http://jbx.sagepub.com/ Journal of Biomolecular Screening http://jbx.sagepub.com/content/early/2014/10/16/1087057114555307 The online version of this article can be found at: DOI: 10.1177/1087057114555307 published online 16 October 2014 J Biomol Screen J. Pierce, Christophe Romier and Manfred Jung Matthias Schiedel, Martin Marek, Julien Lancelot, Berin Karaman, Ingrid Almlöf, Johan Schultz, Wolfgang Sippl, Raymond Schistosoma mansoni -Dependent Histone Deacetylase smSirt2 from + Fluorescence-Based Screening Assays for the NAD Published by: http://www.sagepublications.com On behalf of: Journal of Biomolecular Screening can be found at: Journal of Biomolecular Screening Additional services and information for http://jbx.sagepub.com/cgi/alerts Email Alerts: http://jbx.sagepub.com/subscriptions Subscriptions: http://www.sagepub.com/journalsReprints.nav Reprints: http://www.sagepub.com/journalsPermissions.nav Permissions: What is This? - Oct 16, 2014 OnlineFirst Version of Record >> at INSTITUT PASTEUR DE LILLE on October 24, 2014 jbx.sagepub.com Downloaded from at INSTITUT PASTEUR DE LILLE on October 24, 2014 jbx.sagepub.com Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

http://jbx.sagepub.com/Journal of Biomolecular Screening
http://jbx.sagepub.com/content/early/2014/10/16/1087057114555307The online version of this article can be found at:
DOI: 10.1177/1087057114555307
published online 16 October 2014J Biomol ScreenJ. Pierce, Christophe Romier and Manfred Jung
Matthias Schiedel, Martin Marek, Julien Lancelot, Berin Karaman, Ingrid Almlöf, Johan Schultz, Wolfgang Sippl, RaymondSchistosoma mansoni
-Dependent Histone Deacetylase smSirt2 from +Fluorescence-Based Screening Assays for the NAD
Published by:
http://www.sagepublications.com
On behalf of:
Journal of Biomolecular Screening
can be found at:Journal of Biomolecular ScreeningAdditional services and information for
http://jbx.sagepub.com/cgi/alertsEmail Alerts:
http://jbx.sagepub.com/subscriptionsSubscriptions:
http://www.sagepub.com/journalsReprints.navReprints:
http://www.sagepub.com/journalsPermissions.navPermissions:
What is This?
- Oct 16, 2014OnlineFirst Version of Record >>
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

Journal of Biomolecular Screening 1 –10© 2014 Society for LaboratoryAutomation and ScreeningDOI: 10.1177/1087057114555307jbx.sagepub.com
Original Research
Introduction
Epigenetics is the study of inheritable changes in gene expression without any changes in underlying DNA sequence. Major epigenetic mechanisms are alterations of the methylation state of the DNA base cytosine, the interac-tion of chromatin with small noncoding RNAs (ncRNAs), and posttranslational histone modifications. The latter mainly occur at the basic amino acids lysine and arginine of unstructured N-terminal histone tails that protrude out of the histone octamer.1 Threonine or tyrosine residues and amino acids of the nucleosomal core, however, can be mod-ified as well.2 The different modifications are diverse, and in their manifold combinations they generate the epigenetic code. The epigenetic code is much more complex than the genetic code, and it is far from being deciphered completely. Nevertheless, specific modifications, or a combination of single modifications, can be associated with a well-defined change in gene expression. Chemical groups that can be transferred to the above-mentioned sites are the methyl, acetyl, and phosphoryl groups, as well as bigger molecules [e.g., ubiquitin, fatty acids, and adenosine diphosphate (ADP)-ribose].3 Enzymes that catalyze the transfer of these
groups to the N-terminal histone tails are called writer enzymes. They take part in a dynamic interplay with eraser enzymes, which are able to restore the unmodified state.
555307 JBXXXX10.1177/1087057114555307Journal of Biomolecular ScreeningSchiedel et al.research-article2014
1Institute of Pharmaceutical Sciences, Albert-Ludwigs-Universität Freiburg, Freiburg, Germany2Département de Biologie Structurale Intégrative, Institut de Génétique et Biologie Moléculaire et Cellulaire (IGBMC), Université de Strasbourg (UDS), CNRS, INSERM, Illkirch, France3Center for Infection and Immunity of Lille (CIIL), INSERM U1019—CNRS UMR 8204, Université Lille Nord de France, Institut Pasteur de Lille, Lille, France4Institute of Pharmacy, Martin-Luther-Universität Halle-Wittenberg, Halle, Germany5Kancera AB, Solna, Sweden
Received Jun 5, 2014, and in revised form Sep 9, 2014. Accepted for publication Sep 10, 2014.
Supplementary material for this article is available on the Journal of Biomolecular Screening Web site at http://jbx.sagepub.com/supplemental.
Corresponding Author:Manfred Jung, Institute of Pharmaceutical Sciences, Albert-Ludwigs-Universität Freiburg, Albertstr. 25, 79104 Freiburg, Germany. Email: [email protected]
Fluorescence-Based Screening Assays for the NAD+-Dependent Histone Deacetylase smSirt2 from Schistosoma mansoni
Matthias Schiedel1, Martin Marek2, Julien Lancelot3, Berin Karaman4, Ingrid Almlöf5, Johan Schultz5, Wolfgang Sippl4, Raymond J. Pierce3, Christophe Romier2, and Manfred Jung1
AbstractSirtuins are NAD+-dependent histone deacetylases (HDACs) that cleave off acetyl but also other acyl groups from the ε-amino group of lysines in histones and other substrate proteins. Five sirtuin isoforms are encoded in the genome of the parasitic pathogen Schistosoma mansoni. During its life cycle, S. mansoni undergoes drastic changes in phenotype that are associated with epigenetic modifications. Previous work showed strong effects of hSirt2 inhibitors on both worm life span and reproduction. Thus, we postulate smSirt2 as a new antiparasite target. We report both the optimization of a homogeneous fluorescence-based assay and the development of a new heterogeneous fluorescence-based assay to determine smSirt2 activity. The homogeneous assay uses a coumarin-labeled acetyl lysine derivative, and the heterogeneous version is using a biotinylated and fluorescence-labeled oligopeptide. Magnetic streptavidin-coated beads allow higher substrate loading per well than streptavidin-coated microtiter plates and make it possible to screen for inhibitors of either smSirt2 or its human isoform (hSirt2) for selectivity studies. We also present hits from a pilot screen with inhibitors showing an IC50 lower than 50 µM. Binding of the hits to their targets is rationalized by docking studies using a homology model of smSirt2.
Keywordsepigenetics, histone modifications, in vitro assay, sirtuin, Schistosoma mansoni
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

2 Journal of Biomolecular Screening
Sirtuins belong to the class of eraser enzymes and repre-sent a specific NAD+-dependent class of histone deacety-lases (HDACs), also referred to as class III HDACs. These enzymes catalyze the deacetylation of acetylated ε-amino groups of lysines in histones and other proteins such as p53, tubulin, FOXO proteins, and p300.4 Because an increasing number of nonhistone substrates have been discovered in recent years, it is more accurate to term HDACs as protein deacetylases, and maybe the sirtuins are more signal trans-duction targets than epigenetic modifiers, at least in the human context. Sirtuins have been found in species from bacteria to humans in a highly conserved manner. The hall-mark of the family is a domain of approximately 260 amino acids with a high degree of sequence similarity in all sirtu-ins.5 Sirtuins can be grouped into 5 classes. The 7 mamma-lian sirtuins belong to classes I–IV, whereas sirtuins of the fifth class, termed class U, are present in only bacteria and archaea.6 Human sirtuins (hSirt1–7) differ in their subcel-lular localization and their catalytic activity. Sirtuins can act as amidohydrolases and/or mono-ADP-ribosyltransferases. hSirt1 shows a nuclear and cytosolic localization, and deacetylates histones as well as transcription factors (e.g., p53 and FoxO).4 hSirt2 is a cytosolic protein7 with a major impact on cell cycle regulation and apoptosis by deacetylat-ing substrates like α-tubulin,8 p53,9 and BubR1.10 The mito-chondrial sirtuins are hSirt3–5. hSirt4, for example, downregulates glutamine dehydrogenases by mono-ADP-ribosylation.11 Moreover, hSirt5 controls the activity of carbamoyl-phosphate synthase 1 (CPS-1) via desuccinyl-ation.12 This underlines the effect of mitochondrial sirtuins on cellular metabolism. hSirt6 and hSirt7 are located exclu-sively in the nucleus. hSirt6 has been reported to have only low deacetylase activity, whereas it efficiently hydrolyzes long-chain fatty acyl groups from ε-amino groups of lysine residues.13 The sirtuin PfSir2A from Plasmodium falci-parum also hydrolyzes long-chain acid amides.14
Keeping the variety of sirtuin substrates and the effects of substrate conversion in mind, such as regulation of apoptosis, the cell cycle, metabolism, and aging, it is obvious that sirtuins have emerged as potential drug targets in recent years. Although a remarkable number of sirtuin inhibitors have been discovered, there is still a need for subtype-selective and drug-like sirtuin inhibitors.15 To date, there is only one compound, Selisistat (or SN196), which is a hSirt1 inhibitor, in Phase II clinical trials for Huntington’s disease.16 Nevertheless, the bio-logical effects of sirtuin inhibitors highlight these inhibitors’ potential, especially in cancer therapy.15,17 The use of epi-genetic drugs as anti-infectives is a rather new approach that is particularly usable on eukaryotic parasites due to their chromosomal genome organization. This anti-infective strategy is even more promising for metazoan parasites, which undergo drastic changes in phenotype during their life cycle. Changes in phenotype without an alteration of genotype are caused by epigenetic modifications. An interruption of the
fine-tuned epigenetic interplay should have significant effects on parasites that are at the point of switching phenotype, whereas host cells may remain rather unaffected. The develop-ment of species-selective modulators of epigenetic regulators, however, should be the main goal of epigenetic drug discovery in the field of anti-infectives. Because of a high sequence homology of epigenetic regulators (e.g., HDACs from proto-zoa to humans), this is quite challenging. Nevertheless, Marek et al. identified small-molecule inhibitors of the Schistosoma mansoni HDAC8 (smHDAC8) with a reduced affinity for human HDACs by a structure-based approach. Crystal struc-tures of smHDAC8 with generic HDAC inhibitors reveal spe-cific structural changes in the active site on ligand binding, which cannot be accommodated by human HDACs. These species-selective changes were exploited to develop the smHDAC8 inhibitor J1075, which is capable of inducing apoptosis and mortality in schistosomes.18 Another example for a smHDAC8 selective inhibitor is the thiol-based inhibitor discovered by Stolfa et al.19 Schistosomal histone acetyltrans-ferases SmGCN5 and SmCBP1 have been identified as poten-tial drug targets as well.20 The blood fluke S. mansoni is the main pathogen of schistosomiasis, a disease affecting more than 200 million people in tropical and subtropical countries. Due to the absence of a vaccine, praziquantel is currently the only way of controlling schistosomiasis, and it is distributed mainly through mass administration programs to millions of people every year. Even though praziquantel shows a number of positive features, especially with regard to safety, efficacy, cost, and ease of distribution, a major problem is its lack of efficacy against the immature stages of the parasite. Also, in view of praziquantel’s massive and exclusive use on a large number of individuals, the development of drug resistance is a much-feared possibility. Furthermore, the mechanism of action is still unclear, which does not favor the development of derivatives or alternatives. A large number of compounds have been tested as potential antischistosomal drugs, some with promising results (e.g., oxamniquine, derivatives of antimalarial drug artemisinin, furoxan, and the kinase inhibi-tor imatinib), but none so far represents a suitable substitute for praziquantel.21,22 The current situation emphasizes the urgent need for novel therapeutic strategies to discover new antischistosomal agents. To evaluate schistosomal sirtuins and their potential as therapeutic drug targets, Lancelot et al. identi-fied and characterized 5 sirtuins encoded in the S. mansoni genome and established their homology relationship to human isoforms. Phylogenetic analysis showed that they are orthologs of mammalian Sirt1, Sirt2, Sirt5, Sirt6, and Sirt7. Initial experiments with adult worms and schistosomula showed strong effects of hSirt2 inhibitors on both life span and reproduction.23 In this study, we present two novel com-plementary in vitro assays to determine smSirt2 activity as well as hits from our screening campaign with IC50 values lower than 50 µM. The heterogeneous assay protocol was transferred to and optimized for hSirt2 as well.
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

Schiedel et al. 3
Materials and Methods
Proteins
A truncated version of smSirt2 (smST1-N1C1; amino acid residues 21 to 322) was amplified using the forward primer smSirT2-N1 (5′-GGATATCCATATGGAACTTAAGTCTC TCAATATAGAG-3′) and the reverse primer smSirT2-C1 (5′-CGCGGATCCTCAATTTAAGCGAGAGTCCGT TTC-3′) inserted into the pnEA-tH vector.24 Overexpression was carried out in Escherichia coli BL21(DE3) cells con-taining the pRARE vector (Novagen, Darmstadt, Germany) in lysogeny broth medium. Induction was done at 25 °C by adding isopropyl-1-thio-β-D-galactopyranoside (IPTG; Euromedex, Souffelweyersheim, France) in a final assay concentration of 0.5 mM, in the presence of 100 µM ZnCl2. Harvested bacteria were resuspended in lysis buffer (10 mM Tris-HCl, 400 mM NaCl, pH 8.0) and lysed by sonica-tion. The lysate was clarified by centrifugation at 17,000 rpm for 1 h. The supernatant was loaded onto Talon Metal affinity resin (Clontech, Mountain View, CA) pre-equilibrated with the lysis buffer. The 3C Protease treatment was used to remove His-tags from the recombinant protein, which was subsequently loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (Amersham Bioscience, Amersham, Little Chalfont, UK) equilibrated in 400 mM NaCl, 10 mM Tris-HCl pH 8.0, and 2 mM DTT. Finally, the protein was con-centrated with Amicon Ultra centrifugal filter units (Millipore, Billerica, MA) to reach a final concentration of 1.5 mg/mL as assayed by the Bio-Rad Protein Assay reagent (Bio-Rad, Hercules, CA). The Coomassie-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel of the purified smSirt2 protein and the chro-matogram of the size-exclusion chromatography are depicted in Supplementary Figure S1. Human Sirt2225–389 was expressed N-terminally tagged with His6
25, as previ-ously described, and purified according to standard proce-dures.26 Trypsin from porcine pancreas (13,000–20,000 BAEE units/mg) was purchased from Sigma-Aldrich (St. Louis, MO).
Inhibitors
The Sirt2 reference inhibitor nicotinamide and Tyrphostin AG-825 were purchased from Sigma-Aldrich, Vatalanib 2HCl was purchased from Selleckchem (Houston, TX), and resvera-trol from TCI (Tokyo, Japan). PU92,27 MS3,28 and MS1028 were synthesized in our lab according to literature procedures.
Homogeneous Fluorescence-Based Assay for smSirt2
To determine smSirt2 activity, we optimized a homoge-neous fluorescence-based deacetylase assay, which was developed in our group for human sirtuins.29 The assay was performed in black 384-well nonbinding plates (Greiner
Bio-One, Monroe, NC) with a reaction volume of 20 µL per well. All tests were performed at least in duplicate using Z-(Ac)Lys-AMC (ZMAL) as a substrate. ZMAL was syn-thesized according to published procedures.29 14 µL SmSirt2 (86 ng/µL, final assay concentration 60 ng/µL) in assay buffer (50 mM Tris, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 0.015% Triton X-100, pH 8.0) was mixed with 2.5 µL of a ZMAL solution [prepared from ZMAL stock solution (12.6 mM) in DMSO diluted with assay buffer; final assay concentration, 10.5 µM], and the inhibitor dissolved in DMSO at various concentrations or DMSO as a control [final DMSO concentration, 5% (v/v)]. Deacetylation was started by addition of NAD+ (final assay concentration, 500 µM). After an incubation of 1 h at 37 °C and 140 rpm, deacetylation was stopped by adding 20 µL of a solution containing trypsin and nicotinamide [50 mM Tris, 100 mM NaCl, 6.7% (v/v) DMSO, trypsin 1 mg/mL, 8 mM nicotinamide, pH 8.0]. The microplate was further incubated for 20 min at 37 °C and 140 rpm. Finally, fluores-cence intensity was measured in a microplate reader (BMG Polarstar; λEx = 390 nm, λEm = 460 nm; BMG Labtech, Ortenberg, Germany). Rates of inhibition were calculated using the controls, containing no inhibitor, as a reference for 0% inhibition. The value for 100% inhibition was generated by blank controls containing only ZMAL, NAD+, DMSO, and assay buffer. Graphpad Prism software (Graphpad Software, La Jolla, CA) was used to determine IC50 values.
Heterogeneous Fluorescence-Based Assay for smSirt2
The heterogeneous fluorescence-based assay for smSirt2 was performed at least in duplicate in Hard-Shell low- profile thin-wall 96-well skirted PCR plates (Bio-Rad) using Ac-Glu-Glu-Lys(biotin)-Gly-Gln-Ser-Thr-Ser-Ser-His-Ser-Lys(Ac)-Nle-Ser-Thr-Glu-Gly-Lys(5/6-carboxytetr- a methylrhodamine)-Glu-Glu-NH2, hereafter called Ac-p53-TAMRA, as a substrate (final assay concentration, 250 nM).30 It was custom synthesized (PSL, Heidelberg, Germany). The reaction volume was adjusted to 50 µL per well. Twenty microliters of SmSirt2 (250 ng/µL, final assay concentration 100 ng/µL) in assay buffer (50 mM Tris, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0) were mixed with inhibitor dissolved in DMSO at various concentrations or DMSO as a control [final DMSO concentration, 5% (v/v)]. The enzymatic reaction was started by adding 10 µL of start solution (1.25 µM Ac-p53-TAMRA, 750 µM NAD+ in assay buffer). Then, the plate was covered with a TopSeal-A cover (PerkinElmer, Waltham, MA) and incu-bated for 2 h at 37 °C and 140 rpm. Deacetylation was stopped by adding 20 µL of a stop solution containing tryp-sin and nicotinamide [50 mM Tris, 100 mM NaCl, 11.7% (v/v) DMSO, trypsin 35 µg/mL, 14 mM nicotinamide, pH 8.0]. The reaction mixture was incubated for 20 min at 37 °C and 140 rpm. The next step consisted of the addition
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

4 Journal of Biomolecular Screening
of streptavidin-coupled magnetic beads (Dynabeads MyOne™ Streptavidin T1, 10 mg/mL stock solution; Life Technologies, Carlsbad, CA), 10 µg per well (1 µL bead stock solution), sus-pended in 30 µL assay buffer, and a further incubation of 20 min at 37 °C and 140 rpm. Afterward, the microplate was placed on a DynaMag™-96 side magnet (Life Technologies, Carlsbad, CA) for 3 min to pull the magnetic beads to the side of the PCR tubes. The supernatant was discarded, and the beads were washed with washing buffer [80 mM Tris, 20 mM Tris-HCl, 150 mM NaCl, Tween-20 0.1% (v/v), pH 7.5, 80 µL per well, 4 times]. Subsequently, the magnet was removed, and the samples were resuspended in 80 µL washing buffer and transferred to a black 96-well plate (OptiPlate-96 F, PerkinElmer). Fluorescence intensity was measured in a microplate reader (EnVision; PerkinElmer; λEx = 531 nm, λEm = 591 nm). Rates of inhibition were determined by using controls, containing no inhibitor, as a reference for 0% inhibition. The top of the assay window was defined by samples containing 750 µM nicotinamide as a point of reference for 100% inhibition. Graphpad Prism software (Graphpad Software) was used to determine IC50 values.
Heterogeneous Fluorescence-Based Assay for hSirt2
To determine hSirt2 activity in a heterogeneous fluores-cence-based assay, we used the same protocol as we used for smSirt2 with minor modifications. hSirt2 was adjusted to a final assay concentration of 38 ng/µL, and incubation time of deacetylation was prolonged to 2.5 h.
Counterscreening for Trypsin Inhibition
Because the presented assay protocols rely on trypsin cleav-age of the converted substrate, a counterscreening for tryp-sin inhibition was performed (see Suppl. Material).
Km Determination
For the characterization of substrate affinity, Km values for both substrates were determined (see Suppl. Material).
Z-Factor (Z’-Factor) Measurement
Z-factors (Z’-factors) were determined by measurements of signals for 0% and 100% inhibition (n = 24).31
Computational Studies
See supplementary material.
Results
Screening Procedure
For this initial screen, we preselected known sirtuin inhibi-tors of the human isoforms, nicotinamide analogs and
kinase inhibitors from our internal libraries. Nicotinamide analogs were selected due to their similarity to the endoge-neous sirtuin inhibitor nicotinamide. Kinase inhibitors were tested because adenosine triphosphate mimetics could fit in the NAD+-binding pocket of sirtuins as well, because both enzymes use cofactors that bear an adenosine moiety.32 In the initial screen, all compounds were tested at concentra-tions of 20 and 100 µM. Because this is the first inhibitor screen with smSirt2, we initially chose a rather high thresh-old to definitely identify potential inhibitors. Hits of initial screening (inhibitor @ 100 µM > 50%) from the homoge-neous fluorescence-based assay for smSirt2 were tested for trypsin inhibition (Suppl. Fig. S4) and further confirmed by retesting using our new heterogeneous fluorescence assay format. Rationalization of biological studies was conducted by homology modeling and docking (see Supplement). Confirmed hits were counterscreened for their hSirt2 activ-ity (Suppl. Table S1). Overall, we tested 230 compounds, detected 9 compounds with an inhibition higher than 50% at 100 µM in the initial screen by means of the homogeneous fluorescence-based assay for smSirt2, and characterized 4 compounds with an IC50 lower than 50 µM using our het-erogeneous fluorescence-based assay for smSirt2 (Suppl. Table S2).
Homogeneous Fluorescence-Based Assay for smSirt2
To determine smSirt2 activity, we optimized a homogeneous fluorescence-based deacetylase assay, which was developed in our group for human sirtuins.29 Under assay conditions, which were used for the human isoform, smSirt2 showed less conversion of the substrate ZMAL. In the course of a buffer optimization, we identified Triton X-100 to have beneficial effects on smSirt2 activity (Fig. 1A). Triton X-100 is a non-ionic surfactant that prevents adsorption of proteins to the assay plate surface. We adjusted a final assay concentration of 0.015% (v/v) to take full advantage of the surface-blocking capability without exceeding the critical micellar concentra-tion of 0.0155%. By adding Triton X-100 to the assay buffer and a miniaturization of assay volume, we were able to reduce enzyme consumption per measurement by a factor of 50. A Km value of 59 µM was determined, and the ZMAL concen-tration was adjusted to 10.5 µM to be sensitive to substrate competitive inhibitors (Fig. 1B). Measuring enzyme kinetics enabled us to reduce the incubation time from initially 4 h to 1 h (Fig. 1C). With 1 h of incubation, we are already at the upper limit of linearity. We use these conditions to maximize the signal window, however. In Supplementary Figure S5, we show that the IC50 values for nicotinamide after an incuba-tion of 0.5 h and 1 h do not differ significantly. Z’-factor deter-mination resulted in a Z’-factor of 0.77, which is considered to be excellent (Suppl. Fig. S7A). Variations in DMSO concen-tration revealed a decrease of smSirt2 activity by increasing DMSO concentration (Fig. 1D). Therefore, we adjusted the
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

Schiedel et al. 5
DMSO concentration to 5% (v/v) to facilitate good compound solubility while maintaining good enzyme activity. Higher DMSO concentrations, however, are also tolerated. IC50 determination of the endogenous sirtuin inhibitor nicotin-amide exhibited an IC50 of 32 µM ± 2 µM (Fig. 1E). We then applied our new homogeneous assay to a focused compound library of 230 potential smSirt2 inhibitors and identified 3 hits showing an IC50 lower than 50 µM. The thiobarbiturate MS3 showed the strongest inhibition of smSirt2 with an IC50 of 9 µM ± 1 µM. The kinase inhibitors Vatalanib 2HCl and Tyrphostin AG-825 were detected with similar IC50 values of, respectively, 30 µM ± 2 µM and 31 µM ± 1 µM. To face the controversy surrounding sirtuin assays using a fluorescence-labeled substrate, we tested resveratrol, a known artifact acti-vator, with our heterogeneous fluorescence-based assay. We detected no activation of smSirt2 and only a very weak inhibi-tion with an IC50 value higher than 500 µM (Table 1).
Heterogeneous Fluorescence-Based Assay for smSirt2
By means of our novel homogeneous fluorescence-based assay, we are not able to evaluate the inhibitory effects on
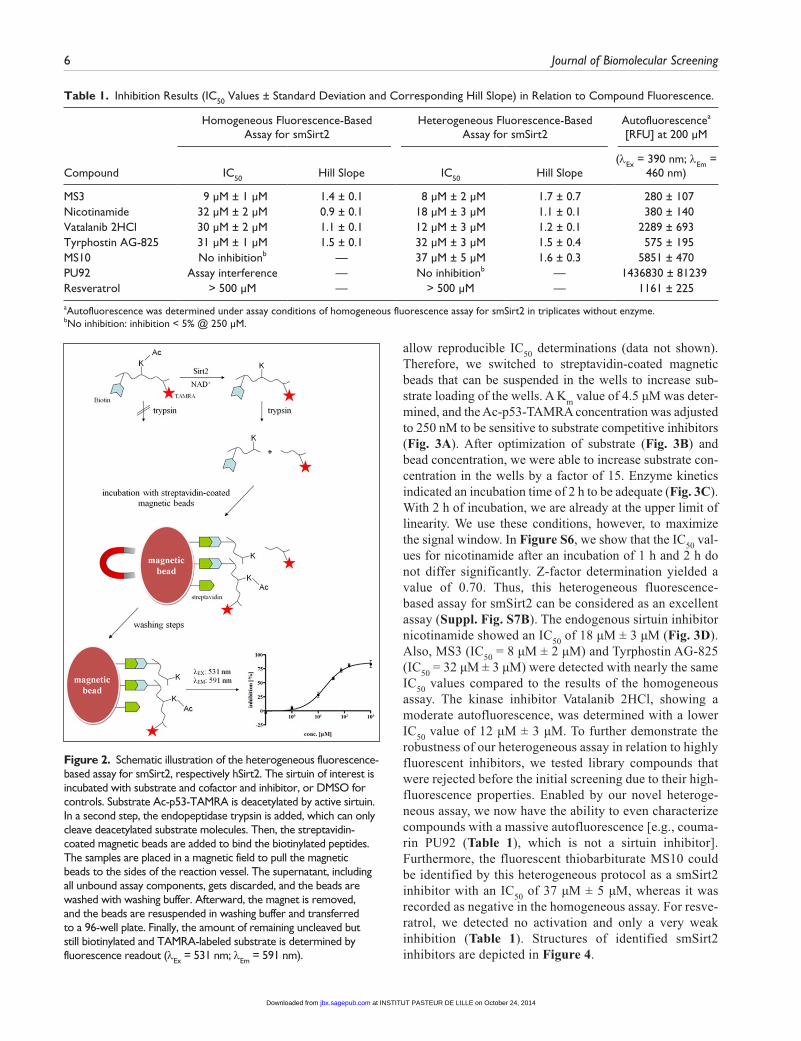
smSirt2 activity of library compounds showing high autofluo-rescence or quenching effects under given assay conditions. Therefore, we developed a heterogeneous fluorescence-based assay for smSirt2 that is not prone to assay interference by compound properties. As a substrate, we choose Ac-p53-TAMRA, a 20–amino acid substrate derived from the sequence of p53, which is a substrate for both hSirt1 and hSirt2.30 The peptide is N-terminally linked to biotin and C-terminally mod-ified with a fluorescent rhodamine-derived tag. These modifi-cations allow immobilization on streptavidin-coated surfaces via biotin and fluorescence detection due to the fluorescence label TAMRA. The reaction for monitoring enzyme activity is a coupled enzymatic reaction. The first reaction, the deacety-lation reaction of Lys(Ac)-12, catalyzed by smSirt2, is detected by coupling to a cleavage at the newly exposed lysine residue by the endopeptidase trypsin.30 Finally, after washing away all unbound assay components, the amount of remaining uncleaved but still biotinylated and TAMRA-labeled substrate is determined by measuring fluorescence intensity (Fig. 2).
Initial experiments with streptavidin-coated microplates showed that Ac-p53-TAMRA is accepted by smSirt2 as a sub-strate. Because of the low binding capacity of streptavidin-coated microplates, the assay window was too small to
Figure 1. Assay development of homogeneous fluorescence-based assay for smSirt2. (A) Beneficial effects of Triton X-100 on smSirt2 activity. (B) Michaelis–Menten plot shows the Z-(Ac)Lys-AMC (ZMAL)-dependent activity of smSirt2. (C) Time course of conversion in the deacetylation reaction with the smSirt2 substrate ZMAL. (D) DMSO tolerance of smSirt2-catalyzed ZMAL conversion was investigated in a range from 0% to 15% (v/v). (E) IC50 determination of endogenous smSirt2 inhibitor nicotinamide resulted in an IC50 of 32 µM ± 2 µM.
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from
6 Journal of Biomolecular Screening
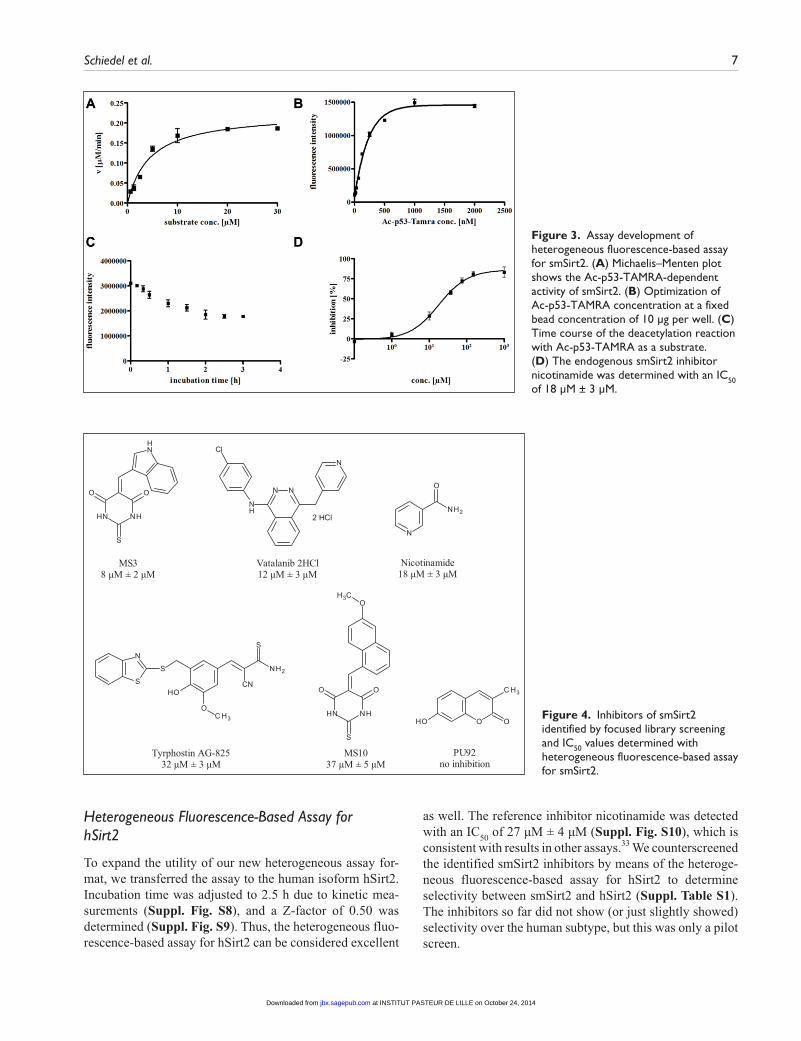
allow reproducible IC50 determinations (data not shown). Therefore, we switched to streptavidin-coated magnetic beads that can be suspended in the wells to increase sub-strate loading of the wells. A Km value of 4.5 µM was deter-mined, and the Ac-p53-TAMRA concentration was adjusted to 250 nM to be sensitive to substrate competitive inhibitors (Fig. 3A). After optimization of substrate (Fig. 3B) and bead concentration, we were able to increase substrate con-centration in the wells by a factor of 15. Enzyme kinetics indicated an incubation time of 2 h to be adequate (Fig. 3C). With 2 h of incubation, we are already at the upper limit of linearity. We use these conditions, however, to maximize the signal window. In Figure S6, we show that the IC50 val-ues for nicotinamide after an incubation of 1 h and 2 h do not differ significantly. Z-factor determination yielded a value of 0.70. Thus, this heterogeneous fluorescence-based assay for smSirt2 can be considered as an excellent assay (Suppl. Fig. S7B). The endogenous sirtuin inhibitor nicotinamide showed an IC50 of 18 µM ± 3 µM (Fig. 3D). Also, MS3 (IC50 = 8 µM ± 2 µM) and Tyrphostin AG-825 (IC50 = 32 µM ± 3 µM) were detected with nearly the same IC50 values compared to the results of the homogeneous assay. The kinase inhibitor Vatalanib 2HCl, showing a moderate autofluorescence, was determined with a lower IC50 value of 12 µM ± 3 µM. To further demonstrate the robustness of our heterogeneous assay in relation to highly fluorescent inhibitors, we tested library compounds that were rejected before the initial screening due to their high-fluorescence properties. Enabled by our novel heteroge-neous assay, we now have the ability to even characterize compounds with a massive autofluorescence [e.g., couma-rin PU92 (Table 1), which is not a sirtuin inhibitor]. Furthermore, the fluorescent thiobarbiturate MS10 could be identified by this heterogeneous protocol as a smSirt2 inhibitor with an IC50 of 37 µM ± 5 µM, whereas it was recorded as negative in the homogeneous assay. For resve-ratrol, we detected no activation and only a very weak inhibition (Table 1). Structures of identified smSirt2 inhibitors are depicted in Figure 4.
Table 1. Inhibition Results (IC50 Values ± Standard Deviation and Corresponding Hill Slope) in Relation to Compound Fluorescence.
Homogeneous Fluorescence-Based Assay for smSirt2
Heterogeneous Fluorescence-Based Assay for smSirt2
Autofluorescencea [RFU] at 200 µM
Compound IC50 Hill Slope IC50 Hill Slope(λEx = 390 nm; λEm =
460 nm)
MS3 9 µM ± 1 µM 1.4 ± 0.1 8 µM ± 2 µM 1.7 ± 0.7 280 ± 107Nicotinamide 32 µM ± 2 µM 0.9 ± 0.1 18 µM ± 3 µM 1.1 ± 0.1 380 ± 140Vatalanib 2HCl 30 µM ± 2 µM 1.1 ± 0.1 12 µM ± 3 µM 1.2 ± 0.1 2289 ± 693Tyrphostin AG-825 31 µM ± 1 µM 1.5 ± 0.1 32 µM ± 3 µM 1.5 ± 0.4 575 ± 195MS10 No inhibitionb — 37 µM ± 5 µM 1.6 ± 0.3 5851 ± 470PU92 Assay interference — No inhibitionb — 1436830 ± 81239Resveratrol > 500 µM — > 500 µM — 1161 ± 225
aAutofluorescence was determined under assay conditions of homogeneous fluorescence assay for smSirt2 in triplicates without enzyme.bNo inhibition: inhibition < 5% @ 250 µM.
Figure 2. Schematic illustration of the heterogeneous fluorescence-based assay for smSirt2, respectively hSirt2. The sirtuin of interest is incubated with substrate and cofactor and inhibitor, or DMSO for controls. Substrate Ac-p53-TAMRA is deacetylated by active sirtuin. In a second step, the endopeptidase trypsin is added, which can only cleave deacetylated substrate molecules. Then, the streptavidin-coated magnetic beads are added to bind the biotinylated peptides. The samples are placed in a magnetic field to pull the magnetic beads to the sides of the reaction vessel. The supernatant, including all unbound assay components, gets discarded, and the beads are washed with washing buffer. Afterward, the magnet is removed, and the beads are resuspended in washing buffer and transferred to a 96-well plate. Finally, the amount of remaining uncleaved but still biotinylated and TAMRA-labeled substrate is determined by fluorescence readout (λEx = 531 nm; λEm = 591 nm).
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from
Schiedel et al. 7
Heterogeneous Fluorescence-Based Assay for hSirt2
To expand the utility of our new heterogeneous assay for-mat, we transferred the assay to the human isoform hSirt2. Incubation time was adjusted to 2.5 h due to kinetic mea-surements (Suppl. Fig. S8), and a Z-factor of 0.50 was determined (Suppl. Fig. S9). Thus, the heterogeneous fluo-rescence-based assay for hSirt2 can be considered excellent
as well. The reference inhibitor nicotinamide was detected with an IC50 of 27 µM ± 4 µM (Suppl. Fig. S10), which is consistent with results in other assays.33 We counterscreened the identified smSirt2 inhibitors by means of the heteroge-neous fluorescence-based assay for hSirt2 to determine selectivity between smSirt2 and hSirt2 (Suppl. Table S1). The inhibitors so far did not show (or just slightly showed) selectivity over the human subtype, but this was only a pilot screen.
MS38 µM ± 2 µM
MS1037 µM ± 5 µM
PU92no inhibition
2 HCl
Vatalanib 2HCl12 µM ± 3 µM
Nicotinamide18 µM ± 3 µM
Tyrphostin AG-82532 µM ± 3 µM
C H3
OOOH
CH3O
O
S
O
NHNH
NH2
O
N
NH2
S
C H3
O
OHS
N
S
O
S
O
NHNH
NH
N
NN
NH
Cl
CN
Figure 3. Assay development of heterogeneous fluorescence-based assay for smSirt2. (A) Michaelis–Menten plot shows the Ac-p53-TAMRA-dependent activity of smSirt2. (B) Optimization of Ac-p53-TAMRA concentration at a fixed bead concentration of 10 µg per well. (C) Time course of the deacetylation reaction with Ac-p53-TAMRA as a substrate. (D) The endogenous smSirt2 inhibitor nicotinamide was determined with an IC50 of 18 µM ± 3 µM.
Figure 4. Inhibitors of smSirt2 identified by focused library screening and IC50 values determined with heterogeneous fluorescence-based assay for smSirt2.
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

8 Journal of Biomolecular Screening
Computational Studies
To rationalize the interaction of the identified inhibitors with smSirt2, we docked MS3 and Vatalanib into a gener-ated homology model of smSirt2 (smSirt2-HM). Comparison of smSirt2-HM with the human Sirt2 crystal structure (PDB ID 3ZGV) reveals that smSirt2-HM shows the classical α–β scaffold folding of human sirtuins, and all major secondary structure elements are well matched. The largest deviation can be observed for the additional α helix, α13 (human Sirt2 numbering), that packs against the out-side of the large domain containing the catalytic site (Suppl. Fig. S11). The smSirt2-HM structure reflects a similar NAD+ binding pocket, as seen in the crystal structure of Sirt2 bound to ADP-ribose. Moreover, smSirt2-HM shares a similar acetyl lysine channel as seen in the crystal struc-tures of human sirtuins (Fig. 5A). The comparison of human Sirt2 and the smSirt2-HM shows that the peptide binding groove consists of the same amino acid residues with few exceptions. Ile169–hSirt2 is changed to Val132, Ile232–hSirt2 is changed to Val193, and Phe234–hSirt2 is changed to Leu195 in smSirt2 (Fig. 5A).
Both docked inhibitors show favorable hydrogen bond interactions with the conserved residue Val194 in the peptide
binding pocket (Fig. 5B and 5C), which is the interaction site for the amide group of the substrate. The NH group of the thio-barbiturate ring of MS3 as well as the amino group present in Vatalanib show favorable H-bonds to the backbone carbonyl of Val194. The aromatic rings of both inhibitors are making van der Waals interactions with the hydrophobic residues and fit nicely to the substrate pocket (Phe59, Phe82, Val132, His150, Val193, and Phe196) (Fig. 5D).
Discussion
The development of biochemical in vitro assays is one of the most important steps in the identification and character-ization of novel modifiers of epigenetic targets, not only for drug discovery but also to elucidate biochemical func-tions of a specific enzyme. Here, we present a convergent assay platform, including the first two assays for smSirt2, a schistosomal sirtuin that is crucial for reproduction and sur-vival of the parasite. This target-based approach will not guarantee to furnish compounds with in vivo activity on Schistosoma mansoni due to several reasons (e.g., pharma-cokinetics). Nevertheless, the presented assay platform allows a rational pre-selection of the compounds showing in vitro activity, which will be further characterized in vivo.
Figure 5. (A) Homology model of smSirt2 (dark green ribbon) in complex with docked acetyl-lysine peptide (carbon atoms colored magenta, gold ribbon). Residues that are different in the smSirt2 acetyl lysine channel compared to human Sirt2 are shown in green. Only interacting amino acid residues are shown for clarity. Hydrogen bonds are shown as dashed lines. (B) Homology model of smSirt2 (dark green ribbon) in complex with docked Vatalanib (carbon atoms colored cyan). Residues that are different in the smSirt2 acetyl lysine channel compared to human Sirt2 are shown in green. Only interacting amino acid residues are shown for clarity. Hydrogen bonds are shown as dashed lines. (C) Docking result obtained for MS3 (carbon atoms are shown in orange). (D) Docking pose of Vatalanib (carbon atoms colored cyan) compared to the bound acetyl-lysine group (carbon atoms colored magenta). The molecular surface of the peptide binding pocket is displayed and colored according to the hydrophobicity. Polar regions are shown in magenta, and hydrophobic regions in green.
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

Schiedel et al. 9
The homogeneous assay enables us, by using the small-molecule substrate ZMAL, to screen large compound librar-ies in a cost-efficient, high-throughput manner. IC50 values are highly reproducible and display minor standard devia-tions. This assay, however, is vulnerable with regard to compounds with strong fluorescence or quenching proper-ties. Thus, the heterogeneous assay protocol is an ideal extension of the assay platform, because its detection is not influenced by compound interference. This allows us to characterize compounds exhibiting very high autofluores-cence and helps us to identify inhibitors, which would be registered as false negatives in the homogeneous assay. We were able to show that the presented homogeneous and het-erogeneous assays are not prone to a prototypical activator that produces artifacts with fluorescently labeled substrates (i.e., resveratrol). Furthermore, the heterogeneous protocol facilitates the confirmation of data of initial screening, using a peptide substrate derived from the physiological sir-tuin substrate p53. The assay procedure of the heteroge-neous assay is more complex and more costly due to the streptavidin-coated beads. Nevertheless, the assay is in principle suitable for high-throughput screening, and com-pared to other heterogeneous assays, which are often anti-body based, the costs are rather low. In addition, we are not dependent on the availability of a highly selective antibody. We have shown that it might also be expanded to other sir-tuins, as exemplified with human Sirt2. Our initial hits either show affinity to the hSirt2 (Suppl. Table S1) or are potent inhibitors of human kinases as well (Vatalanib). We expect virtual screening campaigns based on our smSirt2 homology model, or larger library screens will identify can-didates for selective smSirt2 inhibitors. We are currently not able to evaluate selectivity among the five schistosomal sir-tuin isoforms due to the lack of available protein prepara-tions and in vitro assays. Notwithstanding, the inhibitor MS3 shows strong effects on both the life span and repro-duction of S. mansoni.23 The most important goal is to achieve selectivity over human sirtuins. Hence, further screening and hit optimization are required to yield smSirt2-selective inhibitors, and our assays provide an ideal starting point for this work.
Acknowledgments
We thank K. Schmidtkunz, University of Freiburg, for technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This
work and the authors of this manuscript received funding from the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agree-ments nos. 241865 (SEtTReND) and 602080 (A-ParaDDisE). Research on human sirtuins was funded by the Deutsche Forschungsgemeinschaft (DFG, Ju295/8-1, Si868/6-1).
References
1. Jung, M.; Sippl, W. Epigenetic Targets in Drug Discovery. Wiley-VCH: Weinheim, 2009.
2. Tropberger, P.; Pott, S.; Keller, C.; et al. Regulation of Transcription through Acetylation of H3K122 on the Lateral Surface of the Histone Octamer. Cell. 2013, 152, 859–872.
3. Biel, M.; Wascholowski, V.; Giannis, A. Epigenetics: An Epicenter of Gene Regulation: Histones and Histone-Modifying Enzymes. Angew. Chem. 2005, 117, 3248–3280.
4. Trapp, J.; Jung, M. The Role of NAD Dependent Histone Deacetylases (Sirtuins) in Ageing. Curr. Drug Targets. 2006, 7, 1553–1560.
5. North, B. J.; Verdin, E. Sirtuins: Sir2-Related NAD-Dependent Protein Deacetylases. Genome Biol. 2004, 5, 224.
6. Greiss, S.; Gartner, A. Sirtuin/Sir2 Phylogeny, Evolutionary Considerations and Structural Conservation. Mol. Cell. 2009, 28, 407–415.
7. Michishita, E.; Park, J. Y.; Burneskis, J. M.; et al. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell. 2005, 16, 4623–4635.
8. North, B. J.; Marshall, B. L.; Borra, M. T.; et al. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell. 2003, 11, 437–444.
9. van Leeuwen, I. M.; Higgins, M.; Campbell, J.; et al. Modulation of p53 C-Terminal Acetylation by mdm2, p14ARF, and Cytoplasmic SirT2. Mol. Cancer Ther. 2013, 12, 471–480.
10. North, B. J.; Rosenberg, M. A.; Jeganathan, K. B.; et al. SIRT2 Induces the Checkpoint Kinase BubR1 to Increase Lifespan. EMBO J. 2014, e201386907. [Epub ahead of print]
11. Haigis, M. C.; Mostoslavsky, R.; Haigis, K. M.; et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic Beta Cells. Cell. 2006, 126, 941–954.
12. Du, J.; Zhou, Y.; Su, X.; et al. Sirt5 Is an NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science. 2011, 334, 806–809.
13. Jiang, H.; Khan, S.; Wang, Y.; et al. Sirt6 Regulates TNF Secretion through Hydrolysis of Long-Chain Fatty Acyl Lysine. Nature. 2013, 496, 110–113.
14. Zhu, A. Y.; Zhou, Y.; Khan, S.; et al. Plasmodium falciparum Sir2A Preferentially Hydrolyzes Medium and Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2012, 7, 155–159.
15. Uciechowska, U.; Schemies, J.; Scharfe, M.; et al. Binding Free Energy Calculations and Biological Testing of Novel Thiobarbiturates as Inhibitors of the Human NAD+ Dependent Histone Deacetylase Sirt2. Med. Chem. Commun. 2012, 3, 167–173.
16. Smith, M. R.; Syed, A.; Lukacsovich, T.; et al. A Potent and Selective Sirtuin 1 Inhibitor Alleviates Pathology in Multiple
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from

10 Journal of Biomolecular Screening
Animal and Cell Models of Huntington’s Disease. Hum. Mol. Genet. 2014, 23, 2995–3007.
17. Peck, B.; Chen, C. Y.; Ho, K. K.; et al. SIRT Inhibitors Induce Cell Death and p53 Acetylation through Targeting Both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855.
18. Marek, M.; Kannan, S.; Hauser, A. T.; et al. Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645.
19. Stolfa, D. A.; Marek, M.; Lancelot, J.; et al. Molecular Basis for the Antiparasitic Activity of a Mercaptoacetamide Derivative That Inhibits Histone Deacetylase 8 (HDAC8) from the Human Pathogen Schistosoma mansoni. J. Mol. Biol. 2014, http://dx.doi.org/10.1016/j.jmb.2014.03.007.
20. Carneiro, V. C.; de Moraes Maciel, R.; de Abreu da Silva, I. C.; et al. The Extracellular Release of Schistosoma man-soni HMGB1 Nuclear Protein Is Mediated by Acetylation. Biochem. Biophys. Res. Commun. 2009, 390, 1245–1249.
21. Cioli, D.; Pica-Mattoccia, L.; Basso, A.; et al. Schistosomiasis Control: Praziquantel Forever? Mol. Biochem. Parasitol. 2014, 195, 23–29.
22. Buro, C.; Beckmann, S.; Oliveira, K. C.; et al. Imatinib Treatment Causes Substantial Transcriptional Changes in Adult Schistosoma mansoni In Vitro Exhibiting Pleiotropic Effects. PLoS Negl. Trop. Dis. 2014, 8, e2923.
23. Lancelot, J.; Caby, S.; Dubois-Abdesselem, F.; et al. Schistosoma mansoni Sirtuins: Characterization and Potential as Chemotherapeutic Targets. PLoS Negl. Trop. Dis. 2013, 7, e2428.
24. Diebold, M. L.; Fribourg, S.; Koch, M.; et al. Deciphering Correct Strategies for Multiprotein Complex Assembly by
Co-Expression: Application to Complexes as Large as the Histone Octamer. J. Struct. Biol. 2011, 175, 178–188.
25. North, B. J.; Schwer, B.; Ahuja, N.; et al. Preparation of Enzymatically Active Recombinant Class III Protein Deacetylases. Methods. 2005, 36, 338–345.
26. Neugebauer, R. C.; Uchiechowska, U.; Meier, R.; et al. Structure-Activity Studies on Splitomicin Derivatives as Sirtuin Inhibitors and Computational Prediction of Binding Mode. Med. Chem. 2008, 51, 1203–1213.
27. Leonetti, F.; Favia, A.; Rao, A.; et al. Design, Synthesis, and 3D QSAR of Novel Potent and Selective Aromatase Inhibitors. J. Med. Chem. 2004, 47, 6792–6803.
28. Schmitt, M. L. Synthese von Arylidenthiobarbitursäuren als Leitstrukturen neuartiger Sirtuin-Inhibitoren. Diploma thesis, University of Freiburg, 2009.
29. Heltweg, B.; Trapp, J.; Jung, M. In Vitro Assays for the Determination of Histone Deacetylase Activity. Methods. 2005, 36, 332–337.
30. Milne, J. C.; Lambert, P. D.; Schenk, S.; et al. Small Molecule Activators of SIRT1 as Therapeutics for the Treatment of Type 2 Diabetes. Nature. 2007, 450, 712–716.
31. Zhang, J. H.; Chung, T. D.; Oldenburg, K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73.
32. Trapp, J.; Jochum, A.; Meier, R.; et al. Adenosine Mimetics as Inhibitors of NAD+-Dependent Histone Deacetylases, from Kinase to Sirtuin Inhibition. J. Med. Chem. 2006, 49, 7307–7316.
33. Tervo, A. J.; Kyrylenko, S.; Niskanen, P.; et al. An In Silico Approach to Discovering Novel Inhibitors of Human Sirtuin Type 2. J. Med. Chem. 2004, 47, 6292–6298.
at INSTITUT PASTEUR DE LILLE on October 24, 2014jbx.sagepub.comDownloaded from
Related Documents











