ESTUDIO: ENFERMEDADES RARAS, HUÉRFANAS Y OLVIDADAS

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ESTUDIO: ENFERMEDADES
RARAS, HUÉRFANAS Y
OLVIDADAS

Rare, orphan and neglected diseases 1
Enfermedades raras,
huérfanas y olvidadas
2011

Rare, orphan and neglected diseases 2
Tabla de Contenido
Resumen 5 Introducción 5 Definiciones 6 Epidemiología 7 La investigación clínica en las enfermedades raras 9 La economía de las enfermedades raras 12 Umbrales de costo-efectividad 13 Rule of rescue 14 Legislación para Colombia 15 Legislación para Perú 16 Incentivos y regulación 16 Centros de evaluación de tecnología 17 Análisis bibliométrico de las enfermedades raras 21 Conclusiones 22 Referencias 23 Anexos 25

Rare, orphan and neglected diseases 3
Índice de Tablas Tabla 1. Prevalencia máxima de una enfermedad para cumplir con el criterio de ser considerada “rara”, en diferentes países. 6
Tabla 2. Jerarquía de los estudios clínicos 11
Tabla 3. Ejemplos de tamaños de muestra de estudios clínicos en enfermedades de depósito lisosomal. 12
Tabla 4. Ejemplos de enfermedades huérfanas aprobadas por NICE, con RCEI hasta 10 veces superiores al estándar convencional. 20
Tabla 5. Línea de tiempo de las enfermedades huérfanas. 22
Índice de Figuras
Figura 1. Medicamentos huérfanos aprobados en Estados Unidos y la Unión Europea desde la aprobación del Orphan Drug Act (1983) y la Regulation EC N° 141 (2000), 8
Figura 2. Distribución de los medicamentos huérfanos según especialidad. 16
Figura 3. Distribución mundial de la investigación en enfermedades raras. 21
Figura 4. Número de artículos publicados anualmente con la palabra clave “Rare 21 diseases”.

Rare, orphan and neglected diseases 4
Índice de Recuadros
Recuadro 1. Paradoja de la rareza 5
Recuadro 2. Características generales de las enfermedades raras 6
Recuadro 3. Enfermedades olvidadas 7
Recuadro 4. Enfermedades de depósito lisosomal 8
Recuadro 5. Fases de los estudios clínicos 9
Recuadro 6. El nivel de evidencia del paracaídas 10
Recuadro 7. Razón de costo-efectividad incremental (RCEI) 12
Recuadro 8. El concepto del QALY o avac 12
Recuadro 9. El valor de un medicamento 13
Recuadro 10. La enfermedad más rara del mundo 15

Rare, orphan and neglected diseases 5
-
-
-
Resumen
Las enfermedades “huérfanas” agrupan a las enfermedades “olvidadas” y las enfermedades “raras”. Entre todas, terminan afectando una proporción elevada de la población, por lo que son un problema de salud pública. Los pacientes con enfermedades huérfanas enfrentan numerosos problemas, como las dificultades para un diagnóstico certero, por desconocimiento de los médicos, y como el acceso a tratamientos efectivos, que en muchos casos no existen, y si los hay son difíciles de conseguir, muchas veces a un alto costo. Además de inequitativa, la atención de estos pacientes suele ser de baja calidad por estas y otras razones, como la escasa investigación del tema. Muchos países han creado diferentes incentivos para desarrollar investigación y para mejorar la atención en salud de los pacientes con enfermedades huérfanas. Este escrito revisa las características generales de las enfermedades huérfanas, con énfasis en las enfermedades raras, y discute los dilemas éticos y económicos del manejo de estos pacientes.
Introducción
A pesar de la baja prevalencia individual, las enfermedades raras, en su conjunto, constituyen un serio problema para la salud pública. Según algunos estimados (Eurodis, 2005), las enfermedades raras afectan, si todas ellas se reúnen, a entre el 6% y 8% de la población. En 25 países de la Unión Europea se calcula que hay cerca de 30 millones de personas afectadas por enfermedades genéticas consideradas raras, lo que equivaldría a la población total de Holanda, Bélgica y Luxemburgo. (Eurodis, 2005). Los datos de incidencia y prevalencia para los países de América Latina apenas se pueden inferir.
Las enfermedades raras merecen una atención especial por numerosas razones. Recuadro 1 Para comenzar, los pacientes con una enfermedad rara enfrentan serios problemas.
Paradoja de la rareza Los médicos, por desconocimiento de la enfermedad, no aciertan en el diagnóstico; (paradox of rarity) y si lo hacen, suele ser en forma tardía. Así como algunos pacientes pueden quedar
Las enfermedades raras relegados al olvido por médicos que se despreocupan de ellos, otros pueden ser
son infrecuentes, pero son sometidos a múltiples procedimientos innecesarios; las dos circunstancias llevan a
muchos los que las padecen. pérdida de confianza en el respectivo sistema de salud y a un uso inadecuado de
Esto es porque no es inusual recursos (Eurodis, 2005). La demora en el diagnóstico puede tener otras consecuencias, además del retardo en la iniciación de un tratamiento o de una orientación
tener alguna de entre tan adecuados, como el consecuente avance de la discapacidad y otras consecuencias tas enfermedades raras. Es sociales. Si la enfermedad es genética, existe la posibilidad de que nazcan otros hijos difícil encontrar una familia afectados, durante el tiempo que transcurre mientras que se llega a un diagnóstico. donde ninguno de sus miem En general, sobre las enfermedades raras suele haber menos información disbros haya sido afectado por ponible, tanto para pacientes y cuidadores como para personal de salud. La baja una enfermedad rara. (Euro incidencia de estas condiciones médicas crea un desincentivo a la investigación que, dis nov 2005) por razones justificables, prioriza aquellas condiciones que generan alta “carga de la
enfermedad”. Todos estos factores confluyen para hacer que la atención a pacientes con enfermedades raras sea de baja calidad.
Los sistemas de salud no están preparados para enfrentar el reto de la atención de los pacientes con enfermedades raras, lo que se refleja en inequidad. Los que poseen información para moverse a lo largo del sistema de salud reciben mejor atención que los menos informados, que suelen provenir de poblaciones desprotegidas. Muchas de estas enfermedades raras tienen, además, otras consecuencias de tipo social: como estigmatización, rechazo o exclusión. La estigmatización a estos pacientes los torna vulnerables desde la perspectiva social, económica y psicológica (Vinha da Souza, 2007).

Rare, orphan and neglected diseases 6
•
-
•
• --
-
Definiciones
Aunque hay numerosas enfermedades que son raras y no revisten mayor gravedad, la atención, y a veces la denominación misma de enfermedad rara, se ha dedicado a aquellas enfermedades degenerativas, crónicas y progresivas, que constituyen un peligro para la vida, y cuyo tratamiento debe ser continuo (Boy & Schramm, 2009; Barrera, 2010).
Tabla 1. Prevalencia máxima de una enfermedad para cumplir con el criterio de ser considerada “rara”, en diferentes países.
No hay un consenso establecido sobre qué es y qué no es una “enfermedad rara”. Las definiciones cambian tanto de una legislación a otra como de uno a otro investigador. Por razones obvias, la baja prevalencia es un factor determinante en toda definición de enfermedad rara. Con este criterio de prevalencia, en Suecia una enfermedad se con-sidera rara si afecta a menos de 10 de cada 100.000 habitantes mientras que en la Unión Europea el umbral es cinco veces más alto. La Tabla 1 muestra los diferentes criterios empleados por distintos países.
Un ejemplo de esta disparidad de criterios ocurre en Turquía, en donde se considera rara una enfermedad cuya prevalencia sea menor de 1 en 100.000 habitantes; en Inglaterra, en cambio, ese umbral corresponde al de una enfermedad “ultra rara”. El problema con una definición variable entre los diferentes países es que los estudios clínicos, epidemiológicos y económicos no son comparables. Más grave aún, la disparidad de criterios crea dificultades para generar políticas públicas, y para legislar sobre la protección de estos pacientes. (Wiest, 2010; Eurodis, 2005)
Para complicar un poco las cosas, hay países que no usan la tasa de prevalencia en su definición de enfermedad rara; utilizan en cambio el número total de personas con la enfermedad. Dinamarca, por ejemplo, considera como rara una enfermedad que afecte a menos de 500 daneses (Wiest, 2010).
No se debe confundir el concepto de enfermedad rara con el de enfermedad olvidada (neglected disease), nombre con el que se designan algunas condiciones, muchas de ellas frecuentes, casi siempre transmisibles, que en su mayoría se encuentran en países en vías
Recuadro 2 Características generales de las enfermedades raras
(Eurodis, 2005) Las enfermedades raras
tienen un espectro entre severo y muy severo, un curso crónico, fre cuentemente degenerativo y ponen en peligro la vida. • El inicio de la enfermedadocurre en la infancia en un 50% de las enfermedades raras. Producengrandiscapacidad,
la calidad de vida de los pacientes con enfermedades raras está frecuentemente comprometida y se caracteriza por una disminución o pérdida de la autonomía. • Representan una cargapsicosocial alta para el paciente y su familia, que se agrava por la desesperanza y la falta de soporte para la vida diaria. • La mayoría no tienen untratamiento efectivo, el tratamiento es sintomático, buscando mejorar la calidad o la expectativa de vida. Por lo general, las enfer
medades raras son de difícil mane jo, y las familias advierten serios tropiezos para encontrar el trata miento adecuado.

Rare, orphan and neglected diseases 7
-
-
-
-
--
-
-
--
de desarrollo. Por no representar una prioridad para los países desarrollados, hay muy pocos recursos invertidos en investigación, y para la industria farmacéutica, ese mercado es percibido como poco rentable (Eurodis, 2005).
Aunque a veces se emplea el título de enfermedad huérfana como un sinónimo de enfermedad rara: las enfermedades huérfanas realmente agrupan tanto las enfermedades raras como las olvidadas. Son “huérfanas” tanto desde la perspectiva de los Estados y sus políticas públicas, como de los intereses del mercado. (Eurodis, 2005)
En la misma línea de pensamiento, los medicamentos huérfanos son medicamentos orientados al diagnóstico, la prevención o el tratamiento de las enfermedades huérfanas. Estos medicamentos, casi por definición, se encuentran por debajo de las condiciones normales del mercado; al ser desarrollados para un grupo minoritario de pacientes no suelen ser costo-efectivos, ni en su elaboración, ni en su comercialización. Es fácil que, en condiciones usuales de mercado, una compañía farmacéutica no recupere el costo de entregar un medicamento al mercado con las ventas del mismo. Esta razón, junto con la presión de las organizaciones de pacientes con enfermedades raras, ha llevado a varios gobiernos a plantear incentivos económicos para estimular el desarrollo de este tipo de fármacos. (Eurodis, 2005).
La Figura 1 muestra el número de medicamentos huérfanos aprobados tanto en Estados Unidos como en la Unión Europea desde la aprobación del Orphan Drug Act (1983) y la Regulation EC N° 141 (2000), respectivamente.
Epidemiología
¿Cuál es el número total de pacientes con enfermedades raras en el mundo? Orphanet un portal de información de referencia en enfermedades raras y medicamentos huérfanos, dirigido a todos los públicos, tiene como objetivo contribuir a mejorar el diagnóstico, el cuidado y el tratamiento de los pacientes con enfermedades raras (Orphanet, 2010). Orphanet estima que existen entre 6000 y 7000 enfermedades raras. Por otra parte NORD (National Organization for Rare Disorders, 2011), de los Estados Unidos, reporta 7000 enfermedades diferentes. Otro estimado más es el de Garau y Ferrendis, que proponen un rango que va de 5000 a 8000 enfermedades raras (Wiest, 2010).
Recuadro 3 Enfermedades olvidadas
(neglected diseases) (Bey rer et als, 2007)
El nombre de enfermedades olvida das se ha acuñado para un grupo de enfermedades infecciosas tropi cales, prevalentes en los países menos desarrollados del mundo, y que incluyen infecciones por proto zoarios y helmintos, así como otras infecciones como tracoma y lepra. Las enfermedades por protozoarios incluyen la leishmaniasis, la enfer medad de Chagas y la tripanoso miasis africana. Las helmintiasis son la filariasis linfática, ascariasis, oncocercosis, dracunculiasis y es quistosomiasis. Los cálculos de la carga de enfer medad en el mundo ubican a estas enfermedades, en su conjunto, en un cuarto lugar, después de infecciones respiratorias bajas, VIH/sida y enfer medad diarreica, y por encima de tu berculosis y malaria. El principal problema con estas enfermedades es que afectan a poblaciones marginadas, a los más pobres de los pobres, y son excepcionales en los países desarrollados; por esa razón son en gran medida ignoradas por la ciencia.

Rare, orphan and neglected diseases 8
-
-
-
-
Figura 1. Medicamentos huérfanos aprobados en Estados Unidos y la Unión Europea desde la aprobación del Orphan Drug Act (1983) y la Regulation EC N° 141 (2000),
Recuadro 4 - Enfermedades de depósito lisosomal (Staretz Chacham et al, 2009)
Las enfermedades de depósito lisosomal son un subgrupo de entidades de baja prevalencia, clasificadas dentro del grupo denominado errores innatos del metabolismo. Se presentan por la acumulación de sus tratos intracelulares en los lisosomas. La vulnerabilidad de los lisosomas radica en los numerosos pasos de sus procesos bioquímicos; el déficit de una enzima lleva a que se depositen en los tejidos algunos sustratos intermedios. Clásicamente se considera que no son alteraciones del neonato, porque muchas de ellas toman meses y hasta años para manifestarse, ello se explica por el tiempo necesario para alcan zar concentraciones intracitoplasmáticas que alteren el funcionamiento celular (Futerman, 2004; Vellodi, 2005). El diagnóstico temprano y el inicio oportuno de terapia de remplazo enzimático son esenciales para disminuir el deterioro y para evitar que se afecte la funcionalidad del individuo. El patrón de herencia es autosómico recesivo con tres excepciones que son: enfermedad de Fabry, el síndrome de Hunter y la enfermedad de Danon. Hay que tener consideraciones especiales con algunas minorías étnicas, donde su prevalencia puede ser mayor, como ejemplos se encuentra las enfermedades de Gaucher y Tay-Sachs en judíos Ashkenazi (Kaback et al., 1993), o la mucopolisacaridosis VI en minorías étnicas indígenas co lombianas(Rossellietal.,enprensa).

Rare, orphan and neglected diseases 9
• -
-
•
-
•-
•
•
•
Según Eurodis (2005) las enfermedades raras corresponden a 3% o 4% de todos los nacimientos y se estima que semanalmente se identifican cinco nuevas enfermedades en el mundo.
Del total de enfermedades incluidas en estos listados, dos terceras partes corresponden a enfermedades discapacitantes graves, asociadas a déficit motor, senso rial o autónomo; una proporción similar tiene un inicio temprano. En general, las enfermedades raras tienen un pronóstico desfavorable, y son las responsables del 35% de las muertes en niños menores de un año, y de una de cada diez muertes entre el primer año y los 15 años de edad (Direcção-Geral da Saúde, Portugal, 2008).
Se reconoce que la mayoría (alrededor de 80%) de las enfermedades raras son causadas por trastornos genéticos, pero también hay algunas que se asocian a infecciones y agentes tóxicos o a diferentes formas de neoplasias; un gran número de ellas son de etiología desconocida.
La investigación clínica en las enfermedades raras y huérfanas
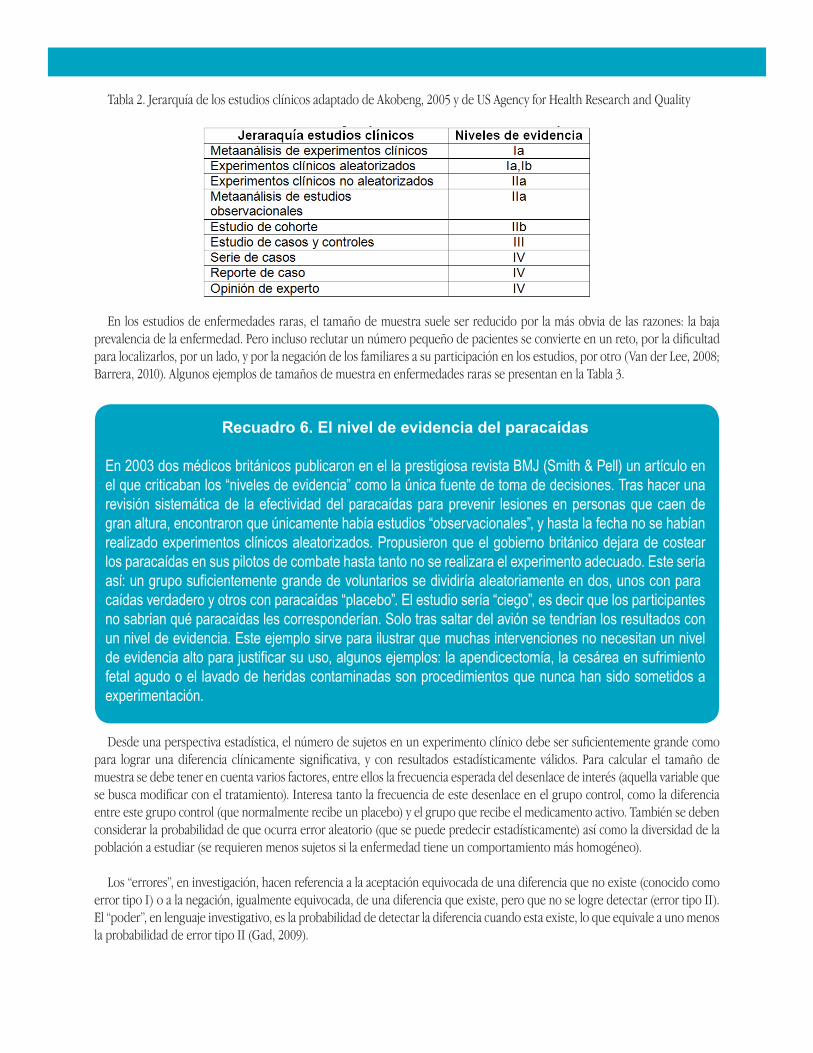
El desarrollo de nuevas moléculas para el tratamiento de enfermedades huérfanas, tanto raras como olvidadas, es uno de los intereses investigativos que ha ido en aumento en los últimos años. Ello ha implicado, un reto grande (Pariser, 2011). Todos los medicamentos, para enfermedades raras o no, deben haber demostrado su efectividad y seguridad en estudios clínicos antes de su aprobación por entes gubernamentales (Miyamoto, 2011). Pero la realización de experimentos clínicos en enfermedades raras implica un problema por varias razones, entre ellas el tamaño de muestra, los dilemas éticos, y la dificultad en el reclutamiento de pacientes. En ocasiones el número es tan limitado que las autorizaciones se logran con estudios observacionales (ver Tabla 2).
Recuadro 5 Fases de los estudios clínicos
Los estudios clínicos comprenden varias fases que son esenciales en el proceso de aprobación de un nuevo medicamento, estas fases son las siguientes: Fasepreclínica:sonestudioshechosenloslabo
ratorios en los que se evalúan las nuevas moléculas in vitro e in vivo (en animales de experimentación), no se hacen en humanos, es una etapa de experi mentación en la que se evalúa eficacia, toxicidad y la farmacocinética del nuevo medicamento (Meinert & Tonascia, 1986). Fase 0: empieza la experimentación en grupos
pequeños de seres humanos, se usan microdosis; el objetivo es evaluar la farmacodinamia y farmaco cinética (Anónimo, 2009). Fase1:serealizaenvoluntariossanos,seevalúa
seguridad, tolerabilidad, farmacocinética y farmaco dinamia. Usualmente es un seguimiento continuo de los individuos, en su gran mayoría se reclutan en centros especializados y se evalúa estrictamente el efecto del medicamento. En esta etapa se determina la dosis tolerable. Fase2:estudiosenpoblaciónobjetivo,seevalúa
la dosificación, así como la eficacia. El objetivo es definir los esquemas de prescripción. Fase 3: son aleatorizados, multicéntricos, secaracterizan por su planeación rigurosa, el tamaño depende claramente de la prevalencia de la condición y de la frecuencia de los desenlaces evaluados, el objetivo es definir la efectividad por medio de la comparación ya sea contra placebo, o con el mejor medicamento disponible hasta ese momento. Son los más costosos, requieren un gran número de personas. Son los que se publican en las grandes revistas. Fase4:seguimientoa lospacientesquerecibenel medicamento para observar en condiciones no ideales cuáles efectos secundarios se presentan. Normalmente se denomina fase de farmacovigilancia.
Rare, orphan and neglected diseases 10
-
Tabla 2. Jerarquía de los estudios clínicos adaptado de Akobeng, 2005 y de US Agency for Health Research and Quality
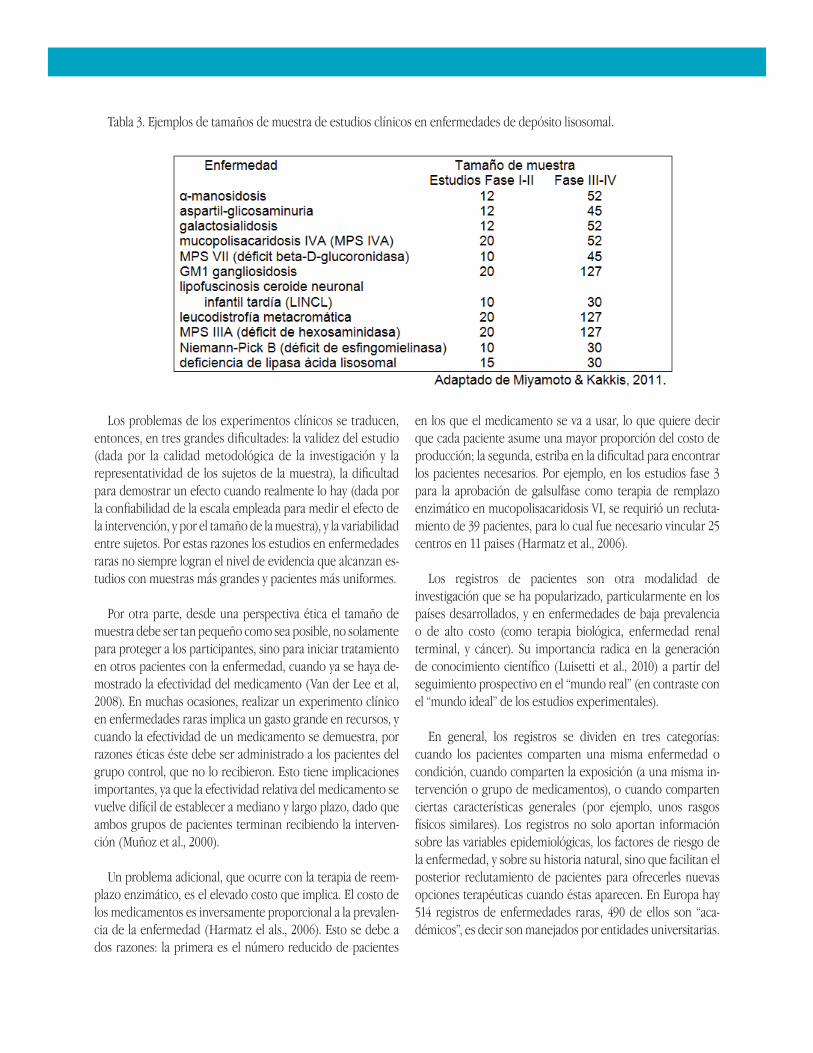
En los estudios de enfermedades raras, el tamaño de muestra suele ser reducido por la más obvia de las razones: la baja prevalencia de la enfermedad. Pero incluso reclutar un número pequeño de pacientes se convierte en un reto, por la dificultad para localizarlos, por un lado, y por la negación de los familiares a su participación en los estudios, por otro (Van der Lee, 2008; Barrera, 2010). Algunos ejemplos de tamaños de muestra en enfermedades raras se presentan en la Tabla 3.
Desde una perspectiva estadística, el número de sujetos en un experimento clínico debe ser suficientemente grande como para lograr una diferencia clínicamente significativa, y con resultados estadísticamente válidos. Para calcular el tamaño de muestra se debe tener en cuenta varios factores, entre ellos la frecuencia esperada del desenlace de interés (aquella variable que se busca modificar con el tratamiento). Interesa tanto la frecuencia de este desenlace en el grupo control, como la diferencia entre este grupo control (que normalmente recibe un placebo) y el grupo que recibe el medicamento activo. También se deben considerar la probabilidad de que ocurra error aleatorio (que se puede predecir estadísticamente) así como la diversidad de la población a estudiar (se requieren menos sujetos si la enfermedad tiene un comportamiento más homogéneo).
Los “errores”, en investigación, hacen referencia a la aceptación equivocada de una diferencia que no existe (conocido como error tipo I) o a la negación, igualmente equivocada, de una diferencia que existe, pero que no se logre detectar (error tipo II). El “poder”, en lenguaje investigativo, es la probabilidad de detectar la diferencia cuando esta existe, lo que equivale a uno menos la probabilidad de error tipo II (Gad, 2009).
Recuadro 6. El nivel de evidencia del paracaídas
En2003dosmédicosbritánicospublicaronenellaprestigiosarevistaBMJ(Smith&Pell)unartículoenel que criticaban los “niveles de evidencia” como la única fuente de toma de decisiones. Tras hacer una revisión sistemática de la efectividad del paracaídas para prevenir lesiones en personas que caen de gran altura, encontraron que únicamente había estudios “observacionales”, y hasta la fecha no se habían realizadoexperimentosclínicosaleatorizados.Propusieronqueelgobiernobritánicodejaradecostearlos paracaídas en sus pilotos de combate hasta tanto no se realizara el experimento adecuado. Este sería así: un grupo suficientemente grande de voluntarios se dividiría aleatoriamente en dos, unos con para caídas verdadero y otros con paracaídas “placebo”. El estudio sería “ciego”, es decir que los participantes no sabrían qué paracaídas les corresponderían. Solo tras saltar del avión se tendrían los resultados con un nivel de evidencia. Este ejemplo sirve para ilustrar que muchas intervenciones no necesitan un nivel de evidencia alto para justificar su uso, algunos ejemplos: la apendicectomía, la cesárea en sufrimiento fetal agudo o el lavado de heridas contaminadas son procedimientos que nunca han sido sometidos a experimentación.
Rare, orphan and neglected diseases 11
Tabla 3. Ejemplos de tamaños de muestra de estudios clínicos en enfermedades de depósito lisosomal.
Los problemas de los experimentos clínicos se traducen, entonces, en tres grandes dificultades: la validez del estudio (dada por la calidad metodológica de la investigación y la representatividad de los sujetos de la muestra), la dificultad para demostrar un efecto cuando realmente lo hay (dada por la confiabilidad de la escala empleada para medir el efecto de la intervención, y por el tamaño de la muestra), y la variabilidad entre sujetos. Por estas razones los estudios en enfermedades raras no siempre logran el nivel de evidencia que alcanzan estudios con muestras más grandes y pacientes más uniformes.
Por otra parte, desde una perspectiva ética el tamaño de muestra debe ser tan pequeño como sea posible, no solamente para proteger a los participantes, sino para iniciar tratamiento en otros pacientes con la enfermedad, cuando ya se haya demostrado la efectividad del medicamento (Van der Lee et al, 2008). En muchas ocasiones, realizar un experimento clínico en enfermedades raras implica un gasto grande en recursos, y cuando la efectividad de un medicamento se demuestra, por razones éticas éste debe ser administrado a los pacientes del grupo control, que no lo recibieron. Esto tiene implicaciones importantes, ya que la efectividad relativa del medicamento se vuelve difícil de establecer a mediano y largo plazo, dado que ambos grupos de pacientes terminan recibiendo la intervención (Muñoz et al., 2000).
Un problema adicional, que ocurre con la terapia de reemplazo enzimático, es el elevado costo que implica. El costo de los medicamentos es inversamente proporcional a la prevalencia de la enfermedad (Harmatz el als., 2006). Esto se debe a dos razones: la primera es el número reducido de pacientes
en los que el medicamento se va a usar, lo que quiere decir que cada paciente asume una mayor proporción del costo de producción; la segunda, estriba en la dificultad para encontrar los pacientes necesarios. Por ejemplo, en los estudios fase 3 para la aprobación de galsulfase como terapia de remplazo enzimático en mucopolisacaridosis VI, se requirió un reclutamiento de 39 pacientes, para lo cual fue necesario vincular 25 centros en 11 paises (Harmatz et al., 2006).
Los registros de pacientes son otra modalidad de investigación que se ha popularizado, particularmente en los países desarrollados, y en enfermedades de baja prevalencia o de alto costo (como terapia biológica, enfermedad renal terminal, y cáncer). Su importancia radica en la generación de conocimiento científico (Luisetti et al., 2010) a partir del seguimiento prospectivo en el “mundo real” (en contraste con el “mundo ideal” de los estudios experimentales).
En general, los registros se dividen en tres categorías: cuando los pacientes comparten una misma enfermedad o condición, cuando comparten la exposición (a una misma intervención o grupo de medicamentos), o cuando comparten ciertas características generales (por ejemplo, unos rasgos físicos similares). Los registros no solo aportan información sobre las variables epidemiológicas, los factores de riesgo de la enfermedad, y sobre su historia natural, sino que facilitan el posterior reclutamiento de pacientes para ofrecerles nuevas opciones terapéuticas cuando éstas aparecen. En Europa hay 514 registros de enfermedades raras, 490 de ellos son “académicos”, es decir son manejados por entidades universitarias.

Rare, orphan and neglected diseases 12
-
--
-
La economía de las enfermedades raras
La evaluación económica es una herramienta para racionalizar el uso de recursos en un sistema de salud. Para ello se elaboran modelos de la enfermedad a estudio y se analizan tanto los resultados clínicos como los costos comparativos de dos o más posibles intervenciones. Estos modelos pueden adoptar varias formas. En los modelos de costo-beneficio se comparan los costos de una intervención, medidos en unidades monetarias, con los beneficios financieros que la intervención genera. Los estudios de costo beneficio son útiles en aquellas contadas intervenciones en salud que resultan en un ahorro para el sistema, al evitar, por ejemplo, hospitalizaciones o complicaciones costosas. Hay que tener cuidado con este tipo de análisis ya que pueden generar conflictos éticos, cuando el ahorro de dinero para el sistema se logra creando barreras de acceso que limiten el número de consultas o procedimientos o, peor aún cuando este ahorro se logra a través de la muerte prematura de un paciente de “alto costo”.
En los modelos de costo-efectividad, los costos (siempre medidos en unidades monetarias) se comparan con unas medidas de efectividad que varían de acuerdo con cada enfermedad. En enfermedades genéticas se puede plantear, por ejemplo, el costo por paciente detectado, si se están comparando dos estrategias de tamizaje. Aquélla que permite detectar casos nuevos al costo más bajo sería la más “costo-efectiva”. Aquí también se pueden encontrar dilemas éticos si, para seguir con el ejemplo anterior, hay una estrategia de tamizaje barata pero que solo detecta la mitad de los casos, al compararla con una de mayor costo pero que los detecta a todos, fácilmente la más económica va a resultar más costo-efectiva. En un caso como éste, el análisis económico ayuda a estimar cuánto más se debería estar preparado a pagar por cada paciente adicional detectado con la tecnología de mayor costo. Ello se conoce como la razón de costo-efectividad incremental (ver Recuadro 7).
Finalmente, están los modelos de costo-utilidad. El indicador más empleado para medir “utilidad” es el QALY (o, en español, avac) (ver Recuadro 8).
Recuadro 7 La “razón de costo
efectividad incremen tal” (RCEI)
La medida más empleada para determinar la costo efectividad de una interven ción que es más costosa y más efectiva que otra se conoce como RCEI. Se define como cuál es el costo adicional que se debe pagar por cada unidad adicional de efectividad que se gana.

Rare, orphan and neglected diseases 13
Umbrales de costo-efectividad y costo-utilidad
En estudios de costo-efectividad o costo-utilidad, el umbral (threshold value) equivale al máximo costo que se estaría dispuesto a pagar por cada unidad adicional de efectividad, (o, en estudios de costo-utilidad, por cada “avac” ganado). Son pocos los países que fijan umbrales explícitos, y la fundamentación teórica (ya sean éstos explícitos o implícitos) es controvertida. (Cleemput et al., 2008). Inglaterra y Gales usan un umbral para la razón de costo-utilidad incremental (RCEI) de £20,000 a £30,000 por avac ganado. Este es el umbral que, de manera implícita, implementa el Instituto de Excelencia Clínica (NICE) para aprobar o rechazar las intervenciones que son cubiertas por el sistema de salud británico. Países como Australia, Nueva Zelanda y Canadá no lo definen, pero el cálculo por avac ganado se ha estimado indirectamente en AU$69,900, NZ$20,000 y CAN$31,000 a CAN$137,000, respectivamente. Otros países como Finlandia, Suecia, Noruega, Dinamarca y Bélgica no sugieren un valor exacto. (Cleemput et al., 2011). Incluso los países que son más enfáticos en justificar sus intervenciones con argumentos de costo efectividad dan un trato diferente a las enfermedades raras. Una de las justificaciones más claras para ello es el elevado costo de un medicamento cuya investigación, desarrollo, aprobación y mercadeo deben ser pagados por un número limitado de pacientes (ver Recuadro 9).

Rare, orphan and neglected diseases 14
Rule of rescue
La “regla de salvamento” (o “Rule of Rescue” en inglés), se denomina a esa obligación humana por salvarle la vida a una persona que se encuentre en peligro inminente de muerte. Salvar a las personas que corren un peligro mortal, o de sufrir un daño serio, es algo que la sociedad valora. Son muchos los ejemplos que se pueden citar de la aplicación en la práctica de la regla de salvamento en contextos tanto médicos como no médicos. Entre los primeros se encuentran los esfuerzos terapéuticos que se hacen en pacientes en unidad de cuidado intensivo, incluso sabiendo que la probabilidad de recuperación es muy baja. Los recursos que se despliegan para rescatar a un náufrago o a un montañista perdido son otros ejemplos, no médicos. Un ejemplo reciente de la regla de salvamento, con una amplia cobertura mediática, fue la inversión de varios millones de dólares en el rescate de los 33 mineros chilenos que en agosto de 2010 quedaron atrapados cientos de metros bajo tierra.
En esas circunstancias, continúa el argumento, son pocas las personas que objetarían el rescate aduciendo, quizás con lógica financiera, que esa misma suma de dinero invertida, por ejemplo, en programas de salud infantil podría haber salvado muchas más vidas. En la naturaleza individual y colectiva del ser humano está el esfuerzo por rescatar a alguna persona cercana o conocida cuando está en peligro de muerte, sin tener en cuenta los costos que implique, y sin importar que a su vez se afecte la salud de muchísimas personas desconocidas. Un resumen de los argumentos éticos y económicos a favor y en contra de la regla de salvamento se presentan en Osborne y Evans (1994, p 779).
La regla de salvamento prioriza la gravedad de la enfermedad, tanto sobre la efectividad como sobre los costos; en ella domina la naturaleza humana sobre la lógica o sobre la ética utilitaristas. En economía de la salud, la regla de salvamento choca con los argumentos que defienden escoger solo aquellas intervenciones costo-efectivas, para maximizar la eficiencia del uso de recursos.
En la misma línea de la regla de salvamento el Australian Pharmaceutical Benefits Advisory Committee (PBAC) definió que un tratamiento debe cumplir las siguientes condiciones para que la regla de salvamento sea aplicable:
• No debe existir en el país un tratamiento alterno (esta condición es de obligatorio cumplimiento). • La condición debe ser severa progresiva, y en la que se espera una muerte prematura. Mientras más joven sea la per
sona, y más severa su condición, más justificación hay. • La condición debe afectar a un reducido número de pacientes, mientras menos sean éstos, mayor consideración re
ciben. (Cookson et al., 2008)
Y siguiendo con la regla de salvamento, NICE, el instituto de excelencia clínica del Reino Unido, creó un Consejo de Ciudadanos conformado por 30 personas de la comunidad, para discutir la aplicabilidad de la regla de salvamento en el sistema de salud británico. Este Consejo de Ciudadanos se planteó dos preguntas: ¿Hay alguna preferencia por salvar la vida de personas que se encuentran en inminente peligro de fallecer sobre la vida de otras personas que no se encuentran en riesgo inminente o sobre la vida de muchas personas indeterminadas en el futuro? Si la respuesta fuera no, se ignoraría la regla de salvamento, y el proceso terminaría ahí. En caso contrario, el Consejo debía dejar explícito bajo qué condiciones y hasta qué limites se debía aplicar la regla de salvamento (NICE, 2006).
En esta reunión de este Consejo se llegó a un consenso con la siguiente conclusión: en ciertas circunstancias la sociedad en general repudiaría la no aplicación de la regla de salvamento y la consideraría como inhumana. Concluyó también que para aplicar la regla de salvamento se debían tener en cuenta las siguientes consideraciones:
• ¿Es necesaria la intervención para evitar la muerte del individuo de forma inmediata o a corto plazo? • ¿Qué tanta es la probabilidad de incrementar la calidad de vida del individuo? • ¿Los eventos adversos que se pueden presentar con el tratamiento no cambiarán el beneficio del mismo? • ¿Hay alguna probabilidad demostrada de incrementar la expectativa de vida del individuo? • ¿Qué consecuencias se esperan si el tratamiento no se suministra?

Rare, orphan and neglected diseases 15
---
-
• ¿Cuáles son los tratamientos alternos y cómo se comparan? • ¿Suministrar el tratamiento puede traer beneficios al conocimiento científico de la enfermedad? • ¿El sistema de salud está en capacidad de pagarlo? • ¿Hasta qué punto es demostrable su efectividad? • ¿Hay razones para creer que este tratamiento podría marcar un precedente en otros pacientes con esta misma
condición, que lleve a una reducción futura de los precios? • ¿El tratamiento de esta condición ayuda a la salud pública? (NICE, 2006).
La regla de rescate fue uno de los criterios explícitos empleados por Chile cuando se diseñó su reforma de salud y se establecieron las prioridades en su plan de beneficios por patologías. De las 56 patologías incluidas en la primera versión del AUGE chileno, en 25 de ellas se invocó la “regla de salvamento”. Fue así como incluyó, junto al infarto de miocardio y la diabetes mellitus, trastornos congénitos mucho más infrecuentes como la fibrosis quística o el labio y paladar hendidos (Vargas & Poblete, 2008).
Legislación para Colombia
En 2010 se promulgó en Colombia la Ley 1392 en la cual se define y se regula la atención de las enfermedades huérfanas en el país (ver Anexo). Con esta norma legal se definió una enfermedad huérfana como aquélla que afecta a menos de una persona por cada 5000 habitantes (la versión original de la Ley establecía 1 por cada 2000 personas, pero esta prevalencia fue modificada mediante la Ley 1438 de 2011 a 1 por cada 5000 personas). Dentro de esta definición se incluyen las enfermedades raras, las ultrahuérfanas y las olvidadas. La responsabilidad sobre la actualización de la lista en la que se incluyen todas las enfermedades huérfanas recae sobre la Comisión de Regulación en Salud (CRES); esta actualización debe realizarse cada dos años. El objetivo de la Ley es garantizar la cobertura de todos los colombianos que sufran de estas enfermedades y se rige bajo los principios de universalidad, corresponsabilidad, solidaridad e igualdad.
Según la Ley 1392, la financiación para el diagnóstico, tratamiento, medicamentos, procedimientos y cualquier otro servicio requerido por las personas que sufran de enfermedades huérfanas, se obtendrá de recursos dispuestos por la Ley 715 de 2001, los recursos faltantes se financiarán a partir de la subcuenta de Eventos Catastróficos y Accidentes de Tránsito (subcuenta ECAT), del Fosyga. Además, se autoriza al Gobierno Nacional para que en un año adopte un sistema de negociación y compra, que podrá ser centralizado. El seguimiento y control de lo dispuesto por la Ley será función de la Superintendencia Nacional de Salud, que se encargará de la inspección, vigilancia y control sobre acceso, prestación de servicios y manejo de los datos.
Las enfermedades raras son también un punto de consideración en la sentencia T760 de 2008 de la Corte Constitucional colombiana, que ha hecho precisiones importantes sobre el sistema de salud colombiano. Entre éstas, obliga al Estado a modificar el plan de beneficios (o POS, plan obligatorio de salud). En particular, en su Auto de Seguimiento número 226 asevera: “…las actualizaciones interiores del POS incentivarán el acceso a servicios médicos para aquellas personas que sufren de enfermedades catastróficas, huérfanas y raras.” (Numeral iv del punto 2,3).
Recuadro 10 - ¿La enfermedad más rara del mundo? La que podría ser quizás la enfermedad más rara en el mundo, descrita en un único paciente, es el déficit de la enzima ribosa 5 fosfato isomerasa. En la revista American Journal of Human Genetics, en 2004, se publicó el caso de un paciente, sin antecedentes familiares e hijo de padres no consanguí neos, que para la fecha de la publicación tenía 20 años. Desde temprana edad el paciente presentó re tardo psicomotor, y desarrolló epilepsia a los 4 años. A los 7 presentó regresión neurológica con ataxia prominente, espasticidad, atrofia óptica y neuropatía sensoriomotora leve. La resonancia magnética mostró compromiso extenso de la sustancia blanca, y mediante espectroscopía se demostraron niveles elevados de pentitol ribitol y D arabitol, productos intermedios de la vía de las pentosas.

Rare, orphan and neglected diseases 16
Legislación para Perú
En Perú, en el 2011, se promulgó la ley de enfermedades raras o huérfanas (ver Anexo). En la que se definen las enfermedades raras o huérfanas (sin distinción) como aquellas enfermedades que comprometen seriamente la vida, cuya prevalencia es baja (no hay criterio de prevalencia) y que tienen problemas es su diagnóstico y seguimiento. En esta Ley se da importancia a la enseñanza en los centros de educación superior, para aumentar así el diagnóstico oportuno. Además, se establece el registro nacional de pacientes que padecen enfermedades raras o huérfanas. Para el financiamiento de las “enfermedades huerfanas o raras”, al ser un gasto prioritario, se genera la Unidad Ejecutora Fondo Intangible Solidario de Salud (Fissal) como administradora de fondos de aseguramiento.
Incentivos y regulación
La primera iniciativa por regular la problematica de las enfermedades huérfanas para el sistema de salud apareció en Estados Unidos en 1983, en la que se dejó explícita la legislación que regulaba el ingreso de medicamentos para enfermedades de baja prevalencia. Esta fue la primera ley del mundo que reguló el comercio de los medicamentos huérfanos, se le denominó ODA “Orphan Drug Act”. En este documento se estableció que una enfermedad que afectara a menos de 200.000 habitantes era clasificada como enfermedad huérfana (para la época equivalía a una prevalencia de 7,5 por 10.000 habitantes).
Uno de los problemas propios en este tipo de patologías es la dificultad para conseguir voluntarios para los experimentos clínicos sumado a la dificultad para su diagnóstico por la falta de infraestructura así como el desconocimiento por parte del cuerpo médico. Para aminorar esta desventaja en el mercado de la producción de medicamentos huérfanos, en el Acta de Medicamentos Huérfanos se propuso que la industria farmacéutica incentivara el diagnóstico oportuno y promoviera la producción de nuevos medicamentos. (Wiest, 2010)
Figura 2. Distribución de los medicamentos huérfanos según especialidad.

Rare, orphan and neglected diseases 17
Iniciativas de otras partes del mundo que han ayudado al desarrollo de estos medicamentos: la Regulación 141 del 2000 (ver Anexo 3), y la Regulación 847 del 2000 ambas aprobadas por la Comisión Europea de Regulación en Productos Médicos Huérfanos (Westermark, 2007; Parlamento Europeo, 2000).
A partir de estas normas se generan incentivos para la elaboración de nuevas moléculas. El primer incentivo es la asistencia en los protocolos de investigación, esto significó asistencia técnica por la EMA (European Medicines Agency) en la conducción de los experimentos clínicos (eficacia y seguridad) para completar los requerimientos propuestos por las regulaciones. El siguiente incentivo es el de centralizar el procedimiento de aceptación, lo cual significa autorización para todos los países de la Unión Europea incluyendo a Noruega e Islandia, lo que representa para las empresas disminuciones arancelarias. La exclusividad de mercado por 10 años protegiéndolo de la entrada al mercado de moléculas similares para la misma indicación. De este último incentivo se desprenden tres excepciones, consentimiento del patrocinador, ausencia del medicamento, y por último la superioridad en cuanto a seguridad y efectividad clínica de otra molécula. También se obliga a los miembros de la Unión Europea a comunicar constantemente qué acciones están llevando a cabo para proteger estos medicamentos. El último incentivo es en investigación para conocer la historia natural de la enfermedad, la patofisiología, y el desarrollo de intervenciones preventivas diagnóstica y terapéuticas (Westermark et al., 2011).
Centros de evaluación de tecnología
Introducción
El Instituto Nacional para la Excelencia Clínica, NICE (Nacional Institute for Health Clinical Excellence) hace parte del Servicio Nacional de Salud del Reino Unido (NHS, National Health Service) y se conformó en 1999. Sus objetivos principales incluyen la realización de guías de práctica clínica orientadas a los cuidadores y los pacientes, la evaluación de guías de práctica clínica, la evaluación de tecnologías, y la evaluación de procedimientos y programas de salud pública.
NICE comprende tres grandes ramas en la investigación: la salud pública, la evaluación de tecnologías y la práctica clínica. Las tres ramas de investigación convergen para guiar al NHS. Dado el rigor y la transparencia de los procesos, NICE se ha convertido en centro de referencia en la evaluación de procesos en salud en el mundo entero.
Cómo opera NICE
Cualquier persona puede sugerir un tema para que NICE elabore una guía, y sugiera sus recomendaciones de acuerdo con la evidencia disponible. Los temas de las guías elaboradas por NICE son seleccionados por el Departamento de Salud, que identifica las áreas sobre las cuales hay interés. Si el tema se considera pertinente, se organiza un documento específico, y se establece de antemano el alcance específico de la guía.
El propósito al definir el alcance de la guía es:
• Examinar qué va a incluir y excluir la guía • Identificar los aspectos claves del cuidado que deben ser incluidos • Delimitar el trabajo, y establecer prioridades de acuerdo a NICE y el Departamento de Salud • Informar el desarrollo de la pregunta clínica así como la estrategia empleada en la búsqueda • Informar a los profesionales y al público acerca de los contenidos que debe tener la guía • Delimitar la cantidad de trabajo, de tal forma que pueda realizarse en un periodo de no más de 18 meses
Para ayudar al personal que está determinando el alcance de la guía, NICE ha desarrollado un formato en el cual se identifica lo que debe ser incluido en una guía, este formato puede consultarse en la página de NICE (2011).
El proceso de elaboración del primer borrador es de alrededor de 4 semanas. Durante este lapso, el alcance de la guía y

Rare, orphan and neglected diseases 18
sus modificaciones pueden ser evaluadas en la página web de NICE. Se invita a las organizaciones que tienen intereses, y a los páneles de revisión de guías. Cuando se habla de organizaciones con intereses se hace referencia a profesionales de la salud, pacientes y cuidadores del NHS así como a las empresas con un interés particular.
Con los comentarios y las modificaciones sugeridas en los pasos anteriores, los miembros del panel de revisión y el revisor jefe hacen los últimos ajustes y es el jefe del panel de revisión el encargado de hacer un reporte formal. En este momento el alcance de la guía está definido y el grupo de elaboración de guías no puede hacerle cambios sin antes consultarlo con NICE; los cambios solo se harían en circunstancias excepcionales. Este nuevo documento se publica en la página web de NICE.
El plan de trabajo
El propósito del plan de trabajo es especificar los métodos, definir tiempos y costos. Se redacta un documento interno, que se convierte en referencia para evaluar el progreso de elaboración de las guías. Se espera que los métodos descritos en el plan de trabajo reflejen lo consignado en este documento.
El tiempo de reclutamiento de las personas que realizarán la guía debe ser entre tres y cuatro meses, en algunas ocasiones puede empezarse antes de la realización del plan de trabajo.
El plan de trabajo debe contener información de los miembros del grupo elaborador de guías, identificación de la evidencia, aproximación a la efectividad clínica y al costeo, detalles del proceso empleado, tanto para escribir la guía como para hacer el proceso de revisión, métodos empleados para determinar recursos, costos y tiempos.
Formación del grupo desarrollador de la guía
Formar el grupo desarrollador es la tarea más difícil. Este grupo es el encargado de evaluar la pregunta clínica, buscar la evidencia y generar las recomendaciones. El grupo debe ser multidisciplinario, y debe incluir médicos, cuidadores, expertos técnicos y pacientes. Además, pueden ser de ayuda a personas con conocimientos en los temas tratados, así como representantes de casas farmacéuticas. El número de personas que normalmente conforman el grupo desarrollador de la guía es de 10 a 12 personas, más el grupo técnico. Este último grupo incluye una persona encargada de la revisión sistemática, un especialista en información, un economista de la salud y un administrador de proyectos.
Como estás guías se aplican a Inglaterra y Gales, los expertos son seleccionados para tener representatividad geográfica, dependiendo de la disponibilidad. Se contactan incluso personas de fuera del Reino Unido, cuando no se cuenta con disponibilidad regional.
El siguiente paso es identificar los conflictos de intereses; hay un formato de conflicto de intereses que deben llenar todas las personas que hacen parte de la guía. Y es el jefe desarrollador de la guía quien determina cuáles de estos conflictos son relevantes. Si se determina que un miembro puede tener un conflicto significativo en alguna parte de la guía, se le comisionará el resto y se excluirá en este aspecto específico. Todos estos procesos deben hacerse abiertos y de conocimiento público. Todos los participantes deberán seguir el código de conducta y confidencialidad.
Los miembros externos pueden asistir como observadores o expertos, previamente deben completar el código de conducta de los desarrolladores de las guías así como el formulario de confidencialidad.
Desarrollo de la pregunta clínica
Después de haber determinado el alcance de la guía, el siguiente paso es la elaboración de la pregunta clínica. El número de preguntas puede variar de acuerdo con la guía que se esté desarrollando, pero en guías de 18 meses el número aproximado de preguntas clínicas es de 30.

Rare, orphan and neglected diseases 19
Identificación de la evidencia
Es importante garantizar que, en lo posible, el proceso de selección de la evidencia esté libre se sesgos. La evidencia se obtiene de dos maneras: a partir de la búsqueda sistemática en las bases de datos electrónicas, pero también de la información que las partes interesadas quieran que se revise. Es importante aclarar que no se espera que el NCC revise literatura gris.
La evaluación económica en las guías
La evaluación económica cumple un papel importante en las guías elaboradas por NICE; el economista en salud debe estar en todo momento en la preparación de la guía y debe estar presente en todas las reuniones que se realicen. Es importante dar a conocer que lo que se están evaluando son costos y consecuencias, de cursos de acción alternativos, por lo cual los estudios de carga de la enfermedad o de costos de la enfermedad no tienen cabida en los análisis económicos de las guías. La razón es que el propósito es la maximización de los recursos en salud ganados. Las decisiones se toman en dos escenarios, el primero cuando la efectividad es superior y el costo necesario también lo es, y la segunda cuando la efectividad es menor con una disminución sustancial de los costos.
Las evaluaciones económicas de las guías desarrolladas por NICE generalmente son estudios de costo-efectividad, y la medida de efectividad más usada son los avac (años de vida ajustados por calidad o QALY en inglés). En algunas ocasiones cuando el cálculo de QALY no puede ser estimado se pueden usar otras medidas de efectividad como los años de vida ganados, casos evitados, o desenlaces específicos de la enfermedad. En general la modelación se hace de tres formas: árboles de decisión, modelos de Markov, y simulación de eventos discretos.
Para la evaluación económica se parte de los siguientes supuestos:
• Todos los efectos en salud en los individuos son estimados • La perspectiva empleada es la del NHS o la del sector social gubernamental. • Se descuentan tanto costos como desenlaces, a una tasa empleada de 3,5% anual • El horizonte temporal se selecciona de acuerdo con la posible evaluación de costos y desenlaces de interés. • La calidad de vida es evaluada a partir de métodos de escogencia basados en preferencias, aplicada a una muestra de
la población y validada por expertos.
Si se va a usar caso referencia se deben explicar las razones del uso de esta metodología.
Cuando se obtienen los resultados y una opción es dominante (con costos menores y efectividad mayor), la recomendación está hecha. Pero cuando existen varias alternativas que resultan ser más costosas pero más efectivas se debe calcular la magnitud de la razón de costo-efectividad incremental (RCEI) (ver Recuadro 7), y de acuerdo con esto se toma la decisión. Como consenso general de NICE se dice que una alternativa es costo-efectiva cuando la RCEI es menor de £20.000 por avac ganado, y se consideran una a una aquéllas que se estima tienen una RCEI comprendida entre £20.000 y £30.000 por QALY ganado.
Cuando se determina una opción como costo-efectiva, el siguiente paso es ver el impacto en recursos de esta nueva intervención, se hace entonces un análisis de impacto presupuestal, simultáneamente con el periodo de validación de la guía con el NCC.
La creación de recomendaciones para las guías
El siguiente paso es traducir la evidencia encontrada en recomendaciones puntuales para la práctica clínica. Esta información debe ser presentada de forma clara y concisa, de tal manera que al leerla, no haya que revisar otras referencias.
Las recomendaciones en cuanto a medicamentos deben contener tres aspectos: 1) El nombre del medicamento: debe presen

Rare, orphan and neglected diseases 20
tarse en la denominación genérica. 2) Dosis: no se cita normalmente en la recomendación exceptuando diferentes dosis para diferentes terapéuticas. 3) Uso off label: si la evidencia demuestra que un medicamento no aceptado tiene un efecto benéfico se hará una recomendación y se espera que su uso se individualice.
La presentación de las recomendaciones se debe hacer siguiendo el formato de preguntas “PICO” (population, intervention, control, outcome), seguido de la explicación de por qué esta pregunta es de relevancia. Esta relevancia debe ser explicada a partir de las prioridades tanto de NICE, como de los pacientes, del NHS, de las prioridades nacionales, del tipo de información encontrada, además de consideraciones éticas y económicas.
El desarrollo de criterios de auditoría clínica
Para todo el proceso de la elaboración de guías, NICE contrata con Clinical Accountability, Service Planning and Evaluation (CASPE) Research Unit y Health Quality Service (HQS) para la auditoría interna del proceso.
La escritura de la guía
Cuando todo el proceso se ha cumplido, se realiza la publicación, que contiene, además de la guía completa, la guía de NICE, la guía de consulta rápida, y la guía para pacientes y cuidadores. Se enfatiza en que la información contenida en la guía completa sea de fácil comprensión para médicos no especialistas y para pacientes que conocen de su enfermedad.
Consulta y negociación con los grupos de interés
Para NICE, la información que se remite de las partes interesadas es de mucha utilidad ya que sirve de revisión por pares. Los comentarios dirigidos a NICE deben seguir el siguiente formato:
• Organización: de donde se remite la información. • Capítulo de interés: localización del texto a comentar. • Comentarios. • Respuesta: lugar donde los desarrolladores de la guía responden las inquietudes.
Si los comentarios vienen de las partes de interés registradas se publican en la página en internet con las aclaraciones necesarias. Antes de imprimir el documento final se envía a evaluación al revisor del grupo desarrollador de guías con todos los cambios sugeridos y aceptados.
En el “appraising orphan drugs”, un documento publicado por NICE en 2006, se hace un análisis de la problemática de las enfermedades huérfanas al ser evaluadas desde los umbrales de costo-efectividad tradicionales, y se concluye que las enfermedades cuya prevalencia es menor de 1 por 50,000 habitantes en el Reino Unido deben tener ciertas consideraciones especiales, con umbrales de costo efectividad diferentes a otras condiciones médicas. La explicación está fundamentada en el principio de equidad que claramente perjudica a los pacientes que sufren de este tipo de enfermedades. La conclusión a la que llegan es que hay que generar un nuevo centro que se especialice en la evaluación de tecnologías en enfermedades raras. Según los cálculos de las RCEI, los umbrales podrían llegar a ser hasta 10 veces mayores. Ejemplos de esto son la enfermedad de Fabry, Gaucher y la mucopolisacaridosis I y III, cuyo manejo se encuentra aprobado, y cuyas RCEI por QALY adicional ganado se encuentran entre £100.000- £400.000 (ver Tabla 4).
Tabla 4. Ejemplos de enfermedades huérfanas aprobadas por NICE, con RCEI hasta 10 veces superiores al estándar convencional.
Rare, orphan and neglected diseases 21
Análisis bibliométrico de las enfermedades raras
El análisis bibliométrico es una herramienta que combina la matemática y la estadística, analizando el número de publicaciones, los temas generales abordados y la distribución geográfica de los documentos científicos (Gonzalez de Dios et al., 2007).
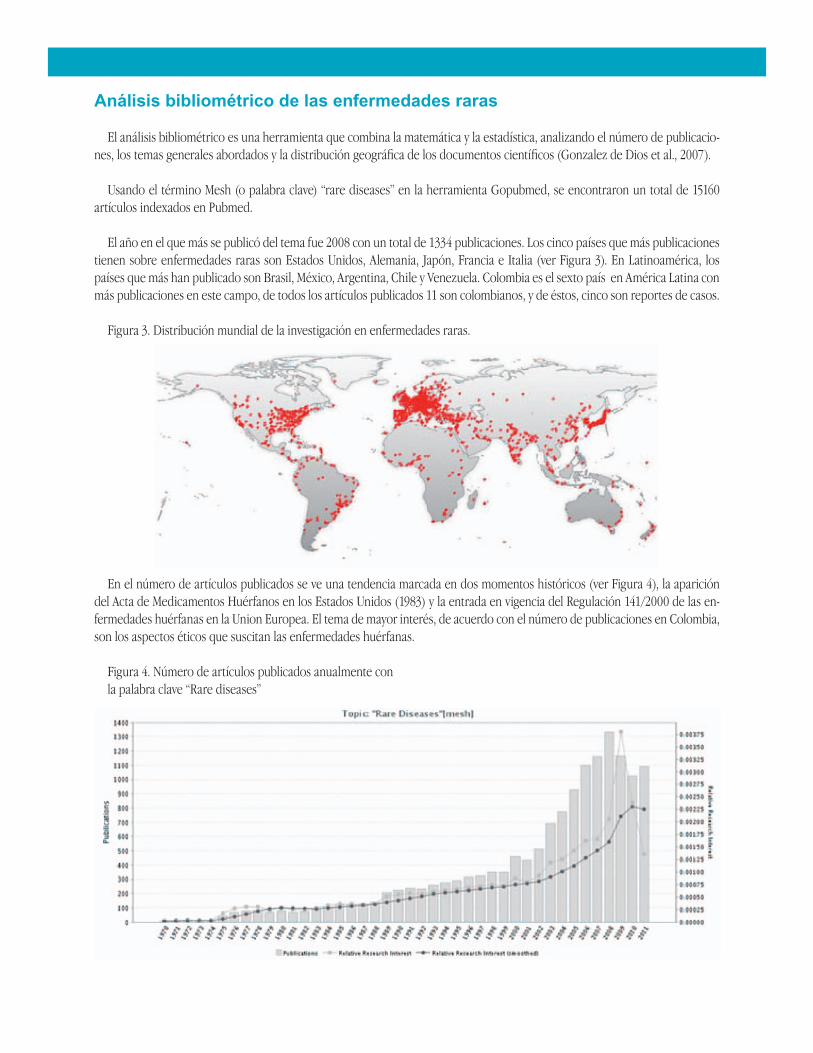
Usando el término Mesh (o palabra clave) “rare diseases” en la herramienta Gopubmed, se encontraron un total de 15160 artículos indexados en Pubmed.
El año en el que más se publicó del tema fue 2008 con un total de 1334 publicaciones. Los cinco países que más publicaciones tienen sobre enfermedades raras son Estados Unidos, Alemania, Japón, Francia e Italia (ver Figura 3). En Latinoamérica, los países que más han publicado son Brasil, México, Argentina, Chile y Venezuela. Colombia es el sexto país en América Latina con más publicaciones en este campo, de todos los artículos publicados 11 son colombianos, y de éstos, cinco son reportes de casos.
Figura 3. Distribución mundial de la investigación en enfermedades raras.
En el número de artículos publicados se ve una tendencia marcada en dos momentos históricos (ver Figura 4), la aparición del Acta de Medicamentos Huérfanos en los Estados Unidos (1983) y la entrada en vigencia del Regulación 141/2000 de las enfermedades huérfanas en la Union Europea. El tema de mayor interés, de acuerdo con el número de publicaciones en Colombia, son los aspectos éticos que suscitan las enfermedades huérfanas.
Figura 4. Número de artículos publicados anualmente con la palabra clave “Rare diseases”

Rare, orphan and neglected diseases 22
Conclusiones
En los últimos años ha aumentado el interés en las enfermedades raras, huérfanas y olvidadas, tanto por los gobiernos, centros de investigación y evaluación de tecnologías, industria farmacéutica y comunidad médica. La magnitud del problema se ha visto tanto desde la perspectiva de la salud pública como de las dificultades que enfrentan los pacientes con una enfermedad de difícil diagnóstico y todo tipo de barreras para un tratamiento eficaz. Esperamos que este escrito cumpla la función de contribuir a la divulgación de estas enfermedades.
Tabla 5. Línea de tiempo de las enfermedades huérfanas.

Rare, orphan and neglected diseases 23
Referencias
Akobeng, A. K. (2005). Principles of evidence based medicine. Archives of Disease in Childhood 90: 837-840.
Anónimo. (2009). Phase 0 trials: a platform for drug development? Lancet, 374(9685): 176.
Barrera, L. A., Cely Galindo, G. (2010). Ethical aspects on rare diseases. Advances in Experimental Medicine and Biology, 686:493-511.
Beyrer, C., Villar, J. C., Suwanvanichkij, V., Singh, S., Baral, S. D. & Mills, E. J. (2007). Neglected diseases, civil conflicts, and the right to health. Lancet, 370(9587):619-627.
Boy, R. & Schramm, R. F. (2009). Bioética da proteção e trata-mento de doenças genéticas raras no Brasil: o caso das doenças de depósito lisossomal. Caderno de Saúde Pública do Rio de Janeiro, 25(6):1276-1284.
Cleemput, I., Neyt, M., Thiry, N., De Laet, C. & Leys, M. (2008). Threshold values for cost-effectiveness in health care. Brussels: Belgian Health Care Knowledge Centre. HTA reports. Obtenido en octubre 26 de 2011. http://kce.fgov.be/?ID=1405
Cleemput, I., Neyt, M., Thiry, N., De Laet, C. & Leys, M. (2011). Using threshold values for cost per quality-adjusted life-year gained in healthcare decisions. International Journal of Technology Assessment in Health Care, 27(1): 71-76.
Cookson, R., McCabe, C. & Tsuchiya, A. (2008). Public healthcare resource allocation and the Rule of Rescue. Journal of Medical Ethics, 34(7): 540-544.
Direcção-Geral da Saúde, Portugal. (2008). Programa Nacional para Doenças Raras (PNDR). Obtenida en agosto 12 de 2011. http://www.min-saude.pt/NR/rdonlyres/555DD3B3-45F0-4F74-B633-28889E721BF1/0/i010420.pdf
Eurodis (2005) Rare Diseases: understanding this public health priority. Obtenida en 3 de octubre de 2011. http://www. orpha.net/consor/cgi-bin/Education.php?lng=ES
European Parliament and the Council of the European Union. (2000). Regulation (EC) No. 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. Official J. Eur. Communities L18/1 – L18/5
Futerman, A. H. & van Meer, G. (2004). The cell biology of lysosomal storage disorders. Nature Reviews Molecular Cell Biology, 5(7):554–565.
Gad, S. C. (2009). Clinical Trials Handbook. London: John Wiley & Sons.
González de Dios, J., Sempere, A. P. & Aleixandre-Benavent, R. (2007) Las publicaciones biomédicas en España a debate (I): estado de las revistas neurológicas. Revista de Neurología, 44(1):32-42.
Harmatz, P., Giugliani, R., Schwartz, I., et al. (2006). Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. Journal of Pediatrics, 148:533-539.
Kaback, M., Lim-Steele, J., Dabholkar, D., Brown, D., Levy, N. & Zeiger, K. (1993). Tay-Sachs disease--carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970 to 1993. JAMA, 270(19):2307-2315.
Luisetti, M., Campo, I., Scabini, R., Zorzetto, M., Kadija, Z., Mariani, F. & Ferrarotti, I. (2010). The problems of clinical trials and registries in rare diseases. Respiratory Medicine, 104 Suppl1: S42-44.
Meinert, C., & Tonascia, S. (1986). Clinical trials: design, conduct, and analysis. New York: Oxford University Press.
Miyamoto, B. E. & Kakkis, E. D. (2011). The potential investment impact of improved access to accelerated approval on the development of treatments for low prevalence rare diseases. Orphanet Journal of Rare Diseases. 6:49.
Moore, D. F., Forget, E. L., Ries, M. & Schiffmann, R. (2007) Enzyme replacement therapy in orphan and ultra-orphan diseases: the limitations of standard economic metrics as exemplified by Fabry-Anderson disease. Pharmacoeconomics. 25(3):201-208.
Muñoz, R. & Bangdiwala, S.I. (2000). Análisis interino en ensayos clínicos: una guía metodológica. Revista Médica de Chile 128(8): 935-941. Obtenida en octubre 8 de 2011. Disponible en: http://www.scielo.cl/scielo.php?script=sci_ arttext&pid=S0034-98872000000800014&lng=es. doi: 10.4067/S0034-98872000000800014.

Rare, orphan and neglected diseases 24
National Organization for Rare Disorders. (2011). About NORD. Obtenido en agosto 14 de 2011. http://www.rarediseases.org/about
National Institute for Excelence and Clinical Evidence - NICE (2006). Citizens Council Report - Rule of Rescue.Obtenida en septiembre 22 de 2011. http://www.nice.org.uk/aboutnice/ whoweare/board/boardmeetings/2006/19july2006agm/nice_ citizens_council_report__rule_of_rescue.jsp
National Institute for Excelence and Clinical Evidence - NICE (2006). Appraising Orphan Drugs. Obtenida en noviembre 25 de 2011. http://www.nice.org.uk/niceMedia/pdf/smt/120705item4. pdf
National Institute for Excelence and Clinical Evidence - NICE (2011) Developing NICE clinical guidelines. Obtenida en octubre 2 de 2011. http://www.nice.org.uk/aboutnice/howwework/ how_we_work.jsp
Orphanet. (2010). About rare diseases. Obtenido en septiembre 22 de 2011. http://www.orpha.net/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN
Oostenbrink, R., Derksen-Lubsen, G., Essink-Bot, M. L., Grobbee, D.E, Moll, H. A., Moons, K. G. M., Oostenbrink, J. B & Redekop, W. K. (2002). Cost-utility analysis of patient care in children with meningeal signs. International Journal of Technology Assessment in Health Care. 18(4):485-496.
Osborne, M. & Evans, T. W. (1994). Allocation of resources in intensive care: a transatlantic perspective. Lancet 343:778–780.
Pariser, A. R., Xu, K., Milto, J. & Coté, T. R. (2011). Regulatory considerations for developing drugs for rare diseases: orphan designations and early phase clinical trials. Discovery Medicine. 11(59): 367-375.
Purmonen, T. T. (2011). Short-course adjuvant trastuzumab therapy in early stage breast cancer in Finland: Cost-effectiveness and value of information analysis based on the 5-year follow-up results of the FinHer Trial. Acta Oncologica 50(3):344-352.
Richards, R. J. & Hammitt, J. K. (2002). Timing of prophylactic surgery in prevention of diverticulitis recurrence: a cost-effectiveness analysis. Digestive Disease & Sciences 47(9):1903-1908.
Rosselli, D., Rueda, J. D. & Solano, M. (2012) Ethical and economic considerations of rare diseases in ethnic minorities: a
case report of mucopolysaccharidosis VI in Colombia. Journal of Medical Ethics (submitted)
Smith, G. C & Pell, JP. (2003). Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ 327(7429):1459-1461.
Staretz-Chacham, O., Lang, T. C., LaMarca, M. E., Krasnewich, D. & Sidransky, E. (2009). Lysosomal storage disorders in the newborn. Pediatrics. 123(4): 1191-1207.
Svatek, R. S. (2010). Cost utility of prostate cancer chemoprevention with dutasteride in men with an elevated PSA. Cancer Prevention Research (Philadelphia) 4(2):277-283:
Van der Lee, J. H., Wesseling, J., Tanck, M. W. & Offringa, M. (2008). Efficient ways exist to obtain theoptimal sample size in clinical trials in rare diseases. Journal of Clinical Epidemiology 61(4): 324-330.
Vargas V, Poblete S. (2008). Health prioritization: the case of Chile. Health Affairs (Millwood) 27(3): 782-92.
Vellodi, A. (2005). Lysosomal storage disorders. British Journal of Haematology 128(4):413-431.
Vinhas de Souza, M., Corrêa, B., Dornelles, P. & Doederlein Schwartz, I. V., (2007) Medicamentos de alto custo para doenças raras no Brasil: o exemplo das doenças lisossômicas. Ciência & Saúde Coletiva:Obtenida en 27 de octubre de 2011. http://redalyc.uaemex.mx/src/inicio/ArtPdfRed.jsp?iCve=63015154015#
Westermark, K. (2007). The regulation of orphan medicines in the EU: objectives reached and main challenges when facing the future. Pharmaceutical Policy & Law 9: 327–342 (2007).
Wiest, R. (2010). A economía das doenças raras: Teoria, evidências e politicas públicas. Monografia apresentada ao departamento de Ciências Econômicas da Facultade de Ciências Econômicas da Universidade Federal do Rio Grande do Sul, como requisito parcial para a obtençâo do título de bacharel em Economia. Porto Alegre, Universidade Federal do Rio Grande do Sul.
Westermark, K., Holm, B. B., Söderholm, M., Llinares-Garcia, J., Rivière, F., Aarum, S. et al. (2011). European regulation on orphan medicinal products: 10 years of experience and future perspectives. Nature Review Drug Discovery 10(5):341-349.

Rare, orphan and neglected diseases 25
Anexo 1 LEY 1392 DE 2010
(julio 2) Diario Oficial No. 47.758 de 2 de julio de 2010
CONGRESO DE LA REPÚBLICA
Por medio de la cual se reconocen las enfermedades huérfanas como de especial interés y se adoptan normas tendientes a garantizar la protección social por parte del Estado colombiano a la población que padece de enfermedades huérfanas y sus cuidadores.
EL CONGRESO DE COLOMBIA
DECRETA:
ARTÍCULO 1o. OBJETO DE LA LEY. La presente ley tiene como objeto reconocer que las enfermedades huérfanas, representan un problema de especial interés en salud dado que por su baja prevalencia en la población, pero su elevado costo de atención, requieren dentro del SGSSS un mecanismo de aseguramiento diferente al utilizado para las enfermedades generales, dentro de las que se incluyen las de alto costo; y unos procesos de atención altamente especializados y con gran componente de seguimiento administrativo.
Para tal efecto el Gobierno Nacional, implementará las acciones necesarias para la atención en salud de los enfermos que padecen este tipo de patologías, con el fin de mejorar la calidad y expectativa de vida de los pacientes, en condiciones de dis ponibilidad, equilibrio financiero, accesibilidad, aceptabilidad y estándares de calidad, en las fases de promoción, prevención, diagnóstico, tratamiento, rehabilitación e inclusión social, así como incorporar los demás componentes de la protección social, más allá de los servicios de salud, para pacientes, cuidadores y familias, dándole un enfoque integral al abordaje y manejo de estas patologías.
ARTÍCULO 2o. DENOMINACIÓN DE LAS ENFERMEDADES HUÉRFANAS. Las enfermedades huérfanas son aquellas crónicamente debilitantes, graves, que amenazan la vida y con una prevalencia menor de 1 por cada 2.000 personas , comprenden, las enfermedades raras, las ultrahuérfanas y olvidadas. Las enfermedades olvidadas son propias de los países en desarrollo y afectan ordinariamente a la población más pobre y no cuentan con tratamientos eficaces o adecuados y accesibles a la población afectada.
PARÁGRAFO. Con el fin de mantener unificada la lista de denominación de las enfermedades huérfanas, el Ministerio de la Protección Social emitirá y actualizará esta lista cada dos años a través de acuerdos con la Comisión de Regulación en Salud (CRES), o el organismo competente.
ARTÍCULO 3o. RECONOCIMIENTO DE LAS ENFERMEDADES HUÉRFANAS COMO ASUNTO DE INTERÉS NACIONAL. El Gobierno Nacional reconocerá de interés nacional las enfermedades huérfanas para garantizar el acceso a los servicios de salud y tratamiento y rehabilitación a las personas que se diagnostiquen con dichas enfermedades, con el fin de beneficiar efectivamente a esta población con los diferentes planes, programas y estrategias de intervención en salud, emitidas por el Ministerio de la Protección Social.
ARTÍCULO 4o. PRINCIPIOS RECTORES. Se tendrán como principios rectores de interpretación para la protección efectiva de las personas que padecen enfermedades huérfanas:
Esta prevalencia fue modificada luego mediante la Ley 1438 de 2011 de 1 por cada 2000 personas a 1 por cada 5000 personas.

Rare, orphan and neglected diseases 26
Universalidad: El Estado deberá garantizar la atención en salud de todas las personas que padecen enfermedades huérfanas en condiciones de calidad, accesibilidad y oportunidad.
Solidaridad: Se creará un mecanismo para coordinar las acciones de la sociedad en general, las organizaciones públicas y privadas, los entes especializados nacionales e internacionales, con miras a potenciar y maximizar el efecto de las acciones tendientes a prevenir, promover, educar sobre las enfermedades huérfanas y proteger los derechos de todas las personas que padecen dichas enfermedades.
Corresponsabilidad: La familia, la sociedad y el Estado son corresponsables en la garantía de los derechos de los pacientes que padecen enfermedades huérfanas y propiciarán ambientes favorables para ellos, con el fin de generar las condiciones adecuadas, tanto en el ámbito público como privado, que permitan su incorporación, adaptación, interacción ante la sociedad.
Igualdad: El Gobierno Nacional, promoverá las condiciones para que la igualdad, sea real y efectiva y adoptará medidas en favor de todas las personas que padezcan enfermedades huérfanas, para que estas gocen de los mismos derechos, libertades y oportunidades sin ninguna discriminación en el acceso a los servicios.
CAPÍTULO II. DE LA FINANCIACIÓN.
ARTÍCULO 5o. FINANCIACIÓN DE LAS ENFERMEDADES HUÉRFANAS. Las personas con enfermedades huérfanas que requieran con necesidad diagnósticos, tratamientos, medicamentos, procedimientos y cualquier otra prestación en salud no incluída en los planes obligatorios de salud, que no tengan capacidad de pago serán financiados en el Régimen Subsidiado con cargo a los recursos señalados en la Ley 715 de 2001 y las demás normas que financien la atención de la población pobre no asegurada y de los afiliados al Régimen Subsidiado en lo no cubierto con subsidios a la demanda. Si las fuentes anteriores no son suficientes, se podrá disponer de manera excepcional de los recursos excedentes de la subcuenta de Eventos Catastróficos y Accidentes de Tránsito–ECAT, del Fosyga. En el Régimen Contributivo, las prestaciones en salud no incluídas en el plan obligatorio serán financiadas con cargo a los recursos de la Subcuenta de Compensación del Fondo de Solidaridad y Garantía–Fosyga, que no afecten los destinados al aseguramiento obligatorio en salud. Para efectos del presente artículo, se faculta al Gobierno Nacional para establecer un régimen especial de condiciones y tarifas máximas al cual deberá sujetarse el reconocimiento y pago de los costos de la atención de dichas enfermedades.
CAPÍTULO III. DE LOS DEBERES Y OBLIGACIONES.
ARTÍCULO 6o. DEBERES POR PARTE DEL GOBIERNO NACIONAL. Dentro de los deberes que estarán a cargo del Gobierno Nacional, se determinan los siguientes:
Deberes del Gobierno Nacional:
1. Establecer a través de las guías de atención que para esto emita el Ministerio de la Protección Social, con la metodología aprobada y basadas en evidencia las directrices, criterios y procedimientos de diagnóstico y tratamiento de los pacientes que padezcan enfermedades huérfanas, identificadas como tal, de acuerdo a los criterios de selección.
2. Evaluar y definir a través del proceso definido con la Comisión de Regulación en Salud (CRES), los servicios de pruebas diagnósticas que es necesario incluir en el plan de beneficios con su respectivo ajuste de UPC, para que las aseguradoras de planes de beneficios puedan garantizar el estudio y diagnóstico.
3. Estudiar, coordinar y promover e implementar con organismos especializados públicos y privados, del orden nacional e internacional, el desarrollo de investigaciones en procura de estudiar las enfermedades huérfanas, buscando la posibilidad de

Rare, orphan and neglected diseases 27
diagnósticos tempranos en pro de una mejor calidad y expectativa de vida.
4. El Gobierno Nacional a través del Ministerio de la Protección Social y los Entes Territoriales, en conjunto con las diferentes asociaciones de pacientes y científicas, entre otros grupos interesados, establecerá una serie de acciones tendientes a la divulgación de las enfermedades huérfanas, con el objetivo de crear sensibilidad y conciencia social en razón de dichas enfermedades.
ARTÍCULO 7o. REGISTRO NACIONAL DE PACIENTES QUE PADECEN ENFERMEDADES HUÉRFANAS. El Gobierno Nacional implementará un sistema de información de pacientes con enfermedades huérfanas.
Con el registro de pacientes se busca generar un sistema de información básico sobre enfermedades huérfanas que proporcione un mayor conocimiento sobre la incidencia de los casos, la prevalencia, la mortalidad o en su defecto el número de casos detectados en cada área geográfica, permitiendo identificar los recursos sanitarios, sociales y científicos, que se requieren, neutralizar la intermediación en servicios y medicamentos, evitar el fraude y garantizar que cada paciente y su cuidador o familia en algunos casos, recibe efectivamente el paquete de servicios diseñado para su atención con enfoque de protección social.
PARÁGRAFO. El Ministerio de la Protección Social contará con 6 meses a partir de la promulgación de la presente ley para reglamentar el presente artículo.
ARTÍCULO 8o. MEDICAMENTOS PARA ENFERMEDADES HUÉRFANAS. Con el fin de mejorar el acceso de los pacientes a los medicamentos huérfanos y una mejor administración de los recursos financieros, se faculta al Gobierno Nacional para que en un plazo de un año adopte un sistema de negociación y compra, que podrá ser centralizado, con las farmacéuticas y laboratorios productores e importadores de medicamentos y tecnologías diagnósticas, para la atención de este tipo de patologías, que permita el acceso equitativo para todos los pacientes.
CAPÍTULO IV. INCENTIVOS PARA CONSOLIDAR LA ATENCIÓN Y EL DESARROLLO DEL CONOCIMIENTO CIENTÍFICO DE LAS ENFER
MEDADES HUÉRFANAS.
ARTÍCULO 9o. CENTROS PARA EL MANEJO DE ENFERMEDADES HUÉRFANAS. El Ministerio de la Protección Social reglamentará la conformación de una red de centros de referencia para la atención de los pacientes que padezcan enfermedades huérfanas, en la cual participarán los distintos actores del Sistema General de Seguridad Social en Salud, según sus competencias. La red estará conformada por 3 subredes:
1. Red de Centros de Diagnóstico
2. Red de Centros de Tratamiento
3. Red de Farmacias para suministro y seguimiento a tratamientos farmacológicos.
Los Centros de Referencia deberán acreditar experiencia, además de contar con el personal idóneo y calificado, de acuerdo a los criterios que para cada subred defina el Ministerio de la Protección Social, con los cuales se seleccionarán, evaluarán y rehabilitarán periódicamente.
PARÁGRAFO. A partir de la expedición de la presente ley, el Ministerio de la Protección Social contará con un término de seis meses para reglamentar el presente artículo.
ARTÍCULO 10. CAPACITACIÓN Y DIVULGACIÓN DEL CONOCIMIENTO SOBRE ENFERMEDADES HUÉRFANAS AL TALENTO HUMANO EN SALUD. Además de los criterios académicos ya desarrollados por el Ministerio de la Protección Social para la capacitación del personal de talento humano en salud, en concordancia a lo establecido con la Ley 1164 de 2007, a través

Rare, orphan and neglected diseases 28
del Consejo Nacional de Talento Humano en Salud, impulsará las acciones tendientes a promover la capacitación a nivel de pregrado, posgrado y docente asistencial que permitan la capacitación y divulgación del conocimiento de las enfermedades huérfanas, a todas las ocupaciones y profesiones de la salud.
ARTÍCULO 11. DE LA INVESTIGACIÓN. El Gobierno Nacional estimulará a través de los mecanismos que para esto expida el Ministerio de la Protección Social, bajo la asesoría del Consejo Nacional de Talento Humano en Salud, de acuerdo con la Ley 1164 de 2007, los mecanismos de promoción y participación, para la investigación científica de los diagnósticos tempranos y posibles medicamentos, tratamientos preventivos, aspectos sicológicos y psiquiátricos asociados con estas enfermedades no solo desde el punto de vista de los pacientes sino de sus familiares.
ARTÍCULO 12. INSERCIÓN SOCIAL. El Gobierno Nacional diseñará estrategias que propendan la inclusión e integración social de la población de pacientes con enfermedades huérfanas, tales como: acceso a bienes y servicios, a educación y al mercado laboral; identificando las barreras de acceso y las prácticas institucionales de discriminación con el fin de establecer mecanismos para su eliminación.
CAPÍTULO V. INSPECCIÓN, VIGILANCIA Y CONTROL.
ARTÍCULO 13. DE LA INSPECCIÓN, VIGILANCIA Y CONTROL. La Superintendencia Nacional de Salud, en ejercicio de sus atribuciones de inspección, vigilancia y control, se encargará del seguimiento y la vigilancia de las acciones que los actores del sistema deban cumplir para la atención de los pacientes que padecen enfermedades huérfanas.
ARTÍCULO 14. ESTÁNDARES DEL SISTEMA DE INSPECCIÓN, VIGILANCIA Y CONTROL DE LA SUPERINTENDENCIA NACIONAL DE SALUD. Para cumplir con las funciones de inspección, vigilancia y control, la Superintendencia Nacional de Salud ejercerá sus funciones teniendo como base los siguientes estándares:
1. Acceso a la atención. Se encargará de velar por el efectivo cumplimiento de los derechos que tiene la población que padece las enfermedades huérfanas.
2. Prestación de servicios de atención en salud. Su objetivo es vigilar que la prestación de los servicios de atención en salud individual y colectiva a los pacientes con enfermedades huérfanas se haga en condiciones de disponibilidad, accesibilidad, aceptabilidad y estándares de calidad, en las fases de promoción, prevención, diagnóstico, tratamiento y rehabilitación.
3. Información. Vigilar que los actores del Sistema garanticen la producción de los datos con calidad, cobertura, pertinencia, oportunidad y transparencia.
ARTÍCULO 15. COOPERACIÓN INTERNACIONAL. El Gobierno Nacional podrá establecer estrategias de Cooperación Internacional, para facilitar el logro de los fines de la presente ley, así como implementar mecanismos que permitan el desarrollo de proyectos estratégicos con otros Estados para promover el tratamiento integral para las personas que padecen enfermedades huérfanas.
ARTÍCULO 16. VIGENCIA. La presente ley rige a partir de su sanción y publicación.
El Presidente del honorable Senado de la República,JAVIER CÁCERES LEAL.
El Secretario General del honorable Senado de la República,EMILIO OTERO DAJUD.

Rare, orphan and neglected diseases 29
El Presidente de la honorable Cámara de Representantes, ÉDGAR ALFONSO GÓMEZ ROMÁN.
El Secretario General de la honorable Cámara de Representantes, JESÚS ALFONSO RODRÍGUEZ CAMARGO.
REPÚBLICA DE COLOMBIA – GOBIERNO NACIONAL Publíquese y cúmplase.
Dada en Bogotá, D. C., a 2 de julio de 2010.
ÁLVARO URIBE VÉLEZ
El Ministro de Hacienda y Crédito Público,ÓSCAR IVÁN ZULUAGA ESCOBAR
El Ministro de la Protección Social,DIEGO PALACIO BETANCOURT

Rare, orphan and neglected diseases 30
Anexo 3
REGULATION (EC) - No 141/2000 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 December 1999 on orphan medicinal products
THE EUROPEAN PARLIAMENT AND THE COUNCIL OF THE EUROPEAN UNION, Having regard to the Treaty establishing the European Community, and in particular Article 95 thereof, Having regard to the proposal from the Commission, Having regard to the opinion of the Economic and Social Committee, Acting in accordance with the procedure laid down in Article 251 of the Treaty, Whereas:
. (1) some conditions occur so infrequently that the cost of developing and bringing to the market a medicinal product to diagnose, prevent or treat the condition would not be recovered by the expected sales of the medicinal product; the pharmaceutical industry would be unwilling to develop the medicinal product under normal market conditions; these medicinal products are called ‘orphan’;
. (2) patients suffering from rare conditions should be entitled to the same quality of treatment as other patients; it is therefore necessary to stimulate the research, development and bringing to the market of appropriate medications by the pharmaceutical industry; incentives for the development of orphan medicinal products have been available in the United States of America since 1983 and in Japan since 1993;
. (3) in the European Union, only limited action has been taken so far, whether at national or at Community level, to stimulate the development of orphan medicinal products; such action is best taken at Community level in order to take advantage of the widest possible market and to avoid the dispersion of limited resources; action at Community level is preferable to uncoordinated measures by the Member States which may result in distortions of competition and barriers to intra-Community trade;
. (4) orphan medicinal products eligible for incentives should be easily and unequivocally identified; it seems most ap propriate to achieve this result through the establishment of an open and transparent Community procedure for the designation of potential medicinal products as orphan medicinal products;
. (5) objective criteria for designation should be established; those criteria should be based on the prevalence of the condition for which diagnosis, prevention or treatment is sought; a prevalence of not more than five affected persons per 10 thousand is generally regarded as the appropriate threshold; medicinal products intended for a life-threatening, seriously debilitating or serious and chronic condition should be eligible even when the prevalence is higher than five per 10 thousand;
. (6) a Committee composed of experts appointed by the Member States should be established to examine applications for designation; this Committee should also include three representatives of patients’ associations, designated by the Commission, and three other persons, also designated by the Commission, on a recommendation from the European Agency for the Evaluation of Medicinal Products (hereinafter referred to as ‘the Agency’); the Agency should be responsible for the adequate coordination between the Committee on orphan medicinal products and the Committee on proprietary medicinal products;
. (7) patients with such conditions deserve the same quality, safety and efficacy in medicinal products as other patients; orphan medicinal products should therefore be submitted to the normal evaluation process; sponsors of orphan medicinal products should have the possibility of obtaining a Community authorisation; in order to facilitate the granting or the maintenance of a Community authorisation, fees to be paid to the Agency should be waived at least in part; the Community budget should compensate the Agency for the loss in revenue thus occasioned;
. (8) experience in the United States of America and Japan shows that the strongest incentive for industry to invest in the development and marketing of orphan medicinal products is where there is a prospect of obtaining market exclusivity for

Rare, orphan and neglected diseases 31
a certain number of years during which part of the investment might be recovered; data protection under Article 4 of Council Directive 65/65/EEC of 26 January 1965 on the approximation of provisions laid down by law, regulation or administrative action relating to medicinal products is not a sufficient incentive for that purpose; Member States acting independently cannot introduce such a measure without a Community dimension as such a provision would be contradictory to Directive 65/65/EEC; if such measures were adopted in an uncoordinated manner by the Member States, this would create obstacles to intraCommunity trade, leading to distortions of competition and running counter to the single market; market exclusivity should however be limited to the therapeutic indication for which orphan medicinal product designation has been obtained, without prejudice to existing intellectual property rights; in the interest of patients, the market exclusivity granted to an orphan medicinal product should not prevent the marketing of a similar medicinal product which could be of significant benefit to those affected by the condition;
. (9) sponsors of orphan medicinal products designated under this Regulation should be entitled to the full benefit of any incentives granted by the Community or by the Member States to support the research and development of medicinal products for the diagnosis, prevention or treatment of such conditions, including rare diseases;
. (10) the specific programme Biomed 2, of the fourth framework programme for research and technological develop ment (1994 to 1998), supported research on the treatment of rare diseases, including methodologies for rapid schemes for the development of orphan medicinal products and inventories of available orphan medicinal products in Europe; those grants were intended to promote the establishment of cross national cooperation in order to implement basic and clinical research on rare diseases; research on rare diseases continues to be a priority for the Community, as it has been included in the fifth framework programme for research and technological development (1998 to 2002); this Regulation establishes a legal framework which will allow the swift and effective implementation of the outcome of this research;
. (11) rare diseases have been identified as a priority area for Community action within the framework for action in the field of public health; the Commission, in its communication concerning a programme of Community action on rare diseases within the framework for action in the field of public health has decided to give rare diseases priority within the public health framework; the European Parliament and the Council have adopted Decision No 1295/1999/EC of 29 April 1999 adopting a programme of Community action on rare diseases within the framework for action in the field of public health (1999 to 2003), including actions to provide information, to deal with clusters of rare diseases in a population and to support relevant patient organisations; this Regulation implements one of the priorities laid down in this programme of action,
HAVE ADOPTED THIS REGULATION:
Article 1 Purpose
The purpose of this Regulation is to lay down a Community procedure for the designation of medicinal products as orphan medicinal products and to provide incentives for the research, development and placing on the market of designated orphan medicinal products.
Article 2 Definitions
For the purposes of this Regulation: . (a) ‘medicinal product’ means a medicinal product for human use, as defined in Article 2 of Directive 65/65/EEC; . (b) ‘orphan medicinal product’ means a medicinal product designated as such under the terms and conditions of this
Regulation; . (c) ‘sponsor’ means any legal or natural person, established in the Community, seeking to obtain or having obtained
the designation of a medicinal product as an orphan medicinal product; . (d) ‘Agency’ means the European Agency for the Evaluation of Medicinal Products.

Rare, orphan and neglected diseases 32
Article 3 Criteria for designation
1. A medicinal product shall be designated as an orphan medicinal product if its sponsor can establish: (a) that it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition
affecting not more than five in 10 thousand persons in the Community when the application is made, or that it is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the Community and that without incentives it is unlikely that the marketing of the medicinal product in the Community would generate sufficient return to justify the necessary investment;
and (b) that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been
authorised in the Community or, if such method exists, that the medicinal product will be of significant benefit to those affected
by that condition.
2. The Commission shall adopt the necessary provisions for implementing this Article in the form of an implementing Regulation in accordance with the procedure laid down in Article 72 of Council Regulation (EEC) No 2309/93.
Article 4 Committee for Orphan Medicinal Products
1. A Committee for Orphan Medicinal Products, hereinafter referred to as ‘the Committee’, is hereby set up within the Agency.
2. The task of the Committee shall be:. (a) to examine any application for the designation of a medicinal product as an orphan medicinal prod
uct which is submitted to it in accordance with this Regulation; . (b) to advise the Commission on the establishment and development of a policy on orphan medicinal
products for the European Union; . (c) to assist the Commission in liaising internationally on matters relating to orphan medicinal products,
and in liaising with patient support groups;. (d) to assist the Commission in drawing up detailed guidelines.
3. The Committee shall consist of one member nominated by each Member State, three members nominated by the Commission to represent patients’ organisations and three members nominated by the Commission on the basis of a recommendation from the Agency. The members of the Committee shall be appointed for a term of three years, which shall be renewable. They may be accompanied by experts.
4. The Committee shall elect its Chairman for a term of three years, renewable once.
5. The representatives of the Commission and the Executive Director of the Agency or his representative may attend all meetings of the Committee.
6. The Agency shall provide the secretariat of the Committee.
7. Members of the Committee shall be required, even after their duties have ceased, not to disclose any information of the kind covered by the obligation of professional secrecy.

Rare, orphan and neglected diseases 33
Article 5 Procedure for designation and removal from the register
1. In order to obtain the designation of a medicinal product as an orphan medicinal product, the sponsor shall submit an application to the Agency at any stage of the development of the medicinal product before the application for marketing authorisation is made.
2. The application shall be accompanied by the following particulars and documents: (a) name or corporate name and permanent address of the sponsor; (b) active ingredients of the medicinal product; (c) proposed therapeutic indication; (d) justification that the criteria laid down in Article 3(1) are met and a description of the stage of development,
including the indications expected.
3. The Commission shall, in consultation with the Member States, the Agency and interested parties, draw up detailed guidelines on the required format and content of applications for designation.
4. The Agency shall verify the validity of the application and prepare a summary report to the Committee. Where appro- priate, it may request the sponsor to supplement the particulars and documents accompanying the application.
5. The Agency shall ensure that an opinion is given by the Committee within 90 days of the receipt of a valid application.
6. When preparing its opinion, the Committee shall use its best endeavours to reach a consensus. If such a consensus cannot be reached, the opinion shall be adopted by a majority of two-thirds of the members of the Committee. The opinion may be obtained by written procedure.
7. Where the opinion of the Committee is that the applica- tion does not satisfy the criteria set out in Article 3(1), the Agency shall forthwith inform the sponsor. Within 90 days of receipt of the opinion, the sponsor may submit detailed grounds for appeal, which the Agency shall refer to the Committee. The Committee shall consider whether its opinion should be revised at the following meeting.
8. The Agency shall forthwith forward the final opinion of the Committee to the Commission, which shall adopt a decision within 30 days of receipt of the opinion. Where, in exceptional circumstances, the draft decision is not in accord- ance with the opinion of the Committee, the decision shall be adopted in accordance with the procedure laid down in Article 73 of Regulation (EEC) No 2309/93. The decision shall be notified to the sponsor and communicated to the Agency and to the competent authorities of the Member States.
9. The designated medicinal product shall be entered in the Community Register of Orphan Medicinal Products.
10. Each year the sponsor shall submit to the Agency a report on the state of development of the designated medicinal product.
11. To have the designation of an orphan medicinal product transferred to another sponsor, the holder of the designation shall make specific application to the Agency. In consultation with the Member States, the Agency and interested parties, the Commission shall draw up detailed guidelines on the form in which applications for transfer shall be made and the content of such applications and all the particulars of the new sponsor.

Rare, orphan and neglected diseases 34
12. A designated orphan medicinal product shall be removed from the Community Register of Orphan Medicinal Products: . (a) at the request of the sponsor; . (b) if it is established before the market authorisation is granted that the criteria laid down in Article 3 are no longer
met in respect of the medicinal product concerned; . (c) at the end of the period of market exclusivity as laid down in Article 8.
Article 6 Protocol assistance
1. The sponsor of an orphan medicinal product may, prior to the submission of an application for marketing author isation, request advice from the Agency on the conduct of the various tests and trials necessary to demonstrate the quality, safety and efficacy of the medicinal product, in accordance with Article 51(j) of Regulation (EEC) No 2309/93.
2. The Agency shall draw up a procedure on the development of orphan medicinal products, covering regulatory assistance for the definition of the content of the application for authorisation within the meaning of Article 6 of Regulation (EEC) No 2309/93.
Article 7 Community marketing authorisation
1. The person responsible for placing on the market an orphan medicinal product may request that authorisation to place the medicinal product on the market be granted by the Community in accordance with the provisions of Regulation (EEC) No 2309/93 without having to justify that the medicinal product qualifies under Part B of the Annex to that Regulation.
2. A special contribution from the Community, distinct from that provided for in Article 57 of Regulation (EEC) No 2309/93, shall be allocated every year to the Agency. The contribution shall be used exclusively by the Agency to waive, in part or in total, all the fees payable under Community rules adopted pursuant to Regulation (EEC) No 2309/93. A detailed report of the use made of this special contribution shall be presented by the Executive Director of the Agency at the end of each year. Any surplus occurring in a given year shall be carried forward and deducted from the special contribution for the following year.
3. The marketing authorisation granted for an orphan medicinal product shall cover only those therapeutic indications which fulfil the criteria set out in Article 3. This is without prejudice to the possibility of applying for a separate marketing authorisation for other indications outside the scope of this Regulation.
Article 8 Market exclusivity
1.Medicinal product is granted pursuant to Regulation (EEC) No 2309/93 or where all the Member States have granted marketing authorisations in accordance with the procedures for mutual recognition laid down in Articles 7 and 7a of Directive 65/65/ EEC or Article 9 of Council Directive 75/319/EEC of 20 May 1975 on the approximation of provisions laid down by law, regulation or administrative action relating to medicinal products, and without prejudice to intellectual property law or any other provision of Community law, the Community and the Member States shall not, for a period of 10 years, accept another application for a marketing authorisation, or grant a marketing authorisation or accept an application to extend an existing marketing authorisation, for the same therapeutic indication, in respect of a similar medicinal product.
2. This period may however be reduced to six years if, at the end of the fifth year, it is established, in respect of the medicinal product concerned, that the criteria laid down in Article 3 are no longer met, inter alia, where it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity. To that end, a Member State shall inform the Agency that the criterion on the basis of which market exclusivity was granted may not be met and the Agency

Rare, orphan and neglected diseases 35
shall then initiate the procedure laid down in Article 5. The sponsor shall provide the Agency with the information necessary for that purpose.
3. By way of derogation from paragraph 1, and without prejudice to intellectual property law or any other provision of Com munity law, a marketing authorisation may be granted, for the same therapeutic indication, to a similar medicinal product if:
. (a) the holder of the marketing authorisation for the original orphan medicinal product has given his consent to the second applicant, or
. (b) the holder of the marketing authorisation for the original orphan medicinal product is unable to supply sufficient quantities of the medicinal product, or
. (c) the second applicant can establish in the application that the second medicinal product, although similar to the orphan medicinal product already authorised, is safer, more effective or otherwise clinically superior.
4. The Commission shall adopt definitions of ‘similar medicinal product’ and ‘clinical superiority’ in the form of an implementing Regulation in accordance with the procedure laid down in Article 72 of Regulation (EEC) No 2309/93.
5. The Commission shall draw up detailed guidelines for the application of this Article in consultation with the Member States, the Agency and interested parties. Where a marketing authorisation in respect of an orphan
Article 9 Other incentives
1. Medicinal products designated as orphan medicinal products under the provisions of this Regulation shall be eligible for incentives made available by the Community and by the Member States to support research into, and the development and availability of, orphan medicinal products and in particular aid for research for small- and medium-sized undertakings provided for in framework programmes for research and technological development.
2. Before 22 July 2000, the Member States shall communicate to the Commission detailed information concerning any measure they have enacted to support research into, and the development and availability of, orphan medicinal products or medicinal products that may be designated as such. That information shall be updated regularly.
3. Before 22 January 2001, the Commission shall publish a detailed inventory of all incentives made available by the Community and the Member States to support research into, and the development and availability of, orphan medicinal products. That inventory shall be updated regularly.
Article 10 General report
Before 22 January 2006, the Commission shall publish a general report on the experience acquired as a result of the application of this Regulation, together with an account of the public health benefits which have been obtained.
Article 11 Entry into force
This Regulation shall enter into force on the day of its publication in the Official Journal of the European Communities. It shall apply as from the date of adoption of the implementing Regulations provided for in Article 3 and Article 8.This Regulation shall be binding in its entirety and directly applicable in all Member States. Done at Brussels, 16 December
1999. For the European Parliament The President N. FONTAINE For the Council The President K. HEMILÄ
Related Documents











