IOP PUBLISHING NANOTECHNOLOGY Nanotechnology 23 (2012) 105601 (10pp) doi:10.1088/0957-4484/23/10/105601 Facile one-pot preparation, surface functionalization, and toxicity assay of APTS-coated iron oxide nanoparticles Mingwu Shen 1 , Hongdong Cai 1 , Xifu Wang 2 , Xueyan Cao 1 , Kangan Li 2 , Su He Wang 3 , Rui Guo 1 , Linfeng Zheng 2 , Guixiang Zhang 2 and Xiangyang Shi 1 ,4 1 College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, People’s Republic of China 2 Department of Radiology, First People’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200080, People’s Republic of China 3 Michigan Nanotechnology Institute for Medicine and Biological Sciences, University of Michigan, Ann Arbor, MI 48109, USA 4 CQM-Centro de Qu´ ımica da Madeira, Universidade da Madeira, Campus da Penteada, 9000-390 Funchal, Portugal E-mail: [email protected] and [email protected] Received 25 October 2011, in final form 23 December 2011 Published 21 February 2012 Online at stacks.iop.org/Nano/23/105601 Abstract We report a facile approach to synthesizing 3-aminopropyltrimethoxysilane (APTS)-coated magnetic iron oxide (Fe 3 O 4 @APTS) nanoparticles (NPs) with tunable surface functional groups for potential biomedical applications. The Fe 3 O 4 NPs with a mean diameter of 6.5 nm were synthesized by a hydrothermal route in the presence of APTS. The formed amine-surfaced Fe 3 O 4 @APTS NPs were further chemically modified with acetic anhydride and succinic anhydride to generate neutral (Fe 3 O 4 @APTS·Ac) and negatively charged (Fe 3 O 4 @APTS·SAH) NPs. These differently functionalized NPs were extensively characterized by x-ray diffraction, transmission electron microscopy, Fourier transform infrared spectroscopy, thermogravimetry analysis, zeta potential measurements, and T 2 relaxometry. The cytotoxicity of the particles was evaluated by in vitro 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide colorimetric viability assay of cells along with microscopic observation of cell morphology. The hemocompatibility of the particles was assessed by in vitro hemolysis assay. We show that the hydrothermal approach enables an efficient modification of APTS onto the Fe 3 O 4 NP surfaces and the formed NPs with different surface charge polarities are water-dispersible and colloidally stable. The acetylated Fe 3 O 4 @APTS·Ac NPs displayed good biocompatibility and hemocompatibility in the concentration range of 0–100 μg ml -1 , while the pristine Fe 3 O 4 @APTS and Fe 3 O 4 @APTS·SAH particles started to display slight cytotoxicity at a concentration of 10 μg ml -1 . The findings from this study suggest that the Fe 3 O 4 @APTS NPs synthesized by the one-pot hydrothermal route can be surface modified for various potential biomedical applications. (Some figures may appear in colour only in the online journal) 1. Introduction Magnetic iron oxide (Fe 3 O 4 ) nanoparticles (IONPs) have been extensively investigated for various applications in biomedical sciences including but not limited to detoxification of biological fluids [1, 2], anti-cancer drug delivery [3–5], hyperthermia [6, 7], and magnetic resonance (MR) imag- ing [8–12]. For a particular biomedical application, it 1 0957-4484/12/105601+10$33.00 c 2012 IOP Publishing Ltd Printed in the UK & the USA

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

IOP PUBLISHING NANOTECHNOLOGY
Nanotechnology 23 (2012) 105601 (10pp) doi:10.1088/0957-4484/23/10/105601
Facile one-pot preparation, surfacefunctionalization, and toxicity assay ofAPTS-coated iron oxide nanoparticles
Mingwu Shen1, Hongdong Cai1, Xifu Wang2, Xueyan Cao1, Kangan Li2,Su He Wang3, Rui Guo1, Linfeng Zheng2, Guixiang Zhang2 andXiangyang Shi1,4
1 College of Chemistry, Chemical Engineering and Biotechnology, Donghua University,Shanghai 201620, People’s Republic of China2 Department of Radiology, First People’s Hospital, School of Medicine, Shanghai Jiao Tong University,Shanghai 200080, People’s Republic of China3 Michigan Nanotechnology Institute for Medicine and Biological Sciences, University of Michigan,Ann Arbor, MI 48109, USA4 CQM-Centro de Quımica da Madeira, Universidade da Madeira, Campus da Penteada,9000-390 Funchal, Portugal
E-mail: [email protected] and [email protected]
Received 25 October 2011, in final form 23 December 2011Published 21 February 2012Online at stacks.iop.org/Nano/23/105601
AbstractWe report a facile approach to synthesizing 3-aminopropyltrimethoxysilane(APTS)-coated magnetic iron oxide (Fe3O4@APTS) nanoparticles (NPs) withtunable surface functional groups for potential biomedical applications. The Fe3O4 NPs with amean diameter of 6.5 nm were synthesized by a hydrothermal route in the presence of APTS.The formed amine-surfaced Fe3O4@APTS NPs were further chemically modified with aceticanhydride and succinic anhydride to generate neutral (Fe3O4@APTS·Ac) and negativelycharged (Fe3O4@APTS·SAH) NPs. These differently functionalized NPs were extensivelycharacterized by x-ray diffraction, transmission electron microscopy, Fourier transform infraredspectroscopy, thermogravimetry analysis, zeta potential measurements, and T2 relaxometry.The cytotoxicity of the particles was evaluated by in vitro 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide colorimetric viability assay of cells along with microscopicobservation of cell morphology. The hemocompatibility of the particles was assessedby in vitro hemolysis assay. We show that the hydrothermal approach enables an efficientmodification of APTS onto the Fe3O4 NP surfaces and the formed NPs with different surfacecharge polarities are water-dispersible and colloidally stable. The acetylated Fe3O4@APTS·AcNPs displayed good biocompatibility and hemocompatibility in the concentrationrange of 0–100 µg ml−1, while the pristine Fe3O4@APTS and Fe3O4@APTS·SAHparticles started to display slight cytotoxicity at a concentration of 10 µg ml−1.The findings from this study suggest that the Fe3O4@APTS NPs synthesized by the one-pothydrothermal route can be surface modified for various potential biomedical applications.
(Some figures may appear in colour only in the online journal)
1. Introduction
Magnetic iron oxide (Fe3O4) nanoparticles (IONPs) havebeen extensively investigated for various applications in
biomedical sciences including but not limited to detoxificationof biological fluids [1, 2], anti-cancer drug delivery [3–5],hyperthermia [6, 7], and magnetic resonance (MR) imag-ing [8–12]. For a particular biomedical application, it
10957-4484/12/105601+10$33.00 c© 2012 IOP Publishing Ltd Printed in the UK & the USA

Nanotechnology 23 (2012) 105601 M Shen et al
Scheme 1. Schematic illustration of the hydrothermal synthesis of Fe3O4@APTS NPs and the subsequent functionalization of theFe3O4@APTS NPs to form Fe3O4@APTS·Ac and Fe3O4@APTS·SAH NPs.
is crucial to have an ability to synthesize IONPs withcontrollable sizes and surface functionalities, since thein vivo biomedical performance, especially the blood half-life,opsonization, pharmacokinetics, and biodistribution of theIONPs are closely associated with the particle size andsurface modification [1]. A range of different approacheshave been employed to synthesize IONPs with controllablesize ranging from a few to hundreds of nanometers. Thesesynthetic routes include controlled coprecipitation of Fe(II)and Fe(III) ions at an elevated temperature [13], a successivereduction–oxidation process in a reverse micelle system [14],a thermal decomposition route [15–17], and a hydrothermalmethod under a higher pressure [18]. These syntheticapproaches can be mainly divided into two categories:aqueous phase synthesis and organic oil phase synthesis. Inboth cases, the IONPs synthesized either in aqueous solutionor in the oil phase have to be surface modified to generatethe desired surface functionalities and aqueous dispersitybefore they can be applied for biomedical applications.Therefore, synthesis of various surface-functionalized IONPswith desired surface functionalities still remains a greatchallenge.
In order to make IONPs with good water dispersity,which is essential for their biomedical applications, surfactantmolecules have been added into the reaction mixture, therebygenerating IONPs with the desired water dispersity [19]. Inmost cases, preformed IONPs have been post-functionalizedby surfactant molecules [20], silane agent [21, 22] or othersmall molecular ligands [23–25] to make the IONPs bearreactive functional groups, enabling further bioconjugationchemistry for biomedical applications. There are only a fewstudies related to the in situ functionalization of IONPs withreactive functional ligands by addition of the agents into thereaction mixture for formation of the IONPs [8, 26–28]. Forinstance, polyethylene glycol (PEG) derivatives have beenadded into the reaction mixture, which enables the formationof IONPs with carboxyl reactive surface groups [8, 26]. Xieet al added another small molecular ligand 4-methylcatecholinto the reaction mixture for the decomposition formationof IONPs to generate functionalized IONPs that enabledconjugation with peptide for MR imaging applications [27].Pramanik and coworkers developed an approach to form-ing 3-aminopropyltriethoxysilane (NH2(CH2)3–Si–(OCH3)3,APTS)-coated IONPs by adding APTS into the mixture so-lution of Fe(II) and Fe(III) ions for controlled coprecipitationformation of IONPs [28]. In general, the post-modification ofpreformed IONPs is time-consuming and requires additional
reactions or steps to process the particles. For the reportedone-pot approaches to synthesizing IONPs with reactivesurface functional groups, the investigations are mainlylimited to high-temperature decomposition formation andcontrolled coprecipitation formation of IONPs.
In our previous work, we have demonstrated a facilehydrothermal approach to synthesizing Fe3O4 IONPs withrelatively uniform size and size distribution under an elevatedtemperature and pressure [18]. The size of the formedIONPs with high crystallinity can be readily tuned from15 to 31 nm through variation of the reaction conditions.For biomedical applications, the formed IONPs shouldbe surface functionalized. It is reasonable to deduce thata one-pot approach to synthesizing IONPs with reactivesurface functional groups may be realized by addition ofa small molecular ligand into the reaction mixture for thehydrothermal synthesis of the IONPs. It is well known thatFe3O4 NPs synthesized by controlled coprecipitation of Fe(II)and Fe(III) ions can be further silanized by APTS by takingadvantage of the rich –OH groups on the Fe3O4 particlesurfaces [21, 22]; therefore it is hypothesized that by additionof the APTS molecules inside the hydrothermal reactionmixture for the synthesis of IONPs, Fe3O4 NPs with surfaceamine groups can be easily synthesized.
In this study, we present a simple one-step APTS-assistedhydrothermal approach to synthesizing APTS-coated Fe3O4NPs (Fe3O4@APTS) with reactive surface amine groups.The APTS modification endowed the Fe3O4 NPs with anexcellent water dispersibility and colloidal stability. Thesurface-reactive amine groups of Fe3O4 NPs were furtheracetylated and carboxylated by reacting with acetic anhydrideand succinic anhydride respectively to generate neutralizedand negatively charged particles (scheme 1), which may beused for different biomedical applications. The as-synthesizedproducts were characterized by x-ray diffraction (XRD),transmission electron microscopy (TEM), Fourier transforminfrared spectrometry (FTIR), thermogravimetry analysis(TGA), and zeta potential measurements. The T2 relaxometryof the Fe3O4@APTS NPs was measured using a standardmultiple spin-echo pulse sequence. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viabilityassay along with cell morphology observation was usedto assess the cytotoxicity of the differently functionalizedIONPs, while hemolysis assay was carried out to evaluatethe hemocompatibility of the particles. To the best of ourknowledge, this is the first report related to the facile one-stepsynthesis of APTS-coated IONPs with surface-reactive
2

Nanotechnology 23 (2012) 105601 M Shen et al
amines and the systemic acylation modification of aminatedIONPs with different surface charge polarities, which may beused for various biomedical applications.
2. Experimental section
2.1. Materials
All chemicals were analytical grade and used without furtherpurification from commercial sources. Ferrous chloridetetrahydrate (FeCl2·4H2O > 99%), ammonia (28–30% NH3in water solution), triethylamine, acetic anhydride, anddimethyl sulfoxide (DMSO) were purchased from SinopharmChemical Reagent Co., Ltd. APTS and succinic anhydridewere obtained from Acros Organics. The KB cells (ahuman epithelial carcinoma cell line) were from Instituteof Biochemistry and Cell Biology, the Chinese Academy ofScience (Shanghai, China). RPMI-1640 medium, fetal bovineserum (FBS), penicillin, and streptomycin were purchasedfrom Hangzhou Jinuo Biomedical Technology (Hangzhou,China). MTT was acquired from Shanghai Sangon BiologicalEngineering Technology & Services Co., Ltd. The water usedin all experiments was purified using a Milli-Q Plus 185water purification system (Millipore, Bedford, MA) with aresistivity higher than 18.2 M� cm.
2.2. Synthesis of APTS-coated Fe3O4 NPs
Fe3O4@APTS NPs were synthesized using a hydrothermalapproach described in our previous work with slightmodification [18]. Typically, FeCl2·4H2O (1.25 g) wasdissolved in 7.75 ml water. Under vigorous stirring,ammonium hydroxide (6.25 ml) was added, and thesuspension was continuously stirred in air for 10 min,allowing the Fe(II) to be oxidized. Then, 2.5 ml of APTSwas added and the reaction mixture was autoclaved (KH-50Autoclave, Shanghai Yuying Instrument Co., Ltd, Shanghai)in a sealed pressure vessel with a volume of 50 ml at134 ◦C. After 3 h, the reaction mixture was cooled downto room temperature. The black precipitate was collectedand purified with water five times and ethanol twice viaa centrifugation–dispersion process (5000 rpm, 10 min) toremove excess reactants. Finally, the obtained Fe3O4@APTSNPs were dispersed in ethanol with a concentration of15 mg ml−1.
2.3. Acetylation and carboxylation of Fe3O4@APTS NPs
The amine groups on the surface of the Fe3O4@APTSNPs were further acetylated and carboxylated by reactingwith acetic anhydride and succinic anhydride respectivelyaccording to the protocols described in our previouswork [29]. Typically, 1 ml of triethylamine was added tothe Fe3O4@APTS (6 mg) solution dispersed in ethanol(5 ml), and the solution was thoroughly mixed. A DMSOsolution (5 ml) containing acetic anhydride (1 ml) wasdropwise added into the solution of Fe3O4@APTS mixedwith triethylamine while it was being stirred vigorously. Themixture was allowed to react for 24 h. The DMSO, excess
reactants, and byproduct were removed from the mixture byfive times of centrifugation/washing/dispersion steps to obtainthe Fe3O4@APTS·Ac NPs finally dispersed in methanol. Forcarboxylation of the surface amines of the Fe3O4@APTSNPs, Fe3O4@APTS NPs (6 mg) dispersed in ethanol (5 ml)was mixed with succinic anhydride (800 mg) dissolved inDMSO (5 ml) under vigorous stirring. The mixture wasallowed to react for 24 h. The DMSO and the excessreactants and byproduct were removed from the mixtureby five centrifugation/washing/dispersion steps to obtain theFe3O4@APTS·SAH NPs that finally dispersed in methanol.
2.4. Characterization techniques
The crystalline structure and the size of the products weredetermined using XRD by step scan in a D/max 2550PC x-ray diffractometer (Japan, Rigaku Cop.) with Cu Kαradiation (λ = 0.1541 nm) at 40 kV and 200 mA. The scanrange (2θ ) was from 25 to 70 ◦C. The morphology of theformed Fe3O4 NPs was confirmed by TEM imaging usinga JEOL 2010F analytical electron microscope operating at200 kV. A TEM sample was prepared by placing one dropof diluted Fe3O4 NP suspension (5 µl) onto a 200 meshcarbon-coated copper grid and air-dried before measurement.The Feret diameter of the NPs was measured using theimage analysis software ImageJ 1.40G (http://rsb.info.nih.gov/ij/download.html). At least 200 randomly selected NPsin different TEM images were analyzed for each sampleto acquire the size distribution histogram. FTIR spectrawere recorded using a Nicolet 5700 FTIR spectrometer(Thermo Nicolet Corporation, US) at the wavenumber rangeof 4000–400 cm−1 at ambient conditions. TGA was carriedout using a TG 209 F1 (NETZSCH Instruments Co., Ltd,Germany) thermogravimetric analyzer with a heating rateof 20 ◦C min−1 and temperature range of 30–900 ◦C undernitrogen gas. Zeta potential measurements and dynamic lightscattering were performed using a Malvern Zetasizer NanoZS model ZEN3600 (Worcestershire, UK) equipped witha standard 633 nm laser. T2 relaxometry was performedusing an NMI20-Analyst NMR Analyzing & Imaging system(Shanghai Niumag Corporation). The instrumental parameterswere set as follows: a 0.5 T magnet, point resolution =156 mm×156 mm, section thickness = 0.6 mm, TE = 60 ms,TR = 4000 ms, number of acquisitions = 1. The T2 relaxivitywas calculated from the linear slope of the inverse T2 (1/T2)relaxation time versus the Fe molar concentration. The Feconcentration of the Fe3O4@APTS NPs was analyzed usinga PRODIGY inductively coupled plasma-atomic emissionspectroscope (ICP-AES) (Teledyne Leeman Labs, USA) afteraqua regia treatment. The stability of the functionalized Fe3O4NPs was assessed by dispersing them in water, phosphatebuffered saline (PBS), and FBS solution for a period of timeof up to one month.
2.5. Cytotoxicity assay
The KB cells were continuously grown in a 50 ml cultureflask in regular RPMI-1640 medium supplemented with 10%
3
Nanotechnology 23 (2012) 105601 M Shen et al
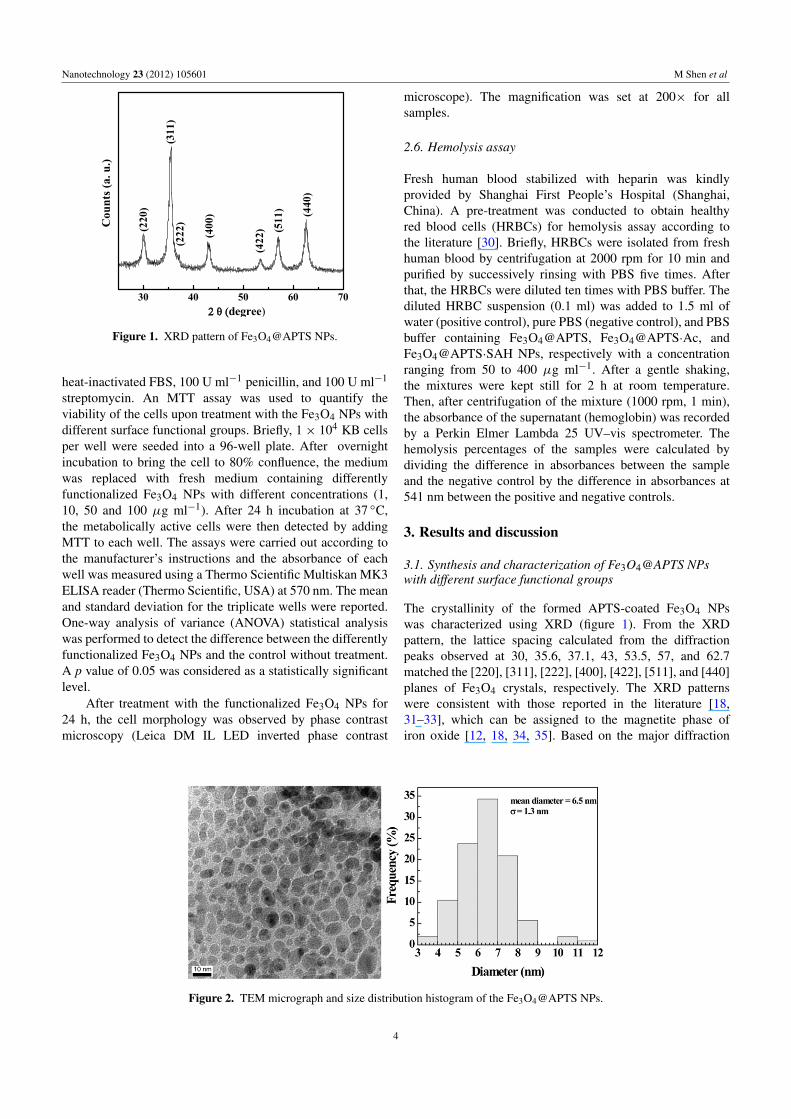
Figure 1. XRD pattern of Fe3O4@APTS NPs.
heat-inactivated FBS, 100 U ml−1 penicillin, and 100 U ml−1
streptomycin. An MTT assay was used to quantify theviability of the cells upon treatment with the Fe3O4 NPs withdifferent surface functional groups. Briefly, 1 × 104 KB cellsper well were seeded into a 96-well plate. After overnightincubation to bring the cell to 80% confluence, the mediumwas replaced with fresh medium containing differentlyfunctionalized Fe3O4 NPs with different concentrations (1,10, 50 and 100 µg ml−1). After 24 h incubation at 37 ◦C,the metabolically active cells were then detected by addingMTT to each well. The assays were carried out according tothe manufacturer’s instructions and the absorbance of eachwell was measured using a Thermo Scientific Multiskan MK3ELISA reader (Thermo Scientific, USA) at 570 nm. The meanand standard deviation for the triplicate wells were reported.One-way analysis of variance (ANOVA) statistical analysiswas performed to detect the difference between the differentlyfunctionalized Fe3O4 NPs and the control without treatment.A p value of 0.05 was considered as a statistically significantlevel.
After treatment with the functionalized Fe3O4 NPs for24 h, the cell morphology was observed by phase contrastmicroscopy (Leica DM IL LED inverted phase contrast
microscope). The magnification was set at 200× for allsamples.
2.6. Hemolysis assay
Fresh human blood stabilized with heparin was kindlyprovided by Shanghai First People’s Hospital (Shanghai,China). A pre-treatment was conducted to obtain healthyred blood cells (HRBCs) for hemolysis assay according tothe literature [30]. Briefly, HRBCs were isolated from freshhuman blood by centrifugation at 2000 rpm for 10 min andpurified by successively rinsing with PBS five times. Afterthat, the HRBCs were diluted ten times with PBS buffer. Thediluted HRBC suspension (0.1 ml) was added to 1.5 ml ofwater (positive control), pure PBS (negative control), and PBSbuffer containing Fe3O4@APTS, Fe3O4@APTS·Ac, andFe3O4@APTS·SAH NPs, respectively with a concentrationranging from 50 to 400 µg ml−1. After a gentle shaking,the mixtures were kept still for 2 h at room temperature.Then, after centrifugation of the mixture (1000 rpm, 1 min),the absorbance of the supernatant (hemoglobin) was recordedby a Perkin Elmer Lambda 25 UV–vis spectrometer. Thehemolysis percentages of the samples were calculated bydividing the difference in absorbances between the sampleand the negative control by the difference in absorbances at541 nm between the positive and negative controls.
3. Results and discussion
3.1. Synthesis and characterization of Fe3O4@APTS NPswith different surface functional groups
The crystallinity of the formed APTS-coated Fe3O4 NPswas characterized using XRD (figure 1). From the XRDpattern, the lattice spacing calculated from the diffractionpeaks observed at 30, 35.6, 37.1, 43, 53.5, 57, and 62.7matched the [220], [311], [222], [400], [422], [511], and [440]planes of Fe3O4 crystals, respectively. The XRD patternswere consistent with those reported in the literature [18,31–33], which can be assigned to the magnetite phase ofiron oxide [12, 18, 34, 35]. Based on the major diffraction
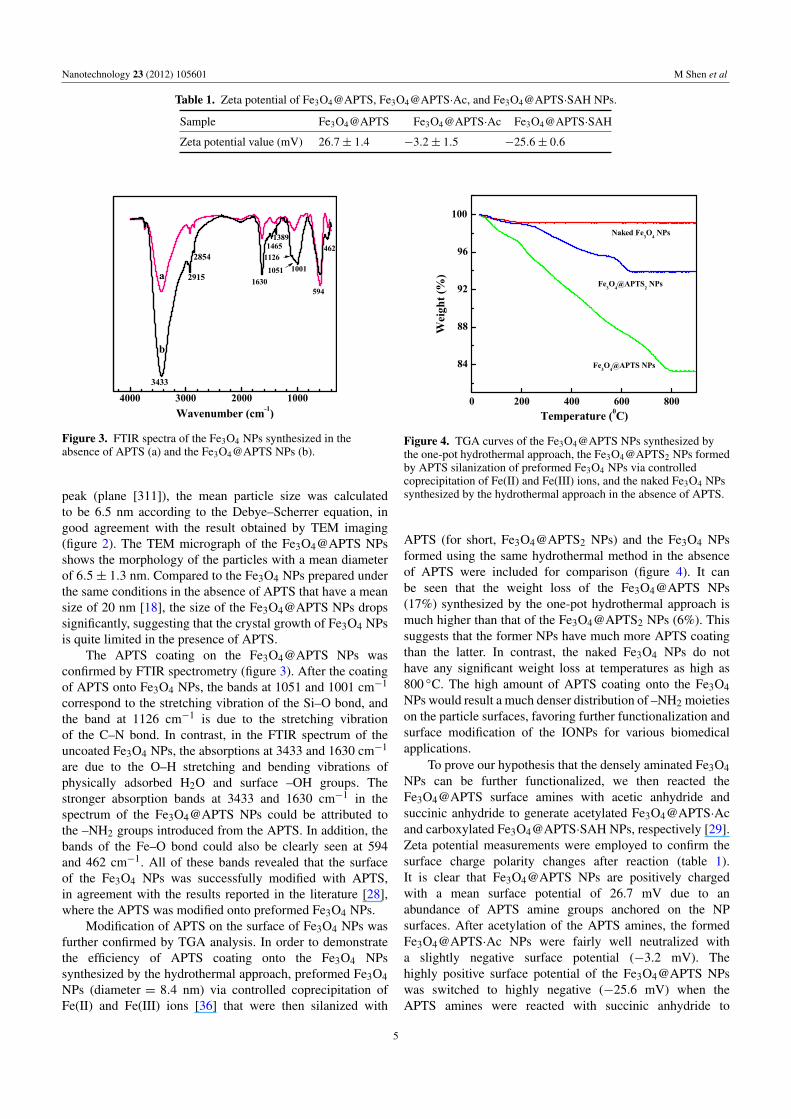
Figure 2. TEM micrograph and size distribution histogram of the Fe3O4@APTS NPs.
4
Nanotechnology 23 (2012) 105601 M Shen et al
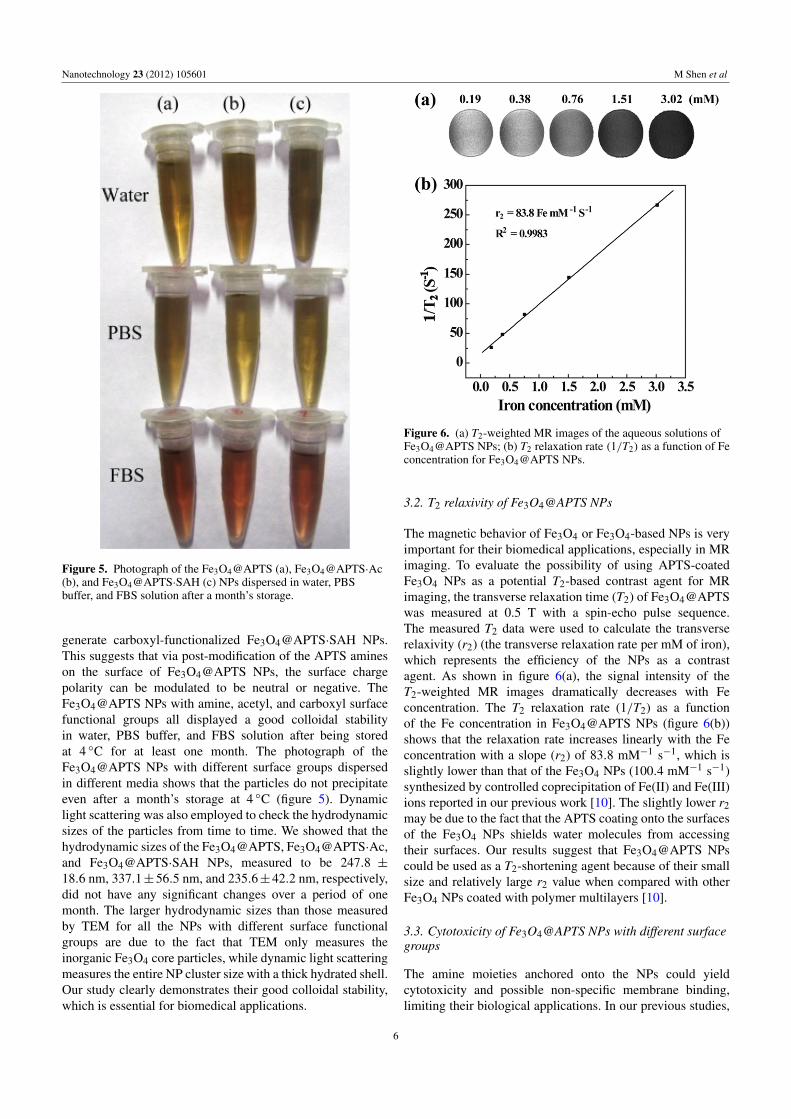
Table 1. Zeta potential of Fe3O4@APTS, Fe3O4@APTS·Ac, and Fe3O4@APTS·SAH NPs.
Sample Fe3O4@APTS Fe3O4@APTS·Ac Fe3O4@APTS·SAH
Zeta potential value (mV) 26.7± 1.4 −3.2± 1.5 −25.6± 0.6
Figure 3. FTIR spectra of the Fe3O4 NPs synthesized in theabsence of APTS (a) and the Fe3O4@APTS NPs (b).
peak (plane [311]), the mean particle size was calculatedto be 6.5 nm according to the Debye–Scherrer equation, ingood agreement with the result obtained by TEM imaging(figure 2). The TEM micrograph of the Fe3O4@APTS NPsshows the morphology of the particles with a mean diameterof 6.5± 1.3 nm. Compared to the Fe3O4 NPs prepared underthe same conditions in the absence of APTS that have a meansize of 20 nm [18], the size of the Fe3O4@APTS NPs dropssignificantly, suggesting that the crystal growth of Fe3O4 NPsis quite limited in the presence of APTS.
The APTS coating on the Fe3O4@APTS NPs wasconfirmed by FTIR spectrometry (figure 3). After the coatingof APTS onto Fe3O4 NPs, the bands at 1051 and 1001 cm−1
correspond to the stretching vibration of the Si–O bond, andthe band at 1126 cm−1 is due to the stretching vibrationof the C–N bond. In contrast, in the FTIR spectrum of theuncoated Fe3O4 NPs, the absorptions at 3433 and 1630 cm−1
are due to the O–H stretching and bending vibrations ofphysically adsorbed H2O and surface –OH groups. Thestronger absorption bands at 3433 and 1630 cm−1 in thespectrum of the Fe3O4@APTS NPs could be attributed tothe –NH2 groups introduced from the APTS. In addition, thebands of the Fe–O bond could also be clearly seen at 594and 462 cm−1. All of these bands revealed that the surfaceof the Fe3O4 NPs was successfully modified with APTS,in agreement with the results reported in the literature [28],where the APTS was modified onto preformed Fe3O4 NPs.
Modification of APTS on the surface of Fe3O4 NPs wasfurther confirmed by TGA analysis. In order to demonstratethe efficiency of APTS coating onto the Fe3O4 NPssynthesized by the hydrothermal approach, preformed Fe3O4NPs (diameter = 8.4 nm) via controlled coprecipitation ofFe(II) and Fe(III) ions [36] that were then silanized with
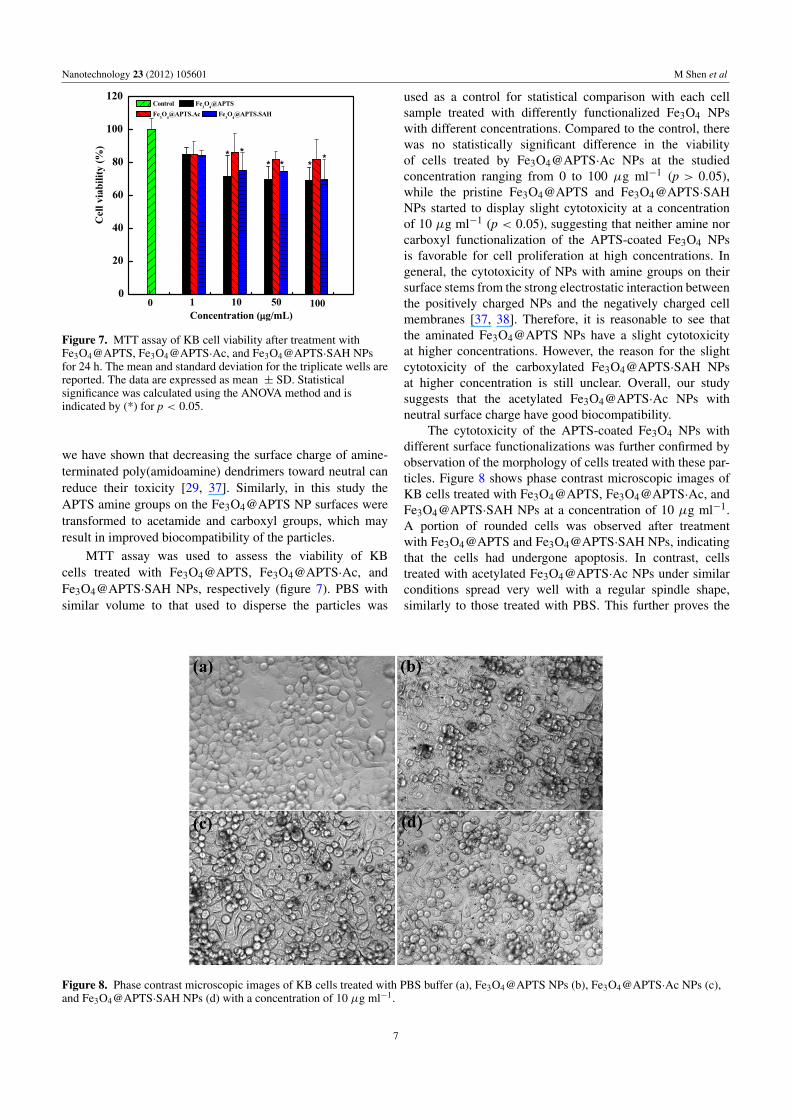
Figure 4. TGA curves of the Fe3O4@APTS NPs synthesized bythe one-pot hydrothermal approach, the Fe3O4@APTS2 NPs formedby APTS silanization of preformed Fe3O4 NPs via controlledcoprecipitation of Fe(II) and Fe(III) ions, and the naked Fe3O4 NPssynthesized by the hydrothermal approach in the absence of APTS.
APTS (for short, Fe3O4@APTS2 NPs) and the Fe3O4 NPsformed using the same hydrothermal method in the absenceof APTS were included for comparison (figure 4). It canbe seen that the weight loss of the Fe3O4@APTS NPs(17%) synthesized by the one-pot hydrothermal approach ismuch higher than that of the Fe3O4@APTS2 NPs (6%). Thissuggests that the former NPs have much more APTS coatingthan the latter. In contrast, the naked Fe3O4 NPs do nothave any significant weight loss at temperatures as high as800 ◦C. The high amount of APTS coating onto the Fe3O4NPs would result a much denser distribution of –NH2 moietieson the particle surfaces, favoring further functionalization andsurface modification of the IONPs for various biomedicalapplications.
To prove our hypothesis that the densely aminated Fe3O4NPs can be further functionalized, we then reacted theFe3O4@APTS surface amines with acetic anhydride andsuccinic anhydride to generate acetylated Fe3O4@APTS·Acand carboxylated Fe3O4@APTS·SAH NPs, respectively [29].Zeta potential measurements were employed to confirm thesurface charge polarity changes after reaction (table 1).It is clear that Fe3O4@APTS NPs are positively chargedwith a mean surface potential of 26.7 mV due to anabundance of APTS amine groups anchored on the NPsurfaces. After acetylation of the APTS amines, the formedFe3O4@APTS·Ac NPs were fairly well neutralized witha slightly negative surface potential (−3.2 mV). Thehighly positive surface potential of the Fe3O4@APTS NPswas switched to highly negative (−25.6 mV) when theAPTS amines were reacted with succinic anhydride to
5
Nanotechnology 23 (2012) 105601 M Shen et al
Figure 5. Photograph of the Fe3O4@APTS (a), Fe3O4@APTS·Ac(b), and Fe3O4@APTS·SAH (c) NPs dispersed in water, PBSbuffer, and FBS solution after a month’s storage.
generate carboxyl-functionalized Fe3O4@APTS·SAH NPs.This suggests that via post-modification of the APTS amineson the surface of Fe3O4@APTS NPs, the surface chargepolarity can be modulated to be neutral or negative. TheFe3O4@APTS NPs with amine, acetyl, and carboxyl surfacefunctional groups all displayed a good colloidal stabilityin water, PBS buffer, and FBS solution after being storedat 4 ◦C for at least one month. The photograph of theFe3O4@APTS NPs with different surface groups dispersedin different media shows that the particles do not precipitateeven after a month’s storage at 4 ◦C (figure 5). Dynamiclight scattering was also employed to check the hydrodynamicsizes of the particles from time to time. We showed that thehydrodynamic sizes of the Fe3O4@APTS, Fe3O4@APTS·Ac,and Fe3O4@APTS·SAH NPs, measured to be 247.8 ±18.6 nm, 337.1±56.5 nm, and 235.6±42.2 nm, respectively,did not have any significant changes over a period of onemonth. The larger hydrodynamic sizes than those measuredby TEM for all the NPs with different surface functionalgroups are due to the fact that TEM only measures theinorganic Fe3O4 core particles, while dynamic light scatteringmeasures the entire NP cluster size with a thick hydrated shell.Our study clearly demonstrates their good colloidal stability,which is essential for biomedical applications.
Figure 6. (a) T2-weighted MR images of the aqueous solutions ofFe3O4@APTS NPs; (b) T2 relaxation rate (1/T2) as a function of Feconcentration for Fe3O4@APTS NPs.
3.2. T2 relaxivity of Fe3O4@APTS NPs
The magnetic behavior of Fe3O4 or Fe3O4-based NPs is veryimportant for their biomedical applications, especially in MRimaging. To evaluate the possibility of using APTS-coatedFe3O4 NPs as a potential T2-based contrast agent for MRimaging, the transverse relaxation time (T2) of Fe3O4@APTSwas measured at 0.5 T with a spin-echo pulse sequence.The measured T2 data were used to calculate the transverserelaxivity (r2) (the transverse relaxation rate per mM of iron),which represents the efficiency of the NPs as a contrastagent. As shown in figure 6(a), the signal intensity of theT2-weighted MR images dramatically decreases with Feconcentration. The T2 relaxation rate (1/T2) as a functionof the Fe concentration in Fe3O4@APTS NPs (figure 6(b))shows that the relaxation rate increases linearly with the Feconcentration with a slope (r2) of 83.8 mM−1 s−1, which isslightly lower than that of the Fe3O4 NPs (100.4 mM−1 s−1)synthesized by controlled coprecipitation of Fe(II) and Fe(III)ions reported in our previous work [10]. The slightly lower r2may be due to the fact that the APTS coating onto the surfacesof the Fe3O4 NPs shields water molecules from accessingtheir surfaces. Our results suggest that Fe3O4@APTS NPscould be used as a T2-shortening agent because of their smallsize and relatively large r2 value when compared with otherFe3O4 NPs coated with polymer multilayers [10].
3.3. Cytotoxicity of Fe3O4@APTS NPs with different surfacegroups
The amine moieties anchored onto the NPs could yieldcytotoxicity and possible non-specific membrane binding,limiting their biological applications. In our previous studies,
6
Nanotechnology 23 (2012) 105601 M Shen et al
Figure 7. MTT assay of KB cell viability after treatment withFe3O4@APTS, Fe3O4@APTS·Ac, and Fe3O4@APTS·SAH NPsfor 24 h. The mean and standard deviation for the triplicate wells arereported. The data are expressed as mean ± SD. Statisticalsignificance was calculated using the ANOVA method and isindicated by (*) for p < 0.05.
we have shown that decreasing the surface charge of amine-terminated poly(amidoamine) dendrimers toward neutral canreduce their toxicity [29, 37]. Similarly, in this study theAPTS amine groups on the Fe3O4@APTS NP surfaces weretransformed to acetamide and carboxyl groups, which mayresult in improved biocompatibility of the particles.
MTT assay was used to assess the viability of KBcells treated with Fe3O4@APTS, Fe3O4@APTS·Ac, andFe3O4@APTS·SAH NPs, respectively (figure 7). PBS withsimilar volume to that used to disperse the particles was
used as a control for statistical comparison with each cellsample treated with differently functionalized Fe3O4 NPswith different concentrations. Compared to the control, therewas no statistically significant difference in the viabilityof cells treated by Fe3O4@APTS·Ac NPs at the studiedconcentration ranging from 0 to 100 µg ml−1 (p > 0.05),while the pristine Fe3O4@APTS and Fe3O4@APTS·SAHNPs started to display slight cytotoxicity at a concentrationof 10 µg ml−1 (p < 0.05), suggesting that neither amine norcarboxyl functionalization of the APTS-coated Fe3O4 NPsis favorable for cell proliferation at high concentrations. Ingeneral, the cytotoxicity of NPs with amine groups on theirsurface stems from the strong electrostatic interaction betweenthe positively charged NPs and the negatively charged cellmembranes [37, 38]. Therefore, it is reasonable to see thatthe aminated Fe3O4@APTS NPs have a slight cytotoxicityat higher concentrations. However, the reason for the slightcytotoxicity of the carboxylated Fe3O4@APTS·SAH NPsat higher concentration is still unclear. Overall, our studysuggests that the acetylated Fe3O4@APTS·Ac NPs withneutral surface charge have good biocompatibility.
The cytotoxicity of the APTS-coated Fe3O4 NPs withdifferent surface functionalizations was further confirmed byobservation of the morphology of cells treated with these par-ticles. Figure 8 shows phase contrast microscopic images ofKB cells treated with Fe3O4@APTS, Fe3O4@APTS·Ac, andFe3O4@APTS·SAH NPs at a concentration of 10 µg ml−1.A portion of rounded cells was observed after treatmentwith Fe3O4@APTS and Fe3O4@APTS·SAH NPs, indicatingthat the cells had undergone apoptosis. In contrast, cellstreated with acetylated Fe3O4@APTS·Ac NPs under similarconditions spread very well with a regular spindle shape,similarly to those treated with PBS. This further proves the
Figure 8. Phase contrast microscopic images of KB cells treated with PBS buffer (a), Fe3O4@APTS NPs (b), Fe3O4@APTS·Ac NPs (c),and Fe3O4@APTS·SAH NPs (d) with a concentration of 10 µg ml−1.
7
Nanotechnology 23 (2012) 105601 M Shen et al
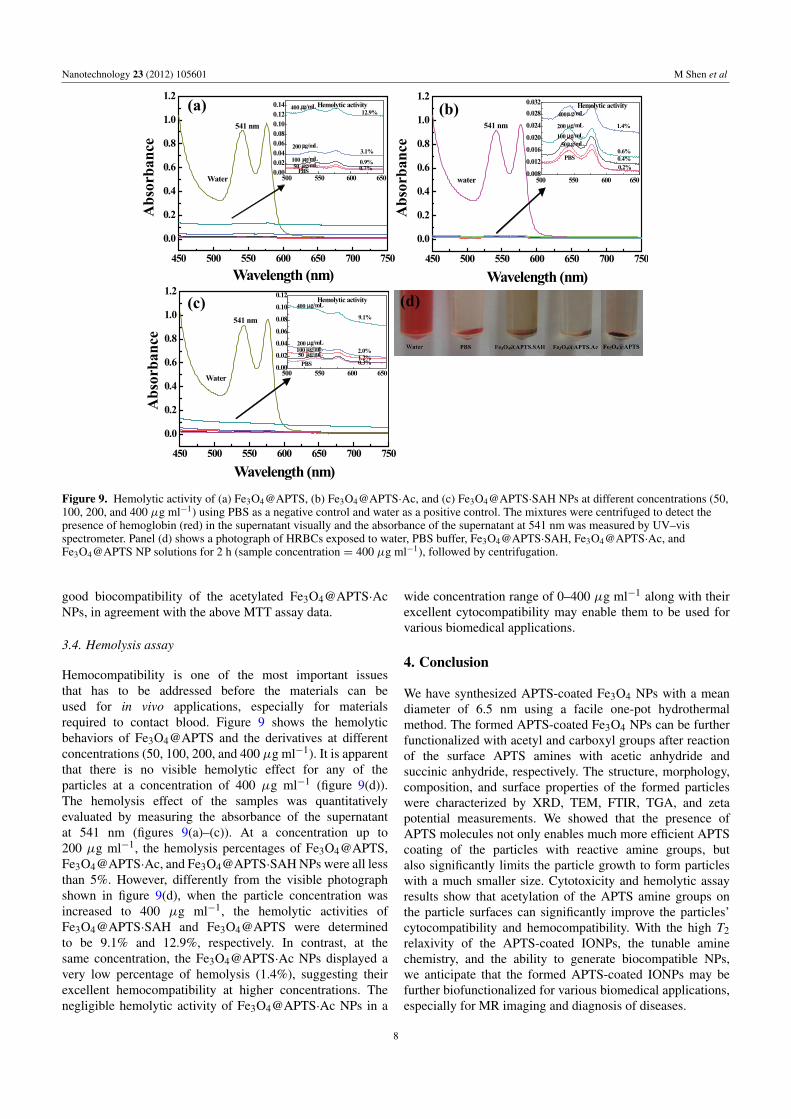
Figure 9. Hemolytic activity of (a) Fe3O4@APTS, (b) Fe3O4@APTS·Ac, and (c) Fe3O4@APTS·SAH NPs at different concentrations (50,100, 200, and 400 µg ml−1) using PBS as a negative control and water as a positive control. The mixtures were centrifuged to detect thepresence of hemoglobin (red) in the supernatant visually and the absorbance of the supernatant at 541 nm was measured by UV–visspectrometer. Panel (d) shows a photograph of HRBCs exposed to water, PBS buffer, Fe3O4@APTS·SAH, Fe3O4@APTS·Ac, andFe3O4@APTS NP solutions for 2 h (sample concentration = 400 µg ml−1), followed by centrifugation.
good biocompatibility of the acetylated Fe3O4@APTS·AcNPs, in agreement with the above MTT assay data.
3.4. Hemolysis assay
Hemocompatibility is one of the most important issuesthat has to be addressed before the materials can beused for in vivo applications, especially for materialsrequired to contact blood. Figure 9 shows the hemolyticbehaviors of Fe3O4@APTS and the derivatives at differentconcentrations (50, 100, 200, and 400 µg ml−1). It is apparentthat there is no visible hemolytic effect for any of theparticles at a concentration of 400 µg ml−1 (figure 9(d)).The hemolysis effect of the samples was quantitativelyevaluated by measuring the absorbance of the supernatantat 541 nm (figures 9(a)–(c)). At a concentration up to200 µg ml−1, the hemolysis percentages of Fe3O4@APTS,Fe3O4@APTS·Ac, and Fe3O4@APTS·SAH NPs were all lessthan 5%. However, differently from the visible photographshown in figure 9(d), when the particle concentration wasincreased to 400 µg ml−1, the hemolytic activities ofFe3O4@APTS·SAH and Fe3O4@APTS were determinedto be 9.1% and 12.9%, respectively. In contrast, at thesame concentration, the Fe3O4@APTS·Ac NPs displayed avery low percentage of hemolysis (1.4%), suggesting theirexcellent hemocompatibility at higher concentrations. Thenegligible hemolytic activity of Fe3O4@APTS·Ac NPs in a
wide concentration range of 0–400 µg ml−1 along with theirexcellent cytocompatibility may enable them to be used forvarious biomedical applications.
4. Conclusion
We have synthesized APTS-coated Fe3O4 NPs with a meandiameter of 6.5 nm using a facile one-pot hydrothermalmethod. The formed APTS-coated Fe3O4 NPs can be furtherfunctionalized with acetyl and carboxyl groups after reactionof the surface APTS amines with acetic anhydride andsuccinic anhydride, respectively. The structure, morphology,composition, and surface properties of the formed particleswere characterized by XRD, TEM, FTIR, TGA, and zetapotential measurements. We showed that the presence ofAPTS molecules not only enables much more efficient APTScoating of the particles with reactive amine groups, butalso significantly limits the particle growth to form particleswith a much smaller size. Cytotoxicity and hemolytic assayresults show that acetylation of the APTS amine groups onthe particle surfaces can significantly improve the particles’cytocompatibility and hemocompatibility. With the high T2relaxivity of the APTS-coated IONPs, the tunable aminechemistry, and the ability to generate biocompatible NPs,we anticipate that the formed APTS-coated IONPs may befurther biofunctionalized for various biomedical applications,especially for MR imaging and diagnosis of diseases.
8

Nanotechnology 23 (2012) 105601 M Shen et al
Acknowledgments
This research is financially supported by the National NaturalScience Foundation of China (81101150, 20974019), theNano Specialized Research Fund of Shanghai Science andTechnology Commission (1052nm05800, 11nm0506400), theFundamental Research Funds for the Central Universities(for MS, RG, XC, and XS), the Shanghai NaturalScience Foundation (11ZR1429300), and the InnovationFunds of Donghua University Doctorate Dissertation ofExcellence (BC201104 for HC). MS acknowledges thesupport from Shanghai Bai Yu Lan foundation (2010B003).KL acknowledges the Shanghai Songjiang Medical ClimbingProgram (2011PD04). XS gratefully acknowledges theFundacao para a Ciencia e a Tecnologia (FCT) and Santanderbank for the Chair in Nanotechnology.
References
[1] Gupta A K and Gupta M 2005 Synthesis and surfaceengineering of iron oxide nanoparticles for biomedicalapplications Biomaterials 26 3995–4021
[2] Samra Z Q, Shabir S, Rehmat Z, Zaman M, Nazir A,Dar N and Athar M A 2010 Synthesis ofcholesterol-conjugated magnetic nanoparticles forpurification of human paraoxonase 1 Appl. Biochem.Biotechnol. 162 671–86
[3] Ling Y, Wei K, Luo Y, Gao X and Zhong S 2011 Dualdocetaxel/superparamagnetic iron oxide loadednanoparticles for both targeting magnetic resonanceimaging and cancer therapy Biomaterials 32 7139–50
[4] Chen D, Jiang M, Li N, Gu H, Xu Q, Ge J, Xia X andLu J 2010 Modification of magnetic silica/iron oxidenanocomposites with fluorescent polymethacrylic acid forcancer targeting and drug delivery J. Mater. Chem.20 6422–9
[5] Hu F X, Neoh K G and Kang E T 2006 Synthesis and in vitroanti-cancer evaluation of tamoxifen-loadedmagnetite/PLLA composite nanoparticles Biomaterials27 5725–33
[6] Purushotham S and Ramanujan R V 2010 Thermoresponsivemagnetic composite nanomaterials for multimodal cancertherapy Acta Biomater. 6 502–10
[7] Regmi R, Bhattarai S R, Sudakar C, Wani A S,Cunningham R, Vaishnava P P, Naik R, Oupicky D andLawes G 2010 Hyperthermia controlled rapid drug releasefrom thermosensitive magnetic microgels J. Mater. Chem.20 6158–63
[8] Hu F Q, Wei L, Zhou Z, Ran Y L, Li Z and Gao M Y 2006Preparation of biocompatible magnetite nanocrystals forin vivo magnetic resonance detection of cancer Adv. Mater.18 2553–6
[9] Shen M W and Shi X 2010 Dendrimer-basedorganic/inorganic hybrid nanoparticles in biomedicalapplications Nanoscale 2 1596–610
[10] Shi X Y, Wang S H, Swanson S D, Ge S, Cao Z Y,Van Antwerp M E, Landmark K J and Baker J R Jr 2008Dendrimer-functionalized shell-crosslinked iron oxidenanoparticles for in vivo magnetic resonance imaging oftumors Adv. Mater. 20 1671–8
[11] Wang S H, Shi X Y, Van Antwerp M, Cao Z Y, Swanson S D,Bi X D and Baker J R Jr 2007 Dendrimer-functionalizediron oxide nanoparticles for specific targeting and imagingof cancer cells Adv. Funct. Mater. 17 3043–50
[12] Yang H, Zhuang Y, Sun Y, Dai A, Shi X, Wu D, Li F,Hu H and Yang S 2011 Targeted dual contrast T1- and
T2-weighted magnetic resonance imaging of tumors usingmultifunctional gadolinium-labeled superparamagnetic ironoxide nanoparticles Biomaterials 32 4584–93
[13] Gass J, Poddar P, Almand J, Srinath S and Srikanth H 2006Superparamagnetic polymer nanocomposites with uniformFe3O4 nanoparticle dispersions Adv. Funct. Mater. 16 71–5
[14] Iida H, Nakanishi T, Takada H and Osaka T 2006 Preparationof magnetic iron-oxide nanoparticles by successivereduction–oxidation in reverse micelles: effects of reducingagent and atmosphere Electrochim. Acta 52 292–6
[15] Woo K, Hong J, Choi S, Lee H W, Ahn J P, Kim C S andLee S W 2004 Easy synthesis and magnetic properties ofiron oxide nanoparticles Chem. Mater. 16 2814–8
[16] Yu W W, Falkner J C, Yavuz C T and Colvin V L 2004Synthesis of monodisperse iron oxide nanocrystals bythermal decomposition of iron carboxylate salts Chem.Commun. 2306–7
[17] Sun S H and Zeng H 2002 Size-controlled synthesis ofmagnetite nanoparticies J. Am. Chem. Soc. 124 8204–5
[18] Ge S, Shi X Y, Sun K, Li C P, Uher C, Baker J R Jr,Banaszak Holl M M and Orr B G 2009 Facile hydrothermalsynthesis of iron oxide nanoparticles with tunable magneticproperties J. Phys. Chem. C 113 13593–9
[19] Feng J, Mao J, Wen X G and Tu M J 2011 Ultrasonic-assistedin situ synthesis and characterization of superparamagneticFe(3)O(4) nanoparticles J. Alloys Compounds 509 9093–7
[20] Xu Y L, Qin Y, Palchoudhury S and Bao Y P 2011Water-soluble iron oxide nanoparticles with high stabilityand selective surface functionality Langmuir 27 8990–7
[21] Wu W, He Q and Jiang C 2008 Magnetic iron oxidenanoparticles: synthesis and surface functionalizationstrategies Nanoscale Res. Lett. 3 397–415
[22] Giri S, Trewyn B G, Stellmaker M P and Lin V S Y 2005Stimuli-responsive controlled-release delivery system basedon mesoporous silica nanorods capped with magneticnanoparticles Angew. Chem. Int. Ed. 44 5038–44
[23] Cheng L, Yang K, Li Y, Chen J, Wang C, Shao M,Lee S-T and Liu Z 2011 Facile preparation ofmultifunctional upconversion nanoprobes for multimodalimaging and dual-targeted photothermal therapy Angew.Chem. Int. Ed. 50 7385–90
[24] Song H-T, Choi J-s, Huh Y-M, Kim S, Jun Y-w, Suh J-S andCheon J 2005 Surface modulation of magnetic nanocrystalsin the development of highly efficient magnetic resonanceprobes for intracellular labeling J. Am. Chem. Soc.127 9992–3
[25] Huh Y-M et al 2005 In vivo magnetic resonance detection ofcancer by using multifunctional magnetic nanocrystals J.Am. Chem. Soc. 127 12387–91
[26] Hu F Q, Li Z, Tu C F and Gao M Y 2007 Preparation ofmagnetite nanocrystals with surface reactive moieties byone-pot reaction J. Colloid Interface Sci. 311 469–74
[27] Xie J, Chen K, Lee H-Y, Xu C, Hsu A R, Peng S, Chen X andSun S 2008 Ultrasmall c(RGDyK)-coated Fe3O4nanoparticles and their specific targeting to integrin alpha vbeta3-rich tumor cells J. Am. Chem. Soc. 130 7542–3
[28] Mohapatra S, Pramanik N, Mukherjee S, Ghosh S K andPramanik P 2007 A simple synthesis of amine-derivatisedsuperparamagnetic iron oxide nanoparticles forbioapplications J. Mater. Sci. 42 7566–74
[29] Shen M W, Wang S H, Shi X Y, Chen X S, Huang Q G,Petersen E J, Pinto R A, Baker J R Jr and Weber W J Jr2009 Polyethyleneimine-mediated functionalization ofmultiwalled carbon nanotubes: synthesis, characterization,and in vitro toxicity assay J. Phys. Chem. C 113 3150–6
[30] Hu H, Zhou H, Du J, Wang Z, An L, Yang H, Li F, Wu H andYang S 2011 Biocompatiable hollow silica microspheres asnovel ultrasound contrast agents for in vivo imagingJ. Mater. Chem. 21 6576–83
9

Nanotechnology 23 (2012) 105601 M Shen et al
[31] Harris L A, Goff J D, Carmichael A Y, Riffle J S, Harburn J J,St Pierre T G and Saunders M 2003 Magnetite nanoparticledispersions stabilized with triblock copolymers Chem.Mater. 15 1367–77
[32] Maoz R, Frydman E, Cohen S R and Sagiv J 2000Constructive nanolithography: site-defined silverself-assembly on nanoelectrochemically patternedmonolayer templates Adv. Mater. 12 424–9
[33] Sun S H, Zeng H, Robinson D B, Raoux S, Rice P M,Wang S X and Li G X 2004 Monodisperse MFe2O4(M = Fe, Co, Mn) nanoparticles J. Am. Chem. Soc.126 273–9
[34] Huang J-H, Parab H J, Liu R-S, Lai T-C, Hsiao M, Chen C-H,Sheu H-S, Chen J-M, Tsai D-P and Hwu Y-K 2008Investigation of the growth mechanism of iron oxidenanoparticles via a seed-mediated method and itscytotoxicity studies J. Phys. Chem. C 112 15684–90
[35] Schneeweiss O, Zboril R, Pizurova N, Mashlan M,Petrovsky E and Tucek J 2006 Novel solid-state synthesisof alpha-Fe and Fe3O4 nanoparticles embedded in a MgOmatrix Nanotechnology 17 607–16
[36] Shi X, Thomas T P, Myc L A, Kotlyar A and Baker J R Jr2007 Synthesis, characterization, and intracellular uptake ofcarboxylterminated poly(amidoamine) dendrimer-stabilizediron oxide nanoparticles Phys. Chem. Chem. Phys.9 5712–20
[37] Shi X, Wang S, Sun H and Baker J R Jr 2007 Improvedbiocompatibility of surface functionalizeddendrimer-entrapped gold nanoparticles Soft Matter 3 71–4
[38] Hong S, Bielinska A U, Mecke A, Keszler B, Beals J L, Shi X,Balogh L, Orr B G, Baker J R Jr and Banaszak Holl M M2004 Interaction of poly(amidoamine) dendrimers withsupported lipid bilayers and cells: hole formation and therelation to transport Bioconjug. Chem. 15 774–82
10
Related Documents











