HIGHLIGHT Engineering Functional Materials by Halogen Bonding PIERANGELO METRANGOLO, 1 GIUSEPPE RESNATI, 1 TULLIO PILATI, 2 ROSALBA LIANTONIO, 1 FRANCK MEYER 1 1 Laboratory of Nanostructured Fluorinated Materials (NFMLab), Department of Chemistry, Materials, and Chemical Engineering ‘‘Giulio Natta’’, Politecnico di Milano, I-20131 Milano, Italy 2 Institute of Molecular Science and Technology, CNR, Department of Chemistry, University of Milan, I-20133 Milano, Italy Received 15 June 2006; accepted 19 June 2006 DOI: 10.1002/pola.21725 Published online in Wiley InterScience (www.interscience.wiley.com). ABSTRACT: Engineering fu- nctional materials endowed with un- precedented properties require the ex- ploitation of new intermolecular inter- actions, which can determine the characteristics of the bulk materials. The great potential of Halogen Bond- ing (XB), namely any noncovalent interaction involving halogens as electron acceptors, in the design of new and high-value functional materi- als is now emerging clearly. This Highlight will give a detailed over- view on the energetic and geometric features of XB, showing how some of them are quite constant in most of the formed supramolecular complexes (e.g. , the angle formed by the covalent and the noncovalent bonds around the halogen atom), while some others depend strictly on the nature of the interacting partners. Then, several specific examples of halogen-bonded supramolecular architectures, whose structural aspects as well as applica- tions in fields as diverse as enantiom- ers’ separation, crystal engineering, liquid crystals, natural, and synthetic receptors, will be fully described. V V C 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 1–15, 2007 Keywords: halogen bonding; molecular recognition; self-as- sembly; structure-property rela- tions; supramolecular structures Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 45, 1–15 (2007) V V C 2006 Wiley Periodicals, Inc. Correspondence to: P. Metrangolo (E-mail: pierangelo.metrango- [email protected]) or G. Resnati (E-mail: [email protected]) 1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

HIGHLIGHT
Engineering Functional Materials by Halogen Bonding
PIERANGELO METRANGOLO,1 GIUSEPPE RESNATI,1 TULLIO PILATI,2
ROSALBA LIANTONIO,1 FRANCK MEYER1
1Laboratory of Nanostructured FluorinatedMaterials (NFMLab), Department of Chemistry,Materials, and Chemical Engineering ‘‘Giulio Natta’’, Politecnico di Milano, I-20131Milano, Italy
2Institute of Molecular Science and Technology, CNR, Department of Chemistry,University of Milan, I-20133 Milano, Italy
Received 15 June 2006; accepted 19 June 2006DOI: 10.1002/pola.21725Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: Engineering fu-
nctional materials endowed with un-
precedented properties require the ex-
ploitation of new intermolecular inter-
actions, which can determine the
characteristics of the bulk materials.
The great potential of Halogen Bond-
ing (XB), namely any noncovalent
interaction involving halogens as
electron acceptors, in the design of
new and high-value functionalmateri-
als is now emerging clearly. This
Highlight will give a detailed over-
view on the energetic and geometric
features of XB, showing how some of
them are quite constant in most of the
formed supramolecular complexes
(e.g., the angle formed by the covalentand the noncovalent bonds around the
halogen atom), while some others
depend strictly on the nature of the
interacting partners. Then, several
specific examples of halogen-bonded
supramolecular architectures, whose
structural aspects as well as applica-
tions in fields as diverse as enantiom-
ers’ separation, crystal engineering,
liquid crystals, natural, and synthetic
receptors, will be fully described.VVC 2006 Wiley Periodicals, Inc. J Polym Sci
Part A: PolymChem 45: 1–15, 2007
Keywords: halogen bonding;
molecular recognition; self-as-
sembly; structure-property rela-
tions; supramolecular structures
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 45, 1–15 (2007)VVC 2006 Wiley Periodicals, Inc.
Correspondence to:P.Metrangolo (E-mail: [email protected]) orG.Resnati (E-mail: [email protected])
1

Giuseppe Resnati (left) was born in 1955 in Monza (Italy) and
obtained his Ph.D. in Industrial Chemistry in 1986 at the University of
Milan. After two years in pharmaceutical companies, he moved to the
Politecnico di Milano where initially he was a researcher of the
National Research Council, then associate professor (1998), and finally
full professor (2001). Since 2001, he has been a member of the Edito-
rial Board of the Journal of Fluorine Chemistry. He has been a NATO
Fellow at the University of Clemson (USA, 1990), a visiting Professor
at the Universite Paris XI (France, 1993), a Fellow of the Japan Soci-
ety for the Advancement of Science, Nagoya University (Japan, 2001).
He is the author and co-author of more than 165 original research
papers and 10 reviews or book chapters. His research interests are flu-
orine chemistry, synthetic methodologies, and self-assembly processes.
Franck Meyer (right) was born in 1972 in Pontoise (France) and
obtained his PhD in Organic Chemistry in 2000 at the local university.
After two years of temporary position in a pharmaceutical company
(2001–2003), he moved to the Politecnico di Milano in 2004 for a
postdoctoral appointment under the supervision of Prof. Resnati (EU
fellow). Since 2005, he has been appointed as a research assistant pro-
fessor at the Politecnico di Milano. His research interests are in the
synthesis of fluorinated tectons for recognition processes.
Tullio Pilati was born in 1946 in Muggio (Italy). He obtained his Lau-
rea in Chemistry in 1971 at the University of Milan. He is a Senior
Researcher of the National Research Council at the University of Mi-
lan. His research interests are in the crystallography of perfluorocar-
bon-hydrocarbon hybrid materials and bioactive compounds.
Pierangelo Metrangolo (left) was born in 1972 in Lecce (Italy). He obtained
his Laurea in Pharmaceutical Chemistry and Technology at the University of
Milan (1997), a master in Synthetic Organic Chemistry (1999), and then the
Ph.D. in Industrial Chemistry at the Politecnico di Milano (2002). Since
2002, he has been an assistant professor at the Politecnico di Milano where
he was promoted to associate professor in 2005. He was a recipient of the
2005 ‘‘G. Ciamician’’ medal of the Organic Chemistry Division of the Italian
Chemical Society and of the 2005 ‘‘Journals Grants for International
Authors’’ of the Royal Society of Chemistry. He has been a EU Fellow at the
University of Toulouse (France, 2001), and visiting Professor at the Univer-
sity of York (UK, 2005). He is the author and co-author of more than 60
original research papers and 6 book chapters. His research interests are in the
field of supramolecular and material chemistry.
Rosalba Liantonio (right) was born in 1973 in Palo del Colle (Italy) and
obtained her Laurea in Pharmaceutical Chemistry and Technology at the
University of Bari (1999). Then, she moved to the Politecnico di Milano in
2001 where she obtained her PhD in Industrial Chemistry in 2005. Since
2005, she has been appointed for postdoctoral work at the Politecnico di
Milano under the supervision of Prof. Resnati. Her research interests are in
the field of self-assembly processes by using halogen bonding.
TULLIO PILATI
PIERANGELOMETRANGOLO (left) AND
GIUSEPPE RESNATI(right)
ROSALBALIANTONIO (left)
ANDFRANCK MEYER (right)
2 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

INTRODUCTION
The physical and chemical properties of bulk materials
are not the cumulative result of the properties of the par-
ticles they are compounded of. Some characteristics of
the bulk spring out of its nano- and microscopic archi-
tecture and are related to the noncovalent interactions
that determine the architecture. Materials with unprece-
dented characteristics can thus result from the availabil-
ity of new intermolecular interactions.
The focus of this Highlight is on halogen bonding
(XB), which is the attractive interaction where halogen
atoms work as electron density acceptors. The functional
properties of materials exerted by the presence of these
interactions will also be discussed. The formation of do-
nor–acceptor complexes between halogens, or halocar-
bons, and lone pair-possessing atoms or anions, was
seminally emphasized by O. Hassel in his Nobel lecture.
In recent years, the use of haloperfluorocarbons as elec-
tron acceptor species allowed the successful engineering
of crystalline systems to be achieved.1 This boosted the
awareness of the great potential of interactions wherein
halocarbons work as electron density acceptors, and use-
ful applications in fields as diverse as organic semicon-
ductors, liquid crystals, and substrate–enzyme binding
optimization are appearing.
The term halogen bonding (XB) was introduced to
name any D���X��Y interaction in which X is the halo-
gen (Lewis acid, XB-donor), D is any donor of electron
density (Lewis base, XB-acceptor), and Y is carbon,
nitrogen, halogen, etc. (Chart 1). This term stresses the
numerous similarities existing between this interaction
and hydrogen bonding (HB).2 Indeed, most of the ener-
getic and geometric features found in hydrogen-bonded
complexes are encountered in halogen-bonded com-
plexes as well.
GENERAL CHARACTERISTICS OF XB
The term XB comprehensively encompasses a vast class
of noncovalent interactions, from the weak
C��X1���X2��C interaction (so named type-I halogen-
��halogen contacts)3 to the very strong I����I2 interaction(forming the tri-iodide anion). It is thus not surprising
that XB interaction energy spans over a very wide range
of values, from 5 to 180 kJ/mol. In particular, the
strength of D���X XB increases with the electron density
on D; this is the reason why anions are usually stronger
XB-acceptors than neutral species. Conversely, the
strength of D���X interactions increases with the
decrease of electron density around X. As a result of
this, the tendency to form strong interactions decreases
moving from C(sp)��X to C(sp2)��X to C(sp3)��X
groups. Fluorine atoms and perfluorinated residues are
very strong electron-withdrawing groups; for this rea-
son, haloperfluorocarbons (halo-PFCs) are stronger XB-
donors than corresponding halocarbon parents. The
tendency to form strong interactions decreases moving
from iodo- to bromo- to chlorocarbons and only occa-
sional reports describe the possible involvement of also
fluorocarbons in XB interactions. As a consequence of
their effectiveness in giving strong XB, iodo-PFCs will
be used as the unifying motif around which this High-
light is organized, whereas other XB-donors will be
considered only within examples of particular rele-
vance.
Both p and n electrons can be involved in the forma-
tion of XB and usually the former give weaker interac-
tions than the latter.4 Nitrogen (e.g., amine and pyridine
derivatives) gives stronger XB than oxygen and sulfur
(e.g., ethers and thioethers). However, a very rigorous
scale wherein XB-donors (or acceptors) are listed
according to their strength cannot be filed as pairing of
complementary sites after HSBA theory is favored so
that the effectiveness of a given XB-donor (or acceptor)
may also depend on the used XB-acceptor (or donor,
respectively).
The attractive nature of XB causes nonminor inter-
penetration of donor and acceptor orbitals. D���X distan-
ces are between the sum of van der Waals radii and
covalent bonds, the stronger the interaction is the shorter
the D���X distance is. Consistent with the n (or p) ? r*nature of this interaction, the X��Y covalent bond
lengthens on XB formation. The angle between covalent
and noncovalent bonds around the halogen in D���X��Y
is approximately 1808.5 This is consistent with the theo-
retical prediction that the anisotropic distribution of
electron density around the halogen atom has a mini-
mum along the extended C��X bond axis. The shorter
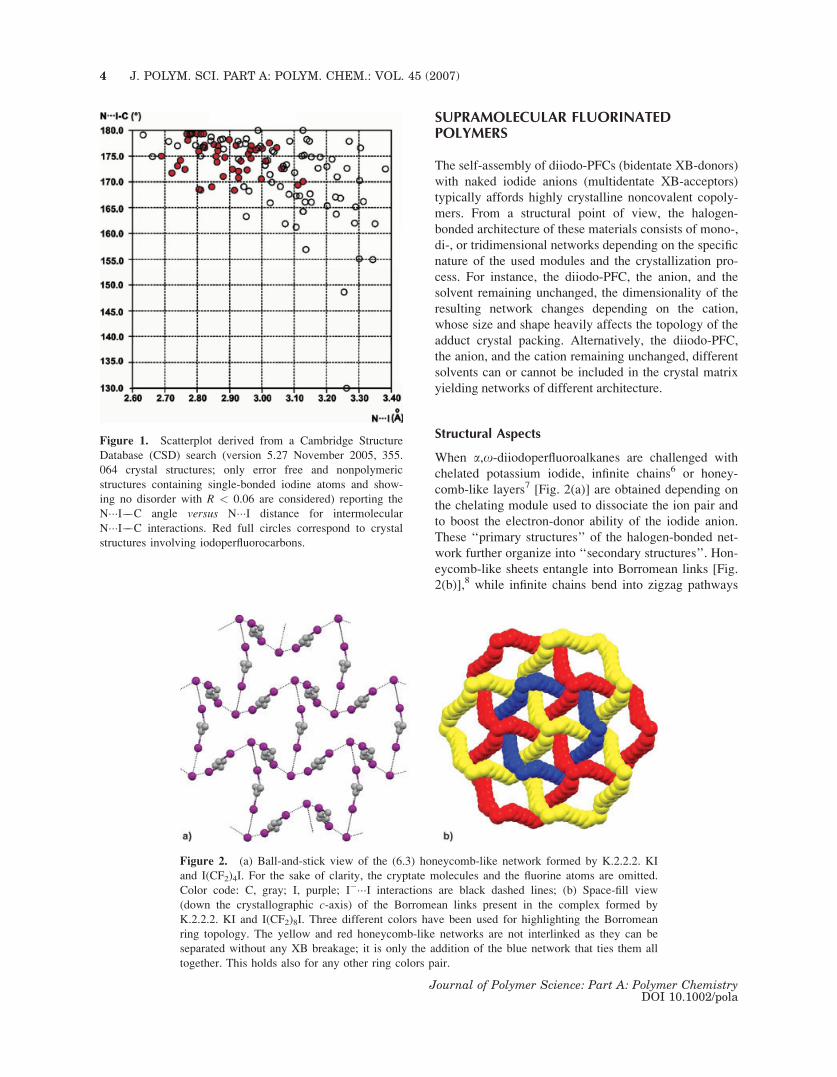
the XB is, the more linear the angle is (Fig. 1).
When n electron-donors are used, XB occurs along
the axis of the orbital containing the donor lone-pair
(Scheme 1).
Chart 1. Schematic representation of halogen bonding.
Donors (D) are neutral or anionic species while acceptors (X)
are halogen atoms linked to a wide variety of molecular scaf-
folds (Y). [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
HIGHLIGHT 3
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
SUPRAMOLECULAR FLUORINATEDPOLYMERS
The self-assembly of diiodo-PFCs (bidentate XB-donors)
with naked iodide anions (multidentate XB-acceptors)
typically affords highly crystalline noncovalent copoly-
mers. From a structural point of view, the halogen-
bonded architecture of these materials consists of mono-,
di-, or tridimensional networks depending on the specific
nature of the used modules and the crystallization pro-
cess. For instance, the diiodo-PFC, the anion, and the
solvent remaining unchanged, the dimensionality of the
resulting network changes depending on the cation,
whose size and shape heavily affects the topology of the
adduct crystal packing. Alternatively, the diiodo-PFC,
the anion, and the cation remaining unchanged, different
solvents can or cannot be included in the crystal matrix
yielding networks of different architecture.
Structural Aspects
When a,x-diiodoperfluoroalkanes are challenged with
chelated potassium iodide, infinite chains6 or honey-
comb-like layers7 [Fig. 2(a)] are obtained depending on
the chelating module used to dissociate the ion pair and
to boost the electron-donor ability of the iodide anion.
These ‘‘primary structures’’ of the halogen-bonded net-
work further organize into ‘‘secondary structures’’. Hon-
eycomb-like sheets entangle into Borromean links [Fig.
2(b)],8 while infinite chains bend into zigzag pathways
Figure 1. Scatterplot derived from a Cambridge Structure
Database (CSD) search (version 5.27 November 2005, 355.
064 crystal structures; only error free and nonpolymeric
structures containing single-bonded iodine atoms and show-
ing no disorder with R < 0.06 are considered) reporting the
N���I��C angle versus N���I distance for intermolecular
N���I��C interactions. Red full circles correspond to crystal
structures involving iodoperfluorocarbons.
Figure 2. (a) Ball-and-stick view of the (6.3) honeycomb-like network formed by K.2.2.2. KI
and I(CF2)4I. For the sake of clarity, the cryptate molecules and the fluorine atoms are omitted.
Color code: C, gray; I, purple; I����I interactions are black dashed lines; (b) Space-fill view
(down the crystallographic c-axis) of the Borromean links present in the complex formed by
K.2.2.2. KI and I(CF2)8I. Three different colors have been used for highlighting the Borromean
ring topology. The yellow and red honeycomb-like networks are not interlinked as they can be
separated without any XB breakage; it is only the addition of the blue network that ties them all
together. This holds also for any other ring colors pair.
4 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

[Fig. 3(a)]6 or roll up into infinite helixes, which, in their
turn, may pair into homochiral double helixes9 [Fig.
3(b)]. A similar behavior is observed when dibromoper-
fluoroalkanes are used as XB-donors.10
The poor affinity perfluoroalkane derivatives for or-
ganic and inorganic compounds determines have mod-
ules’ segregation, which contributes substantially to the
arrangement of formed architectures organized into hier-
archic structures over different length scales. Separate
hydrocarbon (HC) and PFC domains are formed where
the cations are embedded in the HC domains while
anions are at the interface and bind by XB the perfluori-
nated modules. In these examples, XB and nanophase
separation operate in tandem in producing materials in
which structuring at the molecular level (Angstrom
length scale) grows up to supramolecular assemblies
(nanometer length scale). Perfluoroalkanes are endowed
with useful and unique properties (e.g., particularly low
dielectric constants, refractive indexes, surface tensions
and quite high density, and compressibility). Materials
possessing PFC-HC phase-separated domains can thus
be endowed with unique electrical, optical, and elec-
tronic properties.
Bromide, chloride, and fluoride anions are less prone
than iodide anions to give rise to halogen-bonded
adducts but their behaviors are similar.11 The bromide
anion/dibromo-PFCs self-assembly allowed a useful
application to be realized. Racemic 1,2-dibromohexa-
fluoropropane (1) was challenged with (�)-sparteinium
bromide (2), yielding the 1:1 crystalline adduct 3, made
up of 2 and (S)-1, exclusively.10a Each bromide ion
bridges a primary and a secondary bromine of two dis-
tinct and following dibromo-PFC units 1, each of which
is well ordered through bonding to two bromide ions.
Enantiopure, infinite twofold helices are formed and 1 is
resolved into the pure (S) enantiomer; thanks to its
highly specific inclusion in a chiral crystal with a halo-
gen-bonded helical arrangement (Fig. 4). This process
maximizes the transfer of information from the HC to
the PFC units.
The resolution of chiral and racemic protic acids with
chiral and enantiopure bases through HB-driven forma-
tion of diastereomeric salt is a well-established protocol.
The example discussed earlier shows how XB may be
equally effective in driving enantiomer resolution pro-
cesses. This is but an example of the opportunities
opened by this kind of interaction.
Conformational Aspects
Perfluoroalkanes have a very poor tendency to form
crystals suitable for single crystal X-ray analysis as a
consequence of the weak intermolecular interactions
perfluoroalkyl chains give rise to. This accounts for the
high disorder that characterizes perfluoroalkyl chains
also in the solid state. A lack of structural information
on perfluoroalkane molecules and polymers thus ensues.
When long-chain iodo- and bromo-perfluoroalkanes are
halogen-bonded to anionic or neutral lone-pair possess-
ing species, highly crystalline materials develop and sin-
gle crystal X-ray diffraction methods can be used rou-
tinely for their analysis. This has helped in filling the
above-mentioned lack of information on the structural
properties of PFCs. For instance, it was proven that PFC
chains undergo a nonminor widening of the C��C��C
angles (mean value 116.2, mean 6 SE 0.2) and a less re-
markable narrowing of the F��C��F angles (mean value
110.7, mean6 SE 0.3). Other interesting pieces of infor-
mation concerning the preferred conformation of PFC
chains were obtained. While the helical distorted-trans
conformation is most frequently encountered in long-
chain diiodoperfluoroalkanes, the zigzag all-trans is not
Scheme 1. Geometric features shown by a selection of hal-
ogen-bonded complexes involving a variety of N and O elec-
tron donor species. The D���I��C angles (D¼¼N, O) remain
close to 1808 and almost independent on the nature of the
donor species. (a) Supramol Chem 2002, 14, 47–55; (b) Tet-
rahedron 2000, 56, 5535–5550; (c) Chem Commun 2004,
1492–1493; (d) Tetrahedron 2002, 58, 4023–4029; (e) Acta
Crystallogr 2003, E59, o1332–o1333; (f) J Am Chem Soc
2001, 123, 11069–11070; (g) J Mol Struct 2005, 737, 103–
107. [Color figure can be viewed in the online issue, which
is available at www.interscience.wiley.com.]
HIGHLIGHT 5
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

so uncommon. Moreover, in a single PFC molecule, the
helical arrangement always has the same handedness
(Fig. 5).
XB-BASED RECEPTORS
As it is the case for HB and other noncovalent interactions,
XB has been effectively used to direct the selective recog-
nition of ions and neutral molecules in solution and in the
solid state.12
Heteroditopic Receptors for Alkali Metal Halides
Anions are effective electron-donors to halogens. XB
can thus be anticipated as a promise in anion binding
processes. The first confirmation of this expectation
came recently from the binding of halide anions by an
iodotetrafluorophenyl substituted receptor designed apriori.13
Ion pairing in a salt can dramatically reduce receptor/
anion binding affinities and alter binding selectivities.14
For this reason a heteroditopic receptor tailored to the si-
multaneous binding of both the ions of alkali halides
was developed. A careful design resulted in the synthesis
of tris-{2-[2-(4-iodotetrafluorophenoxy)-ethoxy]-ethyl}-
amine 4 [Fig. 6(a)], which coordinates selectively
iodide, bromide, and chloride anions in the solid, liquid,
and gas phases.
The polyoxyethylene core mimics the kriptofix1
2.2.2. (K.2.2.2.) moiety and secures cation binding; the
iodotetrafluorobenzene pendants are the motifs for the
XB-based anion recognition. NMR experiments on 4/
NaI solutions showed that the binding constant of 4 for
the sodium cation increases by approximately 20 times
Figure 3. The self-assembly of naked anions with telechelic diiodoperfluoroalkanes gives rise
to infinite halogen-bonded zigzag chains (a) or to infinite supramolecular helixes, which in their
turn pair into homochiral double helixes (b). Color code: F, green; I, purple; I����I interactionsare black dashed lines. [Color figure can be viewed in the online issue, which is available at
www.interscience.wiley.com.]
Figure 4. Space-fill representation, viewed down the crys-
tallographic a-axis, of the enantiopure halogen-bonded heli-
ces 3 made of (S)-1,2-dibromohexafluoropropane 1 and (�)-
sparteinium bromide 2. For the sake of clarity, all the spartei-
nium cations but one per chain have been removed. Color
code: C, gray; H, white; F, green; Br, red; Br����Br interac-
tions are black dashed lines. [Color figure can be viewed in
the online issue, which is available at www.interscience.
wiley.com.]
6 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

with respect to its pentafluorophenyl analogue. This
proves the boosting effect of the XB-mediated anion
binding on cation complexation. X-ray analysis of a sin-
gle crystal of the 4/NaI complex showed that the ion pair
of the salt is separated; thanks to the simultaneous bind-
ing of I� and Na+ by the two different recognition arrays
of atoms of 4. Na+ is surrounded by the three arms of the
receptor and is separated by 5.585 A from the nearest I�
anion, which is engaged in strong I����I��C XB
[Fig. 6(b)]. I� anions bridge three iodotetrafluorophenyl
rings of three different podand molecules so that rolled-
up polymeric noncovalent chains are formed. Notewor-
thy, although the 4/NaI complex is composed of intrinsi-
cally achiral components, it crystallizes in the chiral
space group P212121.Crystalline complexes of the receptor with Br� and
Cl� ions were not obtained. This is consistent with the
common observation that in solution the formation of
XB between halide anions and iodo-PFCs becomes dis-
favored moving from I� to Br� to Cl� (the 19F NMR
chemical shift changes shown by iodo-PFCs on XB for-
mation decreases moving from I� to Br� to Cl�). Thegreater affinity of 4 toward iodide than bromide and
chloride ions was also confirmed by mass experiments
(ESI-MS). Competitive experiments were performed by
analyzing, in the negative ion polarity mode, mixtures
containing 4 and equimolar amounts of I�, Br�, or Cl�
anions. The signals of [4 + I�] were particularly strong,
while the signals for [4 + Br�] and [4 + Cl�] complexes
were very weak.
Molecularly Imprinted Polymers
When a monomer with tailored functional groups and a
crosslinker are copolymerized in the presence of an
imprinting agent (namely, a template molecule whose
functionalities are complementary binding sites for the
monomer functional groups), a crosslinked copolymer is
obtained and its architecture contains properly posi-
tioned recognition sites for the imprinting agent.15 In
fact, spontaneous monomer preorganization occurs;
thanks to the formation of monomer/imprinting agent
Figure 6. Schematic (a) and ball-and-stick view (b) of the complex tripodand 4/NaI. The so-
dium cation is embedded in the polyoxyethylene core, while the iodide counterion coordinates
three iodine atoms belonging to three different podand molecules. Color code: C, gray; H, white;
F, green; I, purple; O, red; N, blue; Na, orange.
Figure 5. All-trans distorted helical arrangement of mono-
iodoperfluoro-octane (a) and -decane (b) exhibiting the same
handedness when bound to naked iodide ions. For the sake
of clarity, the ammonium cations and fluorine atoms have
been omitted. Color code: C, gray; I, purple; I����I interac-
tions are black dashed lines. [Color figure can be viewed in
the online issue, which is available at www.interscience.
wiley.com.]
HIGHLIGHT 7
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
complexes. Copolymerization translates this preorgani-
zation into a rigid crosslinked matrix, which, upon re-
moval of the imprinting agent, presents cavities whose
size, shape, and electronic features are complementary
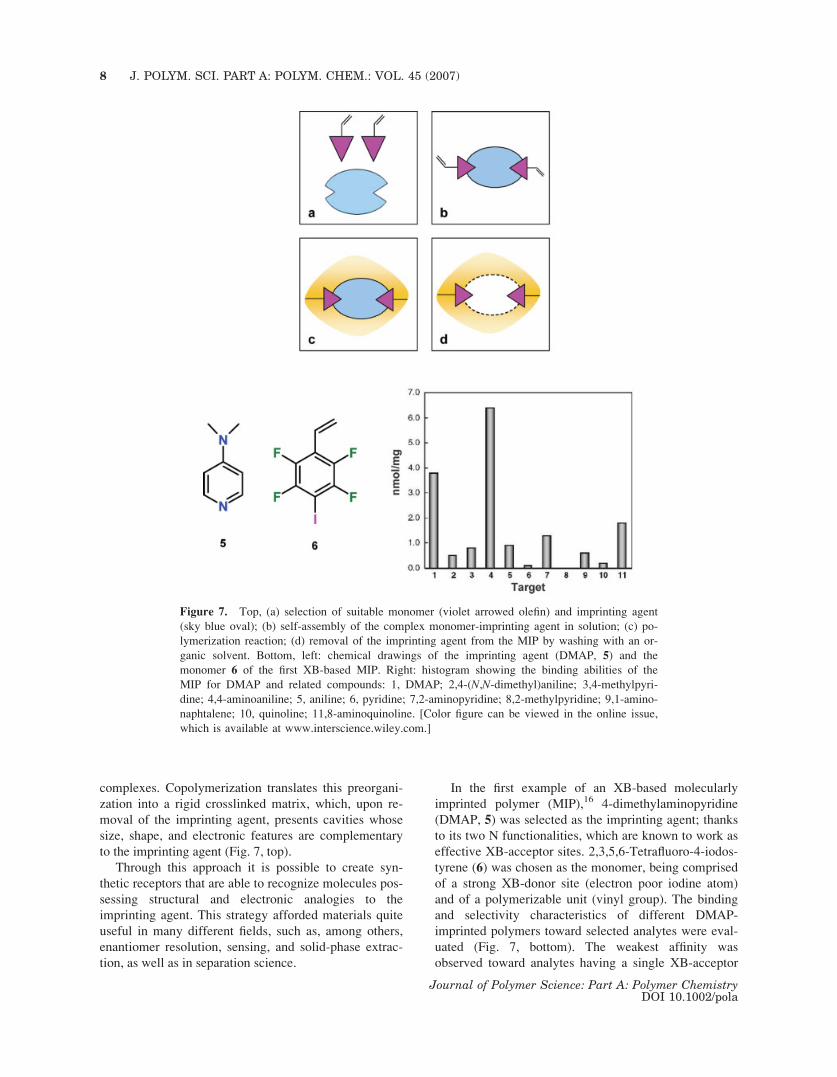
to the imprinting agent (Fig. 7, top).
Through this approach it is possible to create syn-
thetic receptors that are able to recognize molecules pos-
sessing structural and electronic analogies to the
imprinting agent. This strategy afforded materials quite
useful in many different fields, such as, among others,
enantiomer resolution, sensing, and solid-phase extrac-
tion, as well as in separation science.
In the first example of an XB-based molecularly
imprinted polymer (MIP),16 4-dimethylaminopyridine
(DMAP, 5) was selected as the imprinting agent; thanks
to its two N functionalities, which are known to work as
effective XB-acceptor sites. 2,3,5,6-Tetrafluoro-4-iodos-
tyrene (6) was chosen as the monomer, being comprised
of a strong XB-donor site (electron poor iodine atom)
and of a polymerizable unit (vinyl group). The binding
and selectivity characteristics of different DMAP-
imprinted polymers toward selected analytes were eval-
uated (Fig. 7, bottom). The weakest affinity was
observed toward analytes having a single XB-acceptor
Figure 7. Top, (a) selection of suitable monomer (violet arrowed olefin) and imprinting agent
(sky blue oval); (b) self-assembly of the complex monomer-imprinting agent in solution; (c) po-
lymerization reaction; (d) removal of the imprinting agent from the MIP by washing with an or-
ganic solvent. Bottom, left: chemical drawings of the imprinting agent (DMAP, 5) and the
monomer 6 of the first XB-based MIP. Right: histogram showing the binding abilities of the
MIP for DMAP and related compounds: 1, DMAP; 2,4-(N,N-dimethyl)aniline; 3,4-methylpyri-
dine; 4,4-aminoaniline; 5, aniline; 6, pyridine; 7,2-aminopyridine; 8,2-methylpyridine; 9,1-amino-
naphtalene; 10, quinoline; 11,8-aminoquinoline. [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
8 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

site (e.g., aniline and its N,N-dimethyl derivative, 1-
naphthylamine, pyridine, 2- and 4-picoline, quinoline).
The binding affinity increased when the analyte pre-
sented two binding sites (e.g., DMAP, 2- and 4-amino-
pyridine, 8-aminoquinoline). Molecular recognition
being affected by the matching between analyte shape
and MIP cavity shape, the binding abilities of 4-substi-
tuted derivatives were larger than those of 2-substituted
analogues. Noteworthy, 4-aminopyridine showed the
strongest binding to DMAP-imprinted polymers (even
stronger than DMAP).
Biological Receptors
The relevance of XB in the context of natural systems
can be easily predicted. Thousands of halogen-contain-
ing bioactive compounds, vancomycin and chloram-
phenicol, just to name two of them, are currently
known17 and XB may contribute to their binding at the
receptor sites. Forceful signals in this direction come
from the thyroid hormones, which are naturally occur-
ring compounds containing iodine.18 A great number of
short I���O contacts from thyroxine and its derivatives to
their associated proteins have been identified.
When 5-bromouridine substitutes for uridine or thy-
mine in a DNA sequence, a four-stranded Holliday junc-
tion substitutes for the standard duplexes because of
short Br���O contacts (3.0 A, 12% shorter than the sum
of their van der Waals radii for Br and O).19 An ultra-
high resolution structure (0.66 A) of the enzyme aldose
reductase complex with a halogenated inhibitor revealed
Br���O contacts of similar length.20 Moreover, the X-ray
crystallography of some tubulin-bound brominated tax-
anes revealed a role played by XB in the association
with the protein (Fig. 8).21 The potential role of halogens
in taxane-tubulin binding suggests novel possibilities for
the design of other microtubule-stabilizing compounds.
All these results have prompted quantum mechanical
calculations to compare the polarizability of halogen
atoms within the context of functional groups relevant to
biological molecules. A survey of short C��X���O inter-
actions in proteins and nucleic acids data base revealed
clearly the potential significance of XB in ligand binding
and recognition, as well as in molecular folding. The
survey also demonstrated that the XB geometries in bio-
logical systems conform generally to those seen in small
molecules. A similar survey involving p electron-donor
sites in proteins is also available.22
SOLID-STATE SYNTHESIS
Supramolecular photochemistry23 in the solid state pro-
vides an efficient way for accessing stereo- and regio-
controlled syntheses. The advantages of this approach
come from the preorganization of the reacting modules
in the crystal matrix, but the engineering of such a preor-
ganization remains a challenge. Different strategies have
been explored to control modules’ topochemical
arrangement by using in a rational way the noncovalent
interactions they can give rise to. HB,24 p���p stacking,25
and metal–ligand interactions26 were all used success-
fully.
[2 + 2] Template-Assisted Photodimerizationof Olefins
The requirements for the [2 + 2] photocyclization in the
solid state to occur, as identified by Schmidt,27 entail
that two double bonds are oriented in a parallel manner
and 3.5–4.2 A away from each other. Self-organization
and template-assisted organization of the olefins (Chart
2) are the two main strategies used to meet these require-
ments.
The latter strategy requires that olefinic substrate(s)
are organized around linear templates by means of
directing noncovalent interactions. The preorganization
of the template dictates the alignment of the double
bonds to meet the Schmidt’s requirements. The supra-
molecular control in the covalent bond-formation pro-
cess has been recently achieved through the organization
of olefinic modules by XB-based templates.28 The sys-
tem consists of a tetratopic XB-donor module, which
works as the template, and a ditopic XB-acceptor, as the
olefinic substrate. The pentaerythritol ether 7, carrying
four iodotrafluorophenyl rings, was challenged with
trans-1,2-bis(4-pyridyl)ethylene (8), a ditopic XB-
acceptor. The N���I XB being quite a strong interaction,
the template 7 and the substrate 8 cocrystallized in a 1:2
ratio to maximize the formation of halogen bonds. Infi-
nite 1D halogen-bonded ribbons were formed (Fig. 9).
In these ribbons the four arms of 7 points two by two to
opposite sides and the tetrafluorophenyl rings lying on
the same side of the molecule are paired in a quasi-paral-
lel fashion; thanks to face-to-face p���p intramolecular
interactions. Particularly, strong and directional N���Ihalogen bonds (N���I distances 2.795–2.819 A, N���I��C
angles 177.09 and 176.728) transfer this topochemical
information to the olefinic substrates 8 that are pinned in
the ribbons in a parallel fashion with a distance of less
than 4.5 A between the olefins’ centroids. Irradiation of
the powdered cocrystal afforded the corresponding tetra-
kis(4-pyridyl)cyclobutane with only the rctt stereochem-
istry in quantitative yields.
With the aim of applying the tools of supramolecular
chemistry to control the solid-state synthesis of complex
heterotopic tectons tailored to XB-based crystal engi-
neering, we have synthesized the 1D self-complemen-
HIGHLIGHT 9
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

tary tecton 9. Molecules wherein an electron-donor site
and an electron poor halogen atom are linked by an ole-
finic spacer appear promising substrates.29 N,N-Dime-
thylaniline and iodotetrafluorophenyl residues30 were
selected as self-complementary groups for XB and (E)-1-[4-(N,N-dimethylamino)phenyl]-2-(4-iodotetrafluoro-
phenyl)ethylene (9) was prepared (Scheme 2, top).31
It was expected that self-complementary XB induced
the formation of infinite chains, while p���p stacking
interactions between aromatics having opposite quadru-
polar moment paired these chains after a prereactive
arrangement. Single crystal X-ray analysis of 6 thor-
oughly confirmed the expectations. Modules 9 are
assembled head-to-tail and form infinite chains with a
zigzag geometry due to the presence of N���I interactionswith the standard geometrical features. The N���I dis-
tance is 3.093 A (13% shorter than the sum of van der
Waals radii), the N���I��C and C��N���I angles are
169.82 and 97.388, respectively. p���p stacking between
the PFC and HC arenes secures the antiparallel coupling
of the dipolar moments of modules 9 into head-to-tail
dimers while XB connects these dimers into infinite zig-
zag ribbons. As a result of this crystal packing, pairs of
modules 9 in adjacent chains have the double bonds sep-
arated by a distance of 3.761 A (just meeting Schmidt’s
requirements for solid-state photodimerization). A regio-
and stereospecific cycloaddition reaction occurs on the
irradiation of powdered crystals of 9 and the 1,3-bis(4-
iodotetrafluorophenyl)-2,4-bis[4-(N,N-dimethylamino)-
phenyl]cyclobutane (10) having only the rctt stereo-
chemistry is isolated in 80% yield (Scheme 2, bottom).
The packing of this cyclobutane in the crystal is a nice
example of the robustness of the N���I supramolecular
synthon and its ability to prevail over other noncovalent
interactions in directing crystal architecture. The cyclo-
butane 10 possesses two XB-donor sites and two XB-
Figure 9. Single crystal X-ray structure of the complex between pentaerythritol derivative 7,
which functions as the template, and trans-1,2-bis(4-pyridyl)ethylene (8). The distance between
the olefins’ centroids is in the range required for the [2 + 2] photoreaction in the solid state.
Color code: C, black; F, green; N, blue; O, red; I, purple; N���I interactions are black dashed
lines.
Chart 2. Schematic diagram showing the preorganization
of olefins for solid-state photoreaction, as induced by the
self-assembly around linear templates by means of comple-
mentary intermolecular interactions.
Figure 8. O���Br halogen bonds within the tubulin–taxane binding complex. (Courtesy of
Y. Jiang, J.-M. Chen and J. P. Snyder)
10 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

acceptor sites and, on crystallization, it forms all possi-
ble N���I halogen bonds. The resulting architecture
shows large columnar voids, which are filled by solvent
molecules, and no p���p stacking occurs (10 could give
up to eight such interactions) (Fig. 10).
Topochemical Polymerization in the Solid State
Like all topochemical reactions, 1,4-polymerization of
diynes requires a precise prealignment of the reactant
molecules within the crystal. Experiments show that the
preferred monomer repeat distances dm is about 4.9 A
and the angle h is 458, so that the distance R1,4 (between
the reacting atoms C1 and C40) is 3.5 A (Chart 3).
Most diacetylenes do not crystallize in accordance to
these precise structural requirements. To circumvent this
limitation, diynes have been assembled by means of non-
covalent interactions with templates that reliably dictate
the formation of complexes with crystallographic features
corresponding to the targeted alignment needed for reac-
tion.32 Specifically, telechelic diiododiynes are good XB-
donors; thanks to the polarizability of iodine atoms and
the strong electron withdrawing ability of the sp-hybri-
dized carbon atom. On the other hand, Fowler, Lauher,
and coworkers have demonstrated that oxalamides form
self-complementary HB networks with a repeat distance
(4.9–5.1 A) that matches well the 4.9 A distance require-
ment for diyne 1,4-polymerization. When bispyridyl oxa-
lamides cocrystallize with telechelic diiododiynes, 1D in-
finite chains are formed wherein the strength and direc-
tionality of N���I XB translates the packing features of
oxalamide units into the packing arrangement of the diio-
dodiynes units [Chart 4(a)]. Some of the obtained com-
plexes display dm and R1,4 distances and alignment angles
h close to the desired values but unfortunately up to now,
all attempts of polymerization in the solid state have
failed. However, from a methanol solution of diiodobuta-
diyne with a bis(nitrile)oxalamide, a unique poly(diiodo-
diacetylene) [Chart 4(b)] has been isolated, probably as a
result of the spontaneous topochemical polymerization of
the starting diiodobutadiyne in solution.33
LIQUID CRYSTALS
XB has proven successful in determining the self-assem-
bly of supramolecular mesogens and forming new fami-
lies of materials showing liquid-crystalline behavior.
The same had already been the case for HB,34 quadrupo-
lar,35 and charge-transfer36 interactions.
Halogen-Bonded Low Molar Mass Liquid Crystals
Alkoxystilbazoles have been shown to be versatile mod-
ules for the construction of molecular materials, and in
particular metallomesogens and hydrogen-bonded meso-
gens. A series of liquid-crystalline materials resulted on
HB-driven self-assembly of alkoxystilbazole with vari-
ous substituted phenols, neither starting component
Scheme 2. Schematic diagram showing the preorganization
of olefins for solid-state photoreaction, as induced by the
self-assembly around linear templates by means of complemen-
tary intermolecular interactions. [Color figure can be viewed in the
online issue, which is available at www.interscience.wiley.com.]
Figure 10. Mercury 1.4. view down the crystallographic a-axis of the crystal packing of 1,3-bis(4-iodotetrafluorophenyl)-
2,4-bis[4-(N,N-dimethylamino)phenyl]cyclobutane (10). Color
code: C, gray; H, white; F, light green; Cl, green; I, purple; N,
blue. Space-fill representation has been used for clathrated
chloroform. [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
HIGHLIGHT 11
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
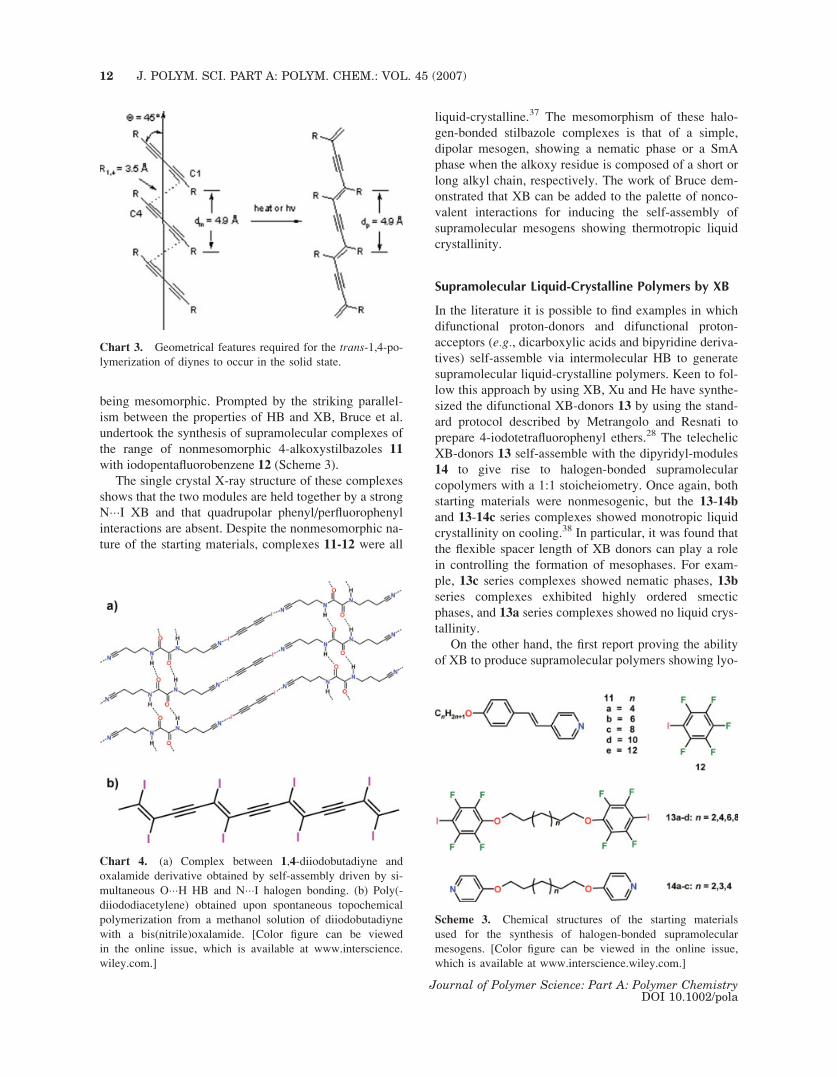
being mesomorphic. Prompted by the striking parallel-
ism between the properties of HB and XB, Bruce et al.
undertook the synthesis of supramolecular complexes of
the range of nonmesomorphic 4-alkoxystilbazoles 11
with iodopentafluorobenzene 12 (Scheme 3).
The single crystal X-ray structure of these complexes
shows that the two modules are held together by a strong
N���I XB and that quadrupolar phenyl/perfluorophenyl
interactions are absent. Despite the nonmesomorphic na-
ture of the starting materials, complexes 11-12 were all
liquid-crystalline.37 The mesomorphism of these halo-
gen-bonded stilbazole complexes is that of a simple,
dipolar mesogen, showing a nematic phase or a SmA
phase when the alkoxy residue is composed of a short or
long alkyl chain, respectively. The work of Bruce dem-
onstrated that XB can be added to the palette of nonco-
valent interactions for inducing the self-assembly of
supramolecular mesogens showing thermotropic liquid
crystallinity.
Supramolecular Liquid-Crystalline Polymers by XB
In the literature it is possible to find examples in which
difunctional proton-donors and difunctional proton-
acceptors (e.g., dicarboxylic acids and bipyridine deriva-
tives) self-assemble via intermolecular HB to generate
supramolecular liquid-crystalline polymers. Keen to fol-
low this approach by using XB, Xu and He have synthe-
sized the difunctional XB-donors 13 by using the stand-
ard protocol described by Metrangolo and Resnati to
prepare 4-iodotetrafluorophenyl ethers.28 The telechelic
XB-donors 13 self-assemble with the dipyridyl-modules
14 to give rise to halogen-bonded supramolecular
copolymers with a 1:1 stoicheiometry. Once again, both
starting materials were nonmesogenic, but the 13-14b
and 13-14c series complexes showed monotropic liquid
crystallinity on cooling.38 In particular, it was found that
the flexible spacer length of XB donors can play a role
in controlling the formation of mesophases. For exam-
ple, 13c series complexes showed nematic phases, 13b
series complexes exhibited highly ordered smectic
phases, and 13a series complexes showed no liquid crys-
tallinity.
On the other hand, the first report proving the ability
of XB to produce supramolecular polymers showing lyo-
Chart 3. Geometrical features required for the trans-1,4-po-lymerization of diynes to occur in the solid state.
Chart 4. (a) Complex between 1,4-diiodobutadiyne and
oxalamide derivative obtained by self-assembly driven by si-
multaneous O���H HB and N���I halogen bonding. (b) Poly(-
diiododiacetylene) obtained upon spontaneous topochemical
polymerization from a methanol solution of diiodobutadiyne
with a bis(nitrile)oxalamide. [Color figure can be viewed
in the online issue, which is available at www.interscience.
wiley.com.]
Scheme 3. Chemical structures of the starting materials
used for the synthesis of halogen-bonded supramolecular
mesogens. [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
12 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

tropic liquid crystallinity and made by self-assembly of
nonmesomorphic species came as early as in 2002.39
The waxy adducts obtained starting from oligomeric 4-
vinylpyridine and 1,6-diiodoperfluorohexane when stud-
ied under polarized light optical microscope showed
many spherulitic textures with a Maltese extinction cross
dispersed in a birefringent matrix, as typical for a smec-
tic-type liquid crystal. This macroscopic organization
remained unchanged upon heating, suggesting a lyo-
tropic character of this system.
Highly Fluorinated Low Molar Mass LiquidCrystals by XB
It is well known that liquid crystals can derive their mo-
lecular dipole moment from terminal perfluoroalkyl (Rf)
chains. However, the introduction of long Rf chains in
mesogens enhances the smectic character of a liquid
crystal, as a consequence of the microphase separation
that occurs between the Rf chains and HC segments of
the molecule (fluorophobic effect). Considering the abil-
ity of a single XB to form dimeric liquid crystals from
nonmesogenic units and the ability of diiodoperfluoroal-
kanes to work as effective XB-donors, a low molar mass
approach to trimeric liquid crystals was pursued. Specifi-
cally, a diiodoperfluoroalkane modules self-assemble
with two molecules of alkoxystilbazole 11 to give a
range of trimeric adducts 15 (Scheme 4).
Single crystal X-ray diffraction of 15d confirms the
expected 2:1 ratio between the stilbazole and diiodoper-
fluorohexane modules in the crystal. The trimeric adduct
is characterized by a stepped arrangement between the
two stilbazoles consequent on the antiperiplanar arrange-
ment of the perfluoromethylene groups of the XB-donor
(Fig. 11, top).
The stilbazole and the perfluorinated modules are
bound together by strong N���I XB and stack in separated
columns parallel to the a crystallographic axis, probably
as a consequence of the fluorophobic effect. On heating,
all the trimers 15 melted directly to the isotropic liquid,
but on cooling, a nematic phase forming from the iso-
tropic liquid was seen, as it is evident from its character-
istic schlieren texture (Fig. 11, bottom). Indeed, the sys-
tematic presence of a nematic phase in all of the com-
plexes 15 is quite surprising, as it is an uncommon
feature for liquid crystals containing long Rf chains. The
expected microphase separation associated with per-
fluoroalkyl chains is surprisingly absent.40
PERSPECTIVES IN MATERIALS SCIENCE
Halogen-Bonded Paramagnetic Complexes
Persistent nitroxide radicals interact with 1,4-diiodo-tet-
rafluorobenzene (DITFB) via XB.41 Specifically, 4-phe-
nyl-2,2,5,5-tetramethyl-3-imidazolin-1-yloxyl radical
and 4-amino-2,2,6,6-tetramethyl(piperidin-1-yloxyl)
radical cocrystallize with DITFB giving rise to 1D coor-
dination polymers consisting of alternating electron-do-
nor and acceptor modules in a 1:1 ratio; thanks to direc-
tional NO����I interactions. The occurrence of NO����Iintermolecular interactions also in the liquid phase was
proven by ESR.42 The technique allowed to establish
that the interaction strength is close to that of a strong
HB. For instance, the DH8 of the TEMPO-1-iodoper-
fluorooctane XB is –7.0 6 0.4 kcal/mol.
Halogen-Bonded Organic Semiconductors
As early as 1995, Imakubo and Kato introduced for the
first time the use of XB to direct intermolecular interac-
Scheme 4. Halogen-bonded trimeric complexes formed by
alkoxystilbazoles and a,x-diiodoperfluoroalkanes and showing
liquid crystal behavior. [Color figure can be viewed in the online
issue, which is available at www.interscience.wiley.com.]
Figure 11. Space-fill representation of the stilbazole-diiodo-
perfluorohexane complex 15d (top) and schlieren texture of
the corresponding monotropic nematic phase as obtained on
cooling from the isotropic phase (bottom). [Color figure can
be viewed in the online issue, which is available at www.
interscience.wiley.com.]
HIGHLIGHT 13
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

tions in crystalline molecular conductors based on tetra-
thiafulvalene (TTF) derivatives.43 In particular, while
XB interactions have been observed only incidentally in
neutral halogenated TTFs, they have been extensively
encountered in their radical cation salts. Since the semi-
nal work of Imakubo and Kato, XB has been fully inves-
tigated as a first choice noncovalent interaction to con-
trol the solid-state structures of organic molecular con-
ductors and hence to influence their electronic
properties.44
XB at the Outer Sphere of Metal–LigandComplexes
Keen to extend his work on the study of the behavior of
inorganic halogen species (Metal-X) as directional
Lewis bases in the formation of hydrogen bonds,
Brammer and coworkers have demonstrated the great
potential of XB in stabilizing the outer sphere of metal–
ligand complexes.45 A clear understanding of the nature
of M��X���X0��C XB provides an impetus for a wide
range of applications to supramolecular construction of
organic–inorganic hybrid materials and has the potential
to yield applications in the control of conformations in
metal complexes, and of substrate binding in catalysis.
CONCLUSIONS
XB is a strong, specific, and directional interaction that
can frequently prevail over other interactions (e.g., HB46,47
and p–p stacking31) in systems of interest to chemical,
biopharmacological, and material sciences. Specific exam-
ples have been discussed where XB plays a key role in
enantiomer separation,10a crystal engineering,1 selective
binding of small molecules to synthetic13,16 and natural17–21
receptors, formation of supramolecular liquid crys-
tals,37–40 and of molecular conductors.43,44 Indeed, XB
impacts in all the fields where the design and manipula-
tion of aggregation phenomena play a key role and seri-
ous problems may arise if its occurrence is neglected.2a
In general, halogen-bonded adducts can be consid-
ered as prereactive complexes (or intermediates) formed
prior to significant charge-transfer or chemical reaction.
The strongest interactions can evolve into different mo-
lecular species if concentration, temperature, solvent po-
larity, or other parameters are changed. In general, com-
plexes between halogens and Lewis bases have been
observed in, or invoked for, many reaction mechanisms
in solution. The 1:1 complex that dihalogen molecules
form with alkenes is a particularly noteworthy case. It
lays on the way to the addition/substitution products’
formation and its evolution depends on the reaction con-
ditions.48 Other well-known examples come from the
chemical reactivity of iodine. This reactivity is generally
greater in solvents that give yellowish solutions (elec-
tron-donating solvents) than in solvent giving violet sol-
utions (noncoordinating solvents).49
Several analytical techniques (solution calorimetry; UV,
IR, and Raman spectroscopy, 1H, 13C, 14N, 19F NMR;
dielectric polarization; nuclear quadrupole resonance; sin-
gle crystal X-ray analysis; ESR) have been used to detect
the formation of XB, to define its nature, to establish its
energetic and geometric characteristics, and to reveal the
striking similarities between XB and HB. All the aggrega-
tion phenomena controlled by HB may have a parallel
under the control of XB. The exploitation of the similarities
between these two interactions can be expected to favor
the flourishing of numerous and successful applications
where the presence of XB is crucial. The molecular struc-
tures of modules that can be involved in self-assembly
processes driven by XB are obviously different from those
involved in HB-driven ones, and this allows new adducts
with new properties to be obtained.
A full utilization of the bottom-up approach to func-
tional materials continuously requires new and effective
noncovalent interactions for assembling molecules into
supramolecular architectures associated with pre-estab-
lished functions. The great potential of XB in the design
of new and high-value functional materials is now
emerging clearly.
REFERENCES AND NOTES
1. Metrangolo, P.; Resnati, G. Chem Eur J 2001, 7,2511.
2. (a) Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati,G. Acc Chem Res 2005, 38, 386; (b) Metrangolo, P.;Resnati, G. In Encyclopedia of SupramolecularChemistry; Atwood, J. L.; Steed, J. W., Eds.; MarcelDekker Inc., New York 2004; pp 628.
3. Broder, C. K.; Howard, J. A. K.; Keen, D. A.; Wil-son, C. C.; Allen, F. H.; Jetti, R. K. R.; Nangia, A.;Desiraju, G. R. Acta Crystallogr B 2000, 56, 1080.
4. Legon, A. C. Angew Chem Int Ed 1999, 38, 2686.5. Hassel, O. Science 1970, 170, 497.6. Gattuso, G.; Liantonio, R.; Metrangolo, P.; Meyer,
F.; Pappalardo, A.; Parisi, M. F.; Pilati, T.; Pisa-gatti, I.; Resnati, G. Supramol Chem 2006, 18, 235.
7. Liantonio, R.; Metrangolo, P.; Pilati, T.; Resnati,G. Cryst Growth Des 2003, 3, 355.
8. Liantonio, R.; Metrangolo, P.; Meyer, F.; Pilati, T.; Nav-arrini, W.; Resnati, G. ChemCommun 2006, 1819.
9. Casnati, A.; Liantonio, R.; Metrangolo, P.; Res-nati, G.; Ungaro, R.; Ugozzoli, F. Angew ChemInt Ed 2006, 45, 1915.
10. (a) Farina, A.; Meille, S. V.; Messina, M. T.;Metrangolo, P.; Resnati, G.; Vecchio, G. Angew
14 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 45 (2007)
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

Chem Int Ed 1999, 38, 2433; (b) Logothetis, T. A.;Meyer, F.; Metrangolo, P.; Pilati, T.; Resnati, G.New J Chem 2004, 28, 760.
11. Farnham, W. B.; Dixon, D. A.; Calabrese, J. C.J Am Chem Soc 1988, 110, 8453.
12. (a) Bowman-James, K. Acc Chem Res 2005, 38,671 and references therein; (b) Sessler, J. L.;Davis, J. M. Acc Chem Res 2001, 34, 989.
13. Mele, A.; Metrangolo, P.; Neukirch, H.; Pilati, T.;Resnati, G. J Am Chem Soc 2005, 127, 14972.
14. Mahoney, J. M.; Beatty, A. M.; Smith, B. D. J AmChem Soc 2001, 132, 5847.
15. Lubke, M.; Whitcombe, M. J.; Vulfson, E. N.J Am Chem Soc 1998, 120, 13342.
16. Takeuchi, T.; Minato, Y.; Masayoshi, M.; Shin-mori, H. Tetrahedron Lett 2005, 46, 9025.
17. Auffinger, P.; Hays, F. A.; Westhof, E.; Ho, P. S.Proc Natl Acad Sci USA 2004, 101, 16789.
18. Wojtczak, A.; Cody, V.; Luft, J. R., Pangborn, W.Acta Crystallogr D 2001, 57, 1061.
19. Hays, F. A.; Vargason, J. M.; Ho, P. S. Biochemis-try 2003, 42, 9586.
20. Howard, E. I.; Sanishvili, R.; Cachau, R. E.;Mitschler, B.; Bart, P.; Lamour, V.; Van Zandt, M.;Sibley, E.; Bon, C.; Moras, D. Schneider, T. R. Joa-chimiak, A.; Podjarny, A. Proteins 2004, 55, 792.
21. (a)Jiang, Y.; Alcaraz, A. A.; Chen, J. M.; Kobayashi,H.; Lu, Y. J.; Snyder, J. P. J Med Chem 2006, 49,1891; (b) Jiang, Y.; Lin, H.-X.; Chen, J.-M.; Chen,M.-Q. Bioorg Med Chem Lett 2005, 15, 839.
22. Saraogi, I.; Vijay, V. G.; Das, S.; Sekar, K.; GuruRow, T. N. Cryst Eng 2003, 6, 69.
23. Huang, C.-H.; Bassani, D. M. Eur J Org Chem2005, 4041.
24. Feldman, K. S.; Campbell, R. F. J Org Chem1995, 60, 1924.
25. Coates, G. W.; Dunn, A. R.; Henling, L. M.; Ziller,J. W.; Lobkovsky, E. B.; Grubbs, R. H. J AmChem Soc 1998, 120, 3641.
26. (a) Papaefstathiou, G. S.; Zhong, Z.; Geng, L.;MacGillivray, L. R. J Am Chem Soc 2004, 126,9158; (b) Chu, Q.; Swenson, D. C.; MacGillivray,L. R. Angew Chem Int Ed 2005, 44, 3569.
27. Schmidt, G. M. J Pure Appl Chem 1971, 27, 647.28. Caronna, T.; Liantonio, R.; Logothetis, T. A.;
Metrangolo, P.; Pilati, T.; Resnati, G. J Am ChemSoc 2004, 126, 4500.
29. Lucassen, A. C. B.; Vartanian, M.; Leitus, G.; Vander Boom, M. E. Cryst Growth Des 2005, 5, 1671.
30. Liantonio, R.; Luzzati, S.; Metrangolo, P.; Pilati,T.; Resnati, G. Tetrahedron 2002, 58, 4023.
31. Marras, G.; Metrangolo, P.; Meyer, F.; Pilati, T.; Res-nati, G.; Vij, A. New J Chem 2006, 1397.
32. (a) Goroff, N. S.; Curtis, S. M.; Webb, J. A.;Fowler, F. W.; Lauher, J. W. Org Lett 2005, 7,1891; (b) Curtis, S. M.; Le, N.; Fowler, F. W.;Lauher, J. W. Cryst Growth Des 2005, 5, 2313.
33. Sun, A.; Goroff, N. S.; Lauher, J. W. Science 2006,312, 1030.
34. (a) Kato, T. In Handbook of Liquid Crystals; Demus,D.; Gray, G. W.; Goodby, J.; Spiess, H.-W.; Vill, V.,Eds.; Wiley-VCH:Weinheim, 1998; (b) Tsiourvas, D.;Paleos, C. Angew Chem Int Ed Engl 1995, 34, 1696.
35. Boden, N.; Bushby, R. J.; Lu, Z.; Lozman, O. R.Liq Cryst 2001, 28, 657.
36. Praefcke, K.; Holbrey, J. D. J Incl Phenom MolRecogn Chem 1996, 24, 19.
37. Nguyen, H. L.; Horton, P. N.; Hursthouse, M. B.;Legon, A. C.; Bruce, D. W. J Am Chem Soc 2004,126, 16.
38. Xu, J.; Liu, X.; Lin, T.; Huang, J.; He, C. Macro-molecules 2005, 38, 3554.
39. Bertani, R.; Metrangolo, P.; Moiana, A.; Perez, E.;Pilati, T.; Resnati, G.; Rico-Lattes, I.; Sassi, A.Adv Mater 2002, 14, 1197.
40. Metrangolo, P.; Prasang, C.; Resnati, G.; Lianto-nio, R.; Whitwood, A. C.; Bruce, D. W. ChemCommun 2006, 3290.
41. Boubekeur, K.; Syssa-Magale, J.-L.; Palvadeauc,P.; Schollhorn, B. Tetrahedron Lett 2006, 47, 1249.
42. Mugnaini, V.; Punta, C.; Liantonio, R.; Metran-golo, P.; Recupero, F.; Resnati, G.; Pedulli, G. F.;Lucarini, M. Tetrahedron Lett 2006, 47, 3265.
43. Imakubo, T.; Sawa, H.; Kato, R. Synth Met 1995,73, 117.
44. Fourmigue, M.; Batail, P. Chem Rev 2004, 104,5379.
45. Espallargas, G. M.; Brammer, L.; Sherwood, P.Angew Chem Int Ed 2006, 45, 435.
46. Corradi, E.; Meille, S. V.; Messina, M. T.; Metran-golo, P.; Resnati, G. Angew Chem Int Ed 2000,39, 1782.
47. Di Paolo, T.; Sandorfy, C. Nature 1974, 252,471.
48. Lenoir, D.; Chiappe, C. Chem Eur J 2003, 9,1030.
49. Reid, C.; Mulliken, R. S. J Am Chem Soc 1954,76, 3869.
HIGHLIGHT 15
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
Related Documents




![Insights into the crystallisation process from anhydrous ... · bonding and halogen bonding.[11] Hydrogen bonding has been found to stabilise crystal structures and to play a major](https://static.cupdf.com/doc/110x72/5f26ffaf57bb03333a7ca42d/insights-into-the-crystallisation-process-from-anhydrous-bonding-and-halogen.jpg)






