130 Journal of Magnetism and Magnetic Materials 98 (1991) 130-140 North-Holland Electronic structure calculations for CeAg Patrizia Monachesi ’ Institut de Physique Theorique, Uniuersite de Lausanne, CH-IOI5 Lausanne, Switzerland Samuele Fraizzoli Institut Romand de Recherche NumPrique en Physique des Materiaux (IRRMA), PHB-Ecublens, CH-I015 Lausanne, Switzerland and Scuola Normale Superiore, I-561 00 Piss, Italy Elio G. Moroni Institut de Physique ExpPrimentale, Unioersite de Lausanne, CH-IO15 Lausanne, Switzerland and Institut Romand de Recherche Numerique en Physique des MatMaux (IRRMA), PHB-Ecublens, CH-IOI5 Lausanne, Switzerland Received 26 November 1991 Self-consistent linear muffin-tin orbital calculations are performed for CeAg. The electronic structure is calculated in the paramagnetic and ferromagnetic phase, for both the cubic and tetragonal symmetry. The presence.of hybridization relevant to the Anderson lattice behavior, invoked to interpret the low-temperature properties of this compound, is confirmed. The comparison of the electronic structures of CeAg and of its f-less analog LaAg allows us to determine the localized part of the Ce 4f charge, amounting to - 0.94 e/Cc, to be identified with the average f-occupation number in the Anderson model. A saturation magnetic moment of = 0.78~~ is found, which is essentially due to the Ce f electrons. 1. Introduction Intermetallic CeAg has been raising interest for more than a decade for two main reasons: its structural transition and its low-temperature mag- netic phase. The former was first interpreted as a band Jahn-Teller effect on the basis of the elec- tronic structure of the isostructural compound LaAg [1,2] and seems now to be well accounted for by a phenomenological model which includes quadrupolar interactions [3]. The latter, instead, is still an open problem since the nature of the 1 Permanent address: Dipartimento di Fisica, Universita dell’Aquila, I-67100 L’Aquila, Italy. ordered ground state is not completely understood 13941. The high-temperature phase of CeAg is cubic with CsCl structure [4]. The paramagnetic suscept- ibility and the effective magnetic moment meas- ured in polycrystals [5] agree with the crystal field scheme deduced from neutron scattering data [4] giving a rs ground state separated by about 300 K from the high I, multiplet. At T = 16 K CeAg undergoes a structural transition to a tetragonal phase, detected by neutron spectroscopy [4] on polycrystalline samples, resistivity [3], elastic stiff- ness [6] specific heat [7] and muon spin-rotation [8] measurements in single crystals. The tetragonal 0304-8853/91/$03.50 0 1991 - Elsevier Science Publishers B.V. (North-Holland)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

130 Journal of Magnetism and Magnetic Materials 98 (1991) 130-140
North-Holland
Electronic structure calculations for CeAg
Patrizia Monachesi ’
Institut de Physique Theorique, Uniuersite de Lausanne, CH-IOI5 Lausanne, Switzerland
Samuele Fraizzoli Institut Romand de Recherche NumPrique en Physique des Materiaux (IRRMA), PHB-Ecublens, CH-I015 Lausanne, Switzerland
and Scuola Normale Superiore, I-561 00 Piss, Italy
Elio G. Moroni Institut de Physique ExpPrimentale, Unioersite de Lausanne, CH-IO15 Lausanne, Switzerland
and Institut Romand de Recherche Numerique en Physique des MatMaux (IRRMA), PHB-Ecublens, CH-IOI5 Lausanne, Switzerland
Received 26 November 1991
Self-consistent linear muffin-tin orbital calculations are performed for CeAg. The electronic structure is calculated in the paramagnetic and ferromagnetic phase, for both the cubic and tetragonal symmetry. The presence.of hybridization relevant to
the Anderson lattice behavior, invoked to interpret the low-temperature properties of this compound, is confirmed. The comparison of the electronic structures of CeAg and of its f-less analog LaAg allows us to determine the localized part of the
Ce 4f charge, amounting to - 0.94 e/Cc, to be identified with the average f-occupation number in the Anderson model. A
saturation magnetic moment of = 0.78~~ is found, which is essentially due to the Ce f electrons.
1. Introduction
Intermetallic CeAg has been raising interest for more than a decade for two main reasons: its structural transition and its low-temperature mag- netic phase. The former was first interpreted as a band Jahn-Teller effect on the basis of the elec- tronic structure of the isostructural compound LaAg [1,2] and seems now to be well accounted for by a phenomenological model which includes quadrupolar interactions [3]. The latter, instead, is still an open problem since the nature of the
1 Permanent address: Dipartimento di Fisica, Universita dell’Aquila, I-67100 L’Aquila, Italy.
ordered ground state is not completely understood 13941.
The high-temperature phase of CeAg is cubic with CsCl structure [4]. The paramagnetic suscept- ibility and the effective magnetic moment meas- ured in polycrystals [5] agree with the crystal field scheme deduced from neutron scattering data [4] giving a rs ground state separated by about 300 K from the high I, multiplet. At T = 16 K CeAg undergoes a structural transition to a tetragonal phase, detected by neutron spectroscopy [4] on polycrystalline samples, resistivity [3], elastic stiff- ness [6] specific heat [7] and muon spin-rotation [8] measurements in single crystals. The tetragonal
0304-8853/91/$03.50 0 1991 - Elsevier Science Publishers B.V. (North-Holland)

P. Monachesi et al. / Electronic structure calculations for CeAg 131
distortion, amounting to a lattice constant ratio [4] c/a = 1.019 at 1.8 K seems not to affect the magnetic susceptibility [5,6] and the X-ray photo- emission spectra [9] in single crystals. This indi- cates that the tetragonal splitting of the I’, state must not exceed a few K. A ferromagnetic phase transition occurs at T, - 5 K with the moments aligned along the [OOl]-direction as initially ob- served by Schmitt et al. [4] and recently confirmed by Morin [3] in a thorough investigation of the magnetization vs. applied field. However, these authors have found a saturation magnetic moment (1.25~~ in ref. [3]) which is much smaller than the value of 2.0~~ expected for the r, crystal field ground state. This reduction of the saturation moment, together with the peculiar behavior of the resistivity [5] and of the Curie temperature under pressure [lO,ll], points to the possible for- mation of a Kondo-like [5,6] ground state compet- ing with the exchange interaction. Therefore CeAg may be a good candidate for the study of the crossover from magnetic to Kondo-lattice ground state, representing an intermediate case between these two extremes. Competition or coexistence of these two regimes have been reported also in other binary Ce-compounds like CeAl, [12], CeCu 2 [13], and solid solutions like Ce, _,La,Ru *Si2 as a function of La substitution [14].
The coupling between localized f and extended conduction states can lead to both magnetism or Kondo compensation of the magnetic moments [15,16]. In the former case the magnetic moments originating from the crystal field ground state of Ce order at the Curie (NCel) temperature under the effect of the indirect exchange and exhibit characteristic susceptibility and magnetization curves in agreement with the crystal field levels scheme. In the latter regime the local moments on Ce are partially or totally screened out by the antiparallel polarization of the conduction elec- trons with suppression of magnetic order and oc- currence of resistivity anomalies at the Kondo temperature. The onset of either regime has been studied within a one-dimensional model by Doniach [17] who found it to depend crucially on the interplay between the Anderson hybridization and the density of states at the Fermi energy. However, no exhaustive, consistent theory for the
magnetic to Kondo regime crossover exists so far even in the diluted impurity limit. Therefore a detailed knowledge of the electronic structure of such materials in the framework of the band the- ory is of paramount importance.
In this paper we present self-consistent elec- tronic structure calculations for CeAg. A short communication with partial results has been pre- sented elsewhere [18]. Our aim is manifold. First of all we want to characterize the hybridization of the Ce 4f states with other conduction states since this is the basic mechanism for the indirect ex- change and the Kondo effect. Second, we wish to provide ourselves with electronic structure calcula- tions as an input for the ab initio determination of the parameters of the Anderson Hamiltonian [19], and eventually to use this model Hamiltonian to clarify our understanding of the low-T properties of CeAg. The Anderson Hamiltonian relying on actual electronic structure calculations has been used quite successfully by several authors [20] to explain microscopically magnetic properties and specific heat of metallic and insulating materials. Finally we want to compare the predictions of a spin-polarized calculation with the experimental result for the saturation magnetic moment.
Existing band structure calculations of iso- structural LaAg [1,21], LaCd [l], DyZn, DyCu, and DyRh [22] all show an overall similar aspect marked by the presence of the Sd-derived conduc- tion bands of the lanthanide at the Fermi energy. As shown below, the situation in Ce compounds is quite different since the narrow 4f bands hybridize with the conduction states near the Fermi level. This feature is also observed in the electronic structure calculations of the cubic Laves Ce com- pounds by Jarlborg et al. [23] and by Yanase [24].
The paper is organized as follows. In the next section we present the calculation of the para- magnetic electronic structure of CeAg in the cubic and tetragonal symmetry. Moreover we perform the same calculation also for the related com- pound LaAg to use it as the f-less analog of CeAg. We analyze the region of energy where the f bands occur, comparing the character of the wave func- tions in CeAg and LaAg. In section 3 we present the calculation for CeAg in the ferromagnetic, tetragonal phase. In particular the various contri-
132 P. Monachesi et al. / Electronic structure calculations for CeAg
butions to the saturation magnetic moment are analyzed. Finally, in section 4 we summarize the results and their relevance to the experiments.
2. Paramagnetic calculation
The band structure of paramagnetic CeAg was determined using the linear muffin-tin orbital (LMTO) method originally devised by Andersen [25] and implemented by Jarlborg and Arbman [26]. The local density approximation (LDA) to the exchange and correlation potential was used in the form given by Hedin and Lundqvist [27]. The initial potential was constructed from atomic Ce with external configuration 4f’, 5d’, 6s’ and atomic Ag with external configuration 4d”, 5s’. The inner shells were treated as core states, but no frozen core approximation has been used during the self-consistency cycle. The shells of the exter- nal atomic configuration, including the Ce 4f one, have been treated as valence states. The full Dirac equation was used for the core states, whereas for the valence states the spin-orbit coupling was neglected (semirelativistic approach). Due to the presence of f states on Ce, the basis set includes angular momenta up to I = 3 on both atoms. The unit cell of CeAg in the cubic phase (CsCl struc- ture), is a cube of edge a which accommodates the two inequivalent atoms with Ce at the origin. The Wigner-Seitz (WS) radii used in the determina- tion of the structure constants were taken equal for the two atoms. The calculations were per- formed in the irreducible wedge (l/48) of the Brillouin Zone (BZ), with a mesh of 220 k-points. Convergence was achieved within 1 mRy in the potential.
2.1. Electronic structure in the cubic symmetry
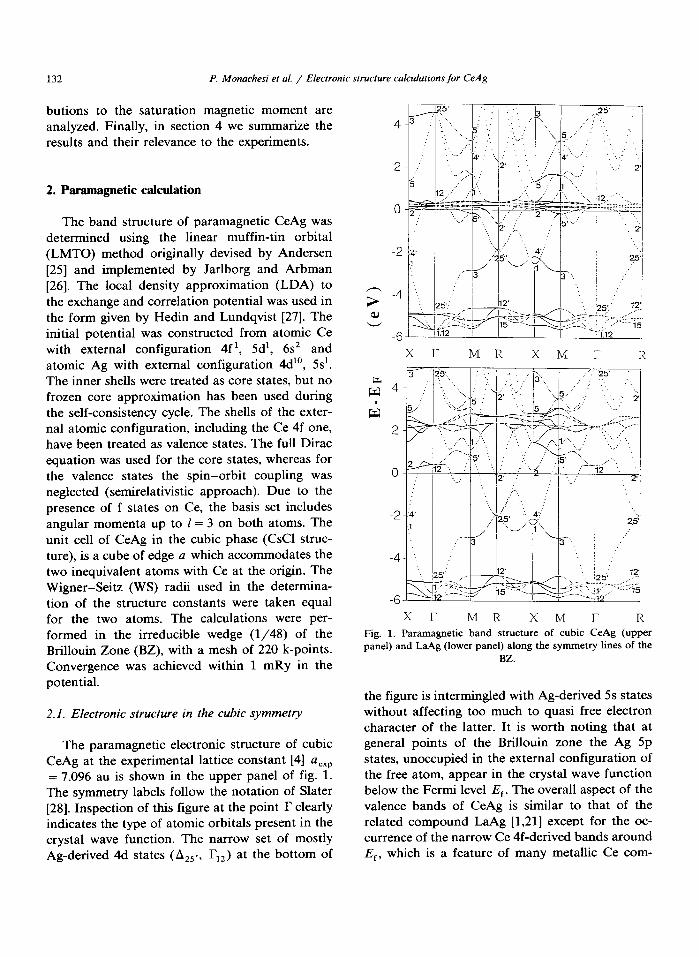
The paramagnetic electronic structure of cubic CeAg at the experimental lattice constant [4] aexp = 7.096 au is shown in the upper panel of fig. 1. The symmetry labels follow the notation of Slater [28]. Inspection of this figure at the point I? clearly indicates the type of atomic orbitals present in the crystal wave function. The narrow set of mostly Ag-derived 4d states (A25,, I,,) at the bottom of
5 1 XI- MR XM I- R
XI- MR XM I- R Fig. 1. Paramagnetic band structure of cubic CeAg (upper panel) and LaAg (lower panel) along the symmetry lines of the
BZ.
the figure is intermingled with Ag-derived 5s states without affecting too much to quasi free electron character of the latter. It is worth noting that at general points of the Brillouin zone the Ag 5p states, unoccupied in the external configuration of the free atom, appear in the crystal wave function below the Fermi level E,. The overall aspect of the valence bands of CeAg is similar to that of the related compound LaAg [1,21] except for the oc- currence of the narrow Ce 4f-derived bands around E,, which is a feature of many metallic Ce com-

P. Monachesi et al. / Electronic structure calculations for CeAg 133
pounds [21-231. The bands at higher energy originate essentially from the Ce Sd and 4f orbitals. The d-doublet I,, is slightly above the f-bands and is well separated ( = 4 eV) from the higher d-triplet I,,,.
In order to extract quantitative information on the properties of the Ce f-orbitals, in the next subsection we will use LaAg as the f-less analog of CeAg. This requires an accurate comparison of the electronic structures of the two compounds. To this end we have calculated the band structure of LaAg with the same lattice constant of CeAg, which corresponds to a slight hydrostatic com- pression of the former. The resulting band struc- ture is shown in the lower panel of fig. 1 and may be compared with the previous calculations of LaAg by Niksch et al. [21]. Although the two calculations do not correspond to the same lattice constant, the agreement is good except for the Ce Sd-derived X, state that we find above the Fermi level in contrast to ref. [21]. The main common features of the electronic structures of CeAg and LaAg are the filled core-like Ag 4f-derived states far below the Fermi energy and the dispersion of the occupied bands up to = -1 eV. The slight shift of the electronic levels towards higher energy with respect to the Ag 4d bands is simply due to the compression of LaAg in our calculations. In both compounds we find at the Fermi energy contributions from Ag 5p and lanthanide 5d orbitals. In CeAg, however, the occurrence of the Ce 4f-derived narrow bands in the same energy region quite changes the character of the wave functions, as we have found by inspection of the eigenvectors.
More insight into the binding properties of the two compounds is gained by looking at the partial charges per atom and per angular momentum component I reported in table 1 for CeAg and LaAg. The corresponding entries in the table are practically the same for the two compounds ex- cept for the presence of one additional f-electron on Ce. The interpretation of the f-component pro- jected charge within the picture of localized atomic orbitals must take into account the effect of the tails of orbitals centered on neighboring atoms. In the LMTO formalism these tails are resolved in components of the angular momentum and give a large contribution to the partial charge densities in the boundary region of the WS sphere. In both compounds considered here, charge with p char- acter is present on Ag and on the lanthanide, but is lacking in the external atomic configuration of the component atoms. Most of the charge with p character is due to the tails and only a minor part represents the effective occupation of the corre- sponding atomic orbital, due to the hybridization in the crystal.
Further information is given by the density of states (DOS) that we have plotted for both com- pounds. In figs. 2a and b we report the total DOS for CeAg and LaAg, respectively, where the fea- tures of the electronic structure sketched above are clearly shown as a function of the energy. The f-component resolved partial DOSS relevant to the comparison between the two compounds are dis- played in figs. 3a and b for CeAg and LaAg, respectively. Comparing the total and partial DOSS on either atom in figs. 2 and 3 one finds again striking analogies among the two compounds. The
Table 1
I-decomposed charge per atom (e/atom) within the WS spheres for CeAg (top) and LaAg (bottom) in the paramagnetic, cubic phase. The WS radius is rws = 3.494 au
S P d f total
CeAg
Ag(WS) Ce(WS)
LaAg
Ag(WS) La(WS)
1.19 1.04 9.66 0.04 11.93
0.39 0.33 1.23 l.li 3.07
1.21 1.01 9.67 0.04 11.93
0.38 0.31 1.20 0.18 2.07
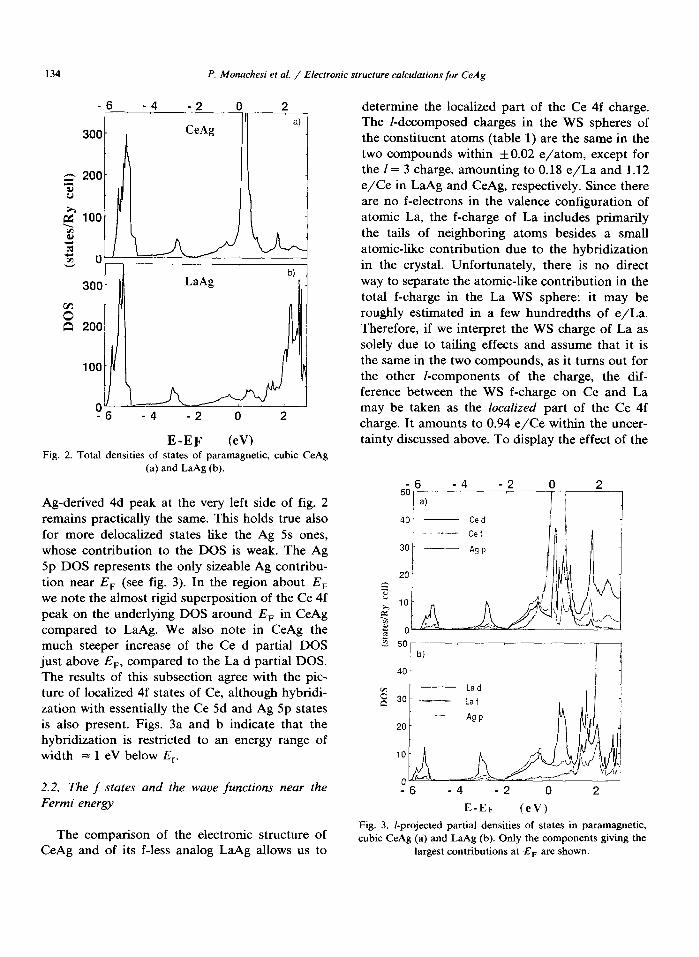
134 P. Monachesi et al. / Electronic structure calculations for CeAg
-6 +
300
!
116 -4 -2 0 2
E-EF (eW Fig. 2. Total densities of states of paramagnetic, cubic CeAg
(a) and LaAg (b).
Ag-derived 4d peak at the very left side of fig. 2 remains practically the same. This holds true also for more delocalized states like the Ag 5s ones, whose contribution to the DOS is weak. The Ag 5p DOS represents the only sizeable Ag contribu- tion near E, (see fig. 3). In the region about E, we note the almost rigid superposition of the Ce 4f peak on the underlying DOS around E, in CeAg compared to LaAg. We also note in CeAg the much steeper increase of the Ce d partial DOS just above E,, compared to the La d partial DOS. The results of this subsection agree with the pic- ture of localized 4f states of Ce, although hybridi- zation with essentially the Ce 5d and Ag 5p states is also present. Figs. 3a and b indicate that the hybridization is restricted to an energy range of width = 1 eV below E,.
2.2. The f states and the wave functions near the Fermi energy
The comparison of the electronic structure of CeAg and of its f-less analog LaAg allows us to
determine the localized part of the Ce 4f charge. The l-decomposed charges in the WS spheres of the constituent atoms (table 1) are the same in the two compounds within f0.02 e/atom, except for the I = 3 charge, amounting to 0.18 e/La and 1.12 e/Cc in LaAg and CeAg, respectively. Since there are no f-electrons in the valence configuration of atomic La, the f-charge of La includes primarily the tails of neighboring atoms besides a small atomic-like contribution due to the hybridization in the crystal. Unfortunately, there is no direct way to separate the atomic-like contribution in the total f-charge in the La WS sphere: it may be roughly estimated in a few hundredths of e/La. Therefore, if we interpret the WS charge of La as solely due to tailing effects and assume that it is the same in the two compounds, as it turns out for the other Z-components of the charge, the dif- ference between the WS f-charge on Ce and La may be taken as the localized part of the Ce 4f charge. It amounts to 0.94 e/Cc within the uncer- tainty discussed above. To display the effect of the
40 - Ced
30
E-EF (eV1
Fig. 3. /-projected partial densities of states in paramagnetic, cubic CeAg (a) and LaAg (b). Only the components giving the
largest contributions at E, are shown.
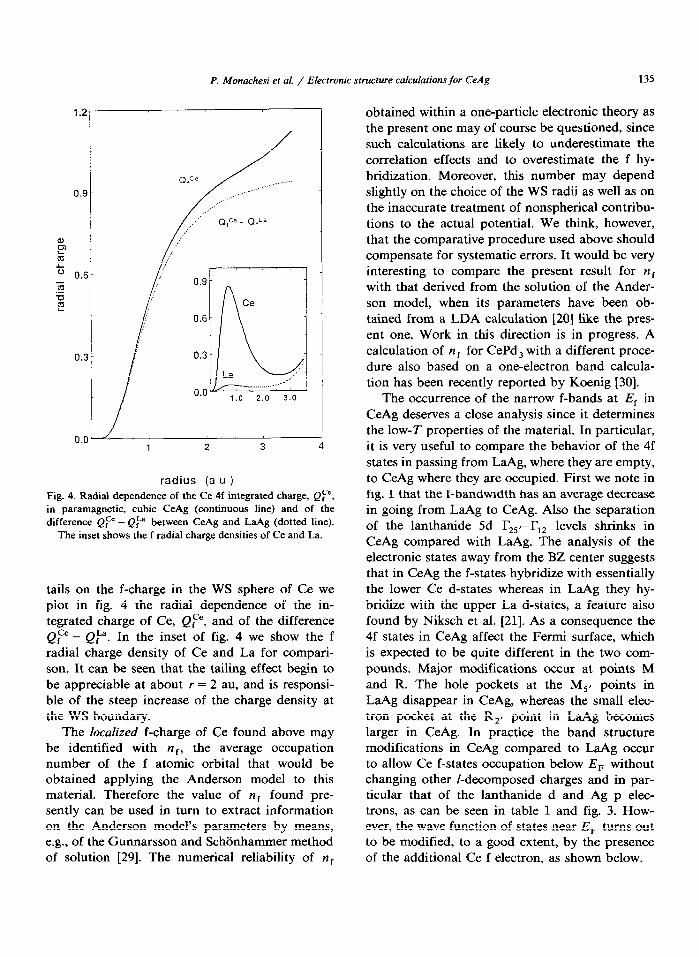
P. Monachesi et al. / Electronic structure calculations for CeAg 135
1.21
radius (a u ) Fig. 4. Radial dependence of the Ce 4f integrated charge, Qp. in paramagnetic, cubic CeAg (continuous line) and of the difference Q,“e - Q:” between CeAg and LaAg (dotted line).
The inset shows the f radial charge densities of Ce and La.
tails on the f-charge in the WS sphere of Ce we plot in fig. 4 the radial dependence of the in- tegrated charge of Ce, Q,“‘, and of the difference QF - Q,““. In the inset of fig. 4 we show the f radial charge density of Ce and La for compari- son. It can be seen that the tailing effect begin to be appreciable at about r = 2 au, and is responsi- ble of the steep increase of the charge density at the WS boundary.
The localized f-charge of Ce found above may be identified with n,, the average occupation number of the f atomic orbital that would be obtained applying the Anderson model to this material. Therefore the value of nr found pre- sently can be used in turn to extract information on the Anderson model’s parameters by means, e.g., of the Gunnarsson and Schonhammer method of solution [29]. The numerical reliability of n,
obtained within a one-particle electronic theory as the present one may of course be questioned, since such calculations are likely to underestimate the correlation effects and to overestimate the f hy- bridization. Moreover, this number may depend slightly on the choice of the WS radii as well as on the inaccurate treatment of nonspherical contribu- tions to the actual potential. We think, however, that the comparative procedure used above should compensate for systematic errors. It would be very interesting to compare the present result for n, with that derived from the solution of the Ander- son model, when its parameters have been ob- tained from a LDA calculation [20] like the pres- ent one. Work in this direction is in progress. A calculation of n, for CePd, with a different proce- dure also based on a one-electron band calcula- tion has been recently reported by Koenig [30].
The occurrence of the narrow f-bands at E, in CeAg deserves a close analysis since it determines the low-T properties of the material. In particular, it is very useful to compare the behavior of the 4f states in passing from LaAg, where they are empty, to CeAg where they are occupied. First we note in fig. 1 that the f-bandwidth has an average decrease in going from LaAg to CeAg. Also the separation of the lanthanide 5d I,,,-I’,, levels shrinks in CeAg compared with LaAg. The analysis of the electronic states away from the BZ center suggests that in CeAg the f-states hybridize with essentially the lower Ce d-states whereas in LaAg they hy- bridize with the upper La d-states, a feature also found by Niksch et al. [21]. As a consequence the 4f states in CeAg affect the Fermi surface, which is expected to be quite different in the two com- pounds. Major modifications occur at points M and R. The hole pockets at the M,, points in LaAg disappear in CeAg, whereas the small elec- tron pocket at the R,/ point in LaAg becomes larger in CeAg. In practice the band structure modifications in CeAg compared to LaAg occur to allow Ce f-states occupation below E, without changing other Z-decomposed charges and in par- ticular that of the lanthanide d and Ag p elec- trons, as can be seen in table 1 and fig. 3. How- ever, the wave function of states near E, turns out to be modified, to a good extent, by the presence of the additional Ce f electron, as shown below.

136 P. Monachesi et al. / Electronic structure calculations for CeAg
Table 2
Wave function composition (%) according to I-character within 1 eV below E, along the symmetry lines of the BZ for CeAg
(top) and LaAg (bottom) in the paramagnetic, cubic phase. The atomic origin is indicated in parentheses. Not reported components are less than 10%
CeAg s(Cc) + s(Ag) p(Ag) d(Ce) f(Cc)
A 18 20 14 36 B 6 19 13 55
A 5 21 21 48
S 8 24 22 42
T 5 22 11 51
2 4 20 5 65
LaAg s(La) + s(Ag) p(Ag) d(La) f(La)
A 25 27 25 4
z 15 33 37 5
A 12 34 35 12
s 12 3-l 35 9 T 11 40 26 13 Z 16 42 20 6
Let us now consider the hybridization of the f electrons in CeAg. In the present context we char- acterize it by the mixing of the different I-compo- nents in the wave functions near E, where the f DOS is concentrated. From the band structure and from the DOSS of fig. 3, we choose an energy range AE = 1 eV below E, and sample the eigen- function I-character along the BZ symmetry lines in this energy range. The results for CeAg are reported in table 2 (upper panel) along with the results for LaAg (lower panel) for comparison. For each symmetry line the entries of table 2 represent the percentage of each l-component/ atom in the wave-function, averaged on the states whose energy falls within a region of width AE below E,. These results confirm quantitatively that important modifications in the wave func- tions have occurred because of the hybridization of the f electrons in passing from LaAg to CeAg, in the whole range AE. In CeAg the largest contri- bution to the hybridization of Ce 4f states comes from Ce 5d and Ag 5p conduction states. The contribution of the s states is very low, as well as that of Ag 4d and Ce 6p states. The hybridization is anisotropic since it depends on the direction of the symmetry line.
From this analysis we conclude that the effect of the Ce 4f electrons on the occupied electronic states of CeAg is restricted to an energy range of about 1 eV below E,, where they hybridize with the extended states. This fact, together with the evidence of the localized behavior of the Ce 4f states obtained by the analysis of the tailing ef- fects indicates that the electronic structure of CeAg may fit in well with the picture of the periodic Anderson model.
2.3. Tetragonal phase
Before closing this section we want to discuss the effect of the observed tetragonal distortion on the Anderson hybridization of the f electrons and their average occupation number n r. We per- formed self-consistent calculations for tetragonal CeAg with the same method described above, assuming the experimental [4] lattice constant a = 7.066 au and ratio c/a = 1.019, which give a slightly larger unit-cell volume than in the cubic phase. Due to the lower symmetry, a larger irre- ducible wedge, equal to l/16 of the tetragonal BZ, has to be taken. To keep the same precision as in the previous calculation a mesh of 550 k-points in this wedge was taken. By this procedure we found no relevant changes in the electronic structure of
the tetragonal phase compared to the cubic one. The analysis of the valence charge distribution per atom and per I-component gives the same results reported in table 1 for the cubic case, within the given precision. In particular, there are no changes in the f partial charge on Ce, whence we conclude that the value of nF found in the cubic phase is not modified by the tetragonal distortion.
In fig. 5 we present the relevant partial DOSS of tetragonal CeAg for comparison with that of cubic CeAg shown in fig. 3a. They are very similar and therefore no dramatic effects of the tetragonal distortion on the Anderson hybridization are ex- pected. In fact, by comparing the components of wave functions of the states near E, in the tetrago- nal phase with the corresponding entries in table 2 we find negligible changes induced by the lower symmetry. These results on the effect of the te- tragonal distortion on the electronic properties of CeAg are supported by XPS measurements [9], in
P. Monachesi et al. / Electronic structure calculations for CeAg 137
the range 300-3.7 K, showing the negligible effect of the distortion on the DOS.
Our calculations provide us also with total en- ergy results but we did not carry out a systematic study of the relative stability of the two structures. This would require to sample the possible differ- ent geometries of the distorted cell, which is out- side the scope of the present work. Moreover, due to the presence of the f electrons in this com- pound, LDA may not be accurate enough to draw definite conclusions on the stability of their struc- ture. For the sake of completeness, however, we have performed also a self-consistent calculation of the total energy of CeAg in the cubic structure, keeping the same volume of the unit cell as in the tetragonal structure and the same number of k- points in the same wedge of the BZ (equal to l/16 of its volume). The difference between the result- ing values of the total energy for the two struc- tures is less than 1 mRy, i.e., comparable to the precision of the present calculation.
A conclusion which may be drawn from the present calculations in the tetragonal symmetry is that the band Jahn-Teller mechanism, invoked by many authors [1,2,21], is not responsible for the structural transition in CeAg.
3. Ferromagnetic calculation
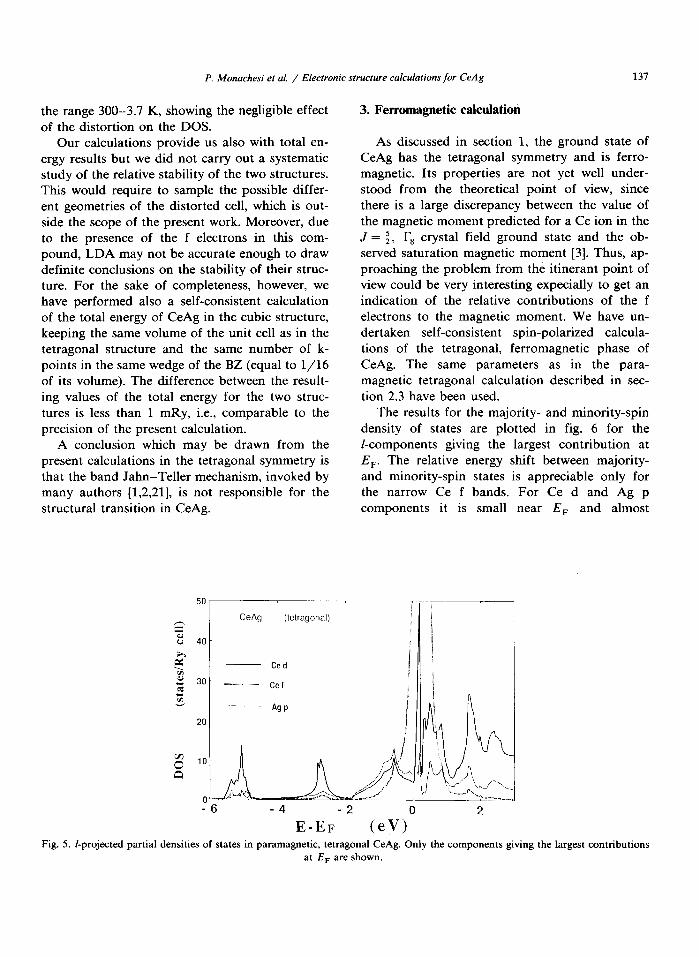
As discussed in section 1, the ground state of CeAg has the tetragonal symmetry and is ferro- magnetic. Its properties are not yet well under- stood from the theoretical point of view, since there is a large discrepancy between the value of the magnetic moment predicted for a Ce ion in the J = $, l?, crystal field ground state and the ob- served saturation magnetic moment [3]. Thus, ap- proaching the problem from the itinerant point of view could be very interesting expecially to get an indication of the relative contributions of the f electrons to the magnetic moment. We have un- dertaken self-consistent spin-polarized calcula- tions of the tetragonal, ferromagnetic phase of CeAg. The same parameters as in the para- magnetic tetragonal calculation described in sec- tion 2.3 have been used.
The results for the majority- and minority-spin density of states are plotted in fig. 6 for the l-components giving the largest contribution at E,. The relative energy shift between majority- and minority-spin states is appreciable only for the narrow Ce f bands. For Ce d and Ag p components it is small near E, and almost
-6 -4 -2 2 E-EF (eV”
Fig. 5. I-projected partial den&es of states in paramagnetic, tetragonal CeAg. Only the components giving the largest contributions
at E, are shown.
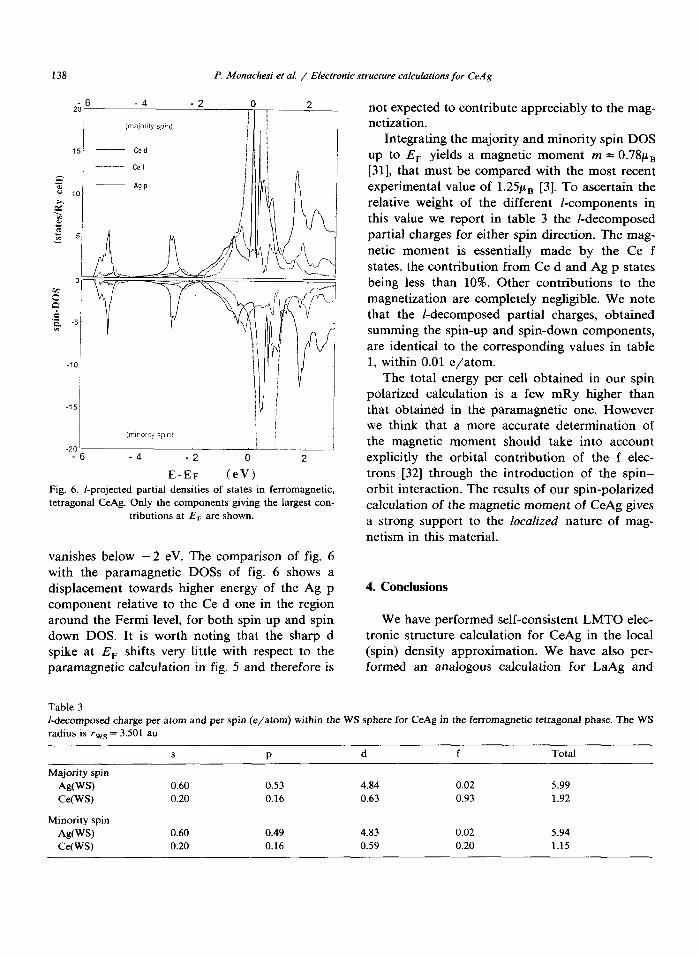
138 P. Mona&vi et al. / Electronic structure cafculations far C&g
-15
(mlnorlty spni
-20
-6 -4 -2 2
E-Et=
Fig. 6. I-projected partial densities of states in ferromagnetic,
tetragonal CeAg. Only the components giving the largest con-
tributions at E, are shown.
vanishes below -2 eV. The comparison of fig. 6 with the paramagnetic DOSS of fig. 6 shows a displacement towards higher energy of the Ag p component relative to the Ce d one in the region around the Fermi level, for both spin up and spin down DOS. It is worth noting that the sharp d spike at E, shifts very little with respect to the paramagnetic calculation in fig. 5 and therefore is
not expected to contribute appreciably to the mag- netization.
Integrating the majority and minority spin DOS up to E, yields a magnetic moment m = 0.78~~ [31], that must be compared with the most recent experimental value of 1.25~~ [3]. To ascertain the relative weight of the different I-components in this value we report in table 3 the I-decomposed partial charges for either spin direction. The mag- netic moment is essentially made by the Ce f states, the contribution from Ce d and Ag p states being less than 10%. Other contributions to the magnetization are completely negligible. We note that the I-decomposed partial charges, obtained summing the spin-up and spin-down components, are identical to the corresponding values in table 1, within 0.01 e/atom.
The total energy per cell obtained in our spin polarized calculation is a few mRy higher than that obtained in the paramagnetic one. However we think that a more accurate determination of the magnetic moment should take into account explicitly the orbital contribution of the f elec- trons [32] through the introduction of the spin- orbit interaction. The results of our spin-polarized calculation of the magnetic moment of CeAg gives a strong support to the localized nature of mag- netism in this material.
4. Conclusions
We have performed self-consistent LMTO elec- tronic structure calculation for CeAg in the local (spin) density approximation. We have also per- formed an analogous calculation for LaAg and
Table 3 l-decomposed charge per atom and per spin (e/atom) within the WS sphere for CeAg in the ferromagnetic tetragonal phase. The WS
radius is rws = 3.501 au
S I, d f Total
Majority spin Ag(WS)
Ce(WS)
0.60 0.53 4.84 0.02 5.99
0.20 0.16 0.63 0.93 1.92
Minority spin Ag(WS)
Ce(WS)
0.60 0.49 4.83 0.02 5.94
0.20 0.16 0.59 0.20 1.15

P. Monachesi et al. / Electronic structure calculations for CeAg 139
used it as the f-less analog of CeAg. Our main findings can be summarized in the following points.
(i) The calculated electronic structure of CeAg seems to fit well into the picture of localized Ce 4f-states hybridized with the more extended con- duction states near E,, in particular with Ce 5d and Ag 5p states. To this conclusion leads not only the inspection of the band structure and of the DOS, but also the comparative analysis of the wave function composition of CeAg and LaAg in the energy region where the f DOS is con- centrated.
(ii) The use of LaAg as the f-less analog of CeAg has turned out extremely effective for the interpretation of the electronic properties of the solid. In particular the comparison of the two electronic structures allows us to determine the localized, atomic-like part of the Ce 4f charge. The identification of this with the average 4f occupa- tion number, n,, in the periodic Anderson model establishes an important link between this many- body Hamiltonian and a single-particle calcula- tion, as the present one. This may be useful to accomplish the ab initio determination of the parameters of the Anderson model suited for de- scribing a real material like CeAg.
(iii) The calculation of the electronic structure of CeAg in the tetragonal symmetry shows that the tetragonal distortion introduces negligible dif- ferences in the partial DOSS and in particular in the f-state occupation. Therefore the conclusion (i) and (ii) apply, with only minor quantitative dif- ferences, also to the tetragonal phase. Moreover, an explanation of the distortion with a band Jahn-Teller mechanisms is excluded by the pres- ent calculation.
(iv) The magnetic moment in the ground state of CeAg originates essentially from the f electrons. This gives a strong support to the localized nature of magnetism in CeAg.
The theoretical description of CeAg deriving from the above points agrees well with the availa- ble experimental results. The localized nature of the 4f states in CeAg and their location near the Fermi level is supported by spectroscopic, resistiv-
ity and magnetic measurements. In a short com- munication Takigawa et al. [ 91 reported a XPS spectrum of CeAg at T = 4 K indicating the possi- ble presence of a Kondo peak. Unfortunately those data are poorly resolved and more extended mea- surements are desirable. Also the resistivity meas- urements [5] on the solid solutions La,_,Ce,,-
Ag,In, --x support the idea of a Kondo behavior related to the presence of Ce in the La-based samples. Concerning the magnetic properties, the paramagnetic susceptibility of CeAg [3] and of Ce impurities in a LaAg matrix [6] agrees well with the crystal field level scheme [4], supporting the localized picture of Ce 4f states at least at high temperature. On the other hand, the reduced value of the magnetic moment measured at low temper- ature indicates a more complex behavior than a Ce ion in the I, crystal field state. This points to the hybridization of the f electrons with the other conduction states and to the related Kondo effect as a possible mechanism of magnetic moment reduction. The value of 0.78~~ obtained in our ferromagnetic calculation is lower than the ob- served one, but its overwhelming Ce f origin indi- cates that a more detailed calculation should in- clude the orbital contribution to the magnetic moment. In this context it is interesting to note the contrasting behavior of the Pauli-like suscept- ibility of LaAg [6] compared to that of CeAg just mentioned. This can be taken as an experimental confirmation of the picture of LaAg as the f-less analog of CeAg. Finally, the negligible effect of the tetragonal distortion on the electronic struc- ture of CeAg is confirmed by the experimental results that photoemission spectra [9] and mag- netic susceptibility [5,6] are not affected by the structural transition.
Acknowledgements
The authors wish to thank T. Jarlborg for pro- viding us with his LMTO code and for helpful discussions. Fruitful discussions with L.C. Andreani and H. Beck are also acknowledged. One of the authors (P.M.) wishes to thank the kind hospitality of IRRMA in Lausanne. This work was supported in part by the Swiss National

140 P. Monachesi et al. / Electronic structure calculations for CeAg
Science Foundation under Grants no. 2000-5.295 and no. 20-5446.87.
References
[l] A. Hasegawa, B. Bremicker and J. Kiibler, Z. Phys. B 22
(1975) 231.
[2] H. Ihrig and S. Methfessel, Z. Phys. B 24 (1976) 381.
[3] P. Morin, J. Magn. Magn. Mater. 71 (1988) 151.
[4] D. Schmitt, P. Morin and J. Pierre, J. Magn. Magn.
Mater. 8 (1978) 249.
[5] H. Ihrig and S. Methfessel, Z. Phys. B 24 (1976) 385.
[6] R. Takke, N. Dolezal, W. Assmus and B. Ltthi, J. Magn.
Magn. Mater. 23 (1981) 247.
[7] P. Morin, J. Rouchy, Y. Miyako and T. Nishioka, J.
Magn. Magn. Mater. 76 & 77 (1988) 319.
[8] K. Hyomi et al., J. Magn. Magn. Mater 76 & 77 (1988)
462.
[9] Y. Takigawa, S. Noguchi and K. Okuda, J. Magn. Magn.
Mater. 76 & 77 (1988) 345.
[lo] A. EiIing and J.S. Schilling, Phys. Rev. Lett. 46 (1981)
364.
[ll] M. Kurisu, H. Kadomatsu and H. Fujiwara, Phys. Lett. A
84 (1981) 496; J. Phys. Sot. Jpn. 52 (1983) 4349.
[12] B. Barbara et al., J. Appl. Phys. 50 (1979) 2300.
B. Barbara et al., Phys. Lett. A 113 (1986) 381.
[13] E. Gratz et al., J. Phys. F 15 (1985) 1975.
[14] S. Quezel et al., J. Magn. Magn. Mater. 76 & 77 (1988)
403.
R. Djerbi et al., J. Magn. Magn. Mater. 76 & 77 (1988)
260.
[15] J.R. Schrieffer and P.A. Wolff, Phys. Rev. 149 (1966) 491.
[16] B. Coqblin and J.R. Schrieffer, Phys. Rev. 185 (1969) 847.
[17] S. Doniach, in: Valence Instabilities and Related
Narrow-Band Phenomena, ed. R.D. Parks (Plenum, New
York, 1977).
J.S. Schilling, Adv. Phys. 28 (1979) 657.
[18] S. Fraizzoli, P. Monachesi and E.G. Moroni, Proc. 35th
MMM-90 Conference, San Diego, California, to appear in J. Appl. Phys. (1991).
[19] P.W. Anderson, Phys. Rev. 124 (1961) 41.
[20] H. Takahashi and T. Kasuya, J. Phys. C 18 (1985) 2755.
J.M. Wills and B.R. Cooper, Phys. Rev. B 36 (1987) 3809.
B.E. Larson, K.C. Hass and H. Ehrenreich, Phys. Rev. B
37 (1988) 4137.
A.K. McMahan, R.M. Martin and S. Satpathy, Phys. Rev.
B 38 (1988) 6650.
R. Monnier, L. Degiorgi and B. Delley, Phys. Rev. B 41
(1990) 573.
[21] M. Niksch, B. Ltithi and J. Ktbler, Z. Phys. B 68 (1987)
291.
[22] M. Belakhovsky, J. Pierre and D.K. Ray, J. Phys. F 5
(1975) 2274.
1231 T. Jarlborg, A.J. Freeman and D.D. Koelling, J. Magn.
Magn. Mater. 60 (1986) 291.
[24] A. Yanase, J. Magn. Magn. Mater. 52 (1985) 403.
[25] O.K. Andersen, Phys. Rev. B 12 (1975) 3060.
[26] T. Jarlborg and G. Arbman, J. Phys. F 7 (1977) 1635.
T. Jarlborg, J. Phys. F 9 (1979) 283. [27] L. Hedin and B.I. Lundqvist, Solid State Commun. 9
(1971) 507.
[28] J.C. Slater, in: Quantum Theory of Molecules and Solids,
vol. 2 (McGraw-Hill, New York, 1965) pp. 57, 408, 430.
[29] 0. Gunnarsson and K. Schiinhammer, Phys. Rev. B 28
(1983) 4315.
[30] C. Koenig, Solid State Commun. 68 (1988) 727.
[31] This value of the magnetic moment corrects the one
quoted previously in P. Monachesi and E.G. Moroni,
Solid State Commun. 74 (1990) 1349.
[32] M.S.S. Brooks and P.J. Kelly, Phys. Rev. Lett. 51 (1983)
1708.
0. Eriksson, L. Nordstrom, M.S.S. Brooks and B. Johans-
son, Phys. Rev. Lett. 60 (1988) 2523.
Related Documents











