Eileen M Burd Ph D D(ABMM) Eileen M. Burd Ph.D., D(ABMM) Director, Clinical Microbiology Emory University Hospital Emory University Hospital Atlanta, GA

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Eileen M Burd Ph D D(ABMM)Eileen M. Burd Ph.D., D(ABMM)Director, Clinical MicrobiologyEmory University HospitalEmory University Hospital
Atlanta, GA


In vitro diagnostic devices (IVDs)g ( )
Substantial equivalence to existing devicel d FDA‐cleared
Novel agent, new method, public health threat (e.g., Mycobacterium tuberculosis)
FDA‐approvedFDA approved
Labeled: For in vitro diagnostic use

FDA‐cleared or –approved tests that have been ppmodified Changes in test components, procedural parameters, cutoff values, specimen types, collection devices, etc.
Tests currently not subject to FDA‐clearance or approval Standardized textbook procedures Tests developed in the laboratoryp y FDA has authority to regulate but has exercised ‘enforcement discretion’ and has chosen not to do so
Test systems in which performance specifications are Test systems in which performance specifications are not provided by the manufacturer ASRs (analyte‐specific reagents)

For research only, not for diagnostic purposesFor research only, not for diagnostic purposes
Not medical devices essentially unregulated
M t b l b l d “F h l N t f Must be labeled “For research use only. Not for use in diagnostic procedures.”
B i i h Basic science research
Research related to product development
Can be used as components of LDTs if FDA approved/cleared or ASRs are not available

Required clinical investigation before a manufacturer can submit an application to FDAmanufacturer can submit an application to FDA
May be distributed only for use in well‐controlled clinical trials to establish performance clinical trials to establish performance characteristics – monitored and documented
Label: “CAUTION‐ Investigational Device Limited by Label: CAUTION‐ Investigational Device. Limited by Federal (or U.S.) law to investigational use.”

VALIDATION VERIFICATIONVALIDATION FDA and ISO: “confirmation by examination and provision of objective evidence that the
VERIFICATION FDA and ISO: “confirmation through the provision of objective evidence, that specified requirements have been fulfilled” j
particular requirements for a specific intended use can be consistently fulfilled"
CLIA does not use the term
• CLIA: Verification of performance specifications. Each laboratory that introduces an unmodified FDA cleared or approved test must demonstrate and document “that it can obtain performance CLIA does not use the term
“validation” but refers to “establishment of performance specifications.”
at the time of assay development
document that it can obtain performance specifications comparable to those established by the manufacturer for the following performance characteristics: accuracy, precision, reportable range of t t lt f th t t t if th t at the time of assay development
prior to reporting patient results. includes determination of accuracy,
precision, reportable range, reference interval, analytical
iti it d l ti l ifi it
test results for the test system, verify that reference intervals (normal values) are appropriate for the laboratory’s patient population.”
sensitivity and analytical specificity.
ongoing quality control and quality assessment

Performance Characteristic
FDA approved/cleared LDTCharacteristic
Reportable Range‐ linearity study (quant assays)
+ +Analytical Sensitivity N/A – not required by CLIAAnalytical Sensitivity‐ limit of detection study
N/A – not required by CLIA +Precision‐ replication experiment
+ +p p
Analytical Specificity‐ interference study
N/A – not required by CLIA +Accuracy (trueness) + +Accuracy (trueness)‐ comparison of methods study
+ +Reference Interval + +
For details:Burd, E.M. Validation of Laboratory‐Developed Molecular Assays for Infectious Diseases. Clin. Microbiol. Rev. 23:50‐576, 2010.

To avoid diagnostic use of laboratory tests with To avoid diagnostic use of laboratory tests with unproven performance characteristics
Use of such tests may mislead healthcare providers Use of such tests may mislead healthcare providers and could cause serious adverse health consequences to patients, who are not aware that co seque ces to pat e ts, o a e ot a a e t atthey are being diagnosed with tests without established performance characteristics

Definition Span of test result values over which the laboratory can establish or verify the Span of test result values over which the laboratory can establish or verify the
accuracy of the instrument or test system measurement response. Does not apply to qualitative tests May only report results within the reportable rangeSynonyms: Measuring interval, analytical measurement range(AMR), linear rangey y g , y g ( ), g
Study suggestionsLinearity experiment: series of samples (n=7‐11) of known concentration – or dilution of known concentration, 2‐4 replicates to check for imprecisionconcentration, 2 4 replicates to check for imprecision
Sample SourcesReference panels, standard solutions, characterized patient samplesMatrix for each specimen type
Data analysismeasured values compared to assigned valueslinear regressionpolynomial regression (preferred)

7 concentrations of analyte prepared by dilution of a high‐concentration standard tested in triplicate
Assigned values (converted to log 10) plotted on the x axis
Measured values (converted to log 10) l tt d th iplotted on the y axis
Linear regressiony=0.9613x+0.1286 r2 = 0.9932
Second order polynomialy= 0 028x2 +1 1937x 0 2667 r2 = 0 9954y=‐0.028x2 +1.1937x ‐ 0.2667 r2 = 0.9954
Third order polynomialy=‐0.0009x3 0.1388x2 +1.5994x ‐ 0.6948 r2 = 0.9958
Second and third order polynomials not Second and third order polynomials not significantly different from linear equation
Reportable range; 30 copies/ml through 3,000,000 copies/ml

DefinitionThe ability of the assay to detect very low concentrations of target in a The ability of the assay to detect very low concentrations of target in a specimen
Qualitative and quantitative assays “limit of detection (LOD)” – consistently detected (not necessarily
quantified) with acceptable precisionquantified) with acceptable precision
Study suggestionsStatistical: signal distinguished from background – limit of blank (plus testing of low‐level positive)(plus testing of low level positive)Empirical: test serial dilutions of known concentration in range of expected detection limit
Sample Sourcesi d d Q i l fi i l llKnown concentration: standards, QC materials, proficiency samples, cell
lines containing target etc.
Data analysisProbit analysis commonly usedProbit analysis commonly used
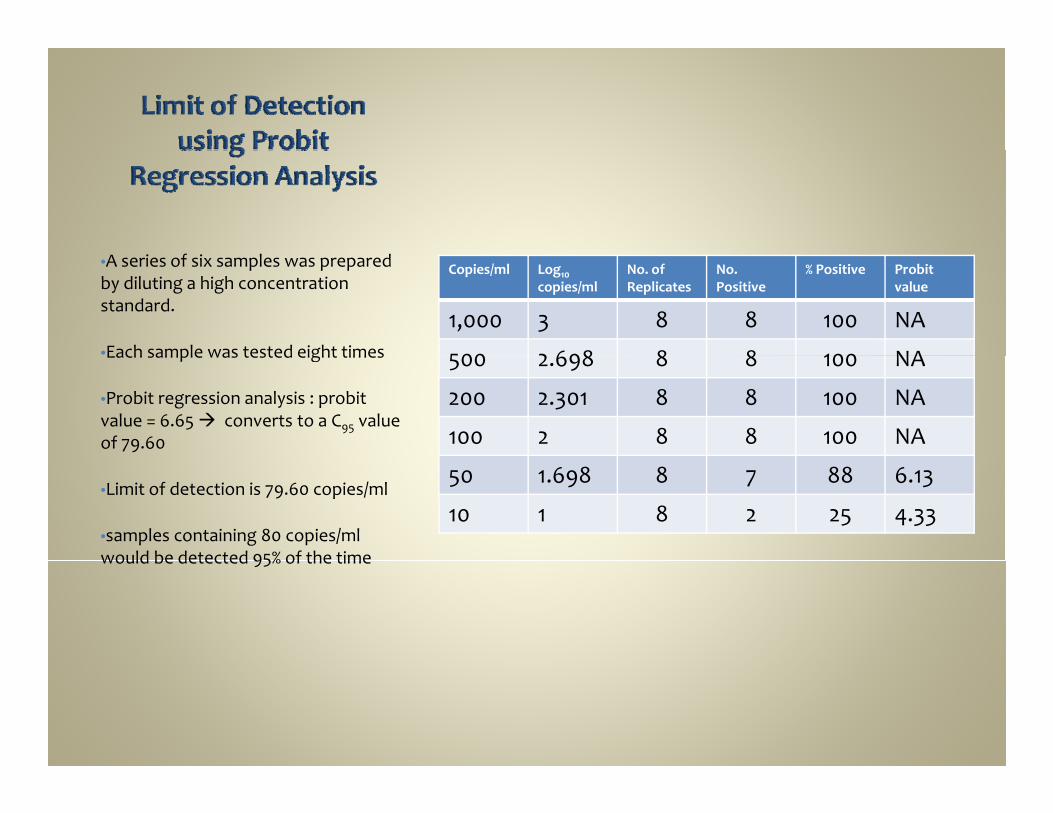
Copies/ml Log10 copies/ml
No. of Replicates
No. Positive
% Positive Probitvalue
1,000 3 8 8 100 NA
500 2 698 8 8 100 NA
•A series of six samples was prepared by diluting a high concentration standard.
•Each sample was tested eight times 500 2.698 8 8 100 NA
200 2.301 8 8 100 NA
100 2 8 8 100 NA
Each sample was tested eight times
•Probit regression analysis : probitvalue = 6.65 converts to a C95 value of 79.60
50 1.698 8 7 88 6.13
10 1 8 2 25 4.33•Limit of detection is 79.60 copies/ml
•samples containing 80 copies/ml would be detected 95% of the timewould be detected 95% of the time

Definition How well a given measurement can be reproduced when a test is applied How well a given measurement can be reproduced when a test is applied
repeatedly to a single homogeneous sample. Random analytical error
Remember: A measurement may be very precise but not very accurate
St d gg tiStudy suggestionsReplication experiment
within runrun‐to‐run*day‐to‐dayday to dayconsider different operators, multiple reagent lots, multiple laboratories
Qualitative: 20% below LOD, LOD, 2o% above LOD in replicates up to 40Quantitative: high, low, LOD – in duplicate over 20 days
Sample Sources standards, QC materials, proficiency testing samples, patient specimens
Data analysisd d i t l d idepends on experimental designstandard deviation, coefficient of variation

l i i
Conccopies/ml
Log10 conc
%CVtotal
SD 95% CILog10viral
Log change 95% CI
Fold change 95%CI
Example: CMV Quantitative Assay
•3 samples spanning the reportable range
In duplicate one run per day over 20 load
9 9
250,000 5.39 1.93 0.104 5.11‐5.67
0.56 3.6
6
•In duplicate, one run per day over 20 days, 2 operators
•Overall %CV ‐ Less precise at lower 5,000 3.69 1.75 0.130 3.43‐3.95
0.53 3.3
300 2.48 6.91 0.164 2.15‐2.81
0.66 4.6
•Overall %CV ‐ Less precise at lower concentrations
•5‐fold change may represent imprecision rather than a true biological differencebiological difference

DefinitionAbility of an assay to detect only the intended targetCross‐reactivity – related or interfering nucleic acids
organisms with similar genetic structurenormal floraorganisms that cause similar disease states or clinically‐relevant co‐infectionsorganisms that cause similar disease states or clinically relevant co infections
Interfering substances – specimen related (e.g., hemolyzed, icteric, lipemic)
Study suggestionsInterference screen:Interference screen:
Add potentially interfering substance to specimens containing the analyte of interest – analyze specimens with and without
Comparative measurement:E l bi i i i i h i l i f i b d Evaluate bias: patient specimens containing the potential interfering substance and analyte of interest – test using new method and comparator method
Data analysisP i d t t t t d t t i d diff t tPaired t test, repeated measures test, paired‐difference testDifference between means and controls with allowable error that is clinically significant for the test

DefinitionCloseness of agreement between the average value from a large series of Closeness of agreement between the average value from a large series of measurements and the true (or accepted) value
Study suggestionsComparison of Methods Study
i t t d b th d d lid ti th dspecimens tested by new method and valid comparative methodin duplicate is recommended – singly OK if close agreement is expected
Recovery Studyif no comparison method is availablespecimens with known amount of analytespecimens with known amount of analytecan also assess proportional error that can occur as concentration of
analyte increases
Sample Sourcesresidual patient specimens positive and negative over range of assayresidual patient specimens, positive and negative, over range of assayNo fewer than 20, typically 40‐50 or more
Data analysisscatter diagram regression analysis (slope, y‐intercept)Bl d Alt diff l tBland‐Altman difference plot

Quantitative real time PCR assays – new extraction methodsA‐B•72 patient samples tested by old and new extraction methods. 43 specimens had numerical results•Linear regression: y=0.9834x+0.0576R2 = 0.9449 (good)•Slope close to 1.0 – no proportional bias•Y intercept close to origin (0.0) – no constant systematic bias•Bland‐Altman mean bias ‐0.01 log (no systematic bias))
C‐D•46 positive specimens•Linear regression: y=0.9698x + 0.3398R2 = 0.9543 (good)R 0.9543 (good)•Slope close to 1.0 – no proportional bias•Y intercept significantly away from origin (0.0) –> constant systematic bias•Bland‐Altman mean bias 0.41 log (about 2.5 fold) –not statistically significant since 95% confidence not statistically significant since 95% confidence interval contains zero, but ? clinically significant

Definitionl f l i ll f d iNormal range – range of values typically found in
individuals who do not have the diseaseStudy suggestionsy gg
Qualitative assays: If the nucleic acid target is always absent in a healthy individual – normal range is “negative” or “not detected” – no study – cite literature or other pertinent not detected no study cite literature or other pertinent information
Quantitative assays: < LOD, LLOQ or clinical decision limit (asymptomatic vs disease)limit (asymptomatic vs disease).
Reference Interval Study: test healthy subjects in intended population (n=120) – use results to determine normal rangerange

LOD
LLOQ
Upper Limit of Linearity
Analyte concentration Low Medium High1 2 3 4 5 6 7 8 9 10 11
Reportable Range(for Quantitative Assays)
X X X X X X X X X X X
Analytical SensitivityAnalytical Sensitivity(LOD)
X X X X X
Precision(qualitative assay)
X X X
Precision(quantitative assay)
X X X(q y)
Analytical Specificity
Accuracy
Reference IntervalReference Interval
Comments:Reportable range: 7‐11 concentrations across anticipated measuring range; 2‐4 replicates on same dayAnalytical Sensitivity: 8‐12 replicates of 4‐5 samples at the low concentration end over 5 days.Precision ‐ Qualitative: Use concentrations at LOD, 20% above and 20% below. Test in duplicate over 15 days (include data from Analytical sensitivity runs to provide data over 20 days).Precision ‐ Quantitative: Use high, low and LOD concentrations. Test in duplicate over 19 days (include data from Reportable Range study as Day 1 to provide data over 20 days).



Table of Contents Subpart K_Quality System for Nonwaived Testing Sec. 493.1256 Standard: Control procedures.
F h t t t th l b t i ibl f h i t l d th t it th d (a) For each test system, the laboratory is responsible for having control procedures that monitor the accuracy and precision of the complete analytic process.
(b) (b) The laboratory must establish the number, type, and frequency of testing [[Page 578]] control materials using, if applicable, the performance specifications verified or established by the laboratory as specified in Sec. 493.1253(b)(3).
(c) (c) The control procedures must‐‐ (1) Detect immediate errors that occur due to test system failure, adverse environmental conditions, and operator performance. (2) Monitor over time the accuracy and precision of test performance that may be influenced by changes in test system performance and environmental conditions, and p y y g y p ,variance in operator performance. (d) Unless CMS approves a procedure, specified in Appendix C of the State Operations Manual (CMS Pub. 7), that provides equivalent quality testing, the laboratory must‐‐ (1) Perform control procedures as defined in this section unless otherwise specified in the additional specialty and subspecialty requirements at Sec. Sec. 493.1261 through 493.1278. (2) For each test system, perform control procedures using the number and frequency specified by the manufacturer or established by the laboratory when they meet or exceed the requirements in paragraph (d)(3) of this section. (3) At least once each day patient specimens are assayed or examined perform the following for‐‐ (i) Each quantitative procedure include two control materials of different concentrations; perform the following for‐‐ (i) Each quantitative procedure, include two control materials of different concentrations; (ii) Each qualitative procedure, include a negative and positive control material; (iii) Test procedures producing graded or titered results, include a negative control material and a control material with graded or titered reactivity, respectively; (iv) Each test system that has an extraction phase, include two control materials, including one that is capable of detecting errors in the extraction process; and (v) Each molecular amplification procedure, include two control materials and, if reaction inhibition is a significant source of false negative results, a control material capable of detecting the inhibition. ...
(ii) I l d t l t t l t i l h l t d li bl hi h t b d th gh h t f (ii) Include at least one control material on each plate or card, as applicable, which must be processed through each step of patient testing, including extraction processes…(ii) The laboratory may use the stated value of a commercially assayed control material provided the stated value is for the methodology and instrumentation employed by the laboratory and is verified by the laboratory.(iii) Statistical parameters for unassayed control materials must be established over time by the laboratory through concurrent testing of control materials having previously determined statistical parameters……(g) The laboratory must document all control procedures performed. (g) y p p(h) If control materials are not available, the laboratory must have an alternative mechanism to detect immediateerrors and monitor test system performance over time. The performance of alternative control proceduresmust be documented.[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003

POSITIVE CONTROL(S) NEGATIVE CONTROLPOSITIVE CONTROL(S)To see if the PCR has worked Obtain commercially, prepare in‐house, other
sourcesA ifi l i id f t t i i th
NEGATIVE CONTROLTo detect contamination Blank control (e.g., water or buffer) Specimen containing known nontarget
A specific nucleic acid fragment containing the entire sequence to be amplified including the primer binding site (e.g., previously amplified and or a cloned DNA fragment that has been sequenced for confirmation
Purified total nucleic acid from the organism
p g gnucleic acid (preferred)
Purified total nucleic acid from the organism containing the sequence of interest
Patient specimens containing the target nucleic acid
Patient specimens spiked with target The whole organism Do not use calibrators (Note: CLIA allows use of
calibrators if other control material is not available – use different lot number
Matrix that matches specimens as closely as Matrix that matches specimens as closely as possible
For each specimen type

QUALITATIVE ASSAYS QUANTITATIVE ASSAYSQUALITATIVE ASSAYS
Positive for each analyte
QUANTITATIVE ASSAYS
Positive ‐ at least 2 controls of different values (generally
for each specimen type
Negative
(ghigh and low)
Negative
Frequency: DailyControl testing is not necessary on days when testing is not performedMultiplex assays: controls for each analyte are included in each run or rotated so that all analytes are tested periodically (MIC.63264)

How do you define a low positive control?
) Wi hi l f LLOD/LLOQ1.) Within 1 log of LLOD/LLOQ
2.) CT value between 30 ‐35
3.) Within 1,2, 3 standard deviations of LLOD/LLOQ
4.) Other4 )

FDA cleared/approved, not modified/ pp ,
Quantitative: 2 levels, run daily
Qualitative: run dailyQua a e u da y
Validation studies: comparison of external and built‐in controls, 25 sample minimum.
External surrogate controls are run for each new lot number or shipment, after major system maintenance and after software upgrades also as recommended by and after software upgrades – also as recommended by the manufacturer or at least every 30 days, whichever is more frequent.

Detect errors in the extraction processp Whole bacteria or virus as it would appear in a patient sample (best)( )
Purified nucleic acid can be used Seed into appropriate matrix at low levelSeed into appropriate matrix at low level Run in parallel with patient samples* if the positive extraction control is taken through all steps of * if the positive extraction control is taken through all steps of the assay it can dually serve as an extraction control and an amplification control

CLIA requires in assays in which reaction inhibition is a q ysignificant source of false‐negative results
Testing for inhibition may be relaxed or discontinued if
Sufficient data (100‐500 samples)
Inhibition rates are found to be within acceptable limits pconsidering the medical implications of a false‐negative test
f dd d b f l l• If added before sample preparation can also serve as an extraction control
• Must be detected for a negative result to be considered valid• Must be detected for a negative result to be considered valid

EXTRINSIC INTERNAL CONTROLS INTRINSIC INTERNAL CONTROLSEXTRINSIC INTERNAL CONTROLS Homologous extrinsic controls Unmodified intended target
Small amount added to second aliquot of i
INTRINSIC INTERNAL CONTROLSHeterologous intrinsic controls
“housekeeping genes” – host genome present in patient specimens in low copy specimen
Easy to use Cost and space occupied by a second reaction
Modified intended target Can be amplified with same primers as intended
present in patient specimens in low copy number
Different set of primers Same or separate reaction β‐globin, β‐actin, gamma‐interferon,
target Contain inserts that can be distinguished from
the intended target by size (100‐200 bp longer) or unique sequence
Can be co‐amplified with intended target in
glyceraldehyde‐3‐phosphate dehydrogenase(GAPDH), etc.
Can also be used to establish the presence of cellular material in a samplesC b f i b h
p gsame reaction vessel
Heterologous extrinsic controls
Non‐target controlsDiff i / b i d
Concern: cumber of copies may be much higher than target – amplification advantage – not accurately test for inhibition
Different primers/probes required

Minimum frequency – at least each dayMinimum frequency at least each day
To qualify each shipment or lot‐to‐lot change of reagentsreagents
Each time there is a major preventive maintenance or replacement of a critical part that may influence or replacement of a critical part that may influence test performance
For assays that contain electronic/procedural/built‐in For assays that contain electronic/procedural/built‐in controls, are FDA‐approved and not modified by the laboratory – new lot and shipment onlyy p y

Negative controls – at the end of the run – the last sample to which reagents are addedsample to which reagents are added
For large runs – place evenly throughout the run (e g every 50 samples) – or – distribute randomly(e.g., every 50 samples) or distribute randomly
For many FDA‐approved/cleared assays, the position of the controls is established by the manufacturer of the controls is established by the manufacturer and cannot be changed

Tolerance and acceptability limits must be defined and monitored for all control proceduresand monitored for all control procedures
Stay tuned

CLIA definition: “the assaying of calibration y gmaterials in the same manner as patient specimens to confirm that the calibration of the instrument, kit, or test system has remained stable throughout the or test system has remained stable throughout the laboratory’s reportable range for patient test results”.
According to CAP, to fulfill this requirement requires two processes: 1. calibration verification2. verification of the analytical measurement range
(AMR, reportable range in CLIA terminology)

This section of the checklist only applies to quantitative s sect o of t e c ec st o y app es to qua t tat etests for which appropriate external materials exist.
Materials must have assigned valuesg
When: At changes of reagent lots, unless the laboratory can demonstrate
h h f diff l d ff h fthat the use of different lots does not affect the accuracy of patient/client test results and the range used to report patient/client test data
QC fails to meet established criteria QC fails to meet established criteria After major maintenance or service When recommended by the manufacturer At least every 6 monthsAt least every 6 months

If standard curve is run with every runIf standard curve is run with every run First point, slope and intercept statistics
f d d i d If standard curve is stored Two points are run with every run statistics

**NEW** 06/15/2009MIC 64915MIC.64915Quantitative Cut‐OffPhase I
For qualitative tests that use a cut‐off value to distinguish positive from negative, q g p g ,the cut‐off value is established initially, and verified with every change in lot or at least every 6 months.NOTE: The limit of detection that distinguishes a positive from a negative result should be established or verified when the test is initially placed in service, and verified with every change in lot (e g new master mix) instrument maintenance or at least every every change in lot (e.g. new master mix), instrument maintenance, or at least every six months thereafter. Note that a low‐positive control that is close to the limit of detection can satisfy this checklist requirement, but must be external to the kit (e.g. weak‐positive patient sample or reference material prepared in appropriate matrix).Evidence of Compliance:pWritten procedure for initial establishment and verification of the cut‐off value ANDRecords of initial establishment and verification documented at defined frequency

**NEW** 06/15/2009MIC.64886AMR Validation CriteriaPhase II
Criteria are established for validating the analytical measurement range and compliance is documented.NOTE: If the materials used for calibration or for calibration verification include low, midpoint and high values that are near the stated AMR and if calibration verification midpoint, and high values that are near the stated AMR, and if calibration verification data are within the laboratory's acceptance criteria, the AMR has been validated; no additional procedures are required. If the calibration and/or calibration verification materials do not include the full AMR, or the laboratory extends the AMR beyond the manufacturer's stated range, the AMR must be validated by assaying materials f g , y y greasonably near the lowest and highest values of the AMR.Evidence of Compliance:Written procedure defining the method, frequency and acceptability criteria for AMR validation
Related Documents











