BMLT INTERNSHIP RECORDS HALDIA INSTITUTE OF HEALTH SCIENCES (AN INSTITUTE OF ICARE) HALDIA AFFILIATED TO VIDYASAGAR UNIVERSITY Main Training Place: INSTITUTE OF POSTGRADUATE MEDICAL EDUCATION & RESEARCH Name: SWARNA SAIN University Roll No.: 000359 Regd. No.: PM/BMLT/VIS/61, NO07035

Final Project 3
Oct 26, 2014
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ð BMLT INTERNSHIP RECORDS
HALDIA INSTITUTE OF HEALTH SCIENCES (AN INSTITUTE OF ICARE)
HALDIA AFFILIATED TO VIDYASAGAR UNIVERSITY
Main Training Place: INSTITUTE OF POST-‐GRADUATE MEDICAL EDUCATION & RESEARCH
Name: SWARNA SAIN
University Roll No.: 000359 Regd. No.: PM/BMLT/VI-‐S/61, NO-‐-‐-‐07035
BMLT INTERNSHIP RECORD 2
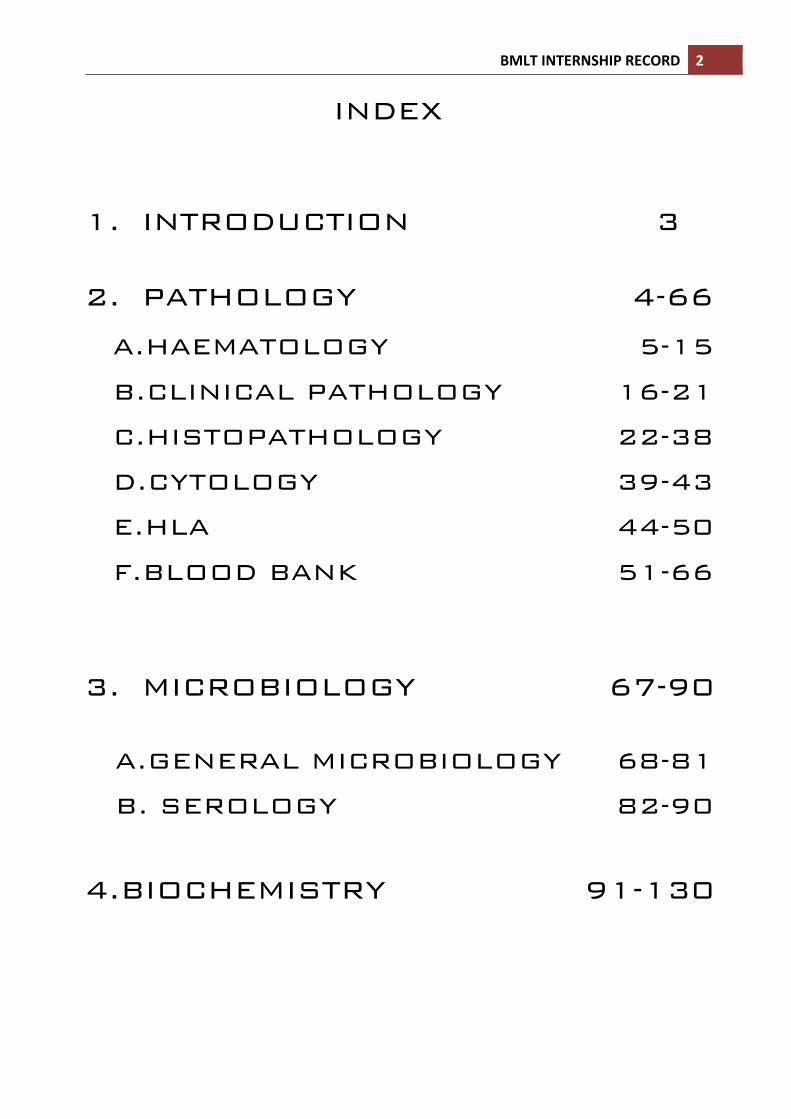
INDEX 1. INTRODUCTION 3
2. PATHOLOGY 4-66
A.HAEMATOLOGY 5-15
B.CLINICAL PATHOLOGY 16-21
C.HISTOPATHOLOGY 22-38
D.CYTOLOGY 39-43
E.HLA 44-50
F.BLOOD BANK 51-66
3. MICROBIOLOGY 67-90
A.GENERAL MICROBIOLOGY 68-81
B. SEROLOGY 82-90
4.BIOCHEMISTRY 91-130

BMLT INTERNSHIP RECORD 3
Introduction
These are the records, which I have kept during my pursuit of gathering knowledge in S.S.K.M Institution and Hospital, working in Pathology, Microbiology and Biochemistry department. As a student of sixth semester graduation course of Medical Laboratory Technology it was my duty to acquire deep knowledge and skills to become professionally sound and competent in necessary techniques through the various theoretical and practical classes that have taken place in the myriad of well equipped laboratories, along with the hospital duties that we have been a part of. After completion of those, I earned an Internship for 6 months in S.S.K.M Kolkata. During this period I have gained vast knowledge and mastered various skills and techniques from the learned and talented eminent teachers and seniors who I have found in SSKM.I believe myself to be fortunate enough to be a part of such engaging and pleasant experience. With this brief overview I now proceed to stay brief with the work done by me during the Internship period in which, I have tried to reflect the knowledge I have acquired during the internship period at SSKM Hospital.

BMLT INTERNSHIP RECORD 4
PATHOLOGY
o HAEMATOLOGY
o CLINICAL PATHOLOGY
o HITOPATHOLOGY
o CYTOLOGY
o HLA
o BLOOD BANK

BMLT INTERNSHIP RECORD 5
HAEMATOLOGY
o INTRODUCTION OF HAEMATOLOGY
o CLEARING OF EQUIPMENT
o ANTICOAGULANT
o COLLECTION OF BLOOD
o STUDY OF BLOOD SMEAR FOR DIFFERENTIAL COUNT AND CELL MORPHOLOGY
o DETERMINATION OF ERYTHROCYTE SEDIMENTATION RATE BY WESTERGREN’S METHOD
o DETERMINATION OF HAEMOGLOBIN CONCENTRATION BY CYANMETHAEMOGLOBIN METHOD
o DETERMINATION OF ERYTHROCYTE SEDIMENTATION RATE BY WESTERGREN’S METHOD
o DETERMINATION OF PROTHROMBIN TIME
o DETERMINATION OF ACTIVATED PARCIAL
THROMBOPLASTIN TIME

BMLT INTERNSHIP RECORD 6
INTRODUCTION OF HAEMATOLOGY
Haematology ( “haima” means blood and “logos” means study ) is the study of blood. It is
concerned primarily with the study of the formed elements of blood which include erythrocytes or red blood cells (RBC) , leukocytes or white blood cells (WBC) and thrombocytes or platelets (PLT) . The enumeration of cells in circulation , haemoglobin concentration, and differential count of leukocytes based on the study of the stained blood smear. Study of the blood smear also helps in detecting the morphological abnormalities of various cells seen in the peripheral blood circulation. The haematology laboratory also helps to investigate the causes of bleeding disorders. The technician working in the haematology laboratory should be aware of with the structure, functions, production and development of various cellular elements of circulating blood in order to comprehend the clinical significance of different haematological tests and can identify his mistakes.

BMLT INTERNSHIP RECORD 7
CLEARING OF EQUIPMENT:
(A)Pipettes: (1)Once a pipette is used, it is rinsed immediately in a steam of cold water to remove blood, urine, serum, reagents etc. If the pipettes is used to measure infected material. They are left in a bowl full of disinfectant solution (1% Hypochlorite solution)
(2)Then they are soaked in a bowl of mild detergent solution 2-‐3 hours and then rinsed by tap water.
(B)Slide:
(1)The slides are kept in a bowl of detergent solution. If they contain infected material they should be first kept in a disinfectant solution.
(2)Rinsing is Done by tap water one by one.
(C)Westergren:
They are rinsed in water and then they are left to soak in clean water. Then they are kept incubator at 370c for drying.
ANTICOAGULANT
EDTA: (Ethelene Diamine Tetraacitic Acid)
Does: 2mg /ml of blood. Action: It acts as a Powerful calcium enclating agent. It convert the ionic calcium to unionised form. Tests which are done by EDTA blood:=>
(i)Haemoglobin. (ii) WBC count. (iii) RBC count. (iv) PCV determination. (v) ESR by Wintrobe`s method. (vi) Platelet count.
(vii)Differential WBC count.
Advantage or merits: (i) It gives the best preservative of cellular morphology. Good morphology is observed even
after 2-‐3 hours of blood collection. (ii) Since platelet clumping is inhibited, for platelet count by using this anticoagulant is
preferred.
Disadvantage: (i) Excess of EDTA affects both red cell and leukocytes causing shrinkage and degenerative
changes. (ii) Excess of EDTA may cause significant decrease in packed cell volume(PCV) and increase in
mean cell haemoglobin concentration(MCHC).

BMLT INTERNSHIP RECORD 8
(iii) Platelets swell and disintegrate due to excess of EDTA and false high platelet count may be obtained due to disintegrate platelet.
(iv) It is not suitable for use in coagulation study (mainly in the determination of Prothrombin time).
(v) EDTA inhibits enzyme such as alkaline phosphatage and creative kinage. It is not suitable for calcium and ions analysis.
Potasium oxalate + calcium = calcium oxalate. Sodium citrate:
It solution act a concentration of 3.8% (gm/dl) is used for ESR by Westergren method in a ratio of 1:4, that is 1 part of sodium citrate and 4 part of blood. In Prothrombin time test 1 part of sodium citrate and 9 part of blood. Action: Citrate prevent the blood from coagulation by chelating with calcium (sodium-‐calcium citrate). This is best anticoagulant for coagulation study because the chelating effect of calcium is easily reversible by addition of calcium. It prevent labile procoagulants. Advantage: (i) It is used in ESR estimation (Westergren method). (ii) It is used in blood banking as a acid citrate dextrose (nutrition of RBC).
Disadvantage: (i) It is not used as routine anticoagulant. (ii) It inhibits enzyme activities such as scrot , SGPT and alkaline phosphate and interferes
in the determinations of calcium and inorganic phosphorus.
COLLECTION OF BLOOD Blood is collected by two methods, one is capillary puncture method and another is
venipuncture method. Latter is discussed here: • BLOOD COLLECTION BY VENIPUNCTURE: The volume of blood obtained by
venipuncture is sufficient to carry out multiple tests. REQUIREMENTS: Container for blood collection
Needle Syringe Tourniquet Disinfectant (methylated spirit or 70% ethyl alcohol) Cotton swab Anticoagulant

BMLT INTERNSHIP RECORD 9
PROCEDURE: (1) All the requirements are assembled before blood collection. (2) The patient is identified and the total amount of blood for all the tests is
decided. (3) The blood collection container with appropriate anticoagulant is selected and
they are labelled with the patient’s identification. (4) The patient is sited on the chair comfortably and his arm is laid on the table,
palm upwards. (5) The puncture site is selected after inspecting both arms. (6) Tourniquet is applied just above the bend in the elbow to get hinder venous
return. (7) The puncture site is disinfected with a swab wet by methanol or 70% ethyl
alcohol. (8) The syringe is removed from the protective wrap. (9) It is checked to make sure that the needle is sharp, the syringe moves
smoothly, the needle is not blocked and there is no air left in the syringe. (10) The needle is positioned with bevel uppermost and it is pushed firmly and
steadily, into the centre of the vein. (11) To enter the blood in the barrel the piston is pulled back slowly. (12) Then tourniquet is released. (13) A swab of cotton wool is placed over the hidden point of the needle. (14) The needle is withdrawn in one rapid movement from under the swab. (15) The patient is advised to press firmly on the cotton wool swab for 3 to 5
minutes. (16) The needle is removed from the syringe and gently expelled the blood into the
appropriate container. (17) Then the blood is mixed gently with appropriate anticoagulant. (18) Before the patient leaves, the venipuncture site is re-‐inspected, if the bleeding
has stopped, an adhesive tape is applied over the wound.

BMLT INTERNSHIP RECORD 10
STUDY OF BLOOD SMEAR FOR DIFFERENTIAL COUNT AND CELL MORPHOLOGY
The smear is primarily used in reporting differential count and any abnormal morphology of white cells and red cells. Differential count is the percent distribution of various white cells in peripheral blood by polychromatic stain (Leishman’s stain). Blood smear examination is related to specific type of disorders like infection (in viral infection lymphocyte are increased), leukaemias (myelogenous, lymphocytic, monocytic) or their fall, the presence of blood parasites, abnormal cells rouleaux formation in case of multiple myeloma, and estimation of the cell count.
• NORMAL VALUE: Neutrophil: 40 to 75% Eosinophils: 1 to 4% Basophils: 0 to 1% Lymphocytes: 20 to 40% Monocytes: 2 to 8%
• EQUIPMENT: Glass slides Spreader slide Applicator stick Staining rack
• REAGENT: 1) Leishman’s stain: Leishman’s powder-‐ 0.2 g Eosin Methylene blue
Methanol (acetone free): up to 100ml After mixing, the solution is warmed at 500c with occasional shaking. Now the solution is filtered and it is ready to use.
2) Buffered water: Disodium hydrogen phosphate-‐ 3.76g Potassium dihydrogen phosphate-‐ 2.10g Distilled water-‐ up to 1000ml
• PRINCIPLE: Three major steps are involved in differential count-‐ preparation of smear, staining of smear and examination of smear. Smear is made directly from skin puncture or form anticoagulated venous blood. Staining is done by polychromic stain that includes Methylene blue and eosin. Polychromic stain gives multiple colours to the cells. Stain is dissolved in methanol, acts as a fixative. Examination of smear is done by oil immersion objective.
• PROCEDURE: Step-‐1: Preparation of smear a) A grease free slide is taken on a plane surface, and it is marked with
identification number. b) One drop of well mixed blood or anticoagulated blood is taken on this slide
1cm apart from the end, about 5mm diameter with applicator stick.

BMLT INTERNSHIP RECORD 11
c) A spreader slide is placed in front of the drop of blood at 450 angle between the two slides.
d) The spreader is drawn back until it touches the drop, after the blood run along the spreader, then the spreader is push to the end of the slide with a smooth quick movement.
e) The blood smear is dried quick by moving it rapidly in the air. Step-‐2: Staining of smear a) Smear slide is placed on a staining rack. b) Slide is covered with Leishman’s stain for 2 minutes. c) Buffer water is added on the slide about double the volume of stain. d) Then mixing is done and it is allowed to continue the stain for 10 minutes. e) After 10 minutes, the stain is washed away from the smear slide with a stream
of buffer water. f) Then the stained smear is air dried.
Step-‐3: Examination of stained blood smear a) After staining, the slide is placed on the stage of microscope. b) At first, by high power objective (40x), the portion of blood smear, where only
slight touching of the red cell is selected. c) Then one drop of immersion oil is placed on this portion. d) Blood smear is observed by oil immersion objective (100x).
• OBSERVATION:
1. White cell: on the basis of their characteristics-‐ Granulocytes:
Neutrophils-‐ Pale pink cytoplasm with fine mauve coloured granules, include band and segment forms nucleus, normally 3 to 4 lobed. Eosinophils-‐ faint pink cytoplasm containing red-‐orange granules. Nucleus is usually 2-‐3 lobed. Basophils-‐ cytoplasmic granules are large, dark, and blue-‐black which fill the cell and obscure the nucleus.
Agranulocyte: Large lymphocyte-‐ large in size, have clear blue cytoplasm on the margin of the nucleus. Small lymphocyte-‐ small in size, dark violet coloured nucleus almost fills the entire cell and has a rim of cytoplasm. Monocyte-‐ large in size of all white cells, wavy margin of cytoplasm, grey-‐blue cytoplasm, kidney shaped nucleus.
2) Red cell-‐ pink red, small sized cell. 3) Platelet-‐ smallest in sized, stain mauve-‐pink.

BMLT INTERNSHIP RECORD 12
• CELL COUNT AND ABNORMAL CELL FINDING:
I) The smear is examined under oil immersion objective, moving the slide as shown in the figure. Individual types of white cells are counted under a differential counter.
II) Any blast cells or other juvenile cells or pathogenic organisms are seen is mentioned in differential count report.
III) Platelets (at least10 microscopic fields) is less than 5, platelet deficiency is reported.
DETERMINATION OF HAEMOGLOBIN
CONCENTRATION BY CYANMETHAEMOGLOBIN METHOD
Haemoglobin is a conjugated protein present in red blood cells. It carries oxygen from the lungs to the tissue cells, and carbon dioxide-‐from the tissue to the lungs. Defects in haemoglobin are called haemoglobinopathies, such as sickle cell disease. Haemoglobin concentration is decreased in case of anaemia, pregnancy, etc. Increased due to haemoconcentration (severe diarrhoea).
• NORMAL VALUE: Male: 13-‐18 gm/dl Female: 12-‐16.5 gm/dl Children (up to 1 year): 11-‐13 gm/dl Children (10-‐12 year): 11.5-‐14.5 gm/dl Infants (new born): 13.5-‐19.5 gm/dl
• PRINCIPLE: When blood is mixed with Drabkins reagent containing potassium cyanide and potassium ferricyanide, haemoglobin reacts with ferricyanide to form methaemoglobin which is converted to stable cyanmethaemoglobin by the cyanide. The intensity of the colour is directly proportional to the haemoglobin concentration and it is compared with a known cyanmethaemoglobin standard at 540 nm (green filter) in the colorimeter.
• EUQIPMENT: Test tube and rack 5 ml graduated pipette Micropipette with tips Colorimeter with 540nm filter

BMLT INTERNSHIP RECORD 13
• REAGENTS: 1. Drabkin’s reagent: Potassium cyanide (HCN)-‐50 mg
Potassium ferricyanide (KFeCN6)-‐200 mg Distilled water-‐1000 ml The reagent is stored at room temperature in a brown bottle. It is stable for several months. It should not be freezed, as this can result in decolourization with reduction of the ferricyanide.
2. Cyanmathaemoglobin standard: 60 mg/dl • SAMPLE: EDTA anticoagulated venous blood. • PROCEDURE:
1. All the reagent and sample are to be assembled before the test is performed.
2. Test tubes are label as blank (B), and test (T). 3. Reagent and sample are dispenced as follows:
Despenced Blank Test
Drabkin’s reagent 5 ml 5 ml Sample -‐ 20 µl
4. Mixing is done, and it is kept for 10 minutes at room temperature. 5. Reading is taken by colorimeter at 540 nm, against the blank.
• CALCULATION:
Blood is diluted with Drabkin’s reagent about 1:250 dilution, but standard is not. So standard concentration 60 mg/dl is multiplied with 250 and obtained 15 g/dl. Here 15 is the standard concentration.
DETERMINATION OF ERYTHROCYTE SEDIMENTATION RATE BY WESTERGREN’S
METHOD Changes in ESR are not diagnostic for any specific condition. ESR has prognostic value.ESR
is increased in all conditions where there is tissue breakdown or where there is entry of foreign proteins in the blood, except for localized mild infections. He determination is useful to check the progress of the disease. If the patient is improving the ESR tends to fall.If the patient’s condition is getting worse the ESR tends to rise.

BMLT INTERNSHIP RECORD 14 NORMAL VALUES Male : 0 -‐ 5mm after 1st hour. Female : 0 -‐ 20mm after 1st hour. METHOD Westergren’s method PRINCIPLE When anticoagulated blood is allowed to stand undisturbed for a period of time, the erythrocytes tend to sink to the bottom. The rate at which the red cells fall is known as Erythrocyte Sedimentation Rate. SPECIMEN 12 to 16 hours fasting serum REQUIREMENTS i -‐ Westergren tube with rack. Ii -‐ 3.8 sodium citrate. Iii -‐ Stop watch. Iv -‐ Blood drawer. V -‐ Test tube with test tube rack. PROCEDURE
1. The Westergren’s tube filled by proper mixing blood and 3.8% sodium citrate as 4:1 ratio, exactly upto 0 mark (air bubble is avoided)
2. The tube is placed upright position into the westergren stand for 1 hour.
OBSERVATION The level of red cell column has fallen is noted after first hour.Result is reported as mm after first hour.
DETERMINATION OF PROTHROMBIN TIME Prothrombin time determination is the preferred method for presurgical screening, as a liver function test, determination of congenital deficiency of factors II, VII, IX, X, and for monitoring of patients on oral anticoagulant therapy.
• NORMAL VALUES: 11-‐15 seconds. • PRINCIPLE: Tissue thromboplastin in the presence of calcium activates the
extrinsic pathway of human blood coagulation mechanism. When calcified thromboplastin reagent is added to normal citrated plasma, the clotting mechanism is initiated, forming a solid gel clot within a specified period of time.
• REAGENT: Lyophilized calcified thromboplastin reagent. It is stored at 2-‐8oc. The reagent is reconstituted prior the test is performed as the direction of manufacfacturer.

BMLT INTERNSHIP RECORD 15
• TEST PROCEDURE: 1. 0.1ml of plasma is placed into a 12 x 75mm tube and the tube is placed at
370c for 3 to 5 minutes. 2. 0.2ml reagent (prewarmed at 370c for atleast 3 minutes) is mixed with
plasma and simultaneously a stopwatch is started. 3. Clot or gel formation of the mixture is observed by gently tilt and time is
recorded as second. • REPORT: The result is reported as Prothrombin time of the test plasma.
DETERMINATION OF ACTIVATED PARCIAL
THROMBOPLASTIN TIME Activated partial thromboplastin time is prolonged by a deficiency of coagulation factors of the intrinsic pathway of the human coagulation mechanism such as factor XII, XI, IX, VIII, X, V, II and fibrinogen. And also monitoring heparin therapy. • NORMAL VALUE: 23-‐33 seconds. • PRINCIPLE: Phospholipid activates the coagulation factors of the intrinsic
pathway of the coagulation mechanism in presence of calcium ions and forming a solid gel clot within a specified time.
• REAGENT:
1. Cephaloplastin reagent-‐ phospholipid. 2. Calcium chloride.
Both reagents are stored at 2-‐80c. • SAMPLE: Citrated plasma, (one part of tri sodium citrate & nine part of
patients blood. • TEST PROCEDURE:
1. All the reagents are to be brought at room temperature before the test is performed.
2. 0.1 ml citrated plasma is placed into a 12x75 mm test tube and 0.1 ml Cephaloplastin reagent (prewarmed at 370c for 5-‐10 minutes.) is mixed and placed at 370c for 3-‐5 minutes.
3. After incubation period, 0.1 ml of prewarmed calcium chloride is added into the mixture.
4. The final mixture is kept at 370c for 20 minutes. 5. After 20 minutes, the observation for the gel formation into the test
tube is done by gently tilt and the time is recorded in second. o REPORT: The result is reported as thromboplastin time of the test plasma.

BMLT INTERNSHIP RECORD 16
CLINICAL PATHOLOGY
o EXAMINATION OF URINE
o ROUTINE EXAMINATION OF URINE
o REPORT OF ROUTINE URINE EXAMINATION
o ROUTINE EXAMINATION OF FEACES
o LABORATORY INVESTIGATION
o BENZIDINE TEST
o REPORT OF ROUTINE STOOL EXAMINATION

BMLT INTERNSHIP RECORD 17 EXAMINATION OF URINE
Urine analysis is primarily requested for the diagnosis of renal disorders. In addition, endocrine disorders, genetic abnormalities like inborn error of amino acid metabolism, pregnancy, parasitic infection, jaundice and drug over doses are also investigated through examination.
• Collection of urine: Random freshly voided sample is used for most of the test. Early morning urine is usually preferred because not only it is most concentrated but also it has a low PH , which pressures the form elements of the urine.
• Preservative for urine: No preservative is required if urine is examined within 1-‐2 hours after voiding. Preservative is required for 24 hours collection. Some of the important preservatives are as follows:
1. Toluene 2. Thymol, Formaline or chloroform 3. Concentrated HCL 4. Sodium Carbonate
ROUTINE EXAMINATION OF URINE Routine urine examination is consist of following tests:-‐
1. Physical examination 2. Microscopical examination 3. Chemical examination
• PHYSICAL EXAMINATION: QUANTITY-‐ Average urine output is 1500 ml/day. Polyuria is increased urine output. Oliguria is less urine output. Anuria is complete cessation of urine. COLOUR-‐ Normal colour of the urine is light straw colour, but colour may sometime change due to various conditions. Such as, red colour due to haematuria, deep brown colour due to haemoglobinuria, dark colour due to poisoning or toxicity, pale white colour due to chyluria, etc. SEDIMENTATION-‐ sediment may also sometime present due to some cast or crystal. Normally urine is clear. SPECIFIC GRAVITY-‐ Specific gravity of urine is normally 1010-‐1025. ODOUR-‐ A fine aromatic odour passed through urine. But sometime thire have some different odour come from urime.
• MICROSCOPIC EXAMINATION: Microscopic examination of urine sediment is done in order to diagnose renal disorders, kidney lesions, haemorrhage and other pathological conditions. PROCEDURE:
1. At first urine should be well mixed and 10ml of sample is poured into a conical centrifuge tube of about 12ml capacity.

BMLT INTERNSHIP RECORD 18
2. Centrifugation is done at 1500 rpm for 5 minutes. 3. After centrifugation supernatant part is discarded by Pasteur pipette. 4. One drop of well mixed sediment is placed on a clean glass slide and a clean
cover slip is placed on the sediment. 5. Identification is done with low power objective (10x) or high power objective
(40x).
MICROSCOPIC FINDINGS: 1. Pus cell-‐ 2-‐3 cells/h.p.f. 2. Epithelial cells-‐ 3-‐5 cells/h.p.f. 3. Erythrocyte-‐ very rera 4. Cast-‐ the renal tubules secrete a mucoprotein called Tamm-‐Horsfall protein
which is believed to form the basic matrix of all the case. Various cast found in urine-‐ granular cast, hyaline cast, red cell cast, white cell cast, epithelial cell cast, waxy cast and fatty cast.
5. Crystals-‐ many of the crystals that are found in the urine have little clinical significance although they may be found in calculus formation, metabolic disorders and in the regulation of medication. A) Crystal present in acid urine: Uric acid crystal, calcium oxalate crystal and
cholesterol crystal. B) Crystal found in alkaline urine: Triple phosphate.
6. Oval fat bodies and fat droplets-‐ diabetes mellitus, fat embolism. 7. Spermatozoa-‐ after coitus they may be present in the urine of both sexes. 8. Yeast cells-‐ it may be present in diabetic patient.
• CHEMICAL EXAMINATION: Glucose: The test spot for glucose contains a buffer enzyme preparation (glucose oxidase and peroxidise) and a chromogen (o-‐tolidine) that shows colour change in the presence of glucose only . The colour shade is compared against the standard colour chart provided by the manufacture which makes the test semiquantitative. Protein: The colour change on the protein test spot is based on the principle of “protein error of indicator”. The indicator tetrabromophenol blue changes its colour differently in the presence or absence of protein at PH 3. Citrate is used as the buffer to keep the PH at 3.0. Indicator turns to green when the urine contains albumin and in case of normal urine (absence of albumin), the colour stays yellow. Reaction or PH : The test spot is impregnated with methyl red and bromothymol blue. The colour change of the indicator can differentiate PH values within the range of PH 5 to 9.

BMLT INTERNSHIP RECORD 19
ROUTINE EXAMINATION OF FEACES
Stool or fecal material is an important specimen for the diagnosis of diseases of gastrointestinal tract, diarrhoea, dysentery, parasite infection, gastrointestinal bleeding, peptic ulcer, carcinoma and malabsorption syndromes including steatorrhoea.
COLLECTION OF SPECIMEN: The specimen of the feces is collected in a wide mouthed dry disposable plastic container at the morning. Random sample is also collected in the same procedure. Middle part of the stool is collected for the examination.
PRECAUTION TAKEN AFTER COLLECTION: 1. The collection vial should be labelled and identified with particular
identification number. 2. Stool specimen should not be left uncovered. It is necessary to prevent
drying. 3. Stool specimen should be examined within in one hour. 4. Specimen should be disposed properly after the examination.
LABORATORY INVESTIGATION
The stool is examined in the laboratory by three steps: 1. Physical examination 2. Chemical examination 3. Microscopic examination
PHYSICAL EXAMINATION A. QUANTITIES: Normally 100 to 200 gm of stool is passes per day.
B. COLOUR: Normal stool is light to dark brown in colour due to the presence of the
bile pigments. Clay colour stool occur in the obstructive jaundice due to the absence of bile pigments in the stool. Tarry or black colour stool occur in the upper gastroinrestinal haemorrhage due to altered blood.
C. CONSISTENCY: Normal stool is well formed. Watery stool occur in diarrhoea. Hard stool suggest constipation.
D. ODOUR: Normal odour of the stool is foul due to presence of indole and skatole which is stranger after a meat diet.
E. BLOOD & MUCOUS: small amount of mucus may be normally present. When large amount of mucus are present especially with blood, it suggests lesion of large gut causes amoebic or bacillary dysentery.

BMLT INTERNSHIP RECORD 20
F. PARASITES: Stool contains warms and segments of warm e.g. round warm, tap warm thread warm etc. But normally they are not found.
MICROSCOPIC EXAMINATION: Microscopic examination is done mainly for observation of pus cell, epithelial cell, RBC, ova, cyst etc. It is done by two ways:
1. Direct method 2. Concentration method
In the hospital, we are done direct method. It has four types: a. Saline preparation b. Iodine preparation c. Wet mount preparation d. Hanging drop preparation
Generally we done saline preparation and iodine preparation SALINE PREPARETION:
A. One drop of normal saline is placed at the one end of a clean and dry slide. B. A little fecal material is taken by an applicator stick and mixed with the saline. C. A cover slip is placed over it.
IODINE PREPARETION: A. One drop of Lugol’s iodine solution is placed at the another end of the same slide. B. A little amount of fecal material is mixed with iodine solution. And a cover slip is
placed over it.
OBSERVATION: Observation is done under 10x or 40x objective in the microscope. By direct preparation , the morphology of the ova, cyst, RBC, pus cell, epithelial cell, fat globules, vegetables cell are observed. CHEMICAL EXAMINATION The chemical examination consists of two tests. These are as follows-‐-‐-‐
1. REACTION: Normal stool is slightly acidic or alkaline in reaction. Acidity is due to the carbohydrate in diet and alkalinity is due to the protein in diet.
2. OCCULT BLOOD TEST (OBT): For the occult blood test of stool the following test are done-‐
a. Orthotoludine test b. Benzedrine test
In the hospital, Benzedrine test is performed.

BMLT INTERNSHIP RECORD 21
BENZIDINE TEST PRINCIPLE: peroxidise activity of the haemoglobin converts hydrogen peroxide into nascent oxygen and water. This nascent oxygen oxidized the benzidine powder in presence of acidic media and produced green to blue colour. PROCEDURE:
a. A pinch of benzidine powder is taken into a clean dry test tube. b. Acidified with 2-‐3 drops of glacial acetic acid, and properly mixed. c. 1.0ml of H2O2 is added and mixing done properly. d. A small quantity of stool specimen is placed on a clean dry new glass slide and one
or two drops of the mixed benzidine solution is mixed with stool specimen. e. Observation is done to see colour the change.
OBSERVATION: No change in colour-‐-‐-‐-‐-‐-‐-‐occult blood absent. Colour change green to blue-‐-‐-‐-‐occult blood present. Green-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐+ Greenish blue-‐-‐-‐-‐-‐-‐-‐-‐++ Blue-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐+++ Deep blue-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐++++

BMLT INTERNSHIP RECORD 22
HISTOPATHOLOGY
o INTRODUCTION OF HISTOPATHOLOGY
o GROSSING
o FIXATION
o DECALCIFICATION
o DEHYDRATION
o Impregnation
o Embedding
o SECTION CUTTING
o STAINING
o PAS (Periodic acid-‐ Schiff technique)
o RETICULIN STAIN
o IMMUNOHISTOCHEMISTRY

BMLT INTERNSHIP RECORD 23
INTRODUCTION OF HISTOPATHOLOGY Histopathology means study of pathogenic or diseased tissue. Histopathology is one of the most effective tools in diagnosing tissue abnormalities and cancerous conditions. In histopathological laboratory the specimens are submitted mostly come operation theatre in form of small pieces of tissue or whole organ, they are either fresh (unfixed) or immersed in a fixative fluid. A histotechnician is responsible for the handling and preparation of specimens to facilitate their gross and microscopic examination which are done by the histopathologist. The basic steps of specimen processing include fixing, embedding, microtomy, staining and mounting. It is expected that the histotechnologist will be sufficiently trained to prepare the specimens according to the specifications, to recognize satisfactory preparation, identify and remedy the causes when unsatisfactory results are obtained.

BMLT INTERNSHIP RECORD 24 Histology : Study of the tissue is called histology. Histopathology : Study of the abnormal tissue under microscope is called histopathology.
Source of the Histopathology : 1) Surgical biopsy
2) Major resections
3) Autopsy
4) Exudate and transudate
For receiving purpose : i) Registered book
1. Patient’s name, 2. Doctor’s name, 3. Operation date, 4. Time, 5. Age, 6. Description of the specimen
ii) Effective fixative iii) Case history (Patient’s identification)
iv) Inspection of tissue
v) Efficient machinery
vi) Putrefaction free
vii) Technician’s precaution
Duty of the technician : i) Specimen preservation in preservatives that is fixative,
ii) Specimen logging that is should be entry in a log book with an identification number,
iii) Preparation of the specimen are grossing that is gross examination which is performed by histotechnician.

BMLT INTERNSHIP RECORD 25
Basic Steps for Tissue Processing Receving of specimen/ tissue Fixation with 10% Formalin
Grossing Preservation/ fixation with 10% formalin
70% Methanol (1 hour) 90% Methanol (1 hour) Methanol (1 hour) Methanol (1 hour) Methanol (1 hour) Methanol (1 hour) Xylene (over night) Paraffin (2 hrs) Paraffin (2 hrs)
Block Section cutting
Staining Mounting by D.P.X.
GROSSING
The routine surgical laboratory receives many different tissue specimens ranging from small biopsies (e.g. of breast, bladder, bone marrow) to complete resections (e.g. larynx, uterus, large bowel). Small amounts of tissue can be unidentifiable as to their anatomical source and thus, gross descriptions are important. The features of the biopsy should be described including the colour and consistency of the tissue and the presence of blood clot or foreign material. There are seven major components in grossing a specimen : i) Reliable and rapid transfer of the specimen from surgery to pathology,
ii) Accurate identification of the specimen,
iii) Description of additional specimens received from the same patient,
iv) Gross description of the specimens normal and abnormal features,
v) Recording the sites from which blocks of the tissue are taken,
vi) Recording markers (e.g. sutures) that help with the correct orientation,
vii) Identifying special studies requested and/or needed.
Techniques of Grossing : The core biopsy should be taken with the lesion at its center. Larger core biopsies (=4mm) should be bisected eccentrically, perhaps 2/3 or 1/3, and the specimen embedded with cut surfaces down. The cutting section of the tissue is processed in the cassettes with few drop of eosin or color substance.

BMLT INTERNSHIP RECORD 26 Small specimens should not be cut, bisected, or inked while fresh and unfixed. Small specimens are processed in cassettes either with a fine mesh in lens paper, or in a ‘tea bag’ so they are not lost during processing. Small specimens are packed in filter paper with few drops of eosin (color indicator), then it is processed in cassettes.
FIXATION Processing cells and tissue components with minimal distortion is the most important step of the processing of the tissue samples. Normally 10% Formalin is used, except bone (70%Formic acid/ 5% Aqueous nitric acid. Advantages of fixation :
i) Prevent autolysis,
ii) Prevent putrefaction,
iii) Preservation of cells and tissue constituents,
iv) Hardening of soft tissues,
v) Stabilized proteins.
Fixation must be complete before subsequent steps in the processing schedule are initiated. The factors that govern diffusion of a fixative into tissue were investigated by Medawar (1941). He found that the depth(d) reached by a fixative is directly proportional to the square root of duration of fixation(t) and expressed this relation as: d = k√t The constant (k) is the coefficient of diffusability, which is specific for each fixative.
DECALCIFICATION
When preparing sections of bone and other calcified tissues decalcification is necessary in order to facilitate cutting. The calcified hard tissues should be first cut into small pieces (2-‐4mm) with thin blade hacksaw to minimize the tearing of surrounding tissues. Acid Method of Decalcification There are several other methods of decalcification (ion-‐ exchange, chelation, electrical ionization) but the acid method of decalcification is the most widely used and will be described here. The acid present in the decalcifying fluid removes the calcium salt present in the tissue, thereby render the tissue soft enough for sectioning. Mostly three decalcifying solutions are – 5% aqueous nitric acid, 70% formic acid, formic acid with hydrochloric acid. Principle : The acid present in the decalcifying fluid removes the calcium salt present in the hard tissue and makes the tissue soft enough for sectioning. Procedure : 1) Suspend the sliced tissue in the decalcifying solution by means of a gauze bag, tied with a
string (The quantity of decalcifying solution should be more than 20 times the volume of the tissue).
2) Stir the fluid occasionally and change the fluid daily till calcium is completely removed from the tissue section. It is tested as follow :

BMLT INTERNSHIP RECORD 27
a. Take about 5ml of decalcifying solution in a test tube.
b. Add conc. ammonia solution drop by drop until it becomes alkaline (test by using litmus paper).
c. If the solution becomes turbid, continue decalcification.
d. If it is clear, add 0.5ml of saturated ammonium oxalate solution. If calcium is present, the solution will become turbid. The continue decalcification. If there is no turbidity, the specimen is free of calcium and ready for further processing.
3) Wash the decalcified specimen in running tap water for 48 hours. The decalcifying solution should be completely removed before dehydration and embedding.
DEHYDRATION The first stage of processing is the removal of unbound water and aqueous fixatives from the tissue components by diffusion. Many dehydrating reagents are hydrophilic (water loving), possessing strong polar groups that interact with the water molecules in the tissue. Note: Excessive dehydration may cause the tissue to become hard, brittle. Incomplete dehydration will prohibit the penetration of the clearing reagents into the tissue, leaving the specimen soft and non-‐receptive to infiltration. Dehydrating agents:
i) Ethanol
ii) Ethanol acetone
iii) Methanol
iv) Propan-‐2-‐ol, isopropyl alcohol
v) Butyl alcohol
vi) Acetone
Procedure:
70% Methanol (1 hour) 90% Methanol (1 hour) Methanol (1 hour) Methanol (1 hour) Methanol (1 hour) Methanol (1 hour)
For delicate tissue it is recommended that the processing start in 30% methanol.

BMLT INTERNSHIP RECORD 28
Chloroform/ Xylene It is a clearing reagent acts as an intermediary between the dehydration and infiltration solutions. It should be miscible with both solutions. When the dehydrating agent has been entirely replaced by most of these solvents the tissue has a translucent appearance hence the term ‘clearing agent’.
Impregnation Impregnation is the process by which the molten wax enters the tissue and replaces xylene. The above procedure is carried out in the paraffin oven for 2-‐3 hours keeping the temperature in the oven between 58-‐ 600C.
Embedding It is the process of blocking where the infiltrated and impregnated tissue is placed in warm paraffin (60-‐ 620C)which forms a firm block after cooling. Procedure : Leuckhard embedding box prepared by pressing 2 ‘L’ shaped pieces of heavy metal (brass) over
the brass plate.
Molten paraffin wax (60-‐ 620C ) pour into the cavity of box.
The specimen is placed on the bottom of the box containing identification number in proper space
without any air.
Then cover the box by cold filter paper or any other cloth.
The box placed inside a container which containing cold water or kept in refrigerator for harding
time require 10-‐ 20 minutes.
The harden block is ready for cutting by microtome.

BMLT INTERNSHIP RECORD 29
SECTION CUTTING Types of Microtome :
i) Rotary microtome,
ii) Rocking microtome,
iii) Sliding microtome,
iv) Freezing microtome,
v) Sledge microtome,
vi) Cryostat microtome,
vii) Ultra microtome.
Rotary microtome : Microtome is used for cutting paraffin tissue sections of uniform thickness. This instrument is designed to cut 1 to 60 µ thin sections. The basic mechanism of rotary microtome is required the rotation of a fine advance hand-‐wheel through 3600 moving the specimen vertically past the cutting surface and returning it to the starting position. The rotary microtome may be manual (completely manipulated by the operator), Semi-‐automated (one motor to advance either the fine or coarse hand-‐wheel), or fully automated (two motors that drive both the fine and the coarse advance hand-‐ wheel). Advantages include, the ability to cut thin 3-‐ 20µ sections and easy adaptation to all types of tissue (hard, fragile or fatty) sectioning. Technological advance in the automation of microtomy have improved section quality increased productivity, and improved occupational safely for the technologist. The section of paraffin embedded tissue:
Requirements :
i) Microtome with sharp knife,
ii) Paraffin block with tissue,
iii) Warm water,
iv) Needle,
v) Brush,
vi) Microscopic slide,
vii) Forceps,
viii) Ice tray,
ix) Diamond pen,

BMLT INTERNSHIP RECORD 30
a. Adhesive : Egg albumin :
1.Albumin part of egg, 2.Glycerol 3.Thymol bit (2 pieces)
Procedure : (1) Trimming the tissue block : The paraffin block may be faced or ‘rough cut’
by setting the micrometer at 15-‐ 30mm .
(2) Attaching of the trimmed paraffin block to the microtome :
(3) Cutting section : Block should be arranged in numerical order on a cooling device, cooling both the tissue and tissue and the paraffin giving them a similar consistency. Routine surgical material should be cut at 3-‐ 4µ. Successive sections will stick edge-‐to-‐edge due to local pressure with each stroke, forming a ribbon. When a ribbon of several sections has been cut, the first section is held by forceps.
(4) Floating out sections :
The ribbon sections are floated in the water.
Take clean microscopic slide, a drop of adhesive (egg albumin) is placed on
a slide and smear done.
By keeping about 1cm gap between the sections these are arranged on the
microscopic slide by using forceps.
Then this slide with sections dipped in the warm water to remove the
creases.
The slide is placed in upright position to drain the water.
Careful writing of identification number on the slide is confirmed before
the staining by diamond pen.

BMLT INTERNSHIP RECORD 31
STAINING
Harris’s haematoxylin and Eosin stain The hematoxylin and eosin stain is the most widely used histological stain. The hematoxylin component stains the cell nuclei blue-‐black, with good intranuclear detail, while the eosin stains cell cytoplasm and most connective tissue fibers in varying shades and intensities of pink, orange, and red. Purpose : Stain of nucleus, cytoplasm, collagen, RBC, etc. Reagent : A. Harris’s hematoxylin i) Hematoxylin – 2.5gm ii) Absolute alcohol – 25ml iii) Potassium alum – 50gm iv) D/W – 500ml v) Mercuric oxide – 1.25gm vi) Glacial acetic acid – 20ml Deep purple color solution. B. Eosin i) Eosin power – 1gm ii) D/W – 100ml C. 1% Hcl. Procedure :
1. Deparaffinised of the section by warm stage for few minutes,
2. Place in xylol for 3 minutes,
3. Another change of fresh xylol for 2 minutes,
4. Then another change of fresh xylol for 2 minutes,
5. Transfer the section to absolute alcohol for 30 sec,
6. Transfer 90% alcohol for 30 sec,
7. Then transfer 70% alcohol for 30sec,
8. Hydrated under tap water for 2 minutes,
9. Place the section of the tissue in harris’s hematoxylin for 4 minutes,
10. Wash in running tap water,

BMLT INTERNSHIP RECORD 32
11. Bluing the section,
12. Decolorised by 1% Hcl for few sec,
13. Wash in running tap water rapidly,
14. Placed the section running tap water upto bluing the section,
15. Counter stain with 1% watery yellow eosin for 2 minutes,
16. Washing at the rever side of the tissue,
17. Dehydration with 70% -‐80% -‐ 90% -‐ absolute alcohol,
18. Clearing with xylol,
19. Mounting with D.P.X.
Result : Nucleus – Blue Colagen – Pink RBC – Brick red Cytoplasm -‐ Red
PAS (Periodic acid- Schiff technique)
Demonstration of carbohydrates or glycoconjugates. [Polysaccharides (Glycogen), connective tissue glycoconjugates (Proteoglycans, Hyaluronic acid),Mucins. The PAS technique may aid in the differential diagnosis of tumors through the detection of mucins or glycogen. Common fungal species that are PAS reactive include Candida albicans, Histoplasma capsulatum, Cryptococcus. Principle : The tissue structures like liver, heart, striated muscles, etc. are studied
by periodic acid – Schiff stain. Periodic acid reacts with aldehyde group of the carbohydrates and afterwards reaction with schiff’s reagent to form a bright red or purple red colour.
Reagent : 1. 0.5 % periodic acid solution
2. Schiff’s reagent
I. 1 gm basic fuchsin dissolved in 100 ml of boiling distilled water.
II. 20ml of 1(N) Hcl is added and then cool further at room tempareture.
III. 1gm of Sodium metabisulfite is added and mixed well.

BMLT INTERNSHIP RECORD 33
IV. Kept in dark place for 24 -‐ 48 hrs.
V. When the solution becomes straw coloured, then 300mg of activated charcoal is added and shake.
VI. Filter and store at 2 – 80C.
3. 1(N) HCl.
4. 0.1gm of light green in 100ml of 0.1% acetic acid.
5. Harris’s hematoxylin stain.
Procedure : I. Deparaffinised of section.
II. Hydration with decending graded of alcohol.
III. Oxidise in periodic acid solution for 5 – 10 minutes.
IV. Wash in tap water for 5 – 10 minutes.
V. Rinse distilled water.
VI. Place in Schiff reagent at a dark place for 20 – 30 minutes.
VII. Place the section directly SO2 solution for 5 minutes.
VIII. Another change fresh SO2 water for 5 minutes.
IX. Counter staining by ½ diluted harris’s hematoxylin for 30sec.
X. Wash in running tap water.
XI. Dehydration with absolute alcohol.
XII. Clean with xylene.
XIII. Mounting by D.P.X.
Result : Nucleus : Blue Glycogen, mucin, hyaluronic acid reticulin, amyloid infiltration, colloid
pituitary stalk etc. : Purple red.

BMLT INTERNSHIP RECORD 34
RETICULIN STAIN Demonstration of the reticulin fibers. These are the fine and delicate fibers that are found connected to coarser and stronger collagenous fibers. They provide the bulk of the supporting framework of the more cellular organs, exp. Spleen, liver, lymphnodes, etc. where they arranged in a three dimensional network to provide a system of individual cell support. Principle : Reticulin fibres are collagen type III fibres with fine branching. These are usually seen best in lymphoid tissue and liver. The most successful way to identify reticulin fibres is to make use of their argyrophilia following Gordon. Sweet’s technique which is also known as reticulin staining. In the staining process, silver is selectively deposited on the fibres in the alkaline medium. This is subsequently converted to reduced (black) silver by suitable reducing agent, allowing visualization of fibres. Procedure : i. Deparaffinised of section.
ii. Bring the section in the water.
1) Absolute alcohol.
2) 90% alcohol.
3) 80% alcohol.
4) 70% alcohol.
5) 30% alcohol.
6) Water
iii. Acidify KMnO4 Solution for 2 – 3minutes.
1) 0.25% KMnO4 – 19ml
2) 3% H2SO4 – 1ml
iv. Rinse in distilled water.
v. 1% Oxalic acid.
vi. Rinse in D/W.
vii. 2% Iron alum for 15-‐20 minutes.

BMLT INTERNSHIP RECORD 35 viii. Rinse in distilled water.
ix. Silver bath for 30 sec to 1 minute.
x. Rinse in distilled water.
xi. Neutral formalin for 2-‐3 minutes.
xii. Rinse in distilled water.
xiii. 0.2% Gold chloride for 2-‐3 minutes.
xiv. Rinse in distilled water.
xv. 5% Na2S2O2 solution for 1-‐ 2 minutes.
xvi. Rinse in distilled water.
xvii. Air dry.
xviii. Clean with xylene.
xix. Mounting by D.P.X.
Result: Reticulin fibres : Black Nuclei : Gray Others tissues : According to counter stain.

BMLT INTERNSHIP RECORD 36
IMMUNOHISTOCHEMISTRY Immunohistochemistry refers to the process of detecting antigens (proteins) in cells of a tissue section by exploiting the principle of antibodies binding specifically to antigens in biological tissues. Immunohistochemical staining is widely used in the diagnosis of abnormal cells such as those found in cancerous tumors. Specific molecular markers are characteristic of particular cellular events such as proliferation or cell death (apoptosis). Immunohistochemistry (IHC) is also widely used in basic research to understand the distribution and localization of biomarkers and differentially expressed proteins in different parts of a biological tissue. In the most common instance, an antibody is conjugated to an enzyme, such as peroxidase, that can catalyse a colour-‐ producing reaction (immunoperoxidase staining). Alternatively, the antibody can also be tagged to a fluorophore, such as fluorescein or rhodamine. Principle : The demonstration of antigens in tissue and cells by immunostaining is a
two-‐step process involving first, the binding of an antibody to the antigen of interest, and second, the detection and visualization of bound antibody by one of a variety of enzyme chromogenic systems. The choice of detection systems will dramatically impact the sensitivity, utility, and ease-‐of-‐use of the method. The NovoLink Polymer Detection Systems utilize a noval controlled polymerization technology to prepare polymeric HRP – linker antibody conjugates. As the system is not based on the biotin-‐ avidin system, problems associated with endogenous biotin are completely eliminated.
Tissues or cell preparations are frozen or fixed, sectioned, and attached to slides. The sections are then dewaxed if paraffin-‐embedded, treated with an antigen retrieval solution if required, blocked with a proteinaceous blocking solution and then incubated with a primary antibody. The bound primary antibody is detected by the addition of secondary antibody conjugated with horseradish peroxidase polymer and DAB substrate. When adequate color development is seen, the slides are washed in water to stop the reaction, counterstained with Novocastra Hematoxylin and covered with a mounting medium.
Reagents : I. Poly-‐L-‐Lysine (adhesive),
II. Peroxidase Block. (3% Hydrogen peroxide.)
III. Protein Block. (0.4% casein in phosphate-‐ buffered saline, with stabilizers, surfactant, and 0.2% Bronidox L as a preservative.)

BMLT INTERNSHIP RECORD 37
IV. Post Primary Block. (Polymer penetration enhancer containing 10% animal serum in tris-‐ buffered saline.)
V. DAB chromogen. 1.74% W/V 3,3’ – diaminobenzidine, in a stabilizer solution.)
VI. NovoLink Polymer. (Anti-‐ mouse/rabbit IgG-‐Poly-‐HRP.)
VII. NovoLink DAB substrate Buffer (Polymer). (Buffer solution containing 0.05% hydrogen peroxide and preservative.)
VIII. Hematoxylin.
Specimen: The recommended fixative is 10% neutrat buffered formalin for paraffin – embedded tissue sections.
Procedure : Put all the poly L –Lysine coated slides in incubator for over night (370C)
and 650C for 30-‐40 minutes. Xylene I for 10 minutes. Xylene II for 10 minutes. Alcohol 100% for 5 minutes.
Alcohol 70% for 5 minutes.
Alcohol 50% for 5 minutes.
D/W for 5 minutes.
Dip all slides in hot citrate buffer and start antigen retrival.
Then wait for one whisttes, then cool it.
Then blot dry.
Pour all the slides in TRIS Buffer (PH 7.4 ) for 7 minutes.
Blot dry.

BMLT INTERNSHIP RECORD 38 Apply Peroxide Block to cover the specimen according to tissue size or
autostaining slide parameters for 8 minutes at room temperature.
Blot dry.
Wash in TRIS Buffer for 5 minutes.
Blot dry.
Protein Block for 10 minutes.
Wipe the slides clearly.
Primary antibody Exp. ER,PR, Her-‐2/neu for 45 minutes.
Wash in TRIS Buffer for 5 minutes for 2 times.
Incubate slide in Post Primary Block for 30 minutes.
Wash in TRIS Buffer for 5 minutes for 2 times.
Novolink Polymer incubate for 30 minutes.
Wash in TRIS Buffer for 5 minutes for 2 times.
Incubate in DAB chromofen for 3 minutes.
Rinse it properly in tap water for 3 minutes.
Counter stain Haematoxilene for 2 minutes.
Rinse it properly in tap water.
Dehydrated it 70% -‐ 100% alcohol.
Then Xylene 10 minutes for 2 times.
Mount it properly and examined under microscope.
Result :

BMLT INTERNSHIP RECORD 39
Cytology
o Advantages of FNAC o Disadvantages of FNAC o Branches of Cytology o Procedure
o Leishman-‐Giemsa Stain o Apanicoloau Stain

BMLT INTERNSHIP RECORD 40
The primary goal of the study of cytology is the early detection of cancer. The cells shed into the body fluid and secretions and these provide materials for diagnosis. Cytology is that study of diagnostic medicine which deals with the study of individual cells and/or tissue fragments spread on laboratory slide and stained appropriately.
Cytology, more commonly known as cell biology, study of cell structure, cell composition, and the interaction of cells with other cells and the larger environment in which they exist. Cytology can also refer to cytopathology, which analyzes cell structure to diagnose. Microscopic and molecular studies of cells can focus on either multi celled or single celled organisms.
Cytological diagnosis is an important part of cervical lesions, accessible mucosal lesions and soft tissue tumors palpable superficially or else approached under fluoroscopic guidance. Advantages of FNAC There are so many advantages in FNAC procedure like it is a quick, convenient, economic and almost painless procedure which can be practiced on an outpatient basis. Local anesthesia is not required, can be attempted multiple sites and repeated times. Malignancy can be confirmed or excluded in potentially. Operable lesions suspicious of malignancy and the extent of surgery can be planned well in advance. It is good diagnostic aid prior to application of radiation in inoperable cases or where surgery is contraindicated. By way of evacuation of a cyst content, it helps as a therapeutic aid in addition to providing diagnosis. Aspirated material can be used for immunological, cytochemical, cytogenetical and microbiological studies. Disadvantages of FNAC False negative results may be obtained if there is extensive fibrosis and sclerosis in a tumor. If the tumor is highly vascular, tumor necrosis. Currently cytology has following branches:-‐
A. exfoliative cytology
B. Aspiration cytology
C. Imprint cytology Exfoliative cytology:-‐-‐ This is the study of cells which are spontaneously shed off from epithelial surface into body cavities or fluid. The cells can also be obtained by scraping,

BMLT INTERNSHIP RECORD 41 brushing or wash of body surfaces, the principle of the technique is that in diseased states rate of exfoliation of cells is increased. Exfolitive cytology is applied in diagnosing diseases of the following-‐ a. female genital tract b. respiratory tract c. gastro intestinal tract d. urinary tract e. body fluid ( plural, peritoneal, csf, semen etc.) f. buccal smear for sex chromatin
Aspiration cytology:-‐ In this study, samples are obtained from diseased tissue by fine needle aspiration (FNA) or aspiration biopsy cytology (ABC). Aspiration cytology is applied for diagnosis of palpable or non-‐ palpable lesions.
1.palpable mass lesion in-‐ a. lymph noes b. breast c. thyroid d. salivary gland e. soft tissue masses f. bones
2. non-‐ palpable mass lesions in-‐
a. abdominal cavity b. thoracic cavity c. retro peritoneum
Imprint cytology :-‐ In imprint cytology touch preparations from cut surfaces or fresh unfixed surgically excised tissue are prepared on clean glass sides. These are fixed, stained and examine immediately. It is considered complementary to frozen section.
PROCEDURE: -‐ 20 ml plastic disposable syringe with 21 to 23 gauze fine needles of variable length, depending upon the site of tumor, are used for aspiration. The syringe is fitted with a specially designed handle which permits a single hand operation during aspiration. The skin is cleaned with antiseptic solution. No local anesthesia is required. The tumor mass is fixed with one hand and with the other hand aspiration is carried out. When the needle enters the tumor, the plunger of the syringe is retracted to create a vacuum in the barrel and the needle is moved to and fro 3 to 4 times. For adequate sampling, the needle may be moved in 3 to 4 different direction. After completion of aspiration, the

BMLT INTERNSHIP RECORD 42 plunger is released before taking out the needle in order to equalize the pressure. The needle is disconnected and after filling the syringe with air, it is reconnected. The content of the needle is expressed on clean glass slides. Smears are made by applying a gentle pressure with the flat surface of another glass slide and allowed to air dry. Smears are routinely fixed in methanol for MGG staining. Whenever PAP staining is required for better nuclear clarity, wet fixation in absolute alcohol is recommended. Smears may be fixed appropriately for various cytochemical stains when fluid aspirated is discharged into a clean tube and centrifuged at 1500 rpm. Smears are made from the deposits when the aspiration fluid is admixed with blood or frankly haemorrhagic. Haematocrit method or lymphoprep can be used to separate the tumor cells from RBCs.
LEISHMAN-GIEMSA STAIN Reagents:-‐
1. Leishman stain: Leishman powder : 0.15gm Methanol ; 100 ml. 2. Giemsa stain: Giemsa powder : 0.75gm Methanol : 65 ml Glycerol : 35 ml
Procedure:-‐
1. fixed dry smear with leishman stain for 2 min.
2. then double volume of buffer is added and mixed well.
3. the slide is washed off under tap water after 10 min.
4. then with giemsa stain flood the slide which is prepared by diluting the stain with distilled water in 1;10 dilution.
5. wait for another 10 min
6. the slide is washed off
,
7. air dried and mount is done RESULT Nuclei : bluish-‐red
Acidophilic granules : pink to red
Basophilic : blue

BMLT INTERNSHIP RECORD 43 PAPANICOLOAU STAIN REAGENTS:
1. Alcohol ether mixture
2. harrish haematoxilin stain
3. 1% acid alcohol
4. 70% ,95% alcohol, absolute alcohol
5. orange green -‐6 (og-‐6) 6. Eosin azide -‐36 (EA-‐36) 7. xylol 8. DPX
STAINING PROCEDURE 1. Fix the smear in alcohol ether mixture for 20-‐30 min. 2. rinse in distilled water 3. stain in harrish haematoxilin for 4 min 4. wash in tap water for 1-‐2 min 5. differentiate in 1% acid alcohol 6. blue in running tap water 7. rinse in tap water 8. transfer the smear in 70% alcohol then 95% alcohol for few second 9. stain in orange green -‐6 or 1-‐2 min 10. rinse in 3 changes of 95% alcohol for few second in each 11. stain in eosine azide -‐36 for 1-‐2 min 12. rinse in 3 changes of 95% alcohol for few second in each 13. dehydrated in absolute alcohol 14. clear in xylol 15. mount by dpx
RESULT Nucleus : blue Acidophilic cell : rec to orange Basophilic cell : green to bluish green Cratenised cell or penetrated by blood: varington of red

BMLT INTERNSHIP RECORD 44
HLA
o INTRODUCTION
o ABOUT HLA
o CLASSIFICATION OF HLA
o IMPORTANCE OF HLA
o ASSOCIATION BETWEEN HLA B27 AND ANKYLOSING
SPONDYLITIS
o HLA TYPING
o HLA CROSS MATCHING
o HLA B27 TYPING TEST

BMLT INTERNSHIP RECORD 45
INTRODUCTION
uman leukocyte Antigen (HLA) is a polymorphic cell surface glycoprotein which is present on the surface of nucleated cell including circulating and tissue cell. The HLA genetic
system is the major histocompatibility complex in human. For several years they are of interest primarily to transplantation immunologists. The human leukocyte antigen (HLA) test also known as HLA typing or tissue typing. Identifies antigens on the white blood cells (WBC’s) that determine tissue compatibility for organ transplantation. Unlike most blood group antigens which are inherited as products of two alleles (typing of gene that occupy the same site on a chromosome). Many different alleles (typing of gene that occupy the same site on a chromosome). Many different alleles can be inherited at each of the HLA loci. There are defined by antibodies (antisera) that recognized specific HLA antigens, or by DNA probes that recognize the HLA allele. Using specific antibodies,26 HLA-A alleles,59 HLA –B alleles, 10 HLA-C alleles, 26 HLA-D alleles, 22HLA-DR alleles, nine HLA-DQ alleles, and six HLA –DP alleles can be recognized. This high degree of genetic variability makes finding compatible organs more difficult than finding compatible blood for transfusion. ABOUT HLA : - The HLA is the name of the major histocompatibility complex (MHC) in humans. It is the cluster of genes on the short arm of chromosome 6. Approximately 40% of the expressed genes are estimated to have an immune system function. HLA are glycoprotein expressed on the surface of more or less every in the body and the molecules form part of system of immune recognition essentially by their ability to recognize the self from non-self.
CLASSIFICATION OF HLA:- Based on the two principal sources of antigenic proteins and the characteristics of T-cell recognition, there are two classes of HLA antigens-
HLA-Class I HLA-Class II
MHC class I genes encoded glycoprotein expressed on the surface of nearly all nucleated cells, major function of the class I gene product is presentation of peptide antigens to Tc-cells. Major class I genes in HLA are-
HLA-A HLA-B HLA-C
MHC class I I genes generally encode glycoprotein expressed on antigen presenting cells (macrophages, dendritic cells , B cells).Where they present processed antigenic peptides to TH-cells. Major class II genes HLA are-
HLA-DP HLA-DQ HLA-DR
IMPORTANCE OF HLA
H

BMLT INTERNSHIP RECORD 46
1. In Transplantation: Any cell displaying some other HLA type is non-self, resulting in the ejecting of the tissue (organ) bearing those cells. By matching as closely as possible, the donor to the patients for heir HLA type, the risk of rejection is significantly decreased.
2. In Infectious disease:- When a foreign pathogen enters the body, specific cells
called antigen-presenting cells(APCs) engulf the pathogen through a process called phagocytosis. Proteins from the pathogen are digested into small pieces (peptides) and loaded onto HLA antigens (specially MHC class II). They are then displayed by the antigen presenting cells for certain cells of the immune system called T cells. Which then produce a variety of effects to eliminate the pathogen.
Through a similar process, proteins ( both native and
foreign, such as the proteins of viruses) produced inside most cells are displayed on HLA antigens (specially MHC classI) on the cell surface. Infected cells can be recognized
and destroyed by components of the immune system (specially CD8+ T cells). 3. In Population Studies: It has been demonstrated that HLA types can occur with
different frequencies in different racial groups. Knowledge of these antigen frequencies can help to identify a population.
4. In Paternity and Forensic testing: HLA genes are inherited from each parent and
are expressed co-dominantly. They are candidates for use in paternity and forensic testing.
5. In Autoimmunity: Some of HLA types are associated with autoimmune disease.
People with certain HLA antigens are more likely to develop certain autoimmune diseases. For e.g. first HLA haplotype association with inflammatory disease was discovered in 1972. Correlating HLA-B27 with autoimmune disease Ankylosing Spondylit is (AS)
HLA & Autoimmune diseases
Disease HLA Relative risk
Ankyosing spondylitis B27 87.4
Uveitis B27 10
Goodpasture syndrome DR2 15.9
Multiple sclerosis DR2 4.8
Graves-Basedow disease DR3 3.7
Systemic lupus erythromatodes DR3 5.8
Myesthenia gravis DR3 2.5
Pemphigus DR4 14.4
Rheumatoid arthritis DR4 4.2
Hashimoto thyreoiditis DR5 3.2

BMLT INTERNSHIP RECORD 47
Association between HLA B27 and Ankylosing spondylitis
Small minority of people with this disease suffer antigen, even taking family with ankylosing spondylitis. Most people in whom it develops ankylosing spondylitis are previously healthy, however, other diseases with similar behavior called spondyloarthropathies, may occur in people with inflammatory bowl disorder. Urinary tract infections or illness of the skin called proriasis.
Ankylosing spondylitis is a rare disease. It appears in young people. Specially in male between 20 and 30.
However, some cases may begin in childhood or adolescence and affect women, although in these the disease is usually milder and is often more difficult to diagnose. Signs that may indicate the condition of ankylosing spondylitis:-
1. Pain onset before age 35. 2. Spine stiffness in the morning, on rising from bed. 3. Improvement of symptoms with activity. 4. Inflammation of the spinal joints. 5. Can lead to severe, chronic pain and discomfort.
The first thing to note the person with ankylosing spondylitis is usually lumber or low back pain, which is produced by inflammation of the joints sacroiliac and vertebral. This pain is inflammatory, and manifest itself insidiously, slowly, unable to pinpoint the moment that the symptom started. Low back pain occurs when the patient is at rest, improving physical activity. The pain is obtain hard in the final hours o the night and early in the morning, when the patient takes a long time in bed. In these circumstances, this symptom requires the person to get up and walk to notice a relief and even disappearance pain. Given this inflammatory pain of ankylosing spondylitis is another lumbago mechanical origin of sudden onset that is either located by the patient in a particular area of the spine, improves with rest and worsens with. The human leukocyte antigen (HLA B27) is found in 5-10% of the U.S population and is often associated with various autoimmune diseases. The most common is ankylosing spondylitis ( AS). About 90% of patients with AS are HLA B27 positive. Other autoimmune diseases are associated juvenile rheumatoid arthritis (80%), and Reiter’s syndrome or reactive arthritis (50-80%). The HLA B27 is also present in 50% of patients with inflammatory bowl disease and psoriasis with ankylosing spondylitis. The HLA B27 is not the cause of these pathologies, but there is a higher prevalence of tis antigen in affected patients.

BMLT INTERNSHIP RECORD 48
HLA TYPING
The human leukocyte antigen (HLA) test, also known as HLA typing or tissue typing, identifies antigens on the white blood cells (WBCs) that determine tissue compatibility for organ transplantation (that is, histocompatibility testing). There are six loci on chromosome 6, where the genes that produce HLA antigens are inherited: HLA-A, HLA-B, HLA-C, HLA-DR, HLA-DQ, HLA-DP. Serological HLA typing test Principle: For the determination of HLA antigens the HLA antibodies with known specificity must be incubated with a lymphocyte suspension of the sample in the presence of complement. After this addition the lymphocytes should be lysed in the presence of the corresponding Ab and complement. This can be made visible using a stain. The assessment of lysed and non-lysed lymphocytes can be carried out using an inverse phase contrast microscope. Reagents:
1. 72 well tray with predropped anti HLA ABC reagents (Biotest). 2. 72 well tray with predropped anti HLA DR/DQ reagents (Biotest). 3. Lymphoprep; density gradient media. 4. Phosphate buffer solution 5. RPMI-1650 6. Bovine serum. 7. Rabbit complement HLA class I & II 8. Nylon Wool 9. Eosin. 10. Formalin.
Equipments:
1. Syringe-20 ml 2. Pasteur pipette 3. 6 channel Teraski pipette 4. Glass tube 5. Centrifuge machine 6. Incubator 7. Inverted microscope.
Methodology: The commercially available kits “Biotest lymphotype HLA” was used for the typing. The steps of “microlymphocyte Toxicity test” was as follows-
1. 20ml of blood samples were taken from all subjects by venipuncture from the anticubital vein.
2. Anticoagulant heparin was added to the blood in test tube. 3. The blood was layered onto the liquid cushion, lymphoprep. The density is greater than
mononuclear cells but less than polymorph nuclear leukocyte and red cells. 4. Lymphocytes were isolated by centrifuging the blood for 20 min at 2000 rpm. As being
less dense, mononuclear leukocytes were found as a white ring at the boundary between the plasma and lymphoprep.
5. Lymphocyte were taken in a separate test tube and same volume of phosphate buffer solution was added.

BMLT INTERNSHIP RECORD 49
6. Lymphocytes were then passed into nylon wool column to separate the T&B cells. 7. The lymphocytes were then placed on 72 well tray with predropped anti HLA reagents. 8. The cells were first incubated with Ab. 9. the complement was added to each well. 10. The tray was allowed to incubate at room temperature. 11. Ab binding was detected by complement dependent cytotoxicity. If any Ab was present,
the complement was fixed to cell, the membrane attack complex would be assembled and this would lead to cell death.
12. This was detected by adding a dye which cannot enter intact cells but stain the interior of the cells whose membrane had been damaged. The cells were then examined by inverted phase microscope and the pattern of the reaction was noted
HLA CROSS MATCHING
1. 20ml of blood samples were taken from all subjects by venipuncture from the anticubital vein.
2. Anticoagulant heparin was added to the blood in test tube. 3. The blood was layered onto the liquid cushion, lymphoprep. The density is greater than
mononuclear cells but less than polymorph nuclear leukocyte and red cells. 4. Lymphocytes were isolated by centrifuging the blood for 20 min at 2000 rpm. As being
less dense, mononuclear leukocytes were found as a white ring at the boundary between the plasma and lymphoprep.
5. Lymphocyte were taken in a separate test tube and same volume of phosphate buffer solution was added to give up another cells rather than lymphocytes.
6. Cross matching plates are taken and filled with2ụl liquid paraffin as a medium. 7. 1-6 rows in A,B column is filled with (2ụl) Commercially available supplied positive
control. 8. 1-6 rows in C,D column is filled with(2ụl) negative control (RPMI MEDIA 1640). 9. 1-6 rows in E,F column is filled with (2ụl) Donors serum. 10. Lymphocyte cell suspension 1ụl in each well. 11. 5ụl rabbit complement in each wells. And wait for 30 mins. 12. Then put 1ụl eosine and wait for 10 min. 13. Lastly we put formalin as a tissue persevere and freeze it for 1 day.

BMLT INTERNSHIP RECORD 50
HLA B27 TYPING TEST It is a test to look for specific protein called HLA-B27 found on the surface of WBCs.HLA-B27 is a class I surface antigen encode by the B locus in the MHC on chr6 and presents microbial antigens to T-cells. This antigen confers susceptibility to certain diseases like AS. MATERIALS & METHOD:
Reagents:
• 72 well tray with predropped anti-HLA-B27 reagent (Biotest) • Lymphoprep. • Phosphate buffer. • Eosin. • Complement class I. • Formalin.
Equipments: Same as serological HLA typing. Method: Same as serological HLA typing. The only difference is only T-cell is required.

BMLT INTERNSHIP RECORD 51
BLOOD BANK o SELECTION OF DONOR o REJECTION OF DONOR o REGISTRATION OF THE DONOR
o TECHNIQE OF THE BLOOD COLLECTION o PRESERVATION OF BLOOD o ANTI HUMAN GLOBULIN (AHG) OR COOMBS TEST
o QUALITATIVE TEST FOR ‘ABO’ GROUPING, WITH ANTISERA (FORWARD GROUPING)
o QUALITATIVE TEST FOR [ Rh TYPING WITH ANTISERA] o COMPATABILITY TESTING OR CROSSMATCHING o SEROLOGICAL TET FOR HIV ( STRIP METHOD ) o THE RAPID VISUAL TEST FOR THE QUALITATIVE DETERMINE OF HbsAg
o SEROLOGICAL TEST FOR HEPATITIS-C ANTIBODY
o SEOLOGICIAL TEST FOR SYPHILIS (VDRL TEST) o SEROLOGICAL TEST OF MALARIA ANTIGEN o PREPARATION AND USE OF BLOOD COMPONENTS

BMLT INTERNSHIP RECORD 52
In the BLOOD BANK of the s.s.k.m. Hospital we perform several works. These are as follows:-‐ • Collection of blood • Preservation of blood • ABO blood grouping and Rh typing. • Serological tests for HIV, Hepatitis-‐ B, Hepatitis C, Syphilis, Malaria. • Cross matching. • Component separation.
Before the collection of blood some informations are require to assess whether a person is a high risk donor and should not therefore donate blood. This proforma is called Selection and Rejection of Donor.
SELECTION OF DONOR:-‐ • In the last 6 month if he/she had sex with someone unsure about? • In the last year if he given any injection? • If given any blood transfusion? • Inject drug or sharer needle & syringes with others? • A women who is pregnant should not donate blood.
Sometimes older people are not sure of their age but it will obvious wheather the person is an acceptable age & sufficient fit to donate blood.
REJECTION OF DONOR:-‐ • Check for swollen glands, skin rashes, sign of intravenous drug use or abdominal
bleeding (purpura).
• Persons weight 50 kgs or more can safety donate 350 ml of blood. ,
• A donor should not give blood when body temperature is raised.
• A donor should not have an abnormally low blood pressure or a high blood pressure. The upper acceptable limits are diastolic is 100 mm/hg and systolic pressure of 160 mm/hg. The minimum acceptable blood pressure is 90/50 mm/hg.
• The pulse rate be regular and less than 100/minute.
• Measurement of Hb is 12 mg/dl (level using the WHO Hb colour scale).

BMLT INTERNSHIP RECORD 53
REGISTRATION OF THE DONOR
This is a must for the blood banks to keep complete record of the donor, so the donor could be traced for medico legal purposes. The following records are to be maintained
1. Name of the donor 2. Age 3. Sex 4. Date of donation 5. Date of last donation 6. Occupation 7. Complete residential address with phone number 8. Blood group.
TECHNIQE OF THE BLOOD COLLECTION:-‐ 1. The donor lie down under aseptic condition in a well air lightening room.
2. A deflated pressure cuff applied to the upper arm about the elbow. The pressure raised between 60-‐80 mm Hg to enable the veins to be seen and felt. A large well situated vein selected for the venipuncture, usually near the bend of the elbow.
3. The required part of the arm cleaned very well with cotton & 70% ethanol.
4. The identity number of the donor written clearly on the blood pack &pilot tube.
5. A blood collection Pack is taken and the blood bag is suspended on a stand about 30 cm below the donor’s arm.
6. The needle guard is unclamped and skin is stretched below near the choose vein. 7. A venipuncture made with the upward bevel toward the vein. To secure the needle in place
with a small strip of adhesive tape.
8. When the blood flowed through the tube, the pressure of the cuff reduced to 40-‐60 mmHg, and asked the donor to squeeze slowly a small object.
9. The blood is mixed with the anticoagulant by lifting & tilting the bag when blood is entered.
,
10. When the bag’s weight is approximately 450-‐500 gms,then the donation is completed. 11. The pressure is reduced to zero, and needle is removed from donor’s hand. Clamed off the
tube 10-‐15 cm from the needle.
12. A knot is tied tightly and also sealed with clip in about 20 cm from the needle. 13. The tube is cut down between the clamp and knot. The blood between the cutting part is
poured it into the pilot tube. Then reclamed the tube.

BMLT INTERNSHIP RECORD 54
14. The venipuncture site is pressed with cotton and when the bleeding is stop sealed the place with colloidal solution.
PRESERVATION OF BLOOD:-‐
For preservation of the collecting blood is mixed with CPDA ( Citrate Phosphate Dextrose Adenine) anticoagulant and store in freeze at 2-‐8c temperature for 35 days.
Composition of the CPDA anticoagulant
Citric acid 3.2 gm
Sodium citrate 25.8 gm
Glucose 25.0 gm
Sodium Phosphate 2.18 gm
Distilled water 1000 ml
27.5 mg of Adenine is mixed with 1000 ml of CPD solution.
Other anticoagulants used for blood preservation
Name of the anticoagulant Stability time of blood
1. Acid citrate Dextrose 14 days
2. Citrate Phosphate Dextrose 21 days
3. Heparin 48 hours
4. RBC suspension in saline , adenine, glucose-‐manitol.
49 days

BMLT INTERNSHIP RECORD 55
ANTI HUMAN GLOBULIN (AHG) OR COOMBS TEST Specimen:-‐ Clotted blood is preferred over whole (citrated) is required for direct antihuman globulin test. In case of indirect antihuman globulin test for antibody screening, serum specimen will be needed.
Principle:-‐ Antihuman globulin test (also called antiglobulin test or coombs test) detects “sensitized red cells” where the red cells get coated with IgG (Anti-‐body) or globulin but do not agglutinate. When the sensitized red cells come in contact with antihuman globulin reagent (or antiglobulin or coombs reagent) they agglutinate.
Reagents:-‐ Antihuman globulin (AHG) reagent Presensitized red cells (coombs control cells) Saline Procedure( Direct coomb):-‐
1. Wash the red cells suspension of being sensitized, 3-‐4 times in large volumes of saline. Complete removal of free globulin is important.
2. Decant completely at the end of the last washing. ,
3. Add 2 drops of antihuman globulin serum to the sediment cells remaining (buttons). Notes follow the manufacturer’s instruction regarding the use of AHG.
4. Tube is mixed well centrifuged at 1500 rpm for 1 min.
5. Examine for agglutination by holding against a lighted background and tapped the bottom of the tube. A small hand lens or magnifying mirror attached to a spotlight may be used optical aids. The tube is holed at an angle, shake gently until all cells are dislodged, then tilted the tube back and forth, gently until an even suspension of cells or agglutinates is observed.
6. In case of no agglutination tube is left at room temperature for 10 mins then recentrifuged and read. A weaker reacting antibody will show delayed reacting. Consider this as positive.
7. In case of no haemagglutination then one drop of presensitized red blood cells indicating that the AHG is reactive and the result is valid. This is an important step of quality control

BMLT INTERNSHIP RECORD 56
because it is the only way to monitor with saline incomplete, antiglobulin will be neutralized by the free globulin before reacting with the coated –globulin.
Interpretation:-‐
Haemagglutination of red cells with the addition of AHG (positive) indicates that the cells are sensitized inside the body. If the antibodies are to be identified, they are eluted and then tested in the same way as the serum. This will be further discusse
QUALITATIVE TEST FOR ‘ABO’ GROUPING, WITH ANTISERA
( FORWARD GROUPING)
PRINCIPLE:- The procedure used with the antisera(Anti-A & Anti-B) are based on principle of agglutination. Normal human red cells possessing antigen, will clump in the presence of corresponding antibody. METHOD:- Slide Method PROCEDURE;-
1. Taken a clean and dry glass slide and divided it into two halves by a lead pencil and leveled them as ‘A’ and ‘B’.
2. One drop of anti-A on the one half of slide marked A; and one drop of Anti-B on the other half is placed.
3. One drop of blood to each half is added. 4. Mixed thoroughly the blood and antiserum with a stick and spread to form a 2cm circle. 5. Tilte the slide back and forth to complete the mixing. 6. Examined for agglutination within 2 mins. Under bright light.
INTERPRETATION:-
Reaction Interpretation group
Anti-A Anti-B
+ – A
– + B
+ + AB
– – O
‘–’ = No agglutination ‘+’ = Haemagglutination

BMLT INTERNSHIP RECORD 57
QUALITATIVE TEST FOR [ Rh TYPING WITH ANTISERA PRINCIPLE:- The procedure is based on the principle of agglutination. Normal human red blood cells, possessing ‘D’ antigen(‘Rh’ antigen), will clump in the presence of blood typing serum containing Anti-D antibody. METHOD:- Slide Method PROCEDURE:- 1. Taken a clean and dry glass slide, and placed one drop of anti-D serum. 2. Added one drop of blood. 3. Mixed thoroughly the blood and antisera with a stick and spread to form a 2cm
circle. 4. Tilted the slide back and forth to complete the mixing. 5. Examined for agglutination within 2 mins. Under bright light INTERPETATION:- BLOOD Anti-D antibody Reaction Interpretation
(‘Rh’typing) 1 drop 1drop Haemagglutination Positive
1drop 1 drop No Haemagglutination Negative
Compatability testing or crossmatching Principle:-‐ Serum of the recipient is tested against the red cells of the donor under different conditions in order to establish their compatibility or non-‐agglutination. Agglutination in any of the conditions indicates the presence of incompatible antibody in patient, natural or immune. The three phases of compatibility testing are done.
1. Saline Phase:-‐ Where the immunologic reaction between red cells suspended in saline medium and the antibody occurs at room temperature.

BMLT INTERNSHIP RECORD 58
2. Thermophase with protein:-‐ Where the immunologic reaction between red cells suspended in saline medium and the antibody occurs at room temperature. <
3. Antihuman globulin(AHG) Phase:-‐ Where the incubated cells are washed (toremove free globulin) and reacted with antihuman globulin serum (coombs reagent or antiglobulin).
ABO incompatibility is recognized in the saline phase while agglutination in other phases indicate the presence of immune, incomplete or irregular antibodies. If agglutination is not seen in any of the above phases, donor’s and recipient’s blood are considered to be compatible.
Specimen:-‐ Donor’s clotted blood specimen is available from the pilot tube. Donor’s red cell are taken out of the clot, repeatedly washed with saline and a 5% v/v suspension is made in saline (0.1 ml packed red cells mixed with 1.9 ml of saline). Patient’s blood is drawn fresh and collected in a sterile prelabelled dry container without any anticoagulant. The serum is separated promptly. Patient’s serum is used for major crossmatching and cell suspension for autocontrol.
Procedure:-‐
1. Two small tubes are taken and marked as them 1 and 2. Tube 1 for crossmatching and 2 for autocontrol.
2. 2 drops of patients serum is added in both tubes. ,,
3. 1 drop of 5% saline is added in tube 2.
4. Mixed and centrifuged them T 1500 RPM for 1 min. 5. After dislodge the cell button and examined for agglutination and haemolysis. If
agglutionation is noted in tube 1, ABO incompatibility is suspected. 6. In both tubes a drop of 22% bovine albumin is added, mixed and incubated at 37c for
30 min.
7. Centrifuged at 1500 rpm for 1 min. Examined for agglutination and haemolysis. Result of agglutination is recorded with grading.
8. Then washed the cells 3 to 4 times with saline, decant completely after each wash and added 2 drops of antihuman globulin serum to the sediment cells. Mixed them properly and centrifuged at 1500 rpm for 1 min. Examined for agglutination and grade the agglutination reaction. Some of antibodies bind with the complement and bring haemolysis which considered as an evidence of immunologic reaction(positive).

BMLT INTERNSHIP RECORD 59
9. If there is no agglutination reaction (negative), added a drop of presensitized cells. The sensitized or check cells must agglutinate if the cells are adequately washed and AHG is reactive.
Interpretation:-‐
1. Agglutination never visible in any phase, including the autocontrol.
2. Incompatibility in saline phase should be investigate in the line of ABO grouping. Haemolysis means tha presence of cold antibodies.
3. Haemolysis in thermophase with protein may indicate the presence of immune antibodies of A and B, anti-‐Le and Rh antibodies.
,
4. Haemagglutination at the AHG phase detects such antibodies as anti-‐Fy, Anti-‐jk, anti-‐K, and others. Some of the hard-‐to-‐detect Rh Antibodies and antibodies of haemolytic anaemia are also in the AHG phase.
SEROLOGICAL TET FOR HIV ( STRIP METHOD )
1. All the reagnts are brought to room temperature. 2. Determined the number of strip comb is required.
3. Dilute the washing buffer. 4. Two drops of sample diluents added in micro test wells.
5. Added 2 drops of sample and controls into each micro test tube well containing sample
diluents. 6. Placed the combs into respective wells.
7. Incubate for 10 mins at room temperature. . 8. Added 4 drops of colloidal gold signal reagent in required number of micro test wells.
9. Washed the combs by moving the comb forward and backward 10 times in the washing
solution containing in the wash trap. 10. Placed the comb micro test wells containing colloidal gold signal reagent.

BMLT INTERNSHIP RECORD 60
11. Incubate for 10 mins at room temperature.
12. Wash the comb by removing the comb forward and backward 10 times in the washing solution containing in the wash trap.
13. Allow the combs air dry and the developed color is noted on the supported area on the
tip of teeth of the comb. INTERPRETATION:- Pink color = positive No color = negative
THE RAPID VISUAL TEST FOR THE QUALITATIVE DETERMINE OF HBsAg
Introduction:-‐ The antigenic determinant of the HBsAg protein moiety is antigenically heterogeneous and it determines specific HBV serotypes and provides a basis of immunodetection. The assay is intended to be used as an aid in the recognition and diag(HBV). Method:-‐ One step immunoassay for the qualitative detection of Hepatitis B surface antigen (HBsAg). Principle of the test:-‐ This is a one step immunoassay based on the antigen capture, or ‘sandwich’ principle. The method uses monoclonal antibodies conjugated to colloidal gold and polyclonal antibodies immobilized on a nitrocellulose strip in a thin line. The test sample is introduced to and flows laterally through an absorbent pad where it mixes with the signal reagent. If the sample contains HBsAg, the colloidal gold-‐antibody conjugate binds to the antigen, forming an antigen-‐antibody-‐ colloidal gold complex. The complex then migrates through the nitrocellulose strip by capillary action. When the complex meets the line of immobilized antibody (Test-‐line)”T”, the complex is trapped forming an antibody-‐antigen-‐antibody colloidal gold complex. This forms a pink band indicating the sample is reactive for HBsAg. To serve as a procedural control, an additional line of anti-‐mouse antibody (control-‐line) “C” has been immobilized at a distance from the test line on the strip. If the test is performed correctly, this will result in the formation of a pink band upon contact with the conjugate.

BMLT INTERNSHIP RECORD 61 Specimen collection & storage:-‐
This test should be performed on human serum or plasma only immediately after collection.
Requirements & Reagents:-‐
Test device (Kit provided) Dessicant pouch (Kit provided). Dropper (Kit provided)
PROCEDURE:-
1. The required number of HEPACARD foil pouches and specimen are brought to room temperature prior to testing.
2. Taken out HEPACARD devices from the foil pouch. In case of reseal make it tightly with clamp and rod so that devices are protected from moisture otherwise the device will get deteriorated thus giving erratic results.
3. Label the test card with patient’s name or identification number. 4. Added 2 drops of human serum specimen into the ample well using the dropper. 5. Allow reaction to occur during the next 20 mints. 6. Read result at 20 mints. 7. After the test reading discard the HEPACARD because it to be potentially infectious.
INTERPRETATION;-
• REACTIVE: Appearance of pink colored line, one each in test region “T” and control region “C” indicates that sample is reactive.
• NON-REACTIVE: Appearance of one distinct pink line in the control region “C’’
only, indicates that the sample is NON-REACTIVE for HbsAg. • INVALID: When neither control line nor the test line appears on te membrane the
test should be treated as invalid.

BMLT INTERNSHIP RECORD 62
SEROLOGICAL TEST FOR HEPATITIS-C ANTIBODY PRINCIPLE: Recombinant antigens (NS3, NS4, NS5, &Core) of Hepatitis C virus are spotted onto the membrane of the test device. When antibodies to HCV are presence in the test serum or plasma, they react with these antigens and attach to the solid-phase. Non-reactive antibodies are filtered through by the wash buffer. HCV antibodies bound on to the membrane are visualized by reacting with colloidal gold protein-A signal reagent. Appearance of two magenta red dots/ spots (one corresponding to the procedural control and other to the recombinant HCV antigen) indicating positive reaction. A built in control, immobilized separately on porous membrane, serves as a procedural control and a dot /spot will always appear at the control region regardless o the presence of anti-HCV. PROCEDURE:
1. Label the test device with patient identification code & keep it onto a horizontal surface. 2. Aded 2 drops (100ụl) of wash buffer to the control of the Test device and allow it to soak
in completely. 3. after 30 secs, hold the dropper vertically and added 2 drop (100ụl) of patients sample
with the disposable plastic dropper and allowed to soak in completely. 4. after 30 secs , 2 drops of wash buffer is added and allow to soak in completely. 5. After 30 secs drops of signal reagent is added and allow to soak in completely. 6. After 30 secs, added 3 drops of wash buffer and allow to soak in completely. 7. Read the result with in 2 mints for earliest interpretation. 8. Final test result should be read after 10 mints.
INTERPRETATION: POSITIVE: If two magenta red dots/spots, one for control and other for test appears. The sample is reactive for Abs. to HCV. NEGATIVE: If only one magenta red dots/spot, only the control, appears , the sample is non-reactive for Abs. to HCV.
SEOLOGICIAL TEST FOR SYPHILIS (VDRL TEST) The full name of VDRL is Veneral Disease Research Laboratory or RPR is Rapid Plasma Reagin test. It is used for detection of Veneral disease like syphilis which is caused by Treponema pallidum.

BMLT INTERNSHIP RECORD 63 PROCEDURE
1. One Drop of positive control, negative control and test sample is transferred at separate circle of the disposable test card.
2. Added one drop of RPR Ag(antigen) individual circle using the Ag dropper. 3. Mixed well and spread the mixture in the entire area of the test card. 4. Tilted the card gently back and forth for 8 mints and observed under bright light.
INTERPRETATION OF RESULT No agglutination indicate--- Negative result Any agglutination indicate--- Positive result.
SEROLOGICAL TEST OF MALARIA ANTIGEN
This test is done by rapid malaria antigen test strip. Which consist of HRP-2 antibody with p-LDH antibody. If blood sample contains of any type of malarial antigen it will detect within 15-20 mints. Generally there are two types of malaria species in the west Bengal zone, one is Plasmodium vivax and another is plasmodium falcifarum. Before transfusion of blood it is also an important test to detect is there any malarial species present in donors blood. If it is in the donor’s blood, then immediately it will reject. TEST STPIR DIAMED rapid malaria antigen test. PROCEDURE
1. All the test strip and sample arebrought to the room temperature. 2. Blood sample should be mixed with EDTA. 3. 0.02 ml of blood is put at the sample area and wait for 5mints to spreading. 4. Then 0.02 ml of buffer solution is added over it and weight for 15 mints. 5. After 15 mints result is observed for interpretation.
INTERPRETATION OF RESULT RESULT C Line Pv Line PF Line
Negative show Nil Nil Plasmodium vivax positive Show Show Nil
Plasmodium falcifarum positive show show show

BMLT INTERNSHIP RECORD 64
• C line:- control line • Pv ine:- plasmodium vivax • Pf line:- plasmodium falcifarum.
PREPARATION AND USE OF BLOOD COMPONENTS
Whole blood is an admixture of many different cellular and noncellular components, each having a different and important physiological function to play. Various components of blood are used in transfusion therapy with the goal of correcting the deficiency of the particular component in the patient. INDICATION FOR COMPONENT SEPERATION:- ˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜˜
1. Donor must be healthy. 2. Hb% should be 12.5 gm /dl. 3. Weight within 55-70 kg. 4. Normal blood pressure and no medication. 5. Component is separated within 6 hours of collection. 6. Donors blood Selected for component separation is collected in double and triple bag.
From whole blood mainly two products are obtained 1. packed red cell 2. Plasma preparation .
1. Packed red cell : - Red blood cells are the primary cellular component used for the transfusion therapy of anaemic patients with low haematocrit. When red cells are separated from the plasma and used for transfusion, they are called packed cells. Packed red cells are obtained from the whole blood by centrifugation to give a haematocrit of 70 to 80%.plasma is removed and the resulting red blood cell preparation is transfused to the patient after diluting with sterile saline. Transfusion of packed cells is reflected by the improvement of haematocrit value. 2. PLASMA PREPARATION:-
Fresh Frozen Plasma (FFP) : - FFP is separated from the whole blood within two hours after the collection of blood from the donor. It is rich in all coagulation factors and is given to patients with general bleeding disorders, burn case and acute dehydrated persons. Administration of FFP requires blood compatibility testing for ABO.
Whole blood 300ml packet is centrifuged at 4000 r.p.m in centrifuge machine.

BMLT INTERNSHIP RECORD 65
Platelet Concentration:-It is separated from whole blood. After centrifugation when plasma is separatation completed then from Buffy coat platelet is collected. The survival time of the platelets within the body is 2 to 6 days and daily transfusions are usually needed. Platelet concentrate is given to thrombocytopaenic patients with a history of bleeding. ABO compatibility is desirable but not essential in case of platelet transfusion. When platelets are given to a bleeding patient, the therapeutic effect is measured by improved haemostasis, and not by the improvement in laboratory values of the platelet count.
Cryoprecipitate:- It is derived from fresh plasma and is rich in factor VIII and
is administered to haemophilic patients (haemophilia A) and patients with Von willebrand’s disease (Vascular haemophilia). Administration of cryoprecipitate does not require any blood compatibility testing. ABO group compatibility is, however, important in infants.
Separation Procedure:: - - -
1. Double and triple bags are centrifuge in CRYOFUSE machine at 3880 rpm/5min.
2. Temperature should be 20-30˚c in case of platelet/plasma/pack cell And 4˚c in case of cryoprecipitate.
3. Centrifuged bag is placed in base of expresser machine with most care. 4. Machine liver is till on upto plasma and Buffy coat level. Just when the
Buffy coat is separated into another bag the liver is off. 5. The junction tube is sealed with sealing machine and cut off. 6. The separated platelet rich plasma (PRP) bag is again centrifuge in 3880
rpm/5 min. 7. With expresser machine the supernatant is transferred to another bag as
fresh frozen plasma (FFP) and sealed it. 8. Sediment is platelet bag.
Storage:-
FFP = –38 to –40˚c Platelet=20 to 25˚c with azitation.in 72 rpm. Packed red cell= 2 to 8 ˚c
DEPARTMENT OF BLOOD BANK Sl No.
Patient’s name
Age (yrs)
Sex ABO Rh HIV HBsAg HCV VDRL MP
1 Anima Maity 22 F B + ve NR NR NR NR NF 2 Rupasri
Panda 23 F B + ve NR NR NR NR NF
3 Rebati Kapat 24 F B + ve NR NR NR NR NF

BMLT INTERNSHIP RECORD 66 4 Sabera Bibi 23 F B + ve NR NR NR NR NF 5 Kanchan
Patra 25 F B + ve NR NR NR NR NF
6 Kajal Senapati
60 M O + ve NR NR NR R NF
7 Gouri Pradhan
42 F B + ve NR R NR NR F
8 Dipali Mondal
33 F O + ve NR NR NR NR NF
9 Rabindra Das
36 M A + ve NR R NR NR NF
10 Ameresh Kumar
35 M AB + ve NR NR NR NR NF
11 Moriyam Bibi
23 F B + ve NR NR NR NR NF
12 Biswajit Gupta
22 M O + ve NR NR NR NR NF
13 Rasbehari Bag
20 M O + ve NR R NR NR NF
14 Sagarika Das 30 F O + ve NR NR NR NR NF 15 Arup
Mondal 24 M AB + ve NR NR NR NR NF
16 Moumita das
23 F A + ve NR NR NR NR F
17 Manju Mahato
30 F O + ve NR R NR NR NF
18 Supiya Bibi 31 F A + ve NR NR NR NR NF 19 Soumitra
Das 38 M B + ve NR NR NR NR NF
20 Kabita Sing 39 F B + ve NR NR NR NR F 21 Shikha
Gayen 26 F B + ve NR R NR NR NF
22 Arati Paul 50 F AB + ve NR NR NR NR NF 23 Sandhya
Maity 46 F O + ve NR NR NR NR NF
24 Abdul Wahab
38 M O + ve NR NR NR NR F
25 Susma Dey 26 F B + ve NR NR NR NR F 26 Rigia Khatun 27 F B + ve NR NR R NR NF 27 Tarani
Sarkar 22 F A + ve NR NR NR NR NF
28 Bundey Paul 24 M AB + ve NR NR NR NR NF 29 Satish Bag 26 M A + ve NR NR NR NR NF 30 Bundey Paul 28 M B + ve NR NR NR NR NF

BMLT INTERNSHIP RECORD 67
MICROBIOLOGY
o GENERAL MICROBIOLOGY
o SEROLOGY

BMLT INTERNSHIP RECORD 68
GENERAL MICROBIOLOGY
o THE BASIC STEPS OF BACTERIOLOGY
o GRAM’S STAIN
o ZIEHL – NEELSEN STAINING
o DIFFERENT TYPES OF MEDIA AND
THEIR PREPARATION
o ESCHERICHIA COLI
o KLEBSIELLA
o PROTEUS SP.
o SENSITIVITY TEST (ANTIBIOTICS TEST)

BMLT INTERNSHIP RECORD 69
o Microbiology is the science of living organisms that are not directly visible to the
necked eye but only under the microscope.
Medical microbiology deals with the causative agents of infectious diseases, the way in which they produce diseases in the human body and essential information for diagnosis and treatment. The basic steps of bacteriology-‐ (I) Specimen collection
(II) Registration (III) Direct smear and staining and media preparation (IV) Culture/inoculation. Smear and staining (V) Biochemical test (VI) Antibiotic sensitivity test
(i) Specimen collection-‐ Sputum, Throat swab, Urine, Pus, Stool and Blood.
Sputum: 1 . Before collect the sample the mouth should be rinsed. 2 . Always collect the first morning sample; it represents the pulmonary secretion accumulated overnight. 3 . In case of children Nasopharyngeal swab may be taken which can be represents the bronchial pathogens. 4 . The sample must be taken in a cotton plug sterile in a cotton plug sterile test tube with cotton plug broom steak.
Throat swab 1 . The sample is collected in the morning before brush. 2 . Throat swab should be collected with a sterile swab from the tonsils, tonsilar fossa, and posterior pharyngeal wall. 3 . The sample must be taken in a cotton plug sterile test tube with cotton plug broom steak.
Urine 1 . First morning sample should be collected. 2 . Mid stream urine sample must be collected. 3 . The sample should be collected 3/4th of a sterile test tube.
Pus
Pus sample should be collected from the wounds of the patients, in a sterile cotton plug test tube with a cotton plug broom steak.

BMLT INTERNSHIP RECORD 70
Stool
First morning stool sample are collected in a sterile container.beside this all fluid sample (ascetic fluid etc.) are collected in a sterile test tube.
Blood 1 . Great care must be taken to prevent contamination of the specimen during its collection. 2 . Blood should be collected with sterile syringe and needle.First the skin over the site is disinfected with 70% alcohol +1% iodine. The operator’s hand must be cleaned, dry and covered with sterile gloves. 3. After drawing the required amount of blood the needle must be replaced by a fresh one (sterile one) for inoculating the blood into the blood culture bottle.
(ii) Registration-‐ After receiving the specimen maintaining of log book or register is a vital step for the other steps. It will be maintain by registration of date of specimen receiving, patient’s identity, diagnosis etc.
(iii) Staining:
GRAM’S STAIN It is the most widely used stain in medical used stain in medical bacteriology . The stan was originally devised by Christian Gram (1884) as a technique of staining bacteria in tissues . There are four steps in the technique .
Reagents: A . Crystal violet solution Crystal violet …… 0.5gm Distilled water to make …... 100ml B . Gram’s iodine Iodine …… 1gm Potassium iodide ……2gm Distilled water to make ……100ml
At first dissolved potassium iodide in 50 ml water. Then add iodine and dissolve in it C . Acetone or alcohol D . Safranin Safranin …… 100ml
PRINCIPAL When bacteria are stained , some bacteria are got the primary stain and when they treated
with decolouring agent they do not lose their original colour . They are called Gram – Positive bacteria and they appear in violet colour because of Crystal Violet dye . Other bacteria lose their primary colour during decolourisation and got the counterstain at last time, they are called Gram – negative bacteria . They are appear in pink colour for the counterstain Saffaranin .

BMLT INTERNSHIP RECORD 71
METHOD: 1 . Primary staning of heat fixed smear of specimen or bacterial culture is made with a pararosaniline dye , e g. crystal violet solution for one minute. Usually the smear is fully covered with crystal violet solution . 2 . Pour off crystal violet and add dilute solution of iodine, such as Gram’s iodine, keep for one minute . 3 . Wash with water. 4 . Decolourisation with an organic solvent (alcohol or acetone) – 10 to 30 seconds . 5 . wash with water immediately to remove decolourisation . 6 . Counterstain with a dye of contrasting colour (dilute carbol fuchsin , safranin or neutral red) for ½ to 1 minute . 7 . The smear is washed with water and then blot dried . 8 . Examine under oil-‐immersion objective (x100).
OBSERVATION: Gram-‐ positive bacteria resist decolourisation and stain violet .Gram-‐ negative and other cells (pus cells) are decolourised and stain pink with counterstaion . The Gram-‐ positive bacteria may sometimes appear Gram-‐negative under certain situations, such as in ageing cultures and when the cell wall is damaged . On the basis of Gram’s staining , bacteria are divided into two categories : 1 . Gram-‐positive bacteria : All cocci except Neisseria and Branhamella are gram – positive . 2 . Gram – negative bacteria : All bacilli are Gram – negative except Corneybacterium , Mycobacterium , Bacillus , Clostridium , Actinomyces , Listeria and Erysepalothrix which are Gram -‐ positive
ZIEHL – NEELSEN STAINING
Ziehl – Neelsen staining , the procedure is named after Ziehl and Neelsen , who had modified it , but the procedure was ultimately discovered by Ehrlich . It has an other name , Acid – fast staining .
REAGENTS 1 . Ziehl – Neelsen’s Carbol fuchsin Basic fuchsin 1gm Phenol (crystalline) 5gm Alcohol (95% or absolute) 10ml Distilled water to make 100ml Dissolve the dye in the alcohol and add the same to phenol 5% plunol solution . 2 . Methylene blue Satured solution of Methylene blue in alcohol 30ml KOH , 0.01% in water 100ml

BMLT INTERNSHIP RECORD 72
PRINCIPLE: Mycobacterium Tuberculosis is very difficult to identify and staining the organism as
organism is coated with lipid containing cell wall . They bind carbol fuchsin tightly and resist re – staining with strong decolourising agents such as alcohol and strong acid . Acid fast negative bacteria readily loss the stain when they treated with alcohol and strong acid . Heat is applied in Ziehl – Neeisen or stain method detect mycobacterium laprae , Nocardia asteroids etc . All these methods used carbol fuchsin as the primary stain and phenol as mordant . Following the counter stain by methylene blue or malachite green . De-‐ colorized acid fast , negative bacilli and other cells kept blue colour in contrast with the red colour acid fast bacilli .
PROCEDURE 1 . Smear is gently heated by flaming from underneath and thus the smear is fixed . 2 . Carbol fuchsin solution is poured on the slide to cover it completely . 3 . The slide is gently heated from underneath by means of a spirit lamp flame until the steam first commences steam . 4 . The steam is left on the slide for 5 – 10 minutes. See that the smear does not dry . To counteract drying , more stain is stain is added on the slide and reheating of the stain is necessary for penetration of the in the bacterial cell wall . 5 . The smear is decolourised with 20% sulphuric acid for half to one minute . Alternatively, acid-‐alcohal (3ml HCL and 97ml ethanol) may be used as a decolourising agent . Decolourisation is continued until the smear is pinkish on washing . However, M . Leprae , shold be strictly decolourised by 5% sulphuric acid only as it is less acid-‐ fast . 6 . The smear is counterstained with 2% methylene blue or malachite green for 2 minutes and washed in water .
OBSERVATION Acid – fast bacilli appear red in blue background of pus cells and epithelial cells.The slide is seen under oil – imersion objective (x100)of microscope . Acid-‐ fast organisms 1 . Mycobacterium – Tubercle and leprae bacilli . 2 . Others – Bacterial spores , ascospores of some yeasts , Actinomyces clubs (in animal tissues), some Nocardia Species and Cryptosporidium spores . The spores are weakely acid – fast where the decolourising agent used is 0.5% H2 SO4 . (iv) Media preparation-‐ Media are important ingredients for a laboratory to initiate growth
of micro-‐organisms for the purpose of diagnoss, research etc. So preparation of media is a vital part of laboratory activities.

BMLT INTERNSHIP RECORD 73 DIFFERENT TYPES OF MEDIA AND THEIR PREPARATION: (1) MacConkey’s Agar media –
1st part -‐-‐-‐-‐-‐-‐ by autoclaving at 15 lb pressure Ditilled water -‐-‐-‐-‐-‐-‐ 100 ml Peptone -‐-‐-‐-‐-‐-‐ 2 gms Sodium Chloride -‐-‐-‐-‐-‐-‐ 0.5 gm Sodium Tauracholate -‐-‐-‐-‐-‐-‐ 0.5% Or Bile salt (0.5)gms
Agar agar powder -‐-‐-‐-‐-‐-‐ 2.5 gm Heat the mixture in a water to make a homogeneous solution and then check the pH of the solution. Then autoclave at 15 lbs pressure for 30 minutes. 2nd part -‐-‐-‐-‐-‐-‐ by steaming Add 1% Lactose and 1% Nutral red solution (indicator) to the 1st part of Macconkey’s Agar and then steam for 30 minutes in an autoclave. Finally pour in a sterile Petridis and after 15 minutes keep them in a refrigerator.
(2) Nutrient Agar Media – Distilled water -‐-‐-‐-‐-‐-‐ 100 ml Peptone -‐-‐-‐-‐-‐-‐ 1 gm (%) Sodium chloride -‐-‐-‐-‐-‐-‐ 0.5 gm (%) Beef heart extract -‐-‐-‐-‐-‐-‐ 1 gm (%) Agar agar -‐-‐-‐-‐-‐-‐ 2.5gm (%) Heat is done to make a homogeneous solution . Check the pH of the solution 7.6 and then autoclave it at 15lbs pressure for 30 minutes then pour it in a sterile petridish.
(3) Nutrient broth Media – Distilled water -‐-‐-‐-‐-‐-‐ 100 ml Peptone -‐-‐-‐-‐-‐-‐ 1 gm (%) Sodium chloride -‐-‐-‐-‐-‐-‐ 0.5 gm (%) Beef heart extract -‐-‐-‐-‐-‐-‐ 1 gm (%) Heat to make a homogeneous solution. Check the pH of solution 7.6 . Then make the tubesof 3 to 4 ml and then plug the tubes with cotton . Then autoclave at 15 lbs pressure for 30 minutes
(4) Peptone water-‐ Distilled water -‐-‐-‐-‐-‐-‐100 ml Peptone -‐-‐-‐-‐-‐-‐ 1 gm (%) Sodium chloride -‐-‐-‐-‐-‐-‐ 0.5 gm (%)

BMLT INTERNSHIP RECORD 74 Heat to make a homogeneous solution. Check the pH of thesolution 7.6 . Make tbbes of 3 to 4 ml . Then autoclave at 15 ibs pressure for 30 minutes.
(5) Blood Agar Media – Sterile nutrient Agar -‐-‐-‐-‐-‐-‐ 100 ml Sterile defrinated -‐-‐-‐-‐-‐-‐ 10 ml Procedure-‐ The temperature of the nutrient agar should be 54 to 56 C . Then add the sheep blood, Which should be at room temperature. So the final temperature are should not below 45 c and the final temperature are should not below 45 c and the final maximum temperature 48 to 52 c . Then stir well the solution to make a homogeneous. Then pour it in a petridish . Then put the plate inside an incubator for 24 hrs to check the agar plate for growth (i.e; weather the plate contaminated or not.)
(6) Preparation of Blood Culture media – Blood Culture Media with glucose broth: Sterile nutrientbroth glucose -‐-‐-‐-‐-‐ 0.2 % Add the nutrient broth the glucose near the flame. Stir to make a solution put in bottles,100 ml each and then autoclave for 30 minutes.
(7) Salmonella shigella (S.S.) media-‐(Indicator:Neutral red) : It is a prepared media. No need to check the pH gm. of S.S. media for 1000 ml.of distilled water. 6.0 gm S.S. media is needed for 100 ml. of distilled water. Take sterile distilled water and add the media. Heat in a water bath to make a solution. Then pour in a sterile plate. No need to autoclave the above media.
(8) Bile Salt Agar media:-‐ Distilled Water -‐-‐ 100 ml. Peptone -‐-‐ 1 gm % Nacl -‐-‐ 0.5 gm (5%) Beef heart extract -‐-‐ 1 gm % Agar agar -‐-‐ 2.5 gm % Sodium Taurocholate or bile salt -‐-‐ 0.5 gm (0.5%) Heating is done in a water bath to make a homogeneous solution. Check the pH of the solution between (8.0 – 8.6) and autoclave at 15 lbs pressure for 30 minutes. Then pour in a sterile Petri dish. Cool and keep in a refrigerator.

BMLT INTERNSHIP RECORD 75
ESCHERICHIA COLI
This genus was named after Escherchia who was the first to describe the colon bacillus . E . Coli lives in the human or animal intestine . When it is voided in faeces it remains in the environment for some days . Detection of E . coli in drinking water is taken as evidence of recent pollution with human or animal faeces .
Morphology E. Coli is a Gram-‐negative , straight rod . it is motile by peritrichate flagella though some strains may be non-‐ motile . Capsules and fimbriae are found in some strains . spores are not formed Many strains have a combination of character . this is because of conjugation and transduction between bacterial strains .
Pathogenicity Escherichia coli is associated with four diseases : 1 . Urinary tract infections. 2 . Diarrhoea / Gastroenteritis . 3 . Pyogenic infections . 4 . Septicemia
Name of the culture media provided : Mc Conkey’s agar Colony- chractrristics : Confluent colonies are smooth , moist , shiny , transluscent , non – mucoid
and pink in colour indicating that the organism is a ‘lactose – fermenter’ . ( Discrete colonies are small , circular , low convex and have other common characteristics ) .
Gram – staining finding : Gram stained smear shows gram –ve , non – sporing bacilli arranged haphazardly .
Test for motility : Hanging drop preparation shows motile organisms . Bio – chemical reactions : Indole production test ------ +ve
: Glucose , Mannitol , lactose , Sucrose all are fermented to produce both acid and gas . : Urease production test ------ - ve
Provisional diagnosis : From the above findings we can provisionally diagnose it as Escherchia coli .
Confirmatory Tests : Catalase test ------ +ve , MR test ----- +ve : Oxidase test ------ - ve , VP test ----- - ve : Nitrate reduction test ------- + ve : Citrate utilization test ------- - ve : H 2S production test ------- - ve : Serotyping can be done with high titre antiserum Final diagnosis : From the above parametere we can finally diagnose , the given organism is
Escherichia coli

BMLT INTERNSHIP RECORD 76
KLEBSIELLA The genus Klebsiella consists of non – motile , capsulated rods which grow very well on ordinary media forming large dome shaped mucoid colonies which are sticky . Klebsiella are widely distributed in nature occurring both as commensals in intestine and as saprophytes in soil and water . They are classified into three species .
• Klebsiella pneumoniae .
• Klebsiella ozaenae . • Klebsiella rhinoscleromatis .
The outcome is poor , case fatality is 80% . There is involvement of one or more lobes of the lung and massive mucoid inflammatory exudates . Necrosis and abscess formation is more common than pneumococcal pneumonia . Urinary tract infection – Klebsiella is a common cause of UTI and they a red resistant to most antibiotics . It also causes pyogenic infections such as abscess , meningitis and septicemia .Rarely it can cause diarroea .
Colony character :Confluent colonies are smooth , moist , shiny , translucent , mucoid and pink in colour indicating the organisms are ‘lactose fermenter’ . ( ( Discreat colonies are large , circular , dome – shaped and having all other above characters )
Gram – staining finding :Smear shows gram – ve , non – sporing plump bacilli arranged hapazaardly .
Motility test :Hanging drop preparation shows non – motile organisms . Bio – chemical reactions : Indole production test - -ve
: Glucose , Lactose , Manitol - all are fermented to Produce acid and gas .
Provisional diagnosis : From the above findings we can provisionally diagnose the organism as Klebsiella sp.
Other tests : Catalase test ------ +ve : Oxidase test ------ -ve : Nitrate reduction test ------ -ve : MR test ------ -ve : VP test ------ -ve : Citrate utilization test ------ +ve : Growth in KCN media ------ +ve
Confirmatory test : Serotyping with specific high titre antisera Final diagnosis : From the parameter given above we can finally diagnose the organism as
Klebsiella sp.

BMLT INTERNSHIP RECORD 77
PROTEUS SP. Proteus is Gram – negative bacilli . They are called proteus after the Greek God Proteus who could assume any shape . Proteus is characterized by spreading growth on agar .
Morphology
(i) Pleomorphism exists with long filaments and granular forms . (ii) They have peritrichate flagella and exhibit swimming motility , best seen at
20 C .They are non – capsulated and non sporing .
Colony character : Both confluentand discrete colonies are present . Confluent colonies are smooth , moist , shiny , translucent having a fishy smell and pale in colour indicating the organism is Lactose non – fermenter . Discrete colonies are small , circular , low convex and have all other above characteristics . they are grayish – white and swarming in nature .
Gram staining findings: Gram stained smear showed gram –ve non – sporing bacilli arranged haphazardly .
Motility test : Hanging drop preparation showed motile organisms . Bio – chemical tests for identification : Indole production test …. +ve
: Glucose fermentation test …. Producing acid And gas : Lactose fermentation test …. Not fermented : Mannitol fermentation test …. Not fermented : Urease production test …. +ve
Provisional diagnosis: From the above the findings we can provisionally diagnose the organism as Proteus sp.
Other test : Catalase test -‐-‐-‐-‐-‐ +ve Nitrate reduction test -‐-‐-‐-‐-‐ +ve Oxidase test -‐-‐-‐-‐-‐ -‐ve MR test -‐-‐-‐-‐-‐ +ve VP test -‐-‐-‐-‐-‐ -‐ve H2S production test -‐-‐-‐-‐-‐ +ve
Confirmatory test : Serotyping with specific high titre antisera. Final diagnosis : From all the above parameters we can finally diagnose the given organism as
Proteus sp.

BMLT INTERNSHIP RECORD 78 (v) Bio-‐Chemical Test
1. Indole Test:
Reagent: Kava’s reagent i. Para-‐dimethyl Amino benzaldehyde ii. Amyl alcohol iii. Concentration Hcl
Principle: This is a test to find out whether bacteria breakdown protein or not. Procedure: Some bacteria are able to convert tryptophan to Indole in presence of Kovas reagent. If the test is positive a pink colour ring is formed at the junction. E.g. …E.coli.
2. Citrate Test:
This is a biochemical test to find out the bacteria which utilizes citrate as sole of carbon source. Procedure: A positive test is indicated by the growth of bacteria and a deep colour due to the presence of indicator Bromothymol blue. E.g….Citobacter Klebsiella.
3. Triple Sugar-‐iron Test (TSI): Principle:
This is a biochemical test to know whether the bacterium utilizes both lactose and glucose or only glucose.
Procedure: In a test tube containing both lactose and glucose a loop of bacteria is inserted. Then it is
kept over night incubation. The result may be of two types-‐-‐-‐-‐-‐ a) If only glucose is fermented yellow colour is showed along with a black ring if H2S
is present. This is to be represented as A/A +_ H2S.
b) If only glucose is fermented pink colour occurs in slant and yellow in both with a black ring if H2S is present. This is to be represented as K/A +_ H2S.
E.g Salmonella typhil, Proteus vulgaris, Proteus mirabilis.
4. Urease Test: To find out presence of enzyme (urase) in the bacteria.

BMLT INTERNSHIP RECORD 79
Urea -‐-‐-‐-‐ Ammonia (urease) + Carbon – Di – Oxide in presence of indicator phenol red turns into deep pink colour.
This is a biochemical test for identifying bacteria. This test indicates whether a bacterium contains enzyme urease or not. This is a positive urease test.
E.g Klebsiella sp.,Proteus sp.
5. Oxidase Test: Tetramethyl , Paraphenyllene , Diamine , Di-‐HCL are the Reagent of this test.
This is a test to find out whether a bacterium contains cytocrome oxidase. 1:1% solution of fresh solution of this reagent is made. Take filter paper and pour the reagent, with a glass rod and platinum loop. We take a loopfulls of bacteria and break it on filter paper. A positive reaction indicates a deep purple colour in 10 seconds. E.g. Pseudomonas, Vibrio, Neisseria.
6. Catalase Test: We take 35 of H2O2 in a test tube and mix bacteria full loop in it. The developments of
bubbles on the slide indicate the positive catalase test. E.g…………Staphylococcus Aures. Streptococcus (Negative) 6. Coagulase Test:
Pick up a few colonies of the bacteria from an ager culture and emulsify in two drops of saline placed on a slide / tube. If the coagulation appear then it indicates the positive coagulase test. E.g……Staphylococcus Aures.
Sensitivity Test (Antibiotics Test):
After identifying the bacteria we must have done the antibiotic test. It is very important thing for the treatment. Procedure:-‐
1 At first the prepared Molar Hilton Agar (MHA) in a Petri dish is taken. 2 Then the antibiotic discs are taken and bring it to the room temperature. 3 Then the pick up a colony from the growth by a loop and put it in a prepared Nutrient
broth, then keep in an incubator for 1hr. 4 Then bacteria mixed nutrient broth poured on the Molar Hilton agar media and allow
drying. 5 After drying the media the antibiotic discs are put on the media one by one line wise.
Then the resistant area is observed for the report.
For Urine Sample (Sensitivity of Gram Negative Bacilli):-‐

BMLT INTERNSHIP RECORD 80 Ampicillin (A) Cotrimoxasole (CO) Nitrofurantoin (FD) Norfloxacin (NX) Ciprofloxasin (CF) Ceftrazidime (Ca) Ceftriaxone (Ci) Levofloxacin (Le) Ofloxasin (Ox) For Staphylococcus in Urine:-‐ Cloxacillin (Cx) Gentamycin (Ge) Azithromycin (Az) Norfloxacin (Nx) Vancomycin (V) Linezolid (LZ) Ciprofloxacin (CF) Clindamycin (Cl) Erithromycin (E) For Staphylococcus in Pus:-‐ Oxacillin (Ox) Cephalexin (Cp) Chloramphenicol (C) Erythromycin (E) Azithromycin (Az) Doxicillin (Do) Ciprofloxacin (CE) Amikacin (Ak) Linezolid (Lz) Getifloxacin (Gf) For Salmonella Typhi in Blood Culture:-‐ Chloramphenicol (C) Ampicillin (A) Amoxacillin (Am) Cotrimoxasole (CO) Ciprofloxacin (CE) Ofloxacin (Ox) Azithromycin (Az) Amikacin (Ak) Ceftrazidime (Ca)

BMLT INTERNSHIP RECORD 81 For Enterococcus in Urine:-‐ Penicillin (P) Ampicillin (A) Gentamycin (GE) Vancomycin (Va) Merophenem (MR) Azithromycin (Az) Amikacin (Ak) For Pseudomonus in any Fluid:-‐ Amikacin (Ak) Ciprofloxacin (CE) Ceftrizidime (Ca) Gentamycin (GE) Levofloxacin (Le)
\

BMLT INTERNSHIP RECORD 82
SEROLOGY
o RAPID PLASMA REAGIN TEST
o QUANTITATIVE DETECTION OF HEPATITIS B SURFACE ANTIGEN
o MANTOUX TEST
o WIDAL TEST
o ESTIMETION OF C-‐REACTIVE PROTEIN
o DETERMINATION OF ANTI STREPTOLYSIN O
o DETERMINETION OF RHEUMATOID FACTORS

BMLT INTERNSHIP RECORD 83
RAPID PLASMA REAGIN TEST
RPR(Rapid plasma regain) is a non triponemal test that detects “regain”(IgG and IgM) produced against the lipoidal material released by damaged host cells as well as to lipoprotein like material and possibly cardiolipin released from treponemas. RPR test uses a nontreponemal antigen containing cardiolipin which has been modified by addition of choline chloride, ethylenediaminetetraacetate(EDTA) and charcoal. Choline chloride inactivates inhibitors thereby eliminating need to heat inactivate the sample, EDTA enhacing the stability of the suspension and charcoal ease up the visualisation of the clumps.
• PRINCIPLE: The RPR reagent containing modified antigen and microparticulate carbon
particles, flocculate when mixed with sample containe “regain”. A reactive sample is indicated by macroscopically visible black clumps against white background and nonreactive specimen appear to have smooth uniform light gray colour.
• SPECIMEN: Plasma or serum.
• REAGENT: RPR kit content three type of reagent: 1. RPR antigen suspension. 2. Positive control serum. 3. Negative control serum. All the reagent should be stored at 2-‐8oc. Do not freeze. And should be protected from direct sunlight and elevated temperature.
• REQUERMENTS: 1. Disposable plastic cards. 2. Disposable plastic droppers. 3. Disposable applicator sticks. 4. Rubber teats.
• QUALITATIVE PROCEDURE: 1. All the reagents and sample are assembled at the room temperature before the
test is performed. 2. Reagents and sample are dispensed at the disposable plastic card as follow: DISPENSE TEST POSITIVE CONTROL NEGATIVE CONTROL
A. RPR antigen 01 Drop -‐ -‐
B. Positive Control -‐ 01 drop -‐
C. Negative Control -‐ -‐ 01 Drop
D. Serum 01 Drop
O1 Drop
01 Drop
3. Then the card is placed on mechanical rotator at 100±2 rpm for 8 minutes.

BMLT INTERNSHIP RECORD 84
4. Observation is done after 8 minutes .
• INTERPRETATION: A. REACTIVE: Visible black clamp at the test and positive control region. B. NON-‐REACTIVE: Black clump show only at the positive control region.
QUANTITATIVE DETECTION OF HEPATITIS B
SURFACE ANTIGEN
Viral hepatitis is a systemic disease primarily involving the liver. The complex antigen found on the surface of the HBV is called HBsAg. The presence of HBsAg in serum or plasma is an indication of an active hepatitis B infection, either acute or chronic. In a typical Hepatitis B infection, HBsAg will be detected 2 to 4 weeks before the ALT level becomes abnormal and 3 to 5 weeks before symptoms or jaundice develop.
• PRINCIPLE: One step HBsAg test is a colloidal gold enhanced immunoassay for the detection of HBV surface antigen in human whole blood, serum or plasma. Goat anti-‐HBsAg antibody is immobilized in the test region on nitrocellulose membrane. During the assay specimen is allowed to react with the coloured conjugate (antibody-‐colloidal gold conjugate), the mixture then migrates chromatographically on the membrane by the capillary action. An HBsAg positive specimen produces a distlnct color band in the test region, formed by the specific antibody-‐HBsAg-‐colored conjugate complex.absence of this colored band in the test region suggests a negative result. A coloured band always appears in the control region serving as procedural control regardless of the test result.
• SPECIMEN: serum or plasma. • REQUREMENTS: 1. Test card individually foil pouched with a desiccant.
2. Disposable plastic dropper The test kit should be stored at 2-‐300c in the sealed pouch and under dry condition.
• ASSAY PROCEDURE: a) All the reagents and sample are brought to the room temperature before the test is
performed. b) The test card is removed from the foil pouch and placed on a clean dry surface. c) 100µl specimen is dispensed into the sample well on the card. d) Interpretation is done after 15 minutes.
• INTERPRETATION: I. REACTIVE: Pink band appear on the test and control region. II. NON-‐REACTIVE: Pink band appear only on the control region. III. INVALID: No pink band appear on the test and control region.

BMLT INTERNSHIP RECORD 85
MANTOUX TEST
The tuberculin skin test is the most commonly used test to detect exposure to mycobacterium tuberculosis, being used in epidemiological surveys, clinical evaluation of patients with suspected active tuberculosis, and assessment of anti-‐tuberculous drug therapy. An intradermal mantoux test is performed with different dilutions of tuberculin PPD. In developing countries, such as India, with high prevalence of tuberculosis, 1 TU is the recommended dose as per the WHO guidelines.
• PRINCIPLE: Infection with mycobacterium tuberculosis results in hypersensitivity to tuberculoprotein. Intradermally injected Tuberculin PPD produces erythema and induration of the skin around the point of injection. The diameter of induration is directly proportional to the degree of sensitization.
• TEST PROCEDURE: A. The preferred site for the test is the flexor or dorsal surface of the forearm about 4
inches below the elbow joint. B. The skin at the chosen site and stopper of the PPD solution is cleaned with 70%
alcohol. C. 0.1 ml of Tuberculin PPD solution is drawn into the sterile tuberculin syringe fitted with
a short 26-‐gauge needle. D. Then the tuberculin PPD is injected intradermally.
As the solution is injected, a pale white bleb, 6 to 10 mm in diameter will rise at the needle point. This will be quickly absorbed and hence no dressing is required.
• RESULT: Result of the mantoux test should read between 48 to 72 hours after the injection.
• INTERPRITATION: a) POSITIVE: Induration measuring 10mm or more. This indicates hypersensitivity to
tuberculoprotein and indicates past or present infection with mycobacterium tuberculosis.
b) DOUBTFUL: Induration measuring between 5 and 9 mm. Retesting is indicated at the another site.
c) NEGATIVE: Induration of less than 5 mm. This indicates lack of hypersensitivity totuberculoprotein and tuberculous infection is highly unlikely.

BMLT INTERNSHIP RECORD 86
WIDAL TEST
This test is basically screening test is performed for the diagnosis of typhoid and
paratyphoid fever. The specific antibodies are usually detectable in the patient’s blood after 6 days of infection.
• Principle: This test is based on the principle of direct agglutination reaction. Antibodies in serum, produced in response to exposure to salmonella organisms will agglutinate bacterial suspension which carry homologous antigens. Thus in the test, four specific antigenic suspension are used-‐
1. Salmonella typhi ‘o’. 2. Salmonella tyfhi ‘H’. 3. Salmonella paratyfhi ‘AH’. 4. Salmonella paratyfhi ‘BH’.
• Sample: Fresh fasting serum or plasma is preferable. • Requirements:
a) Glass plates with ceramic rings. b) Test tube. c) Disposable droppers. d) WIDAL kit e) Timer f) Applicator stick
• Procedure: Rapid slide test or qualitative screening test:-‐ a) All the reagent and sample are brought to the room temperature before the test is
performed. b) Circle of the glass plate are levelled as ‘O’, ‘H’, ‘AH’, ‘BH’, ‘PC’ and ‘NC’. c) One drop of test sample are taken in each of the first four circle and one drop
positive control in circle 5 and 6. d) One drop of antigen ‘O’, ‘H’, ‘AH’ and ‘BH’ in circle 1,2,3 and 4 respectively and ‘O’
antigen in circle 5 and any one of the ‘H’ antigen in circle 6. e) Then contents of each circle are mixed with separate applicator stick to fill the whole
area of the individual circle. f) Then the slide is rotated for one minute.

BMLT INTERNSHIP RECORD 87
Observation: observation is to be make for agglutination. Quantitative slide test: This test is done when qualitative screening test show positive result.
a) 80µl, 40µl, 20µl, 10µl and 5µl of undiluted serum should be taken in 1st, 2nd, 3rd, 4th and 5th circle respectively on the slide. b) One drop of the appropriate antigen suspension whish show agglutination in the rapid slide test is added in each 5 circle. c) The contents of each circle are mixed and spread with separate applicator stick. d) The slide is slowly rotated for one minute and observation is done for agglutination. e) The titre of the Ab is the highest dilution of serum upto which there is clear agglutination.
Test tube 1 2 3 4 5
Titre 1/20 1/40 1/80 1/160 1/320
Serum volume 80µl 40µl 20µl 10µl 5µl
Appropriate antigen 1 drop 1 drop 1 drop 1 drop 1 drop
Inference:
1) only a titre above 1:80 should be considered significant. 2) Increase titre after few days of the 1st test, it suggest of an active infection. 3) Single test result above 160 is considered diagnostic. 4) A simultaneous rise in the titre of all ’H’ Ab suggest recent typhoid vaccination.
ESTIMETION OF C-REACTIVE PROTEIN
The C-‐reactive protein (CRP) is a serum protein, synthesised in the liver. Its rate of
synthesis and secretion increases within hours of an acute injury or the onset of inflammation and may reach as high as 20 times 0f the normal levels. CRP measurement helps in management of neonatal septicaemia and meningitis. CRP levels invariably rise after major surgery but fall to normal within 7-‐10 days. Absence of this fall is indicative of possible septic or inflammatory postoperative complications. CRP measurement also provides useful information in patient with myocardial infarction.
Principle: The slide test for detection of CRP is based on the principle of agglutination. The test specimen is mixed with CRP latex reagent and allowed to react. If CRP level is greater than 0.6 mg/dl, a visible agglutination is observed. If CRP concentration is less than 0.6 mg/dl, then no agglutination is observed.
Specimen: Serum is used for CRP test.

BMLT INTERNSHIP RECORD 88
Reagent: CRP latex reagent, positive control, negative control. Requirement: CRP test card, applicator stick, rubber teat, dispensing pipettes, stopwatch,
test tube and high intensity direct light. Test procedure: Reagent and sample are brought at room temperature before the test is
done. Qualitative method:
I. one drop of serum, positive control and negative control is dispensed at first three circle correspondingly.
II. One drop of CRP latex reagent are dispensed in each circle. III. The contains of the three circle are mixed with separate applicator stick. IV. The slide is rotated for two minutes And then observation is done.
Interpretation: I. Positive: Agglutination is observed at test and positive control
region. II. Negative: No agglutination is observed at three circle.
Semiquantitative method: this procedure is done when qualitative method show positive result.
I. Specimen is diluted as 1:2, 1:4, 1:8, 1:16 and so on. II. One drop of each dilute sample is dispensed at separate circle. III. One drop of CRP latex reagent is dispensed in each circle. IV. Mixing is done by separate applicator stick. V. The slide is rotated for two minutes and then observation is done for
agglutination. Calculation: CRP (mg/dl)= S x D
S= sensitivity of the reagent, i.e. 0.6 mg/dl. D= Highest dilution of the sample which show agglutination.
DETERMINATION OF ANTI STREPTOLYSIN O
The group-‐a haemolytic streptococci produce various exotoxins such as streptolysin-‐o and streptolysin-‐s that can act as antigen. The affected individuals produce specific antibody against streptolysin-‐o, namely anti streptolysin-‐o (ASO). Determination of this antibody is very useful for the diagnosis of streptococcal infections and their relative effects such as rheumatic fever and acute glomarulonephritis. An elevated ASO titre of more than 200IU/ml may indicate an acute streptococcal infection.
• Principle: The ASO slide test for determination of antibody to streptolysin-‐o is based on the principle of agglutination. The test specimen is mixed with ASO latex reagent and allowed to react. If antibody to streptolysin-‐o are present in concentrations more than 200IU/ml, but less than 4000IU/ml, then a visible agglutination is observed. If antibody to sterptolysin-‐o are not present or are in concentrations less than 200IU/ml, then no agglutination will be observed.
• Sample: Serum is used for ASO determination.

BMLT INTERNSHIP RECORD 89
• Reagent: ASO latex reagent, positive control, negative control. • Additional requirement: Stopwatch, high intensity light source, isotoic saline, pipettetes,
test tubes. • Test procedure:
a) Qualitative method: I. One drop of test sample, positive control (PC) and negative control
(NC) are placed on the glass slide by using a disposable pipette. II. And one drop of ASO latex reagent is added with the sample on the slide . III. Then mixing is done by separate applicator stick. IV. Then the slide is rotated for two minutes and observation is done for
agglutination. Interpritetion:
a. Positive: Agglutination is found in specimen and PC region. b. Negative: Agglutination is found only in the PC region.
b) Semiquantitative method: I. The dilution of the sample is prepared as 1:2, 1:4, 1:8, 1:16 and so on. II. Each diluted sample is taken into the separate circle of the slide. III. One drop of reagent are added to each drop of sample. IV. Mixing is done with separate applicator stick. V. Observation is done after two minutes rotation of the slide.
Calculation: ASO (IU/ml)= S X D S= Sensitivity of the reagent, i.e. 200 IU/ml. D= Highest dilution of the serum which show agglutination.
DETERMINETION OF RHEUMATOID FACTORS Sometimes autoantibodies are produced by the human body against self-‐antigens and causes pathogenesis of certain rheumatic disease. IgM class RF(Rheumatoid factors) with specificity to human IgG (FC) is the most useful prognostic marker of RF. The clinical signification of RF determinations consists in differentiation between rheumatoid arthritis, in which RF have been demonstrated in serum of approximately 80 percent of cases examined and rheumatic fever, in which RF are almost always absent.
• Principle: RF slide test for determination of rheumatoid factors is based on the principle of agglutination. The test specimen is mixed with RF latex reagent and allowed to react. If RF is present 10IU/ml or more then visible agglutination is observed. If RF is absent below detectable levels, then no agglutination is observed.
• Sample: Serum is used for RF determination. • Reagent: RF latex reagent, positive control, negative control. • Additional requirements: stopwatch, test tube, high intensity direct light source, glass
slide, rubber teat, disposable dropper and applicator stick.

BMLT INTERNSHIP RECORD 90
• Test procedure: a) Qualitative method:
I. One drop of serum, positive control (PC), negative control (NC) is dispensed into separate circle of the glass slide by separate dropper.
II. One drop of RF latex reagent is dispensed in each three circle. III. The material of each three circle is mixed by separate applicator stick. IV. Then the slide is rotated for two minutes and observation is done for any
agglutination. Interpretation:
c. Positive: Agglutination is found in specimen and PC region. d. Negative: Agglutination is found only in the PC region.
b) Semiquantitative method: This procedure is done when qualitative method show
agglutination. I. The dilution of the sample is prepared as 1:2, 1:4, 1:8, 1:16 and so on. II. Each diluted sample is taken into the separate circle of the slide. III. One drop of reagent is added to each drop of sample. IV. Mixing is done with separate applicator stick. V. Observation is done after two minutes rotation of the slide.
Calculation: RF (IU/ml)= S X D S= Sensitivity of the reagent, i.e. 200 IU/ml. D= Highest dilution of the serum which show agglutination.

BMLT INTERNSHIP RECORD 91
BIOCHEMISTRY
o ESTIMATION OF BLOOD GLUCOSE
o ESTIMATION OF MICROPROTEIN
o ESTIMATION OF CALCIUM
o ESTIMATION OF PHOSPHORUS
o ESTIMATION OF CREATINE KINASE
o DETERMINETION OF SERUM UREA LEVE
o DERTERMINETION OF CREATININE LEVEL OF SERUM OR
PLASMA
o DERTERMINETION OF URIC ACID
o DERTERMINETION OF CREATININE LEVEL OF SERUM OR
PLASMA
o DERTERMINETION OF URIC ACID
o ESTIMETION OF SERUM TRIGLYCEROID LEVEL

BMLT INTERNSHIP RECORD 92
o ESTIMETION OF SERUM OR PLASMA CHOLESTEROL LEVEL
o DETERMINATION OF HDL CHOLESTEROL IN SERUM
o DETERMINATION OF LDL CHOLESTEROL IN SERUM
o DETERMINATION OF SGPT(ALAT) ACTIVITY IN SERUM o DETERMINATION OF SGOT(ASAT) ACTIVITY IN SERUM
o DETERMINATION OF ALKALINE PHOSPHATE
ACTIVITY IN SERUM
o DETERMINATION OF TOTAL PROTEINS IN SERUM AND
PLASMA
o DETERMINATION OF ALBUMIN IN SERUM OR PLASMA
o DETERMINATION OF ALBUMIN IN SERUM OR PLASMA
o DETERMINATION OF SERUM T3
o DETERMINATION OF SERUM T4
o DETERMINATION OF SERUM TSH

BMLT INTERNSHIP RECORD 93 ESTIMATION OF BLOOD GLUCOSE PRINCIPLE: The aldehyde group of glucose is oxidized by glucose oxidase to form gluconic acid and H2O2. In subsequent peroidase catalysed reaction, the oxygen liberated is accepted by colour chromogen system to give a red colour compound. The colour developed is measured colorimetrycally at 505 n m and is directly proportion to glucose concentration of the sample.
REACTION: Glucose (C6H12O6) + O2 + H2O Glucose Oxidase Gluconic acid +H2O2 H2O2 + Phenol + 4-‐aminoantipyrine Peroxidase Quinoneimmine complex + H2O.
NORMAL REFERENCE VALUES: Serum (fasting) : 70 -‐110 mg/dl (2 hrs. P.P) : upto 150 mg/dl
METHOD: Glucose oxidase-‐peroxidase method.
REAGENT: Reagent are supplied in akit which contained Reagent-‐1 Enzyme reagent. Reagent-‐2 Glucose diluent. Reagent-‐3 Glucose standard. (100 mg/dl)
PREPARATION OF WORKING REAGENT: One vial of enzyme reagent (R-‐1) is dissolved in 50 ml or required quantity (indicated on the kit) of glucose diluent (R-‐2) by mixing sloly.
SAMPLE: Serum (Non Haemolysed) or Plasma. PROCEDURE : Pipetted into clean dry test tubes labelled Blank(B), Standard(S) and Test(T) as follows :-‐ ADDITION SEQUENCE BLANK(ml) STANDARD(ml) TEST(ml) Working glucose reagent 1.0 1.0 1.0 Distilled water 0.01 -‐ -‐ Glucose standard -‐ 0.01 -‐ Sample -‐ -‐ 0.01 Mixed well and incubated at 37oc for 10 minutes. Now absorbance of standard and test are read against blank at 505 n.m. (Green filter).

BMLT INTERNSHIP RECORD 94 CALCULATION: Glucose concentration (mg/dl) in the sample = O.D. of the T -‐ O.D. of the B X Conc. Of STD. O.D. of the STD – O.D. of the B = O.D. of the T X Conc. Of the STD [O.D. of BLANK=0] O.D. of STD ESTIMATION OF MICROPROTEIN PRINCIPLE : Proteins from a blue purple complex when reacted with a combination of pyrogallal red dye
and molybdic acid at pH 2.2 . The concentration of the protein in the sample is proportional to the intensity of the colour when measured at 600 n.m.
REACTION:Proteins + Pyrogallol red + Molbdate Acidic medium Blue purple colour complex NORMAL REFERENCE VALUES: CSF: 10 -‐50 mg/dl METHOD : Pyrogallol red method. REAGENT : REAGENT -‐1: Dry reagent Pyrogallol red 0.067 m.mol/l Ammonium molybdy 0.026 m.mol/l Glycine buffer 0.1 m.mol/l REAGENT -‐2 Microprotein STD (100 mg/dl) REAGENT PREPARATION : Reagents are ready to use. Reagent-‐1 is photosensitive should be protected
from strong light. SAMPLE : Fresh sample of C.S.F free from haemolysis.

BMLT INTERNSHIP RECORD 95 PROCEDURE : Pipetted into clean dry test tubes labelled as Blank (B), Standard (S) and Test(T) as follows ADDITION SEQUENCE BLANK (ml) STANDARD (ml) TEST (ml) Micro protein reagent 1.0 1.0 1.0 Distilled water 0.01 -‐ -‐ Micro protein standard -‐ 0.01 -‐ Sample -‐ -‐ 0.01 Mixed well incubated at 37oc for 10 minutes. Now measured absorbance of standard and test against blank at 606 n.m. within 60 minutes. CALCULATION : Micro protein concentration (mg/dl) in the sample = O.D. of the T -‐ O.D. of the B X Conc. Of STD. O.D. of the STD – O.D. of the B = O.D. of the T X Conc. Of the STD [O.D. of BLANK=0] O.D. of STD ESTIMATION OF CALCIUM PRINCIPLE: Calcium in an alkaline medium combined with O-‐cresolphthalein complexone form a purple
coloured complex. Intensity of the coloured formed is directly proportionalto the amount of calcium present in the sample.
REACTION: Calcium + OCPC -‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐ Purple coloured complex NORMAL REFERENCE VALUES: Serum: 9 – 11 mg/dl METHOD: O-‐Cresolphthalein Complexone REAGENT: Reagent are supplied in a kit which contained Reagent-‐1 Buffer reagent Reagent-‐2 Colour reagent

BMLT INTERNSHIP RECORD 96 Reagent-‐3 Calcium standard (10 mg/dl) REAGENT PREPARATION: Reagents are ready to use. Reagent should be protected from strong light. SAMPLE: Serum or Heparinized plasma. PROCEDURE: Pipetted into clean and dry test tubes labelled as Blank (B), Standard (S) and Test (T) as follows:-‐ ADDITION SEQUENCE BLANK (ml) STANDARD (S) TEST (T) Buffer reagent 0.5 0.5 0.5 Colour reagent 0.5 0.5 0.5 Distilled water 0.02 -‐ -‐ Calcium standard -‐ 0.02 -‐ Sample -‐ -‐ 0.02 Mixed well and incubated at room temperature for 5 minutes.
Now measured the absorbance of the standard and test against blank at 570 n.m within 60 minutes. CALCULATION : Calcium concentration (mg/dl) in the sample = O.D. of the T -‐ O.D. of the B X Conc. Of STD. O.D. of the STD – O.D. of the B = O.D. of the T X Conc. Of the STD [O.D. of BLANK=0] O.D. of STD ESTIMATION OF PHOSPHORUS PRINCIPLE: Phosphate ions in acidic medium reacted with ammonium molybdate to form a phosphomolybdate complex. The complex has an absorbance in the ultraviolet range and is measured at 340 nm. Intensity of the complex formed is directly proportional to the amount of inorganic phosphorus present in the sample. REACTION: Phosphorus + Ammonium Molybdate-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐ phosphomolybdate complex

BMLT INTERNSHIP RECORD 97 NORMAL REFERENCE VALUES : Serum: 2.5-‐ 4.5 mg/dl METHOD: Molybdate method REAGENT: Reagent-‐1 Acid reagent Reagent-‐2 Molybdate reagent Reagent-‐3 Phosphorus Standard (5 mg/dl) PREPARATION OF WORKING REAGENT: Poured 1 part of Molybdate reagent (R-‐2) into 4 part of Acid reagent (R-‐1). This working reagent is stable for at least 6 months when at stored at 2-‐8 degree centigrade. SAMPLE: Serum or Heparinized plasma PROCEDURE: Pipetted into clean and dry test tubes labelled as Blank (B), Standard (S) and Test (T) as follows:-‐ ADDITION SEQUENCE BLANK (ml) STANDARD(ml) TEST (ml) Working reagent 1.0 1.0 1.0 Distilled water 0.01 -‐ -‐ Phosphorus Standard -‐ 0.01 -‐ Sample -‐ -‐ 0.01 Mixed well and incubated at room temperature for 5 minutes. Now measured the absorbance of the standard and test against blank at 340 n.m within 60 minutes CALCULATION : Phosphorus concentration (mg/dl) in the sample = O.D. of the T -‐ O.D. of the B X Conc. Of STD. O.D. of the STD – O.D. of the B = O.D. of the T X Conc. Of the STD [O.D. of BLANK=0] O.D. of STD ESTIMATION OF CREATINE KINASE PRINCIPLE: Creatine kinase catalysed the reaction between creatinine phosphate and ADP to formed creatine and ATP. The ATP formed along with glucose is catalysed by hexokinase to form glucose 6 phosphate. The glucose 6 phosphate reduces NADP to NADPH in the presence of glucose 6 phosphate dehydrogenase. The rate of reduction of NADP to NADPH is measured as an increase in absorbance which is proportional to the CK activity in the sample. REACTION: Creatine Phosphate + ADP Creatine kinase Creatine + ATP

BMLT INTERNSHIP RECORD 98 Glucose + ATP Hexokinase Glucose 6 Phosphate + ADP Glucose 6 Phosphate + NADP G-‐6-‐PDH Gluconate-‐6-‐P + NADPH + H NORMAL REFERENCE VALUES: Serum (male) : 24-‐195 U/L (female) : 24-‐170 U/L METHOD: Mod. IFCC method REAGENT : Reagent-‐1 Enzyme reagent Reagent-‐2 Stater reagent PREPARATION OF WORKING REAGENT: Poured 1 part of starter reagent (R-‐2) into 4part of enzyme reagent (R-‐1). This working reagent is stable for at least 10 days when stored at 2-‐8 degree centigrade. PROCEDURE: Pipetted into a clean and dry test tube labelled as Test (T) as follows:-‐
ADDITION SEQUENCE TEST(ml) Working reagent 1.0 Sample 0.05
Mixed well and read the initial absorbance A0 after 1 minute and repeated the absorbance reading after every 1,2 and 3 minutes. Calculated the mean absorbance change per minute. CALCULATION: CK activity (U/L) in the sample = (Mean absorbance/Min) X 8095

BMLT INTERNSHIP RECORD 99
DETERMINETION OF SERUM UREA LEVEL Principle: Urea is the end product of protein metabolism. It is synthesized in the liver from the ammonia produced by the catabolism of amino acids. It is transported by the blood to the kidney from where it is excreted. Increased levels are found in renal diseases, urinary obstructions, shock, congestive heart failure and burns. Decreased Levels are found in liver failure and pregnancy. Principle:-‐Urea hydrolysed urea to ammonia and CO2 . The ammonia formed further reacts with a phenolic chromogen and hypochorite to forma green coloured complex. Intensity of the colour formed is directly proportional to the amount of urea present ion the sample. Urea+H2O Ammonia + c2O Ammonia + Phenolic chromogen Green coloured complex + Hypochlorite Normal reference values:-‐ Serum /Plasma 14-‐ 40 mg/dl It is recommended that each laboratory establish its own normal range representing its patient population. Reagent Preparation:-‐ Reagents are ready to use for the given procedure. Working Enzyme Reagent:-‐ For the flexibility and convenience in performing large assay series, a working enzyme reagent may be made by pouring 1 bottle of L2 (Enzyme Reagent) into 1 bottle of L1(Buffer Reagent). For smaller series combine 10 parts of L1 (Buffer Reagent) and 1 Part of L2(Enzyme Reagent) . Use 1 ml of the working reagent per assay instead of 1 ml of L1 and 0.1 ml of L2 as given in the procedure. The working enzyme reagent is stable for at least 4 weeks when stored at 2-‐80c Working Chromogen Reagent:-‐ For larger volume cuvettes, dilute 1 part of L3 (Chromogen Reagent) with 4 parts of fresh ammonia free distilled /deionised water. Use 1 ml of working chromogen instead of 0.2ml in the assay. The working chromogen reagent is stable for atleast 8 weeks when stored at 2-‐80c In a tightly stopped of plastic bottle. Sample material:-‐ Serum, plasma, Urine, Dilute urine 1+49 with distilled water before the assay(Results x 50). Urea is reported to be stable in the serum for 5 days when stored at 2-‐80c Procedure:-‐ Wavelength /filter : 570nm/(Hg 578 nm)/Yellow

BMLT INTERNSHIP RECORD 100 Temperature : 370c/R.T. Light path : 1 cm Pipette into clean dry test tubes labeled as Blank(B), Standard(S), and Test(T): Addition Sequence
B (ml)
S (ml)
T (ml)
Buffer Reagent (L1) Enzyme Reagent (L2) Distilled water Urea Standard (S) Sample
1.0 0.1 0.01 -‐ -‐
1.0 0.1 -‐ 0.01 -‐
1.0 0.1 -‐ -‐ -‐
Mix well & incubate for 5min at 370c or 10 min at R. T. (250c)
Chromogen Reagent(L3)
0.2 0.2 0.2
Mix well and incubate for 5 min at 370c or 10 min at R. T. (250c). Measure the Absorbance of the Standard (Abs. S), and Test Sample (Abs. T) against the Blank, within 60 Min. Calculation:-‐ Urea in mg/dl = Abs. T X Concentration of S. Abs. Linearity:-‐ This procedure is linear upto 250 mg/dl. Using the working chromogen reagent (1 ml) the linearity in increased to 400mg/dl. If values exceed this limit, dilute the serum with normal saline (Nacl) 0.9% and repeat the assay. Calculate the value using the proper dilution factor.

BMLT INTERNSHIP RECORD 101 DERTERMINETION OF CREATININE LEVEL OF SERUM
OR PLASMA
Summary:-‐ Creatinine is the catabolic product creatinine phosphate which is used by the skeletal muscle. The daily production dependends on muscular mass and it is excreted out of the body entirely by the kidneys. Elevated are found in renal dysfunction, reduced renal blodd flow(Shock, dehydration, congestive heart failure) diabetes acromegaly. Decreased levels are found in muscular dystrophy. Principle:-‐ Picric acid in alkaline medium reacts with creatinine to form a orange coloured complex with the alkaline picrate. Intensity of the colour formed during the fixed time is directly proportional to the amount of creatinine present in the sample. Creatinnine + Alkaline Picrate Orange Coloured Complex Reference Values:-‐ Males :0.6-‐1.2mg% Female : 0.5-‐1.1mg% It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1:Picric Acid Reagent L2:Buffer Reagent S: Creatinine Standard (2mg/dl) Storage/ Stability:-‐ All reagents stable a R. T. till the expiry mentioned on the label. Reagent Preparation:-‐ Reagents are ready to use. Do not pipette with mouth. Working reagent: For larger assay series a working reagent may be prepared by mixing equal volumes of picnic Acid Reagent and Buffer Reagent. The Working reagent is stable of R.T. (25-‐300c) for at least one week. Sample material:-‐ Serum or plasma. Creatinine is stable in serum for 1 day at 2-‐80c Procedur Wavelength/liter : 520nm(Hg 492 nm)/Green Temperature : 300c/370c Light Path : 1cm

BMLT INTERNSHIP RECORD 102 Pipette into a clean dry test tube labeled Standard(S) for Test (T) :-‐
Mix well and read the initial absorbance A1 for the Standard and Test after exactly 30 seconds. Read another absorbance A2 of the Standard and Test exactly 60 second later. Calculate the change in absorbance A for the both the Standard and Test. For Standard AS = A2S – A1S For Test AT = A2T – A1T CALCULATION:-‐ AT Creatinine in mg/dl = X 2.0 AT Linearity:-‐-‐The procedure is linear upto 20 mg/dl of creatinine. If values this limit, dilute the sample with distilled water and repeat the assay. Calculate the value using the proper dilution factor.
DERTERMINETION OF URIC ACID
Principle:-‐ Uric acid is the end product of purine mnetaboloism. Uric acid is excreted to a large degree by the kidneys and to smaller degree in the intestinal tract by microbial degradation. Increased levels are found in Gouf, arthritis, impaired renal functional and starvation. Decreased levels are found in Wilson’s disease. Fanconis syndrome and yellow atrophy of the liver. Principle:-‐ Uricase converts uric acid to allantoin and hydrogen peroxide. The hydrogen peroxide formed further reacts with a phenolic compound and 4 aminoantipyridine by the catalytic action of peroxidase to form a red coloured quinoneimine dye complex. Intesity of the colour formed is directly proportional to the amount of uric present in the sample. Uricase Uric Acid+H2O Allantoin+ H2o2 Peroxidase H2o2 + 4 Aminoantipyride Red Quinoneimine dye + H2O
Addition Sequence
(S) / (T) 300c/370c
Picric Acid Reagent(L1) Buffer reagent(L2)
0.5ml 0.5ml
Bring reagents to the assay temperature and add Creating Standard (S)/ Sample/diluted Urine 0.1ml

BMLT INTERNSHIP RECORD 103 + Phenolic Compound Normal Reference Value:-‐ Serum/Plasma (Male) :3.4 – 7.0 mg/dl (Female) : 2.5 – 6.0 mg/dl It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1 : Buffer Reagent L2 : Enzyme Reagent S : Uric Acid Standard(8 mg/dl ) Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Working reagent:-‐ Pour the contents of 1 bottle of L2(Enzyme Reagent ) into 1 bottle of L1 (Buffer Reagent). This working reagent is stable for at least 4 weeks when stored at 2-‐80c . Upon storage the working reagent may develop a slight pink colour however this does not affect the performance of the reagent. Alternative for flexibility as much of working reagent may be made as and when desired by mixing together 4 parts of L1 (Buffer Reagent) and 1 part of L2 (Enzyme Reagent) Alternatively 0.8 ml of L1 and 0.2ml of L2 may also be used instead of 1ml of the working reagent directly during the assay. Sample material:-‐ Serum, plasma, Uric Acid is reported to be stable in the sample for 3-‐5 days when stored at 2-‐80c Procedure:-‐ Wavelength/filter :520nm(Hg 546 nm)/ Yellow Green Temperature : 370c Light Path : 1 cm Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Mix well and incubate at 370c for 5 min or at R.T. (250c) for 15 min. Measure the absorbance of the Standard (Abs.S) and Test Sample (Abs.T) against the Blank, within 30 Min.
Addition Sequence
B (ml)
S (ml)
T (ml)
Working reagent Distilled water Uric Acid Standard (S) Sample
1.0 0.02 -‐ -‐
1.0 -‐ 0.02 -‐
1.0 -‐ -‐ 0.02

BMLT INTERNSHIP RECORD 104 Calculations:-‐
Abs.T Uric Acid in mg/dl = X.STDcontration Abs.T Linearity :-‐ This procedure is linear up to 20 mg/dl. If values exceed this limit ,dilute the serum with normal saline (NaCl 0.9%)and repeat the assay. Calculate the value using the proper dilution factor.
ESTIMETION OF SERUM TRIGLYCEROID LEVEL
Principle:-‐ Lipoprotine lipase hydrolyses triglycerides to glycerol and free fatty acids. The glycerol formed with ATP in the presence of glycerol kinase forms glycerol 3 phosphate which is oxidi9sed by the enzyme glycerol phosphate oxidase to form hydrogen peroxide. The hydrogen peroxide further reacts with phenolic compound and 4-‐ aminoantipyrine by the catalytic action of peroxidase to form a red coloured quinoneimine dye complex. Intensity of the colour formed is directlyproporational to the amount of triglycerides present in the sample. Lipoprotein Lipase Triglyceride Glycerol + Free fatty acids Glycerol Kinase Glycerol +ATP Glycerol 3 Phosphate +ADP Glycerol 3 PO Glycerol 3 Phosphate Dihydroxyacetone phos. + H2o2 H2o2 +Aminoantipyrine Red Quinonelimine dye + H2O2 + Phenol Normal Reference Value:-‐ Serum/Plasma (Suspicious) :150 mg/dl and above (Elevated) : 200 mg/dl and above It is recommended that each laboratory establish its own normal range representing its patient population.

BMLT INTERNSHIP RECORD 105 Contents: L1 : Enzyme Reagent 1 L2 : Enzyme Reagent2 S : Triglycerides Standard (200 mg/dl) Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Reagents are ready to use. Working reagent:-‐ Pour the contents of 1 bottle of L2(Enzyme Reagent 2) into 1 bottle of L1 (Buffer Reagent). This working reagent is stable for at least 8 weeks when stored at 2-‐80c . Upon storage the working reagent may develop a slight pink colour however this does not affect the performance of the reagent. Alternative for flexibility as much of working reagent may be made as and when desired by mixing together 4 parts of L1 (Enzyme Reagent) & 1 part of L2 (Enzyme Reagent). Alternatively 0.8 ml of L1 and 0.2ml of L2 may also be used instead of 1ml of the working reagent directly during the assay. Sample material:-‐ Serum, plasma, Triglycerides is reported to be stable in the sample for 5 days when stored at 2-‐80c Procedure:-‐ Wavelength/filter :505nm(Hg 546 nm)/ Green Temperature : 370c Light Path : 1 cm Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Mix well and incubate at 370c for 5 min or at R.T. (250c) for 15 min. Measure the absorbance of the Standard (Abs.S) and Test Sample (Abs.T) against the Blank, within 60 Min. Calculations:-‐
Abs.T Uric Acid in mg/dl = X 200
Abs.T
Addition Sequence
B (ml)
S (ml)
T (ml)
Working reagent Distilled water Uric Acid Standard (S) Sample
1.0 0.01 -‐ -‐
1.0 -‐ 0.01 -‐
1.0 -‐ -‐ 0.01

BMLT INTERNSHIP RECORD 106 Linearity :-‐ This procedure is linear up to 1000 mg/dl. If values exceed this limit ,dilute the serum with normal saline (NaCl 0.9%)and repeat the assay. ESTIMETION OF SERUM OR PLASMA CHOLESTEROL LEVEL Principle:-‐ Cholesterol esterase hydrolyses esterified cholesterol. The free cholesterol us oxidized to form hydrogen peroxide which further reacts which further reacts with phenol and 4-‐ aminoantipyrine by the catalytic action pf peroxidase to form and red colourd quinoneimine dye complex.Intesity of the colour formed is directly proporational to the amount of cholesterol present in the sample
Cholesterol Esterase Cholesterol Ester + H2O Cholesterol + Fatty Acids Cholesterol oxidase Cholesterol + o2 Glycerol 3 Phosphate +ADP Peroxidase H2o2 +Aminoantipyrine Red Quinonelimine dye + H2o2 + Phenol Normal Reference Value:-‐ Serum/Plasma (Suspicious) :220 mg/dl and above (Elevated) : 260 mg/dl and above It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1 : Enzyme Reagent 1 L2 : Enzyme Reagent2 S : Triglycerides Standard (200 mg/dl) Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Reagents are ready to use. Working reagent:-‐ Pour the contents of 1 bottle of L2(Enzyme Reagent 2) into 1 bottle of L1 (Buffer Reagent). This working reagent is stable for at least 8 weeks when stored at 2-‐80c . Upon storage the working reagent may develop a slight pink colour however this does not affect the performance of the reagent. Alternative for flexibility as much of working reagent may be made as and when desired by mixing together 4 parts of L1 (Enzyme Reagent) & 1 part of L2 (Enzyme Reagent). Alternatively 0.8 ml of L1 and 0.2ml of L2 may also be used instead of 1ml of the working reagent directly during the assay.

BMLT INTERNSHIP RECORD 107 Sample material:-‐ Serum,EDTA, plasma, Cholesterol is reported to be stable in the sample for 7 days when stored at 2-‐80c.The sample should be preferably be of 12 to 14 hours fasting. Procedure:-‐ Wavelength/filter :505nm(Hg 546 nm)/ Green Temperature : 370c/R.T. Light Path : 1 cm. Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Mix well and incubate at 370c for 5 min or at R.T. (250c) for 15 min. Measure the absorbance of the Standard (Abs.S) and Test Sample (Abs.T) against the Blank, within 60 Min. Calculations:-‐
Abs.T Cholesterol in mg/dl = X 200
Abs.T Linearity :-‐ This procedure is linear up to 1000 mg/dl. If values exceed this limit ,dilute the serum with normal saline (NaCl 0.9%)and repeat the assay. DETERMINATION OF HDL CHOLESTEROL IN SERUM
HDL particles serve to transport lipoproteins in the blood stream. HDL is known as “ good cholesterol” because high levels are thought to lower the risk of heart disease and coronary artery disease. Low HDL cholesterol levels, are considered a greater heart disease risk. Clinical diagnosis should not be made on a single test result but should integrate clinical and other laboratory data.
Addition Sequence
B (ml)
S (ml)
T (ml)
Working reagent Distilled water Uric Acid Standard (S) Sample
1.0 0.01 -‐ -‐
1.0 -‐ 0.01 -‐
1.0 -‐ -‐ 0.01

BMLT INTERNSHIP RECORD 108 Principle:-‐ Direct determination of serum HDLc (high-‐ density lipoprotein cholesterol) level without the need for any pretreatment or centrifugation of the sample. The method depends on the properties of detergent which solubilizes only the HDL so that the HDLc is released to react with the cholesterol esterase, cholesterol oxidase and chromogens to give colour. The non HDL lipoproteins LDL, VLDL and chylomicrons are inhibited form reacting with the enzymes due to absorption of the detergents on their surfaces. The intensity of the color formed is proportional to the HDLc concentration in the sample. Normal reference values:-‐ Male Female Low Risk >50mg/dl >60mg/dl Normal Risk 35-‐50mg/dl 45-‐60mg/dl High Risk <35mg/dl <45mg/dl It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1 : HDL-‐D Reagent 1 L2 :HDL-‐D Reagent2 C : CALIBRATOR (for 1 ml) Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Reagents L1 and L2 are ready to use. Cap the bottles immediately after use and avoid contamination. Calibrator:-‐ Reconstitute with 1 ml of D.W. Mix gently to dissolve the contents. Once reconstituted the calibrator is stable for 2 weeks at 2-‐80c .or 3 months at -‐200c. Do not repeatedly thaw and refreeze. Sample material:-‐ Serum or Heparinized plasma. Serum should be separated from the clot, as soon as possible. HDLis reported to be stable in the sample for 1 week when stored at2-‐80c Procedure:-‐ Wavelength :578 nm Temperature : 370c Light Path : 1 cm Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Addition Sequence
B (µl)
S (µl)
T (µl)

BMLT INTERNSHIP RECORD 109
Mix well and incubate at 370c for 5 min and read the absorbance A2 of the Calibrator and Test against Blank. Calculate the change in absorbance A for both the Calibrator and Test.. Calculation:-‐ For Calibrator Δ AC = A2C -‐ A2C For Test Δ AT = A2C – A1C Linearity:-‐ The procedure is linear up to 150mg/dl.If values exceed this limit dilute the srum 1:1 with normal saline and repeat the assay(results x 2).
DETERMINATION OF LDL CHOLESTEROL IN SERUM
HDL particles are lipoproteins that transport cholesterol to the cells. Often called “ bad cholesterol” because high levels are risk factor for coronary heart disease and are associated with obesity, diabetes and nephrosis. Clinical diagnosis should be made on a single test result; it should integrate clinical and other laboratory data., Principle:-‐ Direct determination of serum LDLc (Low-‐ density lipoprotein cholesterol) level without the need for any pre-‐treatment or centrifugation steps. The assay takes place in two steps. First by the elimination of lipoprotein non-‐ LDL Cholesterol and then the measurement of LDL c. The intensity of the color formed is proportional to the LDLc concentration in the sample. Elimination of non-‐ LDL Cholesterol
Cholesterol Esterase Cholesterol Ester + H2O Cholesterol + Fatty Acids Cholesterol oxidase Cholesterol + o2 4-‐Cholestenon + H2o2 Catalase 2H2o2 2 H2O + o2 Measurement of LDL Cholesterol
Cholesterol Esterase
Cholesterol Ester + H2O Cholesterol + Fatty Acids Cholesterol oxidase
HDL-‐D Reagent 1 (L1) Calibrator Sample
375 -‐ -‐
375 5.0 -‐
375 -‐ 5.0
Mix and incubate for 5 min. at 370c and read the abs. A1 of the Calibrator and the Test agai8nst
Blank and add. HDL-‐D Reagent 2 (L2) 125 125 125

BMLT INTERNSHIP RECORD 110 Cholesterol + o2 4-‐Cholestenon + H2o2 Catalase 2H2o2 + TODS 2 H2O + o2 + 4 Aminiantipyrine Normal reference values:-‐ Low Risk : >100mg/dl Normal Risk : 139-‐160mg/dl High Risk : <160mg/dl It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1 : LDL-‐D Reagent 1 L2 :LDL-‐D Reagent2 C : CALIBRATOR (for 1 ml) Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Reagents L1 and L2 are ready to use. Cap the bottles immediately after use and avoid contamination. Calibrator:-‐ Reconstitute with 1 ml of D.W. Mix gently to dissolve the contents. Once reconstituted the calibrator is stable for 2 weeks at 2-‐80c .or 3 months at -‐200c. Do not repeatedly thaw and refreeze. Sample material:-‐ Serum or Heparinized plasma. Serum should be separated from the clot, as soon as possible. LDLis reported to be stable in the sample for 1 week when stored at2-‐80c Procedure:-‐ Wavelength :546 nm Temperature : 370c Light Path : 1 cm Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Addition Sequence
B (µl)
S (µl)
T (µl)
HDL-‐D Reagent 1 (L1) Calibrator Sample
375 -‐ -‐
375 5.0 -‐
375 -‐ 5.0
Mix and incubate for 5 min. at 370c and read the abs. A1 of the Calibrator and the Test agai8nst Blank
and add. LDL-‐D Reagent 2 (L2) 125 125 125

BMLT INTERNSHIP RECORD 111 Mix well and incubate at 370c for 5 min and read the absorbance A2 of the Calibrator and Test against Blank. Calculation:-‐ LDLc in mg/dl = Abs.T X Conc. Of Calibrator
Abs.T Linearity:-‐ The procedure is linear up to 1000mg/dl. If values exceed this limit dilute the serum 1:1 with norma saline and rrepeat the assay(results x 2) DETERMINATION OF SGPT(ALAT) ACTIVITY IN SERUM
SGPT is found in a variety of tissues but is mainly found in the liver. Increased levels are found in hepatitis, cirrhosis, obstructive jaundice and other hepatic diseases. Slight elevation of the enzymes is also seen in myocardial infarction.
Principle:-‐ SGPT (ALAT) catalyzes the transfer of amino group between L-‐alanine and ∞Ketoglutarate to form Pyruvate and Glutamate. The Pyruvate formed reacts with NADH in the presence of Prsence of Lactate Dehydrogenase to form NAD. The rate of oxidation of NADH to NAD is measured as a decrease in absorbance whichis proportional to the SGPT (ALAT) activity in the sample. SGPT L-‐ Alanine + ∞ Ketoglutarate Pyrivate +L-‐Glutamate Pyrivate +NADH + H+ Lactate +NA D+ Normal Reference Value:-‐ Serum/Plasma (Male) :up to 40 U/L at 370c (Female) :up to 31 U/L at 370c It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1 :Enzyme Reagent L2 : Starter Reagent Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels.

BMLT INTERNSHIP RECORD 112 Reagent Preparation:-‐ Reagents are ready to use. Working reagent:-‐ For sample start assays a single reagents is required. Pour the contents of1 bottle of L2 (Starter Reagent )into 1 bottle of L1 of L1 (Enzyme Reagent).This working reagent is stable for at least 3 weeks when stored at 2-‐80c Alternatively for flexibility as much of working reagent may be made as and when desired by mixing together 4 parts of L1(Enzyme Reagent) and 1 part of L2 (Starter Reagent) Alternatively 0.8 ml of L1 and 0.2ml of L2 may also be used instead of 1ml of the working reagent directly during the assay. Sample material:-‐ Serum Free from hemolysis, SGPT (ALAT) is reported to stable in serum for 3 days at 2-‐80c Procedure:-‐ Wavelength/filter :340nm Temperature : 370c/ 300c/250c Light Path : 1 cm Substrate Start Assay:-‐ Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Mix well and read the initial absorbance a0 & repeat the absorbance reading the absorbance reading after every 1,2, & 3 minute. Calculate the mean absorbance change per minute ( A/min ) Sample Start Assay:-‐ Pipette into a clean dry test tube labeled as Test (T):
Mix well and read the initial absorbance a0 & repeat the absorbance reading the absorbance reading after every 1,2, & 3 minute. Calculate the mean absorbance change per minute ( A/min ) Calculations:-‐ Substrate/Sample start:-‐ SGPT(ALAT) Activity in U/L 250c/300c = A/min. X 952 SGPT(ALAT) Activity in U/L 370c = A/min. X 1746
Addition Sequence
T 300c/250c
T 370c
Enzyme Reagent(L1) Sample
0.8ml 0.2ml
0.8ml 0.1ml
Incubate at the assay temperature for 1 minute and add Start Reagent(L2) 0.2ml 0.2ml
Addition Sequence
T 300c/250c
T 370c
Working Reagent 1.0ml
1.0ml
Incubate at the assay temperature for 1 minute and add Sample 0.2ml 0.ml

BMLT INTERNSHIP RECORD 113
Linearity:-‐ The procedure is linear up to 500 U/L at 370c . If the absorbance change( A min/) exceeds o.250, use only the value of the first two minutes to calculate the result, or dilute the sample 1+9 with normal saline (NaCl) 0.9% and repeat the assay (ResultsX10)
DETERMINATION OF SGOT(ASAT) ACTIVITY IN SERUM
SGOT is an enzyme found in hurt muscle , liver cells skeletal muscle and kidneys. Injury to these tissues results in the release of the enzyme blood. Elevated levels are found in myocardial infarction., Cardiac operation, Hepatitis, acute pancreatitis, acute renal disease, primary muscle disease. Decreased levels may be found in pregnancy, Beri Beri and Diabetic Ketoacidosis Principle:-‐ SGOT (ASAT) catalyzes the transfer of amino group between L-‐Aspartate and ∞Ketoglutarate to form Oxaloacetate and Glutamate. TheOxaloacetate formed reacts with NADH in the presence of Malate Dehydrogenase to form NAD. The rate of oxidation of NADH to NAD is measured as a decrease in absorbance which is proportional to SCOT (ASAT) activity in the Sample. SGOPT L-‐ Aspartate + ∞ Ketoglutarate Oxalotoacetate + Glutamate Oxalotoacetate +NADH + H+ Lactate +NA D+ Normal Reference Value:-‐ Serum/Plasma (Male) :up to 37 U/L at 370c (Female) :up to 31 U/L at 370c It is recommended that each laboratory establish its own normal range representing its patient population. Contents: L1 :Enzyme Reagent L2 : Starter Reagent Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Reagents are ready to use. Working reagent:-‐For sample start assays a single reagents is required. Pour the contents of1 bottle of L2 (Starter Reagent )into 1 bottle of L1 of L1 (Enzyme Reagent).This working reagent is stable for at least 3 weeks when stored at 2-‐80c
Assay Temperature Desired Reporting Temperature 250c 300c 370c
250c 1.00 1.32 1.82 300c 0.76 1.00 1.38
370c 0.55 0.72 1.00

BMLT INTERNSHIP RECORD 114 Alternatively for flexibility as much of working reagent may be made as and when desired by mixing together 4 parts of L1(Enzyme Reagent) and 1 part of L2 (Starter Reagent) Alternatively 0.8 ml of L1 and 0.2ml of L2 may also be used instead of 1ml of the working reagent directly during the assay. Sample material:-‐ Serum Free from hemolysis, SGOT (ASAT) is reported to stable in serum for 3 days at 2-‐80c Procedure:-‐ Wavelength/filter :340nm Temperature : 370c/ 300c/250c Light Path : 1 cm Substrate Start Assay:-‐ Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Mix well and read the initial absorbance a0 & repeat the absorbance reading the absorbance reading after every 1,2, & 3 minute. Calculate the mean absorbance change per minute ( A/min ) Sample Start Assay:-‐ Pipette into a clean dry test tube labeled as Test (T):
Mix well and read the initial absorbance a0 & repeat the absorbance reading the absorbance reading after every 1,2, & 3 minute. Calculate the mean absorbance change per minute (Δ A/min ) Calculations:-‐ Substrate/Sample start:-‐ SGOT(ASAT) Activity in U/L 250c/300c = ΔA/min. X 952 SGOT(ASAT) Activity in U/L 370c = ΔA/min. X 1746
Addition Sequence
T 300c/250c
T 370c
Enzyme Reagent(L1) Sample
0.8ml 0.2ml
0.8ml 0.1ml
Incubate at the assay temperature for 1 minute and add Start Reagent(L2) 0.2ml 0.2ml
Addition Sequence
T 300c/250c
T 370c
Working Reagent 1.0ml
1.0ml
Incubate at the assay temperature for 1 minute and add Sample 0.2ml 0.ml
Assay Temperature Desired Reporting Temperature 250c 300c 370c
250c 1.00 1.37 2.08 300c 0.73 1.00 1.54
370c 0.48 0.65 1.00

BMLT INTERNSHIP RECORD 115 Linearity:-‐ The procedure is linear up to 500 U/L at 370c . If the absorbance change(ΔAmin/) exceeds o.250, use only the value of the first two minutes to calculate the result, or dilute the sample 1+9 with normal saline (NaCl) 0.9% and repeat the assay (ResultsX10).
DETERMINATION OF ALKALINE PHOSPHATE ACTIVITY IN SERUM
Alkaline Phosphate(ALP) is an enzyme of the Hydrolase class of enzyme and acts in an alkaline medium . It is found in high concentrations in the liver, biliary tract epithelium and in the bones. Normal levels are age depended and increase during bone development. Increased levels are associated mainly with liver and bone disease. Moderate increases are seen in Hodgkins disease and congestive heart failure. Principle:-‐ALP at an alkaline pH hydrolyses p-‐Nitrphenylphosphate to form p-‐ Nitrophenol and phosphate. The rate of formation of p-‐Nitrophenol is measured as an increase in absorbance which is proportional to the ALP activity in the sample. ALP p-‐Nitrophenylphosphate p-‐Nitrophenol +Phosphate Normal reference values:-‐ Serum (Adults) : 80-‐290 U/L at 370c (Children) : 245-‐ 770 U/L at 370c It is recommended that each laboratory establish its own normal range representing its patient population Contents 10X3ml 5X15ml 20X15ml L1:Bufffer Reagent 35mml 80ml 2X150ml S: Substrate Reagent 10No. 5No. 2X 10 No Storage/ Stability:-‐ Contents are stable at 2-‐80c till the expiry mentioned on the labels. Reagent Preparation:-‐ Working reagent: Dissolve 1 substance tablet in 3.2 ml (10X3 ml pack) or15 ml (5X15ml pack) of buffer reagent. This working reagent is stable for at least 15 days when stored at 2-‐80c The substance is light and temperature sensitive. Take adequate care, especially after reconstitution. Sample material:-‐ serum Procedure:-‐ Wavelength/filter :4500nm Temperature : 370c/ 300c/250c Light Path : 1 cm
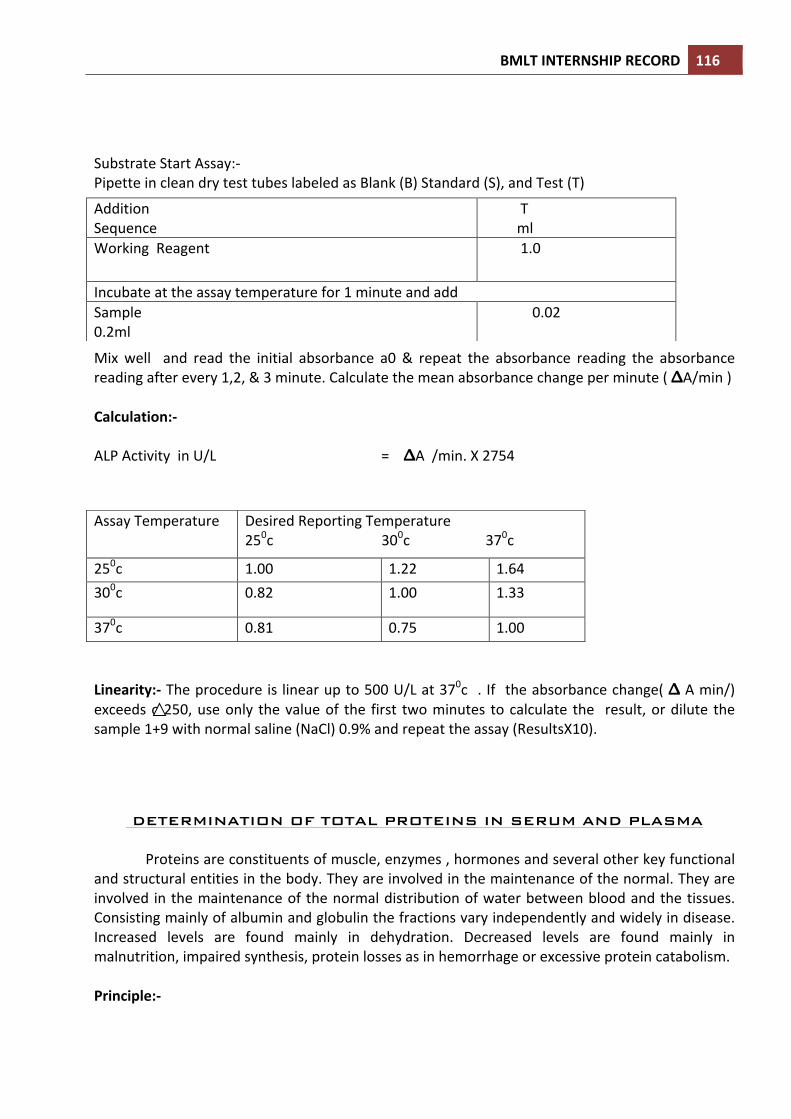
BMLT INTERNSHIP RECORD 116 Substrate Start Assay:-‐ Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T)
Mix well and read the initial absorbance a0 & repeat the absorbance reading the absorbance reading after every 1,2, & 3 minute. Calculate the mean absorbance change per minute ( ΔA/min ) Calculation:-‐ ALP Activity in U/L = ΔA /min. X 2754
Linearity:-‐ The procedure is linear up to 500 U/L at 370c . If the absorbance change( Δ A min/) exceeds o.250, use only the value of the first two minutes to calculate the result, or dilute the sample 1+9 with normal saline (NaCl) 0.9% and repeat the assay (ResultsX10).
DETERMINATION OF TOTAL PROTEINS IN SERUM AND PLASMA
Proteins are constituents of muscle, enzymes , hormones and several other key functional and structural entities in the body. They are involved in the maintenance of the normal. They are involved in the maintenance of the normal distribution of water between blood and the tissues. Consisting mainly of albumin and globulin the fractions vary independently and widely in disease. Increased levels are found mainly in dehydration. Decreased levels are found mainly in malnutrition, impaired synthesis, protein losses as in hemorrhage or excessive protein catabolism. Principle:-‐
Addition Sequence
T ml
Working Reagent
1.0
Incubate at the assay temperature for 1 minute and add Sample 0.2ml
0.02
Assay Temperature Desired Reporting Temperature 250c 300c 370c
250c 1.00 1.22 1.64 300c 0.82 1.00 1.33
370c 0.81 0.75 1.00
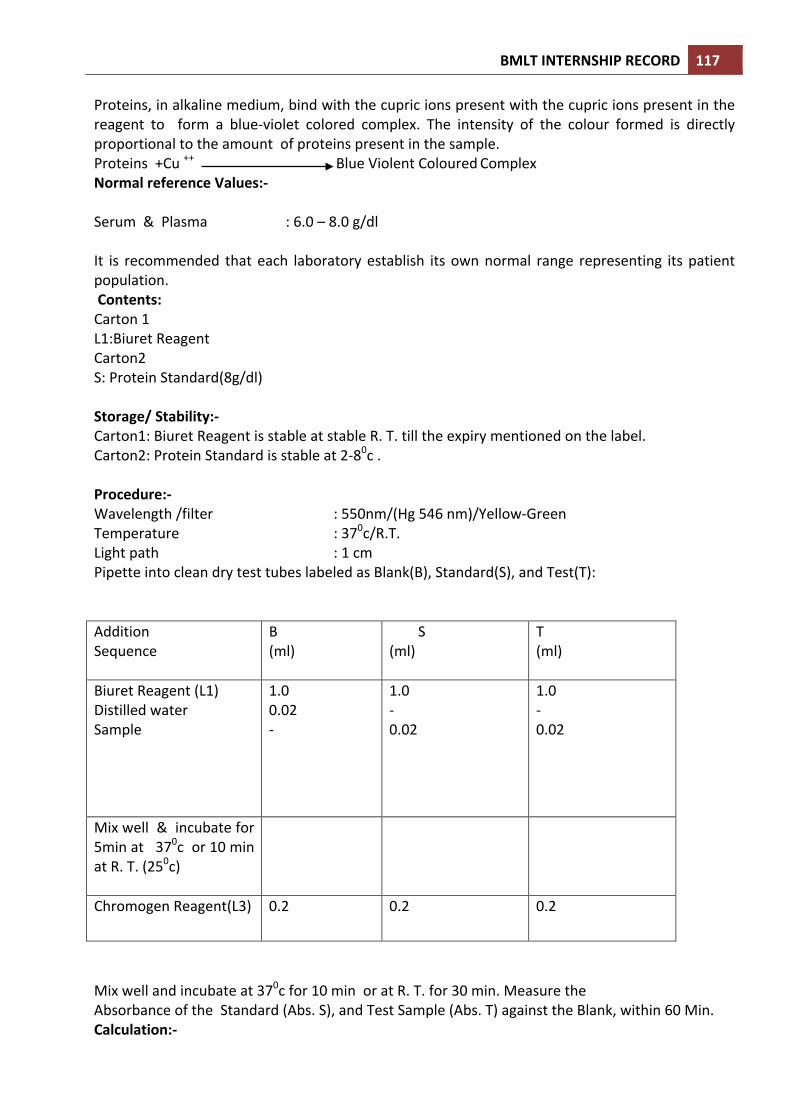
BMLT INTERNSHIP RECORD 117 Proteins, in alkaline medium, bind with the cupric ions present with the cupric ions present in the reagent to form a blue-‐violet colored complex. The intensity of the colour formed is directly proportional to the amount of proteins present in the sample. Proteins +Cu ++ Blue Violent Coloured Complex Normal reference Values:-‐ Serum & Plasma : 6.0 – 8.0 g/dl It is recommended that each laboratory establish its own normal range representing its patient population. Contents: Carton 1 L1:Biuret Reagent Carton2 S: Protein Standard(8g/dl) Storage/ Stability:-‐ Carton1: Biuret Reagent is stable at stable R. T. till the expiry mentioned on the label. Carton2: Protein Standard is stable at 2-‐80c . Procedure:-‐ Wavelength /filter : 550nm/(Hg 546 nm)/Yellow-‐Green Temperature : 370c/R.T. Light path : 1 cm Pipette into clean dry test tubes labeled as Blank(B), Standard(S), and Test(T): Addition Sequence
B (ml)
S (ml)
T (ml)
Biuret Reagent (L1) Distilled water Sample
1.0 0.02 -‐
1.0 -‐ 0.02
1.0 -‐ 0.02
Mix well & incubate for 5min at 370c or 10 min at R. T. (250c)
Chromogen Reagent(L3) 0.2 0.2 0.2
Mix well and incubate at 370c for 10 min or at R. T. for 30 min. Measure the Absorbance of the Standard (Abs. S), and Test Sample (Abs. T) against the Blank, within 60 Min. Calculation:-‐

BMLT INTERNSHIP RECORD 118 Total protein in mg/dl = Abs. T X 8 Abs. S Linearity:-‐The procedure is linear upto 15g/dl. If values exceed this limit, dilute the sample with distilled water and repeat the assay. Calculate the value using the proper dilution factor.
DETERMINATION OF ALBUMIN IN SERUM OR PLASMA Principle:-‐Albumin binds with the dye Bromocresol Green ion a buffer medium to form a green coloured complex. The intensity of the colour formed is directly proportional to the amount of albumin present in the sample. Albumin + Bromocresol Green Green Albumi8n BCG Complex Normal reference Values:-‐ Serum, Plasma (Albumin) : 3.7 – 5.3 g/dl Globulin : 2.3 – 3.6 g/dl A/G Ratio : 1.0 – 2.3 It is recommended that each laboratory established its own normal range representing its patient population. Contents Carton 1 L1:BCG Reagent Carton2 S: Albumin Standard(4g/dl) Reagent Preparation:-‐ Reagents are ready to use. Protect from bright light. Sample material:-‐ Serum, EDTA Plasma , Albumin is reported to be stable in the sample for 6 days at 2-‐80c . Storage/ Stability:-‐ Carton1: BCG Reagent is stable at stable R. T. till the expiry mentioned on the label. Carton2: Albumin Standard is stable at 2-‐80c . till the expiry mentioned on the label. Procedure:-‐ Wavelength /filter : 630nm/(Hg 623 nm)/Red Temperature : R.T. Light path : 1 cm

BMLT INTERNSHIP RECORD 119 Pipette into clean dry test tubes labeled as Blank(B), Standard(S), and Test(T): Addition Sequence
B (ml)
S (ml)
T (ml)
BCG Reagent (L1) Distilled water Albumin Standard(S) Sample
1.0 0.01 -‐ -‐
1.0 -‐ 0.01 -‐
1.0 -‐ -‐ 0.01
Mix well and incubate at R.T. for 5 min. Measured absorbance of the Standard (Abs. S) and Test Sample (Abs. T) against the Blank. Calculation: Abs. T Albumin in mg/dl = Abs. S X4 Globulin in g/dl = (Total Proteins) -‐ (Albumin) (in g/dl) (in g/dl) Albumin in g/dl A/G Ratio = Globulin in g/dl Linearity:-‐The procedure is linear up to 7 g/dl. If values exceed this limit, dilute the sample with distilled water and repeat the assay. Calculate the value using the proper dilution factor.
DETERMINATION OF BILIRUBIN IN SERUM Principle: Bilirubin is mainly formed from the heme portion of aged or damaged RBC’s. It then combines with albumin to form a complex which is not water soluble. The is referred to as indirect or unconjugated Bilirubin. In the liver this Bilirubin complex is combined with glucuronic acid into a water soluble conjugate. This is referred to as conjugated or direct Bilirubin. Elevated levels of bilirubin are found in liver disease(Hepatitis, cirrhosis), excessive hemolysis/destruction of RBC(hemolytic jaundice) obstruction of the biliary tract (obstructive jaundice) and in drug induced

BMLT INTERNSHIP RECORD 120 reaction. The differentiation between the direct and indirect bilirubin is important in diagnosing the cause of hyperbilirubinemia. Bilirubin + Diazotized Sulpanilic acid Azobilirubin Compound Normal Reference Values:-‐ Serum (Direct) : upto 0.2 mg/dl (Total) : upto 1.0 mg/dl It is recommended that each laboratory established its own normal range representing its patient population. Contents:-‐ L1: Direct Bilirubin Reagent . L2: Direct Nitrite Reagent. L1: Total Bilirubin reagent. L2: Total Nitrite Reagent. S : Artificial Standard(10mg/dl) Storage/ Stability:-‐ All reagents are stable R.T. till the expiry mention on the label. Reagent Preparation:-‐ Reagents are ready to use .Do not pipette with mount. Sample material:-‐ Serum, Bilirubin is reported to be stable in the sample for 4 days 2-‐80c from light as it is photosensitive Procedure:-‐ Wavelength/filter :546nm/Yellow-‐Green Temperature :R.T. Light Path : 1 cm Direct Bilirubin Assay:-‐ Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T) Addition Sequence
B (ml)
T (ml)
Direct Bilirubin Reagent(L1) Direct Nitrite Reagent(L2) Sample
1.0 -‐ 0.1
1.0 0.05 0.1
Mix well and incubate at R.T. for 5 min. Measured absorbance of the Standard (Abs. S) and Test Sample (Abs. T) against the Blank.

BMLT INTERNSHIP RECORD 121 Total Bilirubin Assay:-‐ Pipette in clean dry test tubes labeled as Blank (B) Standard (S), and Test (T) Addition Sequence
B (ml)
T (ml)
Total Bilirubin Reagent(L1) Total Nitrite Reagent(L2) Sample
1.0 -‐ 0.1
1.0 0.05 0.1
Mix well and incubate at R.T. for 10 min. Measured absorbance of the Standard (Abs. S) and Test Sample (Abs. T) against their respective Blank. Calculations:-‐ Total or Direct Bilirubin in mg/dl = Abs. T X 13 Linearity:-‐The procedure is linear up to 20 g/dl. If values exceed this limit, dilute the sample with distilled water and repeat the assay. Calculate the value using the proper dilution factor.

BMLT INTERNSHIP RECORD 122
DETERMINATION OF SERUM T3
The human thyroid gland is a major component of the endocrine system. Thyroid hormones perform many important functions. They exert powerful and essential regulatory influences on growth, differentiation, cellular metabolism and general hormonal balance of the body, as well as on the maintenance of metabolic activity and the development of the skeletal and organ system. The determination of T3 levels in serum is essential in assessing thyroid functions. In most hyperthyroid patients, both serum T3 and T4 are elevated. However a condition known as T3 thyrotoxicosis exhibits only elevated T3 concentrations. Serum T3 levels are also an excellent indicator of the effectiveness of thyroid therapy. TEST PRINCIPLE: Competitive EIA(Quantitative) In a competitive EIA , there exists a competitive reaction between native antigen and enzyme antigenconjugate for a limited number of insolubilized binding sites on the antibody coated in the microwell. After the antigen, antibody reaction has taken place, the fraction of the antigen in the conjugate or native antigen from the sample, which does not bind to the coated well , is washer away. The enzyme activity in the antibody bound function which is inversely proportional to the native antigen concentration is measured by addition of the substrate. By utilizing calibrators of known antigen values, a dose response curve may be generated from which the antigen concentration of an unknown can be obtained. KIT CONTENTS:
1. T3 Reaction Coated Microplate(96 wells)
One 96 –well microplate coated with sheep anti-‐T3 serum and packaged in an aluminium bag with dessicant. Store at 2 – 80C.
2. T3 – Enzyme Conjugate(1.5ml/vial)
One vial of T3 – horseradish peroxidise (HRP) conjugate in a bovine albumin – stabilizing matrix. A preservative has been added. Store at 2 -‐ 80C.
3. T3 Conjugate Buffer (13 ml) Redy to use. One bottle reagent containing buffer, red dye, preservative, and binding protein inhibitors. Store at 2 -‐ 80C.
4. T3 Calibrators (1 ml/vial)-‐ Six vials of serum reference for thyroxine at concentration of O ,2., 5.0, 10.0, 15.0 and 25.0 mg/dl. Store at 2 -‐ 80C. A preservative has been added.
5. Substrate A (7 ml/vial)
One bottle containing tetramethylbenzidine (TBM) in buffer. Store at 2 – 80C. 6. SubstrateB (7ml/vial)
One bottle containing hydrogen peroxide(H2O2) in buffer. Store at 2 – 80C. 7. Wash Solution Concentrate (20ml)-‐
One vial containing a surfactant in buffered saline.A preservative has been added. Store at 2 – 80C.
8. Stop Solution (8ml/vial)

BMLT INTERNSHIP RECORD 123
One bottle containing a strong acid (1N HCL). Store at 2 – 80C METERIAL, TOOLS AND EQUIPMENT REQYIRED: 1. Micro plate reader with 450 nm and 620 nm wave length absotbance capability (the 620 nm
filter is optional). 2. Micro plate washer or a squeeze bottle (optional). 3. Timer. 4. Micro plate cover for incubation steps. 5. Storage container for storage of wash buffer. 6. Vacuum aspirator (optional) for wash steps. 7. De ionized water. 8. Pipettes capable of delivering 25ml to 100ml. 9. Dispenser(s) for repetitive deliveries of 0.1 ml and 0.3 ml volumes with a precision of better
than 1.5% (optional). 10. Containers for mixing reagents.
PREPARTION OF REAGENTS AND STORAGE: 1. Working conjugate solution
Prepare the working conjugate solution by mixing 160 ul of conjugate concentration with 1.6 ml of the conjugate buffer (1:11) . The prepared reagents should be stored at 2-‐8®C and must be used within 24 hrs of preparation.
2. Wash Solution Prepare a 1:50 dilution of washing solution by mixing 1 ml of concentrated wash solution with 49 ml of distilled or deionised water. The prepared diluted wash solution can be preserved at 21 -‐25 0C for 60 days.
3. Working Substrate Solution Determine the amount of reagent needed and prepare by mixing equal portions of substrate A and substrate B in a suitable container. Note: the working substrate is not in use if it look blue.
ASSAY PROTOCOL: Before starting the assay, allow all the reagents, serum references & controls to reach room temperature.
1. Format the micro plate wells for each calibrator and patient specimen to be assayed. Replace the unused micro well strips back in to the aluminium foil, seal and store at 2 -‐ 80C.
2. 25ml of the calibrator and the patient specimen is added to the assigned wells. 3. 100ml of the prepared enzyme conjugate solution is added to each micro well. 4. Care should be taken to dispense the entire reagent to the bottom of the coated well. 5. Shake the microplate gently for 20 – 30 seconds & cover. 6. Incubate for 60min. At controlled room temperature (21 -‐250C). 7. Aspirated the contents of the wells & fill them completely (approximately 300 ml) with diluted
washing solution. Repeat the aspiration and washing procedure two more times. After the last wash, blot the micro plate on absorbent tissue to remove excess liquid from wells.

BMLT INTERNSHIP RECORD 124
8. 100 ml of working substrate solution is added to the wells. Always add reagent in the same order to minimize reaction time difference between wells.
9. Incubate at controlled room temperature (21-‐250C) for fifteen minutes. 10. 50ml of stop solution is added to each well and mix well for 15-‐20 seconds. 11. Read the absorbance in each well at 450nm (using a reference wave length of 620-‐630nm to
minimize well imperfections) in a micro plate reader.
CALCULATION OF RESULTS: A. Calculate the absorbance value of calibrators & samples at 450nm.(use 620nm filter as reference
filter, if available). B. Plot a point to point curve by plotting the absorbance of each calibrator on y-‐axis against
concentration of each calibrator on x-‐axis. C. Using the absorbance value of each sample determine the corresponding concentration of
triiodothyronine in mg/dl.from the standard curve obtained.
RANGE: A euthyhroid adult population was studied and the generalized range obtained is as given below: Expected ranges 0.52 – 1.85 ng/ml.

BMLT INTERNSHIP RECORD 125
DETERMINATION OF SERUM T4
CLINICAL SIGNIFICANCE:-‐ Thyroxine Hormone is secreted in the thyroid gland which is most important component of the endocrine system. Over 99% thyroxine secreted in the blood is bound to Thyroxine binding globulin (TBG), albumin and pre-‐albumin. The total Thyroxine(T4) content in blood is important in detecting thyroid disorders. In hyperthyroidism, like in those with Grave’s disease, the T4 level is increased in blood, while in hypothyroidism, like in those with myxedema, the T4 level is decreased. TEST PRINCIPLE: Competitive EIA(Quantitative) In a competitive EIA ,enzyme linked antigen competes with the antigen from the specimen for a limited number of binding sites on the immobilized antibody coated on the micro wells. Unbound antigen fraction is then washed away. The enzyme activity in the antibody bound function which is inversely proportional to the native antigen concentration, is measured by addition of the substrate. By utilizing calibrators of known antigen values, a dose response curve may be generated from which the antigen concentration of an unknown can be obtained. KIT CONTENTS:
9. T4 Reaction Coated Microplate(96 wells)-‐[1]
One 96 –well microplate coated with sheep anti-‐thyroxine serum and packaged in an aluminium bag with dessicant. Store at 2 – 80C.
10. T4 – Enzyme Conjugate(1.5ml/vial)[2]
One vial of thyroxine – horseradish peroxidise (HRP) conjugate in a bovine albumin – stabilizing matrix. A preservative has been added. Store at 2 -‐ 80C.
11. T4 Conjugate Buffer (13 ml) –[3] Redy to use. One bottle reagent containing buffer, red dye, preservative, and binding protein inhibitors. Store at 2 -‐ 80C.
12. T4 Calibrators (1 ml/vial)-‐[4A]-‐[4F] Six vials of serum reference for thyroxine at concentration of O [4A],2.0[4B],5.0[4C], 10.0[4D],15.0[4E] and 25.0[4F]mg/dl. Store at 2 -‐ 80C. A preservative has been added.
13. Substrate A (7 ml/vial)-‐[5]
One bottle containing tetramethylbenzidine (TBM) in buffer. Store at 2 – 80C. 14. SubstrateB (7ml/vial)-‐[6]
One bottle containing hydrogen peroxide(H2O2) in buffer. Store at 2 – 80C. 15. Wash Solution Concentrate (20ml)-‐[7]
One vial containing a surfactant in buffered saline.A preservative has been added. Store at 2 – 80C.
16. Stop Solution (8ml/vial)-‐[8] One bottle containing a strong acid (1N HCL). Store at 2 – 80C
17. Product Insert-‐01 No.

BMLT INTERNSHIP RECORD 126
METERIAL, TOOLS AND EQUIPMENT REQYIRED: 11. Micro plate reader with 450 nm and 620 nm wave length absotbance capability (the 620 nm
filter is optional). 12. Micro plate washer or a squeeze bottle (optional). 13. Timer. 14. Micro plate cover for incubation steps. 15. Storage container for storage of wash buffer. 16. Vacuum aspirator (optional) for wash steps. 17. Deionized water. 18. Pipettes capable of delivering 25ml to 100ml. 19. Dispenser(s) for repetitive deliveries of 0.1 ml and 0.3 ml volumes with a precision of better
than 1.5% (optional). 20. Containers for mixing reagents.
PREPARTION OF REAGENTS AND STORAGE: 4. Working conjugate solution
Prepare the working conjugate solution by mixing 160 ul of conjugate concentration with 1.6 ml of the conjugate buffer (1:11) . The prepared reagents should be stored at 2-‐8®C and must be used within 24 hrs of preparation.
5. Wash Solution Prepare a 1:50 dilution of washing solution by mixing 1 ml of concentrated wash solution with 49 ml of distilled or deionised water. The prepared diluted wash solution can be preserved at 21 -‐25 0C for 60 days.
6. Working Substrate Solution Determine the amount of reagent needed and prepare by mixing equal portions of substrate A and substrate B in a suitable container. Note: the working substrate is not in use if it look blue.
ASSAY PROTOCOL: Before starting the assay, allow all the reagents, serum references & controls to reach room temperature.
12. Format the micro plate wells for each calibrator and patient specimen to be assayed. Replace the unused micro well strips back in to the aluminium foil, seal and store at 2 -‐ 80C.
13. Add 25ml of the calibrator and the patient specimen to the assigned wells. 14. Add 100ml of the prepared enzyme conjugate solution to each micro well. 15. Care should be taken to dispense the entire reagent to the bottom of the coated well. 16. Shake the microplate gently for 20 – 30 seconds & cover. 17. Incubate for 60min. At controlled room temperature (21 -‐250C). 18. Aspirate the contents of the wells & fill them completely (approximately 300 ml) with diluted
washing solution. Repeat the aspiration and washing procedure two more times. After the last wash, blot the micro plate on absorbent tissue to remove excess liquid from wells.

BMLT INTERNSHIP RECORD 127
19. Add 100 ml of working substrate solution to the wells. Always add reagent in the same order to minimize reaction time difference between wells.
20. Incubate at controlled room temperature (21-‐250C) for fifteen minutes. 21. Add 50ml of stop solution to each well and mix well for 15-‐20 seconds. 22. Read the absorbance in each well at 450nm (using a reference wave length of 620-‐630nm to
minimize well imperfections) in a micro plate reader.
CALCULATION OF RESULTS: D. Calculate the absorbance value of calibrators & samples at 450nm.(use 620nm filter as reference
filter, if available). E. Plot a point to point curve by plotting the absorbance of each calibrator on y-‐axis against
concentration of each calibrator on x-‐axis. F. Using the absorbance value of each sample determine the corresponding concentration of
thyroxine in mg/dl.from the standard curve obtained. INTERPRETATION OF RESULTS: 1. The concentration obtained of the calibrators should be within + 10% of the assigned values
in mg/dl. 2. Total serum thyroxine concentration obtained is regulated by many factors like the thyroid
gland function and its regulation, thyroxine binding globulin (TBG) concentration and the binding of thyroxine to (TGB). Thus total thyroxine concentration alone is not assess clinical status.
3. It has been observed that a decreasing in total triiodothyonine is found in protein wasting diseases’, certain lever diseases and administration of testosterone , diphenylhydantoin or salicylates.
4. It has been observed that an increase in total thyroxine is found in conditions such as pregnancy or administration of oral contraceptives. RANGE: A euthyhroid adult population was studied and the generalized range obtained is as given below: Expected ranges (male) 4.4 – 10.8 mg/dl Expected ranges (female) 4.8 – 11.6 mg/dl

BMLT INTERNSHIP RECORD 128
DETERMINATION OF SERUM TSH
The Thyroid stimulating hormone (thyrotropin or TSH) is a glycoprotein with a molecular weight of 28000, secreted by the adenohypophysis. TSH is controlled by negative feedback from circulating T3, T4 and TRH(thyrotropin Releasing Hormone). The measurement of serum TSH has proven to be one of the most sensitive methods for the detection of primary hypothyroidism. In primary hypothyroidism the production of thyroid hormones is impaired and the TSH levels are observed to be higher. However, in secondary and tertiary hypothyroidism, the TSH levels are low because of pituitary or hypothalamic lesions. In hyperthyroidism the circulating levels of TSH is usually subnormal. In some instances, however this condition may result from hyperstimulation of the thyroid, due to hypothalamic or pituitary lesions and in this case the TSH levels is increased. Measurement of circulating levels of TSH may be used as a screening or confirmatory test for hypothyroidism and hyperthyroidism, and as an aid to differential diagnosis of these conditions.TSH levels have been used to monitor the adequacy of thyroid hormone replacement therapy. TEST PRINCIPLE: Competitive EIA(Quantitative) In a quantitative EIA ,high affinity antibodies react with antigen to form an insoluble sandwich complex on the surface of a coated microplate. The antigen from the specimen gets linked at the surface of the well through interaction of reactive IgG coated on the well and affinity purified x-‐ antigen IgG conjugated with HRP.The fraction of the X-‐antigen IgG conjugated with enzyme that does not bind to the coated well is washed away. The enzyme activity , which is proportional to the native antigen concentration, is measured by addition of the substrate. By utilizing calibrators of known antigen values, a dose response curve may be generated from which the antigen concentration of an unknown can be obtained. KIT CONTENTS:
18. TSH Reaction Coated Microplate(96 wells)
One 96 –well microplate coated with sheep anti-‐thyroxine serum and packaged in an aluminium bag with dessicant. Store at 2 – 80C.
19. Enzyme TSH Reagent(13ml/vial)
One vial containing – horseradish peroxidise (HRP) labelled reactive X-‐TSH IgG in buffer, dye and preservative. Store at 2 -‐ 80C.
20. TSH Calibrators (1 ml/vial)
Seven vials of serum reference for TSH at concentration of O, 0.5, 2.5 5.0, 10.0, 20.5 and 40.0 µIu/ml. Store at 2 -‐ 80C. A preservative has been added.
21. Substrate A (7 ml/vial)
One bottle containing tetramethylbenzidine (TBM) in buffer. Store at 2 – 80C. 22. SubstrateB (7ml/vial)
One bottle containing hydrogen peroxide(H2O2) in buffer. Store at 2 – 80C. 23. Wash Solution Concentrate (20ml)

BMLT INTERNSHIP RECORD 129 One vial containing a surfactant in buffered saline.A preservative has been added. Store at 2 – 80C.
24. Stop Solution (8ml/vial) One bottle containing a strong acid (1N HCL). Store at 2 – 80C METERIAL, TOOLS AND EQUIPMENT REQYIRED: 21. Micro plate reader with 450 nm and 620 nm wave length absotbance capability (the 620 nm
filter is optional). 22. Micro plate washer or a squeeze bottle (optional). 23. Timer. 24. Micro plate cover for incubation steps. 25. Storage container for storage of wash buffer. 26. Vacuum aspirator (optional) for wash steps. 27. Deionized water. 28. Pipettes capable of delivering 25ml to 100ml. 29. Dispenser(s) for repetitive deliveries of 0.1 ml and 0.3 ml volumes with a precision of better
than 1.5% (optional). 30. Containers for mixing reagents.
PREPARTION OF REAGENTS AND STORAGE 7. Wash Solution
Prepare a 1:50 dilution of washing solution by mixing 1 ml of concentrated wash solution with 49 ml of distilled or deionised water. The prepared diluted wash solution can be preserved at 21 -‐25 0C for 60 days.
8. Working Substrate Solution Determine the amount of reagent needed and prepare by mixing equal portions of substrate A and substrate B in a suitable container. Note: the working substrate is not in use if it look blue.
ASSAY PROTOCOL: Before starting the assay, allow all the reagents, serum references & controls to reach room temperature.
23. Format the micro plate wells for each calibrator and patient specimen to be assayed. Replace the unused micro well strips back in to the aluminium foil, seal and store at 2 -‐ 80C.
24. 50µl of the calibrator and the patient specimen is added to the assigned wells. 25. 100ml of the prepared enzyme conjugate solution is added to each micro well. 26. Shake the microplate gently for 20 – 30 seconds & cover. 27. Incubate for 60min. At controlled room temperature (21 -‐250C). 28. Aspirated the contents of the wells & fill them completely (approximately 300 ml) with diluted
washing solution. Repeat the aspiration and washing procedure two more times. After the last wash, blot the micro plate on absorbent tissue to remove excess liquid from wells.

BMLT INTERNSHIP RECORD 130
29. 100 ml of working substrate solution is added to the wells. Always add reagent in the same order to minimize reaction time difference between wells.
30. Incubate at controlled room temperature (21-‐250C) for fifteen minutes. 31. 50ml of stop solution is added to each well and mix well for 15-‐20 seconds. 32. Read the absorbance in each well at 450nm (using a reference wave length of 620-‐630nm to
minimize well imperfections) in a micro plate reader.
CALCULATION OF RESULTS: G. Calculate the absorbance value of calibrators & samples at 450nm.(use 620nm filter as reference
filter, if available). H. Plot a point to point curve by plotting the absorbance of each calibrator on y-‐axis against
concentration of each calibrator on x-‐axis. I. Using the absorbance value of each sample determine the corresponding concentration of
thyroxine in µiu/ml.from the standard curve obtained. INTERPRETATION OF RESULTS: 5. The concentration obtained of the calibrators should be within + 10% of the assigned values . 6. Total serum TSH concentration obtained is regulated by many factors like the
TRH,hypothalamus gland infection etc.This concentration of hTSH alone is not sufficient to know the clinical status of a sample.
7. A decrease in serum hTSH concentration is observed with administration of propenolol, methimazol etc. Similarly an increase in serum hTSH concentration is observed by pharmacological intervention, domperiodone etc.
8. Any genetic variation or degradation of intact TSH into sub units affect the binding characteristic of the antibodies and may affect the final reult. RANGE: A euthyhroid adult population was studied and the generalized range obtained is as given below: Low normal ranges 0.39 µIu/ml High normal ranges 6.16 µIu/ml
Related Documents











