Evidence of Tubular Hypoxia in the Early Phase in the Remnant Kidney Model KRISSANAPONG MANOTHAM,* TETSUHIRO TANAKA,* MAKIKO MATSUMOTO,* TAKAMOTO OHSE,* TOSHIO MIYATA, † REIKO INAGI, † KIYOSHI KUROKAWA, † TOSHIRO FUJITA,* and MASAOMI NANGAKU* *Division of Nephrology and Endocrinology, University of Tokyo School of Medicine, Tokyo, Japan; and † Molecular and Cellular Nephrology, Institute of Medical Sciences and Department of Medicine, Tokai University School of Medicine, Tokyo, Japan. Abstract. The remnant kidney model is a mainstay in the study of progressive renal disease. The earliest changes in this model result from glomerular hemodynamic alterations. Given that progressive renal disease is the result of subsequent interstitial damage initiated by undetermined pathogenic factors, the au- thors investigated the role of hypoxia as a pathogenic factor in tubulointerstitial damage after renal ablation in rats. Cortical tissue hypoxia in the early phase (4 and 7 d) in remnant kidney rats, sham-operated rats, and animals treated with the angio- tensin II receptor blocker (ARB) olmesartan (10 mg/kg per d) was assessed by uptake of a hypoxic probe, pimonidazole, expression of HIF-1, and by increased transcription of hy- poxia-responsive genes. Physiologic perfusion status of the postglomerular peritubular capillary network was evaluated by lectin perfusion and Hoechst 33342 diffusion techniques. Re- sults showed that the number of hypoxic tubules was markedly increased 4 and 7 d after nephron loss. These findings ante- dated any histologic evidence of tubulointerstitial damage. The hypoxic state persisted until interstitial damage developed. These results were confirmed using HIF-1 immunoprecipita- tion and increase of hypoxia-responsive genes. Pathologic studies of the vasculature demonstrated significant functional changes that generated a hypoxic milieu. ARB treatment pre- vented vascular changes and ameliorated tubular hypoxia. These results suggest that the initial tubulointerstitial hypoxia in remnant kidney model plays a pathogenic role in the sub- sequent development of tubulointerstitial injury. The initial hypoxia in this model was dependent on activation of the renin-angiotensin system and hemodynamic alterations after nephron loss. The remnant kidney is a representative model in the study of progressive renal disease. The earliest response in this model consists of intrarenal vasodilation with relatively poor dilation of efferent arterioles, which in turn generates glomerular hy- perfiltration and hypertension (1). Glomerular hyperfiltration, which depends on activation of the renin-angiotensin system (RAS), eventually leads to glomerulosclerosis (1). However, the deterioration of renal function is well correlated with the progressive interstitial damage, which evolves in the late phase, rather than glomerular lesions (2– 4), and the mecha- nism(s) that initiate(s) tubulointerstitial injury in the context of early hemodynamic alterations remain(s) to be elucidated. Chronic tissue hypoxia is regarded as a contributing factor in the progression of renal failure (5). Loss of peritubular capil- laries in the late phase of various disease models has been recently reported (6 –11), giving indirect evidence of renal hypoxia. Hypoxia itself can initiate the inflammatory process and eventually lead to tissue fibrosis (12–17). In a remnant kidney model, Kang et al. (11) reported that the loss of peri- tubular capillaries occurred around the second week, at which time severe tubulointerstitial injury started to develop. Preven- tion of this loss by VEGF administration abolished subsequent tubulointerstitial damage. These data strongly support the no- tion that hypoxia might be a cause, rather than an epiphenom- enon, of tubulointerstitial damage. However, evidence of hyp- oxia in the early phase of this representative model before the development of histologic damage remains to be clarified. We have investigated the hypothesis that hemodynamic alterations might lead to tubulointerstitial hypoxia in the very early phase in this model, which might in turn induce tubulointerstitial damage and lead to eventual kidney failure. To prove this hypothesis, we designed the current study to look for evidence of hypoxia within the first week after renal mass reduction. At this time point, hemodynamic alterations are well established but tubulointerstitial damage is negligible (18). Here, findings showed that many tubules suffer from hypoxic conditions in the early phase of remnant kidney. Furthermore, hypoxia in the tubulointerstitial compartment preceded traditional evidence of tubulointerstitial damage and persisted until the damage developed. Investigations utilizing angiotensin II receptor blockage (ARB) suggested that the tubular hypoxia was dependent on the activation of RAS. Received July 29, 2003. Accepted February 26, 2004. Correspondence to Dr. Masaomi Nangaku, University of Tokyo School of Medicine, Division of Nephrology and Endocrinology, 7–3-1 Hongo Bunkyo- Ku, Tokyo, Japan. Phone: 03-3815-5411 ext. 33004; Fax: 03-5800-8806; E-mail [email protected] 1046-6673/1505-1277 Journal of the American Society of Nephrology Copyright © 2004 by the American Society of Nephrology DOI: 10.1097/01.ASN.0000125614.35046.10 J Am Soc Nephrol 15: 1277–1288, 2004

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Evidence of Tubular Hypoxia in the Early Phase in theRemnant Kidney Model
KRISSANAPONG MANOTHAM,* TETSUHIRO TANAKA,* MAKIKO MATSUMOTO,*TAKAMOTO OHSE,* TOSHIO MIYATA,† REIKO INAGI,† KIYOSHI KUROKAWA,†
TOSHIRO FUJITA,* and MASAOMI NANGAKU**Division of Nephrology and Endocrinology, University of Tokyo School of Medicine, Tokyo, Japan; and†Molecular and Cellular Nephrology, Institute of Medical Sciences and Department of Medicine, TokaiUniversity School of Medicine, Tokyo, Japan.
Abstract. The remnant kidney model is a mainstay in the studyof progressive renal disease. The earliest changes in this modelresult from glomerular hemodynamic alterations. Given thatprogressive renal disease is the result of subsequent interstitialdamage initiated by undetermined pathogenic factors, the au-thors investigated the role of hypoxia as a pathogenic factor intubulointerstitial damage after renal ablation in rats. Corticaltissue hypoxia in the early phase (4 and 7 d) in remnant kidneyrats, sham-operated rats, and animals treated with the angio-tensin II receptor blocker (ARB) olmesartan (10 mg/kg per d)was assessed by uptake of a hypoxic probe, pimonidazole,expression of HIF-1�, and by increased transcription of hy-poxia-responsive genes. Physiologic perfusion status of thepostglomerular peritubular capillary network was evaluated bylectin perfusion and Hoechst 33342 diffusion techniques. Re-
sults showed that the number of hypoxic tubules was markedlyincreased 4 and 7 d after nephron loss. These findings ante-dated any histologic evidence of tubulointerstitial damage. Thehypoxic state persisted until interstitial damage developed.These results were confirmed using HIF-1� immunoprecipita-tion and increase of hypoxia-responsive genes. Pathologicstudies of the vasculature demonstrated significant functionalchanges that generated a hypoxic milieu. ARB treatment pre-vented vascular changes and ameliorated tubular hypoxia.These results suggest that the initial tubulointerstitial hypoxiain remnant kidney model plays a pathogenic role in the sub-sequent development of tubulointerstitial injury. The initialhypoxia in this model was dependent on activation of therenin-angiotensin system and hemodynamic alterations afternephron loss.
The remnant kidney is a representative model in the study ofprogressive renal disease. The earliest response in this modelconsists of intrarenal vasodilation with relatively poor dilationof efferent arterioles, which in turn generates glomerular hy-perfiltration and hypertension (1). Glomerular hyperfiltration,which depends on activation of the renin-angiotensin system(RAS), eventually leads to glomerulosclerosis (1). However,the deterioration of renal function is well correlated with theprogressive interstitial damage, which evolves in the latephase, rather than glomerular lesions (2–4), and the mecha-nism(s) that initiate(s) tubulointerstitial injury in the context ofearly hemodynamic alterations remain(s) to be elucidated.
Chronic tissue hypoxia is regarded as a contributing factor inthe progression of renal failure (5). Loss of peritubular capil-laries in the late phase of various disease models has beenrecently reported (6–11), giving indirect evidence of renal
hypoxia. Hypoxia itself can initiate the inflammatory processand eventually lead to tissue fibrosis (12–17). In a remnantkidney model, Kang et al. (11) reported that the loss of peri-tubular capillaries occurred around the second week, at whichtime severe tubulointerstitial injury started to develop. Preven-tion of this loss by VEGF administration abolished subsequenttubulointerstitial damage. These data strongly support the no-tion that hypoxia might be a cause, rather than an epiphenom-enon, of tubulointerstitial damage. However, evidence of hyp-oxia in the early phase of this representative model before thedevelopment of histologic damage remains to be clarified. Wehave investigated the hypothesis that hemodynamic alterationsmight lead to tubulointerstitial hypoxia in the very early phasein this model, which might in turn induce tubulointerstitialdamage and lead to eventual kidney failure.
To prove this hypothesis, we designed the current study tolook for evidence of hypoxia within the first week after renalmass reduction. At this time point, hemodynamic alterationsare well established but tubulointerstitial damage is negligible(18). Here, findings showed that many tubules suffer fromhypoxic conditions in the early phase of remnant kidney.Furthermore, hypoxia in the tubulointerstitial compartmentpreceded traditional evidence of tubulointerstitial damage andpersisted until the damage developed. Investigations utilizingangiotensin II receptor blockage (ARB) suggested that thetubular hypoxia was dependent on the activation of RAS.
Received July 29, 2003. Accepted February 26, 2004.Correspondence to Dr. Masaomi Nangaku, University of Tokyo School ofMedicine, Division of Nephrology and Endocrinology, 7–3-1 Hongo Bunkyo-Ku, Tokyo, Japan. Phone: 03-3815-5411 ext. 33004; Fax: 03-5800-8806;E-mail [email protected]
1046-6673/1505-1277Journal of the American Society of NephrologyCopyright © 2004 by the American Society of Nephrology
DOI: 10.1097/01.ASN.0000125614.35046.10
J Am Soc Nephrol 15: 1277–1288, 2004

Materials and MethodsAnimals
Male Sprague-Dawley rats weighing 260 to 280 g were purchasedfrom Nippon Seibutsu Zairyo Center Co. (Saitama, Japan). They werehoused in an animal room maintained at a constant temperature witha 12-h light-dark cycle with free access to water and standard diet.When urine was collected, rats were housed in metabolic cages underthe same conditions. BP was measured under conscious conditionsusing the volume-oscillometric method (Ueda, Tokyo, Japan). Allstudies conformed to the principles of the Guide for Animal Experi-mentation at the University of Tokyo.
AntibodiesMonoclonal mouse anti-human �–smooth muscle actin (�-SMA)
1:1000 (Boehringer Mannheim, Mannheim, Germany), mouse antipi-monidazole 1:200 (Chemicon, Temecula, CA), mouse anti-rat vimen-tin 1:1000 (Dako, Glostrup, Denmark), biotinylated anti-mouse Ig1:400 (Vector Laboratories, Burlingame, CA), and alkaline phos-phatase-conjugated anti-mouse IgG 1:1000 (Promega, Madison, WI)were used in immunohistochemical studies. Monoclonal mouse anti-human HIF-1� 1:1000 (Novus Biologic, Littleton, CO) was used inimmunoprecipitation studies.
Disease Model and Experimental ProtocolEight rats served as a sham-operated control (sham group), and 36
underwent 5/6 nephrectomy as described previously (19). In brief,after right subcapsular nephrectomy, infarction of approximately twothirds of the left kidney was accomplished by ligation of the posteriorand one or two anterior extrarenal branches of the main left renalartery. Sixteen of these animals received an ARB, olmesartan (Phar-macology and Molecular Biology Research Laboratories, SankyoPharmaceutical, Tokyo, Japan), at a dosage of 10 mg/kg per d (20) bygastric gavage (RK�ARB group). The remaining animals were nottreated with this reagent (RK group, n � 20). Treatment was startedthe day after operation and continued until the day of sacrifice. Eightrats each from the RK and RK�ARB groups were sacrificed at 4 and7 d (early phase) after the operation and designated as the 4d RK, 7dRK, 4d RK�ARB, and 7d RK�ARB groups, respectively. Shamanimals were sacrificed on day 7. The four remaining rats in the RKgroup were killed at 2 wk to examine the persistence of findings(extended RK). Twenty-four–hour urine and tail vein blood werecollected before sacrifice. To detect hypoxia, 60 mg/kg of hypoxicprobe, pimonidazole (Chemicon), was injected intraperitoneally 2 hbefore sacrifice (21–23). To demonstrate functioning vessels, 250 �gof biotinylated lectin (Lycopersicon esculentum Lectin, Vector Lab-oratories) was injected via the tail vein exactly 4 min before sacrifice(24). To study the tissue perfusion properties of each vessel, 15 mg/kgof bisbezamine (Hoechst 33342; Sigma Chemical, St. Louis, MO) wasinjected via the inferior vena cava just below the left renal vein.Exactly 30 sec after injection, the renal pedicle was clamped and thekidney was promptly removed (25). To maintain the most represen-tative perfusion patterns of vessels, all materials were intentionallyinjected without the use of a pressure pump. Kidney tissues foranalysis were carefully selected to exclude necrotic areas. One sampleof kidney tissue was fixed in methyl Carnoy’s fixative and thenembedded in paraffin. All specimens were stored in light-protectedcontainers. A second piece of the kidney was dissected in ice-coldPBS, and the medulla was removed, which was then snap-frozen inliquid nitrogen before transfer to storage at �80°C until furtheranalysis.
To validate our methodology of Hoechst dye perfusion, 12 addi-tional rats were studied as described below in Nuclear FluorescenceStudies of Tissue Perfusion Properties of this section. Three of theseanimals underwent renal artery stenosis according to a model de-scribed previously (17), three underwent 6-wk 5/6 nephrectomy, threeserved as a sham-operated control, and three were examined withoutadministration of the Hoechst dye.
Double and Triple Immunohistochemical AnalysisImmunohistochemical staining was performed as described previ-
ously (26). Avidin HRP was used to stain biotinylated lectin-perfusedvessels. Chromogenic color was developed with 3,3'diaminobenzidinetetrahydrochloride (DAB). After staining the first antigen, the remain-ing peroxidase activity was extinguished with 3% H2O2 in methanolfor 10 min. The remaining biotin was blocked by incubation withavidin solution (Vector Laboratories), after which the first antibodiesfor the second antigen were applied, followed by suitable secondaryantibodies (biotinylated or alkaline phosphatase-conjugated). Colorwas developed with DAB plus 0.8% nickel or Vector Red (VectorLaboratories). When alkaline phosphatase reaction was used, endog-enous alkaline phosphatase was blocked with levamisole solution(Vector Laboratories). The processes were repeated for the thirdantigen staining. Negative controls without first antibodies were care-fully examined for each reaction. All histologic slides were examinedby light microscopy using an Olympus BX51 (Tokyo, Japan), andpictures were taken with the Olympus DP12 system. In some exper-iments, nuclei were counter-stained with methyl green (VectorLaboratories).
Nuclear Fluorescence Studies of Hoechst Dye toInvestigate Tissue Perfusion Properties
For indirect comparison of tissue perfusion properties of Hoechstdye, 4-�m sections of paraffin-embedded tissues were stepwise rehy-drated in a light-protected container. Rehydrated tissues weremounted and immediately photographed at 600� magnification(Eclipse E600, Nikon, Tokyo, Japan; connected to a cool charge-couple device [CCD], Micromax, Princeton Instruments, Trenton, NJ,and Metamorph, Downingtown, PA) avoiding any part of the glomer-uli. In this step, the exposure time was fixed at 50 msec and the CCDtemperature was set below �10°C.
Semiquantitative Scoring SystemAll scoring was done in a blinded manner. Photographs were
examined in gray scale. Tubulointerstitial injuries in the cortex werescored in a blinded manner on 20 randomly selected non-overlapping200� fields per rat on periodic acid-Schiff (PAS) and TrichromeMasson staining sections as described previously (11). Semiquantita-tive scores for pimonidazole adduction, �-SMA expression, and tu-bular vimentin expression were evaluated from 20 nonoverlappingfields at 200� magnification from each rat according to the followingscoring method: 0, no involvement; 1, involvement of �10% of thecortex; 2, involvement of 10% to 25% of the cortex; 3, involvementof 26% to 50% of the cortex; 4, involvement of 51% to 75% of thecortex; and 5, involvement of �75% of the cortex.
The amount of postglomerular peritubular capillaries was eval-uated by the vascular rarefaction index and the number of capillarylumina per 100 tubules by detection of perfused lectin as describedpreviously (7,11). Measurement of capillary luminal areas wasdone using 10 randomly selected 400� pictures per rat (334.2 �55 lumina/animal) in a blinded manner with the Image J software(http://rsb.info.nih.gov/ij/).
1278 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 1277–1288, 2004

Perfusion status with the nucleus-staining Hoechst dye was evalu-ated using Photoshop, version 7.0 (Adobe, San Jose, CA), as follows.Pictures were normalized by decreasing background signals ofautofluorescence of tubular cell cytoplasm to undetectable levels(27,28). Nuclei were selected with the Wand tool of Photoshop, whichautomatically selects areas of nuclei. A mean log scale of luminosityof selected areas from each picture was then measured by the histo-gram function. Our preliminary studies of a renal artery stenosismodel (n � 3) showed almost undetectable parenchymal nuclearfluorescence. Mean nuclear luminosity of 6-wk remnant kidney ani-mals (n � 3) was 63 � 8.6. Nuclear luminosity of sham-operatedanimals (n � 3) by this method was 121.9 � 4.3, and backgroundluminosity of non-injected animals (n � 3) was 0.34 � 0.2.
Proliferation of endothelial cells was evaluated on double-stainingof perfused lectin and PCNA by counting PCNA-positive nuclei perthousand visualized cortical capillary lumina from each rat.
Immunoprecipitation of HIF-1�To confirm that remnant kidneys were hypoxic, 20 mg of protein of
homogenized cortical tissues of each animal was subjected to proteinA Sepharose immunoprecipitation (Amersham International, Buck-ingham, UK) according to the manufacturer’s protocol (29). Theprecipitated protein was separated by electrophoresis in 8% SDS-PAGE gel or 4 to 20% SDS-PAGE gradient gel (Diaichi Pure Chem-ical, Tokyo, Japan), followed by electrotransfer to PVDF membranes(Amersham International). Transfer membranes were blocked with5% nonfat milk in TBS, 0.01% Tween-20, for 30 min at roomtemperature. The membranes were then blotted with anti-HIF1� an-tibody (Novus Biologic) at 1:100 at room temperature for 2 h. Thebound antibodies were detected with alkaline phosphatase-conjugatedsecondary antibodies. 5-Bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium tablets (Sigma Fast; Sigma Chemical) were used asa chromogenic substrate.
Total RNA Isolation and Real-Time RT-PCRTotal RNA from cortical tissues from each rat was isolated with
Isogen according to the manufacturer’s protocol (Wako Chemical,Osaka, Japan). cDNA synthesis was carried out with 2 �g of isolatedRNA at 42°C for 1 h with an oligoDT Inpromp-II kit (Promega). Onemicrogram of cDNA was subjected to real-time PCR (iCycler iQ;Bio-Rad, Hercules, CA) utilizing Syber Green Supermix (Bio-Rad)with the correspondence primers as follows: �-actin: 5'-CTTTCTA-CAATGAGCTGCGTG-3', 5'-TCATGAGGTAGTCTGTCAGG-3';erythropoietin (EPO): 5'-TACGTAGCCTCACTTCACTGCTT-3', 5'-GCAGAAAGTATCCGCTGTGAGTGTTC-3'; Glucose transporter-1(GLUT-1): 5'-CAGTTCGGCTATAACACCGGTGTC-3', 5'-AT-AGCGGTGGTTCCATGTTT-3'; and vascular endothelial growthfactor (VEGF): 5'-TTACTGCTGTACCTCCAC -3', 5'-ACAGGACG-GCTTGAAGATA -3' (30). The PCR was amplified for forty cycles.
Each PCR product was subjected to melting curve analysis after PCRwas completed. Threshold cycles for each primer were separatelyanalyzed. The amount of target mRNA/�-actin mRNA were calcu-lated according to the following equation: target mRNA/�-actinmRNA � 2�(tc � bc) where tc and bc are threshold cycles of the targetgene and �-actin, respectively. Representative data are presented interms of fold mRNA changes compared with sham.
Statistical AnalysesAll numerical data are presented as mean � SD. Distributions of
some data are shown by histogram. One-way ANOVA followed bythe least significant difference (LSD) method was used to determinedifferences among groups for all continuous parameters. The Mann-Whitney test was applied for noncontinuous parameters. Correlationsbetween two parameters were analyzed with Pearson correlation test.The significance level was set at P � 0.05 for all tests (SPSS forWindows 10.0).
ResultsAbsence of Histologic Interstitial Injury in the EarlyPhase in the Remnant Kidney Model
Remnant kidney animals (RK) showed a significant increasein urinary protein excretion, at 34.2 � 8.7 and 51.5 � 8.6 mg/din the 4d RK and 7d RK rats, respectively, compared with 8.2� 0.5 mg/d in the control rats. ARB decreased urinary proteinexcretion to 20.3 � 3.7 and 20.3 � 4.5 mg/d in the 4dRK�ARB and 7d RK�ARB rats, respectively (P � 0.05versus RK). BUN concentrations were 16.1 � 1.2, 32.8 � 9.1,36.5 � 11.7.4, and 62.5 � 6.7 mg/dl for sham, 4d RK, 7d RK,and Extended RK animals, respectively. BP was higher in RKanimals than in the control. ARB treatment significantly de-creased BP (Table 1).
Examination of PAS and Trichrome Masson samples in theearly phase of remnant kidneys, at 4 and 7 d after operation,showed hypertrophic glomeruli and tubules. No significantinterstitial changes were seen at this time point (Figure 1, a ande, b and f). In contrast, rats sacrificed at 2 wk showed someatrophic tubular cells, thickening of tubular basement mem-brane, and mild interstitial expansion (data not shown). Inter-stitial damage scores did not differ between sham and early RKgroups ( Table 2). The extended RK group had a higherinterstitial damage score, although damage in this group wasnot prominent even at this time point. In the ARB-treatedgroup, glomerular hypertrophy was less prominent.
Table 1. Basic biological data
Sham 4d RK 4d RK � ARB 7d RK 7d RK � ARB Extended RK
Systolic BP (mmHg) 118 � 6.3 146 � 5.9a 113 � 5.9b 166 � 9.2a 128 � 7.5b 187 � 4.9a
Diastolic BP (mmHg) 79 � 12.9 105 � 10.7a 80 � 12.4b 120 � 3.6a 90 � 15b 127 � 3.8a
Blood urea nitrogen (mg/dl) 16.1 � 1.2 32.8 � 9.1a 27.6 � 10.1a 36.5 � 11.7a 31.7 � 5.4a 62.5 � 6.7a
24-h urine protein (mg) 8.2 � 0.5 34.2 � 8.7a 20.3 � 3.7a,b 51.5 � 8.6a 20.3 � 4.5a,b 86.5 � 8a
a P � 0.05 versus sham.b P � 0.05 versus matched time point RK (ANOVA).
J Am Soc Nephrol 15: 1277–1288, 2004 Tubular Hypoxia in Remnant Kidney 1279

Development of Tubular Hypoxia in the Early Phase inthe Remnant Kidney Model
In the sham group, pimonidazole, which serves as an indicatorof hypoxic cells (21–23), was detected in medullary tubules (Fig-ure 2, a and d), which as previously reported are normally in a
state of borderline hypoxia (22,23). The number of pimonidazole-positive tubules in the cortex was markedly increased at 4 and 7 dafter nephron loss (Figure 2, b and e). Semiquantitative scores ofcortical pimonidazole uptake were 2.6 � 0.4 and 3.1 � 0.3 for 4dRK and 7d RK, respectively, while that of the control was 0.6 �
Figure 1. Histologic and immunohistochemical analysis of remnant kidneys in the early phase. Periodic acid-Schiff (PAS) and TrichromeMasson staining of normal kidneys (sham group) showed no evidence of tubulointerstitial damage (a and b). PAS and Trichrome Massonstaining of the early phase of remnant kidneys (7d RK) (e and f) also showed no interstitial damage. Double-staining with perfused lectin(brown) and vimentin (black) showed that expression of vimentin in tubules could not be detected in either sham (c) or 7d RK (g).Immunohistochemical studies of �-smooth muscle actin (�-SMA) showed lack of interstitial myofibroblasts in sham (d) and 7d RK (h).
Table 2. Semiquantitative scores of histological analysis
Sham 4d RK 4d RK � ARB 7d RK 7d RK � ARB Extended RK
PASTubular atrophy 0.3 � 0.1 0.3 � 0.2 0.3 � 0.2 0.4 � 0.1 0.4 � 0.1 0.9 � 0.2a
Interstitial infiltration 0.1 � 0.1 0.1 � 0.1 0.1 � 0.1 0.2 � 0.1 0.2 � 0.1 1.0 � 0.1a
Trichrome MassonInterstitial expansion 0.1 � 0.1 0.2 � 0.1 0.2 � 0.1 0.1 � 0.1 0.1 � 0.1 1.3 � 0.2a
Pimonidazole adduction 0.6 � 0.2 2.6 � 0.4a 0.9 � 0.3b 3.1 � 0.3a 1.4 � 0.6b 3.0 � 0.2a
Vimentin 0.1 � 0.1 0.1 � 0.1 0.1 � 0.1 0.1 � 0.1 0.1 � 0.1 0.4 � 0.1a
�-SMA 0 � 0.1 0.1 � 0.1 0.1 � 0.1 0.1 � 0.1 0.1 � 0.1 0.2 � 0.1
a P � 0.05 versus sham.b P � 0.05 versus matched time point RK (Mann-Whitney U test).
1280 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 1277–1288, 2004
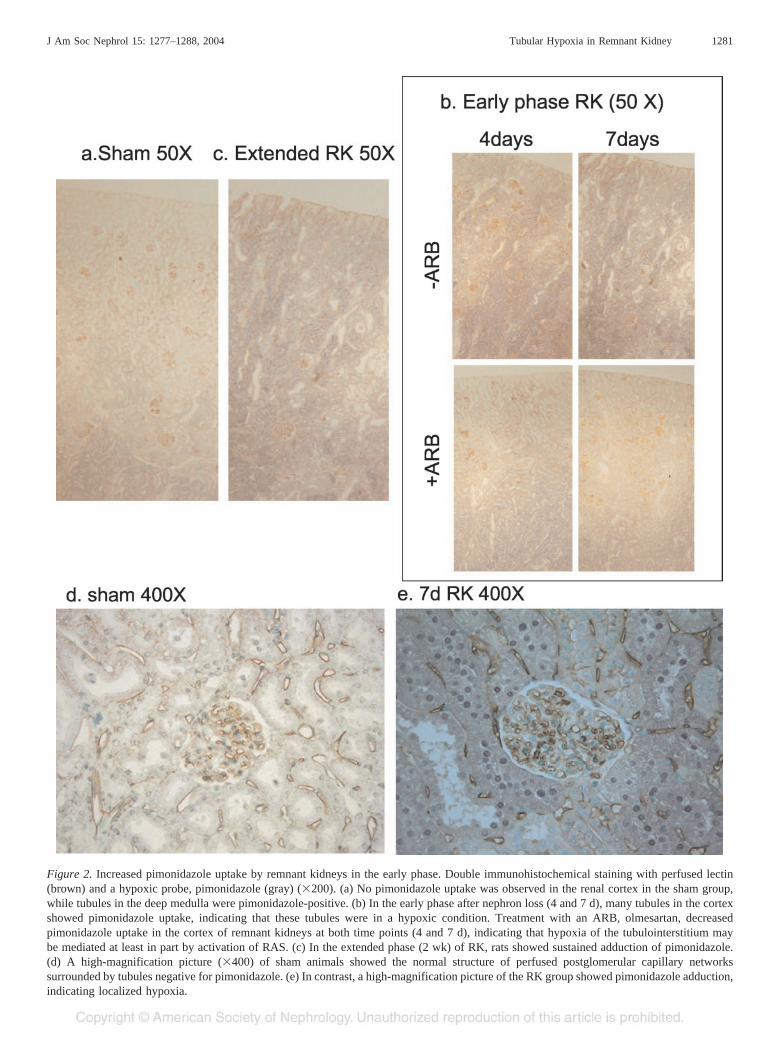
Figure 2. Increased pimonidazole uptake by remnant kidneys in the early phase. Double immunohistochemical staining with perfused lectin(brown) and a hypoxic probe, pimonidazole (gray) (�200). (a) No pimonidazole uptake was observed in the renal cortex in the sham group,while tubules in the deep medulla were pimonidazole-positive. (b) In the early phase after nephron loss (4 and 7 d), many tubules in the cortexshowed pimonidazole uptake, indicating that these tubules were in a hypoxic condition. Treatment with an ARB, olmesartan, decreasedpimonidazole uptake in the cortex of remnant kidneys at both time points (4 and 7 d), indicating that hypoxia of the tubulointerstitium maybe mediated at least in part by activation of RAS. (c) In the extended phase (2 wk) of RK, rats showed sustained adduction of pimonidazole.(d) A high-magnification picture (�400) of sham animals showed the normal structure of perfused postglomerular capillary networkssurrounded by tubules negative for pimonidazole. (e) In contrast, a high-magnification picture of the RK group showed pimonidazole adduction,indicating localized hypoxia.
J Am Soc Nephrol 15: 1277–1288, 2004 Tubular Hypoxia in Remnant Kidney 1281

0.2 (P � 0.05). Pimonidazole staining intensity was still high inthe cortex in the extended phase at 2 wk after the renal massreduction (Figure 2c). ARB treatment decreased the pimonida-zole-positive score to 0.9 � 0.3 and 1.4 � 0.6 at 4 and 7 d,respectively (Figure 2b).
To confirm the development of hypoxia in the early phase, weinvestigated expression levels of HIF-1� protein (Figure 3a). Noexpression of HIF-1� was detectable in the cortex in the shamgroup. In contrast, increased HIF-1� expression in the cortex wasobserved in 7d RK animals, which was consistent with the resultsof the pimonidazole uptake studies. RT-PCR analysis of a seriesof hypoxia-responsive genes also supported the presence of hyp-oxia in the kidney in the early phase. EPO, GLUT-1, and VEGFwere markedly increased at this time point in the remnant kidneys,and ARB attenuated these changes (Figure 3b).
Tubulointerstitial Damage after Tubular HypoxiaTo evaluate tubulointerstitial injury in detail, vimentin was
employed as markers of tubular damage. �-SMA was em-
ployed to detect interstitial myofibroblasts, which are associ-ated with interstitial damage and fibrosis. No difference be-tween the sham and early remnant groups was seen (Figure 1,c, d, g, and h). In contrast, an increase in the number ofvimentin-positive tubular cells as well as in �-SMA expressionwas seen in the extended phase (Table 2). These findingsindicated that hypoxia during the early study period (within thefirst week) was unlikely to have been the result of tubuloin-terstitial damage.
Association of Hypoxia with Decreased Perfusion ofthe Peritubular Capillary Beds in the Early Phase
Because injected lectin binds to perfused endothelial cellsunder physiologic conditions, this method allows evaluation ofboth the amount of and morphologic changes in functioningpostglomerular peritubular capillaries. The number of peritu-bular capillaries per 100 tubules and rarefaction index in 4d RKand 7d RK did not differ from that in the sham rats (Table 3).In contrast, the number of peritubular capillaries in the ex-
Figure 3. Upregulation of hypoxia-responsive proteins in remnant kidneys. (a) Imunoprecipitation of HIF-1� using cortical tissues of sham andremnant kidney rats. HIF-1� was undetectable in sham kidneys, but was increased in remnant kidneys. (b) Transcription of hypoxia-responsivegenes. Quantitative RT-PCR of the renal cortex of sham and remnant kidneys showed that all genes, including EPO, GLUT-1, and VEGF, wereincreased in remnant kidneys. Olmesartan-treated animals showed a decrease in all hypoxia-responsive genes. A, P � 0.05 versus sham; B,P � 0.05 versus RK.
1282 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 1277–1288, 2004

tended RK clearly decreased, to 137.8 � 12.5 per 100 tubules,with rarefaction of 12 � 3.7% (P � 0.05 versus sham). On thisbasis, hypoxia in the early phase could not be explained by anystructural loss of peritubular capillary beds, in contrast to theextended phase, when histologic loss of peritubular capillarieswas established. Notwithstanding this, significant morphologicchanges in peritubular capillaries in the early stage were ob-served. In normal kidneys, peritubular capillaries are elliptical,circular, or triangular in cross-section, which preserves thepatency of the lumina (Figure 4a). In contrast, peritubularcapillaries in remnant kidney animals showed a distorted out-line with relatively narrow capillary lumina (Figure 4b). Anal-ysis of cross-sectional capillaries showed that the mean ofcapillary lumina area in remnant kidney animals was signifi-cantly lower than that of the control rats (Table 3). The histo-gram demonstrated that remnant kidney animals harboredabundant narrow capillaries (Figure 4d). Double immunostain-ing analysis using pimonidazole and lectin showed co-local-ization of narrow capillaries and hypoxic tubules (Figure 2e).This result was supported by correlation analysis (Figure 4e).Furthermore, ARB treatment preserved the structural integrityof postglomerular capillaries as well as luminal patency (Fig-ure 4c and Table 3). These data suggested that the abnormalityin peritubular capillaries might have been mediated at least inpart by the activation of RAS.
Decreased Tissue Perfusion in Remnant Kidneys WasObserved by Administration of Hoechst 33342
When injected, the nuclear dye Hoechst 33342 rapidly dif-fuses from capillaries and is taken up by nuclei of the sur-rounding tissues. The nuclear fluorescence signals of Hoechst33342 in a short perfusion time thus indirectly represent tissueperfusion states, which are determined by a number of factors,including capillary density, capillary permeability, and totalblood flow of capillary networks. Uptake of Hoechst 33342 inglomeruli appeared to be higher than that of tubules in bothnephrectomized and sham rats. The remnant kidney groupsshowed less and weaker nuclear fluorescence signals in thetubulointerstitial area compared with the control animals, fur-ther suggesting the poor tissue perfusion in this area (Figure 5,a and b; Table 3). Again, ARB treatment restored fluorescencesignals of nuclei in tubular cells (Figure 5c; Table 3), indicat-ing the role of RAS activation in the poor perfusion of peritu-bular capillaries.
Endothelial Cell Proliferation Occurred in HypoxicAreas
An increase was seen in the number of PCNA-positiveendothelial cells at day 7 after nephron loss (Figure 6, b and f).In contrast, PCNA-positive endothelial cells were hardly de-tected in the control (61.7 � 14.7 versus 3 � 2 per 1000visualized capillary lumina; P � 0.05) (Figure 6, a and f). Thenumber of PCNA-positive endothelial cells in the extendedphase was slightly decreased to 51.5 � 7.7 per 1000 visualizedcapillary lumina, although this value was still higher than thatin the sham-operated animals (Figure 6f). ARB significantlydecreased PCNA-positive endothelial cells at Day 7 of nephron
ablation to 18.0 � 3 per 1000 visualized capillary lumina (P �0.05 versus 7d RK) (Figure 6, c and f). Triple immunostainingwith lectin, pimonidazole, and PCNA indicated that endothelialproliferation occurred in the hypoxic area (Figure 6, d, e, andg). These findings suggested that endothelial proliferation maybe one path to neoangiogenesis as a response to hypoxicstimuli.
DiscussionThis current study demonstrates for the first time that per-
fusion is decreased in peritubular capillaries in the early phaseof remnant kidney models, and that this decrease is associatedwith hypoxia in the tubulointerstitium. The hypoxic milieu inthe tubulointerstitium precedes any pathologic changes in thecorresponding region, suggesting a pathogenic role of ischemiaat early time points.
We intended to perform this study at the early phase afternephron loss at an equivalent time point to that used in thelandmark study of Hostetter and Brenner (1), when tubuloin-terstitial injury is negligible despite significant changes in renalhemodynamics. On this basis, our results were accompanied byglomerular hyperfiltration but were unlikely to be the result oftubulointerstitial damage. Increased cellular adduction of pi-monidazole in remnant kidney tubules suggested that thesetubular cells were hypoxic. The mechanism of cellular adduc-tion of pimonidazole specifically depends on a hypoxia-in-duced redox state, and previous studies have demonstrated agood correlation between adduction and tissue O2 tension (31).However, to exclude the possibility that hypertrophic tubulesmight adduct pimonidazole without hypoxia via unidentifiedmechanisms, we measured HIF-1� expression.
HIF-1� is the master hypoxia response regulator, the amountof which is determined by oxygen-dependent degradation (32).Rosenberger et al. (33) found no detectable expression ofHIF-1� in the normal kidney cortex. Here, we demonstrated anincreased amount of this protein in the renal cortex in remnantkidneys, confirming that the remnant kidney milieu was indeedhypoxic. We also measured changes in mRNA levels of hy-poxia-responsive genes, including EPO, GLUT-1, and VEGF.All these genes contain hypoxia-response elements (HRE) intheir cis-regulatory promoter regions (30) and increase theirtranscription in the presence of hypoxia. Although the possi-bility of non-hypoxic transcriptional regulation of HIF-1 can-not be excluded (34), the upregulation of HIF and these hy-poxia-responsive genes, together with the results using ahypoxia marker and the evaluation of tissue perfusion status,all point to the establishment of local hypoxia in our studies.
Previous studies suggest that kidney injury in the early phaseis dependent on hemodynamic changes, whereas tubulointer-stitial damage in the late phase is mediated by massive pro-teinuria (35). Development of tubulointerstitial hypoxia in thevery early phase suggests its possible pathogenic role in tubu-lointerstitial injury at this stage. Basile et al. (22) reported thatkidneys did not fully recover after severe ischemic acute renalfailure and that this lack of recovery was associated with theloss of peritubular capillaries and an increase in tubular pi-monidazole adduction. Suga et al. (23) also showed increased
J Am Soc Nephrol 15: 1277–1288, 2004 Tubular Hypoxia in Remnant Kidney 1283

Figure 4. Change in pattern of postglomerular peritubular capillaries in remnant kidneys. Immunohistochemical studies were conducted on bindingof injected lectin to endothelial cells of functioning capillaries. (a) In sham-operated animals, peritubular capillaries were arranged in a regular patternwith patent capillary lumina. (b) In early remnant kidneys (RK) (representative picture from 4d RK), most capillary lumina were distorted and hadrelatively narrow lumens. (c) ARB treatment restored the normal capillary morphology (representative picture from 7d RK�ARB). (d) Histogram ofluminal area demonstrated that remnant kidneys harbored more narrow peritubular capillaries. Treatment with ARB prevented this change. (e)Correlation between mean capillary luminal area and pimonidazole score in each animal showed that narrowing of capillaries correlated with hypoxia.
1284 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 1277–1288, 2004

adduction of pimonidazole in a hypokalemic nephropathymodel with tubulointerstitial injury. Using BOLD MRI, Ries etal. (36) reported hypoxia in the renal cortex of diabetic rats inthe early hyperfiltration phase. Recent studies by Johnson et al.(37) demonstrated that chronic systemic hypoxia causes renalinterstitial damage. Previous work by Truong group (38) andour group (17) on kidneys subject to renal artery stenosis alsosupported a role of hypoxia in tubulointerstitial injury. Incontrast to these previous and our present findings, Priyadarshi
et al. (39) performed direct measurement of tissue oxygentensions by microelectrode in the late phase of a remnantkidney model and reported a relative increase in tissue oxygentension. The reason for this discrepancy is unclear, but themicroelectrode method may have detected regional hyperoxia.An alternative explanation may be the difference in study’stime point.
The finding of tubular hypoxia in the early phase stimulatedus to investigate the responsible mechanisms. Intravenous in-
Table 3. Analysis of the vasculature in the early phase
Sham 4d RK 4d RK � ARB 7d RK 7d RK � ARB
Capillary lumina/100 tubules 187.3 � 8.9 180.9 � 16.9 181.9 � 11.1 180.2 � 12.5 188.8 � 15.9Capillary rarefaction index (%) 1.1 � 0.8 2.3 � 1.5 1.1 � 1.1 2.8 � 1.2 2.8 � 1.8Luminal area (microns2) 84.2 � 0.6 63.3 � 7.5a 73.7 � 7.6a,b 57.7 � 8.4a 73.5 � 5.7a,b
Nuclear fluorescent signal density 121.9 � 4.3 88.8 � 3.7a 106.5 � 8.9a,b 82.5 � 5.2a 104.3 � 10.5a,b
a P � 0.05 versus sham.b P � 0.05 versus matched time point RK (ANOVA).
Figure 5. Tissue perfusion studies with Hoechest 33342 dye. Tissue perfusion status was assessed by injection of Hoechest 33342 dye exactly30 s before removal of the kidney. In sham animals, nuclear fluorescence was intense in every area (a), whereas nuclear fluorescence signalsin the RK group were distinctly decreased in the tubulointerstitial area (b). Treatment with ARB increased nuclear fluorescence (c), indicatingthat ARB restored net perfusion in remnant kidneys. Semiquantitative measurement of nuclear luminosity supported these results (Table 3).
J Am Soc Nephrol 15: 1277–1288, 2004 Tubular Hypoxia in Remnant Kidney 1285

jection of biotinylated lectin under physiologic conditions,which allows evaluation of the amount and structure of func-tioning capillaries (24), showed no changes in the number ofcapillaries per tubule or vascular rarefaction at our early timepoints, but it did reveal striking morphologic changes in thecapillary network pattern. Many peritubular capillaries in rem-nant kidney animals were distorted and collapsed, showing astreak-like appearance. Quantitative analysis showed that rem-nant kidney harbored a large number of narrow peritubularcapillaries. Furthermore, correlation analysis showed that thedegree of capillary narrowing was related to that of pimonida-zole adduction. Of interest, the narrowing of peritubular cap-illaries had been previously reported in several human diseases(6,40). It should be emphasized that the flow in individualcapillary beds is dynamic but under the tight regulation of an
active feedback system and that the diameter of a vessel is theprimary determinant of its flow (41); we therefore speculatethat these peritubular capillary changes may be related to poortissue perfusion, leading to hypoxia. A second experimentprovided indirect support for this speculation; on measurementof tissue perfusion by injection of nuclear dye, a well-estab-lished method in oncology research (25,27,28,42), remnantkidney animals showed a decrease in tubulointerstitial nuclearfluorescence signals.
Neoangiogenesis is a common response to chronic hypoxia.Kang et al. (11) and Pillebout et al. (43) reported transientendothelial proliferation, which may be considered as a com-ponent of neoangiogenesis, in a remnant kidney model whileOhashi et al. (7) observed this phenomenon in a unilateralureteral obstruction (UUO) model. A recent study by Rosen-
Figure 6. Hypoxia-induced endothelial proliferation. Double-staining with lectin (brown) and proliferative cell nuclear antigen (PCNA) (red)showed that endothelial proliferation was increased in remnant kidneys around day 7. (a) In sham animals, endothelial proliferation was rarelydetected. (b) PCNA-positive endothelial cells were markedly increased in the 7d RK group. (c and f) ARB treatment decreased PCNA-positiveendothelial cells at this time point. (d and e) Triple staining with lectin (brown), PCNA (red), and pimonidazole (gray) of 7d RK animals showedthat endothelial proliferation in remnant kidneys was related to hypoxia and capillary pattern changes (representative pictures were taken fromthe same slide). In the hypoxic area of RK rats, tubules were pimonidazole-positive with distorted peritubular capillaries and expression ofPCNA in capillary endothelium (e), while PCNA-positive endothelial cells were rarely seen in the nonhypoxic area, and tubules wereaccompanied by capillaries with preserved morphology. (g) The percentage of PCNA-positive endothelial cells per capillary lumina innonhypoxic areas (pimonidazole score 0 or 1) was significantly lower than that in hypoxic areas (pimonidazole score �1). A, P � 0.05 versussham; B, P � 0.05 versus RK; C, P � 0.05
1286 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 1277–1288, 2004

berger et al. (44) suggested that hypoxia might stimulate peri-tubular capillary endothelial proliferation. Our results also re-vealed that endothelial proliferation co-localized with areas ofhypoxia, suggesting that endothelial proliferation (in thismodel) might be another response to hypoxia.
Activation of RAS is the earliest humoral response afternephron loss (45), and several clinical studies have shown thatangiotensin-converting enzyme inhibitors (ACEI) and ARBeffectively attenuate the progression of kidney disease (46,47).Our results revealed that ARB attenuated pimonidazole adduc-tion in tubular cells and preserved peritubular capillary net-work structure. The expression of hypoxia-responsive geneswas also decreased in ARB-treated animals, suggesting thattubulointerstitial hypoxia in remnant kidneys is mediated bythe activation of RAS, possibly via their vasoconstrictive ef-fects on efferent arterioles. This speculation is supported byrecent findings by Norman et al. (48), who showed that treat-ment with ACEI and ARB increased cortical tissue O2 tensionand proposed that this might be another renoprotective role ofthese drugs. In addition, Fujimoto et al. (49) reported thatchronic angiotensin infusion resulted in a decrease in peritu-bular blood flow, which was accompanied by adduction ofpimonidazole. Taken together, these and the present resultsemphasize the role of RAS activation in the disturbance oftubulointerstitial microcirculation and subsequent hypoxia.Moreover, they support the notion that improvement of corticaltissue oxygenation might be another renoprotective property ofARB and ACEI.
In conclusion, we have demonstrated that the hypoxic milieuin the tubulointerstitium precedes any pathologic change in thecorresponding region, suggesting a pathogenic role of ischemiaat early time points. Our results suggested that perfusion wasdecreased in peritubular capillaries in association with hypoxiain the tubulointerstitium in the early phase of remnant kidneymodels, and these changes may be mediated by the activationof RAS.
AcknowledgmentWe acknowledge research grants from the Japanese Ministry of
Health, Labor and Welfare, NOVARTIS Foundation (Japan) for thePromotion of Science, and Kanae Foundation for Life and Socio-Medical Science (Japan). We thank Dr. Ryoji Sassa (Okazaki KitaClinic, Nagoya, Japan), Dr. Taichi Ezaki (Tokyo Women’s MedicalUniversity, School of Medicine, Tokyo, Japan), and Dr. Kriang Tung-sanga (Division of Nephrology, Chulalongkorn University Hospital,Bangkok, Thailand).
References1. Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Bren-
ner BM: Hyperfiltration in remnant nephrons: a potential adverseresponse to renal abrasion. Am J Physiol 241: F85–F93, 1981
2. Nath KA: Tubulointerstitial changes as a major determinant inprogression of renal damage. Am J Kidney Dis 1: 1–17, 1992
3. Remuzzi G, Bertani T: Pathophysiology of progressive nephrop-athies. N Engl J Med 339: 1448–1456, 1998
4. Eddy AA: Molecular insights into renal interstitial fibrosis. J AmSoc Nephrol 7: 2495–2508, 1996
5. Fine LG, Bandyopadhay D, Norman JT: Is there a commonmechanism for the progression of different types of renal diseaseother than proteinuria? Towards the unifying theme of chronichypoxia. Kidney Int 57[Suppl 75]: S22–S26, 2000
6. Bohle A, Mackensen-Haen S, Wehrmann M: Significance ofpostglomerular capillaries in the pathogenesis of chronic renalfailure. Kidney Blood Press Res 19: 191–195, 1996
7. Ohashi R, Shimizu A, Masuda Y, Kitamura H, Ishizaki M,Sugisaki Y, Yamanaka N: Peritubular capillary regression duringthe progression of experimental obstructive nephropathy. J AmSoc Nephrol 13: 1795–1805, 2002
8. Ohashi R, Kitamura H, Yamanaka N: Peritubular capillary injuryduring the progression of experimental glomerulonephritis inrats. J Am Soc Nephrol 11: 47–56, 2000
9. Nangaku M, Yamada K, Gariepy CE, Miyata T, Inagi R, Kuro-kawa K, Yanagisawa M, Fujita T, Johnson RJ: ET(B) receptorprotects the tubulointerstitium in experimental thrombotic mi-croangiopathy. Kidney Int 62: 922–928, 2002
10. Kang DH, Anderson S, Kim YG, Mazzalli M, Suga S, JeffersonJA, Gordon KL, Oyama TT, Hughes J, Hugo C, Kerjaschki D,Schreiner GF, Johnson RJ: Impaired angiogenesis in the agingkidney: vascular endothelial growth factor and throm-bospondin-1 in renal disease. Am J Kidney Dis 37: 601–611,2001
11. Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL,Mazzali M, Jefferson JA, Hughes J, Madsen KM, Schreiner GF,Johnson RJ: Impaired angiogenesis in the remnant kidney model:I. Potential role of vascular endothelial growth factor and throm-bospondin-1. J Am Soc Nephrol 12: 1434–1447, 2001
12. Norman JT, Clark IM, Garcia P: Hypoxia promotes fibrogenesisin human renal fibroblast. Kidney Int 58: 2351–2366, 2000
13. Sodhi CP, Phadke SA, Batlle D, Sahai A: Hypoxia and highglucose cause exaggerated mesangial cell growth and collagensynthesis: Role of osteopontin. Am J Physiol Renal Physiol 280:F667–F674, 2001
14. Orphanides C, Fine LG, Norman JT: Hypoxia stimulates proxi-mal tubular cell matrix production via TGF-�1 independentmechanism. Kidney Int 52: 637–647, 1997
15. Tanaka T, Hanafusa N, Ingelfinger JR, Ohse T, Fujita T, Nan-gaku M: Hypoxia induces apoptosis in SV40-immortalized ratproximal tubular cells through the mitochondrial pathways, de-void of HIF1-mediated upregulation of Bax. Biochem BiophysRes Commun 309: 222–231, 2003
16. Tanaka T, Miyata T, Inagi R, Kurokawa K, Adler S, Fujita T,Nangaku M: Hypoxia-induced apoptosis in cultured glomerularendothelial cells: Involvement of mitochondrial pathways. Kid-ney Int 64: 2020–2032, 2003
17. Manotham K. Tanaka T, Matsumoto M, Ohse T, Inagi R, MiyataT, Kurokawa K, Fujita T, Ingelfinger JR, Nangaku M: Transdif-ferentiation of cultured tubular cell induced by hypoxia. KidneyInt 65: 871–880, 2004
18. Johnson TS, Griffin M, Thomas GL, Skill J, Cox A, Yang B,Nicholas B, Birckbichler PJ, Muchaneta-Kubara C, El NahasAM: The role of transglutaminase in the rat subtotal nephrec-tomy model of renal fibrosis. J Clin Invest 99: 2950–2960, 1997
19. Kliem V, Johnson RJ, Alpers CE, Yoshimura A, Couser WG,Koch KM, Floege J: Mechanisms involved in the pathogenesis oftubulointerstitial fibrosis in 5/6-nephrectomized rats. Kidney Int49: 666–678, 1996
20. Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K,Fujita T: Imbalance of T-cell subsets in angiotensin II-infused
J Am Soc Nephrol 15: 1277–1288, 2004 Tubular Hypoxia in Remnant Kidney 1287

hypertensive rats with kidney injury. Hypertension 42: 31–38,2003
21. Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG: Chronicenteral ethanol treatment causes hypoxia in rat liver tissue. Hepa-tology 25: 920–926, 1997
22. Basile DP, Donohoe DL, Roethe K, Mattson DL: Chronic renalhypoxia after ischemic injury: effects of L-arginine on hypoxiaand secondary damage. Am J Physiol Renal Physiol 284: F338–F348, 2002
23. Suga SI, Phillips MI, Ray PE, Raleigh JA, Vio CP, Kim Y,Mazzali M, Gordon KL, Hughes J, Johnson RJ: Hypokalemiainduces renal injury and alterations in vasoactive mediators thatfavor salt sensitivity. Am J Physiol Renal Physiol 281: F620–F629, 2001.
24. Ezaki T, Baluk P, Thurston G, La Barbara A, Woo C, McDonaldDM: Time course of endothelial cell proliferation and microvas-cular remodeling in chronic inflammation. Am J Pathol 158:2043–2055, 2001
25. Busch TM, Wileyto EP, Emanuele MJ, Del Piero F, MarconatoL, Glatstein E, Koch CJ: Photodynamic therapy creates fluencerate-dependent gradients in intratumoral spatial distribution ofoxygen. Cancer Res 62: 7273–7279, 2002
26. Nangaku M, Quigg RJ, Shankland SJ, Okada N, Johnson RJ,Couser WG: Over expression of Crry protects mesangial cellsfrom complement–mediated injury. J Am Soc Nephrol 8: 223–233, 1997
27. Kostourou V, Robinson SP, Whitley GS, Griffiths JR: Effects ofover expression of dimethylarginine dimethylaminohydrolase ontumor angiogenesis assessed by susceptibility magnetic reso-nance imaging. Cancer Res 63: 4960–4966, 2003
28. Goertz DE, Yu JL, Kerbel RS, Burns PN, Foster FS: High-frequency doppler ultrasound monitors the effects of antivasculartherapy on tumor blood flow. Cancer Res 62: 6371–6375, 2002
29. Freeburg PB, Robert B, St John PL, Abrahamson DR: Podocyteexpression of hypoxia-inducible factor (HIF)–1 and HIF–2 duringglomerular development. J Am Soc Nephrol 14: 927–938, 2003
30. Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N,Noiri E, Fujita T, Nangaku M: Induction of renoprotective geneexpression by cobalt ameliorate ischemic injury of the kidney inrats. J Am Soc Nephrol 14: 1825–1832, 2003
31. Arteel GE, Thurman RG, Raleigh JA: Reductive metabolism ofhypoxic marker pimonidazole is regulate by oxygentension, in-dependent of pyridine redox state. Eur J Biochem 253: 743–750,1998
32. Safran M, Kaelin WG: HIF hydroxylation and the mammalianoxygen-sensing pathway. J Clin Invest 111: 779–783, 2003
33. Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS,Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S,Eckardt KU: Expression of hypoxia-inducible factor-1alpha and-2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol13: 1721–1732, 2002
34. Richard DE, Berra E, Pouysségur J: Nonhypoxic pathway me-diates the induction of hypoxia-inducible factor 1 in vascularsmooth muscle cells. J Biol Chem 275: 26765–26771, 2000
35. Nangaku M, Pippin J, Couser WG: C6 mediates chronic pro-gression of tubulointerstitial damage in rats with remnant kid-neys. J Am Soc Nephrol 13: 928–936, 2002
36. Ries M, Basseau F, Tyndal B, Jones R, Deminiere C, Catargi B,Combe C, Moonen CW, Grenier N: Renal diffusion and BOLDMRI in experimental diabetic nephropathy. J Magn Reso Imag-ing 17: 104–113, 2003
37. Mazzali M, Jefferson JA, Ni Z, Vaziri ND, Johnson RJ: Micro-vascular and interstititial injury associated with Chronic hypoxiainduced hypertension. Kidney Int 16: 2088–2093, 2003
38. Truong LD, Farhood A, Tasby J, Gillum D: Experimentalchronic renal ischemia: Morphological and immunohistochemi-cal studies. Kidney Int 41: 1676–1689, 1992
39. Priyadarshi A, Periyasamy S, Burke TJ, Britton SL, Malhotra D,Shapiro JI: Effects of reduction of renal mass on renal oxygentension and erythropoietin production in the rat. Kidney Int 61:542–546, 2002
40. Choi YJ, Chakraborty S, Nguyen V, Nguyen C, Kim BK, ShimSI, Suki WN, Truong LD: Peritubular capillary loss is associatedwith chronic tubulointerstitial injury in human kidney: Alteredexpression of vascular endothelial growth factor. Hum Pathol 31:1491–1497, 2000
41. Pries AR, Reglin B, Secomb TW: Structural response of micro-circulatory networks to changes in demand: information transferby shear stress. Am J Physiol Heart Circ Physiol 284: H2204–H2212, 2003
42. Yu JL, Rak JW, Carmeliet P, Nagy A, Kerbel RS, Coomber BL:Heterogenous vascular dependence of tumor cell population.Am J Pathol 158: 1325–1334, 2003
43. Pillebout E, Burtin M, Yuan HT, Briand P, Woolf AS, Fried-lander G, Terzi F: Proliferation and remodeling of peritubularmicrocirculation after nephron reduction associate with the pro-gression of renal lesions. Am J Pathol 159: 547–560, 2001
44. Rosenberger C, Griethe W, Gruber G, Wiesener M, Frei U,Bachmann S, Eckardt KU: Cellular responses to hypoxia afterrenal segmental infarction. Kidney Int 64: 874–886, 2003
45. Rosenberg ME, Correa-Rotter R, Inagami T, Kren SM, HostetterTH: Glomerular renin synthesis and storage in the remnantkidney in the rat. Kidney Int 40: 677–683, 1991
46. Gruppo Italiano di Studi Epidemiologici in Nepfrologia(GISEN): Randomized placebo-control trial of effect of ramiprilin glomerular filtration rate and risk of terminal renal failure inproteinuric non-diabetic nephropathy. Lancet 349: 1857–1863,1997
47. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE,Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S:Effects of losartan on renal and cardiovascular out come inpatients with type 2 diabetes and nephropathy. N Engl J Med345: 861–869, 2001
48. Norman JT, Stidwill R, Singer M, Fine LG: Angiotensin IIblockage augment renal cortical microvascular pO2 indicating anovel potential renoprotective action. Nephron Physiol 94: 39–46, 2003
49. Fujimoto S, Satoh M, Ozeki M, Kobayashi S, Haruna Y, Ara-kawa S, Horike H, Sasaki T, Kashihara N: A long acting calciumchannel blocker, azelnidipine, ameliorates angiotensin II inducedrenal damage in rats [Abstract]. J Am Soc Nephrol 10: 609A,2003
1288 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 1277–1288, 2004
Related Documents











