JOURNAL OF VIROLOGY, Feb. 2007, p. 1961–1971 Vol. 81, No. 4 0022-538X/07/$08.000 doi:10.1128/JVI.01981-06 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Evolutionary Mechanisms of Persistence and Diversification of a Calicivirus within Endemically Infected Natural Host Populations Karen P. Coyne, 1 * Rosalind M. Gaskell, 2 Susan Dawson, 1 Carol J. Porter, 2 and Alan D. Radford 1 Departments of Veterinary Clinical Science 1 and Veterinary Pathology, 2 University of Liverpool, Leahurst, Chester High Road, Neston, South Wirral CH64 7TE, United Kingdom Received 12 September 2006/Accepted 24 November 2006 In order to understand the evolutionary mechanisms of persistence and diversification within the Calici- viridae, we have been exploiting endemic infection of feline calicivirus within five geographically distinct household groups of cats. By sequencing immunodominant and variable regions of the capsid gene, we identified the relative contribution of the different evolutionary processes employed by the virus to ensure its long-term survival in the host population. Such strategies included progressive evolution of a given variant of a strain through mutation accumulation within an individual, sequential reinfection with either a variant of the same strain or with a different strain, and mixed infection. Recombination between different strains in this study has been reported in detail elsewhere (K. P. Coyne et al., J. Gen. Virol. 87:921–926, 2006). Here, we provide evidence to suggest that true long-term persistent infection in individuals is relatively rare, with the majority of apparent viral carriers undergoing a combination of progressive evolution and cyclical reinfection. Progressive evolution at the individual level and variant reinfection at both the individual and population levels were associated with positive selection. Two measures of evolution rate were determined; for a virus progres- sively evolving within an individual (1.32 10 2 to 2.64 10 2 substitutions per nucleotide per year, i.e., no transmission) and for a strain circulating within a population (3.84 10 2 to 4.56 10 2 substitutions per nucleotide per year, i.e., including transmission). Reiteration of both progressive evolution and variant reinfection appeared to lead to a gradual increase in the diversity of a given strain of virus, both in the individual and in the population, until eventually new strains emerged. The Caliciviridae are an important group of human and animal RNA pathogens, causing a wide range of diseases, including acute outbreaks of gastroenteritis in humans (noro- viruses and sapoviruses) and vesicular and other diseases in animals (vesiviruses and lagoviruses) (19). The Caliciviridae are a highly variable family of small positive-strand RNA vi- ruses, demonstrating considerable antigenic and genetic diver- sity both between and within genera. Such variability has been associated with the emergence of highly transmissible globally dominant strains of human caliciviruses (36) and hypervirulent animal caliciviruses that are frequently lethal (9, 33, 39). De- spite the obvious significance of such variation, the origins of this diversity and the evolutionary mechanisms by which new, and possibly more virulent strains evolve remain unclear. Feline calicivirus (FCV) belongs to the genus Vesivirus and is a highly infectious oral and respiratory pathogen of domestic cats (18). Following acute infection, some cats remain persis- tently infected with the virus (11, 40, 48, 59). Such carriers appear to be relatively common in the general population, with prevalences ranging from 15 to 91%, and they play an impor- tant role in the epidemiology of the disease (1, 3, 7, 8, 21, 22, 42). Persistent infections are also recognized in other calici- viruses, although in general these are not as well characterized as in feline calicivirus, and their role in the epidemiology of the disease is not as clear (15, 17, 35, 63). The mechanisms of viral persistence in the Caliciviridae, both within an individual and at the population level, are not fully elucidated. Persistence within an individual is thought to be largely via immune-mediated positive selection (29, 35, 48), although other mechanisms may also play a role. Key questions that need to be resolved are the following: what contribution do such carriers make to the long-term survival of the virus at the population level, and to what extent do they drive virus evolution and diversification. In order to address these issues, we have been exploiting endemic infection of feline calicivirus within stable household groups of cats. We have previously reported that such house- holds tend to be infected with a small number of distinct strains of feline calicivirus (8) and provide a useful model system to monitor calicivirus evolution and persistence over long periods of time, in a defined geographic space within a natural popu- lation. In contrast, in other caliciviruses, most sampling is lim- ited to investigation of disease outbreaks over relatively short periods, where opportunities for virus evolution are likely to be limited (15, 16, 32, 57, 64). The shape of resulting phylogenies therefore tends to be a reflection of the sampling strategy employed and, thus, intermediate evolutionary steps can only be conjectured (20). Here we use structured long-term sam- pling of endemically infected groups of cats to identify the relative contribution of the different evolutionary processes by which caliciviruses generate viral diversity and ensure their long-term survival in the host population. * Corresponding author. Mailing address: Department of Veteri- nary Clinical Sciences, University of Liverpool, Leahurst, Chester High Road, South Wirral CH64 7TE, United Kingdom. Phone: 44 0151 795 6033. Fax: 44 0151 794 6005. E-mail: [email protected]. Published ahead of print on 6 December 2006. 1961 on January 28, 2015 by guest http://jvi.asm.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF VIROLOGY, Feb. 2007, p. 1961–1971 Vol. 81, No. 40022-538X/07/$08.00�0 doi:10.1128/JVI.01981-06Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Evolutionary Mechanisms of Persistence and Diversification of aCalicivirus within Endemically Infected Natural
Host Populations�
Karen P. Coyne,1* Rosalind M. Gaskell,2 Susan Dawson,1 Carol J. Porter,2 and Alan D. Radford1
Departments of Veterinary Clinical Science1 and Veterinary Pathology,2 University of Liverpool, Leahurst,Chester High Road, Neston, South Wirral CH64 7TE, United Kingdom
Received 12 September 2006/Accepted 24 November 2006
In order to understand the evolutionary mechanisms of persistence and diversification within the Calici-viridae, we have been exploiting endemic infection of feline calicivirus within five geographically distincthousehold groups of cats. By sequencing immunodominant and variable regions of the capsid gene, weidentified the relative contribution of the different evolutionary processes employed by the virus to ensure itslong-term survival in the host population. Such strategies included progressive evolution of a given variant ofa strain through mutation accumulation within an individual, sequential reinfection with either a variant of thesame strain or with a different strain, and mixed infection. Recombination between different strains in thisstudy has been reported in detail elsewhere (K. P. Coyne et al., J. Gen. Virol. 87:921–926, 2006). Here, weprovide evidence to suggest that true long-term persistent infection in individuals is relatively rare, with themajority of apparent viral carriers undergoing a combination of progressive evolution and cyclical reinfection.Progressive evolution at the individual level and variant reinfection at both the individual and population levelswere associated with positive selection. Two measures of evolution rate were determined; for a virus progres-sively evolving within an individual (1.32 � 10�2 to 2.64 � 10�2 substitutions per nucleotide per year, i.e., notransmission) and for a strain circulating within a population (3.84 � 10�2 to 4.56 � 10�2 substitutions pernucleotide per year, i.e., including transmission). Reiteration of both progressive evolution and variant reinfectionappeared to lead to a gradual increase in the diversity of a given strain of virus, both in the individual and in thepopulation, until eventually new strains emerged.
The Caliciviridae are an important group of human andanimal RNA pathogens, causing a wide range of diseases,including acute outbreaks of gastroenteritis in humans (noro-viruses and sapoviruses) and vesicular and other diseases inanimals (vesiviruses and lagoviruses) (19). The Caliciviridaeare a highly variable family of small positive-strand RNA vi-ruses, demonstrating considerable antigenic and genetic diver-sity both between and within genera. Such variability has beenassociated with the emergence of highly transmissible globallydominant strains of human caliciviruses (36) and hypervirulentanimal caliciviruses that are frequently lethal (9, 33, 39). De-spite the obvious significance of such variation, the origins ofthis diversity and the evolutionary mechanisms by which new,and possibly more virulent strains evolve remain unclear.
Feline calicivirus (FCV) belongs to the genus Vesivirus and isa highly infectious oral and respiratory pathogen of domesticcats (18). Following acute infection, some cats remain persis-tently infected with the virus (11, 40, 48, 59). Such carriersappear to be relatively common in the general population, withprevalences ranging from 15 to 91%, and they play an impor-tant role in the epidemiology of the disease (1, 3, 7, 8, 21, 22,42). Persistent infections are also recognized in other calici-viruses, although in general these are not as well characterized
as in feline calicivirus, and their role in the epidemiology of thedisease is not as clear (15, 17, 35, 63).
The mechanisms of viral persistence in the Caliciviridae,both within an individual and at the population level, are notfully elucidated. Persistence within an individual is thought tobe largely via immune-mediated positive selection (29, 35, 48),although other mechanisms may also play a role. Key questionsthat need to be resolved are the following: what contributiondo such carriers make to the long-term survival of the virus atthe population level, and to what extent do they drive virusevolution and diversification.
In order to address these issues, we have been exploitingendemic infection of feline calicivirus within stable householdgroups of cats. We have previously reported that such house-holds tend to be infected with a small number of distinct strainsof feline calicivirus (8) and provide a useful model system tomonitor calicivirus evolution and persistence over long periodsof time, in a defined geographic space within a natural popu-lation. In contrast, in other caliciviruses, most sampling is lim-ited to investigation of disease outbreaks over relatively shortperiods, where opportunities for virus evolution are likely to belimited (15, 16, 32, 57, 64). The shape of resulting phylogeniestherefore tends to be a reflection of the sampling strategyemployed and, thus, intermediate evolutionary steps can onlybe conjectured (20). Here we use structured long-term sam-pling of endemically infected groups of cats to identify therelative contribution of the different evolutionary processes bywhich caliciviruses generate viral diversity and ensure theirlong-term survival in the host population.
* Corresponding author. Mailing address: Department of Veteri-nary Clinical Sciences, University of Liverpool, Leahurst, Chester HighRoad, South Wirral CH64 7TE, United Kingdom. Phone: 44 0151 7956033. Fax: 44 0151 794 6005. E-mail: [email protected].
� Published ahead of print on 6 December 2006.
1961
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

MATERIALS AND METHODS
Virus origins and isolation. The origins of the 144 FCV isolates used in thisstudy have been described elsewhere (8). Briefly, virus isolates were sequentiallycollected from cats within five different geographically located communities(designated A to E) over a 15- to 46-month study period. Oropharyngeal swabswere collected into 2 ml of virus transport medium. Viruses were isolated asdescribed previously on feline embryo A cells and Crandall Reese feline kidneycells (27, 61) and stored at �80°C for further analysis.
RNA extraction, reverse transcription-PCR, and consensus sequencing.RNA was extracted from positive samples (second passage or less; QIAmp viralRNA mini kit; QIAGEN) and transcribed into cDNA (Superscript III; Invitro-gen) using 500 ng of specific reverse primer (Table 1) as the first-strand primeraccording to the manufacturers’ instructions. A 529-nucleotide region of thecapsid gene, equivalent to residues 6406 to 6934 of the FCV strain F9 (5) andincorporating immunodominant region E (34, 49, 53), was amplified using PfuDNA polymerase (Stratagene) according to the manufacturer’s instructions.Each 50-�l reaction mixture contained 2 �l of cDNA and 100 ng of each PCRprimer (Table 1). Thermal cycling conditions consisted of DNA denaturation at95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 30 seconds,primer annealing at 50°C for 30 seconds, and primer extension at 72°C for 90seconds. A final extension was performed at 72°C for 5 min.
Amplicons were purified (QIAquick PCR purification kit; QIAGEN Ltd.) andsequenced bidirectionally using the corresponding PCR primers (Table 1) ac-cording to standard protocols (ABI Prism BigDye terminators version 3.0 cyclesequencing kits; Applied Biosystems). For each amplicon, a consensus sequencewas produced using ChromasPro version 1.32 (Technelysium Ptl. Ltd.). Ambi-guities identified consistently in both strands of sequence were included in theconsensus sequence. All primer sites were removed prior to sequence analysis,resulting in a final useable sequence of 420 nucleotides corresponding to codons383 to 522 of the FCV capsid (53) and including variable regions C and E. Inorder to determine the impact of cell culture, virus isolation, passage, and reversetranscription-PCR on consensus sequences, we compared the cell culture-pas-saged sequence to the sequence determined directly from the oropharyngealswab for six viruses. These repeat sequences were 99.3 to 100% identical (datanot presented).
Cloning of PCR products. In order to characterize the diversity of FCV withinindividual cats and to attempt to identify transmission events, multiple clones of20 FCV isolates were obtained from three representative cats for which a num-ber of serial samples were available. Cloning of PCR products was performedusing the Zero Blunt TOPO PCR cloning kit (Invitrogen life technologies)essentially according to the manufacturer’s instructions. Plasmids were purifiedusing a Hamilton Microlab Star robot running the Millipore plasmid extractionkit (Montage Plasmid MiniprepB96B kit) as per the manufacturers’ recommen-dations. Plasmids containing an insert of the appropriate size were sequencedusing standard protocols.
Nucleotide sequence and phylogenetic analysis. Nucleotide distance calcula-tions and sequence alignments were performed using the programs Distance andPileup from version 8 of the Wisconsin package, Genetics Computer Group (13),using the Jukes-Cantor model. This model accounts for multiple substitutionsand ambiguities in the sequence data, which for FCV are attributable to thequasispecies nature of the virus (48). All distance values are corrected unlessotherwise specified. Where appropriate, uncorrected analyses were also carriedout to allow comparison with previous epidemiological studies (44, 45). Phylo-genetic analysis was performed using the program GROWTREE (Jukes-Cantordistance analysis and neighbor joining) also available in the Genetics ComputerGroup package and viewed using TREEVIEW (38). Support for individualnodes was sought by bootstrap analysis using 1,000 repetitions (MEGA version3.0) (30).
Clonal sequence analysis. For those isolates for which clonal sequences wereavailable, phylogenetic analyses and average pair-wise genetic diversity wereperformed using the Jukes-Cantor and neighbor-joining models (MEGA version3.0) (30). No ambiguous sites were present in the cloned sequence data. Supportfor individual nodes was sought by bootstrap analysis using 1,000 repetitions(MEGA version 3.0) (30).
Simpson’s equitability index. Simpson’s equitability index (E) was used inaddition to the average pair-wise genetic diversity to give a further measure ofintraisolate quasispecies diversity (2, 54).
E �1
�i�1
S
Pi2
�1S
where Pi is the proportion of identical clones making up the population and S isthe total number of clones within the population. This returns a value of 1 formaximum diversity (all clones different) and tends toward zero as the diversitydecreases and the clonal population size increases.
Statistical analysis. Statistical analysis was carried out using Minitab for Win-dows 14.1 (Minitab Inc.). Comparisons between nucleotide distance values wereanalyzed using the Mann-Whitney test. The critical probability was taken as a Pvalue of �0.05 for a two-sided alternative hypothesis.
Estimating evolution rates. Where appropriately sized subdata sets of se-quences were available, evolution rates were calculated using Tipdate v. 1.01(50). These data sets were based on consensus sequences from those cats thatwere persistently infected with their own strain of virus (progressive evolvers;group D, cat 4 and cat 2) (8) and from those virus strains that arrived in the groupafter the start of the study and for which the group ancestor sequence wasavailable (strains D2 and E2).
Evidence for positive selection. In order to identify the mechanisms underlyingthe diversification of strains within individual groups, evidence of positive selec-tion was sought using a codon-based approach as implemented in Datamonkey(41). These offer several advantages over previously described methods, includ-ing not needing to assume equal synonymous substitution rates throughout thesequence and allowing the user to choose the most appropriate model fornucleotide substitution from the original data set. Because of the different sizesof the data sets, a combination of three approaches was used. For larger datasets, a single likelihood ancestor counting (SLAC) analysis was performed suinga P value of 0.1. For smaller data sets, where type I errors may be more common,an integrative approach was taken using SLAC, fixed effects likelihood (FEL),and random effects likelihood (REL) (P � 0.25 for SLAC and FEL; Bayes Factorof 10 for REL). In all cases, ambiguities in the consensus sequence were aver-aged in the analysis.
Predicted structure studies. The regions of the FCV capsid predicted to beevolving under positive selection were mapped onto a predicted structure for theFCV F9 capsid protein (GenBank accession number M86379), which was mod-eled against the known structures for San Miguel sea lion virus (SMSV), whichalso belongs to the Vesivirus genus (PDB entry 2gh8.pdb) (6) and human noro-virus (PDB entry1ihm.pdb) (43) using SWISS-MODEL via the ExPASy webserver (52). The predicted FCV capsid was viewed and manipulated using PyMol0.99 (12).
Nucleotide sequence accession numbers. All consensus sequences have beensubmitted to GenBank (accession numbers DQ397674 to DQ397814). Eachsequence is identified by the following strain identification key: group and strainnumber, cat number, and time in months, e.g., b1c7t09 indicates group B, strain1, from cat 7, 9 months after the start of the study.
TABLE 1. Primers used for amplification and sequencing of variable regions C and E of the FCV capsid gene
Household RT primer PCR primer Sequence (5�–3�) Binding sitea
A A2 (reverse) A1 (forward) CCCTTCGTCTTTCAGGCCAACCG 6406–6428C A2 (reverse) A2 (reverse) CCTCGCCAATCCCAGTGTAGCC 6934–6913D P2 (reverse) P1 (forward) CCGTTTGTGTTTCAAGCAAACCG 6406–6428E P2 (reverse) P2 (reverse) CCTCACCTATACCAGTGTAACC 6934–6913B A2/P2 A1/A2 or P1/P2 As above
a Binding sites relate to FCV strain F9.
1962 COYNE ET AL. J. VIROL.
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
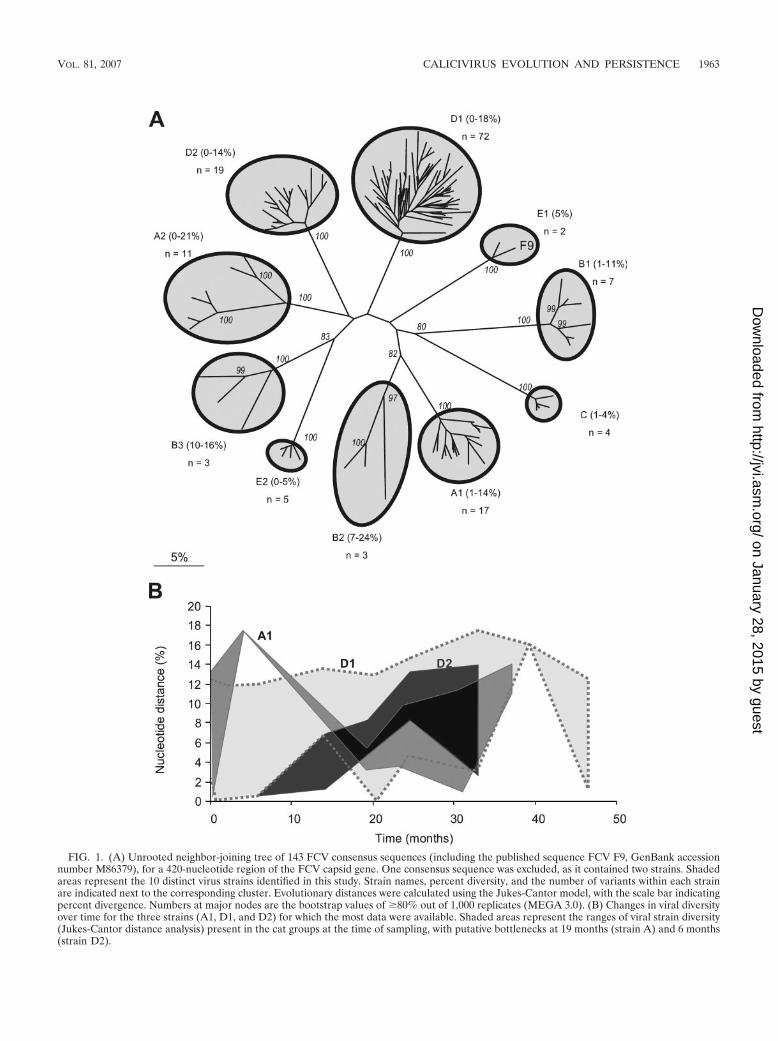
FIG. 1. (A) Unrooted neighbor-joining tree of 143 FCV consensus sequences (including the published sequence FCV F9, GenBank accessionnumber M86379), for a 420-nucleotide region of the FCV capsid gene. One consensus sequence was excluded, as it contained two strains. Shadedareas represent the 10 distinct virus strains identified in this study. Strain names, percent diversity, and the number of variants within each strainare indicated next to the corresponding cluster. Evolutionary distances were calculated using the Jukes-Cantor model, with the scale bar indicatingpercent divergence. Numbers at major nodes are the bootstrap values of �80% out of 1,000 replicates (MEGA 3.0). (B) Changes in viral diversityover time for the three strains (A1, D1, and D2) for which the most data were available. Shaded areas represent the ranges of viral strain diversity(Jukes-Cantor distance analysis) present in the cat groups at the time of sampling, with putative bottlenecks at 19 months (strain A) and 6 months(strain D2).
VOL. 81, 2007 CALICIVIRUS EVOLUTION AND PERSISTENCE 1963
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

RESULTS
Viral prevalence. The mean prevalence of FCV observedfrom the five households sampled ranged from 6 to 75% (over-all mean FCV prevalence, 35%): further details of prevalencedynamics are reported elsewhere (8). Within each household,cats were broadly divided into three categories: those that shedvirus consistently for prolonged periods (“consistent shedders”),those that shed intermittently (“intermittent shedders”), andthose that appeared to be resistant to infection over the studyperiod (“nonshedders”) (8).
Strain identification and variation. Phylogenetic analysisbased on consensus sequences of variable immunodominantregions of the FCV capsid gene identified a total of 10 distinctviral strains among the five households. Each of these strainswas separated by distance values of 26 to 51% (Fig. 1A). Theiruncorrected distance values (which allow direct comparisonwith previous studies) ranged from 21 to 39% (data not shown),as previously reported for distinct epidemiologically unrelatedstrains of FCV (44). Within households, one to three distinctstrains of FCV were identified. The maximum variability withineach strain ranged from 5 to 24% (Fig. 1A) (with uncorrecteddistances of 4 to 20% [data not shown]). This is in contrast toprevious studies where variability within a strain, during eitherdisease outbreaks or persistent infection, was generally relativelylow (up to 6% uncorrected distance) (44, 46). Viral strainspresent within households generally sustained considerable levelsof diversity over time (Fig. 1B), though an occasional reduction indiversity (i.e., a putative transmission bottleneck), followed by anincrease, also occurred (e.g., strain A1 at 19 months [Fig. 1B]). Ineach of two households (D and E), new strains were identifiedduring the course of the study. Virus variability in these twostrains increased from 0 to 5% (strain E2) (Fig. 1A) and from 0to 14% (strain D2) (Fig. 1A and B) over 6 and 27 months,respectively, showing that in this situation viral diversity tends toincrease over time.
Mechanisms of persistence. Having established the geneticidentities of the strains within each household, patterns ofevolution, both within individuals and at the population level,were characterized. A total of 130 partial FCV capsid consen-sus sequences were compared from 31 individual cats whichshed virus over prolonged periods (8). Within these individu-als, two mechanisms for this persistent virus shedding wereidentified. The first mechanism, characterized by mutation ac-cumulation in a given variant of a strain within an individual(“progressive evolution”), was only evident in four cats. Here,all consecutive consensus sequences isolated from an individ-ual cat clustered together phylogenetically (e.g., cat 4) (Fig.2A), consistently demonstrating low interisolate variability be-tween consecutive samples and clear evidence of temporalevolution (Fig. 2A and B, cat 4). Clonal samples from thisindividual cat showed that low interisolate variability was cou-pled with relatively high intraisolate variability along with lowbootstrap support for the clustering of clones from individualisolates at each time point (Fig. 2C, cat 4).
In contrast, for the remaining 27 cats, progressive evolutionwas interrupted by relatively high interisolate variability overshort periods of time, where consecutive isolates from individ-ual cats largely mixed together phylogenetically with virusesfrom other cats (e.g., cat 1 at 33 and 39 months and cat 6 at all
time points except 20 and 25 months) (Fig. 2A and B). Clonalsamples from these two individual cats demonstrated high in-terisolate variability, coupled with low intraisolate variability,with high bootstrap support for the clustering of clones fromindividual isolates at these time points (Fig. 2C, cats 1 and 6).This suggested a second mechanism of persistent infectionwithin individuals, in which cats were being periodically rein-fected at some time points from within the household (“se-quential reinfection”). These transmission events were eitherwith a diverse variant of the same strain (cats 1 and 6 [Fig. 2])or with a distinct strain (cat 1 at 14 months [Fig. 2]).
Interestingly, consensus and cloned sequence analyses showedthat at least one cat was infected with two distinct strains of virussimultaneously (e.g., cat 1 at 6 months [Fig. 2C]).
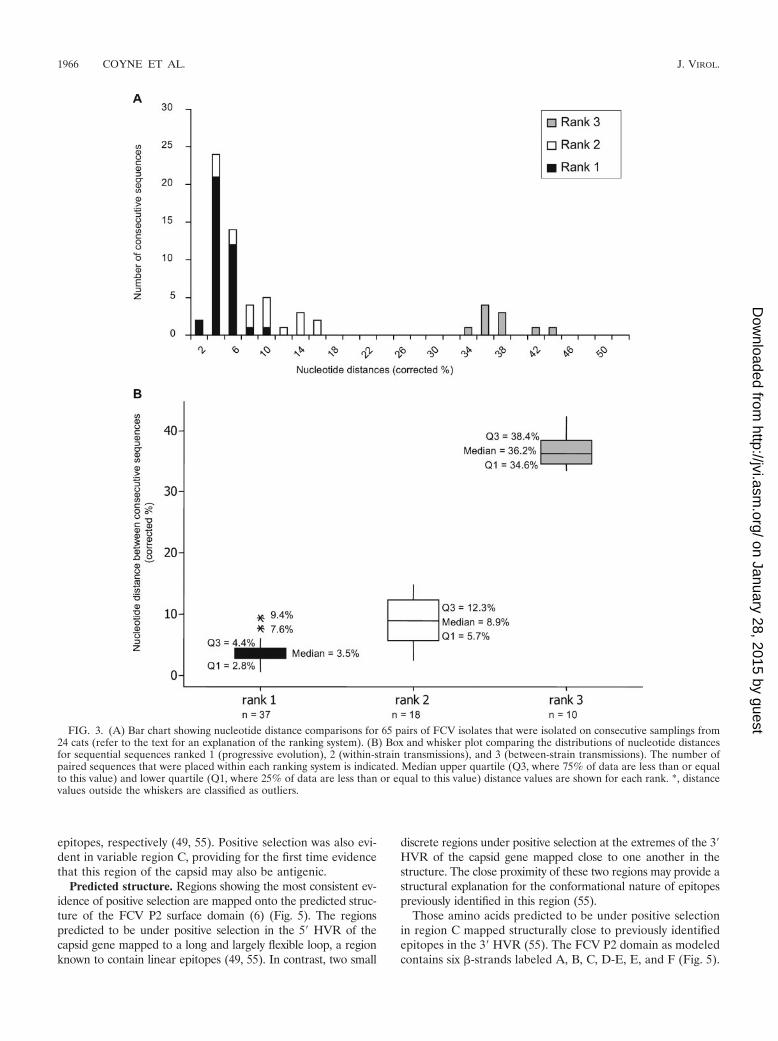
Further evidence was sought for these two mechanisms ofpersistence by ranking interisolate distances between consec-utive pairs of isolates from individual cats. Pairs were ranked as1 if they were more closely related to each other than to anyother viruses cocirculating in the household, suggestive of pro-gressive evolution of a given variant within an individual. Pairswere ranked as 2 if they were more closely related to otherviruses of the same strain cocirculating in the household thanto each other, suggestive of transmission of variants within astrain. Pairs were ranked as 3 if they were distinct strains,suggesting between-strain transmission (Fig. 3). Significant dif-ferences were found between all three categories (P � 0.001).For the four cats that we hypothesized to be persistently in-fected by progressive evolution, interisolate distances wereconsistently ranked one (e.g., cat 4 [Fig. 2c and 3]). In contrast,for the remaining 27 cats which we hypothesized to be persis-tently infected by the processes of progressive evolution as wellas sequential reinfection, their interisolate ranks were a mix-ture of 1, 2, and 3 (e.g., cats 1 and 6 [Fig. 2c and 3]).
Evolution rates. From these data it was possible to deter-mine two measures of FCV strain evolution rate: progressiveevolution firstly within individual cats (no transmission) andsecondly within the population (including transmission). Fortwo of the four cats putatively undergoing progressive evolu-tion of the same strain of virus (cat 4 [Fig. 2] and cat 2 [ref-erence 8]), evolution rates were 1.32 � 10�2 and 2.64 � 10�2
substitutions per nucleotide per year, respectively. For twostrains (D2 and E2) that were putatively introduced into theirhouseholds during the course of the study, thereby allowinganalyses of the founding sequence, evolution rates within thepopulation were 3.84 � 10�2 and 4.56 � 10�2 substitutions pernucleotide per year, respectively.
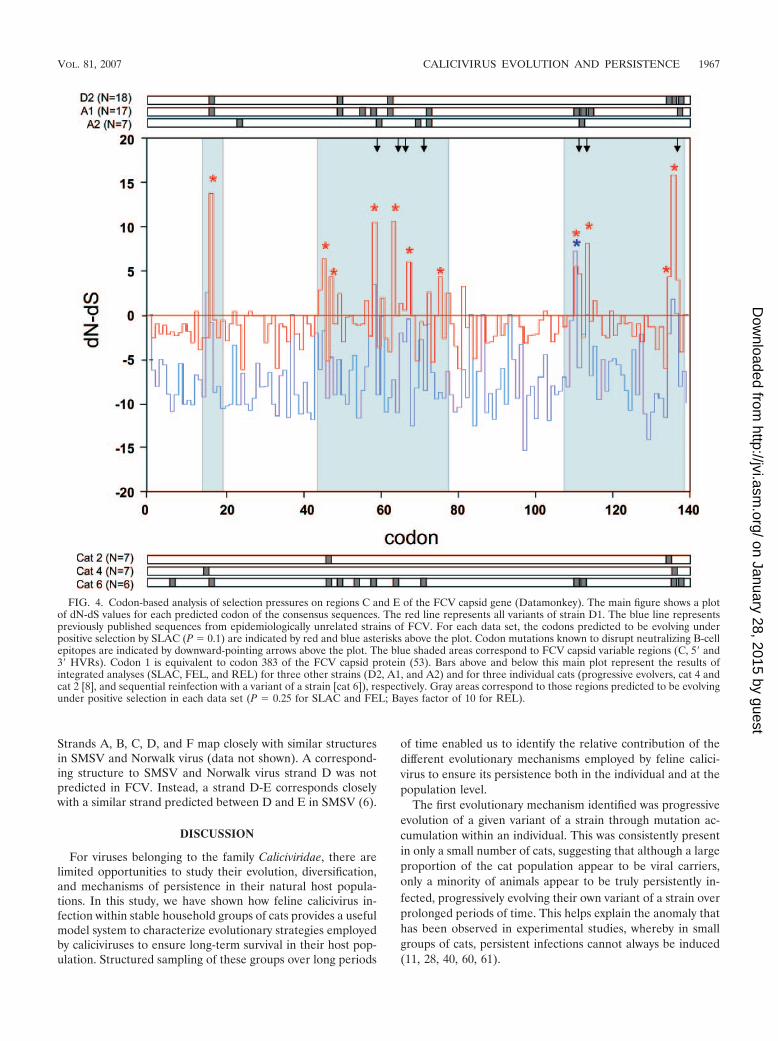
Analysis of positive selection. Patterns of evolution wereclearly associated with positive selection at known antigenicsites, suggesting an immune-mediated mechanism for viralevolution and persistence (Fig. 4). This was clear not only inthe small number of individuals progressively evolving virus(e.g., cats 2 and 4), but also in those cats undergoing variantreinfection (e.g., cat 6) and for all variants of a strain at thepopulation level (strains D2, A1, and A2) (Fig. 4). In contrast,when published sequences from epidemiologically unrelatedFCV strains were compared, there was little evidence for pos-itive selection (Fig. 4). Codons predicted to be under positiveselection mapped to the 5� and 3� hypervariable regions(HVRs) of region E of the FCV capsid (Fig. 4), which havebeen shown to contain linear and conformational B-cell
1964 COYNE ET AL. J. VIROL.
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
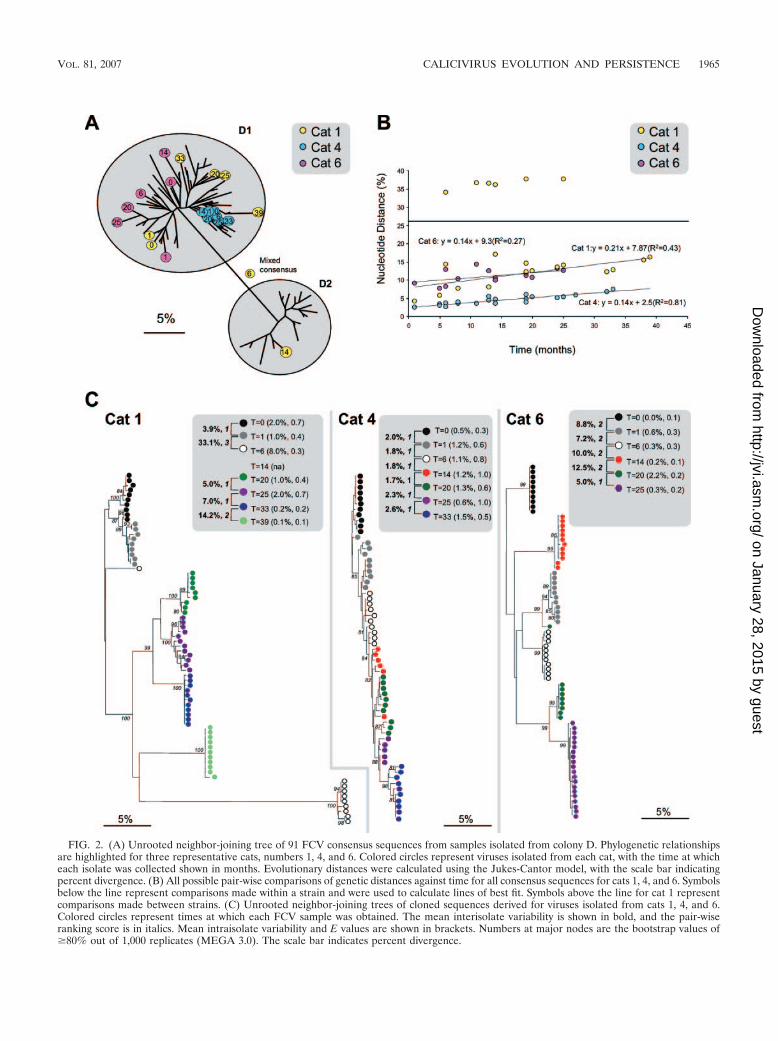
FIG. 2. (A) Unrooted neighbor-joining tree of 91 FCV consensus sequences from samples isolated from colony D. Phylogenetic relationshipsare highlighted for three representative cats, numbers 1, 4, and 6. Colored circles represent viruses isolated from each cat, with the time at whicheach isolate was collected shown in months. Evolutionary distances were calculated using the Jukes-Cantor model, with the scale bar indicatingpercent divergence. (B) All possible pair-wise comparisons of genetic distances against time for all consensus sequences for cats 1, 4, and 6. Symbolsbelow the line represent comparisons made within a strain and were used to calculate lines of best fit. Symbols above the line for cat 1 representcomparisons made between strains. (C) Unrooted neighbor-joining trees of cloned sequences derived for viruses isolated from cats 1, 4, and 6.Colored circles represent times at which each FCV sample was obtained. The mean interisolate variability is shown in bold, and the pair-wiseranking score is in italics. Mean intraisolate variability and E values are shown in brackets. Numbers at major nodes are the bootstrap values of�80% out of 1,000 replicates (MEGA 3.0). The scale bar indicates percent divergence.
VOL. 81, 2007 CALICIVIRUS EVOLUTION AND PERSISTENCE 1965
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
epitopes, respectively (49, 55). Positive selection was also evi-dent in variable region C, providing for the first time evidencethat this region of the capsid may also be antigenic.
Predicted structure. Regions showing the most consistent ev-idence of positive selection are mapped onto the predicted struc-ture of the FCV P2 surface domain (6) (Fig. 5). The regionspredicted to be under positive selection in the 5� HVR of thecapsid gene mapped to a long and largely flexible loop, a regionknown to contain linear epitopes (49, 55). In contrast, two small
discrete regions under positive selection at the extremes of the 3�HVR of the capsid gene mapped close to one another in thestructure. The close proximity of these two regions may provide astructural explanation for the conformational nature of epitopespreviously identified in this region (55).
Those amino acids predicted to be under positive selectionin region C mapped structurally close to previously identifiedepitopes in the 3� HVR (55). The FCV P2 domain as modeledcontains six �-strands labeled A, B, C, D-E, E, and F (Fig. 5).
FIG. 3. (A) Bar chart showing nucleotide distance comparisons for 65 pairs of FCV isolates that were isolated on consecutive samplings from24 cats (refer to the text for an explanation of the ranking system). (B) Box and whisker plot comparing the distributions of nucleotide distancesfor sequential sequences ranked 1 (progressive evolution), 2 (within-strain transmissions), and 3 (between-strain transmissions). The number ofpaired sequences that were placed within each ranking system is indicated. Median upper quartile (Q3, where 75% of data are less than or equalto this value) and lower quartile (Q1, where 25% of data are less than or equal to this value) distance values are shown for each rank. *, distancevalues outside the whiskers are classified as outliers.
1966 COYNE ET AL. J. VIROL.
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
Strands A, B, C, D, and F map closely with similar structuresin SMSV and Norwalk virus (data not shown). A correspond-ing structure to SMSV and Norwalk virus strand D was notpredicted in FCV. Instead, a strand D-E corresponds closelywith a similar strand predicted between D and E in SMSV (6).
DISCUSSION
For viruses belonging to the family Caliciviridae, there arelimited opportunities to study their evolution, diversification,and mechanisms of persistence in their natural host popula-tions. In this study, we have shown how feline calicivirus in-fection within stable household groups of cats provides a usefulmodel system to characterize evolutionary strategies employedby caliciviruses to ensure long-term survival in their host pop-ulation. Structured sampling of these groups over long periods
of time enabled us to identify the relative contribution of thedifferent evolutionary mechanisms employed by feline calici-virus to ensure its persistence both in the individual and at thepopulation level.
The first evolutionary mechanism identified was progressiveevolution of a given variant of a strain through mutation ac-cumulation within an individual. This was consistently presentin only a small number of cats, suggesting that although a largeproportion of the cat population appear to be viral carriers,only a minority of animals appear to be truly persistently in-fected, progressively evolving their own variant of a strain overprolonged periods of time. This helps explain the anomaly thathas been observed in experimental studies, whereby in smallgroups of cats, persistent infections cannot always be induced(11, 28, 40, 60, 61).
FIG. 4. Codon-based analysis of selection pressures on regions C and E of the FCV capsid gene (Datamonkey). The main figure shows a plotof dN-dS values for each predicted codon of the consensus sequences. The red line represents all variants of strain D1. The blue line representspreviously published sequences from epidemiologically unrelated strains of FCV. For each data set, the codons predicted to be evolving underpositive selection by SLAC (P � 0.1) are indicated by red and blue asterisks above the plot. Codon mutations known to disrupt neutralizing B-cellepitopes are indicated by downward-pointing arrows above the plot. The blue shaded areas correspond to FCV capsid variable regions (C, 5� and3� HVRs). Codon 1 is equivalent to codon 383 of the FCV capsid protein (53). Bars above and below this main plot represent the results ofintegrated analyses (SLAC, FEL, and REL) for three other strains (D2, A1, and A2) and for three individual cats (progressive evolvers, cat 4 andcat 2 [8], and sequential reinfection with a variant of a strain [cat 6]), respectively. Gray areas correspond to those regions predicted to be evolvingunder positive selection in each data set (P � 0.25 for SLAC and FEL; Bayes factor of 10 for REL).
VOL. 81, 2007 CALICIVIRUS EVOLUTION AND PERSISTENCE 1967
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

The second evolutionary mechanism, sequential reinfection,which in a number of cases was interspersed with periods ofprogressive evolution, accounted for the majority of animalsthat appeared to be persistently infected. In this situation in-dividual animals were undergoing reinfection from within thehousehold either with a diverse variant of the same strain orwith a distinct strain of virus. Reinfection with distinct strainsof FCV is consistent with previous experimental studies whichhave shown that cats do appear to eliminate virus and that theyremain susceptible to reinfection with a different strain (28,60). However, this is the first report where sequential reinfec-tion has been shown to occur with closely related variants ofthe same strain. This has important implications for both viralevolution and the epidemiology of the disease, particularlywithin groups of cats. Clearly, however, we cannot exclude thepossibility that the original virus may still be present in suchcats and is being out-competed at intervals by a new incomingvirus.
Mixed infection with two distinct strains within an individualcat was also identified in one household, and although putativemixed infections have previously been reported (47), this is thefirst time this has been confirmed for caliciviruses by analysis ofa cloned sequence. Such mixed infections are a prerequisite forthe generation of recombinant viral strains, which togetherwith the putative parent viruses have also been identified inthis household and have been reported in detail elsewhere(10). Recombination in other calicivirus genera has also beendescribed, but parental strains have not been identified (4, 25,26, 37, 58). Recombination between variants of the same strainmay also occur, but because of the close relationship between
variants within a strain, such events would be more difficult todetect.
This study also demonstrated the high levels of viral diversitythat can occur within a strain circulating within a populationover extended periods of time. Using a corrected substitutionmodel, levels of up to 24% (20% uncorrected) were observedwithin a strain within a household and up to 21% (19% un-corrected) was observed within individual animals that werepersistently infected over extended periods of time. This sug-gests that within the overlapping population of viral variantspresent, new strains of FCV appear to be emerging, presum-ably through a combination of the evolutionary mechanismsdescribed above. In cross-sectional studies, levels of diversity ofup to 16% (uncorrected distance values) have been reportedpreviously in some of these households (45). It is clear thatsuch closed or semiclosed communities of endemically infectedanimals provide ideal conditions for the generation of viralbiodiversity, and possible increased virulence, and may haveparallels in other caliciviruses.
How long these strains had been circulating in each house-hold and how this contributed to viral diversity are not known.However, in two colonies it appeared that a new strain wasintroduced during the course of the study, thus providing anatural opportunity to monitor viral evolution and diversifica-tion from a presumed single ancestor. In these two cases, virusvariability increased from 0 to 5% and from 0 to 14% over 6and 27 months, respectively. Thus, it appears that transmissionof a new strain of virus into an individual within a householdmay represent a bottleneck to virus variability. Such reductionsin variability may be caused either by low-dose virus transmis-
FIG. 5. Predicted structure of the FCV F9 capsid P2 surface domain (6), highlighting regions identified in this study that are predicted to beunder positive selection. The yellow region corresponds to the 5� HVR, known to contain linear epitopes. The magenta and green regionscorrespond to the left and right boundaries of the 3� HVR, known to contain conformational epitopes. The cyan region contains variable regionC and maps close to the 3� HVR. The FCV P2 domain as modeled contains six �-strands, labeled A, B, C, D-E, E, and F.
1968 COYNE ET AL. J. VIROL.
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

sion or by preferential selection of the fittest variant from theviral population at the time of transmission. Reductions in viraldiversity by transmission bottlenecks have not previously beenreported for caliciviruses but have been shown during naturaltransmission events for other RNA viruses (62, 65).
This study also allowed the calculation of viral evolutionrates for the partial capsid region analyzed; the rate appearedto be higher (3.84 � 10�2 to 4.56 � 10�2 substitutions pernucleotide per year) for a strain circulating within a household(i.e., including transmission) compared to that for an individ-ual cat (1.32 � 10�2 to 2.64 � 10�2 substitutions per nucleo-tide per year, i.e., progressive evolver, no transmission). Therate at which feline calicivirus evolved within an individual catwas lower than has been previously reported (48), althoughthis latter study was based on the 5� hypervariable regionalone. There are no other estimates available for the rate atwhich caliciviruses evolve within populations, although esti-mates have been made for other RNA viruses (24). Evolutionrates depend on a number of factors, including the region ofthe genome selected and the various selection pressures thatmay be operating. In this study, the evolution rates for felinecalicivirus appear to be relatively high compared to other RNAviruses, which may be in part a reflection of the impact ofpositive selection acting on these regions of the genome.
Positive selection in the immunodominant 5� and 3� hyper-variable regions of the capsid gene was found within strains atboth the individual and population level. Positive selection inthe 5� HVR has been shown previously in an individual in-fected cat, where it was also demonstrated that such changesalter the neutralization profile (48). In other caliciviruses, suchas human norovirus, immune-mediated positive selection inthe P2 domain (analogous to the hypervariable regions of theFCV capsid gene) has also been suggested as a mechanism forfacilitating persistent infection in an individual (35).
Positive selection within a strain at the population level hasnot previously been reported but can probably occur becauseof the closely matched immune response within the endemi-cally infected host population. In contrast, there was littleevidence of positive selection between distinct FCV strains atthe population level, which is similar to the situation in otherRNA viruses (31). It has been suggested that viral evolution atthe population level may be a reflection of the demographicand spatial history of transmission within such populationsrather than positive immune selection (23, 51). However, suchconclusions may to some extent be a consequence of the sam-pling strategy used (20). Where in-depth sampling of hostpopulations is carried out, as in this study, the intermediateevolutionary steps associated with positive immune selectioncan be clearly identified.
Interestingly, evidence of positive selection was also found inregion C of the capsid, which has not previously been reported,and may suggest a possible antigenic role for this region. Pre-vious studies have failed to identify any linear epitopes inregion C (49), suggesting that if this region is antigenic it maycontain either conformational B-cell or T-cell epitopes. Fur-ther support for the conformational nature of such epitopeswas found in the predicted structural studies, where the codonspredicted to be under positive selection within this regionmapped structurally close to other known conformationalepitopes in the 3� HVR (49, 55).
This study has generated insights into the evolutionarymechanisms of FCV persistence and diversification, which arelikely to be generalizable to other caliciviruses. It appears thattrue long-term persistent infection in an individual, character-ized by continuous progressive evolution, is relatively rare andthat the majority of apparent carriers in the population areundergoing a combination of progressive evolution and cyclesof reinfection. In human norovirus infections, persistently in-fected people also appear to be relatively rare, and populationpersistence is largely thought to be through environmentalcontamination and epidemic spread (14, 56). It may be, how-ever, that in some situations the two viruses employ a similarrange of evolutionary strategies and that reinfection with vari-ants of the same strain or with a different strain of norovirusmight be more common than was previously thought. Never-theless, the minority of individuals that are true carriers mayprovide an important fail-safe mechanism to ensure the virus’slong-term survival within a population. It will be critical, there-fore, to identify these individuals to control disease and furtherunderstand the evolution of the Caliciviridae.
ACKNOWLEDGMENTS
This work was funded by the PetPlan Charitable Trust.We thank the cat owners for allowing us to sample their cats, Chris
McCracken and Ruth Ryvar for technical assistance, and SergeiKosakovsky Pond (University of California), Dan Haydon (Universityof Glasgow), and Bryan Grenfell (Pennsylvania State University) forhelpful discussions.
REFERENCES
1. Bannasch, M. J., and J. E. Foley. 2005. Epidemiologic evaluation of multiplerespiratory pathogens in cats in animal shelters. J. Feline Med. Surg. 7:109–119.
2. Begon, M., J. L. Harper, and C. R. Townsend. 1990. Ecology: individuals,populations and communities, 2nd ed. Blackwell Scientific Publications,Cambridge, Mass.
3. Binns, S. H., S. Dawson, A. J. Speakman, L. E. Cuevas, C. A. Hart, C. J.Gaskell, K. L. Morgan, and R. M. Gaskell. 2000. A study of feline upperrespiratory tract disease with reference to prevalence and risk factors forinfection with feline calicivirus and feline herpesvirus. J. Feline Med. Surg.2:123–133.
4. Bull, R. A., G. S. Hansman, L. E. Clancy, M. M. Tanaka, W. D. Rawlinson,and P. A. White. 2005. Norovirus recombination in ORF1/ORF2 overlap.Emerg. Infect. Dis. 11:1079–1085.
5. Carter, M. J., I. D. Milton, J. Meanger, M. Bennett, R. M. Gaskell, and P. C.Turner. 1992. The complete nucleotide sequence of a feline calicivirus.Virology 190:443–448.
6. Chen, R., J. D. Neill, M. K. Estes, and B. V. Prasad. 2006. X-ray structure ofa native calicivirus: structural insights into antigenic diversity and host spec-ificity. Proc. Natl. Acad. Sci. USA 103:8048–8053.
7. Coutts, A. J., S. Dawson, K. Willoughby, and R. M. Gaskell. 1994. Isolationof feline respiratory viruses from clinically healthy cats at UK cat shows. Vet.Rec. 135:555–556.
8. Coyne, K. P., S. Dawson, A. D. Radford, P. J. Cripps, C. J. Porter, C. M.McCracken, and R. M. Gaskell. 2006. Long term analysis of feline calicivirusprevalence and viral shedding patterns in naturally infected colonies ofdomestic cats. Vet. Microbiol. 118:12–25.
9. Coyne, K. P., B. R. D. Jones, A. Kipar, J. Chantrey, C. J. Porter, P. Barber,S. Dawson, R. M. Gaskell, and A. D. Radford. 2006. Lethal outbreak ofdisease associated with feline calicivirus infection in cats. Vet. Rec. 158:544–550.
10. Coyne, K. P., F. C. Reed, C. J. Porter, S. Dawson, R. M. Gaskell, and A. D.Radford. 2006. Recombination of feline calicivirus within an endemically-infected cat colony. J. Gen. Virol. 87:921–926.
11. Dawson, S., N. R. Smyth, M. Bennett, R. M. Gaskell, C. M. McCracken, A.Brown, and C. J. Gaskell. 1991. Effect of primary-stage feline immunodefi-ciency virus infection on subsequent feline calicivirus vaccination and chal-lenge in cats. AIDS 5:747–750.
12. DeLano, W. L. 2002. The PyMOL molecular graphics system. Scientific, SanCarlos, Calif.
VOL. 81, 2007 CALICIVIRUS EVOLUTION AND PERSISTENCE 1969
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

13. Deveraux, J., P. Haeberli, and O. Smithies. 1984. A comprehensive set ofsequence analysis programs for the VAX. Nucleic Acids Res. 12:387–395.
14. de Wit, M. A., M. P. Koopmans, L. M. Kortbeek, N. J. van Leeuwen, J. Vinje,and Y. T. van Duynhoven. 2001. Etiology of gastroenteritis in sentinel gen-eral practices in The Netherlands. Clin. Infect. Dis. 33:280–288.
15. Dingle, K. E. 2004. Mutation in a Lordsdale norovirus epidemic strain as apotential indicator of transmission routes. J. Clin. Microbiol. 42:3950–3957.
16. Fankhauser, R. L., J. S. Noel, S. S. Monroe, T. Ando, and R. I. Glass. 1998.Molecular epidemiology of “Norwalk-like viruses” in outbreaks of gastro-enteritis in the United States. J. Infect. Dis. 178:1571–1578.
17. Forrester, N. L., B. Boag, S. R. Moss, S. L. Turner, R. C. Trout, P. J. White,P. J. Hudson, and E. A. Gould. 2003. Long-term survival of New Zealandrabbit haemorrhagic disease virus RNA in wild rabbits, revealed by RT-PCRand phylogenetic analysis. J. Gen. Virol. 84:3079–3086.
18. Gaskell, R. M., A. D. Radford, and S. Dawson. 2006. Feline infectiousrespiratory disease, p. 145–154. In E. A. Chandler, C. J. Gaskell, and R. M.Gaskell (ed.), Feline medicine and therapeutics, 3rd ed. Blackwell Publish-ing, Ames, Iowa.
19. Green, K. Y., T. Ando, M. S. Balayan, T. Berke, I. N. Clarke, M. K. Estes,D. O. Matson, S. Nakata, J. D. Neill, M. J. Studdert, and H. J. Thiel. 2000.Taxonomy of the caliciviruses. J. Infect. Dis. 181(Suppl. 2):S322–S330.
20. Grenfell, B. T., O. G. Pybus, J. R. Gog, J. L. Wood, J. M. Daly, J. A.Mumford, and E. C. Holmes. 2004. Unifying the epidemiological and evo-lutionary dynamics of pathogens. Science 303:327–332.
21. Harbour, D. A., P. E. Howard, and R. M. Gaskell. 1991. Isolation of felinecalicivirus and feline herpesvirus from domestic cats 1980 to 1989. Vet. Rec.128:77–80.
22. Helps, C. R., P. Lait, A. Damhuis, U. Bjornehammar, D. Bolta, C. Brovida,L. Chabanne, H. Egberink, G. Ferrand, A. Fontbonne, M. G. Pennisi, T.Gruffydd-Jones, D. Gunn-Moore, K. Hartmann, H. Lutz, E. Malandain, K.Mostl, C. Stengel, D. A. Harbour, and E. A. Graat. 2005. Factors associatedwith upper respiratory tract disease caused by feline herpesvirus, felinecalicivirus, Chlamydophila felis and Bordetella bronchiseptica in cats: expe-rience from 218 European catteries. Vet. Rec. 156:669–673.
23. Holmes, E. C. 2004. The phylogeography of human viruses. Mol. Ecol.13:745–756.
24. Jenkins, G. M., A. Rambaut, O. G. Pybus, and E. C. Holmes. 2002. Rates ofmolecular evolution in RNA viruses: a quantitative phylogenetic analysis. J.Mol. Evol. 54:156–165.
25. Jiang, X., C. Espul, W. M. Zhong, H. Cuello, and D. O. Matson. 1999.Characterization of a novel human calicivirus that may be a naturally occur-ring recombinant. Arch. Virol. 144:2377–2387.
26. Katayama, K., T. Miyoshi, K. Uchino, T. Oka, T. Tanaka, N. Takeda, andG. S. Hansman. 2004. Novel recombinant sapovirus. Emerg. Infect. Dis.10:1874–1876.
27. Knowles, J. O., S. Dawson, R. M. Gaskell, C. J. Gaskell, and C. E. Harvey.1990. Neutralisation patterns among recent British and North Americanfeline calicivirus isolates from different clinical origins. Vet. Rec. 127:125–127.
28. Knowles, J. O., F. McArdle, S. Dawson, S. D. Carter, C. J. Gaskell, and R. M.Gaskell. 1991. Studies on the role of feline calicivirus in chronic stomatitis incats. Vet. Microbiol. 27:205–219.
29. Kreutz, L. C., R. P. Johnson, and B. S. Seal. 1998. Phenotypic and genotypicvariation of feline calicivirus during persistent infection of cats. Vet. Micro-biol. 59:229–236.
30. Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: integrated software formolecular evolutionary genetics analysis and sequence alignment. BriefBioinform. 5:150–163.
31. Lemey, P., I. Derdelinckx, A. Rambaut, K. Van Laethem, S. Dumont, S.Vermeulen, E. Van Wijngaerden, and A. M. Vandamme. 2005. Molecularfootprint of drug-selective pressure in a human immunodeficiency virustransmission chain. J. Virol. 79:11981–11989.
32. McCarthy, M., M. K. Estes, and K. C. Hyams. 2000. Norwalk-like virusinfection in military forces: epidemic potential, sporadic disease, and thefuture direction of prevention and control efforts. J. Infect. Dis. 181(Suppl.2):S387–S391.
33. Moss, S. R., S. L. Turner, R. C. Trout, P. J. White, P. J. Hudson, A. Desai,M. Armesto, N. L. Forrester, and E. A. Gould. 2002. Molecular epidemiologyof Rabbit haemorrhagic disease virus. J. Gen. Virol. 83:2461–2467.
34. Neill, J. D. 1992. Nucleotide sequence of the capsid protein gene of twoserotypes of San Miguel sea lion virus: identification of conserved and non-conserved amino acid sequences among calicivirus capsid proteins. VirusRes. 24:211–222.
35. Nilsson, M., K. O. Hedlund, M. Thorhagen, G. Larson, K. Johansen, A.Ekspong, and L. Svensson. 2003. Evolution of human calicivirus RNA invivo: accumulation of mutations in the protruding P2 domain of the capsidleads to structural changes and possibly a new phenotype. J. Virol. 77:13117–13124.
36. Noel, J. S., R. L. Fankhauser, T. Ando, S. S. Monroe, and R. I. Glass. 1999.Identification of a distinct common strain of “Norwalk-like viruses” having aglobal distribution. J. Infect. Dis. 179:1334–1344.
37. Oliver, S. L., D. W. Brown, J. Green, and J. C. Bridger. 2004. A chimeric
bovine enteric calicivirus: evidence for genomic recombination in genogroupIII of the Norovirus genus of the Caliciviridae. Virology 326:231–239.
38. Page, R. D. 1996. TreeView: an application to display phylogenetic trees onpersonal computers. Comput. Appl. Biosci. 12:357–358.
39. Pedersen, N. C., J. B. Elliott, A. Glasgow, A. Poland, and K. Keel. 2000. Anisolated epizootic of hemorrhagic-like fever in cats caused by a novel andhighly virulent strain of feline calicivirus. Vet. Microbiol. 73:281–300.
40. Pedersen, N. C., and K. F. Hawkins. 1995. Mechanisms for persistence ofacute and chronic feline calicivirus infections in the face of vaccination. Vet.Microbiol. 47:141–156.
41. Pond, S. L., and S. D. Frost. 2005. Datamonkey: rapid detection of selectivepressure on individual sites of codon alignments. Bioinformatics 21:2531–2533.
42. Povey, R. C., R. C. Wardley, and H. Jessen. 1973. Feline picornavirus infec-tion: the in vivo carrier state. Vet. Rec. 92:224–229.
43. Prasad, B. V., M. E. Hardy, T. Dokland, J. Bella, M. G. Rossmann, andM. K. Estes. 1999. X-ray crystallographic structure of the Norwalk viruscapsid. Science 286:287–290.
44. Radford, A. D., M. Bennett, F. McArdle, S. Dawson, P. C. Turner, M. A.Glenn, and R. M. Gaskell. 1997. The use of sequence analysis of a felinecalicivirus (FCV) hypervariable region in the epidemiological investigationof FCV related disease and vaccine failures. Vaccine 15:1451–1458.
45. Radford, A. D., S. Dawson, R. Ryvar, K. Coyne, D. R. Johnson, M. B. Cox,E. F. Acke, D. D. Addie, and R. M. Gaskell. 2003. High genetic diversity ofthe immunodominant region of the feline calicivirus capsid gene in endem-ically infected cat colonies. Virus Genes 27:145–155.
46. Radford, A. D., L. Sommerville, R. Ryvar, M. B. Cox, D. R. Johnson, S.Dawson, and R. M. Gaskell. 2001. Endemic infection of a cat colony with afeline calicivirus closely related to an isolate used in live attenuated vaccines.Vaccine 19:4358–4362.
47. Radford, A. D., L. M. Sommerville, S. Dawson, A. M. Kerins, R. Ryvar, andR. M. Gaskell. 2001. Molecular analysis of isolates of feline calicivirus froma population of cats in a rescue shelter. Vet. Rec. 149:477–481.
48. Radford, A. D., P. C. Turner, M. Bennett, F. McArdle, S. Dawson, M. A.Glenn, R. A. Williams, and R. M. Gaskell. 1998. Quasispecies evolution of ahypervariable region of the feline calicivirus capsid gene in cell culture andin persistently infected cats. J. Gen. Virol. 79:1–10.
49. Radford, A. D., K. Willoughby, S. Dawson, C. McCracken, and R. M.Gaskell. 1999. The capsid gene of feline calicivirus contains linear B-cellepitopes in both variable and conserved regions. J. Virol. 73:8496–8502.
50. Rambaut, A. 2000. Estimating the rate of molecular evolution: incorporatingnon-contemporaneous sequences into maximum likelihood phylogenies.Bioinformatics 16:395–399.
51. Rambaut, A., D. Posada, K. A. Crandall, and E. C. Holmes. 2004. The causesand consequences of HIV evolution. Nat. Rev. Genet. 5:52–61.
52. Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: anautomated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385.
53. Seal, B. S., J. F. Ridpath, and W. L. Mengeling. 1993. Analysis of felinecalicivirus capsid protein genes: identification of variable antigenic determi-nant regions of the protein. J. Gen. Virol. 74:2519–2524.
54. Simpson, E. H. 1949. Measurement of diversity. Nature 163:688.55. Tohya, Y., N. Yokoyama, K. Maeda, Y. Kawaguchi, and T. Mikami. 1997.
Mapping of antigenic sites involved in neutralization on the capsid protein offeline calicivirus. J. Gen. Virol. 78:303–305.
56. Tompkins, D. S., M. J. Hudson, H. R. Smith, R. P. Eglin, J. G. Wheeler,M. M. Brett, R. J. Owen, J. S. Brazier, P. Cumberland, V. King, and P. E.Cook. 1999. A study of infectious intestinal disease in England: microbio-logical findings in cases and controls. Commun. Dis. Public Health 2:108–113.
57. van Duynhoven, Y. T., C. M. de Jager, L. M. Kortbeek, H. Vennema, M. P.Koopmans, F. van Leusden, W. H. van der Poel, and M. J. van den Broek.2005. A one-year intensified study of outbreaks of gastroenteritis in TheNetherlands. Epidemiol. Infect. 133:9–21.
58. Vidal, R., P. Roessler, V. Solari, J. Vollaire, X. Jiang, D. O. Matson, N.Mamani, V. Prado, and M. L. O’Ryan. 2006. Novel recombinant noroviruscausing outbreaks of gastroenteritis in Santiago, Chile. J. Clin. Microbiol.44:2271–2275.
59. Wardley, R. C. 1976. Feline calicivirus carrier state. A study of the host/virusrelationship. Arch. Virol. 52:243–249.
60. Wardley, R. C., and R. C. Povey. 1977. The clinical disease and patterns ofexcretion associated with three different strains of feline caliciviruses. Res.Vet. Sci. 23:7–14.
61. Wardley, R. C., and R. C. Povey. 1977. The pathology and sites of persistenceassociated with three different strains of feline calicivirus. Res. Vet. Sci.23:15–19.
62. Weiner, A. J., M. M. Thaler, K. Crawford, K. Ching, J. Kansopon, D. Y.Chien, J. E. Hall, F. Hu, and M. Houghton. 1993. A unique, predominanthepatitis C virus variant found in an infant born to a mother with multiplevariants. J. Virol. 67:4365–4368.
63. White, P. J., R. C. Trout, S. R. Moss, A. Desai, M. Armesto, N. L. Forrester, E. A.Gould, and P. J. Hudson. 2004. Epidemiology of Rabbit haemorrhagic disease
1970 COYNE ET AL. J. VIROL.
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

virus in the United Kingdom: evidence for seasonal transmission by both viru-lent and avirulent modes of infection. Epidemiol. Infect. 132:555–567.
64. Widdowson, M. A., E. H. Cramer, L. Hadley, J. S. Bresee, R. S. Beard, S. N.Bulens, M. Charles, W. Chege, E. Isakbaeva, J. G. Wright, E. Mintz, D.Forney, J. Massey, R. I. Glass, and S. S. Monroe. 2004. Outbreaks of acutegastroenteritis on cruise ships and on land: identification of a predominant
circulating strain of norovirus—United States, 2002. J. Infect. Dis. 190:27–36.
65. Wolinsky, S. M., C. M. Wike, B. T. Korber, C. Hutto, W. P. Parks, L. L.Rosenblum, K. J. Kunstman, M. R. Furtado, and J. L. Munoz. 1992. Selec-tive transmission of human immunodeficiency virus type-1 variants frommothers to infants. Science 255:1134–1137.
VOL. 81, 2007 CALICIVIRUS EVOLUTION AND PERSISTENCE 1971
on January 28, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
Related Documents











