Factores Patogénicos de Helicobacter pylori en Cáncer Gástrico Integrantes: Javiera González Pablo Gutiérrez Javier Rojas Paulina Tirado Tutora: Ileana González Talca, 29 de junio de 2012

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Factores Patogénicos de
Helicobacter pylori en
Cáncer Gástrico
Integrantes: Javiera González Pablo Gutiérrez Javier Rojas Paulina Tirado
Tutora: Ileana González
Talca, 29 de junio de 2012

Factores patogénicos de H. pylori en Cáncer Gástrico Página 1
INDICE
Resumen ....................................................................................................................................................... 2
Introducción .................................................................................................................................................. 3
Factores patogénicos de Helicobacter pylori ................................................................................................ 4
Motilidad ................................................................................................................................................... 4
Ureasa ....................................................................................................................................................... 4
Mucinasa y Fosfolipasas ............................................................................................................................ 5
Catalasa y Superóxido Dismutasa ............................................................................................................. 5
Factores de Adherencia ............................................................................................................................ 6
OIPa ....................................................................................................................................................... 6
BabA ...................................................................................................................................................... 8
SabA ...................................................................................................................................................... 8
Implicancia de los receptores para los productos de glicosilación avanzada (RAGE) .......................... 8
VacA: Citotoxina vacuolizante ................................................................................................................... 9
CagA e Islote de Patogenicidad (PAI) ...................................................................................................... 10
Sistema de secreción tipo IV (T4SS) .................................................................................................... 10
Motivos EPIYA ..................................................................................................................................... 11
Fosforilación CagA y SPH-2 ................................................................................................................. 12
Efectos de CagA independientes a la fosforilación. ............................................................................ 13
Papel del CagA en la carcinogénesis gástrica. ..................................................................................... 13
Helicobacter pylori y Cáncer Gástrico ......................................................................................................... 13
Conclusiones ............................................................................................................................................... 15
Referencias .................................................................................................................................................. 17

Factores patogénicos de H. pylori en Cáncer Gástrico Página 2
RESUMEN
En 1994, Helicobacter pylori es reconocido por la International Agency for Research on Cancer y la Organización Mundial para la Salud como un carcinógeno categoría I. Actualmente la infección por H. pylori afecta al 50% de la población mundial y en Chile la prevalencia corresponde al 74.5% y posee tasas de incidencia de cáncer gástrico de 30 x 100.000 habitantes. El desarrollo de cáncer gástrico es condicionado por diversas características del huésped, tales como la genética, desnutrición, infección temprana en la vida, entre otros componentes, los cuales interactúan con los factores de patogenicidad de H. pylori. Entre estos factores se encuentra la motilidad que posee H. pylori, que junto con la presencia de catalasa y Superóxido Dismutasa le permite evadir la respuesta inmune. La Ureasa, la protege del pH ácido del estómago. Las Mucinasa y Fosfolipasas participan en la modificación de la mucosa gástrica y las proteínas de adherencia HopH, BabA y SabA, juegan un rol fundamental en la colonización bacteriana de la mucosa gástrica y en la inflamación. Además los receptores para los productos de glicosilación avanzada (RAGE) también participan en la inflamación y adherencia. La citotoxina VacA induce la vacuolización citoplasmática de células epiteliales. El Islote de patogenicidad codifica para la proteína CagA y para un conjunto de proteínas que conforman el sistema de secreción tipo IV de la bacteria, permitiéndole la transferencia horizontal de genes, la importación de peptidoglicano y de la proteína CagA al interior de la célula epitelial, desencadenando alteraciones en el citoesqueleto. Palabras Claves: Helicobacter pylori, Cáncer Gástrico, Patogenicidad
ABSTRACT
In 1994, Helicobacter pylori was recognized by the International Agency for Research on Cancer and the World Health Organization as a category I carcinogen. At present, the infection by H. pylori affects 50% of world population; in Chile the prevalence of infection by H. pylori is 74,5%, and it has gastric cancer incidence rates of 30 per 100.000 people. The development of gastric cancer depends of infection by H. pylori, several characteristics of the host such as genetics, nutrition, early infection, among other components, which interacts with the patogenicity factors of H. pylori. These factors include motility of H. pylori, and combined with the presence of Catalase and Superoxide Dismutase allows it to evade the immune response. Urease, another enzyme of H. pylori, protects the bacteria from stomach acid pH. Mucinase and Phospholipases involved in alteration of the gastric mucosa, and the adhesion proteins HopH (OipA), BabA and SabA, play an important role in the bacterial colonization of gastric mucosa and inflammation. In addition, the Receptors for Advanced Glycation Endproducts (RAGE) are also involved in inflammation and adhesion. The vacuolating cytotoxin VacA induces cytoplasmatic vacuolization of epithelial cells. The pathogenicity Island of H. pylori encodes the CagA protein, but also encode for a group of proteins that forms the type IV secretion system, allowing horizontal transference of genes, the peptidoglycan and CagA importing into epithelial cells, triggering alterations in the cytoskeleton, morphology and the motility of the host cell. Keyword: Helicobacter pylori, Gastric cáncer, Pathogenicity.

Factores patogénicos de H. pylori en Cáncer Gástrico Página 3
INTRODUCCIÓN
Helicobacter pylori es una bacteria espirilada gram-negativa multiflagelada y su nicho ecológico es el estómago humano. El estómago, por el bajo pH de su mucosa, posee una potente actividad bactericida, pese a esto H. pylori logra evadir esta actividad con la producción de ureasa, creando un nicho neutro protector. Además, sus flagelos le facilitan penetrar y anidarse bajo la capa de mucus evadiendo la respuesta inmune del huésped y la adhesión a las células epiteliales mediadas por adhesinas le confiere la facultad de colonizar la mucosa gástrica y establecer una permanencia de largo plazo condicionando distintas lesiones inflamatorias [1]. La vía predominante de transmisión de esta bacteria aún no está aclarada, pero se ha propuesto una ruta gastro-oral o fecal-oral como posible vía de transmisión. Otros medios de infección son la ingesta de agua y alimentos contaminados [2].
En el año 1983 Marshall y Warren publicaron influyentes artículos en The Lancet sugiriendo que el cáncer gástrico podía estar relacionado con H. pylori abriendo un debate científico internacional [3]. Estos científicos fundaron el concepto de que la infección por H. pylori podía conducir a una variedad de trastornos gastrointestinales superiores, tales como inflamación gástrica (gastritis), enfermedad de úlcera péptica, adenocarcinoma gástrico distal y de la mucosa gástrica asociada a tejido linfoide (MALT) [4]. En 1994, H. pylori fue reconocido por la International Agency for Research on Cancer y la Organización Mundial para la Salud (OMS) como un carcinógeno categoría I, por lo que
fue recomendado que todos los pacientes H. pylori positivo debían recibir terapia de erradicación [5].
Actualmente la infección por H. pylori constituye probablemente la infección crónica más extensamente difundida en la especie humana, afectando al 50% de la población mundial y hasta el 90% de la que vive en países subdesarrollados. Un pequeño grupo de países desarrollados, presenta prevalencias que oscilan entre el 20 y 40%, mientras un gran grupo de países, pertenecientes al mundo en desarrollo, presentan frecuencias entre el 70 y 90% [6]. La mayoría de las poblaciones muestran una incidencia dos o tres veces superior en las clases socioeconómicas bajas respecto de las altas [7], sin embargo en Chile, esta diferencia en la incidencia según clases sociales no se ha podido objetivar [8]. En Chile la prevalencia de infección por H. pylori es de 74.5% [9]. Además nuestro país, históricamente se ha encontrado entre los países con el mayor riesgo de cáncer gástrico del mundo [10], mantiene tasas de incidencia de 30 x 100.000 habitantes y la región que tiene los más altos índices de cáncer gástrico es la Séptima Región del Maule que cuenta con tasas de mortalidad que llegan hasta 48.2 casos por cada 100,000 habitantes en la comuna de Molina [11].
Ha llamado la atención que en lugares donde hay una alta prevalencia de la infección existe una baja incidencia de cáncer gástrico, como en el llamado “Enigma Africano”. [12] Esta diferencia en la incidencia de cáncer gástrico puede ser explicada porque H. pylori en cada persona no actúa de la misma manera, está determinado por la interacción de: a) los factores patogénicos de la bacteria; b) la respuesta inmunológica del huésped (condicionada además por la susceptibilidad genética); c) la convergencia de ambos factores, que puede ser influenciada por

Factores patogénicos de H. pylori en Cáncer Gástrico Página 4
condiciones ambientales tales como la infección temprana en la vida, desnutrición, ingesta de compuestos nitrosos, déficit en vitamina C, tabaquismo, entre otros factores.[1]
En esta revisión se tratarán los diversos factores patogénicos que posee H. pylori y su asociación con el cáncer gástrico.
FACTORES PATOGÉNICOS DE HELICOBACTER PYLORI
MOTILIDAD
H. pylori posee un haz apical de 3 a 5 flagelos, los cuales están compuestos de tres elementos estructurales: el cuerpo basal, el codo y el filamento. El filamento actúa como un propulsor cuando rota en su base y es un copolímero de flagelinas FlaA y FlaB, donde FlaA es predominante [13]. Al contrario de los flagelos de las bacterias entéricas, el flagelo de H. pylori posee una vaina, extensión de la membrana exterior, que protege a la flagelina de la digestión proteica [14]. Una cualidad interesante de FlaA es la baja actividad intrínseca para activar el receptor tipo Toll 5 (TLR5), lo que contribuye a la evasión de la respuesta inmune que ejecuta el huésped.
La bacteria, a su vez, se mueve desde el ambiente ácido del estómago hacia la superficie epitelial, atravesando la mucina, donde el pH es más alcalino [15]. Incluso se piensa que existe un mecanismo quimiotáctico similar al de las bacterias entéricas, donde éstas son atraídas por la urea. [14]
Así, la motilidad parece ser un factor esencial para el éxito de la colonización gástrica y una contribución a la carcinogénesis.
UREASA
La ureasa de H. pylori tiene una masa molecular de 550 kDa aproximadamente y es una proteína con un residuo de Níquel consistente de seis copias cada una con dos subunidades, UreA de 30 kDa y UreB de 62 kDa. El cluster del gen de la ureasa está formado por 7 genes: los genes UreA y UreB que originan las subunidades principales antes mencionadas, y los genes accesorios ureI, ureE, ureF, ureG, y ureH. [16,17] De estos, UreA, UreG y UreH son genotipos predominantes encontrados en los tejidos neoplásicos gástricos [18]. Posee una constante de Michaelis baja, lo que le permite a esta enzima ser catalíticamente eficiente incluso a concentraciones mínimas de urea [16].
Diversos estudios precisan que uno de los roles de esta enzima en la patogénesis es la regulación del pH durante el tiempo en el que H. pylori está presente en el estómago,
[19,20] generando una tolerancia al medio ácido hostil donde la bacteria se encuentra habitando. Cuando el pH del ambiente es reducido desde 6,5 hacia un pH de 2,5; la actividad enzimática de la ureasa aumenta entre 10-20 veces[17]. La exposición prolongada de H. pylori en pH ácido (pH menor a 4) sin la urea como sustrato disminuye la síntesis proteica [16].
La distinción clave entre H. pylori y presumiblemente otras especies de Helicobacter gástricas y otros neutralófilos acido-resistentes es la capacidad para mantener un pH periplasmático a un nivel de generar una fuerza Protón-Motriz que permita el crecimiento celular continuo en el ambiente gástrico [17]. Este efecto proliferativo estimulante de la ureasa asociada a H. pylori, puede ser considerada un potencial carcinógeno del cáncer gástrico [18,20].

Factores patogénicos de H. pylori en Cáncer Gástrico Página 5
MUCINASA Y FOSFOLIPASAS
Estas dos enzimas cumplen funciones similares: modifican la mucosidad gástrica. La mucinasa de H. pylori se origina a partir de un gen parecido al de la enzima en Vibrio cholerae, el cual participa en la ruptura de la barrera de mucus gástrico [21]. En la misma línea, la fosfolipasa A forma parte en la degradación lipídica del mucus y en la hidrólisis de los fosfolípidos gástricos, causando un daño de la mucosa [22].
Por otra parte, se ha descrito que H. pylori puede espontáneamente y reversiblemente modificar su composición lipídica de la membrana. Es clave en este proceso la acción de la fosfolipasa A de membrana externa (OMPLA, sigla en ingles de outer membrane phospholipase A). Este cambio reversible en la configuración de la membrana lipídica es ocasionada por una variación en el gen HP0499 (gen pldA), el cual podría contribuir a la sobrevida de la bacteria en pH ácido [22].
CATALASA Y SUPERÓXIDO DISMUTASA
Ambas proteínas, catalasa y Superóxido Dismutasa, cumplen el mismo rol en la virulencia de H. pylori: evadir la actividad fagocítica. Esta bacteria es particularmente vulnerable a los efectos nocivos del oxígeno y el estrés oxidativo [15].
La catalasa (KatA) protege a H. pylori contra los efectos dañinos del peróxido de hidrógeno, una de las especies reactivas de oxígeno que son liberadas durante los procesos inflamatorios donde se involucran principalmente los polimorfonucleares (PMNs), para mantener un ambiente estable para el crecimiento del microorganismo [16,20,23,24]. Es una proteína homotetramérica, predominantemente citosólica (1% de la
proteínas de H. pylori), aunque existe información de una localización periplásmica y de superficie [24, 25]. El punto isoeléctrico de la catalasa de H. pylori es entre 9-9,3, el cual es una característica única de ella, agregando también que la catalasa no es estable en el jugo gástrico [23,
26]. Además, es capaz de inhibir la fusión intracelular de los fagosomas con lisosomas, agregando que la sobrevida de H. pylori en los fagosomas de macrófagos depende de la actividad de la catalasa [24]. Otra investigación sugiere que la tasa protectiva de la catalasa natural puede alcanzar un 80%, mientras que con catalasa recombinante puede alcanzar un 90%, basado en experimentos animales, lo que indica que la catalasa es un nuevo antígeno para la preparación de vacunas contra H. pylori [27].
Por otra parte, la enzima Superóxido Dismutasa (SOD) se encuentra presente en al menos dos formas: como superóxido dismutasa cobre/zinc citoplasmática y superóxido dismutasa manganeso mitocondrial. Esta enzima cataliza la dismutación del anión superóxido (O2-) a Peróxido de Hidrógeno, la cual luego es descompuesta por la catalasa o por otra enzima llamada glutatión peroxidasa [28]. La inflamación de la mucosa gástrica está acompañada de un incremento en la concentración y la actividad de la SOD manganeso, por lo tanto H. pylori es considerado el agente causal de la carcinogénesis gástrica [28]. Se cree que uno de los mecanismos de la carcinogénesis inducida por H. pylori depende del daño oxidativo acumulativo en el DNA. Esto se sustenta en diversos reportes de la producción de 8-hidroxidesoxiguanosina, el principal producto oxidativo modificado del DNA, encontrado en pacientes con carcinoma gástrico en niveles elevados [29]. Agregado a esto, la acumulación de especies

Factores patogénicos de H. pylori en Cáncer Gástrico Página 6
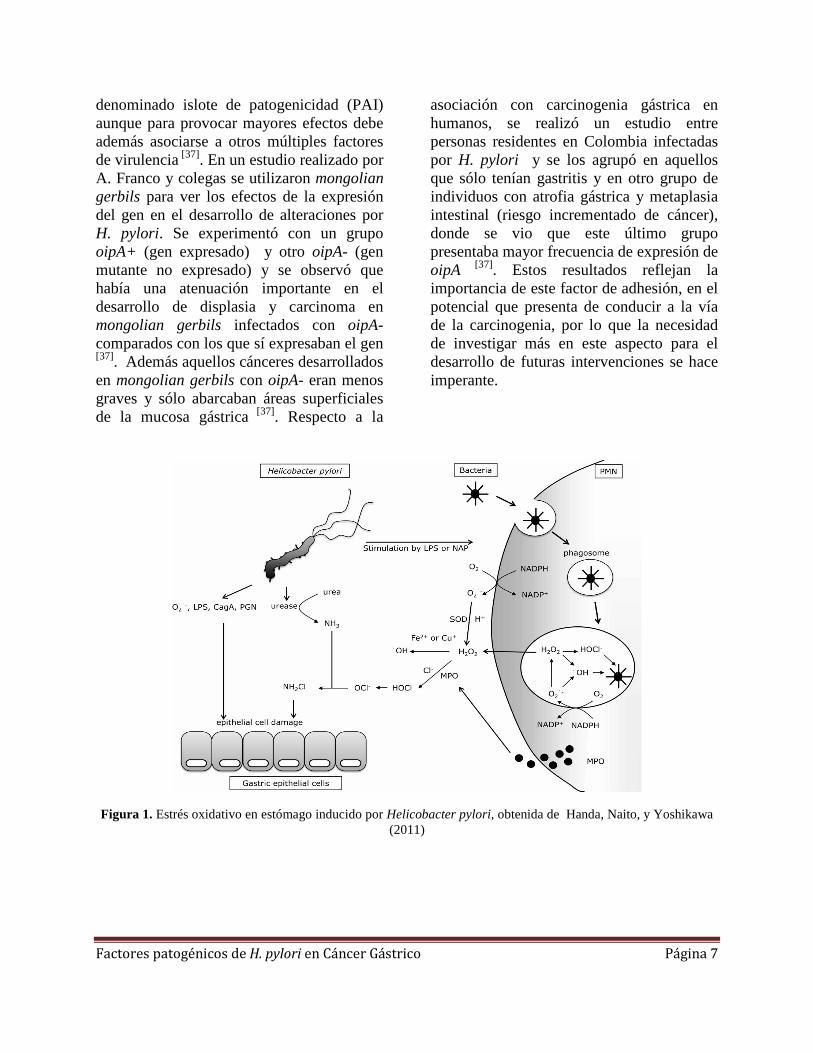
reactivas del oxígeno (EROs) y especies reactivas del nitrógeno (ERNs) pueden inducir mutaciones en el DNA, contribuyendo a la pérdida de expresión y función de diversos genes supresores de tumores como el p53 [29]. También, se ha demostrado que el estrés oxidativo inducido por H. pylori modifica el recambio epitelial en el estómago, descrito como un incremento en la proliferación de células epiteliales y la muerte celular por apoptosis, en respuesta a la infección (Figura 1) [29].
En primer lugar, las células T activadas matan las células epiteliales gástricas directamente, y la respuesta del huésped incrementa la expresión de receptores para H. pylori, y esto incrementa la adhesión bacterial y la inducción apoptótica por la bacteria. Luego, la muerte celular inducida por H. pylori puede estimular la respuesta proliferativa de precursores de células epiteliales [29].
Por lo tanto, debido a la producción elevada de EROs en la mucosa, existe un incremento en la producción de enzimas antioxidantes, se originan fuerzas opuestas entre oxidación y reducción para inducir o prevenir respectivamente una transformación neoplásica. Todo esto hace suponer que el estrés oxidativo puede representar el camino común final hacia la carcinogénesis mediada por H. pylori [30].
FACTORES DE ADHERENCIA
La adherencia por parte de H. pylori al epitelio gástrico del huésped ocurre a través de interacciones entre adhesinas bacterianas y receptores de la célula representadas por algunas proteínas de la matriz extracelular. Esto es importante, ya que hay lesiones inducidas por la bacteria al epitelio hacen perder las características propias de esta zona y se crean uniones estrechas entre
ambas, provocando lisis y destrucción celular, lo que a su vez permite el ingreso de la bacteria y sus productos (factores de virulencia como VacA o CagA) [31] y la posterior formación de procesos inflamatorios en el epitelio gástrico. Las proteínas de membrana externa (OMPs) están incorporadas en la membrana externa de H. pylori y son muy importantes para la adherencia. Existen de múltiples tipos, pero aca nos enfocaremos principalmente a HopH (OipA), además de BabA y SabA. Posteriormente se revisarán nuevas aristas respecto al papel que puede el receptor RAGE en la adhesión de H. pylori a la mucosa gástrica.
OIPA
OipA es una proteína que forma parte de la membrana bacteriana externa y que pertenece a la familia de proteínas Hop [32]. Desarrolla su función como adhesina y juega un rol central en la colonización bacteriana de la mucosa gástrica [33]. El gen que codifica para este factor de virulencia está presente en todas las cepas del H. pylori pero su expresión está modificada por variación de fase, causada por un número variable de repeticiones de dinucleótidos CT en la región 5 ' [32]. OipA es una proteína de respuesta proinflamatoria, que actúa también como adhesina [34]. Su expresión se correlaciona con un incremento en la producción de IL-8 [35], citocina con alto poder quimiotáctico de linfocitos T y polimorfonucleares, que amplifica la respuesta inflamatoria. Además se ha mostrado involucrada en la activación de la quinasa de adhesión focal (FAK) y en reorganización del citoesqueleto, lo que provoca un fenotipo morfológico celular alterado en el huésped. [36] La expresión de oipA está asociada a la presencia de un factor de virulencia
Factores patogénicos de H. pylori en Cáncer Gástrico Página 7
denominado islote de patogenicidad (PAI) aunque para provocar mayores efectos debe además asociarse a otros múltiples factores de virulencia [37]. En un estudio realizado por A. Franco y colegas se utilizaron mongolian gerbils para ver los efectos de la expresión del gen en el desarrollo de alteraciones por H. pylori. Se experimentó con un grupo oipA+ (gen expresado) y otro oipA- (gen mutante no expresado) y se observó que había una atenuación importante en el desarrollo de displasia y carcinoma en mongolian gerbils infectados con oipA- comparados con los que sí expresaban el gen [37]. Además aquellos cánceres desarrollados en mongolian gerbils con oipA- eran menos graves y sólo abarcaban áreas superficiales de la mucosa gástrica [37]. Respecto a la
asociación con carcinogenia gástrica en humanos, se realizó un estudio entre personas residentes en Colombia infectadas por H. pylori y se los agrupó en aquellos que sólo tenían gastritis y en otro grupo de individuos con atrofia gástrica y metaplasia intestinal (riesgo incrementado de cáncer), donde se vio que este último grupo presentaba mayor frecuencia de expresión de oipA [37]. Estos resultados reflejan la importancia de este factor de adhesión, en el potencial que presenta de conducir a la vía de la carcinogenia, por lo que la necesidad de investigar más en este aspecto para el desarrollo de futuras intervenciones se hace imperante.
Figura 1. Estrés oxidativo en estómago inducido por Helicobacter pylori, obtenida de Handa, Naito, y Yoshikawa (2011)

Factores patogénicos de H. pylori en Cáncer Gástrico Página 8
BABA
BabA es una proteína externa de membrana de 78 kd que actúa como adhesina bacteriana de unión a antígeno del grupo sanguíneo que se adhiere específicamente a antígenos glicosilados del grupo sanguíneo Lewis b (Le(b)), presentes en la mucosa gástrica [38]. La adherencia de H. pylori al huésped a través de la adhesión por BabA facilita la colonización, induce inflamación gástrica y potencia la acción de otro factor de adherencia denominado SabA [31]. Cuando la inflamación del huésped se ha incrementado, la expresión de sialyl-Lewis x también se incrementa y con ello H. pylori puede seguir adhiriéndose a la mucosa gástrica a través de SabA. El gen babA es polimórfico y presenta alelos A1 y A2, donde el único funcional es este último. Se ha visto que cepas con expresión del gen babA2 tienen capacidad de adherencia incrementada, al contrario de las cepas babA2 negativas que se adhieren de manera más débil [39]. Por tanto, el gen babA2 podría ser un marcador molecular útil para identificar pacientes con mayor riesgo de presentar patologías severas asociadas a infecciones por H. pylori [40].
Un estudio llevado a cabo por I. González y colegas (2011) en la Región del Maule, Chile (región con más alta mortalidad por cáncer gástrico del país) mostró una alta presencia del gen babA2 (97,4%) dentro de una muestra de 78 pacientes atendidos en la unidad de gastroenterología del Hospital Regional de Talca, cuestión que, según los autores, sólo es comparable a países asiáticos que tienen una alta tasa de mortalidad por cáncer gástrico. [41]
SABA
Similar a BabA, SabA, proteína de unión al ácido siálico (presente en epitelio gástrico inflamado), genera íntima adherencia, pudiendo potenciar la respuesta inflamatoria y a la vez mediar la utilización de nutrientes del huésped. [31] Cuando la respuesta inflamatoria ha alcanzado un máximo, la expresión de esta adhesina se elimina, de manera que la bacteria pueda movilizarse evadiendo el contacto estrecho dentro del medio inflamado, promoviendo la persistencia de la infección [42].
IMPLICANCIA DE LOS RECEPTORES PARA LOS PRODUCTOS DE
GLICOSILACIÓN AVANZADA (RAGE)
Se ha observado que pacientes con hiperglicemia tienen mayor riesgo de cáncer gástrico si están infectados con H. pylori [43]. RAGE juega un rol relevante, puesto que está implicado en numerosos procesos como la contribución a la inflamación, el cáncer, daños renales, cardíacos y nerviosos en pacientes diabéticos [44,45]. En un estudio realizado por A. Rojas y colegas (2011) se obtienen importantes resultados. Lo que se sabía hasta el momento era que pacientes con diabetes tenían un mayor riesgo de infección y a la vez de reinfección por H. pylori; con los resultados obtenidos se observó que RAGE contribuye a la adhesión de H. pylori a las células epiteliales gástricas interactuando directamente. Además, la estimulación celular mediada por RAGE incrementa la expresión del mismo receptor mediante retroalimentación positiva, haciendo persistente la inflamación y amplificando la cascada inflamatoria, provocando disrupción de paredes vasculares, favoreciendo extravasación de líquido y células intravasculares y actuando como potente citoquina para polimorfonucleares, monocitos y macrófagos [46]. Esto último es de relevancia

Factores patogénicos de H. pylori en Cáncer Gástrico Página 9
en el posible paso de una lesión inflamatoria a un proceso con carácter de malignidad, especialmente en pacientes diabéticos.
VACA: CITOTOXINA VACUOLIZANTE
VacA constituye un importante factor de virulencia, un producto de H. pylori que provoca alteraciones específicas, cuyo gen está presente en todas las cepas de la bacteria y el cual se ha visto induce vacuolización citoplasmática en cultivos de células epiteliales y con ello muerte celular, daño epitelial, ulceración de la mucosa gástrica y destrucción de la integridad epitelial [31]. Se forma a partir de una gran molécula, una pre-protoxina la cual incluye un péptido señal y un extremo carboxilo terminal que una vez secretada es dividida en monómeros de toxina [47,48] los cuales una vez maduros se liberan o quedan retenidos en la membrana bacteriana [49]. Además de producir vacuolización, VacA puede inducir otras actividades como formación de canales de membrana anión-selectivos cuando los monómeros se agregan en complejos oligoméricos que se insertan en las bicapas lipídicas y que permiten la salida de aniones y urea; esto es relevante ya que la hidrólisis de la urea, catalizada por la ureasa de H. pylori, protege a la bacteria de la acidez gástrica [50,51]; además de liberación de citocromo c desde la mitocondria y con ello muerte celular, y también influir respuestas proinflamatorias [52]. Sumado a esto puede también inhibir la activación y proliferación de células T e interferir con la presentación de antígenos y con la fagocitosis, lo que lo llevaría a tener el poder de evadir el sistema inmune y perpetuar la infección [53,54].
Los polimorfismos de los alelos de VacA generan distintos niveles de citotoxicidad [31] a través de variaciones en las región señal (s) y en la región media (m) [55]. Respecto de
la región señal se conocen dos tipos: s1 y s2. El tipo s1 es completamente activo, mientras que el tipo s2 tiene un extremo amino corto que bloquea la formación de vacuolas y disminuye la formación de poros en las membranas eucariotas [51]. La región media de VacA codifica para parte del dominio de unión de la toxina a la célula. Sobre la base de la secuencia de nucleótidos de esta región, las cepas se clasifican en dos familias de alelos: el alelo tipo m1 para las cepas de H. pylori que tienen actividad citotóxica y el alelo m2 para las cepas que carecen de ésta [55].
Las formas s1/m1 vacuolizan e inducen la mayor cantidad de daño a las células, las s1/m2 vacuolizan en un menor nivel de células e inducen un menor daño, sin embargo estas últimas también actúan eficientemente en la formación de poros en las membranas e incrementan la permeabilidad celular; las formas s2/m2 carecen de actividad vacuolizante [51,55]. En poblaciones occidentales, VacA s1/m1 puede estar asociado con úlceras gástricas o duodenales y cáncer gástrico [55-57], pero en poblaciones del este asiático (en que la abundan las formas s1/m1) esto no se asocia con las características descritas [31].
Rhead y colegas identificaron otra variante polimórfica en cepas de individuos occidentales, es una región ubicada entre la región (s) y la región (m) que pasó a denominarse región intermedia (i) [58]. Según este estudio, se vio que existen dos variantes alélicas de esta región; i1 (vacuolizante) e i2 (no vacuolizante). La forma i1 se asoció a las formas s1/m1, la i2 a la s2/m2, y ambas podían estar presentes junto con las formas s1/m2. Existen diferencias respecto al real valor que tiene VacA tipo i1 en ser indicador de riesgo de cáncer gástrico puesto que se ha visto en regiones del oriente medio (Irán) este factor está asociado fuertemente al

Factores patogénicos de H. pylori en Cáncer Gástrico Página 10
desarrollo de estas alteraciones y constituiría un buen indicador [59], pero por otra parte en las regiones este y sur de Asia, donde el cáncer gástrico es altamente prevalente, no se ha podido confirmar la relación del factor de virulencia mencionado en esa categoría [60]. Estos resultados encontrados llevan a plantear la necesidad de futuros estudios de este factor de virulencia y cómo influye en ciertas poblaciones y no en otras, o poder investigar otros factores asociados a las características propias del huésped y que lo hacen resistir frente a tales condiciones desfavorables sin mostrar cambios clínicos.
CAGA E ISLOTE DE PATOGENICIDAD (PAI)
La evolución bacteriana no es un proceso continuo, puede producirse adquiriendo segmentos de DNA de origen desconocido que se integran al cromosoma bacteriano mediante recombinación homóloga. La nueva porción de DNA integrado se conoce con el nombre de isla, cuyo DNA puede codificar para varias proteínas involucradas en el metabolismo y sistema patógeno de la bacteria. [61]
La isla de patogenicidad (PAI) de H. pylori inicialmente se denominó cag (gen asociado a citotoxina) ya que se pensaba que estaba asociado con la expresión de VacA; sin embargo, posteriormente se observó que los dos factores VacA y PAI son independientes. [62]
Hay dos tipos de cepas clínicamente aisladas de H. pylori: las productores de CagA (CagA positivas) y las cepas no productoras de CagA (CagA negativas). Las que portan el gen son consideradas más virulentas, ya que la integridad de esta estructura genética es esencial para el transporte óptimo de la proteína bacteriana al interior de las células
del epitelio gástrico. Un H. pylori CagA positivo está asociada con una mayor respuesta inflamatoria gástrica, al igual que con la enfermedad ulcero péptica, atrofia gástrica, metaplasia y adenocarcinoma gástrico [63].
SISTEMA DE SECRECIÓN TIPO IV (T4SS)
El PAI de H. pylori aparte de codificar para la proteína CagA, también posee alrededor de 18 genes que codifican para un conjunto de proteínas que conforman el sistema de secreción tipo IV de la bacteria o “Type 4 Secretion Systems” (T4SS). Éste permite no solo la transferencia horizontal de genes sino también la importación de peptidoglicano y de la proteína CagA al interior de la célula epitelial [65].
Se ha identificado al T4SS como un organelo filamentoso localizado en un polo de la superficie bacteriana e inducido por contacto. El modelo de organización del sistema de secreción tipo IV propone que sus proteínas se agrupan en: proteínas citoplasmáticas o de la membrana interna, proteínas que forman el complejo central o core localizadas en el periplasma y proteínas que hacen parte del pili o de la estructura superficial que se proyecta más allá de la membrana externa [63].
Una vez que el microorganismo entra en contacto con la célula epitelial gracias a adhesinas accesorias comienza el ensamblaje del T4SS, inicialmente con las proteínas que son parte de la membrana interna como CagE/HPO544 (VirB4) y HPO525 (VirB11), las cuales poseen actividad ATPasa para permitir la translocación del sustrato, una proteína adicional, HPO524 homóloga a VirD4 es la encargada de entregar el sustrato a la maquinaria de secreción de T4SS [66].

Factores patogénicos de H. pylori en Cáncer Gástrico Página 11
El ensamblaje de las proteínas que conforman el complejo central o core del T4SS, dentro de las cuales se encuentran CagT/12 (HPO532) que se ubica en la base del organelo y permite el ensamblaje del resto del éste; y CagV/10, CagX/8 (HPO528) y CagY/7 (HPO527) homólogas a VirB7, VirB8, VirB9, y VirB10 respectivamente, que hacen parte del complejo central propiamente tal. Aunque estas proteínas han sido descritas como parte del complejo central, se ha demostrado que tres de estas proteínas, CagY/7, CagX/8, CagT/12 están asociadas con el pili que se forma entre H. pylori y la célula epitelial; además, se ha propuesto que otras proteínas Cag como CagM/16, CagI/19, CagG/21 y CagF/22 podrían representar componentes del complejo core de H. pylori específicos. Debido a que las proteínas que forman el core deben atravesar el peptidoglicano se han identificado proteínas adicionales como HPO523 homóloga a VirB1 que podría actuar como una transglycosilasa, encargada de lisar la capa de mureína de la pared bacteriana y así facilitar el ensamblaje de T4SS a través de la pared bacteriana, la cual también es necesaria para la maduración de CagY/7, CagT/12 y de HPO539, una posible chaperona de estas proteínas [66].
Finalmente, se ensamblan las proteínas que conforman el pili, dentro de las que su mayor componente es CagC homólogo a VirB2, la cual a la vez puede estar recubierta total o parcialmente por CagY y CagL, esta última actúa como adhesina y permite la conexión entre T4SS y la célula blanco [63].
MOTIVOS EPIYA
La proteína CagA, gracias al sistema de secreción tipo IV (Figura 2) es translocada ("inyectada") a la célula huésped y posteriormente fosforilada en sitios específicos conocidos como EPIYA y de
esta manera interactúa con diversas vías de señalización, desencadenando cambios en el citoesqueleto, en la morfología y en la movilidad de la célula huésped [63, 67]. Los motivos EPIYA están constituidos por Glu-Pro-lle-Tyr-Ala los cuales se pueden repetir hasta cinco veces en la mitad del carbono terminal de CagA. Se han denominado cuatro motivos EPIYA distintos (A, B, C, D), que se distinguen por las diferentes secuencias de aminoácidos que los rodean [63, 68]. Éstos tipos de EPIYA son de gran importancia ya que podrían explicar la variabilidad en el número de sitios en CagA, así como diferencias en la patogenicidad de las cepas de H. pylori. El número de repeticiones, de los sitios EPIYA son de especial importancia en cáncer gástrico, cuya incidencia es significativamente más alta en pacientes infectados con cepas de H. pylori que presenten múltiples repeticiones; sin embargo, un inconveniente en la capacidad de estas cepas con múltiples repeticiones para contribuir en desarrollo de cáncer gástrico es su escasa supervivencia en pH ácido por lo que se ha sugerido que estas se manifiestan después de que los pacientes desarrollan atrofia e hipocloridria [63, 69]. En las cepas occidentales, un mayor número de CagA motivo EPIYA-C es un indicador importante del riesgo de desarrollar cáncer gástrico [70, 71]. La mayoría de las proteínas CagA de H. pylori aisladas en países de Asia Oriental como Japón, Corea y China (Asia oriental CagA) poseen también los segmentos EPIYA-A y EPIYA-B, pero no el segmento repetible de EPIYA-C. En cambio, tienen el segmento EPIYA-D, que es única para las especies de CagA de Asia oriental, por lo tanto son consideradas como "CagA tipo AB-D”, donde el motivo EPIYA-D constituye el principal sitio de fosforilación de la tirosina de CagA Asia oriental. [72]

Factores patogénicos de H. pylori en Cáncer Gástrico Página 12
FOSFORILACIÓN CAGA Y SPH-2
La fosforilación es mediada por kinasas conocidas como familia Src (SFK) tales como c-Src, Fyn, Lyn y Yes. [73] Cuya función en las células normales es controlar procesos en citoesqueleto, proliferación celular y diferenciación pero también se han descrito en procesos de carcinogénesis [63]. La fosforilación de CagA por SFK se produce en ausencia de cualquier estímulo, lo que indica que son SFK activados constitutivamente en las células epiteliales gástricas. En general, la fosforilación juega un papel crucial en la transmisión de señales intracelulares que participan en el crecimiento, movilización o la diferenciación celular. Esto ha planteado la
posibilidad de que, en la fosforilación de tirosina, la proteína bacteriana perturba la transducción de señales y de ese modo provoca disfunción celular que eventualmente conduce a la transformación celular [74].
Una vez CagA es tirosin fosforilada por estas kinasas se une específicamente y activa tirosin fosfatasas SHP-2, una oncoproteína cuya mutación está asociada con procesos de malignidad en humanos. CagA desregula a SHP-2 perturbando a la ERK MAP quinasa así como desfosforilando FAK kinasas involucradas en adhesión focal induciendo cambios en la morfología celular, la cual adopta un fenotipo conocido como célula en forma de colibrí [63].
Figura 2. la interacción física y funcional entre el H. pylori CagA y SHP-2. CagA es inyectado de H. pylori a células del epitelio gástrico a través del sistema de secreción tipo IV bacteriana. La proteína CagA translocada se localiza en la superficie interna de la membrana plasmática y se somete a la fosforilación de tirosina quinasas de la familia por Src. Tras la fosforilación de tirosina, CagA se une específicamente al dominio SH2 que contiene tirosina fosfatasa SHP-2 y activa la actividad de la fosfatasa. El CagA-SHP-2 activado potencia la actividad de ERK MAP quinasa, que regula el crecimiento celular y la morfología de las células. SHP-2, también desfosforila un sustrato celular (mostrado como X en la figura), que está implicado en la inducción del fenotipo colibrí que está asociado con la motilidad celular elevada. [74]

Factores patogénicos de H. pylori en Cáncer Gástrico Página 13
EFECTOS DE CAGA INDEPENDIENTES A LA FOSFORILACIÓN.
CagA también daña la interacción célula-célula de manera independiente a la fosforilación. CagA destruye las uniones estrechas y causa pérdida de la polaridad en las células epiteliales, también desestabiliza el complejo E-caderina / β-catenina [75, 76]. Esto implica que H. pylori puede tener un efecto oncogénico directo sobre las células del epitelio gástrico por su oncoproteína CagA, y no solamente de manera indirecta produciendo inflamación persistente e hiperproliferación con el riesgo de que radicales libres lesionen el DNA de estas células con rápido crecimiento [76].
Se ha demostrado que el colesterol es un factor importante para la acción de CagA, ya que al haber una disminución de colesterol celular se reduce la translocación y fosforilación, además se produce un bloqueo de la respuesta celular inducida por la proteína, incluyendo el fenotipo en forma de colibrí y la inducción de IL-8 [77].
PAPEL DEL CAGA EN LA CARCINOGÉNESIS GÁSTRICA.
El desarrollo de la carcinogénesis gástrica es un proceso de múltiples pasos que requiere tanto de alteraciones cualitativas como cuantitativas en la expresión de oncogenes y genes supresores de tumores, y que tienen una duración de varias décadas.
Se ha demostrado en estudios “in vivo”, que CagA es una oncoproteína bacteriana cuya expresión es suficiente para desarrollar neoplasias, independiente a su fosforilación [76]. Las cepas CagA+ aumentan notablemente la expresión de IL-8, induciendo por tanto una respuesta inflamatoria más acentuada en la mucosa
gástrica, que conduce al cáncer gástrico: gastritis superficial, gastritis atrófica, metaplasia intestinal, displasia, carcinoma. Debido a que el complejo CagA-SHP-2 se detecta principalmente en la mucosa atrófica, éste puede ser involucrado en el desarrollo de la gastritis atrófica y en la transición de la atrofia a metaplasia intestinal [78].
Igualmente el CagA causa un aumento inicial en la expresión de las proteínas p53 y p21, seguido de un descenso. Sabiendo que el gen p53 pertenece a la serie de genes de supresión tumoral, permite mostrar una clara evidencia de su participación en procesos carcinogénicos, además, la expresión de Bcl-2 está aumentada, traduciéndose esto en una disminución de la apoptosis y un incremento persistente de la proliferación celular. [79]
HELICOBACTER PYLORI Y CÁNCER GÁSTRICO
El cáncer gástrico ocupa un lugar importante entre las neoplasias de diagnóstico frecuente a nivel mundial, pese a esto en países en vías de desarrollo el diagnóstico se realiza tardíamente, por lo que en estas regiones se asocia a una alta tasa de mortalidad [80]. El H. pylori es un elemento importante en el desarrollo de un cáncer gástrico, puesto que su infección está asociada a un incremento en el riesgo cercano al doble de padecer esta patología respecto a quienes no estén infectados [81].
El adenocarcinoma gástrico puede clasificarse en dos tipos histológico; uno intestinal y otro difuso. El primero predomina en poblaciones de alto riesgo y es más común en personas de edad avanzada. Es precedido por continuos cambios histológicos, tales como gastritis activa, atrofia intestinal, metaplasia y displasia. El

Factores patogénicos de H. pylori en Cáncer Gástrico Página 14
tipo difuso es menos común, con lesiones precancerosas no bien definidas con mayor frecuencia poblaciones con menor riesgo (jóvenes). La evolución histológica del adenocarcinoma gástrico de tipo intestinal, trae cambios histológicos descritos en las infecciones por H. pylori por lo que se le atribuye a este agente principalmente [80]. La infección con H. pylori se asocia con un 70% de los casos de gastritis crónica activa, con el 90 a 95% de las úlceras duodenales y con un 60% de pacientes con cáncer [84].
El H. pylori genera un daño progresivo que está inducido por la presencia prolongada de la bacteria, donde se manifiestan lesiones que evolucionan desde una gastritis superficial, a una gastritis crónica o gastritis atrófica. En ésta última existe infiltración inflamatoria importante con agregados foliculares linfoides que destruyen la mucosa a tal grado, que ocurre pérdida de la función y se induce una metaplasia intestinal, displasia y eventualmente cáncer [82]. (figuras 3-4)
La metaplasia intestinal puede ser considerada como una estrategia defensiva
contra H. pylori, ya que esta bacteria sólo coloniza el epitelio gástrico [80]. H. pylori no sólo tiende a disminuir los niveles de ácido ascórbico (alterando la protección que produce en la depuración de radicales libres) sino que además la gastritis crónica atrófica causada por la infección resulta en una hipoclorhidria que conduce a un sobrecrecimiento bacteriano [83]. Luego de la erradicación exitosa de la bacteria los niveles de ácido gástrico retornan a niveles normales. En un pequeño porcentaje de tumores gástricos del tipo linfoma, la infección con H. pylori también se encuentra fuertemente relacionada. Se ha demostrado una elevada incidencia de tejido linfoide asociado a mucosa (“MALT”) en regiones con una elevada prevalencia de cáncer gástrico e infección con esta bacteria. El tejido linfoide no es normal a nivel del estómago y la erradicación exitosa de la bacteria conduce a una completa regresión de la neoplasia, reforzando aún más la asociación de esta patología con H. pylori [80].

Factores patogénicos de H. pylori en Cáncer Gástrico Página 15
Figura 3. Historia natural de la infección por H. pylori tomada de Romo y Coria 2010
CONCLUSIONES
H. pylori es una bacteria espirilada, gram negativa, con múltiples flagelos, en donde su nicho ecológico es la región gástrica del ser humano. Diversos estudios han implicado a esta bacteria un rol patogénico en el desarrollo de trastornos gastrointestinales como inflamación gástrica, ulceras pépticas y adenocarcinoma gástrico. Los factores patogénicos intrínsecos de esta bacteria, en conjunto con otros factores externos como el ambiente, la genética e infección temprana, para el desarrollo de cáncer gástrico hacen importante su estudio, para entender los mecanismos que explican la alta patogenicidad de H. pylori.
En primer lugar, es relevante el uso de la estructura motil de H. pylori, ya que gracias a esto la bacteria puede moverse como un propulsor desde el ambiente ácido hacia la cercanía del epitelio gástrico de un pH más alcalino, cruzando por la capa de mucina que es degradada y por lo tanto dañada por las mucinasas y fosfolipasasas como la fosfolipasa A que tiene como maquinaria de avance, sin olvidar también un tipo de fosfolipasa A ubicada en la membrana exterior (OMPLA) que participa en un cambio reversible de la configuración de la membrana lipídica que contribuye a la supervivencia de H. pylori. Asociado a esto, la contribución de la ureasa en la regulación del pH para el correcto funcionamiento de las actividades de síntesis proteica, la mejora y tolerancia al ambiente ácido donde reside H. pylori que permite el crecimiento celular normal y continuo del patógeno, además de entregar un potencial de carcinogénesis.

Factores patogénicos de H. pylori en Cáncer Gástrico Página 16
En segundo lugar, es importante el rol evasivo de la actividad fagocítica por parte de dos enzimas: la Catalasa y la Superóxido Dismutasa. La primera, protegiendo a H. pylori contra los efectos nocivos de las EROs, como el Peróxido de Hidrógeno (H2O2) que se liberan durante el proceso inflamatorio en el que está inmerso el microorganismo, proporcionando el ambiente adecuado para su normal crecimiento. Por otro lado, la segunda enzima es partícipe de la dismutación del anión superóxido a H2O2. Agregado a esto,
se asocia el aumento de la concentración de estas enzimas en los procesos inflamatorios de la mucosa gástrica, lo cual lleva a la evidencia que H. pylori es el agente causal del cáncer gástrico. Cabe resaltar el daño al DNA por efecto del daño oxidativo acumulado debido a la presencia constante del patógeno en el epitelio gástrico, demostrado por la producción de 8-hidroxidesoxiguanosina; y el permanente recambio epitelial en el estómago en respuesta a la infección por H. pylori.
Figura 4. Mecanismos que participan en la inducción del cáncer gástrico. (tomada de Romo y Coria 2010 en base a adaptación de Blaser y Atherton 2004)
En tercer lugar, importantes factores a considerar dentro de la patogenicidad propia de H. pylori los constituyen los factores de adherencia. Dentro de las proteínas de adherencia oipA destaca por su relevante efecto en el aumento en la producción de IL-8 y lo que ello conlleva; marcada relevancia
adquiere también BabA pues favorece la colonización por parte de H. pylori y según la documentación obtenida, la presencia del alelo babA2 en pacientes podría utilizarse para identificar aquellos con un mayor riesgo de presentar patologías severas asociadas; SabA, muy relacionado a BabA,

Factores patogénicos de H. pylori en Cáncer Gástrico Página 17
potencia la respuesta inflamatoria generando una íntima unión al epitelio gástrico y generando una persistencia de la infección al regular su propia presencia; por otra parte, nuevos descubrimientos respecto a la implicancia de RAGE en la infección por H. pylori dejan en manifiesto que este receptor contribuye a la adhesión de la bacteria, perpetuando su acción e incrementando con ello las posibilidades de generar una condición de malignidad, aunque aún queda mucho por esclarecer respecto a esta importante interacción en estudios futuros.
La acción vacuolizante de VacA y con ello la pérdida de integridad del epitelio gástrico, además de las múltiples acciones mencionadas anteriormente, revela la preponderancia que adquiere este factor de virulencia en la capacidad de H. pylori para provocar daño. La identificación de las variantes polimórficas del gen permitiría cuantificar cuan dañina será la bacteria en un determinado huésped y por ende su riesgo de desarrollar cáncer, sin embargo, resultados concretos obtenidos generan discrepancia y hacen evidente la necesidad de estudiar más respecto a otros posibles elementos intervinientes.
Pero, sin duda, uno de los más estudiados factores de virulencia de H. pylori son los islotes de patogenicidad y CagA. Existen numerosas poblaciones humanas que han sido infectados con H pylori CagA + y éstos tienen un mayor riesgo de desarrollar carcinoma gástrico en el corto o mediano plazo.
La interacción de CagA con SHP-2, actúa como promotor en tumores malignos humanos y es uno de los principales factores determinantes para el desarrollo de carcinoma gástrico asociado con la infección de H pylori CagA +.
CagA se divide en dos tipos principales: CagA Asia oriental y CagA occidental, diferenciándose en los distintos motivos EPIYA de cada uno, ésta diferencia pudiera ser parte a la respuesta de la interrogante del porqué no en todas las personas infectadas con H. pylori desarrollan enfermedad y posteriormente cáncer gástrico. Desde el punto de vista clínico, el esclarecimiento de cepas de H. pylori CagA + con mayor potencial de desarrollo de carcinoma gástrico será de suma importancia. Estudios recientes han demostrado que la erradicación de H. pylori en humanos parece disminuir el riesgo de desarrollar carcinoma gástrico. Por lo tanto, la erradicación sistemática del oncongen de H. pylori en una población de alto riesgo reduciría drásticamente la incidencia mundial de cáncer gástrico. Por otra parte, la comprensión de los mecanismos moleculares en la interacción de H. pylori y células gástricas no sólo es importante en el desarrollo de terapias más eficaces para el cáncer gástrico, también lo es en la comprensión de la génesis de los cánceres asociados con la inflamación.
Por tanto, H. pylori con todos sus factores de virulencia, es necesario pero no suficiente para producir la mayoría de los adenocarcinomas gástricos en las personas genéticamente predispuestas.
REFERENCIAS
1. Valenzuela E, J. (2004). Helicobacter pylori: 20 años después. Revista médica de Chile, 132(11),1339-1344.
2. Feldman, R. A., Eccersley, A. J. P., & Hardie, J. M. (1998). Epidemiology of Helicobacter pylori: acquisition, transmission, population prevalence and

Factores patogénicos de H. pylori en Cáncer Gástrico Página 18
disease-to-infection ratio. British medical bulletin, 54(1), 39-53.
3. Warren, J. R., & Marshall, B. (1983). Unidentified curved bacilli on gastric epithelium in active chronic gastritis. The Lancet, 321(8336), 1273-1275.
4. Parsonnet, J., Hansen, S., Rodriguez, L., Gelb, A. B., Warnke, R. A., Jellum, E., et al. (1994). Helicobacter pylori infection and gastric lymphoma. New England Journal of Medicine, 330(18), 1267-1271.
5. Ahmed, N. (2005). 23 years of the discovery of Helicobacter pylori: Is the debate over?. Annals of clinical microbiology and antimicrobials, 4(1), 17.
6. Rodríguez, D. A. R. (2008). Infección por Helicobacter pylori. Producción, 113.
7. Kelley, J. R., & Duggan, J. M. (2003). Gastric cancer epidemiology and risk factors. Journal of clinical epidemiology, 56(1), 1-9.
8. Medina, E. (1970). Epidemiología del cáncer gástrico en Chile. Rev Méd Chile, 98, 477-81.
9. Ferreccio, C., Rollán, A., Harris, P. R., Serrano, C., Gederlini, A., Margozzini, P., et al. (2007). Gastric cancer is related to early Helicobacter pylori infection in a high-prevalence country. Cancer Epidemiology Biomarkers & Prevention, 16(4), 662-667.
10. Ferlay, J. (2001). Globocan 2000 [: Cancer Incidence, Mortality and Prevalence Worldwide. IARC press.
11. República de Chile, Ministerio de Salud Chile. Departamento de estadísticas. Bases de dato de mortalidad 1985-2002, Santiago, Chile.
12. Holcombe, C. (1992). Helicobacter pylori: the African enigma. Gut, 33(4), 429-431.
13. Wroblewski, L. E., Peek Jr, R. M., & Wilson, K. T. (2010). Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clinical microbiology reviews, 23(4), 713-739.
14. Nakazawa, T. (2002). Growth cycle of Helicobacter pylori in gastric mucous layer. The Keio journal of medicine, 51, 15.
15. Seyler Jr, R. W., Olson, J. W., & Maier, R. J. (2001). Superoxide Dismutase-Deficient Mutants of Helicobacter pylori Are Hypersensitive to Oxidative Stress and Defective in Host Colonization. Infection and immunity, 69(6), 4034-4040.
16. Bauerfeind, P., Garner, R., Dunn, B., & Mobley, H. (1997). Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut, 40(1), 25-30.
17. Sachs, G., Wen, Y., & Scott, D. R. (2009). Gastric infection by Helicobacter pylori. Current gastroenterology reports, 11(6), 455-461.
18. Wu, C., Huang, M., Yeh, C., Wang, J., Cheng, T., & Lin, S. (2007). Overexpression of Helicobacter pylori-associated urease mRNAs in human gastric cancer. DNA and cell biology, 26(9), 641-648.
19. Stingl, K., Altendorf, K., & Bakker, E. P. (2002). Acid survival of Helicobacter pylori: how does urease activity trigger cytoplasmic pH homeostasis?. Trends in microbiology, 10(2), 70-74.
20. Mori, M., Suzuki, H., Suzuki, M., Kai, A., Miura, S., & Ishii, H. (1997). Catalase and superoxide dismutase secreted from Helicobacter pylori. Helicobacter, 2(2), 100-105.
21. Smith, A. W., Chahal, B., & French, G. L. (1994). The human gastric pathogen Helicobacter pylori has a gene encoding an enzyme first classified as a mucinase in Vibrio cholerae. Molecular microbiology, 13(1), 153-160.
22. Tannæs, T., Dekker, N., Bukholm, G., Bijlsma, J. J., & Appelmelk, B. J. (2001). Phase variation in the Helicobacter pylori phospholipase A gene and its role in acid adaptation. Infection and immunity, 69(12), 7334-7340.

Factores patogénicos de H. pylori en Cáncer Gástrico Página 19
23. Hazell, S. L., Evans Jr, D. J., & Graham, D. Y. (1991). Helicobacter pylori catalase. Journal of general microbiology, 137(1), 57-61.
24. Basu, M., Czinn, S. J., & Blanchard, T. G. (2004). Absence of Catalase Reduces Long‐Term Survival of Helicobacter pylori in Macrophage Phagosomes. Helicobacter, 9(3), 211-216.
25. Harris, A. G., Wilson, J. E., Danon, S. J., Dixon, M. F., Donegan, K., & Hazell, S. L. (2003). Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology, 149(3), 665-672.
26. Bulbuloglu, E., Inanc, F., Bakaris, S., Kantarceken, B., Cetinkaya, A., Cağlar, R., et al. (2005). Association of adenosine deaminase, superoxide dismutase, and catalase activities with Helicobacter pylori. Digestive diseases and sciences, 50(12), 2296-2299.
27. Bai, Y., Zhang, Y., Jin, J., Wang, J., Zhang, Z., & Zhou, D. (2003). Recombinant Helicobacter pylori catalase. World Journal of Gastroenterology, 9(5), 1119-1122.
28. Götz, J., Thio, J., Verspaget, H., Offerhaus, G., Biemond, I., Lamers, C., et al. (1997). Treatment of Helicobacter pylori infection favourably affects gastric mucosal superoxide dismutases. Gut, 40(5), 591-596.
29. Handa, O., Naito, Y., & Yoshikawa, T. (2011). Redox biology and gastric carcinogenesis: the role of Helicobacter pylori. Redox Report, 16(1), 1-7.
30. Farkas, R., Pronai, L., Tulassay, Z., & Selmeci, L. (2005). Relationship between eradication of Helicobacter pylori and gastric mucosal superoxide dismutase activity. Anticancer research, 25(6C), 4763-4767.
31. Wen, S., & Moss, S. F. (2009). Helicobacter pylori virulence factors in
gastric carcinogenesis. Cancer letters, 282(1), 1-8.
32. Bauer, B., & Meyer, T. F. (2011). The Human Gastric Pathogen Helicobacter pylori and Its Association with Gastric Cancer and Ulcer Disease. Ulcers, 2011.
33. Yamaoka, Y. (2010). Mechanisms of disease: Helicobacter pylori virulence factors. Nature Reviews Gastroenterology and Hepatology, 7(11), 629-641.
34. Yamaoka Y, Kwon DH, Graham DY. (2000) "A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori," Proceedings of the National Academy of Sciences of the United States of America, vol. 97, no. 13, pp. 7533‐7538.
35. Tabassam FH, Graham DY, Yamaoka Y. (2008) "OipA plays a role in Helicobacter pylori‐induced focal adhesion kinase activation and cytoskeletal re‐organization," Cellular Microbiology, vol. 10, no. 4, pp. 1008‐1020.
36. Calam J. (1999) "Helicobacter pylori modulation of gastric acid," The Yale Journal of Biology and Medicine, vol. 72, no. 2‐3, pp. 195‐202.
37. Franco, A. T., Johnston, E., Krishna, U., Yamaoka, Y., Israel, D. A., Nagy, T. A., et al. (2008). Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer research, 68(2), p.379.
38. Ramirez, A., Sánchez, R. (2009) Helicobacter pylori 25 años después (1983-2008): Epidemiología, Microbiología, Patogenia, Diagnóstico y Tratamiento, Rev. Gastroenterol. Perú; 29-2: 158-170.
39. Boren T, Normark S, Falk P. (1994) Helicobacter pylori: molecular basis for host recognition and bacterial adherence. Trends Microbiol; 2: 221-8.
40. Yu J, Leung Wk, Go Myy, Cjhan Mcw, To Kf, Ng Ekw Et Al. (2002) Relationship between Helicobacter pylori babA2 status

Factores patogénicos de H. pylori en Cáncer Gástrico Página 20
with gastric epithelial cell turnover and premalignant gastric lesions. Gut; 51: 480-4.
41. González, I., Romero, J., Rodríguez, B., Llanos, J., Morales, E., Figueroa, H., Perez-Castro, R., Valdés, E., Cofre, C., Rojas, A. (2011) High prevalence of virulence-associated genotypes in Helicobacter pylori clinical isolates in the Region del Maule, Chile. Scandinavian Journal of Infectious Diseases. Aug;43(8):652-5
42. Mahdavi, J., Sondén, B., Hurtig, M., Olfat, F. O., Forsberg, L., Roche, N., et al. (2002). Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science, 297(5581), 573-578.
43. Ikeda, F., Doi, Y., Yonemoto, K., Ninomiya, T., Kubo, M., Shikata, K., et al. (2009). Hyperglycemia increases risk of gastric cancer posed by Helicobacter pylori infection: a population-based cohort study. Gastroenterology, 136(4), 1234-1241.
44. S.F. Yan, R. Ramasamy, A.M. Schmidt (2008). Mechanisms of disease: advanced glycation end products and their receptor in inflammation and diabetes complications, Nat. Clin. Pract. Endocrinol. Metab. 4 285-293
45. Rojas, A., Mercadal, E., Figueroa, H., & Morales, M. A. (2008). Advanced glycation and ROS: a link between diabetes and heart failure. Current vascular pharmacology, 6(1), 44-51.
46. Rojas A., González I., Rodríguez, B., Romero, J., Figueroa, H., Llanos, J., Morales, E., Pérez-Castro, R. (2011). Evidence of involvement of the receptor for advanced glycation end-products (RAGE) in the adhesion of Helicobacter pylori to gastric epithelial cells. Microbes and Infection, Volume 13, Issue 10, 818-823.
47. Telford, J. L., Ghiara, P., Dell'Orco, M., Comanducci, M., Burroni, D., Bugnoli, M., et al. (1994). Gene structure of the Helicobacter pylori cytotoxin and evidence
of its key role in gastric disease. The Journal of experimental medicine, 179(5), 1653.
48. Cover, T., & Blaser, M. (1992). Purification and characterization of the vacuolating toxin from Helicobacter pylori. Journal of Biological Chemistry, 267(15), 10570-10575.
49. Ilver, D., Barone, S., Mercati, D., Lupetti, P., & Telford, J. L. (2004). Helicobacter pylori toxin VacA is transferred to host cells via a novel contact‐dependent mechanism. Cellular microbiology, 6(2), 167-174.
50. Cover, T. L., & Blanke, S. R. (2005). Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nature Reviews Microbiology, 3(4), 320-332.
51. López, A., Delgado, M., Jaramillo, C., Amézquita, A., Parra, G., & Echeverry, M. (2009). Caracterización del gen de la citotoxina vacuolizante de Helicobacter pylori a partir de biopsias gástricas de pacientes residentes en Tolima, Colombia. Revista argentina de microbiología, 41(1), 4-10.
52. Kusters, J. G., Van Vliet, A. H., & Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clinical microbiology reviews, 19(3), 449-490.
53. Boncristiano, M., Paccani, S. R., Barone, S., Ulivieri, C., Patrussi, L., Ilver, D., et al. (2003). The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. The Journal of experimental medicine, 198(12), 1887.
54. Gebert, B., Fischer, W., Weiss, E., Hoffmann, R., & Haas, R. (2003). Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science's STKE, 301(5636), 1099.
55. Atherton, J. C., Cao, P., Peek Jr, R. M., Tummuru, M. K., Blaser, M. J., & Cover, T. L. (1995). Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori.

Factores patogénicos de H. pylori en Cáncer Gástrico Página 21
Journal of Biological Chemistry, 270(30), 17771-17777.
56. Atherton, J., Peek Jr, R., Tham, K., Cover, T., & Blaser, M. (1997). Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology, 112(1), 92-99.
57. Miehlke, S., Kirsch, C., Agha‐Amiri, K., Günther, T., Lehn, N., Malfertheiner, P., et al. (2000). The Helicobacter pylori vacA s1, m1 genotype and cagA is associated with gastric carcinoma in Germany. International journal of cancer, 87(3), 322-327.
58. Rhead, J. L., Letley, D. P., Mohammadi, M., Hussein, N., Mohagheghi, M. A., Eshagh Hosseini, M., et al. (2007). A New Helicobacter pylori Vacuolating Cytotoxin Determinant, the Intermediate Region, Is Associated With Gastric Cancer. Gastroenterology, 133(3), 926-936.
59. Basso, D., Zambon, C., Letley, D. P., Stranges, A., Marchet, A., Rhead, J. L., et al. (2008). Clinical Relevance of Helicobacter pylori cagA and vacA Gene Polymorphisms. Gastroenterology, 135(1), 91-99.
60. Churin, Y., Al-Ghoul, L., Kepp, O., Meyer, T. F., Birchmeier, W., & Naumann, M. (2003). Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. The Journal of cell biology, 161(2), 249-255.
61. Mobley Harry LT, Mendz George L, Hazell Stuart L. (2001). Helicobacter pylori. Physiology and Genetics. Chapter 31. The cagPathogenecy Island, 608.
62. Tummuru, M., Cover, T., & Blaser, M. (1993). Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infection and immunity, 61(5), 1799-1809.
63. Backert, S., & Selbach, M. (2008). Role of type IV secretion in Helicobacter pylori
pathogenesis. Cellular microbiology, 10(8), 1573-1581.
64. Ishihara S, Fukuda R, Kawashima K, Moriyama N, Suotsugu H, Ishimura N, et al. T cell-mediated cytotoxicity via Fas/FasL signalling in H. pylori infected gastric corpus. Helicobacter. 2001; 6 (4): [283-93].
65. Yamazaki, S., Yamakawa, A., Ito, Y., Ohtani, M., Higashi, H., Hatakeyama, M., et al. (2003). The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to SHP-2 in human gastric mucosa. Journal of Infectious Diseases, 187(2), 334-337.
66. Busler, V. J., Torres, V. J., McClain, M. S., Tirado, O., Friedman, D. B., & Cover, T. L. (2006). Protein-protein interactions among Helicobacter pylori cag proteins. Journal of bacteriology, 188(13), 4787-4800.
67. Churin, Y., Al-Ghoul, L., Kepp, O., Meyer, T. F., Birchmeier, W., & Naumann, M. (2003). Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. The Journal of cell biology, 161(2), 249-255.
68. Hatakeyama, M. (2004). Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nature Reviews Cancer, 4(9), 688-694.
69. Handa, O., Naito, Y., & Yoshikawa, T. (2007). CagA protein of Helicobacter pylori: A hijacker of gastric epithelial cell signaling. Biochemical pharmacology, 73(11), 1697-1702.
70. Basso, D., Zambon, C., Letley, D. P., Stranges, A., Marchet, A., Rhead, J. L., et al. (2008). Clinical Relevance of Helicobacter pylori cagA and vacA Gene Polymorphisms. Gastroenterology, 135(1), 91-99.
71. Wroblewski, L. E., Peek Jr, R. M., & Wilson, K. T. (2010). Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clinical microbiology reviews, 23(4), 713-739.

Factores patogénicos de H. pylori en Cáncer Gástrico Página 22
72. Higashi, H., Tsutsumi, R., Fujita, A., Yamazaki, S., Asaka, M., Azuma, T., et al. (2002). Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proceedings of the National Academy of Sciences, 99(22), 14428.
73. Selbach, M., Moese, S., Hauck, C. R., Meyer, T. F., & Backert, S. (2002). Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. Journal of Biological Chemistry, 277(9), 6775-6778.
74. Hatakeyama, M., & Higashi, H. (2005). Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis. Cancer science, 96(12), 835-843.
75. Handa, O., Naito, Y., & Yoshikawa, T. (2007). CagA protein of Helicobacter pylori: A hijacker of gastric epithelial cell signaling. Biochemical pharmacology, 73(11), 1697-1702.
76. Ohnishi, N., Yuasa, H., Tanaka, S., Sawa, H., Miura, M., Matsui, A., et al. (2008). Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proceedings of the National Academy of Sciences, 105(3), 1003.
77. Lai, C., Chang, Y., Du, S., Wang, H., Kuo, C., Fang, S., et al. (2008). Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infection and immunity, 76(7), 3293-3303.
78. Yamazaki S, Yamakawa A, Ito Y et al. The CagA protein of H. pylori is translocated into epithelial cells and binds to SHP-2 in human gastric mucosa. J Infect Dis 2003; 187: 334–7.
79. Rivera, M., Contreras, F., & Terán, A. (2004). Helicobacter Pylori: Enteropatógeno frecuente del ser humano. 23(2), 109-117.
80. Rivas-Traverso, F., & Hernández, F. (2000). Factores de virulencia, patología y diagnóstico. Rev Biomed, 11, 187-205
81. Eslick, G. D. (2006). Helicobacter pylori infection causes gastric cancer? A review of the epidemiological, meta-analytic, and experimental evidence. World journal of gastroenterology: WJG, 12(19), 2991.
82. Connor, F., Buckley, M., & O'Morain, C. (1996). Helicobacter pylori: the cancer link. Journal of the Royal Society of Medicine, 89(12), 674.
83. Cárdenas, Y., Castro, V., Nevett, A., Patiño, P., & Urdaneta, G. (2006) Apoptosis and Helicobacter pylori: a new model for infectious oncogenesis. VITAE Academia Biomédica Digital, (29).
Related Documents











