andrea RHorimoto
Bowen Wang
Bruce Psaty
Daniela Witten
Debby Tsuang
Elizabeth Blue*
Ellen Wijsman*
Harkirat Sohi
Hiep Nguyen
Joshua C. Bis
Kenneth Rice
Lisa Brown
MichaelDorschner
Mohamad Saad
Pat Navas
Rafael Nafikov
TimothyThornton*
Tyler Day
Continued
14 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Received February 6, 2018. Accepted in final form October 3, 2018.
References1. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol 2018;25:59–70.2. Gatz M, Pedersen NL, Berg S, et al. Heritability for Alzheimer’s disease: the
study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci 1997;52:M117–M125.
3. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261:921–923.
4. Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individualsidentifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013;45:1452–1458.
5. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP,CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet2011;43:436–441.
6. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl JMed 2013;368:117–127.
7. Reitz C, Mayeux R. Alzheimer’s Disease Genetics C. TREM2 and neurodegenerativedisease. N Engl J Med 2013;369:1564–1565.
8. Le Guennec K, Nicolas G, Quenez O, et al. ABCA7 rare variants and Alzheimerdisease risk. Neurology 2016;86:2134–2137.
9. Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimerdisease. Ann Neurol 2015;77:215–227.
10. Beecham GW, Bis JC, Martin ER, et al. The Alzheimer’s disease sequencing project:study design and sample selection. Neurol Genet 2017;3:e194.
11. Preston MD, Dudbridge F. Utilising family-based designs for detecting rare variantdisease associations. Ann Hum Genet 2014;78:129–140.
12. Vardarajan BN, Barral S, Jaworski J, et al. Whole genome sequence analysis of ca-ribbean hispanic families with late-onset Alzheimer’s disease. Ann Clin Transl Neurol2018;5:406–417.
13. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadian EM. Clinicaldiagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group underthe auspices of department of Health and human services task force on Alzheimer’sdisease. Neurology 1984;34:939–944.
14. Naj AC, Lin H, Vardarajan BN, et al. Quality control and integration of genotypesfrom two calling pipelines for whole genome sequence data in the Alzheimer’s diseasesequencing project. Genomics Epub 2018 May 29.
15. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheelertransform. Bioinformatics 2009;25:1754–1760.
16. Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exomenext-generation sequencing data. BMC Bioinformatics 2012;13:8.
17. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principalcomponents analysis corrects for stratification in genome-wide association studies.Nat Genet 2006;38:904–909.
18. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of densegenetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101.
19. Kunkle BW, Jaworski J, Barral S, et al. Genome-wide linkage analyses of non-Hispanicwhite families identify novel loci for familial late-onset Alzheimer’s disease. Alz-heimers Dement 2016;12:2–10.
20. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a toolset for whole-genome asso-ciation and population-based linkage analysis. Am J Hum Genet 2007;81:559–575.
21. Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpretingand reporting linkage results. Nat Genet 1995;11:241–247.
22. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A generalframework for estimating the relative pathogenicity of human genetic variants. NatGenet 2014;46:310–315.
23. MauranoMT,Haugen E, SandstromR, et al. Large-scale identification of sequence variantsinfluencing human transcription factor occupancy. Nat Genet 2015;47:1393–1400.
24. The 1000 Genomes Project Consortium. A global reference for human genetic var-iation. Nature 2015;526:68–74.
25. Butkiewicz M, Blue E, Leung F, et al. Functional annotation of genomic variants instudies of Late-onset Alzheimer’s disease. Bioinformatics 2018;34:2724–2731.
26. Yan Q, Tiwari HK, Yi N, et al. A sequence kernel association test for dichotomoustraits in family samples under a generalized linear mixed model. HumHered 2015;79:60–68.
27. Kholi MA, Cukier HN, Hamilton-Nelson KL, et al. Segregation of a rare TTC3variant in an extended family with late-onset Alzheimer disease. Neurol Genet2016;2:e41.
28. Bis JC, Jian X, Kunkle BW, et al. Whole exome sequencing study identifies novel rareand common Alzheimer’s-Associated variants involved in immune response andtranscriptional regulation. Mol Psychiatry Epub 2018 Aug 14.
29. Mele M, Ferreira PG, Reverter F, et al. Human genomics: the human transcriptomeacross tissues and individuals. Science 2015;348:660–665.
30. Beisiegel U, Weber W, Ihrke G, Herz J, Stanley KK. The LDL-receptor-relatedprotein, LRP, is an apolipoprotein E-binding protein. Nature 1989;341:162–164.
31. Vazquez-Higuera JL, Mateo I, Sanchez-Juan P, et al. Genetic interaction between tauand the apolipoprotein E receptor LRP1 Increases Alzheimer’s disease risk. DementGeriatr Cogn Disord 2009;28:116–120.
32. Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH. CAPON: a proteinassociated with neuronal nitri oxide synthase that regulates its interactions withPSD95. Neuron 1998;20:115–124.
33. Galimberti D, Scarpini E, Venturelli E, et al. Association of a NOS1 promoter repeatwith Alzheimer’s disease. Neurobiol Aging 2008;29:1359–1365.
34. Villarreal-MolinaMT, Aguilar-Salinas CA, Rodrıguez-Cruz M, et al. The ATP-bindingcassette transporter A1 R230C variant affects HDL cholesterol levels and BMI in theMexican population: association with obesity and obesity-related comorbidities. Di-abetes 2007;56:1881–1887.
35. Rhyne J, Mantaring MM, Gardner DF, Miller M. Multiple splice defects in ABCA1cause low HDL-C in a family with hypoalphalipoproteinemia and premature coronarydisease. BMC Med Genet 2009;10:1.
36. Cenarro A, Artieda M, Castillo S, et al. A common variant in the ABCA1 gene isassociated with a lower risk for premature coronary heart disease in familial hyper-cholesterolaemia. J Med Genet 2003;40:163–168.
37. Brooks-Wilson A,Marcil M, Clee SM, et al. Mutations in ABC1 in Tangier disease andfamilial high-density lipoprotein deficiency. Nat Genet 1999;22:336–345.
38. Bodzioch M, Orso E, Klucken J, et al. The gene encoding ATP-binding cassettetransporter 1 is mutated in Tangier disease. Nat Genet 1999;22:347–351.
39. Reitz C. Dyslipidemia and the risk of Alzheimer’s disease. Curr Atheroscler Rep 2013;15:307.
40. KimWS, Hill AF, Fitzgerald ML, et al. Wild type and Tangier disease ABCA1mutantsmodulate cellular amyloid-β production independent of cholesterol efflux activity.J Alzheimers Dis 2011;27:441–452.
APPENDIX 1 Co-investigators (continued)
Affiliation Full Name Contributions
Washington University St.Louis
Carlos Cruchaga The Washington University St. Louis site contributed expertise to the study design, sequencing ofsamples, bioinformatics analyses, quality control, data management, structural variation workinggroup, as well as both case-control and family study working groups
Daniel C. Koboldt
David E. Larson
ElizabethAppelbaum
JasonWaligorski
LucindaAntonacci-Fulton
Richard K. Wilson
Robert S. Fulton
Asterisks (*) indicate coinvestigators whose contributions were sufficient for authorship.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 15

41. Pervaiz MA, Gau G, Jaffe AS, Saenger AK, Baudhuin L, Ellison J. A non-classical pre-sentation of tangier disease with three ABCA1 mutations. JIMD Rep 2012;4:109–111.
42. Fan J1, Donkin J, Wellington C. Greasing the wheels of Abeta clearance in Alzheimer’sdisease: the role of lipids and apolipoprotein E. Biofactors 2009;35:239–248.
43. Fitz NF, Cronican AA, Saleem M, et al. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice.J Neurosci 2012;32:13125–13136.
44. Shahim P1, Bochem AE, Mattsson N, et al. Plasma amyloid-β in patients with Tangierdisease. J Alzheimers Dis 2013;35:307–312.
45. Palomer X1, Mauricio D, Rodrıguez-Espinosa J, et al. Evaluation of two nonisotopicimmunoassays for determination of glutamic acid decarboxylase and tyrosine phos-phatase autoantibodies in serum. Clin Chem 2004;50:1378–1382.
46. Yu L, Chibnik LB, Srivastava GP, et al. Association of Brain DNA methylation inSORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis ofAlzheimer disease. JAMA Neurol 2015;72:15–24.
47. Mather CA,Mooney SD, Salipante SJ. CADD score has limited clinical validity for theidentification of pathogenic variants in noncoding regions in a hereditary cancer panel.Genet Med 2016;18:1269–1275.
16 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

ARTICLE OPEN ACCESS
Mutation in POLR3K causes hypomyelinatingleukodystrophy and abnormal ribosomal RNAregulationImen Dorboz, PhD, Helene Dumay-Odelot, PhD, Karima Boussaid, MD, Yosra Bouyacoub, PhD,
Pauline Barreau, MD, Simon Samaan, PharmD, PhD, Haifa Jmel, PhD, Eleonore Eymard-Pierre, MDIng,
Claude Cances, MD, Celine Bar, MD, Anne-Lise Poulat, MD, Christophe Rousselle, MD, Florence Renaldo, MD,
Monique Elmaleh- Berges, MD, Martin Teichmann, PhD, and Odile Boespflug-Tanguy, MD, PhD
Neurol Genet 2018;4:e289. doi:10.1212/NXG.0000000000000289
Correspondence
Dr. Boespflug-Tanguy
AbstractObjectiveTo identify the genetic cause of hypomyelinating leukodystrophy in 2 consanguineous families.
MethodsHomozygosity mapping combined with whole-exome sequencing of consanguineous familieswas performed. Mutation consequences were determined by studying the structural change ofthe protein and by the RNA analysis of patients’ fibroblasts.
ResultsWe identified a biallelic mutation in a gene coding for a Pol III–specific subunit, POLR3K(c.121C>T/p.Arg41Trp), that cosegregates with the disease in 2 unrelated patients. Patientsexpressed neurologic and extraneurologic signs found in POLR3A- and POLR3B-related leu-kodystrophies with a peculiar severe digestive dysfunction. The mutation impaired thePOLR3K-POLR3B interactions resulting in zebrafish in abnormal gut development. Functionalstudies in the 2 patients’ fibroblasts revealed a severe decrease (60%–80%) in the expression of5S and 7S ribosomal RNAs in comparison with control.
ConclusionsThese analyses underlined the key role of ribosomal RNA regulation in the development andmaintenance of the white matter and the cerebellum as already reported for diseases related togenes involved in transfer RNA or translation initiation factors.
From the INSERM UMR 1141 PROTECT (I.D., P.B., S.S., O.B.-T.), Universite Paris Diderot- Sorbonne Paris Cite; INSERM U1212-CNRS UMR 5320 (H.D.-O., M.T.), Universite de Bordeaux;Neurologie Pediatrique et Maladies Metaboliques (K.B., F.R., O.B-.T.), Centre de reference des leucodystrophies et leucoencephalopathies de cause rare (LEUKOFRANCE), CHU APHPRobert-Debre, Paris, France; LR11IPT05, Biomedical Genomics and Oncogenetics Laboratory (H.J., Y.B.), Institut Pasteur de Tunis; Department of Medical Genetics, UF MolecularGenetics (S.S.), CHU APHP Robert-Debre Paris; Service de Cytogenetique Medicale (E.E.P.), CHU Clermont-Ferrand; Neurologie Pediatrique (C.C.), Endocrinologie Pediatrique (C.B.),CHU Hopital des Enfants, Toulouse; Hopital FemmeMere Enfant, Neurologie Pediatrique (A.L.P., C.R.), Hospices Civils de Lyon, Bron; Department of Pediatric Radiology (M.E.-B.), CHUAPHP Robert-Debre, Paris, France.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

RNA polymerase III (Pol III) mutations have been impli-cated in autosomal recessive hypomyelinating leukodystro-phies (HLD7 [MIM 607694] and HLD8 [MIM 614381]).HLDs are characterized by a normal T1 abnormal hyper T2/fluid-attenuated inversion recovery (FLAIR) signal of thewhite matter on magnetic resonance imaging (MRI).1,2 Theclinical presentation is variable from infantile to juvenile/adult-onset forms with motor decline manifest as progressivecerebellar dysfunction and mild cognitive regression. Otherfeatures may include hypo/oligodontia, myopia, dysmorphia,and hypogonadotropic hypogonadism.
Pol III, composed of 17 subunits, is the largest eukaryoticRNA polymerase. It transcribes small untranslated RNAs in-volved in cellular processes including the regulation of tran-scription (7SK RNA; Alu RNA), RNA processing (U6 RNA;H1 RNA), and translation (tRNA; 5S RNA).3 Promotersdriving transcription of these genes have been identified, cloned,and characterized.4,5 Mutations causing HLD have been repor-ted first in the POLR3A1 (MIM 614258) and POLR3B2,6
(MIM 614366) genes. More recently, mutations in thePOLR1C gene (MIM 616494) encoding a subunit shared byPol I and Pol III complexes have been reported as HLD11.7
In addition, a homozygous mutation of POLR1A (NM_616404) encoding the largest subunit of Pol I, RPA194, hasbeen described in a family affected by a demyelinating formof leukodystrophy.8
Here, we report a homozygous mutation of POLR3K (NM_606007) in 2 unrelated HLD-affected patients. POLR3K enc-odes the RPC11 subunit of Pol III, which has been implicated inthe processes of transcription termination and reinitiation.9,10
We demonstrate that the mutation affected the POLR3K-POLR3B interactions and decreased the 5S and 7SLRNA levels.
MethodsStandard protocol approvals, registrations,and patient consentsConsent was obtained from patients and their parentsaccording to the LEUKOFRANCE research program forundetermined leukodystrophies (authorization CPP AU788;CNIL 1406552; AFSSAPS B90298-60).
PatientsPatients were referred to the French reference center forleukodystrophies, LEUKOFRANCE, for diagnosis andfollow-up. DNA was extracted from white blood cells of theaffected patients and unaffected family members. Fibroblastswere obtained from skin biopsy according to our previouslyreported protocol.11
DNA analysisWe performed homozygosity mapping in all family mem-bers using GeneChip Human Mapping 250K Nsp Array,and whole-exome sequencing (IntegraGen, Evry, France)using the SureSelect V4 capture kit (Agilent, Massy,France) and the HighSeq2000 sequencer (Illumina, SanDiego, CA).12
Structural modelTo get insight on the mutation effect on POLR3K protein(UniProtKB: Q9Y2Y1) structure, we performed a molecularmodeling analysis. The Protein Data Bank (PDB) files and 2Dstructures were predicted using the PSIPRED server (bioinf.cs.ucl.ac.uk/psipred/).13
To predict the interaction between POLR3K and POLR3B,we used Phyre 2 (sbg.bio.ic.ac.uk/phyre2) to have the PDBfile of POLR3B (UniProtKB: Q9NW08), and we per-formed the protein-protein docking with ClusPro server(cluspro.org).14–16
Fibroblasts analysis
Cell cultureCell lines were grown in Dulbecco Modified Eagle Mediumsupplemented with 15% fetal bovine serum (Invitrogen, Ill-kirch, France), 1% minimum essential medium nonessentialamino acid solution (Sigma, Saint-Quentin-Fallavier, France),100 U/ml penicillin, and 100 μg/mL streptomycin (Invi-trogen). Cell lines were maintained at 37°C in a humidified5% CO2 atmosphere. All extracts were made from subcon-fluent cells in the exponential phase of growth.
RNA extractionAfter several passages (<8) under tissue culture conditions,total RNA was extracted using TRIzol reagent (Invitrogen,Illkirch, France), according to the manufacturer’s protocol.Genomic DNA was removed using the turboDNA free kit(Ambion). RNA concentrations were determined usinga NanoDrop spectrophotometer (Nanodrop Technologies,Wilmington, DE). RNAs integrity was determined with anAgilent 2100 bioanalyzer (Palo Alto, CA). RNA measure-ments are automatically submitted to an algorithm that allowsstandardized control of RNA quality and the calculation of anRNA integrity number.17
Quantitative RT-PCRFor each sample, 2 μg of total RNA was reverse transcribedusing Scientific Maxima Reverse transcriptase (Fisher Scientific,Illkirch, France) and random hexamer primers (Fermentas,Fisher Scientific, Illkirch, France). Expression of POLR3DRNA,POLR3K RNA, and U2 RNA, and that of RNA polymerase
GlossaryHLD = hypomyelinating leukodystrophy.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
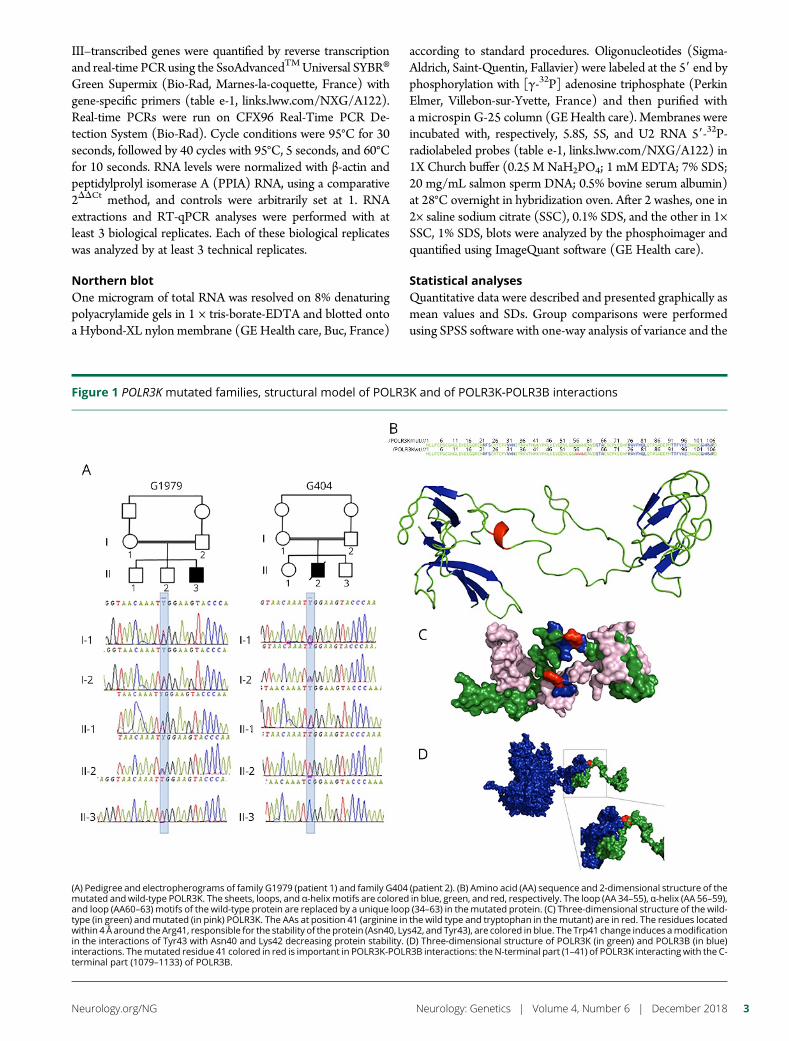
III–transcribed genes were quantified by reverse transcriptionand real-time PCR using the SsoAdvancedTMUniversal SYBR®Green Supermix (Bio-Rad, Marnes-la-coquette, France) withgene-specific primers (table e-1, links.lww.com/NXG/A122).Real-time PCRs were run on CFX96 Real-Time PCR De-tection System (Bio-Rad). Cycle conditions were 95°C for 30seconds, followed by 40 cycles with 95°C, 5 seconds, and 60°Cfor 10 seconds. RNA levels were normalized with β-actin andpeptidylprolyl isomerase A (PPIA) RNA, using a comparative2DDCt method, and controls were arbitrarily set at 1. RNAextractions and RT-qPCR analyses were performed with atleast 3 biological replicates. Each of these biological replicateswas analyzed by at least 3 technical replicates.
Northern blotOne microgram of total RNA was resolved on 8% denaturingpolyacrylamide gels in 1 × tris-borate-EDTA and blotted ontoa Hybond-XL nylon membrane (GE Health care, Buc, France)
according to standard procedures. Oligonucleotides (Sigma-Aldrich, Saint-Quentin, Fallavier) were labeled at the 59 end byphosphorylation with [γ-32P] adenosine triphosphate (PerkinElmer, Villebon-sur-Yvette, France) and then purified witha microspin G-25 column (GEHealth care). Membranes wereincubated with, respectively, 5.8S, 5S, and U2 RNA 59-32P-radiolabeled probes (table e-1, links.lww.com/NXG/A122) in1X Church buffer (0.25 M NaH2PO4; 1 mM EDTA; 7% SDS;20 mg/mL salmon sperm DNA; 0.5% bovine serum albumin)at 28°C overnight in hybridization oven. After 2 washes, one in2× saline sodium citrate (SSC), 0.1% SDS, and the other in 1×SSC, 1% SDS, blots were analyzed by the phosphoimager andquantified using ImageQuant software (GE Health care).
Statistical analysesQuantitative data were described and presented graphically asmean values and SDs. Group comparisons were performedusing SPSS software with one-way analysis of variance and the
Figure 1 POLR3K mutated families, structural model of POLR3K and of POLR3K-POLR3B interactions
(A) Pedigree and electropherograms of family G1979 (patient 1) and family G404 (patient 2). (B) Amino acid (AA) sequence and 2-dimensional structure of themutated andwild-type POLR3K. The sheets, loops, and α-helix motifs are colored in blue, green, and red, respectively. The loop (AA 34–55), α-helix (AA 56–59),and loop (AA60–63) motifs of the wild-type protein are replaced by a unique loop (34–63) in themutated protein. (C) Three-dimensional structure of the wild-type (in green) andmutated (in pink) POLR3K. The AAs at position 41 (arginine in the wild type and tryptophan in themutant) are in red. The residues locatedwithin 4 Å around the Arg41, responsible for the stability of the protein (Asn40, Lys42, and Tyr43), are colored in blue. The Trp41 change induces amodificationin the interactions of Tyr43 with Asn40 and Lys42 decreasing protein stability. (D) Three-dimensional structure of POLR3K (in green) and POLR3B (in blue)interactions. Themutated residue 41 colored in red is important in POLR3K-POLR3B interactions: the N-terminal part (1–41) of POLR3K interactingwith the C-terminal part (1079–1133) of POLR3B.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
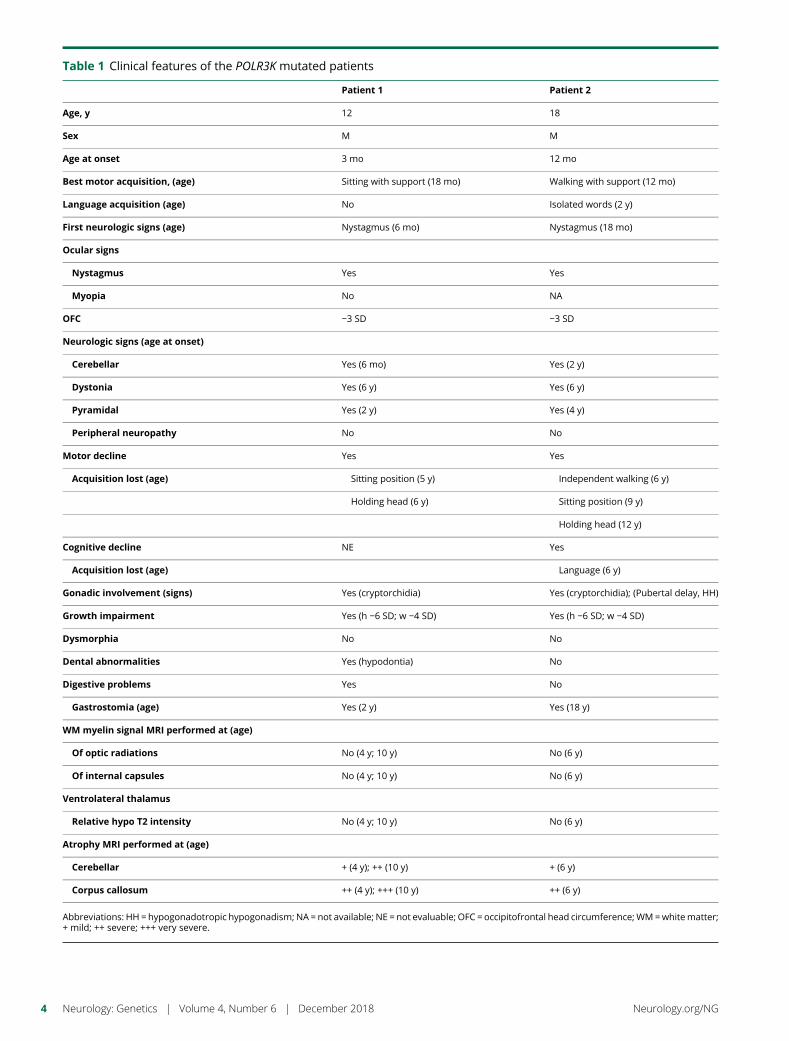
Table 1 Clinical features of the POLR3K mutated patients
Patient 1 Patient 2
Age, y 12 18
Sex M M
Age at onset 3 mo 12 mo
Best motor acquisition, (age) Sitting with support (18 mo) Walking with support (12 mo)
Language acquisition (age) No Isolated words (2 y)
First neurologic signs (age) Nystagmus (6 mo) Nystagmus (18 mo)
Ocular signs
Nystagmus Yes Yes
Myopia No NA
OFC −3 SD −3 SD
Neurologic signs (age at onset)
Cerebellar Yes (6 mo) Yes (2 y)
Dystonia Yes (6 y) Yes (6 y)
Pyramidal Yes (2 y) Yes (4 y)
Peripheral neuropathy No No
Motor decline Yes Yes
Acquisition lost (age) Sitting position (5 y) Independent walking (6 y)
Holding head (6 y) Sitting position (9 y)
Holding head (12 y)
Cognitive decline NE Yes
Acquisition lost (age) Language (6 y)
Gonadic involvement (signs) Yes (cryptorchidia) Yes (cryptorchidia); (Pubertal delay, HH)
Growth impairment Yes (h −6 SD; w −4 SD) Yes (h −6 SD; w −4 SD)
Dysmorphia No No
Dental abnormalities Yes (hypodontia) No
Digestive problems Yes No
Gastrostomia (age) Yes (2 y) Yes (18 y)
WM myelin signal MRI performed at (age)
Of optic radiations No (4 y; 10 y) No (6 y)
Of internal capsules No (4 y; 10 y) No (6 y)
Ventrolateral thalamus
Relative hypo T2 intensity No (4 y; 10 y) No (6 y)
Atrophy MRI performed at (age)
Cerebellar + (4 y); ++ (10 y) + (6 y)
Corpus callosum ++ (4 y); +++ (10 y) ++ (6 y)
Abbreviations: HH = hypogonadotropic hypogonadism; NA = not available; NE = not evaluable; OFC = occipitofrontal head circumference; WM=whitematter;+ mild; ++ severe; +++ very severe.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
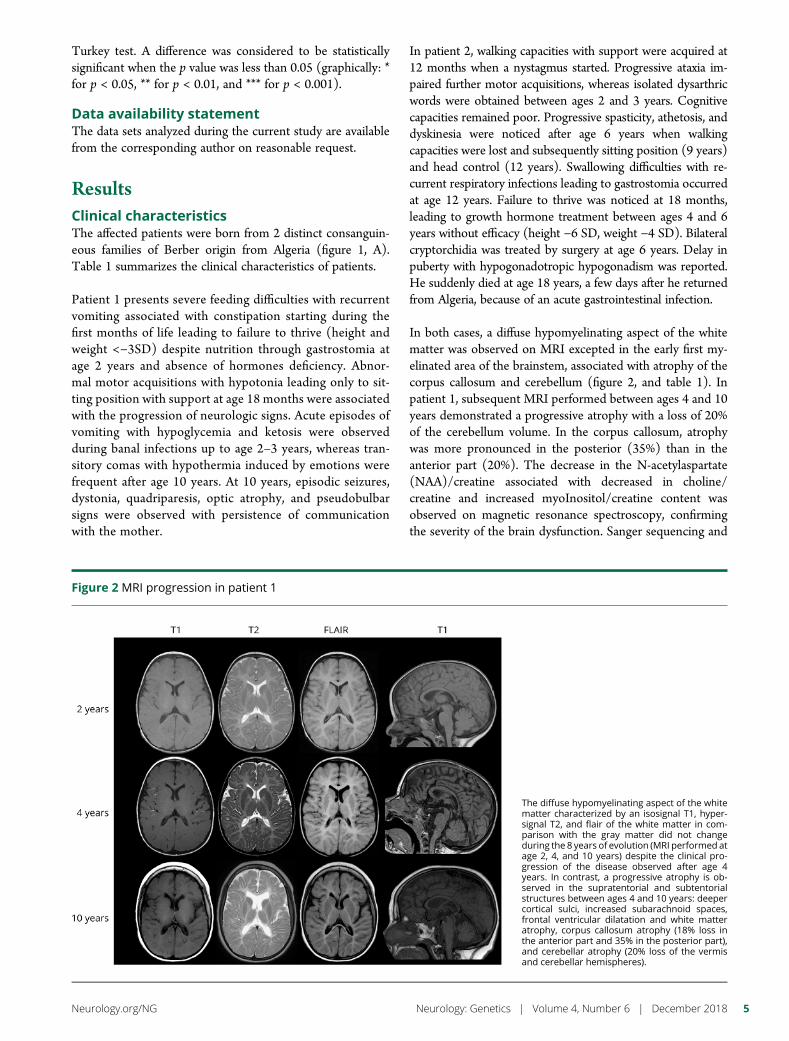
Turkey test. A difference was considered to be statisticallysignificant when the p value was less than 0.05 (graphically: *for p < 0.05, ** for p < 0.01, and *** for p < 0.001).
Data availability statementThe data sets analyzed during the current study are availablefrom the corresponding author on reasonable request.
ResultsClinical characteristicsThe affected patients were born from 2 distinct consanguin-eous families of Berber origin from Algeria (figure 1, A).Table 1 summarizes the clinical characteristics of patients.
Patient 1 presents severe feeding difficulties with recurrentvomiting associated with constipation starting during thefirst months of life leading to failure to thrive (height andweight <−3SD) despite nutrition through gastrostomia atage 2 years and absence of hormones deficiency. Abnor-mal motor acquisitions with hypotonia leading only to sit-ting position with support at age 18 months were associatedwith the progression of neurologic signs. Acute episodes ofvomiting with hypoglycemia and ketosis were observedduring banal infections up to age 2–3 years, whereas tran-sitory comas with hypothermia induced by emotions werefrequent after age 10 years. At 10 years, episodic seizures,dystonia, quadriparesis, optic atrophy, and pseudobulbarsigns were observed with persistence of communicationwith the mother.
In patient 2, walking capacities with support were acquired at12 months when a nystagmus started. Progressive ataxia im-paired further motor acquisitions, whereas isolated dysarthricwords were obtained between ages 2 and 3 years. Cognitivecapacities remained poor. Progressive spasticity, athetosis, anddyskinesia were noticed after age 6 years when walkingcapacities were lost and subsequently sitting position (9 years)and head control (12 years). Swallowing difficulties with re-current respiratory infections leading to gastrostomia occurredat age 12 years. Failure to thrive was noticed at 18 months,leading to growth hormone treatment between ages 4 and 6years without efficacy (height −6 SD, weight −4 SD). Bilateralcryptorchidia was treated by surgery at age 6 years. Delay inpuberty with hypogonadotropic hypogonadism was reported.He suddenly died at age 18 years, a few days after he returnedfrom Algeria, because of an acute gastrointestinal infection.
In both cases, a diffuse hypomyelinating aspect of the whitematter was observed on MRI excepted in the early first my-elinated area of the brainstem, associated with atrophy of thecorpus callosum and cerebellum (figure 2, and table 1). Inpatient 1, subsequent MRI performed between ages 4 and 10years demonstrated a progressive atrophy with a loss of 20%of the cerebellum volume. In the corpus callosum, atrophywas more pronounced in the posterior (35%) than in theanterior part (20%). The decrease in the N-acetylaspartate(NAA)/creatine associated with decreased in choline/creatine and increased myoInositol/creatine content wasobserved on magnetic resonance spectroscopy, confirmingthe severity of the brain dysfunction. Sanger sequencing and
Figure 2 MRI progression in patient 1
The diffuse hypomyelinating aspect of the whitematter characterized by an isosignal T1, hyper-signal T2, and flair of the white matter in com-parison with the gray matter did not changeduring the 8 years of evolution (MRI performed atage 2, 4, and 10 years) despite the clinical pro-gression of the disease observed after age 4years. In contrast, a progressive atrophy is ob-served in the supratentorial and subtentorialstructures between ages 4 and 10 years: deepercortical sulci, increased subarachnoid spaces,frontal ventricular dilatation and white matteratrophy, corpus callosum atrophy (18% loss inthe anterior part and 35% in the posterior part),and cerebellar atrophy (20% loss of the vermisand cerebellar hemispheres).
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
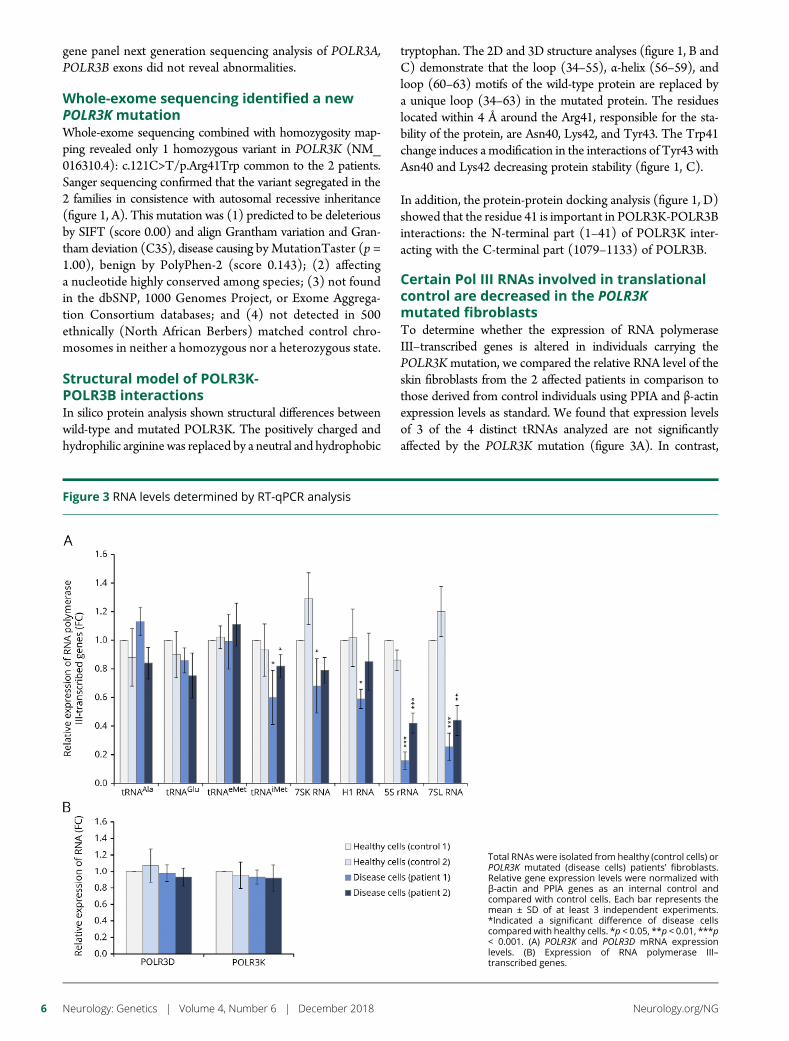
gene panel next generation sequencing analysis of POLR3A,POLR3B exons did not reveal abnormalities.
Whole-exome sequencing identified a newPOLR3K mutationWhole-exome sequencing combined with homozygosity map-ping revealed only 1 homozygous variant in POLR3K (NM_016310.4): c.121C>T/p.Arg41Trp common to the 2 patients.Sanger sequencing confirmed that the variant segregated in the2 families in consistence with autosomal recessive inheritance(figure 1, A). This mutation was (1) predicted to be deleteriousby SIFT (score 0.00) and align Grantham variation and Gran-tham deviation (C35), disease causing by MutationTaster (p =1.00), benign by PolyPhen-2 (score 0.143); (2) affectinga nucleotide highly conserved among species; (3) not foundin the dbSNP, 1000 Genomes Project, or Exome Aggrega-tion Consortium databases; and (4) not detected in 500ethnically (North African Berbers) matched control chro-mosomes in neither a homozygous nor a heterozygous state.
Structural model of POLR3K-POLR3B interactionsIn silico protein analysis shown structural differences betweenwild-type and mutated POLR3K. The positively charged andhydrophilic arginine was replaced by a neutral and hydrophobic
tryptophan. The 2D and 3D structure analyses (figure 1, B andC) demonstrate that the loop (34–55), α-helix (56–59), andloop (60–63) motifs of the wild-type protein are replaced bya unique loop (34–63) in the mutated protein. The residueslocated within 4 Å around the Arg41, responsible for the sta-bility of the protein, are Asn40, Lys42, and Tyr43. The Trp41change induces a modification in the interactions of Tyr43 withAsn40 and Lys42 decreasing protein stability (figure 1, C).
In addition, the protein-protein docking analysis (figure 1, D)showed that the residue 41 is important in POLR3K-POLR3Binteractions: the N-terminal part (1–41) of POLR3K inter-acting with the C-terminal part (1079–1133) of POLR3B.
Certain Pol III RNAs involved in translationalcontrol are decreased in the POLR3Kmutated fibroblastsTo determine whether the expression of RNA polymeraseIII–transcribed genes is altered in individuals carrying thePOLR3Kmutation, we compared the relative RNA level of theskin fibroblasts from the 2 affected patients in comparison tothose derived from control individuals using PPIA and β-actinexpression levels as standard. We found that expression levelsof 3 of the 4 distinct tRNAs analyzed are not significantlyaffected by the POLR3K mutation (figure 3A). In contrast,
Figure 3 RNA levels determined by RT-qPCR analysis
Total RNAs were isolated from healthy (control cells) orPOLR3K mutated (disease cells) patients’ fibroblasts.Relative gene expression levels were normalized withβ-actin and PPIA genes as an internal control andcompared with control cells. Each bar represents themean ± SD of at least 3 independent experiments.*Indicated a significant difference of disease cellscompared with healthy cells. *p < 0.05, **p < 0.01, ***p< 0.001. (A) POLR3K and POLR3D mRNA expressionlevels. (B) Expression of RNA polymerase III–transcribed genes.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
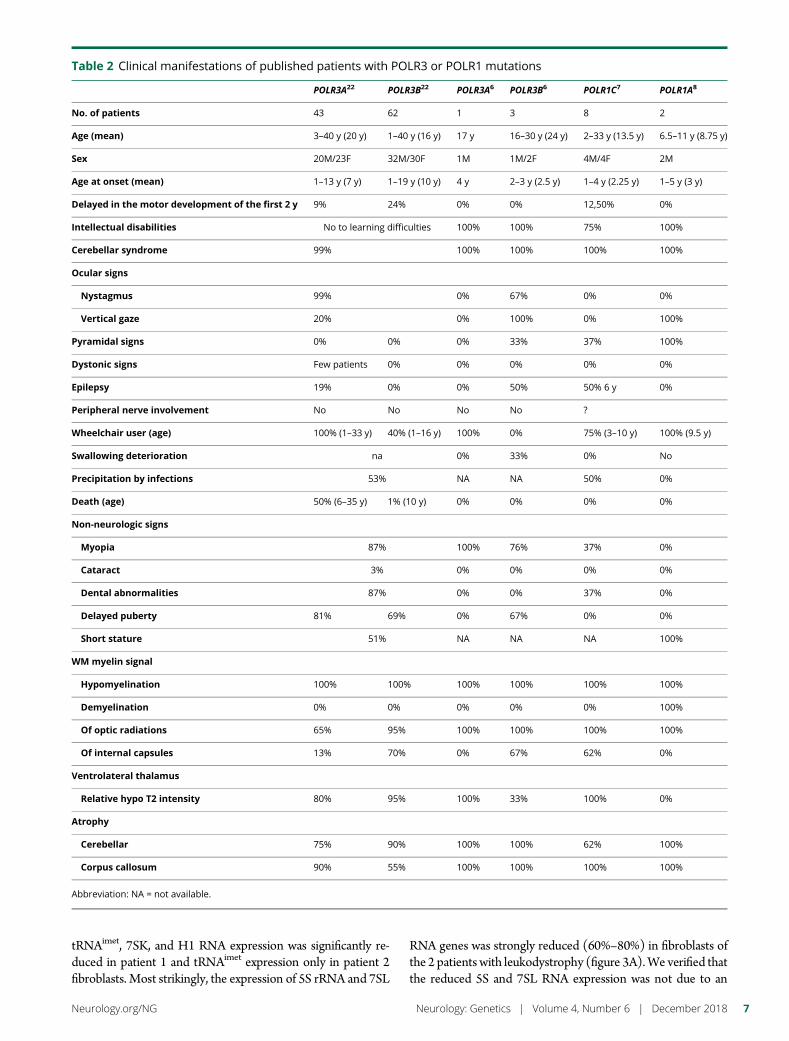
tRNAimet, 7SK, and H1 RNA expression was significantly re-duced in patient 1 and tRNAimet expression only in patient 2fibroblasts. Most strikingly, the expression of 5S rRNA and 7SL
RNA genes was strongly reduced (60%–80%) in fibroblasts ofthe 2 patients with leukodystrophy (figure 3A).We verified thatthe reduced 5S and 7SL RNA expression was not due to an
Table 2 Clinical manifestations of published patients with POLR3 or POLR1 mutations
POLR3A22 POLR3B22 POLR3A6 POLR3B6 POLR1C7 POLR1A8
No. of patients 43 62 1 3 8 2
Age (mean) 3–40 y (20 y) 1–40 y (16 y) 17 y 16–30 y (24 y) 2–33 y (13.5 y) 6.5–11 y (8.75 y)
Sex 20M/23F 32M/30F 1M 1M/2F 4M/4F 2M
Age at onset (mean) 1–13 y (7 y) 1–19 y (10 y) 4 y 2–3 y (2.5 y) 1–4 y (2.25 y) 1–5 y (3 y)
Delayed in the motor development of the first 2 y 9% 24% 0% 0% 12,50% 0%
Intellectual disabilities No to learning difficulties 100% 100% 75% 100%
Cerebellar syndrome 99% 100% 100% 100% 100%
Ocular signs
Nystagmus 99% 0% 67% 0% 0%
Vertical gaze 20% 0% 100% 0% 100%
Pyramidal signs 0% 0% 0% 33% 37% 100%
Dystonic signs Few patients 0% 0% 0% 0% 0%
Epilepsy 19% 0% 0% 50% 50% 6 y 0%
Peripheral nerve involvement No No No No ?
Wheelchair user (age) 100% (1–33 y) 40% (1–16 y) 100% 0% 75% (3–10 y) 100% (9.5 y)
Swallowing deterioration na 0% 33% 0% No
Precipitation by infections 53% NA NA 50% 0%
Death (age) 50% (6–35 y) 1% (10 y) 0% 0% 0% 0%
Non-neurologic signs
Myopia 87% 100% 76% 37% 0%
Cataract 3% 0% 0% 0% 0%
Dental abnormalities 87% 0% 0% 37% 0%
Delayed puberty 81% 69% 0% 67% 0% 0%
Short stature 51% NA NA NA 100%
WM myelin signal
Hypomyelination 100% 100% 100% 100% 100% 100%
Demyelination 0% 0% 0% 0% 0% 100%
Of optic radiations 65% 95% 100% 100% 100% 100%
Of internal capsules 13% 70% 0% 67% 62% 0%
Ventrolateral thalamus
Relative hypo T2 intensity 80% 95% 100% 33% 100% 0%
Atrophy
Cerebellar 75% 90% 100% 100% 62% 100%
Corpus callosum 90% 55% 100% 100% 100% 100%
Abbreviation: NA = not available.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

impaired expression of Pol III subunits by quantifying in par-allel POLR3K and POLR3D mRNA expression levels (figure3B). To verify the decrease in Pol III RNA expression levels byan independent method, we performedNorthern blot analyses.By comparing 5.8S rRNA with U2 snRNA levels, we observeda relative increase in U2 snRNA levels, indicating that theoverall levels of 5.8S rRNA were slightly lower in patients withleukodystrophy than in controls (figure e-1A, links.lww.com/NXG/A121). Comparing 5S rRNA levels with U2 snRNAlevels confirmed significantly lower expression of 5S rRNA inpatient fibroblasts compared with control fibroblasts (figuree-1B). These results clearly indicate that the (p.Arg41Trp)POLR3Kmutation reduces 5S and 7SL RNA levels, potentiallycontributing to the development of the disease.
DiscussionIn this article, we reported a novel homozygous missense variantin the POLR3K gene in 2 HLD-affected patients from 2 con-sanguineous families using whole exome sequencing and func-tional analysis. Following the latest the American College ofMedical Genetics and Genomics and the Association for Mo-lecular Pathology guidelines,18 this variant has 1 strong (func-tional analysis), at least 2 moderate (highly conserved and verylow allele frequency), and 2 supporting (bioinformatics andsegregation analysis) criteria of pathogenicity. The variant is notfound in polymorphisms databases and in controls of the sameBerber ethnic background. The p.Arg41Trp substitution foundis located within domain II of the POLR3K protein, a highlyconserved region from yeast to human.9 Docking analysis of thePOLR3K missense substitution p.Arg41Trp suggested less sta-bility in the interactions of the RPC128 subunits encoded byPOLR3B and RPC11 encoded by POLR3K. Of interest, a mu-tation of the POLR3B gene has been reported in zebrafish withimpaired proliferation of digestive organs. The zebrafish muta-tion reduced RPC11 association with Pol III and the tran-scription of tRNA and 7SL genes. Overexpression of RPC11 inthis model system could rescue some of these defects.19
Our patients expressed neurologic signs classically found inpatients mutated for POLR3A, POLR3B, or POLR1C (table 2)as early hypotonia, nystagmus, ataxia associated with hypo-myelinated white matter, and cerebellar/corpus callosum at-rophy. Extraneurologic signs such as dental abnormalities, shortstature, and hypogonadism are also frequently reported. Dis-ease progression for both patients appeared in the most severerange of Pol III–related leukodystrophies in terms of age atonset (<18 months) and death (<20 years) and motor andcognitive development (no independent walking) and degra-dation (age 4–6 years). However, the progressivity of the mi-crocephaly and the severe spasticity and dystonia observedbefore age 10 years, particularly in patient 1 with low NAA,reflect the severity of the neurodegenerative process. In addi-tion, the severity of the upper and lower digestive dysfunctionsleading to early gastrostomia or cachexia has not yet beenreported in POLR3A, POLR3B, and POLR1Cmutated patientsbut could be related to the impaired proliferation of digestive
organs reported in the zebrafish POLR3B mutant affecting theRPC128-RPC11 interaction.
To determine the effects of the variant on Pol III transcriptionalactivity, we compared the expression levels of some Pol III–transcribed RNAs in patient and control fibroblasts: tran-scription of both 5S rRNA and 7SL RNA was most severelyreduced. 5S rRNA is a component of the large subunit of theribosome and therefore important for ribosomal functioning.7SL RNA is part of the signal recognition particle required forassociating the ribosome nascent peptide chain with the en-doplasmic reticulum. Of interest, a reduction in 7SL RNA wasalso reported in zebrafish with a POLR3B mutation affectingthe RPB128-RPC11 interaction.19 Disruption in ribosomalregulation ofmRNA translationmay contribute to whitematterdevelopmental abnormalities observed in our patients. 7SLRNA seems to play a role in the expression of myelin basicprotein, which is tightly needed for myelin development andstability.20 Abnormal RNA regulation has also been reported inleukodystrophies related to mutations in the mitochondrial orcytoplasmic tRNA synthetases21 and in the 5 subunits of theeukaryotic initiation factor EIF2B (childhood ataxia with cen-tral nervous system hypomyelination/vanishing white mat-ter).11 Stress-induced acute neurologic distress has particularlybeen reported in this latter form of leukodystrophy, whereasneurologic degradation has been also reported after infectionsin 50% of patients with POLR3A, POLR3B, and POLR1Cmutations (table 2), suggesting that altered tRNA and rRNAsynthesis associated common dysfunctional pathways.
Here, we demonstrated the involvement of a hitherto un-known RNA polymerase III mutation of the POLR3K gene inthe development of HLD, supporting the evidence that RNApolymerase III plays a crucial role in white matter and cere-bellar integrity.
Author contributionsI. Dorboz: study concept and design, data analysis, andmanuscript writing. H. Dumay-Odelot: acquisition and in-terpretation of data and critical revision of the manuscript. K.Boussaid: collection and analysis of clinical data. Y. Bouya-coub, P. Barreau, S. Samaan, and H. Jmel: acquisition andinterpretation of molecular data. E. Eymard-Pierre: fibroblastcultures and DNA biobank. C. Cances, C. Bar, A.-L. Poulat, C.Rousselle, and F. Renaldo: acquisition of clinical data. M.Elmaleh-Berges: analysis and interpretation of radiologic data.M. Teichmann: acquisition and interpretation of data andcritical revision of the manuscript. O. Boespflug-Tanguy:study concept and design, analysis and interpretation of data,and manuscript writing.
AcknowledgmentThe authors thank the cytogenetic department of the CHUde Clermont-Ferrand responsible for the LEUKOFRANCEBiobank. They also thank Pr Judith Melki (Inserm UMR-1169, Le Kremlin Bicetre) for her help in homozygositymapping.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Study fundingThis study was supported by the European LeukodystrophyAssociation (ELA), grant number ELA 2009-007I4, and bythe European Union FP7 RD Connect project.
DisclosureI. Dorboz has received research support from the EuropeanLeukodystrophy Association (ELA). H. Dumay-Odelot hasreceived research support from INSERM, CNRS UMR5320, and Ligue contre le cancer. K. Boussaid, Y. Bouyacoub, P.Barreau, S. Samaan, H. Jmel, E. Eymard-Pierre, C. Cances, C.Bar, A. Poulat, C. Rousselle, F. Renaldo, and M. Elmaleh-Berges report no disclosures. M. Teichmann has received re-search support from INSERM, CNRS UMR 5320, and Liguecontre le cancer. O. Boespflug-Tanguy reports no disclosures.Full disclosure form information provided by the authors isavailable with the full text of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics May 22, 2018. Accepted in final formSeptember 5, 2018.
References1. Bernard G, Chouery E, Putorti ML, et al. Mutations of POLR3A encoding a catalytic
subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystro-phy. Am J Hum Genet 2011;89:415–423.
2. Tetreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encodingthe second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy.Am J Hum Genet 2011;89:652–705.
3. Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A. The expanding RNApolymerase III transcriptome. Trends Genet 2007;23:614–622.
4. Dumay-Odelot H, Durrieu-Gaillard S, Da Silva D, Roeder RG, Teichmann M. Cellgrowth- and differentiation-dependent regulation of RNA polymerase III transcrip-tion. Cell Cycle 2010;9:3687–3699.
5. Dumay-Odelot H, Durrieu-Gaillard S, El Ayoubi L, Parrot C, Teichmann M. Con-tributions of in vitro transcription to the understanding of human RNA polymerase IIItranscription. Transcription 2014;5:e27526.
6. Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encodingRNA Polymerase III subunits cause an autosomal-recessive hypomyelinating leu-koencephalopathy. Am J Hum Genet 2011;89:644–651.
7. Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C causea leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun2015;6:7623.
8. Kara B, Koroglu Ç, Peltonen K, et al. Severe neurodegenerative disease inbrothers with homozygous mutation in POLR1A. Eur J Hum Genet 2017;25:315–323.
9. Chedin S, Riva M, Schultz P, Sentenac A, Carles C. The RNA cleavage activity of RNApolymerase III is mediated by an essential TFIIS-like subunit and is important fortranscription termination. Genes Dev 1998;12:3857–3871.
10. Landrieux E, Alic N, Ducrot C, Acker J, Riva M, Carles C. A subcomplex of RNApolymerase III subunits involved in transcription termination and reinitiation. EMBOJ 2006;25:118–128.
11. Huyghe A, Horzinski L, Henaut A, et al. Developmental splicing deregulation inleukodystrophies related to EIF2B mutations. PLoS One 2012;7:e38264.
12. Barbier M, Gross MS, Aubart M, et al. MFAP5 loss-of-function mutations underscorethe involvement of matrix alteration in the pathogenesis of familial thoracic aorticaneurysms and dissections. Am J Hum Genet 2014;95:736–743.
13. Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Scalable web servicesfor the PSIPRED protein analysis workbench. Nucleic Acids Res 2013;41:W349–W357.
14. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal forprotein modeling, prediction and analysis. Nat Protoc 2015;10:845–858.
15. Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking.Nat Protoc 2017;12:255–278.
16. Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algo-rithm for protein-protein docking. Nucleic Acids Res 2004;32:W96–W99.
17. Schroeder A, Mueller O, Stocker S et al. The RIN: an RNA integrity number forassigning integrity values to RNA measurements. BMC Mol Biol 2006;7:3.
18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation ofsequence variants: a joint consensus recommendation of the American College ofMedical genetics and genomics and the Association for Molecular Pathology. GenetMed 2015;17:405–424.
19. Yee NS, Gong W, Huang Y, et al. Mutation of RNA Pol III subunit rpc2/polr3b leadsto deficiency of subunit Rpc11 and disrupts zebrafish digestive development. PLoSBiol 2007;5:e312.
20. Tretiakova A, Gallia GL, Shcherbik N, et al. Association of Puralpha with RNAshomologous to 7 SL determines its binding ability to the myelin basic protein pro-moter DNA sequence. J Biol Chem 1998;273:22241–22247.
21. Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyltRNA syn-thetase deficiency causes leukoencephalopathy with brain stem and spinal cord in-volvement and lactate elevation. Nat Genet 2007;39:534–539.
22. Wolf NI, Vanderver A, van Spaendonk RM et al. Clinical spectrum of 4H leuko-dystrophy caused by POLR3A and POLR3B mutations. Neurology 2014;83:1898–1905.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

ARTICLE OPEN ACCESS
Amyloid- and tau-PET imaging in a familialprion kindredDavid T. Jones, MD, Ryan A. Townley, MD, Jonathan Graff-Radford, MD, Hugo Botha, MB, ChB,
David S. Knopman, MD, Ronald C. Petersen, MD, PhD, Clifford R. Jack, Jr., MD, Val J. Lowe, MD,
and Bradley F. Boeve, MD
Neurol Genet 2018;4:e290. doi:10.1212/NXG.0000000000000290
Correspondence
Dr. Jones
AbstractObjectiveTo study the in vivo binding properties of 18F-AV-1451 (tau-PET) and Pittsburgh compound B(PiB-PET) in a unique kindred with a familial prion disorder known to produce amyloidplaques composed of prion protein alongside Alzheimer disease (AD)–like tau tangles.
MethodsA case series of 4 symptomatic family members with the 12-octapeptide repeat insertion in thePRNP gene were imaged with 3T MRI, PiB-PET, and tau-PET in their fourth decade of life.
ResultsThere was significant neocortical uptake of the tau-PET tracer in all 4 familial prion cases.However, PiB-PET images did not demonstrate abnormally elevated signal in neocortical orcerebellar regions for any of the patients.
ConclusionsIn vivo detection of molecular hallmarks of neurodegenerative diseases will be a prerequisite towell-conducted therapeutic trials. Understanding the in vivo behavior of these PET biomarkersin the setting of various neurodegenerative processes is imperative to their proper use in suchtrials and for research studies focused on the basic neurobiology of neurodegeneration. Thisstudy supports the high specificity of neocortical 18F-AV-1451 binding to AD-like tau and thelack of PiB binding to PrP plaques. It is uncertain how early in the disease course tau pathologyappears in the brains of individuals who carry this PRNP gene mutation or how it evolvesthroughout the disease course, but future longitudinal 18F-AV-1451 imaging of symptomaticand asymptomatic individuals in this kindred will help address these uncertainties.
From the Department of Neurology (D.T.J., R.A.T., J.G.-R., H.B., D.S.K., R.C.P., B.F.B.) and Department of Radiology (D.T.J., C.R.J., V.J.L.), Mayo Clinic, Rochester, MN.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Familial prion diseases are associated with prion protein(PrP) deposits and genetic mutations in the PRNP gene. Inaddition to the PrP deposits seen in all subtypes, othercharacteristic pathologies include the following: spongiformchanges in familial Creutzfeldt-Jakob disease, PrP amyloidplaques in the cerebellum in Gerstmann-Straussler-Scheinkersyndrome, and thalamic neuronal loss seen in fatal familialinsomnia.1 We have reported on a familial prion kindred witha 12-octapeptide repeat insertion in the PRNP gene charac-terized by a frontotemporal dementia (FTD)-like pre-sentation with mixed pathologic features.2 These cases werepathologically characterized by cerebellar plaques that werenegative for Aβ40 and Aβ42 but positive for PrP. The neo-cortical pathology consisted of Alzheimer disease (AD)-likeneurofibrillary tangles (NFT) that were present in addition tothe PrP pathology. The tau pathology found in these cases isunusual for sporadic prion disease, but it has been reported inother familial prion cases,3–5 where it was composed of thesame AD-like tau isoforms. We have recently shown that exvivo6 and in vivo7 binding of 18F-AV-1451 (tau-PET)8 isspecific for AD-like tau isoforms and that the regional distri-bution is distinct for AD dementia relative to familial FTDwith mutations in the MAPT gene.7
Pittsburgh compound B (PiB) is a derivative of thioflavin.9
Therefore, it is possible that PiB-PET could bind PrP plaquesin this prion kindred. The cerebellar predominance of PrPplaques is distinct from the neocortical NFTs, allowing theseaggregates to be differentiated spatially in vivo. In this study,we report the tau-PET and PiB-PET binding patterns in thiskindred that has both tau aggregates and PrP plaques.
MethodsStandard protocol approvals, registrations,and patient consentsAll participants or their designees provided written informedconsent with approval of the Mayo Clinic Foundation andOlmsted Medical Center Institutional Review boards.
Participants and imagingParticipants were part of the Mayo Clinic AD ResearchCenter in Rochester, Minnesota. Four symptomatic familymembers of the 12-octapeptide repeat insertion kindred wereimaged with 3T MRI, amyloid-PET with (PiB-PET),9 andAV-1451 (tau-PET)8 in their fourth decade of life. Thesepatients were all symptomatic for 1–6 years and had a be-havioral variant FTD-like presentation with minimal parkin-sonism. Case 1 also had a prominent nonfluent aphasia.Detailed demographic information is not relevant to the ob-jective of this study and may allow for patient identification.Therefore, we do not report additional metadata that can be
linked to the cases. One patient with young-onset ADwas alsoevaluated for comparison purposes. Given the potential foron-target PET tracer uptake in the cerebellum, which iscommonly used as a reference region for quantification, weused visual assessment as the gold standard without normal-izing signal intensity to a reference region. Once we verifiedthat there was no regional cerebellar signal on either PETmodality, we created intensity-normalized images using thegray matter in the cerebellar crus for display purposes only.
Data availabilityThe datasets analyzed in the current study are not publiclyavailable due to restricted access, but further informationabout the datasets are available from the corresponding au-thor on reasonable request.
ResultsThere was significant neocortical uptake of the tau-PET tracerin all 4 familial prion cases (figure 1, left). However, PiB-PETimages did not demonstrate abnormally elevated signal inneocortical regions for any of the patients (figure 1, right).There was no abnormally elevated signal in the cerebellum oneither PET modality. The global distribution of the elevatedtau-PET signal (figure 2A)matched the known distribution ofNFT in AD (figure 2B) in that the homotypic isocortex wasseverely affected with a stark sparing of the idiotypic cortex.However, there did seem to be relatively greater involvementof the frontal lobe compared with the precuneus in the familialprion participant.
DiscussionThis study demonstrates 2 important properties of thesewidely used molecular PET ligands: (1) in vivo neocorticaltau-PET binding can detect AD-like NFT in the setting offamilial prion disease and (2) PiB-PET did not detect am-yloid plaques composed of prion protein in this kindred.This has important implications for the use of these ligandsin the study of AD. These findings also indicate that the tau-PET is a promising tool for investigating the molecularbasis of neurodegeneration in this familial prion kindred andother hereditary prion diseases with coexisting AD-like NFTpathology.2–5
Four members of the 12-octapeptide repeat insertion in thePRNP gene kindred did not have any abnormally elevatedPiB-PET signal. The fact that no elevated in vivo PiB-PETsignal was seen in a kindred known to harbor amyloid pla-ques composed of PrP suggests that PiB-PETmay be specificfor amyloid plaques composed of beta-amyloid. In the
GlossaryAD = Alzheimer disease; FTD = frontotemporal dementia; NFT = neurofibrillary tangles; PiB = Pittsburgh compound B.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
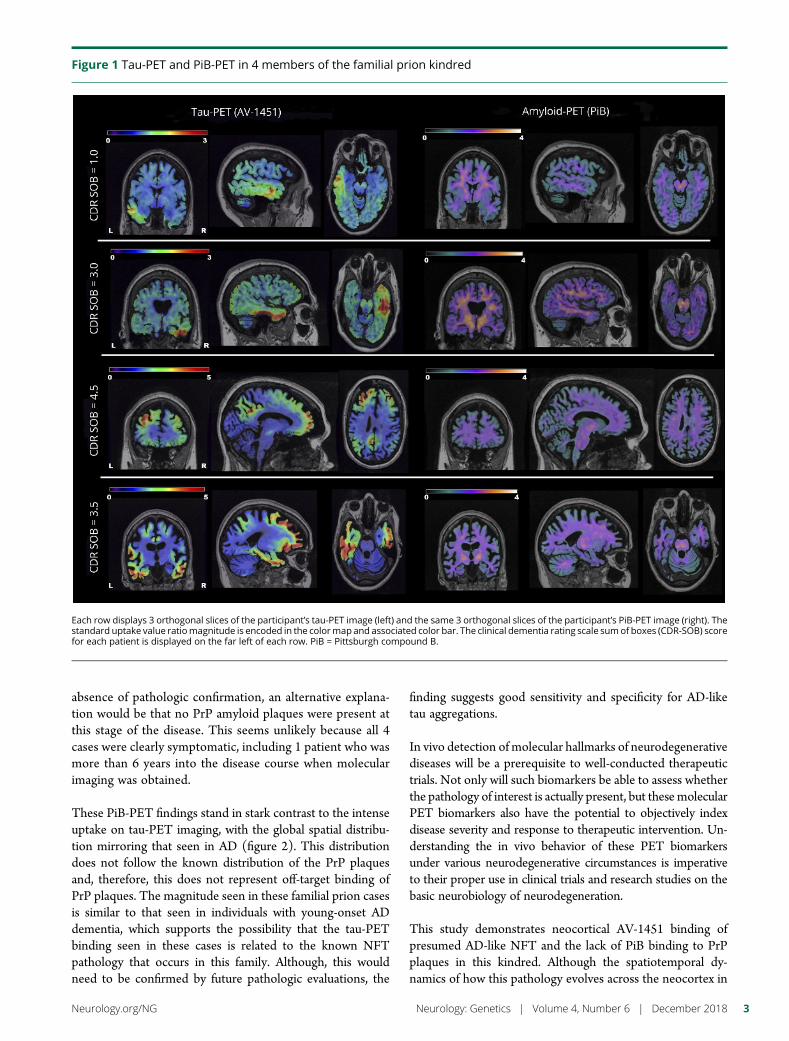
absence of pathologic confirmation, an alternative explana-tion would be that no PrP amyloid plaques were present atthis stage of the disease. This seems unlikely because all 4cases were clearly symptomatic, including 1 patient who wasmore than 6 years into the disease course when molecularimaging was obtained.
These PiB-PET findings stand in stark contrast to the intenseuptake on tau-PET imaging, with the global spatial distribu-tion mirroring that seen in AD (figure 2). This distributiondoes not follow the known distribution of the PrP plaquesand, therefore, this does not represent off-target binding ofPrP plaques. The magnitude seen in these familial prion casesis similar to that seen in individuals with young-onset ADdementia, which supports the possibility that the tau-PETbinding seen in these cases is related to the known NFTpathology that occurs in this family. Although, this wouldneed to be confirmed by future pathologic evaluations, the
finding suggests good sensitivity and specificity for AD-liketau aggregations.
In vivo detection of molecular hallmarks of neurodegenerativediseases will be a prerequisite to well-conducted therapeutictrials. Not only will such biomarkers be able to assess whetherthe pathology of interest is actually present, but thesemolecularPET biomarkers also have the potential to objectively indexdisease severity and response to therapeutic intervention. Un-derstanding the in vivo behavior of these PET biomarkersunder various neurodegenerative circumstances is imperativeto their proper use in clinical trials and research studies on thebasic neurobiology of neurodegeneration.
This study demonstrates neocortical AV-1451 binding ofpresumed AD-like NFT and the lack of PiB binding to PrPplaques in this kindred. Although the spatiotemporal dy-namics of how this pathology evolves across the neocortex in
Figure 1 Tau-PET and PiB-PET in 4 members of the familial prion kindred
Each row displays 3 orthogonal slices of the participant’s tau-PET image (left) and the same 3 orthogonal slices of the participant’s PiB-PET image (right). Thestandard uptake value ratiomagnitude is encoded in the colormap and associated color bar. The clinical dementia rating scale sumof boxes (CDR-SOB) scorefor each patient is displayed on the far left of each row. PiB = Pittsburgh compound B.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

this kindred remains unknown, future longitudinal tau-PETstudies of symptomatic and asymptomatic individuals will beable to address this question directly.
Author contributionsD.T. Jones: acquisition of data, analysis and interpretation of data,andmanuscript preparation. R.A. Townley, J. Graff-Radford,H. Botha, D.S. Knopman, R.C. Petersen, C.R. Jack, V.J.Lowe, and B.F. Boeve: acquisition of data, analysis and in-terpretation of data, and critical revision of the manuscriptfor intellectual content.
AcknowledgmentThe authors greatly thank AVIDRadiopharmaceuticals, Inc., fortheir support in supplying 18F-AV-1451 precursor, chemistryproduction advice and oversight, and FDA regulatory cross-filing permission and documentation needed for this work. Theauthors are particularly grateful to our patients and the extendedfamily for participating in this research.
Study fundingThis research was supported by NIH grants R01 AG011378,R01 AG041581, U01 AG006786, P50 AG016574, The Liston
Figure 2 Global tau-PET distribution in familial prion disease mirrors the distribution seen in Alzheimer disease
The tau-PET signal intensity is overlaid onthe patient’s own brain MRI rendering us-ing MRIcroGL (nitrc.org/projects/mri-crogl). (A) The tau-PET imaging data fromthe familial prion participant in the bottomrowof figure 1 is displayed. (B) The tau-PETimaging data from a patient with young-onset Alzheimer disease is displayed forcomparison purposes.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Family Foundation, The GHR Foundation, Foundation Dr.Corinne Schuler, the Mayo Foundation, and by the Robert H.and Clarice Smith and Abigail van Buren Alzheimer’s DiseaseResearch Program.
DisclosureD.T. Jones, R.A. Townley, J. Graff-Radford, and H. Bothareport no disclosures. D.S. Knopman receives research sup-port from the NIH and the Robert H. and Clarice Smith andAbigail Van Buren Alzheimer’s Disease Research Program ofthe Mayo Foundation; he serves on a Data Safety MonitoringBoard for Lundbeck Pharmaceuticals and for the DIAN study;and is an investigator for clinical trials sponsored by Biogen,TauRX Pharmaceuticals, Lilly Pharmaceuticals, and the Alz-heimer’s Disease Treatment and Research Institute, Univer-sity of Southern California. R.C. Petersen serves on datamonitoring committees for Pfizer Inc and Janssen AlzheimerImmunotherapy; works as a consultant for Merck Inc, RocheInc, Biogen Inc, Eli Lilly and Company, and Genentech Inc;receives publishing royalties for Mild Cognitive Impairment(Oxford University Press, 2003); and receives research sup-port from the NIH and the Robert H. and Clarice Smith andAbigail Van Buren Alzheimer’s Disease Research Program ofthe Mayo Foundation. C.R. Jack receives research fundingfrom the NIH and the Alexander Family Alzheimer’s DiseaseResearch Professorship at Mayo Clinic. V.J. Lowe is a con-sultant for Bayer Schering Pharma, Merck Research, andPiramal Imaging Inc and receives research support from GEHealthcare, Siemens Molecular Imaging, AVID Radio-pharmaceuticals, the NIH (NIA, NCI), the Elsie andMarvinDekelboumFamily Foundation, the Liston Family Foundation,and the MN Partnership for Biotechnology and Medical
Genomics. B.F. Boeve served as an investigator for clinical trialssponsored by GE Healthcare and FORUM Pharmaceuticals.He receives royalties from the publication of a book entitledBehavioral Neurology of Dementia (Cambridge Medicine,2009). He serves on the scientific advisory board of the TauConsortium. He has consulted for Isis Pharmaceuticals. Hereceives research support from the NIH, the Robert H. andClarice Smith and Abigail Van Buren Alzheimer’s DiseaseResearch Program of the Mayo Foundation, and the Man-gurian Foundation. Full disclosure form information providedby the authors is available with the full text of this article atNeurology.org/NG.
Publication historyReceived by Neurology: Genetics October 17, 2017. Accepted in finalform October 5, 2018.
References1. Mastrianni JA. The genetics of prion diseases. Genet Med 2010;12:187–195.2. Kumar N, Boeve BF, Boot BP, et al. Clinical characterization of a kindred with a novel
12-octapeptide repeat insertion in the prion protein gene. Arch Neurol 2011;68:1165–1170.
3. Ishizawa K, Komori T, Shimazu T, et al. Hyperphosphorylated tau deposition parallelsprion protein burden in a case of Gerstmann-Straussler-Scheinker syndrome P102Lmutation complicated with dementia. Acta Neuropathol 2002;104:342–350.
4. Jayadev S, Nochlin D, Poorkaj P, et al. Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol 2011;69:712–720.
5. Tranchant C, Sergeant N, Wattez A, Mohr M, Warter JM, Delacourte A. Neurofi-brillary tangles in Gerstmann-Straussler-Scheinker syndrome with the A117V priongene mutation. J Neurol Neurosurg Psychiatry 1997;63:240–246.
6. Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 TauPET in dementia. Acta Neuropathol Commun 2016;4:58.
7. Jones DT, Knopman DS, Graff-Radford J, et al. In vivo (18)F-AV-1451 tau PET signalin MAPT mutation carriers varies by expected tau isoforms. Neurology 2018;90:e947–e954.
8. Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomog-raphy imaging agent for Alzheimer’s disease. Alzheimers Dement 2013;9:666–676.
9. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s diseasewith Pittsburgh Compound-B. Ann Neurol 2004;55:306–319.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

ARTICLE OPEN ACCESS
Copy number loss in SFMBT1 is commonamong Finnish and Norwegian patientswith iNPHVille E. Korhonen, BMed, Seppo Helisalmi, PhD, Aleksi Jokinen, BMed, Ilari Jokinen, BMed,
Juha-Matti Lehtola, BMed, Minna Oinas, MD, PhD, Kimmo Lonnrot, MD, PhD, Cecilia Avellan, MD,
Anna Kotkansalo, MD, Janek Frantzen, MD, PhD, Jaakko Rinne, MD, PhD, Antti Ronkainen, MD, PhD,
Mikko Kauppinen, MD, Antti Junkkari, MD, PhD, Mikko Hiltunen, PhD, Hilkka Soininen, MD, PhD,
Mitja Kurki, PhD, Juha E. Jaaskelainen, MD, PhD, Anne M. Koivisto, MD, PhD, Hidenori Sato, MD, PhD,
Takeo Kato, MD, PhD, Anne M. Remes, MD, PhD, Per Kristian Eide, MD, PhD, and Ville Leinonen, MD, PhD
Neurol Genet 2018;4:e291. doi:10.1212/NXG.0000000000000291
Correspondence
Mr. Korhonen
AbstractObjectiveTo evaluate the role of the copy number loss in SFMBT1 in a Caucasian population.
MethodsFive hundred sixty-seven Finnish and 377 Norwegian patients with idiopathic normal pressurehydrocephalus (iNPH) were genotyped and compared with 508 Finnish elderly, neurologicallyhealthy controls. The copy number loss in intron 2 of SFMBT1 was determined using quan-titative PCR.
ResultsThe copy number loss in intron 2 of SFMBT1was detected in 10% of Finnish (odds ratio [OR]= 1.9, p = 0.0078) and in 21% of Norwegian (OR = 4.7, p < 0.0001) patients with iNPHcompared with 5.4% in Finnish controls. No copy number gains in SFMBT1 were detected inpatients with iNPH or healthy controls. The carrier status did not provide any prognostic valuefor the effect of shunt surgery in either population. Moreover, no difference was detected in theprevalence of hypertension or T2DM between SFMBT1 copy number loss carriers andnoncarriers.
ConclusionsThis is the largest and the first multinational study reporting the increased prevalence of thecopy number loss in intron 2 of SFMBT1 among patients with iNPH, providing furtherevidence of its role in iNPH. The pathogenic role still remains unclear, requiring further study.
From the Department of Neurosurgery (V.E.K., A. Jokinen, I.J., J.-M.L., A. Junkkari, J.E.J., V.L.), Kuopio University Hospital and University of Eastern Finland; Institute of Clinical Medicine-Neurology (S.H., M.H., H. Soininen, A.M.K.), University of Eastern Finland, Kuopio; Department of Neurosurgery (M.O., K.L.), University of Helsinki and Helsinki University Hospital;Clinical Neurosciences (C.A., A.K., J.F., J.R.), Department of Neurosurgery, University of Turku and Turku University Hospital; Department of Neurosurgery (A.R.), Tampere UniversityHospital; Unit of Clinical Neuroscience (M. Kauppinen, V.L.), Neurosurgery, University of Oulu and Medical Research Center, Oulu University Hospital; Institute of Biomedicine (M.H.),University of Eastern Finland, Kuopio; Analytical and Translational Genetics Unit (M. Kurki), Department of Medicine, Massachusetts General Hospital; Program in Medical andPopulation Genetics (M. Kurki), Broad Institute of MIT and Harvard; Stanley Center for Psychiatric Research (M. Kurki), Broad Institute for Harvard andMIT; Department of Neurology(H. Sato, T.K.), Hematology, Metabolism, Endocrinology and Diabetology, Yamagata University Faculty of Medicine, Japan; Medical Research Center (A.M.R.), Oulu University Hospital,Finland; Unit of Clinical Neuroscience (A.M.R.), Neurology, University of Oulu, Finland; Department of Neurosurgery (P.K.E.), Oslo University Hospital-Rikshospitalet; and Institute ofClinical Medicine (P.K.E.), Faculty of Medicine, University of Oslo, Norway.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing charge was funded by the Authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Idiopathic normal pressure hydrocephalus (iNPH) is a late-onset progressive neurologic disease presenting typically withgait difficulties together with enlarged ventricles and tight-ened parasagittal cortical sulci, whereas other symptoms suchas cognitive impairment and urinary incontinence are oftenpresent.1,2 Recently, the 2 available diagnostic guidelines1,2
have been criticized, and urgent revision and/or unification ofthe diagnostic manual have been requested.3–5
Patients with iNPH are characterized by abnormal CSFcirculation and evidence of delayed cerebral clearance, asrecently shown in an MRI CSF tracer study.6 Althoughthere are no biomarkers to aid in the diagnostics, and theetiology of iNPH remains unclear, there is an increasingamount of proof indicating a potential genetic componentin iNPH.7–12 The prevalence of familial iNPH, i.e., at least2 patients with iNPH, in the first-degree relatives hasbeen reported to range from 4.8% to 7%.10,11 Data alsocontain a pair of identical twins having iNPH11 and a familyin which an autosomal dominant inheritance pattern isobserved.9
A segmental copy number loss in the SFMBT1 gene wasreported in Japan to be present in 50% of patients, whopresent concomitantly with clinical features of iNPH andenlarged ventricles,13 and in 26% of patients with iNPH, whoexperienced a positive shunt response.14 These results havepreviously not been confirmed outside of Japan and withsufficiently large cohorts. Our aim was to evaluate the prev-alence of the copy number loss of SFMBT1 in patients withiNPH of Caucasian origin.
MethodsStandard protocol approvals, registrations,and patient consentsThis study was conducted in the Department of Neurosurgeryin Kuopio University Hospital, the Brain Research Unit of theUniversity of Eastern Finland, and the Department of Neu-rosurgery, Oslo University Hospital, Rikshospitalet, Oslo,Norway, according to the Declaration of Helsinki. KuopioUniversity Hospital Research Ethics Board approved thestudy (5/2008 and 276/2016). In Norway, the study wasapproved by the Regional Committee for Medical and HealthResearch Ethics (REK) of Health Region South-East, Norway(2015/1313), and the Institutional Review Board of OsloUniversity Hospital (2015/8128).
The study consisted of 567 Finnish (mean age 70 years; SD8.3; men: n = 254; table 1) and 377 Norwegian (mean age68 years; SD 11; men: n = 190 (50%); table 2) patients with
possible iNPH. All patients and controls provided their writteninformed consent.
All Finnish andNorwegian patients suspected of having iNPHhave been clinically evaluated by a neurologist and a neuro-surgeon. The patients were referred to a neurosurgeon bya neurologist if the patients were observed to have at leastone of the core symptoms associated with iNPH, whichinclude gait difficulties, cognitive impairment, and urinaryincontinence together with enlarged ventricles (Evans Index>0.30)15 disproportionate to the size of the sulci of the ce-rebral convexities in MRI or CT imaging scans. In addition,most Finnish and Norwegian patients have undergone, asa prognostic test for shunt benefit, a 24-hour intraventricularpressure monitoring, spinal tap, extended lumbar drainage,or lumbar infusion test.
The Norwegian patients were diagnosed, and the decisionto perform the shunt surgery was done following a pre-viously published protocol,16 which included a clinicaljudgment of the severity of iNPH symptoms using a OsloiNPH Grading Scale (ranging from 3 to 15),16 MRI or CTimaging scans for the evaluation of the ventriculomegaly,evaluation of comorbidities, and finally a diagnostic over-night ICP monitoring.
All the Finnish and Norwegian patients fulfilled the clinicaldiagnostic criteria for possible iNPH.1,2 The control groupconsisted of 508 (mean age 70 years; SD 5.1; male: n = 207)Finnish subjects acquired for neurogenetic studies. Allsubjects have undergone clinical and neuropsychological
Table 1 Clinical characteristics of the Finnish iNPH cohort
Variables Mean or no. of cases SD or %
Cases 567
Sex (female) 312 54.9
Comorbidities
High arterial blood pressure 339/564 59.7
Diabetes 169/565 29.8
Age at shunta 71 7.9
Positive subjective shuntresponseb
458/528 86.8
Positive objective shunt responsec 97/203 49
a Available for 551 patients.b Clinical evaluation at 3-month follow-up.c Calculated using the modified 12-point Kubo scale, in which 1-point de-crease is considered to be clinically important.
GlossaryCI = confidence interval; iNPH = idiopathic normal pressure hydrocephalus; OR = odds ratio.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

testing lead by an experienced neurologist specialized inneurodegenerative conditions to exclude any signs ofcognitive impairment (table 3).
Genomic DNA was extracted from venous blood samplesusing QIAamp DNA blood mini extraction kit (QIAGEN).The copy number loss/gain in the intron 2 region of SFMBT1was detected using quantitative PCR (qPCR) and the delta-delta method as previously described.14 Our collaborator (T.Kato) provided a positive control sample, and a commercialnegative control was used.
All statistical analyses were performed using SPSS statisticversion 23 (IBM corp. USA). The χ2 test was used for cate-gorical dichotomous variables for groups >2. The Fisher exacttest was used for pairwise comparison. p < 0.05 was consid-ered statistically significant.
Power calculations were completed before the execution ofthe research and were based on the results of the study by Satoet al.14 in which the expected prevalence was divided bya factor of 2 (26/2% = 13%). With a power of 0.85, p = 0.05,the sample size was determined to be n ≥ 362. Both theFinnish and Norwegian iNPH cohorts and Finnish controlsfulfill this requirement.
Data availability statementAccording to Finnish law, the full individual clinical data setcannot be shared publicly. However, the data set can/will beshared by academic collaboration agreement upon request.
ResultsThe copy number loss in SFMBT1 was detected in 9.9% ofthe Finnish patients with iNPH, in 21% of the Norwegianpatients with iNPH, and in 5.4% of the Finnish controls(figures 1 and 2). Statistically significant difference wasdetected between Finnish and Norwegian patients withiNPH (p < 0.0001). No copy number gains in SFMBT1 weredetected in Finnish or Norwegian patients with iNPH orhealthy controls. In the Finnish iNPH cohort, 9/71 sus-pected familial patients, who are carriers for the copy num-ber loss, were detected, and therefore, no aggregation of thecopy number loss variant was observed in familial iNPH(12.1% vs 10%, p = 0.5). There was no significant differencein age at onset, sex, shunting prevalence, or shunt responsebetween Finnish and Norwegian patients. In addition, nodifference was detected in the prevalence of hypertension orT2DM between SFMBT1 copy number loss carriers andnoncarriers (table 4) or in the frequency of the genetic var-iant between shunt-responsive and nonresponsive patients ineither Finnish (odds ratio [OR] = 1.0, confidence interval [CI]95% 0.44–2.4, p = 0.93) or Norwegian (OR = 0.68, CI 95%0.31–1.5, p = 0.33) cohort.
DiscussionThis is the first study on the prevalence of the copy numberloss in SFMBT1 among patients with iNPH of non-Asianorigin. Although there are studies describing familial aggre-gation11 and even in some rare cases autosomal dominantinheritance pattern,9 SFMBT1 is the first gene to be associatedwith iNPH. Our findings are principally in line with the Jap-anese results providing compelling further evidence on therole of the copy number loss in SFMBT1 in iNPH.
The copy number loss in SFMBT1 is detected only in10%–20% of the patients with iNPH, which would suggest thatiNPH has both a polygenetic and multifactorial origin. Eth-nicity seems to modify the prevalence of this genetic variantbetween Finnish and Norwegian iNPH patients but surpris-ingly is similar between selected Norwegian and Japanesepopulations. In addition, a small percentage of cognitively in-tact controls carry the genetic variant providing further evi-dence on the copy number loss in SFMBT1 of being onlya possible risk-increasing genotype. In the Japanese study,a small percentage of patients and controls were found to carrya copy number gain variant, but no statistically significant dif-ference between patients with iNPH and controls was detec-ted.14 Of interest, in the Nordic cohorts, no copy number gainvariants were detected in either patients with iNPH or healthycontrols. Therefore, it appears that the copy number loss in
Table 2 Clinical characteristics of the Norwegian iNPHcohort
Variables Mean or no. of cases SD or %
Cases 377
Sex (female) 187/377 49.6
Shunt 297/377 78.8
Comorbidities
Arterial hypertension 156/377 41.4
Diabetes 58/377 15.4
Age at shunt 69 9.9
Positive shunt responsea 258/297 86.95
No shunt response 39/297 13.1
Lost to follow-up 7/297 2.4
Abbreviation: iNPH = idiopathic normal pressure hydrocephalus.a Clinical evaluation at 6–12months after surgery. Calculated using the OsloNPH scale.16
Table 3 Finnish control characteristics
Variables Mean or no. of cases SD or %
Controls 508
Sex (female) 301 59.3
Age at inclusion 69.8 5.1
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
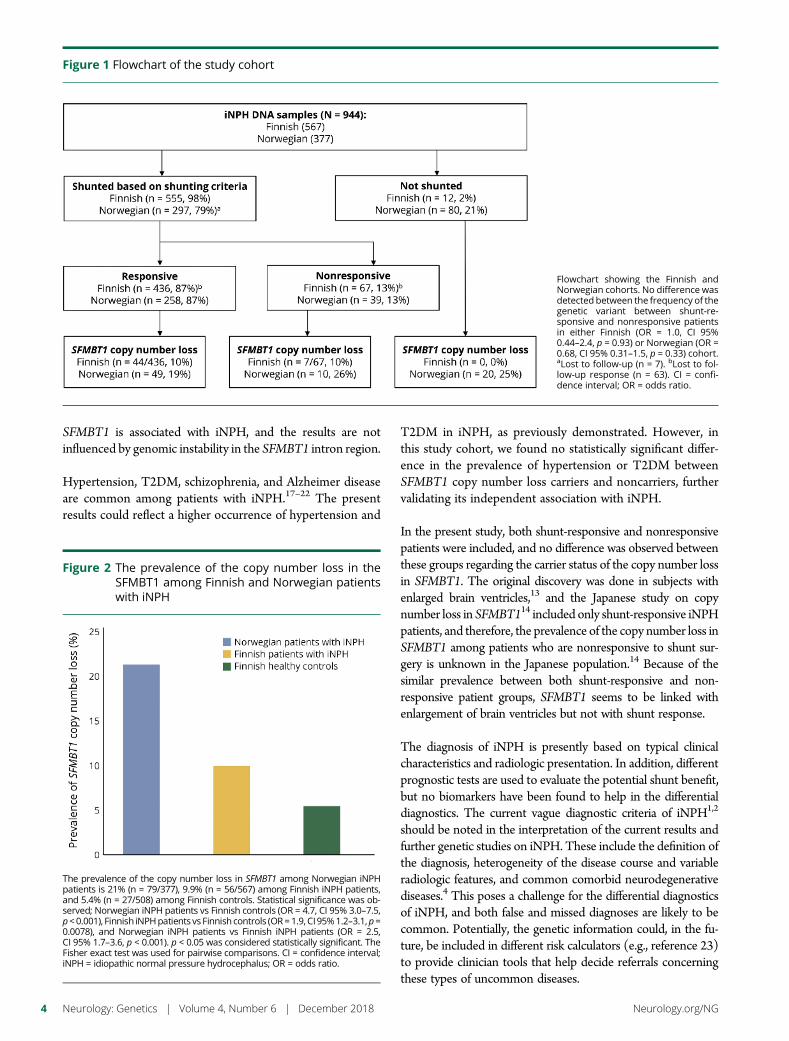
SFMBT1 is associated with iNPH, and the results are notinfluenced by genomic instability in the SFMBT1 intron region.
Hypertension, T2DM, schizophrenia, and Alzheimer diseaseare common among patients with iNPH.17–22 The presentresults could reflect a higher occurrence of hypertension and
T2DM in iNPH, as previously demonstrated. However, inthis study cohort, we found no statistically significant differ-ence in the prevalence of hypertension or T2DM betweenSFMBT1 copy number loss carriers and noncarriers, furthervalidating its independent association with iNPH.
In the present study, both shunt-responsive and nonresponsivepatients were included, and no difference was observed betweenthese groups regarding the carrier status of the copy number lossin SFMBT1. The original discovery was done in subjects withenlarged brain ventricles,13 and the Japanese study on copynumber loss in SFMBT114 included only shunt-responsive iNPHpatients, and therefore, the prevalence of the copy number loss inSFMBT1 among patients who are nonresponsive to shunt sur-gery is unknown in the Japanese population.14 Because of thesimilar prevalence between both shunt-responsive and non-responsive patient groups, SFMBT1 seems to be linked withenlargement of brain ventricles but not with shunt response.
The diagnosis of iNPH is presently based on typical clinicalcharacteristics and radiologic presentation. In addition, differentprognostic tests are used to evaluate the potential shunt benefit,but no biomarkers have been found to help in the differentialdiagnostics. The current vague diagnostic criteria of iNPH1,2
should be noted in the interpretation of the current results andfurther genetic studies on iNPH. These include the definition ofthe diagnosis, heterogeneity of the disease course and variableradiologic features, and common comorbid neurodegenerativediseases.4 This poses a challenge for the differential diagnosticsof iNPH, and both false and missed diagnoses are likely to becommon. Potentially, the genetic information could, in the fu-ture, be included in different risk calculators (e.g., reference 23)to provide clinician tools that help decide referrals concerningthese types of uncommon diseases.
Figure 1 Flowchart of the study cohort
Flowchart showing the Finnish andNorwegian cohorts. No difference wasdetected between the frequency of thegenetic variant between shunt-re-sponsive and nonresponsive patientsin either Finnish (OR = 1.0, CI 95%0.44–2.4, p = 0.93) or Norwegian (OR =0.68, CI 95% 0.31–1.5, p = 0.33) cohort.aLost to follow-up (n = 7). bLost to fol-low-up response (n = 63). CI = confi-dence interval; OR = odds ratio.
Figure 2 The prevalence of the copy number loss in theSFMBT1 among Finnish and Norwegian patientswith iNPH
The prevalence of the copy number loss in SFMBT1 among Norwegian iNPHpatients is 21% (n = 79/377), 9.9% (n = 56/567) among Finnish iNPH patients,and 5.4% (n = 27/508) among Finnish controls. Statistical significance was ob-served; Norwegian iNPH patients vs Finnish controls (OR = 4.7, CI 95% 3.0–7.5,p<0.001), Finnish iNPHpatients vs Finnish controls (OR=1.9, CI95%1.2–3.1,p=0.0078), and Norwegian iNPH patients vs Finnish iNPH patients (OR = 2.5,CI 95% 1.7–3.6, p < 0.001). p < 0.05 was considered statistically significant. TheFisher exact test was used for pairwise comparisons. CI = confidence interval;iNPH = idiopathic normal pressure hydrocephalus; OR = odds ratio.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
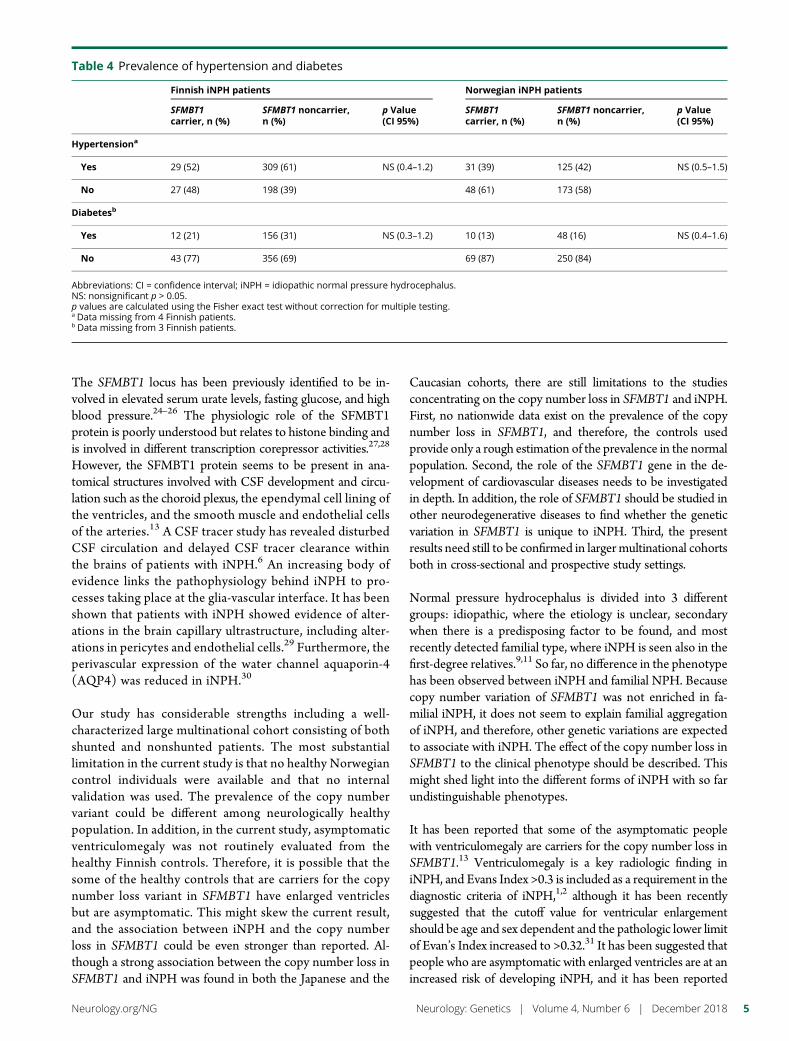
The SFMBT1 locus has been previously identified to be in-volved in elevated serum urate levels, fasting glucose, and highblood pressure.24–26 The physiologic role of the SFMBT1protein is poorly understood but relates to histone binding andis involved in different transcription corepressor activities.27,28
However, the SFMBT1 protein seems to be present in ana-tomical structures involved with CSF development and circu-lation such as the choroid plexus, the ependymal cell lining ofthe ventricles, and the smooth muscle and endothelial cellsof the arteries.13 A CSF tracer study has revealed disturbedCSF circulation and delayed CSF tracer clearance withinthe brains of patients with iNPH.6 An increasing body ofevidence links the pathophysiology behind iNPH to pro-cesses taking place at the glia-vascular interface. It has beenshown that patients with iNPH showed evidence of alter-ations in the brain capillary ultrastructure, including alter-ations in pericytes and endothelial cells.29 Furthermore, theperivascular expression of the water channel aquaporin-4(AQP4) was reduced in iNPH.30
Our study has considerable strengths including a well-characterized large multinational cohort consisting of bothshunted and nonshunted patients. The most substantiallimitation in the current study is that no healthy Norwegiancontrol individuals were available and that no internalvalidation was used. The prevalence of the copy numbervariant could be different among neurologically healthypopulation. In addition, in the current study, asymptomaticventriculomegaly was not routinely evaluated from thehealthy Finnish controls. Therefore, it is possible that thesome of the healthy controls that are carriers for the copynumber loss variant in SFMBT1 have enlarged ventriclesbut are asymptomatic. This might skew the current result,and the association between iNPH and the copy numberloss in SFMBT1 could be even stronger than reported. Al-though a strong association between the copy number loss inSFMBT1 and iNPH was found in both the Japanese and the
Caucasian cohorts, there are still limitations to the studiesconcentrating on the copy number loss in SFMBT1 and iNPH.First, no nationwide data exist on the prevalence of the copynumber loss in SFMBT1, and therefore, the controls usedprovide only a rough estimation of the prevalence in the normalpopulation. Second, the role of the SFMBT1 gene in the de-velopment of cardiovascular diseases needs to be investigatedin depth. In addition, the role of SFMBT1 should be studied inother neurodegenerative diseases to find whether the geneticvariation in SFMBT1 is unique to iNPH. Third, the presentresults need still to be confirmed in largermultinational cohortsboth in cross-sectional and prospective study settings.
Normal pressure hydrocephalus is divided into 3 differentgroups: idiopathic, where the etiology is unclear, secondarywhen there is a predisposing factor to be found, and mostrecently detected familial type, where iNPH is seen also in thefirst-degree relatives.9,11 So far, no difference in the phenotypehas been observed between iNPH and familial NPH. Becausecopy number variation of SFMBT1 was not enriched in fa-milial iNPH, it does not seem to explain familial aggregationof iNPH, and therefore, other genetic variations are expectedto associate with iNPH. The effect of the copy number loss inSFMBT1 to the clinical phenotype should be described. Thismight shed light into the different forms of iNPH with so farundistinguishable phenotypes.
It has been reported that some of the asymptomatic peoplewith ventriculomegaly are carriers for the copy number loss inSFMBT1.13 Ventriculomegaly is a key radiologic finding iniNPH, and Evans Index >0.3 is included as a requirement in thediagnostic criteria of iNPH,1,2 although it has been recentlysuggested that the cutoff value for ventricular enlargementshould be age and sex dependent and the pathologic lower limitof Evan’s Index increased to >0.32.31 It has been suggested thatpeople who are asymptomatic with enlarged ventricles are at anincreased risk of developing iNPH, and it has been reported
Table 4 Prevalence of hypertension and diabetes
Finnish iNPH patients Norwegian iNPH patients
SFMBT1carrier, n (%)
SFMBT1 noncarrier,n (%)
p Value(CI 95%)
SFMBT1carrier, n (%)
SFMBT1 noncarrier,n (%)
p Value(CI 95%)
Hypertensiona
Yes 29 (52) 309 (61) NS (0.4–1.2) 31 (39) 125 (42) NS (0.5–1.5)
No 27 (48) 198 (39) 48 (61) 173 (58)
Diabetesb
Yes 12 (21) 156 (31) NS (0.3–1.2) 10 (13) 48 (16) NS (0.4–1.6)
No 43 (77) 356 (69) 69 (87) 250 (84)
Abbreviations: CI = confidence interval; iNPH = idiopathic normal pressure hydrocephalus.NS: nonsignificant p > 0.05.p values are calculated using the Fisher exact test without correction for multiple testing.a Data missing from 4 Finnish patients.b Data missing from 3 Finnish patients.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

that in the Japanese population, 25% of people with asymp-tomatic ventriculomegaly will eventually develop iNPH.32
However, the cohort sizes have been limited in the studies inwhich the connection between AVIM and iNPH has beenproposed. Therefore, no universal consensus on this matterexists.
The prevalence of the copy number loss in SFMBT1 could beincreased among the families in which iNPH is frequentlyobserved, and therefore, first-degree relatives of patients withiNPH should be included in future genetic studies. It might bethat the copy number loss in SFMBT1 is a risk gene for iNPHbut requires other unknown triggering risk factors to result inclinical disease.
Future studies are still urgently needed to elucidate the ge-netics of iNPH. Understanding even some of the pathologicprocesses causing iNPH provides possibilities to developtargeted or even preventive therapies in the future. This studyencourages functional studies on SFMBT1 to clarify the rolein the disease process of iNPH.
Author contributionsV.E. Korhonen: study concept and design, data acquisition,data analysis and interpretation, and drafting of the manu-script. S. Helisalmi: study concept and design, data acquisi-tion, and critical revision of the manuscript for importantintellectual content. A. Jokinen, I. Jokinen, J.-M. Lehtola,M. Oinas, K. Lonnrot, C. Avellan, A. Kotkansalo, J. Frantzen,J. Rinne, A. Ronkainen, M. Kauppinen, and A. Junkkari: dataacquisition and critical revision of the manuscript for impor-tant intellectual content. M. Hiltunen andH. Soininen: criticalrevision of the manuscript for important intellectual content.M. Kurki: critical revision of the manuscript for importantintellectual content (statistics). J.E. Jaaskelainen, A.M. Koi-visto, H. Sato H, and T. Kato: critical revision of the manu-script for important intellectual content. A.M. Remes: studyconcept and design and critical revision of the manuscript forimportant intellectual content. P.K. Eide and V. Leinonen:study concept and design, data acquisition, critical revision ofthe manuscript for important intellectual content, and studysupervision.
AcknowledgmentThe authors acknowledge Marjo Laitinen for her help insetting up the qPCR method and RN Marita Parviainen formanaging the KUH iNPH register.
Study fundingThis work was supported by the Academy of Finland (no307866), the Sigrid Juselius Foundation, the Kuopio Uni-versity Hospital Research Fund, the Kuopio University Hos-pital VTR fund, the Emil Aaltonen Foundation, the CulturalFoundation of Finland, the North Savo Regional Fund, theOlvi Foundation, and the Finnish Medical Association. InNorway, the study was supported by grants from HealthSouth-East, Norway (grant 2011067).
DisclosureV.E. Korhonen has received research support from the Kuo-pio University Hospital Research Fund, the Maire TaponenFoundation, the University of Eastern Finland, the OlviFoundation, the Emil Aaltonen Foundation, the FinnishCultural Foundation, the North Savo Regional fund, and theFinnish Medical Association. S. Helisalmi, A. Jokinen, I.Jokinen, J.-M. Lehtola, M. Oinas, and K. Lonnrot report nodisclosures. C. Avellan has received research support from theMaire Taponen Foundation, Lastentautien tutkimussaatio,Stiftelsen Dorothea Olivia, Karl Walter och Jarl WalterPerklens minne, and Svenska kulturfonden. A.E. Kotkansaloreports no disclosures. J. Frantzen serves/has served on thescientific advisory board of and as a consultant to BonaliveBiomaterials Ltd. J. Rinne, A. Ronkainen, and M. Kauppinenreport no disclosures. A. Junkkari has received research sup-port from the state research fund (VTR), Kuopio UniversityHospital (KUH), the University of Eastern Finland (UEF),the Finnish Cultural Foundation, and the Maire TaponenFoundation. M. Hiltunen serves/has served on the editorialboards of the Journal of Alzheimer’s Disease, Journal of Alz-heimer’s Disease & Parkinsonism, and The Scientific WorldJournal. H. Soininen serves/has served on the scientific ad-visory board of AC Immune and serves/has served on theeditorial board of the Journal of Alzheimer’s Disease. M. Kurkireports no disclosures. J.E. Jaaskelainen serves/has served onthe editorial board of Acta Neurochirurgica; has received re-search support from the Finnish Academy of Sciences, theJuho Vainio Foundation Paivikki, the Sakari Sohlberg Foun-dation, and Kuopio University Hospital. A.M. Koivisto, H.Sato, T. Kato, A.M. Remes, and P.K. Eide report no dis-closures. V. Leinonen has received funding for travel and/orspeaker honoraria from B. Braun Aesculap; serves/has servedon the editorial board of the Journal of Alzheimer’s Disease; andhas received research support from NeuroVision Imaging,LLC, and Kuopio University Hospital. Full disclosure forminformation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics May 30, 2018. Accepted in final formOctober 9, 2018.
References1. Relkin N, Marmarou A, Klinge P, Bergsneider M, Black PM. INPH guidelines, part II:
diagnosing idiopathic normal-pressure hydrocephalus. Neurosurgery 2005;57:4–16.2. Mori E, Ishikawa M, Kato T, et al. Guidelines for management of idiopathic normal
pressure hydrocephalus: second edition. Neurol Med Chir (Tokyo) 2012;52:775–809.
3. Williams MA, Relkin NR. Diagnosis and management of idiopathic normal-pressurehydrocephalus. Neurol Clin Pract 2013;3:375–385.
4. Espay AJ, Da Prat GA, Dwivedi AK, et al. Deconstructing normal pressure hydro-cephalus: ventriculomegaly as early sign of neurodegeneration. Ann Neurol 2017:503–513.
5. Andersson J, Rosell M, Kockum K, Soderstrom L, Laurell K. Challenges in diagnosingnormal pressure hydrocephalus: evaluation of the diagnostic guidelines. eNeur-ologicalSci 2017;7:27–31.
6. Ringstad G, Vatnehol SAS, Eide PK. Glymphatic MRI in idiopathic normal pressurehydrocephalus. Brain 2017;140:2691–2705.
7. Portenoy RK, Berger A, Gross E. Familial occurrence of idiopathic normal-pressurehydrocephalus. Arch Neurol 1984;41:335–337.
8. Cusimano MD, Rewilak D, Stuss DT, Barrera-Martinez JC, Salehi F, Freedman M.Normal-pressure hydrocephalus: is there a genetic predisposition? Can J Neurol Sci2011;38:274–281.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

9. Takahashi Y, Kawanami T, Nagasawa H, Iseki C, Hanyu H, Kato T. Familial normalpressure hydrocephalus (NPH) with an autosomal-dominant inheritance: a novelsubgroup of NPH. J Neurol Sci 2011;308:149–151.
10. McGirr A, Cusimano MD. Familial aggregation of idiopathic normal pressure hy-drocephalus: novel familial case and a family study of the NPH triad in an iNPHpatient cohort. J Neurol Sci 2012;321:82–88.
11. Huovinen J, Kastinen S, Komulainen S, et al. Familial idiopathic normal pressurehydrocephalus. J Neurol Sci 2016;368:11–18.
12. Korhonen V, Solje E, Suhonen NM, et al. Frontotemporal dementia as a comorbidityto idiopathic normal pressure hydrocephalus (iNPH): a short review of literature andan unusual case. Fluids Barriers CNS 2017;14:10.
13. Kato T, Sato H, EmiM, et al. Segmental copy number loss of SFMBT1 gene in elderlyindividuals with ventriculomegaly: a community-based study. Intern Med 2011;50:297–303.
14. Sato H, Takahashi Y, Kimihira L, et al. A segmental copy number loss of the SFMBT1gene is a genetic risk for shunt-responsive, idiopathic normal pressure hydrocephalus(iNPH): a case-control study. PLoS One 2016;11:e0166615.
15. Shprecher D, Schwalb J, Kurlan R. Normal pressure hydrocephalus: diagnosis andtreatment. Curr Neurol Neurosci Rep 2008;8:371–376.
16. Eide PK, Sorteberg W. Diagnostic intracranial pressure monitoring and surgicalmanagement in idiopathic normal pressure hydrocephalus: a 6-year review of 214patients. Neurosurgery 2010;66:80–90.
17. Leinonen V, Koivisto AM, Savolainen S, et al. Post-mortem findings in 10 patientswith presumed normal-pressure hydrocephalus and review of the literature. Neuro-pathol Appl Neurobiol 2012;38:72–86.
18. Leinonen V, Koivisto AM, Alafuzoff I, et al. Cortical brain biopsy in long-termprognostication of 468 patients with possible normal pressure hydrocephalus. Neu-rodegener Dis 2012;10:166–169.
19. Malm J, Graff-Radford NR, Ishikawa M, et al. Influence of comorbidities in idiopathicnormal pressure hydrocephalus—research and clinical care. A report of the ISHCSFtask force on comorbidities in INPH. Fluids Barriers CNS 2013;10:22.
20. Israelsson H, Carlberg B, Wikkelso C, et al. Vascular risk factors in INPH. Neurology2017;88:577–585.
21. Eide P, Pripp A. Increased prevalence of cardiovascular disease in idiopathic normalpressure hydrocephalus patients compared to a population-based cohort from theHUNT3 survey. Fluids Barriers CNS 2014;11:19.
22. Vanhala V, Junkkari A, Korhonen VE, et al. Prevalence of Schizophrenia in idiopathicnormal pressure hydrocephalus. Neurosurgery Epub 2018 May 8.
23. Luikku AJ, Hall A, Nerg O, et al. Multimodal analysis to predict shunt surgeryoutcome of 284 patients with suspected idiopathic normal pressure hydrocephalus.Acta Neurochir (Wien) 2016;158:2311–2319.
24. Yang HC, Liang YJ, Chen JW, et al. Identification of igf1, slc4a4, wwox, and sfmbt1 ashypertension susceptibility genes in han Chinese with a genome-wide gene-basedassociation study. PLoS One 2012;7:e32907.
25. Kottgen A, Albrecht E, Teumer A, et al. Genome-wide association analyses identify 18new loci associated with serum urate concentrations. Nat Genet 2012;45:145–154.
26. Chung RH, Chiu YF, Hung YJ, et al. Genome-wide copy number variation analysisidentified deletions in SFMBT1 associated with fasting plasma glucose in a HanChinese population. BMC Genomics 2017;18:591.
27. Wu S, Trievel RC, Rice JC. Human SFMBT1 is a transcriptional repressor protein thatselectively binds the N-terminal tail of histone H3. FEBS Lett 2007;581:3289–3296.
28. Zhang J, Bonasio R, Strino F, et al. SFMBT1 functions with LSD1 to regulate ex-pression of canonical histone genes and chromatin-related factors. Genes Dev 2013;27:749–766.
29. Eidsvaag VA, Hansson HA, Heuser K, Nagelhus EA, Eide PK. Brain capillary ultra-structure in idiopathic normal pressure hydrocephalus: relationship with static andpulsatile intracranial pressure. J Neuropathol Exp Neurol 2017;76:1034–1045.
30. Eide PK, Hansson HA. Astrogliosis and impaired aquaporin-4 and dystrophin systemsin idiopathic normal pressure hydrocephalus. Neuropathol Appl Neurobiol 2018;44:474–490.
31. Brix MK, Westman E, Simmons A, et al. The Evans’ Index revisited: new cut-off levelsfor use in radiological assessment of ventricular enlargement in the elderly. Eur JRadiol 2017;95:28–32.
32. Iseki C, Kawanami T, Nagasawa H, et al. Asymptomatic ventriculomegaly with fea-tures of idiopathic normal pressure hydrocephalus on MRI (AVIM) in the elderly:a prospective study in a Japanese population. J Neurol Sci 2009;277:54–57.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

ARTICLE OPEN ACCESS
Duplication and deletion upstream of LMNB1 inautosomal dominant adult-onset leukodystrophyNaomi Mezaki, MD, PhD, Takeshi Miura, MD, PhD, Kotaro Ogaki, MD, PhD, Makoto Eriguchi, MD, PhD,
Yuri Mizuno, MD, Kenichi Komatsu, MD, PhD, Hiroki Yamazaki, MD, Natsuki Suetsugi, MD,
Sumihiro Kawajiri, MD, Ryo Yamasaki, MD, PhD, Takanobu Ishiguro, MD, Takuya Konno, MD, PhD,
Hiroaki Nozaki, MD, PhD, Kensaku Kasuga, MD, PhD, Yasuyuki Okuma, MD, PhD, Jun-Ichi Kira, MD, PhD,
Hideo Hara, MD, PhD, Osamu Onodera, MD, PhD, and Takeshi Ikeuchi, MD, PhD
Neurol Genet 2018;4:e292. doi:10.1212/NXG.0000000000000292
Correspondence
Dr. Ikeuchi
AbstractObjectiveTo characterize the genetic and clinical features of patients with autosomal dominant adult-onset demyelinating leukodystrophy (ADLD) carrying duplication and deletion upstream oflamin B1 (LMNB1).
MethodsNinety-three patients with adult-onset leukoencephalopathy of unknown etiology were ge-netically analyzed for copy numbers of LMNB1 and its upstream genes. We examined LMNB1expression by reverse transcription-qPCR using total RNA extracted from peripheral leuko-cytes. Clinical and MRI features of the patients with ADLD were retrospectively analyzed.
ResultsWe identified 4 patients from 3 families with LMNB1 duplication. The duplicated genomicregions were different from those previously reported. The mRNA expression level of LMNB1in patients with duplication was significantly increased. The clinical features of our patients withLMNB1 duplication were similar to those reported previously, except for the high frequency ofcognitive impairment in our patients. We found 2 patients from 1 family carrying a 249-kbgenomic deletion upstream of LMNB1. Patients with the deletion exhibited relatively earlieronset, more prominent cognitive impairment, and fewer autonomic symptoms than patientswith duplication. The presence of cerebellar symptoms and lesions may be characteristic in ourpatients with the deletion compared with the previously reported family with the deletion.Magnetic resonance images of patients with the deletion exhibited a widespread distribution ofwhite matter lesions including the anterior temporal region.
ConclusionsWe identified 4 Japanese families with ADLD carrying duplication or deletion upstream ofLMNB1. There are differences in clinical and MRI features between the patients with theduplication and those with the deletion upstream of LMNB1.
From the Department of Molecular Genetics (N.M., T.M., T. Ishiguro, K. Kasuga, T. Ikeuchi) and Department of Neurology (N.M., T.M., T. Ishiguro, T.K., O.O.), Brain Research Institute,Niigata University; Department of Neurology (K.O., S.K., Y.O.), Juntendo University Shizuoka Hospital; Division of Neurology, Department of Internal Medicine (M.E., N.S., H.H.),Faculty of Medicine, Saga University; Department of Neurology (Y.M., R.Y., J.-I.K.), Neurological Institute, Graduate School of Medical Sciences, Kyushu University; Department ofNeurology (K. Komatsu, H.Y.), Kitano Hospital, The Tazuke Kofukai Medical Research Institute; and Medical Technology (H.N.), Graduate School of Health Sciences, Niigata University.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing charge was funded by AMED.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Autosomal dominant adult-onset demyelinating leukodys-trophy (ADLD; OMIM #169500) is a slowly progressiveadult-onset leukoencephalopathy that predominantly affectsthe cerebral white matter. The onset of ADLD is typically inthe fourth and fifth decade of life.1 Patients with ADLD areclinically characterized by early development of autonomicsymptoms such as bladder and/or bowel impairment andorthostatic hypotension. Autonomic symptoms usually pre-cede or occur together with other accompanying clinicalfeatures such as pyramidal signs and cerebellar ataxia. Cog-nitive impairment is observed in some patients.2,3 Charac-teristic MR images include diffuse and symmetrical lesions inthe cerebral white matter and cerebellar peduncles. Theperiventricular rim adjacent to the lateral ventricle in the ce-rebral white matter is spared or less affected.1
Duplication of lamin B1 (LMNB1) encoding lamin B1 hasbeen identified as a cause of ADLD.4 To date, 26 pedigreeswith ADLD carrying LMNB1 duplication have been reportedfrom different ethnic backgrounds.3,5,6 The regions of dupli-cation commonly include the entire LMNB1, but differed insize among the pedigrees, ranging from 128 to 475 kb.7 In2015, it was reported that a deletion of 660 kb in the regionupstream of LMNB1 was identified as the cause of ADLD inan Italian pedigree.8 In both mutations, i.e., duplication anddeletion upstream of LMNB1, the expression mRNA levelof LMNB1 was elevated.4,7,8 The key mechanism involvedin the ADLD pathogenesis seems to be lamin B1 over-production; however, it has not been fully understood howlamin B1 overproduction causes demyelination, leading toADLD.
In this study, we analyzed the copy number variation (CNV)of LMNB1 and the upstream genes to identify LMNB1-related ADLD in families with adult-onset leukoencephalo-pathies of unknown etiology. By this analysis, we identified4 ADLD pedigrees including 3 families with LMNB1 dupli-cation and 1 family with the upstream deletion of LMNB1. Wehere report the genetic and clinical characteristics of Japanesefamilies with LMNB1-related ADLD.
MethodsStandard protocol approvals, registrations,and patient consentsThis study was conducted in accordance with the Helsinkideclaration and approved by the Institutional Review Board of
Niigata University. Written informed consent was obtainedfrom the patients or their caregivers.
PatientsOne-hundred ten patients clinically suspected of having anadult-onset leukoencephalopathy, whose etiologies have notbeen determined, were referred to our institute for geneticanalysis between September 2015 and April 2017. Geneticanalysis was performed by PCR-based Sanger sequenc-ing analysis of genes including colony-stimulating factor 1receptor (CSF1R), AARS2, NOTCH3, and SNORD118. Bythis analysis, 11 patients who were found to harbor CSF1Rmutations, 2 patients with AARS2 mutations, 2 patients withNOTCH3 mutations, 1 patient with SNORD118 mutations,and 1 patient with neuronal intranuclear inclusion diseasewere excluded. The remaining 93 patients were included inthis study.
Genetic analysisGenomic DNA was extracted from peripheral leukocytes us-ing a QIAamp DNA Blood Maxi kit (QIAGEN, Hilden,Germany). CNV was analyzed by real-time PCR assay usingTaqMan probes (Thermo Fischer Scientific, Waltham, MA)designed for exons 3 (Hs02537023_cn), 6 (Hs00696436_cn), and 10 (Hs00579415_cn) of LMNB1. TaqMan probeswere also designed for exon 5 of phosphorylated adaptorfor RNA export (PHAX) (Hs0092512_cn), exon 8 of alde-hyde dehydrogenase 7 family member A1 (ALDH7A1)(Hs00222439_cn), and exon 2 of GRAM domain containing3 (GRAMD3) (Hs01106540_cn) to examine the CNV of theupstream genomic region of LMNB1. The amount of a PCRproduct was calculated on the basis of the threshold cycle(Ct), namely, the cycle in which fluorescence was detectedabove the baseline on an ABI PRISM 7900HT instrument(Applied Biosystems, Waltham,MA). We analyzed the resultsusing CopyCaller Software v2.0 (Applied Biosystems). Thecontrol DNACEPH 1347-02 was used as a reference genomicDNA sample, and the endogenous control was calculated byTaqMan copy number reference assay. The range of genomicCNVs around LMNB1 was examined by microarray-basedcopy number profiling using an Affymetrix CytoScan HDarray (Thermo Fischer Scientific).
LMNB1 expression assayTotal RNA was extracted from patients with LMNB1-relatedADLD using a PAXgene blood RNA kit (QIAGEN). RNAintegrity number (RIN) was determined using Bioanalyzer2100. Complementary DNA was synthesized by SuperScript
GlossaryADC = apparent diffusion coefficient; ADLD = adult-onset demyelinating leukodystrophy; ALDH7A1 = aldehydedehydrogenase 7 family member A1; CSF1R = colony-stimulating factor 1 receptor; CNV = copy number variation; DWI =diffusion-weighted imaging; FLAIR = fluid-attenuated inversion recovery; GRAMD3 = GRAM domain containing 3;LMNB1 = lamin B1; MCP = middle cerebellar peduncle; PHAX = phosphorylated adaptor for RNA export; RIN = RNAintegrity number; T1WI = T1-weighted imaging; VWMD = vanishing white matter disease.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
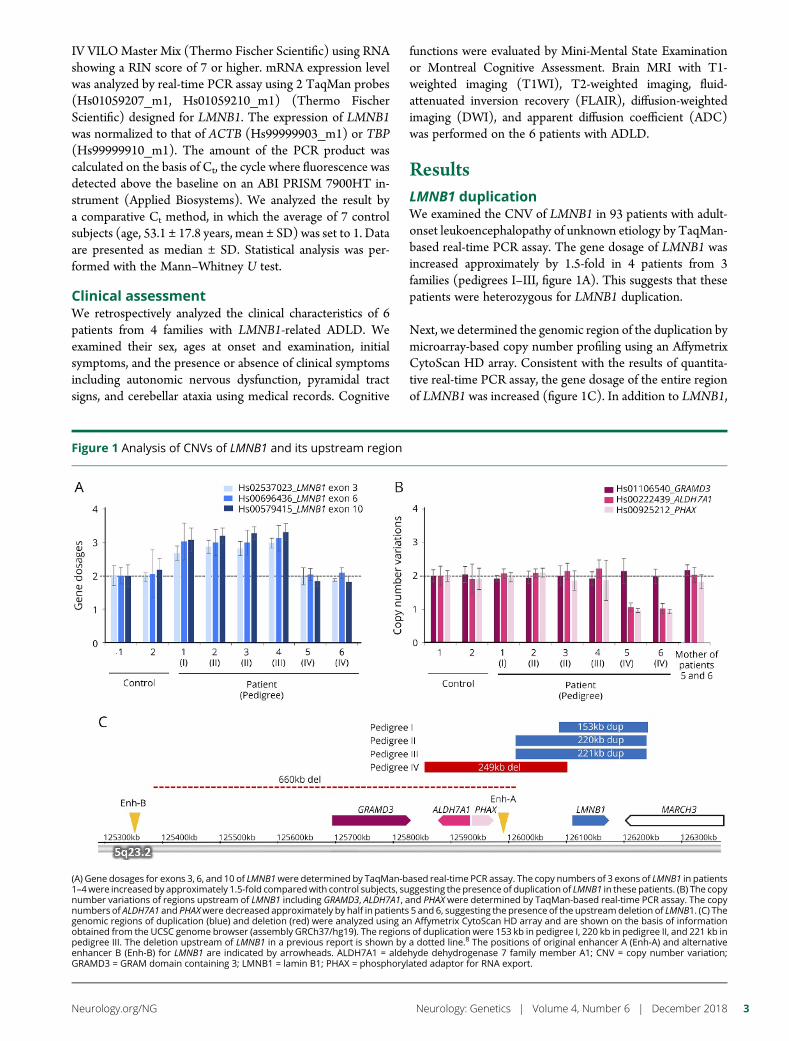
IV VILOMaster Mix (Thermo Fischer Scientific) using RNAshowing a RIN score of 7 or higher. mRNA expression levelwas analyzed by real-time PCR assay using 2 TaqMan probes(Hs01059207_m1, Hs01059210_m1) (Thermo FischerScientific) designed for LMNB1. The expression of LMNB1was normalized to that of ACTB (Hs99999903_m1) or TBP(Hs99999910_m1). The amount of the PCR product wascalculated on the basis of Ct, the cycle where fluorescence wasdetected above the baseline on an ABI PRISM 7900HT in-strument (Applied Biosystems). We analyzed the result bya comparative Ct method, in which the average of 7 controlsubjects (age, 53.1 ± 17.8 years, mean ± SD) was set to 1. Dataare presented as median ± SD. Statistical analysis was per-formed with the Mann–Whitney U test.
Clinical assessmentWe retrospectively analyzed the clinical characteristics of 6patients from 4 families with LMNB1-related ADLD. Weexamined their sex, ages at onset and examination, initialsymptoms, and the presence or absence of clinical symptomsincluding autonomic nervous dysfunction, pyramidal tractsigns, and cerebellar ataxia using medical records. Cognitive
functions were evaluated by Mini-Mental State Examinationor Montreal Cognitive Assessment. Brain MRI with T1-weighted imaging (T1WI), T2-weighted imaging, fluid-attenuated inversion recovery (FLAIR), diffusion-weightedimaging (DWI), and apparent diffusion coefficient (ADC)was performed on the 6 patients with ADLD.
ResultsLMNB1 duplicationWe examined the CNV of LMNB1 in 93 patients with adult-onset leukoencephalopathy of unknown etiology by TaqMan-based real-time PCR assay. The gene dosage of LMNB1 wasincreased approximately by 1.5-fold in 4 patients from 3families (pedigrees I–III, figure 1A). This suggests that thesepatients were heterozygous for LMNB1 duplication.
Next, we determined the genomic region of the duplication bymicroarray-based copy number profiling using an AffymetrixCytoScan HD array. Consistent with the results of quantita-tive real-time PCR assay, the gene dosage of the entire regionof LMNB1 was increased (figure 1C). In addition to LMNB1,
Figure 1 Analysis of CNVs of LMNB1 and its upstream region
(A) Gene dosages for exons 3, 6, and 10 of LMNB1were determined by TaqMan-based real-time PCR assay. The copy numbers of 3 exons of LMNB1 in patients1–4were increasedby approximately 1.5-fold comparedwith control subjects, suggesting the presence of duplication of LMNB1 in these patients. (B) The copynumber variations of regions upstream of LMNB1 including GRAMD3, ALDH7A1, and PHAX were determined by TaqMan-based real-time PCR assay. The copynumbers of ALDH7A1 and PHAXwere decreased approximately by half in patients 5 and 6, suggesting the presence of the upstreamdeletion of LMNB1. (C) Thegenomic regions of duplication (blue) and deletion (red) were analyzed using an Affymetrix CytoScan HD array and are shown on the basis of informationobtained from the UCSC genome browser (assembly GRCh37/hg19). The regions of duplication were 153 kb in pedigree I, 220 kb in pedigree II, and 221 kb inpedigree III. The deletion upstream of LMNB1 in a previous report is shown by a dotted line.8 The positions of original enhancer A (Enh-A) and alternativeenhancer B (Enh-B) for LMNB1 are indicated by arrowheads. ALDH7A1 = aldehyde dehydrogenase 7 family member A1; CNV = copy number variation;GRAMD3 = GRAM domain containing 3; LMNB1 = lamin B1; PHAX = phosphorylated adaptor for RNA export.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
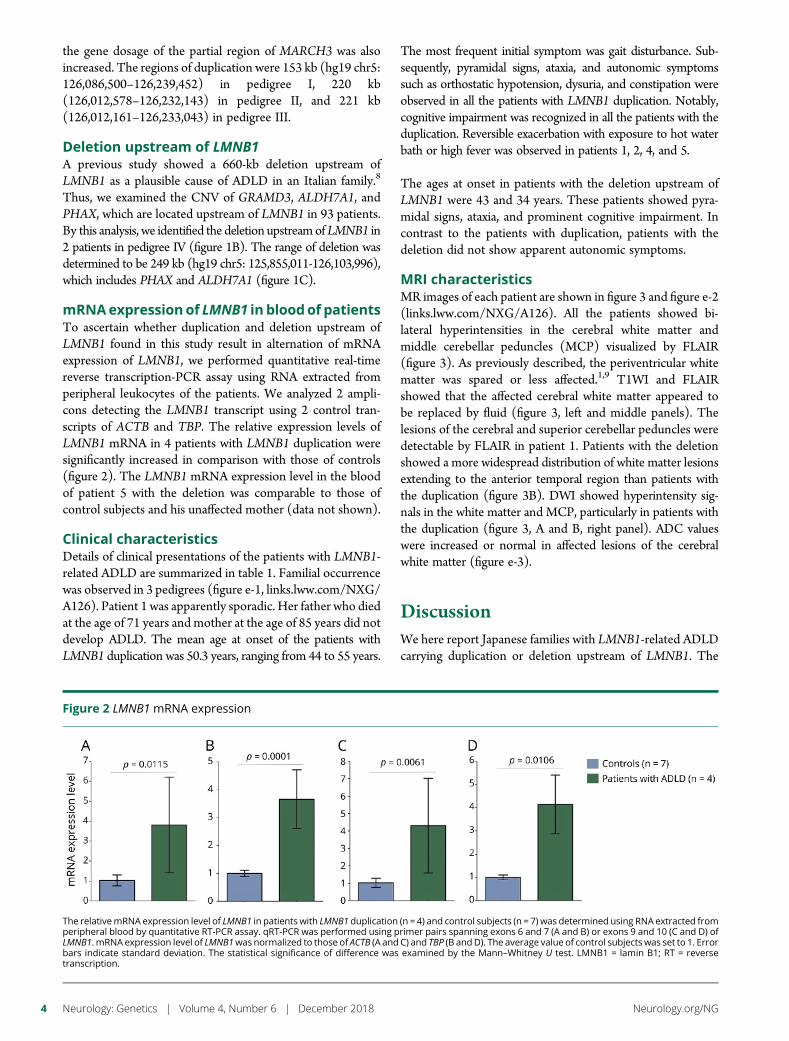
the gene dosage of the partial region of MARCH3 was alsoincreased. The regions of duplication were 153 kb (hg19 chr5:126,086,500–126,239,452) in pedigree I, 220 kb(126,012,578–126,232,143) in pedigree II, and 221 kb(126,012,161–126,233,043) in pedigree III.
Deletion upstream of LMNB1A previous study showed a 660-kb deletion upstream ofLMNB1 as a plausible cause of ADLD in an Italian family.8
Thus, we examined the CNV of GRAMD3, ALDH7A1, andPHAX, which are located upstream of LMNB1 in 93 patients.By this analysis, we identified the deletion upstreamofLMNB1 in2 patients in pedigree IV (figure 1B). The range of deletion wasdetermined to be 249 kb (hg19 chr5: 125,855,011-126,103,996),which includes PHAX and ALDH7A1 (figure 1C).
mRNAexpressionof LMNB1 in bloodof patientsTo ascertain whether duplication and deletion upstream ofLMNB1 found in this study result in alternation of mRNAexpression of LMNB1, we performed quantitative real-timereverse transcription-PCR assay using RNA extracted fromperipheral leukocytes of the patients. We analyzed 2 ampli-cons detecting the LMNB1 transcript using 2 control tran-scripts of ACTB and TBP. The relative expression levels ofLMNB1 mRNA in 4 patients with LMNB1 duplication weresignificantly increased in comparison with those of controls(figure 2). The LMNB1 mRNA expression level in the bloodof patient 5 with the deletion was comparable to those ofcontrol subjects and his unaffected mother (data not shown).
Clinical characteristicsDetails of clinical presentations of the patients with LMNB1-related ADLD are summarized in table 1. Familial occurrencewas observed in 3 pedigrees (figure e-1, links.lww.com/NXG/A126). Patient 1 was apparently sporadic. Her father who diedat the age of 71 years and mother at the age of 85 years did notdevelop ADLD. The mean age at onset of the patients withLMNB1 duplication was 50.3 years, ranging from 44 to 55 years.
The most frequent initial symptom was gait disturbance. Sub-sequently, pyramidal signs, ataxia, and autonomic symptomssuch as orthostatic hypotension, dysuria, and constipation wereobserved in all the patients with LMNB1 duplication. Notably,cognitive impairment was recognized in all the patients with theduplication. Reversible exacerbation with exposure to hot waterbath or high fever was observed in patients 1, 2, 4, and 5.
The ages at onset in patients with the deletion upstream ofLMNB1 were 43 and 34 years. These patients showed pyra-midal signs, ataxia, and prominent cognitive impairment. Incontrast to the patients with duplication, patients with thedeletion did not show apparent autonomic symptoms.
MRI characteristicsMR images of each patient are shown in figure 3 and figure e-2(links.lww.com/NXG/A126). All the patients showed bi-lateral hyperintensities in the cerebral white matter andmiddle cerebellar peduncles (MCP) visualized by FLAIR(figure 3). As previously described, the periventricular whitematter was spared or less affected.1,9 T1WI and FLAIRshowed that the affected cerebral white matter appeared tobe replaced by fluid (figure 3, left and middle panels). Thelesions of the cerebral and superior cerebellar peduncles weredetectable by FLAIR in patient 1. Patients with the deletionshowed a more widespread distribution of white matter lesionsextending to the anterior temporal region than patients withthe duplication (figure 3B). DWI showed hyperintensity sig-nals in the white matter and MCP, particularly in patients withthe duplication (figure 3, A and B, right panel). ADC valueswere increased or normal in affected lesions of the cerebralwhite matter (figure e-3).
DiscussionWe here report Japanese families with LMNB1-related ADLDcarrying duplication or deletion upstream of LMNB1. The
Figure 2 LMNB1 mRNA expression
The relativemRNA expression level of LMNB1 in patients with LMNB1 duplication (n = 4) and control subjects (n = 7) was determined using RNA extracted fromperipheral blood by quantitative RT-PCR assay. qRT-PCR was performed using primer pairs spanning exons 6 and 7 (A and B) or exons 9 and 10 (C and D) ofLMNB1. mRNA expression level of LMNB1was normalized to those of ACTB (A and C) and TBP (B andD). The average value of control subjects was set to 1. Errorbars indicate standard deviation. The statistical significance of difference was examined by the Mann–Whitney U test. LMNB1 = lamin B1; RT = reversetranscription.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
duplicated genomic regions in these Japanese patients weredifferent from those of the reported families with duplica-tion including the Japanese K4975 family.4,7,10 The genomicregions of duplication in pedigrees II and III were similar,suggesting that they may share a common founder of theduplication.We showed that the LMNB1mRNA expressionlevel was significantly elevated, as determined by the anal-ysis using RNA extracted from peripheral blood leukocytesfrom the patients with LMNB1 duplication (figure 2).Consistent with our results, previous reports have shownthat the levels of lamin B1 protein were increased in leuko-cytes or brains of patients with LMNB1 duplication.4,7,9,11,12
Thus, lamin B1 overproduction seems to be a keymechanismunderlying the pathogenesis of ADLD. However, the path-omechanism by which LMNB1 mRNA overexpression cau-ses ADLD has not been fully elucidated.
Lamin B1 is a component of nuclear lamina and ubiquitouslyexpressed in all cells with nuclei. Lamin B1 plays a role inregulating gene expression during DNA replication.13 It hasbeen demonstrated that transgenic mice overexpressingLMNB1 showed age-dependent demyelination similar toADLD.14,15 Comprehensive mRNA expression analysis inthese mice revealed decreased mRNA expression levels ofgenes involved in the synthesis of lipid and cholesterol, whichare the major components of myelin.14 This suggests thatmaintenance and repair of myelin may be impaired by laminB1 overproduction, leading to the development of ADLD.
Previous studies have shown that autonomic symptomsprecede or occur together with gait disturbance and motorsymptoms in patients with LMNB1 duplication.1,16–19 Themost frequent initial symptom was gait disturbance in ourpatients with duplication. Thus, it should be noted thatpatients with ADLD may initially show motor symptoms.Cognitive impairment was observed in all the patients withthe duplication in this study, although the frequency ofcognitive impairment was reported to be 63% in patientswith the duplication.2,3
The characteristics of MR images in previous reportsshowed changes in the cerebral white matter and middlecerebellar peduncles, which were similarly observed in ourpatients with the duplication.1,9 T1WI and FLAIR revealedthe white matter degeneration, which is replaced by fluid inour patients (figure 3). These findings are similarly observedin patients with vanishing white matter disease (VWMD).20
Patients 5 and 6 were initially suspected as having VWMD,and genetic testing of eIF2B was performed with negativeresults. Thus, differential diagnosis between ADLD andVWMDmay be required in patients with such MRI findings.
We identified new patients with ADLD in pedigree IVcarrying the deletion upstream of LMNB1. The region ofthe deletion in this study was 249 kb, which was narrowerthan that of the 660-kb deletion found in the previousItalian family (figure 1C).8 It was demonstrated that theTa
ble
1Clin
ical
featuresofpatients
withLM
NB1-relatedADLD
Patien
tPed
igre
eLM
NB1
muta
tions
Sex
Family
histo
ryAge
at
onse
t,y
Age
at
examination,y
Initials
ympto
ms
Auto
nomic
distu
rbance
Pyr
amidal
sign
sAta
xia
Cogn
itive
impairment
1I
Duplication
F−
4456
Dizzines
s+
++
+MMSE
23
2II
Duplication
M+
5557
Spas
ticga
it+
++
+MoCA21
3II
Duplication
M+
5267
Gaitdisturb
ance
++
++M
MSE
15
4III
Duplication
F+
5070
Gaitdisturb
ance
++
++M
MSE
22
5IV
Deletionof
enhan
cer
F+
4350
Dysarthria,
muscle
wea
knes
s,co
gnitivedec
line
−+
++M
MSE
9
6IV
Deletionof
enhan
cer
M+
3442
Gaitdisturb
ance
−+
++M
oCA10
Abbreviations:
ADLD
=ad
ult-onse
tdem
yelin
atingleuko
dystrophy;
MMSE
=Mini-M
entalS
tate
Exam
ination;M
oCA=Montrea
lCogn
itiveAsses
smen
t;LM
NB1=lamin
B1.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
LMNB1 mRNA expression level was increased in brains ofpatients with the deletion.8 The deletion in the Italian pedi-gree contains a putative enhancer region (Enh-A) regulatingLMNB1 expression and the insulator between PHAX andALDH7A1 (figure 1C). They speculated that another up-stream enhancer (Enh-B)may alternatively work and enhanceLMNB1 expression if the Enh-A and the insulator are deleted.Findings in our patients with the deletion supported thisnotion because the 188-kb deleted region shared by ourpatients and the previously reported patients commonly in-cluded the insulator and Enh-A. However, the mRNA ex-pression level of LMNB1 derived from peripheral leukocytesdid not increase in our patient with the deletion. The reasonwhy the mRNA expression level was not apparently altered inour patient may be explained by the difference in tissues usedfor mRNA examination. The alternative enhancer (Enh-B)was reported to work in a forebrain-specific manner8; thus, itsdeletion may not cause the overexpression of LMNB1 inperipheral blood in patients. The mRNA expression level ofLMNB1 may be altered in brain tissues, especially in oligo-dendrocytes. It would be important to analyze the LMNB1mRNA expression level in brains of patients with the deletionwhen the autopsied brain samples become available.
In previous reports, the patients with the deletion wereclinically characterized by later age at onset (age at onset:
47.2 ± 6.4 years) and the absence of autonomic symptoms atonset and cerebellar ataxia, as compared with the patients withduplication.21,22 Similarly, our patients with the deletion alsolacked autonomic symptoms. In contrast to a previous re-port,22 our patients with the deletion showed relatively severecognitive decline and the presence of cerebellar ataxia (tablee-1, links.lww.com/NXG/A126). Cerebellar lesions were alsonoticeable on MR images of our patients (figure 3B). Thepresence of cerebellar symptoms and the cerebellar lesionsrevealed byMRI may be characteristic in our patients with thedeletion because the Italian family lacked cerebellar symp-toms and rarely exhibited cerebellar lesions onMRI.22 A novelMR finding of this study is that the cerebral white matterlesions extended to the anterior temporal region in patientswith the deletion. The temporal white matter lesions werealso observed in other white matter diseases including cerebralautosomal dominant arteriopathy with subcortical infarcts andleukoencephalopathy and cerebral autosomal recessive arterio-pathy with subcortical infarcts and leukoencephalopathy.23,24
ADLD should be considered as a differential diagnosis forpatients exhibiting anterior temporal lobe white matter lesionson MRI.
In this study, we identified 6 patients with ADLD of 93patients with adult-onset leukoencephalopathy of unknownetiology. There were differences in clinical and MRI features
Figure 3 MRI findings in patients with ADLD with duplication and those with deletion upstream of LMNB1
(A) Findings ofMRIwith T1WI (left panel), FLAIR (middle panel), andDWI (right panel) of patient 1with LMNB1 duplication at the age of 56 years. Arrows point tothe MCP lesion. (B) Findings of MRI with T1WI (left panel), FLAIR (middle panel), and DWI (right panel) of patient 5 with the upstream deletion of LMNB1 at theage of 50 years. ADLD = adult-onset demyelinating leukodystrophy; DWI = diffusion-weighted imaging; FLAIR = fluid-attenuated inversion recovery; MCP =middle cerebellar peduncle; LMNB1 = lamin B1.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

between patients with ADLD with duplication and those withdeletion upstream of LMNB1. The characteristic clinical andimaging features in patients with LMNB1-related ADLD mayprovide the clue for efficient molecular diagnosis in patientswith adult-onset leukoencephalopathies.
Author contributionsN. Mezaki: drafting of the manuscript, study concept, acqui-sition of data, and analysis of data. T. Miura: study concept,acquisition of data, and analysis of data. K. Ogaki, M. Eriguchi,Y. Mizuno, K. Komatsu, H. Yamazaki, N. Suetsugu,S. Kawajiri, R. Yamasaki, T. Ishiguro, T. Konno, H. Nozaki,K. Kasuga, Y. Okuma, J.-I. Kira, and H. Hara: acquisition ofdata. O. Onodera: acquisition of data and study supervision.T. Ikeuchi: drafting of the manuscript, study concept, in-terpretation of data, and obtained the funding.
Study fundingSupported in part by grant-in-aid (JP16H01331 and26117506 to T.Ikeuchi) from the Japan Society for the Pro-motion of Science, a grant-in-aid for Research on IntractableDisease from the Japanese Ministry of Health, Labour andWelfare, Japan (12103055 to T.Ikeuchi and O.O.), a grant-in-aid (18kk0205009 to T.Ikeuchi) from Japan Agency forMedical Research and Development, and a grant from theTsubaki Memorial Foundation (to N.M.).
DisclosureN. Mezaki has received foundation and society researchsupport from the Tsubaki Memorial Foundation and JANiigata Kouseiren grant. T. Miura, K. Ogaki, M. Eriguchi,Y. Mizuno, K. Komatsu, H. Yamazaki, N. Suetsugi, andS. Kawajiri report no disclosure. R. Yamasaki serves on theeditorial board of Clinical and Experimental Neuroimmunology.T. Ishiguro, T. Konno, and H. Nozaki report no disclosure.K. Kasuga has received research support from the Japan So-ciety for the Promotion of Science. Y. Okuma reports nodisclosures. J.-I. Kira has served on the editorial boards ofMultiple Sclerosis, BMC Medicine, Journal of the NeurologicalSciences, Multiple Sclerosis and Related Disorders, PLoS One,Acta Neuropathologica Communications, and Clinical and Ex-perimental Neuroimmunology; has provided consultancy forBiogen Idec Japan; and has received research support fromgovernmental entities the Ministry of Health, Labour andWelfare, Japan Agency for Medical Research and De-velopment, and the MEXT KAKENHI program. H. Hara hasreceived funding for travel from Novartis and for speakerhonoraria from Eisai and has received Grant-in-Aid for Sci-entific Research. O. Onodera has received funding for speakerhonoraria from Kyowa Hakko Kirin Co., Ltd., Bristol-MyersSquibb, Ono Pharmaceutical Co., Ltd., Mitsubishi TanabePharma, Takeda, Daiichi-Sankyo, Fujifilm, Sanofi, and FP-Pharma and has received governmental entity research sup-port for Scientific Research from a Grant-in-Aid from theResearch Committee for Hereditary Cerebral Small VesselDiseases, and from the Ministry of Health, Labour and Wel-fare of Japan. T. Ikeuchi has served on the editorial board of
Parkinsonism and Related Disorders; has acted as a consultantfor Janssen Pharmaceuticals Inc. and Chugai PharmaceuticalCo. Ltd.; serves on the speakers’ bureaus of Daiichi Sankyo,Eisai, Ono Pharmaceutical Co. Ltd., Novartis, and Takeda;and has received governmental entity research support fromKAKENHI and the Ministry of Health. Full disclosure forminformation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics April 10, 2018. Accepted in final formSeptember 10, 2018.
References1. Finnsson J, Sundblom J, Dahl N, Melberg A, Raininko R. LMNB1-related autosomal-
dominant leukodystrophy: clinical and radiological course. Ann Neurol 2015;78:412–425.
2. Schwankhaus JD, Katz DA, Eldridge R, Schlesinger S, McFarland H. Clinical andpathological features of an autosomal dominant, adult-onset leukodystrophy simu-lating chronic progressive multiple sclerosis. Arch Neurol 1994;51:757–766.
3. Sandoval-Rodrıguez V, Cansino-Torres MA, Saenz-Farret M, Castañeda-Cisneros G,Moreno G. Autosomal dominant leukodystrophy presenting as Alzheimer’s-typedementia. Mult Scler Relat Disord 2017;17:230–233.
4. Padiath QS, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomaldominant leukodystrophy. Nat Genet 2006;38:1114–1123.
5. Nahhas N, Sabet Rasekh P, Vanderver A, Padiath Q. Autosomal Dominant Leuko-dystrophy With Autonomic Disease. Seattle: GeneReviews (R); 1993.
6. Dai Y, Ma Y, Li S, et al. An LMNB1 Duplication caused adult-onset autosomaldominant leukodystrophy in Chinese family: clinical manifestations, neuroradiologyand genetic diagnosis. Front Mol Neurosci 2017;10:215.
7. Giorgio E, Rolyan H, Kropp L, et al. Analysis of LMNB1 duplications in autosomaldominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum Mutat 2013;34:1160–1171.
8. Giorgio E, Robyr D, Spielmann M, et al. A large genomic deletion leads to enhanceradoption by the lamin B1 gene: a second path to autosomal dominant adult-onsetdemyelinating leukodystrophy (ADLD). Hum Mol Genet 2015;24:3143–3154.
9. Melberg A, Hallberg L, Kalimo H, Raininko R. MR characteristics and neuropa-thology in adult-onset autosomal dominant leukodystrophy with autonomic symp-toms. AJNR Am J Neuroradiol 2006;27:904–911.
10. Asahara H, Yoshimura T, Suda S, Furuya H, Kobayashi T. A Japanese family withprobably autosomal dominant adult-onset leukodystrophy. Rinsho Shinkeigaku 1996;36:968–972.
11. Schuster J, Sundblom J, Thuresson AC, et al. Genomic duplications mediate over-expression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD)with autonomic symptoms. Neurogenetics 2011;12:65–72.
12. Columbaro M, Mattioli E, Maraldi NM, et al. Oct-1 recruitment to the nuclearenvelope in adult-onset autosomal dominant leukodystrophy. Biochim Biophys Acta2013;1832:411–420.
13. Tang CW, Maya-Mendoza A, Martin C, et al. The integrity of a lamin-B1-dependentnucleoskeleton is a fundamental determinant of RNA synthesis in human cells. J CelSci 2008;121:1014–1024.
14. Rolyan H, Tyurina YY, Hernandez M, et al. Defects of lipid synthesis are linked to theage-dependent demyelination caused by lamin B1 overexpression. J Neurosci 2015;26:12002–12017.
15. Heng MY, Lin ST, Verret L, et al. Lamin B1 mediates cell-autonomous neuropa-thology in a leukodystrophy mouse model. J Clin Invest 2013;123:2719–2729.
16. Brussino A, Vaula G, Cagnoli C, et al. A novel family with Lamin B1 duplicationassociated with adult-onset leukoencephalopathy. J Neurol Neurosurg Psychiatry2009;80:237–240.
17. Dos Santos MM, Grond-Ginsbach C, Aksay SS, et al. Adult-onset autosomaldominant leukodystrophy due to LMNB1 gene duplication. J Neurol 2012;259:579–581.
18. Eldridge R, Anayiotos CP, Schlesinger S, et al. Hereditary adult-onset leukodys-trophy simulating chronic progressive multiple sclerosis. N Engl J Med 1984;311:948–953.
19. Brunetti V, Ferilli MA, Nociti V, Silvestri G. Teaching neuroImages: autosomaldominant leukodystrophy in a sporadic case. Neurology 2014;83:e121.
20. van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. LancetNeurol 2006;5:413–423.
21. Quattrocolo G, Leombruni S, Vaula G, et al. Autosomal dominant late-onset leu-koencephalopathy. Clinical report of a new Italian family. Eur Neurol 1997;37:53–61.
22. Brussino A, Vaula G, Cagnoli C, et al. A family with autosomal dominant leukodys-trophy linked to 5q23.2-q23.3 without lamin B1 mutations. Eur J Neurol 2010;17:541–549.
23. Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil LancetNeurol 2009;8:643–653.
24. Nozaki H, Sekine Y, Fukutake T, et al. Characteristic features and progression ofabnormalities on MRI for CARASIL. Neurology 2015;85:459–463.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

ARTICLE OPEN ACCESS
Atrial fibrillation genetic risk differentiatescardioembolic stroke from other stroke subtypesSara L. Pulit, PhD, Lu-ChenWeng, PhD, Patrick F.McArdle, PhD, Ludovic Trinquart, PhD, SeungHoanChoi, PhD,
Braxton D. Mitchell, PhD, Jonathan Rosand, MD, Paul I. W. de Bakker, PhD, Emelia J. Benjamin, MD, ScM,
Patrick T. Ellinor, MD, PhD, Steven J. Kittner, MD, Steven A. Lubitz, MD, MPH,*
and Christopher D. Anderson, MD*, on behalf of the Atrial Fibrillation Genetics Consortium and the
International Stroke Genetics Consortium
Neurol Genet 2018;4:e293. doi:10.1212/NXG.0000000000000293
Correspondence
Dr. Lubitz
or Dr. Anderson
AbstractObjectiveWe sought to assess whether genetic risk factors for atrial fibrillation (AF) can explain car-dioembolic stroke risk.
MethodsWe evaluated genetic correlations between a previous genetic study of AF and AF in thepresence of cardioembolic stroke using genome-wide genotypes from the Stroke GeneticsNetwork (N = 3,190 AF cases, 3,000 cardioembolic stroke cases, and 28,026 referents). Wetested whether a previously validated AF polygenic risk score (PRS) associated with car-dioembolic and other stroke subtypes after accounting for AF clinical risk factors.
ResultsWe observed a strong correlation between previously reported genetic risk for AF, AF in thepresence of stroke, and cardioembolic stroke (Pearson r = 0.77 and 0.76, respectively, acrossSNPs with p < 4.4 × 10−4 in the previous AFmeta-analysis). An AF PRS, adjusted for clinical AFrisk factors, was associated with cardioembolic stroke (odds ratio [OR] per SD = 1.40, p = 1.45× 10−48), explaining ;20% of the heritable component of cardioembolic stroke risk. The AFPRS was also associated with stroke of undetermined cause (OR per SD = 1.07, p = 0.004), butno other primary stroke subtypes (all p > 0.1).
ConclusionsGenetic risk of AF is associated with cardioembolic stroke, independent of clinical risk factors.Studies are warranted to determine whether AF genetic risk can serve as a biomarker for strokescaused by AF.
*These authors contributed equally to supervision of this study.
From the Department of Genetics (S.L.P., P.I.W.d. B.), University Medical Center Utrecht, Utrecht University, The Netherlands; P.I.W.d.B. is now with Computational Genomics, VertexPharmaceuticals, Boston, MA; Li Ka Shing Centre for Health Information and Discovery (S.L.P.), The Big Data Institute, University of Oxford, United Kingdom; Program in Medical andPopulation Genetics (S.L.P., L.-C.W., S.H.C., J.R., P.T.E., S.A.L., C.D.A.), Broad Institute, Cambridge, MA; Cardiovascular Research Center (L.-C.W., P.T.E., S.A.L.), Center for GenomicMedicine (J.R., C.D.A.), J.P. Kistler Stroke Research Center (J.R., C.D.A.), and Cardiac Arrhythmia Service (P.T.E., S.A.L.), Massachusetts General Hospital, Boston; Department ofMedicine (P.F.M., B.D.M.), Program for Personalized and Genomic Medicine, University of Maryland School of Medicine, Baltimore; National Heart, Lung, and Blood Institute’s andBoston University’s Framingham Heart Study (L.T., E.J.B.); Department of Biostatistics (L.T.) and Department of Epidemiology (E.J.B.), Boston University School of Public Health, MA;Geriatrics Research and Education Clinical Center (B.D.M.), Baltimore Veterans AdministrationMedical Center,MD; Cardiology PreventiveMedicine Sections (E.J.B.), Evans Departmentof Medicine, Boston University School of Medicine; Department of Neurology (S.J.K.), University of Maryland School of Medicine; andDepartment of Neurology (S.J.K.), Veterans AffairsMedical Center, Baltimore, MD.
The list of Atrial Fibrillation Genetics (AFGen) Consortium and Stroke Genetics Network (SiGN) Consortium members can be found at https://links.lww.com/NXG/A123.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the Authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Atrial fibrillation (AF) affects nearly 34 million individualsworldwide1 and is associated with a fivefold increased risk ofischemic stroke,2 a leading cause of death and disability.3,4 AFpromotes blood clot formation in the heart, which can emb-olize distally, and is a leading cause of cardioembolism. Sec-ondary prevention of cardioembolic stroke is directed atidentifying AF as a potential cause and initiating anti-coagulation to prevent recurrences. Yet, AF can remain occulteven after extensive workup owing to the paroxysmal natureand fact that it can be asymptomatic. Because both AF andstroke are heritable, and because there is a compelling clinicalneed to determine whether stroke survivors have AF as anunderlying cause, we sought to determine whether geneticrisk of cardioembolic stroke can be approximated by mea-suring genetic susceptibility to AF.
Recent genome-wide association studies (GWAS) havedemonstrated that both AF5 and ischemic stroke6,7 arecomplex disorders with polygenic architectures. The top locifor cardioembolic stroke, on chromosome 4q25 upstream ofPITX2 and on 16q22 near ZFHX3, are both leading risk locifor AF.8–10 Despite overlap in top risk loci, the genetic sus-ceptibility to both AF and cardioembolic stroke is likely toinvolve the aggregate contributions of hundreds or thousandsof loci, consistent with other polygenic conditions.11
To understand whether genetic risk of AF is an important andpotentially useful determinant of overall cardioembolic strokerisk, we analyzed 13,390 ischemic stroke cases and 28,026referents from theNINDS-Stroke Genetics Network (SiGN)12
with genome-wide genotyping data. First, we assessed whetherpatients with stroke with AF have a genetic predisposition toarrhythmia, leveraging additional GWAS data from the AFGenetics (AFGen) Consortium. Second, we compared geneticrisk factors for AF and stroke to ascertain the extent to whichheritable risk of cardioembolic stroke is explained by geneticrisk factors for AF.
MethodsThe Stroke Genetics NetworkThe SiGN was established with the aim of performing thelargest genome-wide association study (GWAS) of ischemicstroke to date. The study design has been previously de-scribed12 (e-Methods). Briefly, subjects in SiGNwere classifiedinto stroke subtypes using the Causative Classification System(CCS), which subtypes cases through an automated, web-based system that accounts for clinical data, test results, andimaging information.13,14 Within the CCS, there are 2
subcategories: CCS causative, which does not allow for com-peting subtypes in a single sample, and CCS phenotypic, whichdoes. In addition, ;74% of samples were subtyped using theTrial of ORG 10172 in Acute Stroke Treatment (TOAST)subtyping system.15 After quality control, the SiGN data setcomprised 16,851 ischemic stroke cases and 32,473 stroke-freecontrols (e-Methods and table e-1, links.lww.com/NXG/A123). In this study, we analyze only the European- andAfrican-ancestry samples (13,390 cases and 28,026 controls).
Standard protocol approvals, registrations,and patient consentsAll cohorts included in the SiGN data set received approvalfrom the cohort-specific ethical standards committee. Cohortsreceived written informed consent from all patients or guard-ians of patients for participation in the study, where applicable.Details on sample collection have been previously described.12
Identifying AF cases and controlsWe defined AF in SiGN on the basis of 5 variables available inthe CCS phenotyping system: (1) AF, (2) paroxysmal AF, (3)atrial flutter, (4) sick sinus syndrome, and (5) atrial thrombus.This definition yielded 3,190 AF cases for analysis. We alsodefined a strict case set based on “AF” only (N = 1,751 cases)for sensitivity analyses (e-Methods and figure e-1, links.lww.com/NXG/A123).
From the 28,026 controls, we established a set of 3,861control individuals in whom AF was indicated as not present.For the remaining subjects, we assumed that individuals didnot have AF because AF status for most control samples inSiGN is unknown.
Genome-wide association testing of ischemicstroke subtypes and AF in SiGNWe merged genotype dosages together and kept single nucle-otide polymorphisms (SNPs) with imputation quality >0.8 andminor allele frequency (MAF) >1% (e-Methods, links.lww.com/NXG/A123). We performed association testing usinga linear mixed model (LMM) implemented in BOLT-LMM.16
We adjusted the model for the top 10 principal components(PCs) and sex, in addition to the genetic relationship matrix(GRM; e-Methods).16We performedGWAS inAF and each ofthe stroke subtypes available in SiGN. Results were unadjustedfor age because adjusting for age in the AF GWAS gave resultshighly concordant with age-unadjusted results (e-Results).
Heritability calculationsWe calculated additive SNP-based heritability estimates forischemic stroke, stroke subtypes, and AF using restricted
GlossaryAF = atrial fibrillation;AFGen = AFGenetics;CCS =Causative Classification System;CI = confidence interval;GRM = geneticrelationship matrix;GWAS = genome-wide association studies;MAF =minor allele frequency;OR = odds ratio; PC = principalcomponent; PRS = polygenic risk score; SiGN = Stroke Genetics Network.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
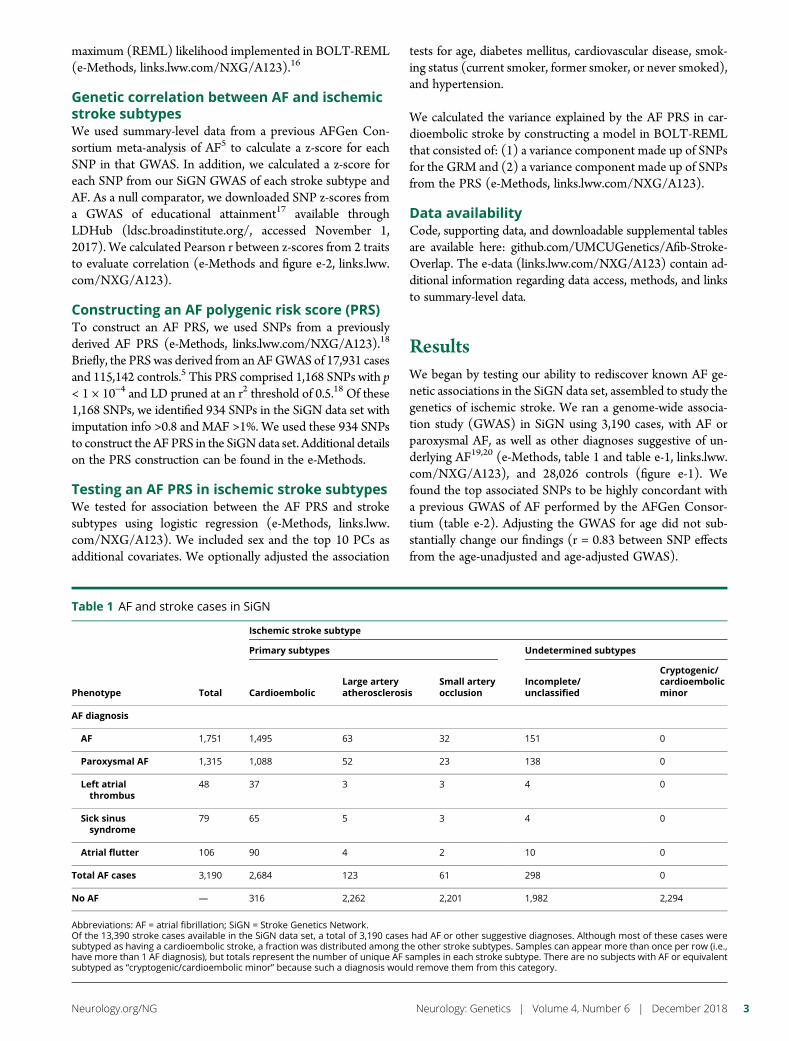
maximum (REML) likelihood implemented in BOLT-REML(e-Methods, links.lww.com/NXG/A123).16
Genetic correlation between AF and ischemicstroke subtypesWe used summary-level data from a previous AFGen Con-sortium meta-analysis of AF5 to calculate a z-score for eachSNP in that GWAS. In addition, we calculated a z-score foreach SNP from our SiGN GWAS of each stroke subtype andAF. As a null comparator, we downloaded SNP z-scores froma GWAS of educational attainment17 available throughLDHub (ldsc.broadinstitute.org/, accessed November 1,2017). We calculated Pearson r between z-scores from 2 traitsto evaluate correlation (e-Methods and figure e-2, links.lww.com/NXG/A123).
Constructing an AF polygenic risk score (PRS)To construct an AF PRS, we used SNPs from a previouslyderived AF PRS (e-Methods, links.lww.com/NXG/A123).18
Briefly, the PRSwas derived from an AFGWAS of 17,931 casesand 115,142 controls.5 This PRS comprised 1,168 SNPs with p< 1 × 10−4 and LD pruned at an r2 threshold of 0.5.18 Of these1,168 SNPs, we identified 934 SNPs in the SiGN data set withimputation info >0.8 and MAF >1%. We used these 934 SNPsto construct the AF PRS in the SiGNdata set. Additional detailson the PRS construction can be found in the e-Methods.
Testing an AF PRS in ischemic stroke subtypesWe tested for association between the AF PRS and strokesubtypes using logistic regression (e-Methods, links.lww.com/NXG/A123). We included sex and the top 10 PCs asadditional covariates. We optionally adjusted the association
tests for age, diabetes mellitus, cardiovascular disease, smok-ing status (current smoker, former smoker, or never smoked),and hypertension.
We calculated the variance explained by the AF PRS in car-dioembolic stroke by constructing a model in BOLT-REMLthat consisted of: (1) a variance component made up of SNPsfor the GRM and (2) a variance component made up of SNPsfrom the PRS (e-Methods, links.lww.com/NXG/A123).
Data availabilityCode, supporting data, and downloadable supplemental tablesare available here: github.com/UMCUGenetics/Afib-Stroke-Overlap. The e-data (links.lww.com/NXG/A123) contain ad-ditional information regarding data access, methods, and linksto summary-level data.
ResultsWe began by testing our ability to rediscover known AF ge-netic associations in the SiGN data set, assembled to study thegenetics of ischemic stroke. We ran a genome-wide associa-tion study (GWAS) in SiGN using 3,190 cases, with AF orparoxysmal AF, as well as other diagnoses suggestive of un-derlying AF19,20 (e-Methods, table 1 and table e-1, links.lww.com/NXG/A123), and 28,026 controls (figure e-1). Wefound the top associated SNPs to be highly concordant witha previous GWAS of AF performed by the AFGen Consor-tium (table e-2). Adjusting the GWAS for age did not sub-stantially change our findings (r = 0.83 between SNP effectsfrom the age-unadjusted and age-adjusted GWAS).
Table 1 AF and stroke cases in SiGN
Phenotype Total
Ischemic stroke subtype
Primary subtypes Undetermined subtypes
CardioembolicLarge arteryatherosclerosis
Small arteryocclusion
Incomplete/unclassified
Cryptogenic/cardioembolicminor
AF diagnosis
AF 1,751 1,495 63 32 151 0
Paroxysmal AF 1,315 1,088 52 23 138 0
Left atrialthrombus
48 37 3 3 4 0
Sick sinussyndrome
79 65 5 3 4 0
Atrial flutter 106 90 4 2 10 0
Total AF cases 3,190 2,684 123 61 298 0
No AF — 316 2,262 2,201 1,982 2,294
Abbreviations: AF = atrial fibrillation; SiGN = Stroke Genetics Network.Of the 13,390 stroke cases available in the SiGN data set, a total of 3,190 cases had AF or other suggestive diagnoses. Although most of these cases weresubtyped as having a cardioembolic stroke, a fraction was distributed among the other stroke subtypes. Samples can appear more than once per row (i.e.,have more than 1 AF diagnosis), but totals represent the number of unique AF samples in each stroke subtype. There are no subjects with AF or equivalentsubtyped as “cryptogenic/cardioembolic minor” because such a diagnosis would remove them from this category.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
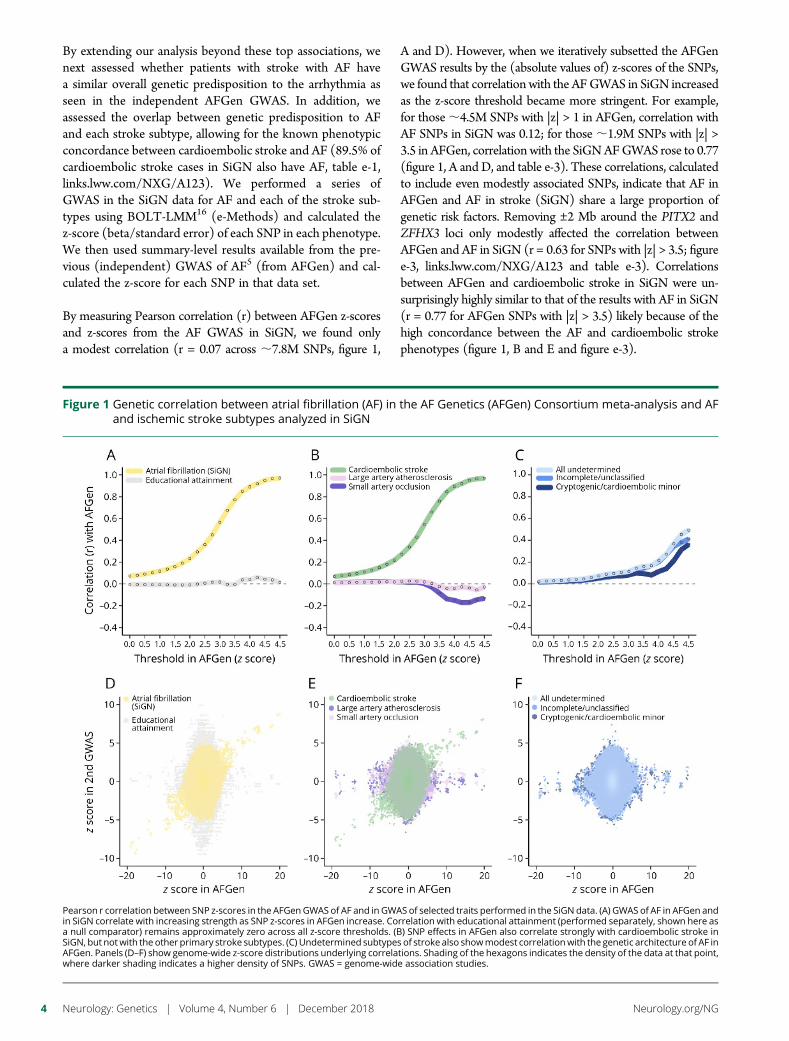
By extending our analysis beyond these top associations, wenext assessed whether patients with stroke with AF havea similar overall genetic predisposition to the arrhythmia asseen in the independent AFGen GWAS. In addition, weassessed the overlap between genetic predisposition to AFand each stroke subtype, allowing for the known phenotypicconcordance between cardioembolic stroke and AF (89.5% ofcardioembolic stroke cases in SiGN also have AF, table e-1,links.lww.com/NXG/A123). We performed a series ofGWAS in the SiGN data for AF and each of the stroke sub-types using BOLT-LMM16 (e-Methods) and calculated thez-score (beta/standard error) of each SNP in each phenotype.We then used summary-level results available from the pre-vious (independent) GWAS of AF5 (from AFGen) and cal-culated the z-score for each SNP in that data set.
By measuring Pearson correlation (r) between AFGen z-scoresand z-scores from the AF GWAS in SiGN, we found onlya modest correlation (r = 0.07 across ;7.8M SNPs, figure 1,
A and D). However, when we iteratively subsetted the AFGenGWAS results by the (absolute values of) z-scores of the SNPs,we found that correlationwith the AFGWAS in SiGN increasedas the z-score threshold became more stringent. For example,for those;4.5M SNPs with |z| > 1 in AFGen, correlation withAF SNPs in SiGN was 0.12; for those;1.9M SNPs with |z| >3.5 in AFGen, correlation with the SiGNAFGWAS rose to 0.77(figure 1, A and D, and table e-3). These correlations, calculatedto include even modestly associated SNPs, indicate that AF inAFGen and AF in stroke (SiGN) share a large proportion ofgenetic risk factors. Removing ±2 Mb around the PITX2 andZFHX3 loci only modestly affected the correlation betweenAFGen and AF in SiGN (r = 0.63 for SNPs with |z| > 3.5; figuree-3, links.lww.com/NXG/A123 and table e-3). Correlationsbetween AFGen and cardioembolic stroke in SiGN were un-surprisingly highly similar to that of the results with AF in SiGN(r = 0.77 for AFGen SNPs with |z| > 3.5) likely because of thehigh concordance between the AF and cardioembolic strokephenotypes (figure 1, B and E and figure e-3).
Figure 1 Genetic correlation between atrial fibrillation (AF) in the AF Genetics (AFGen) Consortium meta-analysis and AFand ischemic stroke subtypes analyzed in SiGN
Pearson r correlation between SNP z-scores in the AFGenGWAS of AF and in GWAS of selected traits performed in the SiGN data. (A) GWAS of AF in AFGen andin SiGN correlate with increasing strength as SNP z-scores in AFGen increase. Correlation with educational attainment (performed separately, shown here asa null comparator) remains approximately zero across all z-score thresholds. (B) SNP effects in AFGen also correlate strongly with cardioembolic stroke inSiGN, but not with the other primary stroke subtypes. (C) Undetermined subtypes of stroke also showmodest correlationwith the genetic architecture of AF inAFGen. Panels (D–F) show genome-wide z-score distributions underlying correlations. Shading of the hexagons indicates the density of the data at that point,where darker shading indicates a higher density of SNPs. GWAS = genome-wide association studies.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
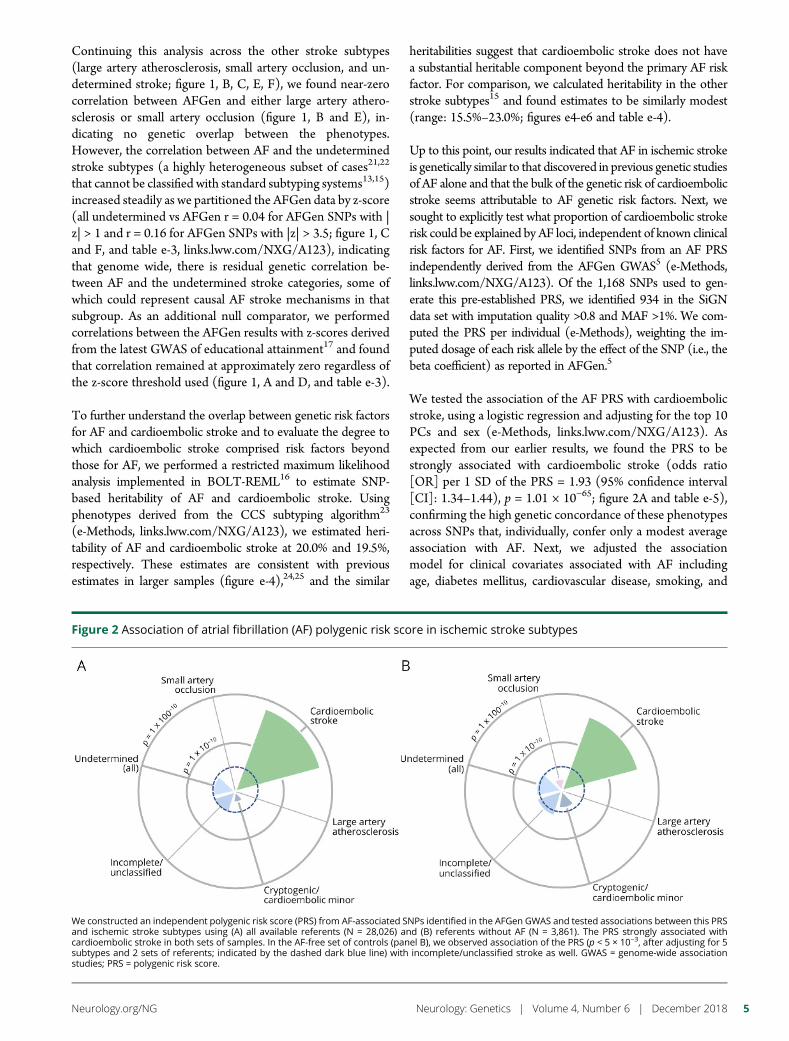
Continuing this analysis across the other stroke subtypes(large artery atherosclerosis, small artery occlusion, and un-determined stroke; figure 1, B, C, E, F), we found near-zerocorrelation between AFGen and either large artery athero-sclerosis or small artery occlusion (figure 1, B and E), in-dicating no genetic overlap between the phenotypes.However, the correlation between AF and the undeterminedstroke subtypes (a highly heterogeneous subset of cases21,22
that cannot be classified with standard subtyping systems13,15)increased steadily as we partitioned the AFGen data by z-score(all undetermined vs AFGen r = 0.04 for AFGen SNPs with |z| > 1 and r = 0.16 for AFGen SNPs with |z| > 3.5; figure 1, Cand F, and table e-3, links.lww.com/NXG/A123), indicatingthat genome wide, there is residual genetic correlation be-tween AF and the undetermined stroke categories, some ofwhich could represent causal AF stroke mechanisms in thatsubgroup. As an additional null comparator, we performedcorrelations between the AFGen results with z-scores derivedfrom the latest GWAS of educational attainment17 and foundthat correlation remained at approximately zero regardless ofthe z-score threshold used (figure 1, A and D, and table e-3).
To further understand the overlap between genetic risk factorsfor AF and cardioembolic stroke and to evaluate the degree towhich cardioembolic stroke comprised risk factors beyondthose for AF, we performed a restricted maximum likelihoodanalysis implemented in BOLT-REML16 to estimate SNP-based heritability of AF and cardioembolic stroke. Usingphenotypes derived from the CCS subtyping algorithm23
(e-Methods, links.lww.com/NXG/A123), we estimated heri-tability of AF and cardioembolic stroke at 20.0% and 19.5%,respectively. These estimates are consistent with previousestimates in larger samples (figure e-4),24,25 and the similar
heritabilities suggest that cardioembolic stroke does not havea substantial heritable component beyond the primary AF riskfactor. For comparison, we calculated heritability in the otherstroke subtypes15 and found estimates to be similarly modest(range: 15.5%–23.0%; figures e4-e6 and table e-4).
Up to this point, our results indicated that AF in ischemic strokeis genetically similar to that discovered in previous genetic studiesof AF alone and that the bulk of the genetic risk of cardioembolicstroke seems attributable to AF genetic risk factors. Next, wesought to explicitly test what proportion of cardioembolic strokerisk could be explained by AF loci, independent of known clinicalrisk factors for AF. First, we identified SNPs from an AF PRSindependently derived from the AFGen GWAS5 (e-Methods,links.lww.com/NXG/A123). Of the 1,168 SNPs used to gen-erate this pre-established PRS, we identified 934 in the SiGNdata set with imputation quality >0.8 and MAF >1%. We com-puted the PRS per individual (e-Methods), weighting the im-puted dosage of each risk allele by the effect of the SNP (i.e., thebeta coefficient) as reported in AFGen.5
We tested the association of the AF PRS with cardioembolicstroke, using a logistic regression and adjusting for the top 10PCs and sex (e-Methods, links.lww.com/NXG/A123). Asexpected from our earlier results, we found the PRS to bestrongly associated with cardioembolic stroke (odds ratio[OR] per 1 SD of the PRS = 1.93 (95% confidence interval[CI]: 1.34–1.44), p = 1.01 × 10−65; figure 2A and table e-5),confirming the high genetic concordance of these phenotypesacross SNPs that, individually, confer only a modest averageassociation with AF. Next, we adjusted the associationmodel for clinical covariates associated with AF includingage, diabetes mellitus, cardiovascular disease, smoking, and
Figure 2 Association of atrial fibrillation (AF) polygenic risk score in ischemic stroke subtypes
We constructed an independent polygenic risk score (PRS) from AF-associated SNPs identified in the AFGen GWAS and tested associations between this PRSand ischemic stroke subtypes using (A) all available referents (N = 28,026) and (B) referents without AF (N = 3,861). The PRS strongly associated withcardioembolic stroke in both sets of samples. In the AF-free set of controls (panel B), we observed association of the PRS (p < 5 × 10−3, after adjusting for 5subtypes and 2 sets of referents; indicated by the dashed dark blue line) with incomplete/unclassified stroke as well. GWAS = genome-wide associationstudies; PRS = polygenic risk score.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

hypertension.26 Using a (smaller) set of cases and controlswith complete clinical risk factor information, we found thatinclusion of these clinical risk factors in the model onlymodestly reduced the PRS signal in cardioembolic stroke (ORper 1 SD = 1.40 [95% CI: 1.34–1.47], p = 1.45 × 10−48; tablese5-e7, links.lww.com/NXG/A123). These results indicatea strong relationship between AFGen risk factors and car-dioembolic stroke risk, independent of the clinical factors thatassociate with AF. Expanding the set of SNPs used to con-struct the PRS to the original 934 SNPs ±25 kb, ±50 kb, and±100 kb (e-Methods) revealed a persistently strong, thoughsomewhat attenuated, association between the PRS and car-dioembolic stroke (PRS including SNPs within 100 kb, p =4.47 × 10−44, table e-6). None of the other stroke subtypeswere significantly associated with the AF PRS (all p > 0.013,figure 2A and figure e-7).
Because AF status was missing for most controls in the SiGNdata set, we performed sensitivity analyses using only the 3,861controls confirmed as having no AF. Although reducing theset of controls to this refined group did not substantially changethe results for the primary stroke subtypes, we found thatthe AF PRS was modestly associated (p < 5 × 10−3, afteradjusting for 5 subtypes and 2 control groups) with the overallundetermined subtype (OR per 1 SD = 1.07 [95% CI:1.02–1.13], p = 4.15 × 10−3) (figure 2B and table e-5, links.lww.com/NXG/A123). Further examination of the 2 mutually ex-clusive subgroups of the undetermined group revealed that thePRS associated significantly with the incomplete/unclassifiedcategorization (OR per 1 SD = 1.09 [95% CI: 1.03–1.16],p = 3.17 × 10−3) (figure 2B) but not with cryptogenic/cardioembolic minor (OR per 1 SD = 1.06 [95% CI:1.00–1.13], p = 5.10 × 10−2). Correcting for clinical cova-riates only modestly changed the signal in the incomplete/unclassified phenotype (p = 9.7 × 10−3, figure 2), supportingthe robustness of the observed association, independent ofclinical risk factors.
Last, we created a model in BOLT-LMM, fitting 2 geneticvariance components: 1 component including SNPs for theGRM and the second component including the original PRSSNPs from the AF PRS (including ±100 kb around these SNPsto include a sufficient number of markers to estimate the var-iance explained) (table e-8). We found that the SNPs from theAF PRS explained 4.1% of the total (20.0%) heritability in AF.In evaluating the variance explained in cardioembolic stroke, wefound a nearly identical result: the component representing theAF risk score explained 4.5% (SE = 1.00%) of the total 19.5%genetic heritability in cardioembolic stroke. Thus, AF geneticrisk accounts for 23.1%, or approximately one-fifth, of the totalheritability of cardioembolic stroke.
DiscussionOur results suggest that individuals with cardioembolicstrokes have an enrichment for AF genetic risk, despite thefact that cardioembolic stroke often affects older adults with
multiple clinical comorbidities27 that could increase risk of AFbecause of nongenetic factors. The fact that cardioembolicstroke and AF share a highly similar genetic architectureextends our understanding of the morbid consequences ofheritable forms of arrhythmia. Furthermore, the observationthat AF genetic risk was only associated with cardioembolicstroke, and (consistently) lacked association in large arteryatherosclerosis or small artery occlusion,28 increases the pos-sibility that AF genetic risk may be informative in the man-agement of ischemic stroke survivors in whom the mechanismmay be unclear.
The use of PRSs for complex traits has proved an efficientmeans of understanding how genetic predisposition to diseasescan overlap. Given the onslaught of genotyping data availablefor common diseases, PRSs can now be used to stratify patientsby risk (e.g., in breast cancer29,30) or predict outcome (e.g., inneuropsychiatric disease29). More recently, PRSs have beenused to identify individuals in the general population witha four-fold risk of coronary disease,31 proposed for inclusion inclinical workups of individuals with early-onset coronary arterydisease,32 and used to identify patients for whom lifestylechanges or statin intervention would be beneficial.33,34 Al-though previous work has also shown an association betweenan AF PRS and cardioembolic stroke,28 we have extended thiswork to formally quantify the extent to which an AF PRScaptures genetic risk of cardioembolic stroke. These findingslay the groundwork for future work that can potentially lever-age this overlap to develop AF PRSs that could be used topredict individuals at highest risk of cardioembolic stroke (toimprove diagnostic resource allocation) or help distinguishbetween clinical subtypes of stroke.
Although our analysis was aimed at understanding the geneticoverlap between cardioembolic stroke and AF, we addition-ally observed genetic correlation between AF and un-determined stroke, a finding not observed in a previousinvestigation of AF PRS in ischemic stroke subtypes, albeit ina smaller sample.28 Perhaps contrary to expectation, we spe-cifically found the AF PRS to bemore strongly associated withthe subset of etiology-undetermined strokes with an in-complete clinical evaluation, as opposed to those with cryp-togenic stroke of a presumed, but not demonstrated, embolicsource. These associations could be due to physician biasesin diagnostic workups, rather than supporting a low preva-lence of occult AF in presumed embolic strokes of un-determined source. Identifying patients with stroke with AF isan important clinical challenge because occult AF is wellknown to cause strokes35,36 and because such patients are athigh risk of recurrent stroke, which is preventable withanticoagulation.37,38 Together, our findings indicate that AFgenetic risk may augment clinical algorithms to determinestroke etiology, but will require further study.
The work presented here benefits from a number ofimprovements, including increased sample size; analysis ofsamples from amulticenter consortium, potentially enhancing
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

the generalizability of the findings; and use of the CCS sub-typing system, which provides more nuanced phenotyping,particularly in the cryptogenic subtype. Nevertheless, somelimitations remain. Stroke is a heterogeneous condition thatoccurs later in life and has a high lifetime prevalence (>15%39),features that can reduce statistical power. Furthermore, samplesizes have lagged behind other GWAS efforts, a challenge fur-ther compounded by subtyping (nearly one-third of all casesare categorized as undetermined23). Reduced sample sizes af-fect power for discovery and make other analyticapproaches—such as standard approaches for measuring traitcorrelation16—unfeasible. Also, our sample primarily com-prised European-ancestry samples, and work in non-Europeans, particularly in African-ancestry samples whererisk of stroke is double that of European samples, is crucial.Finally, the current analysis does not analyze rare variations,which also likely contributes to disease susceptibility.5
We have shown that the cumulative genetic risk of AF inindividuals with a stroke is similar to that reported in a largerpopulation-based cohort.25 Genome-wide variation related toAF is substantially associated with cardioembolic stroke risk.Moreover, AF genetic risk was specific for cardioembolicstroke and was not associated with the other primary strokesubtypes. The observation that AF genetic risk associated withstrokes of undetermined cause supports the notion that un-detected AF underlies a proportion of stroke risk in theseindividuals. Further work will need to incorporate emergingdiscoveries of rare genetic variants in AF and explore thepotential of genetic risk tools, including PRSs performed viaclinical-grade genotyping, to assist in the diagnostic workup ofindividuals with ischemic stroke.
Author contributionsS.L. Pulit: conception of research design, data analysis,drafting of the manuscript, and critical revision of the manu-script. L.-C. Weng: data analysis and critical revision of themanuscript. P.F. McArdle: data acquisition, data analysis, andcritical revision of the manuscript. L. Trinquart and S.H. Choi:data acquisition and critical revision of the manuscript. B.D.Mitchell and J Rosand: data acquisition, study supervision,and critical revision of the manuscript. P.I.W. de Bakker: studysupervision and critical revision of the manuscript. E.J. Ben-jamin, P.T. Ellinor, and S.J. Kittner: data acquisition, studysupervision, and critical revision of the manuscript. S.A. Lubitzand C.D. Anderson: conception of research design, studysupervision, drafting of the manuscript, and critical revision ofthe manuscript.
Study fundingS.L. Pulit, B.D. Mitchell, P.F. McArdle, and S.J. Kittner aresupported by the NIH grant R01NS100178. The NINDS-SiGN Consortium is supported by the NIH grantsR01NS100178 and U01NS069208. S.L. Pulit is supported byVeni Fellowship 016.186.071 (ZonMW) from the DutchOrganization for Scientific Research (Nederlandse Organ-isatie voor Wetenschappelijk Onderzoek, NWO). C.D.
Anderson is supported in part by K23NS086873,R01NS103924, an American Heart Association StrategicallyFocused Research Network in Atrial Fibrillation Award, anda Massachusetts General Hospital Center for GenomicMedicine Catalyst Award. S.A. Lubitz is supported by theNIH grant K23HL114724, an American Heart AssociationStrategically Focused Research Network in Atrial FibrillationAward, and a Doris Duke Charitable Foundation ClinicalScientist Development Award 2014105. L.-C. Weng is sup-ported by an American Heart Association Postdoctoral Fel-lowship Award 17POST33660226. Drs. Ellinor and Benjaminare supported by the NIH grants R01HL092577 andR01HL128914 and an American Heart Association Strategi-cally Focused Research Network in Atrial Fibrillation Award.E.J. Benjamin is additionally supported by the NIH grants1RC1HL101056 and 1R01HL102214. P.T. Ellinor is addi-tionally supported by the NIH grants R01HL104156 andK24HL105780; the National Heart, Lung, and Blood In-stitute (NHLBI); American Heart Association EstablishedInvestigator Award 13EIA14220013; and the FondationLeducq 14CVD01.
DisclosureS.L. Pulit has received speaker honoraria from the commercialentity Illumina. L.-C. Weng has received research supportfrom the American Heart Association. P.F. McArdle has re-ceived research support from the commercial entity Regen-eron and has received governmental research support theNational Institute of Neurological Disorders and Stroke. L.Trinquart and S.H. Choi report no disclosures. B.D. Mitchellhas served on the External Scientific Review Board for theIntegrative Cardiac Health Project (ICHP) at Walter ReedNational Military Medical Center; has served on the editorialboards of Current Genetic Medicine Reports and Frontiers inNeurology; and has received governmental research supportfrom the National Institutes of Health. J. Rosand has servedon the scientific advisory boards of Pfizer and the Data andSafetyMonitoring Board; has served on the editorial boards ofLancet Neurology and Stroke; has received governmental re-search support from the National Institutes of Health; and hasbeen involved in legal proceedings with Boehringer Ingel-heim. P.I.W. de Bakker is employed by Vertex Pharmaceut-icals. E.J. Benjamin has served on the scientific advisoryboards of non-profit organizations, the National Institutes ofHealth, and the National Heart, Lung, and Blood Institute;served on the editorial board of the American Heart Associ-ation; has been employed by Boston University School ofMedicine; has received governmental research support fromthe National Institutes of Health, American Heart Associa-tion, and the Robert Wood Johnson Foundation; and hasreceived foundation/society research support from the Bos-ton University School of Medicine and from the AmericanCouncil on Education. P.T. Ellinor has served on the scientificadvisory board for Bayer AG Novartis Quest Diagnostics; hasreceived research support (commercial) from Bayer AG; hasreceived governmental research support from the NationalInstitutes of Health; and has received foundation/society
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

research support from the American Heart Association andthe Fondation Leducq. S.J. Kittner serves on the editorialboard of Neuroepidemiology and has received governmentalresearch support from the National Institute of NeurologicalDisorders and Stroke. S.A. Lubitz has acted as a consultant forAbbott, Quest Diagnostics, and Bristol-Myers Squibb; hasreceived commercial research support from Bristol-MyersSquibb, Bayer HealthCare, Biotronik, and Boehringer Ingel-heim; has received governmental research support from theNational Institutes of Health; and has received foundation/society research support from the National Institutes ofHealth, the American Heart Association, and the Doris DukeCharitable Foundation. C.D. Anderson has served as a con-sultant for ApoPharma, Inc. and has received governmentalresearch support from the National Institutes of Health andthe American Heart Association. Full disclosure form in-formation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics June 23, 2018. Accepted in final formSeptember 9, 2018.
References1. Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial
fibrillation: a global burden of disease 2010 study. Circulation 2014;129:837–847.2. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for
stroke: the Framingham Study. Stroke 1991;22:983–988.3. World Health Organization. WHO|the Top 10 Causes of Death [online]. World
Health Organization; 2014. Available at who.int/mediacentre/factsheets/fs310/en/.Accessed November 1, 2017.
4. Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide strokeincidence and early case fatality reported in 56 population-based studies: a systematicreview. Lancet Neurol 2009;8:355–369.
5. Christophersen IE, Rienstra M, Roselli C, et al. Large-scale analyses of common and rarevariants identify 12 new loci associated with atrial fibrillation. Nat Genet 2017;57:289.
6. Bevan S, Traylor M, Adib-Samii P, et al. Genetic heritability of ischemic stroke and thecontribution of previously reported candidate gene and genome-wide associations.Stroke 2012;43:3161–3167.
7. Lubitz SA, Ozcan C, Magnani JW, Kaab S, Benjamin EJ, Ellinor PT. Genetics of atrialfibrillation: implications for future research directions and personalized medicine.Circ Arrhythm Electrophysiol 2010;3:291–299.
8. Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrialfibrillation on chromosome 4q25. Nature 2007;448:353–357.
9. Gretarsdottir S, Thorleifsson G, Manolescu A, et al. Risk variants for atrial fibrillationon chromosome 4q25 associate with ischemic stroke. Ann Neurol 2008;64:402–409.
10. Gudbjartsson DF, HolmH,Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22associates with atrial fibrillation and ischemic stroke. Nat Genet 2009;41:876–878.
11. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology,function, and translation. Am J Hum Genet 2017;101:5–22.
12. Pulit SL, McArdle PF, WongQ,Malik R, et al. Loci associated with ischaemic stroke andits subtypes (SiGN): a genome-wide association study. LancetNeurol 2015;15:174–184.
13. Ay H, Benner T, Arsava EM, et al. A computerized algorithm for etiologic classifi-cation of ischemic stroke: the causative classification of stroke system. Stroke 2007;38:2979–2984.
14. Arsava EM, Ballabio E, Benner T, et al. The causative classification of stroke system:an international reliability and optimization study. Neurology 2010;75:1277–1284.
15. Adams HP Jr, Birgitte H, Bendixen P, Jaap Kappelle L, et al. Classification of subtypeof acute ischemic stroke: definitions for use in a multicenter clinical trial. Stroke 1993;24:35–41.
16. Loh PR, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysisincreases association power in large cohorts. Nat Genet 2015;47:284–290.
17. Okbay A, Beauchamp JP, Fontana MA, et al. Genome-wide association study iden-tifies 74 loci associated with educational attainment. Nature 2016;533:539–542.
18. Weng L-C, Preis SR, Hulme OL, et al. Genetic predisposition, clinical risk factorburden, and lifetime risk of atrial fibrillation. Circulation 2018;137:1027–1038.
19. Franz MR, Karasik PL, Li C, Moubarak J, Chavez M. Electrical remodeling of thehuman atrium: similar effects in patients with chronic atrial fibrillation and atrialflutter. J Am Coll Cardiol 1997;30:1785–1792.
20. Lamas GA, Lee KL, SweeneyMO, et al. Ventricular pacing or dual-chamber pacing forsinus-node dysfunction. N Engl J Med 2002;346:1854–1862.
21. Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Ischemicstroke subtypes: a population-based study of incidence and risk factors. Stroke 1999;30:2513–2516.
22. Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU.Epidemiology of ischemic stroke subtypes according to TOAST criteria: incidence,recurrence, and long-term survival in ischemic stroke subtypes: a population-basedstudy. Stroke 2001;32:2735–2740.
23. NINDS Stroke Genetics Network (SiGN), International Stroke Genetics Consor-tium. Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wideassociation study. Lancet Neurol 2015;15:4–7.
24. Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation inadults: national implications for rhythm management and stroke prevention: theAnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001;285:2370–2375.
25. Weng LC, Choi SH, Klarin D, et al. Heritability of atrial fibrillation. Circ CardiovascGenet 2017;10:e001838.
26. Alonso A, Krijthe BP, Aspelund T, et al. Simple risk model predicts incidence of atrialfibrillation in a racially and geographically diverse population: the CHARGE-AFconsortium. J Am Heart Assoc 2013;2:e000102.
27. Henninger N, Goddeau RP Jr, Karmarkar A, Helenius J, McManus DD. Atrialfibrillation is associated with a worse 90-day outcome than other cardioembolic strokesubtypes. Stroke 2016;47:1486–1492.
28. Lubitz SA, Parsons OE, Anderson CD, et al. Atrial fibrillation genetic risk and is-chemic stroke mechanisms. Stroke 2017;48:1451–1456.
29. Lewis CM, Vassos E. Prospects for using risk scores in polygenic medicine. GenomeMed 2017;9:96.
30. Mavaddat N, Pharoah PDP, Michailidou K, et al. prediction of breast cancer risk basedon profiling with common genetic variants. J Natl Cancer Inst 2015;107. doi:10.1093/jnci/djv036.
31. Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores forcommon diseases to identify individuals with risk equivalent to monogenicmutations. Nat Gen 2018;50:1219–1224.
32. Theriault S, Lali R, Chong M, Velianou JL, Natarajan MK, Pare G. Polygenic con-tribution in individuals with early-onset coronary artery DiseaseClinical perspective.Circ Genom Precis Med 2018;11:e001849.
33. Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, andcoronary disease. N Engl J Med 2016;375:2349–2358.
34. Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, andthe clinical benefit of statin therapy: an analysis of primary and secondary preventiontrials. Lancet 2015;385:2264–2271.
35. Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenicstroke. N Engl J Med 2014;370:2467–2477.
36. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrialfibrillation. N Engl J Med 2014;370:2478–2486.
37. ACTIVE Investigators, Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogreladded to aspirin in patients with atrial fibrillation. N Engl J Med 2009;360:2066–2078.
38. Diener H-C, Eikelboom J, Connolly SJ, et al. Apixaban versus aspirin in patients withatrial fibrillation and previous stroke or transient ischaemic attack: a predefinedsubgroup analysis from AVERROES, a randomised trial. Lancet Neurol 2012;11:225–231.
39. Seshadri S, Beiser A, Kelly-Hayes M, et al. The lifetime risk of stroke: estimates fromthe Framingham study. Stroke 2006;37:345–350.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

ARTICLE OPEN ACCESS
Brain somatic mutations in SLC35A2cause intractable epilepsy withaberrant N-glycosylationNam Suk Sim, MD, Youngsuk Seo, MSc, Jae Seok Lim, DDS, PhD, Woo Kyeong Kim, BS, Hyeonju Son, BS,
Heung Dong Kim, MD, PhD, Sangwoo Kim, PhD, Hyun Joo An, PhD, Hoon-Chul Kang, MD, PhD,*
Se Hoon Kim, MD, PhD,* Dong-Seok Kim, MD, PhD,* and Jeong Ho Lee, MD, PhD*
Neurol Genet 2018;4:e294. doi:10.1212/NXG.0000000000000294
Correspondence
Dr. Lee
AbstractObjectiveTo identify whether somatic mutations in SLC35A2 alter N-glycan structures in human braintissues and cause nonlesional focal epilepsy (NLFE) or mild malformation of cortical de-velopment (mMCD).
MethodsDeep whole exome and targeted sequencing analyses were conducted for matched brain andblood tissues from patients with intractable NLFE and patients with mMCD who are negativefor mutations in mTOR pathway genes. Furthermore, tissue glyco-capture and nanoLC/massspectrometry analysis were performed to examine N-glycosylation in affected brain tissue.
ResultsSix of the 31 (19.3%) study patients exhibited brain-only mutations in SLC35A2 (mostlynonsense and splicing site mutations) encoding a uridine diphosphate (UDP)-galactosetransporter. Glycome analysis revealed the presence of an aberrant N-glycan series, includinghigh degrees of N-acetylglucosamine, in brain tissues with SLC35A2 mutations.
ConclusionOur study suggests that brain somatic mutations in SLC35A2 cause intractable focal epilepsywith NLFE or mMCD via aberrant N-glycosylation in the affected brain.
*These authors contributed equally to this work.
From the Graduate School of Medical Science and Engineering (N.S.S., J.S.L., W.K.K., J.H.L.), KAIST; Asia Glycomics Reference Site (Y.S., H.J.A.); Graduate School of Analytical Science &Technology (Y.S., H.J.A.), Chungnam National University, Daejeon, Korea; Department of Biomedical System informatics (H.S., S.K.), Brain Korea 21 PLUS Project for Medical Science,Yonsei University College of Medicine; Division of Pediatric Neurology (H.D.K., H.C.K.), Department of Pediatrics, Pediatric Epilepsy Clinics, Severance Children’s Hospital; EpilepsyResearch Institute (H.D.K., H.C.K.), Yonsei University College of Medicine; Department of Pathology (S.H.K.), Yonsei University College of Medicine, Seoul, Korea; and PediatricNeurosurgery (D.S.K.), Severance Children’s Hospital, Department of Neurosurgery, Yonsei University College of Medicine, Seoul, South Korea.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Epilepsy is a major neurologic disorder estimated to affectapproximately 50 million people worldwide.1 Althoughproper antiepileptic drugs achieve control in 60%–70% ofpatients with epilepsy, more than one-third fail to attainseizure-free status and are diagnosed with intractable epi-lepsy.2 Therein, surgical resection of the epileptic focus in theaffected brain is often required to relieve seizures.
The advent of sequencing technology has enabled in-depthgenetic studies to identify de novo or rare germline muta-tions underlying intractable epilepsy.3,4 We and other groupshave shown that low-level somatic activating mutations witha mutational burden (or variant allele frequency, [VAF]) ofmerely 1% in mTOR pathway genes, which arise only inthe affected brain, are a major genetic etiology for intract-able focal epilepsies with pathologically or radiologicallywell-defined lesions, such as focal cortical dysplasia type II(FCDII) and hemimegalencephaly (HME), exhibiting cyto-megalic neurons and cortical dyslamination.5–9 In contrastto lesional focal epilepsies, such as FCDII and HME, about15%–30% of intractable focal epilepsies show no abnor-malities on MRI, referred to as nonlesional focal epilepsy(NLFE), or mild cortical abnormalities on pathologic ex-amination, referred to as mild malformation of corticaldevelopment (mMCD).10–12 Although these conditions areoften responsive to epilepsy surgery,13,14 the moleculargenetic etiology, especially brain somatic mutations, un-derlying NLFE or mMCD remains elusive.
In this study, we sought to identify somatic mutations inpatients with NLFE using deep whole exome sequencing(WES) and targeted amplicon sequencing and to examine thebiological and pathologic functions of noted mutations inpatient brain tissues.
MethodsStandard protocol approvals, registrations,and patient consentsAll human tissues were obtained with informed consent inaccordance with protocols approved by Severance Hospitaland the KAIST Institutional Review Board and Committee onHuman Research.
We first collected matched brain and peripheral blood tissuesfrom 13 patients with NLFE subjected to epilepsy surgery;none showed an abnormal lesion on MRI (figure 1, A and Band table e-1, links.lww.com/NXG/A124). We performeddeep WES (mean depth, >800x) of extracted genomic DNA
from the paired brain and peripheral tissues (table e-2). Next,to expand the study cohort, we collected matched brainand peripheral tissues from an additional 18 patients withintractable focal epilepsy: 12 with NLFE and 6 with mMCDor nonspecific gliosis in their pathologies (figure e-1A, e-1Band table e-1, links.lww.com/NXG/A124). For these patients,we performed targeted amplicon sequencing (mean depth,1,230X) of SLC35A2 in brain tissues using 12 primers over-lapping at least 10 bp and covering all exonic regions (figure e-2).To investigate the presence of mammalian target of rapamycin(mTOR) pathway mutations potentially causative of intractableepilepsy, we performed deep targeted hybrid capture sequencing(mean depth, 812X) of 10 known mTOR pathway genes(AKT3, DEPDC5, MTOR, PIK3CA, PIK3R2, PTEN, STRADA,TBC1D7, TSC1, and TSC2) in matched brain and peripheraltissues from the patients with mutations identified in SLC35A2(table e-3 and table e-4). Furthermore, to determine whethermTOR pathway hyperactivation occurs, we performed coim-munostaining for phosphorylated S6 and NeuN, a neuronalmarker, in freshly frozen brain tissue from patients carryingSLC35A2 or MTOR mutations.
For all sequencing data, we applied our analysis pipeline, in-house filtering criteria of putative functional impact, andmanual investigation using Integrative Genomic Viewer toidentify potential pathogenic mutations (figure e-1, links.lww.com/NXG/A124). Noted somatic mutations were validatedon a different sequencing platform, such as site-specific ampli-con sequencing (read depth >100,000X). We consideredvariants as true positive when they appeared with a VAFgreater than 1%, 10 times the expected base miscall rate of0.1%.15 Furthermore, we estimated the probability value fortrue positive calls of amplicon sequencing data using a pre-viously described method.16 Briefly, this method calculatesthe discrepancy between expected and observed amounts ofmismatches in amplicon-based, Illumina platform data sets (upto 10,000X) in which 2 independent blood samples withknown single nucleotide polymorphisms (SNPs) were mixedto mimic somatic mutations with 4 different VAFs: 0.5%, 1%,5%, and 10%. Then, the patterns and levels of backgrounderrors generated for the Illumina platform are identified. Basedon these data, we could predict the probability value for truepositive calls of targeted amplicon sequencing by consideringVAFs acquired as background errors and sequencing context.
Finally, to examine whether SLC35A2 mutations affectN-glycosylation status in the affected brain, we performedTissue Glyco-Capture and nano liquid chromatography /mass spectrometry (nanoLC/MS) analysis, a highly sensitive
GlossaryFCDII = focal cortical dysplasia type II; HexNAc = N-acetyl glucosamine; HME = hemimegalencephaly; mMCD = mildmalformation of cortical development;NLFE = nonlesional focal epilepsy;VAF = variant allele frequency;WES = whole exomesequencing.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
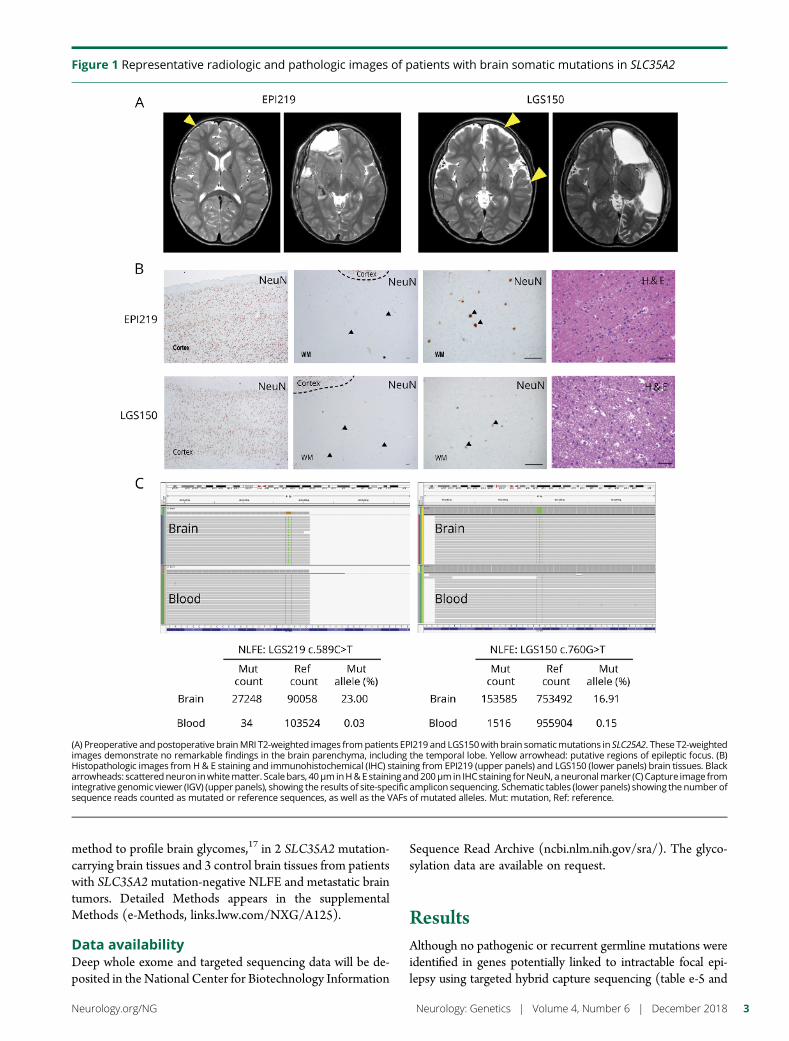
method to profile brain glycomes,17 in 2 SLC35A2 mutation-carrying brain tissues and 3 control brain tissues from patientswith SLC35A2 mutation-negative NLFE and metastatic braintumors. Detailed Methods appears in the supplementalMethods (e-Methods, links.lww.com/NXG/A125).
Data availabilityDeep whole exome and targeted sequencing data will be de-posited in the National Center for Biotechnology Information
Sequence Read Archive (ncbi.nlm.nih.gov/sra/). The glyco-sylation data are available on request.
ResultsAlthough no pathogenic or recurrent germline mutations wereidentified in genes potentially linked to intractable focal epi-lepsy using targeted hybrid capture sequencing (table e-5 and
Figure 1 Representative radiologic and pathologic images of patients with brain somatic mutations in SLC35A2
(A) Preoperative and postoperative brainMRI T2-weighted images frompatients EPI219 and LGS150with brain somaticmutations in SLC25A2. These T2-weightedimages demonstrate no remarkable findings in the brain parenchyma, including the temporal lobe. Yellow arrowhead: putative regions of epileptic focus. (B)Histopathologic images from H& E staining and immunohistochemical (IHC) staining from EPI219 (upper panels) and LGS150 (lower panels) brain tissues. Blackarrowheads: scatteredneuron inwhitematter. Scale bars, 40μminH&Estainingand200μmin IHCstaining forNeuN, aneuronalmarker (C) Capture image fromintegrative genomic viewer (IGV) (upper panels), showing the results of site-specific amplicon sequencing. Schematic tables (lowerpanels) showing the number ofsequence reads counted as mutated or reference sequences, as well as the VAFs of mutated alleles. Mut: mutation, Ref: reference.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
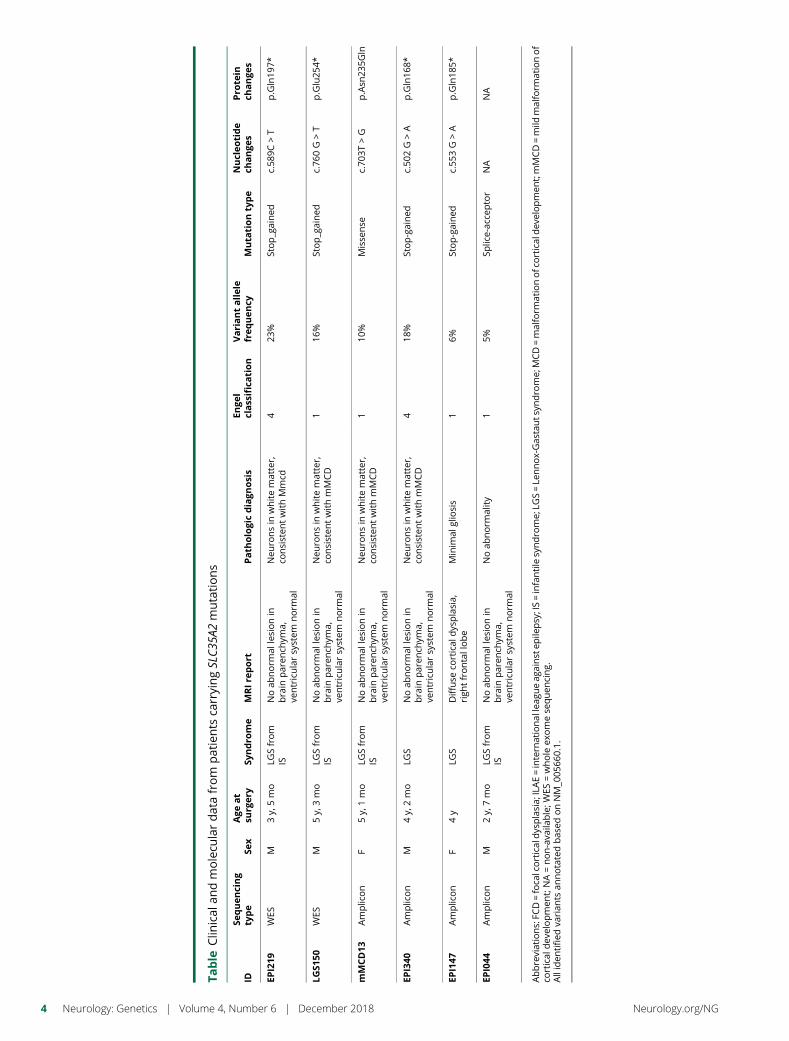
Table
Clin
ical
andmolecu
lardatafrom
patients
carryingSLC3
5A2mutations
IDSe
quen
cing
type
Sex
Age
at
surg
ery
Syndro
me
MRIre
port
Path
ologicdiagn
osis
Enge
lclassification
Variantallele
frequency
Muta
tiontype
Nucleotide
change
sPro
tein
change
s
EPI219
WES
M3y,
5mo
LGSfrom
ISNoab
norm
allesionin
brain
paren
chym
a,ve
ntricularsystem
norm
al
Neu
ronsin
whitematter,
consisten
twithMmcd
423
%Stop_gained
c.58
9C>T
p.Gln19
7*
LGS1
50WES
M5y,
3mo
LGSfrom
ISNoab
norm
allesionin
brain
paren
chym
a,ve
ntricularsystem
norm
al
Neu
ronsin
whitematter,
consisten
twithmMCD
116
%Stop_gained
c.76
0G>T
p.Glu25
4*
mMCD13
Amplicon
F5y,
1mo
LGSfrom
ISNoab
norm
allesionin
brain
paren
chym
a,ve
ntricularsystem
norm
al
Neu
ronsin
whitematter,
consisten
twithmMCD
110
%Misse
nse
c.70
3T>G
p.Asn
235G
ln
EPI340
Amplicon
M4y,
2mo
LGS
Noab
norm
allesionin
brain
paren
chym
a,ve
ntricularsystem
norm
al
Neu
ronsin
whitematter,
consisten
twithmMCD
418
%Stop-gained
c.50
2G>A
p.Gln16
8*
EPI147
Amplicon
F4y
LGS
Diffuse
cortical
dysplasia,
righ
tfrontallobe
Minim
algliosis
16%
Stop-gained
c.55
3G>A
p.Gln18
5*
EPI044
Amplicon
M2y,
7mo
LGSfrom
ISNoab
norm
allesionin
brain
paren
chym
a,ve
ntricularsystem
norm
al
Noab
norm
ality
15%
Splice-ac
ceptor
NA
NA
Abbreviations:FC
D=foca
lcorticaldysplasia;
ILAE=intern
ational
leag
ueag
ainst
epile
psy;IS=infantilesyndrome;
LGS=Le
nnox-Gas
tauts
yndrome;
MCD=malform
ationofcortical
dev
elopmen
t;mMCD=mild
malform
ationof
cortical
dev
elopmen
t;NA=non-ava
ilable;W
ES=whole
exomese
quen
cing.
Alliden
tifie
dva
rian
tsan
notatedbas
edonNM_0
0566
0.1.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

table e-6, links.lww.com/NXG/A124), we surprisingly foundthat brain somatic mutations in SLC35A2 presented as stop-gained variants, SLC35A2 c.589C > T (p.Gln197*) andc.760 G > T (p.Glu254*) on the X chromosome, in 2 of the13 patients, respectively (table and table e-4). Both of these2 patients (LGS150 and EPI219) were male. Their VAFswere 23% for p.Gln197* and 16% for p.Glu254*. Histopatho-logic analysis of mutation-carrying brain tissues revealed theabsence of dysmorphic neurons and balloon cells, as well as anyneuronal abnormality other than scattered neurons, which isconsistent with mMCD (figure 1, A and B). We confirmed thatthe 2 variants were detected only in the brain with a similarmutational burden and not in peripheral tissue by site-specificamplicon sequencing (figure 1, C). According to calculatedprobability values, none of the peripheral tissue sequencing datawere statistically significant (table e-7).
In the additional 18 patients with intractable focal epilepsy,deep targeted sequencing of SLC35A2 in matched brain-blood samples revealed brain somatic mutations in SLC35A2,c.703T > G (p.Asn235Gln), c.502 G > A (p.Gln168*), c.553G > A (p.Gln185*), and chrX:48763821C > A (splice ac-ceptor site mutation) in 4 patients (3 NLFE and 1 mMCDsubjects), respectively (figure e-2A, e-2B, links.lww.com/NXG/A124 and table e-1, links.lww.com/NXG/A124). Twoof the 4 patients were male. Site-specific amplicon sequencingin matched blood samples from these patients confirmed theabsence of the identified somatic variants in peripheral ge-nomic DNA (figure e-2C). The VAFs of these identifiedmutations ranged from 5% to 18%. Calculating the probabilityvalues for amplicon sequencing data for matched peripheraltissue revealed nothing of statistical significance (table e-7).None of the identified variants in SLC35A2 were reported inthe Exome Aggregate Consortium database.18 The mutationswere primarily nonsense and splicing site mutations, likelyacting as loss-of-function mutations (figure 2A).
To rule out the possible involvement of somatic mutations inthemTOR pathway in patients withmutations in SLC35A2, weperformed deep targeted hybrid capture sequencing (meandepth, 812X) of 10 known mTOR pathway genes, in which nosomatic mutations were discovered. In addition, we performedcoimmunostaining for phosphorylated S6 and NeuN, a neuro-nal marker, in freshly frozen brain tissues from 2 SLC35A2mutation-carrying patients and 1 MTOR p.A1459D mutation-carrying patient. The immunostaining results revealed markeddecreases in the numbers of phosphorylated S6 stained neuroncells in patients carrying the SLC35A2 mutation (figures e-4Aand e-4B, links.lww.com/NXG/A124).
SLC35A2 encodes a member of the nucleotide-sugar trans-porter family19 (figure 2A). The encoded protein transportsuridine diphosphate (UDP)-galactose from the cytosol intoGolgi vesicles, where it serves as a glycosyl donor for thegeneration of glycans and plays a crucial role in the gal-actosylation of N-glycans.20 To examine N-glycosylationprofiles in affected brain tissue, we performed TGC and
nanoLC/MS analysis in tissues from 2 patients (EPI219 andLGS150) with SLC35A2 c.589C > T (p.Gln197*) and c.760G > T (p.Glu254*), respectively. The TGC process com-prised selective enrichment steps that were critical to detectingtrace amounts of glycans from brain tissue: (1) Plasmamembrane extraction using ultracentrifugation isolatedmembrane fractions, including glycoproteins, from ho-mogenized tissue samples. After enzymatic N-glycan release,(2) solid-phase extraction based on porous graphitized carbonwas used to remove detergents (e.g., buffer chemicals andnonglycan species) and to capture solely N-glycans.17,21 Theconcentrated N-glycans from each brain tissue could be de-termined by high sensitive nanoLC/MS analysis. The technicalreproducibility and sensitivity of the combined platform ofTGC and nanoLC/MS have already been demonstrated ina previous study on brain glycomes at the microgram level.Using the proven method, we found that mutation-carryingsamples had less galactosylation (associated with truncatedglycans without galactose residues) than control brainsamples, which is consistent with a previous study19(figure2B). In particular, we found that mutation-carrying sampleshad less galactosylation than control brain samples, whichis also consistent with a previous study22(figure 2B).N-glycan structures were unique, showing high degrees ofN-acetylglucosamine (HexNAc), such as Hex3HexNAc7Fuc1and Hex3HexNAc8Fuc1, on high sensitive LC/MS analysis(figure 2, B and C). The glycan representing the ion at m/z1036.90, corresponding to [Hex3HexNAc7Fuc1+2H]
2+, wasidentified by collision-induced dissociationMS/MS (figure 2C).In sequence, the initial loss of HexNAc residues was clearlyobserved from the parent ion, indicating that 5 HexNAc resi-dues were linked to the N-glycan core (i.e., Man3GlcNAc2).These results suggest that somatic loss-of-function muta-tions in SLC35A2 lead to aberrant N-glycan patterns ofHex3HexNAc7Fuc1 in patient brain tissues.
DiscussionThis study suggests that brain somatic mutations in SLC35A2explain 19.3% (6 of 31) of intractable focal epilepsies withNLFE or mMCD and result in aberrant N-glycan patterns inmutation-carrying brain tissues. NLFE or mMCD account for15%–30% of intractable childhood epilepsies. Similar to otherintractable childhood epilepsies, patients with NLFE or mMCDoften undergo surgical intervention; however, only 30% becameseizure free after surgical treatment compared with 62%–80% ofpatients with lesional focal epilepsy.13,14 Althoughmany researchgroups have studied the molecular genetic etiology underlyingintractable childhood epilepsies, these studies have primarilyinvestigated mechanisms underlying lesional epilepsy, such asmalformations of cortical development, tumors, and other cir-cumscribed anomalies. Therefore, the mechanisms underlyingNLFE or mMCD have remained obscure.
An association between SLC35A2 and seizure has been sug-gested. In previous studies, it was reported that de-novo germ-line mutations or postzygotic mosaic mutation in SLC35A2
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
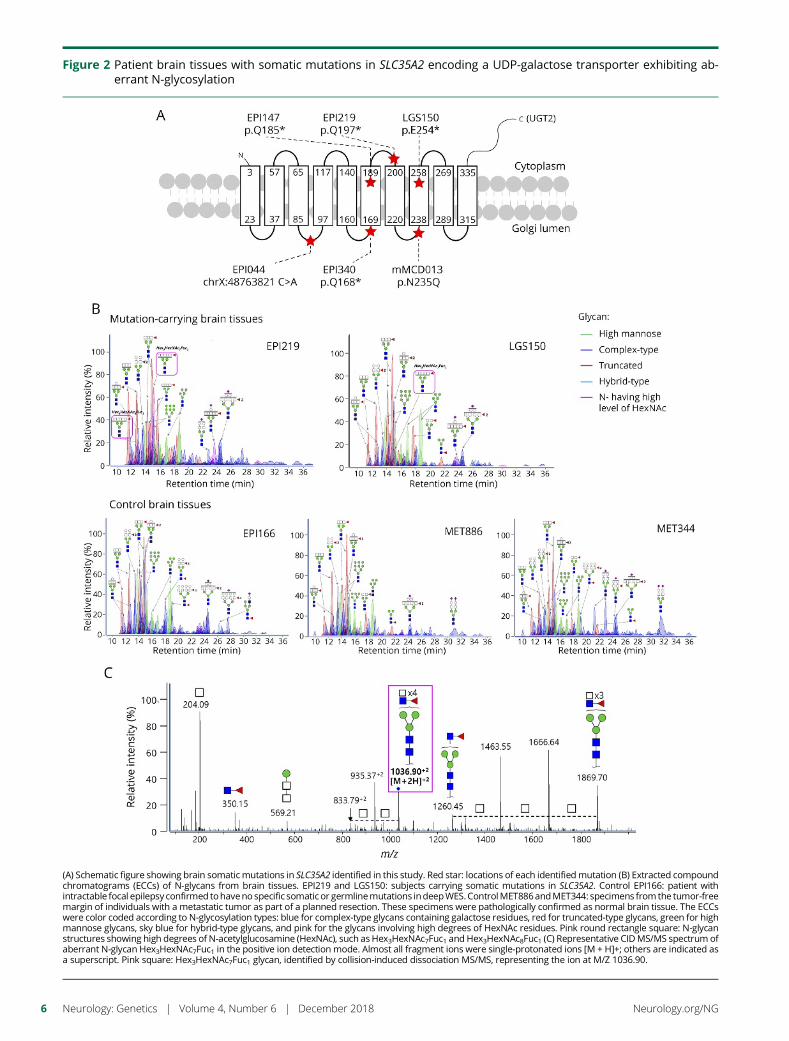
Figure 2 Patient brain tissues with somatic mutations in SLC35A2 encoding a UDP-galactose transporter exhibiting ab-errant N-glycosylation
(A) Schematic figure showing brain somaticmutations in SLC35A2 identified in this study. Red star: locations of each identified mutation (B) Extracted compoundchromatograms (ECCs) of N-glycans from brain tissues. EPI219 and LGS150: subjects carrying somatic mutations in SLC35A2. Control EPI166: patient withintractable focal epilepsy confirmed tohaveno specific somatic or germlinemutations indeepWES.ControlMET886andMET344: specimens from the tumor-freemargin of individuals with a metastatic tumor as part of a planned resection. These specimens were pathologically confirmed as normal brain tissue. The ECCswere color coded according to N-glycosylation types: blue for complex-type glycans containing galactose residues, red for truncated-type glycans, green for highmannose glycans, sky blue for hybrid-type glycans, and pink for the glycans involving high degrees of HexNAc residues. Pink round rectangle square: N-glycanstructures showing high degrees of N-acetylglucosamine (HexNAc), such asHex3HexNAc7Fuc1 and Hex3HexNAc8Fuc1 (C) Representative CIDMS/MS spectrum ofaberrant N-glycan Hex3HexNAc7Fuc1 in the positive ion detection mode. Almost all fragment ions were single-protonated ions [M + H]+; others are indicated asa superscript. Pink square: Hex3HexNAc7Fuc1 glycan, identified by collision-induced dissociation MS/MS, representing the ion at M/Z 1036.90.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

cause a congenital disorder of glycosylation and early-onsetepileptic encephalopathy with epileptic seizures.22–24 Therein,aberrant glycosylation was found in patient fibroblasts andwhole serum.22 Treatment with an oral galactose supple-ment improved clinical symptoms in 1 case.23 Although theysupport our results, these studies are limited in their ability todraw causative links between the SLC35A2 mutation andseizure development. More recently, 1 study reported thatsomatic mutations in SLC35A2 are associated with intractableneocortical epilepsy.25 They found somatic mutations in 3of 18 patients with NLFE and 2 of 38 patients with malfor-mation of cortical development or focal cortical dysplasia.However, no evidence of alterations in glycosylation in af-fected brain tissues was given. The present study providesstronger evidence that brain somatic mutations in SLC35A2cause intractable focal epilepsy with NLFE or mMCD throughaberrant N-glycosylation only in the affected brain and thatSLC35A2 mutations are unlikely to lead to hyperactivation ofthe mTOR pathway using targeted hybrid capture sequencingand immunostaining for phosphorylated S6. Several studieshave explored the effects of N-linked glycosylation on glyco-proteins involved in neural physiology, demonstrating essentialroles of N-glycan structures in neural circuitry.26 N-glycans playspecific modulatory roles controlling neural transmissionand the excitability of neural circuits. Of interest, 1 studyhas shown that N-glycosylation defects elicit epileptic dis-charges.27 Therefore, we suggest that N-glycosylation defectscaused by SLC35A2mutations might alter neural transmissionand the excitability of neural circuits, thereby resulting inseizures. Future studies using matrix-assisted laser desorption/ionization Fourier transform mass spectrometry with non-specific proteolysis and deglycosylation will be necessary toaddress the determination of N-glycosylation sites.28 Throughthese experiments, candidates of functionally altered pro-teins causing intractable epilepsies could be identifiable.Last, identifying target proteins associated with the aberrantN-glycosylation observed in the brain may provide noveltherapeutic targets or molecular diagnostic markers for in-tractable focal epilepsies stemming from NLFE or mMCD.
Author contributionsN.S. Sim and J.H. Lee: organized the project. N.S. Sim and J.S.Lim: performed genetic studies. S.H. Kim: performed path-ologic study. N.S. Sim and J.S. Lim: performed data analysisand bioinformatics analysis. H. Son and S.W. Kim: performedamplicon sequencing data analysis. D.S. Kim, S.H. Kim, andH.C.K.: performed surgeries, collected patient samples, andmanaged patient information and tissues samples, along withS.H. Kim, H.D. Kim, and W.K. Kim. H.J. An and Y. Seo:performed the analysis of glycosylation in patient tissues. N.S.Sim and J.H. Lee: wrote the manuscript. D.S. Kim, S.H. Kim,H.C.K., and J.H. Lee: led the project and oversaw the man-uscript preparation.
Study fundingThis work was supported by grants from the Korean HealthTechnology R&D Project, Ministry of Health & Welfare,
Republic of Korea (H16C0415 to D.S.K and J.H.L;HI15C1601 to H.C.K).
DisclosureN. S. Sim has received research support from the KoreanHealth Technology R&D Project, Ministry of Health &Welfare, Republic of Korea. Y. Seo reports no disclosures. J. S.Lim has received research support from the Korean HealthTechnology R&D Project, Ministry of Health & Welfare,Republic of Korea. W. K. Kim is/has been employed bySoVarGen and has received research support from the KoreanHealth Technology R&D Project, Ministry of Health &Welfare, Republic of Korea. H. Son has received researchsupport from the Korean Health Technology R&D Project,Ministry of Health & Welfare, Republic of Korea. H. D. Kim,S. Kim, and H. J. An report no disclosures. H.C. Kang hasreceived research support from the Ministry of Food andDrug Safety, Republic of Korea. S. H. Kim serves/has servedon the editorial board of the Journal of Pathology and Trans-lational Medicine.D.S. Kim has received research support fromthe Korean Health Technology R&D Project, Ministry ofHealth & Welfare, Republic of Korea. J. H. Lee is/has beenemployed by and receives stock/stock options/board ofdirections compensation from SoVarGen and has receivedresearch support from the Korean Health Technology R&DProject, Ministry of Health &Welfare, Republic of Korea. Fulldisclosure form information provided by the authors isavailable with the full text of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics July 27, 2018. Accepted in final formOctober 3, 2018.
References1. Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet 2006;367:
1087–1100.2. Schuele SU, Luders HO. Intractable epilepsy: management and therapeutic alter-
natives. Lancet Neurol 2008;7:514–524.3. Epi4K consortium; Epilepsy Phenome/Genome Project. Ultra-rare genetic varia-
tion in common epilepsies: a case-control sequencing study. Lancet Neurol 2017;16:135–143.
4. Epi4K consortium; Epilepsy Phenome/Genome Project; Allen AS, Berkovic SF,Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221.
5. Blumcke I, ThomM, Aronica E, et al. The clinicopathologic spectrum of focal corticaldysplasias: a consensus classification proposed by an ad hoc task force of the ILAEdiagnostic methods commission. Epilepsia 2011;52:158–174.
6. Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of thePI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012;44:941–945.
7. Lim JS, Gopalappa R, Kim SH, et al. Somatic mutations in TSC1 and TSC2 causefocal cortical dysplasia. Am J Hum Genet 2017;100:454–472.
8. Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focalcortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400.
9. Nakashima M, Saitsu H, Takei N, et al. Somatic Mutations in the MTOR gene causefocal cortical dysplasia type IIb. Ann Neurol 2015;78:375–386.
10. Kutsy RL. Focal extratemporal epilepsy: clinical features, EEG patterns, and surgicalapproach. J Neurol Sci 1999;166:1–15.
11. Salanova V,MarkandO,Worth R, et al. Presurgical evaluation and surgical outcome oftemporal lobe epilepsy. Pediatr Neurol 1999;20:179–184.
12. Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the corticaldysplasias. Neurology 2004;62:S2–S8.
13. Berkovic SF, McIntosh AM, Kalnins RM, et al. Preoperative MRI predicts outcome oftemporal lobectomy: an actuarial analysis. Neurology 1995;45:1358–1363.
14. Tellez-Zenteno JF, Hernandez Ronquillo L, Moien-Afshari F, Wiebe S. Surgicaloutcomes in lesional and non-lesional epilepsy: a systematic review and meta-analysis.Epilepsy Res 2010;89:310–318.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

15. Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stainscaused by somatic mutation in GNAQ. N Engl J Med 2013;368:1971–1979.
16. Kim J, Kim D, Lim JS, et al. Accurate detection of low-level somatic mutations withtechnical replication for next-generation sequencing. bioRxiv 2017: 179713.
17. Ji IJ, Hua S, Shin DH, et al. Spatially-resolved exploration of the mouse brainglycome by tissue glyco-capture (TGC) and nano-LC/MS. Anal Chem 2015;87:2869–2877.
18. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variationin 60,706 humans. Nature 2016;536:285–291.
19. Hadley B, Maggioni A, Ashikov A, Day CJ, Haselhorst T, Tiralongo J. Structure andfunction of nucleotide sugar transporters: current progress. Comput Struct Bio-technol J 2014;10:23–32.
20. Sosicka P, Jakimowicz P, Olczak T, Olczak M. Short N-terminal region of UDP-galactose transporter (SLC35A2) is crucial for galactosylation of N-glycans. BiochemBiophys Res Commun 2014;454:486–492.
21. An HJ, Gip P, Kim J, et al. Extensive determination of glycan heterogeneityreveals an unusual abundance of high mannose glycans in enriched plasmamembranes of human embryonic stem cells. Mol Cell Proteomics 2012;11:M111.010660.
22. Ng BG, Buckingham KJ, Raymond K, et al. Mosaicism of the UDP-galactose trans-porter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet2013;92:632–636.
23. Dorre K, Olczak M,Wada Y, et al. A new case of UDP-galactose transporter deficiency(SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach.J Inherit Metab Dis 2015;38:931–940.
24. Kodera H, Nakamura K, Osaka H, et al. De novo mutations in SLC35A2 encodinga UDP-galactose transporter cause early-onset epileptic encephalopathy. HumMutat2013;34:1708–1714.
25. Winawer MR, Griffin NG, Samanamud J, et al. Somatic SLC35A2 variants in thebrain are associated with intractable neocortical epilepsy. Ann Neurol 2018;83:1133–1146.
26. Scott H, Panin VM. The role of protein N-glycosylation in neural transmission.Glycobiology 2014;24:407–417.
27. Janz R, Goda Y, Geppert M, Missler M, Sudhof TC. SV2A and SV2B function asredundant Ca2+ regulators in neurotransmitter release. Neuron 1999;24:1003–1016.
28. An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-glycosylation sitesand site heterogeneity in glycoproteins. Anal Chem 2003;75:5628–5637.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

ARTICLE OPEN ACCESS
Ataxia-telangiectasia-like disorder in a familydeficient for MRE11A, caused by a MRE11variantMaryam Sedghi, MSc, Mehri Salari, MD, Ali-Reza Moslemi, PhD, Ariana Kariminejad, MD, Mark Davis, PhD,
Hayley Goullee, MSc, Bjorn Olsson, PhD, Nigel Laing, PhD, and Homa Tajsharghi, PhD
Neurol Genet 2018;4:e295. doi:10.1212/NXG.0000000000000295
Correspondence
Dr. Tajsharghi
AbstractObjectiveWe report 3 siblings with the characteristic features of ataxia-telangiectasia-like disorder as-sociated with a homozygous MRE11 synonymous variant causing nonsense-mediated mRNAdecay (NMD) and MRE11A deficiency.
MethodsClinical assessments, next-generation sequencing, transcript and immunohistochemistryanalyses were performed.
ResultsThe patients presented with poor balance, developmental delay during the first year of age, andsuffered from intellectual disability from early childhood. They showed oculomotor apraxia,slurred and explosive speech, limb and gait ataxia, exaggerated deep tendon reflex, dystonicposture, and mirror movement in their hands. They developed mild cognitive abilities. BrainMRI in the index case revealed cerebellar atrophy. Next-generation sequencing revealed a ho-mozygous synonymous variant in MRE11 (c.657C>T, p.Asn219=) that we show affectssplicing. A complete absence of MRE11 transcripts in the index case suggested NMD andimmunohistochemistry confirmed the absence of a stable protein.
ConclusionsDespite the critical role of MRE11A in double-strand break repair and its contribution to theMre11/Rad50/Nbs1 complex, the absence of MRE11A is compatible with life.
From the Medical Genetics Laboratory (M. Sedghi), Alzahra University Hospital, Isfahan University of Medical Sciences, Isfahan, Iran; Department of Neurology (M. Salari), ShahidBeheshti University of Medical Science, Tehran, Iran; Department of Pathology (A.-R.M.), University of Gothenburg, Sahlgrenska University Hospital, Sweden; Kariminejad-Najmabadi Pathology & Genetics Center (A.K.), Tehran, Iran; Department of Diagnostic Genomics (M.D.), Pathwest, QEII Medical Centre; Centre for Medical Research (H.G., N.L., H.T.),The University of Western Australia and the Harry Perkins Institute for Medical Research, Nedlands, Australia; School of Bioscience (B.O.), University of Skovde; and DivisionBiomedicine (H.T.), School of Health and Education, University of Skovde, Sweden.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by Swedish Research Council.
This is an open access article distributed under the terms of the Creative Commons Attribution License 4.0 (CC BY), which permits unrestricted use, distribution, and reproduction in anymedium, provided the original work is properly cited.
MORE ONLINE
Video
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Neurological defects, including microcephaly, ataxia or neu-rodegeneration, are a hallmark for autosomal recessivemutations in individual genes encoding components of theMre11/Rad50/Nbs1 (MRN) complex. Mutations in theNBS1 encoding gene, NBN, are associated with Nijmegenbreakage syndrome (NBS) (OMIM# 251260) characterizedby microcephaly, immunodeficiency, growth and intellectualdisability, radiosensitivity, and cancer predisposition.1–3 Muta-tions in RAD50 gene RAD50 are related to NBS-like disorder(NBSLD) (OMIM# 613078).4 The clinical features of patientswith NBSLD are very similar to those with NBN, includingmicrocephaly and intellectual disability but not infections, im-munodeficiency, or cancer predisposition. Mutations inMRE11A homolog, double-strand break repair nuclease geneMRE11 are associated with a very rare chromosomal breakagesyndrome (OMIM# 604391). The clinical features are charac-terized by progressive cerebellar degeneration and ionizing ra-diation hypersensitivity, similar to the ataxia telangiectasia(OMIM# 208900) caused by mutations in ATM, encodingataxia telangiectasia mutated (ATM).5,6 The neurologic featureshave a later onset, slower progression and it is referred to asataxia-telangiectasia-like disorder (ATLD) (OMIM# 604391).5,7,8
Unlike patients with ataxia telangiectasia, however, patients withATLD show no telangiectasia or obvious immunodeficiency.7–9
In 1999, first association of ATLD with mutations inMRE11 wasreported,5 followed by additional families with clinical features ofATLD9–16 or NBSLD.17
Here, we report a family with characteristic features of ATLDassociated with a novel homozygous apparently synonymousvariant in exon 7 of MRE11 that ablates normal splicing,induces nonsense-mediated mRNA decay (NMD), and de-ficiency of MRE11A protein.
MethodsStandard protocol approvals, registrations,and patient consentsThe study was approved by the ethical standards of the relevantinstitutional review board, the Ethics Review Committee in theGothenburg Region (Dn1: 842-14), and the Human ResearchEthics Committee of the University of Western Australia. In-formed consent was obtained from the parents included in thisstudy after appropriate genetic counseling. Blood samples wereobtained from patients and their parents.
Clinical evaluationMedical history was obtained and physical examination wasperformed as part of routine clinical workup.
Genetic analysisNext-generation sequencing (NGS) (a targeted neuromus-cular sub-exomic sequencing [NSES] panel and/or wholeexome sequencing [WES]) was performed on patients’DNA.Confirmatory bidirectional Sanger sequencing was performedin the patients and all available unaffected family members(e-Methods, links.lww.com/NXG/A128).
Analyses of muscle biopsyA muscle biopsy was obtained in the index patient (V:2).Morphologic and histochemical analyses of paraffin-embeddedmuscle tissue were performed according to standard protocols.Sections of skeletal muscle tissue were processed for transcript,histologic and immunohistochemical assessments (e-Methods,links.lww.com/NXG/A128).
Transcript analysisTo analyze the impact of the c.657C>T variant in splicingefficiency ofMRE11 exon 7 in the index case (V:2), polymerasechain reaction (PCR) was performed on complementary DNAwith primer pairs covering exon 1 through 8 (e-Methods, links.lww.com/NXG/A128).
Chromosomal assayPeripheral blood samples were obtained from 9 individuals ofthe family (III:1, IV:1, IV:2, IV:8, IV:9, V:1, V:2, V:3, and V:4). Chromosomal breakage tests using mitomycin C (MMC)induction in cultures was carried out according to standardprotocols. Metaphases were stained and scored for sponta-neous chromosomal anomalies. Twenty metaphase spreadswere studied from routine culture, 100 spreads from culturewere prepared with the addition of 2 concentrations of MMC.These were compared with 100 spreads from age-matchednormal controls. Unfortunately, no additional tissue was avail-able for assessment of the effect ofMRE11 c.657C>T variant onsensitivity to ionizing radiation and the impact on NBS1 andRAD50 expression levels or the entire MRN complex.
Data availabilityData not published within the article are available online insupplemental material (links.lww.com/NXG/A128).
ResultsClinical characteristics of patientsThree affected siblings were born to an apparently healthyconsanguineous couple (figure 1). Their clinical presentationswere consistent with the characteristic features of ATLD. The
GlossaryATLD = ataxia-telangiectasia-like disorder; ATM = ataxia telangiectasia mutated; MMC = mitomycin C; NBS = Nijmegenbreakage syndrome; NBSLD = Nijmegen breakage syndrome-like disorder; NGS = Next-generation sequencing; NMD =nonsense-mediated mRNA decay; NSES = neuromuscular sub-exomic sequencing; WES = whole exome sequencing.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

affected individuals presented with developmental delayduring the 1st year of age. They developed poor balance andsuffered frommild intellectual disability from early childhood.The symptoms progressed gradually. Chromosomal studyrevealed a normal 46, XY (V:2 and V:4) or 46, XX (V:3)pattern. There was no history of severe or recurrent infectionsin the index case or in his 2 affected siblings. Follow-up at 23(V:2), 20 (V:3), and 18 (V:4) years of age revealed oculo-motor apraxia, slurred and explosive speech, limb and gaitataxia, exaggerated deep tendon reflexes (+3), dystonic pos-ture, mirror movement in their hands, and poor balance(video 1). They had no sensory deficits. IQ was between 50and 69 in all affected siblings. There was no facial dys-morphism (video 1); however, measurements includingheight, weight, and head circumference were below the 3rdpercentile in all 3 affected siblings. Systemic examinations ofthe affected siblings were normal without any evidence ofskeletal deformity or skin lesions. The laboratory evaluation,including thyroid function, liver function test, and alpha-fetoprotein, revealed levels in the reference range. Echocar-diography of the youngest sibling (V:4) at 16 years of age wasunremarkable. Nerve conduction velocity and EMG re-cording in the index case (V:2) did not show any neuropathicor myopathic features. BrainMRI in the index case (V:2) at 21years of age revealed cerebellar atrophy (figure 2). The cousinof the affected siblings (V:5) (figure 1) was a boy with Downsyndrome, who died at 6 months of age because of tetralogy ofFallot. However, he was not clinically evaluated for ATLD.There is no family history of cancer.
Genetic findingsData from NGS of DNA from 2 affected (V:2 and V:3) and 1unaffected family members (V:1) were analyzed. Targetedsequencing of 336 known neurogenic disease genes, including32 ataxia-associated genes in DNA from the index case V:2identified a MRE11 variant changing the nucleotide 3 basesfrom the 39 end of exon 7 (c.657C>T, rs775017362) in the
homozygous state. The variant did not alter the coded aminoacid (AAC>AAT, p.Asn219=). No rare, likely pathogenicheterozygous or homozygous variants were identified in otherneurogenetic- or ataxia-associated genes included in the targetedpanel, including ATM, APTX, or SETX. Simultaneous WES onDNA samples from individuals V:1, V:2, and V:3 to look forvariants in novel disease genes was performed. The filteringstrategy of initially concentrating on homozygous coding variantsin known neurogenetic disease genes, selected based on variantdatabases Human GenomeMutation Database and ClinVar, and
Figure 1 Pedigree of the family
Pedigree and recessive inheritance of MRE11. Inthe pedigree, squares represent males; circles,females; open symbols, unaffected family mem-bers; and slash, deceased. The affected individualsare represented with shaded symbols. Arrowindicates the proband in the family (V:2). +/− indi-cates heterozygous presence of the variant; +/+indicates homozygous appearance of the variant.
Figure 2 Sagittal T1-weighted MRI of brain
Brain MRI from the index case (V:2) shows significant atrophy of the cere-bellar vermis. The brainstem is relatively preserved.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
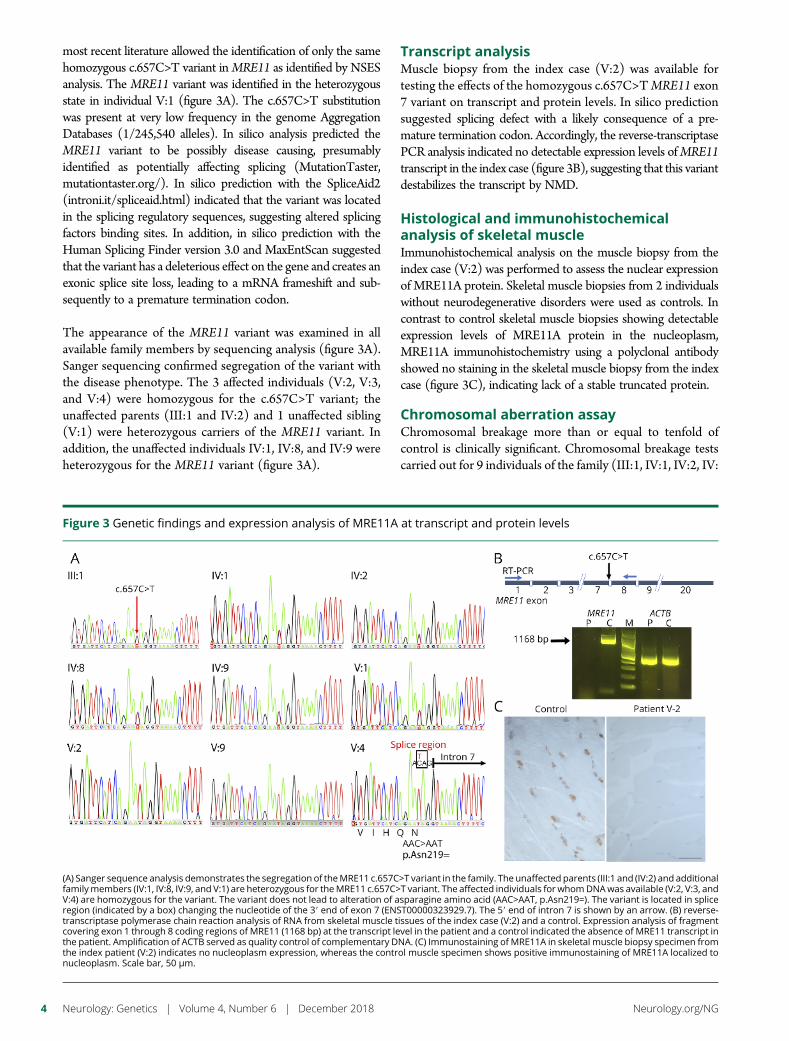
most recent literature allowed the identification of only the samehomozygous c.657C>T variant inMRE11 as identified by NSESanalysis. The MRE11 variant was identified in the heterozygousstate in individual V:1 (figure 3A). The c.657C>T substitutionwas present at very low frequency in the genome AggregationDatabases (1/245,540 alleles). In silico analysis predicted theMRE11 variant to be possibly disease causing, presumablyidentified as potentially affecting splicing (MutationTaster,mutationtaster.org/). In silico prediction with the SpliceAid2(introni.it/spliceaid.html) indicated that the variant was locatedin the splicing regulatory sequences, suggesting altered splicingfactors binding sites. In addition, in silico prediction with theHuman Splicing Finder version 3.0 and MaxEntScan suggestedthat the variant has a deleterious effect on the gene and creates anexonic splice site loss, leading to a mRNA frameshift and sub-sequently to a premature termination codon.
The appearance of the MRE11 variant was examined in allavailable family members by sequencing analysis (figure 3A).Sanger sequencing confirmed segregation of the variant withthe disease phenotype. The 3 affected individuals (V:2, V:3,and V:4) were homozygous for the c.657C>T variant; theunaffected parents (III:1 and IV:2) and 1 unaffected sibling(V:1) were heterozygous carriers of the MRE11 variant. Inaddition, the unaffected individuals IV:1, IV:8, and IV:9 wereheterozygous for the MRE11 variant (figure 3A).
Transcript analysisMuscle biopsy from the index case (V:2) was available fortesting the effects of the homozygous c.657C>TMRE11 exon7 variant on transcript and protein levels. In silico predictionsuggested splicing defect with a likely consequence of a pre-mature termination codon. Accordingly, the reverse-transcriptasePCR analysis indicated no detectable expression levels ofMRE11transcript in the index case (figure 3B), suggesting that this variantdestabilizes the transcript by NMD.
Histological and immunohistochemicalanalysis of skeletal muscleImmunohistochemical analysis on the muscle biopsy from theindex case (V:2) was performed to assess the nuclear expressionof MRE11A protein. Skeletal muscle biopsies from 2 individualswithout neurodegenerative disorders were used as controls. Incontrast to control skeletal muscle biopsies showing detectableexpression levels of MRE11A protein in the nucleoplasm,MRE11A immunohistochemistry using a polyclonal antibodyshowed no staining in the skeletal muscle biopsy from the indexcase (figure 3C), indicating lack of a stable truncated protein.
Chromosomal aberration assayChromosomal breakage more than or equal to tenfold ofcontrol is clinically significant. Chromosomal breakage testscarried out for 9 individuals of the family (III:1, IV:1, IV:2, IV:
Figure 3 Genetic findings and expression analysis of MRE11A at transcript and protein levels
(A) Sanger sequence analysis demonstrates the segregation of theMRE11 c.657C>T variant in the family. The unaffected parents (III:1 and (IV:2) and additionalfamilymembers (IV:1, IV:8, IV:9, and V:1) are heterozygous for theMRE11 c.657C>T variant. The affected individuals for whomDNAwas available (V:2, V:3, andV:4) are homozygous for the variant. The variant does not lead to alteration of asparagine amino acid (AAC>AAT, p.Asn219=). The variant is located in spliceregion (indicated by a box) changing the nucleotide of the 39 end of exon 7 (ENST00000323929.7). The 59 end of intron 7 is shown by an arrow. (B) reverse-transcriptase polymerase chain reaction analysis of RNA from skeletal muscle tissues of the index case (V:2) and a control. Expression analysis of fragmentcovering exon 1 through 8 coding regions of MRE11 (1168 bp) at the transcript level in the patient and a control indicated the absence of MRE11 transcript inthe patient. Amplification of ACTB served as quality control of complementary DNA. (C) Immunostaining of MRE11A in skeletal muscle biopsy specimen fromthe index patient (V:2) indicates no nucleoplasm expression, whereas the control muscle specimen shows positive immunostaining of MRE11A localized tonucleoplasm. Scale bar, 50 μm.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

8, IV:9, V:1, V:2, V:3, and V:4) indicated no significantstructural alterations, such as chromosomal breaks, chromo-somal translocations or gaps, when compared with the con-trols at 450–550 band resolution.
DiscussionIn this study, we describe a family with 3 affected siblings, at 23,20, and 18 years of age, diagnosed with ATLD. Consistent withthis diagnosis, the siblings developed progressive cerebellarataxia, developmental delay, and mild intellectual disability butwith absence of telangiectasia or facial dysmorphism and nohistory of severe infections, immunodeficiency, or cancer.
Next-generation sequencing revealed homozygosity in theaffected individuals for the MRE11 (c.657C>T, p.Asn219=)rs775017362 variant, which is present in population databasesat frequencies compatible with recessive inheritance. Thevariant does not change the amino acid, but in silico analysispredicted that the variant would affect splicing efficiency,most likely resulting in exon 7 skipping, leading to a frameshiftand a premature termination codon (p.Ser183Valfs*31). Thehomozygous variant could only be detected from the se-quencing of genomic DNA. No MRE11 alleles were revealedby transcript assessment, making it likely that this variantdestabilizes the transcript by NMD. This correlated withimmunolabeling findings, demonstrating no MRE11A stain-ing in the skeletal muscle biopsy from the index case, sug-gesting the absence of a stable protein.
So far, several families with MRE11 variants have beenreported.5,9–13,17 A majority of reported variants in MRE11,homozygous or compound heterozygous splicing, nonsenseor missense mutations, have been associated with a spectrumof clinical severity of ATLD5,9–13 (table). However, MRE11variants have been described in 2 unrelated Japanese patientswith characteristic features of NBSLD,17 which is otherwiseassociated withmutations inRAD50.4 In both Japanese patients,variants of close-by nucleotides to the mutated nucleotide in ourpatients were found. One patient was compound heterozygotefor c.658A>C and c.659+1G>A and the other patient wascompound heterozygote c.658A>C and c.338A>G. TheMRE11c.658A>C variant does not lead to alteration of amino acid(p.Arg220=) and the results from reverse-transcriptase PCRanalysis in these patients carrying the transcript of the c.658A>Callele indicated that this variant leads to exon 7 skipping, but thatsome small amount of RNA was correctly spliced.17 Corre-spondingly, a reduced amount of wild-type–sized MRE11Aprotein was detected by immunoblot analysis.17 This indicatesthat c.657C>T alteration in our patients has a greater impact onsplicing efficiency than the c.658A>C variant, leading to com-plete destabilization of the transcript by NMD and subsequentdepletion of MRE11A.
The MRN complex is involved in sensing of DNA double-strand breaks, DNA recombination, and multiple cell-cyclecheckpoints.18,19 Cooperation between the ATM and MRN
complexes is essential in the DNA damage response, which isnot fully determined.20 MRE11A, a member of the MRNcomplex, is involved in homologous and mitotic and meioticrecombination, telomere length maintenance, and DNA double-strand break repair.21 MRE11A possesses DNA exonuclease andendonuclease activities that are highly conserved during evolu-tion.22 The relatively mild impact of MRE11A deficiency, whichpermits viability in our patients, is intriguing. In addition, it is insharp contrast to the early embryonic lethality of nuclease-deficient and null alleles of murine Mre11, associated withmarked genome instability.21,23 However, despite the contribu-tion of MRE11A in the MRN complex and the cooperationbetween the MRN complex and ATM in the DNA double-strand break repair pathways to maintain genomic integrity,20
loss of MRE11A only modestly impaired double-strand breakrepair in the chicken DT40 and human TK6 cell lines.24 Fur-thermore, animal models of NBS1 or MRE11A do not com-pletely recapitulate phenotypes observed in NBS or ATLDpatients, including the neurologic aspects.25–27
Notably, complete absence of ATM kinase, in patients withataxia telangiectasia, a cancer-prone neurodegenerative dis-ease, is not lethal.28 The majority of the variants in ATM inpatients with classic ataxia telangiectasia are biallelic truncat-ing mutations that result in a total loss of destabilized ATMprotein.28 Given the vital role of ATM in the DNA damageresponse for DNA repair, cell cycle checkpoint activation, andapoptosis,29 the viability associated with loss of ATM in ataxiatelangiectasia patients is intriguing. Mouse models deficient inATM recapitulate accurate ataxia telangiectasia diseasephenotypes,30–32 but loss of ATM kinase activity causes earlyembryonic lethality in mice, indicating that inhibition of ATMkinase activity does not equate to loss of the ATMprotein.33 Itwas speculated that embryonic lethality in mice with ATMkinase inactivity was the result of ATM kinase recruitment atDNA breaks, which may impair the function of other proteinsby blocking their access to DNA damage.33
Although MRE11A, NBS1, and RAD50 are components ofthe MRN complex, mutations in MRE11, NBN, and RAD50are associated with different clinical phenotypes, suggestingthat the components have distinct functions and roles in-dependent of the MRN complex. Patients with NBS andataxia telangiectasia have a predisposition to cancer, particu-larly an increased risk of developing lymphoid tumors,20
which may reflect the involvement of ATM and MRN com-plex in DNA damage response. Nevertheless, development ofcancer in ATLD patients associated withMRE11 has not beena frequent finding in reported cases, with only 2 patients sofar,10 and it is not a clinical feature in the family reported here.It thus remains unknown whether patients with ATLD havea predisposition to cancer, given the few patients withMRE11mutation that have been described so far.
Patients with ataxia telangiectasia, NBS, and ATLD usuallyshow an increased level of chromosomal translocation in theperipheral blood involving chromosome 7 and 14.20
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

Table
Clin
ical
findings
ofca
seswithrece
ssiveMRE
11va
rian
ts
Study
Family
Case
Muta
tion
Chro
moso
mal
aber
ration
Inte
llec
tual
disability
Tumor
appea
rance
Eye
move
men
tdisord
ers
Telangiec
tasia
Dys
arthria
Ata
xia
Cere
bellar
atrophy
Short
statu
reMicro
cephaly
Refer
ence
51
1Homozygo
usp.Arg63
3Ter
+−
−+
−+
++
+NA
2+
−−
+−
++
++
NA
23
Compoundheterozygo
us
p.Arg57
1Ter
and
p.Asn
117S
er
+−
−+
−+
++
NA
NA
4+
NA
−+
−+
++
NA
NA
Refer
ence
113
5Compoundheterozygo
us
p.Thr481
Lysan
dp.Arg57
1Ter
+−
−+
−+
++
NA
NA
6+
−−
+−
++
+NA
NA
Refer
ence
124
7Homozygo
usp.Trp
210C
ysNA
NA
−+
NA
++
+NA
NA
8NA
NA
−+
NA
++
NA
NA
NA
9NA
NA
−+
−+
+NA
NA
−
10NA
NA
−+
−+
+NA
NA
−
511
Homozygo
usp.Trp
210C
ys−
+−
+−
++
+NA
+
12−
+−
+−
++
+NA
+
13NA
+−
+−
++
+NA
+
614
Homozygo
usp.Trp
210C
ysNA
NA
−+
−+
++
NA
−
15NA
NA
−+
−+
++
NA
−
16NA
NA
−−
−+
++
NA
+
Refer
ence
107
17Compoundheterozygo
us
p.Trp
243A
rgan
dp.Ile3
40_
Arg36
6del
++
++
−+
++
+−
18+
++
+−
NA
++
+−
Refer
ence
178
19Compoundheterozygo
us
p.Ser18
3ValfsX3
1an
dp.Arg22
0=
++
−−
NA
−−
−−
+
Continued
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
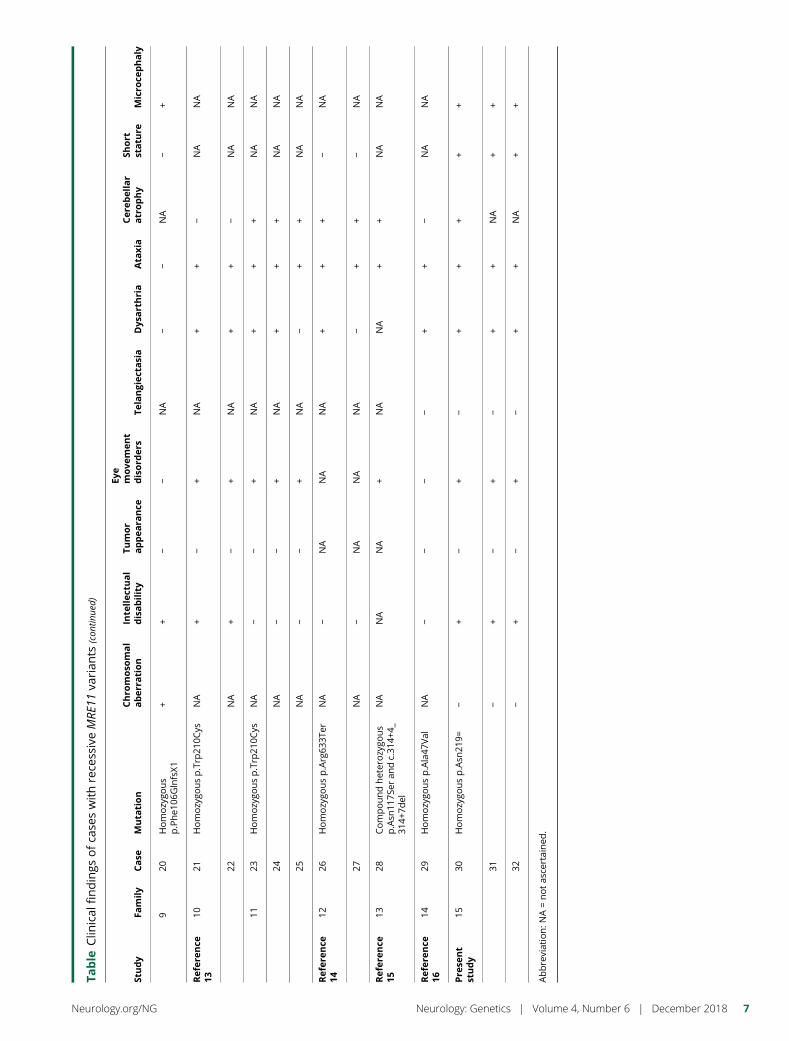
Table
Clin
ical
findings
ofca
seswithrece
ssiveMRE
11va
rian
ts(con
tinue
d)
Study
Family
Case
Muta
tion
Chro
moso
mal
aber
ration
Inte
llec
tual
disability
Tumor
appea
rance
Eye
move
men
tdisord
ers
Telangiec
tasia
Dys
arthria
Ata
xia
Cere
bellar
atrophy
Short
statu
reMicro
cephaly
920
Homozygo
us
p.Phe1
06GlnfsX1
++
−−
NA
−−
NA
−+
Refer
ence
1310
21Homozygo
usp.Trp
210C
ysNA
+−
+NA
++
−NA
NA
22NA
+−
+NA
++
−NA
NA
1123
Homozygo
usp.Trp
210C
ysNA
−−
+NA
++
+NA
NA
24NA
−−
+NA
++
+NA
NA
25NA
−−
+NA
−+
+NA
NA
Refer
ence
1412
26Homozygo
usp.Arg63
3Ter
NA
−NA
NA
NA
++
+−
NA
27NA
−NA
NA
NA
−+
+−
NA
Refer
ence
1513
28Compoundheterozygo
us
p.Asn
117S
eran
dc.31
4+4_
314+
7del
NA
NA
NA
+NA
NA
++
NA
NA
Refer
ence
1614
29Homozygo
usp.Ala47
Val
NA
−−
−−
++
−NA
NA
Pre
sent
study
1530
Homozygo
usp.Asn
219=
−+
−+
−+
++
++
31−
+−
+−
++
NA
++
32−
+−
+−
++
NA
++
Abbreviation:N
A=notas
certained
.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

HomozygousMRE11 variants often alter not only the levels ofMRE11A but also the levels of 2 other components of theMRN complex, NBS1 and RAD50, leading to inactivation of theentire MRN protein complex.5,9,11,17 Although, the effect ofMRE11 c.657C>T variant on sensitivity to ionizing radia-tion and the impact on NBS1 and RAD50 expression levelsor the entire MRN complex in our patients remain un-known, none of the homozygous or heterozygous carriersof theMRE11 variant in our family show any chromosomalabnormalities. This is presumably because of retainedcontribution of damage sensor or mediators involved in theMRN complex and sustained ATM kinase activity inmaintaining chromosomal integrity, as observed in otherATLD patients associated with MRE11 mutation.34 How-ever, absence of chromosomal alterations in this family isintriguing and requires further investigations.
We describe patients with characteristic features of ATLD,associated with a homozygousMRE11 splicing variant leadingto RNA decay and deficiency of MRE11A protein.
AcknowledgmentThe authors thank the family members who provided samplesand clinical information for this study. The authors thank Dr.Mansoor Salehi for providing us with the DNA samples andclinical data of patients.
Study fundingThe study was supported by grants from the EuropeanUnion’s Seventh Framework Programme for research, tech-nological development and demonstration under grant
agreement no. 608473 (H.T.) and the Swedish ResearchCouncil (H.T.). N. Laing is supported by Australian NationalHealth and Medical Research Council Principal ResearchFellowship (APP1117510), N. Laing and H. Goullee byNHMRC EU Collaborative Grant APP1055295. The fundershad no role in the design of the study and collection, analysis,decision to publish, interpretation of data or preparation ofthe manuscript.
DisclosuresThe authors report no disclosures relevant to the manuscript.M. Sedghi, M. Salari, A.-R. Moslemi, A. Kariminejad, M.Davis, H. Goulee, and B. Olsson report no disclosures. N.Laing has received travel funding from the World MuscleSociety, the Asian Oceanian Myology Centre, Sanofi, and theOttawa Neurology Meeting; has served on the editorial boardfor Neuromuscular Disorders Executive Associate Editor andNeuromuscular Disorders; has received publishing royaltiesfor The Sarcomere and Skeletal muscle disease; has receivedresearch support from the governmental entity, the AustralianNational Health, and Medical Research Council; and hasreceived research and support (from foundations and so-cieties) from the U.S. Muscular Dystrophy Association, theAssociation Francaise contre les Myopathies, the Founda-tion Building Strength for Nemaline Myopathy, the MotorNeuron Disease Research Institute of Australia, the WesternAustralian Health and Medical Research Infrastructure Fund,and the Perpetual Foundation. H. Tajsharghi reports no dis-closures. Full disclosure form information provided by theauthors is available with the full text of this article at Neurology.org/NG.
Appendix 1 Author contributions
Name Location Role Contribution
MaryamSedghi
Isfahan University of Medical Sciences, Isfahan,Iran
Author Acquisition of data, analysis and interpretation, clinical assessmentof the patients
Mehri Salari Shahid Beheshti University of Medical Science,Tehran, Iran
Author Acquisition of data, analysis and interpretation, clinical assessmentof the patients
Ali-RezaMoslemi
University of Gothenburg, SahlgrenskaUniversity Hospital, Sweden
Author Acquisition of data, analysis and interpretation
ArianaKariminejad
Kariminejad-Najmabadi Pathology & GeneticsCenter, Tehran, Iran
Author Acquisition of data, analysis and interpretation, clinical assessmentof the patients
Mark Davis Department of Diagnostic Genomics, Pathwest,QEII Medical Centre, Nedlands, WesternAustralia, Australia
Author Acquisition of data, analysis and interpretation
HayleyGoullee
The University of Western Australia, Australia Author Acquisition of data, analysis and interpretation
BjornOlsson
University of Skovde, Skovde, Sweden Author Acquisition of data, analysis and interpretation
Nigel Laing The University of Western Australia, Australia Author Acquisition of data, analysis and interpretation, critical revision ofthe manuscript for important intellectual content
HomaTajsharghi
The University of Western Australia andUniversity of Skovde, Skovde, Sweden
Correspondingauthor
Acquisition of data, analysis and interpretation; critical revision ofthe manuscript for important intellectual content, study concept,and design; and study supervision
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Publication historyReceived by Neurology: Genetics May 25, 2018. Accepted in final formAugust 22, 2018.
References1. Matsuura S, Tauchi H, Nakamura A, et al. Positional cloning of the gene for Nijmegen
breakage syndrome. Nat Genet 1998;19:179–181.2. Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and
Nijmegen breakage syndrome: linkage of double-strand break repair to the cellularDNA damage response. Cell 1998;93:477–486.
3. Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break repairprotein, is mutated in Nijmegen breakage syndrome. Cell 1998;93:467–476.
4. Waltes R, Kalb R, Gatei M, et al. Human RAD50 deficiency in a Nijmegen breakagesyndrome-like disorder. Am J Hum Genet 2009;84:605–616.
5. Stewart GS, Maser RS, Stankovic T, et al. The DNA double-strand break repair genehMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell1999;99:577–587.
6. Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine (Baltimore)1991;70:99–117.
7. Hernandez D, McConville CM, Stacey M, et al. A family showing no evidence oflinkage between the ataxia telangiectasia gene and chromosome 11q22-23. J MedGenet 1993;30:135–140.
8. Klein C, Wenning GK, Quinn NP, Marsden CD. Ataxia without telangiectasiamasquerading as benign hereditary chorea. Mov Disord 1996;11:217–220.
9. Pitts SA, Kullar HS, Stankovic T, et al. hMRE11: genomic structure and a null mu-tation identified in a transcript protected from nonsense-mediated mRNA decay.Hum Mol Genet 2001;10:1155–1162.
10. Uchisaka N, Takahashi N, Sato M, et al. Two brothers with ataxia-telangiectasia-likedisorder with lung adenocarcinoma. J Pediatr 2009;155:435–438.
11. Delia D, Piane M, Buscemi G, et al. MRE11 mutations and impaired ATM-dependentresponses in an Italian family with ataxia-telangiectasia-like disorder. HumMol Genet2004;13:2155–2163.
12. FernetM, GribaaM, SalihMAM, SeidahmedMZ, Hall J, KoenigM. Identification andfunctional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patientswith the ataxia telangiectasia-like disorder. Hum Mol Genet 2005;14:307–318.
13. Bohlega SA, Shinwari JM, Al Sharif LJ, Khalil DS, Alkhairallah TS, Al Tassan NA.Clinical and molecular characterization of ataxia with oculomotor apraxia patients inSaudi Arabia. BMC Med Genet 2011;12:27.
14. Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012;150:533–548.
15. Nemeth AH, Kwasniewska AC, Lise S, et al. Next generation sequencing for moleculardiagnosis of neurological disorders using ataxias as a model. Brain 2013;136:3106–3118.
16. Miyamoto R, Morino H, Yoshizawa A, et al. Exome sequencing reveals a novelMRE11 mutation in a patient with progressive myoclonic ataxia. J Neurol Sci 2014;337:219–223.
17. Matsumoto Y, Miyamoto T, Sakamoto H, et al. Two unrelated patients withMRE11Amutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair(Amst) 2011;10:314–321.
18. D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair andcheckpoint signalling. Nat Rev Mol Cell Biol 2002;3:317–327.
19. Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: definingthe sensors and mediators. Trends Cell Biol 2003;13:458–462.
20. Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinicalpresentation and molecular basis. DNA Repair (Amst) 2004;3:1219–1225.
21. Buis J, Wu Y, Deng Y, et al. Mre11 nuclease activity has essential roles in DNA repairand genomic stability distinct from ATM activation. Cell 2008;135:85–96.
22. Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K.Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meioticrecombination. EMBO J 1998;17:6412–6425.
23. Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase dem-onstrates the requirement for the double-strand break repair Mre11 protein in murineembryonic stem cells. Nucleic Acids Res 1997;25:2985–2991.
24. Hoa NN, Akagawa R, Yamasaki T, et al. Relative contribution of four nucleases, CtIP,Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair byhomologous recombination in both the chicken DT40 and human TK6 cell lines.Genes Cells 2015;20:1059–1076.
25. Theunissen JW, Kaplan MI, Hunt PA, et al. Checkpoint failure and chromosomalinstability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell2003;12:1511–1523.
26. Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murinemodel of Nijmegen breakage syndrome. Curr Biol 2002;12:648–653.
27. Shull ER, Lee Y, Nakane H, et al. Differential DNA damage signaling accounts fordistinct neural apoptotic responses in ATLD and NBS. Genes Dev 2009;23:171–180.
28. Gilad S, Khosravi R, Shkedy D, et al. Predominance of null mutations in ataxia-telangiectasia. Hum Mol Genet 1996;5:433–439.
29. Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM inresponse to DNA damage. Science 1998;281:1674–1677.
30. Barlow C, Hirotsune S, Paylor R, et al. Atm-deficient mice: a paradigm of ataxiatelangiectasia. Cell 1996;86:159–171.
31. Elson A, Wang Y, Daugherty CJ, et al. Pleiotropic defects in ataxia-telangiectasiaprotein-deficient mice. Proc Natl Acad Sci U S A 1996;93:13084–13089.
32. Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted dis-ruption of ATM leads to growth retardation, chromosomal fragmentation duringmeiosis, immune defects, and thymic lymphoma. Genes Dev 1996;10:2411–2422.
33. Daniel JA, Pellegrini M, Lee BS, et al. Loss of ATM kinase activity leads to embryoniclethality in mice. J Cell Biol 2012;198:295–304.
34. Regal JA, Festerling TA, Buis JM, Ferguson DO. Disease-associated MRE11 mutantsimpact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet2013;22:5146–5159.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

ARTICLE OPEN ACCESS
Screening of novel restless legssyndrome–associated genes in French-CanadianfamiliesFulya Akçimen, MSc, Dan Spiegelman, MSc, Alexandre Dionne-Laporte, MSc, Ziv Gan-Or, MD, PhD,
Patrick A. Dion, PhD, and Guy A. Rouleau, MD, PhD, FRCP(C)
Neurol Genet 2018;4:e296. doi:10.1212/NXG.0000000000000296
Correspondence
Dr. Rouleau
AbstractObjectiveTo examine whether any rare, protein-altering variants could be identified across 13 recentlyidentified restless legs syndrome (RLS) loci in familial French-Canadian cases.
MethodsWhole-exome sequences from 7 large French-Canadian families (4–8 affected per family fora total of 38 cases) were examined for variants in any genes located within 1Mb on either side ofeach locus.
ResultsAmong the 43 rare protein-altering variants identified, none segregated with RLS in thefamilies.
ConclusionsOur study does not support a role for causative protein-altering variants in the genes that arelocated either in the previously or newly identified RLS loci. It is therefore possible thatnoncoding regulatory variants within these loci or yet unidentified loci could be the cause ofRLS in our families.
From the Department of Human Genetics (F.A., Z.G.-O., G.A.R.), McGill University; Montreal Neurological Institute (F.A., D.S., A.D.-L., Z.G.-O., P.A.D., G.A.R.), McGill University; andDepartment of Neurology and Neurosurgery (Z.G.-O., P.A.D., G.A.R.), McGill University, Montreal, Quebec, Canada.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Asmuch as 60% of patients with restless legs syndrome (RLS)have a positive family history,1 with a heritability close to 20%.2
Using a cohort of 671 cases (192 probands and 479 affectedrelatives), our team has previously reported that 77.1% ofFrench-Canadian patients had a family history of RLS, sug-gesting an important contribution of genetic factors in thispopulation.3 In an effort to identify coding variants in the 6previously identified RLS loci (MEIS1, BTBD9, PTPRD,MAP2K5/SKOR1, TOX3, and rs6747972), we have pre-viously examined 7 French-Canadian families with an au-tosomal dominant inheritance pattern using whole exomesequencing (WES). Variants were identified in PTPRD andSKOR1, but none of these segregated with the disease in thefamilies studied.4 Recently, a large-scale meta-analysis con-firmed the 6 loci known to be associated with RLS andidentified 13 novel loci.2 In the current study, we reanalyzedWES data from the 7 French-Canadian families to examinewhether coding variants segregating with RLS could beidentified in genes within 1Mb of all 19 loci.
MethodsSamplesSeven French-Canadian families consisting of 32 women(mean age ± SD: 71.44 ± 15.23 years) and 6 men (mean age ±SD: 70.17 ± 17.65 years) were examined using WES (female:male ratio of 5.33:1). All patients were diagnosed according tothe International RLS Study Group criteria.5 Family pedigreesof probands are shown in figures 1–7.
Standard protocol approvals, registrations,and patient consentsAll subjects provided informed consent, and the study wasapproved by the respective institutional review boards.
Whole exome sequencingWES libraries were prepared using the Agilent SureSelectHuman All Exon V4 (Agilent Technologies, Los Angeles,CA) capture kit and sequenced using an Illumina HiSeq2000platform (100 base pair paired-end sequencing). Reads werealigned to the hg19 human reference genome using theBurrows-Wheeler Aligner tool.6 Variant calling was performedusing the HaplotypeCaller tool from the Genome AnalysisToolkit v.3.5.7,8 Finally, variants were annotated for pre-dicted protein alterations and population frequencies usingannotate variation (ANNOVAR).9
Variant filtration and segregation analysisOnly variants that were predicted to be protein-altering(nonsynonymous, splicing, stop-gain) by ANNOVAR wereincluded in the subsequent analysis. Variants were filtered byfrequency using the Exome Aggregation Consortium (ExAC)browser, Cambridge, MA (exac.broadinstitute.org, accessedJanuary 2018). Variants below a threshold of 0.05 allele fre-quency in the non–Finnish European population were in-cluded in the final results.
Data availability statementThe authors confirm that the data necessary for confirmingthe conclusions of this study are available within the articleand its supplementary material. Raw whole exome sequencingdata will be provided freely upon request.
ResultsA total of 71 genes within 1Mb of the 19 loci were found tobe screened in 38 affected individuals and a list of candidatevariants was established (table e-1, links.lww.com/NXG/A131).The average andminimum coverage of genes screenedwere 87x
Figure 1 Pedigree of family 1
*Exome sequencing data available.
GlossaryExAC = exome aggregation consortium; RLS = restless legs syndrome; WES = whole-exome sequencing.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

and 25x, respectively. A total of 43 variants were predicted tobe protein-altering and had a population frequency less than0.05. Among the variants identified, none of them segregatedwell with the disease in pedigrees, which suggests that theyare not disease causing.
DNAH8 p.Val874Met (rs45529837, ExAC MAF = 0.03071)appeared to segregate well in one of the families, and as such,it might explain RLS in this family. However, this particularvariant was also observed in another family (Family 5: IV-5,6,7 and V-8) where it did not segregate with the disease.
DiscussionOur results suggest that nonsynonymous variants within theseloci do not explain RLS in these large families and that it istherefore likely that regulatory (coding or non-coding) variants
are associated with the risk of RLS. While p.Val874Met(rs45529837, ExAC MAF = 0.03071) in DNAH8 (that enc-odes for an axonemal dynein involved in motility of cilia andflagella)10 segregated well in one of the families, it was alsoobserved in another family (Family 5: IV-5,6,7 and V-8)where it did not segregate with the disease, therefore itssegregation should be interpreted with caution. Rare causativevariants, at much lower frequency than the associated commonsingle nucleotide polymorphism (SNP), can create genome-wide associations even when they are megabases away fromthe common variants that tag them.11 A WES approach, likethe one used here, can enable the discovery of novel causativevariants. The likelihood of achieving this increases with the sizeof the pedigrees and the penetrance of the condition examined.Although our study does not support a role of rare protein-altering variants in RLS-associated loci to be a cause of thedisease, further studies in more pedigrees are required to de-termine whether there exist monogenic forms of RLS.
Author contributionsF. Akçimen: design and conceptualized the study; analysis andinterpretation of the data; and drafting the manuscript forintellectual content. D. Spiegelman: analysis of the data. A.Dionne-Laporte: analysis of the data. Z. Gan-Or: drafting orrevising the manuscript for intellectual content. P.A. Dion:design and conceptualized the study; interpretation of thedata; and drafting or revising the manuscript for intellectualcontent. G.A. Rouleau: design and conceptualized the study;interpretation of the data; and drafting or revising the man-uscript for intellectual content.
AcknowledgmentsThe authors thank the patients for their participation in thestudy. GAR holds a Canada Research Chair in Genetics of the
Figure 2 Pedigree of family 2
*Exome sequencing data available.
Figure 3 Pedigree of family 3
*Exome sequencing data available.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

Figure 4 Pedigree of family 4
*Exome sequencing data available.
Figure 5 Pedigree of family 5
*Exome sequencing data available.
Figure 6 Pedigree of family 6
*Exome sequencing data available.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Nervous System and the Wilder Penfield Chair in Neuro-sciences. The authors thank Jay P. Ross andCynthia V. Bourassafor their assistance.
Study fundingNo targeted funding reported.
DisclosureF. Akçimen reports no disclosures. D. Spiegelman reports nodisclosures. A. Dionne-Laporte reports no disclosures. Z.Gan-Or has received funding for travel and/or speaker hon-oraria from Lysosomal Therapeutics Inc. and Idorsia; serves/has served on the editorial board of Parkinsonism & RelatedDisorders; and serves/has served as a consultant for LysosomalTherapeutics Inc., Denali, Prevail Therapeutics, Idorsia, andAllergan. P.A. Dion reports no disclosures. G.A. Rouleau hasreceived research support from the Canadian Institutes ofHealth Research (CIHR), ALS Society of Canada, and theALS Association. Full disclosure form information providedby the authors is available with the full text of this article atNeurology.org/NG.
Publication historyReceived by Neurology: Genetics June 19, 2018. Accepted in final formOctober 3, 2018.
References1. Winkelmann J, Polo O, Provini F, et al. Genetics of restless legs syndrome
(RLS): state-of-the-art and future directions. Mov Disord 2007;22(suppl 18):S449–S458.
2. Schormair B, Zhao C, Bell S, et al. Identification of novel risk loci for restless legssyndrome in genome-wide association studies in individuals of European ancestry:a meta-analysis. Lancet Neurol 2017;16:898–907.
3. Xiong L, Montplaisir J, Desautels A, et al. Family study of restless legs syndrome inQuebec, Canada: clinical characterization of 671 familial cases. Arch Neurol 2010;67:617–622.
4. Gan-Or Z, Zhou S, Ambalavanan A, et al. Analysis of functional GLO1 variantsin the BTBD9 locus and restless legs syndrome. Sleep Med 2015;16:1151–1155.
5. Allen RP, Picchietti DL, Garcia-Borreguero D, et al. Restless legs syndrome/Willis-Ekbom disease diagnostic criteria: updated International Restless Legs SyndromeStudy Group (IRLSSG) consensus criteria—history, rationale, description, and sig-nificance. Sleep Med 2014;15:860–873.
6. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheelertransform. Bioinformatics 2009;25:1754–1760.
7. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery andgenotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
8. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduceframework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303.
9. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of geneticvariants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
10. Neesen J, Koehler MR, Kirschner R, et al. Identification of dynein heavy chain genesexpressed in human and mouse testis: chromosomal localization of an axonemaldynein gene. Gene 1997;200:193–202.
11. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in com-mon disease through whole-genome sequencing. Nat Rev Genet 2010;11:415–425.
Figure 7 Pedigree of family 7
*Exome sequencing data available.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

ARTICLE OPEN ACCESS
Development of a rapid functional assay thatpredicts GLUT1 disease severitySasha M. Zaman, PhD,* Saul A. Mullen, MBBS, PhD,* Slave Petrovski, PhD, Snezana Maljevic, PhD,
Elena V. Gazina, PhD, A. Marie Phillips, PhD, Gabriel Davis Jones, BSc, Michael S. Hildebrand, PhD,
John Damiano, BSc, Stephane Auvin, MD, PhD, Holger Lerche, MD, Yvonne G. Weber, MD,
Samuel F. Berkovic, MBBS, MD, Ingrid E. Scheffer, MBBS, PhD, Christopher A. Reid, PhD,
and Steven Petrou, PhD
Neurol Genet 2018;4:e297. doi:10.1212/NXG.0000000000000297
Correspondence
Dr. Petrou
AbstractObjectiveTo examine the genotype to phenotype connection in glucose transporter type 1 (GLUT1)deficiency and whether a simple functional assay can predict disease outcome from geneticsequence alone.
MethodsGLUT1 deficiency, due to mutations in SLC2A1, causes a wide range of epilepsies. Onepossible mechanism for this is variable impact of mutations on GLUT1 function. To test this,we measured glucose transport by GLUT1 variants identified in population controls andpatients with mild to severe epilepsies. Controls were reference sequence from the NCBI and 4population missense variants chosen from public reference control databases. Nine variantsassociated with epilepsies or movement disorders, with normal intellect in all individuals,formed the mild group. The severe group included 5 missense variants associated with classicalGLUT1 encephalopathy. GLUT1 variants were expressed in Xenopus laevis oocytes, and glu-cose uptake was measured to determine kinetics (Vmax) and affinity (Km).
ResultsDisease severity inversely correlated with rate of glucose transport between control (Vmax = 28± 5), mild (Vmax = 16 ± 3), and severe (Vmax = 3 ± 1) groups, respectively. Affinities of glucosebinding in control (Km = 55 ± 18) and mild (Km = 43 ± 10) groups were not significantlydifferent, whereas affinity was indeterminate in the severe group because of low transport rates.Simplified analysis of glucose transport at high concentration (100 mM) was equally effective atseparating the groups.
ConclusionsDisease severity can be partly explained by the extent of GLUT1 dysfunction. This simpleXenopus oocyte assay complements genetic and clinical assessments. In prenatal diagnosis, thissimple oocyte glucose uptake assay could be useful because standard clinical assessments arenot available.
*These authors contributed equally.
From the Florey Institute of Neuroscience and Mental Health (S.M.Z., S.A.M., S.M., E.V.G., A.M.P., G.D.J., I.E.S., C.A.R., S. Petrou.); Department of Medicine (RMH) University ofMelbourne (S.M.Z., S. Petrovski, M.S.H., J.D., S. Petrou); Department of Medicine (Austin Health) (M.S.H., J.D., S.F.B., I.E.S.), University of Melbourne, Heidelberg; Department ofNeurology and Epileptology (H.L., Y.G.W.), Hertie Institute for Clinical Brain Research, University of Tubingen; School of Biosciences (A.M.P.), University of Melbourne, Parkville,Australia; APHP (S.A.), Hopital Robert Debre, Service de Neurologie Pediatrique; Univ Paris Diderot (S.A.), Sorbonne Paris Cite, INSERM UMR1141, Paris, France; and Department ofPaediatrics (I.E.S.), University of Melbourne, Royal Children’s Hospital, Parkville, Australia.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by National Health and Medical Research Council (NHMRC).
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Failure of the glucose transporter type 1 (GLUT1), coded bythe gene Solute Carrier Family 2 member 1 (SLC2A1), leads toinadequate brain glucose and neurologic disorders.1 ClassicalGLUT1 encephalopathy shows intractable infantile seizures,complex motor disorder, intellectual impairment, low CSFglucose (hypoglycorrhachia), and often microcephaly.2
However, the spectrum of GLUT1 deficiency syndrome ismuch broader. Familial cases frequently have a combinationof epilepsies with absence seizures, normal intellect, and themovement disorder of paroxysmal exertional dyskinesia.3
GLUT1 deficiency has been described as occurring in 10% ofearly-onset absence epilepsy, 5% of epilepsy with myoclonic-atonic seizures, and approximately 1% of genetic generalizedepilepsies.4–7 Focal epilepsies also occur.3–8
One possible mechanism for the wide range of phenotypicseverity in GLUT1 deficiency syndrome is the extent to whichmutations affect GLUT1 function. Deletions and null muta-tions of SLC2A1 are associated with severe encephalopathy,whereas missense mutations can be seen across the wholespectrum of severity.2,3,5,6,9–12 Deletions lead to completehaploinsufficiency, whereas the effects of missense changesare presumed to range from hypomorphic to complete loss offunction. In this study, we examine that the range of residualfunction in missense variants causes GLUT1 deficiency. Wecompare the function of control missense SLC2A1 variantswith patient variants associated with either mild disease orclassical GLUT1 encephalopathy. We hypothesize that re-sidual function of the missense alleles will be greater in thosewith mild disease compared with those with severe diseaseand that the difference will be sufficiently marked to be clin-ically useful in predictive testing.
MethodsVariant selectionThree groups of variants were analyzed (table). Variants weredrawn from the published literature and unpublished casesclinically diagnosed at Austin Health.2,3,7,8,12,13 The controlgroup comprised 4 population variants drawn from the ExACdatabase along with the reference sequence from the NCBIand was analyzed to determine the background variation ofGLUT1 function in the general population. The “mild” groupcomprised 9 variants associated with mild phenotypes beforefunctional assessments (table). Mild GLUT1 deficiency wasdefined as epilepsy or movement disorder with normal in-tellect in all individuals with the variant. Last, the “severe”group comprised 5 missense variants associated with severeGLUT1 encephalopathy before functional assessments. Var-iants in which a mixture of phenotypic severities had beenreported, particularly intellectual disability in some cases,were excluded. GLUT1 deficiency leads to a number of
neurologic disorders with a spectrum of effects, and no ac-cepted rating scale for overall severity exists. By taking theopposite ends of this spectrum, mild disease with normaldevelopment and frank encephalopathy, we can be as confi-dent as possible that the phenotypes are distinct and thatfunctional differences in glucose transport should be present.Experiments were performed blinded to the phenotypic as-sociation (control, mild, and severe) of all genetic variants.
Standard protocol approvals, registrations,and patient consentsThe study was approved by the Austin Health Human Re-search Ethics Committee. All participants provided written,informed consent.
PCR and Sanger sequencingCoding exons and splice sites of the SLC2A1 gene were PCRamplified using specific primers designed to the referencehuman gene transcript (Ref Seq NM_006516). Primersequences are available on request. Amplification reactionswere cycled using a standard protocol on a Veriti ThermalCycler (Applied Biosystems, Carlsbad, CA). Bidirectionalsequencing of all exons and flanking intronic regions in-cluding splice sites was completed with a BigDye v3.1 Ter-minator Cycle Sequencing Kit (Applied Biosystems),according to the manufacturer’s instructions. Sequencingproducts were resolved using a 3730 × l DNA Analyzer(Applied Biosystems). All sequencing chromatograms werecompared with published cDNA sequence; nucleotidechanges were detected using CodonCode Aligner (Codon-Code Corporation, Dedham, MA).
Site-directedmutagenesis and RNA expressionThe GLUT1 coding sequence (NM_006516.2 from NCBI)cloned into the pcDNA3.1 vector was purchased from Gen-Script and subcloned into the oocyte expression vectorpGEMHEmcs, between BamH1 and HindIII sites. Pointmutations were introduced using overlapping PCR (primersare listed in table e-1, links.lww.com/NXG/A129). Constructfidelity was verified by Sanger sequencing. The referenceprotein sequence used in this study was NP_006507.
Plasmids were linearized using the Sph1 restriction enzyme,and cRNA was generated using Message Machine T7 tran-scription kit (Applied Biosciences, Ambion). The concen-tration and integrity of cRNA were determined usingspectrophotometry and gel electrophoresis, respectively.
Oocyte preparation and injectionOocytes (Dumont stage V or VI) were surgically removedfrom Xenopus laevis and incubated in Barth solution (5 mMHEPES, 82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, and pH
GlossaryGLUT1 = glucose transporter type 1.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
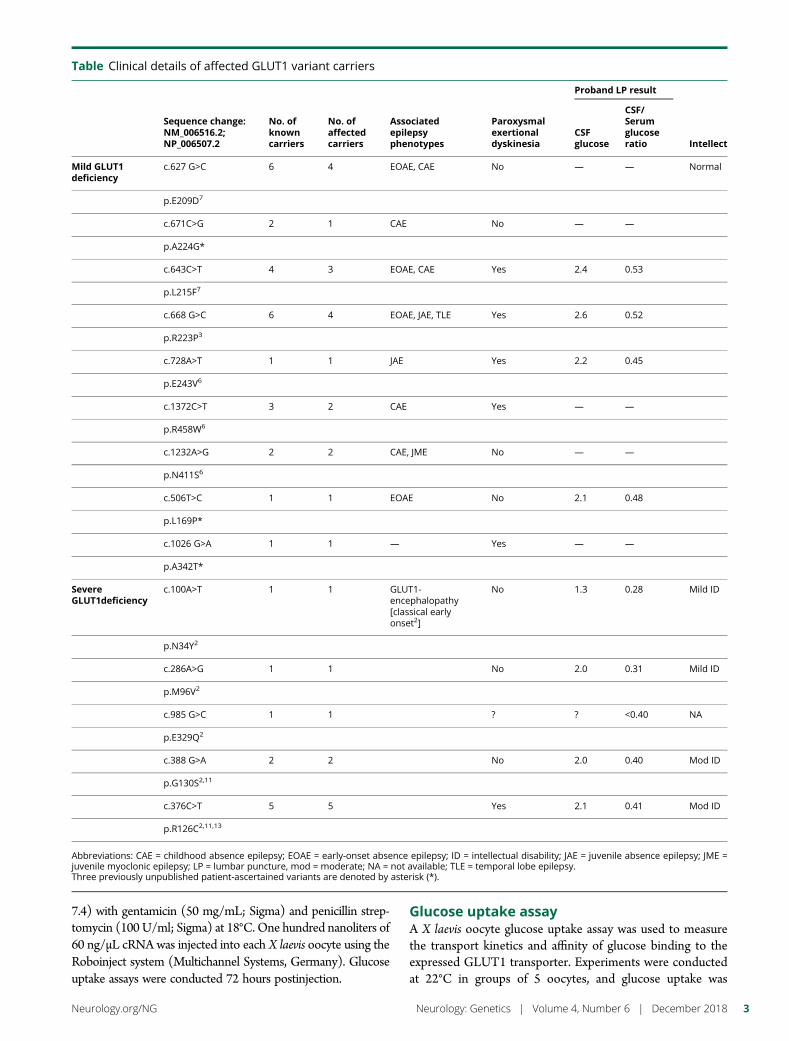
7.4) with gentamicin (50 mg/mL; Sigma) and penicillin strep-tomycin (100 U/ml; Sigma) at 18°C. One hundred nanoliters of60 ng/μL cRNA was injected into each X laevis oocyte using theRoboinject system (Multichannel Systems, Germany). Glucoseuptake assays were conducted 72 hours postinjection.
Glucose uptake assayA X laevis oocyte glucose uptake assay was used to measurethe transport kinetics and affinity of glucose binding to theexpressed GLUT1 transporter. Experiments were conductedat 22°C in groups of 5 oocytes, and glucose uptake was
Table Clinical details of affected GLUT1 variant carriers
Sequence change:NM_006516.2;NP_006507.2
No. ofknowncarriers
No. ofaffectedcarriers
Associatedepilepsyphenotypes
Paroxysmalexertionaldyskinesia
Proband LP result
IntellectCSFglucose
CSF/Serumglucoseratio
Mild GLUT1deficiency
c.627 G>C 6 4 EOAE, CAE No — — Normal
p.E209D7
c.671C>G 2 1 CAE No — —
p.A224G*
c.643C>T 4 3 EOAE, CAE Yes 2.4 0.53
p.L215F7
c.668 G>C 6 4 EOAE, JAE, TLE Yes 2.6 0.52
p.R223P3
c.728A>T 1 1 JAE Yes 2.2 0.45
p.E243V6
c.1372C>T 3 2 CAE Yes — —
p.R458W6
c.1232A>G 2 2 CAE, JME No — —
p.N411S6
c.506T>C 1 1 EOAE No 2.1 0.48
p.L169P*
c.1026 G>A 1 1 — Yes — —
p.A342T*
SevereGLUT1deficiency
c.100A>T 1 1 GLUT1-encephalopathy[classical earlyonset2]
No 1.3 0.28 Mild ID
p.N34Y2
c.286A>G 1 1 No 2.0 0.31 Mild ID
p.M96V2
c.985 G>C 1 1 ? ? <0.40 NA
p.E329Q2
c.388 G>A 2 2 No 2.0 0.40 Mod ID
p.G130S2,11
c.376C>T 5 5 Yes 2.1 0.41 Mod ID
p.R126C2,11,13
Abbreviations: CAE = childhood absence epilepsy; EOAE = early-onset absence epilepsy; ID = intellectual disability; JAE = juvenile absence epilepsy; JME =juvenile myoclonic epilepsy; LP = lumbar puncture, mod = moderate; NA = not available; TLE = temporal lobe epilepsy.Three previously unpublished patient-ascertained variants are denoted by asterisk (*).
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
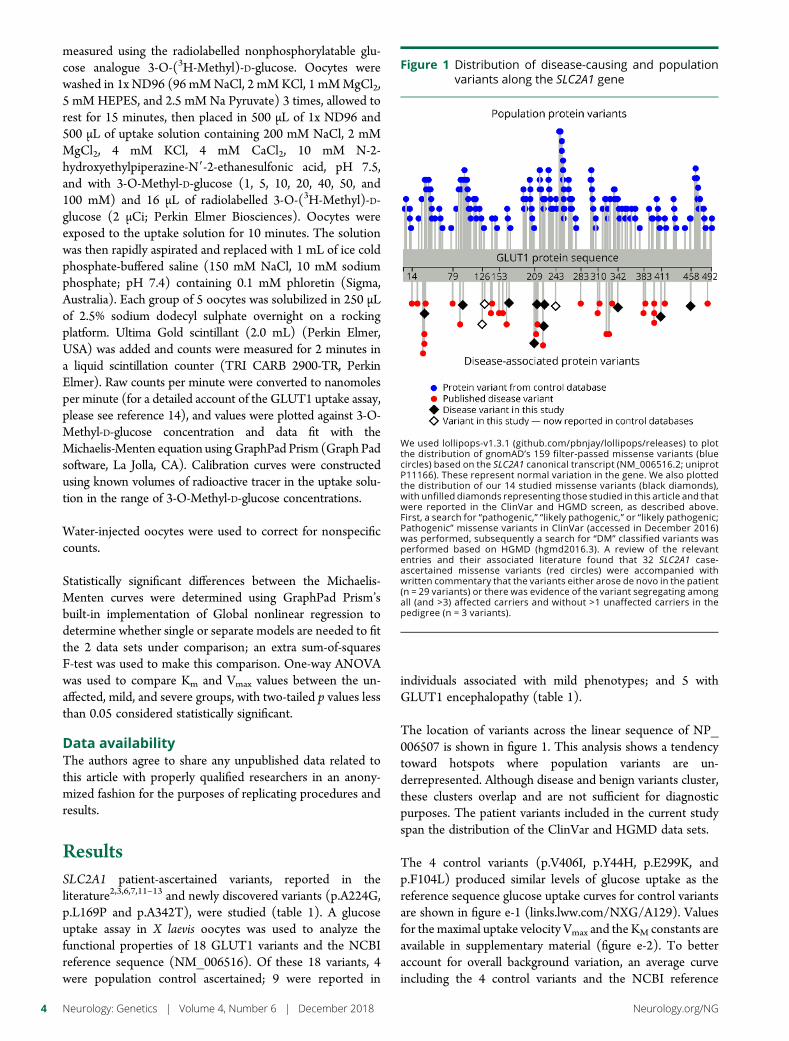
measured using the radiolabelled nonphosphorylatable glu-cose analogue 3-O-(3H-Methyl)-D-glucose. Oocytes werewashed in 1x ND96 (96 mMNaCl, 2 mMKCl, 1 mMMgCl2,5 mM HEPES, and 2.5 mM Na Pyruvate) 3 times, allowed torest for 15 minutes, then placed in 500 μL of 1x ND96 and500 μL of uptake solution containing 200 mM NaCl, 2 mMMgCl2, 4 mM KCl, 4 mM CaCl2, 10 mM N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid, pH 7.5,and with 3-O-Methyl-D-glucose (1, 5, 10, 20, 40, 50, and100 mM) and 16 μL of radiolabelled 3-O-(3H-Methyl)-D-glucose (2 μCi; Perkin Elmer Biosciences). Oocytes wereexposed to the uptake solution for 10 minutes. The solutionwas then rapidly aspirated and replaced with 1 mL of ice coldphosphate-buffered saline (150 mM NaCl, 10 mM sodiumphosphate; pH 7.4) containing 0.1 mM phloretin (Sigma,Australia). Each group of 5 oocytes was solubilized in 250 μLof 2.5% sodium dodecyl sulphate overnight on a rockingplatform. Ultima Gold scintillant (2.0 mL) (Perkin Elmer,USA) was added and counts were measured for 2 minutes ina liquid scintillation counter (TRI CARB 2900-TR, PerkinElmer). Raw counts per minute were converted to nanomolesper minute (for a detailed account of the GLUT1 uptake assay,please see reference 14), and values were plotted against 3-O-Methyl-D-glucose concentration and data fit with theMichaelis-Menten equation usingGraphPad Prism (Graph Padsoftware, La Jolla, CA). Calibration curves were constructedusing known volumes of radioactive tracer in the uptake solu-tion in the range of 3-O-Methyl-D-glucose concentrations.
Water-injected oocytes were used to correct for nonspecificcounts.
Statistically significant differences between the Michaelis-Menten curves were determined using GraphPad Prism’sbuilt-in implementation of Global nonlinear regression todetermine whether single or separate models are needed to fitthe 2 data sets under comparison; an extra sum-of-squaresF-test was used to make this comparison. One-way ANOVAwas used to compare Km and Vmax values between the un-affected, mild, and severe groups, with two-tailed p values lessthan 0.05 considered statistically significant.
Data availabilityThe authors agree to share any unpublished data related tothis article with properly qualified researchers in an anony-mized fashion for the purposes of replicating procedures andresults.
ResultsSLC2A1 patient-ascertained variants, reported in theliterature2,3,6,7,11–13 and newly discovered variants (p.A224G,p.L169P and p.A342T), were studied (table 1). A glucoseuptake assay in X laevis oocytes was used to analyze thefunctional properties of 18 GLUT1 variants and the NCBIreference sequence (NM_006516). Of these 18 variants, 4were population control ascertained; 9 were reported in
individuals associated with mild phenotypes; and 5 withGLUT1 encephalopathy (table 1).
The location of variants across the linear sequence of NP_006507 is shown in figure 1. This analysis shows a tendencytoward hotspots where population variants are un-derrepresented. Although disease and benign variants cluster,these clusters overlap and are not sufficient for diagnosticpurposes. The patient variants included in the current studyspan the distribution of the ClinVar and HGMD data sets.
The 4 control variants (p.V406I, p.Y44H, p.E299K, andp.F104L) produced similar levels of glucose uptake as thereference sequence glucose uptake curves for control variantsare shown in figure e-1 (links.lww.com/NXG/A129). Valuesfor the maximal uptake velocity Vmax and the KM constants areavailable in supplementary material (figure e-2). To betteraccount for overall background variation, an average curveincluding the 4 control variants and the NCBI reference
Figure 1 Distribution of disease-causing and populationvariants along the SLC2A1 gene
We used lollipops-v1.3.1 (github.com/pbnjay/lollipops/releases) to plotthe distribution of gnomAD’s 159 filter-passed missense variants (bluecircles) based on the SLC2A1 canonical transcript (NM_006516.2; uniprotP11166). These represent normal variation in the gene. We also plottedthe distribution of our 14 studied missense variants (black diamonds),with unfilled diamonds representing those studied in this article and thatwere reported in the ClinVar and HGMD screen, as described above.First, a search for “pathogenic,” “likely pathogenic,” or “likely pathogenic;Pathogenic” missense variants in ClinVar (accessed in December 2016)was performed, subsequently a search for “DM” classified variants wasperformed based on HGMD (hgmd2016.3). A review of the relevantentries and their associated literature found that 32 SLC2A1 case-ascertained missense variants (red circles) were accompanied withwritten commentary that the variants either arose de novo in the patient(n = 29 variants) or there was evidence of the variant segregating amongall (and >3) affected carriers and without >1 unaffected carriers in thepedigree (n = 3 variants).
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
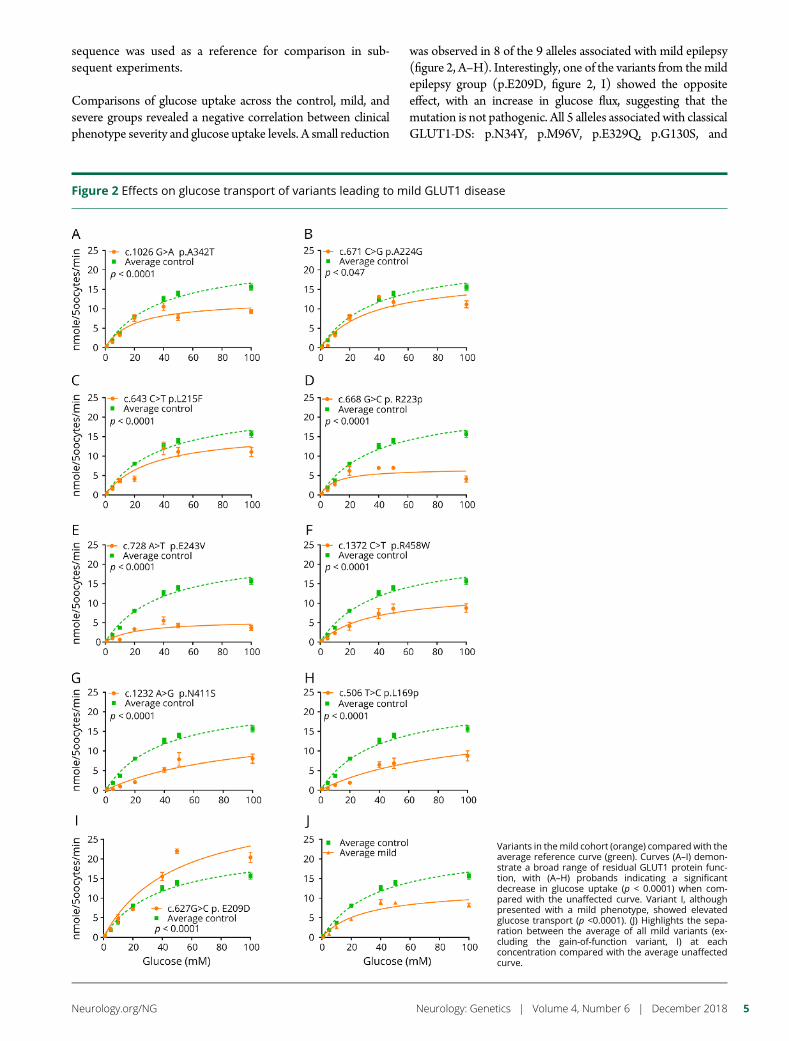
sequence was used as a reference for comparison in sub-sequent experiments.
Comparisons of glucose uptake across the control, mild, andsevere groups revealed a negative correlation between clinicalphenotype severity and glucose uptake levels. A small reduction
was observed in 8 of the 9 alleles associated with mild epilepsy(figure 2, A–H). Interestingly, one of the variants from themildepilepsy group (p.E209D, figure 2, I) showed the oppositeeffect, with an increase in glucose flux, suggesting that themutation is not pathogenic. All 5 alleles associated with classicalGLUT1-DS: p.N34Y, p.M96V, p.E329Q, p.G130S, and
Figure 2 Effects on glucose transport of variants leading to mild GLUT1 disease
Variants in themild cohort (orange) compared with theaverage reference curve (green). Curves (A–I) demon-strate a broad range of residual GLUT1 protein func-tion, with (A–H) probands indicating a significantdecrease in glucose uptake (p < 0.0001) when com-pared with the unaffected curve. Variant I, althoughpresented with a mild phenotype, showed elevatedglucose transport (p <0.0001). (J) Highlights the sepa-ration between the average of all mild variants (ex-cluding the gain-of-function variant, I) at eachconcentration compared with the average unaffectedcurve.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5
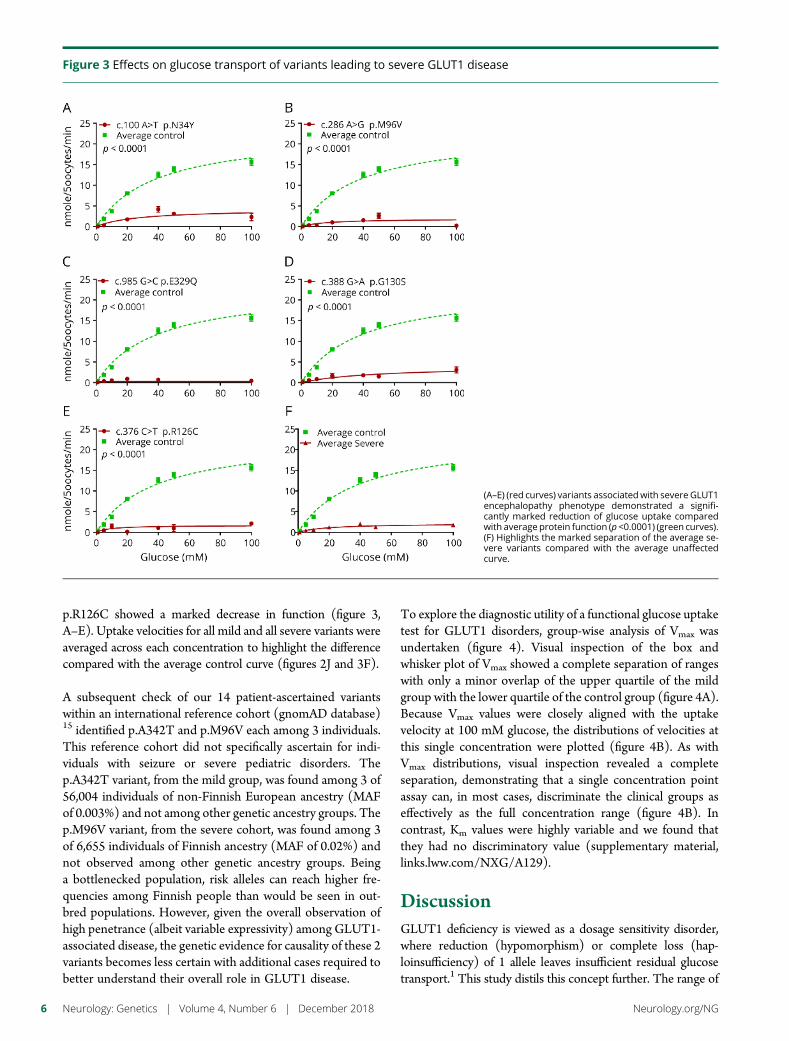
p.R126C showed a marked decrease in function (figure 3,A–E). Uptake velocities for all mild and all severe variants wereaveraged across each concentration to highlight the differencecompared with the average control curve (figures 2J and 3F).
A subsequent check of our 14 patient-ascertained variantswithin an international reference cohort (gnomAD database)15 identified p.A342T and p.M96V each among 3 individuals.This reference cohort did not specifically ascertain for indi-viduals with seizure or severe pediatric disorders. Thep.A342T variant, from the mild group, was found among 3 of56,004 individuals of non-Finnish European ancestry (MAFof 0.003%) and not among other genetic ancestry groups. Thep.M96V variant, from the severe cohort, was found among 3of 6,655 individuals of Finnish ancestry (MAF of 0.02%) andnot observed among other genetic ancestry groups. Beinga bottlenecked population, risk alleles can reach higher fre-quencies among Finnish people than would be seen in out-bred populations. However, given the overall observation ofhigh penetrance (albeit variable expressivity) among GLUT1-associated disease, the genetic evidence for causality of these 2variants becomes less certain with additional cases required tobetter understand their overall role in GLUT1 disease.
To explore the diagnostic utility of a functional glucose uptaketest for GLUT1 disorders, group-wise analysis of Vmax wasundertaken (figure 4). Visual inspection of the box andwhisker plot of Vmax showed a complete separation of rangeswith only a minor overlap of the upper quartile of the mildgroup with the lower quartile of the control group (figure 4A).Because Vmax values were closely aligned with the uptakevelocity at 100 mM glucose, the distributions of velocities atthis single concentration were plotted (figure 4B). As withVmax distributions, visual inspection revealed a completeseparation, demonstrating that a single concentration pointassay can, in most cases, discriminate the clinical groups aseffectively as the full concentration range (figure 4B). Incontrast, Km values were highly variable and we found thatthey had no discriminatory value (supplementary material,links.lww.com/NXG/A129).
DiscussionGLUT1 deficiency is viewed as a dosage sensitivity disorder,where reduction (hypomorphism) or complete loss (hap-loinsufficiency) of 1 allele leaves insufficient residual glucosetransport.1 This study distils this concept further. The range of
Figure 3 Effects on glucose transport of variants leading to severe GLUT1 disease
(A–E) (red curves) variants associatedwith severe GLUT1encephalopathy phenotype demonstrated a signifi-cantly marked reduction of glucose uptake comparedwith average protein function (p <0.0001) (green curves).(F) Highlights the marked separation of the average se-vere variants compared with the average unaffectedcurve.
6 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

missense variants chosen for this study represents the extremesof the GLUT1 deficiency syndrome disease spectrum. Thespectrum of illness associated with GLUT1 deficiency is ex-tremely broad, ranging from disabled individuals with classicalencephalopathy needing lifelong care to high-functioningindividuals with mild, well-controlled epilepsy and movementdisorder. In this study, we compare variants reported to alwayscause severe disease with those reported to always cause milddisease and demonstrate that there is indeed a difference in theresidual function of the mutant protein that predicts thatphenotypic difference. Based on this sampling of variants, weshow clear separation in the distributions of Vmax of variants ineach group, providing a basis for a diagnostic test.
The potential diagnostic value of our approach is highlightedby the analysis of the p.E209D variant. Our GLUT1 uptakeanalysis revealed that p.E209D is unlikely to be pathogenicdespite its absence in population variant databases of normalvariation and affecting a highly conserved amino acid. Wesuggest that a combination of genetic evaluation and func-tional testing is required for improved diagnosis in GLUT1deficiency.
Variable expressivity can be seen in GLUT1 disease, sug-gesting the contribution of additional factors. This is fre-quently observed in familial GLUT1 deficiency wherea spectrum of clinical presentations from unaffected carriersthrough to mild epilepsy or dyskinesia with normal intellect torefractory epilepsy and intellectual disability are seen.3,11,12
Variants from such families were excluded from this analysisbecause it was not possible to assign them to a particular se-verity group. These families highlight the need for futurestudies determining other genetic and environmental influ-ences on disease severity in GLUT1 deficiency.
We show that a simplification of the Xenopus oocyte assay canprovide diagnostic value that is equivalent to complete de-termination of Vmax and Km. Our data show that a singleconcentration of glucose approaching saturation (100 mM/L)is sufficient to distinguish between control, mild, and severecases. The addition of this simple functional test to clinical andgenetic findings has the potential to improve the classificationof patient-ascertained variants, as well as allow rapid
intervention when the functional data suggest risk of seriousdisease. The diagnosis of GLUT1 deficiency syndrome wasinitially based on the combination of seizure phenotype andhypoglycorrhachia.1 After the discovery of SLC2A1 mutations,molecular testing has become routine.1,2 In addition to se-quencing, a well-validated clinical test of GLUT1 function iscurrently available—the red cell glucose uptake assay.12 Thisassay is, however, not available in many centers and requiresfresh, metabolically functional red cells, which can be difficult totransport over distance. The Xenopus oocyte assay used hererequires sequencing data but no other tissue, removing thedifficulty of transport. Prenatal exome screening looking for denovomutations in known disease genes is available and likely tobecome increasingly common.13 The assay used here is possi-ble within the time frames needed to enable a genetic counselorto discuss findings with families after fetal genetic screening.Overall, a simplified Xenopus oocyte assay offers a comple-mentary, sequence-based test for GLUT1 variants of unknownsignificance. The assay helps discriminate background fromcausal alleles and can further give information on anticipatedseverity of disease. We believe that this is an important steptoward multidomain data and advanced pattern recognitionanalysis such as machine learning that could be used to in-tegrate data from clinical, genetic, molecular, structural, andfunctional assays to make faster and more informed diagnosis.
Author contributionsConception and design: S.M. Zaman, S.A. Mullen, S. Maljevic,E.V. Gazina, A.M. Phillips, M.S. Hildebrand, S.F. Berkovic, I.E.Scheffer, C.A. Reid, and S. Petrou. Acquisition and analysis:S.M. Zaman, S.A. Mullen, S. Petrovski, S. Maljevic, E.V. Gazina,A.M. Phillips, G.D. Jones, M.S. Hildebrand, J. Damiano, S.Auvin, H. Lerche, Y.G. Weber, S.F. Berkovic, I.E. Scheffer, C.A.Reid, and S. Petrou. Drafting of the manuscript: S.M. Zaman,S.A. Mullen, S. Petrovski, G.D. Jones, C.A. Reid, and S. Petrou.
AcknowledgmentThe authors thank all the patients and their families forparticipating in our research program.
Study fundingThis work was supported by the National Health and MedicalResearch Council (NHMRC) Program Grant (10915693) to
Figure 4 Comparison of glucose transport across variants seen in the population, mild disease, and severe disease
(A) Box and whisker plot for Vmax values obtained from theMichaelis-Menten curves for the unaffected (n = 5), mild (n =9), and severe (n = 5) variants (single value scatter plotsshown in figure e-1, links.lww.com/NXG/A129). (B) Box andwhisker plot for the velocity at 100 mM glucose concentra-tion for each group of variants. The variant resulting in NP_006507.2 p.E209D has been positioned as an outlier and notincluded in the population analysis.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 7

S.P., I.ES., S.F.B., and C.A.R., Research Fellowship to S.P.,a Postdoctoral Training Fellowship to S.A.M., and an Aus-tralian Postgraduate Award to S.M.Z.. C.A.R is supported bya Dowd Fellowship. The Florey Institute of Neuroscience andMental Health is supported by Victorian State Governmentinfrastructure funds.
DisclosureSasha Zaman has received travel funding in the form of theCaitlin Fund Scholarship; has received governmental researchsupport from the Australian Postgraduate Award Scholarship;and has received academic entity research support from theFlorey Institute of Neuroscience and Mental Health. Saul AMullen has received speaker honoraria from UCB Pharma;has been employed with the Florey Institute of Neuroscienceand Mental Health, Australia Austin Health, and EasternHealth; and has received governmental research support inthe form of the NHMRC Early Career Fellowship and theNHMRC Project Grant. Slave Petrovski serves on the advi-sory board of Pairnomix; has served on the editorial board ofEpilepsia; and has been employed by AstraZeneca. SnezanaMaljevic reports no disclosures. Elena Gazina has receivedgovernmental research support from the National Health andMedical Research Council. A. Marie Phillips has receivedgovernmental research support from the Australian Govern-ment. Gabriel Davis Jones reports no disclosures. MichaelHildebrand has received governmental research support fromthe National Health and Medical Research Council and theNational Health and Medical Research Council. John Dam-iano reports no disclosures. Stephane Auvin has served on thescientific advisory boards of Nutricia and Ultragenyx; hasreceived travel funding or speaker honoraria from Nutricia;and serves on the editorial boards of Epilepsia and EuropeanJournal of Paediatric Neurology. Holger Lerche has served onthe scientific advisory boards of Eisai, UCB, Bial, Telethon,and BioMarin; has received honoraria for speaking on edu-cational activities from Desitin, Esai, UCB, and Bial; serves onthe editorial board of Epilepsia; has received governmentalresearch support from EpiPGX, the Federal Ministry of Ed-ucation and Research (BMBF), and the German ResearchFoundation (Deutsche Forschungsgemeinschaft); and hasreceived foundation/society research support from the HertieFoundation. Yvonne Weber has served on the scientific ad-visory boards of nonprofit entities UCB, Desitin, and EisaiPharma; and has received honoraria from UCB Pharma andfor workshop participation from Eisai and Desitin. SamuelBerkovic has served on the scientific advisory boards of UCBPharma and Eisai; has served on the editorial boards of Brain,Epileptic Disorders, and Lancet Neurology; is one of theinventors listed on a patent held by Bionomics Inc.(WO2006/133508) and is one of the inventors on pendingpatent WO61/010176; has received commercial researchsupport from UCB, SciGen, and Eisai; and has receivedgovernmental research support from the National Health andMedical Research Council of Australia and NINDS. Ingrid E.Scheffer has served on the scientific advisory boards ofNutricia, UCB, and BioMarin; has received travel funding or
speaker honoraria from Zogenix, GSK, Eisai, BioMarin,Athena Diagnostics, and the National Research Foundation inSingapore; has served on the editorial boards of Neurology,Epilepsy Currents, Epileptic Disorders, the Progress in EpilepticDisorders series, and Virtual Neuro Centre; has receivedpatent revenue for diagnostic and therapeutic methods forEFMR and a diagnostic method for epilepsy; holds issuedpatents for methods of treatment and diagnosis of epilepsy bydetecting mutations in the SCN1A gene, a diagnostic methodfor epilepsy (also published as Methods for the Diagnosis andTreatment of Epilepsy), Mutations in Ion Channels, andDiagnostic and Treatment Methods Relating to AutosomalDominant Nocturnal Frontal Lobe Epilepsy; holds pendingpatents for a gene and mutations thereof associated withseizure and movement disorders, and diagnostic and thera-peutic methods for EFMR; has served as a consultant to OvidTherapeutics and UCB; participated in the Epilepsy DrugConsortium as investigator for GW Pharmaceuticals; has re-ceived governmental research support from the NationalHealth and Medical Research Foundation (Australia), theCentre for Research Excellence Grant, the Targeted Call forResearch into Preparing Australia for the Genomics Revolu-tion in Health Care 2016-2020, the National Institute ofHealth’s CentresWithoutWalls, theMedical Research FutureFund, and the Health Research Council (New Zealand);has received academic research support from the Univer-sity of Melbourne School of Health Sciences; has receivedFoundation/Society research support from the March OfDimes Foundation, the Queensland Emergency MedicineResearch Foundation, and the Rebecca L Cooper MedicalResearch Foundation; has received royalty payments fortechnology/inventions for Diagnostic and Therapeutic Meth-ods for EFMR (Epilepsy and Mental Retardation Limited toFemales). Christopher Reid has served on the editorial board ofJournal of Neurochemical Research; and has received govern-mental research support from the National Health andMedicalResearch Council. Steven Petrou has served on scientific ad-visory boards of Pairnomix Inc., RogCon, and Praxis PrecisionMedicines; serves on the editorial boards of PLOS Genetics andNeurobiology of Disease; has served as a consultant for RogConand Praxis; has received ARC, NHMRC, and ARC Centre ofExcellence for Integrative Brain Function (CIBF); has re-ceived academic research support from the University ofMelbourne; has received Foundation/Society research sup-port from the DHB Foundation and the SCN2A ResearchFoundation; and holds stock/stock options from Pairnomix,Praxis Precision Medicine, and RogCon. Full disclosureform information provided by the authors is available withthe full text of this article at Neurology.org/NG.
Publication historyReceived by Neurology: Genetics April 3, 2018. Accepted in final formAugust 9, 2018.
References1. De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI.
Defective glucose transport across the blood-brain barrier as a cause of persistenthypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991;325:703–709.
8 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

2. Leen WG, Klepper J, Verbeek MM, et al. Glucose transporter-1 deficiency syndrome:the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010;133:655–670.
3. Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies withwidely variable onset are a key feature of familial GLUT1 deficiency. Neurology 2010;75:432–440.
4. Suls A, Mullen SA, Weber YG, et al. Early-onset absence epilepsy caused by mutationsin the glucose transporter GLUT1. Ann Neurol 2009;66:415–419.
5. Mullen SA,Marini C, Suls A, et al. Glucose transporter 1 deficiency as a treatable causeof myoclonic astatic epilepsy. Arch Neurol 2011;68:1152–1155.
6. Arsov T, Mullen SA, Rogers S, et al. Glucose transporter 1 deficiency in the idiopathicgeneralized epilepsies. Ann Neurol 2012;72:807–815.
7. Wolking S, Becker F, Bast T, et al. Focal epilepsy in Glucose transporter type 1(Glut1) defects: case reports and a review of literature. J Neurol 2014;261:1881–1886.
8. Arsov T, Mullen SA, Damiano JA, et al. Early onset absence epilepsy: 1 in 10 cases iscaused by GLUT1 deficiency. Epilepsia 2012;53:e204–e207.
9. Schneider SAS, Paisan-Ruiz C, Garcia-Gorostiaga I, et al.. GLUT1 gene mutationscause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord 2009;24:S106.
10. Weber YG, Storch A, Wuttke TV, et al. GLUT1 mutations are a cause of paroxysmalexertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J ClinInvest 2008;118:2157–2168.
11. Suls A, Dedeken P, Goffin K, et al. Paroxysmal exercise-induced dyskinesia andepilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1.Brain 2008;131:1831–1844.
12. Yang H, Wang D, Engelstad K, et al. Glut1 deficiency syndrome and erythrocyteglucose uptake assay. Ann Neurol 2011;70:996–1005.
13. Van den Veyver IB, Eng CM. Genome-wide sequencing for prenatal detection of fetalsingle-gene disorders. Cold Spring Harb Perspect Med 2015;5:a023077.
14. Gould GW, Thomas MT, Jess TJ, et al. Expression of human glucose transporters inXenopus oocytes: kinetic characterization and substrate specificities of the erythro-cyte, liver, and brain isoforms. Biochemistry 1991;30:5139–5145.
15. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variationin 60,706 humans. Nature 2016;536:285–291.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 9

ARTICLE OPEN ACCESS
Leigh syndrome followed by parkinsonism in anadult with homozygous c.626C>T mutation inMTFMTDimitri M. Hemelsoet, MD, Arnaud V. Vanlander, MD, PhD, Joel Smet, BSc, Elise Vantroys, MSc,
Marjan Acou, MD, Ingeborg Goethals, MD, PhD, Tom Sante, MSc, PhD, Sara Seneca, MD, PhD,
Bjorn Menten, MSc, PhD, and Rudy Van Coster, MD, PhD
Neurol Genet 2018;4:e298. doi:10.1212/NXG.0000000000000298
Correspondence
Dr. Hemelsoet
AbstractObjectiveTo report the clinical, radiologic, biochemical, and molecular characteristics in a 46-year-oldparticipant with adult-onset Leigh syndrome (LS), followed by parkinsonism.
MethodsCase description with diagnostic workup included blood and CSF analysis, skeletal muscleinvestigations, blue native polyacrylamide gel electrophoresis, whole exome sequencing tar-geting nuclear genes involved in mitochondrial transcription and translation, cerebral MRI,123I-FP-CIT brain single-photon emission computed tomography (SPECT), and C-11raclopride positron emission tomography (PET).
ResultsThe participant was found to have a defect in the oxidative phosphorylation caused bya c.626C>T mutation in the gene coding for mitochondrial methionyl-tRNA formyltransferase(MTFMT), which is a pathogenic mutation affecting intramitochondrial protein translation.The proband had a normal concentration of lactate in blood and no abnormal microscopicfindings in skeletal muscle. Cerebral MRI showed bilateral lesions in the striatum, mesen-cephalon, pons, and medial thalamus. Lactate concentration in CSF was increased. FP-CITSPECT and C-11 raclopride PET demonstrated a defect in the dopaminergic system.
ConclusionsWe report on a case with adult-onset LS related to a MTFMT mutation. Two years after theonset of symptoms of LS, the proband developed a parkinson-like disease. The c.626C>Tmutation is the most common pathogenic mutation found in 22 patients reported earlier in theliterature with a defect inMTFMT. The age of the previously reported cases varied between 14months and 24 years. Our report expands the phenotypical spectrum of MTFMT-relatedneurologic disease and provides clinical evidence for involvement ofMTFMT in extrapyramidalsyndromes.
From the Department of Neurology (D.M.H.), Ghent University Hospital; Department of Pediatrics (A.V.V., J.S., E.V., R.V.C.), Division of Pediatric Neurology and Metabolism, GhentUniversity Hospital; Department of Radiology (M.A.), Ghent University Hospital; Department of Nuclear Medicine (I.G.), Ghent University Hospital; Center for Medical Genetics Ghent(T.S., B.M.), Ghent University, Belgium; and Center for Medical Genetics (S.S.), UZ Brussel and Reproduction Genetics and Regenerative Medicine, Vrije Universiteit Brussel, Brussels,Belgium.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1

Leigh syndrome (LS) is a devastating neurometabolic disorderoccurring mainly in infants and young children. Affected childreninitially develop normally but present between the age of 2 and 9months with signs of motor regression, weakness, hypotonia, weakcry, and failure to thrive. Shortly thereafter, signs of brainstemdysfunction are seen, including respiratory pattern abnormalities,nystagmus, ptosis, and ophthalmoparesis. Other neurologic man-ifestations include pyramidal tract signs, ataxia, dystonia, tremor,and seizures, eventually progressing to death, usually within 2 yearsafter onset of symptoms. Pathologic hallmarks of the disease arebilaterally symmetrical foci characterized by spongiform necrosiswith capillary proliferation in the midbrain and striatum.1,2
The incidence is 1 in 40,000.3 Rarely, the onset of symptomsis in late childhood, adolescence, or adulthood.
Gene defects associated with LS are located in mitochondrialor nuclear genes involved in the biosynthesis of oxidativephosphorylation (OXPHOS) complexes I, II, III, IV, and V, orinvolved in the biosynthesis of coenzyme Q or pyruvate de-hydrogenase complex. Mutations in the gene coding for mi-tochondrial methionyl-tRNA formyltransferase (MTFMT)were reported earlier as the cause of LS.4–6
In this study, we describe a patient with adult-onset LS caused bya homozygous pathogenic mutation in MTFMT. Our observa-tion broadens the clinical spectrum of MTFMT-related disease.
MethodsCase description with a diagnostic workup included resultsfrom blood and CSF analysis, skeletal muscle investigations,blue native polyacrylamide gel electrophoresis (BN-PAGE),whole exome sequencing (WES) targeting nuclear genes in-volved in mitochondrial transcription and translation, brainMRI(bMRI), 123I-FP-CIT brain single-photon emission computedtomography (SPECT), and C-11 raclopride positron emissiontomography (PET). Written informed consent for research wasobtained from the participant and her guardian.
Data availabilityAnonymized data will be shared by request from any qualifiedinvestigator.
Case descriptionClinical historyA 44-year-old woman was admitted to the department ofneurology after 1 week of dizziness, vomiting, somnolence,
and subfebrility (37.8°C). Before admission, she had complainedof double vision. Divergent eye motility was observed intermit-tently. Her medical history revealed mild cognitive impairment,early-onset bilateral sensorineural hearing loss, autism spectrumdisorder, obsessive-compulsive behavior, depression, osteoporo-sis, obesity, and mild chronic obstructive pulmonary disease.Vitamin B12 hypovitaminosis was diagnosed in the past as thecause of pernicious anemia. Neurologic examination on admis-sion to the hospital was limited because of poor cooperation andsomnolence. Slight nuchal rigidity, gait ataxia, hyperreflexia,plantar reflexes in extension, and absence of sensorimotor dys-function were noticed. Neuroophthalmologic examinationshowed upward gaze palsy, downward gaze paresis, bilateral mi-osis not reacting to light and preserved convergence, and ability toabduct the eyes. Coordination could not be tested because ofinsufficient cooperation. Urinary retention was detected on ar-rival. In the 3 weeks after admission, a deterioration of gait andbalance was seen, as well as lack of initiative. Slight speech diffi-culties (hypophonia and tachylalia) were detected, as well as slowmovements. Differential diagnosis included Wernicke encepha-lopathy, acute demyelinating disease, and LS. Acute stroke wasexcluded with acute brain CT. IV vitamin B-complex, includingthiamin, was started but did not change symptomswithin the firstdays. Later on, her clinical condition stabilized. Gait, balance, andspeech recovered slowly over a period of 1 month, althougha high-pitched tachylalia remained present and appeared to bealready known before admission. Gradual improvement of dip-lopia and bladder dysfunction was seen, although the sensori-neural hearing loss was worsening.
Technical investigationsbMRI showed nonacute lacunar or cystic T2-hyperintenselesions in the basal ganglia (caudate nucleus and putamen)and remarkable acute symmetrical fluid attenuated inversionrecovery (FLAIR)-/T2-hyperintense lesions in the mesen-cephalon, suggestive of Leigh disease (figure, A and B).Follow-up bMRI 1 month later showed that brainstem lesionswere more pronounced in the mesencephalon and pons,extending into the medial thalamus bilaterally. bMRI 3months after symptom onset showed tissue loss and gliosis,with reduced FLAIR hyperintense signals in the mesence-phalic area, in both thalami and new lesions in the middlecerebellar peduncles (figure, C and D). CSF analysis on thefirst admission was normal, except for increased lactate con-centration (38 mg/dL, normal 10–22), confirmed a few dayslater (33 mg/dL). Serum lactate, vitamin B12, and thiaminwere normal. Ophthalmologic examination showed abnormaleye motility but no signs of optic atrophy or of retinitis pig-mentosa. Ultrasound examination of the heart revealed mild
GlossarybMRI = brain magnetic resonance imaging; BN-PAGE = blue native polyacrylamide gel electrophoresis; EPS = extrapyramidalsymptom; FLAIR = fluid attenuated inversion recovery; FP-CIT = N-(3-fluoropropyl)-2beta-carbomethoxy-3beta-(4-iodophenyl)nortropane; LS = Leigh syndrome; OXPHOS = oxidative phosphorylation; PD = Parkinson disease; PET =positron emission tomography; SPECT = single-photon emission computed tomography; WES = whole exome sequencing.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG
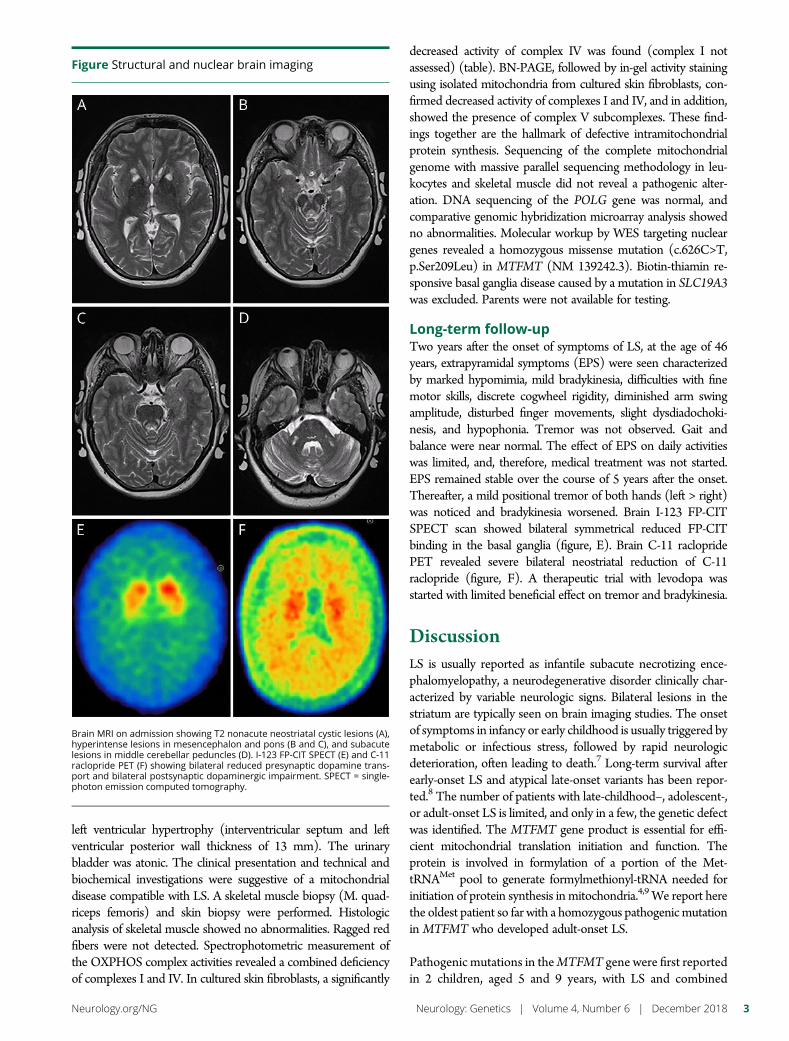
left ventricular hypertrophy (interventricular septum and leftventricular posterior wall thickness of 13 mm). The urinarybladder was atonic. The clinical presentation and technical andbiochemical investigations were suggestive of a mitochondrialdisease compatible with LS. A skeletal muscle biopsy (M. quad-riceps femoris) and skin biopsy were performed. Histologicanalysis of skeletal muscle showed no abnormalities. Ragged redfibers were not detected. Spectrophotometric measurement ofthe OXPHOS complex activities revealed a combined deficiencyof complexes I and IV. In cultured skin fibroblasts, a significantly
decreased activity of complex IV was found (complex I notassessed) (table). BN-PAGE, followed by in-gel activity stainingusing isolated mitochondria from cultured skin fibroblasts, con-firmed decreased activity of complexes I and IV, and in addition,showed the presence of complex V subcomplexes. These find-ings together are the hallmark of defective intramitochondrialprotein synthesis. Sequencing of the complete mitochondrialgenome with massive parallel sequencing methodology in leu-kocytes and skeletal muscle did not reveal a pathogenic alter-ation. DNA sequencing of the POLG gene was normal, andcomparative genomic hybridization microarray analysis showedno abnormalities. Molecular workup by WES targeting nucleargenes revealed a homozygous missense mutation (c.626C>T,p.Ser209Leu) in MTFMT (NM 139242.3). Biotin-thiamin re-sponsive basal ganglia disease caused by a mutation in SLC19A3was excluded. Parents were not available for testing.
Long-term follow-upTwo years after the onset of symptoms of LS, at the age of 46years, extrapyramidal symptoms (EPS) were seen characterizedby marked hypomimia, mild bradykinesia, difficulties with finemotor skills, discrete cogwheel rigidity, diminished arm swingamplitude, disturbed finger movements, slight dysdiadochoki-nesis, and hypophonia. Tremor was not observed. Gait andbalance were near normal. The effect of EPS on daily activitieswas limited, and, therefore, medical treatment was not started.EPS remained stable over the course of 5 years after the onset.Thereafter, a mild positional tremor of both hands (left > right)was noticed and bradykinesia worsened. Brain I-123 FP-CITSPECT scan showed bilateral symmetrical reduced FP-CITbinding in the basal ganglia (figure, E). Brain C-11 raclopridePET revealed severe bilateral neostriatal reduction of C-11raclopride (figure, F). A therapeutic trial with levodopa wasstarted with limited beneficial effect on tremor and bradykinesia.
DiscussionLS is usually reported as infantile subacute necrotizing ence-phalomyelopathy, a neurodegenerative disorder clinically char-acterized by variable neurologic signs. Bilateral lesions in thestriatum are typically seen on brain imaging studies. The onsetof symptoms in infancy or early childhood is usually triggered bymetabolic or infectious stress, followed by rapid neurologicdeterioration, often leading to death.7 Long-term survival afterearly-onset LS and atypical late-onset variants has been repor-ted.8 The number of patients with late-childhood–, adolescent-,or adult-onset LS is limited, and only in a few, the genetic defectwas identified. The MTFMT gene product is essential for effi-cient mitochondrial translation initiation and function. Theprotein is involved in formylation of a portion of the Met-tRNAMet pool to generate formylmethionyl-tRNA needed forinitiation of protein synthesis in mitochondria.4,9 We report herethe oldest patient so far with a homozygous pathogenicmutationin MTFMT who developed adult-onset LS.
Pathogenic mutations in theMTFMT gene were first reportedin 2 children, aged 5 and 9 years, with LS and combined
Figure Structural and nuclear brain imaging
Brain MRI on admission showing T2 nonacute neostriatal cystic lesions (A),hyperintense lesions in mesencephalon and pons (B and C), and subacutelesions in middle cerebellar peduncles (D). I-123 FP-CIT SPECT (E) and C-11raclopride PET (F) showing bilateral reduced presynaptic dopamine trans-port and bilateral postsynaptic dopaminergic impairment. SPECT = single-photon emission computed tomography.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
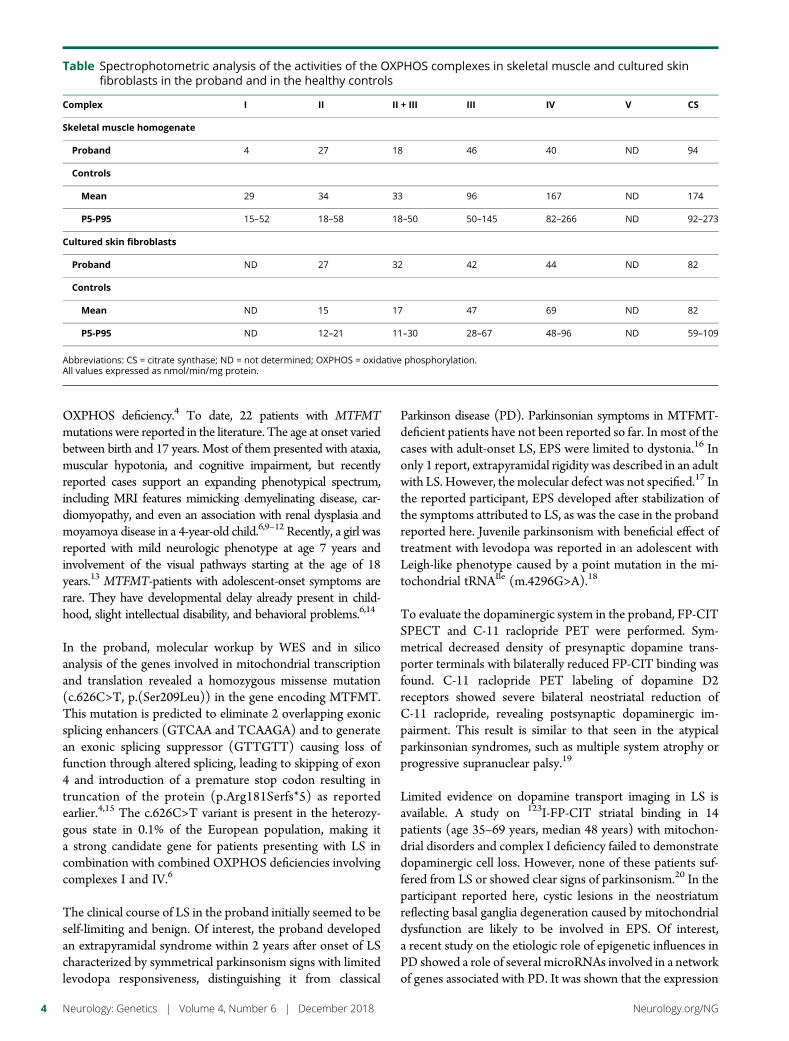
OXPHOS deficiency.4 To date, 22 patients with MTFMTmutations were reported in the literature. The age at onset variedbetween birth and 17 years. Most of them presented with ataxia,muscular hypotonia, and cognitive impairment, but recentlyreported cases support an expanding phenotypical spectrum,including MRI features mimicking demyelinating disease, car-diomyopathy, and even an association with renal dysplasia andmoyamoya disease in a 4-year-old child.6,9–12 Recently, a girl wasreported with mild neurologic phenotype at age 7 years andinvolvement of the visual pathways starting at the age of 18years.13 MTFMT-patients with adolescent-onset symptoms arerare. They have developmental delay already present in child-hood, slight intellectual disability, and behavioral problems.6,14
In the proband, molecular workup by WES and in silicoanalysis of the genes involved in mitochondrial transcriptionand translation revealed a homozygous missense mutation(c.626C>T, p.(Ser209Leu)) in the gene encoding MTFMT.This mutation is predicted to eliminate 2 overlapping exonicsplicing enhancers (GTCAA and TCAAGA) and to generatean exonic splicing suppressor (GTTGTT) causing loss offunction through altered splicing, leading to skipping of exon4 and introduction of a premature stop codon resulting intruncation of the protein (p.Arg181Serfs*5) as reportedearlier.4,15 The c.626C>T variant is present in the heterozy-gous state in 0.1% of the European population, making ita strong candidate gene for patients presenting with LS incombination with combined OXPHOS deficiencies involvingcomplexes I and IV.6
The clinical course of LS in the proband initially seemed to beself-limiting and benign. Of interest, the proband developedan extrapyramidal syndrome within 2 years after onset of LScharacterized by symmetrical parkinsonism signs with limitedlevodopa responsiveness, distinguishing it from classical
Parkinson disease (PD). Parkinsonian symptoms in MTFMT-deficient patients have not been reported so far. In most of thecases with adult-onset LS, EPS were limited to dystonia.16 Inonly 1 report, extrapyramidal rigidity was described in an adultwith LS. However, the molecular defect was not specified.17 Inthe reported participant, EPS developed after stabilization ofthe symptoms attributed to LS, as was the case in the probandreported here. Juvenile parkinsonism with beneficial effect oftreatment with levodopa was reported in an adolescent withLeigh-like phenotype caused by a point mutation in the mi-tochondrial tRNAIle (m.4296G>A).18
To evaluate the dopaminergic system in the proband, FP-CITSPECT and C-11 raclopride PET were performed. Sym-metrical decreased density of presynaptic dopamine trans-porter terminals with bilaterally reduced FP-CIT binding wasfound. C-11 raclopride PET labeling of dopamine D2receptors showed severe bilateral neostriatal reduction ofC-11 raclopride, revealing postsynaptic dopaminergic im-pairment. This result is similar to that seen in the atypicalparkinsonian syndromes, such as multiple system atrophy orprogressive supranuclear palsy.19
Limited evidence on dopamine transport imaging in LS isavailable. A study on 123I-FP-CIT striatal binding in 14patients (age 35–69 years, median 48 years) with mitochon-drial disorders and complex I deficiency failed to demonstratedopaminergic cell loss. However, none of these patients suf-fered from LS or showed clear signs of parkinsonism.20 In theparticipant reported here, cystic lesions in the neostriatumreflecting basal ganglia degeneration caused by mitochondrialdysfunction are likely to be involved in EPS. Of interest,a recent study on the etiologic role of epigenetic influences inPD showed a role of several microRNAs involved in a networkof genes associated with PD. It was shown that the expression
Table Spectrophotometric analysis of the activities of the OXPHOS complexes in skeletal muscle and cultured skinfibroblasts in the proband and in the healthy controls
Complex I II II + III III IV V CS
Skeletal muscle homogenate
Proband 4 27 18 46 40 ND 94
Controls
Mean 29 34 33 96 167 ND 174
P5-P95 15–52 18–58 18–50 50–145 82–266 ND 92–273
Cultured skin fibroblasts
Proband ND 27 32 42 44 ND 82
Controls
Mean ND 15 17 47 69 ND 82
P5-P95 ND 12–21 11–30 28–67 48–96 ND 59–109
Abbreviations: CS = citrate synthase; ND = not determined; OXPHOS = oxidative phosphorylation.All values expressed as nmol/min/mg protein.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

of MTFMT (influenced by miR-488) was downregulated inthe cingulate gyrus of patients with PD as compared tohealthy controls. Dysregulation of 1 or more of the genes inthe PD-genetic network, including MTFMT, might interferewith extrapyramidal neuronal integrity and function.21 Thefindings in the participant described here provide additionalclinical evidence for the hypothesis that MTFMT is involvedin PD-related genetic network and that MTFMT dysregula-tion might be an etiologic factor in parkinsonism.
We cannot rule out that the older age of the proband ascompared to other reported patients may have played a role inthe development of atypical parkinsonism. At present, it isunclear whether parkinsonism is part of the MTFMT phe-notype. Natural history data on patients with MTFMTmutations and advanced age are lacking. Our data suggest thatdopaminergic neurons in patients with LS caused by patho-genic MTFMT mutations may be more susceptible to PDbecause of the impaired OXPHOS system, ultimately leadingto a clinically significant extrapyramidal syndrome.
Adult-onset LS can be seen in patients with c.626C>Tmutation in MTFMT. We showed that the affected patienthad a defect in the dopaminergic system associated withatypical parkinsonism. Further research on the natural historyof patients with pathogenic MTFMT mutations is needed toclarify the full clinical spectrum and underlying physiopath-ologic mechanisms.
Author contributionsEach author listed in the manuscript has participated inediting the manuscript, has seen and approved the submissionof this version of the manuscript, and takes full responsibilityfor the manuscript. In addition, A. V. Vanlander, T. Sante,E. Vantroys, and B. Menten conducted, analyzed, and inter-preted the whole exome sequencing experiments; D. M.Hemelsoet and A. V. Vanlander acquired and analyzed phe-notypic data; J. Smet acquired and processed all patient samplesand analyzed data. A. V. Vanlander, S. Seneca, and B. Mentenperformed and interpreted Sanger sequencing. J. Smet andT. Sante contributed to study design and data analysis.M. Acouand I. Goethals were responsible for analysis of MR, PET, andSPECT imaging data and provided images for figure. D. M.Hemelsoet and R. Van Coster were responsible for primarystudy oversight and design, data acquisition, data analysisand interpretation, and primary writing of the manuscript.D. M. Hemelsoet and R. Van Coster are responsible for theoverall content of the manuscript.
AcknowledgmentsThe authors thank the patient and her caregivers for theirwillingness to participate in this study.
Study fundingA. V. Vanlander was supported by the Ghent University(Ugent, BOF, grant number 01DI2714) and the SteunfondsMarguerite-Marie Delacroix. E. Vantroys was supported by
the Ghent University (Ugent, BOF, grant number24J.2014.0005.02).
DisclosureD. M. Hemelsoet has served on the scientific advisory boardsof Pfizer and Boehringer-Ingelheim and has received com-mercial research support from Sanofi-Genzyme andShire. A. V. Vanlander and J. Smet report no disclosures.E. Vantroys has received governmental research supportfrom Ghent University. M. Acou, I. Goethals, T. Sante,S. Seneca, B. Menten, and R. Van Coster report no dis-closures. Full disclosure form information provided by theauthors is available with the full text of this article atNeurology.org/NG.
Publication historyReceived by Neurology: Genetics April 6, 2018. Accepted in final formOctober 15, 2018.
References1. Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg
Psychiatry 1951;14:216–221.2. Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and bio-
chemical and DNA abnormalities. Ann Neurol 1996;39:343–351.3. Ruhoy IS, Saneto RP. The genetics of Leigh syndrome and its implications for clinical
practice and risk management. Appl Clin Genet 2014;7:221–234.4. Tucker EJ, Hershman SG, Kohrer C, et al. Mutations in MTFMT underlie a human
disorder of formylation causing impaired mitochondrial translation. Cell Metab 2011;14:428–434.
5. Neeve VC, Pyle A, Boczonadi V, et al. Clinical and functional characterization of thecombined respiratory chain defect in two sisters due to autosomal recessive mutationsin MTFMT. Mitochondrion 2013;13:743–748.
6. Haack TB, Gorza M, Danhauser K, et al. Phenotypic spectrum of eleven patients andfive novel MTFMT mutations identified by exome sequencing and candidate genescreening. Mol Genet Metab 2014;111:342–352.
7. Baertling F, Rodenburg RJ, Schaper J, et al. A guide to diagnosis and treatment ofLeigh syndrome. J Neurol Neurosurg Psychiatry 2014;85:257–265.
8. Aulbert W, Weigt-Usinger K, Thiels C, et al. Long survival in Leigh syndrome: newcases and review of literature. Neuropediatrics 2014;45:346–353.
9. Hinttala R, Sasarman F, Nishimura T, et al. An N-terminal formyl methionine onCOX 1 is required for the assembly of cytochrome c oxidase. Hum Mol Genet 2015;24:4103–4113.
10. Pena JA, Lotze T, Yang Y, et al. Methionyl-tNA formyltransferase (MTFMT)deficiency mimicking acquired demyelinating disease. J Child Neurol 2016;31:215–219.
11. Pronicka E, Piekutowska-Abramczuk D, Ciara E, et al. New perspective in diagnosticsof mitochondrial disorders: two years’ experience with whole-exome sequencing at annational paediatric centre. J Transl Med 2016;14:174.
12. Oates A, Brennan J, Slavotinek A, Alsadah A, Chow D, LeeMM. Challenges managingend-stage renal disease and kidney transplantation in a child with MTFMT mutationand moyamoya disease. Pediatr Transplant 2016;20:1000–1003.
13. La Piana R, Weraarpachai W, Ospina LH, et al. Identification and functional char-acterization of a novelMTFMTmutation associated with selective vulnerability of thevisual pathway and a mild neurological phenotype. Neurogenetics 2017;18:97103.
14. Prasun P, Del Mar Pena L. Late onset Leigh syndrome mimicking central nervoussystem vasculitis. Mol Genet Metab Rep 2014;1:280–282.
15. Sinha A, Kohrer C, Weber MH, et al. Biochemical characterization of pathogenicmutations in human mitochondrial methionyl-tRNA formultransferase. J Biol Chem2014;289:32729–32741.
16. Finsterer J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol2008;39:223–235.
17. Maso E, Ferrer I, Herraiz J, Roquer J, Serrano S. Leigh’s syndrome in an adult.J Neurol 1984;231:253–257.
18. Martikainen M, Kytovuori L, Majamaa K. Juvenile parkinsonism, hypogonadism andLeigh-like MRI changes in a patient with m.4296G>A mutation in mitochondrialDNA. Mitochondrion 2013;13:83–86.
19. Schreckenberger M, Hagele S, Siessmeier T, et al. The dopamine D2 receptorligand 18F-desmethoxyfallypride: an appropriate fluorinated PET tracer for thedifferential diagnosis of parkinsonism. Eur J Nucl Med Mol Imaging 2004;31:1128–1135.
20. Minnerop M, Kornblum C, Joe AY, et al. Dopamine transporter SPECT in patientswith mitochondrial disorders. J Neurol Neurosurg Psychiatry 2005;76:118–120.
21. Tatura R, Kraus T, Giese A, et al. Parkinson’s disease: SNCA-, PARK2-, and LRRK2-targeting microRNAs elevated in cingulated gyrus. Parkinsonism Relat Disord 2016;33:115–121.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 5

CLINICAL/SCIENTIFIC NOTES OPEN ACCESS
Homozygous 31 trinucleotide repeats in theSCA2 allele are pathogenic for cerebellar ataxiaMaya Tojima, MD, Gaku Murakami, MD, PhD, Rie Hikawa, Hodaka Yamakado, MD, PhD,
Hirofumi Yamashita, MD, PhD, Ryosuke Takahashi, MD, PhD, and Masaru Matsui, MD, PhD
Neurol Genet 2018;4:e283. doi:10.1212/NXG.0000000000000283
Correspondence
Dr. Yamashita
or Dr. Matsui
Spinocerebellar ataxia type 2 (SCA2), an autosomal dominant cerebellar disorder belonging tothe polyglutamine (polyQ) diseases, is characterized by progressive ataxia, slow saccadic eyemovement, hyporeflexia, peripheral neuropathy, and pyramidal and extrapyramidal signs.1 Thecause of SCA2 is a CAG repeat expansion, sometimes interrupted by CAAwithin, inATXN2 onchromosome 12q24.2,3 Previous reports have shown that the presence of 33 or more hetero-zygous trinucleotide repeats is pathogenic, whereas 14 to 31 repeats is normal.3,4 We reporta case of late-onset SCA2 with homozygous alleles of 31 trinucleotide repeats in ATXN2.
Case reportAn 80-year-old woman visited our hospital with a 1-year history of slowly progressive gaitdisturbance. There was no family history of cerebellar ataxia, although her sister was diagnosedwith amyotrophic lateral sclerosis (ALS) at 68 years of age and died of respiratory failure at72 years (figure, A). Our patient had no history of excessive alcohol drinking. Neurologicexamination findings showed normal eye movement, mild dysarthria, hyporeflexia of the bi-lateral patellar and Achilles tendons, ataxia of the lower extremities, unstable standing withoutaid, and a wide-based ataxic gait. A cognitive examination was normal, with a Mini-Mental StateExamination score of 30/30. Blood test results, including thyroid function, albumin, lipids,vitamin E, tumor markers, and CSF analysis were normal. Cranial MRI revealed bilateralcerebellar atrophy, while N-isopropyl-p-(iodine-123)-iodoamphetamine SPECT showedhypoperfusion of the brainstem and bilateral cerebellar hemispheres (figure, B). Nerve con-duction study and electromyogram findings were normal.
After obtaining informed consent, genomic DNA was extracted from leukocytes. First, we am-plified the region containing the trinucleotide repeats in ATXN2 using the primer pair SCA2-A(59-GGGCCCCTCACCATGTCG-39) and SCA2-B (59-CGGGCTTGCGGACATTGG-39)and found a single amplicon slightly larger than a normal 22 trinucleotide repeat length (figure,C). Other genes for SCAs, including SCA1, 3, and 6, and DRPLA were normal (data notshown). Next, Sanger sequencing for the ATXN2 repeat region was performed, which dem-onstrated homozygous (CAG)13CAA(CAG)8CAA(CAG)8, a total of 31 trinucleotide repeats,and a chromatogram without overlap by another repeat length (figure, D). Finally, we appliedwhole exome sequencing to exclude other genetic causes of ataxia (table e-1, links.lww.com/NXG/A111) and found no causative mutation among the candidate genes except the homo-zygous expansion of trinucleotide repeats in ATXN2 (e-methods, links.lww.com/NXG/A114and table e-2, links.lww.com/NXG/A112).
From the Department of Neurology (M.T., M.M.), Otsu Red Cross Hospital; Department of Neurology (M.T., R.H., H. Yamakado, H. Yamashita, R.T.), Kyoto University Hospital,Otsu, Japan; Murakami Clinic (G.M.), Kyoto, Japan; Department of Neurology (H. Yamashita), Japanese Red Cross Wakayama Medical Center, Wakayama, Japan.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1
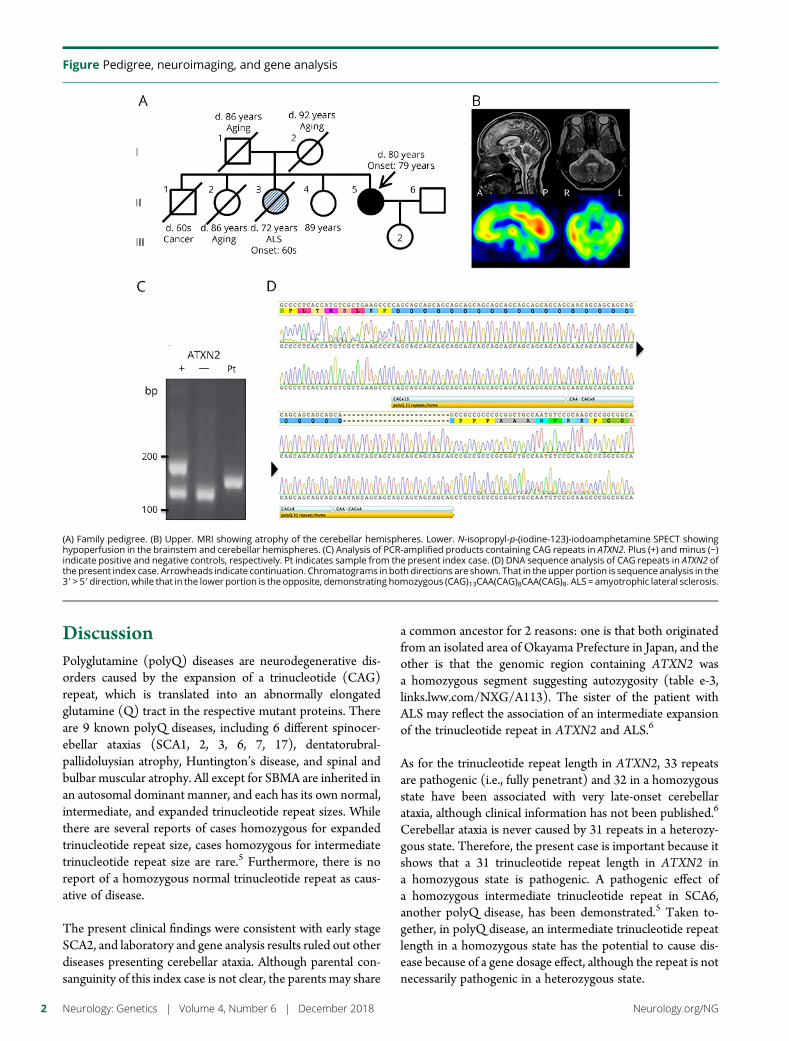
DiscussionPolyglutamine (polyQ) diseases are neurodegenerative dis-orders caused by the expansion of a trinucleotide (CAG)repeat, which is translated into an abnormally elongatedglutamine (Q) tract in the respective mutant proteins. Thereare 9 known polyQ diseases, including 6 different spinocer-ebellar ataxias (SCA1, 2, 3, 6, 7, 17), dentatorubral-pallidoluysian atrophy, Huntington’s disease, and spinal andbulbar muscular atrophy. All except for SBMA are inherited inan autosomal dominant manner, and each has its own normal,intermediate, and expanded trinucleotide repeat sizes. Whilethere are several reports of cases homozygous for expandedtrinucleotide repeat size, cases homozygous for intermediatetrinucleotide repeat size are rare.5 Furthermore, there is noreport of a homozygous normal trinucleotide repeat as caus-ative of disease.
The present clinical findings were consistent with early stageSCA2, and laboratory and gene analysis results ruled out otherdiseases presenting cerebellar ataxia. Although parental con-sanguinity of this index case is not clear, the parents may share
a common ancestor for 2 reasons: one is that both originatedfrom an isolated area of Okayama Prefecture in Japan, and theother is that the genomic region containing ATXN2 wasa homozygous segment suggesting autozygosity (table e-3,links.lww.com/NXG/A113). The sister of the patient withALS may reflect the association of an intermediate expansionof the trinucleotide repeat in ATXN2 and ALS.6
As for the trinucleotide repeat length in ATXN2, 33 repeatsare pathogenic (i.e., fully penetrant) and 32 in a homozygousstate have been associated with very late-onset cerebellarataxia, although clinical information has not been published.6
Cerebellar ataxia is never caused by 31 repeats in a heterozy-gous state. Therefore, the present case is important because itshows that a 31 trinucleotide repeat length in ATXN2 ina homozygous state is pathogenic. A pathogenic effect ofa homozygous intermediate trinucleotide repeat in SCA6,another polyQ disease, has been demonstrated.5 Taken to-gether, in polyQ disease, an intermediate trinucleotide repeatlength in a homozygous state has the potential to cause dis-ease because of a gene dosage effect, although the repeat is notnecessarily pathogenic in a heterozygous state.
Figure Pedigree, neuroimaging, and gene analysis
(A) Family pedigree. (B) Upper. MRI showing atrophy of the cerebellar hemispheres. Lower. N-isopropyl-p-(iodine-123)-iodoamphetamine SPECT showinghypoperfusion in the brainstem and cerebellar hemispheres. (C) Analysis of PCR-amplified products containing CAG repeats in ATXN2. Plus (+) and minus (−)indicate positive and negative controls, respectively. Pt indicates sample from the present index case. (D) DNA sequence analysis of CAG repeats in ATXN2 ofthe present index case. Arrowheads indicate continuation. Chromatograms in both directions are shown. That in the upper portion is sequence analysis in the39 > 59direction, while that in the lower portion is the opposite, demonstrating homozygous (CAG)13CAA(CAG)8CAA(CAG)8. ALS = amyotrophic lateral sclerosis.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

One available mechanism-based treatment for patients withpolyQ disease is reducing the levels of toxic disease-geneproducts by antisense oligonucleotides, short hairpin RNAs,or miRNAs.7 The present case suggests that in polyQ diseaseswith an intermediate to hopefully mildly expanded tri-nucleotide repeat length, the reduction of polyQ proteins by;50% should substantially mitigate the effects of the disease.
Author contributionsM. Tojima: study concept and design, acquisition, analysisand interpretation of data, and drafting the manuscript. G.Murakami: study concept and design. R. Hikawa: acquisitionand analysis of data. H. Yamakado: study concept and design,analysis and interpretation of data. H. Yamashita: study con-cept and design, analysis and interpretation of data, studysupervision, and critical revision of manuscript for intellectualcontent. R. Takahashi: study supervision and critical revisionof manuscript for intellectual content. M. Matsui: studyconcept and design, study supervision, and critical revision ofmanuscript for intellectual content.
Study fundingNo targeted funding reported.
DisclosureThe authors report no disclosures relevant to the manu-script. Full disclosure form information provided by theauthors is available with the full text of this article atNeurology.org/NG.
Received January 17, 2018. Accepted in final form September 26, 2018.
References1. Giunti P, Sabbadini G, Sweeney MG, et al. The role of the SCA2 trinucleotide repeat
expansion in 89 autosomal dominant cerebellar ataxia families. Frequency, clinical andgenetic correlates. Brain 1998;121:459–467.
2. Gispert S, Twells R, Orozco G, et al. Chromosomal assignment of the second locus forautosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat Genet1993;4:295–299.
3. Cancel G, Durr A, Didierjean O, et al. Molecular and clinical correlationsin spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet 1997;6:709–715.
4. Sequeiros J, Seneca S, Martindale J. Consensus and controversies in best practices formolecular genetic testing of spinocerebellar ataxias. Eur J Hum Genet 2010;18:1188–1195.
5. Mariotti C, Gellera C, Grisoli M, Mineri R, Castucci A, Di Donato S. Pathogenic effectof an intermediate-size SCA-6 allele (CAG)(19) in a homozygous patient. Neurology2001;57:1502–1504.
6. Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM. Amyotrophic lateral sclerosisrisk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis.JAMA Neurol 2014;71:1529–1534.
7. Keiser MS, Kordasiewicz HB, McBride JL. Gene suppression strategies for domi-nantly inherited neurodegenerative diseases: lessons from Huntington’s disease andspinocerebellar ataxia. Hum Mol Genet 2016;25:R53–R64.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

CLINICAL/SCIENTIFIC NOTES OPEN ACCESS
Variable penetrance of Andersen-Tawilsyndrome in a family with a rare missenseKCNJ2 mutationReem Deeb, MD, Aravindhan Veerapandiyan, MBBS, Rabi Tawil, MD, and Simona Treidler, MD
Neurol Genet 2018;4:e284. doi:10.1212/NXG.0000000000000284
Correspondence
Dr. Tawil
Andersen-Tawil syndrome is an autosomal dominant potassium channelopathy characterizedby episodic flaccid muscle weakness (periodic paralysis), cardiac abnormalities (ventriculararrhythmias, prolonged QT interval, and prominent U waves), and characteristic skeletalfeatures (low set ears, ocular hypertelorism, small mandible, fifth digit clinodactyly, syndactyly,short stature, scoliosis, and broad forehead).1 Mutations in the KCNJ2 gene on chromosome17q23 are found in about 60% of the patients with Andersen-Tawil syndrome.2 In the absenceof a genetic change in KCNJ2, diagnosis of Andersen-Tawil syndrome is established by thepresence of well-defined clinical findings. The distinctive clinical triad of Andersen-Tawilsyndrome is present in 60%–80% of patients with KCNJ2 mutations.1,2 KCNJ2 encodes thepore-forming subunit of an inward-rectifying potassium channel protein, Kir2.1, helping inskeletal and cardiac muscle resting membrane potential stabilization. Mutations in this genecause loss of function and dominant-negative suppression effects on the Kir2.1 protein, leadingto disruption of the cardiac and skeletal muscle excitability.2,3 KCNJ2 mutations in Andersen-Tawil syndrome affect multiple tissues and results in a wide phenotypic variability causingdiagnostic difficulties and delay.2 We describe the varied clinical characteristics of Andersen-Tawil syndrome in a Caucasian family (mother and her 2 daughters) with a rare missensemutation in KCNJ2.
The 42-year-old mother presented with a 15-year history of episodes of periodic paralysisaffecting her upper or lower extremities. In the absence of a clear diagnosis, she was treatedwith systemic steroids on multiple occasions for presumed polymyositis. At the age of34 years, during the first trimester of her first pregnancy, she had an episode of paralysisassociated with hypokalemia. The weakness recovered after potassium supplementation.Her 7-year-old daughter has episodic muscle weakness, cardiac involvement with prema-ture ventricular contractions and prominent U wave, and distinctive skeletal features whilethe younger daughter has only skeletal features. Both daughters had normal birth anddevelopment. The clinical features of our patients are described in the figure. Targetedsequencing of the KCNJ2 gene showed a heterozygous missense mutation (c. 575C>T;p.Thr192Ile) in exon 2 in the mother and both daughters. The testing was based onidentification of the same mutation in mother’s 21-year-old niece who was noted to haveprolonged QT interval during an evaluation for syncope. She also has short toes andsyndactyly, but no reported episodic weakness. The mother’s brother, an obligate carrier, isasymptomatic.
The missense mutation of p.Thr192Ile found in our patients was previously reported ina single Taiwanese family with Andersen-Tawil syndrome.4 The proband was a 35-year-oldwoman with typical clinical triad of Andersen-Tawil syndrome and fixed extremity andtruncal weakness. In addition, she was also noted to have pyramidal tract signs and major
From the Department of Neurology (R.D.), SUNY Downstate Medical Center, Brooklyn; Department of Neurology (A.V., R.T.), University of Rochester Medical Center; and Departmentof Neurology (S.T.), Stony Brook School of Medicine, NY.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1
Figure Clinical features and pedigree of patients
(A) Clinical features of patients. aLow dose extended release potassium due to history of daily muscle weakness; prophylactic acetazolamide in the peri-menstrual period as menstruation is a trigger for periodic paralysis in her. (B) Pedigree of patients. Black symbols denote family members affected withAndersen-Tawil syndrome with genetic confirmation. Patients described in this report are indicated by arrows.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

depression. Her 8-year-old son, who was also affected withAndersen-Tawil syndrome with long-QT syndrome andcharacteristic skeletal features, was identified to have learn-ing disability. Authors of the study postulated that theatypical neuropsychiatric features noted in this family ex-pand the phenotype of Andersen-Tawil syndrome.4 Suchclinical features (pyramidal tract signs, depression, cognitivedelay, and learning disability) were not noted in our patients.
The pathogenicity of the mutation p.Thr192Ile remains un-proven in vitro. This missense mutation causes replacementof the amino acid threonine by isoleucine at codon 192. Theamino acid substitution is predicted to be “probably damag-ing” by the polymorphism phenotyping tool (Polyphen-2).5
The p.Thr192 residue is located in the highly conserved re-gion of the C-terminal intracellular domain of the Kir2.1protein emphasizing its importance during evolution.4–6
Moreover, replacement of threonine at this position by an-other amino acid alanine (p.Thr192Ala) has been reported tobe causative for Andersen-Tawil syndrome.6,7 We speculatethat the heterozygous missense mutation affecting this highlyconserved region is associated with the Andersen-Tawil syn-drome phenotype in our patients. The kindred described inthis report and the kindred that was previously described4
provide clinical evidence for pathogenicity of this rare mis-sense mutation in KCNJ2. In addition, our kindred demon-strated the wide intra-familial variability in penetrance of theclinical triad seen in Andersen-Tawil syndrome.
We suggest that physicians should be aware of this rare clinicalentity and its high phenotypic variability even within a familywhile evaluating patients with transient muscle weakness. The
subtle clinical and characteristic electrocardiographic featurescan help with early recognition and treatment.
Study fundingNo targeted funding reported.
DisclosureR. Deeb and A. Veerapandiyan report no disclosures. R. Tawilhas served on scientific advisory boards for Fulcrum Thera-peutics and Acceleron Pharma; serves on the editorial boardof the Journal of Neuromuscular Diseases; receives publishingroyalties from Wiley-Blackwell; has been a consultant forAcceleron; and has received research support from FulcrumTherapeutics, NIH, and the FSH Society. S. Treidler reportsno disclosures. Full disclosure form information provided bythe authors is available with the full text of this article atNeurology.org/NG.
Received July 10, 2018. Accepted in final form September 25, 2018.
References1. Veerapandiyan A, Statland JM, Tawil R. Andersen-tawil syndrome. In: Adam MP,
Ardinger HH, Pagon RA, et al, editors. GeneReviews®. Seattle, WA: University ofWashington; 1993.
2. Statland JM, Fontaine B, Hanna MG, et al. Review of the diagnosis and treatment ofperiodic paralysis. Muscle Nerve 2018;57:522–530.
3. PlasterNM,Tawil R, Tristani-FirouziM, et al.Mutations inKir2.1 cause the developmentaland episodic electrical phenotypes of Andersen’s syndrome. Cell 2001;105:511–519.
4. Chan HF, Chen ML, Su JJ, Ko LC, Lin CH, Wu RM. A novel neuropsychiatricphenotype of KCNJ2 mutation in one Taiwanese family with Andersen-Tawil syn-drome. J Hum Genet 2010;55:186–188.
5. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting dam-aging missense mutations. Nat Methods 2010;7:248–249.
6. Haruna Y, Kobori A, Makiyama T, et al. Genotype-phenotype correlations of KCNJ2mutations in Japanese patients with Andersen-Tawil syndrome. HumMutat 2007;28:208.
7. Soom M, Schonherr R, Kubo Y, Kirsch C, Klinger R, Heinemann SH. Multiple PIP2binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett 2001;490:49–53.
Appendix 1 Author contributions
Name Location Role Contribution
Reem Deeb, MD SUNY Downstate Medical Center, New York Author Drafting and revising the manuscript
Aravindhan Veerapandiyan,MBBS
University of RochesterMedical Center, New York Author Study concept and revising the manuscript
Rabi Tawil, MD University of RochesterMedical Center, New York Author Study concept and revising the manuscript
Simona Treidler, MD Stony Brook School of Medicine, New York Author Study concept, drafting, and revising themanuscript
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

CLINICAL/SCIENTIFIC NOTES OPEN ACCESS
A tropomyosin-receptor kinase-fused genemutationassociates with vacuolar myopathyNicolas N. Madigan, MB BCh, PhD, Jennifer A. Tracy, MD, William J. Litchy, MD, Zhiyv Niu, PhD,
Chunhua Chen, PhD, Kun Ling, PhD, and Margherita Milone, MD, PhD
Neurol Genet 2018;4:e287. doi:10.1212/NXG.0000000000000287
Correspondence
Dr. Milone
A 44-year-old Caucasian man presented with a 7-year history of upper and lower limb musclefasciculations and cramps, progressive asymmetric weakness, muscle atrophy, and length-dependent sensory loss. His parents (deceased), 3 older siblings, and 3 children had nohistory of neurologic symptoms, including weakness and sensory symptoms, with the ex-ception of cramps in the mother. He was not taking any medication known to cause neu-ropathy or myopathy. On examination, there was right greater than left shoulder girdlemuscle weakness and atrophy (Medical Research Council grades 3/5 and 4/5), intrinsic lefthand muscle weakness (4+/5) with atrophy, asymmetric pelvic girdle (4/5 right, 3/5 left),and left foot dorsiflexor weakness (4+/5). Strength of neck flexor and extensors, and axialmuscles, was normal. He had bilateral calf muscle atrophy and weakness as suggested by hisinability to stand on toes (figure, A–C). He was areflexic. Sensory examination revealed distalvibration and pinprick deficits in the upper and lower limbs in a length-dependent fashion. Hehad bilateral pes cavus.
Serial measurements of serum creatine kinase ranged between 840 and 2,400 U/L (normal<336 U/L) over the preceding 4 years. MRI revealed thigh muscle atrophy (figure, D).Neurophysiologic studies demonstrated reduced motor conduction amplitudes and diffusefibrillation and fasciculation potentials with large motor unit potentials suggestive of a motorneuronopathy. No small motor unit potentials were recorded. Sensory nerve action potentialswere absent in the upper and lower limbs.
A muscle biopsy of the quadriceps, performed a year prior to presentation in our clinic, showedmixed neurogenic and myopathic changes with several fibers containing rimmed and non-rimmed vacuoles, and perivascular inflammation (figure, E–J). Targeted next generation exomesequencing (NGS) of 95 genes associated with hereditary neuropathy and motor neurondisease detected a known pathogenic heterozygous missense variant in exon 8 (c.854C>T,p.Pro285Leu) of the tropomyosin-receptor kinase-fused gene (TFG), located within a carboxy-terminal proline-glutamine (P/Q)-rich domain. NGS of 104 genes associated with myopathies(appendix e-1, links.lww.com/NXG/A118), including myopathies with rimmed vacuoles,revealed no pathogenic variants. Living relatives declined genetic testing.
DiscussionOur patient presented with a classic clinical phenotype and a known pathologic genotype causedby a heterozygous TFG mutation, along with novel histologic evidence of vacuolar myopathy.TFG mutations were first identified in Okinawa and Kansai prefecture patients with autosomaldominant hereditary motor and sensory neuropathy (HMSN-P) manifesting with proximalweakness and distal sensory loss.1 Two of these kindreds carried the sameTFGmutation detectedin our patient. Limited muscle pathologic findings showing neurogenic changes (fiber type
From the Department of Neurology (N.N.M., J.A.T., W.J.L., M.M.), Department of Laboratory Medicine and Pathology (Z.N.), Department of Clinical Genomics (Z.N.), and Departmentof Biochemistry & Molecular Biology (C.C., K.L.), Mayo Clinic, Rochester, MN.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was funded by the authors.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1
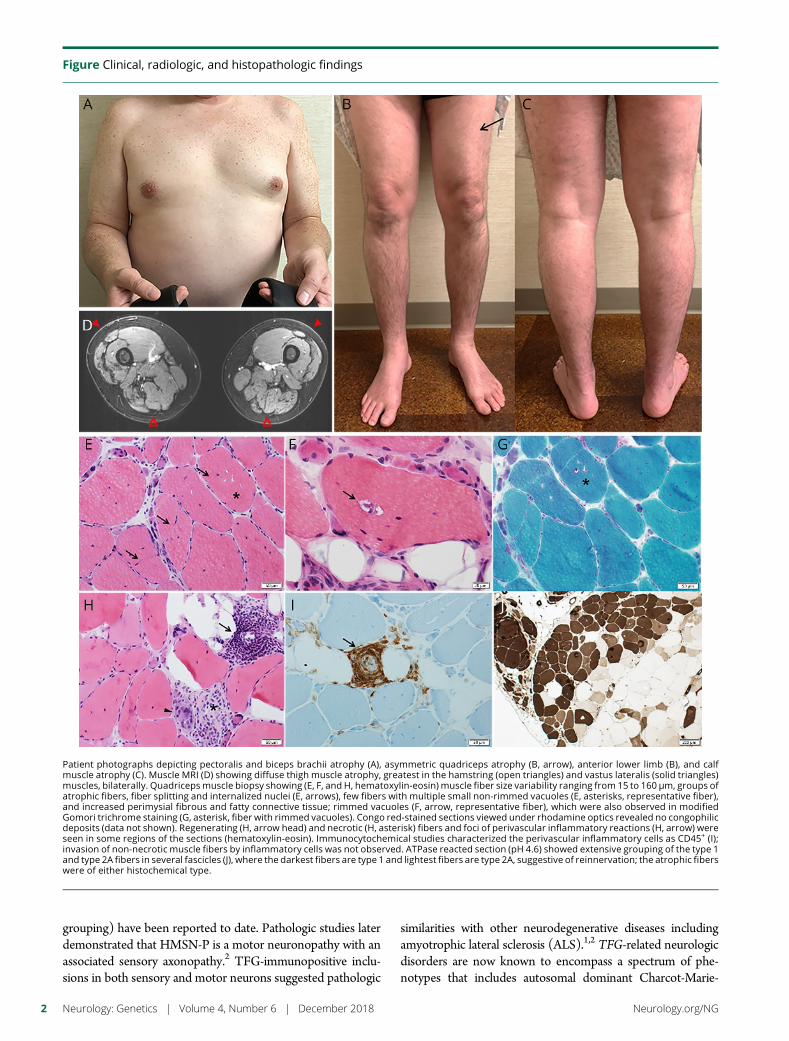
grouping) have been reported to date. Pathologic studies laterdemonstrated that HMSN-P is a motor neuronopathy with anassociated sensory axonopathy.2 TFG-immunopositive inclu-sions in both sensory and motor neurons suggested pathologic
similarities with other neurodegenerative diseases includingamyotrophic lateral sclerosis (ALS).1,2 TFG-related neurologicdisorders are now known to encompass a spectrum of phe-notypes that includes autosomal dominant Charcot-Marie-
Figure Clinical, radiologic, and histopathologic findings
Patient photographs depicting pectoralis and biceps brachii atrophy (A), asymmetric quadriceps atrophy (B, arrow), anterior lower limb (B), and calfmuscle atrophy (C). Muscle MRI (D) showing diffuse thigh muscle atrophy, greatest in the hamstring (open triangles) and vastus lateralis (solid triangles)muscles, bilaterally. Quadricepsmuscle biopsy showing (E, F, and H, hematoxylin-eosin) muscle fiber size variability ranging from 15 to 160 μm, groups ofatrophic fibers, fiber splitting and internalized nuclei (E, arrows), few fibers with multiple small non-rimmed vacuoles (E, asterisks, representative fiber),and increased perimysial fibrous and fatty connective tissue; rimmed vacuoles (F, arrow, representative fiber), which were also observed in modifiedGomori trichrome staining (G, asterisk, fiber with rimmed vacuoles). Congo red-stained sections viewed under rhodamine optics revealed no congophilicdeposits (data not shown). Regenerating (H, arrow head) and necrotic (H, asterisk) fibers and foci of perivascular inflammatory reactions (H, arrow) wereseen in some regions of the sections (hematoxylin-eosin). Immunocytochemical studies characterized the perivascular inflammatory cells as CD45+ (I);invasion of non-necrotic muscle fibers by inflammatory cells was not observed. ATPase reacted section (pH 4.6) showed extensive grouping of the type 1and type 2A fibers in several fascicles (J), where the darkest fibers are type 1 and lightest fibers are type 2A, suggestive of reinnervation; the atrophic fiberswere of either histochemical type.
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

Tooth disease type 2 (CMT2) and autosomal recessive he-reditary spastic paraplegia.3
TFG protein is ubiquitously expressed in a variety of tis-sues, including normal muscle (data not shown). TFGfunctions primarily as a chaperone for vesicle trafficking ofunfolded and misfolded proteins through the endoplasmicreticulum and into the ubiquitin-proteasome system(UPS). The P/Q domain in TFG specifically regulates theinteraction with the UPS, and the TFG p.Pro285Leu vari-ant has been shown to damage the protein quality controlsystem.4 This could lead to accumulation of abnormalproteins in motor neurons as well as in muscle. Variants inheat shock proteins 22 (HSBP8) and 27 (HSPB1),5 whichare also protein chaperones, have been associated withCMT2, motor neuronopathy, and vacuolar myopathy.Variants in valosin-containing protein (VCP), anotherprotein involved in ubiquitin-dependent protein degrada-tion, lead to a similar phenotypic spectrum.6
Low-complexity protein domains enriched for glutaminerepeats, with proline (P/Q) and asparagine (N/Q), are alsofound in prions and are implicated in several degenerativeneuromuscular disease models of DNA, RNA, and proteinprocessing that result in cytoplasmic inclusions and tem-plated protein propagation.7 The p.Glu384Lys mutation inTIA1 cytotoxic granule-associated RNA binding protein, forexample, occurs directly within a glutamine-rich prion-relateddomain and causes distal myopathy with rimmed vacuoles,while an adjacent mutation (pPro362Leu) in the same lowcomplexity domain has been reported in a kindred withALS. Mutations in prion-like domains in hnRNPA2B1 andhnRNPA1 associate with vacuolar myopathy and motor neu-ron disease. Mutations in matrin-3, another RNA bindingprotein involved in stress granule formation, also lead tomyopathy with rimmed vacuoles and ALS. TFG thereforeextends the list of genes associating with vacuolar myopathy,motor neuron disease, and neuropathy.
AcknowledgmentsThe authors thank the patient for the photographs;Dr. A. Kendler at University of Cincinnati for sharingthe muscle biopsy slides; and Invitae for performing theadditional analysis of genes associated with myopathies.
Study fundingThis work was supported by a generous gift from a MayoClinic benefactor to M. Milone and Z. Niu.
DisclosureN.N. Madigan is an employee of the Mayo Clinic (neurologyfellow). J.A. Tracy reports no disclosures. W.J. Litchy hasreceived research funding from Ionis Pharmaceuticals andAlnylam (compensation for travel and training investigators).C. Chen reports no disclosures. K. Ling holds a patent forPhosphatidylinositol phosphate kinase type Igamma regulatesfocal adhesions and cell migration. M. Milone has receivedresearch funding from a Mayo Clinic benefactor, and throughdiscretionary funding from the Department of Neurology.Full disclosure form information provided by the authors isavailable with the full text of this article at Neurology.org/NG.
Received July 3, 2018. Accepted in final form October 3, 2018.
References1. Ishiura H, Sako W, Yoshida M, et al. The TRK-fused gene is mutated in hereditary
motor and sensory neuropathy with proximal dominant involvement. Am J HumGenet 2012;91:320–329.
2. Fujita K, Yoshida M, Sako W, et al. Brainstem and spinal cord motor neuron in-volvement with optineurin inclusions in proximal-dominant hereditary motor andsensory neuropathy. J Neurol Neurosurg Psychiatry 2011;82:1402–1403.
3. Tsai PC, Huang YH, Guo YC, et al. A novel TFG mutation causes Charcot-Marie-Tooth disease type 2 and impairs TFG function. Neurology 2014;83:903–912.
4. Yagi T, Ito D, Suzuki N. TFG-related neurologic disorders: new insights into rela-tionships between endoplasmic reticulum and neurodegeneration. J Neuropathol ExpNeurol 2016;75:299–305.
5. Bugiardini E, Rossor AM, Lynch DS, et al. Homoygouse mutation in HSPB1 causingdistal vacuolar myopathy and motor neuropathy. Neurol Genet 2017;3:e168.
6. Kazamel M, Sorenson EJ, McEvoy KM, et al. Clinical spectrum of valosin containingprotein (VCP)-opathy. Muscle Nerve 2016;54:94–99.
7. An L, Harrison PM. The evolutionary scope and neurological disease linkage of yeast-prion-like proteins in humans. Biol Direct 2016;11:32.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3
Appendix 1 Author contributions
Name Location Role Contribution
Nicolas N. Madigan,MB BCh, PhD
Mayo Clinic,Rochester, MN
Author Study concept and design; acquisition, analysis and interpretation of data; draftingmanuscript;critical revision of final manuscript for intellectual content.
Jennifer A. Tracy, MD Mayo Clinic,Rochester, MN
Author Acquisition, analysis, and interpretation of data; critical revision of final manuscript forintellectual content.
William J. Litchy, MD Mayo Clinic,Rochester, MN
Author Acquisition, analysis, and interpretation of data; critical revision of final manuscript forintellectual content.
Zhiyv Niu, PhD Mayo Clinic,Rochester, MN
Author Analysis and interpretation of data; critical revision of final manuscript for intellectual content.
Chunhua Chen, PhD Mayo Clinic,Rochester, MN
Author Acquisition, analysis, and interpretation of data; critical revision of final manuscript forintellectual content.
Kun Ling, PhD Mayo Clinic,Rochester, MN
Author Acquisition, analysis, and interpretation of data; critical revision of final manuscript forintellectual content.
Margherita Milone,MD, PhD
Mayo Clinic,Rochester, MN
Author Study concept and design; acquisition, analysis, and interpretation of data; draftingmanuscript;critical revision of final manuscript for intellectual content.
4 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

CLINICAL/SCIENTIFIC NOTES OPEN ACCESS
Lysosomal dysfunction in TMEM106Bhypomyelinating leukodystrophyYoko Ito, PhD, Taila Hartley, MSc, Stephen Baird, PhD, Sunita Venkateswaran, MD, Cas Simons, PhD,
Nicole I. Wolf, MD, PhD, Kym M. Boycott, PhD, MD, David A. Dyment, DPhil, MD,*
and Kristin D. Kernohan, PhD*
Neurol Genet 2018;4:e288. doi:10.1212/NXG.0000000000000288
Correspondence
Dr. Dyment
Transmembrane protein 106B (TMEM106B; NM_001134232) was recently identified as a generesponsible for a form of hypomyelinating leukodystrophy (HLD).1,2 All 5 cases identified todate carry the identical c.754 G > A, (p.Asp252Asn) mutation.1,2 Although the exact functionis unknown,3 studies of TMEM106B in the context of frontotemporal lobar degenerationwith 43-kD TAR DNA-binding protein (TDP-43) pathology (FTLD-TDP) indicate thatTMEM106B likely acts as a lysosomal regulator and can modify risk for FTLD-TDP.4 How-ever, the molecular effects of the (p.Asp252Asn) substitution have not yet been reported forTMEM106B-associated HLD. The HLDs are heterogeneous conditions, with the known diseasegenes playing roles in myelin sheath structure (e.g., PLP1) and other cellular functions that arenot oligodendrocyte specific, including protein translation, molecular chaperoning, and cyto-skeletal regulation.5 We set out to assess if this recurrent TMEM106B substitution was affectinglysosome biology or had an alternate role underlying the HLD pathogenesis. Implication oflysosome biology in HLD provides exciting new advances in our understanding of the molecularunderpinnings of this condition and the complexities of neurodevelopment.
Functional analysisUsing patient-derived fibroblasts (patient 4),1 we assessed TMEM106B messenger RNA(mRNA) and protein levels and found that these were unaltered in patient cells compared withcontrols (figure, A). TMEM106B has been shown to affect lysosome number, morphology, andacidification.4,6 LAMP1 staining, which marks lysosomes, showed an increase in the number oflysosomes in patient fibroblasts comparedwith controls, although the average size of the lysosomeremained unchanged (figure, B). Staining with a pH-sensitive fluorescent dye showed that despitethe increased number of lysosomes, a substantial decrease in the number of lysotracker-positivefoci was observed, indicating that patient lysosomes show impaired acidification (figure, B).
Impairment in lysosomal acidification can affect the processing and function of lysosomalenzymes. Therefore, we next examined the levels of cathepsin B, cathepsin L, and dipeptidylpeptidase VII (DPP7) as these are lysosomal proteases previously shown to be decreased inTmem106b null mice.6 The (p.Asp252Asn) variant results in a decrease in the mature form ofcathepsin B protein and, importantly, a concomitant decrease in the activity levels of thisenzyme in patient cells (figure, C and D). This reduction is predicted to not only affect thedegradation of cathepsin B protein substrates but also the regulation of the T-cell transcriptionfactor (TFEB).7 As cathepsin B suppresses lysosomal number in a TFEB-dependent manner,7
it is likely that the reduction in cathepsin B activity contributes to the increased lysosomenumber observed. Patient cells also displayed an accumulation of both intermediate andmatureforms of cathepsin L and no change in DPP7 levels (figure, C, data not shown). Taken together,
*These authors contributed equally to this work as senior author.
From the Children’s Hospital of Eastern Ontario Research Institute (Y.I., T.H., S.B., K.M.B., D.A.D., K.D.K.), Ottawa, Ontario, Canada; Division of Neurology (S.V.), Children’s Hospital ofEastern Ontario, Ottawa, Ontario, Canada; Institute for Molecular Bioscience (C.S.), University of Queensland, St. Lucia, Queensland, Australia; and Department of Child Neurology(N.I.W.), VU University Medical Center, and Amsterdam Neuroscience, Amsterdam, The Netherlands.
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article atNeurology.org/NG.
The Article Processing Charge was paid for by Genome Canada and CIHR.
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND), which permits downloadingand sharing the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1
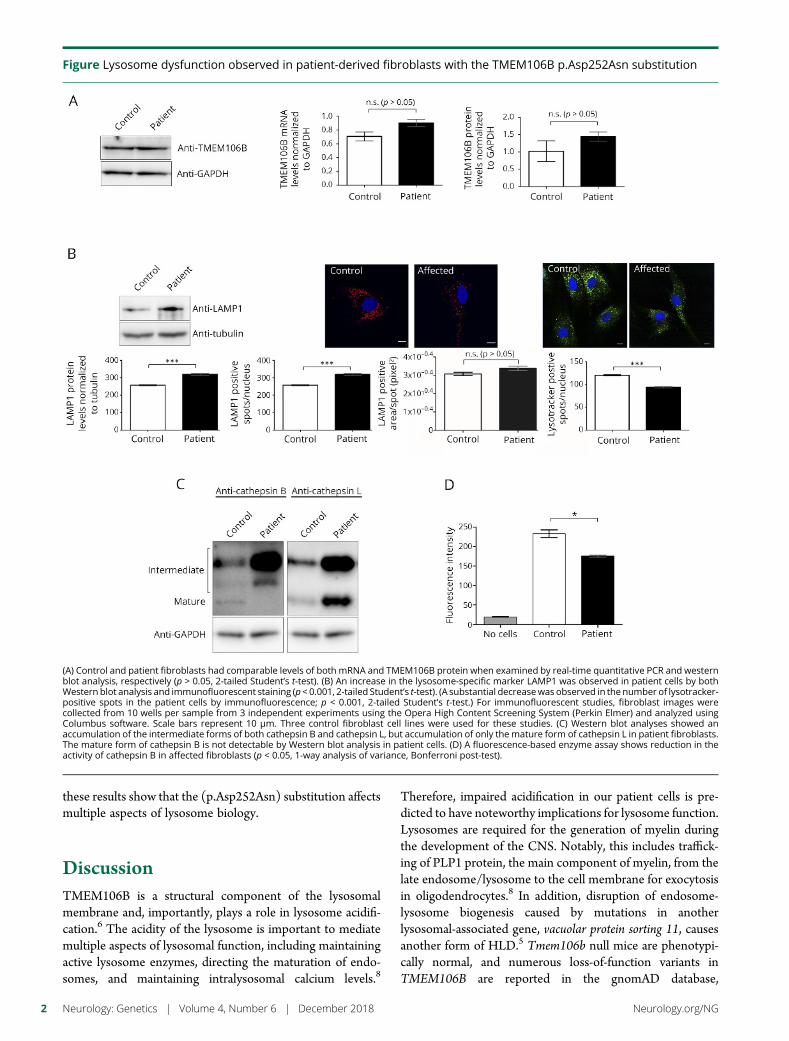
these results show that the (p.Asp252Asn) substitution affectsmultiple aspects of lysosome biology.
DiscussionTMEM106B is a structural component of the lysosomalmembrane and, importantly, plays a role in lysosome acidifi-cation.6 The acidity of the lysosome is important to mediatemultiple aspects of lysosomal function, including maintainingactive lysosome enzymes, directing the maturation of endo-somes, and maintaining intralysosomal calcium levels.8
Therefore, impaired acidification in our patient cells is pre-dicted to have noteworthy implications for lysosome function.Lysosomes are required for the generation of myelin duringthe development of the CNS. Notably, this includes traffick-ing of PLP1 protein, the main component of myelin, from thelate endosome/lysosome to the cell membrane for exocytosisin oligodendrocytes.8 In addition, disruption of endosome-lysosome biogenesis caused by mutations in anotherlysosomal-associated gene, vacuolar protein sorting 11, causesanother form of HLD.5 Tmem106b null mice are phenotypi-cally normal, and numerous loss-of-function variants inTMEM106B are reported in the gnomAD database,
Figure Lysosome dysfunction observed in patient-derived fibroblasts with the TMEM106B p.Asp252Asn substitution
(A) Control and patient fibroblasts had comparable levels of both mRNA and TMEM106B protein when examined by real-time quantitative PCR and westernblot analysis, respectively (p > 0.05, 2-tailed Student’s t-test). (B) An increase in the lysosome-specific marker LAMP1 was observed in patient cells by bothWestern blot analysis and immunofluorescent staining (p< 0.001, 2-tailed Student’s t-test). (A substantial decreasewas observed in the number of lysotracker-positive spots in the patient cells by immunofluorescence; p < 0.001, 2-tailed Student’s t-test.) For immunofluorescent studies, fibroblast images werecollected from 10 wells per sample from 3 independent experiments using the Opera High Content Screening System (Perkin Elmer) and analyzed usingColumbus software. Scale bars represent 10 μm. Three control fibroblast cell lines were used for these studies. (C) Western blot analyses showed anaccumulation of the intermediate forms of both cathepsin B and cathepsin L, but accumulation of only themature form of cathepsin L in patient fibroblasts.The mature form of cathepsin B is not detectable by Western blot analysis in patient cells. (D) A fluorescence-based enzyme assay shows reduction in theactivity of cathepsin B in affected fibroblasts (p < 0.05, 1-way analysis of variance, Bonferroni post-test).
2 Neurology: Genetics | Volume 4, Number 6 | December 2018 Neurology.org/NG

supporting a non-haploinsufficiency mechanism underlyingTMEM106B-related HLD. Further studies are required to elu-cidate the specific dominant negative or gain-of-function effectof the (p.Asp252Asn) mutation that results in decreased lyso-somal acidification. Also, potential effects on other HLD-relatedproteins, such as PLP1, which would have direct consequenceson myelin formation need to be examined. We appreciate thatthese experiments are based on a sample from a single individualand that additional studies from other (p.Asp252Asn)-positiveindividuals are necessary to confirm these findings. Nevertheless,this report provides an important first step in defining the role ofTMEM106B in lysosome function in HLD. TMEM106B nowjoins the catalogue of essential lysosomal proteins implicated inhuman neurologic disease.
Author contributionsY. Ito: study concept and design, acquisition, analysis, and in-terpretation of data, and manuscript preparation. T. Hartley:critical revision of manuscript for intellectual content. S. Baird:acquisition of data and critical revision of manuscript for in-tellectual content. S. Venkateswaran: critical revision of man-uscript for intellectual content. C. Simons: critical revision ofmanuscript for intellectual content. N.I. Wolf: critical revisionof manuscript for intellectual content. K.M. Boycott: criticalrevision of manuscript for intellectual content. D.A. Dyment:analysis and interpretation of data, manuscript preparation, andstudy supervision. K.D. Kernohan: analysis and interpretationof data, manuscript preparation, and study supervision.
AcknowledgmentsThe authors would like to thank the patient and family; withoutwhom this work would not be possible. The authors also wishto acknowledge Dr. Wendy Mears for her tissue cultureexpertise. This work was supported by the Care4Rare CanadaConsortium funded by Genome Canada, the CanadianInstitutes of Health Research, the Ontario Genomics Institute,Ontario Research Fund, Genome Quebec, and the Children’sHospital of Eastern Ontario Foundation.
Study fundingThis work was performed under the Care4Rare Canada Con-sortium funded by Genome Canada, the Canadian Institutes ofHealth Research, the Ontario Genomics Institute, OntarioResearch Fund, Genome Quebec, and Children’s Hospital ofEastern Ontario Foundation.
DisclosureY. Ito, T. Hartley, S. Baird, S. Venkateswaran, and C. Simonsreport no disclosures. N.I Wolf has served on scientific advi-sory boards for European Leukodystrophy Association (ELA)and Mission Massimo Foundation (both without compen-sation); has received travel funding from ELA; has served onthe editorial boards of Neuropediatrics and Neurology; hasreceived research support from Hersenstichting, Metakids,the Stofwisselkracht Foundation, the M.O. Knip Foundation,and Yasho’s Leukodystrophy Foundation; and holds stock/stock options in Aer Beatha. K.M. Boycott, D.A. Dyment, andK.D. Kernohan report no disclosures. Full disclosure forminformation provided by the authors is available with the fulltext of this article at Neurology.org/NG.
Received by Neurology March 23, 2018. Accepted in final form August21, 2018.
References1. Simons C, Dyment D, Bent SJ, et al. A recurrent de novo mutation in TMEM106B
causes hypomyelinating leukodystrophy. Brain 2017;140:3105–3111.2. Yan H, Kubisiak T, Ji H, Xiao J, Wang J, Burmeister M. The recurrent mutation in
TMEM106B also causes hypomyelinating leukodystrophy in China and is a CpG hotspot. Brain 2018;141:e36.
3. Nicholson AM, Zhou X, Perkerson RB, et al. Loss of Tmem106b is unable to ame-liorate frontotemporal dementia-like phenotypes in an AAV mouse model ofC9ORF72 -repeat induced toxicity. Acta Neuropathol 2018;6:1–14.
4. Nicholson AM, Rademakers R. What we know about TMEM106B in neuro-degeneration. Acta Neuropathol 2016;132:639–651.
5. Pouwels PJW, Vanderver A, Bernard G, et al. Hypomyelinating leukodystrophies:translational research progress and prospects. Ann Neurol 2014;76:5–19.
6. Klein ZA, Takahashi H, Ma M, et al. Loss of TMEM106B ameliorates lysosomal andfrontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron2017;95:281–296.e6.
7. Man SM, Kanneganti TD. Regulation of lysosomal dynamics and autophagy byCTSB/cathepsin B. Autophagy 2016;12:2504–2505.
8. Shen Y, Yuan Y, Su W, Gu Y, Chen G. The roles of lysosomal exocytosis in regulatedmyelination. J Neurol Neuromed 2016;1:4–8.
Neurology.org/NG Neurology: Genetics | Volume 4, Number 6 | December 2018 3

CORRECTION
Novel genotype-phenotype andMRI correlations in a large cohort ofpatients with SPG7 mutationsNeurol Genet 2018;4:e300. doi:10.1212/NXG.0000000000000300
In the article, “Novel genotype-phenotype and MRI correlations in a large cohort of patientswith SPG7 mutations” by Hewamadduma, et al.1, first published online on October 24, 2018,the email address for the corresponding author should read as [email protected]. Theauthors regret the error.
ReferenceHewamadduma CA, Hoggard N, O’Malley R, et al. Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7mutations. Neurol Genet 2018;4:e279.
Copyright © 2018 American Academy of Neurology 1
Related Documents











