Extracellular cAMP inhibits D 1 dopamine receptor expression in CAD catecholaminergic cells via A 2a adenosine receptors Thuy Do, Qian Sun, Annie Beuve and Eldo V. Kuzhikandathil Department of Pharmacology and Physiology, UMDNJ-New Jersey Medical School, Newark, New Jersey, USA Abstract The expression of D 1 dopamine (DA) receptor gene is regu- lated during development, aging, and pathophysiology. The extracellular factors and signaling mechanisms that modulate the expression of D 1 DA receptor have not been well char- acterized. Here, we present novel evidence that endogenous D 1 DA receptor expression is inhibited by extracellular cAMP in the Cath.A Derived (CAD) catecholaminergic neuronal cell line. CAD cells express the multi-drug resistance protein 5 transporters and secrete cAMP. Addition of exogenous cAMP decreases D 1 receptor mRNA and protein greater than four- fold in 24 h. The cAMP-induced decrease of D 1 receptor mRNA levels is blocked by cGMP and by 1,3-dipropyl-8-(p- sulfo-phenyl)xanthine, an inhibitor of ecto-phosphodiestrase. Extracellular AMP, a metabolite of cAMP, also independently decreased D 1 receptor mRNA levels. Inhibitors of ecto- nucleotidases, a,b-methyleneadenosine 5¢-di-phosphate and GMP, completely blocked the decrease of D 1 receptor mRNA by extracellular cAMP, but only partially blocked the decrease induced by extracellular AMP. Levamisole, an inhibitor of tis- sue non-specific alkaline phosphatase, completely blocked the AMP-induced decrease of D 1 receptor mRNA. The extracellular cAMP, AMP, and adenosine (ADO)-induced de- crease in D 1 receptor mRNA expression are mediated by A 2a ADO receptor subtype. The results suggest a novel molecular mechanism linking activation of A 2a ADO receptors with inhi- bition of D 1 DA receptor expression. Keywords: development, diabetes, gene expression, hyper- tension, kidney, Parkinson’s disease, signal transduction, striatum, Tourette’s syndrome. J. Neurochem. (2007) 101, 619–631. Secretion of cAMP by mammalian cells in response to stimuli that increase intracellular cAMP was described originally by Davoren and Sutherland (1963). Secretion of cAMP has since been reported in numerous cells and tissues (reviewed in Jackson and Dubey 2001; Bankir et al. 2002). Efflux of cAMP has also been reported in vivo in the brain of awake-behaving rats (Stone and John 1990), liver (Kuster et al. 1973), and kidney (Exton et al. 1971; Levy and Starr 1972). The physiological effects of extracellular cAMP in mammals are primarily mediated by its conversion to adenosine (ADO) via ecto-phosphodiesterases (PDEs) and ecto-nucleotidases (Ecto-Nuc). ADO activates ADO recep- tors which together with the ecto-PDEs and Ecto-Nuc, make up the extracellular cAMP-ADO signaling pathway (Jackson and Dubey 2001; Latini and Pedata 2001). ADO activates four known P 1 ADO receptor subtypes that include the A 1 , A 2a ,A 2b , and A 3 receptors. The A 1 and A 3 ADO receptors exhibit inhibitory coupling to adenylyl cyclase (AC) and decrease cAMP levels, whereas the A 2a and A 2b receptors couple to AC via stimulatory G-proteins (G s /G olf ) and increase cAMP levels (reviewed in Jacobson and Gao 2006). The ADO receptor isoforms are expressed in cells and tissues that have the extracellular cAMP-ADO signaling pathway (Jackson and Dubey 2001; Jacobson and Gao 2006). The neurotransmitter dopamine (DA) through its cell surface receptors plays a significant role in the regulation of a Received July 11, 2006; revised manuscript received October 9, 2006; accepted October 20, 2006. Address correspondence and reprint requests to Eldo V. Kuzhikan- dathil, Department of Pharmacology and Physiology, UMDNJ-New Jersey Medical School, MSB, I-647, 185 South Orange Avenue, Newark, NJ 07103, USA. E-mail: [email protected] Abbreviations used: AC, adenylyl cyclase; ADA, adenosine deami- nase; ADO, adenosine; AMPCP, a,b-methyleneadenosine 5¢-di-phos- phate; BSA, bovine serum albumin; CAD, Cath.A Derived; DA, dopamine; DMEM, Dulbecco’s modified Eagle’s medium; dNTPs, de- oxynucleotide triphosphates; DPCPX, 8-cyclopentyl-1,3-dipropylxan- thine; DPSPX, 1,3-dipropyl-8-(p-sulfo-phenyl)xanthine; Ecto-Nuc, ecto- nucleotidases; HRP, horseradish peroxidase; IBMX, 3-isobutyl-1-meth- ylxanthine; MRP, multi-drug resistance protein; ND, non-differentiated; PDE, phosphoesteridase; RT, reverse transcriptase; TBE, Tris–Borate– EDTA; TNAP, tissue non-specific alkaline phosphatase. Journal of Neurochemistry , 2007, 101, 619–631 doi:10.1111/j.1471-4159.2006.04388.x ȑ 2007 The Authors Journal Compilation ȑ 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631 619

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Extracellular cAMP inhibits D1 dopamine receptor expression inCAD catecholaminergic cells via A2a adenosine receptors
Thuy Do, Qian Sun, Annie Beuve and Eldo V. Kuzhikandathil
Department of Pharmacology and Physiology, UMDNJ-New Jersey Medical School, Newark, New Jersey, USA
Abstract
The expression of D1 dopamine (DA) receptor gene is regu-
lated during development, aging, and pathophysiology. The
extracellular factors and signaling mechanisms that modulate
the expression of D1 DA receptor have not been well char-
acterized. Here, we present novel evidence that endogenous
D1 DA receptor expression is inhibited by extracellular cAMP
in the Cath.A Derived (CAD) catecholaminergic neuronal cell
line. CAD cells express the multi-drug resistance protein 5
transporters and secrete cAMP. Addition of exogenous cAMP
decreases D1 receptor mRNA and protein greater than four-
fold in 24 h. The cAMP-induced decrease of D1 receptor
mRNA levels is blocked by cGMP and by 1,3-dipropyl-8-(p-
sulfo-phenyl)xanthine, an inhibitor of ecto-phosphodiestrase.
Extracellular AMP, a metabolite of cAMP, also independently
decreased D1 receptor mRNA levels. Inhibitors of ecto-
nucleotidases, a,b-methyleneadenosine 5¢-di-phosphate and
GMP, completely blocked the decrease of D1 receptor mRNA
by extracellular cAMP, but only partially blocked the decrease
induced by extracellular AMP. Levamisole, an inhibitor of tis-
sue non-specific alkaline phosphatase, completely blocked
the AMP-induced decrease of D1 receptor mRNA. The
extracellular cAMP, AMP, and adenosine (ADO)-induced de-
crease in D1 receptor mRNA expression are mediated by A2a
ADO receptor subtype. The results suggest a novel molecular
mechanism linking activation of A2a ADO receptors with inhi-
bition of D1 DA receptor expression.
Keywords: development, diabetes, gene expression, hyper-
tension, kidney, Parkinson’s disease, signal transduction,
striatum, Tourette’s syndrome.
J. Neurochem. (2007) 101, 619–631.
Secretion of cAMP by mammalian cells in response tostimuli that increase intracellular cAMP was describedoriginally by Davoren and Sutherland (1963). Secretion ofcAMP has since been reported in numerous cells and tissues(reviewed in Jackson and Dubey 2001; Bankir et al. 2002).Efflux of cAMP has also been reported in vivo in the brain ofawake-behaving rats (Stone and John 1990), liver (Kusteret al. 1973), and kidney (Exton et al. 1971; Levy and Starr1972). The physiological effects of extracellular cAMP inmammals are primarily mediated by its conversion toadenosine (ADO) via ecto-phosphodiesterases (PDEs) andecto-nucleotidases (Ecto-Nuc). ADO activates ADO recep-tors which together with the ecto-PDEs and Ecto-Nuc, makeup the extracellular cAMP-ADO signaling pathway (Jacksonand Dubey 2001; Latini and Pedata 2001). ADO activatesfour known P1 ADO receptor subtypes that include the A1,A2a, A2b, and A3 receptors. The A1 and A3 ADO receptorsexhibit inhibitory coupling to adenylyl cyclase (AC) anddecrease cAMP levels, whereas the A2a and A2b receptorscouple to AC via stimulatory G-proteins (Gs/Golf) andincrease cAMP levels (reviewed in Jacobson and Gao
2006). The ADO receptor isoforms are expressed in cellsand tissues that have the extracellular cAMP-ADO signalingpathway (Jackson and Dubey 2001; Jacobson and Gao2006).
The neurotransmitter dopamine (DA) through its cellsurface receptors plays a significant role in the regulation of a
Received July 11, 2006; revised manuscript received October 9, 2006;accepted October 20, 2006.Address correspondence and reprint requests to Eldo V. Kuzhikan-
dathil, Department of Pharmacology and Physiology, UMDNJ-NewJersey Medical School, MSB, I-647, 185 South Orange Avenue,Newark, NJ 07103, USA. E-mail: [email protected] used: AC, adenylyl cyclase; ADA, adenosine deami-
nase; ADO, adenosine; AMPCP, a,b-methyleneadenosine 5¢-di-phos-phate; BSA, bovine serum albumin; CAD, Cath.A Derived; DA,dopamine; DMEM, Dulbecco’s modified Eagle’s medium; dNTPs, de-oxynucleotide triphosphates; DPCPX, 8-cyclopentyl-1,3-dipropylxan-thine; DPSPX, 1,3-dipropyl-8-(p-sulfo-phenyl)xanthine; Ecto-Nuc, ecto-nucleotidases; HRP, horseradish peroxidase; IBMX, 3-isobutyl-1-meth-ylxanthine; MRP, multi-drug resistance protein; ND, non-differentiated;PDE, phosphoesteridase; RT, reverse transcriptase; TBE, Tris–Borate–EDTA; TNAP, tissue non-specific alkaline phosphatase.
Journal of Neurochemistry, 2007, 101, 619–631 doi:10.1111/j.1471-4159.2006.04388.x
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631 619

number of functions, most notably cognition, motor control,emotion, associative learning, and neuroendocrine functions(reviewed in Berke and Hyman 2000). DA receptors are alsoexpressed in the kidney and in smooth muscle cells of bloodvessels of many organ systems, and affect cardiovascular andrenal functions (Hussain and Lokhandwala 2003). DAreceptors are divided into two broad categories on the basisof their structural and functional properties: the D1-likereceptors (D1R and D5R) and the D2-like receptors (D2R,D3R, and D4R) (Civelli et al. 1993; Sibley et al. 1993;Missale et al. 1998). The D1-like receptors link to stimula-tory Gs/Golf proteins and activate AC to increase intracellularcAMP. In contrast, the D2-like receptors are coupled toinhibitory Gi/Go proteins and decrease cAMP levels(Missale et al. 1998).
Neurochemical and pharmacological studies have demon-strated functional interactions between ADO and DA neu-rotransmitter systems (Ferre et al. 1992). In the striatum, A2a
ADO receptors are largely co-expressed with D2 DAreceptors in striatal–pallidial neurons (Schiffmann et al.1991; Fink et al. 1992). Conversely, only very few A2a
ADO receptor mRNA-containing neurons co-express the D1
DA receptor in the direct pathway (Svenningsson et al.1998). Consistent with these observations, a recent studyshowed that in A2a ADO receptor knockout mice, D1
receptor mRNA was significantly higher in the caudateputamen and nucleus accumbens when compared with theirwild-type littermates (Short et al. 2006). Furthermore, com-parison of postnatal ontogeny of A2a receptors and DAreceptors showed that D1 DA receptor expression levelsbegin to decline as A2a receptor expression levels increase inseveral brain regions (Johansson et al. 1997).
Deregulated DA signaling has been demonstrated inmany disorders, including Parkinson’s disease, Tourette’ssyndrome, Huntington’s disease, schizophrenia, attention-deficit hyperactivity disorder, and drug addiction (Missaleet al. 1998; Vallone et al. 2000). One mechanism thatunderlies the dopaminergic deregulation in the abovedisorders involves alteration in DA receptor gene expres-sion, which can contribute to the long-term changes.Indeed, significant changes in the expression of DAreceptor genes have been reported during normal develop-ment, aging, in diseases such as Parkinson’s, Huntington’s,and following therapeutic treatment of dopaminergic disor-ders (Rogue et al. 1991; Xu et al. 1992; Butkerait andFriedman 1993; Laurier et al. 1994; Schambra et al. 1994;Weeks et al. 1996; Ginovart et al. 1997; Stoessl and de laFuente-Fernandez 2003). Despite the importance for phys-iology and pathophysiology, the molecular mechanisms andfactors that mediate the changes in DA receptor expressionare not clear.
We have previously shown that the mouse Cath.A Derived(CAD) catecholaminergic cell line expresses endogenous D1,D2, D3, and D5 DA receptor subtypes with D1 receptor
mRNA being expressed at the highest level. In addition,CAD cells also express various G-protein alpha subunits andACs that together comprise the DA receptor-AC signaltransduction pathway (Pasuit et al. 2004). In this article, weinvestigated the regulation of D1 DA receptor in CADcatecholaminergic cells and, for the first time, provide directevidence for inhibition of D1 receptor expression by theactivation of the extracellular cAMP-ADO signaling path-way. These results suggest a novel molecular mechanism thatlinks the ADO A2a receptor signaling pathway and D1
receptor mRNA expression. Our results also suggest aputative mechanistic hypothesis to explain the lack of co-expression of D1 and A2a receptor mRNA in striatal neuronsin vivo.
Materials and methods
Cell culture
The CAD cells were maintained in Dulbecco’s modified Eagle’s
medium (DMEM)/F12 media (Cambrex, Walkersville, MD, USA),
8% fetal calf serum, and 100 U/mL penicillin/streptomycin (Invi-
trogen, Carlsbad, CA, USA). Differentiation was induced by treating
cells with serum-free medium consisting of DMEM/F12, 20 lg/mL
transferrin, 50 ng/mL sodium selenite, and 100 U/mL penicillin/
streptomycin for 48 h. In this study, to evaluate the effect of
extracellular cAMP signaling, we primarily used non-differentiated
(ND) CAD cells as differentiation of CAD cells in itself is known to
increase D1 receptor mRNA levels (Pasuit et al. 2004). CAD cells
used in the experiments were plated and grown in either 100 mm
tissue culture plates or T-75 tissue culture flasks (Sarstedt Inc.,
Newton, NC, USA). Cells were treated with 3¢,5¢-cAMP sodium
salt, 3¢,5¢-cGMP sodium salt, 5¢-AMP sodium salt, 5¢-GMP
disodium salt, ADO, forskolin, 8-bromocAMP, 1,3-dipropyl-8-(p-sulfo-phenyl)xanthine (DPSPX), a,b-methyleneadenosine 5¢-di-phosphate (AMPCP), levamisole, 3-isobutyl-1-methylxanthine
(IBMX), probenecid, adenosine deaminase (ADA), 9-chloro-2-(2-
furanyl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amine (CGS15943), 8-
cyclopentyl-1,3-dipropylxanthine (DPCPX), 7-(2-phenylethyl)-5-
amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
(SCH58261), 8-[4-[((4-cyanophenyl)carbamoylmethyl)oxy]phenyl]-
1,3-di(n-propyl)xanthine (MRS1754), 3-propyl-6-ethyl-5-[(ethyl-
thio)carbonyl]-2 phenyl-4-propyl-3-pyridine carboxylate (MRS1523)
(Sigma, St Louis, MO, USA), as described in the appropriate figure
legends.
RNA isolation and reverse transcriptase reaction
RNA isolation was performed using QIAGEN RNeasy� Mini Kit
(Qiagen, Valencia, CA, USA) according to the manufacturer’s
instructions. DNA-free� (Ambion, Austin, TX, USA) was used to
remove DNA contamination from the total RNA sample (50 lg totalRNA, 1 · DNase I Buffer, 0.08 U DNase I). The quality and
integrity of the DNase-treated RNAwas confirmed by running 2 lgof total RNA on a 1.2% Tris–Borate–EDTA (TBE) agarose gel.
To set up the reverse transcriptase (RT) reaction, 5 lg of DNase-
treated total RNA and 300 ng random hexamers (Invitrogen) were
combined. The mixture was denatured by heating at 70�C for
620 T. Do et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631� 2007 The Authors

10 min and quick chilling in ice slurry. After the addition of 1 ·first-strand Superscript III RT Buffer (Invitrogen), the denatured
samples were annealed at 25�C for 10 min. To initiate cDNA
synthesis, 0.5 mmol/L deoxynucleotide triphosphates (dNTPs),
10 mmol/L dithiothreitol, 1 U SUPERase In (Ambion), and 10 U
Superscript III RT (Invitrogen) were added and the reaction
incubated at 42�C for 1 h. The reaction was stopped by heat
inactivation at 70�C for 15 min.
Real-time PCR
Real-time PCR was performed using the Roche Light Cycler
(Roche, Indianapolis, IN, USA) in the SYBR� Green (Invitrogen)
format using primers and conditions described previously (Pasuit
et al. 2004). Briefly, the final 20 lL PCR reaction contained
0.04 lg cDNA, 0.4 lmol/L custom primers (Invitrogen), 0.2 mmol/
L dNTPs (Invitrogen), 0.25 lg bovine serum albumin (BSA) (Idaho
Technology, Salt Lake City, UT, USA), 1.6 lL Enzyme Diluent
(10 mmol/L Tris, pH 8.3, 2.5 mg/mL BSA) (Idaho Technology),
0.075 · SYBR Green I, 1 · Advantage 2 PCR Buffer, and 1 ·Advantage 2 PCR Polymerase Mix (Clontech, Palo Alto, CA,
USA). The optimal PCR amplification conditions for the individual
primer pairs were determined experimentally and have been
previously reported (Pasuit et al. 2004). Two negative and one
positive control were incorporated in each RT-PCR run. Negative
control included a PCR reaction in which water was substituted for
the template. For a second negative control, PCR was run with
products from an RT reaction in which the Superscript III RT
enzyme was omitted. Mouse brain cDNA was used as a positive
control. The primers used to identify the expression of the various
multi-drug resistance proteins (MRP) were MRP1 (5¢-AG-CTGAACCATGAGTGTGCA-3¢ and 5¢-CACACCAAGCCAGCA-TCCTT-3¢); MRP2 (5¢-CCCTGGAATTGGCTCATCTC-3¢ and 5¢-CAATCTTGCCGCTGTCTAGG-3¢); MRP3 (5¢-ACCTGCGCTC-TCTCAACTCACC-3¢ and 5¢-CTAGGCAAGTCCCGCATCCT-3¢);MRP4 (5¢-ATGGATACTGAATTAGCAGA-3¢ and 5¢-CACTGTC-AATGATGGTGTTC-3¢); MRP5 (5¢-TTCTCTGTTGGGGAACG-GCA-3¢ and 5¢-CGATGGGGTGTCAAACTCCA-3¢); MRP6 (5¢-AGGGAGATGACCTGAGCGTG-3¢ and 5¢-GGGCTAGCCTGTA-AAACAGG-3¢); MRP7a (5¢-CACCTGGTCTCCGCTCTACT-3¢and 5¢-GGGGAACGTCACAGGAATAC-3¢); MRP9 (5¢-TTCCT-GAACACACTCACCCC-3¢ and 5¢-TCAGCTTGGTTCGGAGG-TCT-3¢). Quantitative differences in D1 receptor and internal control
b-actin mRNA were measured using TaqMan� Gene Expression
Assays (Applied Biosystems, Foster City, CA, USA). Fold-
differences were determined using crossing point analysis, as
described previously (Pasuit et al. 2004).
Cyclic AMP assay
The CAD cells were grown in 12-well plates with 1 mL medium for
24 h before treatment. After treatment with 1 mmol/L IBMX in the
presence or absence of 0.5 mmol/L probenecid for different time
periods, the medium was first collected and perchloric acid (Fisher,
Fairlawn, NJ, USA) added to a final concentration of 2.5%. The
solution was neutralized by adding potassium hydroxide to a final
concentration of 0.75%, and the supernatant used for extracellular
cAMP assay. For the intracellular cAMP assay, after the media was
removed, the cells on the plate underwent the same treatment with
perchloric acid and potassium hydroxide. Measurement of cAMP by
radioimmunoassay was conducted as reported by Domino et al.(1991) with modifications. Briefly, cAMP in samples and standards
were acetylated with a triethylamine and acetic anhydride mixture
(1 : 1; Fisher). The concentration of cAMP was determined from a
standard curve (5, 10, 20, 40, 50, 100, and 200 fmol). To each
sample and standard, 20 000 cpm of 125I-labeled cAMP and 20 lLof anti-cAMP antibody were added. After overnight incubation at
4�C, the complex formed by cAMP antibody and 125I-labeled cAMP
was precipitated with PEG-8000 and FBS. The radioactivity of the
pellet by centrifuge was determined by a Cobra II-Gamma counter
(PerkinElmer, Wellesley, MA, USA).
Riboprobe synthesis and RNase protection assay
The construction of D1 receptor and b-actin probe template, and the
generation of riboprobes for RNase protection assay have been
described previously (Pasuit et al. 2004). RNase protection assay
was performed with 1–100 lg of total RNA using a modified
Supersignal� RPA III kit (Pierce Biochemicals, Rockford, IL, USA),
as described previously (Pasuit et al. 2004). Final pellets were
resuspended in 2· Gel Loading Buffer, denatured by heating at
95�C for 5 min, electrophoresed through a 5% TBE-Urea Gel, and
transferred onto a Biodyne B Nylon Membrane (Pierce Biochem-
icals). The membrane was UV cross-linked and the signals detected
using Supersignal RPA III Chemiluminescent Detection Kit protocol
(Pierce Biochemicals), using a 1 : 300 dilution of the streptavidin–
horseradish peroxidase (HRP) conjugates. All experiments were
repeated at least three independent times.
Western blot analysis
The CAD cells were harvested from T-75 flasks (Sarstedt) and cell
pellets were lyzed and solubilized using the Cellytic�-M reagent
(Sigma) supplemented with 1 mmol/L phenylmethylsulfonylfluo-
ride and 1% protease inhibitor cocktail (Sigma). Protein amounts in
the lysates were determined using the bicinchoninic acid assay (Bio-
Rad, Hercules, CA, USA) and equal amounts of total cell proteins
were electrophoresed on 10% sodium dodecyl sulfate – polyacryl-
amide gel electrophoresis gels, transferred on to nitrocellulose and
blocked with 10% cow’s milk for 2 h. The blots were probed with
the D1 DA receptor rat monoclonal antibody (1 : 1000 dilution;
Sigma) for 2 h. Signals were detected using films and/or gel imager
by employing chemiluminescent methods (Pierce Biochemicals),
using HRP-conjugated secondary antibodies (1 : 10 000; Pierce
Biochemicals). The signals were detected using the SuperSignal
WestDura substrate chemiluminescence detection kit (Pierce Bio-
chemicals).
Data analysis
To quantitate the intensity of bands in the RNase protection assay
and western blots, photographic films were subjected to densito-
metric analysis using the FluorChem gel documentation system
(Alpha Innotech, San Leandro, CA, USA). For each blot, multiple
film exposures were obtained and scanned to determine the linear
range of exposure. Analysis of variance (ANOVA) and Holm’s
multiple pair-wise comparison tests were performed with Primer of
Biostatistics software (Version 5.0; McGraw-Hill, New York, NY,
USA). Student’s t-test was performed with the SigmaPlot software
(SPSS Inc., Chicago, IL, USA). Data were considered statistically
significant when the probability value (p) was less than 0.05.
Regulation of D1 receptor expression by extracellular cAMP 621
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631
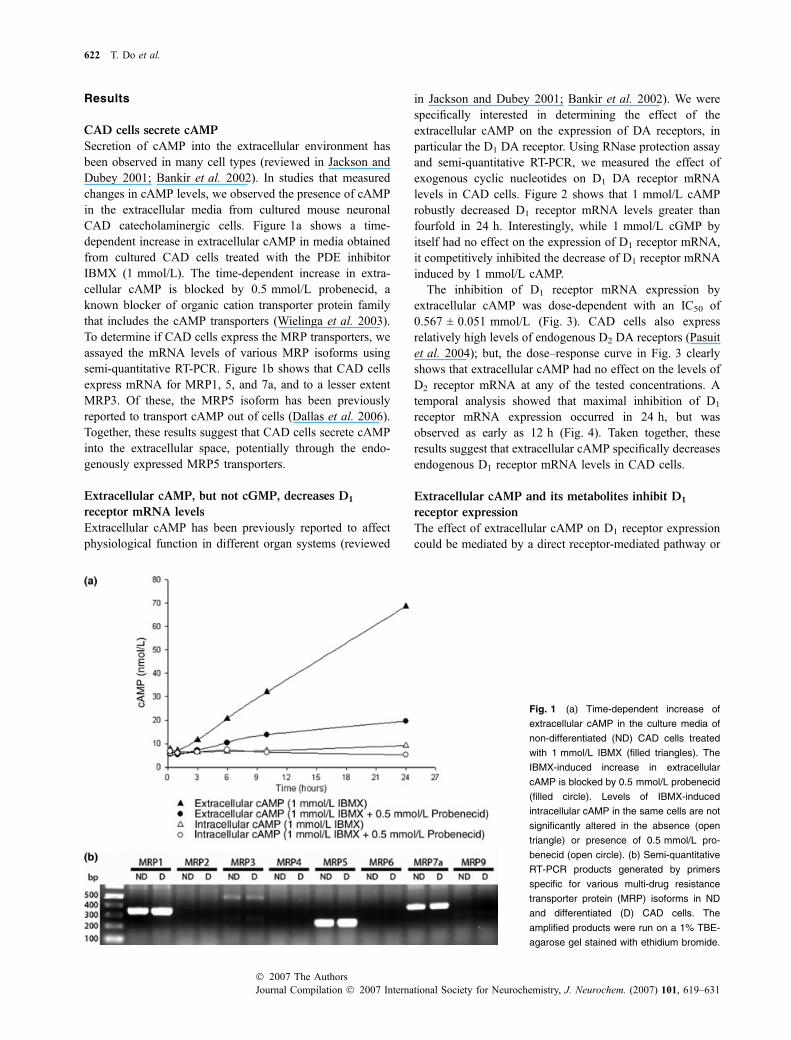
Results
CAD cells secrete cAMP
Secretion of cAMP into the extracellular environment hasbeen observed in many cell types (reviewed in Jackson andDubey 2001; Bankir et al. 2002). In studies that measuredchanges in cAMP levels, we observed the presence of cAMPin the extracellular media from cultured mouse neuronalCAD catecholaminergic cells. Figure 1a shows a time-dependent increase in extracellular cAMP in media obtainedfrom cultured CAD cells treated with the PDE inhibitorIBMX (1 mmol/L). The time-dependent increase in extra-cellular cAMP is blocked by 0.5 mmol/L probenecid, aknown blocker of organic cation transporter protein familythat includes the cAMP transporters (Wielinga et al. 2003).To determine if CAD cells express the MRP transporters, weassayed the mRNA levels of various MRP isoforms usingsemi-quantitative RT-PCR. Figure 1b shows that CAD cellsexpress mRNA for MRP1, 5, and 7a, and to a lesser extentMRP3. Of these, the MRP5 isoform has been previouslyreported to transport cAMP out of cells (Dallas et al. 2006).Together, these results suggest that CAD cells secrete cAMPinto the extracellular space, potentially through the endo-genously expressed MRP5 transporters.
Extracellular cAMP, but not cGMP, decreases D1
receptor mRNA levels
Extracellular cAMP has been previously reported to affectphysiological function in different organ systems (reviewed
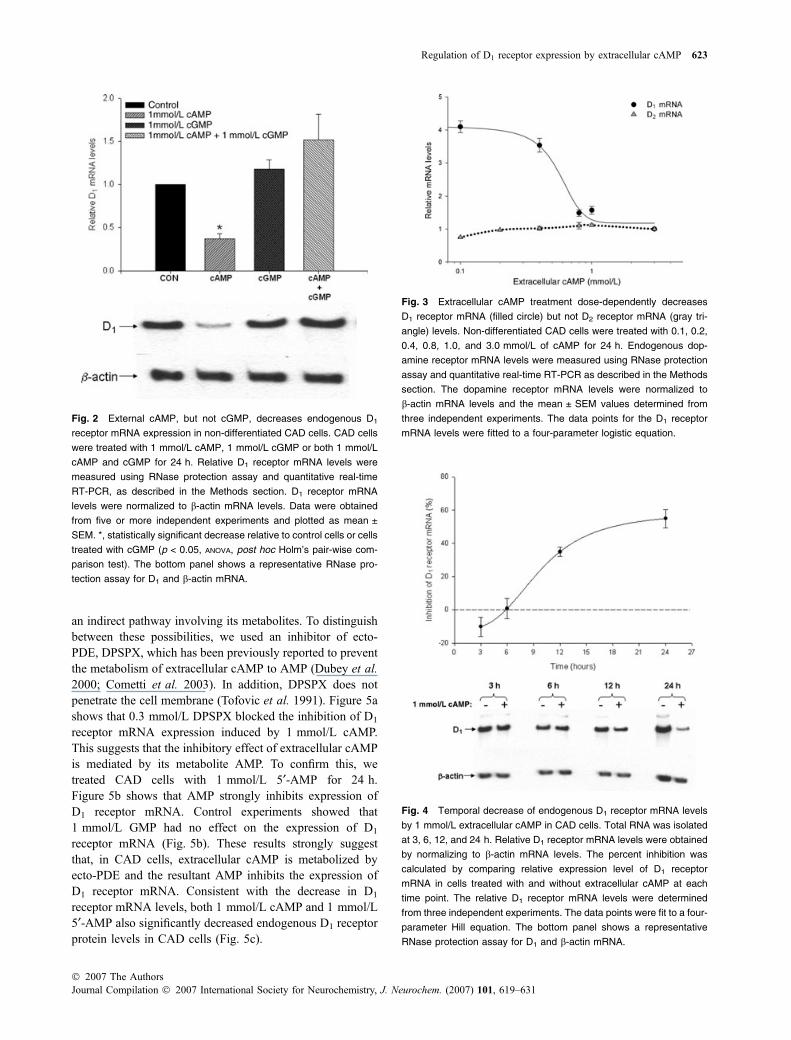
in Jackson and Dubey 2001; Bankir et al. 2002). We werespecifically interested in determining the effect of theextracellular cAMP on the expression of DA receptors, inparticular the D1 DA receptor. Using RNase protection assayand semi-quantitative RT-PCR, we measured the effect ofexogenous cyclic nucleotides on D1 DA receptor mRNAlevels in CAD cells. Figure 2 shows that 1 mmol/L cAMProbustly decreased D1 receptor mRNA levels greater thanfourfold in 24 h. Interestingly, while 1 mmol/L cGMP byitself had no effect on the expression of D1 receptor mRNA,it competitively inhibited the decrease of D1 receptor mRNAinduced by 1 mmol/L cAMP.
The inhibition of D1 receptor mRNA expression byextracellular cAMP was dose-dependent with an IC50 of0.567 ± 0.051 mmol/L (Fig. 3). CAD cells also expressrelatively high levels of endogenous D2 DA receptors (Pasuitet al. 2004); but, the dose–response curve in Fig. 3 clearlyshows that extracellular cAMP had no effect on the levels ofD2 receptor mRNA at any of the tested concentrations. Atemporal analysis showed that maximal inhibition of D1
receptor mRNA expression occurred in 24 h, but wasobserved as early as 12 h (Fig. 4). Taken together, theseresults suggest that extracellular cAMP specifically decreasesendogenous D1 receptor mRNA levels in CAD cells.
Extracellular cAMP and its metabolites inhibit D1
receptor expression
The effect of extracellular cAMP on D1 receptor expressioncould be mediated by a direct receptor-mediated pathway or
Fig. 1 (a) Time-dependent increase of
extracellular cAMP in the culture media of
non-differentiated (ND) CAD cells treated
with 1 mmol/L IBMX (filled triangles). The
IBMX-induced increase in extracellular
cAMP is blocked by 0.5 mmol/L probenecid
(filled circle). Levels of IBMX-induced
intracellular cAMP in the same cells are not
significantly altered in the absence (open
triangle) or presence of 0.5 mmol/L pro-
benecid (open circle). (b) Semi-quantitative
RT-PCR products generated by primers
specific for various multi-drug resistance
transporter protein (MRP) isoforms in ND
and differentiated (D) CAD cells. The
amplified products were run on a 1% TBE-
agarose gel stained with ethidium bromide.
622 T. Do et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631� 2007 The Authors
an indirect pathway involving its metabolites. To distinguishbetween these possibilities, we used an inhibitor of ecto-PDE, DPSPX, which has been previously reported to preventthe metabolism of extracellular cAMP to AMP (Dubey et al.2000; Cometti et al. 2003). In addition, DPSPX does notpenetrate the cell membrane (Tofovic et al. 1991). Figure 5ashows that 0.3 mmol/L DPSPX blocked the inhibition of D1
receptor mRNA expression induced by 1 mmol/L cAMP.This suggests that the inhibitory effect of extracellular cAMPis mediated by its metabolite AMP. To confirm this, wetreated CAD cells with 1 mmol/L 5¢-AMP for 24 h.Figure 5b shows that AMP strongly inhibits expression ofD1 receptor mRNA. Control experiments showed that1 mmol/L GMP had no effect on the expression of D1
receptor mRNA (Fig. 5b). These results strongly suggestthat, in CAD cells, extracellular cAMP is metabolized byecto-PDE and the resultant AMP inhibits the expression ofD1 receptor mRNA. Consistent with the decrease in D1
receptor mRNA levels, both 1 mmol/L cAMP and 1 mmol/L5¢-AMP also significantly decreased endogenous D1 receptorprotein levels in CAD cells (Fig. 5c).
Fig. 3 Extracellular cAMP treatment dose-dependently decreases
D1 receptor mRNA (filled circle) but not D2 receptor mRNA (gray tri-
angle) levels. Non-differentiated CAD cells were treated with 0.1, 0.2,
0.4, 0.8, 1.0, and 3.0 mmol/L of cAMP for 24 h. Endogenous dop-
amine receptor mRNA levels were measured using RNase protection
assay and quantitative real-time RT-PCR as described in the Methods
section. The dopamine receptor mRNA levels were normalized to
b-actin mRNA levels and the mean ± SEM values determined from
three independent experiments. The data points for the D1 receptor
mRNA levels were fitted to a four-parameter logistic equation.
Fig. 2 External cAMP, but not cGMP, decreases endogenous D1
receptor mRNA expression in non-differentiated CAD cells. CAD cells
were treated with 1 mmol/L cAMP, 1 mmol/L cGMP or both 1 mmol/L
cAMP and cGMP for 24 h. Relative D1 receptor mRNA levels were
measured using RNase protection assay and quantitative real-time
RT-PCR, as described in the Methods section. D1 receptor mRNA
levels were normalized to b-actin mRNA levels. Data were obtained
from five or more independent experiments and plotted as mean ±
SEM. *, statistically significant decrease relative to control cells or cells
treated with cGMP (p < 0.05, ANOVA, post hoc Holm’s pair-wise com-
parison test). The bottom panel shows a representative RNase pro-
tection assay for D1 and b-actin mRNA.
Fig. 4 Temporal decrease of endogenous D1 receptor mRNA levels
by 1 mmol/L extracellular cAMP in CAD cells. Total RNA was isolated
at 3, 6, 12, and 24 h. Relative D1 receptor mRNA levels were obtained
by normalizing to b-actin mRNA levels. The percent inhibition was
calculated by comparing relative expression level of D1 receptor
mRNA in cells treated with and without extracellular cAMP at each
time point. The relative D1 receptor mRNA levels were determined
from three independent experiments. The data points were fit to a four-
parameter Hill equation. The bottom panel shows a representative
RNase protection assay for D1 and b-actin mRNA.
Regulation of D1 receptor expression by extracellular cAMP 623
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631

Inhibition of ecto-nucleotidases blocks the extracellular
cAMP effect
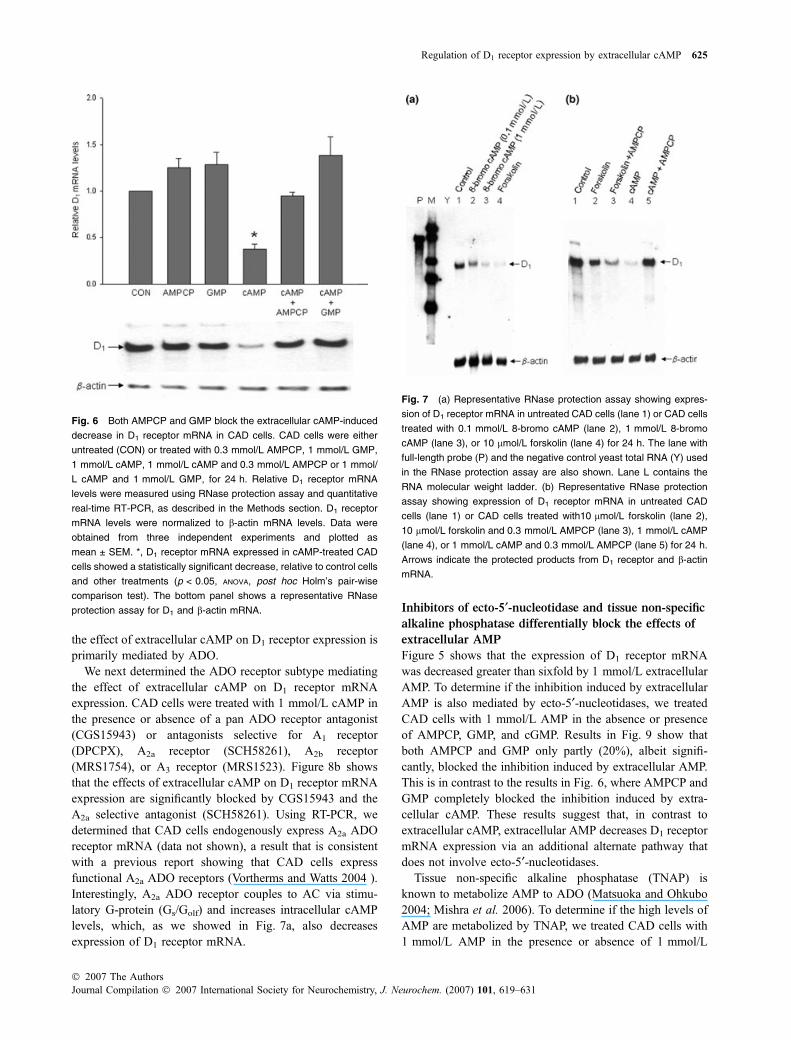
The AMP derived from extracellular cAMP is furthermetabolized to ADO by ecto-5¢-nucleotidase or ecto-alkalinephosphatases (Matsuoka and Ohkubo 2004), which could bepotentially expressed in CAD cells. Indeed, using gene-specific primers, we determined that CAD cells express themRNA for, CD73, the ecto-5¢-nucleotidase (data not shown).To determine if the AMP generated from cAMP is furthermetabolized to ADO, we co-treated CAD cells with 1 mmol/L cAMP and two different inhibitors of Ecto-Nuc (Fig. 6).Previous studies have shown that AMPCP and GMP areinhibitors of ecto-5¢-nucleotidases and prevent the metabo-lism of AMP to ADO (Dubey et al. 2000; Cometti et al.2003; Song and Sladek 2005). Furthermore, AMPCP is aninhibitor of ecto-5¢-nucleotidase, but not cytosolic 5¢-nucle-otidase (Zimmermann 1992). Figure 6 shows that theinhibition of D1 receptor mRNA expression by extracellularcAMP was completely blocked by the co-application ofeither 0.3 mmol/L AMPCP or 1 mmol/L GMP. These resultssuggest that metabolism of extracellular cAMP to AMP, andthen to ADO by ecto-5¢-nucleotidases mediates the inhibitionof D1 receptor mRNA expression.
Increasing extracellular and intracellular cAMP levels
inhibits D1 receptor mRNA expression levels
We next determined if the effect of extracellular cAMP on D1
receptor mRNA expression was different from that obtainedby raising intracellular cAMP levels. Figure 7a shows thatboth 8-bromo cAMP (a cell-permeable cAMP analog) andforskolin decrease D1 receptor mRNA levels in 24 h. Of the
two, forskolin, by activating AC, raises intracellular cAMPlevels without directly increasing extracellular cAMP levels.The results shown in Fig. 7a suggest that raising intracellularcAMP also decreases D1 receptor mRNA levels in CADcells. While previous studies have shown that cAMP doesnot penetrate lipid bilayers (Rosenberg and Dichter 1989;Jackson et al. 2006), it is possible that effects of extracellularcAMP are mediated by directly raising intracellular cAMPthrough simple transport into the cell. However, the controlexperiment in Fig. 7b shows that the inhibitor of ecto-5¢-nucleotidases, AMPCP, only blocks the decrease induced byextracellular cAMP (lanes 1, 4, and 5). The decrease of D1
receptor mRNA expression brought about by forskolin-induced elevation of intracellular cAMP is not blocked byAMPCP (Fig. 7b, compare lanes 1, 2, and 3). These resultsstrongly suggest that the effect of extracellular cAMP is notmediated by a mechanism that involves simple transport intothe cell, but rather by a pathway that involves its extracellularmetabolism to ADO.
Effect of extracellular cAMP is mediated by A2a
adenosine receptor
To directly determine if the effects of extracellular cAMP onD1 receptor mRNA expression is mediated by ADO, wetreated CAD cells with 1 mmol/L cAMP, in the absence orpresence of 8 U/mL ADA. ADA metabolizes ADO toinosine, and previous reports have used ADA as a moleculartool to demonstrate the involvement of ADO (e.g., Bilodeauet al. 2005). Figure 8a shows that ADA significantly blocksthe extracellular cAMP-mediated decrease of D1 receptormRNA expression. This result provides direct evidence that
Fig. 5 (a) Representative RNase protection assay showing that an
inhibitor of ecto-phosphodiesterase, DPSPX, blocks the extracellular
cAMP-induced decrease of D1 receptor mRNA. Control CAD cells
were treated without (lane 1) or with 1 mmol/L extracellular cAMP
(lane 2) for 24 h. In addition, shown are the D1 receptor mRNA levels
in CAD cells treated with 0.3 mmol/L DPSPX in the absence (lane 3)
or presence of 1 mmol/L (lane 4) extracellular cAMP for 24 h. (b)
Representative RNase protection assay showing decrease of D1
mRNA in CAD cells treated with 1 mmol/L cAMP (lane 2) or 1 mmol/L
AMP (lane 3) for 24 h. Untreated CAD cells (lane 1) or cells treated
with 1 mmol/L GMP (lane 4) for 24 h show equivalent expression of D1
receptor mRNA. Lane P contains the full-length D1 riboprobe and lane
L contains the RNA molecular weight ladder. Lane Y contains the
negative control yeast total RNA used in the RNase protection assay.
Arrows indicate the protected products from D1 receptor and b-actin
mRNA. (c) Western blot assay showing relative levels of D1 receptor
protein in control, untreated CAD cells (lane 1) or CAD cells treated
with 1 mmol/L cAMP (lane 2) or 1 mmol/L AMP (lane 3) for 30 h. Equal
amounts of protein lysates were loaded in all lanes, as shown by the
levels of total protein present on the same blot (lower panel). Total
protein levels were detected using the amido black protein stain.
624 T. Do et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631� 2007 The Authors
the effect of extracellular cAMP on D1 receptor expression isprimarily mediated by ADO.
We next determined the ADO receptor subtype mediatingthe effect of extracellular cAMP on D1 receptor mRNAexpression. CAD cells were treated with 1 mmol/L cAMP inthe presence or absence of a pan ADO receptor antagonist(CGS15943) or antagonists selective for A1 receptor(DPCPX), A2a receptor (SCH58261), A2b receptor(MRS1754), or A3 receptor (MRS1523). Figure 8b showsthat the effects of extracellular cAMP on D1 receptor mRNAexpression are significantly blocked by CGS15943 and theA2a selective antagonist (SCH58261). Using RT-PCR, wedetermined that CAD cells endogenously express A2a ADOreceptor mRNA (data not shown), a result that is consistentwith a previous report showing that CAD cells expressfunctional A2a ADO receptors (Vortherms and Watts 2004 ).Interestingly, A2a ADO receptor couples to AC via stimu-latory G-protein (Gs/Golf) and increases intracellular cAMPlevels, which, as we showed in Fig. 7a, also decreasesexpression of D1 receptor mRNA.
Inhibitors of ecto-5¢-nucleotidase and tissue non-specific
alkaline phosphatase differentially block the effects of
extracellular AMP
Figure 5 shows that the expression of D1 receptor mRNAwas decreased greater than sixfold by 1 mmol/L extracellularAMP. To determine if the inhibition induced by extracellularAMP is also mediated by ecto-5¢-nucleotidases, we treatedCAD cells with 1 mmol/L AMP in the absence or presenceof AMPCP, GMP, and cGMP. Results in Fig. 9 show thatboth AMPCP and GMP only partly (20%), albeit signifi-cantly, blocked the inhibition induced by extracellular AMP.This is in contrast to the results in Fig. 6, where AMPCP andGMP completely blocked the inhibition induced by extra-cellular cAMP. These results suggest that, in contrast toextracellular cAMP, extracellular AMP decreases D1 receptormRNA expression via an additional alternate pathway thatdoes not involve ecto-5¢-nucleotidases.
Tissue non-specific alkaline phosphatase (TNAP) isknown to metabolize AMP to ADO (Matsuoka and Ohkubo2004; Mishra et al. 2006). To determine if the high levels ofAMP are metabolized by TNAP, we treated CAD cells with1 mmol/L AMP in the presence or absence of 1 mmol/L
Fig. 7 (a) Representative RNase protection assay showing expres-
sion of D1 receptor mRNA in untreated CAD cells (lane 1) or CAD cells
treated with 0.1 mmol/L 8-bromo cAMP (lane 2), 1 mmol/L 8-bromo
cAMP (lane 3), or 10 lmol/L forskolin (lane 4) for 24 h. The lane with
full-length probe (P) and the negative control yeast total RNA (Y) used
in the RNase protection assay are also shown. Lane L contains the
RNA molecular weight ladder. (b) Representative RNase protection
assay showing expression of D1 receptor mRNA in untreated CAD
cells (lane 1) or CAD cells treated with10 lmol/L forskolin (lane 2),
10 lmol/L forskolin and 0.3 mmol/L AMPCP (lane 3), 1 mmol/L cAMP
(lane 4), or 1 mmol/L cAMP and 0.3 mmol/L AMPCP (lane 5) for 24 h.
Arrows indicate the protected products from D1 receptor and b-actin
mRNA.
Fig. 6 Both AMPCP and GMP block the extracellular cAMP-induced
decrease in D1 receptor mRNA in CAD cells. CAD cells were either
untreated (CON) or treated with 0.3 mmol/L AMPCP, 1 mmol/L GMP,
1 mmol/L cAMP, 1 mmol/L cAMP and 0.3 mmol/L AMPCP or 1 mmol/
L cAMP and 1 mmol/L GMP, for 24 h. Relative D1 receptor mRNA
levels were measured using RNase protection assay and quantitative
real-time RT-PCR, as described in the Methods section. D1 receptor
mRNA levels were normalized to b-actin mRNA levels. Data were
obtained from three independent experiments and plotted as
mean ± SEM. *, D1 receptor mRNA expressed in cAMP-treated CAD
cells showed a statistically significant decrease, relative to control cells
and other treatments (p < 0.05, ANOVA, post hoc Holm’s pair-wise
comparison test). The bottom panel shows a representative RNase
protection assay for D1 and b-actin mRNA.
Regulation of D1 receptor expression by extracellular cAMP 625
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631
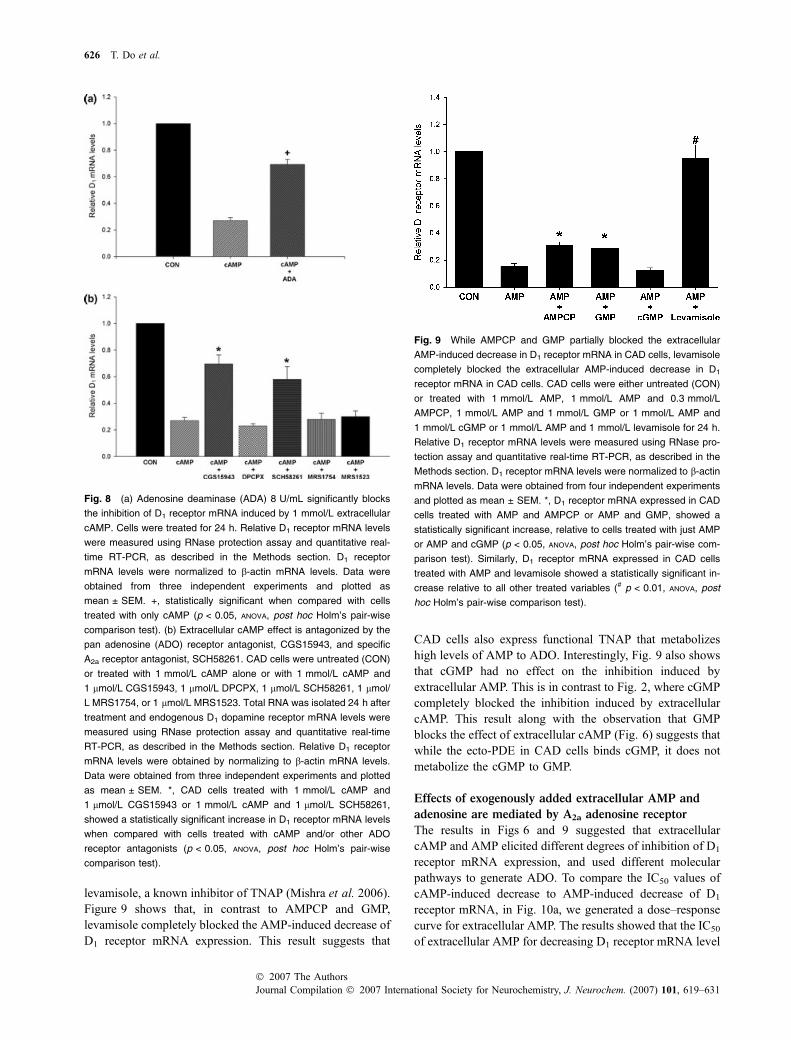
levamisole, a known inhibitor of TNAP (Mishra et al. 2006).Figure 9 shows that, in contrast to AMPCP and GMP,levamisole completely blocked the AMP-induced decrease ofD1 receptor mRNA expression. This result suggests that
CAD cells also express functional TNAP that metabolizeshigh levels of AMP to ADO. Interestingly, Fig. 9 also showsthat cGMP had no effect on the inhibition induced byextracellular AMP. This is in contrast to Fig. 2, where cGMPcompletely blocked the inhibition induced by extracellularcAMP. This result along with the observation that GMPblocks the effect of extracellular cAMP (Fig. 6) suggests thatwhile the ecto-PDE in CAD cells binds cGMP, it does notmetabolize the cGMP to GMP.
Effects of exogenously added extracellular AMP and
adenosine are mediated by A2a adenosine receptor
The results in Figs 6 and 9 suggested that extracellularcAMP and AMP elicited different degrees of inhibition of D1
receptor mRNA expression, and used different molecularpathways to generate ADO. To compare the IC50 values ofcAMP-induced decrease to AMP-induced decrease of D1
receptor mRNA, in Fig. 10a, we generated a dose–responsecurve for extracellular AMP. The results showed that the IC50
of extracellular AMP for decreasing D1 receptor mRNA level
Fig. 8 (a) Adenosine deaminase (ADA) 8 U/mL significantly blocks
the inhibition of D1 receptor mRNA induced by 1 mmol/L extracellular
cAMP. Cells were treated for 24 h. Relative D1 receptor mRNA levels
were measured using RNase protection assay and quantitative real-
time RT-PCR, as described in the Methods section. D1 receptor
mRNA levels were normalized to b-actin mRNA levels. Data were
obtained from three independent experiments and plotted as
mean ± SEM. +, statistically significant when compared with cells
treated with only cAMP (p < 0.05, ANOVA, post hoc Holm’s pair-wise
comparison test). (b) Extracellular cAMP effect is antagonized by the
pan adenosine (ADO) receptor antagonist, CGS15943, and specific
A2a receptor antagonist, SCH58261. CAD cells were untreated (CON)
or treated with 1 mmol/L cAMP alone or with 1 mmol/L cAMP and
1 lmol/L CGS15943, 1 lmol/L DPCPX, 1 lmol/L SCH58261, 1 lmol/
L MRS1754, or 1 lmol/L MRS1523. Total RNA was isolated 24 h after
treatment and endogenous D1 dopamine receptor mRNA levels were
measured using RNase protection assay and quantitative real-time
RT-PCR, as described in the Methods section. Relative D1 receptor
mRNA levels were obtained by normalizing to b-actin mRNA levels.
Data were obtained from three independent experiments and plotted
as mean ± SEM. *, CAD cells treated with 1 mmol/L cAMP and
1 lmol/L CGS15943 or 1 mmol/L cAMP and 1 lmol/L SCH58261,
showed a statistically significant increase in D1 receptor mRNA levels
when compared with cells treated with cAMP and/or other ADO
receptor antagonists (p < 0.05, ANOVA, post hoc Holm’s pair-wise
comparison test).
Fig. 9 While AMPCP and GMP partially blocked the extracellular
AMP-induced decrease in D1 receptor mRNA in CAD cells, levamisole
completely blocked the extracellular AMP-induced decrease in D1
receptor mRNA in CAD cells. CAD cells were either untreated (CON)
or treated with 1 mmol/L AMP, 1 mmol/L AMP and 0.3 mmol/L
AMPCP, 1 mmol/L AMP and 1 mmol/L GMP or 1 mmol/L AMP and
1 mmol/L cGMP or 1 mmol/L AMP and 1 mmol/L levamisole for 24 h.
Relative D1 receptor mRNA levels were measured using RNase pro-
tection assay and quantitative real-time RT-PCR, as described in the
Methods section. D1 receptor mRNA levels were normalized to b-actin
mRNA levels. Data were obtained from four independent experiments
and plotted as mean ± SEM. *, D1 receptor mRNA expressed in CAD
cells treated with AMP and AMPCP or AMP and GMP, showed a
statistically significant increase, relative to cells treated with just AMP
or AMP and cGMP (p < 0.05, ANOVA, post hoc Holm’s pair-wise com-
parison test). Similarly, D1 receptor mRNA expressed in CAD cells
treated with AMP and levamisole showed a statistically significant in-
crease relative to all other treated variables (# p < 0.01, ANOVA, post
hoc Holm’s pair-wise comparison test).
626 T. Do et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631� 2007 The Authors
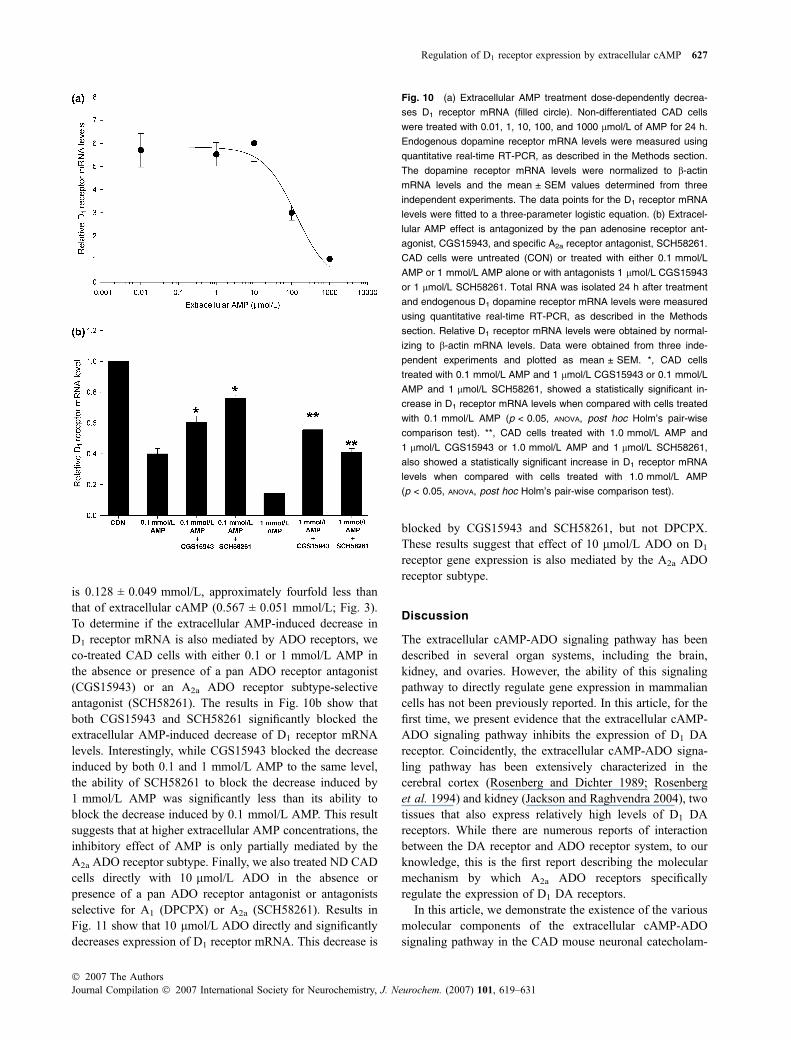
is 0.128 ± 0.049 mmol/L, approximately fourfold less thanthat of extracellular cAMP (0.567 ± 0.051 mmol/L; Fig. 3).To determine if the extracellular AMP-induced decrease inD1 receptor mRNA is also mediated by ADO receptors, weco-treated CAD cells with either 0.1 or 1 mmol/L AMP inthe absence or presence of a pan ADO receptor antagonist(CGS15943) or an A2a ADO receptor subtype-selectiveantagonist (SCH58261). The results in Fig. 10b show thatboth CGS15943 and SCH58261 significantly blocked theextracellular AMP-induced decrease of D1 receptor mRNAlevels. Interestingly, while CGS15943 blocked the decreaseinduced by both 0.1 and 1 mmol/L AMP to the same level,the ability of SCH58261 to block the decrease induced by1 mmol/L AMP was significantly less than its ability toblock the decrease induced by 0.1 mmol/L AMP. This resultsuggests that at higher extracellular AMP concentrations, theinhibitory effect of AMP is only partially mediated by theA2a ADO receptor subtype. Finally, we also treated ND CADcells directly with 10 lmol/L ADO in the absence orpresence of a pan ADO receptor antagonist or antagonistsselective for A1 (DPCPX) or A2a (SCH58261). Results inFig. 11 show that 10 lmol/L ADO directly and significantlydecreases expression of D1 receptor mRNA. This decrease is
blocked by CGS15943 and SCH58261, but not DPCPX.These results suggest that effect of 10 lmol/L ADO on D1
receptor gene expression is also mediated by the A2a ADOreceptor subtype.
Discussion
The extracellular cAMP-ADO signaling pathway has beendescribed in several organ systems, including the brain,kidney, and ovaries. However, the ability of this signalingpathway to directly regulate gene expression in mammaliancells has not been previously reported. In this article, for thefirst time, we present evidence that the extracellular cAMP-ADO signaling pathway inhibits the expression of D1 DAreceptor. Coincidently, the extracellular cAMP-ADO signa-ling pathway has been extensively characterized in thecerebral cortex (Rosenberg and Dichter 1989; Rosenberget al. 1994) and kidney (Jackson and Raghvendra 2004), twotissues that also express relatively high levels of D1 DAreceptors. While there are numerous reports of interactionbetween the DA receptor and ADO receptor system, to ourknowledge, this is the first report describing the molecularmechanism by which A2a ADO receptors specificallyregulate the expression of D1 DA receptors.
In this article, we demonstrate the existence of the variousmolecular components of the extracellular cAMP-ADOsignaling pathway in the CAD mouse neuronal catecholam-
Fig. 10 (a) Extracellular AMP treatment dose-dependently decrea-
ses D1 receptor mRNA (filled circle). Non-differentiated CAD cells
were treated with 0.01, 1, 10, 100, and 1000 lmol/L of AMP for 24 h.
Endogenous dopamine receptor mRNA levels were measured using
quantitative real-time RT-PCR, as described in the Methods section.
The dopamine receptor mRNA levels were normalized to b-actin
mRNA levels and the mean ± SEM values determined from three
independent experiments. The data points for the D1 receptor mRNA
levels were fitted to a three-parameter logistic equation. (b) Extracel-
lular AMP effect is antagonized by the pan adenosine receptor ant-
agonist, CGS15943, and specific A2a receptor antagonist, SCH58261.
CAD cells were untreated (CON) or treated with either 0.1 mmol/L
AMP or 1 mmol/L AMP alone or with antagonists 1 lmol/L CGS15943
or 1 lmol/L SCH58261. Total RNA was isolated 24 h after treatment
and endogenous D1 dopamine receptor mRNA levels were measured
using quantitative real-time RT-PCR, as described in the Methods
section. Relative D1 receptor mRNA levels were obtained by normal-
izing to b-actin mRNA levels. Data were obtained from three inde-
pendent experiments and plotted as mean ± SEM. *, CAD cells
treated with 0.1 mmol/L AMP and 1 lmol/L CGS15943 or 0.1 mmol/L
AMP and 1 lmol/L SCH58261, showed a statistically significant in-
crease in D1 receptor mRNA levels when compared with cells treated
with 0.1 mmol/L AMP (p < 0.05, ANOVA, post hoc Holm’s pair-wise
comparison test). **, CAD cells treated with 1.0 mmol/L AMP and
1 lmol/L CGS15943 or 1.0 mmol/L AMP and 1 lmol/L SCH58261,
also showed a statistically significant increase in D1 receptor mRNA
levels when compared with cells treated with 1.0 mmol/L AMP
(p < 0.05, ANOVA, post hoc Holm’s pair-wise comparison test).
Regulation of D1 receptor expression by extracellular cAMP 627
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631
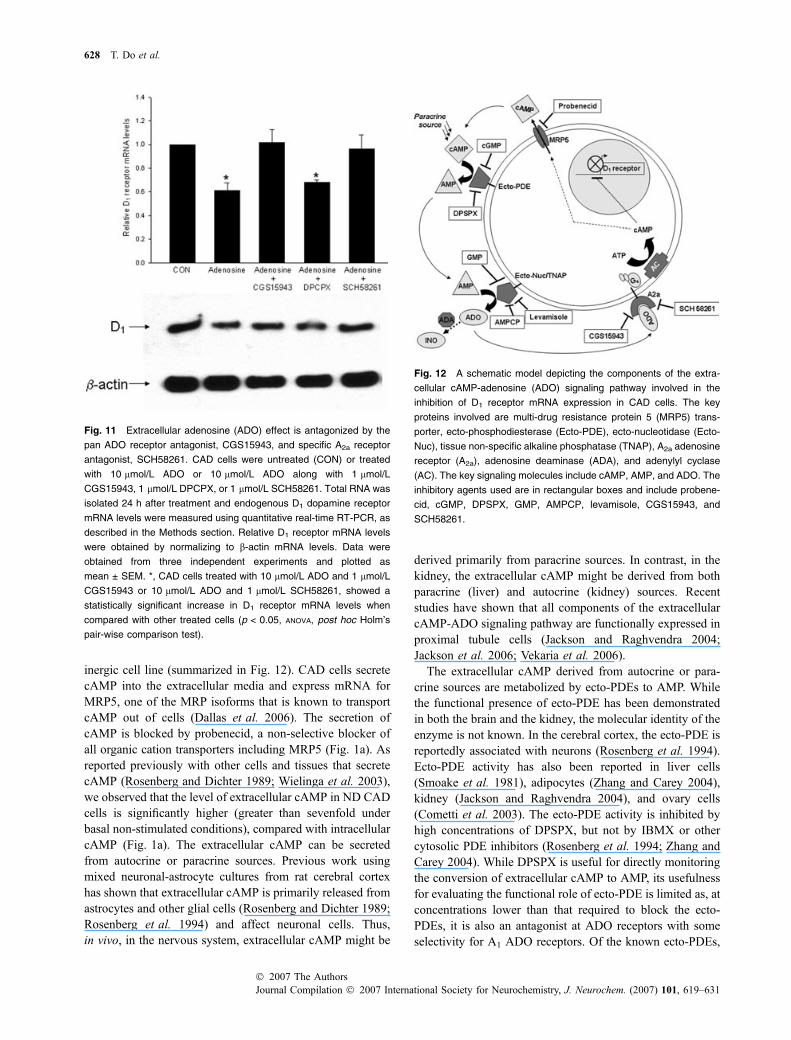
inergic cell line (summarized in Fig. 12). CAD cells secretecAMP into the extracellular media and express mRNA forMRP5, one of the MRP isoforms that is known to transportcAMP out of cells (Dallas et al. 2006). The secretion ofcAMP is blocked by probenecid, a non-selective blocker ofall organic cation transporters including MRP5 (Fig. 1a). Asreported previously with other cells and tissues that secretecAMP (Rosenberg and Dichter 1989; Wielinga et al. 2003),we observed that the level of extracellular cAMP in ND CADcells is significantly higher (greater than sevenfold underbasal non-stimulated conditions), compared with intracellularcAMP (Fig. 1a). The extracellular cAMP can be secretedfrom autocrine or paracrine sources. Previous work usingmixed neuronal-astrocyte cultures from rat cerebral cortexhas shown that extracellular cAMP is primarily released fromastrocytes and other glial cells (Rosenberg and Dichter 1989;Rosenberg et al. 1994) and affect neuronal cells. Thus,in vivo, in the nervous system, extracellular cAMP might be
derived primarily from paracrine sources. In contrast, in thekidney, the extracellular cAMP might be derived from bothparacrine (liver) and autocrine (kidney) sources. Recentstudies have shown that all components of the extracellularcAMP-ADO signaling pathway are functionally expressed inproximal tubule cells (Jackson and Raghvendra 2004;Jackson et al. 2006; Vekaria et al. 2006).
The extracellular cAMP derived from autocrine or para-crine sources are metabolized by ecto-PDEs to AMP. Whilethe functional presence of ecto-PDE has been demonstratedin both the brain and the kidney, the molecular identity of theenzyme is not known. In the cerebral cortex, the ecto-PDE isreportedly associated with neurons (Rosenberg et al. 1994).Ecto-PDE activity has also been reported in liver cells(Smoake et al. 1981), adipocytes (Zhang and Carey 2004),kidney (Jackson and Raghvendra 2004), and ovary cells(Cometti et al. 2003). The ecto-PDE activity is inhibited byhigh concentrations of DPSPX, but not by IBMX or othercytosolic PDE inhibitors (Rosenberg et al. 1994; Zhang andCarey 2004). While DPSPX is useful for directly monitoringthe conversion of extracellular cAMP to AMP, its usefulnessfor evaluating the functional role of ecto-PDE is limited as, atconcentrations lower than that required to block the ecto-PDEs, it is also an antagonist at ADO receptors with someselectivity for A1 ADO receptors. Of the known ecto-PDEs,
Fig. 12 A schematic model depicting the components of the extra-
cellular cAMP-adenosine (ADO) signaling pathway involved in the
inhibition of D1 receptor mRNA expression in CAD cells. The key
proteins involved are multi-drug resistance protein 5 (MRP5) trans-
porter, ecto-phosphodiesterase (Ecto-PDE), ecto-nucleotidase (Ecto-
Nuc), tissue non-specific alkaline phosphatase (TNAP), A2a adenosine
receptor (A2a), adenosine deaminase (ADA), and adenylyl cyclase
(AC). The key signaling molecules include cAMP, AMP, and ADO. The
inhibitory agents used are in rectangular boxes and include probene-
cid, cGMP, DPSPX, GMP, AMPCP, levamisole, CGS15943, and
SCH58261.
Fig. 11 Extracellular adenosine (ADO) effect is antagonized by the
pan ADO receptor antagonist, CGS15943, and specific A2a receptor
antagonist, SCH58261. CAD cells were untreated (CON) or treated
with 10 lmol/L ADO or 10 lmol/L ADO along with 1 lmol/L
CGS15943, 1 lmol/L DPCPX, or 1 lmol/L SCH58261. Total RNA was
isolated 24 h after treatment and endogenous D1 dopamine receptor
mRNA levels were measured using quantitative real-time RT-PCR, as
described in the Methods section. Relative D1 receptor mRNA levels
were obtained by normalizing to b-actin mRNA levels. Data were
obtained from three independent experiments and plotted as
mean ± SEM. *, CAD cells treated with 10 lmol/L ADO and 1 lmol/L
CGS15943 or 10 lmol/L ADO and 1 lmol/L SCH58261, showed a
statistically significant increase in D1 receptor mRNA levels when
compared with other treated cells (p < 0.05, ANOVA, post hoc Holm’s
pair-wise comparison test).
628 T. Do et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631� 2007 The Authors

NPP1 (PC-1) potentially metabolizes cAMP (Kelly andButler 1975); however, recent studies suggest that cAMP isnot a substrate or a poor substrate for NPP1 (Bollen et al.2000). While it is not known if CAD cells express NPP1, theobservation that cAMP is a poor substrate for NPP1 isconsistent with the relatively high IC50 (0.6 mmol/L)required for inhibiting D1 receptor mRNA expression, andalso with our observations that the inhibition induced byextracellular cAMP is less when compared with that inducedby its metabolite AMP. The relatively high IC50 for inhibitingD1 receptor mRNA expression could also be due to lowlevels of expression of the ecto-PDE in CAD cells.Interestingly, both cGMP and GMP block the effect ofextracellular cAMP (Figs 2 and 6); however, the effect ofextracellular AMP is blocked only by GMP (Fig. 9). Theseresults suggest that while cGMP competes with cAMP forthe ecto-PDE in CAD cells, unlike cAMP, cGMP is not asubstrate and is not metabolized by the CAD cell ecto-PDEto GMP.
The effect of extracellular cAMP is mediated by itsmetabolite AMP, as two different inhibitors of ecto-5¢-nucleotidase, AMPCP and GMP, completely blocked theextracellular cAMP-induced inhibition of D1 receptor mRNAexpression in CAD cells (Fig. 6). Interestingly, when extra-cellular AMP was directly added to the media, the inhibitionof D1 receptor mRNA expression was significantly greater(Fig. 10) than that observed with extracellular cAMP (Fig. 3)and only partly blocked by AMPCP and GMP (Fig. 9). Thissuggests that extracellular AMP might also mediate its effectvia a pathway that is independent of ecto-5¢-nucleotidases.Extracellular AMP reportedly binds ADO P1 receptors (Rosset al. 1998) and thus it could inhibit D1 receptor expressionby directly activating the ADO receptors. There is alsoevidence that extracellular AMP is transported into cells(Rosenberg and Dichter 1989) where it is metabolized toADO via cytosolic 5¢-nucleotidases. Thus, it is possible thatin CAD cells, the ADO produced intracellularly fromexogenously added AMP, is transported out by bidirectionalADO transporters and activates ADO receptors (Latini andPedata 2001). Extracellular AMP is also a substrate for theecto-alkaline phosphatase, TNAP, which metabolizes it toADO (Matsuoka and Ohkubo 2004; Mishra et al. 2006).While we have determined that CAD cells express very lowlevels of the ecto-5¢-nucleotidase (CD73) mRNA, we do notknow if CAD cells express TNAP; however, it is known thatin the brain, CD73 is expressed primarily on astrocytes(Jacobson and Gao 2006), whereas TNAP is expressed inneurons (Fonta et al. 2005). Interestingly, TNAP is notinhibited by AMPCP (Ohkubo et al. 2000), raising thepossibility that, in CAD neuronal cells, extracellular AMPmight be metabolized to ADO by TNAP. Indeed, the resultsin Fig. 9 show that an inhibitor of TNAP, levamisole,completely blocked the decrease of D1 receptor mRNAlevels, induced by extracellular AMP. This strongly suggests
that the effect of extracellular AMP on D1 receptor expres-sion in CAD cells is not mediated by direct binding andactivation of ADO P1 receptors, but rather by the conversionof AMP to ADO by TNAP.
Results in Figs 8, 10, and 11 strongly suggest that theeffect of extracellular cAMP, AMP, and ADO on D1 receptormRNA expression is mediated by ADO receptor, specificallythe A2a ADO receptor subtype. A2a ADO receptor isexpressed at high levels in the central nervous system,spleen, thymus, leukocytes, and blood platelets, and atintermediate levels in heart, lung, and blood vessels (Yaaret al. 2005). In the kidney, A2a ADO receptors are signifi-cantly up-regulated in streptozotocin-induced diabetic rats(Pawelczyk et al. 2005). In the central nervous system, highlevels of the A2a receptor are found in the caudate nucleus,putamen, nucleus accumbens, olfactory tubercles, and globuspallidus pars external segment in the rat and human brain(Jarvis and Williams 1989; Parkinson and Fredholm 1990;Martinez-Mir et al. 1991; Svenningsson et al. 1997). BothADO A2a receptor protein and mRNA are highly expressedin the striatum (reviewed in Svenningsson et al. 1999). In thestriatum, A2a receptors are largely co-expressed with DA D2
receptor (Jarvis and Williams 1989; Schiffmann et al. 1991;Fink et al. 1992). Conversely, only 1–3% of neuronsexpressing A2a receptor mRNA co-expressed the D1 DAreceptor mRNA (Svenningsson et al. 1998).
Among all ADO receptor subtypes, A2a receptors play animportant role in the control of motor behavior and inmodulating DA-mediated responses (Durcan and Morgan1989; Ferre et al. 1992, 1997). Both ADO A2a receptors andD1 DA receptors have been implicated in Parkinson’s diseaseand Huntington’s disease. In Tourette’s syndrome, a post-mortem analysis of brain tissue from adult patients showed adecrease in cAMP levels (Singer et al. 1990). Interestingly, arecent study in brain tissue isolated from a young Tourette’spatient showed a 431% and 226% increase in D1 receptorexpression in the putamen and prefrontal cortex, respectively(Minzer et al. 2004). This is consistent with our results that adecrease in cAMP levels will result in increased D1 receptorexpression, and suggests a potential role for the extracellularcAMP-ADO signaling pathway in Tourette’s syndrome.
Our results also provide a novel molecular link betweenthe activation of A2a ADO receptor and the expression of D1
DA receptor. The most parsimonious model would predictthat conditions which increase A2a ADO receptor expression,along with an increased activation of the ADO signalingpathway, will result in the down-regulation of D1 DAreceptor expression. Our model is supported by severalobservations, including the lack of co-expression of A2a andD1 DA receptors in different brain regions (Svenningssonet al. 1998), the decrease of D1 receptor mRNA as A2a
receptor expression increases during development (Johans-son et al. 1997), the increase of D1 DA receptor expressionin the caudate putamen and nucleus accumbens of A2a null
Regulation of D1 receptor expression by extracellular cAMP 629
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631

mice (Short et al. 2006), and the decrease of renal D1 DAreceptor expression (Hussain et al. 1999; Marwaha et al.2004) and increase of A2a ADO receptor (Pawelczyk et al.2005) in rat models of type I and type II diabetes.
Acknowledgements
This work was supported by grants from the F. M. Kirby Foundation
(EVK and AB) and the UMDNJ Deans Biomedical Research
Support Program (EVK).
References
Bankir L., Arloulay M., Devreotes P. N. and Parent C. A. (2002)Extracellular cAMP inhibits proximal reabsorption: are plasmacAMP receptors involved Am. J. Phyisol. Renal Physiol. 282,F376–F392.
Berke J. and Hyman S. (2000) Addiction, dopamine, and the molecularmechanisms of memory. Neuron 25, 515–532.
Bilodeau M. L., Ji M., Paris M. and Andrisani O. M. (2005) Adenosinesignaling promotes neuronal, catecholaminergic differentiation ofprimary neural crest cells and CNS-derived CAD cells. Mol. Cell.Neurosci. 29, 394–404.
Bollen M., Gijsbers R., Ceulemans H., Stalmans W. and Stefan C. (2000)Nucleotide pyrophosphates/phosphodiesterases on the move. Crit.Rev. Biochem. Mol. Biol. 35, 393–432.
Butkerait P. and Friedman E. (1993) Repeated reserpine increases striataldopamine receptor and guanine nucleotide binding protein RNA.J. Neurochem. 60, 566–571.
Civelli O., Bunzow J. R. and Grandy D. K. (1993) Molecular diversity ofthe dopamine receptors. Annu. Rev. Pharmacol. Toxicol. 33, 281–307.
Cometti B., Dubey R. K., Imthurn B., Jackson E. K. and Rosselli M.(2003) Oviduct cells express the cyclic AMP-adenosine pathway.Biol. Reprod. 69, 868–875.
Dallas S., Miller D. S. and Bendayan R. (2006) Multidrug resistance-associated proteins: expression and function in the central nervoussystem. Pharmacol. Rev. 58, 140–161.
Davoren P. R. and Sutherland E. W. (1963) The effect of L-epinephrineand other agents on the synthesis and release of adenosine 3¢,5¢-phosphate by whole pigeon erythrocytes. J. Bio. Chem. 238, 3009–3015.
Domino S. E., Tubb D. J. and Garbers D. L. (1991) Assay of guanylylcyclase catalytic activity. Methods Enzymol. 195, 345–355.
Dubey R. K., Gillespie D. G., Mi Z. and Jackson E. K. (2000) Cardiacfibroblasts express the cAMP-adenosine pathway. Hypertension36, 337–342.
Durcan M. J. and Morgan P. F. (1989) Evidence for adenosine A2
receptor involvement in the hypomobility effects of adenosineanalogues in mice. Eur. J. Pharmacol. 168, 285–290.
Exton J. H., Robison J. A., Sutherland E. W. and Park C. R. (1971)Studies on the role of adenosine 3¢,5¢-monophosphate in the hep-atic actions of glucagon and catecholamines. J. Bio. Chem. 246,6166–6177.
Ferre S., Fuxe K., von Euler G., Johansson B. and Fredholm B. B.(1992) Adenosine-dopamine interaction in the brain. Neuroscience51, 501–512.
Ferre S., Fredholm B. B., Morelli M., Popoli P. and Fuxe K. (1997)Adenosine–dopamine receptor–receptor interactions as an integra-tive mechanism in the basal ganglia. Trends Neurosci. 20, 482–487.
Fink J. S., Weaver D. R., Rivkees S. A., Peterfreund R. A., Pollack A. E.,Adler E. M. and Reppert S. M. (1992) Molecular cloning of the rat
A2 adenosine receptor: selective co-expression with D2 dopaminereceptors in rat striatum. Brain Res. Mol. Brain. Res. 14, 186–195.
Fonta C., Negyessy L., Renaud L. and Barone P. (2005) Postnataldevelopment of alkaline phosphatase activity correlates with thematuration of neurotransmission in the cerebral cortex. J. Comp.Neurol. 486, 179–196.
Ginovart N., Lundin A., Farde L., Halldin C., Backman L., Swahn C. G.,Pauli S. and Sedvall G. (1997) PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative processin Huntington’s disease. Brain 120, 503–514.
Hussain T. and Lokhandwala M. F. (2003) Renal dopamine and hyper-tension. Exp. Biol. Med. 228, 134–142.
Hussain T., Beheray S. A. and Lokhandwala M. F. (1999) Defectivedopamine receptor function in proximal tubules of obese zuckerrats. Hypertension 34, 1091–1096.
Jackson E. K. and Dubey R. K. (2001) Role of the extracellular cAMP-adenosine pathway in renal physiology. Am. J. Phyisol. RenalPhysiol. 281, F597–F612.
Jackson E. K. and Raghvendra D. K. (2004) The extracellular cyclicAMP-adenosine pathway in renal physiology. Annu. Rev. Physiol.66, 571–599.
Jackson E. K., Zacharia L. C., Zhang M., Gillespie D. G., Zhu C. andDubey R. K. (2006) cAMP-adenosine pathway in the proximaltubule. J. Pharmacol. Exp. Ther. 317, 1219–1229.
Jacobson K. A. and Gao Z. G. (2006) Adenosine receptors as therapeutictargets. Nat. Rev. Drug Discov. 53, 247–264.
Jarvis M. F. and Williams M. (1989) Direct autoradiographic localizationof adenosine A2 receptors in the rat brain using the A2-selectiveagonist, [3H]CGS 21680. Eur. J. Pharmacol. 168, 243–246.
Johansson B., Georgiev V. and Fredholm B. B. (1997) Distribution andpostnatal ontogeny of adenosine A2A receptors in rat brain:comparison with dopamine receptors. Neuroscience 80, 1187–1207.
Kelly S. J. and Butler L. G. (1975) Enzymic hydrolysis of phosphonateesters. Biochem. Biophys. Res. Commun. 66, 316–321.
Kuster J., Zap F. J. and Jakob A. (1973) Effects of hormones on cyclicAMP release in perfused rat liver. FEBS Lett. 32, 73–77.
Latini S. and Pedata F. (2001) Adenosine in the central nervous system:release mechanisms and extracellular concentrations. J. Neuro-chem. 79, 463–484.
Laurier L. G., Corrigall W. A. and George S. R. (1994) Dopaminereceptor density, sensitivity and mRNA levels are altered followingself-administration of cocaine in the rat. Brain Res. 634, 31–40.
Levy M. and Starr N. L. (1972) The mechanism of glucagon-inducednatriuresis in dogs. Kidney Int. 2, 76–84.
Martinez-Mir M. I., Probst A. and Palacios J. M. (1991) Adenosine A2receptors: selective localization in the human basal ganglia andalterations with disease. Neuroscience 42, 697–706.
Marwaha A., Banday A. A. and Lokhandwala M. F. (2004) Reducedrenal dopamine D1 receptor function in streptozotocin-induceddiabetic rats. Am. J. Physiol. Renal Physiol. 286, F451–F457.
Matsuoka I. and Ohkubo S. (2004) ATP- and adenosine-mediatedsignaling in the central nervous system: adenosine receptor acti-vation by ATP through rapid and localized generation of adenosineby ecto-nucleotidases. J. Pharmacol. Sci. 94, 95–99.
Minzer K., Lee O., Hong J. J. and Singer H. S. (2004) Increased pre-frontal D2 protein in Tourette syndrome: a postmortem analysis offrontal cortex and striatum. J. Neurol. Sci. 219, 55–61.
Mishra S. K., Braun N., Shukla V. et al. (2006) Extracellular nucleotidesignaling in adult neural stem cells: synergism with growth factor-mediated cellular proliferation. Development 133, 675–684.
Missale C., Nash S. R., Robinson S. W., Jaber M. and Caron M. G.(1998) Dopamine receptors: from structure to function. Physiol.Rev. 78, 189–225.
630 T. Do et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631� 2007 The Authors

Ohkubo S., Kimura J. and Matsuoka I. (2000) Correlation betweenadenine nucleotide-induced cyclic AMP elevation and extracellularadenosine formation in NG108-15 cells. Jpn. J. Pharmacol. 84,325–333.
Parkinson F. E. and Fredholm B. B. (1990) Autoradiographic evidencefor G-protein coupled A2-receptors in rat neostriatum using [3H]-CGS 21680 as a ligand. Naunyn Schmiedebergs Arch. Pharmacol.342, 85–89.
Pasuit J. B., Li Z. and Kuzhikandathil E. V. (2004) Multi-modal regu-lation of endogenous D1 dopamine receptor expression and func-tion in the CAD catecholaminergic cell line. J. Neurochem. 89,1508–1519.
Pawelczyk T., Grden M., Rzepko R., Sakowicz M. and Szutowicz A.(2005) Region-specific alterations of adenosine receptors expres-sion level in kidney of diabetic rat. Am. J. Pathol. 167, 315–325.
Rogue P., Hanauer A., Zwiller J., Malviya A. N. and Vincendon G.(1991) Up-regulation of dopamine D2 receptor mRNA in rat stri-atum by chronic neuroleptic treatment. Eur. J. Pharmacol. 207,165–168.
Rosenberg P. A. and Dichter M. A. (1989) Extracellular cAMP accu-mulation and degradation in rat cerebral cortex in dissociated cellculture. J. Neurosci. 9, 2654–2663.
Rosenberg P. A., Knowles R., Knowles K. P. and Li Y. (1994) Beta-adrenergic receptor-mediated regulation of extracellular adenosinein cerebral cortex in culture. J. Neurosci. 14, 2953–2965.
Ross F. M., Brodie M. J. and Stone T. W. (1998) Adenosine mono-phosphate as a mediator of ATP effects at P1 purinoceptors. Br.J. Pharmacol. 124, 818–824.
Schambra U. B., Duncan G. E., Breese G. R., Fornaretto M. G., CaronM. G. and Fremeau R. T. Jr (1994) Ontogeny of D1A and D2dopamine receptor subtypes in rat brain using in situ hybridizationand receptor binding. Neuroscience 62, 65–85.
Schiffmann S. N., Jacobs O. and Vanderhaeghen J. J. (1991) Striatalrestricted adenosine A2 receptor (RDC8) is expressed by enke-phalin but not by substance P neurons: an in situ hybridizationhistochemistry study. J. Neurochem. 57, 1062–1067.
Short J. L., Ledent C., Drago J. and Lawence A. J. (2006) Receptorcrosstalk: characterization of mice deficient in dopamine D1 andadenosine A2A receptors.Neuropsychopharmacology 31, 525–534.
Sibley D. R., Monsma F. J. Jr and Shen Y. (1993) Molecular neurobi-ology of dopaminergic receptors. Int. Rev. Neurobiol. 35, 391–415.
Singer H. S., Hahn I. H., Krowiak E., Nelson E. and Moran T. (1990)Tourette’s syndrome: a neurochemical analysis of postmortemcortical brain tissue. Ann. Neurol. 27, 443–446.
Smoake J. A., McMahon K. L., Wright R. K. and Solomon S. S. (1981)Hormonally sensitive cyclic AMP phosphodiesterase in liver cells.An ecto-enzyme. J. Biol. Chem. 256, 8531–8535.
Song Z. and Sladek C. D. (2005) Does conversion of ATP to adenosineterminate ATP-stimulated vasopressin release from hypothalamo-neurohypophyseal explants Brain Res. 1047, 105–111.
Stoessl A. J. and de la Fuente-Fernandez R. (2003) Dopamine receptorsin Parkinson’s disease: imaging studies. Adv. Neurol. 91, 65–71.
Stone E. A. and John S. M. (1990) In vivo measurement of extracellularcyclic AMP in the brain: use in studies of beta-adrenoceptorfunction in nonanesthetized rats. J. Neurochem. 55, 1942–1949.
Svenningsson P., Hall H., Sedvall G. and Fredholm B. B. (1997) Dis-tribution of adenosine receptors in the postmortem human brain: anextended autoradiographic study. Synapse 27, 322–335.
Svenningsson P., Le Moine C., Aubert I., Burbaud P., Fredholm B. B.and Bloch B. (1998) Cellular distribution of adenosine A2Areceptor mRNA in the primate striatum. J. Comp. Neurol. 399,229–240.
Svenningsson P., Le Moine C., Fisone G. and Fredholm B. B. (1999)Distribution, biochemistry and function of striatal adenosine A2Areceptors. Prog. Neurobiol. 59, 355–396.
Tofovic S. P., Branch K. R., Oliver R. D., Magee W. D. and JacksonE. K. (1991) Caffeine potentiates vasodilator-induced renin release.J. Pharmacol. Exp. Ther. 256, 850–860.
Vallone D., Picetti R. and Borrelli E. (2000) Structure and function ofdopamine receptors. Neurosci. Biobehav. Rev. 24, 125–132.
Vekaria R. M., Shirley D. G., Sevigny J. and Unwin R. J. (2006)Immunolocalization of ectonucleotidases along the rat nephron.Am. J. Physiol. Renal Physiol. 290, F550–F560.
Vortherms T. A. and Watts V. J. (2004) Sensitization of neuronal A2Aadenosinereceptors after persistent D2 dopamine receptor activa-tion. J. Pharmacol. Exp. Ther. 308, 221–227.
Weeks R. A., Piccini P., Harding A. E. and Brooks D. J. (1996) StriatalD1 and D2 dopamine receptor loss in asymptomatic mutationcarriers of Huntington’s disease. Ann. Neurol. 40, 49–54.
Wielinga P. R., van der Heijden I., Reid G., Beijnen J. H., Wijnholds J.and Borst P. (2003) Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J. Biol.Chem. 278, 17 664–17 671.
Xu S. X., Monsma F. J. Jr, Sibley D. R. and Creese I. (1992) Regulationof D1A and D2 dopamine receptor mRNA during ontogenesis,lesion and chronic antagonist treatment. Life Sci. 50, 383–396.
Yaar R., Jones M. R., Chen J. F. and Ravid K. (2005) Animal models forthe study of adenosine receptor function. J. Cell. Physiol. 202, 9–20.
Zhang X. and Carey G. B. (2004) Plasma membrane-bound cyclic AMPphosphodiesterase activity in 3T3-L1 adipocytes. Comp. Biochem.Physiol. 137, 309–316.
Zimmermann H. (1992) 5¢-Nucleotidase: molecular structure and func-tional aspects. Biochem. J. 285, 345–365.
Regulation of D1 receptor expression by extracellular cAMP 631
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 101, 619–631
Related Documents











