CHANTAL BURELOUT ETUDE DE L'EFFET INHIBITEUR DES PROSTAGLANDINES E 2 SUR L'ACTIVATION DU NEUTROPHILE HUMAIN Caractérisation du mécanisme de signalisation Thèse présentée à la Faculté des études supérieures de l'Université Laval dans le cadre du programme de doctorat en Microbiologie-Immunologie pour l'obtention du grade de Philosophiae Doctor (PhD) DEPARTEMENT DE MICROBIOLOGIE-IMMUNOLOGIE FACULTÉ DE MEDECINE UNIVERSITÉ LAVAL QUÉBEC AVRIL 2007 © Chantai Burelout, 2007

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

CHANTAL BURELOUT
ETUDE DE L'EFFET INHIBITEUR DESPROSTAGLANDINES E2 SUR L'ACTIVATION DU
NEUTROPHILE HUMAINCaractérisation du mécanisme de signalisation
Thèse présentéeà la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat en Microbiologie-Immunologiepour l'obtention du grade de Philosophiae Doctor (PhD)
DEPARTEMENT DE MICROBIOLOGIE-IMMUNOLOGIEFACULTÉ DE MEDECINE
UNIVERSITÉ LAVALQUÉBEC
AVRIL 2007
© Chantai Burelout, 2007

Résumé court
Les polynucléaires neutrophiles sont des cellules effectrices majeures de l'inflammation qui
sont rapidement recrutées vers les tissus lésés. L'activation de leurs fonctions dépend de la
présence de médiateurs pro- ou anti-inflammatoires dans le voisinage du site inflammé.
PGE2 inhibe les fonctions du neutrophile et l'objectif de cette thèse était de caractériser les
mécanismes de signalisation inhibiteurs de ce prostanoïde. Nos travaux montrent que PGE2
inhibe l'activation de la PI3-Ky stimulée par un facteur chimiotactique, le fMLP. Cet effet
inhibiteur de PGE2 est transmis par l'intermédiaire des récepteurs EP2 et de la voie de
l'AMPc/PKA. L'inhibition de la PI3-Ky et de ses effecteurs cellulaires via les récepteurs
EP2 est le principal mécanisme qui permet à PGE2 de réguler les fonctions du neutrophile
activées par le fMLP et montre de quelle façon ce médiateur peut inhiber l'activation des
cellules effectrices de la réaction inflammatoire et de la réponse immune innée.

11
Résumé long
Les polynucléaires neutrophiles sont les premières cellules à infiltrer les tissus en réponse à
des facteurs chimiotactiques lors d'une réaction inflammatoire. Leur recrutement vers les
tissus inflammés et l'activation de leurs fonctions sont régulés par des médiateurs qui ont la
propriété d'élever les niveaux d'AMPc intracellulaire, tels que les prostaglandines E2
(PGE2) ou l'adénosine. L'objectif principal de cette étude était de caractériser les
mécanismes de signalisation intracellulaire qui permettent d'expliquer l'inhibition par
PGE2 de l'activation des neutrophiles humains stimulés par un facteur chiomiotactique
d'origine bactérienne, le fMLP.
Les travaux expérimentaux présentés dans cette thèse montrent que PGE2 réduit
notablement l'accumulation de PtdIns(3,4,5)P3: un second messager primordial pour la
chimiotaxie et la production de radicaux oxygénés stimulées par le fMLP, en bloquant le
recrutement de la PI3-Ky vers les sous-unités G(3y libérées par l'activation du récepteur au
fMLP (FPR). La principale conséquence de la réduction de la génération de PtdIns(3,4,5)P3
par PGE2 est la diminution du recrutement à la membrane des principaux effecteurs de la
PI3-Ky qui sont les sérine/thréonine kinases Akt et PDK1, les tyrosine kinases Tec ainsi
que les petites GTPases Rho et Arf. Le recrutement à la membrane de la PKCa stimulé par
le fMLP est également réduit en présence de PGE2. Rho A, Arf et les isoformes classiques
de PKC étant les trois principaux facteurs d'activation de la PLD, nos résultats montrent
que le mécanisme d'inhibition de la PLD par PGE2 réside principalement dans la
diminution du recrutement à la membrane de ses trois cofacteurs d'activation. Les effets
inhibiteurs de PGE2 sont transmis dans le neutrophile par l'intermédiaire des récepteurs EP2
qui stimulent la formation d'AMPc intracellulaire et nous avons montré que l'activation de
la PKA par la voie EP2 est impliquée dans l'inhibition de l'activation de la PI3-Ky et de ses
effecteurs par PGE2.
Nos travaux ont également permis de mettre en évidence un mécanisme alternatif
d'activation d'Akt par le fMLP en présence de PGE2 ou d'adénosine, caractérisé par une
dissociation entre la phosphorylation activatrice d'Akt et son recrutement à la membrane.

111
Ces travaux permettent de mieux comprendre l'effet modulateur de PGE2 sur les fonctions
des neutrophiles humains.

Avant-ProposAu Pr Sylvain Bourgoin, mon directeur de thèse, j'exprime toute ma reconnaissance pour la
confiance qu'il m'a témoignée en m'acceptant dans son laboratoire. Je le remercie
chaleureusement pour le soutien moral et financier qu'il m'a apporté au long de ces années
d'études ainsi que pour la liberté qu'il m'a accordée pour conduire ce projet de recherche.
Je tiens à remercier le Pr Paul Naccache pour l'intérêt qu'il a bien voulu apporté à mes
travaux de recherche et pour avoir accepté d'être mon co-directeur de thèse. Ses conseils
pertinents ont été d'un grand soutien et nos conversations, tant sur des sujets scientifiques
que culturels, ont toujours été stimulantes et appréciées.
Je remercie tous les membres de l'équipe du Pr Bourgoin pour leur collaboration et leur
soutien. J'exprime tout particulièrement ma gratitude à Danièle Harbour, assistante de
recherche, pour la gentillesse, la patience et la disponibilité qu'elle prodigue sans compter
aux étudiants; travailler à ses côtés pendant ces années aura été un grand plaisir. Je remercie
également Nathalie Thibault et Lynn Davis pour leur collaboration et leurs conseils fort
utiles. J'ai aussi beaucoup apprécié les échanges enrichissants que j 'a i eu avec mes
collègues étudiants, Martin Houle, Valérie Garceau, Chantale Bertnachez, Eric Boilard,
François Chouinard et Aminé El-Azreq, ainsi qu'avec les stagiaires post-doctoraux Andrée
Fortin, Sonya Grenier, Christophe Pivot-Pajot et Chen-qi Zhao et je les en remercie.
J'ai également beaucoup apprécié ma collaboration avec les membres de l'équipe du Pr
Paul Naccache. Leur longue expérience dans la signalisation du neutrophile humain et leur
soutien technique et scientifique ont été particulièrement enrichissants. Je remercie tout
particulièrement Sylvain Levasseur pour sa collaboration sur mon projet ainsi
qu'Emmanuelle Rollet-Labelle pour ses bons conseils. La collaboration technique avec
Guillaume Paré, Sébastien Marois et Isaline Boulven a également été très appréciée; qu'ils
soient ici remerciés pour leur gentillesse et leur serviabilité.
Tout au long de ce doctorat, j 'ai eu aussi l'occasion de collaborer avec d'autres équipes du
Centre de Recherche en Rhumatologie et Immunologie, et je remercie particulièrement

Nicolas Flamand pour ses conseils pertinents ainsi que Serge Picard pour les nombreux
services rendus.
Je tiens également à remercier le Dr. Fawzi Aoudjit et la Dre Reem Al-Dakak pour m'avoir
fait connaître le Centre de Recherche en Rhumatologie et Immunologie et ses chercheurs.
Et bien sûr, je remercie chaleureusement Caroline Gilbert pour son soutien qui m'a été
précieux, tant sur le plan amical et que scientifique.
Finalement, je remercie ma famille pour ses encouragements constants, en particulier ma
mère ainsi que mon mari Lahlou pour ses conseils et son support scientifique.
Ma contribution personnelle pour chacun des articles inclus dans cette thèse est précisée
dans le paragraphe suivant. Une première note, exprimée en pourcentage, est attribuée pour
ma contribution technique et une seconde note représente ma contribution à la conception
du projet et à la rédaction des publications.
Chapitre II. Burelout Chantai, Thibault Nathalie, Levasseur Sylvain, Simard Sébastien,
Naccache Paul H, Bourgoin Sylvain G. (2004). Prostaglandin E2 inhibits the phospholipase
D pathway stimulated by formyl-Methionyl-Leucyl-Phenylalanine in human neutrophils.
Involvement of EP2 receptors and phosphatidylinositol 3-kinase y. Mol Pharmacol 66 (2):
293-301. (50%, 65%)
Chapitre III. Burelout Chantai, Thibault Nathalie, Harbour Danièle, Naccache Paul H and
Bourgoin Sylvain G. The PGE2-induced inhibition of the PLD activation pathway
stimulated by fMLP in human neutrophils is mediated by PKA at the PI3-Ky level. Article
soumis pour publication à Biochem Pharmacol. (70%, 60%)
Chapitre IV. Chantai Burelout, Paul H. Naccache and Sylvain G. Bourgoin. Dissociation
between the translocation and the activation of Akt in fMLP-stimulated human neutrophils.
Effect of prostaglandin E2. Article soumis pour publication à J Leuk Biol. (90%, 90%)

Table des matières
Résumé court iRésumé long iiAvant-Propos ivTable des matières viListe des schémas ixListe des figures 1Liste des abréviations 3CHAPITRE! 6INTRODUCTION 6
1.1 Introduction générale 71.1.1. L'inflammation 71.1.2. La réponse immune innée 9
1.2. Le polynucléaire neutrophile 101.2.1. Introduction 101.2.2. Le recrutement et la migration trans-endothéliale des neutrophiles 111.2.3. La chimiotaxie 131.2.4. La production de radicaux oxygénés par laNADPH oxydase 161.2.5. La dégranulation 191.2.6. La phagocytose 191.2.7. Synthèse de médiateurs lipidiques 201.2.8. Synthèse de cytokines et chimiokines 20
1.3. Réponses du neutrophile au fMLP 221.3.1. Le fMLP 221.3.2. Le récepteur pour le fMLP : FPR 221.3.3. Signalisation induite par le FPR 24
1.3.3.1. Voie de la PLC|3 : mobilisation de calcium et activation de la PKC 241.3.3.2. Formation de PtdIns(3,4,5)P3 par les PI3-K 26
1.3.3.2.1. La génération du PtdIns(3,4,5)P3 261.3.3.2.2. Les PI3-K 281.3.3.2.3. Les Ptdlns(3,4,5)3 phophatases : SHIP et PTEN 341.3.3.2.4. Akt et PDK1 : des protéines régulées par le PtdIns(3,4,5)P3 35
1.3.3.3. Lestyrosine kinases 371.3.3.3.1. Les kinases Src 381.3.3.3.2. Les kinases Tec 381.3.3.3.3. Pyk2 39
1.3.3.4. Les petites GTPases 391.3.3.4.1. Les Rho-GTPases 401.3.3.4.2. Arf 43
1.3.4. La phospholipase D (PLD) 441.3.5. LesMAPK:p38etp42/44 461.3.6. Activation de la voie de l'AMPc/PKA 47
1.4. Régulation de l'activation des neutrophiles par les agents qui élèvent 1 'AMPc 501.4.1. L'adénosine 50

Vil
1.4.2. Les prostaglandines E2 511.5. Objectifs de l'étude 56
CHAPITRE II 572.1 Résumé 582.2 Abstract 592.3. Introduction 602.4. Materials and methods 622.5Results 67
2.5.1. fMLP-induced PLD activity is inhibited by PGE2 via EP2 receptor 672.5.2. PGE2 inhibition of Ca2+ influx is mediated via EP2 receptor 682.5.3. Effect of PGE2 on recruitment of PKCa, Arf and Rho GTPases to membranes.
692.5.4. PGE2 decreases PKCa, Arf and Rho GTPases recruitment to membranesthrough EP2 receptor 692.5.5. Effect of PGE2 on PtdIns(3,4,5)P3 formation and PI3Ky activity 692.5.6. Effect of PGE2on the fMLP-induced pattern of tyrosine phosphorylation 70
2.6. Figures 722.7. Discussion 82
2.8. Acknowledgements 872.9. Abbreviations 872.10. Bibliography 88
CHAPITRE III 933.1. Résumé 943.2. Abstract 953.3. Introduction 963.4. Materials and methods 973.5.Results 101
3.5.1. Effects of fMLP and PGE2 on PKA activation 1013.5.2. Rôle of PKA in the PGE2-mediated inhibitory effect on fMLP-induced PLDactivity 1023.5.3. Rôle of PKA in the PGE2-mediated inhibitory effect on Ca2+ influx induced byfMLP 1023.5.4. Rôle of PKA in the inhibition of PKCa and small GTPases translocation byPGE2 1033.5.5. Rôle of PKA in the PGE2-mediated inhibitory effect on tyrosinephosphorylation 1043.5.6. Rôle of PKA in the inhibition of pi10y translocation by PGE2 1043.5.7. Phosphorylation of PI3-Ky by PKA 105
3.6. Figures 1073.7. Discussion 117
3.8. Acknowledgement 1233.9. Abbreviations 123
3.10. Bibliography 124CHAPITRE IV 132
4.1. Résumé 1334.2. Abstract 134

V l l l
4.3. Introduction 1354.4. Materials and methods 1374.5.Results 140
4.5.1 .EffectofPGE2onfMLP-induced Akt phosphorylation 1414.5.2. Effect of a PKA inhibitor on the phosphorylation of Akt in the présence ofPGE2 1414.5.3. PGE2 inhibits the translocation of PDK1 and Akt to membranes. Effect of H-89
1424.5.4. Effect ofPGE2 on the p38-MAPKAPK2-Akt pathway 1424.5.5. Effect of cAMP elevating agents on the translocation of pi 10y, Akt and PDK1induced by fMLP 1434.5.6. fMLP-induced Akt activation is maintained in the présence of cAMP elevatingagents 1444.5.7. Effect of PGE2 and of the A2A receptor agonist on Akt kinase activity 1444.5.8. Effect of a PI3-KÔ spécifie inhibitor on the fMLP-induced phosphorylation ofAkt in the présence or the absence of PGE2 145
4.6. Figures 1474.7. Discussion 1594.8. Acknowledgements 1654.9. Abbreviations 1654.10. Bibliography 166
CHAPITRE V 174CONCLUSION 174Bibliographie 182

Liste des schémas
Schéma 1. La réaction inflammatoire
Schéma 2. Modèle de recrutement des neutrophiles dans les tissus
Schéma 3. Organisation spatiale des principaux composants impliqués dans la chimiotaxie
Schéma 4. Assemblage du complexe de la NADPH oxydase dans le neutrophile activé
Schéma 5. Biosynthèse des médiateurs lipidiques
Schéma 6. Formation du PtdIns(3,4,5)P3 par la PI3-K
Schéma 7. Métabolisme des PtdlnsP par les PI-Kinases et les PI phosphatases
Schéma 8. Structure des isoformes de PI3-K de la classe I
Schéma 9. Activation des PI3-K IA et IB.
Schéma 10. Mécanisme d'activation des Rho-GTPases
Schéma 11. Schéma récapitulatif des voies de signalisation stimulées par le fMLP dans le
neutrophile.
Schéma 12. Biosynthèse des prostaglandines par la voie des cyclooxygénases
Schéma 13. Voies de signalisation couplées aux récepteurs EP

Liste des figures
Chapitre II
Figure 1: Effect of PGE2 and EP receptor agonists/antagonists on fMLP-induced PLD
activity
Figure 2: Effects of PGE2 and of EP agonists/antagonists on cytoplasmic free calcium
concentration
Figure 3: Effect of PGE2 on the fMLP-induced translocation of PKCa, Arfl and Rho
GTPases to the membranes
Figure 4: Effect of CAY10399, an EP2 agonist, on the fMLP-induced translocation of
PKCa, Arfl and Rho GTPases to the membranes
Figure 5: Effect of PGE2 and CAY10399 on the fMLP-induced accumulation of
PtdIns(3,4,5)P3
Figure 6: Effect of PGE2 on fMLP-induced pi 10y activity and translocation to membranes
Figure 7: Effect of PGE2 and CAY10399 on the global profile of tyrosine phosphorylation
and on the translocation of Tec kinases induced by fMLP
Chapitre III
Figure 1: Activation of PKA by fMLP and PGE2
Figure 2: Effect of H-89 on the PGE2-induced inhibition of PLD activity stimulated by
fMLP
Figure 3: Effect of H-89 on the EP2-mediated inhibition of calcium influx stimulated by
fMLP

Figure 4: Effect of H-89 on the EP2-mediated inhibition of PKCcx, Arf and Rho-GTPases
translocation stimulated by fMLP
Figure 5: Effect of H-89 on the PGE2-induced inhibition of tyrosine phosphorylation of
proteins stimulated by fMLP
Figure 6: Effect of H-89 on the EP2-mediated inhibition of pi 1 Oy translocation stimulated
with fMLP
Figure 7: PKA phosphorylation assay
Chapitre IV
Figure 1: Effect of PGE2 and CAY10399 on fMLP-induced Akt phosphorylation
Figure 2: Effect of H-89 on fMLP-induced Akt phosphorylation
Figure 3: Effect of PGE2 on fMLP-induced Akt and PDK1 translocation
Figure 4: Effect of PGE2 and CAY10399 on p38 and MAPKAPK-2 phosphorylation
Figure 5: Effect of cAMP elevating agents on fMLP-induced pi lOy translocation
Figure 6: Effect of cAMP-elevating agents on fMLP-induced Akt phophorylation
Figure 7: Enzymatic activity of Akt in fMLP-stimulated PMNs
Figure 8: Effect of IC87114 on fMLP-induced Akt phosphorylation in the présence of
PGE2
Figure 9: Model of Akt signaling pathway in the présence of PGE2

Liste des abréviations
AAACADAAINSAKAPAMPcArfARNmATPAH23848
BtkC5aC3biCa2+
CaM-KCAY10399
CBCDCGS-21680
CGS-15943COXCRDAGDFPDHEPERKFADFAKFcyFPRFPR-LfMLPGAPGDIGDPGEFGRKGROGTP
Acide arachidoniqueAdénylate cyclaseAdénosine déaminaseAgent anti-inflammatoire non stéroïdienA- kinase anchoring proteinAdénosine 3'-5'-monophosphate cycliqueADP-rybosilation factorAcide ribonucléique messagerAdénosine triphosphate1 a (Z),2B,5a]-(±)-7-[5-[[(l, 1 '-biphenyl)-4-yl]methoxy]-2-(4-morpholinyl)-3-oxocyclopentyl]-4-heptenoic acidBurton tyrosine kinaseFragment C5a du complémentFragment C3bi du complémentCalciumCalmoduline kinase9-oxo-l l a , 1 S-dihydroxy-17-cyclobutyl-prosta-5Z,13E-dien1 -oie acidCytochalasine BCluster of Differentiation2-p-(2-carboxyethyl)phenethylamino-5'-N-ethylcarboxiamido adénosine hydrochloridechloro-2-(2-furyl)[l,2,4]triazolo[l,5-c]quinazolin-5-amineCyclooxygénaseComplément ReceptorDiacylglycérolDi-isopropylfluorophosphateDbl homologyE prostaglandinExtracellular signal-Regulated KinasesFlavine adénine dinucléotideFocal-adhesion kinaseFragment constant des immuhoglobulines GN-formyl-peptide receptorN-formyl-peptide receptor-likeN-formyl-methionyl-leucyl-phenylalanineGTPase activating proteinGDP dissociation inhibitorGuanine diphosphateGuanine nucleotide exchange factorG-protein-coupled receptor kinaseGrowth-related gène productGuanine triphosphate

GSK-3G-CSFGM-CSFHBSSHMHRH-89
IBMXICAMILIns(l,4,5)P3ITAMJNKLFALFM-A13LPALPSLTBLXA5-LOMAPKMAPKAPK-2MCPM-CSFMIPMMPMPONADPHPAPAFPAKPAMPPButPDEPDK1PEtPI3-KPGPGHSPHPhoxPI(4)P 5-KPKAPKCPLAPLC
Glycogen synthase kinase-3Granulocyte-colony stimulating factorGranulocyte-macrophage colony-stimulating factorHank's balanced sait solutionHydrophobic motifHomology régionN-[2-p-bromo-cinnamylamino)ethyl]-5-isoquinolinesulfonamide3 -isobuty 1-1 -methy lxanthineIntracellular adhésion moléculeInterleukineInositol (l,4,5)phosphate 3Immunoreceptor tyrosine-based activation motifC-Jun N-terminal kinaseLymphocyte function-associated antigenAnalogue Al3 du leflunomideLysophosphatidic acidLipopolysaccharideLeucotriène BLipoxine A5-LipoxygénaseMitogen-activated protein kinaseMitogen-activated protein kinase-activated protein kinase-2Monocyte chemotactic proteinMacrophage-colony stimulating factorMacrophage inflammatory proteinMatrix metalloproteinase (métalloprotéase)MyéloperoxydaseNicotinamide adénine dinucléotide phosphatePhosphatidic acidPlatelet-activating factorp21-activated kinasePathogen-associated molecular patternPhosphatidyl-butanolPhosphodiestérase3-phosphoinositide-dependent kinase-1Phosphatidyl-ethanolPhosphatidylinositol 3-kinaseProstaglandineProstaglandin H synthasePleckstrin homologyPhagocyte oxydasePhosphatidylinositol (4)phosphate 5-kinasecAMP-dependent protein kinase (Protéine kinase A)Protéine kinase CPhospholipase APhospholipase C

PLDPMAPP2
PP2APSPtdlnsPtdlns PPtdIns(3)PPtdIns(3,4,5)P3
PtdIns(3,4)P2
PtdIns(4,5)P2
PTENPXRBDRCPGRLOROCKSHSHIPTGFTLRTNFTHTPAVEGFWASP
Phospholipase DPhorbol-myristate acétate(4-amino-5-(4-chloro-phenyl)-7-(/-butyl)pyrazolo[3,4-</]pyrimidine)Protein phosphatase 2 APho sphatidy 1 serinePhosphatidylinositolPhosphatidylinositol phosphatePhosphatidylinositol 3-phosphatePhosphatidylinositol 3,4,5- triphosphatePhosphatidylinositol 3,4-diphosphatePhosphatidylinositol 4,5-diphosphateTensin homolog deleted on chromosome 10PhoXRas binding domainRécepteur couplé aux protéine G hétérotrimériquesRadicaux libres oxygénésRho-associated kinaseSrc-homologySH2-containing 5' inositol phosphataseTransforming growth factorToll-like receptorTumor necrosis factorTec homology12-O-tetradecanoylphorbol-13-acétateVascular endothelial growth factorWiskott-Aldrich syndrome protein

CHAPITRE I
INTRODUCTION

1.1 Introduction générale
1.1.1. L'inflammationL'inflammation est une réponse des tissus vivants vascularisés à une agression qui peut être
de nature physique (mécanique, radiations, brûlure), chimique, infectieuse (introduction de
pathogènes) ou immunologique (formation de complexes immuns, réactions auto-immunes,
allergie). Le processus inflammatoire est habituellement bénéfique car il permet la
réparation des tissus lésés ou l'élimination d'agents pathogènes. Dès l'antiquité,
l'inflammation a été caractérisée par l'apparition de quatre signes cliniques dits
« cardinaux » : rougeur, douleur, chaleur et gonflement. Ces signes cliniques résultent en
fait d'une cascade de réactions cellulaires et moléculaires provoquant une vasodilatation,
l'augmentation de perméabilité vasculaire et la fuite de protéines plasmatiques, l'activation
de complexes de protéines plasmatiques, la migration de divers types cellulaires vers le site
inflammatoire, la synthèse et la libération de nombreux médiateurs. L'activité protéolytique
des enzymes libérées et la formation de radicaux libres provoquent la dégradation des tissus
inflammés, qui est normalement suivie d'une phase de régénération et de cicatrisation des
tissus lésés.
La reconnaissance des signaux d'initiations (exogènes ou endogènes) provoque l'activation
de quatre systèmes protéolytiques plasmatiques : le système de contact (kinine-
bradykinine), la voie du complément, la coagulation et le système fibrinolytique. La
libération de médiateurs, de nature protéique (cytokines) ou lipidique (PAF, leucotriènes)
active les leucocytes circulants ainsi que les cellules de l'endothélium vasculaire,
permettant la margination et l'adhésion des leucocytes puis le passage de la barrière
endothéliale par diapédèse afin de migrer vers les tissus lésés. La composition en différents
types cellulaires retrouvés au site inflammatoire peut varier selon les tissus et l'agent causal
de l'inflammation, mais les polynucléaires neutrophiles sont en général les premières
cellules qui migrent vers les tissus suivis des monocytes/macrophages, et éventuellement
des lymphocytes. La régulation fine de la libération de médiateurs et de l'activation des
cellules impliquées dans le processus inflammatoire permet la résolution de la réaction
inflammatoire. Dans le cas d'une persistance de l'agent causal ou d'un défaut de régulation
du processus, la réaction inflammatoire évolue vers la chronicité, ce qui conduit à la

destruction des tissus et à l'apparition de pathologies comme par exemple la polyarthrite
rhumatoïde.
LA RÉACTION INFLAMMATOIRE
Signaux d'initiation
Exogène(bactéries, virus, irradiation,
destruction cellulaire)
Endogène(complexes immuns antigène)
Activation
Systèmes protéolytiquesde contact (facteur Hageman, Kinines...)
voie alterne du complément (C3)coagulation (facteurs de la coagulation)
fibrinolytique. (fibronigène)
Cellules endothéliales
AdhésionActlVBtlon Migration
ThromUniLPSUpkto. ILTB4...)IL1.TNF
Chlmloklne. (ILS, MCP1..JLipides (PAF, LTB4...JAutre! (C5a, FLMP...)Pro l«r»t d'tdheslon ( C D « )
ChlrnlohlnBflLipides (PAF, LTB4...)Cytoklnet (IL1. TNF, tFN/l
Réponse cellulaire
Immédiate Retardée
Résolution ou chronicité ?
Schéma 1 : La réaction inflammatoire

1.1.2. La réponse immune innée.
La réponse immunitaire de l'organisme aux micro-organismes pathogènes est
classiquement décrite comme ayant deux composantes : l'immunité innée ou réponse
immune innée, qui n'est pas spécifique d'un pathogène particulier et l'immunité acquise ou
réponse immune acquise, caractérisée par un haut niveau de spécificité ainsi que par la
propriété de « mémoire » immunologique. L'immunité innée, phylogénétiquement plus
ancienne que l'immunité acquise, est présente dans tous les organismes pluricellulaires
alors que l'immunité acquise, plus récente dans l'évolution, n'apparaît que chez les
vertébrés. La réponse immunitaire innée permet une reconnaissance et une réponse rapides
contre les agents pathogènes et elle fait appel à des mécanismes encodés dans les cellules
germinales. Les principales cellules effectrices de la réponse immune innée sont les
polynucléaires neutrophiles ainsi que les monocytes/macrophages, les cellules dendritiques
et les cellules NK. Ces différents types cellulaires possèdent sur leur surface des récepteurs
capables de reconnaître des structures exprimées par les micro-organismes infectieux ainsi
que des substances produites par ces micro-organismes ou provenant de leur
reconnaissance par différents systèmes de protéines solubles, comme par exemple le
complément. La connaissance des mécanismes qui régissent la reconnaissance des
pathogènes au niveau de la réponse immune innée s'est considérablement enrichie ces
dernières années avec la découverte d'une nouvelle classe de récepteurs, les récepteurs
Toll-like ou TLRs (Toll-like receptor) capables de reconnaître des motifs moléculaires
propres aux microorganismes dénommés PAMPs (pathogen-associated molecular patterns)
(1). Suite à cette étape de reconnaissance, les microorganismes sont ingérés et détruits par
les neutrophiles et les monocytes/macrophages par un processus appelé phagocytose qui a
été décrit pour la première fois à la fin du XIXeme siècle par le zoologiste Elie Metchnikoff
comme étant le principal mécanisme de défense et d'élimination des agents pathogènes de
l'organisme. Lors de la réponse innée, de nombreux médiateurs solubles sont synthétisés
(cytokines, chimiokines, médiateurs lipidiques) et leur libération génère une réaction
inflammatoire locale, en général limitée dans le temps.

10
1.2. Le polynucléaire neutrophile
1.2.1. IntroductionLe polynucléaire neutrophile est un leucocyte appartenant au sous-groupe des granulocytes
appelés aussi polynucléaires en raison de leur noyau polylobé. La dénomination
« neutrophile » provient du fait que les granules de ce polynucléaire ont la propriété de
fixer les colorants neutres, alors que les granules du polynucléaire éosinophile fixent
l'éosine et que ceux du polynucléaire basophile fixent les colorants basiques. Les
neutrophiles constituent la plus importante population leucocytaire du sang circulant (60-
70% des leucocytes sanguins) et les principales cellules phagocytaires dans la circulation
sanguine. Les personnes présentant une neutropénie, congénitale ou acquise, souffrent de
pathologies infectieuses récurrentes, ce qui témoigne bien du rôle primordial du neutrophile
dans la défense contre les microorganismes pathogènes. Les neutrophiles sont issus de la
différenciation et de la maturation de cellules souches pluripotentes de la moelle osseuse.
Leur premier précurseur, le myéloblaste, se différencie en promyélocyte, puis en
myélocyte. A ce stade de développement de la lignée myéloïde, la prolifération des
cellules par mitose prend fin et les myélocytes se différencient en cellules non segmentées
(« band cells ») puis en cellules segmentées dans lesquelles apparaît un noyau multilobé et
enfin en polynucléaire neutrophiles (2). Ce processus de différenciation est régulé par des
facteurs de croissance hématopoïétiques pour les granulocytes tels que le GM-CSF, le G-
CSF et l'IL-3. Environ 100 milliards de neutrophiles terminalement différenciés migrent de
la moelle osseuse vers le sang circulant chaque jour. Leur demi-vie dans la circulation
sanguine est relativement courte : environ 12 heures. Tout au long du processus de
différenciation et de maturation des précurseurs du neutrophile apparaissent des granules
cytoplasmiques qui sont une des principales caractéristiques physiologiques du neutrophile.
Ces granules contiennent essentiellement des enzymes cytolytiques permettant la digestion
des microorganismes phagocytés ainsi que des enzymes qui dégradent les tissus (par
exemple des collagénases) pour faciliter la migration des neutrophiles. Les granules ont été
classifiés en quatre catégories en fonction de leur densité et de leur contenu : les granules
primaires (car ils sont les premiers à apparaître) ou azurophiles, les granules secondaires ou
spécifiques, les granules tertiaires ou gélatinase et les vésicules sécrétoires. Plusieurs

Il
excellentes études ont permis de caractériser la composition en protéines de ces différents
types de granules (3-5).
Les fonctions effectrices du neutrophile qui lui confèrent un rôle majeur dans
l'inflammation et la réponse immune innée sont présentées successivement dans les
paragraphes suivants.
1.2.2. Le recrutement et la migration trans-endothéliale des neutrophiles
Les neutrophiles sont les premières cellules sanguines recrutées vers le site d'infection ou
d'inflammation grâce à des substances chimiotactiques qui peuvent être des chimiokines,
des cytokines (l'interleukine 8, IL-8), les fragments du complément C5a, C3a et C4a, des
médiateurs lipidiques (essentiellement le leucotriène B4, LTB4 et le PAF) ou des produits
d'origine bactérienne comme les peptides formylés (fMLP). Certaines substances, bien que
n'étant pas toujours chimiotactiques par elles-même facilitent le processus de recrutement
et pré-activent les neutrophiles par effet de « priming » ou sensibilisation. Ces substances
peuvent être endogènes (le TNFa, le GM-CSF ou l'IL-1) ou exogènes comme le
lipopolysaccharide (LPS) provenant des bactéries Gram-négatives. Le processus
d'extravasation des neutrophiles peut être divisé en quatre étapes successives : 1) le
roulement, 2) l'arrêt et l'adhésion ferme à l'épithélium vasculaire et 3) l'activation des
cellules par les facteurs chimiotactiques et les cytokines et 4) la migration
transendothéliale. Le roulement du neutrophile sur l'endothélium vasculaire est assuré par
les sélectines, des glycoprotéines membranaires possédant un domaine semblable à des
lectines qui se lie avec les chaînes carbohydrates présentes sur des protéines de type mucine
fortement glycosylées. Lors d'une réponse inflammatoire, des cytokines (IL-1, TNFa)
activent l'expression des sélectines E et P par les cellules endothéliales ainsi que
l'expression des sélectines L à la surface des neutrophiles. La liaison de ces trois types de
sélectines avec leurs ligands respectifs permet un attachement souple des neutrophiles à
l'endothélium vasculaire et ralentit leur entraînement dans le flot du sang circulant.
L'activation des neutrophiles et des cellules endothéliales par les cytokines
proinflammatoires (IL-1 et TNF) stimule la conversion des intégrines d'un état inactif à un

12
MODÈLE DE RECRUTEMENT DU NEUTROPHILE (PMN)
FLUX SANGUINMARGINATION
PMN CAPTURE ROULEMENT ACTIVATIONet ADHESION
IL-1 JTNFa
O |O O| o Y I I
U Sélectineso
D Intégrines
çp Particules étrangères
•.*.* Médiateurs pro-inflammatoires
IL-8, MCP1ADHESION
CELL ULEa El \D C/TilËL JAUS '
. PARTICULES - ". . ÉTRANGÈRESO • .
DIAPEDESE
MIGRATION
PHAGOCYTOSEES PARTICULESÉTRANGÈRES
TJ33U
Tiré de Balzog B., News in Physiol. Sciences, 2000
Schéma 2. Modèle de recrutement des neutrophiles dans les tissus.

état actif par un changement de conformation ainsi que le recrutement d'intégrines à la
membrane plasmique du neutrophile à partir des réservoirs des granules cytoplasmiques.
Les intégrines qui permettent l'adhésion ferme des neutrophiles aux cellules endothéliales
appartiennent à la famille des intégrines (32, fortement exprimées dans les neutrophiles
circulants non activés. Elles sont formées de l'association d'une chaîne a (ou CD11) et
d'une chaîne (32 (CD18). Trois types d'intégrines (32 sont exprimés par le neutrophile :
CD 11 a/CD 18 (ou LFA-1), CD 1 le/CD 18 (ou Mac-1) et CD 1 le/CD 18 (ou gp 150/95) (6).
Leur liaison aux ICAMs ou molécules d'adhésion intercellulaires, des protéines de la
superfamille des immunoglobulines exprimées à la surface des cellules endothéliales
permet l'adhésion ferme des neutrophiles qui précède leur transmigration à travers les
jonctions intercellulaires des cellules épithéliales, un phénomène appelé aussi diapédèse.
Au niveau des tissus, le neutrophile activé peut aussi exprimer des intégrines (31 et (33 qui
se lient à des composants de la matrice extracellulaire (collagène, fibronectine,
vitronectine) ce qui permet sa migration vers le foyer inflammatoire ou infectieux (6-8).
Les sélectines et les intégrines ne sont pas seulement des structures moléculaires qui
permettent des phénomènes mécaniques comme l'adhésion, elles peuvent aussi transmettre
des messages de signalisation intracellulaire qui interagissent avec les signaux
intracellulaires induits dans les neutrophiles par les cytokines pro-inflammatoires et les
facteurs chimiotactiques présents (9-12) (voir Schéma 2: Modèle de recrutement des
neutrophiles dans les tissus).
1.2.3. La chimiotaxie
La chimiotaxie désigne la migration orientée de cellules dans un gradient de substance
chimiotactique des concentrations les plus faibles vers les concentrations les plus élevées.
Les leucocytes, et plus particulièrement les neutrophiles, ont développé des mécanismes
finement régulés qui leur permettent de détecter des faibles concentrations de facteurs
chimiotactiques et d'ajuster leur capacité de motilité de façon à migrer dans un gradient
d'agent chimiotactique des concentrations les plus faibles vers les plus fortes. La
chimiotaxie commence avec la protrusion de pseudopodes ou de lamellipodes à l'avant de
la cellule, c'est-à-dire au pôle cellulaire faisant face au gradient chimiotactique grâce à
l'établissement asymétrique de sites de polymérisation de l'actine, ce qui permet une

14
migration directionnelle (13). Alors que la cellule avance, le pseudopode devient de plus en
plus adhérent, il se remplit avec le contenu intracellulaire et s'aplanit. Le corps cellulaire se
contracte et l'arrière de la cellule se rétracte. D'autres pseudopodes se forment et le cycle se
répète, permettant une migration orientée de la cellule. La détection de faibles
concentrations de facteurs chimiotactiques est assurée par des récepteurs membranaires de
haute affinité pour leur ligands qui, pour la plupart des facteurs (excepté pour le TGF|3),
sont des récepteurs à sept domaines transmembranaires couplés aux protéines G
hétérotrimériques (RCPG). La liaison du facteur chimiotactique provoque la dissociation
des sous-unités Ga et G(3y qui est à l'origine d'une cascade de signalisation intracellulaire
aboutissant à un remodelage rapide et polarisé du cytosquelette permettant à la cellule de se
déformer et de se mouvoir rapidement de façon directionnelle. Sur le plan morphologique,
on observe une polarisation de la cellule qui est indispensable pour permettre la motilité, et
qui est maintenue pendant toute la phase de migration. De nombreuses études utilisant des
modèles cellulaires eucaryotes (neutrophiles) ou procaryotes (Dictyoselium Discoideum)
ont été effectuées afin d'analyser les mécanismes moléculaires impliqués dans la
chimiotaxie, et plus particulièrement ceux qui contrôlent la polarisation et l'établissement
du front de migration de la cellule (14). La liaison des facteurs chimiotactiques à leurs
récepteurs n'induit pas de changement de leur répartition sur la membrane plasmique (15,
16), ni de redistribution spatiale des sous-unités G^y (17), ce qui indique que la polarisation
est contrôlée en aval de l'activation des récepteurs. Il est actuellement bien reconnu que le
premier mécanisme moléculaire à l'origine de la polarisation repose sur le recrutement
asymétrique de la phosphoinositol 3-kinase (PI3-K), principalement de la PD-Ky (18), qui
catalyse la phosphorylation de polyphosphoinositides membranaires, le
phosphatidylinositol-4,5-biphosphate (PtdIns(4,5)P2) en phosphatidylinositol-3,4,5-
triphosphate (PtdIns(3,4,5)P3) (19-21). Simultanément, la phosphoinositide phosphatase
PTEN qui déphosphoryle le PtdIns(3,4,5)P3 en position 3 du groupe inositol pour former du
PtdIns-4,5-P2, s'accumule latéralement ainsi qu'au pôle antérieur de la cellule (22, 23).

15
CHIMIOTAXIE
CHEMOATTRACTANT
Schéma 3. Organisation spatiale des principaux composants impliqués dans la chimiotaxie.
Tiré de Charest P. et al, 2006, Current Opinion in Genetics & Gevelopment, 16.

16
Cette distribution asymétrique de la PI3-K et de PTEN permet de limiter l'accumulation de
PtdIns(3,4,5)P3 dans la région du front de migration et empêche la formation latérale de
pseudopodes. La forte concentration de PtdIns(3,4,5)P3 à l'avant de la cellule permet le
recrutement à la membrane plasmique de protéines, et plus particulièrement de kinases qui
contiennent un domaine PH ayant la propriété de reconnaître et de lier les
phosphoinositides avec une spécificité et une affinité variables. La principale kinase
recrutée au front de migration lors de la chimiotaxie est Akt, une serine/threonine kinase
contenant un domaine PH liant le PtdIns(3,4,5)P3 et le PtdIns(3,4)P2 avec une haute affinité
(24, 25). Les petites protéines G monomériques de la famille Rho (Rho-GTPases), qui sont
des régulateurs essentiels du cytosquelette d'actine (26), jouent également un rôle
important dans la phase de polarisation et elles transmettent les signaux au niveau des
protéines du cytosquelette, en particulier Rac et Cdc42 (27, 28). Le recrutement des Rho-
GTPases à la membrane dépend de la formation de PtdIns(3,4,5)P3 et contribue à créer une
boucle de rétro-contrôle positive qui régule la production de PtdIns(3,4,5)P3 par la PI3-K et
contribue au maintien de la polarité (19, 29, 30). Rac et Cdc42 contrôlent la polymérisation
de l'actine filamenteuse (F-actine) et la formation de pseudopodes et des lamellipodes
respectivement alors que RhoA semble plutôt contrôler la rétraction de l'arrière de la
cellule par la formation de complexes contractiles actine-myosine (31-33). La répartition et
l'organisation des différentes molécules impliquées dans la chimiotaxie dans la cellule sont
représentées dans le schéma 3. Les modes d'activation et d'action de la PI3-K et des Rho-
GTPases seront exposés plus en détails dans la partie «Signalisation » de cette thèse.
1.2.4. La production de radicaux oxygénés par la NADPH oxydase
La production de radicaux oxygénés libres par le neutrophile est une des fonctions qui
permet la neutralisation et la destruction des microorganismes pathogènes ingérés par le
neutrophile. L'activation du neutrophile est accompagnée d'une forte augmentation de sa
consommation d'oxygène dénommée l'explosion (ou la bouffée) oxydative (« respiratory
burst »). Le neutrophile, comme les autres phagocytes, contient le complexe enzymatique
de la NADPH oxydase qui transfert un électron à l'oxygène (O2), le réduisant en anion
superoxyde (O2.") selon la réaction suivante : NADPH + O2 -* NADP+ + H+ + O2'. L'anion
superoxyde, très instable, est rapidement dismuté en peroxyde d'hydrogène (H2O2) : 2O2" +

17
2H+ -» O2 + H2O2. H2O2 sert de co-substrat à la myéloperoxidase (MPO) contenue dans les
granules primaires qui catalyse la formation de HOC1 (acide hypochloreux), un agent
microbicide puissant. La réaction H2O2 + HOC1 produit l'oxygène singulet 'C^, une espèce
oxygénée extrêmement réactive qui attaque les doubles liaisons. De plus, la NADPH
oxydase génère le radical hydroxyl (OH') à partir de H2O2 et en présence de métal (Fe2+) :
H2O2 + M+ -» OH" + OH'+ M2+ (9).
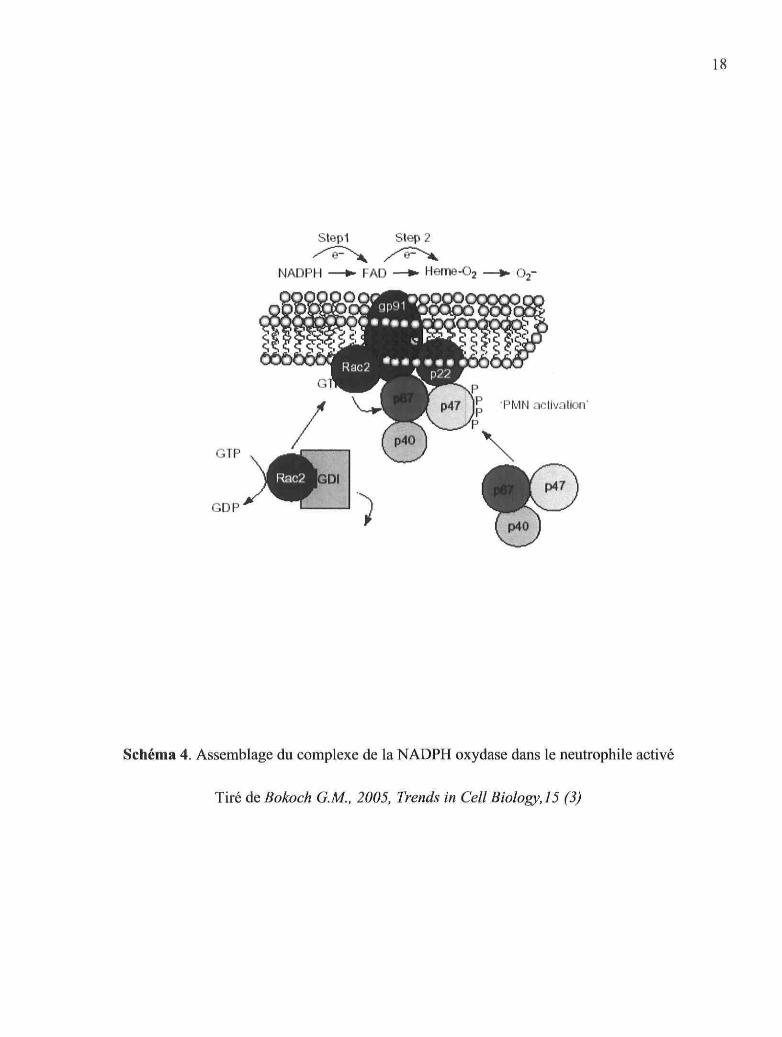
La NADPH oxydase est un complexe enzymatique composé de six éléments : le
cytochrome b558, une flavohémoprotéine dimérique composée de la gp91phox et de la
gp22phox, est associé à la petite protéine G Rapl à la membrane alors que la gp47phox, la
gp67phox, la gp40phox et la petite protéine G monomérique Rac2 sont des éléments
cytosoliques (phox est l'abbréviation de « phagocyte oxydase ») (9). L'activation du
neutrophile par des stimuli chimiotactiques ou phagoeytaires induit la translocation des
composants cytosoliques vers les membranes plasmiques ou granulaires pour former le
complexe enzymatique actif. Le cytochrome b558 peut alors se lier à la NADPH
cytosolique et transférer un électron vers l'oxygène via le FAD et les groupes prosthétiques
de la gp91phox (voir Schéma 4). L'assemblage et l'activation du complexe NADPH oxydase
sont régulés par la phosphorylation de plusieurs de ses composants, notamment de p47phox
(34) et également par des phosphoinositides (PtdIns3P, PtdIns(3,4)P2 et PtdIns(3,4,5)P3) et
des phospholipides acides comme l'acide phosphatidique (PA) (35). Une déficience
génétique de synthèse de l'un des composants du complexe NADPH oxydase est à l'origine
de la maladie granulomateuse chronique. Les patients atteints de cette maladie sont
victimes d'infections récurrentes que leur organisme ne peut combattre en raison de
l'incapacité des granulocytes à produire des radicaux oxygénés, ce qui montre clairement le
rôle essentiel des espèces oxygénées réactives dans la défense anti-microbienne (36).
IX
Stepi
NADPH —*- FAD —*• Heme-O2 —»» o2-
GTP
. . H '
vy
Schéma 4. Assemblage du complexe de la NADPH oxydase dans le neutrophile activé
Tiré de Bokoch G.M., 2005, Trends in Cell Biology, 15 (3)

19
1.2.5. La dégranulation
La libération des enzymes lytiques contenues dans les granules intracytoplasmiques ou
dégranulation est une autre fonction qui participe à l'activité microbicide et
proinflammatoire du neutrophile. Les granules azurophiles (primaires) contiennent une
grande variété d'enzymes microbicides comme le lysozyme, l'élastase, l'al-antitrypsine,
les défensines et la MPO et sont recrutés lors de la phagocytose pour fusionner avec les
phagolysosomes et détruire les microorganismes pathogènes. Les metalloprotéases (MMPs)
(principalement la collagénase, la gélatinase et la leucolysine) contenues dans des granules
spécifiques (secondaires) et gélatinases (tertiaires) sont nécessaires à l'extravasation et à la
migration des neutrophiles dans les tissus. Les granules spécifiques contiennent également
la lactoferrine qui possède une activité microbicide importante en séquestrant le fer
indispensable à la croissance bactérienne. Les membranes des granules spécifiques,
gélatinases et surtout des vésicules sécrétoires contiennent des récepteurs membranaires
comme le récepteur pour le fMLP, des intégrines |32 (CD1 lb/CD18), le CD 14, le récepteur
FcylIIb (CD 16) et servent de réservoir pour ces récepteurs. Lors des premières étapes
d'activation du neutrophile, les vésicules sécrétoires sont recrutées et leur fusion avec la
membrane plasmique permet l'expression des récepteurs à la surface du neutrophile (37).
1.2.6. La phagocytose
La phagocytose est le principal mécanisme qui permet au neutrophile d'ingérer et de
détruire les microorganismes pathogènes. Le neutrophile reconnaît préférentiellement les
pathogènes opsonisés, c'est-à-dire recouvert par des anticorps ou des fragments du
complément, essentiellement C3. Ces opsonines sont reconnues par les récepteurs pour le
fragment Fc des immunoglobulines, qui sont essentiellement les récepteurs FcYRIIa (CD32)
et FcYRlIIb (CD 16) pour le neutrophile et par les récepteurs pour le fragment C3bi du
complément (CR3 ou Macl ou CD1 lb/CD18). Les récepteurs Fcy possèdent cependant une
capacité de phagocytose plus forte que celle des récepteurs du complément. Les récepteurs
FcyRIIa contiennent dans leur partie intracellulaire des motifs ITAM qui sont phosphorylés
par des tyrosine kinases (38). Cette première étape d'une cascade de signalisation aboutit à
un réarrangement du cytosquelette d'actine permettant l'extension de pseudopodes

20
quienglobent la particule dans un phagosome (39) qui est ensuite fusionné avec les granules
spécifiques pour former un phagolysosome dans lequel le microorganisme sera digéré grâce
aux enzymes contenues dans les granules et à la production de radicaux libres oxygénés par
la NADPH oxydase membranaire (40).
1.2.7. Synthèse de médiateurs lipidiques
Le neutrophile activé est capable de générer les trois types de médiateurs lipidiques qui ont
un rôle pro-inflammatoire important : le PAF (41, 42), le LTB4 (43-45) et la prostaglandine
E2 (PGE2) (46). Le PAF et le LTB4 sont essentiellement des facteurs chimiotactiques alors
que PGE2 possède plutôt des propriétés pro-inflammatoires dues à ses effets
vasodilatateurs. La première étape de biosynthèse de ces médiateurs est la libération
d'acide arachidonique (AA) un acide gras en C20 :4 et de lyso-PAF à partir des
phospholipides membranaires par l'activité de la phospholipase A2 (PLA2). L'AA peut être
métabolisé par la 5-lipoxygénase pour générer du LTA4 qui est rapidement hydrolyse en
LTB4 par la LTA4 hydrolase, ou par les cyclooxygénases (COX-1 et COX-2) pour générer
les prostaglandines G2 (PGG2) qui sont transformées en PGE2 par la PGE synthétase. Le
PAF est préférentiellement formé par acétylation du lyso-PAF par une acétylCoA
transférase la biosynthèse des médiateurs lipidiques est illustrée dans le Schéma 5).
1.2.8. Synthèse de cytokines et chimiokines
Bien que le neutrophile ait été considéré pendant longtemps comme une simple cellule
phagocytaire incapable d'activité transcriptionnelle en raison de sa courte durée de vie, de
nombreuses études ont montré depuis une quinzaine d'années qu'il est également capable
de synthétiser des cytokines et des chimiokines jouant un rôle régulateur dans les réponses
inflammatoire et immune, notamment en réponse au LPS. Le neutrophile peut synthétiser
in vitro les principales cytokines pro-inflammatoires : l'IL-la et l'IL-ip, le TNFa, l'IL-6,
l'INF-a et l'INF-y, l'IL-17, PIL-18 ainsi que des cytokines anti-inflammatoires : l'IL-IRA,
le TGF(3 (47). Plusieurs chimiokines peuvent aussi être produites par le neutrophile : l'IL-8,
GRO-a et GRO-p\ MlP-la, MIP-1(3, MCP-1 (48), ainsi que des facteurs de croissance (G-
CSF, M-CSF, GM-CSF) et des facteurs angiogéniques ou fibrogéniques (VEGF, TGF)
(47). La libération de ces facteurs permet le recrutement et l'activation de nouvelles cellules

21
effectrices au site inflammatoire (neutrophiles, macrophages) et constitue un autre
mécanisme par lequel les neutrophiles participent à la défense contre les microorganismes
pathogènes.
.(CH3)
-Choline
1 -hexadécyl,2-arachidonoyl-PC
Activité PLA2
-COOH
Acide Arachidonique
COX-1/COX-2PGE Synthétase
COOH
5-LO/FLAPLTA4 Hydrolase
OH OH
LTB4
HO
,(CH3)
Choline
Lyso-PAF
AcétylCoA:lyso-PAFAcétyltransférase
COOH HOCholine
PAF
Schéma 5. Biosynthèse des médiateurs lipidiques

22
1.3. Réponses du neutrophile au fMLP
1.3.1. Le fMLP
Les bactéries et les mitochondries ont la particularité de synthétiser des peptides dont le
résidu methionine de la partie Nhb-terminale est formylé. L'observation que les bactéries
(E.Coli) peuvent sécréter des agents retrouvés à faible concentration dans les milieux de
cultures et ayant un fort pouvoir chimiotactique a conduit à l'isolement de peptides
formylés qui se sont révélés être les facteurs exerçant un fort effet chimiotactique sur les
neutrophiles ainsi que sur les macrophages (49). Le fMLP (N-formyl-methionyl-leucyl-
phenylalanine) a été identifié comme étant le peptide chimiotactique majeur libéré par
E.Coli (50) et a été le premier agent chimiotactique synthétique. Il a été par la suite utilisé
dans de nombreuses études comme prototype de cette catégorie d'agents chimiotactiques.
Le fMLP, et plus généralement les peptides formylés, sont reconnus spécifiquement par des
récepteurs spécifiques (51) qui appartiennent à la famille des récepteurs à sept domaines
transmembranaires liés aux protéines G hétérotrimériques (RCPG ou récepteur couplé aux
protéines G) et qui transmettent leurs effets cellulaires. Les fonctions cellulaires stimulées
en réponse à la liaison du fMLP à son récepteur spécifique sont l'adhésion par l'activation
des intégrines, la chimiotaxie, la dégranulation, la production de radicaux libres oxygénés,
la synthèse de médiateurs lipidiques et la synthèse de cytokines/chimiokines, bien que
cette dernière fonction soit limitée dans le neutrophile (52).
1.3.2. Le récepteur pour le fMLP : FPR
II existe plusieurs types de récepteurs pour le fMLP, divisés en deux familles : les
récepteurs FPR (N-Formyl Peptide Receptor) et les récepteurs FPRL (N-Formyl Peptide
Receptor-like). Le neutrophile humain exprime le FPR et le FPRL1 qui proviennent de
deux gènes différents (FPR et FPRL1). Le FPR possède une haute affinité pour le fMLP
(Kd = 0,6 + 0,2 nM) et les neutrophiles humains expriment à leur surface environ 53 000
FPRs (53). Le FPRL1 est un récepteur de plus faible affinité pour le fMLP (Kd = 1 \xM) et
peut lier des agonistes très différents des peptides formylés comme par exemple les
lipoxines A4 (LXA4). Ces deux récepteurs activent les mêmes réponses fonctionnelles dans
le neutrophile, bien qu'il existe certaines différences au niveau des voies de signalisation

23
stimulées (54). Le FPR a été le premier RCPG clone dans le neutrophile à partir d'une
banque d'ADN provenant de cellules de la lignée leucémique HL-60 (55). La liaison du
ligand au FPR provoque un changement de conformation du récepteur qui peut alors
s'associer à la sous-unité a des protéines G hétérotrimériques (Ga) qui a la propriété
intrinsèque d'hydrolyser le GTP. L'observation que la toxine pertussique, qui a la propriété
d'inactiver spécifiquement les sous unités ai par ADP-ribosylation, bloquait les fonctions
du neutrophile stimulées par le fMLP a montré que le FPR se lie aux sous-unités de type
Gai (56-59), plus particulièrement à Gai2 et Gai3 (60), qui se dissocient alors des sous-
unités GPY- Cependant les isoformes des sous-unités Gpy associées au FPR n'ont toujours
pas été identifiées.
Suite à la stimulation du FPR par des peptides chimiotactiques, les cellules deviennent
réfractaires à d'autres stimulations par le même agoniste (désensibilisation homologue) ou
d'autres facteurs chimiotactiques (désensibilisation hétérologue). Cette désensibilisation
rapide du FPR peut résulter de plusieurs mécanismes, dont la phosphorylation du récepteur
et son internalisation. L'internalisation du récepteur semble nécessiter une phosphorylation
préalable dans la partie intracellulaire C-terminale (61-63). Le FPR est inactivé
principalement par un mécanisme de désensibilisation homologue par des phosphorylations
séquentielles de plusieurs résidus serine et thréonine par la GRK-2 (G protein receptor
kinase) (64). Le FPR n'est pas phosphorylé par la PKC (65, 66) ni par la PKA (67). Une
fois phosphorylé, le FPR peut ensuite s'associer aux arrestines, ce qui diminue l'affinité du
récepteur pour les protéines G. Cependant, au contraire des autres RCPGs, la liaison des
arrestines au FPR n'est pas indispensable à son internalisation mais elle joue un rôle
important lors de son recyclage (68). Le FPR ne semble pas être sensible à la
désensibilisation hétérologue puisqu'il n'est pas phosphorylé par les kinases activées par
d'autres récepteurs (69) mais il peut contrôler la désensibilisation d'autres RCPGs pour des
facteurs chimiotactiques par un mécanisme de désensibilisation croisée propre à cette classe
de RCPG (70).
L'activation du FPR est aussi régulée par des interactions avec le cytosquelette d'actine.
(71). Le fait que des substances qui inhibent la polymérisation de l'actine comme la
cytochalasine B (72) ou la toxine botulique C2 (73) augmentent la durée et l'intensité de la

24
bouffée oxydative stimulée par le fMLP mais inhibent la désensibilisation (74) indique que
le cytosquelette joue un rôle important dans la régulation de l'activité du FPR et dans son
internalisation. Il semble aussi que la formation de complexes entre le FPR et les protéines
du cytosquelette (actine) régule l'association du récepteur avec les protéines G (71).
1.3.3. Signalisation induite par le FPR.
Suite à la liaison du fMLP avec le FPR, les protéines Gai et les sous-unités G(3y activent
plusieurs enzymes effectrices qui génèrent des seconds messagers intracellulaires dans le
neutrophile, comme par exemple le calcium, le DAG, le PtdIns(3,4,5)P3 et l'AMPc. Les
seconds messagers produits activent des kinases et les cascades de signalisation induites
permettent l'accomplissement des différentes réponses fonctionnelles stimulées par le
fMLP.
1.3.3.1. Voie de la PLCfJ : mobilisation de calcium et activation de la PKC.
La PLC catalyse l'hydrolyse des phosphatidylinositol-4,5-diphosphates membranaires
(PtdIns(4,5)P2) pour générer des inositol 1,4,5-triphosphates (Ins(l,4,5)P3) et le
diacylglycérol (DAG), deux seconds messagers intracellulaires importants (75).
L'Ins(l,4,5)P3 déclenche la libération de Ca2+ des pools intracellulaires alors que le DAG ,
en synergie avec le Ca2+ active les protéine kinases C (PKC). Le fMLP active les PLCP2 et
PLCp3 (76) par une interaction directe entre leur domaine PH et les sous-unités Gfiy (77).
Cependant, l'activité de l'isoforme PLCP2 semble être prédominante puisqu'elle est
responsable de 90% de la production d'Ins(l,4,5)P3 (78). L'activation des PLC(32/3 est
essentielle pour la production d'anions superoxyde en réponse au fMLP, mais ne semble
pas être impliquée dans la chimiotaxie (76, 79). L'activation de la PLC a également été
reliée à la dégranulation (80) et à l'adhésion (81). Une phosphorylation inhibitrice par la
PKA régule l'activité des PLC(32 et PLC(33 (82, 83).
Un des événements les plus précoces stimulé par le fMLP est l'augmentation rapide des
concentrations intracytoplasmiques de calcium ([Ca2+]i) (84, 85), un second messager
nécessaire à la production d'anions superoxyde par la NADPH oxydase (86-88), à la
dégranulation (86, 87), et à l'adhésion (89). Cette augmentation de [Ca2+]i est biphasique

25
et résulte de deux phénomènes distincts et successifs: la libération de Ca2+ à partir des
réservoirs cytoplasmiques qui provoque une augmentation rapide des [Ca2+]i, suivie d'un
influx de calcium extracellulaire par des canaux ioniques transmembranaires (90, 91). La
mobilisation de Ca2+ à partir des réservoirs cytoplasmiques est déclenchée par la liaison des
Ins(l,4,5)P3 libérés par l'activité de la PLC à des récepteurs spécifiques qui forment des
canaux ioniques perméables au Ca2+ (92). Les phénomènes qui régulent l'influx de Ca2+
extracellulaire ne sont pas encore tous connus mais il semble que 1 ouverture des canaux
cationiques soit en grande partie due à l'efflux de Ca2+ des réservoirs cytoplasmiques (93,
94).
Les effets du calcium sont transmis dans la cellule par deux classes de protéines kinases :
principalement les protéine kinases C (PKC) et les calmodulines kinases (CaMK) (95-98).
Les protéine kinases C forment une grande famille de sérine/thréonine kinases comprenant
de nombreuses isoenzymes. Elles sont regroupées en 3 catégories selon les seconds
messagers requis pour leur activation: les PKC « classiques » (ou conventionnelles)
activées par le Ca2+ et le DAG (PKCa, (31, (311, y), les PKC « nouvelles » activées par le
DAG mais insensibles au Ca2+ (PKCe,r|,ô,6) et les PKC « atypiques »(PKCi, Q insensibles
au Ca2+ et ne répondant pas au DAG (99). Cependant, l'activation de toutes les isoformes
de PKC nécessite la présence de phosphatidylsérine (PS), un phospholipide acide situé sur
la face cytoplasmique des membranes (100). Les PKC classiques sont les cibles des esters
de phorbol (PMA, TPA), des promoteurs de tumeur qui stimulent directement l'activité
enzymatique en se liant au domaine Cl à la place du DAG et diminuent la concentration de
Ca2+ requise pour l'activation de ces isoformes. Les isotypes de PKC exprimés par le
neutrophile humain sont les PKCa, PKCpI, PKC|3II, PKCÔ et PKCÇ (101). L'activation
des PKCs est généralement associée avec leur relocalisation vers les membranes (100), ce
qui a été vérifié dans les neutrophiles stimulés au fMLP ou avec du PMA (102, 103). Les
PKCs classiques, plus particulièrement la PKCp, ainsi que la PKC£ jouent un rôle
important dans l'activation de la NADPH oxydase (104, 105) en phosphorylant p47phox
(103, 106, 107). Le fait que la chimiotaxie ne soit pas inhibée lorsque l'augmentation de
calcium cytosolique est abolie dans les neutrophiles (78, 108) indique que les isoformes de
PKC reliées à cette fonction sont insensibles au Ca2+. La PKCô est impliquée dans la

26
polarisation des neutrophiles induite par les facteurs chimiotactiques sans avoir d'effet sur
la polymérisation de l'actine alors que la PKCÇ contrôle la chimiotaxie en agissant au
niveau de la polymérisation de l'actine (109).
1.3.3.2. Formation de PtdIns(3,4,5)P3 par les PI3-K
1.3.3.2.1. La génération du PtdIns(3,4,5)P3
Le phosphatidylinositol (Ptdlns), un phospholipide membranaire est le précurseur d'une
famille de seconds messagers :les polyphosphoinositides (PI). Le Ptdlns est composé d'un
groupe de D-myo-inositol-1-phosphate lié par un pont phosphodiester au DAG. L'anneau
inositol possède 5 groupes hydroxyl (OH) libres dont trois peuvent être phosphorylés (OH
en position 3', 4' et 5') dans les cellules selon différentes combinaisons. Les PI
représentent moins de 10% des phospholipides membranaires et ils sont les substrats de
kinases, de phosphatases et de lipases qui sont recrutées à la membrane lorsque la cellule
est activée (110). Le PtdIns(4,5)P2 est le plus abondant des PtdlnsP doublement
phosphorylés et il est, in vivo, le substrat des PI3-K de classe I qui phosphorylent
l'hydroxyl en position 3' de l'anneau inositol pour former le Ptdlns(3,4,5)p3. La formation
de PtdIns(3,4,5)P3 dans les cellules a été décrite pour la première fois dans les neutrophiles
dans lesquels l'accumulation rapide d'un nouveau phospholipide polaire avait été observée
en réponse au fMLP (111). Les propriétés chromatographiques de ce phospholipide isolé
dans les fractions lipidiques ont conduit à postuler qu'il s'agissait très probablement de
PtdIns(3,4,5)P3 (111, 112), cette hypothèse ayant été confirmée par la suite par Stephens et
al. (113). La génération de PtdIns(3,4,5)P3 est indépendante de la libération de Ca2+
stimulée par le fMLP et de la PKC, elle peut être stimulée par des analogues non-
hydrolysables du GTP (GTPyS) et elle est inhibée par la toxine Pertussis ainsi que par
l'isoprotérénol, un agoniste des récepteurs (3-adrénergiques qui stimule la formation
d'AMPc. Elle peut aussi être stimulée par d'autres agents chimiotactiques (PAF, LTB4,
C5a) avec cependant une plus faible amplitude que le fMLP (112, 114, 115). La formation
de PtdIns(3,4,5)P3 est accompagnée de la génération d'un autre PtdlnsP, le PtdIns(3,4)P2,
qui est moins rapide mais dont l'augmentation suit de quelques secondes celle du
PtdIns(3,4,5)P3, ce qui conduisit à supposer que ce second PtdlnsP était le produit de
déphosphorylation du PtdIns(3,4,5)P3 (113). Il est à noter que les concentrations de ces

27
deux PtdlnsP sont tellement faibles dans les cellules au repos qu'elles sont quasiment
indétectables. Le PtdIns(3,4,5)P3 s'est rapidement révélé être un second messager de
première importance impliqué dans la chimiotaxie et d'autres fonctions des leucocytes,
mais également dans la prolifération et le métabolisme cellulaires (114).
OH 9
=o3
PI(3,4,5)P3
Schéma 6. Formation du PtdIns(3,4,5)P3 par la PI3-K
L'utilisation d'inhibiteurs spécifiques de la PI3-K, la wortmannine, le LY294002, ont
permis d'établir le rôle de la formation de PtdIns(3,4,5)P3 dans les réponses fonctionnelles
des neutrophiles (116-119). Le LY294002 et la wortmannine inhibent la production
d'anions superoxide stimulée par le fMLP mais pas par le PMA dans les neutrophiles (120,
121). La dégranulation induite par le fMLP est également inhibée par la wortmannine
(122). Pour certains, les inhibiteurs de PI3-K n'affectent pas la formation d'actine
filamenteuse (120, 121). Cependant, l'introduction dans les neutrophiles d'un ester de

28
PtdIns(3,4,5)P3 est suivie du développement d'une polarité de la cellule et de
l'accumulation d'actine filamenteuse au niveau des lamellipodes formés (123). De plus, la
wortmannine réduit la polarisation et la capacité de locomotion induite par le fMLP (124),
ce qui indiquait que la PI3K joue un rôle important au niveau de certaines étapes de la
réponse chimiotactique. L'adhésion des neutrophiles sur des plaques recouvertes de KLH
(hémocyanine) stimulée par le fMLP n'est pas inhibée alors que l'adhésion intercellulaire
homotypique et hétérotypique nécessite une activité de la PI3-K sans que l'expression des
intégrines CD1 lb/CD18 ne soit altérée (125). Enfin, le « priming » des neutrophiles par des
agents tels que le GM-CSF, le TNFa ou la CB permet d'augmenter notablement la
production de PtdIns(3,4,5)P3 stimulée par le fMLP dans les neutrophiles humains (126,
127).
1.3.3.2.2. Les PI3-K
Les PI3-K catalysent la phosphorylation en position 3 de l'anneau inositol des PtdlnsP. Les
PI3-K exercent également un rôle de protéine kinase sur des résidus serine, mais la liste des
substrats est encore limitée concernant cette activité enzymatique (128). Les multiples
isoformes de PI3-K sont classées en trois classes selon leur structure, leur mode de
régulation et la spécificité pour leurs substrats. La structure des sous-unités catalytiques des
trois classes montre une certaine homologie: toutes possèdent un domaine catalytique HR1
(Homology Région) lié à un domaine hélicoïdal HR2 (ou domaine PIK pour PI Kinase) et
un domaine C2 (ou HR3) (110). Les PI3-K de la classe I sont des hétérodimères composés
d'une sous-unité catalytique de 110 kDa (pi 10) et d'une sous-unité régulatrice (p85 ou p55
ou p50) qui régule leur localisation cellulaire et leur activation. Bien qu'elles peuvent
phosphoryler différents types de Ptdlns in vitro (Ptdlns, PtdIns(4)P et PtdIns(4,5)P2), leur
substrat dans les cellules est uniquement le PtdIns(4,5)P2 (114). La classe II contient trois
isoformes (a,p\y) composées seulement d'une sous-unité catalytique qui utilise le Ptdlns
comme substrat. La classe III comprend une seule isoforme capable de générer uniquement
du PtdIns(3)P et qui semble impliquée surtout dans le trafic vésiculaire et la phagocytose
(129).

29
PI3-K
PI3-K
MTMPI5-K
PI(3)P
PIKfyve
PI(5)P\ PI 3-K
IPK
PI(4,5)P2 \ PI 3-K PI(3,5)P2
MTM
PTEN
PI(3,4,5)P3 PI(3,4)P2
Schéma 7. Métabolisme des PtdlnsP par les PI-Kinases (rouge) et les PI phosphatases
(vert).
Tiré de Prestwich GD et al, 2004, Chemistry & Biology, 11(5)
La classe I est divisée en deux sous-classes (IA et IB) selon la nature de la sous-unité
régulatrice associée à la sous-unité catalytique, ce qui détermine le mode d'activation de
l'hétérodimère résultant. Les trois isoformes de sous-unités catalytiques (pi 10a, |3, et ô) de
la classe IA sont codées par trois gènes différents et peuvent être associées à cinq sous-
unités régulatrices (p85a, p85(3 ,p55a, p55y ou p50a) codées par trois gènes distincts.
(130). Toutes les sous-unités régulatrices contiennent deux domaines SH2 (Src homology
2) qui lient des résidus tyrosine phosphorylés contenus dans la séquence (P)Y-X-X-Met.
Les p85 se distinguent des autres isoformes (p55 et p50) par un domaine SH3 et un
domaine de liaison pour les petites protéines G de la famille Rho activées (Rac-GTP ou
Cdc42-GTP) appelé aussi domaine BH. De plus, les sous-unités régulatrices possèdent
également des régions riches en proline qui se lient aux domaines SH3, ce qui contribue à
promouvoir des interactions intra- et inter-moléculaires qui participent au mécanisme
d'activation des PI3-K IA. Les trois sous-unités catalytiques pi 10a, pi 10 (3, et pllOÔ

30
possèdent la même structure : elles sont composées d'un domaine de liaison aux sous-
unités régulatrices (p85 binding domain), un domaine de liaison pour la petite protéine G
Ras (Ras-GTP) (RBD ou domaine HR4), un domaine C2 (ou HR3) suivi d'un domaine
hélicoïdal (HR2) et du domaine kinase (HR1) (110). Dans la cellule au repos, la p85 exerce
un rôle inhibiteur sur l'activité catalytique de la pi 10 associée. Lors de l'activation
cellulaire, cette contrainte inhibitrice est supprimée par la liaison de résidus tyrosine
phosphorylés sur les domaines SH2 (131) qui permet le recrutement des PI3-K IA à la
membrane ainsi que par la tyrosine phosphorylation de la p85 (132). L'activation des PI3-
Ks de la classe IA est donc entièrement dépendante de l'activité des tyrosine kinases et elle
est souvent stimulée par les récepteurs aux facteurs de croissance possédant une activité
tyrosine kinase intrinsèque. La classe IB contient une seule isoforme : la PI3-Ky qui est
activée exclusivement par les RCPG. La sous-unité catalytique, pllOy, possède une
structure similaire à celle des pi 10 de la classe IA. L'analyse de sa structure cristalline
montre que le domaine kinase constitué de deux lobes contenant une boucle d'activation et
une boucle catalytique présente une certaine similarité avec le domaine catalytique des
protéine kinases, plus particulièrement des tyrosine kinases de la famille Src, ce qui peut
expliquer l'activité protéine kinase de la pi 10 (133). Cette activité protéine kinase permet
Pautophosphorylation des pi 10. Dans le cas de la pllOy, cette autophosphorylation est
régulée par les sous-unités G|3y niais ne semble pas être impliquée dans la régulation de
l'activité catalytique de la PI3-KY (134). La pllOy peut être associée à deux sous-unités
régulatrices : la plOl (135) ou la p84 (ou p87PI1CAP) (136, 137). Deux domaines fonctionnels
ont été identifiés pour la pi01: un domaine de liaison à la pllOy en N-terminal et un
domaine de liaison aux sous-unités Gfty dans la partie C-terminale (138) qui permet le
recrutement à la membrane de la PI3-K où l'activité catalytique est activée directement par
les sous-unités G(3y (139). L'expression de la pi01 est majoritaire dans les neutrophiles
murins (136, 137) et elle confère la sélectivité pour le PtdIns(4,5)P2 comme substrat de la
PI3-Ky (140). Une fois recrutée à la membrane, l'activité catalytique des pi 10 de la classe
IA aussi bien que de la Classe IB est également stimulée par la petite protéine G Ras qui se
lie au RBD (141-143).

31
Class IA Regulatory Isoforms
Pro. rich
J Racblnding |Ç^ SI
Pro. rich
SH2 ) p55y
Class IA Catalytic Isoforms
Class IB Catalytic and Regulatory Isoforms
pHQyblndlng
p110y
p101
| p84/p87P I K A P
Schéma 8. Structure des isoformes de PI3-K de la classe I
Tiré de Deane J. et al. 2004, Annual Review of Immunology,22

32
La question de déterminer la part respective des deux sous-classes de PI3-K I dans la
génération de PtdIns(3,4,5)P3 en réponse au fMLP dans les neutrophiles est restée un sujet
de controverse pendant quelques années en raison de résultats parfois contradictoires sur le
rôle attribué aux tyrosine kinases dans le mécanisme de formation du PtdIns(3,4,5)P3. L.
Stephens et al. ont initialement montré que l'activité de tyrosine phosphorylation n'est
responsable que d'une faible part de la formation de PtdIns(3,4,5)P3 dans des cellules
myéloïdes (neutrophiles ou lignée monocytaire U937) stimulées par le fMLP (144). Le
même groupe a isolé à partir des cytosols de U937 une nouvelle isoforme de PI3-K activée
spécifiquement par les sous-unités Gffy des RCPG et dont l'activation ne dépend pas d'une
activité tyrosine kinase (145). Le clonage de la sous-unité catalytique de cette nouvelle
isoforme, dénommée pi 10y, a permis de confirmer qu'elle était activée directement par les
RCPG par un mécanisme différent des PI3-K de classe IA (146) et l'identification de la
plOl comme sous-unité régulatrice associée à la pllOy a montré qu'elle permettait
l'activation de la PI3-Ky directement par les sous-unités G(3y (135). Alors que ces faits
expérimentaux indiquaient que le fMLP active principalement la PD-Ky, les résultats
d'autres groupes montraient que des inhibiteurs de tyrosine kinases réduisaient fortement la
production de PtdIns(3,4,5)P3 stimulée par le fMLP dans les neutrophiles (147, 148) et
qu'une activité PI3-K était retrouvée dans des précipités immuns obtenus avec des
anticorps anti-phospho-tyrosine (149), ce dernier résultat contredisant ceux de Vlahos et al.
qui n'ont pas détecté d'activité PI3-K dans les mêmes conditions (150). Finalement, la
génération de souris déficientes en PI3-Ky a clairement montré le rôle prépondérant de
cette isoforme dans la production de PtdIns(3,4,5)P3, la migration directionnelle (24, 76,
151, 152), la production d'ions superoxyde (76, 151, 152) ainsi que l'expression des
intégrines (32 et l'adhésion (153) stimulées par le fMLP dans les neutrophiles murins. Dans
les neutrophiles humains, la PI3-Ky semble également être l'isoforme majeure activée par
le fMLP (et l'IL-8) (154). Cependant, le développement d'inhibiteurs spécifiques pour la
PI3-KÔ, exprimée essentiellement dans les cellules hématopoïétiques, a permis d'attribuer
un rôle primordial pour cette isoforme dans la polarisation et la migration directionnelle des
neutrophiles humains dans un gradient de fMLP (155) ainsi que dans la production d'ions
superoxyde et la dégranulation (156). Comme la PI3-Ky (157-159), la PI3-KÔ est impliquée
dans la migration des neutrophiles vers les tissus inflammés dans des modèles murins de

33
pathologies inflammatoires (156, 160). Il semble néanmoins que l'importance relative des
deux isoformes diffère selon l'espèce étudiée : la PI3-Ky serait nettement prépondérante
dans la réponse au fMLP des neutrophiles murins exposés préalablement au TNFa alors
que dans les neutrophiles humains, l'activation séquentielle de la PD-Ky puis de la PI3-KÔ
est nécessaire pour obtenir une production de PtdIns(3,4,5)P3 et des réponses fonctionnelles
maximales (161).
B
Schéma 9. Activation des PI3-K IA et IB.
Tiré de Hawkins P., 2006, Biochem. Soc. Tram., vol.34(5)

34
Le modèle communément admis pour le moment repose donc sur activation séquentielle
des deux isoformes de PI3-K, la formation de PtdIns(3,4,5)P3 par la PI3-Ky permettant
l'activation ultérieure de la PI3-KÔ (155, 161). Les résultats d'expériences montrant les
effets d'inhibiteurs spécifiques de PI3-Ky récemment développés (162) sur la production de
PtdIns(3,4,5)P3 et les réponses fonctionnelles au fMLP permettraient de clore ce débat
quant à l'importance relative des deux isoformes de PI3-K dans le neutrophile humain.
1.3.3.2.3. Les Ptdlns(3,4,5)3 phophatases : SHIP et PTEN
L'accumulation du PtdIns(3,4,5)P3 est régulée par l'activité de deux PtdlnsP phosphatases :
PTEN (phosphatase and tensin homologue deleted on chromosome 10) qui déphosphoryle
le PtdIns(3,4,5)P3 en position 3' de l'inositol et SHIP (SH2-containing inositol
phosphatase) qui déphosphoryle le PtdIns(3,4,5)P3 en position 5'. PTEN et la PI3-K
forment une boucle de régulation de la formation de Ptdlns(3,4,5)p3 puisque l'activité
enzymatique de PTEN génère du PtdIns(4,5)P2 qui est le substrat des PI3-K. Comme nous
l'avons mentionné dans la description de la chimiotaxie, la distribution de PTEN dans les
régions postérieures et latérales de Dictyostelum Discoideum régule la formation de
PtdIns(3,4,5)P3 de façon localisée et permet l'accumulation de PtdIns(3,4,5)P3 au front de
migration et le développement de la polarité dans un gradient chimiotactique (163). Dans
les cellules de mammifères, le mode d'action de PTEN semble différent puisque la
suppression de l'expression de PTEN par interférence avec TARN dans des cellules HL-60
différenciées en neutrophiles et stimulées par le fMLP diminue le taux de migration et la
polymérisation de l'actine mais n'influence pas la direction de la migration (164).
Cependant, on observe dans ces cellules une re-distribution de PTEN vers la partie
postérieure comme dans D.Discoideum (165). La régulation de l'activité de PTEN est
encore mal connue mais il semble que des déphosphorylations augmenteraient l'affinité
pour les substrats et l'activité lipide phosphatase de l'enzyme.
Il existe deux isoformes de SHIP : SHIP1 et SHIP2 qui catalysent la formation de
PtdIns(3,4)P2 à partir du PtdIns(3,4,5)P2. Ces phosphatases contiennent des domaines SH2
qui leur permettent d'interagir avec des protéines tyrosine phosphorylées (par exemple la
protéine adaptatrice Shc) ce qui détermine sa localisation lors de l'activation de la cellule et
elles peuvent être elle-mêmes phosphorylées sur des résidus tyrosine lors de leur

35
recrutement à la membrane. Cette phosphorylation n'a pas d'effet sur leur activité
phosphatase qui est constitutive mais constitue un indice de leur recrutement. Il est assez
probable que SHIP soit activée en réponse au fMLP puisque l'apparition de PtdIns(3,4)P2 a
été observée en parallèlement à celle de PtdIns(3,4,5)P3 après stimulation des neutrophiles
mais il n'existe pas actuellement de données expérimentales précises publiées sur ce sujet.
1.3.3.2.4. Akt et PDK1 : des protéines régulées par le Ptdlns(3,4,5)P3
Les domaines PH (pleckstrin homology) sont des petits modules d'une centaine d'acides
aminés qui ont la propriété de reconnaître et de lier le groupe polaire des Pis. Certains
domaines PH ont une affinité particulière pour les produits (directs ou indirects) de la PI3-
K, c'est-à-dire le PtdIns(3,4,5)P3 et le PtdIns(3,4)P2 et sont contenus dans des
sérine/thréonine kinases comme PDK1 ou Akt, les tyrosine kinases de la famille Tec, les
facteurs d'échanges et les protéines activatrices des petites protéines G (GEFs et GAPs). La
principale fonction des domaines PH est de permettre le recrutement de ces protéines
effectrices vers les régions enrichies en PtdIns(3,4,5)P3 et en PtdIns(3,4)P2 où elles
pourront être activées (Akt) ou activer leurs substrats (PDK1). Pour certaines protéines, la
liaison des Pis au domaine PH peut induire un changement de conformation favorisant leur
activation (166).
Une des cibles les plus connues de la PI3-K est Akt (ou PKB). Akt est une sérine/thréonine
kinase appartenant à la grande famille des kinases AGC. Elle a été identifiée comme étant
le produit d'expression d'un oncogène murin, v-Akt provenant du retrovirus AKT8, qui
possède une activité sérine/thréonine kinase (167, 168). Par ailleurs, une stratégie de
clonage homologue a permis d'identifier une nouvelle sérine/thréonine kinase dénommée
RAC-PK (Related to A and C Protein-Kinase) en raison de l'homologie retrouvée entre les
domaines catalytiques de ces kinases (169). La RAC-PK s'est révélée être l'homologue du
produit d'expression de v-Akt (168) et la dénomination Akt lui a été attribuée. Il existe trois
isoformes d'Akt (Akt 1-3) (170) qui possèdent une structure commune et qui sont toutes
exprimées dans le neutrophile (171). Akt est constituée d'un domaine PH en N-terminal, un
domaine catalytique central et un domaine régulateur carboxy-terminal qui contient un
motif hydrophobe (HM) caractéristique des kinases de la famille AGC. Le domaine PH
d'Akt lie le PtdIns(3,4,5)P3 et le PtdIns(3,4)P2 avec une haute affinité (172), ce qui rend la

36
liaison proportionnelle à la concentration de ces PtdlnsP dans la membrane et a permis
d'utiliser le recrutement du domaine PH d'Akt à la membrane comme indice de formation
du PtdIns(3,4,5)P3 dans les cellules (19). L'activation d'Akt est régulée par des
phosphorylations qui induisent des changements de conformations nécessaires pour
stimuler l'activité catalytique. Suite à son recrutement à la membrane, Akt est phosphorylée
sur le résidu Thr308 situé dans la boucle d'activation du domaine catalytique et sur le
résidu Ser473 situé dans le motif hydrophobe du domaine régulateur (173). Ces deux
phosphorylations sont dépendantes de l'activité de la PI3-K puisqu'elles sont inhibées par
la wortmannine et elles sont nécessaires et suffisantes pour stimuler l'activité catalytique
d'Akt (173, 174). L'activité d'Akt ne peut pas être directement stimulée par le
PtdIns(3,4,5)P3 et le PtdIns(3,4)P2 (175, 176), mais leur liaison au domaine PH peut induire
un changement de conformation qui module l'activité d'Akt (172, 177). La kinase qui
phosphoryle la Thr308 a été identifiée comme étant PDK1 (phosphoinositide-dependent
kinase) (176, 178). PDK1 appartient également à la famille des kinases AGC et possède un
domaine PH de haute affinité pour les PtdlnsP dérivants de l'activité de la PI3-K. (179). Son
activité est constitutive et son recrutement à la membrane via le domaine PH n'est requis
que pour phosphoryler certains substrats comme Akt (180), la plupart des autres substrats
de PDK1 étant phosphorylés dans le cytosol. La deuxième phosphorylation (Ser473) sur
Akt est attribuée à des « PDK2 » dont la nature peut varier selon le type cellulaire.
L'activité d'Akt peut aussi être régulée par des phosphorylations sur des résidus tyrosine
qui nécessitent au préalable la translocation d'Akt à la membrane et la phosphorylation de
la Thr308 (181-183). Une phosphorylation de la Thr3O8 par la CaM-K kinase a aussi été
observée (184).
L'activité intracellulaire d'Akt est antagonisée par deux phosphatases : la Ser/Thr
phosphatase PP2A qui déphosphoryle Akt (185) et PTEN qui régule négativement l'activité
d'Akt en diminuant les concentrations en PtdIns(3,4,5)P3 (186).
Après avoir été activée par cette double phosphorylation, Akt migre vers le cytosol et le
noyau où elle phosphoryle ses substrats. La plupart des substrats cellulaires d'Akt sont
reliés à des fonctions anti-apoptotiques (Bad, caspase 9, facteurs de transcription) ou
métaboliques (GSK-3) importantes, ce qui en fait une enzyme essentielle dans le contrôle

de la survie et de la prolifération cellulaires (187) et une cible thérapeutique potentielle très
étudiée. Akt semble aussi jouer un rôle important dans la migration cellulaire et
l'angiogénèse (188).
Le fMLP, ainsi que des chimiokines (IL-8 et GROa) stimulent l'activité d'Akt dans les
neutrophiles d'une façon qui dépend de la PI3-K (189). La Ser473 d'Akt est phosphorylée
par la MAPKAPK-2, (ou MK-2), un substrat de p38 dans les neutrophiles humains stimulés
par le fMLP, de façon également dépendante de la PI3-K (171). La voie des CaM-K est
également impliquée dans la phosphorylation et l'activation d'Akt stimulée par le fMLP
(98). Les fonctions du neutrophile reliées à l'activation d'Akt par le fMLP sont la
production d'ions superoxyde via la phosphorylation activatrice de p47phox par Akt et la
chimiotaxie (190, 191). Akt est recrutée au front de migration dans la phase de polarisation
des neutrophiles stimulés au fMLP (24) et semble jouer aussi un rôle important au niveau
de la polymérisation de l'actine dans les pseudopodes (192).
1.3.3.3. Les tyrosine kinases
La phosphorylation des résidus tyrosine de plusieurs protéines a été observée dans les
neutrophiles stimulés par des agents chimiotactiques dont le fMLP (193, 194). Ces
phosphorylations sur des résidus tyrosine étaient abrogées en présence de la toxine
Pertussis et leur inhibition par des inhibiteurs pharmacologiques montrait que leur activité
était nécessaire pour la formation d'ions superoxydes (193), la locomotion des neutrophiles
(195) mais pas pour la dégranulation. Les tyrosine kinases catalysent le transfert d'un
groupe phosphate de l'ATP vers des résidus tyrosine des protéines en présence de cations
divalents. Elles sont divisées en deux catégories : celles dont l'activité est associée à des
récepteurs de façon intrinsèque (le plus souvent des récepteurs pour des facteurs de
croissance) et celles non associées aux récepteurs mais qui sont activées par différents types
de récepteurs dont les RCPG. Les tyrosine kinases qui ont été identifiées en réponse au
fMLP dans les neutrophiles sont de cette dernière catégorie et appartiennent aux familles
des Src kinases, des Tec kinases et de FAK.

38
1.3.3.3.1. Les kinases Src
Les Src kinases sont constituées d'un domaine SH4 en N-terminal sur lequel sont greffés
des groupements myristoyl ou palmitoyl qui permettent leur ancrage à la membrane, d'un
domaine SH2 liant les résidus tyrosine phosphorylés, d'un domaine SH3 liant les régions
riches en prolines et d'un domaine catalytique SH1. Si le mécanisme d'activation des Src
kinases par les récepteurs aux facteurs de croissance a été étudié intensivement depuis la
découverte des tyrosine kinases dans les années 80, leur activation par les RCPGs reste mal
caractérisée. Il a été montré que les sous-unités Gai peuvent activer directement Src et Hck
en se liant au domaine catalytique ce qui induirait un changement de conformation
facilitant l'accessibilité pour les substrats (196). Grâce au développement d'un inhibiteur
spécifique des kinases Src, le PP2, il a été possible de montrer que l'activité de ces kinases
stimulée par le fMLP joue un rôle dans plusieurs réponses fonctionnelles du neutrophile : la
chimiotaxie, la polymérisation de l'actine et l'adhésion au fibrinogène (197). Ces travaux
ont également montré que les Src régulent l'accumulation de PtdIns(3,4,5)P3 stimulée par
le fMLP, et il est probable qu'elles agissent aussi bien au niveau de la formation de
PtdIns(3,4,5)P3 par la PD-Kô qu'au niveau de sa dégradation par la phosphatase SHIP, ces
deux enzymes étant régulées par des phophorylations sur des résidus tyrosine. Trois
membres de la familles des Src sont activés par le fMLP dans les neutrophiles : Lyn (198,
199), Fgr (200) et Hck (201). Ces trois tyrosine kinases sont fortement exprimées dans les
lignées hématopoïétiques (202). L'activité de Lyn est stimulée par autophosphorylation
(198) et l'association de Lyn avec l'adaptateur Shc via les domaines SH2 stimule l'activité
de la PI3-K dans les neutrophiles stimulés par le fMLP (199). La tyrosine kinase Fgr qui est
associée aux granules spécifiques est relocalisée à la membrane plasmique suite à la
stimulation par le fMLP (200) et elle est activée par cet agent seulement dans des
neutrophiles adhérents (203). Fgr et Hck sont impliquées dans l'adhésion et pourraient
jouer un rôle dans la dégranulation induite par le fMLP dans des neutrophiles adhérents
(201).
1.3.3.3.2. Les kinases Tec
Les tyrosine kinases de la famille Tec ont une structure proche de celle des Src kinases,
sauf qu'elles ne possèdent pas de domaine SH4 mais un domaine PH en N-terminal avec

une haute affinité pour le PtdIns(3,4,5)P3 qui permet le recrutement des Tec à la membrane
suite à l'activation de la PI3-K (204). La structure des Tec kinases comprend aussi un
domaine TH (Tec homology) caractéristique de cette famille et adjacent au domaine PH. Le
domaine TH contient deux motifs: un motif Btk qui peut se lier aux sous-unités Ga et un
motif riche en proline favorisant l'interaction avec des domaines SH3 adjacents ou
exprimés sur d'autres protéines commes les Src. Ces interactions permettent une
autophosphorylation ou une transphosphorylation des tyrosines activatrices des Tec. Trois
membres des Tec kinases ont été identifiés dans le neutrophile humain: Tec, Btk et Bmx
(205). Ces trois Tec kinases sont recrutées à la membrane d'une façon dépendante de
l'activité de la PI3-K et sont phosphorylées sur des résidus tyrosine en réponse au fMLP.
L'utilisation d'un inhibiteur spécifique de Btk (Bruton tyrosine kinase)(et probablement des
autres membres de cette famille), le LFM-A13, un analogue du leflunomide, a montré que
l'activation des kinases Tec par le fMLP joue un rôle dans la production d'ions superoxyde,
l'adhésion des neutrophiles au fibrinogène et la chimiotaxie induites par le fMLP (206).
Cette étude a aussi révélé que les kinases Tec augmentent l'accumulation de
PtdIns(3,4,5)P3 stimulée par le fMLP. Ce dernier résultat suggère deux mécanismes
d'action potentiels des kinases Tec sur le métabolisme du PtdIns(3,4,5)P3: soit une
inhibition de la PI phosphatase SHIP qui déphosphoryle le PtdIns(3,4,5)P3 en PtdIns(3,4)P2
ou bien l'activation de la PD-Kô régulée par des phosphorylations sur des résidus tyrosine,
ce qui est plus probable.
1.3.3.3.3. Pyk2
Pyk2 appartient à la famille des FAK (focal-adhesion kinase). Après stimulation par le
fMLP, Pyk est rapidement phosphorylée et se lie aux intégrines (32 qu'elle active dans des
neutrophiles adhérents exposés préalablement au TNFa (207) ou des HL-60 différenciées
en neutrophiles (208).
1.3.3.4. Les petites GTPases
Les petites protéines G monomériques sont des GTPases de faible poids moléculaires (20-
30 kDa) qui ont la propriété de lier les nucléotides cycliques guanylés (GDP et GTP) et
d'hydrolyser le GTP. Elles contrôlent une grande variété de processus cellulaires comme la

40
prolifération et la croissance, le contrôle du cycle cellulaire, la transcription, l'adhésion et la
motilité, le trafic vésiculaire et l'exocytose. Les petites GTPases alternent entre deux
conformations: une conformation active dans laquelle la protéine est liée à un GTP et une
conformation inactive dans laquelle elle est liée à un GDP. Les petites GTPases peuvent
être activées par un grand nombre d'agonistes via des récepteurs du type récepteur aux
facteurs de croissance, des RCPGs ou les intégrines. Le cycle d'activation des petites
GTPases est régulé par des facteurs d'échange (GEF) qui transfèrent un GTP à la place du
GDP et les protéines activatrices (GAP) qui stimulent l'activité d'hydrolyse intrinsèque du
GTP par les GTPases. Des inhibiteurs de dissociation (GDI) maintiennent les GTPases à
l'état inactif dans le cytosol. Les petites GTPases subissent des isoprénylations post-
traductionnelles au niveau du motif CAAX en C-terminal, ce qui permet leur ancrage à la
membrane lors du processus d'activation (209). Dans l'état actif lié au GTP, les petites
GTPases se lient à des protéines effectrices, souvent des protéines kinases, qui médient leur
action au niveau cellulaire. Il existe cinq grandes familles de GTPases monomériques : la
famille Ras, impliquée dans la croissance et la prolifération cellulaire et qui contient la
première petite GTPase découverte, Ras, ainsi que Rap et Rai, la famille Rho, les familles
Rab et Arf impliquées dans le transport vésiculaire et la famille Ran impliquée dans le
transport de protéines et d'ARN vers le noyau. Ras et Rai sont activées en réponse au fMLP
dans les neutrophiles, mais nous allons détailler davantage les mécanismes d'action des
familles Rho et Arf dans les paragraphes suivants puisqu'elles jouent un rôle fondamental
dans plusieurs fonctions du neutrophile et qu'elles ont été étudiées dans nos travaux
expérimentaux.
1.3.3.4.1. Les Rho-GTPases
Cette famille de petites GTPases inclut Rho A-G, Racl-3 et Cdc42. Certaines toxines
bactériennes comme l'exoenzyme C3 de Clostridium Botulinum ou les toxines A et B de
Clostridium Difficile inhibent l'activité des Rho-GTPases, ce qui en a fait des outils très
utiles pour étudier leurs fonctions dans les cellules (210, 211). Les Rho-GTPases peuvent
être activées par de nombreux facteurs d'échange propres à cette famille (212) comme Vav,
exprimé surtout dans les cellules hématopoïétiques (213-215), Tiam (216) ou P-Rex-1
(217). La structure des Rho-GEFs comprend un domaine DH (Dbl Homology) qui assure

41
l'échange GDP-GTP et un domaine PH adjacent qui participe à la fois à la localisation
intracellulaire et à l'activation des GEFs. L'activation des Rho-GEFs est régulée par la
liaision du PtdIns(3,4,5)P3 au domaine PH, par des phosphorylations sur des résidus
tyrosine et des interactions avec d'autres protéines (218). Suite à l'activation de la cellule,
les Rho-GEFs sont recrutés à la membrane où ils activent l'échange du GTP des petites
GTPases (219). Les Rho-GAPs qui hydrolysent le GTP sont régulées également par leur
liaison avec des phosphoinositides, par phosphorylation et par des interactions avec
d'autres protéines (220). Les protéines effectrices des Rho-GTPases activées sont souvent
des protéines kinases ayant un effet au niveau du réarrangement du cytosquelette (par
exemple la ROCK, la PAK ou WASP) (221) et elles sont donc des régulateurs importants
au niveau du contrôle du cytosquelette d'actine (26). Rho régule la formation des filaments
contractiles d'actine-myosine qui forment les fibres de stress et des complexes d'adhésion
focale (222) alors que Rac et Cdc42 sont impliqués dans la formation de lamellipodes et
filopodes respectivement (222, 223). En conséquence, les Rho-GTPases sont impliquées
dans les fonctions du neutrophile nécessitant une réorganisation dynamique du
cytosquelette comme la chimiotaxie (101), la phagocytose (224, 225), l'adhésion (226,
227), la régulation du trafic endocytaire (228, 229) et la dégranulation (230).
RhoA (231, 232) Rac et Cdc42 (233-235) sont activées en réponse au fMLP dans les
neutrophiles. Leur migration vers la membrane est inhibée par la wortmannine, le PP2
(résultats de C.Gilbert et N.Thibault, non publiés) et le LFM-A13 (206), ce qui indique que
leur activation dépend de l'activité des PI3-Ks et des tyrosine kinases des familles Src et
Tec. Les Rho-GTPases jouent un rôle important dans la coordination des événements de la
réponse chimiotactique en agissant à plusieurs niveaux : le maintien de la polarisation (Rac
et Cdc42), la polymérisation du cytosquelette d'actine et l'extension de pseudopodes (Rac
et Cdc42), la réversibilité de l'adhésion via les intégrines (RhoA) ainsi que la rétraction de
la partie postérieure de la cellule grâce la formation de fibres actine/myosine contractiles
(RhoA et Rac) (101, 236-238).

42
cytoskeletal organisationgène expression
celi cycle progressionapoptosls
membrane traffic
Schéma 10. Mécanisme d'activation des Rho-GTPases.
Tiré de Schmidt A. et Hall A., 2002, Gens Dev. 16 (13)
Dans les neutrophiles stimulés par des agents chimiotactiques, RhoA régule les
mécanismes d'adhésion via les intégrines (239). Rac2 est l'isoforme majoritaire dans le
neutrophile (240, 241). Elle est un des composants du complexe de la NADPH oxydase et
elle est nécessaire à la production d'ions superoxyde dans les neutrophiles (242-244). Lors
de l'activation du neutrophile par le fMLP, Rac2 migre vers la membrane où elle s'associe
avec p67phox pour former, avec p67phox-p47phox et les constituants membranaires, le
complexe de la NADPH oxydase fonctionnel (240, 245, 246). Plus récemment, il a été
montré que Rac2 joue un rôle actif dans le processus du transfert d'électrons à travers le
complexe enzymatique (247). Une déficience génétique ou des mutations de Rac2
provoquent des dysfonctions du neutrophile dans la défense contre les agents pathogènes,
notamment en raison de l'incapacité de ces neutrophiles à migrer vers des agents

43
chimiotactiques, à produire efficacement des RLOs et à libérer le contenu des granules
primaires (248-251). L'action de Rac2 au niveau de la NADPH oxydase est antagonisée par
Cdc42, ce qui confère à Cdc42 un rôle régulateur dans la production d'ions superoxyde
(252, 253).
En plus de l'activation des protéines effectrices auxquelles elles se lient lorsqu'elles sont
sous la forme GTP, les Rho-GTPases sont aussi impliquées dans la régulation de
différentes voies de signalisation et le métabolisme des phospholipides: la voie PI3-K (Rac
et Cdc42) (30, 254), la formation de PtdIns(4,5,)P2 par la PIP 5-K (RhoA via la ROCK), les
MAPK (p38 et p42/44) (249, 253), la PLC|32 (Cdc42) (253) et la PLD (RhoA) (255).
1.3.3.4.2. Arf
Les GTPases de la famille Arf (ADP-ribosylation factor) sont régulées comme les autres
petites GTPases par des GEFs (cytohésine, ARNO, Grpl pour en nommer certains
exprimés dans les neutrophiles) et des GAPs propres à cette famille, mais elles ne sont pas
complexées avec des facteurs inhibiteurs dans le cytosol. Les Arf-GEF possèdent dans leur
structure un domaine PH qui montre parfois une grande affinité pour les Pis et plus
particulièrement le PtdIns(3,4,5)P3, comme par exemple le domaine PH de Grpl (256-258).
Il existe six isoformes d'Arf (Arf 1-6), les plus étudiées étant Arfl et Arf6. Les Arf-
GTPases sont impliquées essentiellement dans la régulation du trafic membranaire et elles
jouent donc un rôle important dans l'endocytose, la sécrétion ou la phagocytose, ainsi que
dans l'adhésion (259). Le principal rôle de Arfl et ArO est de recruter les protéines du
manteau (COP) aux vésicules de transport de l'appareil de Golgi. Elles régulent également
l'activation de deux enzymes importantes au niveau du métabolisme des phospholipides : la
PLD et la PI(4)P 5-K qui forme du PtdIns(4,5)P2 (260), un PI important dans la régulation
des protéines du cytosquelette (261). Arf est activée en réponse au fMLP dans les
neutrophiles (ou les HL-60 différenciées) et sa migration vers les membranes est
dépendante de l'activité des tyrosine kinases. Il semble que, dans ces cellules, Arf6 ait un
rôle régulateur dans l'activation de la NADH oxydase par un mécanisme dépendant de la
PLD (262).

44
1.3.4. La phospholipase D (PLD)
La PLD est une enzyme membranaire qui catalyse l'hydrolyse de la phosphatidylcholine
(PC) pour former de l'acide phosphatidique (PA) et de la choline libre. Elle catalyse aussi
une réaction de transphosphatidylation en utilisant des alcools primaires (éthanol, 1-
butanol) comme nucléophile pour générer respectivement du phosphatidyl-éthanol (PEt) ou
du phosphatdyl-butanol (PBut) (263). Le PA peut être métabolisé en DAG par une PA-
phosphohydrolase ou en LPA (lysophosphatidic acid) par une PLA2. L'activité de la PLD
stimulée par le fMLP dans le neutrophile et les HL-60 différenciées est responsable de la
majorité de la génération de PA (264-267). Deux gènes codant pour deux isoformes de
PLD: PLD1 et PLD2 ont été identifiés chez les mammifères et clones (268, 269). Deux
variants (a et b) de chaque isoforme résultent d'un épissage alternatif de l'ARNm (269,
270). Seule la PLD la a été détectée dans les neutrophiles humains (271). Les deux
isoformes possèdent une structure similaire comprenant deux motifs HKD portant l'activité
catalytique, un motif polybasique PIP2 liant le PtdIns(4,5)P2 situé entre les deux motifs
HKD, un domaine PH et un domaine PX (Phox) liant les Pis (préférentiellement le
PtdIns(5)P) dans la partie N-terminale. La PLD1 possède également une boucle située dans
la partie centrale de la protéine qui serait un élément régulateur négatif de l'activité
enzymatique (272).
L'activité de la PLD est régulée par plusieurs facteurs : les Pis, les PKC classiques et les
petites GTPases RhoA et Arf. Le PtdIns(4,5)P2 est un PI indispensable pour l'activation des
deux isoformes de PLD (272). Le domaine PH de la PLD est sélectif pour les PtdlnsP
biphosphorylés ayant deux groupes phosphates adjacents, c'est-à-dire le PtdIns(4,5)P2 et le
PtdIns(3,4)P2. La liaison des ces deux types de PtdInsP2 au domaine PH influence la
localisation de la PLD au niveau de la membrane et participe aussi à la stimulation de
l'activité catalytique (273). De plus, le PtdIns(4,5)P2 stimule l'activité de la PLD en se liant
au domaine spécifique PIP2. Le PtdIns(3,4,5)P3 peut se lier au domaine PX mais il n'est
qu'un faible activateur direct de la PLD (274). Des modifications post-transductionnelles
de la PLD1, notamment la palmytoylation de résidus cystéine dans le domaine PX
influencent sa localisation cellulaire.

45
En plus des Pis, l'activité de la PLD est régulée par sa liaison avec trois cofacteurs
d'activation : les PKC classiques (PKCa et PKCP) et les petites GTPases Arf et RhoA. Les
esters de phorbol tels que le PMA sont des forts activateurs de la PLD, ce qui indique un
rôle important pour la PKC dans l'activation de la PLD. Les isoformes classiques, PKCa et
p\ activent la PLD1 (269, 275-277) par un mécanisme qui ne nécessite pas l'activité
catalytique de la PKC (277-279), bien que la PLD1 puisse aussi être phosphorylée par la
PKC (278). La PKC se lie à la partie N-terminale de la PLD (280). Les niveaux de calcium
cytosolique régulent aussi l'activité de la PLD (281) et permettent à la PLD d'être
fonctionnelle dans les neutrophiles activés par le fMLP (282).
Arf a été initialement identifiée comme un activateur de la PLD dans les HL-60 (283-285).
Arf active la PLD1 par une interaction directe (286) et peut aussi activer faiblement la
PLD2 (268). Le site d'interaction de la PLD avec Arf n'a pas encore été déterminé avec
précision mais il n'est pas inclus dans la partie N-terminale qui lie la PKC (280). RhoA est
l'autre GTPase activant directement la PLD (279, 287). RhoA se lie à la partie C-terminale
de la PLD1 (288, 289). L'effet activateur de RhoA sur l'activité PLD peut aussi être
attribué, de façon indirecte, à la formation de PtdIns(4,5)P2 par la PI(4)P 5-K (290). Il a été
montré que Rac et Cdc42 ont aussi le potentiel d'activer la PLD1(255), mais le rôle de ces
deux Rho-GTPases dans l'activation de la PLD1 n'a pas été observé dans des neutrophiles
stimulés par le fMLP. Les trois facteurs (PKC, Arf et RhoA) agissent en synergie pour
stimuler une activité optimale de la PLD (291-293).
La stimulation par le fMLP des HL-60 différenciées en neutrophiles induit la translocation
des facteurs d'activation de la PLD à la membrane (294). Cette étape de l'activation de la
PLD est régulée par des tyrosine kinases. La wortmannine inhibe aussi fortement l'activité
de la PLD stimulée par le fMLP (295, 296), ce qui indique un rôle important de la PI3-K
dans la régulation de l'activité de la PLD.
Le rôle de la PLD dans un grand nombre de fonctions cellulaires telles que la croissance, la
régulation du cytosquelette et l'exocytose (297), la phagocytose (298), la régulation de la
NADPH oxydase est attribué au second messager PA qu'elle génère (299). Un certain
nombre de protéines peuvent lier le PA qui régule leur activité. Parmi les enzymes

46
activéespar le PA, citons la PtdIns(5)P 4-K de type I dont le produit (le PtdIns(4,5)P2) est
un régulateur important des protéines du cytosquelette, ce qui confère indirectement à la
PLD, en coopération avec Arf, la capacité de réguler le cytosquelette. La PLD régule
également la production d'ions superoxyde stimulée par le fMLP dans les neutrophiles
(300). Les composants p22phox et p47phox peuvent être phosphorylés par une kinase activée
par le PA afin de former le complexe fonctionnel de la NADPH oxydase (301-303). De
plus, le PA peut se lier au domaine PX de la p47phox, mais le rôle précis de cette interaction
n'est pas encore connu (304). La PLD est également impliquée dans la dégranulation
induite par le fMLP (305).
1.3.5. Les MAPK : p38 et p42/44.
L'activation des neutrophiles par des facteurs de croissance (GM-CSF) ou par des agents
chimiotactiques induit la voie des MAPK (Mitogen Activated Protein Kinase), une famille
de sérines/thréonine kinases qui sont classiquement divisées en trois groupes : ERIC 1/2
(Extra-cellular signal-Related Kinase) ou p42/44, les protéines kinases activées par le stress
(c-Jun N-terminal Kinase ou JNK) et p38 MAPK, ces deux dernières catégories étant plus
particulièrement activées par le stress et par des cytokines pro-inflammatoires. L'activité de
ces MAPK est régulée en amont par une cascade de phosphorylations aboutissant à leur
phosphorylation activatrice (306). Le fMLP stimule l'activité des ERKs p42/44 (307-310),
de p38 (311, 312) et de JNK (313) par des voies de signalisation distinctes. Cependant,
l'activation de ERK1/2 aussi bien que celle de p38 par le fMLP dépend de la voie PLC-
Ca2+-PKC (311, 313, 314), de l'activité de la PI3-K (311,315), ainsi que de l'activité des
tyrosine kinases qui phosphorylent p38 et p42/44 (316). Pour certains, la PI3-Ky régulerait
l'activité des MAPK par l'intermédiaire d'une tyrosine kinase, des protéines adaptatrices
Shc et Grb2 ainsi que du GEF Sos et de Ras et Raf (317). Un autre groupe a montré, grâce
à la construction de mutants de PI3-Ky ne possédant que l'activité protéine kinase, que la
régulation des MAPK par la PB-Ky n'implique que son activité protéine kinase (318). Les
mécanismes de régulation des MAPK par la PI3-K restent donc à éclaircir et dépendent en
grande partie du modèle cellulaire et du type de stimulation étudiés.

47
L'utilisation d'inhibiteurs spécifiques pour p38 et p42/44 a permis d'étudier leurs rôles
respectifs dans les fonctions du neutrophile stimulé par le fMLP. Ces études montrent que
p38 joue un rôle important surtout dans la chimiotaxie (319-321), mais aussi dans la
production d'ions superoxyde (313, 321), la dégranulation (316, 322) et l'adhésion (321).
Le rôle des p42/44 dans les différentes fonctions stimulées par le fMLP semble moins
important que celui de p38: ces kinases ne semblent pas être impliquées dans la réponse
chimiotactique (149, 319), ni dans la dégranulation (316), et leur contribution à la
production d'ions superoxyde est controversée (149, 313, 319).
1.3.6. Activation de la voie de l'AMPc/PKA.
La stimulation des neutrophiles par le fMLP induit rapidement la formation d'AMP
cyclique (AMPc), un autre second messager. La concentration intracellulaire d'AMPc (
[AMPc]j) augmente jusqu'à atteindre 15-30 secondes après la stimulation un maximum
équivalent à 2-3 fois le niveau de base puis diminue pour retrouver le niveau de base vers
1-5 minutes (323-325). Le niveau intracellulaire d'AMPc est régulé d'une part par la
biosynthèse d'AMPc à partir de l'ATP par des adénylate cyclases (AC) et d'autre part par
l'hydrolyse de l'AMPc par des phosphodiesterases (PDE). L'augmentation des [AMPc]j par
le fMLP est bien inhibée par des inhibiteurs d'AC (326) et augmentée et prolongée par des
inhibiteurs de PDE comme la théophylline ou l'IBMX (3-isobutyll-methylxanthine) (323,
324, 327) mais le mécanisme d'activation des AC par le fMLP reste encore à déterminer
clairement. Il existe 9 isoformes d'AC régulées par des mécanismes distincts et localisées
dans des compartiments cellulaires variables (cytosoliques ou particulaires) (328). Dans les
neutrophiles, l'augmentation des [AMPcJj a aussi été observée avec le ionophore calcique
A23187 (323, 325) par un mécanisme commun avec le fMLP qui n'impliquait pas l'activité
d'AC associées à la membrane (327). De plus, l'élévation de l'[AMPc]j par le fMLP était
dépendante de la mobilisation de Ca2+ cytosolique (323, 327) et de l'activation préalable de
la PLC (329), ce qui indiquait clairement que une ou plusieurs isoformes d'AC régulées par
la voie du calcium pouvaient être impliquées. La caractérisation des isoformes d'AC
activées par le fMLP dans les neutrophiles humains est très récente (330). Le neutrophile
humain exprime les ARNm des AC III, IV, VII et IX, l'ARNm codant pour l'AC IX étant
le plus abondant. Aucune de ces isoformes n'est sensible à l'inhibition des sous-unités Gai.

L'activité adenylate cyclase stimulée par le fMLP en grande partie insensible à la
forskoline, un agent pharmacologique stimulant l'activité des ACs, excepté celle de l'AC
IX qui est inhibée en présence de cet agent, ce qui indique que cette isoforme est
préférentiellement activée par le fMLP. Cependant, l'AC IX est une isoforme ayant une
activité enzymatique faible comparée aux autres membres de cette famille. L'activité de
l'AC III, régulée par la calmoduline, et celles des AC IV et VII, régulées par la PKC
pourraient être stimulées par le fMLP dans le neutrophile étant donné le rôle de
l'augmentation de [Ca2+] cytosolique dans la formation de l'AMPc.
Les isoformes de PDE détectées dans les neutrophiles sont les PDE 5 et surtout les PDE 4
(331), plus particulièrement la PDE 4B (332) qui sont exprimées de façon prédominante
dans les leukocytes.
Le rôle de l'augmentation des [AMPc]j dans les réponses fonctionnelles du neutrophile
stimulé par le fMLP n'a pas été déterminé clairement. Certains groupes ont observé que
l'élévation des niveaux d'AMPc serait nécessaire pour la chimiotaxie et la production
d'ions superoxyde, bien que ce second messager ne soit pas le plus important (326). Pour
d'autres, l'augmentation des [AMPc]; n'aurait aucun rôle dans ces fonctions (333). Il
semble que la concentration d'[AMPc]j soit un facteur déterminant dans les effets
physiologiques de ce second messager car des faibles [AMPcjj provoquent une migration
non orientée alors que des fortes [AMPc]j inhibent la chimiotaxie stimulée par le fMLP
(334). Le principal effecteur de l'AMPc dans les cellules est la PKA (cAMP-dependent
protein kinase). Cette sérine/thréonine kinase est composée de deux sous-unités
régulatrices (R) et de deux sous-unités catalytiques (C) formant l'holoenzyme inactive. La
liaison de deux molécules d'AMPc sur des sites spécifiques des sous-unités régulatrices
induit la libération des sous-unités catalytiques actives. Le fMLP induit faiblement
l'activité de la PKA dans les neutrophiles humains (335). Cependant, l'inhibition
pharmacologique de la PKA augmente la formation d'actine filamenteuse et inhibe la
chimiotaxie induite par le fMLP (336, 337). Des travaux plus récents montrent qu'une
activité asymétrique de la PKA dans le neutrophile permet d'établir la polarisation du
cytosquelette d'actine et la migration induite par le fMLP (336, 337). Ceci montre que la

relativement faible augmentation des [AMPc]j joue un rôle physiologique dans la
chimiotaxie.
Schéma 11. Schéma récapitulatif des voies de signalisation stimulées par le fMLP dans le
neutrophile
fMLP
MAPK Tec K Akt
Rho-GTPases
Arf-GTPasesPA

50
1.4. Régulation de l'activation des neutrophiles par les agentsqui élèvent l'AMPcII est connu depuis longtemps que l'exposition des neutrophiles à des agents
pharmacologiques qui élèvent les [AMPcji avant stimulation inhibe leurs fonctions. Citons,
entre autres, la réduction de la formation d'ions superoxyde (338, 339), l'inhibition de la
chimiotaxie (340, 341), la diminution de la dégranulation (342-344) et de l'adhésion (345,
346) ainsi que la diminution de la biosynthèse de LTB4 (347, 348) et du PAF (349).
Inversement, une diminution des [AMPc]j due à l'action synergique du TNF et des
intégrines (32 semble faciliter la stimulation des fonctions du neutrophile (350). Les
principales substances connues pour moduler in vivo les fonctions du neutrophile en élevant
les niveaux intracellulaires d'AMPc sont l'adénosine et les prostaglandines E (PGE2,
PGE,).
1.4.1. L'adénosineL'adénosine provient du catabolisme des nucléotides adénylés (ATP, ADP). La libération
de l'ATP et de l'adénosine dans les fluides extracellulaires par les cellules (entre autres les
neutrophiles et les cellules endothéliales) est modulée selon les conditions physiologiques.
L'adénosine endogène est déaminée en inosine par l'adénosine déaminase (ADA).
L'adénosine active des RCPGs dont il existe quatre sous-types : les récepteurs Ai, A?.A, A2B
et A3. Les récepteurs Ai et A3 transmettent des signaux intracellulaires via les protéines
Gai/o et inhibent l'activité des AC, activent les canaux au K+ ou inhibent les canaux
calciques selon le type cellulaire dans lequel ils sont exprimés. Les récepteurs Ai peuvent
également stimuler la mobilisation de Ca2+ cytosolique via la protéine Gai et l'activation de
la PLCp\ Les récepteurs A2A et A2B activent les protéines Gars qAii stimulent l'activité des
AC, le récepteur A2B pouvant aussi être couplé aux protéines Gotq q\ai stimulent la
mobilisation de Ca2+ (351). Le profil d'expression des récepteurs à l'adénosine dans le
neutrophile reste un sujet de controverse en raison de résultats pas toujours concordants
publiés sur ce sujet. Plusieurs études ont rapporté que les quatre types de récepteurs (352-
356) ainsi que leurs ARNm peuvent être exprimés dans les neutrophiles (357-359).

51
Cependant, dans notre laboratoire, seuls les ARNm des récepteurs A2A et A3 ont été
détectés dans nos conditions expérimentales (360).
L'adénosine inhibe la plupart des fonctions du neutrophile stimulées par le fMLP: la
production d'ions superoxyde (339), l'adhésion aux cellules endothéliales (361) ainsi que
les dommages induits par l'activation des neutrophiles sur ces cellules (362), l'expression
de CD1 lc/CD18 (363), la polymérisation de l'actine (364) et la sécrétion de MMP9 (365)
et la biosynthèse du LTB4 (366) et du PAF (367). Etant donné ses effets inhibiteurs sur
l'activation des neutrophiles, l'adénosine est considérée comme un agent anti-
inflammatoire endogène important qui protège les tissus des dommages induits par la
réaction inflammatoire (351, 368). Ces effets inhibiteurs sont transmis par les récepteurs
A 2 A(361 , 363, 365, 369, 370), des RCPG couplés aux protéines Gas qui stimulent l'activité
des AC (371) et donc la formation intracellulaire d'AMPc dans les neutrophiles (372). Par
ailleurs, il a été observé que des souris ayant subi une délétion génétique du récepteur A2A
présentent, en comparaison avec des souris normales, des dommages tissulaires aggravés et
même létaux dans différents modèles d'inflammation, ce qui confirme le rôle protecteur et
modulateur de l'inflammation de ce récepteur (373). Un rôle inhibiteur sur l'activation des
neutrophiles a aussi été décrit pour le récepteur A3 (374), mais cette observation reste à
être confirmée.
1.4.2. Les prostaglandines E2
Les PGE2 ont été initialement découvertes dans les glandes séminales par Von Euler dans
les années 1930, d'où la dénomination « prostaglandines ». Depuis cette époque, les
prostaglandines ont été étudiées intensivement car elles jouent un rôle régulateur important
dans de nombreuses fonctions physiologiques telles que la fertilité et la reproduction, le
maintien de l'intégrité du tractus digestif, l'inflammation, la douleur et la fièvre, la
régulation du tonus cardiovasculaire et de la fonction rénale, ainsi que la genèse de certains
cancer (375). L'identification de leur structure dans les années 1960 (216, 376-378) a
montré que les PG sont des acides carboxyliques de 20 atomes de carbone incluant un
anneau cyclopentane entre C8 et C12. Les PGE2 proviennent du métabolisme de l'acide
arachidonique (AA) par les cyclooxygénases (COX-1 et COX-2, ou PGHS pour

Prostaglandin H synthase) et la PGE synthétase. Leur biosynthèse s'effectue en trois étapes:
1) l'oxygénation de l'AA par la COX permet de former l'anneau cyclopentane et d'insérer
un groupement hydroperoxyde en Cl5, générant la PGG2, 2) la réduction de PGG2 en
PGH2 catalysée par un site différent de la COX et 3) la conversion de PGH2 en PGE2 par la
PGE synthétase (voir illustration de la biosynthèse des prostaglandines, schéma 12). La
PGH2 peut aussi être convertie en d'autres métabolites physiologiquement actifs (les
prostanoïdes) comme PGI2 (la prostacycline), PGD2, PGF2 ou le TXA2 par d'autres
synthétases. Les agents anti-inflammatoires non stéroïdiens (AINS) comme les dérivés de
l'acide salicylique inhibent l'activité enzymatique des COX et donc la biosynthèse des PG.
Il existe deux isoformes de COX : COX-1 et COX-2, partageant environ 60% d'homologie
dans leur séquence et les mêmes propriétés catalytiques, sauf que la COX-1 est régulée par
allostérisme négatif à de faibles concentrations d'AA (379). Une autre différence
structurale réside dans le fait que le site actif de la COX-2 a une taille plus importante que
celui de la COX-1, ce qui a permis le développement d'inhibiteurs pharmacologiques
spécifiques pour la COX-2 (les Coxibs) qui n'ont pas accès au site actif de la COX-1. Mais
la caractéristique qui différencie le plus ces deux isoformes est leur mode d'expression : la
COX-1 est exprimée constitutivement dans la plupart des cellules mammifères alors que
l'expression de la COX-2 est inductible par différents stimulis (agents mitogéniques,
cytokines pro-inflammatoires) dans des conditions de stress physiologique ou mécanique
ou d'inflammation.
Les principales cellules productrices de PGE2 lors de la réaction inflammatoire sont les
monocytes/macrophages, les cellules endothéliales et les fibroblastes. Cependant, une
activité constitutive de la COX-2 a pu être observée dans certains types cellulaires, dont le
neutrophile (380). Les neutrophiles peuvent aussi libérer des PGE2 en réponse à divers
stimuli (particules, GM-CSF,TNFa et fMLP) (46, 381, 382). L'expression de la COX-2
dans les neutrophiles est augmentée par le LPS, le GM-CSF, le TNFa, le fMLP, l'IL-1 et
l'IL-8, les particules opsonisées et les cristaux d'urate (380, 383-385) alors que PIL-4 et
l'IL-10 diminuent son expression induite par le LPS (386).

53
-COOH
Arachidonic acid
• 2 O 2
NSAID COX-1 or COX-2
COOH
PGI synthase Q
PGI2 (prostacyclin)
PGDsynthase
COOH
COOH
COOH
(9a, 11a-PGF2)
Schéma 12. Biosynthèse des prostaglandines par la voie des cyclooxygénases

54
Les effets physiologiques de PGE2 sont médiés par des RCPG : les récepteurs EP (E
prostaglandin) dont il existe quatre sous-types (EP1-EP4) couplés à des voies de
signalisation distinctes. Les récepteurs EPi sont couplés au métabolisme du calcium, les
récepteurs EP2 et EP4 sont couplés aux protéines Gas qui élèvent 1' [AMPc]i en stimulant
l'activité des AC, alors que les différentes isoformes de récepteurs EP3 peuvent soit
diminuer l'[AMPc]i via des protéines Gai, soit élever P[AMPc]i via Gas ou bien stimuler
le métabolisme des Pis et du calcium via des protéines Gaq (387). L'expression
différentielle des ces quatre types de récepteurs EP dans les tissus permet d'expliquer la
grande variété des effets physiologiques de PGE 2.
RÉCEPTEURS EP
EPi EP2 EP4 EP3a, \i and y
ACI fACl 1A
cAMPf cAMPf cAMPJ,
TRENDS in Immunology Vol.23 No.3
Schéma 13. Voies de signalisation couplées aux récepteurs EP.

55
PGE2 est un médiateur important de la réaction inflammatoire puisqu'elle contribue à la
vasodilatation (388) et à l'augmentation de la perméabilité vasculaire (389, 390) ainsi qu'à
la nociception (391). Elle est retrouvée en quantité importante dans les tissus inflammés
(392) et contribue donc à la genèse des signes cardinaux de l'inflammation. De plus,
l'utilisation des dérivés de l'acide salicylique, connus depuis longtemps pour diminuer
l'inflammation (393, 394) et, plus récemment, des inhibiteurs sélectifs de la COX-2 (395)
comme agents thérapeutiques anti-inflammatoires a montré clairement le rôle pro-
inflammatoire de PGE2. Pourtant, un effet inhibiteur de PGEi et PGE2 sur la progression de
l'arthrite et la destruction du cartilage (396, 397), sur la vasculite induite par des complexes
immuns (398) ou sur l'inflammation provoquée par des cristaux (399) a été observé dans
différents modèles animaux. Ces observations suggéraient que les PGE1/2 pouvaient aussi
réguler la réaction inflammatoire en la diminuant dans certaines conditions expérimentales.
Un rôle inhibiteur de PGE2 sur les fonctions des leucocytes activés a finalement été
découvert et pourrait expliquer les résultats obtenus qui paraissaient parfois paradoxaux
(342,400,401).
PGE2 inhibe la plupart des fonctions du neutrophiles stimulées par des agents
chimiotactiques ou particulaires. En particulier, la production d'anions superoxyde (402-
404), la libération d'enzymes cytotoxiques (405), la biosynthèse et la libération de LTB4
(406), l'adhésion aux cellules épithéliales (345) ainsi que la chimiotaxie stimulées par le
fMLP sont inhibées lorsque les neutrophiles sont pré-incubés en présence de PGE2. Ces
effets inhibiteurs sont médiés par le récepteurs EP2 (407-409).

56
1.5. Objectifs de l'étudeAlors que l'effet inhibiteur d'une élévation des niveaux intracellulaires d'AMPc sur les
fonctions du neutrophile est connu depuis assez longtemps, les mécanismes de signalisation
qui permettraient d'expliquer cette inhibition fonctionnelle restent peu connus jusqu'à
présent. Notre objectif principal a donc été de caractériser le mécanisme inhibiteur de PGE2
sur les voies de signalisation induites par le fMLP dans les neutrophiles humains. Un effet
inhibiteur de PGE2 sur l'activité de la PLD stimulée par le fMLP dans les neutrophiles a été
rapporté (410, 411). Le fait que l'activité de la PLD soit inhibée par PGE2 lorsqu'elle est
stimulée par le fMLP mais pas par le PMA indique que la PLD elle-même ne peut pas être
la cible directe de PGE2. La (ou les) cible(s) de l'effet inhibiteur de PGE2 doi(ven)t donc
être située(s) à un niveau plus proche du FPR dans la voie de signalisation induite par le
fMLP. Nous avons donc cherché à caractériser les effets de PGE2 sur les différents facteurs
qui régulent en amont l'activité de la PLD, c'est-dire les petites GTPases des familles Rho
et Arf, la PKC, la libération de calcium, les tyrosine kinases et la PI3-K.
Par ailleurs, l'effet inhibiteur de PGE2 sur l'activation des neutrophiles stimulés par le
fMLP, et plus particulièrement sur l'activation de la PLD, a été attribué à l'augmentation
des niveaux intracellulaires d'AMPc que l'on observe de façon concomittante avec
l'inhibition des fonctions cellulaires. Cependant, les mécanismes de signalisation précis
impliqués dans l'effet inhibiteur de PGE2 ne sont pas connus. Nous avons donc cherché à
déterminer si l'inhibition des voies de signalisation stimulées par le fMLP observée en
présence de PGE2 impliquait le principal effecteur de l'AMPc dans les cellules, c'est-à-dire
laPKA.

CHAPITRE II
Prostaglandin E2 inhibits thc phospholipase D pathway stimulated by formyl-
Methionyl-Leucyl-Phenylalanine in human neutrophils. Involvement of EP2 receptors
and phosphatidylinositol 3-kinase y
L'article présenté dans ce chapitre a été publié dans Molecular Pharmacology, 66 (2): 1-9,
2004.
Copyright © 2004 The American Association For Pharmacology and Expérimental
Therapeutics. Ail right reserved.

58
2.1 RésuméLa prostaglandine E2 (PGE2), découverte à l'origine comme un médiateur pro-
inflammatoire, inhibe également dans le neutrophile plusieurs fonctions stimulées par des
agents chimiotactiques, telles que l'adhérence, la sécrétion d'enzymes cytotoxiques, la
production d'anions superoxides et le chimiotactisme. Dans cette étude, nous avons
examiné les effets de PGE2 et des agonistes/antagonistes spécifiques des récepteurs EP des
prostaglandines E sur différentes étapes de la voie d'activation de la phospholipase D
(PLD) induite par le fMLP dans le neutrophile humain afin d'élucider le mécanisme
d'inhibition par PGE2 PGE2 et les agonistes du récepteur EP2 inhibent, d'une manière dose-
dépendante, l'activité de la PLD stimulée par le fMLP. La translocation aux membranes de
la PKCa et des GTPases Rho et Arf stimulée par le fMLP est diminuée en présence de
PGE2 et des agonistes des récepteurs EP2 De plus, la PGE2 et les agonistes des récepteurs
EP2 diminuent l'activation de la phosphatidylinositol 3-kinase y (PI3K-y) et des Tec kinases
ainsi que de la phosphorylation de protéines sur des résidus tyrosine lorsqu'elles sont
stimulées par le fMLP. Ces résultats apportent de fortes évidences que : 1) les récepteurs
EP2 sont impliqués dans l'effet inhibiteur de PGE2 sur la voie d'activation de la PLD
induite par le fMLP et que 2) la suppression de l'activité de la PI3-Ky représente une étape
cruciale de l'inhibition par PGE2, via les récepteurs EP2, de la cascade de signalisation
induite par le fMLP.

59
2.2 AbstractProstaglandin E2 (PGE2), originally discovered as a pro-inflammatory mediator, also
inhibits several chemoattractant-elicited neutrophil functions including adhésion, sécrétion
of cytotoxic enzymes, production of superoxide anions and chemotaxis. In this study, we
hâve examined the effects of PGE? and prostaglandin E (EP) receptor sélective
agonists/antagonists on several steps of the fMLP-induced phospholipase D (PLD)
activation pathway in human neutrophils in order to elucidate the PGE2 inhibitory
mechanism.
PGE2 and EP2 receptor agonists inhibited the stimulation of the activity of PLD induced by
fMLP in a concentration-dependent manner. The fMLP-stimulated translocation to
membranes of PKCct, Rho and Arf GTPases was diminished in the présence of PGE2 or
EP2 agonists. Moreover, PGE2 and EP2 agonists decreased the activation of
phosphatidylinositol 3-kinase y (P13Ky) and Tec kinases as well as the tyrosine
phosphorylation of proteins stimulated by fMLP. Thèse data provide strong évidence that
1) the inhibitory effects of PGE2 on the fMLP-induced PLD activation pathway were
mediated via EP2 receptors and that 2) the suppression of PI3-Ky activity was the crucial
step in the EP2-mediated inhibition of the fMLP-induced signalling cascade.

60
2.3. IntroductionPhospholipase D (PLD) catalyzes the hydrolysis of choline-containing lipids to produce the
second messenger phosphatidic acid (PA). The PLD pathway is thought to play a critical
rôle in regulating cell responses such as phagocytosis (Lennartz, 1999), sécrétion and
production of superoxide anions by the NADPH oxidase complex (Liscovitch et al., 2000)
in human neutrophils. Two mammalian gènes coding for PLD1 and PLD2 hâve been
cloned (Frohman et al., 1999), however, only the PLD1 isoform has been detected in
human neutrophils (Marcil et al., 1997). PLD1 requires phosphoinositides for activity and
is regulated by direct interactions with conventional PKC isoforms and the small GTPases
RhoA and Arf (ADP-ribosylation factor) (Exton, 1999). fMLP-induced PLD activity is also
modulated by variations in cytosolic calcium and its stimulation is controlled by
phosphatidylinositol 3-kinase (PI3K) and tyrosine kinase-dependent events (Exton, 1999).
Besides their rôle in the régulation of PLD activity, the Rho GTPase family members (Rho,
Rac and Cdc42) also modulate multiple cellular processes by cycling between a GDP-
bound inactive form and a GTP-bound form that directly activâtes several downstream
targets. They are implicated in the régulation of the dynamics of the actin cytoskeleton, in
changes in cell shape and motility (Hall, 1998) and they regulate various leukocyte
functions including adhésion (Laudanna et al., 1996), phagocytosis (Chimini and Chavrier,
2000) and the migratory response to chemoattractants (Servant et al., 2000). In phagocytic
cells, Rac also plays a major rôle in the production.of superoxide anions by the NADPH
oxidase complex (Babior, 1999). Rho GTPases are therefore critical regulators of the
functional responsiveness in neutrophils.
Prostaglandins E (PGEs) are cyclooxygenase (COX) products that exert pleiotropic effects
in a paracrine fashion. The physiological activities of PGEs are mediated via G-protein
linked seven transmembrane domain receptors that hâve been classified into four subtypes,
EPi to EP4, according to their structure and coupling to distinct signalling pathways (Breyer
et al., 2001). EP2 and EP4 stimulate adenylyl-cyclase, whereas EPi triggers intracellular
calcium release. The various EP3 isoforms generated by alternative splicing are connected
either to cAMP or to calcium metabolism. Pro-inflammatory functions hâve been attributed
to PGEs by virtue of their vasodilator and nociceptive properties. However, PGEs also

61
display inhibitory effects on most leukocyte functions and are considered as modulators of
inflammation rather than strictly pro-inflammatory mediators (Tilley et al., 2001). Most
neutrophil functional responses including adhésion to epithelial cells (Bloemen et al.,
1997), motility and chemotaxis (Rivkin et al., 1975), production of superoxide anions
(Fantone and Kinnes, 1983) and the release of cytotoxic enzymes and leukotriene B4 (Ham
et al., 1983; Hecker et al., 1990) are diminished by PGE2 or PGEi. Pharmacological studies
hâve shown that the inhibitory effects of PGE2 on fMLP-induced superoxide production,
enzyme release and chemotaxis are mediated via the EP2 reeeptor (Armstrong, 1995;
Talpain et al., 1995).
The effects of PGE2 on signal transduction pathways induced by chemoattractants in
neutrophils hâve only been incompletely characterized so far. PGE2 has been shown to
inhibit fMLP but not PMA-induced PLD activation in human neutrophils (Agwu et al.,
1991) but the mechanism involved has not been clariiïed. Therefore, we investigated
further the effects of PGE2 (and of spécifie EP reeeptor agonists) on the signalling cascade
that leads to PLD activation upon fMLP stimulation. In this study, we report that PGE2
inhibits the stimulation of the activity of PLD, the translocation of PKCa, Arf and Rho
GTPases and Tec kinases to membranes, the accumulation of Ptdlns(3,4,5)P3 and the
activity of PI3-Ky induced by fMLP. Thèse effects were mediated via EP2 receptors. Our
results suggest that suppression of fMLP-induced PI3-Ky activity by PGE2 plays a
prominent rôle in the inhibition of downstream signalling events such as the activation of
small GTPases, of PKCa and Tec kinases, thereby resulting in decreased PLD activation.

62
2.4. Materials and methodsAntibodies : The anti-PKCa (PI6520) and anti-Cdc42 (C70820) monoclonal Abs were
purchased from BD Transduction Laboratories (Mississauga, ON, Canada). Anti-RhoA
(SC-179), anti-Rac2 (SC-96), anti-ïec (SC-1109) and anti-Btk (SC-1107) polyclonal Abs
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The polyclonal anti-EP2
receptor Ab (#101750) and the EP2 receptor blocking peptide (#301750) were obtained
from Cayman Chemical (Ann Arbor, Ml). The polyclonal anti-Arfl and anti-pl 10y antisera
were raised in rabbits as described previously (Houle et al., 1999; Naccache et al., 2000).
The anti-phosphotyrosine Ab (UBI 05-321, clone 4G10) was purchased from Upstate
Biotechnology Inc. (Lake Placid, NY). Secondary anti-mouse (#NXA931) and anti-rabbit
(#NA934V) Abs were obtained from Amersham Biosciences (Baie d'Urfé, Québec,
Canada) and the secondary anti-goat Ab (SC-2056) from Santa Cruz Biotechnology.
Reagents : Dextran T-500 was purchased from Pharmacia Biotech (Dorval, Québec,
Canada) and Ficoll-Paque from Wisent (St-Bruno, Québec, Canada). Adenosine deaminase
(ADA) was purchased from Roche Diagnostics (Laval, Québec, Canada), and di-
isopropylfluorophosphate (DFP) from Serva (Heidelberg, Germany). fMet-Leu-Phe (fMLP)
and cytochalasin B (CB) were obtained from Sigma-Aldrich Canada (Oakville, Ontario,
Canada). l-O-[3H]alkyl-2-lyso-phosphatidylcholine was obtained from Amersham
Biosciences (Baie d'Urfé, Québec, Canada). PGE2, the butaprost acid-form CAY 10399,
11-deoxy PGEi, sulprostone and AH 23848 were purchased from Cayman Chemical (Ann
Arbor, MI). [32P] orthophosphate (1000 Ci/mmol) and [y-32 P] ATP (3000 Ci/mmol) were
purchased from Perkin-Elmer Life Sciences (Boston, MA). Fura-2/AM was obtained from
Molecular Probes (Eugène, OR).
Isolation of human neutrophils
Venous blood was collected from healthy adult volunteers in isocitrate anticoagulant
solution. Neutrophils were separated as described previously (Marcil et al, 1999). Briefly,
whole blood was centrifuged at 180g for 10 min and the resulting platelet rich plasma was
discarded. Leukocytes were obtained following erythrocytes sédimentation in 2% Dextran

63
T-500. Mononuclear cells were removed by centrifugation on Ficoll-Paque cushions and
contaminating erythrocytes in the neutrophil pellets were removed by a 20 sec hypotonie
lysis in water. Neutrophils were resuspended in Manks Balanced Sait Solution (HBSS), pH
7.4, containing 1.6 mM Ca 2l~ but no Mg2. The cells were sterilely purified to avoid
bacterial contamination of the neutrophil suspensions.
PLD measurements
Neutrophils were labeled with l-O-[3H]alkyl-2-lyso-phosphatidylcholine (2 |a.Ci/107 cells)
for 90 min as described previously (Marcil et al., 1999). The cells were washed and
resuspended at 107 cells/ml in HBSS. Cell suspensions (0.5 ml) were warmed at 37 °C for 5
min and then pre-treated 5 min with 10 uM CB and 0.1 U/ml ADA to eliminate
endogenous adenosine and in the présence or the absence of the indicated concentrations of
agonists or antagonists. Neutrophils were stimulated with 100 nM fMLP for 10 min in the
présence of 1% ethanol to monitor the formation of radiolabeled phosphatidylethanol (PEt)
catalysed by PLD. Incubations were stopped by adding 1.8 ml of chloroform/methanol/HCl
(50:100:l,v/v/v) and unlabeled PEt as a standard. Lipids were extracted and the levels of
[3H]PEt were quantified as described previously (Marcil et al., 1999).
Measurement of cytoplasmic free calcium concentrations
Neutrophils (107 cells/ml) were incubated at 37 °C for 30 min with 1 uM Fura-2/AM. The
cells were washed and resuspended at 5 x 106 cells/ml in HBSS with 1.6 mM Ca2+.
Neutrophils were transferred to the thermostated cuvette compartment of a
spectrofluorimeter (SLM 8000C) and preincubated 5 min at 37 °C with 0.1 U/ml ADA in
the présence of various EP receptor agonists/antagonists or the equal volume of Me2SO.
Cells were stimulated with 100 nM fMLP at the time indicated by the arrow. The
fluorescence of the cells was monitored at an excitation wavelength of 340 nm and an
émission wavelength of 510 nm. The internai calcium concentrations were calculated using
the following formula: 224((y-Fmin)(Fmax-y)"') in which y represents the fluorescence of

64
the samples. Fmax was obtained by disrupting the cells with 1% TritonX-100 and Fmin
was obtained by adding 5 mM of both EGTA and NaOH.
Manganèse influx measurement
Neutrophils were loaded with Fura-2/AM, washed and resuspended in HBSS without
calcium and magnésium just prior incubation with ADA and PGE2 or EP receptor agonists
for 5 min at 37 °C in a thermostated cuvette as described above. The wavelength used for
thèse experiments were 360 and 505 nm for excitation and émission, respectively. After 10
sec, 200 uM MnCl2 were added to the cell suspensions that were then stimulated by 100
nM fMLP at 70 sec.
Translocation assays
Neutrophils (4 x 107 cells/ml) were treated with 1 mM DFP for 10 min at room
température. The cell suspensions were centrifuged once and the cells were resuspended in
HBSS at 107 cells/ml. The cells were warmed 5 min at 37 °C and treated 5 min with 10 uM
CB, 0.1 U/ml ADA and PGE2 or EP receptor agonists or an equal volume of diluent
(Me2SO) as a control. Neutrophils were stimulated with 100 nM fMLP at 37 °C.
Incubations were stopped by diluting the cells five fold with ice cold HBSS and total
membrane proteins were collected as described previously (Marcil et al., 1999). Protein
samples (10-20 [ig) were resolved on a 7.5-20% gradient SDS-PAGE and transferred to
Immobilon PVDF membranes (Millipore Corporation, Bedford, MA, USA). Western blots
were performed using anti-Arfl (1/2500), anti-RhoA (1/1000), anti-PKCa (1/1000), anti-
Rac2 (1/200) and anti-Cdc42 (1/250), anti-pllOy (1/1000), anti-Tec (1/1000) or anti-Btk
(1/1000) Abs. Proteins were revealed with HRP-conjugated secondary anti-mouse or anti-
rabbit Ab (1/20,000) or anti-goat Ab (1/15,000) and the Renaissance détection System
(NEN, Perkin Elmer Life Sciences).
Phosphatidylinositol 3,4,5 -trisphosphate (PtdIns(3,4,5)P3) formation.

65
Neutrophils (5 x 107 cells/ml) were incubated with 0.5 mCi/ml of [32P] orthophosphate for
1 h at 37 °C in a labeling buffer (137 mM NaCl, 5 mM KC1, 10 mM glucose, 20 raM
Hepes, pH 7.4). Unincorporated radioactivity was discarded and the cells were washed
twice in HBSS. The cells were resuspended in HBSS and pre-incubated with PGE2/EP
agonists or diluent in the présence of ADA 0.1 U/ml for 5 min at 37 °C. The cells were
stimulated with 100 nM fMLP for 30 sec and reactions were stopped by addition of 400 \xl
of chloroform-methanol (1:1, v/v). The samples were processed as described previously
(Gilbert et al., 2003) and the final products visualized by exposure of x-ray films at -80 °C
or with a bio-imaging analyzer (Fujifilm BAS-1800).
Measurements of PI3-Ky activity
Cell suspensions (4 x 107 cells/ml) were pre-incubated for 5 min at 37 °C with 10"5 M PGE2
(or an equal volume of Me2SO) and 0.1 U/ml ADA. Cells were stimulated with 100 nM
fMLP and the reaction was stopped by addition of an equal volume ice-cold buffer I
(phosphate-buffered saline containing 1 mM CaCb, 1 mM MgCh, 100 \xM Na3VO4).
Samples were processed as already described for the pi lOy immunoprecipitation and the
PI3-K assays (Naccache et al., 2000).
Tyrosine phosphorylation patterns
Neutrophils (2 x 107 cells/ml) were pre-incubated at room température with 1 mM DFP,
with or without the indicated concentrations of PGE2 or EP agonists for 10 min at 37 °C
prior to stimulation with 100 nM fMLP for the indicated times. The reactions were stopped
by transferring 100 u.1 of the cell suspensions to an equal volume of boiling 2X Laemmli
sample buffer (SB) (lx is 62.5 mM Tris-HCl, pH 6.8, 4% SDS, 5% p-mercaptoethanol,
8.5% glycerol, 2.5 mM orthovanadate, 10 mM paranitro-phenylphosphate, 10 |ag/ml
leupeptin, 10 ug/ml aprotinin, 0.025% bromophenol blue) and boiled for 7 min. The
samples were then subjected to 7.5-20% gradient SDS-PAGE and transferred to Immobilon
PVDF membranes. Immunoblotting was performed using the monoclonal anti-

66
phosphotyrosine Ab 4G10 (1/4000) and revealed as described previously (Gilbert et al.,
2003).
Statistical analysis.
The data were analyzed using the one-sample Student t test for the PLD and translocation
assays data or the Student paired t test (two tailed) for the PI3K assay data. The levels of
significance (* p<0.05 and **p<0.01) were determined between the treated samples and the
appropriate controls.

67
2.5 Results
2.5.1. fMLP-induced PLD activity is inhibited by PGE2 via EP2 receptor.
As is the case for other cAMP elevating substances (adenosine, forskolin, adrenergic pV
agonists), PGE2 has also been shown to inhibit fMLP-induced PLD activity in the présence
of phosphodiesterase (PDE) inhibitors (IBMX or theophylline) (Agwu et al., 1991). We
hâve first investigated the effect of PGE2 on PLD activity in our expérimental conditions,
i.e. in the absence of PDE inhibitor and in the présence of AD A to eliminate the inhibitory
effect of endogenous adenosine that could interfère with that of PGE2 (Thibault et al.,
2000). As shown in Fig. 1A, PGE2 inhibited [3H]PEt formation in a concentration-
dependent manner. The amounts of [3H]PEt formed were 25.89 ± 5.01% (p = 0.0045),
25.44 + 5.74 % (p = 0.0059), 35.27 ± 6.83% (p = 0.011) and 43.3 ± 5.21% (p = 0.0083) of
the fMLP-stimulated control in the présence of 10"5, 10"6, 10"7 and 10"8 M PGE2,
respectively. To détermine which EP receptor subtype mediated the inhibition of PLD
activity by PGE2, [3H]PEt formation was assessed in the présence of EP receptor sélective
agonists and antagonists, ail of which being PGE2 analogues. We tested the effects of
one EP2 receptor sélective agonist, CAY10399, one EP2/EP4 agonist, 11-deoxyPGEi, one
EP1/EP3 agonist, sulprostone, and one sélective EP4 receptor antagonist, AH23848 on PLD
activity. FMLP-induced [3H]PEt formation was signilïcantly reduced to 25.67 + 2.37 (p =
0.01), 40.89 ± 5.8 (p = 0.0095), 65.3 ± 3.68 (p = 0.011) and 88.24 ±2.15 (p = 0.032) of its
level in untreated controls in the présence of 10"5> 10"6 10"7 and 10"8 M 11-deoxyPGEi,
respectively. In the présence of 10"5, 10'6 and 10"7 M CAY10399, [3H]PEt formation was
also reduced to 33.5 ± 4.25 (p = 0.004), 41.8 ± 3.9 (p = 0,004) and 58.75 ± 4,45 (p = 0.011)
of its level in the untreated controls, respectively. The sélective EP1/EP3 receptor agonist,
sulprostone had no effect on the fMLP-induced PLD activity (Fig. 1A). The sélective EP4
receptor antagonist AH23848 did not affect the inhibitory effect of PGE2 on the stimulation
of the activity of PLD by fMLP (Fig. 1B), indicating that PGE2 did not exert its inhibitory
effect via the EP4 receptor. Since the EP1/EP3 agonist sulprostone did not affect fMLP-
stimulated PLD activity and the EP4 antagonist did not modify the PGE2-mediated
inhibition of PLD activity, thèse data suggested that EP2 receptors mediated the inhibitory
effects ofPGE2.

68
The présence of EP2 receptors on neutrophil membranes was assessed next by
immunoblotting of the membrane fractions with an anti-EP2 antibody. We observed a band
at 53 kDa that was no longer detected when the antibody was pre-neutralized with the
immunizing peptide (Fig. 1C).
2.5.2. PGE2 inhibition of Ca 2+ influx is mediated via EP2 receptor.
fMLP induces a rapid increase in free cytoplasmic calcium resulting from the intracellular
release of Ca2+ stores mediated by PLCp-released inositol 3,4,5-trisphosphate followed by
a sustained Ca2+ influx from the extracellular médium. fMLP-induced PLD activity has
been shown to be dépendent on this rise in Ca2+ (Exton, 1999) as well as on extracellular
calcium (Pai et al., 1988). First, we examined the effect of PGE2 on calcium mobilization in
fMLP-stimulated human neutrophils. As already described, fMLP induced a rapid and
transient increase in cytoplamic free calcium as monitored using the fluorescent probe
Fura-2. PGE2 had no effect on the magnitude of the rapid rise in free cytoplasmic calcium,
but reduced the duration of the calcium spike suggesting that the contribution of calcium
influx to the mobilization of calcium could be partially decreased by PGE2 (Fig. 2A). We
next assessed whether the effect of PGE2 on calcium mobilization were also mediated via
EP2 receptors. The EP2 receptor sélective agonist CAY10399 reduced the duration of the
calcium spike in a manner akin to PGE2, (Fig. 2B) while the EP4 receptor antagonist AH
23848 did not modify the réduction of the mobilization of cytosolic calcium induced by
PGE2 (Fig. 2C). Furthermore, the EP1/EP3 receptor agonist, sulprostone, influenced neither
the magnitude nor the duration of the élévation of cytoplasmic calcium elicited by fMLP
(Fig. 2D).
To confirm that PGE2 and EP2 agonists acted on the influx of calcium, we monitored the
quenching of intracellular Fura-2 fluorescence that results from the entry of Mn2+ through
divalent cation channels (Fig. 2E). This experiment showed that a pretreatment of
neutrophils with PGE2 or the EP2 agonist CAY 10399 partially reduced the fMLP-induced
rate of quenching of Fura-2, indicating that the stimulation of the EP2 pathway exerted an
inhibitory effect on the fMLP-stimulated calcium influx.

2.5.3. Effect of PGE2 on recruitment of PKCa, Arf and Rho GTPases tomembranes.
The cytosolic PLD activation factors, PKCa, and the small GTPases Arfl and RhoA
migrate to membranes where they médiate the activation of PLD by fMLP (Houle et al.,
1995). To investigate the mechanism implicated in the inhibition of fMLP-induced PLD
activity by PGE2, we assessed whether PGE2 interfered with the translocation to
membranes of the PLDl activation factors. As shown in Fig. 3, 10"5 M PGE2 alone had no
noticeable effect on the basai membrane levels of PKCa, Arfl and RhoA. In comparison,
PGE2 pre-treatment prior to fMLP stimulation resulted in a significant and dose-dependent
inhibition of the translocation of PKCa, Arfl and RhoA to the membranes (as compared to
the fMLP-stimulated control). The recruitment to membranes of the two other Rho
GTPases family members, Rac2 and Cdc42 was also decreased by PGE2 pre-treatment.
2.5.4. PGE2 decreases PKCa, Arf and Rho GTPases recruitment tomembranes through EP2 receptor.
We next assessed whether EP2 agonists affected the membrane translocation of PKCa and
small GTPases. As was the case with PGE2, the EP2 sélective agonist CAY10399 alone did
not affect the basai level of PKCa, Arf and Rho GTPases associated with membranes, but
pre-incubation of cells with CAY10399 prior to fMLP stimulation significantly decreased
the amounts of membrane-translocated PKCa, Arf and Rho GTPases in a concentration-
dependent manner (Fig. 4). Thèse data hâve been buttressed by the use of another sélective
EP2 agonist: the 19(R)-hydroxy PGE2, which showed effects similar to those of CAY 10399
(data not shown), indicating that the inhibition of the translocation of PLD cofactors by
PGE2 was mediated via the EP2 receptors.
2.5.5. Effect of PGE2 on PtdIns(3,4,5)P3 formation and PI3-Ky activity.
PLDl activation is also regulated by phosphoinositides and pharmacological inhibitors of
PI3-K strongly inhibit PLD activity induced by chemoattractants (Reinhold et al., 1990).
Moreover, PI3-K exerts a regulatory rôle over small GTPase activation, especially on Rac2
and Cdc42 (Benard et al, 1999). Thus, we tested the effects of PGE2 on fMLP-induced
PtdIns(3,4,5)P3 formation in human neutrophils. As shown in Fig. 5A, PGE2 (10'5 to 10"8

70
M) decreased the amounts of PtdIns(3,4,5)P.3 ibrmed following fMLP stimulation. The
amounts of PtdIns(3,4,5)P3 formed in response to fMLP were also monitored in the
présence of CAY10399. As shown in Fig. 5B, CAY 10399 (10"5 and 10"6 M) also inhibited
fMLP-stimulated PtdIns(3,4,5)P3 formation, indicating that PGE2 exerted its inhibitory
effect on PtdIns(3,4,5)P3 formation via the EP2 receptors. Decreased accumulation of
PtdIns(3,4,5)P3 could be due to decreased transformation of PtdIns(4,5)P2 into
PtdIns(3,4,5)P3 or to accelerated dephosphorylation of PtdIns(3,4,5)P3. To distinguish
between thèse possibilities, we monitored the activity of pli0y induced by fMLP in the
présence of PGE2, for pllOy is the major PI3-K isoform activated by chemoattractants in
neutrophils (Hirsch et al., 2000; Naccache et al., 2000). Briefly, PI3-Ky was
immunoprecipitated using an anti-pl lOy Ab at différent times following fMLP stimulation
and PI3-Ky activity was tested using an exogenous substrate (Ptdlns). The results of thèse
experiments (Fig. 6A) indicated that fMLP-induced pllOy activity was maximal at 15-30
sec, and this peak of pi lOy activity was totally abolished by PGE2 (p = 0.0264 at 15 sec and
p = 0.0089 at 30 sec). The activation of pi lOy by chemotactic factors is characterized by its
rapid recruitment to membranes, and we hâve therefore examined the effects of PGE2 and
EP2 agonists on this translocation step. PGE2 and CAY 10399 alone had no effect on the
amounts of membrane-associated pllOy in unstimulated cells, but reduced the levels of
membrane-associated pllOy to that of the unstimulated controls in fMLP-stimulated
neutrophils (Fig. 6B). Thus, the inhibition of the fMLP-stimulated accumulation
PtdIns(3,4,5)P3 by PGE2 appears to be the direct conséquence of the inhibition of pllOy
activity.
2.5.6. Effect of PGE2on the fMLP-induced pattern of tyrosinephosphorylation.
Tyrosine kinases are well-known regulators of PLD activity induced by various agonists
and hâve been shown to act upstream of fMLP-induced recruitment of cytosolic PLD
cofactors to membranes (Houle et al., 1999). We hâve therefore sought évidence for an
effect of PGE2 on the tyrosine phosphorylation of proteins stimulated by fMLP. The pattern
of tyrosine phosphorylation induced by fMLP was examined in the présence or absence of
PGE2 or the EP2 agonist CAY10399. As shown in Fig. 7A, incubation of neutrophils with

71
10"5 M PGE2 or CAY10399 prior to fMLP stimulation decreased the intensity and the
duration of phosphorylation of the band at 120 kDa. For a few donors, the tyrosine
phosphorylation of the 42-44 kDa bands corresponding to the MAPKs as evidenced by
reprobing the membranes with an anti-phospho ERK1/2 Ab (data not shown) was also
reduced by PGE2 (middle panel) but not by CAY10399 (right panel). Taken together, thèse
data indicate that PGE2 reduced the activation of some still unidentified tyrosine kinase(s)
via EP2 receptor.
Several members of the Tec family kinases (Tec, Btk and Bmx) hâve been shown to be
involved in response to chemoattractants in human neutrophils (Lachance et al., 2002). The
activation of Tec kinases is dépendent on PI3-K and characterized by a rapid translocation
to the PtdIns(3,4,5)P3-enriched membranes. We thus examined the effect of PGE2 and EP2
agonist on this index of activation of Tec kinases. The data shown in Fig. 7B indicate that
PGE2 as well as CAY 10399 (10"5-107 M) decreased the amounts of Tec and Btk
translocated to membranes in fMLP-stimulated cells.
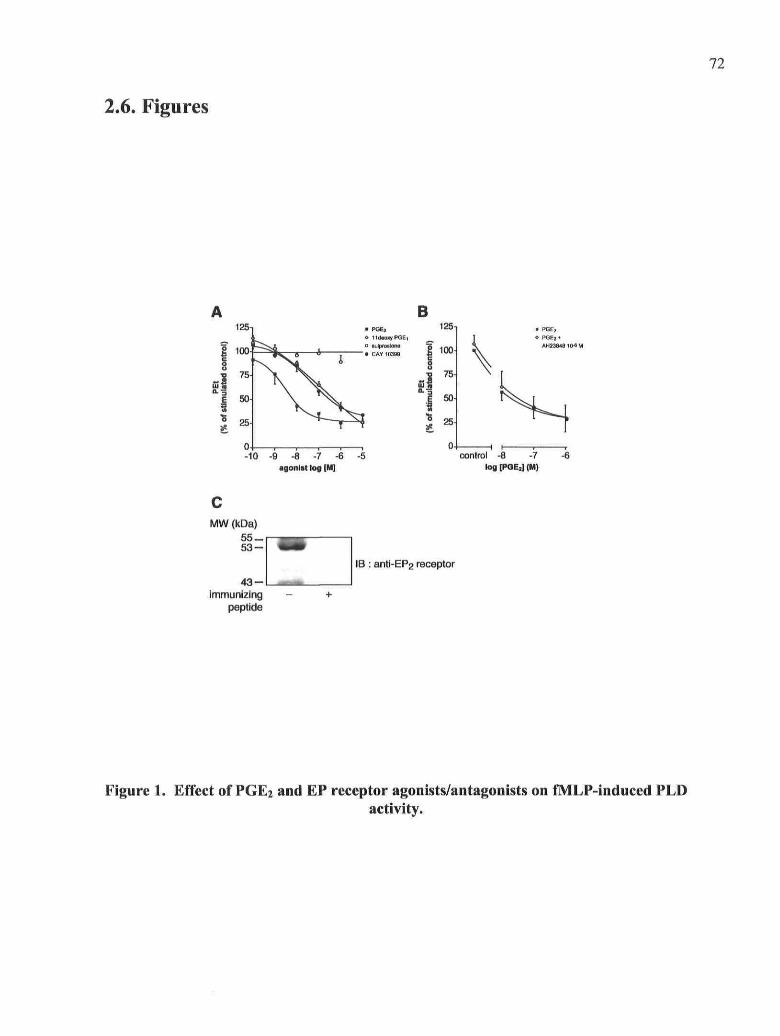
2.6. Figures
72
125n
-10 -9 -8 -7 -6 -5agonist log [M]
B125!
100
75
•s
• PGE2
© PGEa+
AH23848 10 * U
50
control -8 -7log [POE,] (M)
MW (kDa)5 5 -5 3 -
4 3 -immunizing
peptide
IB : anti-EP2 receptor
Figure 1. Effect of PGE2 and EP receptor agonists/antagonists on fMLP-induced PLDactivity.

73
PGE2
BCAY 10399
PGE2 + AH 23848 Suiprostone
.2 1000
S
Si1"
750-
500-
250
n
IMLP
u
1 - -
ConliolAH 23848 10"a M tPGE2 IO-°H
. . . PGEj10-sM
2000
; f 1000
s s R s a gTime (sec)
8 RTime (sec)
Time (sec)
Figure 2: Effects of PGE2 and of EP agonists/antagonists on cytoplasmic free calciumconcentration.
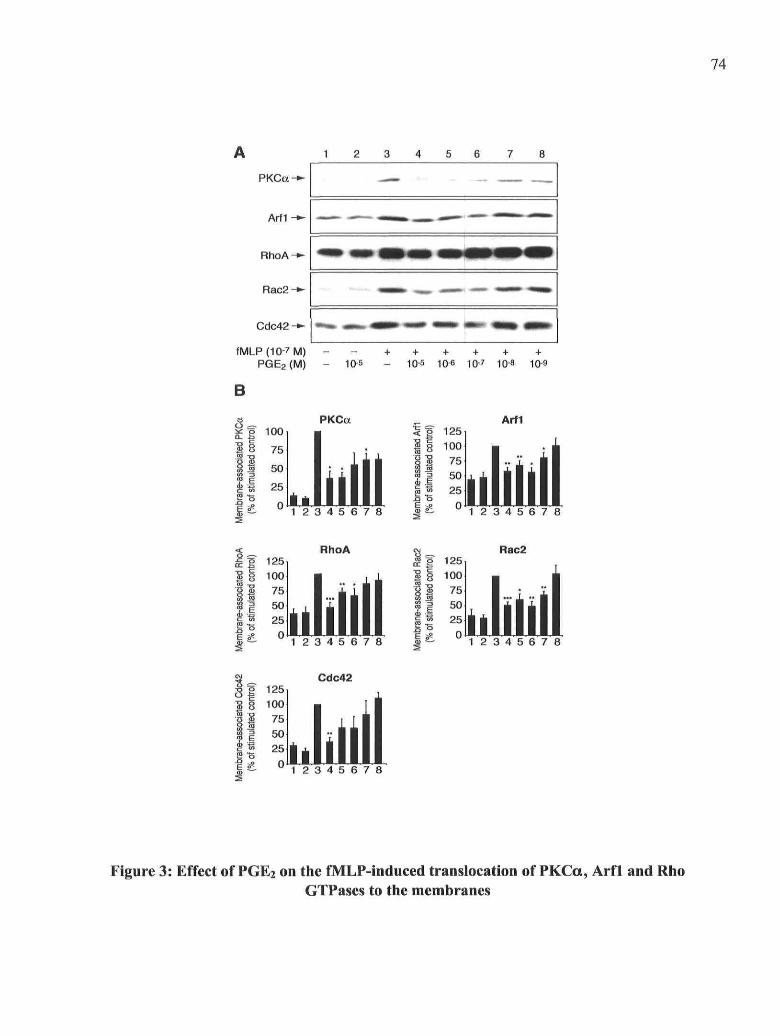
74
1 2 3 4 5 7 8
PKCa
Arf1
RhoA
Cdc42 -*-
fMLP (10-7 M)PGE2 (M)
B
f 10°; § 75
: 1 50
| 1 25I 0
- 10-5 - 10-5 10-6 10-7 10-8 10-9
PKCa
1 2 3 4 5 6 7 8
5f 125
| 8 10°'g S 75| | 50If 25
_Q O
Arfl
1 2 3 4 5 6 7 8
S § 100| | 75
II 25ES? 0
RhoA Rac2
1 2 3 4 5 6 7 8
g 125§ 100S 75m
50250
^
1 2 3 4 5 6 7 8
Cdc42I 125§ 100
I 75
f 50| 255 0 1 2 3 4 5 6 7 8
Figure 3: Effect of PGE2 on the fMLP-induced translocation of PKCa, Arfl and RhoGTPases to the membranes

s
o
VO
and
R]
eOH
an
EP
crooS
o
pilCD 2
(8S"nss* * •
S"ss
Membrane-associated Cdc42(% of stimulated control)
ro 01 vi o roO Ol O Ol O Ol
OI
co
00
Oa
Membrane-associated RhoA(% of stimulated control)
ro 01 vi oO Ol O 01 O
ror -\UJ
Ol
RhoA
ill
^ •
Membrane-associated Rac2(% of stimulated control)
N Ol vl O MO 01 O 01 O 01
Membrane-associated PKCa(% of stimulated control)
ro oi vi o ro oio oi o oi o oi o
QJ
0=
OS
Membrane-associated Arfl(% of stimulated control)
ro oi vi o roo oi o oi o oi
co
w •*co o
- 22I i s. = -
5SBi i
9 iOl
1 +
9 +Ol
9 +->i
9 +œ
9 +
{
i
|
!
|
11
*
1•
i
!
!
i
i
î
11
|111l11
{
1I
s!I

76
PtdlnsP-^
PtdlnsP3-»-
fMLP(10-7M)PGE2 (M)
BPtdlnsP-^
PtdlnsP3-^
1
S
1
•
2
;
10-5
2
•
3
;
3
t
4
;
10-5
4
4
5
;
+
10-6
5
f
6
f+
10-7
6
?
7
f
10-8
7
t
8
f10-9
8
?fMLP(10-7M)
CAY10399(M) - - 10"5 10"7 10"8 10"9
PGE2
= • 100 -
8 75-
S 50-
J 25-
1 2 3 4 5 6 7
= S 75-
• | 50-
: * 0
CAY 10399
1 2 3 4 S 6 7 8
Figure 5: Effect of PGE2 and CAY10399 on the fMLP-induced accumulation ofPtdIns(3,4,5)P3

25 50 75 100 125T i m e (sec)
B cytosoi 1 2 3 4 5 6 7 8 9 1 0
p 1 1 0 y -
M W
(kDa)
- 116
- 97
f M L P ( 1 0 - ? M ) - - - - + + + + + + +
P G E 2 ( M ) - - 10-5 _ _ 10-5 10-610-7
C A Y 10399 (M) - - - 10-5 - - - 10-5 10-6 10-7
77
£=•125-,
Éilll1 2 3 4 5 6 7 8 9 10
Figure 6: Effect of PGE2 on fMLP-induced pllGy activity and translocation tomembranes

m m m•
-M- m r •mm wm —«P aBs *BT sST s » «S S E
««*«*> ****** **Mw **##*• « » • «MM» .«a^*
• * »
_ ^ _ _ i ^ _ _ _ _ _ , ^ _ _
•
. * .
MW(kDa)
-116-97
-66
-55
-43
Time 0 15" 30" 1' 21 51 10' 0 15" 30" 11 21 51 10' 0 15" 30" 1' 2' 5' 10'
B
fMLP fMLP + PGE2 fMLP + CAY 10399
5 6 7 8 9 10
Btk-
Tec-*-
fMLP (10-7 M)PGE2 (M)CAY10399(M)
10-5
5 ~ 100j5
PilS 'S 25S o
+ + + + + +10-5 10-6 10-7
10-5 10-6 10-7
100
iiliÉliklliiil!1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10
Figure 7: Effect of PGE2 and CAY10399 on the global profile of tyrosinephosphorylation and on the translocation of Tec kinases induced by fMLP

79
Figure Legends
Figure 1: Effect of PGE2 and EP receptor agonists/antagonists on fMLP-induced PLD
activity. A, B: Neutrophils (107 cells/ml) were pre-incubated with the indicated
concentrations of PGE2 or/and the EP agonists/antagonists for 5 min at 37 °C in the
présence of 0.1 U/ml ADA and 10 |aM CB. Cells were stimulated with fMLP in the
présence of 1% ethanol for 10 min. The amounts of [3H]PEt formed were evaluated as
described in Material and Methods. The levels of [3H]PEt formed are expressed as the
percentage of the fMLP-stimulated control. The data are from three différent experiments
performed in duplicates and are expressed as the means + SEM. C: Présence of EP2
receptors on neutrophil membrane. Membranes were prepared as described in Material and
Methods and were analyzed by immunoblotting with the anti-EP2 receptor Ab alone (lane
1) or pre-neutralized with the immunizing peptide (lane 2). The immunoblot shown is
représentative of three différent experiments.
Figure 2: Effects of PGE2 and of EP agonists/antagonists on cytoplasmic free calcium
concentration. A, B, C, D: Neutrophils (5 x 106 cells/ml) were preincubated 5 min at 37 °C
with PGE2 or EP agonists/antagonists in the présence of 0.1 U/ml ADA. Cells were
stimulated with fMLP at the time indicated by the arrow and cytosolic calcium
concentrations were monitored as described in Material and Methods. E: Effect of PGE2
and CAY 10399 on fMLP-stimulated Mn2+ influx. Neutrophils resuspended in HBSS
with no Ca2+ were preincubated 5 min at 37 °C with PGE2 or EP agonists/antagonists in the
présence of 0.1 U/ml ADA. 200 JJM MnCb and fMLP was added at the times indicated by
the arrows. The results are représentative of three separate experiments.
Figure 3: Effect of PGE2 on the fMLP-induced translocation of PKCa, Arfl and Rho
GTPases to the membranes. Neutrophils (107 cells/ml) were preincubated with the
indicated concentrations of PGE2 for 5 min at 37 °C, in the présence of 0.1 U/ml ADA and
10 |JM CB. Cell suspensions were stimulated with fMLP for 2 min and the incubations

were stopped as described in Material and Methods. The samples were analyzed for PKCa,
Arfl, RhoA, Rac2 and Cdc42 by immunoblotting. A. The immunoblots shown are
représentative of at least three independent experiments .B. The data of densitometric
analyses are expressed as the percentage of fMLP-stimulated control and are the means +
SD from at least three experiments. The numbers identify the corresponding lanes of the
immunoblots.
Figure 4: Effect of CAY 10399, an EP2 agonist, on the fMLP-induced translocation of
PKCa, Arfl and Rho GTPases to the membranes. Neutrophils (107 cells/ml) were
preincubated with the indicated concentrations of CAY 10399 for 5 min at 37 °C, in the
présence of 0.1 U/ml ADA and 10 uM CB. Cell suspensions were stimulated with fMLP
100 nM for 2 min and the reactions were stopped as described in Material and Methods.
The samples were analyzed for PKCa, Arfl, RhoA, Rac2 and Cdc42 by immunoblotting.
A. The immunoblots shown are représentative of at least three independent experiments. B.
The data of densitometric analyses are expressed as the percentage of fMLP-stimulated
control and are the means + SD from at least three experiments. The numbers identify the
corresponding lanes of the immunoblots.
Figure 5: Effect of PGE2 and CAY 10399 on the fMLP-induced accumulation of
PtdIns(3,4,5)P3. [32P]-labeled neutrophils (5 x 107cells/ml) were pre-incubated 5 min at 37
°C with the indicated concentrations of PGE2 (A) or CAY 10399 (B) in the présence of 0.1
U/ml ADA. Cells were stimulated with fMLP for 30 sec at 37 °C and the amounts of
PtdIns(3,4,5)P3 were measured as described in Material and Methods. The autoradiograms
shown are représentative of three independent experiments. C. The data of the
densitometric analyses are expressed as the percentage of the fMLP-stimulated control and
are the means + SD from three experiments. The numbers identify the corresponding lanes
of the autoradiograms.
Figure 6: Effect of PGE2 on fMLP-induced pllOy activity and translocation to
membranes. A. Neutrophils (107 cells/ml) were preincubated 5 min at 37 °C with 10"5 M
PGE2 in the présence of 0.1 U/ml ADA. Cells were stimulated with fMLP for the indicated
times, lysed and the immunoprecipitated pi 10 y was assayed for lipid kinase

phosphorylation as described in Material and Methods. The results are from four
independent experiments and are expressed as percentage of the unstimulated sample at t =
0. B. Neutrophils (107 cells/ml) were preincubated with PGE2 or CAY 10399 at the
indicated concentrations for 5 min at 37 °C, in the présence of 0.1 U/ml ADA and 10 uM
CB. Cell suspensions were stimulated with fMLP for 30 sec and the reactions were stopped
as described in Material and Methods. The samples were analyzed for pllOy by
immunoblotting. The immunoblot shown is représentative of three independent
experiments. C. The data of densitometric analyses are expressed as the percentage of
fMLP-stimulated control and are the means + SD from at least three experiments. The
numbers identify the corresponding lanes of the immunoblots.
Figure 7: Effect of PGE2 and CAY 10399 on the global profile of tyrosine
phosphorylation and on the translocation of Tec kinases induced by fMLP. A.
Neutrophils (2 x 107/ml) were pre-incubated with 10"5 M PGE2 or 10"5 M CAY 10399 in
the présence of 0.1 U/ml ADA at 37 °C for 10 min. The assay was performed as described
in Material and Methods. The results shown are représentative of at least three independent
experiments. B. Neutrophils (107 cells/ml) were preincubated with the indicated
concentrations of PGE2 or CAY 10399 for 5 min at 37 °C in the présence of 0.1 U/ml ADA
and 10 uM CB. Cell suspensions were stimulated with fMLP for 30 sec and the reactions
were stopped as described in Material and Methods. The samples were analyzed for Btk
and Tec by immunoblotting. The immunoblots shown are représentative of three
independent experiments. C. The data of densitometric analyses are expressed as the
percentage of fMLP-stimulated control and are the means + SD from at least three
experiments. The numbers identify the corresponding lanes of the immunoblots.

82
2.7. DiscussionIn this study, we hâve investigated the mechanisms underlying the inhibition of PLD
activity by PGE2 We demonstrate that PGE2 inhibits several critical events stimulated by
fMLP including the influx of calcium, translocation of PKCa and small GTPases, the
activation of PI3-Ky and tyrosine kinases, and the stimulation of PLD activity as well.
Using various agonists and antagonists of the EP receptors, we demonstrate that EP2 but not
EPi, EP3 or EP4 receptors médiates the inhibitory effect of PGE2 on the fMLP-induced PLD
activation pathway. Taken together, our results are consistent with previous
pharmacological studies attributing a major rôle for EP2 receptors in PGE2-induced
inhibition of neutrophil functional responses (Armstrong, 1995 ; Talpain et al., 1995),
The mechanism underlying the PGE2-induced inhibition of the stimulation of PLD activity
by fMLP has not yet been clarified. The fact that PGE2 inhibited fMLP- but not PMA-
induced PLD activity suggested that PGE2 did not exert its inhibitory effect directly on
PLD but rather at a site more proximal to the fMLP receptors (Agwu et al., 1991). This
hypothesis led us to examine the effect of PGE2 on the signalling steps located upstream of
PLD in the fMLP-stimulated PLD activation pathway. We confirmed that PGE2 did not
influence the mobilization of Ca2+ from intracellular stores but decreased the Ca2+ influx
from the extracellular médium (Ahmed et al., 1995). The inhibitory mechanism of PGE2 on
influx has not yet been elucidated, but several hypothèses can be advanced. An up-
regulating rôle for PI3-K in Ca2+ influx has been reported in chemokines-stimulated cells
(Kansra et al., 2001) but this does not seem to be the case in our model because we and
others (Ahmed et al., 1995) hâve found that wortmannin, a potent PI3-K inhibitor, does not
decrease fMLP-stimulated calcium influx in neutrophils but on the contrary slightly
augments this influx (unpublished results). Therefore, the inhibition of PI3-K by PGE2
cannot explain the reduced calcium influx observed in our expérimental model. Since PGE2
has no impact on the release of Ca2+ from intracellular stores, the capacitative calcium entry
is probably not affected by PGE2 and a relevant hypothesis is that PGE2 rather inhibits the
opening of a receptor-operated calcium channel (Ahmed et al., 1995). Another possibility is
that, as previously shown for adenosine and other cAMP elevating agents, PGE2 could
promote an accelerated clearance of cytosolic calcium by up-regulating the endo-membrane

83
Ca2+-ATPase that recaptures cytosolic calcium (Theron et al., 2002). However, this
hypothesis would account in part for the decreased mobilization of calcium but not for the
inhibition of Mn2+ influx observed in the présence of PGE2. Because the influx of Ca2+ has
been shown to increase PLD activity stimulated by fMLP (Pai et al., 1988), the réduction of
this influx of Ca2+ by PGE2 could possibly downregulates fMLP-induced PLD activity,
although this effect of PGE2 is probably of minor influence on PLD activation since other
factors than calcium directly regulate PLD.
We observed that PGE2 and EP2 agonists significantly decreased the translocation of
PKCa, Arfl and RhoA to fMLP-stimulated neutrophil membranes. Thèse PLD activation
cofactors act synergistically to enhance PLD activity (Exton, 1999) and their simultaneous
inhibition by PGE2 provides a mechanistic explanation for the inhibitory effect of the EP2
signalling pathway on fMLP-induced PLD activity. Thèse results are in accordance with
and further support a previous study from our laboratory indicating that adenosine inhibited
the fMLP-induced PLD pathway via the A2A receptor by diminishing membrane
recruitment of PKCa, Arfl and RhoA (Thibault et al., 2000). The translocation of two other
Rho GTPase family members, Rac2 and Cdc42 that are involved in several functions
inhibited by PGE2 is also reduced by PGE2 via the EP2 receptors.
The concomitant inhibition of Rho and Arf GTPases suggested that PGE2 exerts an
inhibitory effect upstream of both small GTPase families. Although the intermediary
signalling steps linking membrane bound receptors to small GTPase activation are not yet
fully understood, PI3-K and tyrosine kinases seem to play key rôles in membrane
recruitment and activation of small GTPases (Benard et al., 1999).
We report hère that PGE2 significantly reduced the amounts of PtdIns(3,4,5)P3 formed in
response to fMLP via EP2 receptor. Moreover, we observed an almost complète inhibition
of pllOy kinase recruitment and activation by PGE2. Thèse results strongly suggest that
PGE2 inhibited the chemoattractant-induced activation of pi 10y rather than stimulating an
accelerated rate of dephosphorylation of PtdIns(3,4,5)P3. It is therefore unlikely that the
phosphoinositide-specific phosphatases PTEN or SHIP are involved in the inhibitory effect
of PGE2 on PtdIns(3,4,5)P3 accumulation. This inhibitory effect of PGE2 on pllOy adds a

new élément to the mechanism involved in EP2-mediated inhibition of the fMLP-induced
PLD signalling pathway.
The activation of small GTPases is tightly controlled by the opposite actions of guanine
nucleotide exchange factors (GEFs) and guanine nucleotide activating proteins (GAPs) that
stimulate GTP hydrolysis. GEFs for the Rho and Arf family GTPases contain a PH domain
known to bind phosphoinositol lipids, thereby targeting GEF to PtdIns(3,4,5)P3-enriched
membrane régions. Increasing évidence indicates that Rho GTPases are indirectly regulated
by PI3-K via spécifie GEFs (Welch et al., 2003). Of particular relevance to our work is the
observation that the Rho-GEF Vav stimulated by the fMLP receptor médiates the activation
of Rac by PI3-Ky. The relationship between Arf-GEFs and PI3-K in vivo is more
controversial, and différent reports differ according to the GEF and the cellular type
studied. Nevertheless, the PH domain of cytohesin-1, the main Arf-GEF expressed in
human neutrophils, binds to PtdIns(3,4,5)P3 (Corvera and Czech, 1998) and we observed
that Arf translocation to membranes is downregulated by wortmannin (unpublished resuit).
Thèse expérimental lines of évidence linking PI3-K to GEF activation in various cellular
Systems support our hypothesis that the inhibition of pllOy by PGE2 decreases the
translocation of Arf and Rho GTPases.
The inhibition of PKCa translocation by PGE2 may at first seem intriguing because PGE2
neither alters the first peak of cytosolic calcium released from intracellular pools by fMLP-
induced PLC activation nor effectively inhibits DAG formation in fMLP-stimulated
neutrophils (Takenawa et al., 1986). The reduced calcium influx could account partly for
the inhibitory effect of PGE2 on PKCa activation, but PKCa inhibition is more likely
attributable to the decreased activity of PI3-Ky since PKC has been shown to be located
downtream of PI3-K in the fMLP signalling pathway (Vlahos et al., 1995).
PLD and small GTPase activation is also regulated by tyrosine phosphorylation events.
There is no évidence for a direct tyrosine phosphorylation of PLD following fMLP
stimulation in neutrophils (Marcil et al., 1999), but the translocation of PLD cofactors
(PKCa, Arf and RhoA) as well as Rac2 and Cdc42 activation has been shown to occur
downstream of tyrosine phosphorylation events in fMLP-stimulated HL-60 cells, providing

85
a functional link between tyrosine kinases and PLD activation (Benard et al., 1999; Houle
et al, 1999). PGE2 (or CAY10399) reduced the fMLP-stimulated tyrosine phosphorylation
of a set of substrates around 120 kDa by as yet unidentified kinases. This suggests that
tyrosine kinase(s) could be another target for PGE2. As the profile of tyrosine
phosphorylation cannot provide précise information on the putative kinases involved, we
further investigated the effect of PGE2 on Tec kinases that hâve be shown to be activated by
fMLP (Lachance et al., 2002). PGE2 significantly decreased Tec and Btk translocation to
membranes, an effect that can be viewed as a conséquence of PI3-Ky inhibition since the
recruitment and the activation of thèse PH-domain containing kinases are dépendent on
PI3-K. This latter resuit is to be related to a previous study that established a functional link
between Btk (and possibly other Tec kinases) and the activation of Rho, Arf GTPases and
PLD in human neutrophils (Gilbert et al., 2003) and further support the involvement of PI3-
Ky as a crucial target of the EP2 inhibitory pathway.
The mechanism underlying the inhibition of PI3-Ky by PGE2 remains to be further
investigated. PI3-Ky is composed of a pliOy catalytic subunit tightly bound to a plOl
regulatory unit, and upon fMLP-receptor stimulation, this cytosolic complex is activated by
binding to the Gpr units of heterotrimeric G protein. The noncatalytic pi01 subunit is
thought to act as an adaptor that médiates the recruitment of the enzymatic complex to the
membrane where Gpv directly activate the catalytic pi 1 Oy subunit (Brock et al., 2003).
Since PGE2 inhibits the translocation of the pli Oy, we can suggest that PGE2 prevents the
binding of the enzymatic complex to GpY by targeting a site either on the plOl/pllO
complex or on Gpy, and that the decreased amount of plOl/pllOy recruited explains the
suppression of the stimulation of pi lOy activity by the EP2 pathway.
The signalling events linking the EP2 receptor to its fMLP-stimulated targets (possibly PI3-
Ky) also remain to be characterized. The EP2 receptor is coupled to a Gas protein that
increases cAMP levels by stimulating adenylate cyclase (Breyer et al., 2001). Although
several suppressive effects of PGE2 on neutrophil functions hâve been paralleled by an
increase in cAMP (Bloemen et al., 1997; Rivkin et al., 1975; Talpain et al., 1995) others,
e.g. chemotaxis and aggregation triggered by fMLP, hâve not (Armstrong, 1995)

86
suggesting that EP2 could transmit signais through an alternative and still unknown
pathway. Nevertheless, the fact that superoxide production stimulated by fMLP was
inhibited by PGE2 in a cAMP/PKA dépendent manner (Armstrong, 1995) would be in
favor for a rôle of this pathway in the inhibition of PLD activity. A more detailed study of
the involvement of PKA in the inhibition of the fMLP signalling cascades, more
particularly in the inhibition of PI3-K, is presently underway.
The présent study also provides new insights into the signalling mechanisms underlying the
EP2 mediated inhibition of the respiratory burst, chemotaxis and sécrétion elicited by
chemoattractants in human neutrophils. The assembly and activation of the NADPH
complex is tightly regulated at multiple levels and has been shown to require (i) the
phosphorylation of several components by PKC, (ii) the présence of PLD-derived PA
implicated in phosphorylation processes, (iii) the translocation of Rac2 and finally, (iiii) the
activation of PI3-K, mainly pi 10y (Babior, 1999; Hirsch et al., 2000; Vlahos et al., 1995).
The EP2-mediated inhibitory effects on fMLP-induced PI3-K and PLD activities as well as
on the translocation of Rac2 and PKC can account for the strong réduction of the
respiratory burst by PGE2. PI3-Ky and Rho GTPases also play essential rôles in the
polarization of the cell that initiâtes the chemotactic response (Servant et al., 2000), and
their inhibition by the EP2 pathway explains, at least in part, the impairment of chemotaxis
by PGE2. Exocytosis and degranulation processes are regulated by Arf and PLD in
neutrophils (Cockcroft, 2001) and their inhibition by the EP2 pathway might also account
for the inhibition of the fMLP-stimulated release of cytotoxic enzymes by PGE2.
In conclusion, the présent study shows that PGË2 inhibits the chemoattractant-stimulated
PLD pathway by binding to the EP2 receptor in human neutrophils. Moreover, we provide
évidence that this inhibitory pathway decreases pi 10y activation by fMLP, and as a resuit
activation of Tec kinases, small GTPases and PLD. The results of this investigation provide
new insights into the signalling mechanisms implicated in the down-regulation by PGE2 of
the inflammatory response to chemoattractants.

X7
2.8. Acknowledgements
The authors wish to thank Guillaume Paré and Danielle Harbour for their expert help in the
measurement of Mn2 + influx and PLD activity, respectively.
This work was supported in part by grants from the Canadian Institutes of Health Research
.S.G. Bourgoin is the récipient of a Research Scientist Award from the Arthritis Society of
Canada. P.H. Naccache is the récipient of the Canada Research Chair on the Molecular
Physio-pathology of the human neutrophil.
2.9. AbbreviationsAD A, adenosine deaminaseArf, ADP-rybosilation factorDAG, diacylglycerolDFP, di-isopropylfluorophosphateEP receptor, E prostaglandin receptorERK, extracellular signal-regulated kinasesMAPKs, mitogen-activated protein kinasesPA, phosphatidic acidPDE, phosphodiesterasePEt, phosphatidylethanolPH, pleckstrin homologyPKA, cAMP-dependent protein kinasePtdlns,phosphatidylinositolPtdIns(3)P, phosphatidylinositol 3-phosphatePtdIns(3,4,5)P3, phosphatidylinositol 3,4,5- trisphophatePhospholipase D, PLDPhospholipase C, PLC

88
2.10. Bibliography
Agwu DE, McCall CE and McPhail LC (1991) Régulation of phospholipase D-induced
hydrolysis of choline-containing phosphoglycerides by cyclic AMP in human neutrophils. J
Immunol 146:3895-903.
Ahmed MU, Hazeki K, Hazeki O, Katada T and Ui M (1995) Cyclic AMP-increasing
agents interfère with chemoattractant-induced respiratory burst in neutrophils as a resuit of
the inhibition of phosphatidylinositol 3-kinase rather than receptor-operated Ca2+ influx. J
Biol Chem 270:23816-22.
Armstrong RA (1995) Investigation of the inhibitory effects of PGE2 and sélective EP
agonists on chemotaxis of human neutrophils. Br J Pharmacol 116:2903-8.
Babior BM (1999) NADPH oxidase: an update. Blood 93:1464-76.
Benard V, Bohl BP and Bokoch GM (1999) Characterization of rac and cdc42 activation in
chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J
Biol Chem 274:13198-204.
Bloemen PG, van den Tweel MC, Henricks PA, Engels F, Kester MH, van de Loo PG,
Blomjous FJ and Nijkamp FP (1997) Increased cAMP levels in stimulated neutrophils
inhibit their adhésion to human branchial epithelial cells. Am J Physiol 272:L580-7.
Breyer RM, Bagdassarian CK, Myers SA and Breyer MD (2001) Prostanoid receptors:
subtypes and signaling. Annu Rev Pharmacol Toxicol 41:661-90.
Brock C, Schaefer M, Reusch HP, Czupalla C, Michalke M, Spicher K, Schultz G and
Nurnberg B (2003) Rôles of G beta gamma in membrane recruitment and activation of
pi 10 gamma/plOl phosphoinositide 3-kinase gamma. JCell Biol 160:89-99.
Chimini G and Chavrier P (2000) Function of Rho family proteins in actin dynamics during
phagocytosis and engulfment. Nat Cell Biol 2:E191-6.

Cockcroft S (2001) Signalling rôles of mammalian phospholipase Dl and D2. Cell Mol Life
Sci 58:1674-87.
Corvera S and Czech MP (1998) Direct targets of phosphoinositide 3-kinase products in
membrane traffic and signal transduction. Trends Cell Biol 8:442-6.
Exton JH (1999) Régulation of phospholipase D. Biochim Biophys Acta 1439:121-33.
Fantone JC and Kinnes DA (1983) Prostaglandin El and prostaglandin 12 modulation of
superoxide production by human neutrophils. Biochem Biophys Res Commun 113:506-12.
Frohman MA, Sung TC and Morris AJ (1999) Mammalian phospholipase D structure and
régulation. Biochim Biophys Acta 1439:175-86.
Gilbert C, Levasseur S, Desaulniers P, Dusseault AA, Thibault N, Bourgoin SG and
Naccache PH (2003) Chemotactic factor-induced recruitment and activation of Tec family
kinases in human neutrophils. II. Effects of LFM-A13, a spécifie Btk inhibitor. J Immunol
170:5235-43.
Hall A (1998) Rho GTPases and the actin cytoskelelon. Science 279:509-14.
Ham EA, Soderman DD, Zanetti ME, Dougherty HW, McCauley E and Kuehl FA, Jr.
(1983) Inhibition by prostaglandins of leukotriene B4 release from activated neutrophils.
ProcNatlAcadSci USA 80:4349-53.
Hecker G, Ney P and Schror K (1990) Cytotoxic enzyme release and oxygen centered
radical formation in human neutrophils are selectively inhibited by E-type prostaglandins
but not by PGI2. Naunyn Schmiedebergs Arch Pharmacol 341:308-15.
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S,
Mantovani A, Altruda F and Wymann MP (2000) Central rôle for G protein-coupled
phosphoinositide 3-kinase gamma in inflammation. Science 287:1049-53.

90
Houle MG, Kahn RA, Naccache PH and Bourgoin S (1995) ADP-ribosylation factor
translocation correlates with potentiation of GTP gamma S-stimulated phospholipase D
activity in membrane fractions ofHL-60 cells. J Biol Chem 270:22795-800.
Houle MG, Naccache PH and Bourgoin S (1999) Tyrosine kinase-regulated small GTPase
translocation and the activation of phospholipase D in HL60 granulocytes. J Leukoc Biol
66:1021-30.
Kansra V, Groves C, Gutierrez-Ramos JC and Polakiewicz RD (2001) Phosphatidylinositol
3-kinase-dependent extracellular calcium influx is essential for CX(3)CRl-mediated
activation of the mitogen-activated protein kinase cascade. J Biol Chem 276:31831-8.
Lachance G, Levasseur S and Naccache PH (2002) Chemotactic factor-induced recruitment
and activation of Tec family kinases in human neutrophils. Implication of
phosphatidynositol 3-kinases. J Biol Chem 277:21537-41.
Laudanna C, Campbell JJ and Butcher EC (1996) Rôle of Rho in chemoattractant-activated
leukocyte adhésion through integrins. Science 271:981-3.
Lennartz MR (1999) Phospholipases and phagocytosis: the rôle of phospholipid-derived
second messengers in phagocytosis. Int J Biochem Cell Biol 31:415-30.
Liscovitch M, Czarny M, Fiucci G and Tang X (2000) Phospholipase D: molecular and cell
biology of anovel gène family. Biochem J345 Pt 3:401-15.
Marcil J, Harbour D, Houle MG, Naccache PH and Bourgoin S (1999) Monosodium urate-
crystal-stimulated phospholipase D in human neutrophils. Biochem J337 ( Pt 2): 185-92.
Marcil J, Harbour D, Naccache PH and Bourgoin S (1997) Human phospholipase Dl can
be tyrosine-phosphorylated in HL-60 granulocytes. J Biol Chem 272:20660-4.
Naccache PH, Levasseur S, Lachance G, Chakravarti S, Bourgoin SG and McColl SR
(2000) Stimulation of human neutrophils by chemotactic factors is associated with the
activation of phosphatidylinositol 3-kinase gamma. J Biol Chem 275:23636-41.

91
Pai JK, Siegel MI, Egan RW and Billah MM (1988) Phospholipase D catalyzes
phospholipid metabolism in chemotactic peptide-stimulated HL-60 granulocytes. J Biol
Chem 263:12472-7.
Reinhold SL, Prescott SM, Zimmerman GA and Mclntyre TM (1990) Activation of human
neutrophil phospholipase D by three separable mechanisms. Faseb ,74:208-14.
Rivkin I, Rosenblatt J and Becker EL (1975) The rôle of cyclic AMP in the chemotactic
responsiveness and spontaneous motility of rabbit peritoneal neutrophils. The inhibition of
neutrophil movement and the élévation of cyclic AMP levels by catecholamines,
prostaglandins, theophylline and choiera toxin. J lmmunol 115:1126-34.
Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW and Bourne HR (2000) Polarization
of chemoattractant receptor signaling during neutrophil chemotaxis. Science 287:1037-40.
Takenawa T, Ishitoya J and Nagai Y (1986) Inhibitory effect of prostaglandin E2,
forskolin, and dibutyryl cAMP on arachidonic acid release and inositol phospholipid
metabolism in guinea pig neutrophils../ Biol Chem 261:1092-8.
Talpain E, Armstrong RA, Coleman RA and Vardey CJ (1995) Characterization of the PGE
receptor subtype mediating inhibition of superoxide production in human neutrophils. Br J
Pharmacol 114:1459-65.
Theron A, Steel H, Tintinger G and Anderson R (2002) Endogenous adenosine régulâtes
neutrophil pro-inflammatory activities by cyclic AMP-dependent accelerated clearance of
cytosolic calcium. Inflamm Res 51:594 - 602.
Thibault N, Harbour D, Borgeat P, Naccache PH and Bourgoin SG (2000) Adenosine
receptor occupancy suppresses chemoattractant-induced phospholipase D activity by
diminishing membrane recruitment of small GTPases. Blood 95:519-27.
Tilley SL, Coffman TM and Koller BH (2001) Mixed messages: modulation of
inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest
108:15-23.

92
Vlahos CJ, Matter WF, Brown RF, Traynor-Kaplan AE, Heyworth PG, Prossnitz ER, Ye
RD, Marder P, Schelm JA, Rothfuss KJ and et al. (1995) Investigation of neutrophil signal
transduction using a spécifie inhibitor of phosphatidylinositol 3-kinase. J Immunol
154:2413-22.
Welch HC, Coadwell WJ, Stephens LR and Hawkins PT (2003) Phosphoinositide 3-kinase-
dependent activation of Rac. FEBS Lett 546:93-7.

CHAPITRE III
The PGE2-induced inhibition of the PLD activation pathway stimulated by fMLP in
human neutrophils is mediated by PKA at the PI3-Ky level.
L'article présenté dans ce chapitre a été soumis pour publication à Biochemical
Pharmacology.

94
3.1. Résumé
La prostaglandine E2, un eicosanoïde qui module l'inflammation, possède la propriété
d'inhiber dans le neutrophile plusieurs fonctions induites par des facteurs chimiotactiques
telles que le chimiotactisme, la production d'ions superoxide, l'adhésion, la sécrétion
d'enzymes cytotoxiques et la synthèse de leucotriène B4. Nous avons déjà montré que la
PGE2 inhibe la voie de signalisation de la PLD stimulée par le fMLP par l'intermédiaire de
la suppression de l'activité de la PI3-Ky et de la diminution du recrutement aux membranes
des facteurs d'activation de la PLD: la PKCot et les GÏPases Rho et Arf. Cet effet
inhibiteur de PGE2 est transmis via les récepteurs EP2 connus pour stimuler une
augmentation des niveaux intracellulaires d'AMPc dans les cellules. L'inhibition par PGE2
de la plupart des réponses fonctionnelles du neutrophile induites par le fMLP via les
récepteurs EP2 implique l'activation de la PKA, excepté la réponse chimiotactique. Dans
cette étude, nous avons examiné le rôle de la PKA dans l'inhibition, par les récepteurs EP2,
de la voie d'activation de la PLD stimulée par le fMLP. Le H-89, un inhibiteur
pharmacologique spécifique de la PKA, supprime les effets inhibiteurs de la PGE2 à toutes
les étapes de la voie d'activation de la PLD par le fMLP, c'est-à-dire: l'activité de la PLD,
le recrutement de la PKCa et des GTPases Rho et Arf aux membranes, l'influx de calcium,
la phosphorylation de certaines protéines sur des résidus tyrosine, et finalement le
recrutement aux membranes de la sous-unité catalytique pi 10y de la PI3-Ky. Cependant, ni
la PLD ni la PI3-Ky ne sont des substrats pour la PKA. Ces résultats montrent que l'activité
de la PKA stimulée par la PGE2 via les récepteurs EP2 régule la voie de la PLD stimulée par
le fMLP au niveau de la PI3-Ky. L'inhibition par la PKA de l'activation de la PI3-Ky
stimulée par le fMLP demeure un mécanisme complexe qui reste à élucider.

95
3.2. Abstract
Prostaglandin E2 (PGE2), an eicosanoid that modulâtes inflammation, inhibits several
chemoattractant-elicited functions in neutrophils such as chemotaxis, production of
superoxide anions, adhésion, sécrétion of cytotoxic enzymes and synthesis of leukotriene
B4. We previously reported that PGE2 inhibits the fMLP-signaling pathway that leads to
PLD activation through suppression of PI3-Ky activity and the decreased recruitment to
membranes of PLD activation factors, PKC, Rho and Arf-GTPases. This effect is mediated
via the EP2 receptors known to raise cAMP in cells.
The inhibition of most fMLP-induced functional responses by PGE2 via EP2 receptors is
mediated by PKA, except the chemotactic response. We hâve investigated the rôle of PKA
in the EP2-mediated inhibition of the PLD activation pathway. H-89, a spécifie PKA
pharmacological inhibitor suppressed the inhibitory effects of PGE2 at ail stages of the PLD
pathway activated by fMLP, i.e. PLD activity, translocation to membranes of PKCoc, Rho
and Arf-GTPases, calcium influx, tyrosine phosphorylation of proteins and finally
translocation ofpilOy catalytic subunit of PI3-K to membranes. However, neither PLD nor
PI3-Ky are substrates of PKA. Thèse data provide évidence that PGE2-stimulated PKA
activity régulâtes the PLD pathway stimulated by fMLP at the level of PI3-Ky and that the
inhibition of PI3-Ky activation by PKA is a complex mechanism that remains to be
completely elucidated.

96
3.3. Introduction
Prostaglandin E2 (PGE2) is an eicosanoid produced by cyclooxygenases (C0X1/2) that is
usually considered as a potent inflammatory mediator. Uowever, PGE2 has also the
property to down-regulate the activation of leukocytes and plays an immuno-modulatory
rôle at the level of the innate as well as the acquired immune responses [1, 2]. Most
particularly, PGE2 inhibits fMLP-induced functional responses such as superoxide ions
production [3-6], chemotaxis [7], release of cytotoxic enzymes [4, 8], synthesis of
leukotriene B4 (LTB4) [9] and adhésion to epithelial cells [10] in polymorphonuclear
neutrophils (PMNs), important cell effectors of the innate immune response. Thèse
inhibitory effects on PMNs are ail mediated via the E prostaglandin 2 (EP2) receptors that
trigger cAMP élévation in cells [7, 11]. In a previous study on the inhibitory signaling
mechanism of PGE2, we reported that the main target of PGE2 in the pathway that leads to
activation of phospholipase D (PLD) by fMLP was phosphoinositol 3- kinase y (PI3-Ky),
an important upstream regulator of PLD activity [12]. In this study, PGE2 was shown to
abolish the translocation of PI3-Ky to membranes that allows the stimulation of the pi lOy
subunit enzymatic activity by binding to G(3y subunits upon fMLP stimulation. The
inhibition of the translocation of PI3-Ky to G(3y resulted in a decreased formation of
PtdIns(3,4,5)P3 and hence, in a reduced recruitment to membranes of the factors that
activate PLD, i.e. Rho and Arf GTPases as well as PKCa.
Most of the EP2 receptors-mediated inhibitory effects of PGE2 on PMNs fonctions
activated by fMLP are paralleled by an increase in cAMP and hence hâve been attributed to
an élévation of this second messenger. PKA, the main effector of cAMP in cells, has been
shown to médiate the inhibition of fMLP-induced superoxide anion production by PGE2 via
EP2 receptors [7]. However, the inhibition of chemotaxis by PGE2 dépends neither on a
cAMP increase nor on PKA activation [7]. This latter observation suggests that EP2
receptors may activate signaling pathways other than the cAMP/PKA pathway. Therefore,
we hâve investigated the involvement of PKA in the EP2 receptor-mediated inhibition of
the différent components of the PLD pathway induced by fMLP, i.e. PLD, Rho and Arf-

97
GTPases, PKCa and PI3-Ky, as well as calcium mobilization and tyrosine phosphorylation
e vents.
3.4. Materials and methodsAntibodies: The anti-PKCcc (PI6520) and anti-Cdc42 (C70820) monoclonal Abs were
purchased from BD Transduction Laboratories (Mississauga, ON, Canada). Anti-RhoA
(SC-179), anti-Rac2 (SC-96) polyclonal Abs were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA). The polyclonal anti-Akt (#9272) and anti-phospho-PKA
substrate (#9621) Abs were obtained from Cell Signaling Technology (Beverly, MA). The
polyclonal anti-Arfl and anti-pllOy antisera, the polyclonal anti-CD32 (FcyRIIa) Ab were
raised in rabbits as described previously [13-15]. The monoclonal anti-phosphotyrosine Ab
(UBI 05-321, clone 4G10) was purchased from Upstate Biotechnology Inc. (Lake Placid,
NY). The rabbit polyclonal anti-Lyn Ab was obtained from Santa Cruz. The polyclonal
anti-CD32 Ab was raised in rabbits as described previously [15]. Secondary anti-mouse
(#NXA931) and anti-rabbit (#NA934V) Abs were obtained from Amersham Biosciences
(Baie d'Urfé, Québec, Canada).
Reagents: Dextran T-500 was purchased from Pharmacia Biotech (Dorval, Québec,
Canada) and Ficoll-Paque from Wisent (St-Bruno, Québec, Canada). Adenosine deaminase
(ADA) was purchased from Roche Diagnostics (Laval, Québec, Canada), and di-
isopropylfluorophosphate (DFP) from Serva (Heidelberg, Germany). fMet-Leu-Phe (fMLP)
and cytochalasin B (CB) were obtained from Sigma-Aldrich Canada (Oakville, Ontario,
Canada). PGE2 and CAY10399 were purchased from Cayman Chemical (Ann Arbor, MI).
H-89 and recombinant mouse PKA catalytic subunit were obtained from Calbiochem. Fura-
2/AM was obtained from Molecular Probes (Eugène, OR). Recombinant tagged PI3-KyHls"GST (pll0yHis/pl01GST) and pi 10yHis catalytic subunit were purchased from Jena Bioscience
(Jena, Germany). [y-32P] ATP (3000 Ci/mmol) (NEG 502A) was obtained from Perkin
Elmer Lifesciences (Woodridge, ON, Canada)

Isolation of hunian neutrophils
Venous blood was collected from healthy adult volunteers in isocitrate anticoagulant
solution. Neutrophils were separated as described previously [16]. Briefly, whole blood
was centrifuged at 180g for 10 min and the resulting platelet rich plasma was discarded.
Leukocytes were obtained following erythrocytes sédimentation in 2% Dextran T-500.
Mononuclear cells were removed by centrifugation on Ficoll-Paque cushions and
contaminating erythrocytes in the neutrophil pellets were removed by a 20 sec hypotonie
lysis in water. Neutrophils were resuspended in Hanks Balanced Sait Solution (HBSS), pH
7.4, containing 1.6 mM Ca 2+ but no Mg2+.
PLD measurements
Neutrophils were labeled with l-O-[3H]alkyl-2-lyso-phosphatidylcholine (2 \iCif\O1 cells)
for 90 min as described previously [16]. The cells were washed and resuspended at 107
cells/ml in HBSS. Cell suspensions (0.5 ml) were warmed at 37°C for 5 min and then pre-
treated 5 min with 10 uM CB and 0.1 U/ml AD A to eliminate endogeneous adenosine and
in the présence or the absence of the indicated concentrations of PGE2 or EP2 receptor
agonist (CAY10399). Neutrophils were stimulated with 100 nM fMLP for 10 min in the
présence of 1% ethanol. Incubations were stopped by adding 1.8 ml of
chloroform/methanol/HCl (50:100:l,v/v/v) and unlabeled phosphatidylethanol (PEt) as a
standard. Lipids were extracted and the levels of [3H]PEt were quantified as described
previously [16].
Measurement of cytoplasmic free calcium concentrations
Neutrophils (107 cells/ml) were incubated at 37°C for 30 min with 1 |_iM Fura-2/AM. The
cells were washed and resuspended at 5x106 cells/ml in HBSS with 1.6 mM Ca2+.
Neutrophils were transferred to the thermostated cuvette compartment of a
spectrofluorimeter (SLM 8000C) and preincubated 5 min at 37 °C with 0.1 U/ml ADA in
the présence of various EP receptor agonists/antagonists or the equal volume of Me2SO.
Cells were stimulated with 100 nM fMLP at the time indicated by the arrow. The

99
fluorescence of the cells was monitored at an excitation wavelength of 340 nm and an
émission wavelength of 510 nm. The internai calcium concentrations were calculated as
described by Grynkievicz et al. [17].
Translocation assays
Neutrophils (4xlO7 cells/ml) were treated with 1 mM DFP for 10 min at room température,
The cell suspensions were centrifuged once and the cells were resuspended in HBSS at 107
cells/ml. The cells were warmed for 5 min at 37°C and treated for an additional 5 min with
10 uM CB, 0.1 U/ml ADA and PGE2 or EP2 receptor agonist (CAY10399) or an equal
volume of diluent (Me2SO) as a control. Neutrophils were stimulated with 100 nM fMLP at
37° C. Incubations were stopped by diluting the cells five fold with ice cold HBSS and total
membrane proteins were collected as described previously [16]. Protein samples (10-20 ug)
were resolved on a 7.5-20% gradient SDS-PAGE and transferred to Immobilon PVDF
membranes (Millipore Corporation, Bedford, MA, USA). Immunoblots were performed
using anti-Arfl (1/2500), anti-RhoA (1/1000), anti-PKCoc (1/1000), anti-Rac2 (1/200), anti-
Cdc42 (1/250), anti-pl lOy (1/1000) Abs. The PVDF membranes were reprobed with an Ab
against CD-32, a marker of plasma membrane, to assure equal loading of proteins in ail
samples. Proteins were revealed with HRP-conjugated secondary anti-mouse or anti-rabbit
Ab (1/20,000), anti-mouse Ab (1/20,000) and the Renaissance détection System (NEN,
Perkin Elmer Life Sciences).
Tyrosine and PKA phosphorylation patterns
Neutrophils (2 x 107 cells/ml) were pre-incubated at room température with 1 mM DFP,
with or without the indicated concentrations of PGE2 or EP2 receptor agonists for 10 min at
37°C prior to stimulation with 100 nM fMLP for the indicated times. The reactions were
stopped by transferring 100 ju.1 of the cell suspensions to an equal volume of boiling 2X
Laemmli sample buffer (SB) (lx is 62.5 mM Tris-HCl, pH 6.8, 4% SDS, 5% p-
mercaptoethanol, 8.5% glycerol, 2.5 mM orthovanadate, 10 mM paranitro-
phenylphosphate, 10 u,g/ml leupeptin, 10 u-g/ml aprotinin, 0.025% bromophenol blue) and

100
boiled for 7 min. The samples were then subjected to 7.5-20% gradient SDS-PAGE and
transferred to Immobilon PVDF membranes. Immunoblotting was performed using the
monoclonal anti-phosphotyrosine Ab 4G10 (1/4000) or the polyclonal anti-phospho PKA
substrate Ab and revealed with HRP-conjugated secondary anti-mouse or anti-rabbit Ab
(1/20,000) and the Renaissance détection System (NEN, Perkin Elmer Life Sciences).
Phosphorylation of recombinant PI3-Ky
Recombinant PI3-Ky (700 ng p 11 OyHis/p 1010ST or 350 ng pllOy"'s) was incubated in
phosphorylation buffer (20 ul final volume; Tris HC1 pH 7.5, MgCb 5 mM, DTT 1 mM)
with PKA catalytic subunit (50 U), lmM ATP and 2faCi [y-32P]ATP at 30°C for 30 min.
When indicated, PI3-Ky was preincubated with 100 nM wortmannin and PKA was
preincubated with 1 \xM H-89 for 10 min prior to the phosphorylation assay. In each
experiment, histones were used as a substrate to monitor the PKA phosphorylation reaction.
The reactions were stopped by addition of 10 (4.1 SB 3X and 5 min boiling. The samples
were resolved by 12% SDS-PAGE and the dried gels analyzed by autoradiography.

101
3.5. Results
3.5.1. Effects of fMLP and PGE2 on PKA activation.Chemoattractants such as fMLP are known to elicit a rapid rise in intracellular cAMP that
peaks at 15-20 sec followed by a decrease to basai level by 2-5 min in PMNs [18-20], EP2
has been identified as the PGE2 inhibitory receptor on PMNs [7, 12, 21] and has been
shown to stimulate cAMP formation in cells [22]. In PMNs, PGE2 (or PGEi) is able to
stimulate a modest increase in cAMP levels in the présence of phosphodiesterase inhibitors
(IBMX, theophylline) only, but promotes a large and sustained enhancement of
intracellular cAMP levels when PMNs are subsequently stimulated by fMLP [19, 23, 24].
PKA is the main effector of cAMP in cells and its activation is regulated by cAMP binding
on its regulatory subunits. We hâve thus examined the effects of the cAMP élévation
triggerred by fMLP and PGE2 on PKA activity in PMNs. To this purpose, we hâve
analysed whole cell lysates of fMLP-stimulated PMNs with a phospho-PKA substrate Ab
which specifically recognizes the phophorylated PKA consensus motif on PKA substrates.
For ail the experiments presented in this study, PMNs were pre-incubated in the présence of
adenosine deaminase (ADA) in order to dégrade the endogenous adenosine to inosine. We
hâve previously shown that adenosine rapidly accumulâtes in PMNs suspensions and
activâtes the cAMP/PKA pathway via the A2A inhibitory receptors [25, 26].
The data shown in Figure 1 indicate that fMLP stimulated the phosphorylation of several
proteins that are recognized by the anti-phospho-PKA substrate Ab (MW at approximately
47-50, 60-65, 95-97, 120-130 and 140 kDa). Phosphorylation peaked at 1min and gradually
decreased thereafter. In the présence of PGE2, the levels of phosphorylation of PKA
substrates stimulated by fMLP were globally increased, and the increased intensity of
phosphorylation observed at 0.5 and 1 min indicated an earlier onset of the response when
PMNs were preincubated with PGE2 prior to fMLP stimulation. It is worthwhile to notice
that preincubation of cells with PGE2 alone increased the intensity of phosphorylation by
PKA of some substrates at 50, 80-97 and around 120 kDa (fMLP vs fMLP+PGE2, time 0).
PMNs were also preincubated with H-89, a spécifie PKA inhibitor in order to ascertain the
rôle of PKA in the phosphorylation pattern previously obtained. In the présence of H-89,
the intensity of phosphorylation of the prédominant PKA substrates in response to fMLP

102
was attenuated (mainly substrates at 50, 60-65, 97 and 120 kDa) and the stimulating effect
of PGE2 on the PKA phosphorylation profile stimulated by fMLP was attenuated as well.
Taken altogether, thèse data show that the PKA activity stimulated by fMLP is increased in
the présence of PGE2 but, although this eicosanoid has been shown to promote a long-
lasting rise of cAMP in fMLP-stimulated PMNs, it does not seem to prolong the
phosphorylation of proteins by PKA in thèse cells.
3.5.2. Rôle of PKA in the PGE2-mediated inhibitory effect on fMLP-induced PLD activityWe previously reported that PGE2 inhibits PLD activity stimulated by fMLP via EP2
receptors [12]. Since PGE2 also activâtes PKA in fMLP-stimulated cells, we wondered
whether PKA mediated the inhibitory effect of PGE2 on PLD activity. To test this
hypothesis, PMNs were preincubated in the présence of H-89 prior to addition of PGE2 to
cell suspensions and stimulation by fMLP. The data presented in Figure 2 show that H-89
had no effect on PLD activity stimulated by fMLP but completely suppressed the inhibitory
effect of PGE2 on PLD activity. Thèse data clearly indicate that the PGE2 inhibitory effect
on PLD activity is mediated by PKA in fMLP-stimulated PMNs. Since Jang et al hâve
reported that PKA could phosphorylate PLDl[27j, the major PLD isoform expressed in
PMNs, we verified the hypothesis of a direct phosphorylation of PLD1 in PMNs
preincubated with PGE2. PLD1 was immunoprecipitated from PMNs treated with PGE2
and stimulated with fMLP. The PLD1 immunoprecipitates were analyzed by
immunoblotting with the anti-phospho-PKA substrate Ab. Under thèse expérimental
conditions, we were not able to detect a phosphorylation of PLD1 in PMNs (data not
shown).
3.5.3. Rôle of PKA in the PGE2-mediated inhibitory effect on Ca2+ influxinduced by fMLP
The rapid and transient rise in intracellular Ca2+ triggered by fMLP is an important
regulator of PLD activity [28, 29]. We hâve already reported that PGE2 diminishes the late
Ca2+ influx induced by fMLP via EP2 receptors in PMNs [12]. We thus wondered whether

103
PKA was implicated in the inhibition of Ca2+ influx by PGE2. As shown in Figure 3, the
addition of PGE2 reduced the late mobilization of cytoplasmic Ca2+ due to extracellular
Ca2+ influx stimulated by fMLP. This effect of PGE2 was abolished in the présence of H-
89. The EP2 sélective agonist CAY10399 had a similar effect as PGE2 on Ca2+ influx
induced by fMLP and was similarly sensitive to H-89. Thèse results are consistent with an
inhibitory effect of PGE2 and CAY10399 through the cAMP-mediated activation of PKA
on the fMLP-induced Ca2+ influx.
3.5.4. Rôle of PKA in the inhibition of PKCa and small GTPasestranslocation by PGE2.The activation of PLD is regulated by three cytosolic factors that need to translocate to
membranes to be activated: classical PKC isoforms, and the small GTPases RhoA and Arf
[30]. We previously reported that PGE2, via EP2 receptors, decreased the translocation to
membranes of thèse PLD activation cofactors stimulated by fMLP, as well as the
translocation of two other Rho GTPases members: Rac2, which is a cytosolic component of
the NADPH oxidase complex and Cdc42, an important regulator of actin polymérisation
[12]. To further investigate the rôle of PKA in the PGE2 inhibitory mechanism mediated via
EP2 receptors, we tested the effect of H-89 on the fMLP-induced translocation of PKCa
and Arf and Rho GTPases in the présence of PGE2 or of the EP2 receptor agonist
CAY 10399. The results illustrated in Figure 4 show that PGE2 and CAY 10399 decreased
the amounts of PKCa, Rho GTPases (RhoA, Rac2 and Cdc42) and Arfl that translocate to
membranes in response to fMLP. In PMNs pre-treated with H-89 and PGE2 or CAY10399
prior to stimulation with fMLP, the amounts of thèse factors associated to membranes
(lanes 9-10) are similar to those obtained from control fMLP-stimulated PMNs incubated in
the présence (lane 8) or in the absence of H-89 (lanes 5). Thèse data show that H-89
almost completely (PKC, RhoA, Rac2) or completely (Arf, Cdc42) suppressed the
inhibition of the translocation of thèse factors mediated via EP2 receptors, and therefore
indicate that PKA activated by the EP2 pathway decreased, at least in part, the fMLP-
induced recruitment of PKC and small GTPases to membranes.

104
3.5.5. Rôle of PKA in the PGE2-mediated inhibitory effect on tyrosinephosphorylation.
We hâve previously reported that PGE2 decreases the fMLP-induced tyrosine
phosphorylation of a subset of unidentified proteins at 116-120 kDa [12]. To assess
whether PKA is involved in this inhibitory effect of PGE2, PMNs were pre-incubated with
H-89 prior to addition of PGE2 to cell suspensions and then stimulated with fMLP. The
levels of tyrosine phosphorylation were evaluated by analyses of whole cell lysates with an
anti-phospho-tyrosine Ab. As shown in Figure 5, PGE2 decreased the tyrosine
phosphorylation of the proteins in the 116-120 kDa région stimulated by fMLP.
Furthermore, H-89 slightly increased the fMLP-stimulated tyrosine phosphorylation of
thèse proteins. The decreased tyrosine phosphorylation of the 116-120 kDa set of substrates
caused by PGE2 was no longer observed in the présence of H-89. Thèse data indicate that
1) PKA modulâtes the tyrosine phosphorylation reactions triggered by fMLP and that 2) the
inhibition of tyrosine phosphorylations of the 116-120-kDa substrates by PGE2 is mediated
byPKA.
3.5.6. Rôle of PKA in the inhibition of pi 10y translocation by PGE2.
PI3-Ky is the major PI3-K isoform activated in response to fMLP in PMNs [14] and is
responsible for the rapid and massive accumulation of PtdIns(3,4,5)P3 at the leading edge
of the cell exposed to a gradient of chemotactic agent [14, 31]. This accumulation allows
the relocation of proteins containing PH domains, such as the exchange factors for small
GTPases (GEFs), to PtdIns(3,4,5)P3-enriched membrane régions. PI3-Ky is a heterodimer
composed of a pllOy catalytic subunit tightly bound to a plOl regulatory unit, and upon
fMLP-receptor stimulation, this cytosolic complex is activated by binding to the Gpy units
of heterotrimeric G protein. We hâve previously reported that PGE2 inhibits the
translocation of pi lOy to membranes where its catalytic activity is stimulated by binding to
the GpY units after stimulation by fMLP. This inhibition was mediated via EP2 receptors and
consequently inhibited PI3-Ky activity and PtdIns(3,4,5)P3 formation [12]. To assess the
rôle of PKA in the inhibitory effect of PGE2 on PI3-Ky translocation, we hâve evaluated the
amounts of membrane-associated pi 10y in fMLP-stimulated PMNs in the présence of H-89

105
and PGE2. The data presented in Figure 6 show that a pre-incubation of cells with H-89
alone did not significantly modify the translocation of the pllOy catalytic subunit to
membranes in response to fMLP (lane 8, vs lane 5) but, when added prior to PGE2 or
CAY10399, H-89 did notably block the inhibitory effect of PGE2 or CAY10399 on pllOy
translocation (lanes 9-10 vs lane 6-7). Together, thèse data show that H-89 blocks, at least
in part, the EP2-mediated inhibition of the translocation of PI3-Ky induced by fMLP and
indicate that PKA is involved in this PGE2 inhibitory effect.
3.5.7. Phosphorylation of PI3-Ky by PKA.
Since PKA negatively régulâtes the translocation of pi lOy induced by fMLP, we wondered
whether this inhibitory effect could be mediated through a direct phosphorylation of the
PI3-Ky heterodimer (pliOy/p 101) by PKA. First, we analyzed the protein séquences of
plOl and pi 10y by using the Scansite program (http://scansite.mit.edu) in order to identify
putative motifs of phosphorylation by PKA (RxxT or RRxS) in thèse proteins. This
theoretical analysis revealed and that human pi01 contains two such potential sites: Thr-31
(RRST) and Ser-440 (RRDS) as well as two 14-3-3 protein binding groups that contain the
consensus motif of phosphorylation by PKA: Thr-114( RFLTWP) and Thr-577(RSQTPP)
and that human pllOy contains one potential site of phosphorylation by PKA: Ser-257
(KKKS).
To assess whether pi 1 Oy/p 101 could be a substrate of PKA, we performed an in vitro PKA
kinase assay using recombinant heterodimeric pi 10y"'s/pl01GS1 or recombinant pi 10yHls as
potential substrates. Phosphorylation of recombinant pi 10yH'7pl01Gsr and pllOyHls was
monitored using 50 U PKA catalytic subunit and [y-32P]ATP and was analyzed by
autoradiography. The results of this assay, presented in Figure 7A, show that heterodimeric
pll0yH]S/pl01G' T and pll0yHlswere phosphorylated whether or not they were incubated
with PKA. The level of phosphorylation of pllOyHls was much higher than that of the
heterodimer pl i OyHis/p 101GST. To further investigate the rôle of PKA in the in vitro
phosphorylation of PI3-Ky, we preincubated the PKA catalytic subunit with H-89 prior to
the kinase assay. H-89 did not modify either the phosphorylation status of pi 10yHls/pl01GST

106
or that of pi 10yHls. Taken altogether, thèse data indicate that the observed phosphorylation
of pi 10y1Ils/pl01GSI and of pi 10yH's is not the resuit of PKA enzymatic activity but is rather
attributable to the autophosphorylation of the catalytic subunit pi 10y that has been already
reported [32, 33]. To verify the involvement of an autophosphorylation event on pllOy,
recombinant pllOyHls was incubated with wortmannin, a potent PI3-K inhibitor that
covalently links the catalytic domain of pi 10 and then assayed for phosphorylation as
described. As shown in Figure 7B, pllOyHls was phosphorylated whether or not it was
incubated with PKA and wortmannin decreased the level of phosphorylation of pllOyHls.
This resuit indicates that, under our expérimental conditions, the observed pllOy
phosphorylation is indeed due to autophosphorylation.

3. 6. Figures
107
MWkDa
116-
9 7 -
6 6 -
5 5 -
4 3 -
3 5 -
2 6 -
Mit tlt*3= zr
substrate
PKA
Time (min) 0 0.5 1 5 10n 0 0.5 1 5 10M 0 0.5 1 5 10,, 0 0.5 1 5 10,
fMLP fMLP + PGE2 fMLP + H-89 fMLP + H-89 + PGE2
Figure 1: Activation of PKA by fMLP and PGE2

108
140-1
DMSO H-89 PGE2 fMLP fMLP fMLP fMLP+ H-89 + PGE2 + H-B9
+ PGIE2
Figure 2: Effect of H-89 on the PGE2-induced inhibition of PLD activity stimulated byfMLP.
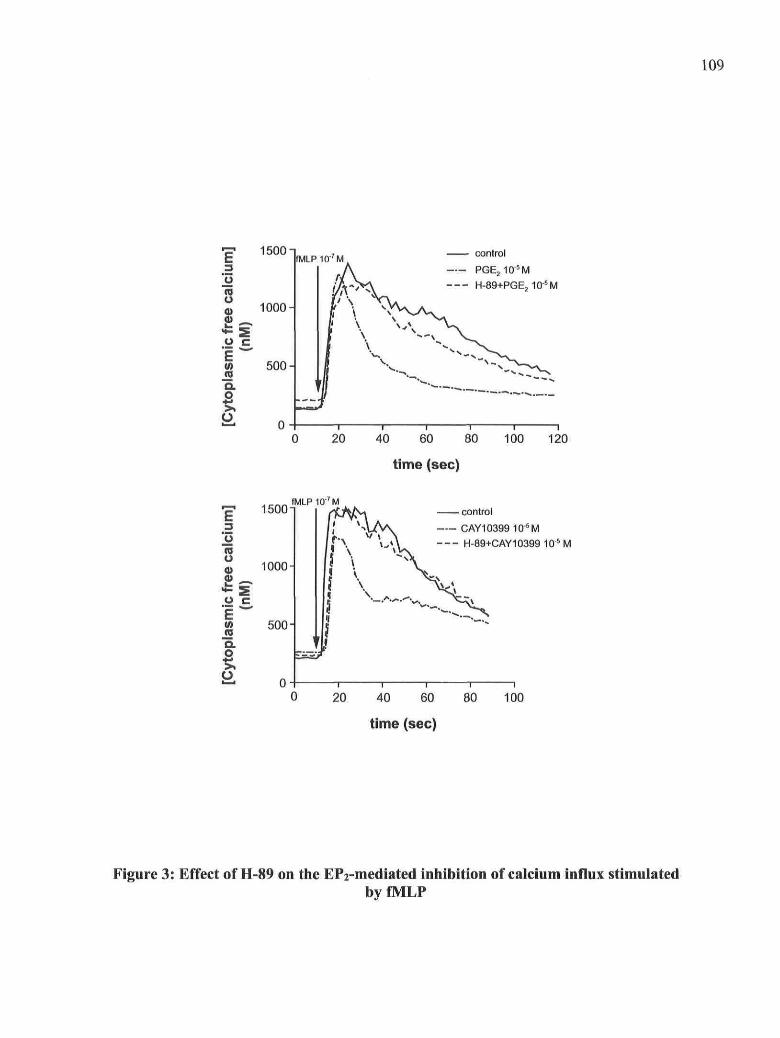
109
o
S
O C
i w
V)
IOUU"
1000-
500-
0 -
fMLP10-7M.
1
! \l v1
control
PGE210-5M
H-89+PGE2105M
r20 40 60 80 100 120
time (sec)
15001fMLPIO7 M
nio
Q)
U
. -(AS.o>.^ 0
1000-
500-
control
CAY10399 10'5M
H-89+CAY10399 1 0 5 M
0 20 40 60 80 100
time (sec)
Figure 3: Effect of H-89 on the EP2-mediated inhibition of calcium influx stimulatedbyfMLP

110
PKCa-»-
RhoA-»
Art-»-
Rac2-»
Cdc42-*
CD-32-+
1 2 3 4 5 6 7 8 9 10fMLP(10"7M)PGE2(10-6M)
CAY10399(10-6M)H-89(10"5M)
B
1 2 3 4 5 8 7 8 9 10
Figure 4: Effect of H-89 on the EP2-mediated inhibition of PKCa, Arf and Rho-GTPases translocation stimulated by fMLP

111
MWkDa
1 5 8 -
116 —97 —
55 —
44 —
3 6 -
2 7 -
55 —
Time (min)
TPO
Lyn
0 0.5 1 5 ,, 0 0.5 1 5 , ,0 0.5 1 0 0.5 1fMLP fMLP + PGE2 fMLP + H-89 fMLP + H-89 + PGE2
Figure 5: Effect of H-89 on thePGE2-induced inhibition of tyrosine phosphorylation ofproteins stimulated by fMLP

112
P 1 1 0 Y -
CD-32-
10fMLP(10"7M)PGE2(10-6M)
CAY10399(10-6M)H-89(10"5M)
1 2 3 4 5 6 7 8 9 10
Figure 6: Effect of H-89 on the EP2-mediated inhibition of pllOy translocationstimulated with fMLP
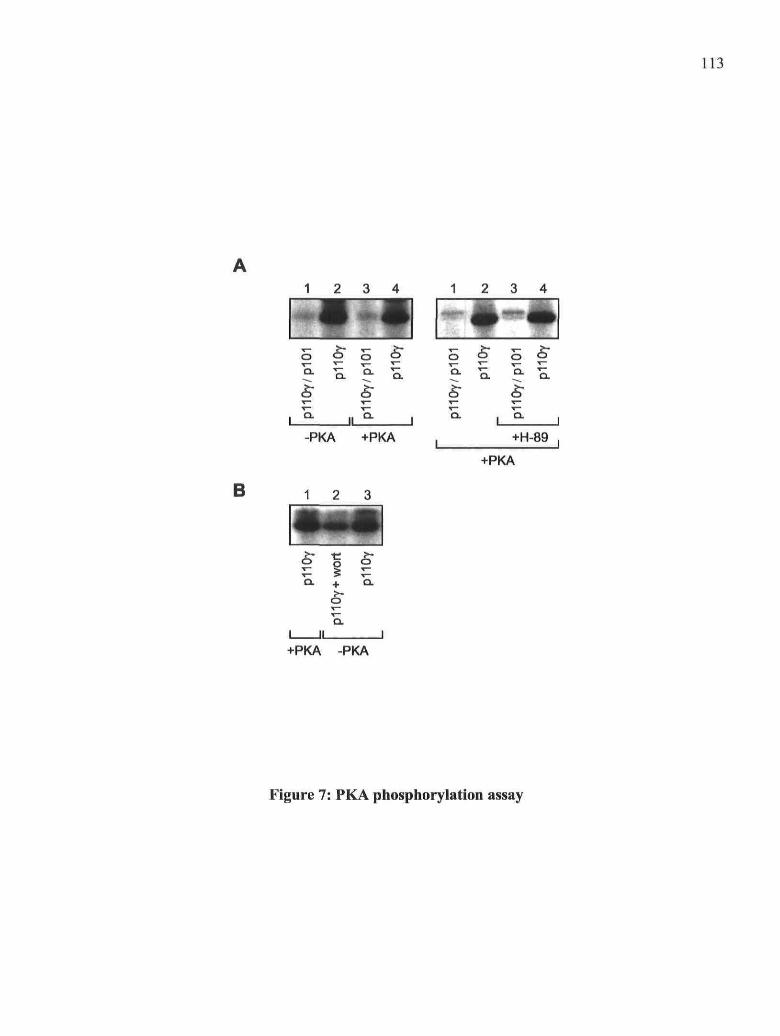
113
1 2 3 4 1 2 3 4
B
I • •!o o S o
a- II û- I
-PKA +PKA
1 2 3
IIIo ^ o
Q. + Q.
X™
II I+PKA -PKA
5 5 o o"
Q. , Q.
+H-89 ,
+PKA
Figure 7: PKA phosphorylation assay

114
FIGURE LEGENDS
Figure 1: Activation of PKA by fMLP and PGE2, PMNs were preincubated with 10'5 M
PGE2 at 37 °C for 10 min in the présence of 0.1U /ml ADA and stimulated with 100 nM
fMLP for the times indicated. Where indicated, PMNs were treated for 10 min with 10"5 M
H-89 prior to incubation with PGE2. Whole cell lysates were analyzed by immunoblotting
with the anti-phospho PKA substrate Ab as described in Material and Methods. As a
control for equal loading, the PVDF membranes were reprobed with an anti-PKA mAb to
monitor amounts of the kinase in each sample. The immunoblot shown is représentative of
at least three independent experiments.
Figure 2: Effect of H-89 on the PGE2-induced inhibition of PLD activity stimulated by
fMLP. Neutrophils (107 cells/ml) were pre-incubated with 10"5 M H-89 for 5 min. in the
présence of 0.1 U/ml ADA and with 10"6 M PGE2 for 5 additional min at 37 °C and 10 uM
CB. Cells were stimulated with 100 nM fMLP in the présence of 1% ethanol for 10 min.
The amounts of [3H]PEt formed were evaluated as described in Material and Methods. The
levels of [3H]PEt formed are expressed as the percentage of the fMLP-stimulated control.
The data are from three différent experiments performed in duplicates and are expressed as
the means + SEM.
Figure 3: Effect of H-89 on the EP2-mediated inhibition of calcium influx stimulated
by fMLP. Neutrophils (5 x 106 cells/ml) were preincubated for 5 min at 37 °C with 10"5 M
H-89 in the présence of 0.1 U/ml ADA and for 5 additional min with PGE2 or CAY10399.
Cells were stimulated with 100 nM fMLP at the time indicated by the arrow and cytosolic
calcium concentrations were monitored as described in Material and Methods. The data
shown are représentative of three independent experiments.
Figure 4: Effect of H-89 on the EP2-mediated inhibition of PKCa, Arf and Rho-
GTPases translocation stimulated by fMLP. A. Neutrophils (107cells/ml) were
preincubated at 37 °C with 10"5 M H-89 for 5 min in the présence of 0.1 U/ml ADA and for
5 additional min with 10"6 M PGE2 or CAY10399 and 10 |aM CB. Cell suspensions were
stimulated with 100 nM fMLP for 2 min. The incubations were stopped and the membrane
fractions were prepared as described in Material and Methods. The samples were analyzed

115
for PKCot, Arfl, RhoA, Rac2 and Cdc42 by immunoblotting. Each membrane was
reprobed with an anti-CD32 Ab to assure equal protein loading in ail samples. The
immunoblots shown are représentative of at least three independent experiments. B.
Densitometric analyses are expressed as the percentage of fMLP-stimulated control and are
the means + SD from at least three experiments. The numbers identify the corresponding
lanes of the immunoblots shown in panel A.
Figure 5: Effect of H-89 on thePGE2-induced inhibition of tyrosine phosphorylation of
proteins stimulated by fMLP. Neutrophils (2 x 107/ml) were pre-incubated with 10"5 M H-
89 for 10 min in the présence of 0.1 U/ml ADA and with 10"5 M PGE2 at 37 °C for 10
additional min. The assay was performed as described in Material and Methods. The results
shown are représentative of at least three independent experiments. Whole cell lysates were
analyzed by immunoblotting with an anti-phospho tyrosine mAb as described in Material
and Methods. As a control to assess equal loading, the PVDF membranes were reprobed
with an anti-Lyn Ab.
Figure 6: Effect of H-89 on the EPî-mediated inhibition of pllOy translocation
stimulated with fMLP. Neutrophils (107 cells/ml) were preincubated for 10 min with 10"5
M H-89 in the présence of 0.1 U/ml ADA and for additional 10 min with 10"5 M PGE2 or
CAY 10399 at 37 °C, 10 uM CB were added 5 min prior to stimulation with fMLP for 30
sec. The reactions were stopped and the membrane fractions were prepared as described in
Material and Methods. The samples were analyzed for pllOy by immunoblotting. Each
membrane was reprobed with an anti-CD-32 Ab to assure equal protein loading in ail
samples. The immunoblot shown is représentative of three independent experiments.
Densitometric analyses are expressed as the percentage of fMLP-stimulated control and are
the means + SD from at least three experiments. The numbers identify the corresponding
lanes of the immunoblots.
Figure 7: PKA phosphorylation assay. Recombinant pi 10yH'7pl01GSr (700 ng) and
pi 10yHls (350 ng) were incubated or not with 50 U PKA catalytic subunit and a mix of 1
mM cold ATP and 2 juCi [y-32PlATP at 30 °C for 30 min. When indicated, PKA catalytic
subunit was preincubated with 1 uM H-89. The phosphorylation of proteins was analyzed

116
by autoradiography. B. Recombinant pi 10yHls was preincubated with 100 nM wortmannin
10 min prior to the onset of the phosphorylation reaction.

117
3.7. DiscussionIn a previous study, we showed that PGE2, via EP2 receptors, inhibits the PLD pathway
induced by fMLP at the level of PI3-Ky, an important upstream regulator of PLD activity.
But the inhibitory signaling mechanism of the EP2 pathway itself has not been
characterized yet. EP2 receptors are linked to a Gas protein known to stimulate adenylyl
cyclase activity and hence the formation of cAMP in cells [22, 34]. The main intracellular
effector of cAMP is PKA, a well known serine/threonine kinase activated by the binding of
cAMP to its regulatory subunit that results in the release of two active catalytic subunits.
However, the activation of EP2 receptors may also signal via pathways independent of
cAMP and of PKA. For example, the activation of EP2 receptors stimulâtes the PI3~K-Akt
pathway in dendritic cells [35-37] and inhibits fMLP-stimulated chemotaxis in PMNs in a
cAMP/PKA independent manner. Thèse observations suggested that EP2 receptors may
stimulate multiple signaling mechanisms. Therefore, in the présent study, we hâve
investigated the rôle of PKA in the EP2-mediated inhibition of the différent components of
the signaling cascade that leads to PLD activation in PMNs stimulated by fMLP.
We assessed first the effect of PGE2 on the PKA phosphorylation pattern triggered by
fMLP in PMNs. The data reported hère show that PGE2 increases the intensity of
phosphorylation of PKA substrates, suggesting that PKA activity stimulated by fMLP is
enhanced in the présence of PGE2. We also noticed that PGE2 alone was able to trigger a
weak phosphorylation of PKA substrates. This latter observation is consistent with a study
reported by Martin et al. that shows the capacity of PGE2 to stimulate PKA activity in
humanPMNs [38].
The inhibitory effect of PGE2 on fMLP-stimulated PLD activity was blocked by the PKA
spécifie inhibitor H-89, indicating that the inhibition of PLD mediated via EP2 receptors
involves PKA activation. We verified that endogenous PLD1 in PMNs was not the direct
target of PKA as reported by others in cells overexpressing this PLD isoform [27]. This
resuit indicates that PKA régulation of PLD activity opérâtes at an upper level than PLD
itself. We thus wondered if PKA inhibited the mobilization of the PLD activation factors
induced by fMLP, i.e. Rho- and Arf-GTPases and PKCot. The fact that H-89 blocked the
EP2-mediated inhibitory effect of PGE2 on the fMLP-induced translocation of small

118
GTPases and PKCoc clearly shows a rôle for PKA in the inhibition of thèse PLD activating
factors. Furthermore, the fact that the translocation of ail the Rho-GTPase family members
(RhoA, Rac2 and Cdc42) as well as that of Arf 1 and PKCa is inhibited by PGE2 in a PKA-
dependent manner suggested that PKA probably acts at a level upstream ot the mobilization
of thèse cofactors in the signaling cascade triggered by fMLP. We therefore examined the
effect of H-89 on the EP2 receptor-mediated inhibition of pi lOy translocation as well as on
the inhibiton of tyrosine phosphorylation events since PI3-Ky and tyrosine kinases are both
important regulators of small GTPases activation (see [12]). According to our results, the
inhibition of fMLP-induced pi lOy translocation by PGE2 was, at least in part, dépendent on
PKA. The inhibition by PGE2 of the tyrosine phosphorylation of the 116-120 kDa set of
substrates stimulated by fMLP was also shown to dépend on PKA. The fact that both the
translocation of pllOy and the tyrosine phosphorylation of this particular set of substrates
dépend on PKA may suggest that the tyrosine-kinase(s) involved in this effect could be
PI3-K-dependent and probably belong(s) to the Tec kinases family, one of the two tyrosine
kinase families activated by fMLP [39], The possible link between the simultaneous
inhibition of PI3Ky and of tyrosine phosphorylation by Tec kinases mediated by PKA in
our expérimental model is supported by the observation that a spécifie inhibitor of Btk (and
possibly of other Tec kinases) decreased the level of phosphorylation of this 116-120-kDa
set of substrates [40]. Taken altogether, our results suggest that PGE2 stimulâtes the
activation of PKA via EP2 receptors and that the inhibition of fMLP-stimulated PI3-Ky
activity by PKA régulâtes several downstream signaling events such as the translocation of
Rho- and Arf-GTPase and PKCa and hence PLD activation. Finally, the fact that PKA has
an impact on the mechanism of activation of PI3-Ky induced by fMLP further confirms the
model we hâve previously proposed to explain the inhibitory effect of PGE2 on the PLD
pathway [12].
We cannot exclude that PKA can also phosphorylate other substrates involved in the fMLP-
induced PLD pathway. For example, RhoA has been shown to be phosphorylated on Ser-
188 by PKA in cell-free Systems [41] as well as in several cell types [27, 42-47]. Amongst
diverse effects, this RhoA phosphorylation inhibits cell motility [42], RhoA translocation
promoted by integrins [48], adhésion of leukocytes induced by chemoattractants [43] as

119
well as the interaction of RhoA with PLD1 [27, 41 ]. It could therefore be possible that PKA
activated by PGE2 also régulâtes PLD activity via its direct inhibitory effect on RhoA.
Another possible substrate for PKA is the Rac exchange factor P-Rexl, which is highly
expressed in PMNs and plays a major rôle in Rac activation by fMLP [49]. The
phosphorylation of P-Rexl by PKA has been reported to inhibit the LPA-stimulated
guanine nucleotide exchange activity of P-Rexl on Rac in HEK293T cells [50]. However,
in our expérimental model, the direct inhibitory effect of PKA on RhoA and P-Rexl would
be additive to the effect mediated by PKA at the P13-Ky level which probably remains the
major mechanism that explains the inhibition of small GTPases activation by PGE2 since
on one hand, Cdc42 is not a substrate for PKA [47] and PKA does not affect Arfl-
mediated activation of PLD in cell-free System [41] and, on the other hand, Cdc42 and Arfl
activation is inhibited in a PKA-dependent fashion in the same way as RhoA. Taken
together, our data suggest that PGE2 inhibits fMLP-induced small GTPases translocation to
PMN membranes through a common mechanism, possibly at the PI3-Ky level.
The rôle of PKA in the régulation of PI3-Ky activity has not yet been reported and the
précise mechanism that underlies the PKA inhibitory effect at this level is still totally
unknown. cAMP is known to inhibit the accumulation of PtdIns(3,4,5)P3 that results from
the activation of p-85 associated class la PI3-K in spécifie cell types. However, a direct
phosphorylation of the pi 10 and p85 subunits by PKA is not involved in this inhibitory
mechanism that remains to be elucidated [51]. We report in this study that PGE2-induced
PKA activity régulâtes the activation of PI3-Ky triggered by fMLP but that the regulatory
plOl and the catalytic pi 10y subunits of PI3-Ky are not phosphorylated by PKA in vitro
and hence, do not appear to be possible substrates of PKA in cells. PI3-Ky activation is
regulated by the binding of the heterodimer plOl/pllOy to OPy subunits released upon
GPCR activation by their ligands. The regulatory pi01 subunit is thought to be involved in
this binding and to play the rôle of an adaptor protein [52, 53]. In lMLP-stimulated PMNs,
GPy subunits bind to and directly activate other downstream effectors such as PLCP2/3 or
the Rac exchange factor P-Rex 1. The fact that the GPy-mediated activation of thèse two
effectors is inhibited as a resuit of their phosphorylation by PKA [50, 54, 55] suggests that
subunits are not likely targets of PKA. Moreover, PKA modulâtes the specifïcity of the

120
coupling of several GPCRs to their GPy subunits through direct phosphorylation of the
receptors, but the phosphorylation of the GPy subunits by PKA has never been described so
far [56], It is of interest to note that the fMLP receptor itself is not phosphorylated by PKA
[57]. Therefore, the targets of PKA in our model are not likely to be either GPy subunits or
the fMLP receptor. Intriguingly and contrary to PLC(32/3, we observed that PKA activated
by fMLP alone has no effect on PI3-Ky translocation to membranes but that a pre-
incubation step with PGE2 is necessary to observe the inhibitory effect of PKA on POKy.
One possible explanation to this observation is that the recruitment of PI3-Ky to membranes
and its activation by GPy, a very early event in the fMLP-signaling cascade, occurs before
the élévation of cAMP and the subséquent activation of PKA triggered by fMLP. Another
possible élément of explanation is that PKA may regulate PLCP2/3 and PI3Ky at différent
stages of their activation process by fMLP: whereas PKA terminâtes the PLCP2/3 signaling
induced by fMLP, it prevents the activating mechanism that précèdes the formation of
PtdIns(3,4,5)P3 by PI3-Ky. This is probably the reason why the pre-activation of PKA by
cAMP elevating agents is required to inhibit the activation of PI3-Ky in fMLP-stimulated
PMNs.
Taken altogether, thèse observations indicate that neither PI3-Ky nor GPy subunits are PKA
substrates and suggest that the inhibitory effect of PKA on PI3-Ky activation is mediated
through the phosphorylation of a yet unidentified protein that prevents the translocation of
plOl/pl lOy to GPy subunits released upon fMLP stimulation by a mechanism that has to be
investigated. The fact that pi01 contains 14-3-3 binding sites for which a function has not
yet been ascribed to in the activation process of PI3Ky is intriguing. 14-3-3 proteins are
small dimers that are able to bind a multitude of proteins, usually phosphorylated on ser/thr
residues, to form signaling complexes that regulate many cellular processes [58]. Although
the 14-3-3 binding site on pi01 does not seem to be phosphorylated by PKA in our
expérimental model, 14-3-3 proteins could be involved in the inhibition of PI3-Ky
translocation mediated by PKA either by forming a molecular complex with the
heterodimer plOl/pllOy that would prevent its translocation to membranes or on the
contrary, by preventing the formation of a complex that would allow the activation of PI3-

121
Ky. The hypothesis of the formation of an inhibitory molecular complex is strengthened by
the fact that the effects of PKA in cells are often mediated by AKAPs (A-kinase anchoring
proteins) which are scaffolding proteins that coordinate PKA signaling with other pathways
by recruiting multiple enzymes and substrates (ref)- For example, some AKAPs such as
AKAP-Lbc, a Rho-GEF, can be phosphorylated by PKA in a manner that promotes the
binding of 14-3-3 proteins to AKAP signaling complex, thereby inhibiting the guanine
nucleotide exchange activity of AKAP-Lbc [59, 60]. This example shows that the
PKA/AKAP and the 14-3-3 signaling pathways can intertwine and that both could possibly
be involved in the PKA-mediated inhibitory mechanism on PI3-Ky.
The inhibition of calcium influx by PGE2 in fMLP-activated PMNs is also mediated by
PKA since this inhibitory effect induced by EP2 receptors was abolished in the présence of
H-89. The metabolism of calcium and the formation of PtdIns(3,4,5)P3 are totally
independent signaling events in PMNs stimulated by fMLP [61], suggesting that PKA
targets différent substrates in both pathways. The calcium influx is thought to be mainly a
capacitive calcium entry (SOCs) through plasma membrane non sélective cation channels
[62, 63]. Since PGE2 does not affect the first rise of cytosolic calcium generated by the
release of calcium from intracellular stores and hence the mechanism that brings about
SOCs, the target of PKA could possibly be at the level of non-selective cation channels at
the plasma membrane. Several types of ion channels hâve been shown to be PKA
substrates and therefore the hypothesis of a direct inhibitory phosphorylation of the non
sélective cation channels by PKA can be advanced. Other mechanisms such as the
régulation of channel opening by the products of PI3-Ky or PLD activities cannot be
excluded.
In summary, the results reported in this study shows that the inhibition of the fMLP-
induced PLD activation pathway by PGE2 via EP2 receptors is mediated by the activation
of PKA. Although some of the components of the PLD pathway can be PKA substrates, the
main inhibitory effect of PKA that explains the inhibition of the whole PLD signaling
cascade occurs more likely at the level of PI3-Ky. The stimulation of PKA activity by PGE2
via EP2 receptors prevents fMLP-induced PI3-Ky translocation to membranes but the
pli Oy/p 101 heterodimer is not a substrate of PKA, at least in vitro. One of the various

122
stratégies that has been explored recently to treat inflammatory diseases consists of the
sélective inhibition by pharmacologie agents of the PI3-K isoforms involved in the
migration of leukocytes, i.e. the y and 8 P13-K isoforms [64-66]. Our study shows that the
inhibition of PI3-Ky by the cAMP/PKA pathway could be a valuable and alternative mean
to achieve the inhibition of PI3-Ky in PMNs.

123
3.8. AcknowledgementThis work was supported by grants from the Canadian Institutes of Health Research
(CIHR).
We thank Sebastien Simard for helpful technical assistance and Sylvain Levasseur for his
valuable suggestions.
3.9. AbbreviationsAD A, adenosine deaminaseAKAP, A-kinase anchoring proteinArf, ADP-rybosilation factorDFP, di-isopropylfluorophosphateEP receptor, E prostaglandin receptorfMLP, N-formyl-methionyl-leucyl-phenylalanineGEF, guanine nucleotide exchange factorPEt, phosphatidylethanolPH, pleckstrin homologyPI3-K, phosphatidylinositol 3-kinasePKA, cAMP-dependent protein kinasePMN, polymorphonuclear neutrophilPtdIns(3,4,5)P3, phosphatidylinositol 3,4,5- trisphophatePhospholipase D, PLDPhospholipaseC, PLC

124
3.10. Bibliography
1. Rocca, B., FitzGerald, G.A. (2002) Cyclooxygenases and prostaglandins: shaping
up the immune response. lut Immunopharmacol 2, 603-30.
2. Tilley, S.L., Coffman, T.M., Koller, B.H. (2001) Mixed messages: modulation of
inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest
108, 15-23.
3. Fantone, J.C., Kinnes, D.A. (1983) Prostaglandin El and prostaglandin 12
modulation of superoxide production by human neutrophils. Biochem Biophys Res
Commun 113,506-12.
4. Lad, P.M., Goldberg, B.J., Smiley, P.A., Oison, C.V. (1985) Receptor-specific
threshold effects of cyclic AMP are involved in the régulation of enzyme release and
superoxide production from human neutrophils. Biochim Biophys Acta 846, 286-95.
5. Sedgwick, J.B., Berube, M.L., Zurier, R.B. (1985) Stimulus-dependent inhibition of
superoxide génération by prostaglandins. Clin Immunol Immunopathol 34, 205-15.
6. Gryglewski, R.J., Szczeklik, A., Wandzilak, M. (1987) The effect of six
prostaglandins, prostacyclin and iloprost on génération of superoxide anions by human
polymorphonuclear leukocytes stimulated by zymosan or formyl-methionyl-leucyl-
phenylalanine. Biochem Pharmacol 36, 4209-13.
7. Armstrong, R.A. (1995) Investigation of the inhibitory effects of PGE2 and
sélective EP agonists on chemotaxis of human neutrophils. Br J Pharmacol 116, 2903-8.
8. Hecker, G., Ney, P., Schror, K. (1990) Cytotoxic enzyme release and oxygen
centered radical formation in human neutrophils are selectively inhibited by E-type
prostaglandins but not by PGI2. Naunyn Schmiedebergs Arch Pharmacol 341, 308-15.
9. Ham, E.A., Soderman, D.D., Zanetti, M.E., Dougherty, H.W., McCauley, E., Kuehl,
F.A., Jr. (1983) Inhibition by prostaglandins of leukotriene B4 release from activated
neutrophils. Proc Natl Acad Sci U S A 80, 4349-53.

125
10. Bloemen, P.G., van den Tweel, M.C., Henricks, P.A., Engels, F., Kester, M.H., van
de Loo, P.G., Blomjous, F.J., Nijkamp, F.P. (1997) Increased cAMP levels in stimulated
neutrophils inhibit their adhésion to human bronchial epithelial cells. Am J Physiol 272,
L580-7.
11. Abdel-Latif, D., Steward, M., Macdonald, D., Francis, G., Dinauer, M., Lacy, P.
(2004) Rac2 is critical for neutrophil primary granule exocytosis. BLOOD 104, 832 - 839.
12. Burelout, C, Thibault, N., Levasseur, S., Simard, S., Naccache, P., Bourgoin, S.
(2004) Prostaglandin E-2 inhibits the phospholipase D pathway stimulated by formyl-
methionyl-leucyl-phenylalanine in human neutrophils. Involvement of EP2 receptors and
phosphatidylinositol 3-kinase gamma. Mol. Pharmacol 66, 293 - 301.
13. Houle, M.G., Naccache, P.H., Bourgoin, S. (1999) Tyrosine kinase-regulated small
GTPase translocation and the activation of phospholipase D in HL60 granulocytes. J
Leukoc Biol 66, 1021-30.
14. Naccache, P.H., Levasseur, S., Lachance, G., Chakravarti, S., Bourgoin, S.G.,
McColl, S.R. (2000) Stimulation of human neutrophils by chemotactic factors is associated
with the activation of phosphatidylinositol 3-kinase gamma. J Biol Chem 275, 23636-41.
15. Barabe, F., Rollet-Labelle, E., Gilbert, C, Fernandes, M.J., Naccache, S.N.,
Naccache, P.H. (2002) Early events in the activation of Fc gamma RUA in human
neutrophils: stimulated insolubilization, translocation to détergent-résistant domains, and
dégradation of Fc gamma RUA. J Immunol 168, 4042-9.
16. Marcil, J., Harbour, D., Houle, M.G., Naccache, P.H., Bourgoin, S. (1999)
Monosodium urate-crystal-stimulated phospholipase D in human neutrophils. Biochem J
337 (Pt 2), 185-92.
17. Grynkiewicz, G., Poenie, M., Tsien, R.Y. (1985) A new génération of Ca2+
indicators with greatly improved fluorescence properties. J Biol Chem 260, 3440-50.
18. Simchowitz, L., Fischbein, L.C., Spilberg, I., Atkinson, J.P. (1980) Induction of a
transient élévation in intracellular levels of adenosine-3',5'-cyclic monophosphate by

126
chemotactic factors: an early event in human neutrophil activation. J Immunol 124, 1482-
91.
19. Smolen, J.E., Korchak, H.M., Weissmann, G. (1980) Increased levels of cyclic
adenosine-3',5'-monophosphate in human polymorphonuclear leukocytes after surface
stimulation. J Clin Invest 65, 1077-85.
20. Jackowski, S., Sha'afi, R.I. (1979) Response of adenosine cyclic 3',5'-
monophosphate level in rabbit neutrophils to the chemotactic peptide formyl-methionyl-
leucyl-phenylalanine. Mol Pharmacol 16, 473-81.
21. Talpain, E., Armstrong, R.A., Coleman, R.A., Vardey, C.J. (1995) Characterization
of the PGE receptor subtype mediating inhibition of superoxide production in human
neutrophils. Br J Pharmacol 114, 1459-65.
22. Regan, J.W., Bailey, T.J., Pepperl, D.J., Pierce, K.L., Bogardus, A.M., Donello,
J.E., Fairbairn, CE., Kedzie, K.M., Woodward, D.F., Gil, D.W. (1994) Cloning of a novel
human prostaglandin receptor with characteristics of the pharmacologically defined EP2
subtype. Mol Pharmacol 46, 213-20.
23. Smolen, J.E., Weissmann, G. (1981) Stimuli which provoke sécrétion of azurophil
enzymes from human neutrophils induce incréments in adenosine cyclic 3'-5'-
monophosphate. Biochim Biophys Acta 672, 197-206.
24. Tyagi, S.R., Oison, S.C., Burnham, D.N., Lambeth, J.D. (1991) Cyclic AMP-
elevating agents block chemoattractant activation of diradylglycerol génération by
inhibiting phospholipase D activation. J Biol Chem 266, 3498-504.
25. Thibault, N., Harbour, D., Borgeat, P., Naccache, P.H., Bourgoin, S.G. (2000)
Adenosine receptor occupancy suppresses chemoattractant-induced phospholipase D
activity by diminishing membrane recruitment of small GTPases. Blood 95, 519-27.
26. Thibault, N., Burelout, C, Harbour, D., Borgeat, P., Naccache, P.H., Bourgoin, S.G.
(2002) Occupancy of adenosine A2a receptors promotes fMLP-induced cyclic AMP

127
accumulation in human neutrophils: impact on phospholipase D activity and recruitment of
small GTPases to membranes. JLeukoc Biol 71, 367-377.
27. Jang, M.J., Lee, M.J., Park, H.Y., Bae, Y.S., Min do, S., Ryu, S.H., Kwak, J.Y.
(2004) Phosphorylation of phospholipase Dl and the modulation of its interaction with
RhoA by cAMP-dependent protein kinase. Exp Mol Med 36, 172-8.
28. Exton, J.H. (1999) Régulation of phospholipase D. Biochim Biophys Acta 1439,
121-33.
29. Exton, J. (2002) Régulation of phospholipase D. FEBS Lett 531, 58 - 61.
30. Houle, M.G., Kahn, R.A., Naccache, P.H., Bourgoin, S. (1995) ADP-ribosylation
factor translocation correlates with potentiation of GTP gamma S-stimulated phospholipase
D activity in membrane fractions of HL-60 cells. J Biol Chem 270, 22795-800.
31. Hirsch, E., Katanaev, V.L., Garlanda, C, Azzolino, O., Pirola, L., Silengo, L.,
Sozzani, S., Mantovani, A., Altruda, F., Wymann, M.P. (2000) Central rôle for G protein-
coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049-53.
32. Maier, U., Babich, A., Nurnberg, B. (1999) Rôles of non-catalytic subunits in
gbetagamma-induced activation of class I phosphoinositide 3-kinase isoforms beta and
gamma. J Biol Chem 21 A, 29311-7.
33. Czupalla, C, Culo, M., Muller, E.C., Brock, C, Reusch, H.P., Spicher, K., Krause,
E., Nurnberg, B. (2003) Identification and characterization of the autophosphorylation sites
of phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem 278, 11536-45.
34. Regan, J.W. (2003) EP2 and EP4 prostanoid receptor signaling. Life Sci 74, 143-53.
35. Jing, H., Yen, J.H., Ganea, D. (2004) A novel signaling pathway médiates the
inhibition of CCL3/4 expression by prostaglandin E2. J Biol Chem 279, 55176-86.
36. Vassiliou, E., Sharma, V., Jing, H., Sheibanie, F., Ganea, D. (2004) Prostaglandin
E2 promotes the survival of bone marrow-derived dendritic cells. J Immunol 173, 6955-64.

128
37. Baratelli, F., Krysan, K., Heuze-Vourc'h, N., Zhu, L., Escuadro, B., Sharma, S.,
Reckamp, K., Dohadwala, M., Dubinett, S.M. (2005) PGE2 confers survivin-dependent
apoptosis résistance in human monocyte-derived dendritic cells. J Leukoc Biol 78, 555-64.
38. Martin, M.C., Dransfield, I., Haslett, C, Rossi, A.G. (2001) Cyclic AMP régulation
of neutrophil apoptosis occurs via a novel protein kinase A-independent signaling pathway.
JBiolChem 276, 45041-50.
39. Lachance, G., Levasseur, S., Naccache, P.H. (2002) Chemotactic factor-induced
recruitment and activation of Tec family kinases in human neutrophils. Implication of
phosphatidynositol 3-kinases. J Biol Chem 277, 21537-41.
40. Gilbert, C, Levasseur, S., Desaulniers, P., Dusseault, A.A., Thibault, N., Bourgoin,
S.G., Naccache, P.H. (2003) Chemotactic factor-induced recruitment and activation of Tec
family kinases in human neutrophils. II. Effects of LFM-A13, a spécifie Btk inhibitor. J
Immunol 170, 5235-43.
41. Kwak, J.Y., Uhlinger, D.J. (2000) Downregulation of phospholipase D by protein
kinase A in a cell-free system of human neutrophils. Biochem Biophys Res Commun 267,
305-10.
42. Lang, P., Gesbert, F., Delespine-Carmagnat, M., Stancou, R., Pouchelet, M.,
Bertoglio, J. (1996) Protein kinase A phosphorylation of RhoA médiates the morphological
and functional effects of cyclic AMP in cytotoxic lymphocytes. Embo J 15, 510-9.
43. Laudanna, C, Campbell, J.J., Butcher, E.C. (1997) Elévation of intracellular cAMP
inhibits RhoA activation and integrin-dependent leukocyte adhésion induced by
chemoattractants. J Biol Chem 272, 24141-4.
44. Dong, J.M., Leung, T., Manser, E., Lim, L. (1998) cAMP-induced morphological
changes are counteracted by the activated RhoA small GTPase and the Rho kinase
ROKalpha. J Biol Chem 273, 22554-62.

129
45. Murthy, K.S., Zhou, H., Grider, J.R., Makhlouf, G.M. (2003) Inhibition of sustained
smooth muscle contraction by PKA and PKG preferentially mediated by phosphorylation
of RhoA. AmJPhysiol Gastrointest Liver Physiol 284, G1006-16.
46. Qiao, J., Huang, F., Lum, H. (2003) PKA inhibits RhoA activation: a protection
mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol
284, L972-80.
47. Ellerbroek, S.M., Wennerberg, K., Burridge, K. (2003) Serine phosphorylation
negatively régulâtes RhoA in vivo. J Biol Chem 278, 19023-31.
48. O'Connor, K.L., Nguyen, B.K., Mercurio, A.M. (2000) RhoA function in lamellae
formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J
Cell Biol 148,253-8.
49. Welch, H.C., Coadwell, W.J., Ellson, CD., Ferguson, G.J., Andrews, S.R.,
Erdjument-Bromage, H., Tempst, P., Hawkins, P.T., Stephens, L.R. (2002) P-Rexl, a
PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac.
Cell 108, 809-21.
50. Mayeenuddin, L.H., Garrison, J.C. (2006) Phosphorylation of P-Rexl by the cyclic
AMP-dependent protein kinase inhibits the phosphatidylinositiol (3,4,5)-trisphosphate and
Gbetagamma-mediated régulation of its activity. J Biol Chem 281, 1921-8.
51. Kim, S., Jee, K., Kim, D., Koh, H., Chung, J. (2001) Cyclic AMP inhibits Akt
activity by blocking the membrane localization of PDK1. J Biol Chem 276, 12864-70.
52. Stephens, L.R., Eguinoa, A., Erdjument-Bromage, H., Lui, M., Cooke, F.,
Coadwell, J., Smrcka, A.S., Thelen, M., Cadwallader, K., Tempst, P., Hawkins, P.T. (1997)
The G beta gamma sensitivity of a PI3K is dépendent upon a tightly associated adaptor,
pl01.Ce//89, 105-14.
53. Brock, C, Schaefer, M., Reusch, H.P., Czupalla, C, Michalke, M., Spicher, K.,
Schultz, G., Nurnberg, B. (2003) Rôles of G beta gamma in membrane recruitment and
activation of pi 10 gamma/plOl phosphoinositide 3-kinase gamma. J Cell Biol 160, 89-99.

130
54. Yue, C, Dodge, K.L., Weber, G., Sanborn, B.M. (1998) Phosphorylation of serine
1105 by protein kinase A inhibits phospholipase Cbeta3 stimulation by Galphaq. J Biol
Chem 273, 18023-7.
55. Liu, M., Simon, M.I. (1996) Régulation by cAMP-dependent protein kinease of a
G-protein-mediated phospholipase C. Nature 382, 83-7.
56. Lefkowitz, R.J., Pierce, K.L., Luttrell, L.M. (2002) Dancing with différent partners:
protein kinase a phosphorylation of seven membrane-spanning receptors régulâtes their G
protein-coupling specificity. Mol Pharmacol 62, 971-4.
57. Ali, H., Richardson, R.M., Tomhave, E.D., DuBose, R.A., Haribabu, B.,
Snyderman, R. (1994) Régulation of stably transfected platelet activating factor receptor in
RBL-2H3 cells. Rôle of multiple G proteins and receptor phosphorylation. J Biol Chem
269, 24557-63.
58. Mackintosh, C. (2004) Dynamic interactions between 14-3-3 proteins and
phosphoproteins regulate diverse cellular processes. Biochem J 381, 329-42.
59. Diviani, D., Baisamy, L., Appert-Collin, A. (2006) AKAP-Lbc: a molecular
scaffold for the intégration of cyclic AMP and Rho transduction pathways. Eur J Cell Biol
85,603-10.
60. Jin, J., Smith, F.D., Stark, C, Wells, CD., Fawcett, J.P., Kulkarni, S., Metalnikov,
P., O'Donnell, P., Taylor, P., Taylor, L., Zougman, A., Woodgett, J.R., Langeberg, L.K.,
Scott, J.D., Pawson, T. (2004) Proteomic, functional, and domain-based analysis of in vivo
14-3-3 binding proteins involved in cytoskeletal régulation and cellular organization. Curr
Biol 14, 1436-50.
61. Ahmed, M.U., Hazeki, K., Hazeki, O., Katada, T., Ui, M. (1995) Cyclic AMP-
increasing agents interfère with chemoattractant-induced respiratory burst in neutrophils as
a resuit of the inhibition of phosphatidylinositol 3-kinase rather than receptor-operated
Ca2+ influx. J Biol Chem 270, 23816-22.

131
62. von Tscharner, V., Prod'hom, B., Baggiolini, M., Reuter, H. (1986) Ion channels in
human neutrophils activated by a rise in free cytosolic calcium concentration. Nature 324,
369-72.
63. Demaurex, N., Monod, A., Lew, D.P., Krause, K.H. (1994) Characterization of
receptor-mediated and store-regulated Ca2+ influx in human neutrophils. Biochem J 291 (
Pt 3), 595-601.
64. Camps, M., Ruckle, T., Ji, H., Ardissone, V., Rintelen, F., Shaw, J., Ferrandi, C,
Chabert, C, Gillieron, C, Francon, B., Martin, T., Gretener, D., Perrin, D., Leroy, D.,
Vitte, P.A., Hirsch, E., Wymann, M.P., Cirillo, R., Schwarz, M.K., Rommel, C. (2005)
Blockade of PDKgamma suppresses joint inflammation and damage in mouse models of
rheumatoid arthritis. Nat Med 11, 936-43.
65. Sadhu, C, Masinovsky, B., Dick, K., Sowell, C.G., Staunton, D.E. (2003) Essential
rôle of phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol 170,
2647-54.
66. Puri, K.D., Doggett, T.A., Douangpanya, J., Hou, Y., Tino, W.T., Wilson, T., Graf,
T., Clayton, E., Turner, M., Hayflick, J.S., Diacovo, T.G. (2004) Mechanisms and
implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into
inflamed tissue. Blood 103, 3448-56.

132
CHAPITRE IV
Dissociation between the translocation and the activation of Akt in fMLP-stimulated
human neutrophils. Effect of prostaglandin E2.
L'aricle présenté dans ce chapitre est soumis pour publication à Journal of Leukocyte
Biology

133
4.1. Résumé
La prostaglandine E2 (PGE2) et d'autres agents élevant les niveaux intracellulaires d'AMPc
sont connus pour réguler négativement la plupart des fonctions stimulées par le fMLP dans
le polynucléaire neutrophile humain. Nous avons déjà montré que le pouvoir inhibiteur de
PGE2 réside dans sa capacité à supprimer l'activation de la phosphatidylinositol-3-kinase y
stimulée par le fMLP via les récepteurs EP2, ce qui diminue en conséquence la formation
des PtdIns(3,4,5)P3. L'activité d'Akt est stimulée par la phosphorylation de la Thr308 et de
la Ser473 par la PDK1 et la MAPKAPK-2 respectivement, d'une manière dépendante de la
PI3-K. Malgré la suppression de l'activation de la PI3-Ky induite par le fMLP observée en
présence de PGE2, nous montrons que Akt est complètement phosphorylée aux résidus
Thr308 et Ser473. Cependant, dans nos conditions expérimentales, le recrutement à la
membrane d'Akt induit par le fMLP est sévèrement diminué. PGE2 n'a pas d'effet sur la
phosphorylation de la MAPKAPK-2, mais elle diminue le recrutement à la membrane de
PDK1 induit par le fMLP. D'autres agents qui augmentent les niveaux intracellulaires
d'AMPc, tels que l'adénosine, bloquent également le processus d'activation de la PI3-Ky
induit par le fMLP, mais n'inhibent pas la phosphorylation d'Akt. Cependant, l'activité de
l'Akt stimulée par le fMLP est légèrement diminuée par PGE2 ou l'adénosine. La PKA est
impliquée dans l'inhibition de la translocation d'Akt par PGE2 mais elle n'intervient pas
dans le maintien de la phosphorylation d'Akt. Par ailleurs, l'inhibition de l'activité de la
PI3-KÔ stimulée par le fMLP par l'inhibiteur sélectif 1C87114 a seulement un effet minime
sur la phase tardive de la phosphorylation d'Akt en présence de la PGE2, ce qui indique que
la PI3-K5 ne joue qu'un rôle mineur dans l'activation d'Akt observée dans notre système
expérimental. Considérés dans leur ensemble, ces résultats suggèrent que les agents qui
augmentent les niveaux intracellulaires d'AMPc, tels que PGE2 ou l'adénosine, sont
capables d'induire un mécanisme alternatif d'activation d'Akt par le fMLP. Ce mécanisme
alternatif est caractérisé par une phosphorylation activatrice d'Akt qui est dissociée de son
recrutement vers les membranes enrichies en PtdIns(3,4,5)P3.

134
4.2. Abstract
Prostaglandin E2 (PGE2) and other cAMP-elevating agents are known to down-regulate
most functions stimulated by fMLP in human polymorphonuclear neutrophils (PMNs). We
previously reported that the inhibitory potential of PGE2 résides in its capacity to suppress
fMLP-stimulated phosphatidylinositol 3-kinase y (PI3-Ky) activation via EP2 receptors, and
hence to decrease PtdIns(3,4,5)P.i formation. Akt activity is stimulated by fMLP through
phosphorylation on Thr308 and Ser473 by PDK1 and MAPKAPK-2 respectively, in a PI3-
K-dependent fashion. Despite the suppression of fMLP-induced PI3-Ky activation observed
in the présence of PGE2, we show that Akt is fully phosphorylated on both Thr3O8 and
Ser473. However, fMLP-induced Akt translocation is markedly decreased in this context.
PGE2 does not affect the phosphorylation of MAPKAPK-2, but decreases the translocation
of PDK1 induced by fMLP. Other cAMP-elevating agents such as adenosine similarly
block the fMLP-induced PI3-Ky activation process but do not inhibit Akt phosphorylation.
However, Akt activity stimulated by fMLP is slightly down-regulated by agonists that
elevate cAMP levels. Whereas PKA is not involved in the maintenance of Akt
phosphorylation, it is required for the inhibition of Akt translocation by PGE2. Moreover,
inhibition of fMLP-stimulated PI3-K8 activity by the sélective inhibitor IC87114 only
partially affects the late phase of Akt phosphorylation in the présence of PGE2. Taken
altogether, thèse results suggest that cAMP-elevating agents, such as PGE2 or adenosine,
are able to induce an alternative mechanism of Akt activation by fMLP in which the
translocation of Akt to Ptdlns(3,4,5)p3 enriched-membranes is not required prior to its
phosphorylation.

135
4.3. Introduction
Polymorphonuclear neutrophils (PMNs) play a prédominant rôle in the innate host immune
defence through phagocytosis of invading microorganisms, release of proteolytic enzymes,
cytokines and chemokines, and génération of reactive oxygen species. Attracted by
chemotactic substances from endogenous origin such as leukotriene B4 (LTB4), interleukin-
8 (IL-8) and the anaphylatoxin C5a, or from bacterial origin such as N-formyl-methionyl-
leucyl-phenylalanine (fMLP), PMNs rapidly migrate towards the site of infection where
they perform their bactericidal activity. Their physiological functions are down-regulated
by agonists such as adenosine (Ado) or prostaglandin E2 (PGE2) that raise the intracellular
level of cyclic AMP (cAMP). PGE2 is an active metabolite derived from arachidonic acid
under the action of cyclooxygenases (COX-1/2) and PGE synthases. Several effector cells
of the innate immune response such as monocytes/macrophages, PMNs or dendritic cells
(DCs) are important sources of endogeneous PGE2 which is synthesized following the
activation of the inducible COX-2 under inflammatory conditions [1]. Several PMNs
functional responses to fMLP are inhibited by PGE2, including motility and chemotaxis [2],
production of superoxide anions [3] as well as the release of cytotoxic enzymes and LTB4
[4, 5]. PGE2 has also been shown to delay constitutive apoptosis in PMNs [6 , 7]. The
physiological activities of PGE2 are mediated via G-protein-linked seven transmembrane
domain receptors classified into four subtypes, EPi to EP4, according to their structure and
coupling to distinct signalling pathways [8]. EP2 and EP4 stimulate adenylyl-cyclase,
whereas EPi triggers intracellular calcium release. The various EP3 isoforms generated by
alternative splicing are connected either to cAMP or to calcium metabolism.
Pharmacological studies hâve shown that the inhibitory effects of PGE2 on fMLP-induced
superoxide production, enzyme release and chemotaxis are mediated via the EP2 receptor in
humanPMNs[9, 10].
Akt, a serine /threonine protein kinase belonging to the "AGC" superfamily of kinases,
plays critical rôles in the régulation of cell survival and prolifération [11, 12]. Its conserved
structure contains a N-terminal pleckstrin homology (PH) domain that binds
phosphoinositides with high affinity, a central catalytic domain and a C-terminal regulatory

136
domain characterized by the présence of a hydrophobic motif (HM) that médiates
interactions with signaling molécules [11]. The PH domain of Akt specifically binds
PtdIns(3,4)P2 and PtdIns(3,4,5)P3, both resulting from phosphoinositide 3-kinase (PI3-K)
activity. Upon activation of PI3-K by various agonists (growth factors, insulin), Akt is
recruited to the plasma membrane where it undergoes phosphorylation on two sites:
threonine 308 (Thr308) in the kinase domain by the phosphoinositide-dependent kinase-1
(PDK1) and serine 473 (Ser473) in the C-terminal domain by a PDK2 the nature of which
may vary according to the cell-type. Thèse two phosphorylation steps are necessary and
sufficient for full activation of Akt [11]. Akt activity can be stimulated in PMNs by
chemoattractants such as fMLP [13] and has been shown to contribute to chemotaxis and to
the respiratory burst [14]. In fMLP-stimulated neutrophils, MAPKAPK-2, a p38 substrate,
phosphorylates the Ser473 of Akt in PI3-K-dependent manner and thus fullfills the rôle of
PDK2[15].
We previously reported that PGE2 interfères with the fMLP-signalling pathway at the PI3-
Ky level in human PMNs. Indeed, PGE2 abolished the stimulation of the pllOy activity
induced by fMLP and consequently decreased PtdIns(3,4,5)P3 formation. This inhibitory
effect of PGE2 is mediated via EP2 receptors. Subsequently, the translocation to membranes
of several components downstream of PI3-K (Tec kinases, Rho and Arf GTPases, PKC) is
severely impaired [16]. In the présent study, we further investigated the conséquences of
the inhibition of the fMLP-induced PI3-Ky activity by PGE2 and we specially focused on
the effect of PGE2 on Akt activation induced by fMLP since Akt is a major downstream
targetofPD-K.

137
4.4. Materials and methodsReagents: fMet-Leu-Phe (fMLP), wortmannin and cytochalasin B (CB) were purchased
from Sigma Chemical Co. (St-Louis, MO, USA). Dextran T-500 was purchased from
Pharmacia Biotech (Dorval, Québec, Canada) and Ficoll-Paque from Wisent (St-Bruno,
Québec, Canada). Adenosine deaminase (ADA) was purchased from Roche Diagnostics
(Laval, Québec, Canada), and di-isopropylfluorophosphate (DFP) from Serva (Heidelberg,
Germany). PGE2 and CAY 10399 (9-oxo-l la,lS-dihydroxy-17-cyclobutyl-prosta-5Z,13E-
dien-1-oic acid) were purchased from Cayman Chemical (Ann Arbor, MI). Forskolin was
obtained from BIOMOL Research Laboratory (Plymouth Meeting, PA). 2-p-(2-
carboxyethyl)phenethylamino-5'-N-ethylcarboxiamido adenosine hydrochloride (CGS-
21680), chloro-2-(2-furyl)[l,2,4]triazolo[l,5-c]quinazolin-5-amine (CGS-15943) were
obtained from RBI (Natick, MA). H-89 was purchased from Calbiochem (La Jolla, CA).
IC87114 was a gift from Dr. Crabbe (UCB, Slough, United Kingdom). The Akt Kinase
Assay Kit was purchased from Cell Signaling Technology (#9840) (Beverly, MA).
Antibodies. The mouse monoclonal antibody (Ab) against phospho-p38 (Thrl80/Tyrl82)
(#9216) and the rabbit polyclonal Ab against p38 (#9212), MAPKAPK-2 (#3042),
phospho-MAPKAPK-2 (Thr222) (#3044), phospho-MAPKAPK-2 (Thr334) (#3041),
PDK1 (#3062), Akt (#9272), phospho-Akt (Ser473) (#9271), phospho-Akt (Thr308)
(#9275), phospho-Bad (Serl36) (#9295) and against the phospho-GSK-3a/p (Ser21/9)
(#9331) were obtained from Cell Signaling Technology (Beverly, MA). The mouse
monoclonal immobilized Ab against Akt (1G1) was purchased from Cell Signaling
Technology (#9279). The anti-flotillin-1 mouse monoclonal Ab was obtained from BD
Biosciences (Mississauga, ON, Canada) (#610820). The polyclonal anti-pllOy Ab was
raised in rabbits as described previously [17].
Isolation of hiiinan neutrophils. Venous blood was collected from healthy adult
volunteers in isocitrate anticoagulant solution. PMNs were separated as described
previously [18]. Briefly, whole blood was centrifuged at 180g for 10 min and the resulting
platelet rich plasma was discarded. Leukocytes were obtained following erythrocytes
sédimentation in 2% Dextran T-500. Mononuclear cells were removed by centrifugation on
Ficoll-Paque cushions and contaminating erythrocytes in the neutrophil pellets were

138
removed by a 20 sec hypotonie lysis in water. PMNs were resuspended in Hanks Balanced
Sait Solution (HBSS), pH 7.4, containing 1.6 raM Ca2+ but no Mg2'.
Immunoblotting analysis of whole cell lysates, PMNs (2xlO7 cells/ml) were pre-
incubated at 37 °C with 1 mM DFP for 10 min. The cells were pre-incubated for 10 min
with 0.1 U/ml ADA to eliminate endogenous adenosine (Ado) and EP2 receptor agonists
(PGE2 and CAY10399) or the A2A receptor agonist CGS-21680 or antagonist CGS-15943
or forskolin or an equal volume of diluent (Me2SO) as a control. When indicated kinase
inhibitors (H-89, wortmannin or IC87114) were added prior to PGE2. PMNs were
stimulated with 100 nM fMLP for the indicated times and the reaction was stopped by
transferring 100 ul of the cell suspensions to an equal volume of boiling 2X Laemmli
sample buffer (SB) (lx is 62.5 mM Tris-HCl, pH 6.8, 4% SDS, 5% p-mercaptoethanol,
8.5% glycerol, 2.5 mM orthovanadate, 10 mM paranitro-phenylphosphate, 10 |ag/ml
leupeptin, 10 ug/ml aprotinin, 0.025% bromophenol blue) and boiled for 7 min. The
samples were then subjected to 7.5-20% gradient SDS-PAGE and transferred to Immobilon
PVDF membranes. Immunoblotting was performed using the indicated Abs and revealed
with the HRP-conjugated secondary anti-rabbit or anti-mouse Abs (1/20 000) and the
Renaissance détection System (NEN, Perkin Elmer Life Sciences).
Translocation assays. PMNs (4xlO7 cells/ml) were treated with 1 mM DFP for 10 min at
room température. The cell suspensions were centrifuged and the cells were resuspended in
HBSS at 107 cells/ml. PMNs were warmed 5 min at 37 °C and preincubated for 10 min
with 0.1 U/ml ADA to eliminate endogeneous Ado. When indicated, PGE2 or the EP2
receptor agonist CAY10399, CGS-21680 (A2A receptor agonist) or CGS-15943 (A2A
receptor antagonist) or forskolin or an equal volume of diluent (Me2SO) as a control were
added to the cell suspensions at the same time as ADA. 10 uM CB was added 5 min prior
to stimulation with fMLP. PMNs were stimulated with 100 nM fMLP at 37 °C for 30 sec.
Incubations were stopped by diluting the cell suspensions five fold with ice-cold HBSS and
total membrane proteins were collected as described previously [18]. Briefly, cell
suspensions were centrifuged as indicated and resuspended in 1 ml ice cold KC1-HEPES
relaxation buffer (100 mM KC1, 50 mM HEPES, 5 mM NaCl, 1 mM MgCl2, 0.5 mM

139
EGTA, 2.5 ng/ml aprotinin, 2.5 ug/ml leupeptin and 2.5 mM PMSF, pH 7.2). Cell
suspensions were sonicated 20 sec and centrifuged 7 min at 1000g. Unbroken cells and
nuclei were discarded and the supernatants ultracentrifuged at 180 000g for 45 min.
Membrane pellets were washed once and resuspended in a small volume of buffer A
containing 0.25 M Na2HPO4, 0.3 M NaCl, 2.5% sodium dodecyl sulfate (SDS), 10 ng/ml
aprotinin, 10 ug/ml leupeptin and 2.5 mM PMSF and samples were assayed for protein
content with the Coomassie Brilliant Blue protein assay (Pierce, Rockford, IL). Protein
samples (30 ug) were resolved on a 8% SDS-PAGE and transferred to Immobilon PVDF
membranes (Millipore Corporation, Bedford, MA, USA). Immunoblotting analyses were
performed using anti-pllOy (1/1000), anti-Akt (1/1000) or anti-PDKl (1/1000) Abs. The
PVDF membranes were reprobed with an anti-flotillin-1 Ab as a control for equal protein
loading. Proteins were revealed with HRP-conjugated secondary anti-rabbit or anti-mouse
Ab and the Renaissance détection system (NEN, Perkin Elmer Life Sciences) as described
above.
Akt immunoprecipitation and kinase assay. PMN suspensions (1 ml at 2 x 107cells/ml)
were warmed at 37°C in the présence of 1 mM DFP and preincubated with 0.1 U/ml ADA
for 5 min of preincubation. 10"5 M H-89 (or an equal volume of Me2SO) was added for an
additional 5 min and 10"5 M PGE2 or CGS-21680 (or an equal volume of Me2SO) for
another 10 min incubation. PMNs were then stimulated with 100 nM fMLP (or Me2SO as a
non-stimulated control) and the reaction was stopped by a quick centrifugation of the cell
suspensions. Cell pellets were lysed by adding 1 ml ice-cold lysis buffer (20 mM Tris pH
7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium
pyrophosphate, 1 mM paranitrophenylphosphate, 1 mM orthovanadate, 1 mM PMSF and
10 ug/ml aprotinin/leupeptin) and were left 10 min on ice. The cell lysates were centrifuged
at 12,000g for 10 min at 4°C. 30 (il of the supernatants were kept aside to ensure that the
same amounts of Akt were présent in cell lysates. Akt was immunoprecipitated in the
clarified lysates (900 \x\) with 20 ul of immobilized Akt Ab bead slurry for 2 hours at 4°C.
The beads were collected and washed twice with cold lysis buffer and twice with kinase
buffer (25 mM Tris pH 7.5, 10 mM MgCl2, 5 mM p-glycerophosphate, 2 mM DTT). The
bead pellets were suspended in 50 ul kinase buffer supplemented with 1 jal of 10 mM ATP

140
and 1 (ag of GSK-3 fusion protein as a substrate and incubated at 30 °C for 25 min. The
reaction was stopped by adding 25 \x\ of 3x Laemmli sample buffer. Proteins were
separated on 7.5-20 % gradient SDS-PAGE and transferred to Immibilon PVDF
membranes. Immunoblottings were performed with anti-Akt (1/1000) and anti-phospho-
GSK-3 (1/1000) Abs.
Statistics. The paired Student t test was used for data analysis.

141
4.5. Results
4.5.1 . Effect of PGE2 on fMLP-induced Akt phosphorylationAkt activation stimulated by various agonists is PI3-K-dependent and its phosphorylation
on Ser473 and Thr308 residues is most often considered as an index of PI3-K activation.
As we previously reported that PGE2, acting via EP2 receptors, inhibited the stimulation of
PI3-Ky activity induced by fMLP in human PMNs, we examined the effect of this
eicosanoid and of the EP2 sélective agonist CAY10399 on the fMLP-stimulated
phosphorylation status of Akt. The data presented in Fig. 1 show that, unexpectedly, the
stimulation of the phosphorylation of Akt on both Ser473 and Thr308 induced by fMLP
was maintained when PMNs were incubated with PGE2 or CAY10399 prior to stimulation.
The level of Akt phosphorylation on both residues was however slightly decreased at 5 min
and 10 min after fMLP stimulation. Thèse data indicate indicate that PGE2 only weakly
modified the Akt phosphorylation time-course induced by fMLP. In PMNs incubated with
PGE2 alone up to 40 min, we did not observe the phosphorylation of Akt.
4.5.2. Effect of a PKA inhibitor on the phosphorylation of Akt in theprésence of PGE2.The binding of PGE2 to EP2 receptors stimulâtes the formation of cAMP by adenylyl-
cyclase [19]. The inhibition of the fMLP-stimulated production of superoxide anions by
PGE2 in human PMNs is mediated via the EP2 receptors and the cAMP/PKA pathway [9].
Moreover, we observed that the inhibition of the translocation of the pllOy catalytic
subunit to membranes by the EP2 pathway was PKA-dependent (unpublished results). It has
also been reported that PKA stimulated Akt activity in a PI3-K-independent manner in 293-
EBNA and COS7 cell lines [20, 21]. In order to examine the rôle of PKA in the stimulation
of Akt phosphorylation induced by fMLP in the présence of PGE2, we examined the effects
of the PKA pharmacological inhibitor H-89 on this response. The data presented in Fig. 2
show that preincubation of PMNs with H-89 did not significantly modify the
phosphorylation of Akt induced by fMLP. Therefore, in our expérimental model, PKA does
not appear to be involved, directly or indirectly, in the phosphorylation of Akt.

142
4.5.3. PGE2 inhibits the translocation of PDK1 and Akt to membranes.Effect of H-89Both PDK1 and Akt rapidly translocate to the plasma membrane in response to PI3K
activation. This membrane localization has been shown to be necessary for the
phosphorylation of Akt by PDK1 and for its full activation [22, 23]. Since, on the one hand,
PGE2 decreased the fMLP-induced formation of PtdIns(3,4,5)P3 and on the other hand, the
PH domains of Akt and PDK1 display a high affmity for polyphosphoinositides
(PtdIns(3,4)P2 and PtdIns(3,4,5)P3) which render them very sensitive to variations in the
concentration of polyphosphoinositides at the plasma membrane, we therefore examined
the effect of PGE2 on the translocation of Akt and PDK1 induced by fMLP. As expected,
we observed that when PMNs were incubated in the présence of PGE2 prior to stimulation
by fMLP, the amounts of membrane-associated Akt and PDK1 were dramatically
decreased in a dose-dependent manner as compared to the fMLP-stimulated controls (Fig.
3A).
In order to examine the rôle of PKA in the inhibition of Akt translocation by PGE2, we
performed the translocation assays in the présence of the PKA inhibitor, H-89. As shown in
Fig. 3B, the inhibitory effect of PGE2 and of the EP2 agonist CAY10399 was almost
completely abolished when PMNs were preincubated with H-89 (lane 6 vs lane 9 and lane
7 vs lane 10). This latter resuit indicates that PKA is involved in the decreased translocation
of Akt to membranes induced by PGE2 via the EP2 receptor.
4.5.4. Effect of PGE2 on the p38-MAPKAPK2-Akt pathway.
MAPKAPK-2, a p38 substrate, has been shown to phosphorylate Akt on Ser473 in human
PMNs in a PI3-K-dependent manner and therefore to fulfill the PDK2 function in this cell
type [15]. We hâve thus examined the effect of PGE2 and the EP2 agonist CAY10399 on
the phosphorylation of MAPKAPK-2 on Thr222 and Thr334 mediated by p38 and the
phosphorylation of p38 as well. Our data show that fMLP induced the phosphorylation of
p38 and of MAPKAPK-2 on both Thr222 and Thr334 and that none of thèse
phosphorylations were affected by PGE2 (Fig. 4). Thus, PGE2 inhibited the fMLP-
stimulated PI3-Ky activity and subsequently the formation of PtdIns(3,4,5)P3 but affected
neither the phosphorylation of p38 nor that of its substrate MAPKAPK-2. Thèse latter

143
results are similar to those observed with the phophorylation of Akt in the présence of
PGE2.
4.5.5. Effect of cAMP elevating agents on the translocation of pllOy, Aktand PDK1 induced by fMLP.
Ail agents that elevate intracellular cAMP (forskolin, perméable cAMP analogs,
phosphodiesterase inhibitors, Ado) share the ability to inhibit PMN functions stimulated by
fMLP as well as to delay apoptosis. The inhibitory effect of endogenous Ado on the fMLP-
stimulated phospholipase D (PLD) pathway in human PMNs is mediated via the A2A
receptors which stimulate an increase of intracellular cAMP [24]. We thus wondered if
thèse agents hâve the same effect as PGE2 on the PD-Ky-Akt signalling pathway. First, the
effect of Ado and of the adenylyl cyclase activator forskolin were tested on the
translocation to membranes of pli Oy, PDK1 and Akt in fMLP-stimulated PMNs. Cells
were preincubated with ADA to suppress the inhibitory effect of Ado, or with ADA and the
A2A receptor agonist CGS-21680 or the A2A receptor antagonist CGS-15943, or with
forskolin prior stimulation with fMLP.
When 0.1 U/ml ADA was added to cell suspensions, the amounts of membrane-associated
pllOy, PDK1 and Akt were markedly increased as compared to the fMLP-stimulated
control (Fig. 5, lanes 6 and 7), thereby suggesting an inhibitory rôle for endogenously
released Ado in the fMLP-induced recruitment to membranes of thèse kinases. Conversely,
in the présence of both ADA and the A2A receptor agonist (CGS-21680), the amounts of
pli0y, PDK1 and Akt translocated to membranes were considerably decreased (Fig. 5, lane
8, as compared to lane 7) and were comparable to those observed in the absence of ADA
(i.e. in the présence of Ado in the suspensions, lane 6). Similarly to the addition of ADA to
the PMN suspensions, the A2A receptor antagonist CGS-15943 increased the recruitment of
pllOy, PDK1 and Akt to the membranes (lane 9, as compared to lane 6). Taken together,
thèse results indicate that Ado, acting via A2A receptors, dramatically diminishes the fMLP-
induced translocation of pi 10y, PDK1 and Akt to the membranes.

144
Forskolin, a direct activator of adenylyl cyclase which élevâtes intracellular levels of
cAMP, also decreased the amounts of membrane-associated pllOy, PDK1 and Akt (lane
10).
4.5.6. fMLP-induced Akt activation is maintained in the présence ofcAMP elevating agents.
S i n c e A d o a n d f o r s k o l i n n e g a t i v e l y r egu l a t e t he t r a n s l o c a t i o n o f p l i O y , A k t a n d P D K 1
induced by fMLP, we wondered if thèse substances were also able to modulate Akt
phosphorylation. PMNs were incubated with ADA alone or with both ADA and CGS-
21680, or with forskolin prior to stimulation with fMLP. Neither the dégradation of
extracellular Ado by ADA nor the binding of the sélective agonist CGS-21680 to A2A
receptor modified the phosphorylation of Akt on Ser473 or Thr308 (Fig. 6). Similarly,
forskolin did not affect the phosphorylation of Akt. Thus, as is the case for PGE2, cAMP
elevating agents inhibit the translocation of pi lOy, Akt and PDK1 to membranes but not the
phosphorylation of Akt.
4.5.7. Effect of PGE2 and of the A2A receptor agonist on Akt kinaseactivity
The data presented above suggest that in the présence of cAMP elevating agonists, such as
PGE2 or Ado, Akt is phosphorylated on Thr308 and Ser473 in response to fMLP although
the mechanism of translocation is severely impaired by thèse agonists. Since the
phosphorylation of Akt on both residues is considered as its main mechanism of activation,
the amount of phosphorylated Akt is often presented as an indicator of Akt activity in cells.
We thus wondered whether, in the présence of cAMP elevating agonists, the kinase activity
of Akt was correlated with its phosphorylation. Using the Akt Kinase Assay Kit, Akt was
immunoprecipitated in PMNs preincubated with PGE2 or CGS-21680 and stimulated with
fMLP (30 sec). The kinase activity of Akt was monitored using the phosphorylation of a
GSK-3 fusion protein, an exogenous substrate. The amount of phosphorylated GSK-3 was
measured by immunoblotting with an anti-phospho-GSK-3a/p(Ser21/9) Ab. The results of
thèse experiments, illustrated in Fig. 7, show that Akt immunoprecipitated from fMLP-
stimulated but not from control PMNs strongly phosphorylates GSK-3. PGE2 and CGS-

145
21680 slightly decrease the amounts of GSK-3 fusion protein phosphorylated by
immunoprecipitated Akt from fMLP-stimulated PMNs. In order to examine the rôle of
PKA in the fMLP-stimulated Akt activity in the présence of PGE2, PMNs were also pre-
incubated with H-89 prior to addition of PGE2 to cell suspensions and stimulation. In thèse
expérimental conditions, we observed a slight increase in the amounts of phosphorylated
GSK-3 fusion protein, whether the PMNs were incubated with PGE2 or not. Taken
altogether, thèse data indicate that PGE2 and CGS-21680 only slightly diminished Akt
activity and that PKA slightly down-regulates Akt activity stimulated by fMLP.
4.5.8. Effect of a PI3-KÔ spécifie inhibitor on the fMLP-inducedphosphorylation of Akt in the présence or the absence of PGE2.
A recently developed sélective inhibitor of the PI3-K8 isoform (IC87114) has been shown
to decrease PtdIns(3,4,5)P3 formation and to partially inhibit several functions stimulated
by fMLP in human PMNs [25, 26]. We thus wondered if fMLP-stimulated PI3-KÔ activity
could be involved in the phosphorylation of Akt and compensate for the inhibition of PI3-
Ky in the présence of PGE2. To address this question, PMNs were preincubated with
IC87114 prior to stimulation by fMLP in the présence or absence of PGE2 and the amounts
of phosphorylated Akt were determined by immunoblotting and densitometric analyses.
The results of thèse studies are depicted in Figure 8. As shown above (Fig. 1), stimulation
of PMNs by fMLP led to a rapid increase in the phosphorylation levels of Akt (on both
Thr308 and Ser473). This response was little affected by PGE2. However, hère again, the
phosphorylation of Akt in the présence of PGE2 seemed to decrease somewhat faster than in
fMLP-stimulated control cells. Preincubation of cells with the PI3-KÔ inhibitor (IC87114)
did not decrease the stimulation of the phosphorylation of Akt although, like PGE2, it led to
a more rapid decrease in the level of phosphorylation than in fMLP-stimulated control cells
(most évident at the 5 minute time point, p = 0.03, lane 4 vs Ianel2 for Thr308
phosphorylation), an effect that was additive with that of PGE2 (p = 0.0132, lane 8 vs lane
16)
Thèse data indicate that: 1) PI3-KÔ is not directly involved in the initiation of the fMLP-
induced stimulation of the phosphorylation of Akt but that it plays a rôle in the late phase of

146
the latter (i.e. the phosphorylation observed at 5 min of stimulation) and 2) PI3-KÔ is, at
least in part, involved in the maintenance of the Ser473 phosphorylation after 5 min of
stimulation by fMLP in the présence of PGE2.

147
4.6. Figures
kDa6 6 -
6 6 -
6 6 -« M *
IB:
: Ser-473
: Thr-308
Akt
Time(min) 0 0.5 1 5 10 0 0.5 1il
10 0ii
0.5 1 10
fMLP fMLP + PGE2 fMLP + CAY10399
Figure 1. Effect of PGE2 and CAY10399 on fMLP-induced Akt phosphorylation

148
kDa6 6 -
6 6 -
6 6 -
•)Akt Ser-473
®Akt Thr-308
Akt
Time (min) 0 0.5 1 5 0 0.5 1v ' i il5 0 0.5 1ii 5 0 0.5 1ii
fMLP fMLP + PGE2 fMLP + H-89 fMLP + PGE2+ H-89
Figure 2. Effect of H-89 on fMLP-induced Akt phosphorylation

149
kDa 1 2 3 4 5 66 6 -
55 -6 6 -
55
Akt
PDK1
Flotillin-1
fMLP(10'7M) - + + + +PGE2(M) - 10-5 - 10"5 10-6 10"7
•o
•8 100-
!
2 25-]
0 ii illo
8V)
1 2 3 4 5 6» 0 il
1 2 3 4 5 6
BA k t -
Flotillin-1 -
1 2 3 4 5 6 7 8 9 10fMLPPGE2
CAY10399H-89
(107
(10-6
(10-6
(10-5
M)M)M)M)
—---
:ed
m
I• <
bra
ma
-+--
125
100-
c 7 5 'C 5 0 -
25-
0-1 2 3 4 5 6 7 8 9 10
Figure 3. Effect of PGE2 on fMLP-induced Akt and PDKl translocation

150
kDa
4 3 -
4 3 -
—•—- •mm' -mm*
^
2 5 10 0 0.5 1il
fMLP
Time (min) _ 0 0.5 1
BkDa
55-
43 -
55-
43-
55-
43 -Time (min) 0 0.5 1 2 5 10 M 0 0.5 1
5 10 0 0.5 111 2 5 10
fMLP + PGE2 fMLP+ CAY 10399
©MAPKAPK2-Thr-222
®MAPKAPK2-Thr-334
MAPKAPK2
2 5 10 0 0.5 111 5 10
fMLP fMLP + PGE2 fMLP+ CAY 10399
Figure 4. Effect of PGE2 and CAY10399 on p38 and MAPKAPK-2 phosphorylation

151
1
« M »
m
mm
2
m*
3
ff»
"mm
•>
4 5
— 4
6
m m
7 8 9
HÉ.- """""' '• • • • " • • " : . -
» —«•ttKDt̂ MHbb, moi
10
- •
pi
fMLP(10"7M)ADA (0.1 U/ml)
CGS-21680(10BM)CGS-15943(10^M)
Forskolin(105M)
IB:
p110y
PDK1
Akt
Flotillin-1
+ +
T3
02 700i•S 600-S ^ 5 0 0cp o 400gJE.3002 200E 100| 0
p110y
MS
1 2 3 4 5 6 7 8 9 10 E
£ 200i
| Q 1 0 0
2
0
50
PDK1
1 2 3 4 5 6 7 8 9 10
Aktj i 250i
8 200
^ 1 0 0 'coJ3
0)
50- ll.l,1 2 3 4 5 6 7 8 9 10
Figure 5. Effect of cAMP elevating agents on fMLP-induced pllOy translocation.

152
6 6 -
6 6 -
6 6 -
®Akt Ser-473
®Akt Thr-308
Akt
Time (min) 0 0.5 1 5 0 0.5il
5 0il 0.5 1 5 0 0.5
ilfMLP fMLP + ADA fMLP + ADA
+ CGS-21680fMLP
+ Forskolin
Figure 6. Effect of cAMP-elevating agents on fMLP-induced Akt phophorylation

153
IPAkt
IB = Akt
IB = P-GSK-3
NS fMLP +PGE2 +CGS- +H-89 +H-8921680
+fMLP
fMLP +PGEO +CGS- +H-89 +H-8921680 +PGEO
+fMLP
Figure 7. Enzymatic activity of Akt in fMLP-stimulated PMNs

154
+ IC87114
fMLPfMLP +PGE, fMLP
fMLP +PGE,
I fMLP +Wortmannin
1 2
mmm
3
'SB
mm
•
4
—
mm
•m
'5
n
mm
6 7
ÎSËPÎSP
«MM
8 9
«M» «a» 11 g p
10
« • •
mm
11
«
m
121
jmm
•i3
—•
n
14
« |
«•»
M*
15
• mm
—
161
—
17 18
— .
®-AktThr-308
®-AktSer-473
Akt
Time (min) 0 0.5 1 5 0 0.5 1 5 0 0.5 1 5 0 0.5 1 5 0 0.5
B
4 *
Phospho-Akt Thr.308
11 -10 -
76
2
n il
p=0,03 *
p=0,053,ns
I
i
{ J
Tf I
i1 2 3 4 5 6 7 8 9 101112131415161718
fMLP fMLP+PGE2
fMLP+IC87114
fMLP+ fMLP+IC87114+ Wort
PGE2
Phospho-Akt Ser.473
p=0,009" p=0,0132*
i1 2 3 4 5 6 7 8 9 1011121314 1516 1718
I || || || u ifMLP fMLP+ fMLP+ fMLP+ fMLP+
PGE2 IC87114 IC87114+ WortPGE2
Figure 8. Effect of IC87114 on fMLP-induced Akt phosphorylation in the présence of
PGE2.
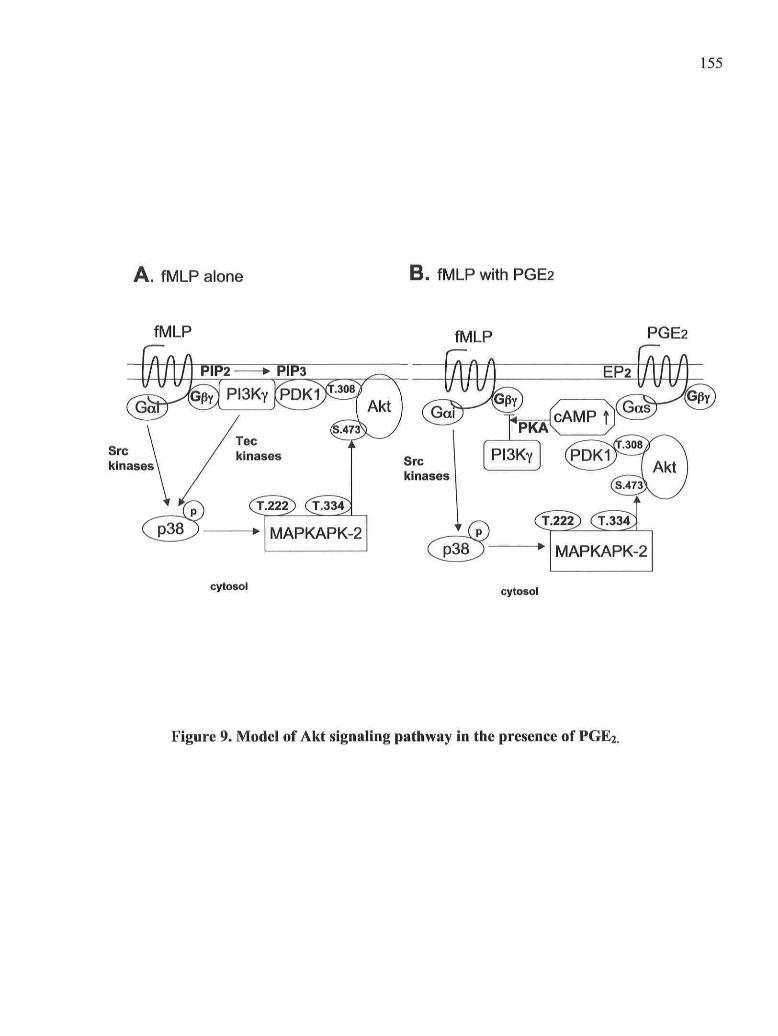
155
A . fMLP alone B . fMLP with PGE2
fMLP fMLP PGE2
Srckinases
cytosol cytosol
Figure 9. Model of Akt signaling pathway in the présence of PGE2.

156
FIGURE LEGENDS
Figure 1. Effect of PGE2 and CAY10399 on fMLP-induced Akt phosphorylation.
PMNs were preincubated for 10 min with 1CT5 M PGE2 or 10"5 M CAY10399 and
stimulated with 100 nM fMLP for the times indicated. The cell lysates were analyzed as
described in Materials and Methods. The membranes were probed with anti-phospho-
Ser473 Akt, anti-phospho-Thr308 Akt or anti-Akt Abs. The data shown are représentative
of four independent experiments. The data shown are représentative of four independent
experiments.
Figure 2. Effect of H-89 on fMLP-induced Akt phosphorylation. PMNs were
preincubated for 5 min with 10"5 M H-89 and for 10 additional min with 10"5 M PGE2 and
stimulated with 100 nM fMLP for the times indicated. The cell lysates were analyzed as
described in Materials and Methods. The membranes were probed with anti phospho-
Ser473 Akt, anti-phospho-Thr308 Akt or anti-Akt Abs. The immunoblots shown are
représentative of four independent experiments.
Figure 3. Effect of PGE2 on fMLP-induced Akt and PDK1 translocation. A. PMNs
were preincubated for 10 min with 10"5 M PGE2 and stimulated with 100 nM fMLP for 30
sec. B. PMNs were preincubated for 5 min with 10 uM H-89 and for 10 additional minutes
with 10"6 M PGE2 or CAY10399 and then stimulated with 100 nM fMLP for 30 sec. The
membrane fractions were prepared as described in Materials and Methods and were
analyzed by immunoblotting with anti-Akt or anti-PDKl Abs, and anti-flotillin-1 Ab as a
control for total protein loading. Densitometric analyses are expressed as percentages of
fMLP-stimulated controls and shown as means of three immunoblots obtained from three
independent experiments.
Figure 4. Effect of PGE2 and CAY10399 on p38 and MAPKAPK-2 phosphorylation.
Neutrophils were preincubated for 10 min with 10-5 M PGE2 or 10-5 M CAY10399 and
stimulated with 100 nM fMLP for the times indicated. The cell lysates were analyzed as
described in Materials and Methods. A. The membranes were probed with anti phospho-

157
p38 and anti-p38 Abs. B. The membranes were probed with anti-phospho-MAPKAPK-2
Thr 222, anti-phospho-MAPKAPK-2 Thr334 or anti-MAPKAPK-2 Abs. The immunoblots
shown are représentative of three independent experiments.
Figure 5. Effect of cAMP elevating agents on fMLP-induced pllOy translocation.
PMNs were preincubated for 10 min with ADA alone (0.1 U/ml) or ADA plus CGS-21680
(10"6 M) or CGS-15943 (10"6 M) or forskolin (10"5 M) prior to stimulation with 100 nM
fMLP. The membrane fractions were prepared as described in Materials and Methods and
were analyzed by immuno-blotting with anti-pllOy, anti-Akt or anti-PDKl Abs. Anti-
flotillin-1 immunoblot is shown as a control for total protein loading. Densitometric
analyses are expressed as percentages of fMLP-stimulated samples and are shown as means
of three immunoblots obtained from three independent experiments.
Figure 6. Effect of cAMP-elevating agents on fMLP-induced Akt phophorylation.
PMNs were preincubated for 10 min with ADA alone (0.1 U/ml) or ADA plus CGS-21680
(10"6 M) or forskolin (10"5 M) prior to stimulation with 100 nM fMLP. The cell lysates
were analyzed as described in Materials and Methods. Membranes were probed with anti-
phospho-Ser473 Akt, anti-phospho-Thr308 Akt or anti-Akt Abs. The data shown are
représentative of three independent experiments.
Figure 7. Enzymatic activity of Akt in fMLP-stimulated PMNs. PMN suspensions
(2xlO7 cell/ml) were preincubated with 0.1 U/ml ADA (5 min), with 10 uM H-89 for an
additional 5 min and with 10"5 M PGE2 or CGS-21680 for 10 min. After stimulation with
100 nM fMLP, Akt was immunoprecipitated in cell lyates as described in Material and
Method. The Akt kinase assay was carried out for 25 min using a GSK-3 fusion protein as
substrate. Proteins were separated with SDS-PAGE, and the level of GSK-3 fusion protein
was monitored using an anti-phospho-GSK-3 Ab. Membranes were also probed with an
anti-Akt Ab to show the amounts of immunoprecipitated Akt. Densitometric analyses of the
phospho-GSK-3 immunoblots are shown. The data shown are représentative of two
independent experiments.
Figure 8. Effect of IC87114 on fMLP-induced Akt phosphorylation in the présence of
PGE2. PMN suspensions containing 0.1 U/ml ADA were preincubated for 10 min with 10

158
uM IC87114 or 100 nM wortmannin and for 10 additional min with 10"5 M PGE2 prior to
stimulation with 100 nM fMLP for the times indicated. Cell lysates were analyzed as
described in Materials and Methods. A. The membranes were probed with anti-phospho-
Ser473 Akt, anti-phospho-Thr308 Akt, or anti-Akt Abs. B. Densitometric analyses of the
immunoblots are shown. The data shown are représentative of five independent
experiments.
Figure 9. Model of Akt signaling pathway in the présence of PGE2. A. fMLP stimulâtes
the formation of PtdIns(3,4,5)P3 by PI3-Ky and the translocation of PDKl and Akt that
allows the phosphorylation of Akt on Thr3O8 by PDKl and on Ser473 by MAPKAPK-2 at
the plasma membrane. B. In the présence of PGE2 (or other cAMP-elevating agents), the
translocation of PI3Ky is inhibited, and as a resuit of the reduced formation of
PtdIns(3,4,5)P3, that of Akt and PDKl as well. However, the phosphorylation of Akt on
Thr308 and Ser473 triggered by fMLP is not inhibited by PGE2 and hence occurs in the
cytosol. The activation of PKA by PGE2 médiates the inhibition of p l i Oy, Akt and PDKl
translocation but does not influence on Akt phosphorylation.

159
4.7. DiscussionIn a previous study, we showed that the main signaling mechanism accounting for the
inhibitory effect of PGE2 on the fMLP signaling in PMNs was the inhibition of the PI3-Ky
activity via the EP2 receptor [16]. As a conséquence of the decreased amount of
PtdIns(3,4,5)P3 produced, we observed that the recruitment to membranes of several factors
that play a major rôle in the activation of PLD, i.e. Arf and Rho GTPases, PKCa and Tec
kinase, was dramatically reduced. Given the high dependency of Akt activation on PI3-K,
we expected that, in the présence of PGE2 which inhibits fMLP-induced PI3-Ky activation
and the formation of its product PtdIns(3,4,5)P3, the translocation of Akt would be greatly
decreased and that subsequently, the activation of Akt through phosphorylation would at
least be partially inhibited. Quite unexpectedly and to the contrary, we found that the
fMLP-induced phosphorylation of Akt on both Ser473 and Thr308, was not significantly
decreased by PGE2. Only minor changes in the kinetics of Akt phosphorylation were
observed. Nevertheless, and as expected, the translocation of Akt to membranes was
dramatically diminished in the présence of PGE2 or the EP2 receptor agonist CAY10399.
However, whereas Akt was fully phosphorylated in the présence of PGE2, its activity
stimulated by fMLP was slightly reduced. Therefore, it seems that in the présence of PGE2,
a very unusual mode of activation of Akt is induced, characterized by a dissociation
between the translocation and the phosphorylation of Akt on Ser473 and Thr3O8.
In PMNs, Akt Ser473 is phosphorylated by MAPKAPK-2, a substrate of p38 that is
activated by phosphorylation on Thr222 and Thr334. The phosphorylation of Akt on
Ser473 by MAPKAPK-2, as well as the phosphorylation of p38 hâve been reported to be
inhibited by wortmannin and thus to dépend on PI3-K activation [15]. Nevertheless, we
observed that the phosphorylation of Akt on Ser473 is not affected when the fMLP-induced
PI3-Ky activity is inhibited by PGE2. The persistence of this phosphorylation is supported
by the évidence that the phosphorylation of p38 and MAPKAPK-2 stimulated by fMLP is
similarly not inhibited in the présence of PGE2. The interconnection between the MAPK
and the cAMP signaling pathways may vary considerably in a cell- and stimulus-dependent
fashion [27, 28]. In fMLP-stimulated PMNs, we observed no inhibitory effect of PGE2 on

160
the MAPK pathway and this resuit was confirmée! in our laboratory by the fact that other
cAMP elevating agents such as the A2A receptor agonist CGS-21680 did not affect the
phosphorylation of p38 in TNFa-primed and fMLP-stimulated PMNs [29], Even though
the phosphorylation of Akt by MAPKAPK-2 has been reported to dépend on
PtdIns(3,4,5)P3 formation [15], p38 and MAPKAPK-2 are cytosolic kinases that do not
possess PH domain and need not to relocate to the plasma membrane to be activated. We
could therefore hypothesize that the p38/MAPKAPK-2/Akt phosphorylation cascade could
occur in the cytosol in the présence of PGE2. An argument in favor of this hypothesis relies
on the observation that Ser473 phosphorylation dépends on PI3-K activity but in a fashion
that does not involve Akt membrane localization [30, 31].
The phosphoinositides generated by PI3-K brings Akt and PDK1 in close proximity at the
plasma membrane and thus allow Akt phosphorylation on Thr308 by PDK1 [22, 23, 32], In
this study, we report that although the formation of PtdIns(3,4,5)P3 is significantly
decreased and, in turn, the re-localization at the membrane of Akt and PDK1 is impaired in
the présence of PGE2, the phosphorylation of Akt on Thr308 is not affected. This suggests
that PGE2 renders the fMLP-stimulated activation of Akt independent of PI3-Ky and
promotes a différent pattern of activation in which Thr3O8 phosphorylation does not require
the translocation of PDK1 and Akt. Indeed, several alternative mechanisms of Akt
activation that are independent of PI3-K and PDK1 hâve been described [33, 34]. Amongst
them, PKA has been reported to stimulate Akt activity in a PI3-K-independent manner and
through a mechanism that results in Akt phosphorylation on Thr308. Since PGE2 stimulâtes
the cAMP/PKA pathway via EP2 receptors, we hypothesized that PKA could promote
Thr308 Akt phosphorylation. However, the PKA inhibitor H-89 had no effect on Akt
phosphorylation either on Thr308 or on Ser473. Therefore, PKA cannot be considered as a
mediator of Akt phosphorylation in our model. Nevertheless, we showed that the inhibition
of Akt translocation by PGE2 is mediated by PKA in fMLP-stimulated PMNs, as is the case
for pllOy (C.Burelout, unpublished data). Moreover, PKA is also involved in the slight
down-regulation of fMLP-stimulated Akt activity observed in the présence of PGE2. A
direct effect of PKA on Akt cannot be considerered as a mechanism of régulation since Akt
is not phosphorylated by PKA in vitro or in cells [20, 21]. Although PKA affects both

161
events (the translocation of Akt and the régulation of its activity), we observed that the
magnitude of the inhibitory effect was much higher on Akt translocation than on Akt
activity (approximately 50 % and 10-20 % of the fMLP-induced responses, respectively).
One possible rôle of PKA in the régulation of Akt activity by PGE2 could réside in its
capacity to inhibit PI3-Ky and Akt translocation and hence the binding of phosphoinositides
to the Akt PH domain since it has been reported that this binding could resuit in a
conformational change that influences Akt activity [35, 36]. The inhibition of Akt by the
cAMP/PKA pathway has already been reported elsewhere [37, 38], Thèse studies showed
that this inhibitory pathway affected upstream regulators of Akt, such as PI3-K and PDK1,
and resulted in a concomitant decrease in the levels of Akt phosphorylation and activity.
The inhibitory mechanism involved in our expérimental model is therefore very différent
since the inhibition of the translocation of Akt mediated by PKA is not coupled to the
inhibition of its phosphorylation.
Another putative kinase that could be involved is the Ca2+/calmodulin-dependent protein
kinase kinase (CaM-KK) that has been shown to promote cell survival by phosphorylating
Akt on Thr308 [34]. We cannot exclude that this signaling pathway could account, at least
in part, for Thr3O8 phosphorylation since 1) CaM-KK is expressed in human PMNs and the
calmodulin inhibitor W7 diminishes the phosphorylation of Akt on both Thr308 and Ser473
[39] and 2) PGE2 decreases only the late influx of calcium triggered by fMLP [16, 40]. Akt
activity stimulated by growth factors can also be regulated by tyrosine phosphorylation,
either on Tyr474 [41] or on Tyr315 by Src [42, 43]. Thèse tyrosine-phosphorylation events
hâve no major impact on the serine/threonine phosphorylation of Akt but require PI3-K
activation. A régulation of Akt activity by tyrosine phosphorylation is thus not likely to
take place in the présence of PGE2 since PI3-Ky is inhibited.
The fact that PGE2 suppressed the translocation of Akt induced by fMLP but not its
phosphorylation also suggests that Akt could be activated by PDK1 directly in the cytosol.
PDK1 is a cytosolic kinase that shows a high constitutive activity and is able to
phosphorylate most of its substrates (such as PKCs or p70S6K) in the cytosol
independently of PI3-K activity [44]. Therefore, we can envision a possible molecular
mechanism induced by PGE2 that would permit the formation of a signaling complex

162
enabling the interaction of Akt with PDK1 (or another as yet unidentified kinase) in the
cytosol. It has already been reported that in heat-shocked cells, Akt can be activated
through the formation of a protein complex in a PI3-K-independent manner [45]. Several
Akt binding partners or adaptor proteins that can modulate Akt or PDK1 activity hâve been
described [46]. Amongst them, TCL1, an oncoprotein that activâtes Akt by
oligomerization, the heat shock proteins Hsp27 and Hsp90 or the more recently discovered
ArgBP2y [46] are putative candidates that could be involved in a possible mechanism to be
explored in further détail.
Another point that needs to be clarified is the relative contribution of the différent PI3-K
isoforms to the lMLP-induced Akt activation pathway. The génération of PI3-Ky-deficient
mice has defined a prédominant rôle for this isoform in the polarization and the directional
migration of murine PMNs in a 1MLP gradient [47-49], Another conséquence of the
genetic deletion of PI3-Ky was the partial [50] or almost complète disappearance [47] of
the Ser473 Akt phosphorylation. In human PMNs, PI3-Ky (plOl/pl 10y) is rapidly activated
in response to fMLP [17] and the first burst of Ptdlns(3,4,5)P3 produced allows the
activation of the class la pi 108 that is associated with the p85 regulatory subunit [25]. The
récent development of pharmacologie inhibitors sélective for the various PI3-K isoforms
has brought expérimental évidence for the involvement of PI3-K.Ô in several functional
responses (ie chemotaxis, superoxide anions production, cytotoxic enzyme release)
stimulated with fMLP in human PMNs [26]. Our data obtained with the PI3-K.Ô sélective
inhibitor (IC87114) suggest that this isoform modulâtes only the late phosphorylation of
Akt (5 min of stimulation by fMLP) and could also partially account for the persistence of
Ser473 Akt phosphorylation (5 min) observed in the présence of PGE2. However, PI3-KÔ
does not seem to be involved either in the early steps of phosphorylation of Akt stimulated
by fMLP or in the maintenance of Akt phosphorylation in the présence of PGE2 (1 min of
stimulation by fMLP). Thèse data are consistent with the model proposed by Sadhu et al.
according to which PI3-K6 is involved in the amplification loop regulating the formation of
PtdIns(3,4,5)P3 but only in late signaling events. This latter resuit also suggests that the
alternative mechanism of Akt activation observed in the présence of PGE2 is mainly

163
independent of PI3-K activation. Several studies hâve reported that Akt activity can be
stimulated by a mechanism independent of PI3-K activation in stress-stimulated cells,
therefore strengthening the pertinence of our observations [45, 51].
It is of interest to note that PGE2 alone has been shown to induce the phosphorylation of
Akt in various cell-types including DCs [52-55], This increased phosphorylation of Akt
was always related to the activation of PI3-K and in some cases was mediated via EP2
receptors [54, 55], Nevertheless, thèse findings do not apply to our model since PGE2 alone
was not able to promote the phosphorylation of Akt in PMNs.
cAMP is the major second messenger formed after EP2 receptor stimulation. We thus
wondered whether Ado which stimulâtes cAMP formation via A2A receptors [56, 57] and
the adenylyl cyclase activator forskolin affected the PI3-Ky/Akt pathway in a manner
similar to PGE2. Our data indicate that the élévation of intracellular cAMP induced by
various stimuli inhibited the translocation of pliOy as well as Akt and PDK1 to
membranes. However, the phosphorylation of Akt was not suppressed in the présence of
thèse cAMP-elevating agents. Therefore, thèse results suggest that the élévation of
intracellular cAMP promûtes a mechanism of activation of Akt that does not require a
translocation step in fMLP-stimulated PMNs. However, as is the case with PGE2, the A2A
agonist slightly reduced Akt activity, further suggesting a common mechanism of
activation of Akt mediated through EP2 or A2A receptors.
A physiological rôle for the persistence of Akt phosphorylation in the présence of PGE2
remains to be found. On the one hand, cAMP-elevating agents delay apoptosis in PMNs [7,
58, 59]. On the other hand, the anti-apoptotic rôle of Akt in various cellular types is well
established. Therefore, it is tempting to speculate that the anti-apoptotic function of PGE2
and other cAMP elevating agents could be mediated through the alternative mode of Akt
activation reported in this study. Nevertheless, such a conclusion could be erroneous since
1) fMLP (or IFN-P) stimulâtes Akt activity without delaying apoptosis in PMNs, indicating

164
that the fMLP-induced activation of Akt is not linked to a pro-survival response [60] and
2) we hâve verified that PGE2 did not significantly affect cell viability when PMNs were
stimulated with fMLP (unpublished data).
Amongst the known Akt substrates, p47phox (a NADPH complex component) [14, 61],
GSK-3 [62] and the FOXO transcription factors [63] hâve been shown to be
phosphorylated in response to fMLP in PMNs. Using a spécifie Akt inhibitor, we found
that GSK-3 is not a substrate of Akt in fMLP-stimulated PMNs (data not shown). Since Akt
activity stimulated by fMLP seems to be primarily involved in the respiratory burst activity
and in chemotaxis in PMNs [64], the exposure of cells to PGE2 or Ado could help maintain
thèse cellular fonctions at a minimal level.
In summary, our data show that although PtdIns(3,4,5)P3 formation by fMLP-activated PI3-
Ky is decreased in the présence of PGE2 or other cAMP-elevating agents, Akt
phosphorylation is not affected, whereas its activity is slightly down-regulated. In this
expérimental model, fMLP-induced Akt phosphorylation does not require its translocation
to membranes, thereby suggesting that cAMP-elevating agents alter compartmentalization
of fMLP-induced Akt signaling. Taken together, thèse observations lead us to hypothesize
that cAMP-elevating agents induce an alternative mechanism for Akt activation in PMNs
stimulated with fMLP. This mechanism is schematically represented in Figure 9 and needs
further characterization.

165
4.8. AcknowledgementsWe are grateful to Nicolas Flamand, PhD and Caroline Gilbert, PhD for their helpful
comments.
This work was supported by grants from the Canadian Institutes of Health Research
(CIHR).
We thank Dr. Tom Crabbe from UCB (Slough, UK) for kindly supplying the PI3KÔ
sélective inhibitor IC87114.
4.9. AbbreviationsAdo, adenosine; cAMP, cyclic AMP; ADA, adenosine deaminase; CB, cytochalasin B;
COX, cyclooxygenase; DC, dendritic cells; fMLP, N-formyl-methionyl-leucyl-
phenylalanine; GSK-3, glycogen synthase kinase-3; MAPK, mitogen-activated protein
kinase; MAPKAPK-2, mitogen-activated protein kinase-activated protein kinase-2; PDK1,
3-phosphoinositide-dependent kinase-1; PI3-K, phosphatidylinositol 3-kinase; PH,
pleckstrin homology; PKA, cAMP-dependent protein kinase; PtdIns(3,4,5)P3,
phosphatidylinositol 3,4,5- triphosphate; PtdIns(3,4)P2, phosphatidylinositol 3,4-
diphosphate; PLD, phospholipase D; PMN, polymorphonuclear neutrophil.

166
4.10. Bibliography
1. Rocca, B., FitzGerald, G.A. (2002) Cyclooxygenases and prostaglandins: shaping
up the immune response. Int Immunopharmacol 2, 603-30.
2. Rivkin, L, Rosenblatt, J., Becker, E.L. (1975) The rôle of cyclic AMP in the
chemotactic responsiveness and spontaneous motility of rabbit peritoneal neutrophil s. The
inhibition of neutrophil movement and the élévation of cyclic AMP levels by
catecholamines, prostaglandins, theophylline and choiera toxin. JImmunol 115, 1126-34.
3. Fantone, J.C., Kinnes, D.A. (1983) Prostaglandin El and prostaglandin 12
modulation of superoxide production by human neutrophils. Biochem Biophys Res
Commun 113, 506-12.
4. Ham, E.A., Soderman, D.D., Zanetti, M.E., Dougherty, H.W., McCauley, E., Kuehl,
F.A., Jr. (1983) Inhibition by prostaglandins of leukotriene B4 release from activated
neutrophils. Proc Natl Acad Sci USA 80, 4349-53.
5. Hecker, G., Ney, P., Schror, K. (1990) Cytotoxic enzyme release and oxygen
centered radical formation in human neutrophils are selectively inhibited by E-type
prostaglandins but not by PGI2. Naunyn Schmiedebergs Arch Pharmacol 341, 308-15.
6. Ottonello, L., Gonella, R., Dapino, P., Sacchetti, C , Dallegri, F. (1998)
Prostaglandin E2 inhibits apoptosis in human neutrophilic polymorphonuclear leukocytes:
rôle of intracellular cyclic AMP levels. Exp Hematol 26, 895-902.
7. Martin, M.C., Dransfield, L, Haslett, C, Rossi, A.G. (2001) Cyclic AMP régulation
of neutrophil apoptosis occurs via a novel protein kinase A-independent signaling pathway.
JBiolChem 276, 45041-50.
8. Breyer, R.M., Bagdassarian, C.K., Myers, S.A., Breyer, M.D. (2001) Prostanoid
receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41, 661-90.

167
9. Armstrong, R.A. (1995) Investigation of the inhibitory effects of PGE2 and
sélective EP agonists on chemotaxis of human neutrophils. Br JPharmacol 116, 2903-8.
10. Talpain, E., Armstrong, R.A., Coleman, R.A., Vardey, C.J. (1995) Characterization
of the PGE receptor subtype mediating inhibition of superoxide production in human
neutrophils. Br J Pharmacol 114, 1459-65.
11. Hanada, M., Feng, J., Hemmings, B.A. (2004) Structure, régulation and function of
PKB/AKT~a major therapeutic target. Biochim Biophys Acta 1697, 3-16.
12. Song, G., Ouyang, G., Bao, S. (2005) The activation of Akt/PKB signaling pathway
and cell survival. J Cell Mol Med 9, 59-71.
13. Tilton, B., Andjelkovic, M., Didichenko, S.A., Hemmings, B.A., Thelen, M. (1997)
G-Protein-coupled receptors and Fcgamma-receptors médiate activation of Akt/protein
kinase B in human phagocytes. J Biol Chem 272, 28096-101.
14. Chen, Q., Powell, D., Rane, M., Singh, S., Butt, W., Klein, J., McLeish, K. (2003)
Akt phosphorylates p47(phox) and médiates respiratory burst activity in human neutrophils.
Jlmmunol 170, 5302-5308.
15. Rane, M.J., Coxon, P.Y., Powell, D.W., Webster, R., Klein, J.B., Pierce, W., Ping,
P., McLeish, K.R. (2001) p38 Kinase-dependent MAPKAPK-2 activation functions as 3-
phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem 276 ,
3517-23.
16. Burelout, C, Thibault, N., Levasseur, S., Simard, S., Naccache, P., Bourgoin, S.
(2004) Prostaglandin E-2 inhibits the phospholipase D pathway stimulated by formyl-
methionyl-leucyl-phenylalanine in human neutrophils. Involvement of EP2 receptors and
phosphatidylinositol 3-kinase gamma. Mol Pharmacol 66, 293 - 301.
17. Naccache, P.H., Levasseur, S., Lachance, G., Chakravarti, S., Bourgoin, S.G.,
McColl, S.R. (2000) Stimulation of human neutrophils by chemotactic factors is associated
with the activation of phosphatidylinositol 3-kinase gamma. J Biol Chem 275, 23636-41.

168
18. Mardi, J., Harbour, D., Houle, M.G., Naccache, P.H., Bourgoin, S. (1999)
Monosodium urate-crystal-stimulated phospholipase D in human neutrophils. Biochem J
337 (Pt 2), 185-92.
19. Regan, J.W., Bailey, T.J., Pepperl, D.J., Pierce, K.L., Bogardus, A.M., Donello,
J.E., Fairbairn, CE., Kedzie, K.M., Woodward, D.F., Gil, D.W. (1994) Cloning of a novel
human prostaglandin receptor with characteristics of the pharmacologically defined EP2
subtype. Mol Pharmacol 46, 213-20.
20. Sable, CL., Filippa, N., Hemmings, B., Van Obberghen, E. (1997) cAMP
stimulâtes protein kinase B in a Wortmannin-insensitive manner. FEBS Lett 409, 253-7.
21. Filippa, N., Sable, CL., Filloux, C, Hemmings, B., Van Obberghen, E. (1999)
Mechanism of protein kinase B activation by cyclic AMP-dependent protein kinase. Mol
Cell Biol 19, 4989-5000.
22. Anderson, K.E., Coadwell, J., Stephens, L.R., Hawkins, P.T. (1998) Translocation
of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein
kinase B. Curr Biol 8, 684-91.
23. Andjelkovic, M., Alessi, D.R., Meier, R., Fernandez, A., Lamb, N.J., Frech, M.,
Cron, P., Cohen, P., Lucocq, J.M., Hemmings, B.A. (1997) Rôle of translocation in the
activation and function of protein kinase B. J Biol Chem 272, 31515-24.
24. Thibault, N., Harbour, D., Borgeat, P., Naccache, P.H., Bourgoin, S.G. (2000)
Adenosine receptor occupancy suppresses chemoattractant-induced phospholipase D
activity by diminishing membrane recruitment of small GTPases. Blood 95, 519-27.
25. Sadhu, C, Masinovsky, B., Dick, K., Sowell, C.G., Staunton, D.E. (2003) Essential
rôle of phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol 170,
2647-54.
26. Sadhu, C, Dick, K., Tino, W.T., Staunton, D.E. (2003) Sélective rôle of PI3K delta
in neutrophil inflammatory responses. Biochem Biophys Res Commun 308, 764-9.

169
27. Houslay, M.D., Kolch, W. (2000) Cell-type spécifie intégration of cross-talk
between extracellular signal-regulated kinase and cAMP signaling. Mol Pharmacol 58,
659-68.
28. Stork, P.J., Schmitt, J.M. (2002) Crosstalk between cAMP and MAP kinase
signaling in the régulation of cell prolifération. Trends Cell Biol 12, 258-66.
29. Flamand, N., Lefebvre, J., Lapointe, G., Picard, S., Lemieux, L., Bourgoin, S.G.,
Borgeat, P. (2006) Inhibition of platelet-activating factor biosynthesis by adenosine and
histamine in human neutrophils: involvement of cPLA2{alpha} and reversai by lyso-PAF.
JLeukoc Biol79, 1043-51.
30. Andjelkovic, M., Maira, S.M., Cron, P., Parker, P.J., Hemmings, B.A. (1999)
Domain swapping used to investigate the mechanism of protein kinase B régulation by 3-
phosphoinositide-dependent protein kinase 1 and Ser473 kinase. Mol Cell Biol 19, 5061-72.
31. Scheid, M.P., Marignani, P.A., Woodgett, J.R. (2002) Multiple phosphoinositide 3-
kinase-dependent steps in activation of protein kinase B. Mol Cell Biol 22, 6247-60.
32. Currie, R.A., Walker, K.S., Gray, A., Deak, M., Casamayor, A., Downes, C.P.,
Cohen, P., Alessi, D.R., Lucocq, J. (1999) Rôle of phosphatidylinositol 3,4,5-trisphosphate
in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-
\.BiochemJ331(?t 3), 575-83.
33. Franke, T.F., Hornik, C.P., Segev, L., Shostak, G.A., Sugimoto, C. (2003) PI3K/Akt
and apoptosis: size matters. Oncogene 22, 8983-98.
34. Yano, S., Tokumitsu, H., Soderling, T.R. (1998) Calcium promûtes cell survival
through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396, 584-7.
35. Frech, M., Andjelkovic, M., Ingley, E., Reddy, K.K., Falck, J.R., Hemmings, B.A.
(1997) High affmity binding of inositol phosphates and phosphoinositides to the pleckstrin
homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol
Chem 272, 8474-81.

170
36. Milburn, C.C., Deak, M., Kelly, S.M., Price, N.C., Alessi, D.R., Van Aalten, D.M.
(2003) Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology
domain of protein kinase B induces a conformational change. Biochem J375, 531-8.
37. Kim, S., Jee, K., Kim, D., Koh, H., Chung, J. (2001) Cyclic AMP inhibits Akt
activity by blocking the membrane localization of PDK1. JBiol Chem 276, 12864-70.
38. Mei, F.C., Qiao, J., Tsygankova, O.M., Meinkoth, J.L., Quilliam, L.A., Cheng, X.
(2002) Differential signaling of cyclic AMP: opposing effects of exchange protein directly
activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B
activation. J Biol Chem 277, 11497-504.
39. Verploegen, S., van, L.C., van, D.H., Lammers, J., Koenderman, L., Coffer, P.
(2002) Rôle of Ca2+/calmodulin regulated signaling pathways in chemoattractant induced
neutrophil effector functions - Comparison with the rôle of phosphotidylinositol-3 kinase.
EurJ Biochem 269, 4625 - 4634.
40. Ahmed, M.U., Hazeki, K., Hazeki, O., Katada, T., Ui, M. (1995) Cyclic AMP-
increasing agents interfère with chemoattractant-induced respiratory burst in neutrophils as
a resuit of the inhibition of phosphatidylinositol 3-kinase rather than receptor-operated
Ca2+ influx. J Biol Chem 270, 23816-22.
41. Conus, N.M., Hannan, K.M., Cristiano, B.E., Hemmings, B.A., Pearson, R.B.
(2002) Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt
protein kinase. J Biol Chem 277, 38021-8.
42. Jiang, T., Qiu, Y. (2003) Interaction between Src and a C-terminal proline-rich
motif of Akt is required for Akt activation. J Biol Chem 278, 15789 - 15793.
43. Chen, R., Kim, O., Yang, J., Sato, K., Eisenmann, K.M., McCarthy, J., Chen, H.,
Qiu, Y. (2001) Régulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem
276,31858-62.
44. Storz, P., Toker, A. (2002) 3'-phosphoinositide-dependent kinase-1 (PDK-1) in PI 3-
kinase signaling. Front Biosci 7, d886-902.

171
45. Konishi, H., Fujiyoshi, T., Fukui, Y., Matsuzaki, H., Yamamoto, T., Ono, Y.,
Andjelkovic, M., Hemmings, B.A., Kikkawa, U. (1999) Activation of protein kinase B
induced by H(2)O(2) and heat shock through distinct mechanisms dépendent and
independent of phosphatidylinositol 3-kinase. J Biochem (Tokyo) 126, 1136-43.
46. Yuan, Z.Q., Kim, D., Kaneko, S., Sussman, M., Bokoch, G.M., Nicosia, S.V.,
Cheng, J.Q. (2005) ArgBP2gamma interacts with Akt and p21-activated kinase-1 and
promotes cell survival. J Biol Chem 280, 21483-90.
47. Hirsch, E., Katanaev, V.L., Garlanda, C, Azzolino, O., Pirola, L., Silengo, L.,
Sozzani, S., Mantovani, A., Altruda, F., Wymann, M.P. (2000) Central rôle for G protein-
coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049-53.
48. Li, Z., Jiang, H., Xie, W., Zhang, Z., Smrcka, A.V., Wu, D. (2000) Rôles of PLC-
beta2 and -beta3 and PDKgamma in chemoattractant-mediated signal transduction. Science
287, 1046-9.
49. Hannigan, M., Zhan, L., Li, Z., Ai, Y., Wu, D., Huang, C.K. (2002) Neutrophils
lacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-
Met-Leu-Phe-induced chemotaxis. Proc Natl AcadSci USA 99, 3603-8.
50. Yang, K., Arcaroli, J., Kupfner, J., Pitts, T., Park, J., Strasshiem, D., Perng, R.,
Abraham, E. (2003) Involvement of phosphatidylinositol 3-kinase gamma in neutrophil
apoptosis. Cell Signal 5, 225 - 233.
51. Konishi, H., Matsuzaki, H., Tanaka, M., Ono, Y., Tokunaga, C, Kuroda, S.,
Kikkawa, U. (1996) Activation of RAC-protein kinase by heat shock and hyperosmolarity
stress through a pathway independent of phosphatidylinositol 3-kinase. Proc Natl Acad Sci
USA 93,7639-43.
52. Buchanan, F.G., Wang, D., Bargiacchi, F., DuBois, R.N. (2003) Prostaglandin E2
régulâtes cell migration via the intracellular activation of the epidermal growth factor
receptor. J Biol Chem 278, 35451-7.

172
53. Jing, H., Yen, J.H., Ganea, D. (2004) A novel signaling pathway médiates the
inhibition of CCL3/4 expression by prostaglandin E2. JBiol Chem 279, 55176-86.
54. Tessner, T.G., Muhale, F., Riehl, T.E., Anant, S., Stenson, W.F. (2004)
Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism
involving AKT activation and bax translocation. J Clin Invest 114, 1676-85.
55. Vassiliou, E., Sharma, V., Jing, IL, Sheibanie, F., Ganea, D. (2004) Prostaglandin
E2 promotes the survival of bone marrow-derived dendritic cells. J Immunol 173, 6955-64.
56. Fredholm, B.B., Abbracchio, M.P., Burnstock, G., Daly, J.W., Harden, T.K.,
Jacobson, K.A., Leff, P., Williams, M. (1994) Nomenclature and classification of
purinoceptors. Pharmacol Rev 46, 143-56.
57. Thibault, N., Burelout, C, Harbour, D., Borgeat, P., Naccache, P.H., Bourgoin, S.G.
(2002) Occupancy of adenosine A2a receptors promotes fMLP-induced cyclic AMP
accumulation in human neutrophils: impact on phospholipase D activity and recruitment of
small GTPases to membranes. JLeukoc Biol 71, 367-377.
58. Rossi, A.G., Cousin, J.M., Dransfield, I., Lawson, M.F., Chilvers, E.R., Haslett, C.
(1995) Agents that elevate cAMP inhibit human neutrophil apoptosis. Biochem Biophys
Res Commun 217, 892-9.
59. Parvathenani, L.K., Buescher, E.S., Chacon-Cruz, E., Beebe, S.J. (1998) Type I
cAMP-dependent protein kinase delays apoptosis in human neutrophils at a site upstream
of caspase-3. J Biol Chem 273, 6736-43.
60. Scheel-Toellner, D., Wang, K., Henriquez, N.V., Webb, P.R., Craddock, R., Pilling,
D., Akbar, A.N., Salmon, M., Lord, J.M. (2002) Cytokine-mediated inhibition of apoptosis
in non-transformed T cells and neutrophils can be dissociated from protein kinase B
activation. Eur J Immunol 32, 486-93.
61. Hoyal, C, Gutierrez, A., Young, B., Catz, S., Lin, J., Tsichlis, P., Babior, B. (2003)
Modulation of p47(PHOX) activity by site-specific phosphorylation: Akt-dependent
activation of the NADPH oxidase. Proc NatlAcadSci USA 100, 5130-5135.

173
62. De Mesquita, D.D., Zhan, Q., Crossley, L., Badwey, J.A. (2001) p90-RSK and Akt
may promote rapid phosphorylation/inactivation of glycogen synthase kinase 3 in
chemoattractant-stimulated neutrophils. FEBS Lett 502, 84-8.
63. Crossley, L.J. (2003) Neutrophil activation by fMLP régulâtes FOXO (forkhead)
transcription factors by multiple pathways, one of which includes the binding of FOXO to
the survival factor Mcl-1. J Leukoc Biol 74, 583-92.
64. Chen, Q., Powell, D.W., Rane, M.J., Singh, S., Butt, W., Klein, J.B., McLeish, K.R.
(2003) Akt phosphorylates p47phox and médiates respiratory burst activity in human
neutrophils. JImmunol 170, 5302-8.

174
CHAPITRE V
CONCLUSION

175
La réaction inflammatoire est une réaction de défense de l'organisme qui doit être finement
régulée par différents médiateurs pour aboutir normalement à une phase de résolution et à
la réparation des tissus lésés. L'infiltration prolongée de neutrophiles dans les tissus
associée à la libération massive d'enzymes cytotoxiques et de RLO par ces neutrophiles
activés peut en effet provoquer des dégâts tissulaires irréversibles entraînant une perte de
fonction que l'on observe dans des pathologies chroniques comme l'arthrite rhumatoïde
(412, 413). Le rôle physiologique du processus de résolution est donc d'éviter l'évolution
de la réaction inflammatoire vers la chronicité. L'élévation des niveaux intracellulaires
d'AMPc par des médiateurs endogènes produits au cours de la réaction inflammatoire
constitue le principal mécanisme physiologique permettant d'atténuer l'activation des
neutrophiles in vivo (414). Il est donc important de mieux comprendre les mécanismes de
signalisation intracellulaire impliqués dans l'inhibition des fonctions du neutrophile par les
principaux autacoïdes physiologiques qui élèvent les [AMPc]i, c'est-à-dire l'adénosine et
PGE2.
Les travaux présentés dans cette étude montrent que PGE2 réduit l'accumulation de
PtdIns(3,4,5)P3, un des premiers seconds messagers intracellulaires produit par les
neutrophiles en réponse aux agents chimiotactiques, dont le fMLP. La diminution de
l'accumulation de PtdIns(3,4,5)P3 est due à l'inhibition par PGE2 du mécanisme
d'activation de la PI3-Ky, la première isoforme de PI3-K activée par le fMLP dans le
neutrophile. Les deux autres événements précoces de signalisation stimulés par le fMLP,
c'est-à-dire la mobilisation de calcium et l'activation des tyrosine kinases sont peu affectés
par PGE2, mais nous avons néanmoins observé une diminution de l'influx tardif de calcium
extracellulaire ainsi que la diminution de la phosphorylation sur des résidus tyrosine d'une
série de substrats aux alentours de 116-120 kDa. La principale conséquence de la réduction
de l'accumulation de PtdIns(3,4,5)P3 est la diminution du recrutement à la membrane des
principaux effecteurs de la PI3-K qui sont les kinases de la famille Tec, les kinases Akt et
PDK1 et les petites GTPases des familles Rho et Arf par l'intermédiaire de leurs facteurs
d'échange dont l'activation dépend en grande partie de l'activité de la PI3-K. Le
recrutement à la membrane de la PKCa qui est une étape nécessaire pour son activation est
aussi fortement diminué suite à la réduction de la formation de PtdIns(3,4,5)P3 en présence
de PGE2. Finalement, la diminution du recrutement à la membrane des trois cofacteurs

176
d'activation de la PLD que nous avons observée en présence de PGE2 explique bien
l'inhibition de l'activité de la PLD stimulée par le fMLP qui avait été rapportée
précédemment et qui constituait le point de départ de notre étude (410, 411). Nos résultats
peuvent être comparés à ceux d'une étude publiée précédemment par notre laboratoire qui
montrait que l'inhibition par l'adénosine de l'activité de la PLD stimulée par le fMLP
résultait d'une diminution du recrutement à la membrane de RhoA, Arf et de la PKCoc
(415). Les résultats présentés dans le Chapitre III permettent d'établir que, de façon
similaire à PGE2, l'adénosine endogène, via le récepteur A2A, inhibe le recrutement à la
membrane de la PI3-Ky induit par le fMLP. Ceci montre que ces deux autacoïdes régulent
l'activation du neutrophile par un mécanisme similaire. Nos résultats nous ont conduits à
proposer un modèle de mécanisme expliquant l'effet inhibiteur de PGE2 sur les voies de
signalisation stimulées par le fMLP dans neutrophile (Chapitre II). Selon ce modèle,
l'inhibition de l'activité de la PI3-Ky, une enzyme ayant un rôle majeur dans les voies de
signalisation stimulées par le fMLP, constitue l'événement essentiel du mécanisme
inhibiteur de PGE2 et explique bien l'inhibition des réponses fonctionnelles du neutrophile
au fMLP observées en présence de PGE2 puisque la PI3-Ky joue un rôle fondamental dans
l'adhésion et surtout le recrutement des neutrophiles vers le site inflammatoire, ainsi que
dans la production de RLO (416).
En utilisant des agonistes sélectifs des différents sous-types de récepteurs EP, nous avons
montré que l'effet inhibiteur de PGE2 est transmis par l'activation des récepteurs EP2. C'est
d'ailleurs le seul récepteur EP que nous avons trouvé exprimé sur la surface des
neutrophiles non stimulés. Le récepteur EP2 a été clone et il est couplé aux protéines Gocs
qui stimulent l'activité des adénylate-cyclases (417). Une augmentation des l'[AMPc]i
modeste mais soutenue dans le temps a été observée lorsque les neutrophiles sont exposés à
PGE2 (ou PGEi) seule (327, 335). Une pré-exposition des neutrophiles à PGEi ou PGE2 a
pour effet d'augmenter et de prolonger nettement la formation d'AMPc stimulée par le
fMLP (324, 325, 403, 405, 411, 418) par un mécanisme qui n'a pas été encore élucidé,
mais il semble que les deux agonistes (fMLP et PGE1/2) n'activent pas les mêmes isoformes
d'AC (327), les isoformes activées par PGE2 étant localisées à la membrane et ne
dépendant pas du calcium. Le principal effecteur de l'AMPc dans les cellules étant la PKA,

177
nous avons cherché à définir le rôle de cette kinase dans le processus inhibiteur activé par
PGE2 via les récepteurs EP2 dans les neutrophiles stimulés par le fMLP. Nous avons
observé que la PKA est impliquée dans l'inhibition par PGE2 de tous les composants des
voies de signalisation stimulées par le fMLP que nous avons étudiés, c'est-à-dire l'activité
de la PLD, la translocation à la membrane des petites GTPases Rho et Arf, de la PKC et
d'Akt, l'influx de calcium et les phosphorylations sur des résidus tyrosines (chapitre II). La
PKA est de plus impliquée dans l'inhibition de la translocation de la pllOy par PGE2.
Cependant, nous avons montré que ni la pi 01 ni la pi lOy ne sont des substrats possibles de
la PKA. Etant donné que la PI3-Ky est située en amont des petites GTPases Rho et Arf et
de la PLD, il semble logique que les différents effecteurs qu'elle régule soient inhibés par
PGE2 d'une façon qui dépend de la PKA. Ces résultats ont donc confirmé le modèle que
nous avons proposé au chapitre II selon lequel l'activation de la PI3-Ky est la cible majeure
de PGE2 dans les neutrophiles activés par le fMLP. Le mécanisme de l'inhibition de
l'activation de la PI3-Ky par la PKA devra cependant être exploré plus en détail et le (ou
les) substrat(s) de la PKA restent à être identifiés. Etant donné l'amplitude de l'inhibition
de la translocation de la PI3-Ky par PGE2, on pourrait envisager que PGE2 induit, via la
PKA, la formation d'un complexe moléculaire qui empêcherait le recrutement de PI3-Ky
vers les sous-unités G(3y. Depuis quelques années, le concept de la compartimentalisation
de la signalisation associée à la PKA a émergé grâce à la découverte des AKAP qui sont
des protéines liant la PKA, ses substrats ainsi que d'autres kinases ou des phosphatases.
Ces AKAP jouent un rôle important dans l'organisation spatio-temporale des signaux
intracellulaires constituent des plateformes permettant d'intégrer plusieurs voies de
signalisation ainsi que de concentrer l'activité de la PKA dans un compartiment cellulaire
précis (419-421). Par exemple, l'association PKA-AKAP régule l'activation des
lymphocytes T (422), la signalisation induite par des RCPG comme le récepteur pV
adrénergique (423), le cytosquelette (424) ainsi que le métabolisme de l'AMPc par les
PDEs (425). Certaines AKAP peuvent être elles-mêmes le substrat de la PKA, comme par
exemple l'AKAP-Lbc qui est aussi un facteur d'échange pour les Rho-GTPases. La
phosphorylation de cette AKAP sur un résidu serine (Serl565) par la PKA promeut le
recrutement des protéines 14-3-3, ce qui a pour conséquence d'inhiber l'activité d'échange
de AKAP-Lbc (426). Les protéines 14-3-3 sont des petits dimères ayant la propriété de lier

178
des motifs phosphorylés sur des résidus serine ou thréonine, bien que des interactions avec
des protéines non phosphorylées aient aussi été décrites (427). Comme les AKAP, les 14-3-
3 constituent une base pour la formation de complexes multimoléculaires qui permettent
d'intégrer différentes voies de signalisation. Or la pi01 possède deux motifs théoriques de
liaison avec les 14-3-3, dont un (Rxx-Thrl 14-xP) qui est situé en N-terminal, dans le
domaine de liaison avec les sous-unités Gp"y. On pourrait donc émettre l'hypothèse d'une
connection entre la voie de l'AMPc/PKA et celle de la PI3-Ky par l'intermédiaire de la
formation d'un complexe PKA-AKAP-14-3-3-PI3-Ky qui empêcherait l'association de la
PI3-Ky avec les sous-unités GPy et qui expliquerait l'inhibition du recrutement de la PI3-Ky
observée en présence de PGE2 ou d'adénosine. En perspective, il serait intéressant de
rechercher quelles sont les protéines associées à la sous-unité catalytique de la PKA, en
particulier les AKAP, afin de préciser le mécanisme inhibiteur de PGE2 et de l'adénosine
sur l'activité de la PI3-Ky stimulée par le fMLP. Une étude protéomique permettrait aussi
d'identifier les substrats de la PKA phosphorylés sur des résidus serine ou thréonine en
présence de PGE2 ou d'adénosine et pourrait aussi apporter de nouveaux éléments utiles à
la compréhension du mécanisme inhibiteur de ces agonistes.
Une des principales cibles effectrices de la PI3-K dans les cellules est la sérine/thréonine
kinase Akt puisqu'elle possède un domaine PH de haute affinité pour le PtdIns(3,4,5)P3 et
le PtdIns(3,4)P2. Il a été clairement établi que l'accumulation de PtdIns(3,4,5)P3 généré par
les PI3-K est primordiale pour l'activation d'Akt via les deux phosphorylations activatrices
sur la Thr308 et la Ser473, si bien que ces deux phophorylations d'Akt sont habituellement
considérées comme des indices de l'activité de la PI3-K dans les cellules. De plus, la
phosphorylation d'Akt sur la Ser473 stimulée par le fMLP est presque totalement abolie
dans les neutrophiles de souris déficientes en PI3-Ky, ce qui indique un rôle prépondérant
pour cette isoforme en amont de l'activation d'Akt par le fMLP (151, 152). Il était donc
attendu que la phosphorylation d'Akt stimulée par le fMLP serait fortement diminuée en
présence de PGE2 en raison de la diminution de la formation de PtdIns(3,4,5)P3 que nous
avions observée. Or nous avons montré que, de façon surprenante, l'élévation d'AMPc
induite par PGE2, l'adénosine ou d'autres agents pharmacologiques ne modifie pas la
phosphorylation d'Akt alors que ces agonistes inhibent tous le recrutement de la PI3-Ky à

179
la membrane stimulé par le fMLP (Chapitre IV). Cependant, le recrutement à la membrane
d'Akt et de PDK1 qui phosphoryle Akt sur la Thr308 est nettement diminué en présence
des agents qui élèvent les niveaux intracellulaires d'AMPc, et cette diminution implique
l'activité de la PKA. De plus, l'activité d'Akt stimulée par le fMLP est légèrement
diminuée sans être franchement inhibée en présence de PGE2 ou d'adénosine, et ceci d'une
façon qui dépend de la PKA. Ces observations expérimentales indiquent que le modèle
classique d'activation d'Akt ne s'applique pas dans le cadre de notre modèle expérimental
et suggèrent qu'un mécanisme alternatif d'activation d'Akt par le fMLP est induit en
présence d'agents qui élèvent les niveaux intracellulaires d'AMPc. Nos résultats soulèvent
donc plusieurs questions sur ce mécanisme alternatif qui reste à préciser. Il serait d'abord
intéressant de savoir si la phosphorylation d'Akt sur la Thr308 est bien assurée par PDK1
et, du fait de la réduction de la translocation des ces deux kinases vers la membrane, il
faudrait vérifier que cette phosphorylation a bien lieu dans le cytosol, peut-être grâce à la
formation de complexes moléculaires avec d'autres protéines induite spécifiquement par
l'élévation d'AMPc. D'autre part, nous avons montré que la voie p38-MAPKAPK-2 qui
stimule la phosphorylation d'Akt sur la Ser473 n'est pas inhibée en présence de PGE2
malgré le lien de dépendance qui existe entre la voie des MAPK et l'activité PI3-K dans les
neutrophiles stimulés par le fMLP (171). Finalement, le rôle physiologique de ce
mécanisme alternatif d'activation d'Akt en présence de PGE2 ou d'adénosine reste à
préciser dans le contexte de la réaction inflammatoire. Nous avons confirmé que le fMLP
n'a pas d'effet notable sur l'apoptose des neutrophiles et que l'activation d'Akt par ce
facteur chimiotactique module principalement la régulation de la production des RLO et de
la chimiotaxie, mais pas dans l'inhibition de l'apoptose comme c'est le cas lors de
l'activation d'Akt par d'autres agonistes comme le GM-CSF. Il est donc donc possible que
l'effet majeur de l'absence d'inhibition de l'activité d'Akt en présence de PGE2 ou
d'adénosine est de maintenir ces fonctions du neutrophile à un niveau sub-optimal sans les
inhiber totalement.
La principale contribution de nos travaux expérimentaux est d'avoir montré que l'activation
de la voie de l'AMPc/PKA par PGE2 ou l'adénosine régule négativement les fonctions du
neutrophile activées par le fMLP par un mécanisme de signalisation qui inhibe l'activation
de la PI3-Ky et des ses effecteurs, excepté Akt. Il serait intéressant de mieux caractériser les

180
étapes de la réaction inflammatoire ciblées par ces agonistes au niveau physiologique. En
effet, deux études ont montré que l'élévation d'AMPc par PGE2 ou d'autres agents inhibe
les fonctions des neutrophiles circulants mais pas celles des neutrophiles infiltrés dans les
tissus, qui deviennent beaucoup moins sensibles à l'effet inhibiteur de PGE2 (428, 429). Par
ailleurs, l'exposition des neutrophiles à PGE2 perturbe la polymérisation de l'actine et
empêche l'acquisition d'une certaine rigidité du cytosquelette nécessaire à leur migration
(335). Il semble donc que ces substances modulent la réaction inflammatoire en limitant la
migration des neutrophiles circulants vers les tissus inflammés et en diminuant en même
temps leur capacité de libérer des substances toxiques comme les RLO ou les enzymes
lysosomiales. Cet effet immédiat sur les fonctions des neutrophiles s'ajoute aux effets
inhibiteurs de PGE2 au niveau de la transcription de cytokines et de chimiokines qui
agissent à plus long terme sur l'évolution de la réaction inflammatoire. En effet, PGE2
inhibe la synthèse de deux cytokines pro-inflammatoires majeures, le TNFa et l'IL-ip,
dans différents types cellulaires impliqués dans l'inflammation tels que les
monocytes/macrophages (430-432), les neutrophiles (433) ou les cellules microgliales du
cerveau (434) alors qu'elle augmente la synthèse de la cytokine anti-inflammatoire IL-10
dans les synoviocytes (435), les macrophages (431) et les cellules dendritiques (436). De
plus, PGE2 inhibe l'expression de chimiokines pro-inflammatoires telles que MCP-1 dans
les fibroblastes synoviaux (437), MlP-la (CCL3) et MIP-lp (CCL4) dans les cellules
dendritiques (438, 439) ou l'IL-8 dans les neutrophiles activés par le zymosan (440 ).
Finalement, les effets inhibiteurs de PGE2 et de l'adénosine sur l'activation de la PI3-Ky
s'ajoutent également à l'inhibition par ces médiateurs de la biosynthèse de médiateurs
lipidiques exerçant un fort effet chimiotactique sur les neutrophiles tels que les LTB4 et le
PAF(348, 367, 441).
Dans cette étude, nous nous sommes attachés à caractériser les effets inhibiteurs de PGE2
sur des neutrophiles en suspension activés par une concentration uniforme d'agent
chimiotactique, le fMLP. PGE2 inhibe également, par l'intermédiaire des récepteurs EP2, la
réponse chimiotactique des neutrophiles dans un gradient de fMLP, mais cependant, l'effet
inhibiteur de PGE2 dans ce type de modèle expérimental ne semble pas dépendre de la voie
AMPc/PKA (409). Une des suites de nos travaux consisterait donc à étudier l'effet

181
inhibiteur de PGE2 dans un modèle expérimental de chimiotaxie et à vérifier, par des
méthodes de microscopie à fluorescence, si la production de PtdIns(3,4,5)P3 par la PI3-Ky
au niveau du front de migration est également inhibée dans ce type de modèle et par quel
mécanisme.
Par ailleurs, PGE2 peut aussi inhiber des fonctions du neutrophiles telle que la production
de RLO lorsqu'elles sont activées par des agents particulaires comme le zymosan (403,
404, 442). De plus, un effet inhibiteur de PGE2 transmis par les récepteurs EP2 a été
observé sur la phagocytose de bactéries par des macrophages alvéolaires (443). Or les
stimulis phagocytaires ne stimulent pas l'activité de la PI3-Ky (151, 152) mais celle des
PI3-K de classe IA dont l'activation dépend des tyrosine kinases. Il serait par conséquent
intéressant de vérifier si PGE2 peut inhiber également la phagocytose dans le neutrophile et
d'explorer les mécanismes de signalisation mis en jeu dans ce modèle expérimental. Une
telle étude permettrait de mieux comprendre l'effet immuno-modulateur de PGE2 dans la
réponse innée.
En conclusion, les travaux présentés dans cette thèse permettent d'établir que l'inhibition
de la PI3-Ky par les autacoïdes qui élèvent les niveaux intracellulaires d'AMPc tels que
PGE2 ou l'adénosine est le mécanisme de signalisation majeur permettant d'expliquer
l'inhibition des fonctions du neutrophile stimulées par un agent chimiotactique.
L'inhibition sélective des isoformes de PI3-Ks exprimées dans les leucocytes, plus
particulièrement de la PI3-Ky, a été ces dernières années une des stratégies thérapeutiques
privilégiée par l'industrie pharmaceutique pour le développement de nouveaux agents anti-
inflammatoires (162). Nos travaux confortent une autre stratégie thérapeutique consistant à
provoquer une augmentation d'AMPc, par exemple par des inhibiteurs de PDE ou des
agonistes des récepteurs A2A actuellement à l'étude dans diverses pathologies
inflammatoires, qui pourraient promouvoir une partie de leurs effets anti-inflammatoires
par l'intermédiaire de l'inhibition de la PI3-Ky. Finalement, l'étude des mécanismes de
signalisation inhibiteurs de PGE2 et de l'adénosine indique que ces médiateurs jouent un
rôle primordial dans la modulation, voire même dans la résolution de l'inflammation.

Bibliographie
1. Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune récognition. Annu RevImmunol 20:197.
2. Skubitz, K. 2003. Neutrophilic leukocytes. In Wintrobe's Clinical Hematology, Vol.Vol.l. G. F. L. R. P. G. . éd. Lippincott Williams & Wilkins, New York, p. 267.
3. Borregaard, N., and J. B. Cowland. 1997. Granules of the human neutrophilicpolymorphonuclear leukocyte. Blood 89:3503.
4. Lollike, K., M. Lindau, J. Calafat, and N. Borregaard. 2002. Compound exocytosisof granules in human neutrophils. J Leuk Biol 71:973
5. Lominadze, G., D. W. Powell, G. C. Luerman, A. J. Link, R. A. Ward, and K. R.McLeish. 2005. Proteomic analysis of human neutrophil granules. Mol CellProteomics 4:1503.
6. Lindbom, L., and J. Werr. 2002. Integrin-dependent neutrophil migration inextravascular tissue. Semin Immunol 14:115.
I. Lang, P., F. Gesbert, M. Delespine-Carmagnat, R. Stancou, M. Pouchelet, and J.Bertoglio. 1996. Protein kinase A phosphorylation of RhoA médiates themorphological and functional effects of cyclic AMP in cytotoxic lymphocytes.EmboJ 15:510.
8. Walzog, B., and P. Gaehtgens. 2000. Adhésion Molécules: The Path to a NewUnderstanding of Acute Inflammation. News Physiol Sci 15:107.
9. Babior, B. 2004. NADPH oxidase. Curr Opin Immunol 16:4210. Berton, G., and C. A. Lowell, 1999. Integrin signalling in neutrophils and
macrophages. Cell Signal 11:621.I1. Crockett-Torabi, E. 1998. Selectins and mechanisms of signal transduction. J
Leukoc Biol 63:1.12. Ley, K. 2002. Intégration of inflammatory signais by rolling neutrophils. Immunol
Rev 186:813. Weiner, O. D., G. Servant, M. D. Welch, T. J. Mitchison, J. W. Sedat, and H. R.
Bourne. 1999. Spatial control of actin polymerization during neutrophil chemotaxis.Nat Cell Biol 1:75.
14. Parent, C. A. 2004. Making ail the right moves: chemotaxis in neutrophils andDictyostelium. Curr Opin Cell Biol 16:4.
15. Servant, G., O. D. Weiner, E. R. Neptune, J. W. Sedat, and H. R. Bourne. 1999.Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis.Mol Biol Cell 10:1163.
16. Xiao, Z., N. Zhang, D. B. Murphy, and P. N. Devreotes. 1997. Dynamic distributionof chemoattractant receptors in living cells during chemotaxis and persistentstimulation. J Cell Biol 139:365.
17. Jin, T., N. Zhang, Y. Long, C. A. Parent, and P. N. Devreotes. 2000. Localization ofthe G protein betagamma complex in living cells during chemotaxis. Science287:1034.
18. Rickert, P., O. D. Weiner, F. Wang, H. R. Bourne, and G. Servant. 2000.Leukocytes navigate by compass: rôles of PDKgamma and its lipid products.Trends Cell Biol 10:466.

183
19. Servant, G., O. D. Weiner, P. Herzmark, T. Balla, J. W. Sedat, and H. R. Bourne.2000. Polarization of chemoattractant receptor signaling during neutrophilchemotaxis. Science 287:1037.
20. Huang, Y. E., M. Iijima, C. A. Parent, S. Funamoto, R. A. Firtel, and P. Devreotes.2003. Receptor-mediated régulation of PI3Ks confines PI(3,4,5)P3 to the leadingedge of chemotaxing cells. Mol Biol Cell 14:1913.
21. Wang, F., P. Herzmark, O. Weiner, S. Srinivasan, G. Servant, and H. Bourne. 2002.Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility inneutrophils. Nat Cell Biol 4:513
22. Funamoto, S., R. Meili, S. Lee, L. Parry, and R. A. Firtel. 2002. Spatial andtemporal régulation of 3-phosphoinositides by PI 3-kinase and PTEN médiateschemotaxis. Cell 109:611.
23. Iijima, M., and P. Devreotes. 2002. Tumor suppressor PTEN médiates sensing ofchemoattractant gradients. Cell 109:599.
24. Hannigan, M., L. Zhan, Z. Li, Y. Ai, D. Wu, and C. K. Huang. 2002. Neutrophilslacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc Natl AcadSci USA 99:3603.
25. Chung, C. Y., and R. A. Firtel. 2002. Signaling pathways at the leading edge ofchemotaxing cells. J Muscle Res Cell Motil 23:773.
26. Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science 279:509.27. Ridley, A. J., M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy,
J. T. Parsons, and A. R. Horwitz. 2003. Cell migration: integrating signais fromfront to back. Science 302:1704.
28. Ridley, A. J. 2006. Rho GTPases and actin dynamics in membrane protrusions andvesicle trafficking. Trends Cell Biol 16:522.
29. Weiner, O., P. Neilsen, G. Prestwich, M. Kirschner, L. Cantley, and H. Bourne.2002. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop régulâtesneutrophil polarity. Nat Cell Biol 4:509
30. Srinivasan, S., F. Wang, S. Glavas, A. Ott, F. Hofmann, K. Aktories, D. Kalman,and H. Bourne. 2003. Rac and Cdc42 play distinct rôles in regulating PI(3,4,5)P-3and polarity during neutrophil chemotaxis. J Cell Biol 160:375
31. Xu, J., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y.Takuwa, N. Sugimoto, T. Mitchison, and H. R. Bourne. 2003. Divergent signais andcytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell114:201.
32. Xu, J., F. Wang, A. Van Keymeulen, M. Rentel, and H. R. Bourne. 2005.Neutrophil microtubules suppress polarity and enhance directional migration. ProcNatl Acad Sci USA 102:6884.
33. Van Keymeulen, A., K. Wong, Z. A. Knight, C. Govaerts, K. M. Hahn, K. M.Shokat, and H. R. Bourne. 2006. To stabilize neutrophil polarity, PIP3 and Cdc42augment RhoA activity at the back as well as signais at the front. J Cell Biol174:437.
34. Sheppard, F. R., M. R. Kelher, E. E. Moore, N. J. McLaughlin, A. Banerjee, and C.C. Silliman. 2005. Structural organization of the neutrophil NADPH oxidase:phosphorylation and translocation during priming and activation. JLeukoc Biol78:1025.

184
35. Perisic, O., M. I. Wilson, D. Karathanassis, J. Bravo, M. E. Pacold, C. D. Ellson, P.T. Hawkins, L. Stephens, and R. L. Williams. 2004. The rôle of phosphoinositidesand phosphorylation in régulation of NADPH oxidase. Adv Enzyme Regul 44:279.
36. Goldblatt, D., and A. J. Thrasher. 2000. Chronic granulomatous disease. Clin ExpImmunol 122:1.
1)1. Faurschou, M., and N. Borregaard. 2003. Neutrophil granules and secretory vesiclesin inflammation. Microbes Infect 5:1317.
38. Cox, D., and S. Greenberg. 2001. Phagocytic signaling stratégies: Fc(gamma)receptor-mediated phagocytosis as a model System. Semin Immunol 13:339
39. Castellano, F., P. Chavrier, and E. Caron. 2001. Actin dynamics duringphagocytosis. Semin Immunol 13:347
40. Lee, W. L., R. E. Harrison, and S. Grinstein. 2003. Phagocytosis by neutrophils.Microbes Infect 5:1299.
41. Lotner, G. Z., J. M. Lynch, S. J. Betz, and P. M. Henson. 1980. Human neutrophil-derived platelet activating factor. J Immunol 124:676.
42. Betz, S. J., and P. M. Henson. 1980. Production and release of platelet-activatingfactor (PAF); dissociation from degranulation and superoxide production in thehuman neutrophil. J Immunol 125:2756.
43. Borgeat, P., and B. Samuelsson. 1979. Metabolism of arachidonic acid inpolymorphonuclear leukocytes. Structural analysis of novel hydroxylatedcompounds. J Biol Chem 254:7865.
44. Borgeat, P., and B. Samuelsson. 1979. Arachidonic acid metabolism inpolymorphonuclear leukocytes: unstable intermediate in formation of dihydroxyacids. Proc Natl AcadSci USA 76:3213.
45. Borgeat, P., and B. Samuelsson. 1979. Transformation of arachidonic acid by rabbitpolymorphonuclear leukocytes. Formation of a novel dihydroxyeicosatetraenoicacid. J Biol Chem 254:2643.
46. Zurier, R. B. 1976. Prostaglandin release from human polymorphonuclearleukocytes. Adv Prostaglandin Thromboxane Res 2:815.
47. Cassatella, M. A. 1999. Neutrophil-derived proteins: selling cytokines by the pound.Adv Immunol 73:369.
48. Scapini, P., J. A. Lapinet-Vera, S. Gasperini, F. Calzetti, F. Bazzoni, and M. A.Cassatella. 2000. The neutrophil as a cellular source of chemokines. Immunol Rev177:195.
49. Schiffmann, E., B. A. Corcoran, and S. M. Wahl. 1975. N-formylmethionylpeptides as chemoattractants for leucocytes. Proc Natl Acad Sci USA 72:1059.
50. Marasco, W. A., S. H. Phan, H. Krutzsch, H. J. Showell, D. E. Feltner, R. Nairn, E.L. Becker, and P. A. Ward. 1984. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factorproduced by Escherichia coli. J Biol Chem 259:5430.
51. Williams, L. T., R. Snyderman, M. C. Pike, and R. J. Lefkowitz. 1977. Spécifiereceptor sites for chemotactic peptides on human polymorphonuclear leukocytes.Proc Natl Acad Sci U S A 74:1204.
52. Prossnitz, E. R., and R. D. Ye. 1997. The N-formyl peptide receptor: a model forthe study of chemoattractant receptor structure and function. Pharmacol Ther74:73.

185
53. Sklar, L. A., D. A. Finney, Z. G. Oades, A. J. Jesaitis, R. G. Painter, and C. G.Cochrane. 1984. The dynamics of ligand-receptor interactions. Real-time analysesof association, dissociation, and internalization of an N-formyl peptide and itsreceptors on the human neutrophil. JBiol Chem 259:5661.
54. Fu, H., J. Karlsson, J. Bylund, C. Movitz, A. Karlsson, and C. Dahlgren. 2006.Ligand récognition and activation of formyl peptide receptors in neutrophils. JLeukoc Biol 79:247.
55. Boulay, F., M. Tardif, L. Brouchon, and P. Vignais. 1990. The human N-formylpeptide receptor. Characterization of two cDNA isolâtes and évidence for anew subfamily of G-protein-coupled receptors. Biochemistry 29:11123.
56. Bokoch, G. M., and A. G. Gilman. 1984. Inhibition of receptor-mediated release ofarachidonic acid by pertussis toxin. Cell 39:301.
57. Lad, P. M., C. V. Oison, I. S. Grewal, and S. J. Scott. 1985. A pertussis toxin-sensitive GTP-binding protein in the human neutrophil régulâtes multiple receptors,calcium mobilization, and lectin-induced capping. Proc Natl Acad Sci USA82:8643.
58. Okajima, F., T. Katada, and M. Ui. 1985. Coupling of the guanine nucleotideregulatory protein to chemotactic peptide receptors in neutrophil membranes and itsuncoupling by islet-activating protein, pertussis toxin. A possible rôle of the toxinsubstrate in Ca2+-mobilizing receptor-mediated signal transduction. J Biol Chem260:6761.
59. Becker, E. L., J. C. Kermode, P. H. Naccache, R. Yassin, M. L. Marsh, J. J. Munoz,and R. I. Sha'afi. 1985. The inhibition of neutrophil granule enzyme sécrétion andchemotaxis by pertussis toxin. J Cell Biol 100:1641.
60. Bokoch, G. M. 1995. Chemoattractant signaling and leukocyte activation. Blood86:1649.
61. Hsu, M. H., S. C. Chiang, R. D. Ye, and E. R. Prossnitz. 1997. Phosphorylation ofthe N-formyl peptide receptor is required for receptor internalization but notchemotaxis. J Biol Chem 272:29426.
62. Paclet, M., C. Davis, P. Kotsonis, J. Godovac-Zimmermann, A. Segal, and L.Dekker. 2004. N-Formyl peptide receptor subtypes in human neutrophils activate L-plastin phosphorylation through différent signal transduction intermediates.BiochemJ 377:469
63. Prossnitz, E. R. 1997. Desensitization of N-formylpeptide receptor-mediatedactivation is dépendent upon receptor phosphorylation. J Biol Chem 272:15213.
64. Prossnitz, E. R., C. M. Kim, J. L. Benovic, and R. D. Ye. 1995. Phosphorylation ofthe N-formyl peptide receptor carboxyl terminus by the G protein-coupled receptorkinase, GRK2. J Biol Chem 270:1130.
65. Tardif, M., L. Mery, L. Brouchon, and F. Boulay. 1993. Agonist-dependentphosphorylation of N-formylpeptide and activation peptide from the fïfthcomponent of C (C5a) chemoattractant receptors in differentiated HL60 cells. JImmunol 150:3534.
66. Ali, H., R. M. Richardson, E. D. Tomhave, J. R. Didsbury, and R. Snyderman.1993. Différences in phosphorylation of formylpeptide and C5a chemoattractantreceptors correlate with différences in desensitization. J Biol Chem 268:24247.
67. Ali, H., R. M. Richardson, E. D. Tomhave, R. A. DuBose, B. Haribabu, and R.Snyderman. 1994. Régulation of stably transfected platelet activating factor receptor

186
in RBL-2H3 cells. Rôle of multiple G proteins and receptor phosphorylation. J BiolChem 269:24557.
68. Vines, C. M., C. M. Revankar, D. C. Maestas, L. L. LaRusch, D. F. Cimino, T. A.Kohout, R. J. Lefkowitz, and E. R. Prossnitz. 2003. N-formyl peptide receptorsinternalize but do not recycle in the absence of arrestins. J Biol Chem 278:41581.
69. Haribabu, B., R. M. Richardson, M. W. Verghese, A. J. Barr, D. V. Zhelev, and R.Snyderman. 2000. Function and régulation of chemoattractant receptors. ImmunolRes 22:271.
70. Ali, H., R. M. Richardson, B. Haribabu, and R. Snyderman. 1999. Chemoattractantreceptor cross-desensitization. J Biol Chem 274:6027.
71. Jesaitis, A. J., and K. N. Klotz. 1993. Cytoskeletal régulation of chemotacticreceptors: molecular complexation of N-formyl peptide receptors with G proteinsand actin. Eur J Haematol 51:288.
72. Jesaitis, A. J., J. O. Tolley, G. M. Bokoch, and R. A. Allen. 1989. Régulation ofchemoattractant receptor interaction with transducing proteins by organizationalcontrol in the plasma membrane of human neutrophils. JCell Biol 109:2783.
73. Jesaitis, A. J., and R. A. Allen. 1988. Activation of the neutrophil respiratory burstby chemoattractants: régulation of the N-formyl peptide receptor in the plasmamembrane. J Bioenerg Biomembr 20:679.
74. Harbecke, O., L. Liu, A. Karlsson, and C. Dahlgren. 1997. Desensitization of thefMLP-induced NADPH-oxidase response in human neutrophils is lacking inokadaic acid-treated cells. JLeukoc Biol 61:753.
75. Berridge, M. J. 1987. Inositol trisphosphate and diacylglycerol: two interactingsecond messengers. Annu Rev Biochem 56:159.
76. Li, Z., H. Jiang, W. Xie, Z. Zhang, A. V. Smrcka, and D. Wu. 2000. Rôles of PLC-beta2 and -beta3 and PDKgamma in chemoattractant-mediated signal transduction.Science 287:1046.
77. Rhee, S. G. 2001. Régulation of phosphoinositide-specific phospholipase C. AnnuRev Biochem 70:281.
78. Jiang, H., Y. Kuang, Y. Wu, W. Xie, M. I. Simon, and D. Wu. 1997. Rôles ofphospholipase C beta2 in chemoattractant-elicited responses. Proc Natl Acad Sci US A 94:7971.
79. Ferretti, M. E., S. Spisani, M. C. Pareschi, M. Buzzi, R. Cavallaro, S. Traniello, E.Reali, I. Torrini, M. P. Paradisi, G. P. Zecchini, and et al. 1994. Two newformylated peptides able to activate chemotaxis and respiratory burst selectively astools for studying human neutrophil responses. Cell Signal 6:91.
80. Smith, R. J., L. M. Sam, J. M. Justen, G. L. Bundy, G. A. Bala, and J. E. Bleasdale.1990. Receptor-coupled signal transduction in human polymorphonuclearneutrophils: effects of a novel inhibitor of phospholipase C-dependent processes oncell responsiveness. J Pharmacol Exp Ther 253:688.
81. Smith, R. J., J. M. Justen, A. R. McNab, C. L. Rosenbloom, A. N. Steele, P. A.Detmers, D. C. Anderson, and A. M. Manning. 1996. U-73122: a potent inhibitor ofhuman polymorphonuclear neutrophil adhésion on biological surfaces andadhesion-related effector functions. J Pharmacol Exp Ther 278:320.
82. Liu, M., and M. I. Simon. 1996. Régulation by cAMP-dependent protein kinease ofa G-protein-mediated phospholipase C. Nature 382:83.

187
83. Ali, H., S. Sozzani, I. Fisher, A. J. Barr, R. M. Richardson, B. Haribabu, and R.Snyderman. 1998. Differential régulation of formyl peptide and platelet-activatingfactor receptors. Rôle of phospholipase Cbeta3 phosphorylation by protein kinaseA. JBiolChem 273:11012.
84. White, J. R., P. H. Naccache, and R. I. Sha'afi. 1982. The synthetic chemotacticpeptide formyl-methionyl-leucyl-phenylalanine causes an increase in actinassociated with the cytoskeleton in rabbit neutrophils. Biochem Biophys ResCommun 108:144.
85. White, J. R., P. H. Naccache, T. F. Molski, P. Borgeat, and R. I. Sha'afi. 1983.Direct démonstration of increased intracellular concentration of free calcium inrabbit and human neutrophils following stimulation by chemotactic factor. BiochemBiophys Res Commun 113:44.
86. Smolen, J. E., H. M. Korchak, and G. Weissmann. 1981. The rôles of extracellularand intracellular calcium in lysosomal enzyme release and superoxide aniongénération by human neutrophils. Biochim Biophys Acta 677:512.
87. Lew, P. D., C. B. Wollheim, F. A. Waldvogel, and T. Pozzan. 1984. Modulation ofcytosolic-free calcium transients by changes in intracellular calcium-bufferingcapacity: corrélation with exocytosis and 02-production in human neutrophils. JCellBiol 99:1212.
88. Foyouzi-Youssefi, R., F. Petersson, D. P. Lew, K. H. Krause, and O. Nusse. 1997.Chemoattractant-induced respiratory burst: increases in cytosolic Ca2+concentrations are essential and synergize with a kinetically distinct second signal.Biochem J322 (Pt3):709.
89. Davies, M. P., T. J. Hallam, and J. E. Merritt. 1990. A rôle for calcium and proteinkinase C in agonist-stimulated adhésion of human neutrophils. Biochem J 267:13.
90. Merritt, J. E., R. Jacob, and T. J. Hallam. 1989. Use of manganèse to discriminatebetween calcium influx and mobilization from internai stores in stimulated humanneutrophils. J Biol Chem 264:1522.
91. Naccache, P. H., H. J. Showell, E. L. Becker, and R. I. Sha'afi. 1979.Pharmacological differentiation between the chemotactic factor inducedintracellular calcium redistribution and transmembrane calcium influx in rabbitneutrophils. Biochem Biophys Res Commun 89:1224.
92. Prentki, M., C. B. Wollheim, and P. D. Lew. 1984. Ca2+ homeostasis inpermeabilized human neutrophils. Characterization of Ca2+-sequestering pools andthe action of inositol 1,4,5-triphosphate. J Biol Chem 259:13777.
93. von Tscharner, V., B. Prod'hom, M. Baggiolini, and H. Reuter. 1986. Ion channelsin human neutrophils activated by a rise in free cytosolic calcium concentration.Nature 324:369.
94. Demaurex, N., A. Monod, D. P. Lew, and K. H. Krause. 1994. Characterization ofreceptor-mediated and store-regulated Ca2+ influx in human neutrophils. Biochem J297 (Pt 3):595.
95. Naccache, P. H., T. F. Molski, T. Alobaidi, E. L. Becker, H. J. Showell, and R. I.Sha'afi. 1980. Calmodulin inhibitors block neutrophil degranulation at a step distalfrom the mobilization of calcium. Biochem Biophys Res Commun 97:62.
96. Elferink, J. G., M. Deierkauf, and J. C. Riemersma. 1982. Involvement ofcalmodulin in granulocyte chemotaxis: the effect of calmodulin inhibitors. ResCommun Chem Pathol Pharmacol 38:77.

188
97. Perianin, A., E. Pedruzzi, and J. Hakim. 1994. W-7, a calmodulin antagonist, primesthe stimulation of human neutrophil respiratory burst by formyl peptides andplatelet-activating factor. FEBS Lett 342:135.
98. Verploegen, S., L. C. van, D. H. van, J. Lammers, L. Koenderman, and P. Coffer.2002. Rôle of Ca2+/calmodulin regulated signaling pathways in chemoattractantinduced neutrophil effector functions - Comparison with the rôle ofphosphotidylinositol-3 kinase. Eur J Biochem 269:4625
99. Mellor, H., and P. J. Parker. 1998. The extended protein kinase C superfamily.Biochem J 332 (Pt2):281.
100. Newton, A. C. 1997. Régulation of protein kinase C. Curr Opin Cell Biol 9:161.101. Niggli, V. 2003. Signaling to migration in neutrophils: importance of localized
pathways. Int J Biochem Cell Biol 35:1619.102. Dang, P. M., S. Rais, J. Hakim, and A. Perianin. 1995. Redistribution of protein
kinase C isoforms in human neutrophils stimulated by formyl peptides and phorbolmyristate acétate. Biochem Biophys Res Commun 212:664.
103. Majumdar, S., M. W. Rossi, T. Fujiki, W. A. Phillips, S. Disa, C. F. Queen, R. B.Johnston, Jr., O. M. Rosen, B. E. Corkey, and H. M. Korchak. 1991. Protein kinaseC isotypes and signaling in neutrophils. Differential substrate specificities of atranslatable calcium- and phospholipid-dependent beta-protein kinase C and aphospholipid-dependent protein kinase which is inhibited by long chain fatty acylcoenzyme A. J Biol Chem 266:9285.
104. Dekker, L. V., M. Leitges, G. Altschuler, N. Mistry, A. McDermott, J. Roes, and A.W. Segal. 2000. Protein kinase C-beta contributes to NADPH oxidase activation inneutrophils. Biochem J 347 Pt 1:285.
105. Korchak, H. M., B. E. Corkey, G. C. Yaney, and L. E. Kilpatrick. 2001. Négativerégulation of ligand-initiated Ca(2+) uptake by PKC-beta II in differentiated HL60cells. AmJPhysiol Cell Physiol 28LC514.
106. Kramer, I. M., A. J. Verhoeven, R. L. van der Bend, R. S. Weening, and D. Roos.1988. Purified protein kinase C phosphorylates a 47-kDa protein in controlneutrophil cytoplasts but not in neutrophil cytoplasts from patients with theautosomal form of chronic granulomatous disease. J Biol Chem 263:2352.
107. Dang, P. M., A. Fontayne, J. Hakim, J. El Benna, and A. Perianin. 2001. Proteinkinase C zêta phosphorylates a subset of sélective sites of the NADPH oxidasecomponent p47phox and participâtes in formyl peptide-mediated neutrophilrespiratory burst. J Immunol 166:1206.
108. Perez, H. D., F. Elfman, S. Marder, E. Lobo, and H. E. Ives. 1989. Formyl peptide-induced chemotaxis of human polymorphonuclear leukocytes does not requireeither marked changes in cytosolic calcium or spécifie granule discharge. Rôle offormyl peptide receptor reexpression (or recycling). J Clin Invest 83:1963.
109. Katanaev, V. L. 2001. Signal transduction in neutrophil chemotaxis. Biochemistry(Mosc) 66:351.
110. Vanhaesebroeck, B., S. J. Leevers, K. Ahmadi, J. Timms, R. Katso, P. C. Driscoll,R. Woscholski, P. J. Parker, and M. D. Waterfield. 2001. Synthesis and function of3-phosphorylated inositol lipids. Annu Rev Biochem 70:535.
111. Traynor-Kaplan, A. E., A. L. Harris, B. L. Thompson, P. Taylor, and L. A. Sklar.1988. An inositol tetrakisphosphate-containing phospholipid in activatedneutrophils. Nature 334:353.

189
112. Traynor-Kaplan, A. E., B. L. Thompson, A. L. Harris, P. Taylor, G. M. Omann, andL. A. Sklar. 1989. Transient increase in phosphatidylinositol 3,4-bisphosphate andphosphatidylinositol trisphosphate during activation of human neutrophils. JBiolChem 264:15668.
113. Stephens, L. R., K. T. Hughes, and R. F. Irvine. 1991. Pathway ofphosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nature351:33.
114. Stephens, L. R., T. R. Jackson, and P. T. Hawkins. 1993. Agonist-stimulatedsynthesis of phosphatidylinositol(3,4,5)-trisphosphate: a new intracellular signallingSystem? Biochim Biophys Acta 1179:27.
115. Dobos, G. J., J. Norgauer, M. Eberle, P. J. Schollmeyer, and A. E. Traynor-Kaplan.1992. C5a reduces formyl peptide-induced actin polymerization andphosphatidylinositol(3,4,5)trisphosphate formation, but not phosphatidylinositol(4,5) bisphosphate hydrolysis and superoxide production, in human neutrophils. JImmunol 149:609.
116. Okada, T., L. Sakuma, Y. Fukui, O. Hazeki, and M. Ui. 1994. Blockage ofchemotactic peptide-induced stimulation of neutrophils by wortmannin as a resuit ofsélective inhibition of phosphatidylinositol 3-kinase. J Biol Chem 269:3563.
117. Thelen, M., M. P. Wymann, and H. Langen. 1994. Wortmannin binds specifically to1-phosphatidylinositol 3-kinase while inhibiting guanine nucleotide-bindingprotein-coupled receptor signaling in neutrophil leukocytes. Proc Natl AcadSci USA 91:4960.
118. Wymann, M. P., G. Bulgarelli-Leva, M. J. Zvelebil, L. Pirola, B. Vanhaesebroeck,M. D. Waterfield, and G. Panayotou. 1996. Wortmannin inactivatesphosphoinositide 3-kinase by covalent modification of Lys-802, a residue involvedin the phosphate transfer reaction. Mol Cell Biol 16:1722.
119. Walker, E. H., M. E. Pacold, O. Perisic, L. Stephens, P. T. Hawkins, M. P.Wymann, and R. L. Williams. 2000. Structural déterminants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, andstaurosporine. Mol Cell 6:909.
120. Vlahos, C. J., W. F. Matter, R. F. Brown, A. E. Traynor-Kaplan, P. G. Heyworth, E.R. Prossnitz, R. D. Ye, P. Marder, J. A. Schelm, K. J. Rothfuss, and et al. 1995.Investigation of neutrophil signal transduction using a spécifie inhibitor ofphosphatidylinositol 3-kinase. J Immunol 154:2413.
121. Arcaro, A., and M. P. Wymann. 1993. Wortmannin is a potent phosphatidylinositol3-kinase inhibitor: the rôle of phosphatidylinositol 3,4,5-trisphosphate in neutrophilresponses. Biochem J 296 (Pi 2):297.
122. Dewald, B., M. Thelen, and M. Baggiolini. 1988. Two transduction séquences arenecessary for neutrophil activation by receptor agonists. J Biol Chem 263:16179.
123. Niggli, V. 2000. A membrane-permeant ester of phosphatidylinositol 3,4, 5-trisphosphate (PIP(3)) is an activator of human neutrophil migration. FEBS Lett473:217.
124. Niggli, V., and H. Keller. 1997. The phosphatidylinositol 3-kinase inhibitorwortmannin markedly reduces chemotactic peptide-induced locomotion andincreases in cytoskeletal actin in human neutrophils. Eur JPharmacol 335:43.
125. Capodici, C, S. Hanft, M. Feoktistov, and M. H. Pillinger. 1998.Phosphatidylinositol 3-kinase médiates chemoattractant-stimulated, CD1 lb/CD18-

190
dépendent cell-cell adhésion of human neutrophils: évidence for an ERK-independent pathway. J Immunol 160:1901.
126. Condliffe, A. M., P. T. Hawkins, L. R. Stephens, C. Haslett, and E. R. Chilvers.1998. Priming of human neutrophil superoxide génération by tumour necrosisfactor-alpha is signalled by enhanced phosphatidylinositol 3,4,5-trisphosphate butnot inositol 1,4,5-trisphosphate accumulation. FEBSLett 439:147.
127. Cadwallader, K. A., A. M. Condliffe, A. McGregor, T. R. Walker, J. F. White, L. R.Stephens, and E. R. Chilvers. 2002. Régulation of phosphatidylinositol 3-kinaseactivity and phosphatidylinositol 3,4,5-trisphosphate accumulation by neutrophilpriming agents. J Immunol 169:3336.
128. Dhand, R., I. Hiles, G. Panayotou, S. Roche, M. J. Fry, I. Goût, N. F. Totty, O.Truong, P. Vicendo, K. Yonezawa, and et al. 1994. PI 3-kinase is a dual specificityenzyme: autoregulation by an intrinsic protein-serine kinase activity. Embo J13:522.
129. Deane, J., and D. Fruman. 2004. Phosphoinositide 3-kinase: Diverse rôles inimmune cell activation. Annu Rev Immunol 22:563
130. Deane, J. A., and D. A. Fruman. 2004. Phosphoinositide 3-kinase: diverse rôles inimmune cell activation. Annu Rev Immunol 22:563.
131. Yu, J., Y. Zhang, J. Mcllroy, T. Rordorf-Nikolic, G. A. Orr, and J. M. Backer. 1998.Régulation of the p85/pl 10 phosphatidylinositol 3'-kinase: stabilization andinhibition of the pi lOalpha catalytic subunit by the p85 regulatory subunit. Mol CellBiol 18:1379.
132. Cuevas, B. D., Y. Lu, M. Mao, J. Zhang, R. LaPushin, K. Siminovitch, and G. B.Mills. 2001. Tyrosine phosphorylation of p85 relieves its inhibitory activity onphosphatidylinositol 3-kinase. J Biol Chem 276:27455.
133. Walker, E. H., O. Perisic, C. Ried, L. Stephens, and R. L. Williams. 1999.Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature402:313.
134. Czupalla, C, M. Culo, E. Muller, C. Brock, H. Reusch, K. Spicher, E. Krause, andB. Nurnberg. 2003. Identification and characterization of the autophosphorylationsites of phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem278:11536
135. Stephens, L. R., A. Eguinoa, H. Erdjument-Bromage, M. Lui, F. Cooke, J.Coadwell, A. S. Smrcka, M. Thelen, K. Cadwallader, P. Tempst, and P. T.Hawkins. 1997. The G beta gamma sensitivity of a PI3K is dépendent upon a tightlyassociated adaptor, pi01. Cell 89:105.
136. Suire, S., J. Coadwell, G. J. Ferguson, K. Davidson, P. Hawkins, and L. Stephens.2005. p84, a new Gbetagamma-activated regulatory subunit of the type IBphosphoinositide 3-kinase pi lOgamma. Curr Biol 15:566.
137. Voigt, P., M. B. Dorner, and M. Schaefer. 2006. Characterization of p87PIKAP, anovel regulatory subunit of phosphoinositide 3-kinase gamma that is highlyexpressed in heart and interacts with PDE3B. J Biol Chem 281:9977.
138. Voigt, P., C. Brock, B. Nurnberg, and M. Schaefer. 2005. Assigning functionaldomains within the pi 01 regulatory subunit of phosphoinositide 3-kinase gamma. JBiol Chem 280:5121.
139. Brock, C , M. Schaefer, H. P. Reusch, C. Czupalla, M. Michalke, K. Spicher, G.Schultz, and B. Nurnberg. 2003. Rôles of G beta gamma in membrane recruitment

191
and activation of pi 10 gamma/plOl phosphoinositide 3-kinase gamma. J Cell Biol160:89.
140. Maier, U., A. Babich, and B. Nurnberg. 1999. Rôles of non-catalytic subunits ingbetagamma-induced activation of class I phosphoinositide 3-kinase isoforms betaand gamma. J Biol Chem 274:29311.
141. Suire, S., A. M. Condliffe, G. J. Ferguson, C. D. Ellson, H. Guillou, K. Davidson,H. Welch, J. Coadwell, M. Turner, E. R. Chilvers, P. T. Hawkins, and L. Stephens.2006. Gbetagammas and the Ras binding domain of pi lOgamma are both importantregulators of PI3Kgamma signalling in neutrophils. Nat Cell Biol 8:1303.
142. Rodriguez-Viciana, P., P. H. Warne, R. Dhand, B. Vanhaesebroeck, I. Goût, M. J.Fry, M. D. Waterfield, and J. Downward. 1994. Phosphatidylinositol-3-OH kinaseas a direct target of Ras. Nature 370:527.
143. Suire, S., P. Hawkins, and L. Stephens. 2002. Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol 12:1068.
144. Stephens, L., A. Eguinoa, S. Corey, T. Jackson, and P. T. Hawkins. 1993. Receptorstimulated accumulation of phosphatidylinositol (3,4,5)-trisphosphate by G-proteinmediated pathways in human myeloid derived cells. Embo J 12:2265.
145. Stephens, L., A. Smrcka, F. T. Cooke, T. R. Jackson, P. C. Sternweis, and P. T.Hawkins. 1994. A novel phosphoinositide 3 kinase activity in myeloid-derived cellsis activated by G protein beta gamma subunits. Cell 77:83.
146. Stoyanov, B., S. Volinia, T. Hanck, I. Rubio, M. Loubtchenkov, D. Malek, S.Stoyanova, B. Vanhaesebroeck, R. Dhand, B. Nurnberg, and et al. 1995. Cloningand characterization of a G protein-activated human phosphoinositide-3 kinase.Science 269:690.
147. Ptasznik, A., E. R. Prossnitz, D. Yoshikawa, A. Smrcka, A. E. Traynor-Kaplan, andG. M. Bokoch. 1996. A tyrosine kinase signaling pathway accounts for the majorityof phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulatedhuman neutrophils. J Biol Chem 271:25204.
148. Thelen, M., and S. A. Didichenko. 1997. G-protein coupled receptor-mediatedactivation of PI 3-kinase in neutrophils. Ann N Y AcadSci 832:368.
149. Coffer, P. J., N. Geijsen, L. M'Rabet, R. C. Schweizer, T. Maikoe, J. A.Raaijmakers, J. W. Lammers, and L. Koenderman. 1998. Comparison of the rôles ofmitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase signaltransduction in neutrophil effector function. BiochemJ 329 (Pt 1):121.
150. Vlahos, C. J., and W. F. Matter. 1992. Signal transduction in neutrophil activation.Phosphatidylinositol 3-kinase is stimulated without tyrosine phosphorylation. FEBSLett 309:242.
151. Hirsch, E., V. L. Katanaev, C. Garlanda, O. Azzolino, L. Pirola, L. Silengo, S.Sozzani, A. Mantovani, F. Altruda, and M. P. Wymann. 2000. Central rôle for Gprotein-coupled phosphoinositide 3-kinase gamma in inflammation. Science287:1049.
152. Sasaki, T., J. Irie-Sasaki, R. G. Jones, A. J. Oliveira-dos-Santos, W. L. Stanford, B.Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, N. Joza, T. W. Mak, P.S. Ohashi, A. Suzuki, and J. M. Penninger. 2000. Function of PDKgamma inthymocyte development, T cell activation, and neutrophil migration. Science287:1040.

192
153. Ferguson, G. J., L. Milne, S. Kulkarni, T. Sasaki, S. Walker, S. Andrews, T.Crabbe, P. Finan, G. Jones, S. Jackson, M. Camps, C. Rommel, M. Wymann, E.Hirsch, P. Hawkins, and L. Stephens. 2006. PI(3)Kgamma has an importantcontext-dependent rôle in neutrophil chemokinesis. Nat Cell Biol.
154. Naccache, P. H., S. Levasseur, G. Lachance, S. Chakravarti, S. G. Bourgoin, and S.R. McColl. 2000. Stimulation of human neutrophils by chemotactic factors isassociated with the activation of phosphatidylinositol 3-kinase gamma. J Biol Chem275:23636.
155. Sadhu, C, B. Masinovsky, K. Dick, C. G. Sowell, and D. E. Staunton. 2003.Essential rôle of phosphoinositide 3-kinase delta in neutrophil directionalmovement. JImmunol 170:2647.
156. Sadhu, C, K. Dick, W. T. Tino, and D. E. Staunton. 2003. Sélective rôle of PI3Kdelta in neutrophil inflammatory responses. Biochem Biophys Res Commun308:764.
157. Camps, M., T. Ruckle, H. Ji, V. Ardissone, F. Rintelen, J. Shaw, C. Ferrandi, C.Chabert, C. Gillieron, B. Francon, T. Martin, D. Gretener, D. Perrin, D. Leroy, P. A.Vitte, E. Hirsch, M. P. Wymann, R. Cirillo, M. K. Schwarz, and C. Rommel. 2005.Blockade of PDKgamma suppresses joint inflammation and damage in mousemodels of rheumatoid arthritis. Nat Med 11:936.
158. Yum, H., J. Arcaroli, J. Kupfner, R. Shenkar, J. Penninger, T. Sasaki, K. Yang, J.Park, and E. Abraham. 2001. Involvement of phosphoinositide 3-kinases inneutrophil activation and the development of acute lung injury. J Immunol167:6601
159. Barber, D. F., A. Bartolome, C. Hernandez, J. M. Flores, C. Redondo, C.Fernandez-Arias, M. Camps, T. Ruckle, M. K. Schwarz, S. Rodriguez, A. C.Martinez, D. Balomenos, C. Rommel, and A. C. Carrera. 2005. PDKgammainhibition blocks glomerulonephritis and extends lifespan in a mouse model ofsystemic lupus. Nat Med 11:933.
160. Puri, K. D., T. A. Doggett, J. Douangpanya, Y. Hou, W. T. Tino, T. Wilson, T.Graf, E. Clayton, M. Turner, J. S. Hayflick, and T. G. Diacovo. 2004. Mechanismsand implications of phosphoinositide 3-kinase delta in promoting neutrophiltrafficking into inflamed tissue. Blood 103:3448.
161. Condliffe, A. M., K. Davidson, K. E. Anderson, C. D. Ellson, T. Crabbe, K.Okkenhaug, B. Vanhaesebroeck, M. Turner, L. Webb, M. P. Wymann, E. Hirsch, T.Ruckle, M. Camps, C. Rommel, S. P. Jackson, E. R. Chilvers, L. R. Stephens, andP. T. Hawkins. 2005. Sequential activation of Class IB and Class IA PI3Ks isimportant for the primed respiratory burst of human but not murine neutrophils.Blood.
162. Ruckle, T., M. K. Schwarz, and C. Rommel. 2006. PDKgamma inhibition: towardsan 'aspirin of the 21 st century'? Nat Rev Drug Discov 5:903.
163. Corner, F. L, and C. A. Parent. 2002. PI 3-kinases and PTEN: how oppositeschemoattract. Cell 109:541.
164. Lacalle, R. A., C. Gomez-Mouton, D. F. Barber, S. Jimenez-Baranda, E. Mira, A.C. Martinez, A. C. Carrera, and S. Mânes. 2004. PTEN régulâtes motility but notdirectionality during leukocyte chemotaxis. J Cell Sci 117:6207.

193
165. Li, Z., X. Dong, Z. Wang, W. Liu, N. Deng, Y. Ding, L. Tang, T. Hla, R. Zeng, L.Li, and D. Wu. 2005. Régulation of PTEN by Rho small GTPases. Nat Cell Biol7:399.
166. Cozier, G. E., J. Carlton, D. Bouyoucef, and P. J. Cullen. 2004. Membrane targetingby pleckstrin homology domains. Curr Top Microbiol Immunol 282:49.
167. Staal, S. P. 1987. Molecular cloning of the akt oncogene and its human homologuesAKT1 and AKT2: amplification of AKT1 in a primary human gastricadenocarcinoma. Proc Natl AcadSci USA 84:5034.
168. Bellacosa, A., J. R. Testa, S. P. Staal, and P. N. Tsichlis. 1991. A retroviraloncogene, akt, encoding a serine-threonine kinase containing an SH2-like région.Science 254:274.
169. Coffer, P. J., and J. R. Woodgett. 1991. Molecular cloning and characterisation of anovel putative protein-serine kinase related to the cAMP-dependent and proteinkinase C families. Eur JBiochem 201:475.
170. Vanhaesebroeck, B., and D. R. Alessi. 2000. The PI3K-PDK1 connection: morethan just a road to PKB. Biochem J 346 Pt 3:561.
171. Rane, M. J., P. Y. Coxon, D. W. Powell, R. Webster, J. B. Klein, W. Pierce, P.Ping, and K. R. McLeish. 2001. p38 Kinase-dependent MAPKAPK-2 activationfonctions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. JBiol Chem 276:3517.
172. Frech, M., M. Andjelkovic, E. Ingley, K. K. Reddy, J. R. Falck, and B. A.Hemmings. 1997. High affinity binding of inositol phosphates andphosphoinositides to the pleckstrin homology domain of RAC/protein kinase B andtheir influence on kinase activity. J Biol Chem 272:8474.
173. Alessi, D. R., M. Andjelkovic, B. Caudwell, P. Cron, N. Motrice, P. Cohen, and B.A. Hemmings. 1996. Mechanism of activation of protein kinase B by insulin andIGF-1. EmboJ 15:6541.
174. Kohn, A. D., F. Takeuchi, and R. A. Roth. 1996. Akt, a pleckstrin homologydomain containing kinase, is activated primarily by phosphorylation. J Biol Chem271:21920.
175. James, S. R., C. P. Downes, R. Gigg, S. J. Grove, A. B. Holmes, and D. R. Alessi.1996. Spécifie binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subséquent activation. Biochem J315 (Pt 3):709.
176. Alessi, D. R., S. R. James, C. P. Downes, A. B. Holmes, P. R. Gaffney, C. B. Reese,and P. Cohen. 1997. Characterization of a 3-phosphoinositide-dependent proteinkinase which phosphorylates and activâtes protein kinase Balpha. Curr Biol 7:261.
177. Milburn, C. C , M. Deak, S. M. Kelly, N. C. Price, D. R. Alessi, and D. M. VanAalten. 2003. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrinhomology domain of protein kinase B induces a conformational change. Biochem J375:531.
178. Stephens, L., K. Anderson, D. Stokoe, H. Erdjument-Bromage, G. F. Painter, A. B.Holmes, P. R. Gaffney, C. B. Reese, F. McCormick, P. Tempst, J. Coadwell, and P.T. Hawkins. 1998. Protein kinase B kinases that médiate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279:710.
179. Storz, P., and A. Toker. 2002. 3'-phosphoinositide-dependent kinase-1 (PDK-1) inPI 3-kinase signaling. Front Biosci 7:d886.

194
180. Anderson, K. E., J. Coadwell, L. R. Stephens, and P. T. Hawkins. 1998.Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 toactivate protein kinase B. Curr Biol 8:684.
181. Chen, R., O. Kim, J. Yang, K. Sato, K. M. Eisenmann, J. McCarthy, H. Chen, andY. Qiu. 2001. Régulation of Akt/PKB activation by tyrosine phosphorylation. J BiolChem 276:31858.
182. Conus, N. M., K. M. Hannan, B. E. Cristiano, B. A. Hemmings, and R. B. Pearson.2002. Direct identification of tyrosine 474 as a regulatory phosphorylation site forthe Akt protein kinase. J Biol Chem 277:38021.
183. Jiang, T., and Y. Qiu. 2003. Interaction between Src and a C-terminal proline-richmotif of Akt is required for Akt activation. J Biol Chem 278:15789
184. Yano, S., H. Tokumitsu, and T. R. Soderling. 1998. Calcium promotes cell survivalthrough CaM-K kinase activation of the protein-kinase-B pathway. Nature 396:584.
185. Bayascas, J. R., and D. R. Alessi. 2005. Régulation of Akt/PKB Ser473phosphorylation. Mol Cell 18:143.
186. Stambolic, V., A. Suzuki, J. L. de la Pompa, G. M. Brothers, C. Mirtsos, T. Sasaki,J. Ruland, J. M. Penninger, D. P. Siderovski, and T. W. Mak. 1998. Négativerégulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell95:29.
187. Song, G., G. Ouyang, and S. Bao. 2005. The activation of Akt/PKB signalingpathway and cell survival../ Cell Mol Med 9:59.
188. Kim, D., and J. Chung. 2002. Akt: versatile mediator of cell survival and beyond. JBiochem Mol Biol 35:106.
189. Tilton, B., M. Andjelkovic, S. A. Didichenko, B. A. Hemmings, and M. Thelen.1997. G-Protein-coupled receptors and Fcgamma-receptors médiate activation ofAkt/protein kinase B in human phagocytes. J Biol Chem 272:28096.
190. Chen, Q., D. W. Powell, M. J. Rane, S. Singh, W. Butt, J. B. Klein, and K. R.McLeish. 2003. Akt phosphorylates p47phox and médiates respiratory burst activityin human neutrophils. J Immunol 170:5302.
191. Hoyal, C, A. Gutierrez, B. Young, S. Catz, J. Lin, P. Tsichlis, and B. Babior. 2003.Modulation of p47(PHOX) activity by site-specific phosphorylation: Akt-dependentactivation of the NADPH oxidase. Proc NatlAcadSci USA 100:5130
192. Chodniewicz, D., and D. V. Zhelev. 2003. Chemoattractant receptor-stimulated F-actin polymerization in the human neutrophil is signaled by 2 distinct pathways.Blood 101:1181.
193. Naccache, P. H., C. Gilbert, A. C. Caon, M. Gaudry, C. K. Huang, V. A. Bonak, K.Umezawa, and S. R. McColl. 1990. Sélective inhibition of human neutrophilfunctional responsiveness by erbstatin, an inhibitor of tyrosine protein kinase. Blood76:2098.
194. Rollet, E., A. C. Caon, C. J. Roberge, N. W. Liao, S. E. Malawista, S. R. McColl,and P. H. Naccache. 1994. Tyrosine phosphorylation in activated humanneutrophils. Comparison of the effects of différent classes of agonists andidentification of the signaling pathways involved. J Immunol 153:353.
195. Gaudry, M., A. C. Caon, C. Gilbert, S. Lille, and P. H. Naccache. 1992. Evidencefor the involvement of tyrosine kinases in the locomotory responses of humanneutrophils. J Leukoc Biol 51:103.

195
196. Ma, Y. C, J. Huang, S. Ali, W. Lowry, and X. Y. Huang. 2000. Src tyrosine kinaseis a novel direct effector of G proteins. Cell 102:635.
197. Desaulniers, P. 2006. Etude des réponses phagocytaires et chimiotactiques duneutrophile humain dans des modèles in vitro. In Département d'Immunologie-Microbiologie, faculté de Médecine. Unibersité Laval, Québec.
198. Gaudry, M., C. Gilbert, F. Barabe, P. E. Poubelle, and P. H. Naccache. 1995.Activation of Lyn is a common élément of the stimulation of human neutrophils bysoluble and particulate agonists. Blood 86:3567.
199. Ptasznik, A., A. Traynor-Kaplan, and G. M. Bokoch. 1995. G protein-coupledchemoattractant receptors regulate Lyn tyrosine kinase.Shc adapter proteinsignaling complexes. J Biol Chem 270:19969.
200. Gutkind, J. S., and K. C. Robbins. 1989. Translocation of the FGR protein-tyrosinekinase as a conséquence of neutrophil activation. Proc Natl Acad Sci USA86:8783.
201. Mocsai, A., E. Ligeti, C. A. Lowell, and G. Berton. 1999. Adhesion-dependentdegranulation of neutrophils requires the Src family kinases Fgr and Hck. JImmunol 162:1120.
202. Bolen, J. B., and J. S. Brugge. 1997. Leukocyte protein tyrosine kinases: potentialtargets for drug discovery. Annu Rev Immunol 15:371.
203. Berton, G., L. Fumagalli, C. Laudanna, and C. Sorio. 1994. Beta 2 integrin-dependent protein tyrosine phosphorylation and activation of the FGR proteintyrosine kinase in human neutrophils. J Cell Biol 126:1111.
204. Mano, H. 1999. Tec family of protein-tyrosine kinases: an overview of theirstructure and function. Cytokine Growth Factor Rev 10:267.
205. Lachance, G., S. Levasseur, and P. H. Naccache. 2002. Chemotactic factor-inducedrecruitment and activation of Tec family kinases in human neutrophils. Implicationof phosphatidynositol 3-kinases. J Biol Chem 277:21537.
206. Gilbert, C , S. Levasseur, P. Desaulniers, A. A. Dusseault, N. Thibault, S. G.Bourgoin, and P. H. Naccache. 2003. Chemotactic factor-induced recruitment andactivation of Tec family kinases in human neutrophils. II. Effects of LFM-A13, aspécifie Btk inhibitor. J Immunol 170:5235.
207. Yan, S. R., and M. J. Novak. 1999. Beta2 integrin-dependent phosphorylation ofprotein-tyrosine kinase Pyk2 stimulated by tumor necrosis factor alpha and fMLP inhuman neutrophils adhèrent to fibrinogen. FEBS Lett 451:33.
208. Miura, Y., Y. Tohyama, T. Hishita, A. Lala, E. De Nardin, Y. Yoshida, H.Yamamura, T. Uchiyama, and K. Tohyama. 2000. Pyk2 and Syk participate infunctional activation of granulocytic HL-60 cells in a différent manner. Blood96:1733.
209. Casey, P. J. 1994. Lipid modifications of G proteins. Curr Opin Cell Biol 6:219.210. Barbieri, J., M. Riese, and K. Aktories. 2002. Bacterial toxins that modify the actin
cytoskeleton. Annu Rev Cell Dev Biol 18:315211. Fiorentini, C , M. Gauthier, G. Donelli, and P. Boquet. 1998. Bacterial toxins and
the Rho GTP-binding protein: what microbes teach us about cell régulation. CellDeathDiffer 5:720.
212. Hoffman, G. R., and R. A. Cerione. 2002. Signaling to the Rho GTPases:networking with the DH domain. FEBS Lett 513:85.

196
213. Collins, T. L., M. Deckert, and A. Altman. 1997. Views on Vav. Immunol Today18:221.
214. Kim, C, C. C. Marchai, J. Penninger, and M. C. Dinauer. 2003. The hemopoieticRho/Rac guanine nucleotide exchange factor Vavl régulâtes N-formyl-methionyl-leucyl-phenylalanine-activated neutrophil functions. J Immunol 171:4425.
215. Abe, K., K. L. Rossman, B. Liu, K. D. Ritola, D. Chiang, S. L. Campbell, K.Burridge, and C. J. Der. 2000. Vav2 is an activator of Cdc42, Racl, and RhoA. JBiolChem 275:10141.
216. Habets, G. G., E. H. Scholtes, D. Zuydgeest, R. A. van der Kammen, J. C. Stam, A.Berns, and J. G. Collard. 1994. Identification of an invasion-inducing gène, Tiam-1,that encodes a protein with homology to GDP-GTP exchangers for Rho-likeproteins. Cell 77:537.
217. Welch, H. C, W. J. Coadwell, C. D. Ellson, G. J. Ferguson, S. R. Andrews, H.Erdjument-Bromage, P. Tempst, P. T. Hawkins, and L. R. Stephens. 2002. P-Rexl,a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factorfor Rac. Cell 108:809.
218. Schmidt, A., and A. Hall. 2002. Guanine nucleotide exchange factors for RhoGTPases: turning on the switch. Gènes Dev 16:1587.
219. Bokoch, G. M., B. P. Bohl, and T. H. Chuang. 1994. Guanine nucleotide exchangerégulâtes membrane translocation of Rac/Rho GTP-binding proteins. J Biol Chem269:31674.
220. Moon, S., and Y. Zheng. 2003. Rho GTPase-activating proteins in cell régulation.Trends Cell Biol 13:13
221. Bishop, A. L., and A. Hall. 2000. Rho GTPases and their effector proteins. BiochemJ 348 Pt 2:241.
222. Ridley, A. J., and A. Hall. 1992. The small GTP-binding protein rho régulâtes theassembly of focal adhésions and actin stress fibers in response to growth factors.Cell 70:389.
223. Nobes, C. D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate theassembly of multimolecular focal complexes associated with actin stress fibers,lamellipodia, and filopodia. Cell 81:53.
224. Caron, E., and A. Hall. 1998. Identification of two distinct mechanisms ofphagocytosis controlled by différent Rho GTPases. Science 282:1717.
225. Chimini, G., and P. Chavrier. 2000. Function of Rho family proteins in actindynamics during phagocytosis and engulfment. Nat Cell Biol 2:El91.
226. Schwartz, M. A., and S. J. Shattil. 2000. Signaling networks linking integrins andrho family GTPases. Trends Biochem Sci 25:388.
227'. Cox, E. A., S. K. Sastry, and A. Huttenlocher. 2001. Integrin-mediated adhésionrégulâtes cell polarity and membrane protrusion through the Rho family ofGTPases. Mol Biol Cell 12:265.
228. Ellis, S., and H. Mellor. 2000. Régulation of endocytic traffic by rho familyGTPases. Trends Cell Biol 10:85.
229. Qualmann, B., and H. Mellor. 2003. Régulation of endocytic traffic by rhoGTPases. Biochem J371:233
230. Lacy, P. 2005. The rôle of Rho GTPases and SNAREs in mediator release fromgranulocytes. Pharmacol Ther 107:358.

197
231. Alblas, J., L. Ulfman, P. Hordijk, and L. Koenderman. 2001. Activation of Rhoaand ROCK are essential for detachment of migrating leukocytes. Mol Biol Cell12:2137.
232. Stasia, M. J., A. Jouan, N. Bourmeyster, P. Boquet, and P. V. Vignais. 1991. ADP-ribosylation of a small size GTP-binding protein in bovine neutrophils by the C3exoenzyme of Clostridium botulinum and effect on the cell motility. BiochemBiophys Res Commun 180:615.
233. Benard, V., B. P. Bohl, and G. M. Bokoch. 1999. Characterization of rac and cdc42activation in chemoattractant-stimulated human neutrophils using a novel assay foractive GTPases. J Biol Chem 274:13198.
234. Geijsen, N., S. van Delft, J. A. Raaijmakers, J. W. Lammers, J. G. Collard, L.Koenderman, and P. J. Coffer. 1999. Régulation of p21rac activation in humanneutrophils. Blood 94:1121.
235. Akasaki, T., H. Koga, and H. Sumimoto. 1999. Phosphoinositide 3-kinase-dependent and -independent activation of the small GTPase Rac2 in humanneutrophils. J Biol Chem 274:18055.
236. Fenteany, G., and M. Glogauer. 2004. Cytoskeletal remodeling in leukocytefunction. Curr Opin Hematol 11:15.
237. Wu, D. 2005. Signaling mechanisms for régulation of chemotaxis. Cell Res 15:52.238. Charest, P. G., and R. A. Firtel. 2006. Feedback signaling controls leading-edge
formation during chemotaxis. Curr Opin Genêt Dev 16:339.239. Laudanna, C, J. J. Campbell, and E. C. Butcher. 1996. Rôle of Rho in
chemoattractant-activated leukocyte adhésion through integrins. Science 271:981.240. Quinn, M. T., T. Evans, L. R. Loetterle, A. J. Jesaitis, and G. M. Bokoch. 1993.
Translocation of Rac correlates with NADPH oxidase activation. Evidence forequimolar translocation of oxidase components. J Biol Chem 268:20983.
241. Heyworth, P. G., B. P. Bohl, G. M. Bokoch, and J. T. Curnutte. 1994. Ractranslocates independently of the neutrophil NADPH oxidase components p47phoxand p67phox. Evidence for its interaction with flavocytochrome b558. J Biol Chem269:30749.
242. Abo, A., E. Pick, A. Hall, N. Totty, C. G. Teahan, and A. W. Segal. 1991.Activation of the NADPH oxidase involves the small GTP-binding protein p21racl.Nature 353:668.
243. Knaus, U. G., P. G. Heyworth, T. Evans, J. T. Curnutte, and G. M. Bokoch. 1991.Régulation of phagocyte oxygen radical production by the GTP-binding protein Rac2. Science 254:1512.
244. Knaus, U. G., P. G. Heyworth, B. T. Kinsella, J. T. Curnutte, and G. M. Bokoch.1992. Purification and characterization of Rac 2. A cytosolic GTP-binding proteinthat régulâtes human neutrophil NADPH oxidase. J Biol Chem 267:23575.
245. Diekmann, D., A. Abo, C. Johnston, A. W. Segal, and A. Hall. 1994. Interaction ofRac with p67phox and régulation of phagocytic NADPH oxidase activity. Science265:531.
246. Dorseuil, O., L. Reibel, G. M. Bokoch, J. Camonis, and G. Gacon. 1996. The Ractarget NADPH oxidase p67phox interacts preferentially with Rac2 rather than Racl.J Biol Chem 271:83.

198
247. Diebold, B. A., B. Fowler, J. Lu, M. C. Dinauer, and G. M. Bokoch. 2004.Antagonistic cross-talk between Rac and Cdc42 GTPases régulâtes génération ofreactive oxygen species. JBiol Chem 279:28136.
248. Roberts, A. W., C. Kim, L. Zhen, J. B. Lowe, R. Kapur, B. Petryniak, A. Spaetti, J.D. Pollock, J. B. Bornéo, G. B. Bradford, S. J. Atkinson, M. C. Dinauer, and D. A.Williams. 1999. Deficiency of the hematopoietic cell-specific Rho family GTPaseRac2 is characterized by abnormalities in neutrophil function and host défense.Immuni ty 10:183.
249. Kim, C, and M. C. Dinauer. 2001. Rac2 is an essential regulator of neutrophilnicotinamide adenine dinucleotide phosphate oxidase activation in response tospécifie signaling pathways. J Immunol 166:1223.
250. Abdel-Latif, D., M. Steward, D. Macdonald, G. Francis, M. Dinauer, and P. Lacy.2004. Rac2 is critical for neutrophil primary granule exocytosis. Blood 104:832
251. Ambruso, D. R., C. Knall, A. N. Abell, J. Panepinto, A. Kurkchubasche, G.Thurman, C. Gonzalez-Aller, A. Hiester, M. deBoer, R. J. Harbeck, R. Oyer, G. L.Johnson, and D. Roos. 2000. Human neutrophil immunodeficiency syndrome isassociated with an inhibitory Rac2 mutation. Proc NatlAcadSci USA 97:4654.
252. Diebold, B. A., and G. M. Bokoch. 2005. Rho GTPases and the control of theoxidative burst in polymorphonuclear leukocytes. Curr Top Microbiol Immunol291:91.
253. Rabiet, M. J., M. Tardif, L. Braun, and F. Boulay. 2002. Inhibitory effects of adominant-interfering form of the Rho-GTPase Cdc42 in the chemoattractant-elicitedsignaling pathways leading to NADPH oxidase activation in differentiated HL-60ccWs.Blood 100:1835.
254. Barber, M. A., and H. C. Welch. 2006. PI3K and RAC signalling in leukocyte andcancer cell migration. Bull Cancer 93.E44.
255. Powner, D., and M. Wakelam. 2002. The régulation of phospholipase D by inositolphospholipids and small GTPases. FEBS Lett 531:62
256. Klarlund, J. K., A. Guilherme, J. J. Holik, J. V. Virbasius, A. Chawla, and M. P.Czech. 1997. Signaling by phosphoinositide-3,4,5-trisphosphate through proteinscontaining pleckstrin and Sec7 homology domains. Science 275:1927.
257. Klarlund, J. K., L. E. Rameh, L. C. Cantley, J. M. Buxton, J. J. Holik, C. Sakelis, V.Patki, S. Corvera, and M. P. Czech. 1998. Régulation of GRPl-catalyzed ADPribosylation factor guanine nucleotide exchange by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273:1859.
258. Klarlund, J. K., W. Tsiaras, J. J. Holik, A. Chawla, and M. P. Czech. 2000. Distinctpolyphosphoinositide binding selectivities for pleckstrin homology domains ofGRPl-like proteins based on diglycine versus triglycine motifs. J Biol Chem275:32816.
259. D'Souza-Schorey, C , and P. Chavrier. 2006. ARF proteins: rôles in membranetraffic and beyond. Nat Rev Mol Cell Biol 7:347.
260. Santarius, M., C. H. Lee, and R. A. Anderson. 2006. Supervised membraneswimming: small G-protein lifeguards regulate PIPK signalling and monitorintracellular PtdIns(4,5)P2 pools. BiochemJ 398:1.
261. Niggli, V. 2005. Régulation of protein activities by phosphoinositide phosphates.Annu Rev Cell Dev Biol 21:57.

199
262. Dana, R. R., C. Eigsti, K. L. Holmes, and T. L. Leto. 2000. A regulatory rôle forADP-ribosylation factor 6 (ARF6) in activation of the phagocyte NADPH oxidase.JBiolChem 275:32566.
263. Frohman, M. A., T. C. Sung, and A. J. Morris. 1999. Mammalian phospholipase Dstructure and régulation. Biochim Biophys Acta 1439:175.
264. Fensome, A., J. Whatmore, C. Morgan, D. Jones, and S. Cockcroft. 1998. ADP-ribosylation factor and Rho proteins médiate fMLP-dependent activation ofphospholipase D in human neutrophils. JBiol Chem 273:13157.
265. Pai, J. K., M. I. Siegel, R. W. Egan, and M. M. Billah. 1988. Activation ofphospholipase D by chemotactic peptide in HL-60 granulocytes. Biochem BiophysRes Commun 150:355.
266. Pai, J. K., M. I. Siegel, R. W. Egan, and M. M. Billah. 1988. Phospholipase Dcatalyzes phospholipid metabolism in chemotactic peptide-stimulated HL-60granulocytes. J Biol Chem 263:12472.
267. Billah, M. M., S. Eckel, T. J. Mullmann, R. W. Egan, and M. I. Siegel. 1989.Phosphatidylcholine hydrolysis by phospholipase D détermines phosphatidate anddiglyceride levels in chemotactic peptide-stimulated human neutrophils.Involvement of phosphatidate phosphohydrolase in signal transduction. J Biol Chem264:17069.
268. Lopez, I., R. S. Arnold, and J. D. Lambeth. 1998. Cloning and initialcharacterization of a human phospholipase D2 (hPLD2). ADP-ribosylation factorrégulâtes hPLD2. J Biol Chem 273:12846.
269. Hammond, S. M., J. M. Jenco, S. Nakashima, K. Cadwallader, Q. Gu, S. Cook, Y.Nozawa, G. D. Prestwich, M. A. Frohman, and A. J. Morris. 1997. Characterizationof two alternately spliced forms of phospholipase Dl. Activation of the purifiedenzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, andRho family monomeric GTP-binding proteins and protein kinase C-alpha. J BiolChem 272:3860.
270. Steed, P. M., K. L. Clark, W. C. Boyar, and D. J. Lasala. 1998. Characterization ofhuman PLD2 and the analysis of PLD isoform splice variants. Faseb J 12:1309.
271. Marcil, J., D. Harbour, M. G. Houle, P. H. Naccache, and S. Bourgoin. 1999.Monosodium urate-crystal-stimulated phospholipase D in human neutrophils.Biochem J 337 (Pt2):185.
272. Jenkins, G. M., and M. A. Frohman. 2005. Phospholipase D: a lipid centric review.Cell Mol Life Sci 62:2305.
273. Hodgkin, M. N., M. R. Masson, D. Powner, K. M. Saqib, C. P. Ponting, and M. J.Wakelam. 2000. Phospholipase D régulation and localisation is dépendent upon aphosphatidylinositol 4,5-biphosphate-specific PH domain. Curr Biol 10:43.
21 A. Powner, D. J., and M. J. Wakelam. 2002. The régulation of phospholipase D byinositol phospholipids and small GTPases. FEBS Lett 531:62.
275. Conricode, K. M., K. A. Brewer, and J. H. Exton. 1992. Activation ofphospholipase D by protein kinase C. Evidence for a phosphorylation-independentmechanism. J Biol Chem 267:7199.
276. Conricode, K. M., J. L. Smith, D. J. Burns, and J. H. Exton. 1994. Phospholipase Dactivation in fibroblast membranes by the alpha and beta isoforms of protein kinaseC. FEBS Lett 342:149.

200
277. Lopez, I., D. J. Burns, and J. D. Lambeth. 1995. Régulation of phospholipase D byprotein kinase C in human neutrophils. Conventional isoforms of protein kinase Cphosphorylate a phospholipase D-related component in the plasma membrane. JBiolChem 270:19465.
278. Hu, T., and J. Exton. 2003. Mechanisms of régulation of phospholipase Dl byprotein kinase Ca. J Biol Chem 278:2348
279. Singer, W. D., H. A. Brown, G. M. Bokoch, and P. C. Sternweis. 1995. Resolvedphospholipase D activity is modulated by cytosolic factors other than Arf. J BiolChem 270:14944.
280. Sung, T. C, Y. Zhang, A. J. Morris, and M. A. Frohman. 1999. Structural analysisof human phospholipase Dl. J Biol Chem 274:3659.
281. Exton, J. H. 1999. Régulation of phospholipase D. Biochim Biophys Acta 1439:121.282. Kessels, G. C, D. Roos, and A. J. Verhoeven. 1991. fMet-Leu-Phe-induced
activation of phospholipase D in human neutrophils. Dependence on changes incytosolic free Ca2+ concentration and relation with respiratory burst activation. JBiol Chem 266:23152.
283. Brown, H. A., S. Gutowski, C. R. Moomaw, C. Slaughter, and P. C. Sternweis.1993. ADP-ribosylation factor, a small GTP-dependent regulatory protein,stimulâtes phospholipase D activity. Cell 75:1137.
284. Brown, H. A., S. Gutowski, R. A. Kahn, and P. C. Sternweis. 1995. Partialpurification and characterization of Arf-sensitive phospholipase D from porcinebrain. J Biol Chem 270:14935.
285. Cockcroft, S., G. M. Thomas, A. Fensome, B. Geny, E. Cunningham, I. Goût, I.Hiles, N. F. Totty, O. Truong, and J. J. Hsuan. 1994. Phospholipase D: adownstream effector of ARF in granulocytes. Science 263:523.
286. Hammond, S. M., Y. M. Altshuller, T. C. Sung, S. A. Rudge, K. Rosé, J.Engebrecht, A. J. Morris, and M. A. Frohman. 1995. Human ADP-ribosylationfactor-activated phosphatidylcholine-specific phospholipase D defines a new andhighly conserved gène family. J Biol Chem 270:29640.
287. Bowman, E. P., D. J. Uhlinger, and J. D. Lambeth. 1993. Neutrophil phospholipaseD is activated by a membrane-associated Rho family small molecular weight GTP-binding protein. J Biol Chem 268:21509.
288. Cai, S., and J. H. Exton. 2001. Détermination of interaction sites of phospholipaseDl for RhoA. BiochemJ355:779.
289. Yamazaki, M., Y. Zhang, H. Watanabe, T. Yokozeki, S. Ohno, K. Kaibuchi, H.Shibata, H. Mukai, Y. Ono, M. A. Frohman, and Y. Kanaho. 1999. Interaction ofthe small G protein RhoA with the C terminus of human phospholipase Dl. J BiolChem 274:6035.
290. Exton, J. 2002. Régulation of phospholipase D. FEBS Lett 531:58291. Singer, W. D., H. A. Brown, X. Jiang, and P. C. Sternweis. 1996. Régulation of
phospholipase D by protein kinase C is synergistic with ADP-ribosylation factorand independent of protein kinase activity. J Biol Chem 271:4504.
292. Ohguchi, K., Y. Banno, S. Nakashima, and Y. Nozawa. 1996. Régulation ofmembrane-bound phospholipase D by protein kinase C in HL60 cells. Synergisticaction of small GTP-binding protein RhoA. J Biol Chem 271:4366.
293. Du, G., Y. M. Altshuller, Y. Kim, J. M. Han, S. H. Ryu, A. J. Morris, and M. A.Frohman. 2000. Dual requirement for rho and protein kinase C in direct activation

201
of phospholipase Dl through G protein-coupled receptor signaling. Mol Biol Cell11:4359.
294. Houle, M. G., P. H. Naccache, and S. Bourgoin. 1999. Tyrosine kinase-regulatedsmall GTPase translocation and the activation of phospholipase D in HL60granulocytes. J Leukoc Biol 66:1021.
295. Naccache, P. H., A. C. Caon, C. Gilbert, M. Gaudry, C. J. Roberge, P. E. Poubelle,and S. Bourgoin. 1993. Inhibition of tyrosine phosphorylation by wortmannin inhuman neutrophils. Dissociation from its inhibitory effects on phospholipase D. LabInvest 69:19.
296. Nakamura, M., S. Nakashima, Y. Katagiri, and Y. Nozawa. 1997. Effect ofwortmannin and 2-(4-morpholinyl)-8-phenyl-4H-l-benzopyran-4-one (LY294002)on N-formyl-methionyl-leucyl-phenylalanine-induced phospholipase D activation indifferentiated HL60 cells: possible involvement of phosphatidylinositol 3-kinase inphospholipase D activation. Biochem Pharmacol 53:1929.
297. Jones, D., C. Morgan, and S. Cockcroft. 1999. Phospholipase D and membranetraffic. Potential rôles in regulated exocytosis, membrane delivery and vesiclebudding. Biochim Biophys Acta 1439:229.
298. Lennartz, M. R. 1999. Phospholipases and phagocytosis: the rôle of phospholipid-derived second messengers in phagocytosis. Int J Biochem Cell Biol 31:415.
299. Steed, P. M., and A. H. Chow. 2001. Intracellular signaling by phospholipase D as atherapeutic target. Curr Pharm Biotechnol 2:241.
300. Bonser, R. W., N. T. Thompson, R. W. Randall, and L. G. Garland. 1989.Phospholipase D activation is functionally linked to superoxide génération in thehuman neutrophil. Biochem J 264:617.
301. McPhail, L. C, D. Qualliotine-Mann, and K. A. Waite. 1995. Cell-free activation ofneutrophil NADPH oxidase by a phosphatidic acid-regulated protein kinase. ProcNatlAcadSci USA 92:7931.
302. Regier, D. S., D. G. Greene, S. Sergeant, A. J. Jesaitis, and L. C. McPhail. 2000.Phosphorylation of p22phox is mediated by phospholipase D-dependent and -independent mechanisms. Corrélation of NADPH oxidase activity and p22phoxphosphorylation. J Biol Chem 275:28406.
303. Waite, K. A., R. Wallin, D. Qualliotine-Mann, and L. C. McPhail. 1997.Phosphatidic acid-mediated phosphorylation of the NADPH oxidase componentp47-phox. Evidence that phosphatidic acid may activate a novel protein kinase. JBiol Chem 272:15569.
304. Karathanassis, D., R. V. Stahelin, J. Bravo, O. Perisic, C. M. Pacold, W. Cho, andR. L. Williams. 2002. Binding of the PX domain of p47(phox) tophosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by anintramolecular interaction. Embo J21.5057.
305. Kanaho, Y., H. Kanoh, K. Saitoh, and Y. Nozawa. 1991. Phospholipase Dactivation by platelet-activating factor, leukotriene B4, and formyl-methionyl-leucyl-phenylalanine in rabbit neutrophils. Phospholipase D activation is involvedin enzyme release. J Immunol 146:3536.
306. Imajo, M., Y. Tsuchiya, and E. Nishida. 2006. Regulatory mechanisms andfunctions of MAP kinase signaling pathways. IUBMB Life 58:312.

202
307. Grinstein, S., and W. Furuya. 1992. Chemoattractant-induced tyrosinephosphorylation and activation of microtubule-associated protein kinase in humanneutrophils. J Biol Chem 267:18122.
308. Worthen, G. S., N. Avdi, A. M. Buhl, N. Suzuki, and G. L. Johnson. 1994. FMLPactivâtes Ras and Raf in human neutrophils. Potential rôle in activation of MAPkinase. J Clin Invest 94:815.
309. Torres, M., F. L. Hall, and K. O'Neill. 1993. Stimulation of human neutrophils withformyl-methionyl-leucyl-phenylalanine induces tyrosine phosphorylation andactivation of two distinct mitogen-activated protein-kinases. J Immunol 150:1563.
310. Thompson, H. L., M. Shiroo, and J. Saklatvala. 1993. The chemotactic factor N-formylmethionyl-leucyl-phenylalanine activâtes microtubule-associated protein 2(MAP) kinase and a MAP kinase kinase in polymorphonuclear leucocytes. BiochemJ290(Pt 2): 483.
311. Krump, E., J. S. Sanghera, S. L. Pelech, W. Furuya, and S. Grinstein. 1997.Chemotactic peptide N-formyl-met-leu-phe activation of p38 mitogen-activatedprotein kinase (MAPK) and MAPK-activated protein kinase-2 in humanneutrophils. J Biol Chem 272:937.
312. El Benna, J., J. Han, J. W. Park, E. Schmid, R. J. Ulevitch, and B. M. Babior. 1996.Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidasecomponent p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys334:395.
313. Rane, M. J., S. L. Carrithers, J. M. Arthur, J. B. Klein, and K. R. McLeish. 1997.Formyl peptide receptors are coupled to multiple mitogen-activated protein kinasecascades by distinct signal transduction pathways: rôle in activation of reducednicotinamide adenine dinucleotide oxidase. J Immunol 159:5070.
314. Zhang, H., C. D. Garlichs, A. Mugge, and W. G. Daniel. 1998. Involvement oftyrosine kinases, Ca2+ and PKC in activation of mitogen-activated protein (MAP)kinase in human polymorphonuclear neutrophils. J Physiol 513 ( Pt 2):359.
315. Kagari, T., D. Tanaka, H. Doi, and T. Shimozato. 2003. Essential rôle of Fc gammareceptors in anti-type II collagen antibody-induced arthritis. J Immunol 170:4318
316. Mocsai, A., Z. Jakus, T. Vantus, G. Berton, C. A. Lowell, and E. Ligeti. 2000.Kinase pathways in chemoattractant-induced degranulation of neutrophils: the rôleof p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol164:4321.
317. Lopez-Ilasaca, M., P. Crespo, P. G. Pellici, J. S. Gutkind, and R. Wetzker. 1997.Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI3-kinase gamma. Science 275:394.
318. Bondeva, T., L. Pirola, G. Bulgarelli-Leva, I. Rubio, R. Wetzker, and M. P.Wymann. 1998. Bifurcation of lipid and protein kinase signais of PDKgamma tothe protein kinases PKB and MAPK. Science 282:293.
319. Zu, Y. L., J. Qi, A. Gilchrist, G. A. Fernandez, D. Vazquez-Abad, D. L. Kreutzer,C. K. Huang, and R. I. Sha'afi. 1998. p38 mitogen-activated protein kinaseactivation is required for human neutrophil function triggered by TNF-alpha orFMLP stimulation. J Immunol 160:1982.
320. Heit, B., S. Tavener, E. Raharjo, and P. Kubes. 2002. An intracellular signalinghierarchy détermines direction of migration in opposing chemotactic gradients. JCell Biol 159:91.

203
321. Nick, J. A., N. J. Avdi, S. K. Young, C. Knall, P. Gerwins, G. L. Johnson, and G. S.Worthen. 1997. Common and distinct intracellular signaling pathways in humanneutrophils utilized by platelet activating factor and FMLP. J Clin Invest 99:975.
322. Ma, A. D., A. Metjian, S. Bagrodia, S. Taylor, and C. S. Abrams. 1998.Cytoskeletal reorganization by G protein-coupled receptors is dépendent onphosphoinositide 3-kinase gamma, a Rac guanosine exchange factor, and Rac. MolCell Biol 18:4744.
323. Jackowski, S., and R. I. Sha'afi. 1979. Response of adenosine cyclic 3',5'-monophosphate level in rabbit neutrophils to the chemotactic peptide formyl-methionyl-leucyl-phenylalanine. Mol Pharmacol 16:473.
324. Simchowitz, L., L. C. Fischbein, I. Spilberg, and J. P. Atkinson. 1980. Induction ofa transient élévation in intracellular levels of adenosine-3',5'-cyclic monophosphateby chemotactic factors: an early event in human neutrophil activation. J Immunol124:1482.
325. Smolen, J. E., H. M. Korchak, and G. Weissmann. 1980. Increased levels of cyclicadenosine-3',5'-monophosphate in human polymorphonuclear leukocytes aftersurface stimulation. J Clin Invest 65:1077.
326. Spisani, S., M. C. Pareschi, M. Buzzi, M. L. Colamussi, C. Biondi, S. Traniello, G.Pagani Zecchini, M. Paglialunga Paradisi, I. Torrini, and M. E. Ferretti. 1996. Effectof cyclic AMP level réduction on human neutrophil responses to formylatedpeptides. Cell Signal 8:269.
327. Verghese, M. W., K. Fox, L. C. McPhail, and R. Snyderman. 1985.Chemoattractant-elicited altérations of cAMP levels in human polymorphonuclearleukocytes require a Ca2+-dependent mechanism which is independent oftransmembrane activation of adenylate cyclase. J Biol Chem 260:6769.
328. Cooper, D. M. 2003. Régulation and organization of adenylyl cyclases and cAMP.BiochemJ 375:517.
329. Ferretti, M. E., M. Nalli, C. Biondi, M. L. Colamussi, B. Pavan, S. Traniello, and S.Spisani. 2001. Modulation of neutrophil phospholipase C activity and cyclic AMPlevels by fMLP-OMe analogues. Cell Signal 13:233.
330. Mahadeo, D. C, M. Janka-Junttila, R. L. Smoot, P. Roselova, and C. A. Parent.2006. A Chemoattractant-mediated Gi-coupled Pathway Activâtes AdenylylCyclase in Human Neutrophils. Mol Biol Cell.
331. Schudt, C, S. Winder, S. Forderkunz, A. Hatzelmann, and V. Ullrich. 1991.Influence of sélective phosphodiesterase inhibitors on human neutrophil functionsand levels of cAMP and Cai. Naunyn Schmiedebergs Arch Pharmacol 344:682.
332. Wang, P., P. Wu, K. M. Ohleth, R. W. Egan, and M. M. Billah. 1999.Phosphodiesterase 4B2 is the prédominant phosphodiesterase species and undergoesdifferential régulation of gène expression in human monocytes and neutrophils. MolPharmacol 56:170.
333. Simchowitz, L., I. Spilberg, and J. P. Atkinson. 1983. Evidence that the functionalresponses of human neutrophils occur independently of transient élévations incyclic AMP levels. J Cyclic Nucleotide Protein Phosphor Res 9:35.
334. Elferink, J. G., and B. M. de Koster. 1993. The effect of cyclic GMP and cyclicAMP on migration by electroporated human neutrophils. Eur J Pharmacol 246:157.

204
335. Downey, G. P., E. L. Eison, B. Schwab, 3rd, S. C. Erzurum, S. K. Young, and G. S.Worthen. 1991. Biophysical properties and microfilament assembly in neutrophils:modulation by cyclic AMP. J Ce// Biol 114:1179.
336. Ydrenius, L., L. Molony, J. Ng-Sikorski, and T. Andersson. 1997. Dual action ofcAMP-dependent protein kinase on granulocyte movement. Biochem Biophys ResCommun 235:445.
337. Jones, S. L., and Y. Sharief. 2005. Asymmetrical protein kinase A activityestablishes neutrophil cytoskeletal polarity and enables chemotaxis. J Leukoc Biol78:248.
338. Ottonello, L., M. P. Morone, P. Dapino, and F. Dallegri. 1995. Cyclic AMP-elevating agents down-regulate the oxidative burst induced by granulocyte-macrophage colony-stimulating factor (GM-CSF) in adhèrent neutrophils. Clin ExpImmunol 101:502.
339. Cronstein, B. N., S. B. Kramer, E. D. Rosenstein, G. Weissmann, and R.Hirschhorn. 1985. Adenosine modulâtes the génération of superoxide anion bystimulated human neutrophils via interaction with a spécifie cell surface receptor.AnnN YAcadSci 451:291.
340. Rivkin, L, J. Rosenblatt, and E. L. Becker. 1975. The rôle of cyclic AMP in thechemotactic responsiveness and spontaneous motility of rabbit peritonealneutrophils. The inhibition of neutrophil movement and the élévation of cyclicAMP levels by catecholamines, prostaglandins, theophylline and choiera toxin. JImmunol 115:1126.
341. Harvath, L., J. D. Robbins, A. A. Russell, and K. B. Seamon. 1991. cAMP andhuman neutrophil chemotaxis. Elévation of cAMP differentially affects chemotacticresponsiveness. J Immunol 146:224.
342. Fantone, J. C, S. L. Kunkel, and P. A. Ward. 1981. Suppression of humanpolymorphonuclear function after intravenous infusion of prostaglandin El.Prostaglandins Med 7:195.
343. Lad, P. M., B. J. Goldberg, P. A. Smiley, and C. V. Oison. 1985. Receptor-specificthreshold effects of cyclic AMP are involved in the régulation of enzyme releaseand superoxide production from human neutrophils. Biochim Biophys Acta 846:286.
344. Zurier, R. B., G. Weissmann, S. Hoffstein, S. Kammerman, and H. H. Tai. 1974.Mechanisms of lysosomal enzyme release from human leukocytes. II. Effects ofcAMP and cGMP, autonomie agonists, and agents which affect microtubulefunction. J Clin Invest 53:297.
345. Bloemen, P. G., M. C. van den Tweel, P. A. Henricks, F. Engels, M. H. Kester, P.G. van de Loo, F. J. Blomjous, and F. P. Nijkamp. 1997. Increased cAMP levels instimulated neutrophils inhibit their adhésion to human bronchial epithelial cells. AmJPhysiol 272:1580.
346. Derian, C. K., R. J. Santulli, P. E. Rao, H. F. Solomon, and J. A. Barrett. 1995.Inhibition of chemotactic peptide-induced neutrophil adhésion to vascularendothelium by cAMP modulators. J Immunol 154:308.
347. Krump, E., and P. Borgeat. 1999. Adenosine. An endogenous inhibitor ofarachidonic acid release and leukotriene biosynthesis in human neutrophils. AdvExp Med Biol 447:107.
348. Flamand, N., S. Boudreault, S. Picard, M. Austin, M. E. Surette, H. Plante, E.Krump, M. J. Vallée, C. Gilbert, P. Naccache, M. Laviolette, and P. Borgeat. 2000.

205
Adenosine, a potent natural suppressor of arachidonic acid release and leukotrienebiosynthesis in human neutrophils. Am J Respir Crit Care Med 161:S88.
349. Fonteh, A. N., J. D. Winkler, T. J. Torphy, J. Heravi, B. J. Undem, and F. H.Chilton. 1993. Influence of isoproterenol and phosphodiesterase inhibitors onplatelet-activating factor biosynthesis in the human neutrophil. JImmunol 151:339.
350. Nathan, C, and E. Sanchez. 1990. Tumor necrosis factor and CD11/CD18 (beta 2)integrins act synergistically to lower cAMP in human neutrophils. J Cell Biol111:2171.
351. Linden, J. 2001. Molecular approach to adenosine receptors: receptor-mediatedmechanisms of tissue protection. Annu Rev Pharmacol Toxicol 41:775.
352. Bazzichi, L., L. Trincavelli, A. Rossi, F. De Feo, A. Lucacchini, S. Bombardieri,and C. Martini. 2005. A2B adenosine receptor activity is reduced in neutrophilsfrom patients with systemic sclerosis. Arthritis Res Ther 7:RI89.
353. Cronstein, B. N., L. Daguma, D. Nichols, A. J. Hutchison, and M. Williams. 1990.The adenosine/neutrophil paradox resolved: human neutrophils possess both Al andA2 receptors that promote chemotaxis and inhibit 02 génération, respectively. JClin Invest 85:1150.
354. Gessi, S., K. Varani, S. Merighi, E. Ongini, and P. A. Borea. 2000. A(2A)adenosine receptors in human peripheral blood cells. Br J Pharmacol 129:2.
355. Gessi, S., K. Varani, S. Merighi, E. Cattabriga, V. Iannotta, E. Leung, P. G. Baraldi,and P. A. Borea. 2002. A(3) adenosine receptors in human neutrophils andpromyelocytic HL60 cells: a pharmacological and biochemical study. MolPharmacol 61:415.
356. Falleni, A., M. L. Trincavelli, M. Macchia, F. Salvetti, M. Hamdan, F. Calvani, V.Gremigni, A. Lucacchini, and C. Martini. 1999. A(l) adenosine receptors in humanneutrophils: direct binding and électron microscope visualization. J Cell Biochem75:235.
357. Fredholm, B. B., Y. Zhang, and I. van der Ploeg. 1996. Adenosine A2A receptorsmédiate the inhibitory effect of adenosine on formyl-Met-Leu-Phe-stimulatedrespiratory burst in neutrophil leucocytes. Naunyn Schmiedebergs Arch Pharmacol354:262.
358. Bouma, M. G., T. M. Jeunhomme, D. L. Boyle, M. A. Dentener, N. N. Voitenok, F.A. van den Wildenberg, and W. A. Buurman. 1997. Adenosine inhibits neutrophildegranulation in activated human whole blood: involvement of adenosine A2 andA3 receptors. J Immunol 158:5400.
359. Murakami, S., T. Hashikawa, T. Saho, M. Takedachi, T. Nozaki, Y. Shimabukuro,and H. Okada. 2001. Adenosine régulâtes the IL-1 beta-induced cellular functionsof human gingival fibroblasts. Int Immunol 13:1533.
360. Fortin, A., D. Harbour, M. Fernandes, P. Borgeat, and S. Bourgoin. 2006.Differential expression of adenosine receptors in human neutrophils: up-regulationby spécifie Thl cytokines and lipopolysaccharide. J Leukoc Biol 79:574.
361. Cronstein, B. N., R. I. Levin, M. Philips, R. Hirschhorn, S. B. Abramson, and G.Weissmann. 1992. Neutrophil adhérence to endothelium is enhanced via adenosineAl receptors and inhibited via adenosine A2 receptors. J Immunol 148:2201.
362. Cronstein, B. N., R. I. Levin, J. Belanoff, G. Weissmann, and R. Hirschhorn. 1986.Adenosine: an endogenous inhibitor of neutrophil-mediated injury to endothelialcells. J Clin Invest 78:760.

206
363. Wollner, A., S. Wollner, and J. B. Smith. 1993. Acting via A2 receptors, adenosineinhibits the upregulation of Mac-1 (Cdl lb/CD18) expression on FMLP-stimulatedneutrophils. Am J Respir Cell Mol Biol 9:179.
364. Zalavary, S., and T. Bengtsson. 1998. Adenosine inhibits actin dynamics in humanneutrophils: évidence for the involvement of cAMP. Eur J Cell Biol 75:128.
365. Ernens, I., D. Rouy, E. Velot, Y. Devaux, and D. R. Wagner. 2006. Adenosineinhibits matrix métalloproteinase-9 sécrétion by neutrophils: implication of A2areceptor and cAMP/PKA/Ca2+ pathway. Cire Res 99:590.
366. Krump, E., S. Picard, J. Mancini, and P. Borgeat. 1997. Suppression of leukotrieneB4 biosynthesis by endogenous adenosine in ligand-activated human neutrophils. JExpMed 186:1401.
367. Flamand, N., J. Lefebvre, G. Lapointe, S. Picard, L. Lemieux, S. G. Bourgoin, andP. Borgeat. 2006. Inhibition of platelet-activating factor biosynthesis by adenosineand histamine in human neutrophils: involvement of cPLA2{alpha} and reversai bylyso-PAF. J Leukoc Biol.
368. Cronstein, B. N. 1994. Adenosine, an endogenous anti-inflammatory agent. JApplPhysiol 76:5.
369. Cronstein, B. N., E. D. Rosenstein, S. B. Kramer, G. Weissmann, and R.Hirschhorn. 1985. Adenosine; a physiologie modulator of superoxide aniongénération by human neutrophils. Adenosine acts via an A2 receptor on humanneutrophils. J Immunol 135:1366.
370. Krump, E., G. Lemay, and P. Borgeat. 1996. Adenosine A2 receptor-inducedinhibition of leukotriene B4 synthesis in whole blood ex vivo. Br J Pharmacol117:1639.
371. Fredholm, B. B., M. P. Abbracchio, G. Burnstock, J. W. Daly, T. K. Harden, K. A.Jacobson, P. Leff, and M. Williams. 1994. Nomenclature and classification ofpurinoceptors. Pharmacol Rev 46:143.
372. Cronstein, B. N., S. B. Kramer, E. D. Rosenstein, H. M. Korchak, G. Weissmann,and R. Hirschhorn. 1988. Occupancy of adenosine receptors raises cyclic AMPalone and in synergy with occupancy of chemoattractant receptors and inhibitsmembrane depolarization. Biochem J 252:709.
373. Ohta, A., and M. Sitkovsky. 2001. Rôle of G-protein-coupled adenosine receptors indownregulation of inflammation and protection from tissue damage. Nature414:916.
374. Gessi, S., K. Varani, S. Merighi, E. Cattabriga, V. Iannotta, E. Leung, P. Baraldi,and P. Borea. 2002. A(3) adenosine receptors in human neutrophils andpromyelocytic HL60 cells: A pharmacological and biochemical study. MolPharmacol 61:415
375. Simmons, D. L., R. M. Botting, and T. Hla. 2004. Cyclooxygenase isozymes: thebiology of prostaglandin synthesis and inhibition. Pharmacol Rev 56:387.
376. Hamberg, M., and B. Samuelsson. 1965. Isolation and structure of a newprostaglandin from human séminal plasma. Prostaglandins and related factors 43.Biochim Biophys Acta 106:215.
377'. Hamberg, M., and B. Samuelsson. 1966. Prostaglandins in human séminal plasma.Prostaglandins and related factors 46. J Biol Chem 241:257.
378. Hamberg, M., and B. Samuelsson. 1967. On the mechanism of the biosynthesis ofprostaglandins E-l and F-1-alpha. J Biol Chem 242:5336.

207
379. Smith, W. L., D. L. DeWitt, and R. M. Garavito. 2000. Cyclooxygenases: structural,cellular, and molecular biology. Annu Rev Biochem 69:145.
380. Gilbert, C, P. Poubelle, P. Borgeat, M. Pouliot, and P. Naccache. 2003. Crystal-induced neutrophil activation - VIII. Immédiate production of prostaglandin E-2mediated by constitutive cyclooxygenase 2 in human neutrophils stimulated byurate crystals. Arthritis Rheum 48:1137
381. McGuire, J. C, and F. F. Sun. 1980. Metabolism of arachidonic acid andprostaglandin endoperoxide by assorted leukocytes. Adv ProstaglandinThromboxane Res 8:1665.
382. Herrmann, F., A. Lindemann, J. Gauss, and R. Mertelsmann. 1990. Cytokine-stimulation of prostaglandin synthesis from endogenous and exogenous arachidonicacids in polymorphonuclear leukocytes involving activation and new synthesis ofcyclooxygenase. Eur JImmunol 20:2513.
383. Pouliot, M., C. Gilbert, P. Borgeat, P. E. Poubelle, S. Bourgoin, C. Creminon, J.Maclouf, S. R. McColl, and P. H. Naccache. 1998. Expression and activity ofprostaglandin endoperoxide synthase-2 in agonist-activated human neutrophils.FasebJ 12:1109.
384. Fasano, M. B., J. D. Wells, and C. E. McCall. 1998. Human neutrophils express theprostaglandin G/H synthase 2 gène when stimulated with bacteriallipopolysaccharide. Clin Immunol Immunopathol 87:304.
385. Maloney, C. G., W. A. Kutchera, K. H. Albertine, T. M. Mclntyre, S. M. Prescott,and G. A. Zimmerman. 1998. Inflammatory agonists induce cyclooxygenase type 2expression by human neutrophils. J Immunol 160:1402.
386. Niiro, H., T. Otsuka, K. Izuhara, K. Yamaoka, K. Ohshima, T. Tanabe, S. Hara, Y.Nemoto, Y. Tanaka, H. Nakashima, and Y. Niho. 1997. Régulation by interleukin-10 and interleukin-4 of cyclooxygenase-2 expression in human neutrophils. Blood89:1621.
387. Breyer, R. M., C. K. Bagdassarian, S. A. Myers, and M. D. Breyer. 2001.Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41:661.
388. Williams, T. J., and M. J. Peck. 1977. Rôle of prostaglandin-mediatedvasodilatation in inflammation. Nature 270:530.
389. Crunkhorn, P., and A. L. Willis. 1969. Actions and interactions of prostaglandinsadministered intradermally in rat and in man. Br J Pharmacol 36.216P+.
390. Moncada, S., S. H. Ferreira, and J. R. Vane. 1973. Prostaglandins, aspirin-like drugsand the oedema of inflammation. Nature 246:217.
391. Ferreira, S. H., B. B. Lorenzetti, and F. M. Correa. 1978. Blockade of central andperipheral génération of prostaglandins explains the antialgic effect of aspirin likedrugs. PolJ Pharmacol Pharm 30:133.
392. Zurier, R. B. 1974. Prostaglandins, inflammation, and asthma. Arch Intern Med133:101.
393. Vane, J. R. 1971. Inhibition of prostaglandin synthesis as a mechanism of action foraspirin-like drugs. Nat New Biol 231:232.
394. Ferreira, S. H., S. Moncada, and J. R. Vane. 1971. Indomethacin and aspirin abolishprostaglandin release from the spleen. Nat New Biol 231:237.
395. Turini, M. E., and R. N. DuBois. 2002. Cyclooxygenase-2: a therapeutic target.Annu Rev Med 53:35.

208
396. Aspinall, R. L., and P. S. Cammarata. 1969. Effect of prostaglandin E2 on adjuvantarthritis. Nature 224:1320.
397'. Zurier, R. B., and F. Quagliata. 1971. Effect of prostaglandin E 1 on adjuvantarthritis. Nature 234:304.
398. Kunkel, S. L., R. S. Thrall, R. G. Kunkel, J. R. McCormick, P. A. Ward, and R. B.Zurier. 1979. Suppression of immune complex vasculitis in rats by prostaglandin. JClin Invest 64:1525.
399. Rossetti, R. G., K. Brathwaite, and R. B. Zurier. 1994. Suppression of acuteinflammation with liposome associated prostaglandin El. Prostaglandins 48:187.
400. Weissmann, G. 1993. Prostaglandins as modulators rather than mediators ofinflammation. J LipidMédiat 6:275.
401. Tilley, S. L., T. M. Coffman, and B. H. Koller. 2001. Mixed messages: modulationof inflammation and immune responses by prostaglandins and thromboxanes. J ClinInvest 108:15.
402. Fantone, J. C, and D. A. Kinnes. 1983. Prostaglandin El and prostaglandin 12modulation of superoxide production by human neutrophils. Biochem Biophys ResCommun 113:506.
403. Sedgwick, J. B., M. L. Berube, and R. B. Zurier. 1985. Stimulus-dependentinhibition of superoxide génération by prostaglandins. Clin Immunol Immunopathol34:205.
404. Gryglewski, R. J., A. Szczeklik, and M. Wandzilak. 1987. The effect of sixprostaglandins, prostacyclin and iloprost on génération of superoxide anions byhuman polymorphonuclear leukocytes stimulated by zymosan or formyl-methionyl-leucyl-phenylalanine. Biochem Pharmacol 36:4209.
405. Hecker, G., P. Ney, and K. Schror. 1990. Cytotoxic enzyme release and oxygencentered radical formation in human neutrophils are selectively inhibited by E-typeprostaglandins but not by PGI2. Naunyn Schmiedebergs Arch Pharmacol 341:308.
406. Ham, E. A., D. D. Soderman, M. E. Zanetti, H. W. Dougherty, E. McCauley, and F.A. Kuehl, Jr. 1983. Inhibition by prostaglandins of leukotriene B4 release fromactivated neutrophils. Proc NatlAcadSci USA 80:4349.
407. Wheeldon, A., and C. J. Vardey. 1993. Characterization of the inhibitory prostanoidreceptors on human neutrophils. Br J Pharmacol 108:1051.
408. Talpain, E., R. A. Armstrong, R. A. Coleman, and C. J. Vardey. 1995.Characterization of the PGE receptor subtype mediating inhibition of superoxideproduction in human neutrophils. Br J Pharmacol 114:1459.
409. Armstrong, R. A. 1995. Investigation of the inhibitory effects of PGE2 and sélectiveEP agonists on chemotaxis of human neutrophils. Br J Pharmacol 116:2903.
410. Agwu, D. E., C. E. McCall, and L. C. McPhail. 1991. Régulation of phospholipaseD-induced hydrolysis of choline-containing phosphoglycerides by cyclic AMP inhuman neutrophils. J Immunol 146:3895.
411. Tyagi, S. R., S. C. Oison, D. N. Burnham, and J. D. Lambeth. 1991. Cyclic AMP-elevating agents block chemoattractant activation of diradylglycerol génération byinhibiting phospholipase D activation. J Biol Chem 266:3498.
412. Smith, J. A. 1994. Neutrophils, host défense, and inflammation: a double-edgedsword. JLeukoc Biol 56:672.
413. Crockett-Torabi, E., and P. A. Ward. 1996. The rôle of leukocytes in tissue injury.Eur J Anaesthesiol 13:235.

209
414. Lawrence, T., D. A. Willoughby, and D. W. Gilroy. 2002. Anti-inflammatory lipidmediators and insights into the resolution of inflammation. Nal Rev Immunol 2:787.
415. Thibault, N., D. Harbour, P. Borgeat, P. H. Naccache, and S. G. Bourgoin. 2000.Adenosine receptor occupancy suppresses chemoattractant-induced phospholipaseD activity by diminishing membrane recruitment of small GTPases. Blood 95:519.
416. Wymann, M. P., S. Sozzani, F. Altruda, A. Mantovani, and E. Hirsch. 2000. Lipidson the move: phosphoinositide 3-kinases in leukocyte function. Immunol Today21:260.
417. Regan, J. W., T. J. Bailey, D. J. Pepperl, K. L. Pierce, A. M. Bogardus, J. E.Donello, C. E. Fairbairn, K. M. Kedzie, D. F. Woodward, and D. W. Gil. 1994.Cloning of a novel human prostaglandin receptor with characteristics of thepharmacologically defined EP2 subtype. Mol Pharmacol 46:213.
418. Smolen, J. E., and G. Weissmann. 1981. Stimuli which provoke sécrétion ofazurophil enzymes from human neutrophils induce incréments in adenosine cyclic3'-5'-monophosphate. Biochim Biophys Acta 672:197,
419. Feliciello, A., M. E. Gottesman, and E. V. Avvedimento. 2001. The biologicalfunctions of A-kinase anchor proteins. J Mol Biol 308:99.
420. Smith, F. D., L. K. Langeberg, and J. D. Scott. 2006. The where's and when's ofkinase anchoring. Trends Biochem Sci 31:316.
421. Michel, J. J., and J. D. Scott. 2002. AKAP mediated signal transduction. Annu RevPharmacol Toxicol 42:235.
422. Williams, R. O. 2002. Cutting edge: A-kinase anchor proteins are involved inmaintaining resting T cells in an inactive state. J Immunol 168:5392.
423. Malbon, C. C, J. Tao, and H. Y. Wang. 2004. AKAPs (A-kinase anchoringproteins) and molécules that compose their G-protein-coupled receptor signallingcomplexes. Biochem J 3 79:1.
424. Diviani, D., and J. D. Scott. 2001. AKAP signaling complexes at the cytoskeleton. JCe II Sci 114:1431.
425. Baillie, G. S., J. D. Scott, and M. D. Houslay. 2005. Compartmentalisation ofphosphodiesterases and protein kinase A: opposites attract. FEBS Lett 579:3264.
426. Diviani, D., L. Baisamy, and A. Appert-Collin. 2006. AKAP-Lbc: a molecularscaffold for the intégration of cyclic AMP and Rho transduction pathways. Eur JCell Biol 85:603.
427. Mackintosh, C. 2004. Dynamic interactions between 14-3-3 proteins andphosphoproteins regulate diverse cellular processes. Biochem J 381:329.
428. al-Essa, L. Y., M. Niwa, K. Kohno, M. Nozaki, and K. Tsurumi. 1995.Heterogeneity of circulating and exudated polymorphonuclear leukocytes insuperoxide-generating response to cyclic AMP and cyclic AMP-elevating agents.Investigation of the underlying mechanism. Biochem Pharmacol 49:315.
429. Kanamori, Y., M. Niwa, K. Kohno, L. Y. Al-Essa, H. Matsuno, O. Kozawa, and T.Uematsu. 1997. Migration of neutrophils from blood to tissue: altération ofmodulatory effects of prostanoid on superoxide génération in rabbits and humans.Life Sci 60:1407.
430. Meja, K. K., P. J. Barnes, and M. A. Giembycz. 1997. Characterization of theprostanoid receptor(s) on human blood monocytes at which prostaglandin E2inhibits lipopolysaccharide-induced tumour necrosis factor-alpha génération. Br JPharmacol 122:149.

210
431. Shinomiya, S., H. Naraba, A. Ueno, I. Utsunomiya, T. Maruyama, S. Ohuchida, F.Ushikubi, K. Yuki, S. Narumiya, Y. Sugimoto, A. Ichikawa, and S. Oh-ishi. 2001.Régulation of TNFalpha and interleukin-10 production by prostaglandins 1(2) andE(2): studies with prostaglandin receptor-deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem Pharmacol 61:1153.
432. Seldon, P. M., K. K. Meja, and M. A. Giembycz. 2005. Rolipram, salbutamol andprostaglandin E2 suppress TNFalpha release from human monocytes by activatingType II cAMP-dependent protein kinase. Pulm Pharmacol Ther 18:277.
433. Yamane, H., Y. Sugimoto, S. Tanaka, and A. Ichikawa. 2000. Prostaglandin E(2)receptors, EP2 and EP4, differentially modulate TNF-alpha and IL-6 productioninduced by lipopolysaccharide in mouse peritoneal neutrophils. Biochem BiophysRes Commun 278:224.
434. Caggiano, A. O., and R. P. Kraig. 1999. Prostaglandin E receptor subtypes incultured rat microglia and their rôle in reducing lipopolysaccharide-inducedinterleukin-lbeta production. J Neurochem 72:565.
435. Min, S. Y., W. U. Kim, M. L. Cho, S. Y. Hwang, S. H. Park, C. S. Cho, J. M. Kim,and H. Y. Kim. 2002. Prostaglandin E2 suppresses nuclear factor-kappaB mediatedinterleukin 15 production in rheumatoid synoviocytes. J Rheumatol 29:1366.
436. Harizi, H., M. Juzan, V. Pitard, J. F. Moreau, and N. Gualde. 2002.Cyclooxygenase-2-issued prostaglandin e(2) enhances the production ofendogenous IL-10, which down-regulates dendritic cell functions. JImmunol168:2255.
437. Largo, R., I. Diez-Ortego, O. Sanchez-Pernaute, M. J. Lopez-Armada, M. A.Alvarez-Soria, J. Egido, and G. Herrero-Beaumont. 2004. EP2/EP4 signallinginhibits monocyte chemoattractant protein-1 production induced by interleukinlbeta in synovial fibroblasts. Ann Rheum Dis 63:1197.
438. Jing, H., E. Vassiliou, and D. Ganea. 2003. Prostaglandin E2 inhibits production ofthe inflammatory chemokines CCL3 and CCL4 in dendritic cells. J Leukoc Biol74:868.
439. Jing, H., J. H. Yen, and D. Ganea. 2004. A novel signaling pathway médiates theinhibition of CCL3/4 expression by prostaglandin E2. J Biol Chem 279:55176.
440. Au, B. T., M. M. Teixeira, P. D. Collins, and T. J. Williams. 1998. Effect of PDE4inhibitors on zymosan-induced IL-8 release from human neutrophils: synergismwith prostanoids and salbutamol. Br J Pharmacol 123:1260.
441. Flamand, N., M. E. Surette, S. Picard, S. Bourgoin, and P. Borgeat. 2002. CyclicAMP-mediated inhibition of 5-lipoxygenase translocation and leukotrienebiosynthesis in human neutrophils. Mol Pharmacol 62:250.
442. Fantone, J. C, W. A. Marasco, L. J. Elgas, and P. A. Ward. 1984. Stimulusspecificity of prostaglandin inhibition of rabbit polymorphonuclear leukocytelysosomal enzyme release and superoxide anion production. Am JPathol 115:9.
443. Aronoff, D., C. Canetti, and M. Peters-Golden. 2004. Prostaglandin E-2 inhibitsalveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediatedincrease in intracellular cyclic AMP. J Immunol 173:559
Related Documents











