Errores innatos del Metabolismo

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Errores innatos del Metabolismo

Errores innatos del Metabolismo
1908 – Archibald Garrod – Alcaptonuria
Caractrizada por: ácido homogentísico AH en orina (alcapton)
Suministro AH, fenilalanina o tirosina =A.H. urinario
Congénita – familiar
Postuló: AH es intermediario metabolismo fenilalanina y tirosina
Excreción AH por déficit enzima hepática. Confirmad luego en Bx hepáticas
Enfermedad recesiva = doble dosis gen alterado
Alkapton denota sustancias de rápida absorción de O2 cuando es expuesta a alcali o aire
“Herencia Mendeliana en el hombre” catálogo de Me-Kusick:
3907 monogénicas, se conoce la localización en 1096 de ellas,
En 250 se ha identificado el defecto bioquímico básico
En 196 se ha identificado anormalidades en las proteínas enzimáticas.

FUNDAMNETOS BUIOQUÍMICOS GENERALES
Expresión cuantitativa de genes mutantes:
-Autosómico recesivo = heterocigotos suficiente el 50% de la enzima.
-Autosómico Dominantes: detectados clínicamente como heterocigotos (vías enzimáticas o proteínas)
Enzimáticas: Control por retroalimentación como Porfiria intermitente agúda: Uroporfibinógeno I sintetasa – Hem
Proteínas: Hipercolesterolemia familiar: Pocos receptores de CHO (lipoproteínas de baja densidad)
- Ligado al X: recesivo o dominante. La mujer homocigota o heterocigota, recesiva o dominante. El hombre por tener un solo cromosoma son homocigotos nomales o deficientes.
G6PD tiene do copias en la mujer y una en el hombre = Lyon “compensación de dosis”

Influencias ambientales
Factores ambientales que modifican la expresión de ciertos EIM: Galactosemia clásica (Galactosa-1-fosfato uridiltransfrerasa). Daño hepático, retardo mental, etc = no galactosa
Heterogeneidad genética
Fenotipo clínico que puede ser resultado de más de un genotipo. Diferentes mutaciones en un solo locus o alelos mutantes en locis diferentes. 40 variantes de Hb con variantes en la cadena beta.
Las proteínas como productos génicos
Hemoglobinopatías y talasemias: Genotipos, proteína, función, celular. Genes estructurales, controladores.
Efectos: catálisis, disminución afinidad por sustratos, menor concentración intracelular de la proteína, disminución de la velocidad de síntesis, degradación rápida.

Consecuencias de los defectos metabólicos
-Deficiencia de precursor: E. Hartnup, triptofano no transportado (defecto) = materia prima de nicotinamida. Ataxia cerebelosa (suministro oral).
-Acumulación metabolitos previos al bloqueo. Galactosemia
-Formación insuficiente de metabolitos post bloqueo: Hiperplasia suprarenal congénita. Déficit de 21-hidroxilasa = insuficiente corticoides y aldosterona desde CHO.
-Utilización de vías alternas: Fenilcetonuria clásica, déficit de FAH. Excreción de A. fenilpirúvico, feniláctico, fenilacético desde fenilalanina.
- Alteraciones en el control de retroalimentación: Hipercolesterolemia familiar. Mutación receptor=No se capta CHO. Síntesis hidroximetilglutaril coenzima A reductasa = limita la velocidad de flujo a través de la vía biosintética. El CHO exógeno regula su síntesis inhibiendo la síntesis de la enzima.
- Sobreproducción de intermediarios: Artritis gotosa: incremento en fosforribosilpirofosfato. Enzima mutada incrementa actividad = incremento de fosforibosilpirofosfato desde purinas y A. úrico

Diagnostico
Pruebas químicas cualitativas
Cromatografia: AA
Pruebas microbiológicas: inhibición bacteriana
Enzimáticos: tamizaje por fluorescencia: Eritrocitos
Confirmatorias: cuantificación metabolitos o actividad enzimática

Orina
Alcaptonuria
Oxidasa del ácido Homogentísico
Más de 400 mutaciones (sinónimas, no sinónimas, deleción, inserción, polimorfismos y de splicing) = diferente grado. Defectos en la síntesis de BH4 (biopterina, cofactor)
FenilcetonuriaAMINOÁCIDOS


FENILCETONURIA
Una pequeña proporción de AH es retenida en el cuerpo porque es eliminado vía renal.
El AH es oxidado a ácido benzoquinoacético por la influiencia de polifenoloxidasa y polimeriza para formar macromoléculas que dañan la espina dorsal y el tejido conectivo incluyendo el cartílago. El cartílago inelástico es visible en las aurículas (Figs 1 and 2).
La pigmentación del ojo es llamada ocrosis porque es negra-gris gruesa, ocre y aparwece en adultos. La espondilopatía orcrótica es típica como calcificaciones en los discos vertebrales con reduccción de los espacios y refracción osteoporótica de los cuerpos vertebralesgFig. 3.
Retardo mentalFrecuencia 1X10-6 nacidos vivos
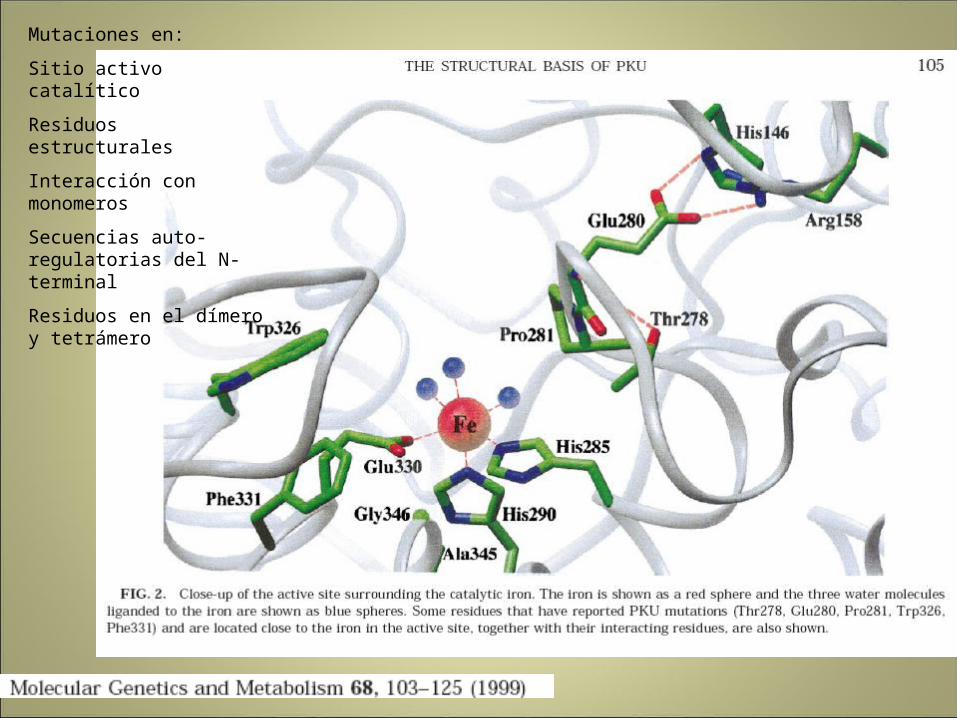
Mutaciones en:
Sitio activo catalítico
Residuos estructurales
Interacción con monomeros
Secuencias auto-regulatorias del N-terminal
Residuos en el dímero y tetrámero

Cistinuria
Aminoácidos transportados a través de células por transporte activo - permeasa
Absorción gástrica y resorción tubular renal
El transporte intestinal específico dado para:
AA dicarboxílicos, Dibásicos, iminoácidos y glicina, aliáticos neutros y aromáticos; beta aminoácidos
La cisteína es similar a los dibásicos:lisina ornitina y arginina; lisina esencial.
Cisteína mediada por transporte compartido: Específico, compartido para AA dibásicos y u no específico para lisina, arginina y ornitina.
Cistinuria = déficit sitema compartido dibásicos y cistina. Absorción intestinal y resorción deficiente: cistina, lisina, arginina y ornitina. Niveles plasmáticos normales con excreción de los mismos.
Cálculos renales de cistina, también en uretra y vejiga,. OPbstrucción, infecciones, IRA.
Frecuencia 1/7000 Recesivo
Dx cristales en orina, nitropusiato orina, transport3 intestinal, demostración AA acompañantes. TTO: alcalinización orina, aumento solubilidad cistina.

Disease Enzyme deficiency Symptoms
Citrullinema Arginosuccinate lyase Lethergy, siezures, reduced muscle tension
Tyrosinemia Various enzymes of tyrosine degradation
Weakness, self-mutilation, liver damage, mental retardation
Albinism Tyrosinase Absence of pigmentation
Homocystinuria Cystathionine β-synthase Scoliosis, muscle weakness, mental retardation, thin blond hair
Hyperlysinemia α-Aminoadipic semialdehyde dehydrogenase
Seizures, mental retardation, lack of muscle tone, ataxia
Errores en el metabolismo de aminoácidos

Disease Name Gene
Symbol Chromosomal
Locus Protein Name
Molecular Genetic Test Availability
Carbamoylphosphate synthetase I deficiency
CPS1 1 2q35 Carbamoyl-phosphate
synthase ammonia
Clinical
Ornithine transcarboxylase deficiency
OTC Xp21.1 Ornithine
carbamoyltransferase
Clinical
Citrullinemia type I ASS 9q34 Argininosuccinate
synthase
Clinical
Argininosuccinicaciduria ASL 7cen-q11.2 Argininosuccinate lyase Clinical
Arginase deficiency ARG1 6q23 Arginase 1 Clinical
NAGS deficiency NAGS 17q21.3 N-acetyl glutamate
synthetase
Clinical
1. Summar et al 2003
Errores innatos del metabolismo de la úrea

Ciclo de la úrea

Alteraciones de la hemoglobina- Enfermedad molecular
TTE Oxígeno, formada por HEM y globina
Porción globínica formada por 4 cadenas polipeptídicas (tetrámeros): alfas (dos cadenas Zeta y 2 alfa, 141 AA, cromosoma 16) y no alfas codificadas por genes específicos (epsilon, gammma, delta y beta, 146 AA, cromosoma 11).
Activos en diferentes estados vida. Forman 6 tipos de HB humnas normales: Gower I y II, Portland, fetal, A2 y adulta.
Anemia Falciforme: HbS difiere de la HbA en que tiene Valina sustituída por glutámico. Actualmente 350 variaciones.
Las hemoglobinopatías estan dadas por: Variantes estructurales de cadenas polipeptídicas, Ausencia de síntesis de cadenas globínicas y que alteran la relación alfa/no alfa = talasemias y Modificación del cambio HbF a HbA.
Mutacones: Sustitución, deleción o entrecruzamiento (meiosis).

HbS más grave – síntomas desde 6 mes.
Eritrocito rígido: daño vascular, hipoxia: pulmón, hueso, riñón, hígado.
Talasemia alfa: El humano posee 4 cadenas alfa y las manifestaciones dependen de los genes afectados (- alfa/alfa alfa) es portador.
Hipocromía, hemólisis, deleción de los 4 genes (fetal) edema y esplenomegalia incompatible con vida. Tetrámeros conocidos como cuerpos de Bart
Talasemias beta: Puede ser beta0 o beta+ . Ausencia o disminución de cadenas beta. Anemia de Coley (primeros meses, progresa y grave). Infecciones, diarrea, detiene desarrollo, cuadro mongoloide, hepato, pigmentación.
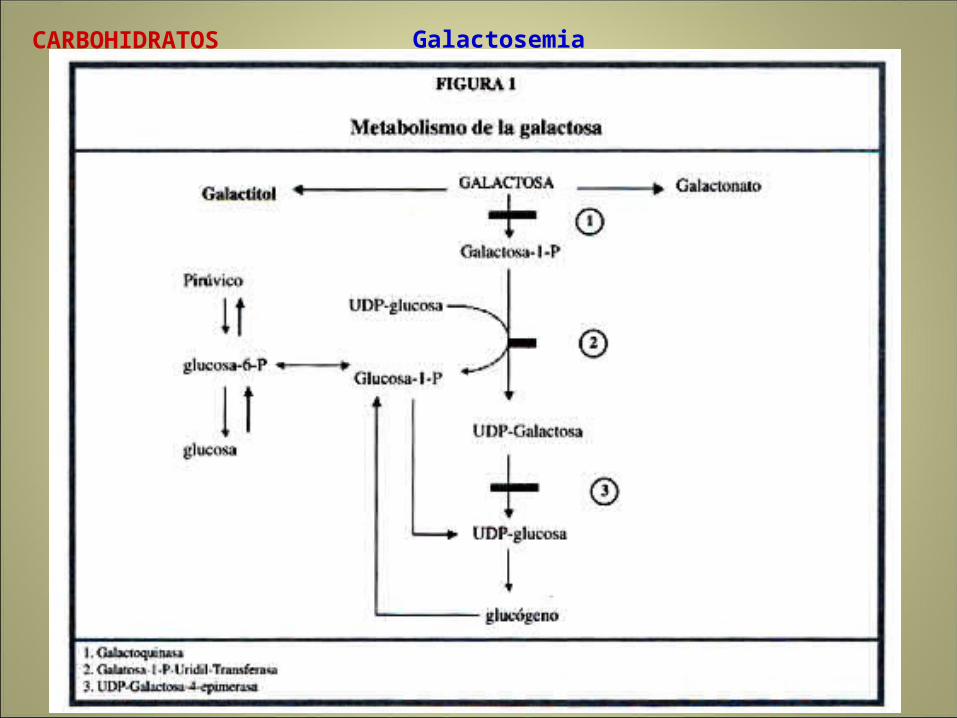
GalactosemiaCARBOHIDRATOS

Galactosemia
Efectos contra dieta normal: Acumulación de metaboliots previos al punto de bloque y aperuras devías metabólicas alternas.
Galactocinasa:
Imposibilita cell par fosforilación oxidativa de galactosa
Acumula en tejidos y excreta en orina
seacumula galactosa-1-fosfato en eritrocitos
Se acumukla en el cristalino, daño en hígado, cerebro y riñón
Fracuencia 1/62.000
Related Documents











