Neurobiology of Aging 25 (2004) 465–474 Evaluation of HFE (hemochromatosis) mutations as genetic modifiers in sporadic AD and MCI Daniel Berlin a,b , George Chong c , Howard Chertkow a,b,d,e , Howard Bergman a,d,e , Natalie A. Phillips a,d,f , Hyman M. Schipper a,b,d,e,∗ a Bloomfield Centre for Research in Aging, Montreal, Que., Canada b Department of Neurology and Neurosurgery, McGill University, Montreal, Canada c Department of Diagnostic Medicine, Lady Davis Institute for Medical Research, S.M.B.D. Jewish General Hospital, 3755 Cote St. Catherine Road, Montreal, Que., Canada H3T 1E2 d Centre for Neurotranslational Research, Montreal, Que., Canada e Division of Geriatric Medicine, Department of Medicine, McGill University, Montreal, Canada f Department of Psychology, Concordia University, Montreal, Canada Received 7 February 2003; received in revised form 13 May 2003; accepted 16 June 2003 Abstract Background: Pathological brain iron deposition has been implicated as a source of neurotoxic reactive oxygen species in Alzheimer disease (AD). Recent reports suggest that heterozygosity for the two common hfe mutations responsible for hereditary hemochromatosis (HH) may be a risk factor for AD, possibly by accelerating brain iron accumulation. Methods: To test this hypothesis, we genotyped 213 sporadic AD, 106 MCI, and 63 normal elderly control (NEC) individuals for the H63D and C282Y hfe mutations by polymerase chain reaction (PCR)/restriction fragment length polymorphism (RFLP) analysis. We determined the relationship of these mutations to the demographic, clinical, and neuropsychological features of AD and MCI, and evaluated whether an interaction existed between hfe and apolipoprotein E (apoE) status in these patients. Results: We observed no significant impact of H63D or C282Y heterozygosity on age at AD symptoms onset or diagnosis, age at onset of cognitive symptoms (AD and MCI combined), rates of MCI-to-AD conversion or specific neuropsychological deficits. No interactions between hfe zygosity and apoE status were discerned. Patients homozygous for H63D exhibited trends towards accelerated MCI-to-AD conversion rates and a subset of younger individuals (aged 55–75) exhibited earlier onset of cognitive symptoms relative to wild-type hfe and H63D heterozygotes. Conclusions: Contrary to earlier reports, the results of the present study do not implicate the common hfe mutations as genetic modifiers of sporadic AD and MCI. Trends towards accelerated cognitive dysfunction in H63D homozygotes warrant further study. © 2003 Elsevier Inc. All rights reserved. Keywords: Alzheimer disease; Hemochromatosis; hfe mutation; Iron; Mild cognitive impairment 1. Introduction Sporadic Alzheimer disease (AD) affects approximately 5–10% of North Americans over the age of 65 and as many as 30–50% of those who survive to the end of their ninth decade [2]. Although the precise mechanisms responsible for neuronal demise in AD remain largely unknown, there ex- ists ample evidence implicating oxidative stress (free radical injury) in the pathogenesis of this condition [16]. Oxidative damage in AD tissues may, in turn, be related to the aug- mented deposition of redox-active iron, an important gener- ator of reactive oxygen species (ROS), in the basal forebrain ∗ Corresponding author. Tel.: +1-514-340-8260; fax: +1-514-340-7502. E-mail address: [email protected] (H.M. Schipper). and association cortices of AD victims [23]. Attempts to delineate metabolic pathways subserving the enhanced se- questration of iron in AD brain have revealed aberrations in the CNS expression of proteins implicated in iron mobiliza- tion and storage, including transferrin receptor [13], ferritin [4], iron response proteins [21], lactoferrin [14], melan- otransferrin [12], and heme oxygenase-1 [24]. Despite these observations, a coherent picture of the mechanism(s) gov- erning pathological brain iron deposition in AD and other aging-related neurodegenerative disorders has yet to emerge. Hereditary hemochromatosis (HH) is the most common autosomal recessive disorder in Caucasians, affecting ap- proximately 1 out of every 200 people of European origin [18]. HH is characterized by excessive absorption of dietary iron and toxic deposition of this transition metal in liver, 0197-4580/$ – see front matter © 2003 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2003.06.008

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Neurobiology of Aging 25 (2004) 465–474
Evaluation of HFE (hemochromatosis) mutations as geneticmodifiers in sporadic AD and MCI
Daniel Berlina,b, George Chongc, Howard Chertkowa,b,d,e, Howard Bergmana,d,e,Natalie A. Phillipsa,d,f , Hyman M. Schippera,b,d,e,∗
a Bloomfield Centre for Research in Aging, Montreal, Que., Canadab Department of Neurology and Neurosurgery, McGill University, Montreal, Canada
c Department of Diagnostic Medicine, Lady Davis Institute for Medical Research, S.M.B.D. Jewish General Hospital,3755 Cote St. Catherine Road, Montreal, Que., Canada H3T 1E2
d Centre for Neurotranslational Research, Montreal, Que., Canadae Division of Geriatric Medicine, Department of Medicine, McGill University, Montreal, Canada
f Department of Psychology, Concordia University, Montreal, Canada
Received 7 February 2003; received in revised form 13 May 2003; accepted 16 June 2003
Abstract
Background: Pathological brain iron deposition has been implicated as a source of neurotoxic reactive oxygen species in Alzheimerdisease (AD). Recent reports suggest that heterozygosity for the two commonhfe mutations responsible for hereditary hemochromatosis(HH) may be a risk factor for AD, possibly by accelerating brain iron accumulation.Methods: To test this hypothesis, we genotyped213 sporadic AD, 106 MCI, and 63 normal elderly control (NEC) individuals for the H63D and C282Yhfe mutations by polymerasechain reaction (PCR)/restriction fragment length polymorphism (RFLP) analysis. We determined the relationship of these mutations tothe demographic, clinical, and neuropsychological features of AD and MCI, and evaluated whether an interaction existed betweenhfe andapolipoprotein E (apoE) status in these patients.Results: We observed no significant impact of H63D or C282Y heterozygosity on ageat AD symptoms onset or diagnosis, age at onset of cognitive symptoms (AD and MCI combined), rates of MCI-to-AD conversion orspecific neuropsychological deficits. No interactions betweenhfe zygosity and apoE status were discerned. Patients homozygous for H63Dexhibited trends towards accelerated MCI-to-AD conversion rates and a subset of younger individuals (aged 55–75) exhibited earlier onsetof cognitive symptoms relative to wild-typehfe and H63D heterozygotes.Conclusions: Contrary to earlier reports, the results of the presentstudy do not implicate the commonhfe mutations as genetic modifiers of sporadic AD and MCI. Trends towards accelerated cognitivedysfunction in H63D homozygotes warrant further study.© 2003 Elsevier Inc. All rights reserved.
Keywords: Alzheimer disease; Hemochromatosis;hfe mutation; Iron; Mild cognitive impairment
1. Introduction
Sporadic Alzheimer disease (AD) affects approximately5–10% of North Americans over the age of 65 and as manyas 30–50% of those who survive to the end of their ninthdecade[2]. Although the precise mechanisms responsible forneuronal demise in AD remain largely unknown, there ex-ists ample evidence implicating oxidative stress (free radicalinjury) in the pathogenesis of this condition[16]. Oxidativedamage in AD tissues may, in turn, be related to the aug-mented deposition of redox-active iron, an important gener-ator of reactive oxygen species (ROS), in the basal forebrain
∗ Corresponding author. Tel.:+1-514-340-8260; fax:+1-514-340-7502.E-mail address: [email protected] (H.M. Schipper).
and association cortices of AD victims[23]. Attempts todelineate metabolic pathways subserving the enhanced se-questration of iron in AD brain have revealed aberrations inthe CNS expression of proteins implicated in iron mobiliza-tion and storage, including transferrin receptor[13], ferritin[4], iron response proteins[21], lactoferrin [14], melan-otransferrin[12], and heme oxygenase-1[24]. Despite theseobservations, a coherent picture of the mechanism(s) gov-erning pathological brain iron deposition in AD and otheraging-related neurodegenerative disorders has yet to emerge.
Hereditary hemochromatosis (HH) is the most commonautosomal recessive disorder in Caucasians, affecting ap-proximately 1 out of every 200 people of European origin[18]. HH is characterized by excessive absorption of dietaryiron and toxic deposition of this transition metal in liver,
0197-4580/$ – see front matter © 2003 Elsevier Inc. All rights reserved.doi:10.1016/j.neurobiolaging.2003.06.008

466 D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474
pancreas, heart, and pituitary gland[11,15]. Two missensemutations in thehfe gene located on chromosome 6p are re-sponsible for the majority of HH cases: (a) a guanine to ade-nine transition at nucleotide position 845 (G845A) causinga cysteine to tyrosine substitution at amino acid position 282(C282Y) of the HFE protein and (b) a cytosine to guaninetransversion at nucleotide 187 (C187G) resulting in a histi-dine to aspartate substitution at amino acid 63 (H63D)[6].The worldwide prevalence of these mutations is estimatedat 1.9% for the more penetrant (“major”) C282Y mutationand 8.1% for the less penetrant (“major”) H63D mutation[18]. The normal HFE protein is thought to interact with thetransferrin receptor and attenuate its capacity to mediate in-tracellular iron delivery. Excessive tissue iron sequestrationmay accrue from abnormal interaction of the transferrin re-ceptor with mutant HFE protein[7].
In 2000, Moalem et al.[19] reported that the pres-ence of mutanthfe alleles may put apolipoprotein E4(apoE4)-negative men at increased risk for manifesting fa-milial AD (FAD). In 2001, Sampietro et al.[22] presentedevidence that AD patients possessing one or two copiesof the H63D mutation develop AD, on average, 5 yearsearlier than those with wild-typehfe. The authors of bothstudies concluded that HFE mutations may contribute topathological brain iron deposition and earlier manifestationof symptoms in AD patients.
In the present study, we set out to determine whether (a)HFE mutations are over-represented or impact disease onsetin a larger, heterogeneous population of patients with spo-radic AD and mild cognitive impairment (MCI) relative tonormal elderly controls (NEC), (b) HFE mutations interactwith apoE status as genetic modifiers of sporadic AD, and(c) HFE mutations are associated with specific neuropsy-chological deficits in patients with sporadic AD.
2. Materials and methods
2.1. Materials
The QIAamp DNA blood minikit and the polymerasechain reaction (PCR) amplification kit were purchased fromQiagen (Mississauga, Ont., Canada). PCR was performed onan MJ Research PTC 200 PCR machine (Watertown, MA,USA). PCR primers and the 100 bp DNA ladder were ob-tained from Invitrogen, Canada.DpnII and RsaI restrictionenzymes were purchased from New England Biolabs (Bev-erly, MA, USA). Agarose was obtained from FMC Bioprod-ucts (Rockland, ME, USA).
2.2. Subjects
This study was approved by the Research Ethics Commit-tee of the Sir Mortimer B. Davis Jewish General Hospital(JGH) in Montreal, Canada. Three hundred and eighty-twoindividuals were enrolled in this study. Patients with sporadic
AD and MCI were recruited from the JGH/McGill Univer-sity Memory Clinic, a tertiary care ambulatory clinic servingan ethnically diverse population in Montreal. Two hundredand thirteen patients (100 men/113 women) met NationalInstitute of Neurological and Communicative Disorders andStroke-Alzheimer’s Disease and Related Disorders Associa-tion (NINCDS-ADRDA) criteria for probable AD[17]. Onehundred and six patients were diagnosed as having MCI[20]. These individuals exhibited subjective memory com-plaints, objective memory loss on mental status testing, andpreservation of functional abilities. They did not meet crite-ria for dementia and had no other medical or neurologicalillnesses to explain their decline. All MCI patients scored0.5 on the Clinical Dementia Rating Scale[10]. Sixty-threeNEC individuals were recruited from the Family MedicineUnit at the same hospital. None had memory complaints andall scored within 1 S.D. of age- and education-standardizednormative values on a series of memory and attention tests(Clinical Dementia Rating Scale= 0.0).
2.3. DNA extraction
Written informed consent was obtained from all individ-uals or their primary caregivers. Whole blood was collectedby phlebotomy in tubes containing EDTA. DNA extractionfrom blood samples was performed in the Molecular Diag-nostics Laboratory of the Jewish General Hospital. Bloodsamples were centrifuged at 1800 rpm for 10 min, leukocytebuffy layers were collected, and DNA was isolated from thisfraction using the QIAamp DNA blood minikit.
2.4. HFE genotyping
Genomic DNA analysis for the C187G and G845Ahfemutations was performed by PCR and restriction fragmentlength polymorphism (RFLP) analysis. The restriction pat-terns were visualized by agarose gel electrophoresis andethidium bromide staining. The primer sequences used forthe DNA region containing the C187G mutation were ACATGG TTA AGG CCT GTT GC (sense) and GCC ACA TCTGGC TTG AAA TT (antisense)[13]. The relevant region ofDNA for the G845A mutation was amplified with the fol-lowing primer sequences: TGG CAA GGG TAA ACA GATCC (sense) and CTC AGG CAC TCC TCT CAA CC (an-tisense; ibid.). The PCR reaction entailed an initial denatur-ing step at 95◦C for 5 min, 35 cycles of denaturing at 94◦Cfor 30 s, annealing at 55◦C for 30 s, and extension at 72◦Cfor 30 s. A final 10-min extension period at 72◦C was per-formed following the final PCR cycle. The PCR productswere incubated overnight with the appropriate restriction en-zymes at 37◦C. TheDpnII restriction enzyme was used todigest the 208 bp product of the C187G-region amplificationand RsaI was employed to digest the 388 bp PCR productof the G845A-region amplification. The digested productswere size-separated on a 3% agarose gel by electrophoresisand visualized by ethidium bromide staining.

D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474 467
2.5. ApoE genotyping
ApoE genotyping was performed in the Molecular Diag-nostics Laboratory of the Jewish General Hospital. The PCRprimers and assay conditions were identical to those previ-ously described by Hixon and Vernier[9].
2.6. Neuropsychological testing
Neuropsychological testing of all subjects was conductedby two psychologists affiliated with the JGH/McGill Uni-versity Memory Clinic. The global severity of cognitiveimpairment was examined using the Folstein Mini-MentalState Examination (MMSE)[8]. Tests of individual cogni-tive domains included clock drawing, delayed verbal recall(WMS-III Logical Memory II Subtest), orthographic (F,S) and semantic (animals) oral fluency, confrontationalnaming (Boston Naming Test), attention and psychomotorspeed (WAIS-III Digit Symbol Subtest), and Trail MakingTest A.
2.7. Database construction
For each individual, sex, year of birth, years of formaleducation, diagnosis, family history of dementia/AD, yearof symptoms onset, year of AD/MCI diagnosis, medicationsused, MMSE scores, neuropsychological test scores, and theresults of apoE and HFE genotyping were documented ina computerized personal data form. The age of symptomonset was defined as the age when memory impairment wasfirst noted by relatives. The age of AD diagnosis denotedthe age at which the patient was first clinically diagnosed ashaving AD by a neurologist or geriatrician. For those ADpatients who were previously diagnosed with MCI, the timespan between the two diagnoses was noted and termed theMCI–AD interval.
2.8. Statistical analyses
Group differences with respect to education levels,MMSE scores, and age at diagnosis were evaluated usinga between-subjects ANOVA with Tukey’s post hoc test forsignificant main effects. A Chi-square test was employedto compare the observed and expected values for the HFEand apoE genotype distributions between the AD, MCI,and NEC groups, the HFE genotype distributions betweenmen and women in the AD group, and whether interac-tions existed between HFE mutations and the apoE4 allele.Kaplan–Meier (KM) survival analysis was used to analyzethe effects of the HFE mutations and apoE4 allele on the ageof AD symptoms onset and the age of AD diagnosis. KMsurvival analysis was also employed to ascertain whetherthe H63D mutation accelerated the progression from MCIto AD. MCI subjects who had not developed AD at the timeof this study were censored in this analysis. The log-ranktest was utilized to determine whether genotype-specific
survival functions were significantly different from oneanother. Between-subject multivariate analysis of variance(MANOVA) was conducted to determine whether over-all neuropsychological performance differed significantlyamong the groups and whether the HFE mutations im-pacted the testing scores of AD patients. If the multivariatetest was significant, then the univariate ANOVA on eachneuropsychological test was evaluated. Significant effectswere followed up using Tukey’s HSD post hoc tests. Inall cases,P-values<0.05 indicated significance. Statisticalanalyses were performed using SPSS Software, version10.1 (Chicago, IL, USA).
3. Results
3.1. HFE genotyping
The DpnII enzyme cuts the wild-type PCR product intotwo fragments of 138 and 70 bp (Fig 1A). In the C187G(H63D) homozygote, the mutation resulted in loss of thenormal restriction site and expression of the full 208 bp PCRsegment. In heterozygotes, three fragments of 208, 138, and70 bp were generated due to the presence of both normal andmutated alleles. Addition ofRsaI to the PCR-amplified DNAcontaining the wild-type allele generated two fragments of248 and 140 bp I (Fig. 1B). In individuals homozygous forthe G845A (C282Y) mutation an additional restriction sitewas created, producing three fragments of 248, 111, and29 bp. In heterozygotes, four fragments of 248, 140, 111,and 29 bp were generated due to the presence of both normaland mutated DNA.
3.2. Patient demographics and MMSE scores
As expected, mean MMSE scores were significantlylower in the AD group (22.8 ± 3.7) than in the MCI(27.9 ± 1.6) and NEC (29.0 ± 1.0) groups,F(2, 364) =165.0, P < 0.001. Years of formal education were also lessin the AD group (10.7 ± 3.9) than in the MCI (11.9 ± 3.8)and NEC (12.8± 2.8) cohorts,F(2, 363) = 9.6, P < 0.001.The mean age at neuropsychological evaluation did notdiffer significantly between the AD (76.5 ± 7.6), MCI(75.4±6.8), and NEC (75.0±5.5) groups,F(2, 348) = 1.4,P < 0.247.
3.3. HFE mutation frequency in AD, MCI,and NEC subjects
When stratified by H63D genotype status, the AD, MCI,and NEC groups displayed almost identical frequency dis-tributions for the wild-type, heterozygous, and homozygousconditions (Table 1). In each group, 66–74% of individualswere wildtype, 25–30% were heterozygous, and 1–4% werehomozygous for the H63D mutation. The proportions ofindividuals possessing each genotype were not significantly
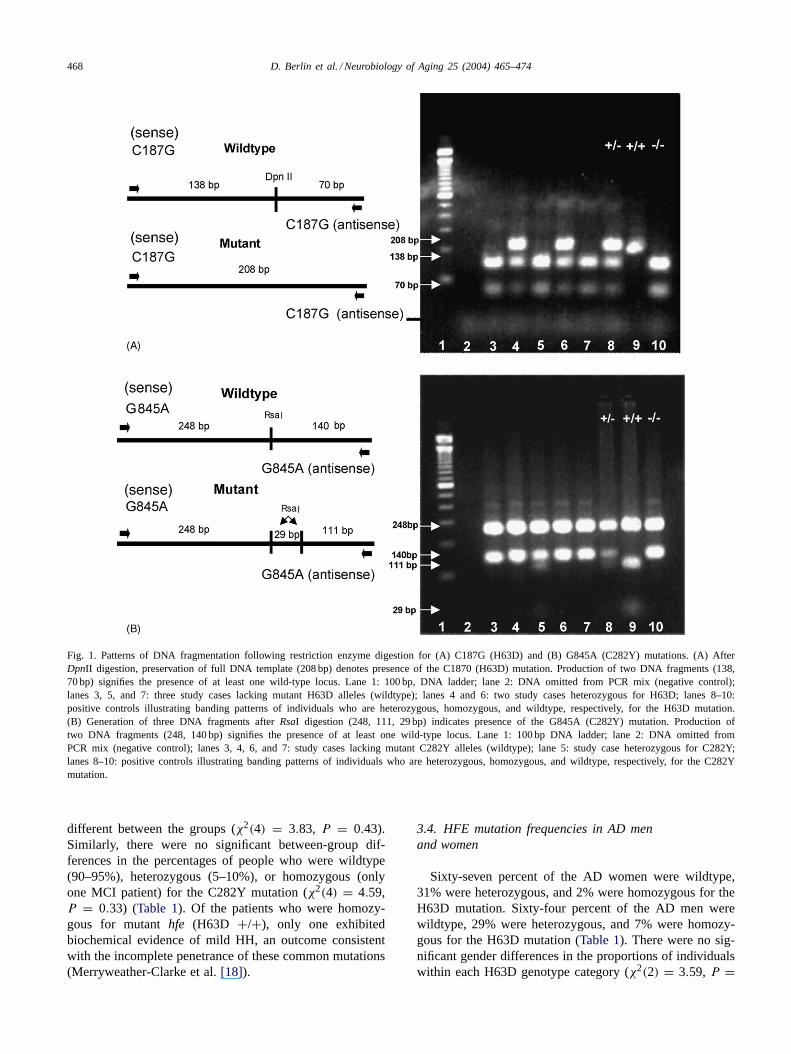
468 D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474
Fig. 1. Patterns of DNA fragmentation following restriction enzyme digestion for (A) C187G (H63D) and (B) G845A (C282Y) mutations. (A) AfterDpnII digestion, preservation of full DNA template (208 bp) denotes presence of the C1870 (H63D) mutation. Production of two DNA fragments (138,70 bp) signifies the presence of at least one wild-type locus. Lane 1: 100 bp, DNA ladder; lane 2: DNA omitted from PCR mix (negative control);lanes 3, 5, and 7: three study cases lacking mutant H63D alleles (wildtype); lanes 4 and 6: two study cases heterozygous for H63D; lanes 8–10:positive controls illustrating banding patterns of individuals who are heterozygous, homozygous, and wildtype, respectively, for the H63D mutation.(B) Generation of three DNA fragments afterRsaI digestion (248, 111, 29 bp) indicates presence of the G845A (C282Y) mutation. Production oftwo DNA fragments (248, 140 bp) signifies the presence of at least one wild-type locus. Lane 1: 100 bp DNA ladder; lane 2: DNA omitted fromPCR mix (negative control); lanes 3, 4, 6, and 7: study cases lacking mutant C282Y alleles (wildtype); lane 5: study case heterozygous for C282Y;lanes 8–10: positive controls illustrating banding patterns of individuals who are heterozygous, homozygous, and wildtype, respectively, for theC282Ymutation.
different between the groups (χ2(4) = 3.83, P = 0.43).Similarly, there were no significant between-group dif-ferences in the percentages of people who were wildtype(90–95%), heterozygous (5–10%), or homozygous (onlyone MCI patient) for the C282Y mutation (χ2(4) = 4.59,P = 0.33) (Table 1). Of the patients who were homozy-gous for mutanthfe (H63D +/+), only one exhibitedbiochemical evidence of mild HH, an outcome consistentwith the incomplete penetrance of these common mutations(Merryweather-Clarke et al.[18]).
3.4. HFE mutation frequencies in AD menand women
Sixty-seven percent of the AD women were wildtype,31% were heterozygous, and 2% were homozygous for theH63D mutation. Sixty-four percent of the AD men werewildtype, 29% were heterozygous, and 7% were homozy-gous for the H63D mutation (Table 1). There were no sig-nificant gender differences in the proportions of individualswithin each H63D genotype category (χ2(2) = 3.59, P =

D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474 469
Table 1Genotype frequencies of the H63D and C282Y mutations among the AD, MCI, and NEC subjects and as a function of gender in the AD cohort (P > 0.05for each comparison)
Type Genotype frequencies (%)
H63D C282Y
Diagnosis Gender Diagnosis Gender
NEC MCI AD Male Female NEC MCI AD Male Female
Wildtype 66 74 67 64 67 90 94 95 95 95Heterozygous 30 25 29 29 31 10 5 5 5 5Homozygous 4 1 4 7 2 0 1 0 0 0
0.17). Similarly, there were no gender influences on the dis-tribution of AD cases among the C282Y genotype categories(χ2(1) = 0.01, P = 0.92) (Table 1). Ninety-five percent ofAD men and women were wildtype and 5% were heterozy-gous for C282Y. There were no C282Y homozygotes in theAD group.
3.5. Effect of HFE mutations on age of AD symptom onsetand diagnosis
There were no statistically significant differences in theage of symptom onset between AD patients who were wild-type (median age 76;n = 140), heterozygous (74;n = 64),or homozygous (76;n = 9) for the H63D mutation (χ2(2) =0.67, P = 0.72; Fig. 2A). Neither were there significantdifferences in the age of AD diagnosis among patients whowere wildtype (median age 79), heterozygous (age 77), orhomozygous (age 80) for H63D (χ2(2) = 1.79, P = 0.41;Fig. 2B). Similarly, there were no significant differences inthe age of symptom onset between individuals who werewildtype (median age 75;n = 202) or heterozygous (75;
Fig. 2. KM survival curves denoting the effect of the H63D mutation on age of AD symptoms onset (A) and age of AD diagnosis (B). (A) Thereare no statistically significant differences in the age of AD symptoms onset between patients who are wildtype (n = 140), heterozygous (n = 64), orhomozygous (n = 9) for the H63D mutation (χ2(2) = 0.67, P = 0.72). (B) There are no significant differences in the age of AD diagnosis amongpatients who are wildtype, heterozygous, or homozygous for the H63D mutation (χ2(2) = 1.79, P = 0.41).
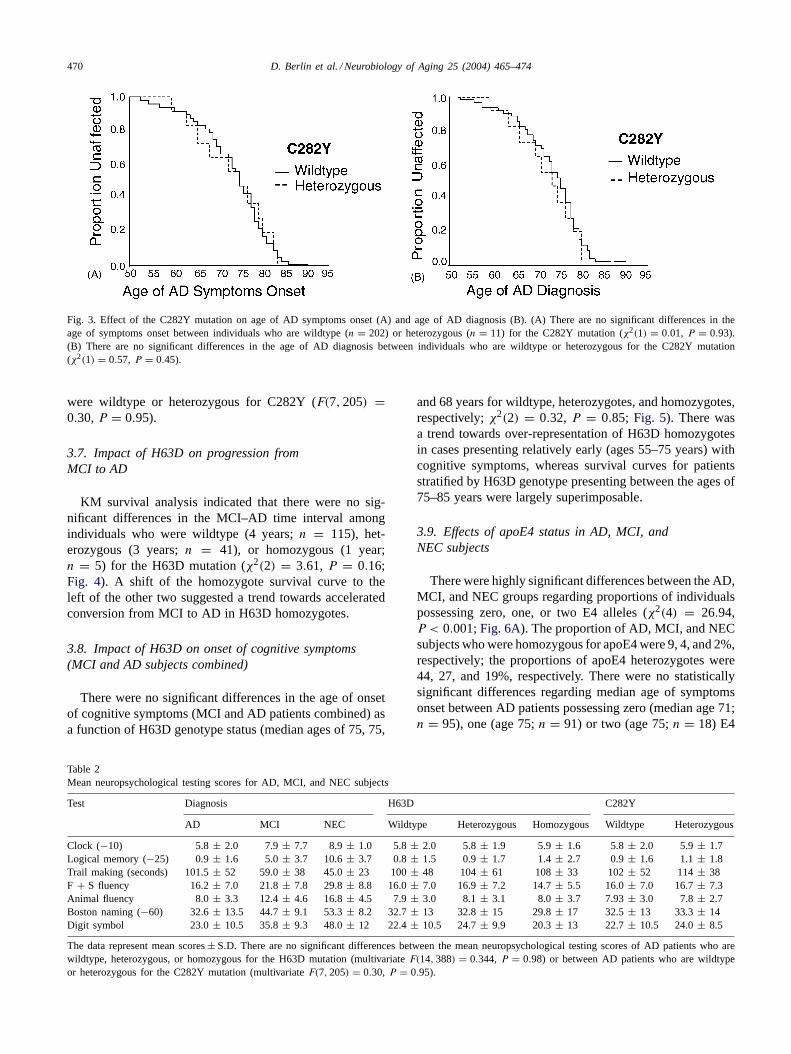
n = 11) for the C282Y mutation (χ2(1) = 0.01, P = 0.93;Fig. 3A). Finally, mean age of AD diagnosis did not dif-fer amongst patients who were wildtype (median age 79) orheterozygous (age 77) for C282Y (χ2(1) = 0.57,P = 0.45;Fig. 3B).
3.6. Effect of HFE mutations on neuropsychologicalscores in AD patients
As expected, mean neuropsychological test scores forthe AD patients were significantly worse overall than thoseof the NEC and MCI cohorts (Wilks’ Lambda= 0.238,F(14, 746) = 55.9, P < 0.001) with MCI scores invari-ably falling intermediate between the other two groups.Follow-up univariate ANOVAs indicated that this patternwas obtained for all individual neuropsychological tests (allP-values<0.05). There were no significant differences inmean neuropsychological testing scores between AD pa-tients who were wildtype, heterozygous, or homozygousfor the H63D mutation (F(14, 388) = 0.344, P = 0.98;Table 2). Similar results were obtained for AD patients who
470 D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474
Fig. 3. Effect of the C282Y mutation on age of AD symptoms onset (A) and age of AD diagnosis (B). (A) There are no significant differences in theage of symptoms onset between individuals who are wildtype (n = 202) or heterozygous (n = 11) for the C282Y mutation (χ2(1) = 0.01, P = 0.93).(B) There are no significant differences in the age of AD diagnosis between individuals who are wildtype or heterozygous for the C282Y mutation(χ2(1) = 0.57, P = 0.45).
were wildtype or heterozygous for C282Y (F(7, 205) =0.30, P = 0.95).
3.7. Impact of H63D on progression fromMCI to AD
KM survival analysis indicated that there were no sig-nificant differences in the MCI–AD time interval amongindividuals who were wildtype (4 years;n = 115), het-erozygous (3 years;n = 41), or homozygous (1 year;n = 5) for the H63D mutation (χ2(2) = 3.61, P = 0.16;Fig. 4). A shift of the homozygote survival curve to theleft of the other two suggested a trend towards acceleratedconversion from MCI to AD in H63D homozygotes.
3.8. Impact of H63D on onset of cognitive symptoms(MCI and AD subjects combined)
There were no significant differences in the age of onsetof cognitive symptoms (MCI and AD patients combined) asa function of H63D genotype status (median ages of 75, 75,
Table 2Mean neuropsychological testing scores for AD, MCI, and NEC subjects
Test Diagnosis H63D C282Y
AD MCI NEC Wildtype Heterozygous Homozygous Wildtype Heterozygous
Clock (−10) 5.8± 2.0 7.9± 7.7 8.9± 1.0 5.8± 2.0 5.8± 1.9 5.9± 1.6 5.8± 2.0 5.9± 1.7Logical memory (−25) 0.9± 1.6 5.0± 3.7 10.6± 3.7 0.8± 1.5 0.9± 1.7 1.4± 2.7 0.9± 1.6 1.1± 1.8Trail making (seconds) 101.5± 52 59.0± 38 45.0± 23 100± 48 104± 61 108± 33 102± 52 114± 38F + S fluency 16.2± 7.0 21.8± 7.8 29.8± 8.8 16.0± 7.0 16.9± 7.2 14.7± 5.5 16.0± 7.0 16.7± 7.3Animal fluency 8.0± 3.3 12.4± 4.6 16.8± 4.5 7.9± 3.0 8.1± 3.1 8.0± 3.7 7.93± 3.0 7.8± 2.7Boston naming (−60) 32.6± 13.5 44.7± 9.1 53.3± 8.2 32.7± 13 32.8± 15 29.8± 17 32.5± 13 33.3± 14Digit symbol 23.0± 10.5 35.8± 9.3 48.0± 12 22.4± 10.5 24.7± 9.9 20.3± 13 22.7± 10.5 24.0± 8.5
The data represent mean scores± S.D. There are no significant differences between the mean neuropsychological testing scores of AD patients who arewildtype, heterozygous, or homozygous for the H63D mutation (multivariateF(14, 388) = 0.344, P = 0.98) or between AD patients who are wildtypeor heterozygous for the C282Y mutation (multivariateF(7, 205) = 0.30, P = 0.95).
and 68 years for wildtype, heterozygotes, and homozygotes,respectively;χ2(2) = 0.32, P = 0.85; Fig. 5). There wasa trend towards over-representation of H63D homozygotesin cases presenting relatively early (ages 55–75 years) withcognitive symptoms, whereas survival curves for patientsstratified by H63D genotype presenting between the ages of75–85 years were largely superimposable.
3.9. Effects of apoE4 status in AD, MCI, andNEC subjects
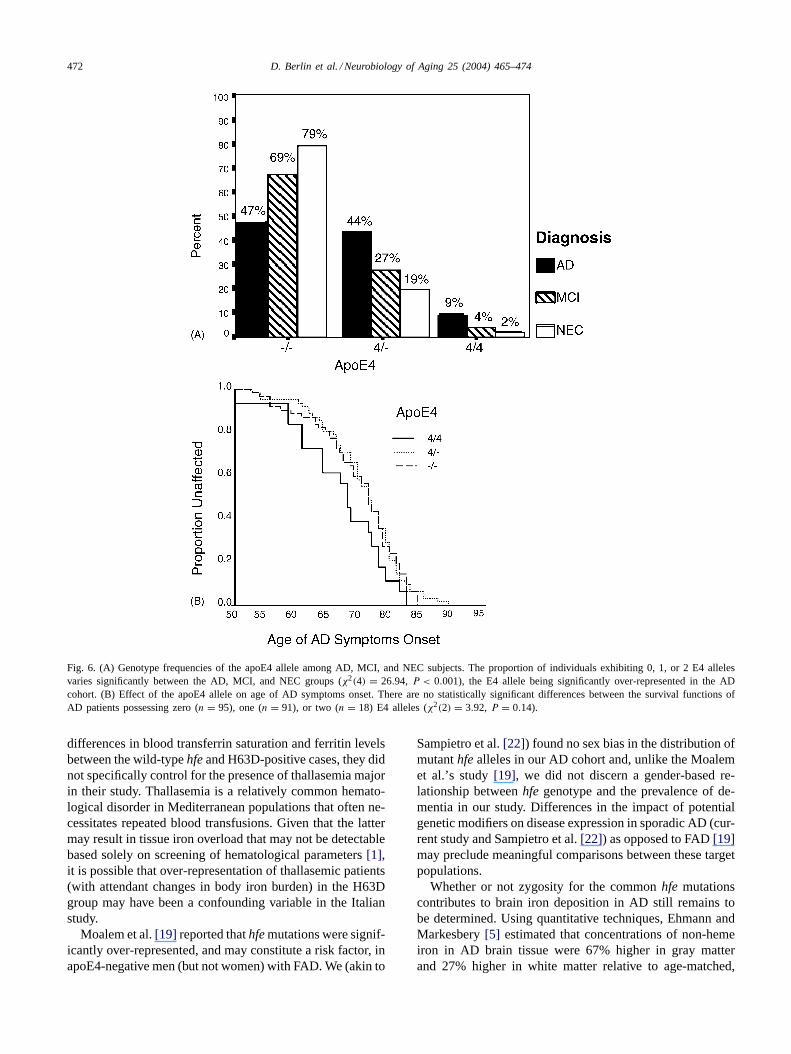
There were highly significant differences between the AD,MCI, and NEC groups regarding proportions of individualspossessing zero, one, or two E4 alleles (χ2(4) = 26.94,P < 0.001;Fig. 6A). The proportion of AD, MCI, and NECsubjects who were homozygous for apoE4 were 9, 4, and 2%,respectively; the proportions of apoE4 heterozygotes were44, 27, and 19%, respectively. There were no statisticallysignificant differences regarding median age of symptomsonset between AD patients possessing zero (median age 71;n = 95), one (age 75;n = 91) or two (age 75;n = 18) E4

D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474 471
Fig. 4. Effect of the H63D mutation on progression from MCI to AD.There are no significant differences in the MCI–AD time interval amongindividuals who are wildtype (n = 115), heterozygous (n = 41), orhomozygous (n = 5) for the H63D mutation (χ2(2) = 3.61, P = 0.16).Patients homozygous for H63D exhibit a trend towards accelerated rateof conversion from MCI to AD. The plus signs denote individuals whowere censored at the time points specified.
Fig. 5. Effect of the H63D mutation on age of symptoms onset in MCIand AD subjects combined. There are no significant differences in theage of symptoms onset between patients who are wildtype, heterozygous,or homozygous for H63D (χ2(2) = 0.32, P = 0.85). A trend towardsearlier cognitive deficits is noted in H63D homozygotes aged 55–75 yearsrelative to the other groups.
alleles (χ2(2) = 3.92, P = 0.14; Fig. 6B). No statisticallysignificant associations were noted between the apoE4 alleleand either the H63D mutation (χ2(2) = 4.04, P = 0.13) orthe C282Y mutation (χ2(2) = 4.80, P = 0.09) in the ADcohort.
4. Discussion
Two previously published case-control studies have raisedthe intriguing possibility that the commonhfe mutations,H63D and C282Y, may be genetic modifiers of AD. In 2000,Moalem et al.[19] reported that the presence of mutanthfe
alleles may put apoE4-negative men at increased risk formanifesting FAD. In the following year, Sampietro et al.[22] presented evidence that sporadic AD patients heterozy-gous for H63D develop AD, on average, 5 years earlier thanthose with wild-typehfe. In that study, prevalence of theH63D mutation was five times higher in AD patients withsymptoms onset before age 70 than in individuals present-ing after the age of 80. The authors of both studies con-cluded thathfe mutations may contribute to pathologicalbrain iron deposition and earlier manifestation of symptomsin AD patients. If affirmed, serial phlebotomy or iron chela-tion therapy may constitute a rational intervention to slowAD progression or forestall the conversion of MCI to ADin patients harboringhfe mutations. In the current study,we attempted to delineate further the role of the H63D andC282Y alleles as potential genetic modifiers of sporadic ADin well-characterized cohorts followed in the JGH/McGillUniversity Memory Clinic. Advantages of our study designcompared to those of Moalem et al.[19] and Sampietro et al.[22] included larger sample sizes, analysis of an MCI group,estimation of MCI-to-AD conversion rates and assessmentof the impact ofhfe genotype on specific cognitive domains.In addition, we and Moalem et al.[19] evaluated ethnicallymixed populations characteristic of major Canadian urbancenters whereas the data set from the Sampietro et al.[22]study was derived from a more genetically restricted Italianpopulation. Attesting to the representativeness of our studypopulations, (a) H63D and C282Y allele frequencies in oursubjects fell within the ranges reported for these mutationsworldwide [10] and (b) as would be anticipated[25], theapoE4 allele was significantly over-represented in the ADpatients relative to the NEC group. A limitation of our studyis that case accrual at a tertiary care facility may not accu-rately reflect disease and genotype distributions in the com-munity at large.
Consistent with the observations of Sampietro et al.[21],the results of the present study indicate that the occurrenceof H63D and C282Y mutations do not differ significantlyamong AD, MCI, and NEC subjects. Moreover, we observedno significant impact of H63D or C282Y heterozygosity onage at AD symptoms onset, age at AD diagnosis, age at on-set of cognitive symptoms (AD and MCI combined), ratesof MCI-to-AD conversion or severity of neuropsychologicaldeficits. Patients homozygous for H63D exhibited trendstowards accelerated MCI-to-AD conversion rates and, in asubset of younger individuals (aged 55–75), earlier onsetof cognitive symptoms relative to wild-typehfe and H63Dheterozygotes. These tendencies failed to reach statisticalsignificance, possibly due to the small numbers of H63Dhomozygotes enrolled in this study.
The results of the present study contest the conclusion ofSampietro et al.[22] that clinical manifestations of sporadicAD are expressed earlier in H63D heterozygotes relative toAD patients with wild-typehfe. Variations in sample size andpopulation demographics may account for these discrepantobservations. Although Sampietro et al.[22] reported no
472 D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474
Fig. 6. (A) Genotype frequencies of the apoE4 allele among AD, MCI, and NEC subjects. The proportion of individuals exhibiting 0, 1, or 2 E4 allelesvaries significantly between the AD, MCI, and NEC groups (χ2(4) = 26.94, P < 0.001), the E4 allele being significantly over-represented in the ADcohort. (B) Effect of the apoE4 allele on age of AD symptoms onset. There are no statistically significant differences between the survival functions ofAD patients possessing zero (n = 95), one (n = 91), or two (n = 18) E4 alleles (χ2(2) = 3.92, P = 0.14).
differences in blood transferrin saturation and ferritin levelsbetween the wild-typehfe and H63D-positive cases, they didnot specifically control for the presence of thallasemia majorin their study. Thallasemia is a relatively common hemato-logical disorder in Mediterranean populations that often ne-cessitates repeated blood transfusions. Given that the lattermay result in tissue iron overload that may not be detectablebased solely on screening of hematological parameters[1],it is possible that over-representation of thallasemic patients(with attendant changes in body iron burden) in the H63Dgroup may have been a confounding variable in the Italianstudy.
Moalem et al.[19] reported thathfe mutations were signif-icantly over-represented, and may constitute a risk factor, inapoE4-negative men (but not women) with FAD. We (akin to
Sampietro et al.[22]) found no sex bias in the distribution ofmutanthfe alleles in our AD cohort and, unlike the Moalemet al.’s study[19], we did not discern a gender-based re-lationship betweenhfe genotype and the prevalence of de-mentia in our study. Differences in the impact of potentialgenetic modifiers on disease expression in sporadic AD (cur-rent study and Sampietro et al.[22]) as opposed to FAD[19]may preclude meaningful comparisons between these targetpopulations.
Whether or not zygosity for the commonhfe mutationscontributes to brain iron deposition in AD still remains tobe determined. Using quantitative techniques, Ehmann andMarkesbery[5] estimated that concentrations of non-hemeiron in AD brain tissue were 67% higher in gray matterand 27% higher in white matter relative to age-matched,

D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474 473
non-demented controls. In both the AD and Parkinsonbrain, regional concentrations of transferrin binding sitesremain unchanged or vary inversely with the elevated ironstores. These and other observations suggest that the trans-ferrin pathway of iron mobilization, important for normaliron delivery to most peripheral tissues, plays little orno role in the pathological accumulation of brain iron inthese aging-related neurodegenerative disorders[23]. Thewild-type HFE protein appears to dampen the affinity ofthe transferrin receptor for diferric transferrin, thereby cur-tailing iron influx into cells. In HH, mutations of the HFEprotein disinhibit this transferrin/transferrin receptor inter-action, promoting excessive tissue iron sequestration[7]. Innormal human brain, immunoreactive HFE protein localizesto capillaries, choroid plexus, and ependymocytes, areas richin transferrin receptors. In AD brain, HFE immunostainingoccurs additionally in the vicinity of neuritic plaques and inperivascular astrocytes[3]. Given the fact that pathologicalbrain iron deposition in AD is largely independent of thetransferrin pathway (vide supra), heterozygosity for H63Dor C282Y may have little impact on CNS iron stores in thiscondition. The possibility remains, however, that mutantHFE may augment sequestration of transferrin-derived ironin AD-affected brain regions by facilitating interactions be-tween diferric transferrin and a normal or even diminishedcomplement of central transferrin receptors.
In contradistinction to earlier reports, the results of thepresent study do not implicate heterozygosity for the com-mon HFE mutations as a genetic modifier of sporadic AD(and MCI). As such, serial phlebotomy or metal chelationtherapy specifically targeting H63D and C282Y heterozy-gotes with MCI or early AD cannot be currently advocated.Further studies, both clinical and fundamental, addressingthe important issue of redox-active metal deposition in ADbrain are clearly warranted.
Acknowledgments
We thank Adrienne Liberman for excellent technical as-sistance, Drs. Nora Kelner and Lennie Babins for neuropsy-chological testing and the JGH/McGill University MemoryClinic staff for help with patient recruitment. We also ac-knowledge the assistance of the Consultation Service of theCentre for Clinical Epidemiology and Community Studies(JGH) in the analysis and presentation of the data. This studywas supported by the Canadian Institutes of Health Research(H.M.S.), Alzheimer’s Society of Canada (H.C., H.B.) andthe Fonds de la Recherche en Santé du Québec (H.M.S.,H.C., H.B.).
References
[1] Brittenham GM, Cohen AR, McLaren C, Martin MB, GriffithPM, Nienhuis AW, et al. Hepatic iron stores and plasma ferritin
concentrations in patients with sickle cell anemia and thallasemiamajor. Am J Hematol 1993;42:81–5.
[2] Chertkow H, Bergman H, Schipper HM, Gauthier S, Bouchard R,Fontaine S, et al. Assessment of suspected dementia. Can J NeurolSci 2001;28(Suppl 1):S28–41.
[3] Connor JR, Milward EA, Moalem S, Sampietro M, Boyer P, PercyME, et al. Is hemochromatosis a risk factor for Alzheimer disease?J Alzheimer’s Dis 2001;3:471–7.
[4] Connor JR, Snyder BS, Arosio P, Loeffler DA, LeWitt P. A quan-titative analysis of isoferritins in select regions of aged, parkinsonian,and Alzheimer’s diseased brains. J Neurochem 1995;65:717–24.
[5] Ehmann WD, Markesbery WR. Brain trace elements in Alzheimer’sdisease. Neurotoxicology 1986;7:197–206.
[6] Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA,Basava A, et al. A novel MHC class 1-like gene is mutated inpatients with hereditary haemochromatosis. Nat Genet 1996;13:399–408.
[7] Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N.The hemochromatosis gene product complexes with the transferrinreceptor and lowers its affinity for ligand binding. Proc Natl AcadSci USA 1998;95:1472–7.
[8] Folstein M, Folstein F, McHugh P. “Mini-mental state”: a practicalmethod for grading the cognitive state of patients for the clinician.J Clin Res 1975;12:189–98.
[9] Hixon JE, Vernier DT. Restriction isotyping of human apolipoproteinE by gene amplification and cleavage withHhaI. J Lipid Res1990;31:545–8.
[10] Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A newclinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–72.
[11] Jazwinska EC. Hemochromatosis: a genetic defect in iron meta-bolism. BioEssays 1998;20:562–8.
[12] Jefferies WA, Food MR, Gabathuler R, Rothenberger S, YamadaT, Yasuhara O, et al. Reactive microglia specifically associatedwith amyloid plaques in Alzheimer’s disease brain tissue expressmelanotransferrin. Brain Res 1996;712:122–6.
[13] Kalaria RN, Sromek SM, Grabovac I, Hank SI. Transferrin receptorsof rat and human brain and cerebral microvessels and their status inAlzheimer’s disease. Brain Res 1992;585:87–93.
[14] Leveugle B, Spik G, Peri DB, Bouras C, Fillit HM, HofPR. The iron binding protein lactotransferrin is present inpathologic lesions in a variety of neurodegenerative disorders: acomparative immunohistochemical analysis. Brain Res 1994;650:20–31.
[15] Lyon F, Frank EL. Hereditary hemochromatosis since discovery ofthe HFE gene. Clin Chem 2001;47:1147–56.
[16] Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease.Free Radic Biol Med 1997;23:134–47.
[17] McKhann G, Drachman D, Fostein M, Katzman R, Price D,Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of theNINCDS-ADRDA work group under the auspices of the Departmentof Health and Human Services Task Force on Alzheimer’s disease.Neurology 1984;34:939–44.
[18] Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJH.Global prevalence of putative haemochromatosis mutations. J MedGenet 1997;34:275–8.
[19] Moalem S, Percy ME, Andrews DF, Knick TPA, Wong S, DaltonAJ, et al. Are hereditary hemochromatosis mutations involved inAlzheimer disease? Am J Med Genet 2000;93:58–66.
[20] Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, KokmenE. Mild cognitive impairment: clinical characterization and out-come. Arch Neurol 1999;56:303–8 [Erratum: Arch Neurol 1999;56:760].
[21] Pinero DJ, Hu J, Connor JR. Alterations in the interaction betweeniron regulatory proteins and their iron responsive element in

474 D. Berlin et al. / Neurobiology of Aging 25 (2004) 465–474
normal and Alzheimer’s diseased brains. Cell Mol Biol 2000;46:761–76.
[22] Sampietro M, Caputo L, Casatta A, Meregalli M, Pellagaffi A,Tagliabue J, et al. The hemochromatosis gene affects the age of onsetof sporadic Alzheimer’s disease. Neurobiol Aging 2001;22:563–8.
[23] Schipper HM. Glial iron sequestration and neurodegeneration.In: Schipper HM, editor. Astrocytes in brain aging andneurodegeneration. Austin: Landes; 1998. p. 235–51.
[24] Schipper HM, Cissé S, Stopa EG. Expression of heme oxygenase-1in the senescent and Alzheimer-diseased brain. Ann Neurol1995;37:758–68.
[25] Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M,Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity bindingto beta-amyloid and increased frequency of type 4 allele in late-onsetfamilial Alzheimer disease. Proc Natl Acad Sci USA 1993;90:1977–81.
Related Documents











