Atherosclerosis Supplements 12 (2011) 221–263 Familial hypercholesterolaemia: A model of care for Australasia Gerald F. Watts a,∗ , David R Sullivan b , Nicola Poplawski c , Frank van Bockxmeer d , Ian Hamilton-Craig e , Peter M. Clifton f , Richard O’Brien g , Warrick Bishop h , Peter George i , Phillip J. Barter j , Timothy Bates a , John R. Burnett k , John Coakley l , Patricia Davidson m , Jon Emery n , Andrew Martin o , Waleed Farid p , Lucinda Freeman q , Elizabeth Geelhoed r , Amanda Juniper a,s , Alexa Kidd t , Karam Kostner u , Ines Krass v , Michael Livingston w , Suzy Maxwell s , Peter O’Leary s , Amal Owaimrin x , Trevor G. Redgrave a , Nicola Reid y , Lynda Southwell a , Graeme Suthers c , Andrew Tonkin z , Simon Towler aa , Ronald Trent q , Familial Hypercholesterolaemia Australasia Network Consensus Group (Australian Atherosclerosis Society) 1 a Lipid Disorders Clinic, Metabolic Research Centre and Department of Internal Medicine, Royal Perth Hospital, School of Medicine and Pharmacology, University of Western Australia, Perth, Western Australia, Australia b Department of Biochemistry and Lipid Clinic, Royal Prince Alfred Hospital, University of Sydney, New South Wales, Australia c South Australia Clinical Genetics Service, Genetics & Molecular Pathology Directorate, Women’s & Children’s Hospital, Adelaide, South Australia, Australia d Cardiovascular Genetics Laboratory, Royal Perth Hospital, University of Western Australia, Western Australia, Australia e Preventive Cardiology and Lipid Clinic, Gold Coast Hospital, Griffith University, Queensland, Australia f Baker IDI Heart and Diabetes Institute, Adelaide, South Australia, Australia g Department of Medicine, Diabetes and Endocrinology, Austin Hospital, University of Melbourne, Victoria, Australia h Department of Cardiology, Calvary Cardiac Centre, Calvary Health Care, Tasmania, Australia i Biochemistry and Pathology, Canterbury Health Laboratories, Lipid Clinic, Christchurch Hospital, University of Otago, Christchurch, New Zealand j Heart Research Institute, University of Sydney, Sydney, New South Wales, Australia k Core Clinical Pathology & Biochemistry, PathWest Laboratory Medicine WA, Lipid Disorders Clinic, Royal Perth Hospital, University of Western Australia, Western Australia, Australia l Department of Paediatrics and Clinical Biochemistry, The Children’s Hospital Westmead, Sydney, New South Wales, Australia m Cardiovascular and Chronic Care, Curtin University, and Nursing Research, St Vincent’s Hospital, Sydney, New South Wales, Australia n School of Primary, Aboriginal and Rural Health Care, University of Western Australia, Western Australia, Australia o Department of Paediatric and Adolescent Medicine, Princess Margaret Hospital, Perth, Western Australia, Australia p FH Family Support Group of Western Australia, Perth, Western Australia, Australia q Department of Molecular and Clinical Genetics, Royal Prince Alfred Hospital, University of Sydney, Australia r School of Population Health, University of Western Australia, Western Australia, Australia s Office of Population Health Genomics, Department of Health, Government of Western Australia, Australia t Clinical Genetics, Canterbury Health Laboratories, Christchurch Hospital, New Zealand u Cardiac Imaging Group, Department of Cardiology, Mater Hospital, University of Queensland, Australia v Department of Pharmacy Practice, Faculty of Pharmacy, University of Sydney, New South Wales, Australia w International Cholesterol Foundation, Sutton-Courtney, Oxfordshire, United Kingdom x Department of Dietetics, Familial Hypercholesterolaemia Clinical Support Service, Auburn Hospital, Sydney, New South Wales, Australia y Cardiovascular Prevention and Lipid Disorders Clinic, Christchurch Hospital, New Zealand z Cardiovascular Research Unit, Monash University, Melbourne, Victoria, Australia aa Health Networks, Department of Health, Government of Western Australia, Australia ∗ Corresponding author at: School of Medicine and Pharmacology, Royal Perth Hospital, University of Western Australia, GPO Box X2213, Perth, Western Australia 6847, Australia. Tel.: +61 8 9224 0245. E-mail address: [email protected] (G.F. Watts). 1 See Appendix 1. 1567-5688/$ – see front matter © 2011 Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.atherosclerosissup.2011.06.001

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

W
1d
Atherosclerosis Supplements 12 (2011) 221–263
Familial hypercholesterolaemia: A model of care for Australasia
Gerald F. Watts a,∗, David R Sullivan b, Nicola Poplawski c, Frank van Bockxmeer d,Ian Hamilton-Craig e, Peter M. Clifton f, Richard O’Brien g, Warrick Bishop h, Peter George i,
Phillip J. Barter j, Timothy Bates a, John R. Burnett k, John Coakley l, Patricia Davidson m,Jon Emery n, Andrew Martin o, Waleed Farid p, Lucinda Freeman q, Elizabeth Geelhoed r,Amanda Juniper a,s, Alexa Kidd t, Karam Kostner u, Ines Krass v, Michael Livingston w,
Suzy Maxwell s, Peter O’Leary s, Amal Owaimrin x, Trevor G. Redgrave a, Nicola Reid y,Lynda Southwell a, Graeme Suthers c, Andrew Tonkin z, Simon Towler aa, Ronald Trent q,
Familial Hypercholesterolaemia Australasia Network Consensus Group(Australian Atherosclerosis Society)1
a Lipid Disorders Clinic, Metabolic Research Centre and Department of Internal Medicine, Royal Perth Hospital, School of Medicine and Pharmacology,University of Western Australia, Perth, Western Australia, Australia
b Department of Biochemistry and Lipid Clinic, Royal Prince Alfred Hospital, University of Sydney, New South Wales, Australiac South Australia Clinical Genetics Service, Genetics & Molecular Pathology Directorate, Women’s & Children’s Hospital, Adelaide,
South Australia, Australiad Cardiovascular Genetics Laboratory, Royal Perth Hospital, University of Western Australia, Western Australia, Australia
e Preventive Cardiology and Lipid Clinic, Gold Coast Hospital, Griffith University, Queensland, Australiaf Baker IDI Heart and Diabetes Institute, Adelaide, South Australia, Australia
g Department of Medicine, Diabetes and Endocrinology, Austin Hospital, University of Melbourne, Victoria, Australiah Department of Cardiology, Calvary Cardiac Centre, Calvary Health Care, Tasmania, Australia
i Biochemistry and Pathology, Canterbury Health Laboratories, Lipid Clinic, Christchurch Hospital, University of Otago, Christchurch, New Zealandj Heart Research Institute, University of Sydney, Sydney, New South Wales, Australia
k Core Clinical Pathology & Biochemistry, PathWest Laboratory Medicine WA, Lipid Disorders Clinic, Royal Perth Hospital,University of Western Australia, Western Australia, Australia
l Department of Paediatrics and Clinical Biochemistry, The Children’s Hospital Westmead, Sydney, New South Wales, Australiam Cardiovascular and Chronic Care, Curtin University, and Nursing Research, St Vincent’s Hospital, Sydney, New South Wales, Australia
n School of Primary, Aboriginal and Rural Health Care, University of Western Australia, Western Australia, Australiao Department of Paediatric and Adolescent Medicine, Princess Margaret Hospital, Perth, Western Australia, Australia
p FH Family Support Group of Western Australia, Perth, Western Australia, Australiaq Department of Molecular and Clinical Genetics, Royal Prince Alfred Hospital, University of Sydney, Australia
r School of Population Health, University of Western Australia, Western Australia, Australias Office of Population Health Genomics, Department of Health, Government of Western Australia, Australia
t Clinical Genetics, Canterbury Health Laboratories, Christchurch Hospital, New Zealandu Cardiac Imaging Group, Department of Cardiology, Mater Hospital, University of Queensland, Australia
v Department of Pharmacy Practice, Faculty of Pharmacy, University of Sydney, New South Wales, Australia
w International Cholesterol Foundation, Sutton-Courtney, Oxfordshire, United Kingdomx Department of Dietetics, Familial Hypercholesterolaemia Clinical Support Service, Auburn Hospital, Sydney, New South Wales, Australiay Cardiovascular Prevention and Lipid Disorders Clinic, Christchurch Hospital, New Zealand
z Cardiovascular Research Unit, Monash University, Melbourne, Victoria, Australia
aa Health Networks, Department of Health, Gov∗ Corresponding author at: School of Medicine and Pharmacology, Royal Perth Hestern Australia 6847, Australia. Tel.: +61 8 9224 0245.
E-mail address: [email protected] (G.F. Watts).1 See Appendix 1.
567-5688/$ – see front matter © 2011 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.atherosclerosissup.2011.06.001
ernment of Western Australia, Australia
ospital, University of Western Australia, GPO Box X2213, Perth,

2
A
au
msa
afff
A©
K
C
tCcEtlDacS
22 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
bstract
Familial hypercholesterolaemia (FH) is a dominantly inherited disorder present from birth that causes marked elevation in plasma cholesterolnd premature coronary heart disease. There are at least 45,000 people with FH in Australia and New Zealand, but the vast majority remainsndetected and those diagnosed with the condition are inadequately treated.
To bridge this major gap in coronary prevention the FH Australasia Network (Australian Atherosclerosis Society) has developed a consensusodel of care (MoC) for FH. The MoC is based on clinical experience, expert opinion, published evidence and consultations with a wide
pectrum of stakeholders, and has been developed for use primarily by specialist centres intending starting a clinical service for FH. This MoCims to provide a standardised, high-quality and cost-effective system of care that is likely to have the highest impact on patient outcomes.
The MoC for FH is presented as a series of recommendations and algorithms focusing on the standards required for the detection, diagnosis,ssessment and management of FH in adults and children. The process involved in cascade screening and risk notification, the backboneor detecting new cases of FH, is detailed. Guidance on treatment is based on risk stratifying patients, management of non-cholesterol riskactors, safe and effective use of statins, and a rational approach to follow-up of patients. Clinical and laboratory recommendations are givenor genetic testing. An integrative system for providing best clinical care is described.
This MoC for FH is not prescriptive and needs to be complemented by good clinical judgment and adjusted for local needs and resources.fter initial implementation, the MoC will require critical evaluation, development and appropriate modification.2011 Elsevier Ireland Ltd. All rights reserved.
eywords: Familial hypercholesterolaemia; model of care; adults; children; adolescents; diagnosis; genetic testing; cascade screening; assessment; treatment
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2232. Summary of recommendations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2253. Overview of algorithms for the model of care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2274. Detection of index cases and diagnosis of FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2275. Assessment of adult patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2286. Diagnosis and assessment of children and adolescents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2307. Management of adults . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 232
7.1. LDL-cholesterol and apoB targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2327.2. Diet and lifestyle modifications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2327.3. Pharmacotherapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 232
7.3.1. Safety monitoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2347.3.2. Medication adherence and tolerance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 234
7.4. Review intervals and shared care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2347.5. Assessing atherosclerosis and CHD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2347.6. FH in women . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 235
8. Management of children and adolescents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2358.1. General and lifestyle considerations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2358.2. LDL-cholesterol targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2368.3. Pharmacotherapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 237
8.4. Assessing atherosclerosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .8.5. Clinical monitoring and continuity of care . . . . . . . . . . . . . . . .9. LDL-apheresis and radical therapy for FH . . . . . . . . . . . . . . . . . . . . . .
Abbreviations: ABI, ankle brachial index; ACE, angiotensin converting enzyme; ALein B; ARMS, amplification refractory mutation system; AST, aspartate aminotraCS, coronary calcium score; CHD, coronary heart disease; CIMT, carotid intimomputerised tomography coronary angiography; CUS, carotid ultrasonography; CBESA, exon by exon sequence analysis; ECG, electrocardiography; EST, exercise
ion; FDA, Food and Drug Administration; GP, general practitioner; HDL, high dow density lipoprotein; LFT, liver function test; Lp(a), lipoprotein(a); MBS, Medieaths; MLPA, Multiplex Ligation Probe Amplification; MoC, model of care; NAT
nd Medical Research Council; NPAAC, National Pathology Accreditation Advionvertase subtilisin/kexin type 9; PGD, pre-implantation genetic diagnosis; PNDHAPE, Screening for Heart Attack Prevention and Education Task Force; TGA, T
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 237. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 237
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 238
T, alanine aminotransferase; ApoA-I, apolipoprotein A-I; apoB, apolipopro-nsferase; BMI, body mass index; CACS, Coronary Artery Calcium Score;a-medial thickness; CK, creatine kinase; CRP, C-reactive protein; CTCA,VD, cardiovascular disease; DLCNS, Dutch Lipid Clinic Network Score;
stress test; FH, familial hypercholesterolaemia; FMD, flow-mediated dilata-ensity lipoprotein; InterChol, International Cholesterol Foundation; LDL,care Benefits Schedule; MEDPED, Make Early Diagnosis to Prevent EarlyA, National Association of Testing Authorities; NHMRC, National Health
sory Council; PBS, Pharmaceutical Benefits Scheme; PCSK9, proprotein, prenatal diagnosis; RCPA, Royal College of Pathologists of Australasia;herapeutic Goods Administration; TSH, thyroid-stimulating hormone.

G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 223
9.1. Indications, patient selection and targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2389.2. Methods for apheresis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2389.3. Monitoring therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2389.4. Cost considerations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2409.5. Other therapeutic options . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 240
10. Cascade screening: testing and risk notification of families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24010.1. Risk notification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 240
10.1.1. Contacting and informing families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24010.2. Co-ordination of cascade screening . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24210.3. Risk notification without consent . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24210.4. Insurance cover and genetic testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242
11. Genetic testing of families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24312. Laboratory approach to genetic testing for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243
12.1. Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24312.2. A protocol for genetic testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24512.3. Assessing the significance of gene variants detected in index cases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 245
13. The web of care for FH: the optimal service model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24513.1. Focus, aims and objectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24713.2. Co-ordination and integration of care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 247
Inter-specialty links . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24813.3. Administrative and information technology support . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24913.4. Clinical governance: audit, education, training . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24913.5. Patient and family support groups . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24913.6. Into the future: chronic care model, commissioning, evaluation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250Appendix 1. FH Australasia Network Consensus Group and Process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250
Chair . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250Writing committee . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250Steering committee . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250Contributors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250Consensus process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251Funding. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251Disclosures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251
Appendix 2. Dutch Lipid Clinic Network Criteria for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251Appendix 3. Simon Broome Criteria for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252Appendix 4. MEDPED Criteria for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252Appendix 5. Typical examination features of FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 253Appendix 6. Hypothetical Pedigree . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 254Appendix 7. ‘Real case’ Pedigree . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 254Appendix 8. Selected websites for clinical services . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 255
. . . . . .
1
atfcitfc[idii
tc1cA
AaZMttp
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
Familial hypercholesterolaemia (FH) is the most commonnd serious form of inherited hyperlipidaemia [1]. FH is dueo dominant mutations of genes predominantly affecting theunction of the low-density lipoprotein (LDL) receptor thatlears LDL particles from plasma [2,3], and hence resultsn marked elevation in plasma LDL-cholesterol concentra-ion. FH is present from birth and accelerates the onset of allorms of atherosclerotic cardiovascular disease (CVD), espe-ially coronary heart disease (CHD), by one to four decades1,4,5]. Opportunistic diagnosis of FH followed by screen-ng of family members, the so-called cascade screening, can
etect individuals at an early stage of FH [4,6–11]. Thiss critically important because it enables early interventionncluding lifestyle measures, cholesterol-lowering medica-pc
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 255
ions (particularly statins), and management of other majorardiovascular risk factors [12–17b]. Alarmingly, less than0% of cases with FH have been diagnosed in most Westernommunities, and only 5% are adequately treated [5,18–20];ustralia and New Zealand being no exception [21–23].To address this demand in coronary prevention, the FH
ustralasia Network established a Consensus Group to devisemodel of care (MoC) for FH from an Australian and Newealand (Australasian) perspective. In the present context, theoC was conceptualised as an overarching system, based on
heoretical, experiential and evidence-based standards, forhe provision of highest quality health care services for allatients with FH [24].
This MoC aims to establish a standard of care for FHatients in a framework within which future evidence andonsensus may be included and developed, thereby extending

224 G.F. Watts et al. / Atherosclerosis Sup
Table 1Grades for recommendations employed for consensus statements.
Grade of recommendation Description
A Recommendation can be trusted toguide practice
B Recommendation can be trusted toguide practice in most situations
C Recommendations may be used toguide practice, but care should betaken in its application
These grades were applied to each of the recommendations in Section 2.Individual members of the Steering Committee were asked to grade the rec-ommendations based on their knowledge of the literature and what theyconsidered best practice in caring for patients with FH. Gradings were dis-cussed and after full consensus of the committee was reached a final gradewas ascribed to each recommendation. All members of the FH AustralasiaN
oAldbiTloE
tmpeMr[
(lcTNstwubrnImrl
etwork Consensus Group approved the final gradings.
ther proposed clinical care programs from Europe and Northmerica [12,13,25–29]. The MoC is intended primarily for
ipid disorder clinics in tertiary centres intending to initiate orevelop a clinical service for FH. The MoC has been informedy published research, clinical experience, expert opinion andnternational guidelines for managing FH [5,12–16,30–34].
he major premises for these recommendations were pub-ished data on clinical efficacy and outcomes, but informationn cost-effectiveness was also employed where available.xpert opinion was sourced from diverse stakeholders from
cep
Diagnostic criteriaAppendices 2, 3 & 4
ProcesFigure
Clinical protocolFigure 9
Laboratory protocolsFigures 10 & 11
Optimal componentsFigure 12
ModelCare fo
Fig. 1. Overview of algorithms f
plements 12 (2011) 221–263
he disciplines of adult medicine, paediatric and adolescentedicine, clinical genetics, clinical biochemistry, nursing,
harmacy, general practice, population health, and healthconomics; a patient support group was also consulted. TheoC significantly extends and consolidates other Australian
ecommendations on the detection and management of FH10,15,32,35–38].
The MoC is presented as a series of recommendationsTable 1, Section 2) and algorithms (Fig. 1) that if fol-owed could provide a cost-effective, standardised system ofare likely to have the highest impact on patient outcomes.he recommendations were graded according to a modifiedHMRC classification [39], and reflected the full consen-
us of an expert committee that was based on knowledge ofhe relevant literature and best clinical practice. Algorithmsere chosen to provide easy visualisation of the conceptsnderpinning the MoC. The algorithms are accompanied byackground information and explanatory text and reflect theecommendations of the Consensus Group. This MoC shouldot be perceived as prescriptive, but as a tool for guidance.t should therefore be complemented by good clinical judg-ent and adjustments made to the model according to local
equirements, protocols and resources. Acknowledging theack of objective evidence supporting its clinical-efficacy and
ost-effectiveness, this MoC for FH should be viewed as anvolving set of diagnostic and care pathways that will neederiodic review, clinical appraisal and modification.Children/AdolescentsFigure 4
AdultsFigure 2 & 3
Children/AdolescentsFigure 6
AdultsFigure 5
s8
of r FH
ProcessFigure 2
LDL-ApheresisFigure 7
or model of care for FH.

2
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 225
. Summary of recommendations
Recommendation I Models of care and components of service Grade
Figs. 1 and 12 a. Models of care for familial hypercholesterolaemia (FH) should focus on detecting, diagnosing,assessing and managing index cases, as well as on risk notification and cascade screening of familymembers.
A
b. Adults and children/adolescents will require different models of care. Ac. All services need to be integrated across several specialties and incorporated into primary care. Ad. Good clinical governance, teaching and training programs, and family support groups are integral to allmodels of care.
A
Recommendation II Identifying index cases Grade
Fig. 2 a. Index cases of FH should be sought amongst adults with premature cardiovascular disease in primaryand secondary care settings.
A
b. In adults a simple clinical tool based on the Dutch Lipid Clinic Network Score should be used. Ac. All patients with possible-to-definite FH should be referred to a lipid disorders clinic for more detailedassessment and institution of cascade screening.
A
Recommendation III Clinical assessment and management allocation of adults Grade
Fig. 3 a. Secondary causes of hypercholesterolaemia should first be excluded. Ab. The diagnosis of FH should be made using both phenotypic and genetic testing. Ac. Patients should be stratified into risk categories according to presence of cardiovascular risk factorsand personal history of cardiovascular disease.
A
d. Risk stratification should guide the intensity of medical management. A
Recommendation IV Clinical assessment and management allocation of children and adolescents Grade
Fig. 4 a. Children (≥ 5 yr) and adolescents should be tested for FH after the diagnosis of FH has been made in aparent.
A
b. Secondary causes of hypercholesterolaemia should first be excluded. Ac. With rare exceptions, children and adolescents should only be genetically tested for FH after apathogenic variant (mutation) has been identified in a parent or first degree relative.
A
d. Age- and gender-specific plasma LDL-cholesterol concentration thresholds should be used to make thephenotypic diagnosis of FH, an LDL-cholesterol ≥ 5.0 mmol/L indicating highly probable/ definite FH;two fasting lipid profiles are recommended.
B
e. Patients should be stratified into risk categories according to age, presence of other cardiovascular riskfactors, prematurity of family history of cardiovascular disease and the level of hypercholesterolaemia atdiagnosis.
A
f. Risk stratification should guide the intensity of medical management. A
Recommendation V Management of FH in adults Grade
Fig. 5 a. All adult patients with FH must receive advice on lifestyle modifications and all non-lipid risk factorsmust be addressed.
A
b. Plasma LDL-cholesterol targets for routine, enhanced and intensive management should be<4 mmol/L, <3 mmol/L, <2 mmol/L, respectively.
C
c. Achieving these targets will require a fat-modified diet, plant sterols (or stanols) and a statin with orwithout ezetimibe.
A
d. Niacin, resins and a fibrate may be required with more intensive strategies. Ae. Plasma levels of hepatic aminotransferases, creatine kinase and creatinine should be measured beforestarting pharmacotherapy. All patients on pharmacotherapy, particularly statins, should have hepaticaminotransferases monitored; creatine kinase should only be measured when musculoskeletal symptomsare reported; creatinine should be monitored in those with kidney disease.
A
f. All women with FH of child-bearing age should have pre-pregnancy counselling Ag. Statins and other systemically absorbed lipid regulating agents should be discontinued 3 months beforeconception and during pregnancy and breast feeding in women with FH.
A
h. Non-invasive testing for coronary heart disease and atherosclerosis should be considered in patientsundergoing standard and enhanced treatment, with a step-up in treatment considered if there is evidenceof progression of disease. Non-invasive testing for atherosclerosis need not be carried out morefrequently than every two years.
C
i. Patients receiving standard or enhanced management should be reviewed every 6–12 months, and thosereceiving intensive management should be reviewed according to clinical context, with appropriateinterval assessment of cardiac function and referral to cardiology.
B
2
(
R
F
R
F
R
F
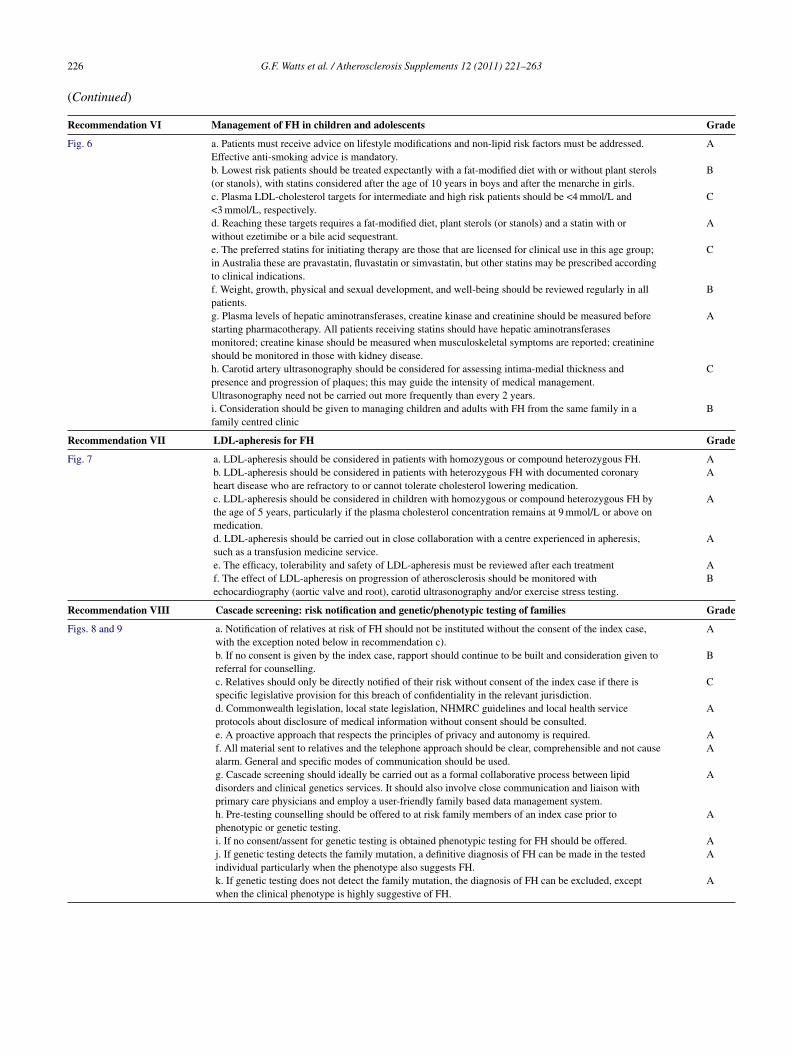
26 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Continued)
ecommendation VI Management of FH in children and adolescents Grade
ig. 6 a. Patients must receive advice on lifestyle modifications and non-lipid risk factors must be addressed.Effective anti-smoking advice is mandatory.
A
b. Lowest risk patients should be treated expectantly with a fat-modified diet with or without plant sterols(or stanols), with statins considered after the age of 10 years in boys and after the menarche in girls.
B
c. Plasma LDL-cholesterol targets for intermediate and high risk patients should be <4 mmol/L and<3 mmol/L, respectively.
C
d. Reaching these targets requires a fat-modified diet, plant sterols (or stanols) and a statin with orwithout ezetimibe or a bile acid sequestrant.
A
e. The preferred statins for initiating therapy are those that are licensed for clinical use in this age group;in Australia these are pravastatin, fluvastatin or simvastatin, but other statins may be prescribed accordingto clinical indications.
C
f. Weight, growth, physical and sexual development, and well-being should be reviewed regularly in allpatients.
B
g. Plasma levels of hepatic aminotransferases, creatine kinase and creatinine should be measured beforestarting pharmacotherapy. All patients receiving statins should have hepatic aminotransferasesmonitored; creatine kinase should be measured when musculoskeletal symptoms are reported; creatinineshould be monitored in those with kidney disease.
A
h. Carotid artery ultrasonography should be considered for assessing intima-medial thickness andpresence and progression of plaques; this may guide the intensity of medical management.Ultrasonography need not be carried out more frequently than every 2 years.
C
i. Consideration should be given to managing children and adults with FH from the same family in afamily centred clinic
B
ecommendation VII LDL-apheresis for FH Grade
ig. 7 a. LDL-apheresis should be considered in patients with homozygous or compound heterozygous FH. Ab. LDL-apheresis should be considered in patients with heterozygous FH with documented coronaryheart disease who are refractory to or cannot tolerate cholesterol lowering medication.
A
c. LDL-apheresis should be considered in children with homozygous or compound heterozygous FH bythe age of 5 years, particularly if the plasma cholesterol concentration remains at 9 mmol/L or above onmedication.
A
d. LDL-apheresis should be carried out in close collaboration with a centre experienced in apheresis,such as a transfusion medicine service.
A
e. The efficacy, tolerability and safety of LDL-apheresis must be reviewed after each treatment Af. The effect of LDL-apheresis on progression of atherosclerosis should be monitored withechocardiography (aortic valve and root), carotid ultrasonography and/or exercise stress testing.
B
ecommendation VIII Cascade screening: risk notification and genetic/phenotypic testing of families Grade
igs. 8 and 9 a. Notification of relatives at risk of FH should not be instituted without the consent of the index case,with the exception noted below in recommendation c).
A
b. If no consent is given by the index case, rapport should continue to be built and consideration given toreferral for counselling.
B
c. Relatives should only be directly notified of their risk without consent of the index case if there isspecific legislative provision for this breach of confidentiality in the relevant jurisdiction.
C
d. Commonwealth legislation, local state legislation, NHMRC guidelines and local health serviceprotocols about disclosure of medical information without consent should be consulted.
A
e. A proactive approach that respects the principles of privacy and autonomy is required. Af. All material sent to relatives and the telephone approach should be clear, comprehensible and not causealarm. General and specific modes of communication should be used.
A
g. Cascade screening should ideally be carried out as a formal collaborative process between lipiddisorders and clinical genetics services. It should also involve close communication and liaison withprimary care physicians and employ a user-friendly family based data management system.
A
h. Pre-testing counselling should be offered to at risk family members of an index case prior tophenotypic or genetic testing.
A
i. If no consent/assent for genetic testing is obtained phenotypic testing for FH should be offered. Aj. If genetic testing detects the family mutation, a definitive diagnosis of FH can be made in the tested A
individual particularly when the phenotype also suggests FH.k. If genetic testing does not detect the family mutation, the diagnosis of FH can be excluded, exceptwhen the clinical phenotype is highly suggestive of FH.A

sis Supplements 12 (2011) 221–263 227
(
R roach Grade
F d to all ‘index cases’ who have a phenotypic diagnosis of FHe)
A
s unlikely (e.g. by Dutch Lipid Clinic Network Score), geneticried out.
C
out in an accredited laboratory. Andex case’, genetic testing may be carried out initially using B
p or kit technology should be confirmed using a different A
s definite or probable (e.g. by Dutch Lipid Clinic Networkby methods that target specific mutations, comprehensive exon
A
boratory report should include an assessment of its significance,e variant is a pathogenic mutation, a previously reported variantof uncertain significance or a benign (normal) variant.
A
detect a mutation, the laboratory report should include a caveatundetected mutations or mutations in untested genes,ongly suggestive of FH
A
T
3
sc2lacrlmMsntdp
4
acfr‘bttiii
owc[Ldsi[bwacmktxan[ro[wnc(m[o
G.F. Watts et al. / Atherosclero
Continued)
ecommendation IX Genetic testing for FH: laboratory app
igs. 10 and 11 a. Genetic testing for FH should be offere(e.g. by Dutch Lipid Clinic Network Scorb. When the phenotypic diagnosis of FH itesting of the ‘index case’ need not be carc. Genetic testing for FH must be carriedd. When searching for a mutation in an ‘icommercial chip or kit technology.e. All abnormal genetic test results by chivalidated method.f. When the phenotypic diagnosis of FH iScore), but no genetic variant is detectedby exon sequencing is recommended.g. If genetic testing detects a variant the laand the report should clearly indicate if thof uncertain significance, a novel varianth. If the genetic testing protocol does notthat the result does not exclude FH due toparticularly if the clinical phenotype is str
he definitions of the grade for the recommendations are given in Table 1.
. Overview of algorithms for the model of care
Fig. 1 provides an overview for the MoC for FH. Theequence of presentation is as follows: detection of indexases and clinical diagnostic criteria (Fig. 2; Appendices–4); diagnosis and assessment of adults, children and ado-escents (Figs. 2–4); management of FH in adults, childrennd adolescents (Figs. 5 and 6); LDL-apheresis and radi-al therapy for FH (Fig. 7); cascade screening, includingisk notification and predictive testing (Fig. 8); clinical andaboratory protocols for genetic testing (Figs. 9–11); opti-
al components of a clinical service for FH (Fig. 12). TheoC has initially been devised for use within a specialised
etting, such as a lipid clinic, run out of departments of inter-al medicine, cardiology or endocrinology in secondary orertiary referral centres. The MoC will need to be furthereveloped to consider current and future potential roles forrimary care providers.
. Detection of index cases and diagnosis of FH
A key challenge facing the care of FH is the system-tic detection of index cases [37,38,40]. The term ‘indexase’ refers to the first individual diagnosed with FH in theamily. Identifying index cases is important because it rep-esents the starting point for family tracing, referred to ascascade screening’, by which the majority of FH cases cane efficiently detected [4,7–9,11]. There are several diagnos-ic tools for diagnosing FH clinically, including those from
he Dutch Lipid Clinic Network [34], Simon Broome Reg-stry [12,41] and the US MEDPED Program [42] (see tablesn Appendices 2–4, respectively). There are, however, nonternationally agreed criteria for the phenotypic diagnosismitd
f FH [8,37,43]. We favour the Dutch Lipid Clinic Net-ork Score (DLCNS) because we consider it simpler for
linical use and the numerically integrated scoring system34,37], which does not fully rely on the plasma level ofDL-cholesterol, can provide a more sensitive method foretecting index cases with FH [8,44]. The Simon Broomeystem is an alternative tool favoured in the UK [12] thats comparable to the DLCNS in predicting an FH mutation,8] but could overlook patients with true FH who may note overtly hypercholesterolaemic. The MEDPED System,hich is based solely on plasma total and LDL-cholesterol
nd has some practical appeal, is less sensitive than the otherriteria in predicting an FH mutation and accurate imple-entation requires that cholesterol measurements be widely
nown in family members [42,45]. Illustrative examples ofhe typical examination features (arcus cornealis, tendonanthomata) of FH are given in Appendix 5. Gender- andge-specific thresholds for plasma LDL-cholesterol for diag-osing first-degree relatives with FH were recently published46], but their value in clinical practice has not yet beeneported. The phenotypic diagnosis of FH should be basedn at least two fasting measures of plasma LDL-cholesterol14,47–49]. The value of taking a family history of CHD isell established [5,12,14–16,33,34,50], but its use is ofteneglected in clinical practice [22,51] and this needs rectifi-ation [52–54]. Secondary causes of hypercholesterolaemiae.g. primary hypothyroidism, proteinuria, cholestasis, andedications such as corticosteroids) must also be excluded
14,15], but it is important to note that FH may co-exist withther cardiovascular risk factors [55–57], most importantlyetabolic syndrome and diabetes [17a]. LDL-cholesterol
s underestimated by the Friedewald equation at plasmariglycerides >4.5 mmol/L [47], above which a well validated,irect assay should be employed to measure LDL-cholesterol

2 sis Sup
[tds
bCaaw[iDcicoLLrotfrIutdHo[
epva[aBtahbcaoocpcfpatot
ftLhgp
cdicpDmoplaotusf[tawcaaotcaa
5
iapsmcvwpaes
28 G.F. Watts et al. / Atherosclero
14,47]; measurement of apoB is an alternative [58,59], buthe diagnostic cut-off for diagnosing FH has not yet beenefined. Patients with FH do not, however, classically exhibitignificantly high plasma triglycerides concentrations.
Fig. 2 indicates that potential index cases of FH shoulde sought amongst patients aged less than 60 years withVD presenting to coronary care, stroke, cardiothoracicnd vascular units [43], as well as amongst similar patientsttending cardiac rehabilitation programs. The greatest yieldill be from screening younger adult patients with CHD
22,38,41,60]. Where the suspicion of FH is high, prelim-nary assessment of patients using either a full or modifiedLCNS should be employed [22,34]. A simplified modifi-
ation of the DLCNS, suitable for use by non-specialists,nvolves estimating a score for family and personal clini-al history and LDL-cholesterol alone [22,37]; for patientsn statins an upwards adjustment of approximately 30% inDL-cholesterol could be employed [61] if the pre-treatmentDL-cholesterol is unknown. The role of this simple tool
equires evaluation. A persuasive case for universal, aspposed to selective, screening of children for hypercholes-erolaemia has been made, since deficiency in obtaining aamily history of CHD and/or hypercholesterolaemia willesult in children not being tested and treated for FH [62].t has been proposed that this could be done at immunizationsing plasma cholesterol alone and subsequent child–parentesting where indicated [63] or by universally screening chil-ren aged 9–11 years with a standard lipid profile [64a].owever, the acceptability, specificity and cost-effectivenessf these universal screening strategies for FH are questionable4,65,66].
Opportunistic application of the DLCNS should also bemployed in specialist clinics and in primary care. Generalractitioners (GPs) are usually the first to encounter indi-iduals who may have unsuspected FH in the community,nd are therefore critically placed for detecting index cases12,67,68]. Planned health assessment in middle age, suchs the 45–49 year old health check assessment (Medicareenefits Schedule (MBS) Item A27) [69] would be an oppor-
une time for GPs to routinely test for FH, but its uptakend yield needs evaluation. Children with a positive familyistory of hypercholesterolaemia or premature CHD shoulde screened for FH [64a], but this is best done as part of ao-ordinated cascade testing process [9,12]. In primary careretrospective search of clinical databases affords another
pportunity for generating new cases of FH [67]. Flaggingf laboratory reports on patients with plasma cholesteroloncentrations > 7 mmol/L is another method for alertingractitioners about the possibility of FH, but its yield andost-effectiveness remain to be reported. There is also a roleor opportunistic application of the DLCNS by communityharmacists to patients attending to collect a prescription for
statin [70], with the possibility of on-site testing of rela-ives with a finger prick cholesterol measurement. Anotherpportunity for detecting FH is amongst patients referredo rheumatologists with tenosynovitis or to plastic surgeons
Pvci
plements 12 (2011) 221–263
or the excision of tendon masses. Certain ethnic groups inhe community, such as Christian Lebanese, Afrikaaners andithuanian Jews, in whom the prevalence of FH is particularlyigh due to a gene founder effect [1], should also be tar-eted for screening for the condition; culturally appropriaterocesses should be followed.
When the DLCNS is ≥3, we recommend referral to a spe-ialised FH service or lipid clinic for confirmation of theiagnosis and advice on management and cascade screen-ng. Health providers working in these clinics should haveompetence and training in both clinical lipidology andrevention of CVD [26,71,72]. FH is unlikely when theLCNS is <3 [8,44]. This needs to be appropriately com-unicated to the patient and their GP. For patients on statins,
r other cholesterol lowering medications, a contemporarylasma LDL-cholesterol concentration will give a falselyow DLCNS and pre-treatment values must be obtained toccurately assess the chance of having FH. Hence, we rec-mmend that index cases of FH should be identified in awo-stage process starting with initial phenotypic assessmentsing the DLCNS followed by referral to a specialist FHervice. Employing a DLCNS as low as 3 as a criterionor referring patients to a lipid clinic may be too sensitive8,38], in which case the score may be increased accordingo workloads and resources. If resources allow, an alternativepproach would be to genetically screen for FH in all patientsith premature CHD (e.g. age < 50 years) presenting to the
ardiac services referred to in Fig. 2. All patients identifieds having a likelihood of FH should be provided with simplend clearly written information explaining the importancef FH and the steps involved in diagnosing the condition inhem and their relatives [8,12,37]. The recommended proto-ols for risk notification and cascade screening of relativesnd for genetic testing are discussed later and given in thelgorithms in Figs. 8–10.
. Assessment of adult patients
Whether classified as having possible, probable or def-nite FH [34], all patients should have a detailed clinicalssessment to investigate other cardiovascular risk factors,resence of symptomatic or subclinical atherosclerosis andecondary causes of hypercholesterolaemia. Clinical assess-ent should ideally be undertaken by a specialist trained in
linical lipidology [26,71], with skills in preventative cardio-ascular medicine [72]. The risk of CVD amongst patientsith FH can vary widely [43,73]. This may relate to there-treatment plasma level of cholesterol, genetic causesffecting lipid metabolism or arterial biology, and the pres-nce of other major cardiovascular risk factors, in particularmoking, obesity, hypertension and diabetes [43,55–57,74].
athogenic genetic variants (mutations) that lead to very ele-ated plasma cholesterol and premature CHD may also beonsidered a major risk factor [66], as should an elevationn plasma lipoprotein(a) (Lp(a)) concentration > 0.5 g/L [75].
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 229
Potential sources for identifying index cases with FH:Coronary Care, Cardiac Rehabilitation, Cardiothoracic Surgery, Stroke Unit, Vascular Surgery, Ward nursing staff, Specialists, GPs, Laboratory report alerts, Pharmacists,
Clinical Genetics Services, Opportunistically in clinics/hospitalsHigh index of suspicion for new cases, Retrospective audit for previous cases, Use
simplified modification of phenotypic criteria for FH (Appendix 2 & 3)
FH unlikely: DLCNS <3FH likely: DLCNS >3
Review by specialist FH clinic
DLCNS
≥3 <3
Follow-up in clinicSee Figure 3
Inform patient and GP
Preliminary clinical assessmentAppendix 2: Dutch Lipid Clinic Network Score (DLCNS)
ex case
B(iatfCfatatpcmct[icr
a
lfnarh(aor[(pzpeCepa
Fig. 2. Detection of ind
ox 1, linked to Fig. 3, gives mandatory, and recommendedbut optional) clinical indices that may be useful in assess-ng patients with FH. Non-invasive tests for atherosclerosisnd CHD should be considered optional and individualisedo specific clinical situations [76–78], being particularly use-ul in FH when the family history of CVD is unclear [79].arotid ultrasonography (CUS) can be a particularly use-
ul test that recognizes that in FH atherosclerosis is alsoccelerated in extra-cranial cerebral arteries and its detec-ion serves as a surrogate for the involvement of coronaryrteries [80–82]. Clinical assessment must take account ofhe psychological, intellectual, social and ethnic status of theatient [8,83–86], and potential need for special methods ofounselling. Inadequate health literacy is not uncommon andust be addressed [87,88]. Detailed exploration and clear
ommunication and discussion of the individual’s family his-ory of CVD are essential for effective management of FH89,90]. After clinical assessment, patients should be dividednto those at lowest, intermediate and highest risk of CVD;ategories should be modified according to regular clinical
eview (see Fig. 5).This stratification of patients is consistent with otherpproaches to cardiovascular risk assessment [14–17a]. At
bii
s and diagnosis of FH.
owest risk are patients with no other cardiovascular riskactors (smoking, obesity, diabetes, and hypertension) andegative tests for subclinical atherosclerosis. At intermedi-te risk are patients with at least one other cardiovascularisk factor or subclinical evidence of early atherosclerosis. Atighest risk are patients with a history of symptomatic CVDcoronary, cerebral or peripheral vascular disease) and/orrevascularization procedure, or with subclinical evidence
f more advanced atherosclerosis. Subclinical atheroscle-osis may be defined according to published guidelines77,78] – no evidence: a carotid intima-medial thicknessCIMT) < 75th percentile (for age and sex) with no carotidlaques or a Coronary Artery Calcium Score (CACS) ofero; early evidence: a CIMT > 75th percentile with nolaques (or stenosis) or a CACS ≥ 1 but <100; more advancedvidence: presence of carotid plaques (or stenosis) or aACS > 100. If electing to do ankle brachial index (ABI),xercise electrocardiography (ECG) testing and/or com-uterised tomography coronary angiography (CTCA) insymptomatic patients on clinical grounds, abnormal results
y recognised criteria [13,77,78,91] may also be employedn allocating patients to the higher risk categories shownn Fig. 3.
230 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Box 1: Information used for clinical assessment of adults with FH
• Mandatory: Age, gender, history: CVD (coronary, cerebral and peripheral arterial disease)/revascularisation, history of major cardiovascular risk factors, psychological and socioeconomic status, family history of hypercholesterolaemia and CVD, BMI, waist circumference, blood pressure, bruits, arcus cornealis, xanthelasma, tendon xanthomata, triglycerides, total cholesterol, HDL-C, Lp(a), apoB, smoking status, menopausal status, reproductive status, drug history
• Recommended/optional: glucose, insulin, CRP, creatinine, TSH, albuminuria, ApoA-I, ABI, EST, carotid and Achilles tendon ultrasound, CTCA/calcium score
• Major CVD Risk factors include very elevated LDL-C (>7 mmol/l), low HDL-C, diabetes, smoking, obesity, hypertension, high Lp(a), family history of very premature CHD.
FH diagnosed
Intensivemanagement
Intermediate risk>1 other major CVD risk factors, early subclinical
CVD
Highest riskSymptomatic CVD,
revascularization, advanced subclinical CVD
Lowest riskNo other major CVD risk
factors, no symptomatic or subclinical CVD
Enhanced management
Standard management
Clinical assessment
ent of
aawtssptmtrapacrcp2Ps[u
6a
c
aclcFa[samp[act
tatacr≥fyfh
Fig. 3. Assessm
This subdivision into lowest, intermediate and highest riskllows management to be classified as standard, enhancednd intensive, respectively. This risk stratification procedureill allow the best use of clinical resources, which can in
urn increase cost-effectiveness. Framingham risk scores, orcores derived from other cardiovascular risk engines, are notufficiently reliable to guide management in FH [14,15,38],articularly in younger patients, in whom a measure of long-erm risk based on imaging of subclinical atherosclerosis
ay be more appropriate [92]. Another option would beo offer intensive management to intermediate and highestisk cases of probable/definite FH. Similarly, enhanced man-gement could be considered for the lowest risk cases withrobable/definite FH and for the possible cases of FH whore at intermediate or highest risk of CVD. The yield andost-effectiveness of all these different therapeutic strategiesequires further evaluation. The importance of assessing otherardiovascular risk factors beyond hypercholesterolaemia isarticularly underscored by the rising tide of obesity, typediabetes and hypertension in our community [17a,93,94].
atients considered to have a homozygous FH phenotypehould evidently be classified at exceptionally high risk43,95] and be referred for consideration for radical therapysing LDL-apheresis (see Fig. 7).
. Diagnosis and assessment of children and
dolescentsOne of the most potentially contentious issues in theare of FH is the identification of the condition in children
[to
adult patients.
nd adolescents [48,96–100]. Efforts are well justified bylear indications that FH remains underdiagnosed particu-arly in the young [18,21,96] and that untreated patients sufferardiovascular damage from an early age [81,96,101,102].urthermore, there are reassuring data concerning the safetynd efficacy of dietary and pharmacological treatments99,103–108], including confirmation that children receivingtatins achieve normal growth and developmental milestonesnd do not exhibit alterations in plasma adrenocortical hor-one levels. While there have been concerns about the
sychological impact of ‘disease labelling’ from an early age8,84,85], formal studies provide reassurance that this is notsignificant issue [7,86,109]. Despite advances in the overallare of patients with FH, medical services for children withhe condition remain particularly underdeveloped [110].
Fig. 4 summarizes the recommended approaches tohe diagnosis, assessment (Box 2), and risk stratificationllocation of FH in children and adolescents. We recommendhat FH screening be offered to all children (aged ≥ 5 years)nd adolescents at risk of having FH, particularly in theontext of a co-ordinated cascade testing process. Otherecommendations suggest screening at risk children earlier at2 years of age [64a,b] and universally screening all children
or dyslipidaemias aged 9–11 years [64a]. Under the age of 5ears screening may be justified where there is a strong desirerom the parents or severe FH (homozygous or compoundeterozygous) is suspected, consistent with other guidelines
12,98]. Because the DLCNS is not applicable to children,he diagnosis of FH must rely either on serial measurementsf a fasting plasma level of LDL-cholesterol [46,48,97], or
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 231
Box 2: Information used for clinical assessment of children and adolescents with FH
Mandatory: Age, gender, history including cardiovascular risk factors, psychological status, family history of hypercholesterolaemia and CVD, BMI, waist circumference, blood pressure, bruits, arcus cornealis, xanthelasma, tendon xanthomata, triglycerides, total cholesterol, LDL-C, HDL-C, Lp(a), apoB, smoking status, reproductive status, drug history
, CUS for
LDL-C <5.0 mmol/L
Genetic + LDL-C testing of child
Pathogenic mutation detected
No
LDL-C < 75 percentileBoys <3.0 mmol/LGirls <3.3 mmol/L
No pathogenic mutation detected
LDL-C >75 percentileBoys >3.0 mmol/LGirls >3.3 mmol/L
LDL-C >5.0 mmol/L
Consent / assent to genetic testing
Pathogenic gene variant (mutation) identified in parent(s)
Clinical assessment
Boys <10yr, Girls before menarche
Boys >10yr, Girls after menarche
1 or more of the following:Early (M <50yr, F <40yr) CVD in family,
>2 CVD risk factors, diabetes, LDL-C >6.0 mmol/L
Lowest risk FHExpectant
management
Intermediate risk FHEnhanced
management
Highest risk FHIntensive management
Reassure patient and family and inform GP
Parent(s) diagnosed with FH
LDL-C testing of child
ildren a
pfcIga[wa[dsrBmiacwmmlmhoemB
apapab(CcmsFapwFpfcFcarts
Recommended/optional: glucose, insulin, CRP, creatinine, TSH, albuminuria, ApoA-I
Fig. 4. Diagnosis and assessment of ch
referably on genetic testing where a recognised mutationor FH has been detected in a parent [7,8,66]. Secondaryauses of hypercholesterolaemia must be excluded [64a,b].rrespective of whether the diagnosis of FH will be madeenetically, plasma LDL-cholesterol must be measured inll patients since the result is essential to guide therapy96–98]. Predictive genetic testing of children for FH is alsoell justified as preventive treatment can be instituted before
dulthood with lifestyle measures and pharmacotherapy96–98,103,108] (see Fig. 6). Genetic counselling includingiscussion of the implications of DNA testing in childrenhould be provided at the time the parent receives the geneticesults confirming the diagnosis of FH [8,85,111–115].ecause of ethical issues involved in genetically testinginors [114,115], it is usual and best practice to first genet-
cally test a phenotypically affected parent [64b]. This canlso circumvent issues related to a non-paternity event. In rareircumstances, such as refusal of a parent to be tested first orhen autosomal recessive FH is suspected [3], genetic testingay first be carried out in the child. Another special situationay arise when the child has significant hypercholestero-
aemia without the detection of the family’s pathogenicutation. In this case, after exclusion of secondary causes of
ypercholesterolaemia and appropriate genetic counselling,ther causative mutations for FH should be sought from the
xtended pedigree. Detecting an FH causing mutation wouldake the child or adolescent eligible for Pharmaceuticalenefits Scheme (PBS) government subsidy for a statin atwbw
early atherosclerosis, FMD of brachial artery for endothelial function.
nd adolescents. *See text for caveats.
n LDL-cholesterol > 4.0 mmol/L when the family history ofremature CVD or tendon xanthomata is unclear or unobtain-ble [116]. When an FH causing mutation is unknown in thearent, the diagnosis of FH in children should be based on agend sex adjusted LDL-cholesterol levels, the 95th percentileeing 3.5 mmol/L for boys and 3.8 mmol/L for girls [37,48]see Fig. 4). If employing the modified Simon Broomeriteria, the universal cut-off for probable FH is LDL-holesterol > 4.0 mmol/L [12,41]. At least two consecutiveeasurements of LDL-cholesterol over 6 months in fasting
amples should be used to make the phenotypic diagnosis ofH [49,98]; a non-fasting lipid profile may be employed asn initial screening test, however. Knowledge of the child’slasma LDL-cholesterol and whether a parent is being treatedith a statin may provide a simple clinical tool for diagnosingH [117]. Most children with plasma LDL-cholesterol >95thercentile for age and sex and an autosomal dominant patternor inherited hypercholesterolaemia, in whom secondaryauses of dyslipidaemia have been excluded, will have anH causing mutation [118]. The thresholds for plasma LDL-holesterol concentration recommended above for makingphenotypic diagnosis of FH are compatible with those
ecently reported to accurately predict an FH causing muta-ion [46]. Whether using phenotypic or genetic approaches,creening children for FH requires expertise in working
ith and giving advice to families [8,84,98,119,120] and isest undertaken in close liaison with paediatric services andhere indicated with genetic counsellors [115,121].
2 sis Sup
sofmtcmaiolcracsertLhtbtfaasasdccFrac
7
cppmaotc
7
c
(tmaCi<tutc
siamttLabtl
7
o[bdrvpaodewetq[tta[2m[
7
32 G.F. Watts et al. / Atherosclero
All individuals with at least a possible diagnosis of FHhould be clinically assessed according to mandatory and rec-mmended (but optional) requirements shown in Fig. 4. Withew exceptions, these are generally similar to those recom-ended previously for adults. Of the available non-invasive
ests for subclinical atherosclerosis in children and adoles-ents, measurement of CIMT with ultrasonography is theost promising at present, but it requires special expertise
nd if used in risk stratification should be carried out accord-ng to recommended protocols [82,122]. With the exceptionf homozygous or compound heterozygous FH, boys agedess than 10 years and girls who have not reached the menar-he should generally be considered to have low risk FH andeceive expectant treatment. Boys over the age of 10 yearsnd girls who have reached the menarche without cardiovas-ular risk factors or objective evidence of increased CIMThould be considered to have moderate risk FH and receivenhanced treatment. Boys over 10 years and girls who haveeached the menarche, with a family history of very prema-ure CVD, two or more major cardiovascular risk factors, orDL-cholesterol > 6 mmol/L, should be considered to haveigh risk FH and receive intensive treatment [12,96–98]. Cer-ain high risk children and adolescents with FH should ideallye managed in a joint adult-paediatric FH clinic [123]. Fac-ors that may be used to triage patients for this clinic includeamily dynamics, current lifestyle of the family and child,dherence to treatment, severity of family history of CHD,nd presence of other major cardiovascular risk factors (obe-ity, hypertension, diabetes and smoking) [123,124]. Whilell children with FH should ideally be referred to a paediatricervice, we consider it feasible for affected parents and chil-ren to be reviewed together by an adult service in a ‘familylinic’ [123], provided staff have the required competen-ies and the environment of the clinic is appropriate. YoungH patients being reviewed in a paediatric clinic should beeferred to an adult clinic around the age of 16 years, withppropriate arrangements made for transitional care and withlose involvement of the GP.
. Management of adults
Fig. 5 shows the protocols for adult patients with FH allo-ated to standard, enhanced and intensive management. Inarallel with lowering elevated plasma cholesterol, appro-riate lifestyle modifications should be emphasised and allajor non-lipid cardiovascular risk factors must be treated
ccording to expert guidelines [14,16,17a,32,33,125–129];ffering advice and support on smoking cessation is manda-ory. Low-dose aspirin should be used in highest risk FH andonsidered in intermediate risk FH [128].
.1. LDL-cholesterol and apoB targets
The recommended therapeutic targets for absolute plasmaoncentrations of LDL-cholesterol and apolipoprotein B
sp
plements 12 (2011) 221–263
apoB) are given in Fig. 5. These targets have been choseno be compatible with other therapeutic guidelines for theanagement of hypercholesterolaemia [13–16,32,59]; ther-
peutic targets should evidently be lower with increasingVD risk. Measuring apoB may not, however, be necessary
n leaner FH patients with plasma triglyceride concentrations2.0 mmol/L. ApoB accurately reflects the plasma concen-
ration of atherogenic LDL particles. ApoB is particularlyseful, and preferable to LDL-cholesterol, when LDL par-icle number increases and both size and density fall as aonsequence of hypertriglyceridaemia [58,59].
Hypertriglyceridaemia is a feature of obesity, metabolicyndrome and type 2 diabetes [17a], all of which are increas-ng in prevalence in the background population and hencemongst patients with FH [93,94]. Use of an apoB targetay be restricted to FH patients who exhibit an elevated
riglyceride level > 2.0 mmol/L [17a,58]. Even with con-emporary treatments, achieving the absolute targets forDL-cholesterol and apoB shown in Fig. 5 may not bettainable by some patients, particularly those with a higheraseline plasma cholesterol [61], in which case a more realis-ic general target of a 40–50% reduction from pre-treatmentevels could be used [12,128].
.2. Diet and lifestyle modifications
Diet and lifestyle modifications are cornerstonesf the management of all types of dyslipidaemias14–17a,32,33,59,130], including FH [13,131]. Diets shoulde low in saturated fat and energy and adjusted to achieveesirable body weight [15,132]. Dietary counselling by aegistered dietician is recommended for all affected indi-iduals and families [133]. Dietary supplementation withlant sterols or stanols should be considered [134]. Moder-te intensity aerobic exercise for at least 30 min on 5 daysf the week should be considered to prevent obesity andiabetes, with advice appropriately adjusted in those withstablished CHD [32]. Several strategies may be employed,here indicated and feasible, for improving long-term adher-
nce to dietary and life-style changes, including involvinghe patient, setting goals, encouraging self-monitoring, fre-uent and prolonged contact, and motivational interviewing135]. However, almost all patients will require medicationo lower the elevation in LDL-cholesterol. Offering effectivereatments and advice on smoking cessation is mandatorynd appropriate management guidelines should be followed32]. Alcohol consumption should be limited to no more than
standard drinks per day. Stress, anxiety and depressionust be considered in all patients and managed accordingly
32,136].
.3. Pharmacotherapy
In Australia, the PBS eligibility criteria for governmentubsidy for lipid modifying agents cover almost all adultatients with FH, particularly if the molecular diagnosis of

G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 233
Enhanced
Diet + Plant Sterols + Statin
± Ezetimibe± Niacin
Diet + Plant Sterols + Statin
± Ezetimibe± Niacin ± Resin
± Fenofibrate
Adherence, tolerability +
safety checks
Adherence, tolerability +
safety checks
Consider interval non-invasive testing for
coronary artery disease / atherosclerosis
Positive result or progression
of disease
Consider interval test of cardiac function:
exercise ECG / Stress echocardiogram /
myocardial perfusion scan
Clinically significant
positive result
IntensiveStandard
Diet + Plant Sterols + Statin
± Ezetimibe
Adherence, tolerability +
safety checks
Consider interval non-invasive testing for
coronary artery disease / atherosclerosis
Positive result
Provide and reinforce information on:Healthy diet, tobacco avoidance, exercise, family and psychological support and medication adherence.
Treat non-lipid risk factors:Diabetes, hypertension, obesity, smoking, consider aspirin
Negative result Negative or stable result
Negative or stable result
Review interval: 12 months Review interval: 3-6 months Review interval: as per clinical context
F lowingr
FZditdttfsaNsrapttsridbh
ndgpt[haaambtapisHai
ig. 5. Management of Adults (see text for more information on action foladical therapy).
H has been confirmed with a DNA test [116]. In Newealand government subsidies are similar, but the spectrum ofrugs is slightly more restricted [137]. HMG CoA reductasenhibitors (or statins) are by far the most common and effec-ive drugs to treat FH [104,107,128,129,138–146]. Statinsecrease the incidence of CHD [143–145] and are estimatedo be cost-effective in treating FH [147,148]. Reduction inhe costs of statins will, however, make the medical careor FH even more cost-effective in the future. All the majortatins are government subsidised in Australia, but onlytorvastatin, simvastatin and pravastatin are subsidised inew Zealand. After initiation of statin therapy, all patients
hould be reviewed at 6–8 weeks to monitor LDL-cholesterolesponse, adherence, safety parameters and tolerability [61],nd 6–12 monthly thereafter if targets are achieved and noroblems documented. The statin should be up-titrated tohe maximally recommended tolerable dose that achieveshe therapeutic targets shown in Fig. 5; patients may requirewitching to more potent statins [61], such as atorvastatin orosuvastatin. Higher risk patients who require greater lower-
ng of plasma LDL-cholesterol and apoB will require otherrugs, especially ezetimibe [149,150], but also niacin, fenofi-rate and bile acid binding resins [151–154]. Patients withigher plasma LDL-cholesterol levels will require a combi-3iei
initiation of therapy, treatment of special groups, shared care with GP and
ation of drugs to achieve therapeutic targets. Combinationrug regimens that target LDL-cholesterol can decrease pro-ression of CHD in patients with FH [155]. Niacin may bearticularly indicated for lowering high plasma Lp(a) concen-ration [75,151,152]; in FH we recommend an Lp(a) < 0.5 g/L75]. Fibrates would be relatively contraindicated with aistory of untreated choleliathisis [156]. For patients notchieving treatment targets, additional agents should gener-lly only be introduced after at least 12 months of testingnd adjusting the statin regimen and confirming adherence toedication. In higher risk patients, additional agents should
e considered after an earlier interval (e.g. 4–6 months) ofesting the statin regimen. Residual hypertriglyceridaemiand low HDL-cholesterol (the so-called ‘atherogenic lipidrofile’) while on a statin is an indication for consider-ng treatment with niacin, a fibrate or higher doses ofupplemental omega-3 fatty acid ethyl esters [152,157].ypertriglyceridaemia in FH patients should be managed
ccording to recently published guidelines [158,159]. Theres outcome evidence supporting use of lower doses of omega-
fatty acids in patients who have sustained a myocardialnfarction [160]. Niacin, fibrates and omega-3 fatty acidthyl esters, together with a very low fat diet, will also bendicated in rare instances of severe hypertriglyceridaemia

2 sis Sup
trcfcr
7
kt[aalquaCrispaiscaseftsnetttss
7
uad[bfiitsabc
ssn[rc
sciswtflshaogmiFtaathsi[
7
rarctirlctrsi
7
e
34 G.F. Watts et al. / Atherosclero
o prevent acute pancreatitis [14,157,161]. There has beenenewed interest from Japan in the therapeutic role of probu-ol [162], a drug formerly used to treat FH and withdrawnrom the European and US markets because of safety con-erns; use of the present formulation of this drug cannot beecommended.
.3.1. Safety monitoringPlasma hepatic aminotransferases (ALT, AST), creatine
inase (CK) and creatinine levels should be measured rou-inely as baseline safety checks prior to starting medications61,163]. Hepatic aminotransferases should be monitoredccording to the approved product information for the drugs,nd checked at least every 3 months if there is a history ofiver disease (e.g. chronic hepatitis or cirrhosis) or more fre-uently if plasma levels rise to three-fold greater than thepper reference limit; measurement of serum bilirubin maylso be used to indicate the severity of liver toxicity. PlasmaK should be measured when musculoskeletal symptoms are
eported. Particular vigilance is required in patients receiv-ng higher doses of a statin, and patients predisposed to statinide-effects, specifically the elderly and those taking multi-le medications including the combination of a statin withfibrate [163,164]. Patients on niacin also require monitor-
ng of plasma glucose and uric acid because of a small, butignificantly increased risk of hyperglycaemia and hyperuri-aemia [165]. Plasma ALT and AST should be measuredbout every 6 months in patients receiving statin–niacin andtatin–fibrate combinations. Evidence of chronic kidney dis-ase, as estimated by an elevation in plasma creatinine andall in estimated glomerular filtration rate, would be a precau-ionary indication to initiate treatment with, or switch to, atatin that is not eliminated by the kidney [61]. Statins shouldot be initiated if the baseline plasma, ALT, AST or CK lev-ls are >3 times the upper reference limit. Discontinuation ofhe statin or revision of the dose or regimen is required whenhe plasma aminotransferase or CK levels rise to >3 timeshe upper reference limit on treatment. Alternative agentsuch as a ezetimibe or bile acid sequestrants may need to beubstituted for a statin.
.3.2. Medication adherence and tolerancePharmacists could play a key role by monitoring patients’
se of therapy, flagging non-adherent patients to GPsnd clinics, reducing therapeutic complexity and by moreirect involvement in improving adherence to medication166–169]. FH patients who are non-adherent to therapy areest reviewed in a dedicated clinic that could involve inputrom pharmacy, clinical pharmacology, psychology and nurs-ng [170]. Action plan interventions may be more effectiven FH than interventions aimed at altering perceptions aboutaking statins [171]. Detailed communication and discus-
ion of an individual’s family history of CVD may improvedherence to treatment [89,90]. Health literacy must alsoe considered [172,173]. The underlying causes of hyper-holesterolaemia and adherence to statin therapy remains apidm
plements 12 (2011) 221–263
ignificant issue amongst many at risk patients who have notpecifically been diagnosed with FH [174,175]. This alsoeeds addressing at a primary care level in the community176–178]. Poor control of hypercholesterolaemia and otherisk factors for atherosclerosis is a particular on-going con-ern in all patients with CHD [179,180].
Patients who are intolerant of medications require specialupport and follow-up [170]. Musculoskeletal side-effectsan be frequently reported with statins and require special-st care [170,181]. Potential drug interactions with statinshould be closely monitored, [163] noting the increased riskith drugs that are metabolised by CYP3A4 with simvas-
atin and atorvastatin and by CYP2C9 with rosuvastatin anduvastatin [61,163]. Ezetimibe is well tolerated and has atatin dose sparing effect [149,150]. Bile acid binding resinsave frequent gastrointestinal side-effects (e.g. constipationnd abdominal discomfort) and can affect the absorption ofther drugs and fat soluble vitamins [153]; tolerability isreatest with colesevelam [182]. Resins should be taken witheals and gastrointestinal side-effects minimised by increas-
ng fluid and fibre intake and use of stool softeners [153].lushing is a problem with niacin, but this is diminished with
he newer formulations (Niacin-ER, Tredaptive) and with co-dministration of aspirin [151,152]; hyperglycaemia can beparticular problem in FH patients with impaired glucose
olerance or diabetes, and hyperuricaemia in those with aistory of gout [165]. Co-administration of fibrates and atatin can increase the risk of myopathy, but this is dimin-shed significantly when a statin is combined with fenofibrate156,163].
.4. Review intervals and shared care
Uncomplicated patients on routine therapy may beeferred back to the GP for long-term follow-up, but shouldlso be reviewed annually in a lipid disorders clinic. Thoseeceiving enhanced care should also be monitored in the lipidlinic at 3–6 monthly intervals until plasma LDL-cholesterolargets are achieved and if stable should be reviewed annuallyn this clinic with 6 monthly follow-up by the GP. Patientseceiving intensive management should be retained in theipid disorders clinic and reviewed at intervals determined bylinical context and requirements. When changing medica-ion and increasing doses, patients may need more frequenteview. Systems for the shared care of FH patients betweenpecialties and primary care need to be further investigatedn order to appropriately inform future MoCs for FH [68].
.5. Assessing atherosclerosis and CHD
CTCA, CUS, ankle-brachial index measurement, stresschocardiography, treadmill ECG and myocardial nuclear
erfusion scanning are all potential options for monitor-ng subclinical atherosclerosis and/or symptomatic coronaryisease [76–78] (see Fig. 5). Consistent with expert recom-endations [76–78,91], we consider that there is potential
sis Sup
vatdossaasratmsodwodacFttscpsc
7
iwtrCtebtbwiasawrstgde
ibtLtAtFbfwpop
fhoaipa[ewstu
8
a
8
biom[rFNhvppii
G.F. Watts et al. / Atherosclero
alue in non-invasive testing in asymptomatic FH patients fortherosclerosis and coronary disease. However, non-invasiveesting has been better validated for assessing target organamage and stratifying risk than for monitoring progressionf disease [13,79]. Nevertheless, clear evidence of progres-ion of atherosclerosis in coronary, carotid or femoral arterieshould be a clinical indication for intensifying treatment andttaining the recommended targets for LDL-cholesterol andpoB [13,77,183]. The intervals for repeat testing shownhould be determined by clinical judgment and availableesources. With CUS, the choice of equipment, technicalspects and operator training must be followed accordingo expert recommendations [78,82,122]; imaging protocols
ust be comprehensive and standardised, and age- and sex-pecific reference values for CIMT and a detailed assessmentf plaques should be employed. CUS is better suited for chil-ren and younger patients owing to absence of radiation risk,hile CTCA may be useful to assess plaque burden andbstructive stenosis in asymptomatic adult patients. Futureevelopments and refinements in cardiac CT imaging mayllow this imaging modality to be fully incorporated intolinical algorithms for assessing atherosclerosis and CHD inH. All asymptomatic patients should proceed to a functional
est, ideally a stress echocardiogram. All ‘positive’ treadmillests, myocardial perfusion scans and stress echocardiogramshould be followed by referral to a cardiologist for theonsideration of invasive coronary angiography and appro-riate further care [13,78,110]. Patients with more than 70%tenosis of the internal carotid artery should be referred foronsideration for revascularization [184,185].
.6. FH in women
Special considerations apply to the management of FHn women [43,186]. CHD risk is lower in women than menith FH [43,73,187]. Women may therefore be less likely
o be treated with a statin at a younger age, but this needsevision according to a detailed family history of prematureVD, the presence of other cardiovascular risk factors and
he rate of progression of CIMT [43,55–57,74,75,188]. Lowstrogen-containing oral agents, intra-uterine devices andarrier methods are the preferred approaches to contracep-ion in women with FH [186]. The latter two options shoulde particularly recommended to women older than 35 yearsho are of childbearing potential. Pre-pregnancy counselling
s recommended for all women [66,129,186,187]. Statinsnd other systemically absorbed lipid regulating medicationshould be discontinued 3 months prior to planned conceptionnd during pregnancy and lactation [186]. However, womenho fall pregnant accidentally while taking a statin could be
e-assured that the likelihood of any foetal complications ismall [189,190]. The risks for future pregnancy and the foe-
us should be discussed at least annually with all women andirls of childbearing age. Controlling hypercholesterolaemiauring pregnancy is particularly important in women withstablished CHD, and it may also decrease the severity of FHcm
d
plements 12 (2011) 221–263 235
n offspring who inherit the condition [189,191]. Bile acidinding resins are the only safe agents to control hypercholes-erolaemia in pregnancy, but only modestly lower plasmaDL-cholesterol levels and poor tolerability related to gas-
rointestinal side-effects remains a major problem [129,153].newer formulation, colesevelam, is more tolerable than
he older resins [182]. Pregnant women with heterozygousH and established CHD, or with homozygous FH, shoulde considered for LDL-apheresis [95,129]. During breasteeding, resins could be employed to lower LDL-cholesterolhere indicated. More data are required on the outcomes ofregnancy in women with FH and on the effect of statinsn the foetus in the first trimester; an appropriate registry ofatients is recommended.
Particular considerations are also required concerninguture pregnancy when one or both members of a coupleave FH. In these circumstances, there are a number ofptions available which enable the couple to avoid havingchild affected by FH. These options include not conceiv-
ng, adoption (local and overseas), using donor gametes,renatal diagnosis (PND) using chorionic villus sampling ormniocentesis and pre-implantation genetic diagnosis (PGD)192,193]. There are a number of issues associated whichach of these choices and couples may find it useful to meetith a counsellor who has expertise in reproductive coun-
elling. Couples considering PND or PGD should be referredo a clinical genetics service for counselling and pre-test workp, ideally prior to conceiving.
. Management of children and adolescents
Fig. 6 summarizes the management of FH in children anddolescents.
.1. General and lifestyle considerations
All patients, and ideally all immediate family mem-ers, should receive expert advice on lifestyle modificationsncluding healthy eating, regular exercise and avoidancef cigarette smoking [12,97,103]. Prevention of obesity,etabolic syndrome and diabetes is paramount in FH
97]. Psychological counselling and social support may beequired in special circumstances in certain families withH [8,84,124]. Parents of affected children must not smoke.on-cholesterol cardiovascular risk factors such as diabetes,ypertension and obesity should be treated according to rele-ant expert guidelines [97]. There is clear value in reviewingatients in a paediatric clinic, but we recommend that theaediatrician has some expertise in clinical lipidology andn the prevention of CVD [72]. A good, practical alternatives a family based clinic in which affected children, adoles-
ents and their parents can be reviewed collectively by aultidisciplinary team [123].Lowest risk FH should be treated with a fat modifiediet and plant sterol supplementation [97,103,134,194],

236 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Provide and reinforce information on:Healthy diet, tobacco avoidance, exercise, family and psychological support and medication adherence.
Treat non-lipid risk factors:Diabetes, hypertension, obesity, smoking
Expectant management Enhanced management Intensive management
Target LDL-C <4.0 mmol/L
Target LDL-C <3.0 mmol/L
TreatmentDiet + plant sterols
TreatmentDiet + plant sterols + statin ± ezetimibe ± bile-acid sequestrant
Statins contraindicated in pregnancy: contraceptive advice in adolescents
Monitoring: 1 – 2 yearlyWeight, growth,
development/well-being, compliance (lifestyle)
MonitoringWeight, growth, well-being, physical & sexual development,
hepatic aminotransferases, creatine kinase (if musculoskeletal symptoms present), adherence (lifestyle, pharmacotherapy)
No
Restratifyas per
assessment protocol
Consider carotid ultrasound 2 – 5 yearly
Consider treating as high risk adult FH
Treat as high risk childhood FH
Consider carotid ultrasound 1 – 3 yearly
Boys <10 yr, Girls before menarche
Boys >10 yr, Girls after menarche
Progression of carotid atherosclerosis (increase in CIMT and/or detection of plaques)
of child
wdecesrbcapahLpmp[p
cbFsabmpoc
8
c
Fig. 6. Management
ith annual or bi-annual monitoring of weight, growth andevelopmental milestones. Increased intake of fruit and veg-tables may compensate for reduction in plasma carotenoidoncentrations in children consuming a plant sterol-esternriched diet [195]. The value and safety of prescribingtatins for children and adolescents with FH is universallyecognised [64a,96–98,105,106,108]. As a general guide,oys older than 10 years and girls who have reached menar-he should be considered for statin therapy. The specific aget which to introduce a statin is not evidence-based and inractice should be determined by good clinical judgmentnd assessment of several factors. These include the familyistory of premature CVD, the prevailing plasma level ofDL-cholesterol, the type of FH mutation identified and theresence of other cardiovascular risk factors (e.g. diabetesellitus) [1,12,43,55–57,66,73–75,188], as well as the
arents’ perceptions and concerns on use of medication123,196]. The LDL-cholesterol treatment targets in youngatients with heterozygous FH will also depend on similar
<ogc
ren and adolescents.
onsiderations (Fig. 6). The health literacy of parents muste addressed when managing children and adolescents withH [88,172,173]. Homozygous patients with FH need to betarted on pharmacotherapy as soon as practicable after birthnd definitely by the age of 10 years [95]. In Australia, toe eligible for PBS subsidy any FH patient aged <18 yearsust have a plasma LDL-cholesterol >4.0 mmol/L with a
ositive family history of premature, symptomatic CVDr tendon xanthomata, or known to be positive for an FHausing mutation [116].
.2. LDL-cholesterol targets
The recommended targets for plasma LDL-cholesteroloncentration for enhanced and intensive management are
4 mmol/L and <3 mmol/L, respectively, consistent withther lipid treatment guidelines [14,15,97,98]. An alternativelobal therapeutic target is a 40% reduction in LDL-holesterol [12,37,64b]. For highest risk FH with diabetes,
sis Sup
o([t
8
g[aowiswatrlsfihtaamtatmrss[havetiBim(Aawtatpmcs[
8
tpsdiCFaaoiroCsagaa[naaWbohwptofahpacp
8
lHit[st
G.F. Watts et al. / Atherosclero
besity or metabolic syndrome and elevated triglycerides>2.0 mmol/L), an apoB target of <1.1 g/L may be used17a,58], noting that there are no published guidelines onhe use of apoB or non-HDL cholesterol in children [97,98].
.3. Pharmacotherapy
The therapeutic targets of LDL-cholesterol in this ageroup may be attained with a fat modified diet and a statin97,98], but the addition of ezetimibe [98,197,198] or a bile-cid sequestrant [98,153,199] may be required. In Australianly fluvastatin, pravastatin and simvastatin are registeredith the Therapeutic Goods Administration (TGA) for use
n adolescents and children older than 10 years [200]; thesetatins should be initiated at the lowest recommended dose,hich should be up-titrated according to cholesterol response
nd tolerability. Simvastatin and pravastatin have been showno improve arterial function and carotid atherosclerosis,espectively, in children with FH [104,201]. The efficacy inowering cholesterol and the short-term safety of more potenttatins, such as atorvastatin and rosuvastatin, has been con-rmed in children with FH [202,203]. Both of these statinsave been approved by the US Food and Drug Administra-ion (FDA) for use in children with FH aged 10 years andbove. Ezetimibe is TGA registered for use in adolescentsnd children older than 10 years [200], with no require-ent for dose adjustment. Bile-acid sequestrants can affect
he absorption of folate and fat soluble vitamins [153], andppropriate supplementation will be required with longererm use of these agents in children. Colesevelam is the
ost tolerable form of these agents [182,199], but is not yetegistered for use in Australasia or New Zealand. Bile-acidequestrants should be taken with meals and gastrointestinalide-effects minimised by increasing fluid and fibre intake153]. Niacin may be indicated for selected patients withomozygous or compound heterozygous FH [98], as wells for heterozygous FH patients with very high Lp(a) and aery early family history of CHD [75]. Niacin should oth-rwise rarely be used to treat paediatric FH owing to poorolerability and concerns about increased risk of glucosentolerance, myopathy, hyperuricaemia and hepatitis [98].y contrast, fibrates are relatively safe and well tolerated
n children, but are rarely indicated except in the treat-ent of FH patients with significant hypertriglyceridaemia
>4 mmol/L) or with a homozygous phenotype [14,97,98].s with fibrates, higher doses of supplemental omega-3 fatty
cid ethyl ester may also be very rarely indicated for patientsith severe hypertriglyceridaemia to prevent acute pancreati-
is [14,161]. Omega-3 fatty acid ethyl ester may also improverterial function in children with FH [204]. The recommenda-ions for monitoring drug safety and tolerability in paediatricatients are generally similar to adult FH, but special require-
ents are noted below and in Fig. 6. More systematicallyollected long-tem safety data are required on the use oftatins and other lipid-regulating agents in paediatric FH205].
o[fc
plements 12 (2011) 221–263 237
.4. Assessing atherosclerosis
CUS, with specific measurement of CIMT and the detec-ion of early plaques, may be useful in assessing therogression of early carotid atherosclerosis and hence in risktratification of children [78,122,206]. Longitudinal data iniverse populations support lowering hypercholesterolaemian children with FH from the age of at least 10 years [206].US could be carried out every two years in moderate riskH and annually in high risk FH. These intervals are onlyguide and should be revised according to clinical context
nd available resources. Evidence of an increase in CIMT,r development of early atherosclerotic plaques, would be anndication for intensifying treatment in moderate and highisk cases of FH [183] (see Fig. 5). CUS should be carriedut by a fully credentialled vascular imaging service andIMT estimated employing well validated edge-detection
oftware [82,122]; this facility may not be routinely avail-ble to many clinics and consequentially other paediatricuidelines do not specifically recommend use of CUS in man-ging FH [64a,b]. Imaging protocols need to be standardisednd age- and sex-specific reference data for CIMT employed82,122]. Contemporary CTCA and calcium scoring shouldot at present be undertaken in children owing to the radi-tion doses involved. Uncontrolled dyslipidaemias in youngdulthood predicts coronary calcium in middle-aged [207].e do not consider that serial measurement of the ankle-
rachial index plays a role in the management of childhoodr adolescent FH, except in very high risk cases includingomozygous patients. Measurement of endothelial function,ith flow mediated dilatation of the brachial artery, is atresent considered a research tool that is not ready for rou-ine clinical practice [122]. Children with a family historyf CHD in early adulthood will require non-invasive testingor CHD [13,97] and referral to a paediatric cardiologist isdvised [110]. For patients with homozygous, or compoundeterozygous FH on LDL-apheresis, regular echocardiogra-hy of the aortic valve and aortic root, with estimation of theortic valve gradient, is recommended [95,97]. These specialases of very high risk FH should be managed jointly with aaediatric cardiologist.
.5. Clinical monitoring and continuity of care
Recommendations for the follow-up of children and ado-escents with FH are broadly similar to adults [14,15,97,98].owever, certain issues require specific attention and review
ntervals may not need to be as frequent as in adults. Drugolerability, interactions and safety need careful surveillance97,98,153,156,163,165]. All children and adolescents ontatins require monitoring of physical growth and puber-al development, as well as when indicated plasma levels
f hepatic aminotransferases, creatine kinase and creatinine97,98]. Before commencing statin therapy in adolescentemales, specific advice should be given regarding contra-eptive choices and the potential risk to the foetus should
2 sis Sup
tstatootamiainepn[abalsamoctf
9
tls[Cfi
9
gwrvLg>pLt[t
Lsmtoe
cthiPmac[w
9
Lha[LPbLartcfissbuasa4b
9
musLt
38 G.F. Watts et al. / Atherosclero
hey fall pregnant while on medication [186]. These issueshould be considered at each review. Adolescence is an oppor-une time to evaluate each patient’s understanding of FHnd provide age-appropriate education as required. Impor-ant issues that should be addressed include: the significancef the autosomal inheritance pattern of FH, with potentialffspring having a 50% chance of inheriting the condition;he value of maintaining a healthy lifestyle; the importance ofdhering to the medication prescribed and attending appoint-ents for clinical review. Adherence to medical management
s a challenge for many individuals with chronic conditionsnd can be a particular issue in adolescence [170,208]. Fam-ly counselling and medication support systems, includingurse or pharmacist co-ordinated programs, to ensure adher-nce with therapy may be useful, particularly in higher riskatients [166,167,170]. Enrolling the assistance of person-el skilled in managing adolescent problems may be useful209]. Pharmacists may be utilised for monitoring adherencend flagging non-adherence to medication in the intervalsetween clinical reviews, as well as for directly improvingdherence to medication [166,168]. All children and ado-escents with FH should be reviewed at least yearly in apecialist clinic. Highest risk patients should be reviewedt least every 6 months and homozygotes on LDL-apheresisore frequently [95,97,210]. Patients who are well controlled
n stable treatment could be managed in primary or sharedare. Appropriate clinical pathways should be developed forransitioning and transferring adolescent patients with FHrom paediatric to adult clinical care services [211].
. LDL-apheresis and radical therapy for FH
LDL-apheresis is a radical form of treatment for FHhat entails the extracorporeal removal of apoB-containingipoproteins from the circulation [212,213]. Its use in treatingevere FH is supported by published international guidelines13,95,214–217] and evidence that LDL-apheresis improvesHD outcomes, progression of atherosclerosis and aorticbrosis, endothelial function and coagulation [212,218,219].
.1. Indications, patient selection and targets
LDL-apheresis is indicated for patients with homozy-ous or compound heterozygous FH, as well as for patientsith heterozygous FH with documented CHD who are
efractory to pharmacotherapy [95,216]. FH patients withery high plasma Lp(a) levels may also benefit fromDL-apheresis [75,217]. Untreated patients with a homozy-ous phenotype typically have plasma LDL-cholesterol12 mmol/L and should be treated with maximally toleratedharmacotherapy for at least 6 months before considering
DL-apheresis [95,220]. Untreated heterozygous patientsypically have plasma LDL-cholesterol from 5 to 12 mmol/L95] and may have true non-responsiveness or intoleranceo cholesterol lowering pharmacotherapy. The recommended
aoLp
plements 12 (2011) 221–263
DL-cholesterol thresholds for selecting patients for aphere-is in Fig. 7 are only an approximate guide and should beodified according to clinical context; an alternative selec-
ion criterion could be a <50% reduction in LDL-cholesteroln maximal pharmacotherapy for both homozygous and het-rozygous patients.
To be suitable for LDL-apheresis patients must be psy-hologically and clinically stable and be committed to thereatment. Haemorrhagic diatheses and hypersensitivity toeparin are contra-indications. Clinical assessment shouldnclude baseline echocardiogram for aortic stenosis [215].atients unsuitable for LDL-apheresis must continue diet andedication. LDL-apheresis is an efficacious, well tolerated
nd safe treatment for children with severe FH and may beommenced after the age of 5 years in affected individuals95,97,210]. It should also be considered for pregnant womenith uncontrolled FH and stable CHD [221,222].
.2. Methods for apheresis
There are five apheresis methods that are selective forDL: membrane differential filtration, immunoadsorption,eparin-induced LDL precipitation, dextran sulphate LDLdsorption and haemoperfusion with direct LDL adsorption95,212,216,217]. All are comparable at acutely loweringDL-cholesterol by 50–70% following a single treatment.lasma exchange (or centrifugal plasma apheresis) may alsoe used, but its major disadvantage is that it is not selective forDL. The choice of method will depend on local expertisend resources. Treatment should be carried out in collabo-ation with a specialty experienced in apheresis, such as aransfusion medicine service, with whom close communi-ation is mandatory. Plasmapheresis and probably cascadeltration should be available via all transfusion medicineervices. A dedicated, fully resourced LDL-apheresis unithould also have a variety of methods of apheresis that cane tailored to maximise efficacy and tolerability in individ-al patients [223]. Vascular access is achieved using bilateralntecubital vein cannulation, but a long-term arteriovenoushunt or central venous catheter may be required. Antico-gulation with heparin and citrate is required. Typically,–6 L exchanges of plasma are carried out for 2–4 h weekly,iweekly or longer according to therapeutic response.
.3. Monitoring therapy
The efficacy, safety and tolerability of LDL-apheresisust be monitored at each treatment and the regimen reg-
lated accordingly [215]. The frequency of LDL-apheresishould be adjusted to achieve a time-average plasmaDL-cholesterol concentration between LDL-apheresis
herapy of 6.5 mmol/L and 2.5 mmol/L for homozygotes
nd heterozygotes, respectively (Fig. 7); a mean reductionf >65% in plasma LDL-cholesterol concentration betweenDL-apheresis relative to no form of treatment is anotherossible target. To achieve these targets will usually require
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 239
Diet and maximal drug therapy
Homozygous or compound heterozygous FH
Heterozygous FH with documented CHD
No No
LDL-C >5 mmol/L
Assess suitability for LDL-apheresis
Liaise with transfusion service to select LDL-apheresis method,
regimen and set efficacy and safety parameters
Apherese 1.5 – 2 plasma volumes for 2 – 4 hours every 2 –
3 weeks, continue statins and other drugs, avoid ACE inhibitors
Review efficacy, tolerability and safety of apheresis at each
treatment
Review adherence and tolerability
LDL-C >7 mmol/L
Therapeutic target Time average
LDL-C <6.5 mmol/L
Revise regimen and method of apheresis; consider need for liver
transplantation
Consider need and suitabilityfor liver transplantation,
enrolment in clinical trial of new treatments, or applying
via special access scheme for drugs in development for FH
Diet and maximal drug therapy
Review adherence and tolerability
Consider enrolment in clinical trial of new treatments or applying via special access scheme for drugs in development for FH
Yes
Not Suitable Homozygous or compound
heterozygous FH
Not SuitableHeterozygous FH
Suitable
Homozygous or compound heterozygous FH
Heterozygous FH with documented CHD
Therapeutic target Time average
LDL-C <2.5 mmol/L
s and ra
aesuctaoeausr
wLidlas
pc
Fig. 7. LDL-apheresi
n acute reduction of ≥70% in LDL-cholesterol duringach procedure. Statins should be continued since theylow the post-exchange rebound in LDL-cholesterol, butse of angiotensin converting enzyme (ACE) inhibitors areontra-indicated with most systems because of a poten-ial bradykinin reaction; [95,217] angiotensin II receptorntagonists are suitable alternatives. The overall incidencef clinical side effects (including hypotension, vasovagalpisodes, symptomatic hypocalcaemia and anaemia) duringpheresis is less than 5%; if present, these may be corrected
sing standard medical therapy. Non-specific symptomsuch as nausea, fatigue, dyspnoea and abdominal painespond to temporary discontinuation of therapy. Patientstpc
dical therapy for FH.
ho are persistently intolerant of a particular method ofDL-apheresis should be trialled on an alternative method,
ncluding plasma exchange if required [212]. Because of theemands of treatment, psychological status and quality ofife should be monitored in all patients. Accordingly, patientsnd parents of affected children undergoing LDL-apheresishould be offered psychological counselling when indicated.
The long-term efficacy of treatment in homozygousatients should be assessed about every 2 years using CUS forarotid atherosclerosis load, and echocardiography for aor-
ic valve and root involvement, proceeding to stress ECG andossibly coronary angiography, if there is evidence of signifi-ant progression of disease [13,78,215,224]. In heterozygous
2 sis Sup
paoLoc
9
tta[thHsf
9
stnasnpCmaPatadcofivtoammts
1o
t
o[u[gtaihbigitpli
1
nitiidugFnia
1
btfiAttCwwna
nfwg
40 G.F. Watts et al. / Atherosclero
atients on LDL-apheresis CUS and exercise testing shouldlso be employed at suitable intervals to monitor progressionf atherosclerosis [13,183,215]. Many patients undergoingDL-apheresis will need to be jointly managed with adultr paediatric cardiologists [215,224] and appropriate lines ofommunication should be established.
.4. Cost considerations
Besides the complexities of the treatment, the main barriero using LDL-apheresis is the financial cost. It is estimatedhat on average each procedure costs AU$2200–2600, so thennual cost of a bi-weekly cycle is approximately AU$53,00095]. This is comparable to haemodialysis, but in absoluteerms would be less than 1% of the national expenditure onaemodialysis. LDL-apheresis is underutilised in Australia.ealth insurance providers, Medicare and tertiary hospitals
hould support it as a routine service for patients with severeorms of FH.
.5. Other therapeutic options
As shown in Fig. 7, homozygous patients who are notuitable for LDL-apheresis should be considered for liverransplantation [225,226] or enrollment in clinical trials ofovel cholesterol lowering treatments [227], such as apoB-ntisense oligonucleotide therapy and proprotein convertaseubtilisin/kexin type 9 (PCSK9) inhibitors [228,229]. Alter-atively, these new treatments could be made available toatients on a compassionate basis via a special access scheme.oronary artery bypass surgery and/or aortic valve replace-ent should be considered prior to liver transplantation
ccording to pre-operative cardiac investigations [215,224].atients with heterozygous FH who are not suitable for LDL-pheresis should also be considered for enrollment in clinicalrials of new cholesterol lowering treatments [228,229], or anpplication should be made via a special access scheme forrugs in development for FH. Partial ileal bypass should beonsidered in heterozygous FH patients who are intolerantf or refractory to pharmacotherapy, noting both the bene-ts and risks of this surgical procedure [230,231]. There isery limited experience in Australasia with liver transplan-ation for severe FH. Portocaval shunting carries a high riskf encephalopathy and has now been superseded by LDL-pheresis and liver transplantation; its use in homozygous FHay, however, be considered in countries where newer andore expensive treatments are not available [220]. Finally,
here may be a role in the future for gene therapy in treatingevere FH [232].
0. Cascade screening: testing and risk notification
f familiesIt is well accepted that the most cost-effective approacho detecting new cases of FH is family cascade screening
mtad
plements 12 (2011) 221–263
f close relatives using a genotypic or phenotypic strategy4,10,31,65,233–236]. Genetic testing of an at-risk individ-al within a family is often referred to as ‘predictive testing’237]; predictive testing on the basis of a known pathogenicenetic variant (i.e. a mutation) is more accurate than predic-ive testing based on an individual’s clinical phenotype alone,nd this also applies to FH [7,8,66]. In a condition like FH,n which the phenotype almost always expresses as hyperc-olesterolaemia, the descriptor ‘predictive’ could be replacedy ‘presymptomatic’ or ‘diagnostic’ to describe genetic test-ng [237]. However, because ‘predictive’ describes a moreeneral context and is more widely accepted in clinical genet-cs it is also used in this document when referring to geneticesting of relatives for FH. Screening for FH using the clinicalhenotype alone is a form of ‘diagnostic’ testing, particu-arly when assessment provides a definite outcome, as in anndividual with, for example, a DLCNS > 8 [8,34,44].
0.1. Risk notification
Integral to effective and ethical cascade screening is riskotification [7,8,119,120]. Risk notification is the process ofnforming relatives that (1) they are at risk of FH, (2) thathis may have implications for their health, and (3) that clin-cal and genetic testing (if applicable) is available to clarifyf they do or do not have FH. Fig. 8 provides recommen-ations, based on expert guidelines and position papers, forndertaking the process of cascade screening whether using aenotypic or phenotypic approach [8,113,119,120,238–240].undamental ethical principles of autonomy, beneficence,on-maleficence and justice are central to this process. Healthlliteracy is not uncommon in individuals in the communitynd must be carefully considered and addressed [87,88,173].
0.1.1. Contacting and informing familiesThe general protocol for cascade screening should begin
y contacting first and then second degree relatives, notinghat cases so detected will then become probands for risk noti-cation of their own first and second degree relatives [6–9].typical pedigree tree showing the autosomal dominant pat-
ern of inheritance of FH (clinical phenotype expressed) andhe yield from cascade screening is shown in Appendix 6.ulturally sensitive strategies may be required when dealingith some ethnic groups [1,241]. Approaching and liaisingith community leaders early on is advised to facilitate riskotification and predictive testing occurring in a culturallyppropriate way.
The index case should be invited to discuss family riskotification with the clinician or nurse, who will construct aamily pedigree and identify first and second degree relativesho should initially be offered testing for FH. Use of a pedi-ree drawing tool [242] and a fully integrated information
anagement system [243] is strongly recommended to facili-ate family assessment, work planning, multidisciplinary carend communication of results to individuals tested and theiroctors [9,12,25]. Appendix 7 shows a pedigree tree for

G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 241
No
No response
Family member consents to screening Family member does not consent to screening
Accept decision not to be tested
But encourage risk notification of their 1st and
2nd degree relatives
Arrange appointment with FH serviceAs per local protocol
Index case consents to risk notification of relatives
Is there a particularly strong clinical
indication for risk notifying relatives without consent?
YesSend 2nd letter / Telephone
family member
No response
Risk notificationSend letter and information sheet to family member and / or give to index
case to distribute to relatives
ResponseReceived by
clinical service
No
notifica
anaiiawibd
etsabafenctctm[
wtt
tatoCdo
appic[cbfEic
vtp[[f
Fig. 8. Cascade screening: risk
n actual family with FH, illustrating the autosomal domi-ant inheritance of hypercholesterolaemia, premature CVDnd a missense mutation in the LDL-receptor gene. Draw-ng and detailing such pedigrees is essential for effectivemplementation of cascade screening, including appropri-te risk notification. Clinical genetics services often haveell-established protocols for managing families with famil-
al disorders and, where possible, cascade screening shoulde implemented as a joint collaborative effort of the lipidisorders and clinical genetics services [12,26,115,121,237].
Comprehensive and well validated resources should bemployed and developed to accurately convey informationo relatives [12,25,37]. The method for risk notificationhould be tailored to each family, so that relatives may bepproached either by the index case, the clinical service, ory both [6,8,119,120,244]. Understanding the reasons whyn index case may not consent to cascade screening of otheramily members is important to provide re-assurance andncourage rapport and disclosure [37,245,246]. Dual riskotification may be the best option: notification by the clini-al service alone may lead to relatives feeling aggrieved thathey were not informed by their related index case; notifi-ation by the index case alone may be taken less seriouslyhan notification by a medical service. Genetic counsellors
ust be involved when sensitive family issues are identified8,25,110,121,238].
Index cases who contact relatives should be provided with
ritten information which explains the predictive/diagnosticesting process and implications, and be encouraged to givehis to their relatives [12,25,37]. Information and letters sent
aPp
tion and approaching families.
o relatives should be written in general language to avoidlarm and concern, while emphasizing the voluntary nature ofesting [119,120], and the health consequences of a diagnosisf FH being missed when a person decides not to be tested.ommunications should also emphasise the health gains ofiagnosis and treatment [12,25], as well as the benefits forffspring who may have FH.
In the absence of a response to the first letter, it may beppropriate to make a second approach by letter or tele-hone call to the family member [37,244]. However, thisolicy should be discussed with relevant local experts on eth-cal and risk management matters. Multiple contact attemptsould constitute a violation of privacy for some individuals119,120] and may be counter-productive to the process ofascade screening. Reasons why a relative does not wish toe tested for FH should be explicitly documented [84,86] toacilitate, where indicated, re-approaching them in the future.lectronic methods of communication (e.g. e-mail) could be
nvestigated for their value in enhancing risk notification andascade screening for FH.
Prior to testing, family members should be given detailederbal and written information about FH. Written informa-ion should again be in clear, non-technical language andreferably accompanied by simplified but explicit diagrams25,37,119]. Health illiteracy should always be addressed87,88,173]. Family members who consent to being testedor FH should be offered a standard plasma lipid profile and
genetic test if the mutation is known in the index case.atients without GPs should be encouraged to visit one asart of the FH counselling process. With consent from the

2 sis Sup
iFm
1
irlobchbobacgfaicchc[
w[EiGf[
cl[hIwtgccs
1
ftvp
ecolo[n
1dgbt(tPtpiplr
itl[bafigwitCatwaic[tlm
1
atC
42 G.F. Watts et al. / Atherosclero
ndividual, the outcome of phenotypic or genetic testing forH should be communicated to their GP and other relevantedical services.
0.2. Co-ordination of cascade screening
Different care pathways may be employed for screen-ng individuals for FH, depending on circumstances andesources [7,25,26,37]. Family members may be tested via theipid disorders clinic, genetic services or their GP. Dependingn knowledge and experience, pre-test counselling may beest undertaken by a specialist medical practitioner, geneticounsellor or specialist nurse. FH specialist nurses shouldave some competency in genetic counselling [121,247], thiseing particularly relevant for efficiently co-ordinating anutreach screening service. Phenotypic screening for FH maye carried out by GPs, noting that use of clinical criterialone may result in the underdiagnosis of FH in a primaryare setting [248]. However, GPs should not directly requestenetic testing without appropriate specialist training. Theuture should see more education of GPs in medical geneticsnd this will increase their role in predictive testing of fam-lies for FH [249]. This is particularly important in primaryare, given the greater accuracy of genetic testing over clini-al criteria in diagnosing FH. FH services that do not employealth practitioners with formal counselling training couldonsider referral to a clinical genetics service for counselling26,121,237].
Referral to genetic services should also be consideredhen contemplating genetic testing in children or adolescents
115,238], and when sensitive family issues are identified.nquiries concerning PND and PGD for FH are a special
ndication for referral to a clinical genetics service [192,193].enetic counsellors should be involved in training staff caring
or FH patients and in supporting GPs and outreach services7,8,12,26,121].
Cascade screening for FH is best carried out ando-ordinated centrally by a specialist service ideally run col-aboratively by lipid disorders and clinical genetics services12,26,71,115,121,237]. However, relatives may choose toave genetic or phenotypic testing in a primary care setting.n this case, close liaison and communication will be requiredith their GP. If genetic testing is required, support in pre-
est counselling should be given by a clinical geneticist or aenetic counsellor [115,121,237]. If the diagnosis of FH isonfirmed genetically, or is highly suspected on phenotypicriteria, GPs should refer the individual to a specialised FHervice for advice on treatment and follow-up [12,25,26,38].
0.3. Risk notification without consent
If the index case does not consent to risk notification of
amily members it is important to explore and understandhe rationale behind this [83,245], and in particular the pre-ailing family dynamics [83,119,120,246]. By maintainingrofessionalism and continuing to patiently and judiciouslytbae
plements 12 (2011) 221–263
xplore and resolve key issues [120,246], an effective rapportan often be built with the index case sufficient to revisit theption of risk notification after 6–12 months, or sometimesonger. Referral to a genetic counsellor [121] and involvementf a patient support group for appropriate lay counselling250–252] may all assist in achieving acceptance of riskotification.
Recent amendments to the Commonwealth’s Privacy Act988 in Australia allow private health practitioners “to use orisclose patients’ genetic information, whether or not theyive consent, in circumstances where there is reasonableelief that doing so is necessary to lessen or prevent a serioushreat to the life, health or safety of their genetic relatives.”Privacy Legislation Amendment Act 2006 (Cth) amendmento the Privacy Act 1988 (Cth)) [253]. This amendment of therivacy Act only applies to the private sector [253]. Prac-
itioners employed in the public sector need to refer to therivacy legislation in their own state or jurisdiction concern-ng the release of information without the consent of theatient [246]. The difference in the privacy laws for pub-ic and private patients in Australia is an issue that requiresesolution.
A decision to risk notify without the consent of thendex case should therefore be very carefully and judiciouslyaken with attention to privacy legislation in different states,ocal health service protocols and the NHMRC guidelines239,246]. Health professionals must understand the geneticasis for FH, take reasonable steps to obtain patient consentnd document the process fully before considering risk noti-cation of relatives without consent. The recent NHMRCuidelines [239] are not prescriptive and we urge cautionhen independently approaching relatives. Nevertheless, FH
s a potentially lethal condition and if there are high risk fea-ures in the family, such as a strong history of prematureVD, contacting of relatives without consent is justifiable,s indicated in Fig. 8. Refusal of an index patient with FHo share genetic information with relatives is unusual, buthen present poses a significant challenge to risk notification
nd cascade screening [119,120,246]. Best clinical practicemplies that disclosure of information without consent duringascade screening for FH should be viewed as a last resort246]. However, when information is disclosed, it should behe minimum required, avoid identifying the patient or theirack of consent, and should only be made available to family
embers no further removed than third-degree relatives.
0.4. Insurance cover and genetic testing
All individuals with potential FH should be made awarend understand the implications of genetic testing for cer-ain types of insurance cover [254–256]. Family history ofVD, a clinical diagnosis of FH, plasma level of choles-
erol and predictive genetic information could all potentiallye employed by the insurance industry to make decisionsbout exclusion of specific conditions from insurance cov-rage and setting an insurance policy premium [256–258].

sis Sup
IcbaarHCiiatp
1
imiraH2csc
iFptdGgwibcfto[
boTcsOasTip
aoihGv[
gifatH[teb
pcPiPtSol
1
1
At(uCNNrcldt
irfa
G.F. Watts et al. / Atherosclero
n Australia premiums for insurance products which includeover for life (term), disability/income protection, trauma,usiness and bank loans (but not private health insurance)re calculated according to the present and past health of thepplicant, their family history and any known genetic testesult(s) in the applicant, their siblings or their parents [256].owever, insurers who are members of the Financial Servicesouncil have agreed that they will not ask an individual who
s applying for insurance coverage to have a genetic test ift has not been done already [259]. Similar conditions applyt present in New Zealand. Better control of hypercholes-erolaemia with statins in FH is likely to lower insuranceremiums [258].
1. Genetic testing of families
FH is an autosomal dominantly inherited disorder, affectedndividuals having a 50% chance of passing the causative
utation to each offspring [1–3]. If a pathogenic mutations identified in an affected index case, predictive testing ofelatives using genetic testing is a cost-effective, accurate andcceptable approach for detecting new cases [66,234,260].owever, genetic mutations may not be detected in at least0% of patients in whom a clinical diagnosis of FH can beonfidently made [3,23,45,66,261,262]. Thus, in a smaller butignificant proportion of families diagnostic testing should bearried out phenotypically [12,38,68].
One of the main advantages of predictive genetic testings its ability to definitively confirm or exclude a diagnosis ofH in the relatives of an index case [66]. Fig. 9 outlines therocess to be followed when employing predictive geneticesting in cascade screening. The majority of cases of FH areue to mutations in the LDLR, APOB and PCSK9 genes [1–3].iven that a mutation is known to be present in the index case,enetic testing should be offered to all at-risk family membersho present for predictive testing [8,66]. Because of ethical
ssues involved in genetically testing minors [114,115], it isest and usual practice to first genetically test a phenotypi-ally affected parent. When no consent, or assent, is obtainedor genetic testing, the diagnosis of FH can be made pheno-ypically according to the DLCNS in an adult [8,34,38,44]r to the plasma LDL-cholesterol concentration in a child46,48,97] (Fig. 9).
Genetic testing of families for FH should be co-ordinatedy a specialist ideally run collaboratively by a lipid dis-rder clinic and clinical genetics [12,26,71,115,121,237].he psychological consequences of genetic testing must bearefully considered and appropriate education and coun-elling offered in all individuals [8,66,84,86,115,121,237].btaining informed consent, or parental consent plus child
ssent in the case of testing children, requires pre-test coun-
elling concerning the implications of the result [115,238].his is particularly important in children because the futuremplications are more difficult to anticipate and the consentrocess via parents or guardians is less direct [115]. Age
itaa
plements 12 (2011) 221–263 243
nd developmentally appropriate assent/consent should bebtained from children and adolescents prior to testing andnformation should be tailored to the child’s level of compre-ension [115,121] and the parent’s level of literacy [87,88].enetic testing in children can be carried out without invasiveenepuncture, for example by using a buccal swab specimen263].
If the family mutation is not identified by a predictiveenetic test, the diagnosis of FH can be excluded in that fam-ly member. However, there may be some situations when theamily member has significant hypercholesterolaemia (andphenotype strongly suggestive of FH) but does not carry
he pathogenic mutation identified in other family members.aving excluded secondary causes of hypercholesterolaemia
14,15], it may then be appropriate to do a full ‘FH muta-ion search’ in that individual, including investigation of thextended pedigree. This mutation search should be precededy appropriate pre-test counselling [115,121,237].
As indicated earlier, when one or both members of a cou-le has FH and there are concerns about having an affectedhild, referral for specialist genetic counselling regardingND and PGD should be considered [192,193]. Couples opt-
ng for pre-natal testing should be seen prior to conception.GD will require detailed assessment by an in vitro fer-
ilization service and a specialised DNA laboratory [264].ervices providing PGD must comply with established lab-ratory requirements and relevant State and Commonwealthegislation [265].
2. Laboratory approach to genetic testing for FH
2.1. Background
Laboratories providing medical genetic testing inustralia must comply with requirements developed by
he National Pathology Accreditation Advisory CouncilNPAAC) [264]. Compliance with these standards is reg-larly assessed in a joint program managed by the Royalollege of Pathologists of Australasia (RCPA) and theational Association of Testing Authorities (NATA) [266].PAAC requirements address the need for appropriate labo-
atory resources, governance and supervision, training andontinuing accreditation of staff, and appropriate clinicaliaison. Medical testing that may be the basis for clinicalecision-making must be performed in an accredited labora-ory.
Accordingly, genetic testing for FH should be carried outn a NATA/RCPA accredited laboratory [266] that will issueesults to the requesting doctor. The process of screeningor genetic mutations, confirming identified genetic variants,ssessing pathogenicity and issuing a formal report should
deally not take longer than three months. Supported by fur-her evaluation of its cost-effectiveness, we recommend thatn application is made for genetic testing for FH to be listeds an MBS item.
244 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Proband or relative carrying gene variant of known pathogenicity (i.e. mutation)
Counseling of family membersPsychological / genetic / lay counseling. Clinical
genetics and paediatric review if required. Informed consent / assent sought
No assent/consent to genetic testing
Assent/consentto genetic testing
Genetic testing Phenotypic testing
Family mutation not present
Family mutation present
Discuss risk notification of close
relativesFigure 8
Offer phenotypic or genetic testing to
relatives
If still concerned about possibility of
FH, continue to build rapport and
reconsider genetic testing
FH unlikelyProbable to Definite
FH confirmed
FH excluded*
f familie
ai(Infamu[d
FafimaiAgcitgn
cgnAafimonp
bFipggCdbnt
Fig. 9. Genetic testing o
In the present context, a gene variant is defined as an alter-tion in the normal sequence of a gene. Genetic testing of anndex case may fail to identify a variant in the genes testedvariant absent) or it may identify a variant (variant present).dentified variants may have been previously reported or beovel. NATA/RCPA accredited laboratories have processesor assessing the likely significance of an identified gene vari-nt and for classifying the variant as clearly pathogenic (autation), clearly non-pathogenic (a benign variant) or of
ncertain significance (a variant of uncertain significance)264]. The assessment of significance is discussed in moreetail in a later section.
FH has significant locus and allelic heterogeneity, i.e.H is caused by mutations in a number of different genesnd many different pathogenic mutations have been identi-ed in each implicated gene [112,261,267–270]. Thus, theain challenges for genetic testing for FH remain the costs
nd feasibility of testing the large numbers of patients clin-cally diagnosed with or suspected to have the condition.s noted earlier, the likelihood of detecting a pathogenicene variant in an individual suspected of being an indexase is directly proportional to the clinical probability of thatndividual having FH [1,3,66,112,118], as assessed pheno-
ypically by, for example, the DLCNS [34,38,44]. Hence,enetic testing of adults with a phenotypic DLCNS < 3 mayot be cost-effective [38,44].moh
s. *See text for caveats.
Currently, mutations in three different genes are known toause FH [1,3,66]. A pathogenic mutation in one of theseenes is identified in about 70% of phenotypically defi-ite and 20% of phenotypically probable/possible FH [8,66].bout 95% of the identified mutations are in the LDLR gene
nd 4–5% in the APOB gene. Mutations are seldom identi-ed in the PCSK9 gene. Detection of a mutation in a familyember allows the definite diagnosis of FH to be made, as
utlined in Fig. 9. However, failure to detect a mutation doesot exclude a diagnosis of FH, particularly if the clinicalhenotype is highly suggestive of FH [12,37,66].
There are several reasons why mutations might note detected in patients with a high clinical suspicion ofH [1,3,66,262]. First, the present laboratory technology
s not sufficiently sensitive nor specific for detecting allathogenic mutations. Second, FH is genetically hetero-eneous (caused by mutations in a number of differentenes) and not all causative genes have been identified.learly, genetic testing is not helpful in families with FHue to mutations in unknown genes as these genes cannote included in genetic testing. Third, the index case mayot have ‘true’ FH, since a family history of hypercholes-erolaemia and early CVD is not sufficiently specific for
aking the diagnosis of FH; some of these patients may havether genetic disorders [66], particularly familial combinedyperlipidaemia [271,272].

sis Sup
i[oePlM(A
1
etfFoFattmWaa
utgnasicitiDttiEfratga
dttcd
Tso
1d
naplt(sasUeaicetti
vpvopfo[
rnvmttitWtn
1m
G.F. Watts et al. / Atherosclero
Most diagnostic laboratories undertaking FH genetic test-ng will utilize a number of mutation detection methods66]. These may include: (1) nucleotide sequence analysisf each of the exons and flanking splice regions (exon byxon sequence analysis, EBESA) of the LDLR, APOB andCSK9 genes [3,112,261,270,273]; (2) methods that detect
arge duplications and deletions in the LDLR gene based onultiplex Ligation Probe Amplification (MLPA) [274,275];
3) methods that screen for specific mutations in the LDLR,POB and PCSK9 genes [260,276–280].
2.2. A protocol for genetic testing
Complete screening for all known FH-causing genes isxpensive and time consuming [66]. A laboratory protocolhat is likely to be cost-effective for genetic testing (searchingor a mutation) an ‘index case’ considered to have phenotypicH is proposed in Fig. 10. This protocol could form the basisf a standardised, national protocol for genetic testing forH. We emphasize that laboratories may choose to vary thepproach to genetic testing for FH shown in Fig. 10 accordingo the available resources: for example, laboratories may opto proceed directly to DNA sequencing for an LDL-receptor
utation of all test samples received [3,66,112,261,270,273].e recognize that technological advances in genetic testing
nd the discovery of new genes causing FH will necessitateppropriate modification to the protocol [66,110] in Fig. 10.
The steps proposed in Fig. 10 are as follows: (1) individ-als with a DLCNS < 3 (unlikely FH) are not offered geneticesting, because the likelihood of identifying a pathogenicene variant is low and with currently available technologyot cost-effective; (2) genetic testing for FH is offered toll individuals with a DLCNS ≥ 3, which includes all pos-ible, probable and definite phenotypic cases of FH; (3) forndividuals with a score of 3–5 (possible FH), to maximizeost-effectiveness, testing could be limited to sequential test-ng using: Screen 1, employing commercial methods thatarget specific mutations; [260,280–282] Screen 2, employ-ng MLPA [274,275,283] (Fig. 10); (4) individuals with aLCNS ≥ 6 could be offered more comprehensive sequen-
ial testing: Screen 1, employing commercial methods thatarget specific mutations; [250,270–272] Screen 2, employ-ng MLPA; [274,275,283] Screen 3, employing sequentialBESA of at least the LDLR gene (the recommended order
or EBESA is LDLR > ABOB > PCSK9) [3,66] (Fig. 10). Allesults from commercial chip or kit technology that identifygene variant as being present should be confirmed using
he second validated testing method. This is a requirement ofood laboratory practice [264,266] and relates to the potentialnalytical errors with the commercial methods.
It should be noted that the protocol in Fig. 10 refers toiagnostic testing (mutation searching) for FH in a pheno-
ypically defined ‘index case’. In a predictive testing settinghe laboratory will already know the mutation in the indexase and there is no need to screen for mutations other thanirectly testing for the one identified in the family [8,66].iTp
plements 12 (2011) 221–263 245
o increase acceptability, genetic testing for FH in childrenhould be available using DNA extracted from either bloodr buccal samples [263].
2.3. Assessing the significance of gene variantsetected in index cases
Various pieces of evidence are used to determine the sig-ificance of an identified gene variant [264] and this clearlylso applies to genetic testing for FH [66]. These include theublished literature (including database entries), epidemio-ogic studies, in vitro and (preferably) in vivo demonstrationhat the variant causes a functional abnormality, and in silicomolecular software) assessment [284,285]. For practical rea-ons, in silico assessment coupled with search of the literaturend established databases [267,286] are the usual primaryources used for assessing the significance of a gene variant.nfortunately, published literature may in general contain
rrors, so careful assessment is required. In silico data arelso variable in quality, particularly when it comes to assess-ng splicing mutations [287]. Patients and their managinglinicians should also be aware that a laboratory interpretsach genetic test result in accordance with the best informa-ion available at the time of the test, and that it is possiblehat new information may emerge which results in a changen how a genetic test is interpreted.
Particular care needs to be taken when classifying a geneariant as a pathogenic mutation, since it may be used inredictive genetic testing of asymptomatic individuals. If aariant of uncertain significance (either previously reportedr novel) is detected, it is not appropriate to regard it asathogenic. Clinical management of the individual and theiramily should be based on the plasma lipid phenotype (andther clinical criteria) and not on the genetic test result12,13,26,38].
Fig. 11 provides a general approach for assessing andeporting the outcome of genetic testing for FH in a phe-otypically defined index case. The significance of a geneariant should be assessed as recommended above. The for-al laboratory report of the FH variant/mutation should detail
he standardised methods used and the evidence supportinghe classification [264]. If a pathogenic variant (mutation) isdentified the report should also make the recommendationhat further relatives be genetically screened for FH [264].
here no gene variant or pathogenic mutation is detected,he report should include the caveat that such a result doesot definitively exclude the diagnosis of FH.
3. The web of care for FH: the optimal serviceodel
Fig. 12 encapsulates the multiple components that shoulddeally comprise a comprehensive healthcare model for FH.he recommendations and MoC described in this documentrovide detailed pathways for the principal clinical compo-
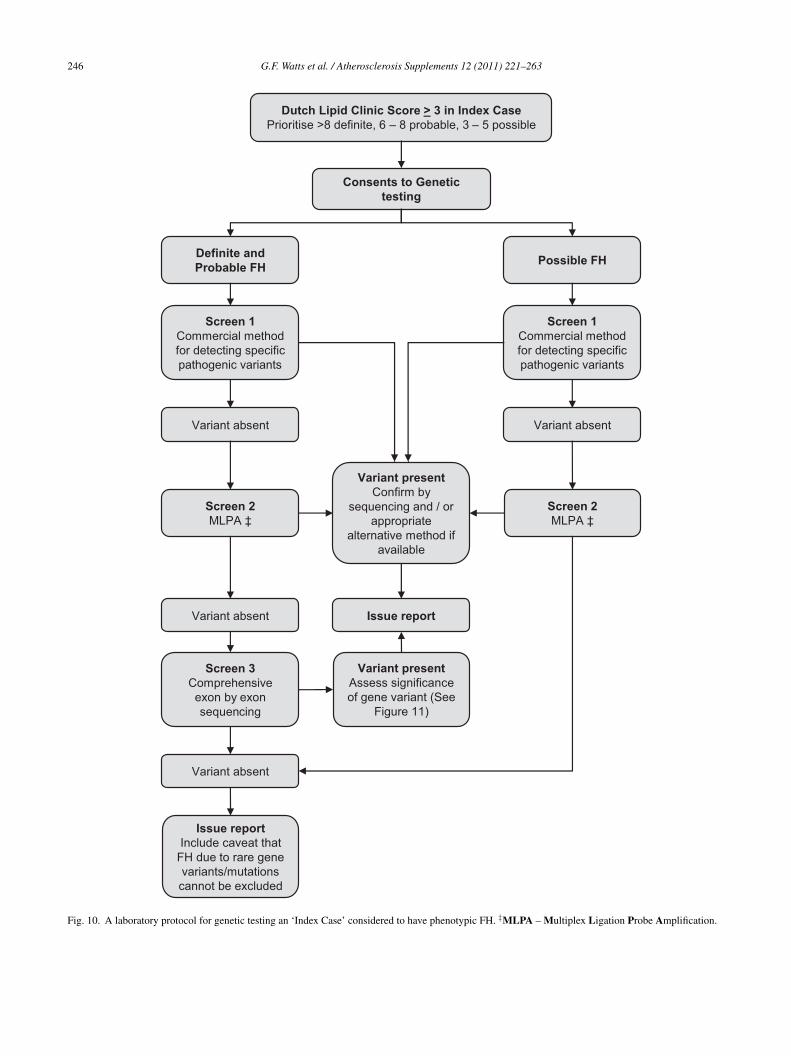
246 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Dutch Lipid Clinic Score > 3 in Index CasePrioritise >8 definite, 6 – 8 probable, 3 – 5 possible
Screen 1Commercial method for detecting specific pathogenic variants
Screen 1Commercial method for detecting specific pathogenic variants
Definite and Probable FH
Possible FH
Variant absent Variant absent
Screen 2MLPA ‡
Screen 2MLPA ‡
Variant absent
Screen 3Comprehensive exon by exon sequencing
Variant presentConfirm by
sequencing and / or appropriate
alternative method if available
Issue report
Consents to Genetic testing
Issue report Include caveat that
FH due to rare gene variants/mutations cannot be excluded
Variant absent
Variant presentAssess significance of gene variant (See
Figure 11)
Fig. 10. A laboratory protocol for genetic testing an ‘Index Case’ considered to have phenotypic FH. ‡MLPA – Multiplex Ligation Probe Amplification.

G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 247
‘Index case’ consents to genetic test
Genetic testing as per Figure 10
Assess significance of variant *
Variant of uncertain significance
Pathogenic variant (mutation)
Issue Reportwith caveat that FH cannot be excluded
Benign variant
Issue ReportWith appropriate recommendation for
testing relatives (See Figure 8 & 9)
No variant detectedVariant detected
enetic t
nss
1
pcabaCrmaaltetsai[
hpat
1
csccnrtvtbrmf
Fig. 11. Assessment and reporting of the outcome of g
ents shown in Fig. 12. This MoC represents an overarchingystem that significantly extends and updates other publishedervice models [12,13,25–28].
3.1. Focus, aims and objectives
Health service provision should focus on three main areas:atient care services, laboratory services, and research andlinical audit. The aims, objectives, expectations, outcomesnd key performance indicators for each of these shoulde clearly identified and documented. Research and auditgendas for FH have been published elsewhere [12,37,288].are pathways for the seamless flow of patients between
elevant specialties and other health providers, including pri-ary care, should be specified and developed. The general
im of clinical care should be to identify, assess, man-ge and treat children and adults with FH to the highestevel of clinical excellence. The primary therapeutic objec-ive is not only to reduce cardiovascular risk related tolevation in plasma LDL-cholesterol [1,2,12,13], but alsoo treat other cardiovascular risk factors including obe-
ity, hypertension, type 2 diabetes, metabolic syndromend smoking [17a,32,33,43,55–57,74,125,127]; psycholog-cal and psychosocial factors must also be addressed8,83–86,124,136,289]. A major long-term objective ofpotp
esting in an ‘Index Case’. *See text for further details.
ealth service provision for FH should be to identify mosteople with the condition in the community, aiming tochieve realistic targets in given time-frame, e.g. 30% detec-ion rate after 3 years of service implementation.
3.2. Co-ordination and integration of care
To achieve the clinical aims and objectives of the health-are model for FH requires a dedicated multidisciplinaryervice [110] that is best co-ordinated by a lipid disorderslinic [26,71]. This service should be managed by suitablyredentialled personnel operating out of departments of inter-al medicine, endocrinology or cardiology. Physicians whoegularly manage patients with FH should have specialistraining in clinical lipidology and competencies in the pre-ention of CVD [26,72], and be credentialled to supervise theraining of junior physicians. Uncomplicated patients coulde referred back to their GPs for long-term care but should beeviewed annually in a lipid disorders clinic. Existing clinicsay need to up-skill their approaches to cascade screening
or FH [9,290]. GPs should actively seek index cases in their
ractice by recognizing the importance of the family historyf CVD and marked hypercholesterolaemia [68]. However,he optimal method for systematic identification of FH inrimary care needs to be defined and a specific MoC devised
248 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Specialist & Primary Care Physicians, Physicians-
in-training. Training,
Credentialing,Professional development
Specialised Adult-Paediatric Service
Family Clinics
Structured Clinical Management Program
Shared between specialist clinics and primary care
Administrative, Secretarial & Information Technology
ServicesSupport for Clinics, Outreach
services and FH Registry
Specialist Nurses & Allied Health Support
Pharmacy / Medication supportNutrition
PsychologyExercise
Lay counsellingTraining & Professional
Development
Cardiac & Imaging Facilities
Stress testingUltrasonography
EchocardiographyCardiac CT Scanning
Medical Laboratory Services
Routine and Specialising in lipids and
lipoproteins
Clinical Genetics, Family & Genetic
CounsellingDefined clinical
pathways
Clinical LiaisonCardiology
Cardiac RehabilitationCardiothoracic Surgery
Stroke unitVascular Surgery
Patient & Family Support Groups
Consultation,Education,Advocacy
Structured Education Program
For community and health providers, multidisciplinary case conferences, journal
clubs, accreditation
Audit & Research Program
Registry, Clinical & basic Science, Clinical trials, Epidemiology &
Health Economics
Specialised Laboratory for
Genetic Testing Service and
research
Influencers & StakeholdersDepartment of Health
Policy MakersHealth Networks
Atherosclerosis AssociationFamily Support Group
National Heart Foundation
or FH: t
abdewcihbabictacsbossecasig
dtsstrAacabfcmlsic
I
wf
Fig. 12. The web of care f
nd tested. Decentralization of the management of FH shoulde a major objective of future developments. As FH servicesevelop further in the community, the specific roles of gen-ral practice and other members of the primary care teamill become clearer and inform future MoCs. Outreach lipid
linics in the community would be a useful first step, but clin-cal protocols need developing and testing. Telehealth mayave a role in the management of FH in Australia, [291]ut its cost-effectiveness remains to be evaluated. Patientsged <16 years should be seen in a paediatric clinic runy a specialist in metabolic medicine; dietetic counsellings essential in this age group. An integrated adult–paediatriclinic may be useful for certain families. Nurses should berained in managing patients and families with FH, and prefer-bly have some experience in the prevention of CVD andompetency in genetic counselling [72,110,121]. Cascadecreening, including risk notification, should be co-ordinatedy a health practitioner or specialised nurse working outf a central FH clinic or a clinical genetics service, andhould include (where required) provision of an outreachervice. With appropriate specialist training, nurses couldffectively administer genetic counselling [121]. Beyond cas-ade screening, FH nurses may be involved in several otherreas. These include the clinical care of patients, medication
upport, education and training, audit and research, enhanc-ng multidisciplinary links and working with a patient supportroup; nurse co-ordinated clinics and multidisciplinary casetcbb
he optimal service model.
iscussions should be integral to the service. By example,he effectiveness of nurse-led interventions has been demon-trated for management of non-cholesterol CVD risk factors,uch as hypertension and diabetes [292,293]. The role ofhe dietician is essential, for dietary management and weightegulation is a cornerstone of treatment [14–17a,32,33,133].dditional input from health and adolescent psychologists
nd exercise physiologists [136,209] may be required in spe-ial circumstances. Health literacy needs to be appropriatelyddressed [172,173]. Suitably trained nurses may have theest overall skills for co-ordinating the workflow necessaryor the optimal care of patients with FH. Clinical pharma-ists can have a special role in managing non-adherence toedication and in liaising with GPs [166–168]. The use of
ay counselling [252], particularly in patients who lack under-tanding of their condition and are non-adherent to treatment,s another promising resource that requires evaluating in theontext of FH.
nter-specialty linksBeyond the lipid disorders clinic strong links are essential
ith departments of clinical genetics in respect of the need foramily and genetic counselling [66,110,115,121,237], par-
icularly during the cascade screening process. A geneticsentred service may be appropriate for cascade screening,ut it may be more effective if genetic counsellors areased in and work out of the lipid disorders clinic. Not all
sis Sup
fbgngbt[
aslwFar
talCosrTatdchi
aLgwC[
1s
adtphiatawptq
ab(c(tbA
1
oeistwvcietRtcsmsrinaaf
1
avwtwirposesa
G.F. Watts et al. / Atherosclero
amilies or patients with FH require genetic counselling,ut some exposure and basic training in the principles ofenetic counselling is important for both physicians andurses who manage FH [72,121]. The general skills of aenetic counsellor may, on the other hand, be also moreroadly utilised in an FH service when dealing with cer-ain clinical issues [121], such as adherence with medication170,208].
Many patients with FH may be diagnosed amongst thosettending departments of cardiology, cardiothoracic surgery,troke medicine and vascular surgery [22,38,60], and closeiaison with all these is essential. At least one in twenty peopleith premature CVD cared for by these specialties may haveH [22,38]. A structured system for the routine screening andccurate detection of FH in these clinical settings is stronglyecommended.
An FH service should also have close links with labora-ory medicine and access to routine and specialised laboratorynd clinical tests [71]. These include routine lipids andipoproteins, apoB, Lp(a), homocysteine and high sensitivity-reactive protein (CRP). Genetic testing should be carriedut in a NATA/RCPA accredited laboratory [266] that cancreen for all the major genes that cause FH. These laborato-ies must comply with the standards set by the NPAAC [264].he clinical assessment of patients with FH also involvesccess to cardiac and imaging facilities, including treadmillesting, myocardial perfusion scanning, CUS and echocar-iography [13,76–78]. Close links with the department ofardiology are essential for the referral of symptomatic, origh risk cases, that will require coronary angiography andnvasive interventions.
Links with a transfusion medicine unit is important fordministering and monitoring LDL-apheresis [215–217].DL-apheresis should be offered to all patients with homozy-ous (or compound heterozygous) FH and to heterozygotesho are not at target LDL-cholesterol and have progressiveHD or are intolerant of cholesterol lowering medications
215–217].
3.3. Administrative and information technologyupport
The clinical service should be underpinned by appropriatedministrative and secretarial support [25,26]. A specialisedatabase for storing clinical and family data and informa-ion technology support systems are essential for effectiverovision of services [25]. Computerised programs shouldave several key capabilities for dealing with pedigree draw-ng, work flow management, production of template letters,rchiving data, clinical audits and research. Other impor-ant requirements include a high level of data confidentialitynd security, and compatibility with other healthcare soft-
are systems. Favourable experience with one such softwarerogram [243] has been reported [294], but requires fur-her evaluation in full clinical service mode. A secure, highuality software system is required to establish and developbci[
plements 12 (2011) 221–263 249
national registry of FH patients that links family mem-ers across states. The International Cholesterol FoundationInterChol) is an integrator and facilitator of communi-ations between clinical services worldwide; its webpagewww.interchol.org/links) provides links to several interna-ional bodies that deal with FH. Details of websites that coulde useful for clinical services caring for FH are given inppendix 8.
3.4. Clinical governance: audit, education, training
Efficient clinical governance is fundamental to all aspectsf an FH service [37,295]. Clinical governance shouldntail holding regular multidisciplinary conferences, focus-ng on the quality and efficiency of all aspects of theervice. Retrospective and ongoing clinical audits are essen-ial for monitoring the efficiency of service delivery. Aell-designed and comprehensive clinical registry can pro-ide invaluable information for improving the quality ofare for FH [296], consistent with requirements for allnherited cardiovascular conditions [110,297]. Continuingducation and training of all healthcare providers is essen-ial for professional development and service improvement.egular journal clubs can inform on recent advances in
he diagnosis and care of patients with FH. Essential forlinical governance is a clearly identifiable managementtructure headed by a medical director and a clinic or nurseanager [71]. Standard operating procedures, staff respon-
ibilities and credentialling, documentation and essentialesources must all be clearly defined. Marketing the clinic,ncreasing the number of referrals, devising an effective busi-ess plan and generating income for the service, as wells promoting collaborative research, are all highly desir-ble requirements for developing an effective clinic serviceor FH.
3.5. Patient and family support groups
Every effort should be made to initiate and sustain anctive association for patients and families affected by FHia a support group [250,251]. This forum provides a net-ork for individuals and families to form an advocacy group
o develop and promote services within the community, asell as a nucleus for learning and sharing information that
s critical for the detection, management and reduction ofisk in families with FH. The perceptions and views ofatients and their families are clearly important in devel-ping and improving health services for FH [110]. Patientupport for families and the raising of community and gov-rnment awareness about FH is an important function of theupport groups. A national patient support organisation couldfford the best means of establishing a national registry of
oth patients and services, including clinics undertaking cas-ade screening. Such an FH registry could be incorporatednto the Australian National Genetic Heart Disease Registry297].
2 sis Sup
1c
FCiehahtsinAcioFdfgmwiaaodcoFehata
AG
C
MMA
W
RRN
aMNlW
S
SCHCAMCTCH
C
SnRWRJHdRJCtPPFiGtJmAtKdIvCMD
50 G.F. Watts et al. / Atherosclero
3.6. Into the future: chronic care model,ommissioning, evaluation
Future design and development of health provision forH needs to take place within the framework of thehronic Care Model [298], and hence in a positive pol-
cy environment [299]. This will require interacting andstablishing partnerships with a wide spectrum of stake-olders, including patient support groups, heart foundationsnd related non-government organizations, health networks,ealth economists, policy makers and health ministers. Inime, the care of FH could be integrated into national and stateervices for inherited cardiovascular conditions [110]. Theres a major need to increase public and health provider aware-ess of FH through continuing education programs [288].cademic health service systems may be ideally suited for
o-ordinating and integrating the health care of FH and othernherited cardiovascular disorders, but these need to be devel-ped in Australia [300]. Publishing guidance on the care ofH is one thing, but effectively implementing recommen-ations is another, a gap well emphasised by a recent reportrom the UK [301]. Addressing the national and internationalaps in the management of hypercholesterolaemia at a com-unity level will benefit the detection and care of patientsith FH [174–178]. Programs that are successful in deliver-
ng high quality services for FH also have an obligation indvising on and supporting the development of such servicest a national and international level. We emphasize that anptimal FH service provides a classical paradigm and stan-ard for the detection and treatment of other disorders thatause premature atherosclerosis that need attention in theirwn right. Finally, we strongly recommend that our MoC forH be commissioned and incorporated into healthcare deliv-ry and prevention strategies in our community with a veryigh level of priority. It should also be subjected to regularuditing and health economic evaluation, thereby allowinghe MoC to grow into a standard of excellence for the care ofll patients with FH.
ppendix 1. FH Australasia Network Consensusroup and Process
hair
Winthrop Prof. Gerald F. Watts (Lipid Disorders Clinic,etabolic Research Centre and Department of Internaledicine, Royal Perth Hospital, University of Westernustralia, Perth, Western Australia).
riting committee
Winthrop Prof. Gerald F. Watts, Associate Prof. DavidSullivan (Department of Biochemistry and Lipid Clinic,
oyal Prince Alfred Hospital, University of Sydney), Dricola Poplawski (SA Clinical Genetics Service, Women’s
PDMH
plements 12 (2011) 221–263
nd Children’s Hospital, North Adelaide, and Genetics andolecular Pathology Directorate, SA Pathology (WCH site),orth Adelaide), Prof. Frank van Bockxmeer (Cardiovascu-
ar Genetics Laboratory, Royal Perth Hospital, University ofestern Australia).
teering committee
Winthrop Prof. Gerald F. Watts, Associate Prof. Davidullivan, Prof. Frank M. van Bockxmeer, Prof. Ian Hamilton-raig (Preventive Cardiology and Lipid Clinic, Gold Coastospital, Griffith University, Queensland), Prof. Peter M.lifton (Baker IDI Heart and Diabetes Institute, Southustralia), Associate Prof. Richard O’Brien (University ofelbourne, Victoria), Dr Warrick Bishop (Department ofardiology, Calvary Cardiac Centre, Calvary Health Care,asmania), Prof. Peter George (Biochemistry and Pathology,anterbury Health Laboratories, Lipid Clinic, Christchurchospital, Christchurch, New Zealand).
ontributors
Prof Phillip J. Barter (Director, Heart Research Institute,ydney), Dr Timothy Bates (Lipid Disorders Clinic, Inter-al Medicine, Royal Perth Hospital), Clinical Prof. John. Burnett (Core Clinical Pathology & Biochemistry, Path-est Laboratory Medicine WA, Lipid Disorders Clinic,oyal Perth Hospital, University of Western Australia), Dr
ohn Coakley (Paediatrics and Biochemistry, The Children’sospital Westmead, Sydney), Prof. Patricia Davidson (Car-iovascular and Chronic Care, Curtin University, and Nursingesearch, St Vincent’s Hospital, Sydney), Winthrop Prof.
on Emery (School of Primary, Aboriginal and Rural Healthare, University of Western Australia), Dr Andrew Mar-
in (Department of Paediatric and Adolescent Medicine,rincess Margaret Hospital, Perth), Dr Waleed Farid (FHatient Support Group of Western Australia), Ms Lucindareeman (Department of Molecular and Clinical Genet-
cs, Royal Prince Alfred Hospital, Sydney), Prof. Elizabetheelhoed (Health Economics and Policy, School of Popula-
ion Health, University of Western Australia), Ms Amandauniper (Office of Population Health Genomics, Depart-ent of Health, Government of Western Australia), Drlexa Kidd (Clinical Genetics, Canterbury Health Labora-
ories, Christchurch Hospital, New Zealand), Associate Prof.aram Kostner (Cardiac Imaging Group, Department of Car-iology, Mater Hospital, University of Queensland), Prof.nes Krass (Pharmacy Practice, Faculty of Pharmacy, Uni-ersity of Sydney), Mr Michael Livingston (Internationalholesterol Foundation, Sutton-Courtney, Oxfordshire, UK),s Suzy Maxwell (Office of Population Health Genomics,epartment of Health, Government of Western Australia),
rof. Peter O’Leary (Office of Population Health Genomics,epartment of Health, Government of Western Australia),s Amal Owaimrin (Department of Dietetics, Familialypercholesterolaemia Clinical Support Service, Auburn
sis Sup
HDiovNRG&HRPDPcS
C
Sawentioidctctptts
F
sAAabd
D
liPN
PBAItR
A
tm
AF
n
Probable FH 6–8Possible FH 3–5Unlikely FH <3
G.F. Watts et al. / Atherosclero
ospital, Sydney), Emeritus Prof. Trevor Redgrave (Lipidisorders Clinic, Royal Perth Hospital, Department of Phys-
ology, School of Medicine and Pharmacology, Universityf Western Australia), Ms Nicola Reid (Cardiovascular Pre-ention and Lipid Disorders Clinic, Christchurch Hospital,ew Zealand), Ms Lynda Southwell (FH Western Australia,oyal Perth Hospital, University of Western Australia), Drraeme Suthers (SA Clinical Genetics Service, GeneticsMolecular Pathology Directorate, Women’s & Children’s
ospital, Adelaide), Prof. Andrew Tonkin (Cardiovascularesearch Unit, Monash University, Melbourne, Victoria),rof. Simon Towler (Chief Medical Officer, Health Networks,epartment of Health, Government of Western Australia),rof. Ronald Trent (Department of Molecular & Clini-al Genetics, Royal Prince Alfred Hospital, University ofydney).
onsensus process
The Steering Committee met three times in Adelaide,ydney and Brisbane, organised and chaired by GFW. A vari-ble number of other contributors attended these meetings orere invited to comment on evolving drafts of the paper via
mail or telephone. The first meeting discussed a prelimi-ary model of care for FH from Western Australia funded byhe Australia Better Health Initiative and based on materialnitially developed by FH Australasia (GFW, DS). The sec-nd meeting reviewed the core evidence on FH (publishedn the English language on PubMed and webaddresses) andiscussed and critically commented on a first draft of theonsensus paper written by GFW. GFW, DS, NP and FvBhen re-drafted sections of the paper, after reviewing furtheromments by all members of the group. At the third meeting,he Steering Committee re-examined a pre-final draft of theaper and reached consensus on the principal recommenda-ions of the model of care. GFW then prepared a final draft ofhe paper. All members approved the final document beforeubmission.
unding
The meetings of the FH Australasia Network Consen-us Group were supported by educational grants to theustralian Atherosclerosis Society from Pfizer, Abbott,stra-Zeneca and MSD. These companies were not present
t the Consensus Panel meetings and made no contri-utions to the design, content or final approval of theocument.
isclosures
Some members of the Consensus Group have received
ecture honoraria, consultancy fees, and/or research fund-ng from: Pfizer (GFW, DS, AT, TB, WB, IHC, PD, PMC,JB, ROB), Astra Zeneca (GFW, DS, AT, TB, WB, IHC,R, PMC, ROB), MSD (GFW, DS, AT, TB, WB, IHC, NR,plements 12 (2011) 221–263 251
D, PJB), Abbott (GFW, DS, AT, TB, IHC, PJB, ROB),oehringer-Ingelheim (GFW, AT, TB, WB, ROB), Sanofi-ventis (GFW, DS, TB, WB, IHC), Novartis (GFW, AT,
HC, PJB, ROB), GlaxoWellcome (GFW, IHC, ROB), Bris-ol Myers Squibb (AT, ROB), Servier (TB, WB, IHC, PMC,OB), Roche (GFW, PJB, DS).
cknowledgements
We thank Jennifer Seabrook from Meetings First andhe Australian Atherosclerosis Society for co-ordinating the
eetings of the FH Australasia Network Consensus Group.
ppendix 2. Dutch Lipid Clinic Network Criteria forH
Dutch Lipid Clinic Network Criteria for making a diag-osis of FH in adults [34].
Criteria Score
Family historyFirst degree relative with known premature coronary
and/or vascular disease (Men <55 years, Females <60years), OR
1
First degree relative with known LDL-cholesterol>95th percentile for age and sex
First degree relative with tendon xanthomata and/orarcus cornealis, OR
2
Children aged <18 years with LDL-cholesterol>95th percentile for age and sex
Clinical historyPatient with premature coronary artery disease (age
as above)2
Patient with premature cerebral or peripheral vasculardisease (age as above)
1
Physical examinationTendon xanthomata 6Arcus cornealis at age <45 years 4
LDL-cholesterol (mmol/L)LDL-C ≥8.5 8LDL-C 6.5–8.4 5LDL-C 5.0–6.4 3LDL-C 4.0–4.9 1
DNA analysis—functional mutation in the LDLR,APOB or PCSK9 gene
8
Stratification Total score
Definite FH >8

2
A
S
A
M
T
52 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
ppendix 3. Simon Broome Criteria for FH
imon Broome Criteria for the diagnosis of FH [12,41].
Definite FHRaised cholesterol:In children (<16years): total cholesterol > 6.7 mmol/L OR LDL-C > 4.0 mmol/LIn adults (>16 years): Total cholesterol > 7.5 mmol/L OR LDL-C > 4.9 mmol/LANDTendon xanthomata in the patient or in a first or second degree relativeORDNA based evidence of a LDL-receptor, familial defective apo B-100 or PCSK9 mutation
Possible FHRaised cholesterol:In children (<16 years): total cholesterol > 6.7 mmol/L OR LDL-C > 4.0 mmol/LIn adults (> 16 years): total cholesterol > 7.5 mmol/L OR LDL-C > 4.9 mmol/LAND one of the following:Family history of premature myocardial infarctionMI at <50 years in second degree,MI at <60 years in first degree relatives.ORFamily history of raised cholesterolIn adult (>16 years), first or second degree relatives: total cholesterol >7.5 mmol/LIn child (<16 years), first degree relatives: total cholesterol >6.7 mmol/L
ppendix 4. MEDPED Criteria for FH
EDPED Criteria for the diagnosis of FH [42].
Age (years) Total and LDL-cholesterol (mmol/L) criteria for Diagnosing Probable Heterozygous Familial Hypercholesterolaemia (FH)
1st degree relative with FHTC (LDL-C)
2nd degree relative with FHTC (LDL-C)
3rd degree relative with FHTC (LDL-C)
General populationTC (LDL-C)
<20 5.7 (4.0) 5.9 (4.3) 6.2 (4.4) 7.0 (5.2)
20–29 6.2 (4.4) 6.5 (4.6) 6.7 (4.8) 7.5 (5.7)30–39 7.0 (4.9) 7.2 (5.2) 7.5 (5.4) 8.8 (6.2)≥40 7.5 (5.3) 7.8 (5.6) 8.0 (5.8) 9.3 (6.7)C = total cholesterol.
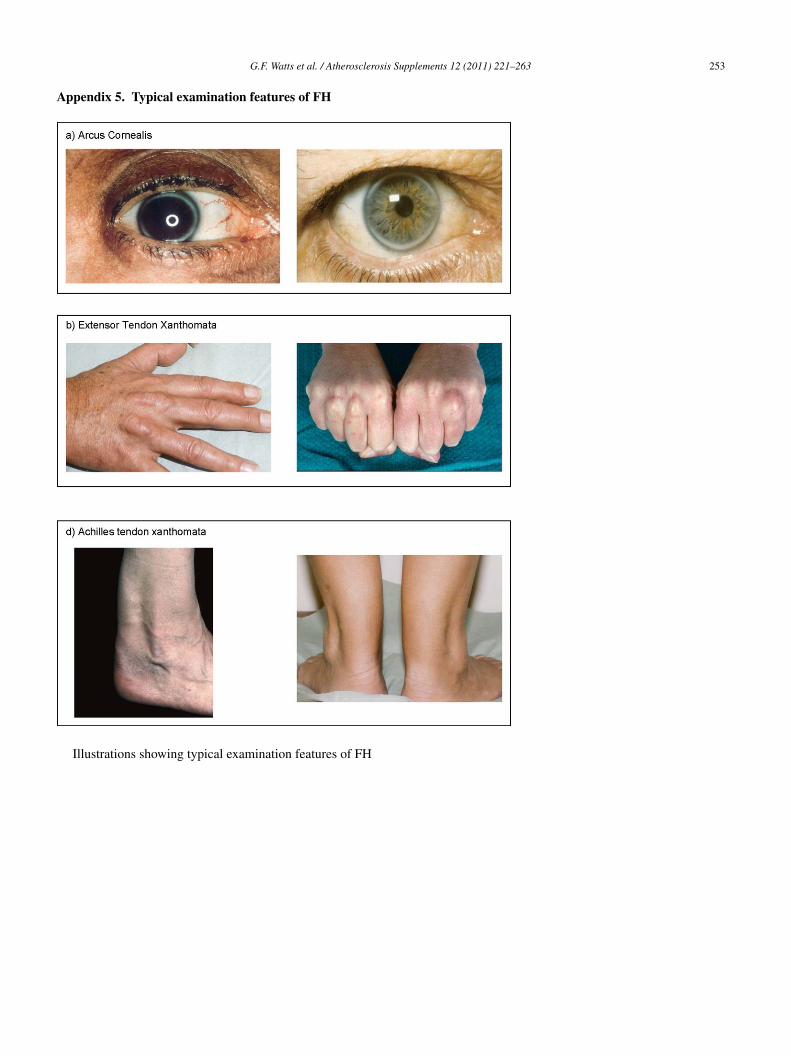
A
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263 253
ppendix 5. Typical examination features of FH
Illustrations showing typical examination features of FH
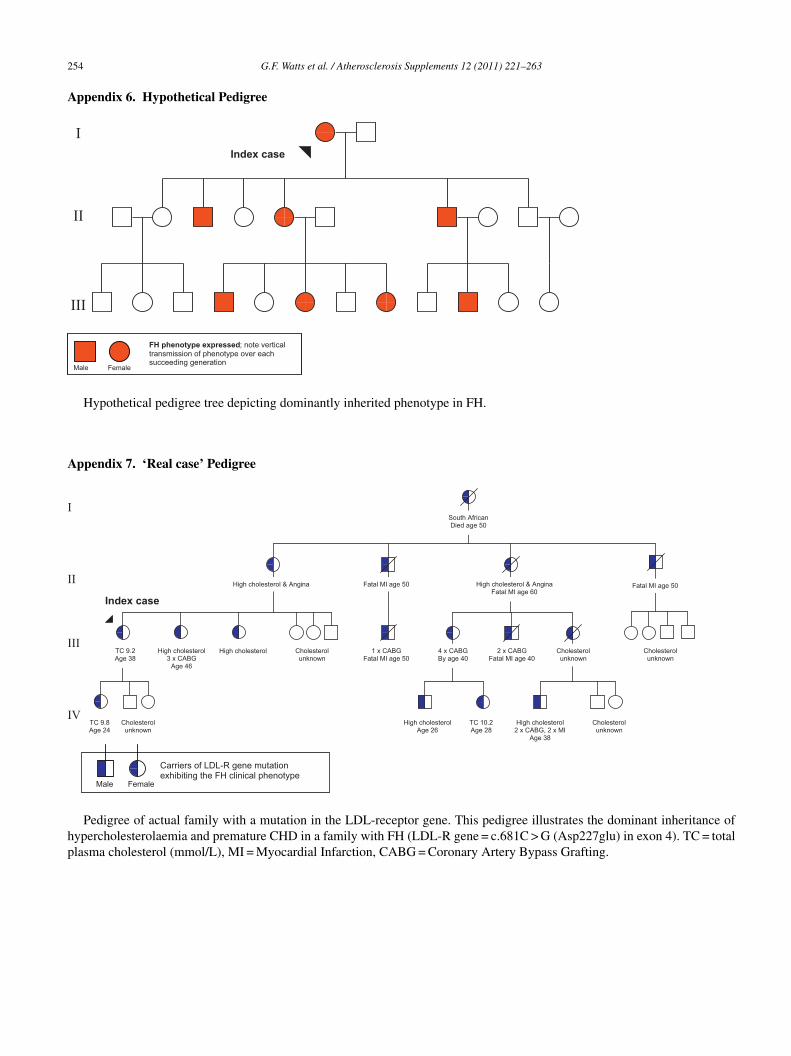
2
A
A
I
I
I
I
hp
54 G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
ppendix 6. Hypothetical Pedigree
Female Male
FH phenotype expressed; note vertical transmission of phenotype over each succeeding generation
Index case
I
II
III
Hypothetical pedigree tree depicting dominantly inherited phenotype in FH.
ppendix 7. ‘Real case’ Pedigree
TC 9.2 Age 38
High cholesterol 3 x CABG
Age 46
High cholesterol
High cholesterol & Angina Fatal MI age 50 High cholesterol & Angina Fatal MI age 60
Fatal MI age 50
Cholesterol unknown
1 x CABG Fatal MI age 50
Cholesterol unknown
High cholesterol 2 x CABG, 2 x MI
Age 38
TC 10.2 Age 28
High cholesterol Age 26
Cholesterol unknown
2 x CABG Fatal MI age 40
4 x CABG By age 40
TC 9.8 Age 24
Cholesterol unknown
Cholesterol unknown
South African Died age 50
Index case
Carriers of LDL-R gene mutation exhibiting the FH clinical phenotype
I
II
V
Male Female
Pedigree of actual family with a mutation in the LDL-receptor gene. This pedigree illustrates the dominant inheritance ofypercholesterolaemia and premature CHD in a family with FH (LDL-R gene = c.681C > G (Asp227glu) in exon 4). TC = totallasma cholesterol (mmol/L), MI = Myocardial Infarction, CABG = Coronary Artery Bypass Grafting.

sis Sup
A
c
R
G.F. Watts et al. / Atherosclero
ppendix 8. Selected websites for clinical services
Selected websites that may be useful for clinical servicesaring for patients with FH.
• International Cholesterol Foundationwww.interchol.orgNew foundation, formed from the merger ofMEDPED-International and HEART-EU, that has a strong focus onFH and provides useful links to international websites of interest topatients, researchers and health professionals.• HEART UKwww.heartuk.org.ukLeading UK cholesterol charity that provides extensive resourcesfor health professionals, patients and families on all aspects of thedetection and management of FH.• Public Health Genomics Foundation, UKwww.phgfoundation.orgInternational foundation that publishes authoritative reports on therole of advances in genomics in health care, and has a particularlyexcellent document on services for inherited cardiovascularconditions.• National Heart Foundation, Australiawww.heartfoundation.org.auLeading Australian charity that provides a wealth of resources forhealth professionals and the community on all aspects of primaryand secondary prevention of cardiovascular disease.• British Heart Foundationwww.bhf.org.ukLeading British foundation provides excellent resources for healthprofessionals and patients, including informative videos on a widespectrum of conditions and risk factors.• Centre for Genetics Education, New South Wales Healthwww.genetics.com.auEducational arm of NSW Genetic Service that provides geneticinformation of individuals and families affected by geneticconditions and health professionals who work with them. Activitiesinclude workshops and training programs.• Human Genetics Society of Australasiawww.hgsa.org.auPremier Australasian society that provides educational materials,training, polices, guidelines and position statements on all aspectsof human genetics.• Lipids Online, Baylor College of Medicinewww.lipidsonline.orgEstablished on-line facility, coordinated by Baylor College ofMedicine (Houston, Texas, USA), providing resources (slides,visual meetings, commentaries), for clinicians, researches andeducators on several aspects of dyslipidaemia, atherosclerosis andcardiovascular disease.• National Lipids Association (NLA), USAwww.lipid.orgUS based multidisciplinary specialty society providing education,training, guidelines and position statements on all aspects of thedetection and management of dyslipidaemia and related disorders.• Learn Your Lipids, NLAwww.learnyourlipids.comInformation for patients with dyslipidaemia, including FH, asprovided by the foundation of the National lipid Association in theUS.• New Zealand Guidelines Groupwww.nzgg.org.nz
New Zealand group of experts that specialises in developing andimplementing guidelines for best clinical practice; excellentresources on the assessment and management of all cardiovascularrisk factors.plements 12 (2011) 221–263 255
• Rational Assessment of Drugs and Research (RADAR),National Prescribing Service (NPS)www.nps.org.au/health professionals/publications/nps radarEvidence based assessment of all new drugs, PBS listings and latestresearch for health professionals provided by the NPS, anindependent organisation funded by the Australian GovernmentDepartment of Health and Ageing.• Make Early Diagnosis Prevent Early Death (MEDPED) FHwww.medped.orgUS based website of the original MEDPED Project coordinated bythe University of Utah School of Medicine (Salt Lake City, UT,USA) focusing on all aspects of the management of FH, includingeducation of patients and families and the first attempt atestablishing a US registry.• Office of Population Genomics, FH Pilot Cascade ScreeningProgram, Western Australiawww.genomics.health.wa.gov.au/fhState funded office that aims to translate genomic knowledge intohealth benefits for WA health; resources provided relevant to thedetection and management of FH• Wales FH Testing Service, Cardiff Universitywww.fhwales.co.ukLeading FH service in the UK that provides useful information andresources for clinical practice, including activities of FH FamilyForum.• FH Support Group of Western Australiawww.fhfamilysupportgroup.websyte.com.auWebsite of the first support group in Australia for families with FH;provides relevant information, communication and support services.• FH Guideline Implementation Team Toolkitwww.heartuk.org.uk/FHToolkitInvaluable resource for implementing the seminal NICE guideline71 on identification and management of FH.• FH Australasia Network, Australian Atherosclerosis Societywww.athero.org.au/FHWebsite of the FH Australasian Network, updated in 2011
eferences
[1] Austin MA, Hutter CM, Zimmern RL, Humphries SE. Geneticcauses of monogenic heterozygous familial hypercholestero-laemia: a HuGE prevalence review. Am J Epidemiol 2004;160:407–20.
[2] Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia:new insights in pathogenesis and treatment. J Clin Invest2003;111:1795–803.
[3] Soutar AK, Naoumova RP. Mechanisms of disease: genetic causesof familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med2007;4:214–25.
[4] Marks D, Thorogood M, Neil HAW, Wonderling D, Humphries SE.Comparing costs and benefits over a 10 year period for strategiesfor familial hypercholesterolaemia screening. J Public Health Med2003;25:47–52.
[5] World Health Organization. Familial hypercholesterolaemia: reportof a WHO consultation. Paris: World Health Organisation;1997.
[6] Marks D, Thorogood M, Humphries S, Neil HAW. Cascade screeningfor familial hypercholesterolaemia: implications of a pilot study fornational screening programmes. J Med Screen 2006;13: 156–9.
[7] Leren TP. Cascade genetic screening for familial hypercholes-
terolemia. Clin Genet 2004;66:483–7.[8] Hadfield SG, Humphries SE. Implementation of cascade testing forthe detection of familial hypercholesterolaemia. Curr Opin Lipidol2005;16:428–33.

2 sis Sup
56 G.F. Watts et al. / Atherosclero[9] Hadfield SG, Horara S, Starr BJ, et al. Family tracing to identifypatients with familial hypercholesterolaemia: the second audit of theDepartment of Health familial hypercholesterolaemia cascade testingproject. Ann Clin Biochem 2009;46:24–32.
[10] Australian and New Zealand Horizon Scanning Unit AustralianGovernment. Genetic screening for familial hypercholesterolaemia.Canberra: Department of Health and Ageing; 2007.
[11] Umans-Eckenhausen MAW, Defesche JC, Sijbrands EJG, ScheerderRLJM, Kastelein JJP. Review of first 5 years of screening forfamilial hypercholesterolaemia in the Netherlands. Lancet 2001;357:165–8.
[12] National Institute for Health and Clinical Excellence, TheNational Collaborating Centre for Primary Care. NICE clinicalguideline 71: identification and management of familial hyperc-holesterolaemia; 2008. Available from: www.nice.org.uk/nicemedia/pdf/CG071NICEGuideline.pdf [2011].
[13] Civeira F. Guidelines for the diagnosis and management ofheterozygous familial hypercholesterolaemia. Atherosclerosis2004;173:55–68.
[14] National Cholesterol Education Program (NCEP). Third report ofthe National Cholesterol Education Program (NCEP) Expert Panelon Detection, Evaluation, and Treatment of High Blood Choles-terol in Adults (Adult Treatment Panel III) final report. Circulation2002;106:3143–421.
[15] Tonkin A, Barter P, Best J, et al. National Heart Foundation ofAustralia and the Cardiac Society of Australia and New Zealand:position statement on lipid management—2005. Heart Lung Circ2005;14:275–91.
[16] Graham I, Atar D, Borch-Johnsen K, et al. European guidelines oncardiovascular disease prevention in clinical practice: executive sum-mary. Eur Heart J 2007;28:2375–414.
[17] (a) Brunzell JD, Davidson M, Furberg CD, et al. Lipoproteinmanagement in patients with cardiometabolic risk. Diabetes Care2008;31:811–22;(b) Reiner Z, Catapano AL, De Backer G, et al. ESC/EASGuidelines for the management of dyslipidaemias. Eur Heart J2011;32:1769–818.
[18] Neil HAW, Hammond T, Huxley R, Matthews DR, HumphriesSE. Extent of underdiagnosis of familial hypercholesterolaemia inroutine practice: prospective registry study. Br Med J 2000;321:148.
[19] Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterollowering treatment of patients with familial hypercholesterolemia:a large cross-sectional study in the Netherlands. Atherosclerosis2010;209:189–94.
[20] Huijgen R, Kindt I, Verhoeven S, et al. Two years after moleculardiagnosis of familial hypercholesterolemia: majority on cholesterol-lowering treatment but a minority reaches treatment goal. PLoS ONE2010;5:e9220.
[21] Hamilton-Craig I. Case-finding for familial hypercholesterolemia inthe Asia-Pacific region. Semin Vasc Med 2004;4:87–92.
[22] Bates TR, Burnett JR, van Bockxmeer FM, Hamilton S, ArnoldaL, Watts GF. Detection of familial hypercholesterolaemia: a majortreatment gap in preventative cardiology. Heart Lung Circ 2008;17:411–3.
[23] Muir LA, George PM, Laurie AD, Reid N, Whitehead L. Preventingcardiovascular disease: a review of the effectiveness of identifyingthe people with familial hypercholesterolaemia in New Zealand. N ZMed J 2010;123:97–102.
[24] Davidson PM, Halcomb EJ, Hickman LD, Phillips JL, Graham B.Beyond the rhetoric: what do we mean by a ‘model of care’? Aust JAdv Nurs 2006;23:47–55.
[25] HEART UK. FH guideline implementation team toolkit; 2010. Avail-
able from: www.heartuk.org.uk/FHToolkit.[26] Datta BN, McDowell IF, Rees A. Integrating provision of specialistlipid services with cascade testing for familial hypercholesterolaemia.Curr Opin Lipidol 2010;21:366–71.
plements 12 (2011) 221–263
[27] Stephenson SH, Larrinaga-Shum S, Hopkins PN. Benefits of theMEDPED treatment support program for patients with familial hyper-cholesterolemia. J Clin Lipidol 2009;3:94–100.
[28] Hayden MR, Josephson R. Development of a program for identi-fication of patients with familial hypercholesterolaemia in BritishColumbia: a model for prevention of coronary disease. Am J Cardiol1993;72:25D–9D.
[29] Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholes-terolemia: screening, diagnosis and management of pediatric andadult patients: clinical guidance from the National Lipid Associa-tion Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol2011;5:133–40.
[30] Wierzbicki AS, Humphries SE, Minhas R, on behalf of the GuidelineDevelopment Group. Familial hypercholesterolaemia: summary ofNICE guidance. Br Med J 2008;337:a1095.
[31] Minhas R, Humphries SE, Qureshi N, Neil HAW, on behalf ofthe NGDG. Controversies in familial hypercholesterolaemia: rec-ommendations of the NICE guideline development group for theidentification and management of familial hypercholesterolaemia.Heart 2009;95:584–7.
[32] National Heart Foundation of Australia, Cardiac Society of Australiaand New Zealand. Reducing risk in heart disease 2007: guidelinesfor preventing cardiovascular events in people with coronary heartdisease; 2008. Available from: www.heartfoundation.org.au/Profes-sional Information/Clinical Practice/Prevention/Pages/default.aspx.
[33] New Zealand Guidelines Group. The assessment and management ofcardiovascular risk. Wellington: NZGG; 2003.
[34] World Health Organization. Familial hypercholesterolaemia. Reportof a second WHO consultation. Geneva: World Health Organization;1999.
[35] Burnett JR, Ravine D, van Bockxmeer FM, Watts GF. Familialhypercholesterolaemia: a look back, a look ahead. Med J Aust2005;182:552–3.
[36] Sullivan D, Members of the CSANZ Cardiovascular Genetics Work-ing Group. Guidelines for the diagnosis and management of familialhypercholesterolaemia. Heart Lung Circ 2007;16:25–7.
[37] Familial Hypercholesterolaemia Western Australia Program Commit-tee. Model of care familial hypercholesterolaemia; 2008. Availablefrom: www.genomics.health.wa.gov.au/fh/index.cfm [cited 28 March2011].
[38] Watts GF, van Bockxmeer FM, Bates T, Burnett JR, Juniper A,O’Leary P. A new model of care for familial hypercholesterolaemiafrom Western Australia: closing a major gap in preventive cardiology.Heart Lung Circ 2010;19:419–22.
[39] National Health and Medical Research Council (NHMRC). NHMRCadditional levels of evidence and grades for recommendations fordevelopers of guidelines. Pilot program 2005–2007; 2005.
[40] Watts GF, Lewis B, Sullivan DR. Familial hypercholesterolaemia: amissed opportunity in preventive medicine. Nat Clin Pract CardiovascMed 2007;4:404–5.
[41] Scientific Steering Committee, Simon Broome Register Group. Riskof fatal coronary heart disease in familial hypercholesterolaemia. Sci-entific Steering Committee on behalf of the Simon Broome Registergroup. Br Med J 1991;303:893–6.
[42] Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing het-erozygous familial hypercholesterolemia using new practical criteriavalidated by molecular genetics. Am J Cardiol 1993;72:171–6.
[43] Marks D, Thorogood M, Neil HAW, Humphries SE. A review on thediagnosis, natural history, and treatment of familial hypercholestero-laemia. Atherosclerosis 2003;168:1–14.
[44] Damgaard D, Larsen ML, Nissen PH, et al. The relationship of molec-ular genetic to clinical diagnosis of familial hypercholesterolemia ina Danish population. Atherosclerosis 2005;180:155–60.
[45] Hopkins PN, Toth PP, Ballantyne CM, Rader DJ. Familial hyper-cholesterolemias: prevalence, genetics, diagnosis and screeningrecommendations from the National Lipid Association Expert Panelon Familial Hypercholesterolemia. J Clin Lipidol 2011;5:S9–17.

sis Sup
G.F. Watts et al. / Atherosclero[46] Starr B, Hadfield G, Hutton BA, et al. Development of sensi-tive and specific age-and gender-specific low-density lipoproteincholesterol cutoffs for diagnosis of first-degree relatives with famil-ial hypercholesterolaemia in cascade testing. Clin Chem Lab Med2008;46:791–803.
[47] Tiyyagura SR, Smith DA. Standard lipid profile. Clin Lab Med2006;26:707–32.
[48] Daniels SR, Greer FR, and the Committee on Nutrition. Lipidscreening and cardiovascular health in childhood. Pediatrics2008;122:198–208.
[49] Freedman DS, Wang YC, Dietz WH, Xu J-H, Srinivasan SR, BerensonGS. Changes and variability in high levels of low-density lipoproteincholesterol among children. Pediatrics 2010;126:266–73.
[50] Chow CK, Islam S, Bautista L, et al. Parental history and myocardialinfarction risk across the world: the interheart study. J Am Coll Cardiol2011;57:619–27.
[51] Emery JD, Walter FM, Ravine D. Family history: the neglected riskfactor in disease prevention. Med J Aust 2010;192:677–8.
[52] Walter FM, Emery J. ‘Coming down the line’—patients’ understand-ing of their family history of common chronic disease. Arch Fam Med2005;3:405–14.
[53] O’Neill SM, Rubinstein WS, Wang C, et al. Familial risk for commondiseases in primary care: the Family HealthwareTM Impact Trial. AmJ Prev Med 2009;36:506–14.
[54] Bates TR, Poulter EB, Van Bockxmeer FM, Watts GF. Family his-tory: the neglected risk factor in disease prevention. Med J Aust2010;193:429–30 [Letter].
[55] Hopkins PN, Stephenson S, Wu LL, Riley WA, Xin Y, Hunt SC. Eval-uation of coronary risk factors in patients with heterozygous familialhypercholesterolemia. Am J Cardiol 2001;87:547–53.
[56] Jansen ACM, van Aalst-Cohen ES, Tanck MW, et al. The con-tribution of classical risk factors to cardiovascular disease infamilial hypercholesterolaemia: data in 2400 patients. J Intern Med2004;256:482–90.
[57] De Sauvage Nolting PRW, Defesche JC, Buirma RJA, Hutten BA,Lansberg PJ, Kastelein JJP. Prevalence and significance of cardio-vascular risk factors in a large cohort of patients with familialhypercholesterolaemia. J Intern Med 2003;253:161–8.
[58] Sniderman A, Couture P, de Graaf J. Diagnosis and treatmentof apolipoprotein B dyslipoproteinemias. Nat Rev Endocrinol2010;6:335–46.
[59] Genest J, McPherson R, Frohlich J, et al. 2009 Canadian Cardiovas-cular Society/Canadian guidelines for the diagnosis and treatmentof dyslipidemia and prevention of cardiovascular disease in theadult—2009 recommendations. Can J Cardiol 2009;25:567–79.
[60] Dorsch MF, Lawrance RA, Durham NP, Hall AS. Familialhypercholesterolaemia is underdiagnosed after AMI. Br Med J2001;322:111.
[61] McKenney JM, Ganz P, Wiggins BS, Saseen JS. Statins. In:Ballantyne CM, editor. Clinical lipidology: a companion to Braun-wald’s heart disease. Philadelphia: Saunders, Elsevier; 2009. p. 253–280.
[62] Ritchie SK, Murphy EC-S, Ice C, et al. Universal versus targeted bloodcholesterol screening among youth: the cardiac project. Pediatrics2010;126:260–5.
[63] Wald DS, Bestwick JP, Wald NJ. Child-parent screening for familialhypercholesterolaemia: screening strategy based on a meta-analysis.Br Med J 2007;335:599.
[64] (a) Daniels SR, Gidding SS, de Ferranti SD. Pediatric aspects of famil-ial hypercholesterolemias: recommendations from the National LipidAssociation Expert Panel on Familial Hypercholesterolemia. J ClinLipidol 2011;5:S30–7;(b) Descamps OS, Tenoutasse S, Stephenne X, et al. Management
of familial hypercholesterolemia in children and young adults: Con-sensus paper developed by a panel of lipidologists, cardiologists,paediatricians, nutritionists, gastroenterologists, general practitionersand a patent organization. Atherosclerosis 2011 (in press).plements 12 (2011) 221–263 257
[65] Marks D, Wonderling D, Thorogood M, Lambert H, HumphriesSE, Neil HAW. Cost effectiveness analysis of different approachesof screening for familial hypercholesterolaemia. Br Med J2002;324:1303–9.
[66] Humphries SE, Norbury G, Leigh S, Hadfield SG, Nair D. What isthe clinical utility of DNA testing in patients with familial hyperc-holesterolaemia? Curr Opin Lipidol 2008;19:362–8.
[67] Gray J, Jaiyeola A, Whiting M, Modell M, Wierzbicki AS. Identi-fying patients with familial hypercholesterolaemia in primary care:an informatics-based approach in one primary care centre. Heart2008;94:754–8.
[68] Qureshi N, Humphries SE, Seed M, Rowlands P, Minhas R, NICEGuideline Development Group. Identification and management offamilial hypercholesterolaemia: What does it mean to primary care?Br J Gen Pract 2009;59:773–8.
[69] Australian Government Department of Health and Ageing.A27 health assessment provided for people aged 45–49 yearswho are at risk of developing chronic disease; 2011. Availablefrom: www9.health.gov.au/mbs/fullDisplay.cfm?type=note&qt=NoteID&q=A27 [9th March 2011].
[70] Peterson G, Fitzmaurice K, Kruup H, Jackson S, Rasiah R. Cardio-vascular risk screening program in Australian community pharmacies.Pharm World Sci 2010;32:373–80.
[71] Mason CM. Development and management of a lipid clinic. In:Davidson MH, Toth PP, Maki KC, editors. Contemporary cardiol-ogy: therapeutic lipidology. Totowa, NJ: Human Press Inc; 2007. p.441–80.
[72] Bairey Merz CN, Alberts MJ, Balady GJ, et al. ACCF/AHA/ACP2009 competence and training statement: a curriculum on preventionof cardiovascular disease. J Am Coll Cardiol 2009;54:1336–63.
[73] Austin MA, Hutter CM, Zimmern RL, Humphries SE. Familial hyper-cholesterolemia and coronary heart disease: a HuGE associationreview. Am J Epidemiol 2004;160:421–9.
[74] Oosterveer DM, Versmissen J, Schinkel AF, Langendonk JG, MulderM, Sijbrands EJ. Clinical and genetic factors influencing cardiovas-cular risk in patients with familial hypercholesterolemia. Clin Lipidol2010;5:189–97.
[75] Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a car-diovascular risk factor: current status. Eur Heart J 2010;31:2844–53.
[76] Taylor AJ, Merz CNB, Udelson JE. Executive summary—canatherosclerosis imaging techniques improve the detection of patientsat risk for ischemic heart disease? J Am Coll Cardiol 2003;41:1860–2.
[77] Naghavi M, Falk E, Hecht HS, et al. From vulnerable plague to vulner-able patient—part III: executive summary of the Screening for HeartAttack Prevention and Education Task Force Report. Am J Cardiol2007;99:1481–2.
[78] Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guide-line for assessment of cardiovascular risk in asymptomatic adults: areport of the American College of Cardiology Foundation/AmericanHeart Association Task Force on Practice Guidelines. Circulation2010;122:2748–64.
[79] Michos ED, Nasir K, Rumberger JA, et al. Relation of family historyof premature coronary heart disease and metabolic risk factors to riskof coronary arterial calcium in asymptomatic subjects. Am J Cardiol2005;95:655–7.
[80] Hutter CM, Austin MA, Humphries SE. Familial hypercholes-terolemia, peripheral arterial disease and stroke: a HuGE minireview.Am J Epidemiol 2004;160:430–5.
[81] Wiegman A, de Groot E, Hutten BA, et al. Arterial intima-media thick-ness in children heterozygous for familial hypercholesterolaemia.Lancet 2004;363:369–70.
[82] Dogan S, Duivenvoorden R, Grobbee DE, et al. Ultrasound proto-
cols to measure carotid intima-media thickness in trials; comparisonof reproducibility, rate of progression, and effect of intervention insubjects with familial hypercholesterolemia and subjects with mixeddyslipidemia. Ann Med 2010;42:447–64.
2 sis Sup
58 G.F. Watts et al. / Atherosclero[83] Andersen LK, Jensen HK, Juul S, Faergeman O. Patients’ attitudestoward detection of heterozygous familial hypercholesterolemia.Arch Intern Med 1997;157:553–60.
[84] Agard A, Bolmsjo I, Hermeren G, Wahlstom J. Familial hyperc-holesterolemia: ethical, practical and psychological problems fromthe perspectives of patients. Patient Educ Couns 2005;57:162–7.
[85] Claassen L, Henneman L, Kindt I, Marteau TM, Timmermans DRM.Perceived risk and representations of cardiovascular disease andpreventive behaviour in people diagnosed with familial hypercholes-terolemia. J Health Psychol 2010;15:33–43.
[86] Marteau T, Senior V, Humphries SE, et al. Psychological impact ofgenetic testing for familial hypercholesterolemia within a previouslyaware population: a randomized controlled trial. Am J Med Genet2004;128A:285–93.
[87] Australian Bureau of Statistics. 4233.0—health literacy,Australia; 2006. Available from: www.abs.gov.au/AUSSTATS/[email protected]/DetailsPage/4233.02006.
[88] von Wagner C, Steptoe A, Wolf MS, Wardle J. Health literacy andhealth actions: a review and a framework from health psychology.Health Educ Behav 2009;36:860–77.
[89] Frich JC, Ose L, Malterud K, Fugelli P. Perceived vulnerability to heartdisease in patients with familial hypercholesterolemia: a qualitativeinterview study. Arch Fam Med 2006;4:198–204.
[90] Walter FM, Emery J, Braithwaite D, Marteau TM. Lay understandingof familial risk of common chronic diseases: a systematic review andsynthesis of qualitative research. Arch Fam Med 2004;2:583–94.
[91] Mark DB, Berman DS, Budoff MJ, et al.ACCF/ACR/AHA/NASCI/SAIP/SCAI/SCCT 2010 expert consensusdocument on coronary computed tomographic angiography: a reportof the American College of Cardiology Foundation Task Force onExpert Consensus Documents. J Am Coll Cardiol 2010;55:2663–99.
[92] Lloyd-Jones DM. Cardiovascular risk prediction: basic concepts, cur-rent status, and future directions. Circulation 2010;121:1768–77.
[93] Dunstan D, Zimmet P, Welborn T, et al. Diabesity & associated disor-ders in Australia—2000: the accelerating epidemic. Melbourne: TheAustralian Diabetes, Obesity and Lifestyle Study; 2001.
[94] Grundy SM. Metabolic syndrome pandemic. Arterioscler ThrombVasc Biol 2008;28:629–36.
[95] Thompson GR, HEART-UK LDL Apheresis Working Group. Rec-ommendations for the use of LDL apheresis. Atherosclerosis2008;198:247–55.
[96] Rodenburg J, Vissers MN, Wiegman A, Trip MD, Bakker HD,Kastelein JJP. Familial hypercholesterolaemia in children. Curr OpinLipidol 2004;15:405–11.
[97] Kavey R-EW, Allada V, Daniels SR, et al. Cardiovascular risk reduc-tion in high-risk pediatric patients: a scientific statement from theAmerican Heart Association Expert Panel on Population and Pre-vention Science; the Councils on Cardiovascular Disease in theYoung, Epidemiology and Prevention, Nutrition, Physical Activ-ity and Metabolism, High Blood Pressure Research. CardiovascularNursing, and the Kidney in Heart disease; and the interdisciplinaryworking group on quality of care and outcomes research. Circulation2006;114:2710–38.
[98] McCrindle BW, Urbina EM, Dennison BA, et al. Drug therapy ofhigh risk lipid abnormalities in children and adolescents. Circulation2007;115:1–20.
[99] van der Graaf A, Kastelein J, Wiegman A. Heterozygous familialhypercholesterolaemia in childhood: cardiovascular risk prevention.J Inherit Metab Dis 2009;32:699–705.
[100] O’Gorman C, Higgins M, O’Neill M. Systematic review and meta-analysis of statins for heterozygous familial hypercholesterolemia inchildren: evaluation of cholesterol changes and side effects. PediatrCardiol 2009;30:482–9.
[101] de Jongh S, Lilien MR, Bakker HD, Hutten BA, Kastelein JJP,Stroes ESG. Family history of cardiovascular events and endothe-lial dysfunction in children with familial hypercholesterolemia.Atherosclerosis 2002;163:193–7.
plements 12 (2011) 221–263
[102] Steinberg D, Glass CK, Witztum JL. Evidence mandating earlierand more aggressive treatment of hypercholesterolemia. Circulation2008;118:672–7.
[103] Obarzanek E, Kimm SY, Barton BA, et al. Long term safetyand efficacy of a cholesterol lowering diet in children with ele-vated low density lipoprotein cholesterol: seven year results of thedietary intervention study in children (DISC). Paediatrics 2001;107:256–64.
[104] de Jongh S, Lilien MR, op’t Roodt J, Stroes ESG, Bakker HD,Kastelein JJP. Early statin therapy restores endothelial function inchildren with familial hypercholesterolemia. J Am Coll Cardiol2002;40:2117–21.
[105] Avis HJ, Vissers MN, Stein EA, et al. A systematic review andmeta-analysis of statin therapy in children with familial hypercholes-terolemia. Arterioscler Thromb Vasc Biol 2007;27:1803–10.
[106] Arambepola C, Farmer AJ, Perera R, Neil HAW. Statin treatmentfor children and adolescents with heterozygous familial hypercholes-terolaemia: a systematic review and meta-analysis. Atherosclerosis2007;195:339–47.
[107] Rodenburg J, Vissers MN, Wiegman A, et al. Statin treatment inchildren with familial hypercholesterolemia: the younger, the better.Circulation 2007;116:664–8.
[108] Vuorio A, Kuoppala J, Kovanen PT, et al. Statins for children withfamilial hypercholesterolemia. Cochrane Database Syst Rev 2010;(7).Art. No.: CD006401.
[109] van Maarle MC, Stouthard MEA, Bonsel GJ. Quality of life in a familybased genetic cascade screening programme for familial hypercholes-terolaemia: a longitudinal study among participants. J Med Genet2003;40, e3–e.
[110] Burton H, Alberg C, Stewart A. Heart to heart: inher-ited cardiovascular conditions services. A needs assessmentand service review. PHG Foundation; 2009. Available from:www.phgfoundation.org/reports/4986/.
[111] Humphries SE, Galton D, Nicholls P. Genetic testing for famil-ial hypercholesterolaemia: practical and ethical issues. Q J Med1997;90:169–81.
[112] Humphries SE, Whittall RA, Hubbart CS, et al. Genetic causes offamilial hypercholesterolaemia in patients in the UK: relation toplasma lipid levels and coronary heart disease risk. J Med Genet2006;43:943–9.
[113] World Health Organization. Review of ethical issues in medical genet-ics. Geneva: World Health Organisation; 2003.
[114] European Society of Human Genetics. Genetic testing in asymp-tomatic minors: recommendations of the European Society of HumanGenetics. Eur J Hum Genet 2009;17:720–1.
[115] Human Genetics Society of Australasia. Position statement: pre-symptomatic and predictive testing in children and young people;2008. Available from: www.hgsa.org.au/2009/12/pre-symptomatic-and-predictive-testing-in-children-and-young-people/ [25 January2011].
[116] Australian Government Department of Health and Ageing.PBS-eligibility criteria for lipid lowering drugs fact sheet; 2006. Can-berra. Available from: www.health.gov.au/internet/main/publishing.nsf/content/lipid eligibilitycriteria.htm [March 2010].
[117] Benlian P, Turquet A, Carrat F, et al. Diagnosis scoring for clinicalidentification of children with heterozygous familial hypercholes-terolemia. J Pediatr Gastroenterol Nutr 2009;48:456–63.
[118] van der Graaf A, Avis HJ, Kusters DM, et al. Molecular basis of auto-somal dominant hypercholesterolemia: assessment in a large cohortof hypercholesterolemic children. Circulation 2011;123:1167–73.
[119] Newson AJ, Humphries SE. Cascade testing in familial hypercholes-terolaemia: How should family members be contacted? Eur J HumGenet 2005;13:401–8.
[120] Suthers GK, Armstrong J, McCormack J, Trott D. Letting thefamily know: balancing ethics and effectiveness when notifying rel-atives about genetic testing for a familial disorder. J Med Genet2006;43:665–70.

sis Sup
G.F. Watts et al. / Atherosclero[121] Human Genetics Society of Australasia. Process of genetic coun-selling; 2008. Available from: www.hgsa.org.au/2009/12/process-of-genetic-counselling/ [25 January 2011].
[122] Urbina EM, Williams RV, Alpert BS, et al. Noninvasive assess-ment of subclinical atherosclerosis in children and adolescents:recommendations for standard assessment for clinical research: a sci-entific statement from the American Heart Association. Hypertension2009;54:919–50.
[123] Ose L. Diagnostic, clinical, and therapeutic aspects of familial hyper-cholesterolemia in children. Semin Vasc Med 2004;4:51–7.
[124] Tonstad S. Stratification of risk in children with familial hypercholes-terolemia with focus on psychosocial issues. Nutr Metab CardiovascDis 2001;11(Suppl 5):64–7.
[125] American Diabetes Association. Standards of medical care indiabetes—2010. Diabetes Care 2010;33:S11–61.
[126] Chobanian AV, Bakris GL, Black HR, et al. Seventh report of thejoint national committee on prevention, detection, evaluation, andtreatment of high blood pressure. Hypertension 2003;42:1206–52.
[127] National Heart Foundation of Australia (National Blood Pressureand Vascular Disease Advisory Committee). Guide to managementof hypertension 2008; 2010. Updated December. Available from:www.heartfoundation.org.au/SiteCollectionDocuments/A HypertGuidelines2008 2010Update FINAL.pdf.
[128] Robinson JG, Goldberg AC. Treatment of adults with familial hyper-cholesterolemia and evidence for treatment: recommendations fromthe National Lipid Association Expert Panel on Familial Hypercholes-terolemia. J Clin Lipidol 2011;5:S18–29.
[129] Ito MK, McGowan MP, Moriarty PM. Management of familialhypercholesterolemias in adult patients: recommendations from theNational Lipid Association Expert Panel on Familial Hypercholes-terolemia. J Clin Lipidol 2011;5:S38–45.
[130] Cooper A, O’Flynn N, on behalf of the Guideline Development Group.Risk assessment and lipid modification for primary and secondaryprevention of cardiovascular disease: summary of NICE guidance. BrMed J 2008;336:1246–8.
[131] Connor WE, Connor SL. Importance of diet in the treatment of famil-ial hypercholesterolemia. Am J Cardiol 1993;72:D42–53.
[132] Lichtenstein AH, Appel LJ, Brands M, et al. Diet and lifestyle recom-mendations revision 2006: a scientific statement from the AmericanHeart Association Nutrition Committee. Circulation 2006;114:82–96.
[133] Gidding SS, Lichtenstein AH, Faith MS, et al. Implementing Amer-ican Heart Association pediatric and adult nutrition guidelines: ascientific statement from the American Heart Association Nutri-tion Committee of the Council on Nutrition, Physical Activity andMetabolism, Council on Cardiovascular Disease in the Young, Coun-cil on Arteriosclerosis, Thrombosis and Vascular Biology, Councilon Cardiovascular Nursing, Council on Epidemiology and Preven-tion, and Council for High Blood Pressure Research. Circulation2009;119:1161–75.
[134] Moruisi KG, Oosthuizen W, Opperman AM. Phytosterols/stanolslower cholesterol concentrations in familial hypercholesterolemicsubjects: a systematic review with meta-analysis. J Am Coll Nutr2006;25:41–8.
[135] Artinian NT, Fletcher GF, Mozaffarian D, et al. Interventions to pro-mote physical activity and dietary lifestyle changes for cardiovascularrisk factor reduction in adults: a scientific statement from the Ameri-can Heart Association. Circulation 2010;122:406–41.
[136] Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factorson the pathogenesis of cardiovascular disease and implications fortherapy. Circulation 1999;99:2192–217.
[137] Pharmaceutical Management Agency (PHARMAC). Pharma-ceutical schedule. 2011. Available from: www.pharmac.govt.nz/2011/03/01/Schedule.pdf.
[138] Baigent C, Keech A, Kearney PM, et al. Efficacy and safety ofcholesterol-lowering treatment: prospective meta-analysis of datafrom 90,056 participants in 14 randomised trials of statins. Lancet2005;366:1267–78.
plements 12 (2011) 221–263 259
[139] Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in peoplewithout established cardiovascular disease but with cardiovascularrisk factors: meta-analysis of randomised controlled trials. Br Med J2009;338:b2376.
[140] Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy andsafety of more intensive lowering of LDL cholesterol: a meta-analysisof data from 170 000 participants in 26 randomised trials. Lancet2010;376:1670–81.
[141] Ward S, Lloyd-Jones M, Pandor A, et al. A systematic review andeconomic evaluation of statins for the prevention of coronary events.Health Technol Assess 2007;11:1–160.
[142] Scientific Steering Committee, Simon Broome Register Group.Mortality in treated heterozygous familial hypercholesterolaemia:implications for clinical management. Atherosclerosis 1999;142:105–12.
[143] Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy ofstatins in familial hypercholesterolaemia: a long term cohort study.Br Med J 2008;337:a2423.
[144] Neil A, Cooper J, Betteridge J, et al. Reductions in all-cause, cancer,and coronary mortality in statin-treated patients with heterozygousfamilial hypercholesterolaemia: a prospective registry study. EurHeart J 2008;29:2625–33.
[145] Harada-Shiba M, Sugisawa T, Makino H, et al. Impact of statintreatment on the clinical fate of heterozygous familial hypercholes-terolemia. J Atheroscler Thromb 2010;17:667–74.
[146] Smilde TJ, van Wissen S, Wollersheim H, Trip MD, KasteleinJJ, Stalenhoef AF. Effect of aggressive versus conventional lipidlowering on atherosclerosis progression in familial hypercholestero-laemia (ASAP): a prospective, randomised, double-blind trial. Lancet2001;357:577–81.
[147] Alonso R, Fernandez de Bobadilla J, Mendez I, Lazaro P, MataN. Cost-effectiveness of managing familial hypercholesterolemiausing atorvastatin-based preventive therapy. Rev Esp Cardiol2008;61:382–93.
[148] Nherera L, Calvert NW, DeMott K, et al. Cost-effectiveness analysisof the use of a high-intensity statin compared to a low-intensity statinin the management of patients with familial hypercholesterolaemia.Curr Med Res Opin 2010;26:529–36.
[149] National Institute for Health and Clinical Excellence. Ezetim-ibe for the treatment of primary (heterozygous-familial andnon-familial) hypercholesterolaemia; 2010. Available from: guid-ance.nice.org.uk/nicemedia/live/11886/38213/38213.doc.
[150] Hamilton-Craig I, Kostner K, Colquhoun D, Woodhouse S. Com-bination therapy of statin and ezetimibe for the treatment offamilial hypercholesterolaemia. Vasc Health Risk Manag 2010;6:1023–37.
[151] Toth PP. Drug treatment of hyperlipidaemia: a guide to the rationaluse of lipid-lowering drugs. Drugs 2010;70:1363–79.
[152] Chapman MJ, Redfern JS, McGovern ME, Giral P. Niacin and fibratesin atherogenic dyslipidemia: pharmacotherapy to reduce cardiovas-cular risk. Pharmacol Ther 2010;126:314–45.
[153] Guyton JR, Goldgerg AC. Bile acid sequestrants. In: Ballantyne CM,editor. Clinical lipidology: a companion to Braunwald’s heart disease.Philadelphia: Saunders, Elsevier; 2009. p. 281–314.
[154] Huijgen R, Abbink EJ, Bruckert E, et al. Colesevelam added to com-bination therapy with a statin and ezetimibe in patients with familialhypercholesterolemia: a 12-week, multicenter, randomized, double-blind, controlled trial. Clin Ther 2010;32:615–25.
[155] Kane JP, Malloy MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ.Regression of coronary atherosclerosis during treatment of familialhypercholesterolemia with combined drug regimens. J Am Med Assoc1990;264:3007–12.
[156] Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety con-
siderations with fibrate therapy. Am J Cardiol 2007;99:S3–18.[157] Watts GF, Karpe F. Triglycerides and atherogenic dyslipidaemia:extending treatment beyond statins in the high-risk cardiovascularpatient. Heart 2011;97:350–6.

2 sis Sup
60 G.F. Watts et al. / Atherosclero[158] Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardio-vascular disease: a scientific statement from the American HeartAssociation. Circulation 2011;123(May):2292–333.
[159] Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-richlipoproteins and high-density lipoprotein cholesterol in patients athigh risk of cardiovascular disease: evidence and guidance for man-agement. Eur Heart J 2011.
[160] Saravanan P, Davidson NC, Schmidt EB, Calder PC. Cardiovasculareffects of marine omega-3 fatty acids. Lancet 2010;376:540–50.
[161] Bays H. Rationale for prescription omega-3-acid ethyl ester therapyfor hypertriglyceridemia: a primer for clinicians. Drugs Today (Barc)2008;44:205–46.
[162] Yamashita S, Matsuzawa Y. Where are we with probucol: a new lifefor an old drug? Atherosclerosis 2009;207:16–23.
[163] Bottorff MB. Statin safety and drug interactions: clinical implications.Am J Cardiol 2006;97:S27–31.
[164] McKenney JM, Davidson MH, Jacobson TA, Guyton JR. Final con-clusions and recommendations of the National Lipid AssociationStatin Safety Assessment Task Force. Am J Cardiol 2006;97:S89–94.
[165] Guyton JR, Bays HE. Safety considerations with niacin therapy. AmJ Cardiol 2007;99:S22–31.
[166] Lee JK, Grace KA, Taylor AJ. Effect of a pharmacy care programon medication adherence and persistence, blood pressure, and low-density lipoprotein cholesterol. J Am Med Assoc 2006;296:2563–71.
[167] Elliott R, Barber N, Clifford S, Horne R, Hartley E. The costeffectiveness of a telephone-based pharmacy advisory service toimprove adherence to newly prescribed medicines. Pharm World Sci2008;30:17–23.
[168] Eussen SR, van der Elst ME, Klungel OH, et al. A pharmaceuticalcare program to improve adherence to statin therapy: a randomizedcontrolled trial. Ann Pharmacother 2010;44:1905–13.
[169] Choudhry NK, Fischer MA, Avorn J, et al. The implications of thera-peutic complexity on adherence to cardiovascular medications. ArchIntern Med 2011:E1–9.
[170] Bates TR, Connaughton VM, Watts GF. Non-adherence to statintherapy: a major challenge for preventive cardiology. Expert OpinPharmacother 2009;10:2973–85.
[171] Senior V, Marteau T, Weinman J. Self-reported adherence tocholesterol-lowering medication in patients with familial hyperc-holesterolaemia: the role of illness perceptions. Cardiovasc DrugsTher 2004;18:475–81.
[172] Scott TL, Gazmararian JA, Williams MV, Baker DW. Health liter-acy and preventive health care use among Medicare enrollees in amanaged care organization. Med Care 2002;40:395–404.
[173] Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patientswith inadequate health literacy. Fam Med 2004;36:588–94.
[174] Roth GA, Fihn SD, Mokdad AH, Aekplakorn W, Hasegawa T, LimSS. High total serum cholesterol, medication coverage and therapeuticcontrol: an analysis of national health examination survey data fromeight countries. Bull World Health Organ 2011;89:92–101.
[175] Simons LA, Oritz M, Calcino G. Long term persistence with statintherapy: experience in Australia 2006–2010. Aust Fam Physician2011;40:319–22.
[176] Carrington MJ, Retegan C, Johnston CI, Jennings GL, Stewart S.Cholesterol complacency in Australia: time to revisit the basics ofcardiovascular disease prevention. J Clin Nurs 2009;18:678–86.
[177] Carrington MJ, Stewart S. Australia’s cholesterol crossroads: ananalysis of 199,331 GP patient records. Melbourne, Australia:Baker IDI Heart and Diabetes Institute; 2011. Available from:www.bakeridi.edu.au/Assets/Files/Australia’s%20Cholesterol%20Crossroads%20Report FINAL.pdf.
[178] Kotseva K, Wood D, De Backer G, et al. Euroaspire III. Managementof cardiovascular risk factors in asymptomatic high-risk patients in
general practice: cross-sectional survey in 12 European countries. EurJ Cardiovasc Prev Rehabil 2010;17:530–40.[179] Vale M, Jelinek MV, Best JD, on behalf of the COACH study group.How many patients with coronary heart disease are not achieving
plements 12 (2011) 221–263
their risk-factor targets? Experience in Victoria 1996–1998 versus1999–2000. Med J Aust 2002;176:211–5.
[180] Kotseva K, Wood D, De Backer G, De Bacquer D, Pyörälä K, KeilU. Euroaspire III. A survey on the lifestyle, risk factors and use ofcardioprotective drug therapies in coronary patients from 22 Europeancountries. Eur J Cardiovasc Prev Rehabil 2009;16:121–37.
[181] Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild tomoderate muscular symptoms with high-dosage statin therapy inhyperlipidemic patients the PRIMO Study. Cardiovasc Drugs Ther2005;19:403–14.
[182] Bays H, Dujovne C. Colesevelam HCl: a non-systemic lipid alteringdrug. Expert Opin Pharmacother 2003;4:779–90.
[183] Spence JD, Hackam DG. Treating arteries instead of risk fac-tors: a paradigm change in management of atherosclerosis. Stroke2010;41:1193–9.
[184] Rothwell PM. Endarterectomy for symptomatic and asymptomaticcarotid stenosis. Neurol Clin 2008;26:1079–97.
[185] Brott TG, Halperin JL, Abbara S, Bacharach JM, Barr JD, BushRL, et al. 2011 ASA/ACCF/AHA/AANN/AANS/ACR/ASNR/CNS/SAIP/SCAI/SIR/SNIS/SVM/SVS guideline on the management ofpatients with extracranial carotid and vertebral artery disease: a reportof the American College of Cardiology Foundation/American HeartAssociation Task Force on Practice Guidelines, and the AmericanStroke Association, American Association of Neuroscience Nurses,American Association of Neurological Surgeons, American Collegeof Radiology, American Society of Neuroradiology, Congress ofNeurological Surgeons, Society of Atherosclerosis Imaging and Pre-vention, Society for Cardiovascular Angiography and Interventions,Society of Interventional Radiology, Society of NeuroInterventionalSurgery, Society for Vascular Medicine, and Society for VascularSurgery. Circulation 2011;123:1–77.
[186] Thorogood M, Seed M, De Mott K, Guideline Development Group.Management of fertility in women with familial hypercholes-terolaemia: summary of NICE guidance. Br J Obstet Gynaecol2009;116:478–9.
[187] van der Graaf A, Hutten BA, Kastelein JJ, Vissers MN. Prema-ture cardiovascular disease in young women with heterozygousfamilial hypercholesterolemia. Expert Rev Cardiovasc Ther 2006;4:345–51.
[188] Nenseter MS, Lindvig HW, Ueland T, et al. Lipoprotein(a) levels incoronary heart disease-susceptible and -resistant patients with familialhypercholesterolemia. Atherosclerosis 2011. March 2 [Epub ahead ofprint].
[189] Avis HJ, Hutten BA, Twickler MT, et al. Pregnancy in women suf-fering from familial hypercholesterolemia: a harmful period for bothmother and newborn? Curr Opin Lipidol 2009;20:484–90.
[190] Kusters DM, Homsma SJM, Hutten BA, et al. Dilemmas in treatmentof women with familial hypercholesterolaemia during pregnancy.Neth J Med 2010;68:299–303.
[191] van der Graaf A, Vissers MN, Gaudet D, et al. Dyslipidemia of moth-ers with familial hypercholesterolemia deteriorates lipids in adultoffspring. Arterioscler Thromb Vasc Biol 2010;30:2673–7.
[192] Geraedts JPM, De Wert GMWR. Preimplantation genetic diagnosis.Clin Genet 2009;76:315–25.
[193] Barlow-Stewart K, Saleh M, Parasivam G. FS17(c): prenataltesting—CVS & amniocentesis. Sydney: Centre for Genetics Edu-cation; 2007 [18 January 2011] www.genetics.com.au/pdf/factsheets/fs17c.pdf.
[194] Amundsen AL, Ose L, Nenseter MS, Ntanios FY. Plant sterol ester-enriched spread lowers plasma total and LDL cholesterol in childrenwith familial hypercholesterolemia. Am J Clin Nutr 2002;76:338–44.
[195] Amundsen AL, Ntanios F, van der Put N, Ose L. Long-term compli-ance and changes in plasma lipids, plant sterols and carotenoids in
children and parents with FH consuming plant sterol ester-enrichedspread. Eur J Clin Nutr 2004;58:1612–20.[196] de Jongh S, Kerckhoffs MC, Grootenhuis MA, Bakker HD, HeymansHSA, Last BF. Quality of life, anxiety and concerns among statin-

sis Sup
G.F. Watts et al. / Atherosclerotreated children with familial hypercholesterolaemia and their parents.Acta Paediatr 2003;92:1096–101.
[197] van der Graaf A, Cuffie-Jackson C, Vissers MN, et al. Efficacy andsafety of coadministration of ezetimibe and simvastatin in adolescentswith heterozygous familial hypercholesterolemia. J Am Coll Cardiol2008;52:1421–9.
[198] Clauss S, Wai K-M, Kavey R-EW, Kuehl K. Ezetimibe treat-ment of pediatric patients with hypercholesterolemia. J Pediatr2009;154:869–72.
[199] Stein EA, Marais AD, Szamosi T, et al. Colesevelam hydrochloride:efficacy and safety in pediatric subjects with heterozygous familialhypercholesterolemia. J Pediatr 2010;156, 231-6.e3.
[200] Monthly Index of Medical Specialities Australia. MIMSonline 2010. (data version: March 2010), New South Wales,Australia; 2010. Available from: www-mimsonline-com-au.rplibresources.health.wa.gov.au.
[201] Wiegman A, Hutten BA, de Groot E, et al. Efficacy and safety of statintherapy in children with familial hypercholesterolemia: a randomizedcontrolled trial. J Am Med Assoc 2004;292:331–7.
[202] Avis HJ, Hutten BA, Gagne C, et al. Efficacy and safety of rosuvastatintherapy for children with familial hypercholesterolemia. J Am CollCardiol 2010;55:1121–6.
[203] McCrindle BW, Ose L, Marais AD. Efficacy and safety of atorvas-tatin in children and adolescents with familial hypercholesterolemia orsevere hyperlipidemia: a multicenter, randomized, placebo-controlledtrial. J Pediatr 2003;143:74–80.
[204] Engler M, Engler M, Malloy M, et al. Docosahexaenoic acid restoresendothelial function in children with hyperlipidemia: results from theearly study. Int J Clin Pharmacol Ther 2004;42:672–9.
[205] Wierzbicki AS, Viljoen A. Hyperlipidaemia in paediatric patients:the role of lipid-lowering therapy in clinical practice. Drug Saf2010;33:115–25.
[206] Juonala M, Magnussen CG, Venn A, et al. Influence of age onassociations between childhood risk factors and carotid intima-media thickness in adulthood: the Cardiovascular Risk in YoungFinns Study, the Childhood Determinants of Adult Health Study, theBogalusa Heart Study, and the Muscatine Study for the InternationalChildhood Cardiovascular Cohort (i3C) Consortium. Circulation2010;122:2514–20.
[207] Pletcher MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipidscommonly present in young adults and coronary calcium later in life:the CARDIA (Coronary Artery Risk Development in Young Adults)Study. Ann Intern Med 2010;153:137–46.
[208] Dawood OT, Izham M, Ibrahim M, Palaian S. Medication complianceamong children. World J Pediatr 2010;6:200–2.
[209] Hill M, Pawsey M, Cutler A, Holt JL, Goldfield SR. Consensus stan-dards for the care of children and adolescents in Australian healthservices. Med J Aust 2011;194:78–82.
[210] Hudgins LC, Kleinman B, Scheuer A, White S, Gordon BR. Long-term safety and efficacy of low-density lipoprotein apheresis inchildhood for homozygous familial hypercholesterolemia. Am J Car-diol 2008;102:1199–204.
[211] Viner R. Barriers and good practice in transition from paediatric toadult care. J R Soc Med 2001;94:2–4.
[212] Thompson GR. LDL apheresis. Atherosclerosis 2003;167:1–13.[213] Thompsen J, Thompson PD. A systematic review of LDL aphere-
sis in the treatment of cardiovascular disease. Atherosclerosis2006;189:31–8.
[214] Hudgins LC, Gordon BR, Parker TS, Saal SD, Levine DM,Rubin AL. LDL apheresis: an effective and safe treatment forrefractory hypercholesterolemia. Cardiovasc Drug Rev 2002;20:271–80.
[215] Thompson GR, Barbir M, Davies D, et al. Efficacy criteria and choles-
terol targets for LDL apheresis. Atherosclerosis 2010;208:317–21.[216] Thompson GR, Catapano A, Saheb S, et al. Severe hypercholestero-laemia: therapeutic goals and eligibility criteria for LDL apheresis inEurope. Curr Opin Lipidol 2010;21:492–8.
plements 12 (2011) 221–263 261
[217] Szczepiorkowski Z, Bandarenko N, Kim H, et al. Guidelines onthe use of therapeutic apheresis in clinical practice—evidence-basedapproach from the Apheresis Applications Committee of the Ameri-can Society for Apheresis. J Clin Apheresis 2007;22:106–75.
[218] Mabuchi H, Koizumi J, Shimizu M, et al. Long-term efficacy of low-density lipoprotein apheresis on coronary heart disease in familialhypercholesterolemia. Am J Cardiol 1998;82:1489–95.
[219] Aengevaeren WRM, Kroon AA, Stalenhoef AFH, Uijen GJH, VanDer Werf T. Low density lipoprotein apheresis improves regionalmyocardial perfusion in patients with hypercholesterolemia andextensive coronary artery disease: the LDL-Apheresis AtherosclerosisRegression Study (LAARS). J Am Coll Cardiol 1996;28:1696–704.
[220] Marais AD, Firth JC, Blom DJ. Homozygous familial hypercholes-terolemia and its management. Semin Vasc Med 2004;4(43):50.
[221] Cashin-Hemphill L, Noone M, Abbott JF, Waksmonski CA, Lees RS.Low-density lipoprotein apheresis therapy during pregnancy. Am JCardiol 2000;86:1160.
[222] Anedda S, Mura S, Marcello C, Pintus P. HELP LDL-apheresis intwo cases of familial hypercholesterolemic pregnant women. TransfusApher Sci 2011;44:21–4.
[223] HEART UK. Lipid Specialist Sub-Committee. LDL apheresis; 2009.Available from: www.heartuk.org.uk/ldl/.
[224] Nemati MH, Astaneh B. Optimal management of familial hyperc-holesterolemia: treatment and management strategies. Vasc HealthRisk Manag 2010;6:1079–88.
[225] Kakaei F, Nikeghbalian S, Kazemi K, et al. Liver transplantation forhomozygous familial hypercholesterolemia: two case reports. Trans-plant Proc 2009;41:2939–41.
[226] Moini M, Mistry P, Schilsky ML. Liver transplantation for inher-ited metabolic disorders of the liver. Curr Opin Organ Tran2010;15:269–76.
[227] Goldberg AC. Novel therapies and new targets of treatment for famil-ial hypercholesterolemia. J Clin Lipidol 2010;4:350–6.
[228] Visser ME, Kastelein JJ, Stroes ES. Apolipoprotein B synthesis inhi-bition: results from clinical trials. Curr Opin Lipidol 2010;21:319–23.
[229] Duff CJ, Hooper NM. PCSK9: an emerging target for treatment ofhypercholesterolemia. Expert Opin Ther Targets 2011;15:157–68.
[230] Moghadasian M, Frohlich J, Saleem M, Hong J, Qayumi K, Scu-damore C. Surgical management of dyslipidemia: clinical andexperimental evidence. J Invest Surg 2001;14:71–8.
[231] Buchwald H, Rudser KD, Williams SE, Michalek VN, Vagasky J,Connett JE. Overall mortality, incremental life expectancy, and causeof death at 25 years in the program on the surgical control of thehyperlipidemias. Ann Surg 2010;251:1034–40.
[232] Al-Allaf F, Coutelle C, Waddington S, David A, Harbottle R, ThemisM. LDLR-gene therapy for familial hypercholesterolaemia: prob-lems, progress, and perspectives. Int Arch Med 2010;3:36.
[233] Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE,Neil HAW. Screening for hypercholesterolaemia versus case findingfor familial hypercholesterolaemia: a systematic review and cost-effectiveness analysis. Health Technol Assess 2000;4:1–123.
[234] Wonderling D, Umans-Eckenhausen MAW, Marks D, Defesche JC,Kastelein JJP, Thorogood M. Cost-effectiveness analysis of thegenetic screening program for familial hypercholesterolaemia in theNetherlands. Semin Vasc Med 2004;4:97–104.
[235] Oliva J, López-Bastida J, Moreno SG, Mata P, Alonso R. Cost-effectiveness analysis of a genetic screening program in the closerelatives of Spanish patients with familial hypercholesterolemia. RevEsp Cardiol 2009;62:57–65.
[236] Marang-van de Mheen PJ, ten Asbroek AHA, Bonneux L, Bonsel GJ,Klazinga NS. Cost-effectiveness of a family and DNA based screeningprogramme on familial hypercholesterolaemia in the Netherlands. EurHeart J 2002;23:1922–30.
[237] Human Genetics Society of Australasia. Presymptomatic andpredictive testing for genetic disorders; 2005. Available from:www.hgsa.org.au/2009/12/presymptomatic-and-predictive-testing-for-genetic-disorders/ [25 January 2011].

2 sis Sup
62 G.F. Watts et al. / Atherosclero[238] Forrest LE, Delatycki MB, Skene L, Aitken M. Communicatinggenetic information in families—a review of guidelines and positionpapers. Eur J Hum Genet 2007;15:612–8.
[239] National Health and Medical Research Council (NHMRC),Office of the Privacy Commissioner. Use and disclosure ofgenetic information to a patient’s genetic relatives under sec-tion 95AA of the Privacy Act 1988 (cth): guidelines for healthpractitioners in the private sector. NHMRC; 2009. Availablefrom: www.nhmrc.gov.au/ files nhmrc/file/publications/synopses/e96.pdf.
[240] Royal College of Physicians, Royal College of Pathologists, BritishSociety for Human Genetics. Consent and confidentiality in geneticpractice: guidance on genetic testing and sharing genetic information.In: Report of the Joint Committee on Medical Genetics. London:RCP/RCPath/BSHG; 2005.
[241] Saleh M, Barlow-Stewart K, Meiser B, Muchamore I. Challengesfaced by genetics service providers’ practicing in a culturally andlinguistically diverse population: an Australian experience. J GenetCouns 2009;18:436–46.
[242] Progeny. Progeny Clinical 8. 2011. Available from:www.progenygenetics.com/clinical.
[243] PASS Clinical. PASS Clinical vascular. Rijswijk, Netherlands; 2011.Available from: www.pass-software.com.
[244] Maxwell SJ, Molster CM, Poke SJ, O’leary P. Communicating famil-ial hypercholesterolemia genetic information within families. GenTest Mol Bioma 2009;13:301–6.
[245] Clarke A, Martin R, Kerzin-Storrar L, et al. Genetic professionals’reports of nondisclosure of genetic risk information within families.Eur J Hum Genet 2005;13:556–62.
[246] Suthers GK, McCusker EA, Wake SA. Alerting genetic relatives toa risk of serious inherited disease without a patient’s consent. Med JAust 2011;194:385–6.
[247] Greco KE, Salveson C. Identifying genetics and genomics nursingcompetencies common among published recommendations. J NursEduc 2009;48:557–65.
[248] Leren TP, Finborud TH, Manshaus TE, Ose L, Berge KE. Diagnosis offamilial hypercholesterolemia in general practice using clinical diag-nostic criteria or genetic testing as part of cascade genetic screening.Community Genet 2008;11:26–35.
[249] Burke W, Emery J. Genetics education for primary-care providers.Nat Rev Genet 2002;3:561–6.
[250] Cardiff University. Wales familial hypercholesterolaemia testing ser-vice: FH family forum; 2010. Available from: www.fhwales.co.uk [17February 2010].
[251] FH Family Support Group. Available from: www.fhfamily-supportgroup.websyte.com.au [31 March 2011].
[252] Fleury J, Keller C, Perez A, Lee S. The role of lay health advisorsin cardiovascular risk reduction: a review. Am J Community Psychol2009;44:28–42.
[253] Australian Government. Privacy Legislation Amendment Act 2006;2006. Available from: www.comlaw.gov.au/Details/C2006A00099 [6April 2011].
[254] Marang-van de Mheen PJ, van Maarle MC, Stouthard MEA. Gettinginsurance after genetic screening on familial hypercholesterolaemia;the need to educate both insurers and the public to increase adherenceto national guidelines in the Netherlands. J Epidemiol CommunityHealth 2002;56:145–7.
[255] Otlowski MFA, Stranger MJA, Barlow-Stewart K, Taylor S, TreloarS. Investigating genetic discrimination in the Australian life insurancesector: the use of genetic test results in underwriting, 1999–2003. JLaw Med 2007;14:367–96.
[256] Human Genetics Society of Australasia. Position statement:genetic testing and life insurance in Australia; 2008. Available
from: www.hgsa.org.au/2009/12/genetic-testing-and-life-insurance-in-australia/ [25 January 2011].[257] Neil HA, Mant D. Cholesterol screening and life assurance. Br MedJ 1991;302:891–3.
plements 12 (2011) 221–263
[258] Neil HAW, Hammond T, Mant D, Humphries SE. Effect of statintreatment for familial hypercholesterolaemia on life assurance: resultsof consecutive surveys in 1990 and 2002. Br Med J 2004;328:500–1.
[259] Financial Services Council. FSC standard no. 11. Genetic testingpolicy; 2005 , www.ifsa.com.au/downloads/file/FSCStandards/11SGeneticTestingPolicy.pdf.
[260] Alonso R, Defesche JC, Tejedor D, et al. Genetic diagnosis of famil-ial hypercholesterolemia using a DNA-array based platform. ClinBiochem 2009;42:899–903.
[261] Taylor A, Wang D, Patel K, et al. Mutation detection rate and spectrumin familial hypercholesterolaemia patients in the UK pilot cascadeproject. Clin Genet 2010;77:572–80.
[262] Lombardi MP, Redeker EJW, van Gent DHM, Smeele KL, Weed-erstein R, Mannens MM. Molecular genetic testing for familialhypercholesterolaemia in the Netherlands: a stepwise screening strat-egy enhances the mutation detection rate. Genet Test 2006;10:77–84.
[263] DNA Genotek. Oragene DNA. Ontario, Canada: Kanata; 2011 ,www.dnagenotek.com [15 March 2011].
[264] National Pathology Accreditation Advisory Council (NPAAC). Draftrequirements for medical testing of human nucleic acids-public con-sultation. 1st ed. 20xx Canberra: Australian Government, Departmentof Health and Ageing; 2010.
[265] Brice P. PGD for familial hypercholesterolaemia approved. London:PHG Foundation; 2007 , www.phgfoundation.org/news/3947/ [25January 2011].
[266] National Association of Testing Authorities Australia (NATA).Medical testing accreditation 2009; 2009. Available from:www.nata.asn.au/types-of-accreditation [25 January 2011].
[267] Leigh SEA, Foster AH, Whittall RA, Hubbart CS, Humphries SE.Update and analysis of the University College London Low DensityLipoprotein Receptor Familial Hypercholesterolemia Database. AnnHum Genet 2008;72:485–98.
[268] Dedoussis GVZ, Schmidt H, Genschel J. LDL-receptor mutations inEurope. Hum Mutat 2004;24:443–59.
[269] Marduel M, Carrié A, Sassolas A, et al. Molecular spectrum ofautosomal dominant hypercholesterolemia in France. Hum Mutat2010;31:E1811–24.
[270] Laurie AD, Scott RS, George PM. Genetic screening of patients withfamilial hypercholesterolaemia (FH): a New Zealand perspective.Atherosclerosis Suppl 2004;5:13–5.
[271] Civeira F, Jarauta E, Cenarro A, et al. Frequency of low-densitylipoprotein receptor gene mutations in patients with a clinical diag-nosis of familial combined hyperlipidemia in a clinical setting. J AmColl Cardiol 2008;52:1546–53.
[272] Kwiterovich Jr PO. Clinical implications of the molecular basis offamilial hypercholesterolemia and other inherited dyslipidemias. Cir-culation 2011;123:1153–5.
[273] Defesche JC, Lansberg PJ, Umans-Eckenhausen MA, Kastelein JJ.Advanced method for the identification of patients with inheritedhypercholesterolemia. Semin Vasc Med 2004;4:59–65.
[274] Taylor A, Martin B, Wang D, Patel K, Humphries SE, Norbury G.Multiplex ligation-dependent probe amplification analysis to screenfor deletions and duplications of the LDLR gene in patients withfamilial hypercholesterolaemia. Clin Genet 2009;76:69–75.
[275] Holla ØL, Teie C, Berge KE, Leren TP. Identification of deletions andduplications in the low density lipoprotein receptor gene by MLPA.Clin Chim Acta 2005;356:164–71.
[276] Chiou K-R, Charng M-J, Chang H-M. Array-based resequencingfor mutations causing familial hypercholesterolemia. Atherosclerosis2011. March 2 [Epub ahead of print].
[277] Dusková L, Kopecková L, Jansová E, et al. An apex-based genotypingmicroarray for the screening of 168 mutations associated with familial
hypercholesterolemia. Atherosclerosis 2011;216:139–45.[278] Mamotte C, van Bockxmeer F. A robust strategy for screening andconfirmation of familial defective apolipoprotein B-100. Clin Chem1993;39:118–21.

sis Sup
national audit of the management of familial hypercholestero-
G.F. Watts et al. / Atherosclero
[279] Newton CR, Graham A, Heptinstall LE, et al. Analysis of any pointmutation in DNA. The amplification refractory mutation system(ARMS). Nucleic Acids Res 1989;17:2503–16.
[280] Taylor A, Tabrah S, Wang D, et al. analysis to detect 13 commonmutations in familial hypercholesterolaemia. Clin Genet 2007;71:561–8.
[281] Progenika. LIPOchip. Vizcaya, Spain; 2011. Available from:www.progenika.com/eu/index.php?option=com content&task=view&id=138&Itemid=18.
[282] Gen-Probe. Elucigene FH20. Abingdon, Oxfordshire, United King-dom; 2011. Available from: www.gen-probe.com/global/products-services/cardiovascular.aspx.
[283] MRC-Holland. SALSA MLPA kit P062. Amsterdam, Netherlands;2011. Available from: www.mlpa.com.
[284] CLC-Bio. CLC Main Workbench. Aarhus, Denmark; 2011. Availablefrom: www.clcbio.com/index.php?id=92.
[285] Interactive Biosoftware. Alamut. Rouen, France; 2011. Availablefrom: www.interactive-biosoftware.com/alamut.html.
[286] University College London. Low density lipoprotein receptorfamilial hypercholesterolemia database. London, United Kingdom;2011. Available from: www.ucl.ac.uk/ldlr/Current/index.php?selectdb=LDLR.
[287] Bourbon M, Duarte MA, Alves AC, Medeiros AM, Marques L, SoutarAK. Genetic diagnosis of familial hypercholesterolaemia: the impor-tance of functional analysis of potential splice-site mutations. J MedGenet 2009;46:352–7.
[288] Goldberg AC, Robinson JG, Cromwell WC, Ross JL, Ziajka PE.Future issues, public policy, and public awareness of familialhypercholesterolemias: recommendations from the National LipidAssociation Expert Panel on Familial Hypercholesterolemia. J ClinLipidol 2011;5:S46–51.
[289] Aatre RD, Day SM. Psychological issues in genetic testing for inher-ited cardiovascular diseases. Circ Cardiovasc Genet 2011;4:81–90.
[290] Hadfield SG, Horara S, Starr BJ, et al. Are patients with familial hyper-cholesterolaemia well managed in lipid clinics? An audit of elevenclinics from the Department of Health familial hypercholesterolaemiacascade testing project. Ann Clin Biochem 2008;45:199–205.
plements 12 (2011) 221–263 263
[291] Neubeck L, Redfern J, Fernandez R, Briffa T, Bauman A, FreedmanSB. Telehealth interventions for the secondary prevention of coronaryheart disease: a systematic review. Eur J Cardiovasc Prev Rehabil2009;16:281–9.
[292] Clark CE, Smith LFP, Taylor RS, Campbell JL. Nurse led inter-ventions to improve control of blood pressure in people withhypertension: systematic review and meta-analysis. Br Med J2010;341:c3995.
[293] Loveman E, Frampton GK, Clegg AJ. The clinical effectiveness ofdiabetes education models for type 2 diabetes: a systematic review.Health Technol Assess 2008;12:1–116.
[294] Haralambos K. “PASS Clinical” software for familial hypercholes-terolaemia (FH): evaluation report including 3 month clinical pilot;2009. Available from: www.heartuk.org.uk/FHToolkit/.
[295] Braithwaite J, Travaglia JF. An overview of clinical governancepolicies, practices and initiatives. Aust Health Rev 2008;32:10–22.
[296] Evans SM, Bohensky M, Cameron PA, McNeil J. A survey of Aus-tralian clinical registries: can quality of care be measured? Intern MedJ 2011;41:42–8.
[297] Ingles J, McGaughran J, Vohra J, et al. Establishment of an Australiannational genetic heart disease registry. Heart Lung Circ 2008;17:463–7.
[298] World Health Organization. Building blocks for action innovative carefor chronic conditions: global report; 2002.
[299] Coleman K, Austin BT, Brach C, Wagner EH. Evidence on thechronic care model in the new millennium. Health Aff (Millwood)2009;28:75–85.
[300] Fisk NM, Wesselingh SL, Beilby JJ, et al. Academic healthscience centres in Australia: let’s get competitive. Med J Aust2011;194:59–60.
[301] Pedersen KMV, Humphries SE, Roughton M, Besford JS. The
laemia 2010: full report. Clinical Standards Department, RoyalCollege of Physicians; 2010. Available from: www.rcplondon.ac.uk/node/2584.
Related Documents








![Assessment report · lipoprotein (a) [Lp(a)] in patients with homozygous familial hypercholesterolaemia (HoFH) and in patients with severe heterozygous familial hypercholesterolaemia](https://static.cupdf.com/doc/110x72/5e32b9f8d3afcb53c94a2775/assessment-report-lipoprotein-a-lpa-in-patients-with-homozygous-familial-hypercholesterolaemia.jpg)


