Effects of p53-Expressing Adenovirus on the Chemosensitivity and Differentiation of Anaplastic Thyroid Cancer Cells MIKHAIL V. BLAGOSKLONNY, PARASKEVI GIANNAKAKOU, MALGORZATA WOJTOWICZ, LARISA Y. ROMANOVA, KENNETH B. AIN, SUSAN E. BATES, AND TITO FOJO Medicine Branch (M.V.B., P.G., M.W., S.B., T.F.) and Laboratory of Genetics (L.Y.R.), National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892; and the Thyroid Cancer Research Laboratory (K.B.A.), Department of Internal Medicine, Veterans Administration Medical Center, University of Kentucky Medical Center, Lexington, Kentucky 40536 ABSTRACT We investigated the p53 status and the ability of exogenous wild- type (wt) p53 to affect chemosensitivity in three anaplastic thyroid carcinoma cell lines (BHT-101, SW-1736, and KAT-4). All three cell lines had nonfunctional p53. Treatment with mitomycin C or adria- mycin did not result in accumulation of p53 or induction of p21 WAF1/CIP1 or Mdm-2 and did not cause Rb dephosphorylation. BHT-101 and KAT-4 cells had mutant p53. SW-1736 cells were func- tionally mutant because of marked down-regulation of wt p53 mes- senger ribonucleic acid, representing a novel mechanism of p53 dys- function. Infection with a p53-expressing adenovirus (Ad-p53) induced high levels of p21 and Mdm-2 proteins. In BHT-101 cells, induction of p21 and Mdm-2 was evident 10 h after infection. In KAT-4 cells, induction of p21 and Mdm-2 was observed 1 day after infection, and continued to increase over the ensuing 24 h. SW-1736 cells dem- onstrated intermediate kinetics. Sensitivity to the cytotoxic effect of Ad-p53 paralleled the kinetics of p21/Mdm-2 induction. BHT-101 cells were most sensitive to killing by Ad-p53, with an IC 50 of less than 2 multiplicity of infection; SW-1736 cells were intermediate in sensi- tivity; KAT-4 cells were resistant. All three cell lines became more sensitive to adriamycin after wt p53 expression, with a 10-fold de- crease in IC 50 values. The latter observation may make a combination of wt p53 and chemotherapeutic drugs an attractive modality for treating anaplastic thyroid cancer. (J Clin Endocrinol Metab 83: 2516 –2522, 1998) A NAPLASTIC thyroid carcinoma, recognized as one of the most aggressive malignant tumors in humans, frequently fails to respond to available chemotherapeutic agents (1, 2). Dedifferentiation of these cancer cells is char- acterized by the absence of expression of thyroid-specific genes (thyroglobulin, thyroid peroxidase, and TSH receptor). This results in their inability to incorporate radioactive io- dine and precludes this tissue-selective modality for treatment. Although the molecular changes responsible for the ag- gressive behavior remain to be elucidated, a consistent ob- servation has been a higher frequency of mutations in the p53 tumor suppressor gene in anaplastic thyroid cancers (3). Thus, in one study using immunohistochemistry, p53 ex- pression was found in 62.5% of undifferentiated carcinomas, but in only 5% of other types of thyroid carcinomas (4); in another report, p53 was detected in 11.1% of well differen- tiated, 40.9% of poorly differentiated, and 63.6% of undif- ferentiated carcinomas (5). As mutations of the p53 gene are associated with the most aggressive histologic types of thy- roid tumors, including anaplastic thyroid cancers, it appears that alterations in p53 represent a late genetic event in human thyroid carcinogenesis (6). Based on these data, it has been proposed that mutations in the p53 gene are responsible for the progression from differentiated into anaplastic carci- noma (7–9). Moreover, it has been demonstrated that intro- duction of wild-type (wt) p53 can induce differentiation in some thyroid cancer cell lines (10, 11). The aggressive clinical behavior of anaplastic thyroid can- cer includes a lack of sensitivity to the majority of available chemotherapeutic agents. Although it is likely this resistance is multifactorial, it may be explained to some extent by the high frequency of p53 mutations, as wt p53 may be important for apoptosis and growth arrest after the administration of DNA-damaging drugs (12, 13). In the present study we report three anaplastic thyroid cancer cells lines with nonfunctional p53 characterized by a lack of induction after exposure to DNA-damaging drugs. To further address the role of p53 in anaplastic thyroid carci- nomas, we employed a p53-expressing adenovirus (Ad-p53). Introduction of wt p53 resulted in the induction of p21 and Mdm-2, confirming that the pathway downstream of p53 was intact. Furthermore, introduction of wt p53 increased sensitivity to adriamycin and cisplatin in all three anaplastic thyroid cancer cell lines. Materials And Methods Cell lines and culture conditions All of the cell lines were derived from primary cultures of human anaplastic thyroid carcinoma tumors. BHT-101 was provided by Istvan Received December 3, 1997. Revision received February 25, 1998. Accepted April 14, 1998. Address all correspondence and requests for reprints to: Dr. Tito Fojo, Medicine Branch, National Institutes of Health, Building 10, Room 12N226, 9000 Rockville Pike, Bethesda, Maryland 20892. E-mail: [email protected]. 0021-972X/98/$03.00/0 Vol. 83, No. 7 Journal of Clinical Endocrinology and Metabolism Printed in U.S.A. Copyright © 1998 by The Endocrine Society 2516 at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.org Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Effects of p53-Expressing Adenovirus on theChemosensitivity and Differentiation of AnaplasticThyroid Cancer Cells
MIKHAIL V. BLAGOSKLONNY, PARASKEVI GIANNAKAKOU,MALGORZATA WOJTOWICZ, LARISA Y. ROMANOVA, KENNETH B. AIN,SUSAN E. BATES, AND TITO FOJO
Medicine Branch (M.V.B., P.G., M.W., S.B., T.F.) and Laboratory of Genetics (L.Y.R.), National CancerInstitute, National Institutes of Health, Bethesda, Maryland 20892; and the Thyroid Cancer ResearchLaboratory (K.B.A.), Department of Internal Medicine, Veterans Administration Medical Center,University of Kentucky Medical Center, Lexington, Kentucky 40536
ABSTRACTWe investigated the p53 status and the ability of exogenous wild-
type (wt) p53 to affect chemosensitivity in three anaplastic thyroidcarcinoma cell lines (BHT-101, SW-1736, and KAT-4). All three celllines had nonfunctional p53. Treatment with mitomycin C or adria-mycin did not result in accumulation of p53 or induction ofp21WAF1/CIP1 or Mdm-2 and did not cause Rb dephosphorylation.BHT-101 and KAT-4 cells had mutant p53. SW-1736 cells were func-tionally mutant because of marked down-regulation of wt p53 mes-senger ribonucleic acid, representing a novel mechanism of p53 dys-function. Infection with a p53-expressing adenovirus (Ad-p53)induced high levels of p21 and Mdm-2 proteins. In BHT-101 cells,induction of p21 and Mdm-2 was evident 10 h after infection. In KAT-4
cells, induction of p21 and Mdm-2 was observed 1 day after infection,and continued to increase over the ensuing 24 h. SW-1736 cells dem-onstrated intermediate kinetics. Sensitivity to the cytotoxic effect ofAd-p53 paralleled the kinetics of p21/Mdm-2 induction. BHT-101 cellswere most sensitive to killing by Ad-p53, with an IC50 of less than 2multiplicity of infection; SW-1736 cells were intermediate in sensi-tivity; KAT-4 cells were resistant. All three cell lines became moresensitive to adriamycin after wt p53 expression, with a 10-fold de-crease in IC50 values. The latter observation may make a combinationof wt p53 and chemotherapeutic drugs an attractive modality fortreating anaplastic thyroid cancer. (J Clin Endocrinol Metab 83:2516–2522, 1998)
ANAPLASTIC thyroid carcinoma, recognized as one ofthe most aggressive malignant tumors in humans,
frequently fails to respond to available chemotherapeuticagents (1, 2). Dedifferentiation of these cancer cells is char-acterized by the absence of expression of thyroid-specificgenes (thyroglobulin, thyroid peroxidase, and TSH receptor).This results in their inability to incorporate radioactive io-dine and precludes this tissue-selective modality fortreatment.
Although the molecular changes responsible for the ag-gressive behavior remain to be elucidated, a consistent ob-servation has been a higher frequency of mutations in the p53tumor suppressor gene in anaplastic thyroid cancers (3).Thus, in one study using immunohistochemistry, p53 ex-pression was found in 62.5% of undifferentiated carcinomas,but in only 5% of other types of thyroid carcinomas (4); inanother report, p53 was detected in 11.1% of well differen-tiated, 40.9% of poorly differentiated, and 63.6% of undif-ferentiated carcinomas (5). As mutations of the p53 gene areassociated with the most aggressive histologic types of thy-roid tumors, including anaplastic thyroid cancers, it appearsthat alterations in p53 represent a late genetic event in human
thyroid carcinogenesis (6). Based on these data, it has beenproposed that mutations in the p53 gene are responsible forthe progression from differentiated into anaplastic carci-noma (7–9). Moreover, it has been demonstrated that intro-duction of wild-type (wt) p53 can induce differentiation insome thyroid cancer cell lines (10, 11).
The aggressive clinical behavior of anaplastic thyroid can-cer includes a lack of sensitivity to the majority of availablechemotherapeutic agents. Although it is likely this resistanceis multifactorial, it may be explained to some extent by thehigh frequency of p53 mutations, as wt p53 may be importantfor apoptosis and growth arrest after the administration ofDNA-damaging drugs (12, 13).
In the present study we report three anaplastic thyroidcancer cells lines with nonfunctional p53 characterized by alack of induction after exposure to DNA-damaging drugs. Tofurther address the role of p53 in anaplastic thyroid carci-nomas, we employed a p53-expressing adenovirus (Ad-p53).Introduction of wt p53 resulted in the induction of p21 andMdm-2, confirming that the pathway downstream of p53was intact. Furthermore, introduction of wt p53 increasedsensitivity to adriamycin and cisplatin in all three anaplasticthyroid cancer cell lines.
Materials And MethodsCell lines and culture conditions
All of the cell lines were derived from primary cultures of humananaplastic thyroid carcinoma tumors. BHT-101 was provided by Istvan
Received December 3, 1997. Revision received February 25, 1998.Accepted April 14, 1998.
Address all correspondence and requests for reprints to: Dr. Tito Fojo,Medicine Branch, National Institutes of Health, Building 10, Room12N226, 9000 Rockville Pike, Bethesda, Maryland 20892. E-mail:[email protected].
0021-972X/98/$03.00/0 Vol. 83, No. 7Journal of Clinical Endocrinology and Metabolism Printed in U.S.A.Copyright © 1998 by The Endocrine Society
2516
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from
Palyi (National Institute of Oncology, Budapest, Hungary). SW-1736was developed by Drs. Leibowitz and McCombs III at the Scott andWhite Memorial Hospital (Temple, TX) in 1977 and was provided byNils-Erik Heldin (Uppsala University, Uppsala, Sweden). KAT-4 wasdeveloped and maintained in one of our laboratories (that of K.B.A.). Thethree cell lines were maintained in RPMI medium containing 10% FBS.
Adenovirus infections
Ad-lacZ, a b-galactosidase expressing replication-deficient adenovi-rus, and Ad-p53, a wild-type p53-expressing replication-deficient ade-novirus, were gifts from Dr. Bert Vogelstein (Johns Hopkins University,Baltimore, MD). Adenovirus titers were determined by plaque forma-tion after infection of 293 cells. The multiplicity of infection (MOI) wasdefined as the ratio of the total number of plaque-forming units used ina particular infection divided by the number of cells. X-Galactosidase
staining of Ad-lacZ-infected tumor cells was performed 1 day afterinfection (14).
PCR amplification and sequence analysis of p53and b-tubulin
RT of 1 mg total ribonucleic acid (RNA) was performed using a primercomplimentary to p53 sequences downstream of exon 9: 59-1022GTTC-CGAGAGCTGAATGAGGC1042-39. PCR amplifications of p53 were per-formed using 2.5 mmol/L MgCl2, an annealing temperature of 55 C for40 cycles, and a primer upstream of exon 5: 59-339TTCTTGCATTCTGG-GACAGCC359-39 together with the primer used in the RT reaction. UsingRNA from SW-1736 cells, a PCR product was not obtained even after 45cycles of PCR amplification. Therefore, for SW-1736 RNA, PCR ampli-fication of p53 was performed using two different sets of primers that
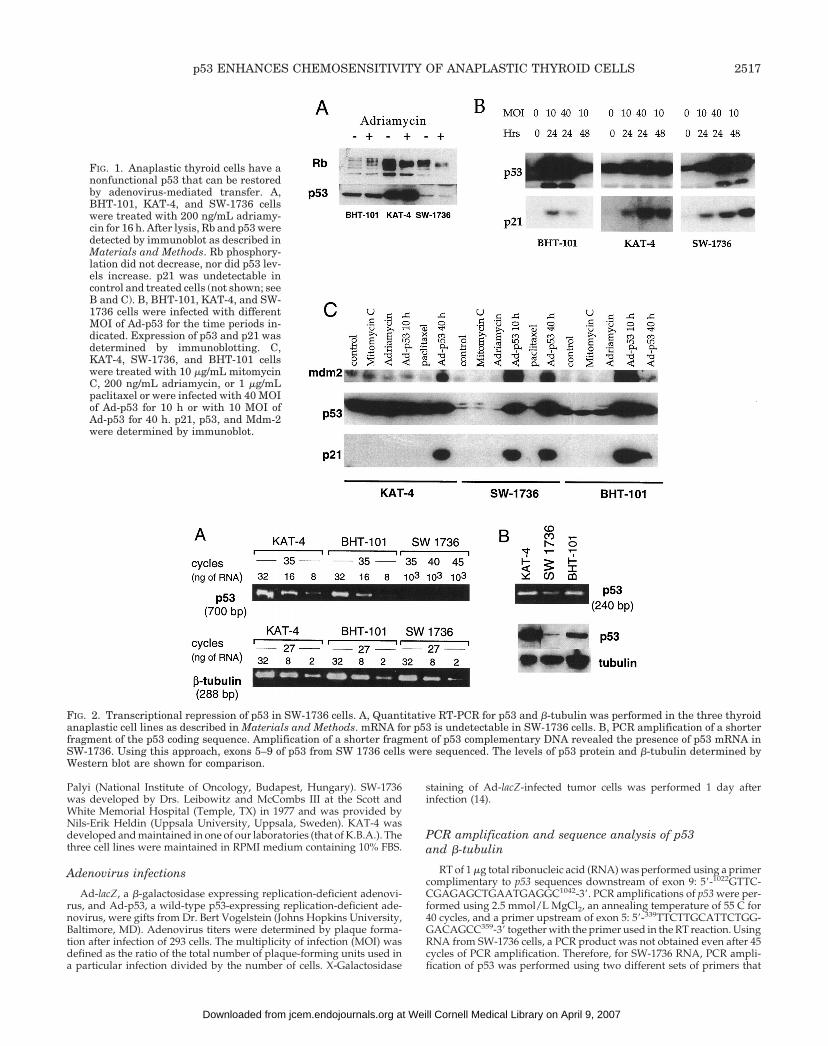
FIG. 1. Anaplastic thyroid cells have anonfunctional p53 that can be restoredby adenovirus-mediated transfer. A,BHT-101, KAT-4, and SW-1736 cellswere treated with 200 ng/mL adriamy-cin for 16 h. After lysis, Rb and p53 weredetected by immunoblot as described inMaterials and Methods. Rb phosphory-lation did not decrease, nor did p53 lev-els increase. p21 was undetectable incontrol and treated cells (not shown; seeB and C). B, BHT-101, KAT-4, and SW-1736 cells were infected with differentMOI of Ad-p53 for the time periods in-dicated. Expression of p53 and p21 wasdetermined by immunoblotting. C,KAT-4, SW-1736, and BHT-101 cellswere treated with 10 mg/mL mitomycinC, 200 ng/mL adriamycin, or 1 mg/mLpaclitaxel or were infected with 40 MOIof Ad-p53 for 10 h or with 10 MOI ofAd-p53 for 40 h. p21, p53, and Mdm-2were determined by immunoblot.
FIG. 2. Transcriptional repression of p53 in SW-1736 cells. A, Quantitative RT-PCR for p53 and b-tubulin was performed in the three thyroidanaplastic cell lines as described in Materials and Methods. mRNA for p53 is undetectable in SW-1736 cells. B, PCR amplification of a shorterfragment of the p53 coding sequence. Amplification of a shorter fragment of p53 complementary DNA revealed the presence of p53 mRNA inSW-1736. Using this approach, exons 5–9 of p53 from SW 1736 cells were sequenced. The levels of p53 protein and b-tubulin determined byWestern blot are shown for comparison.
p53 ENHANCES CHEMOSENSITIVITY OF ANAPLASTIC THYROID CELLS 2517
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from
gave smaller PCR products, allowing for amplification and sequencingof p53 exons 5–9. The primer pairs used in these reactions included: A,the primer corresponding to residues 339–359 as the 59-primer togetherwith a primer complimentary to residues 786–804 as the 39-primer59-786GGTAATCTACTGGGACGGA804-39; and B, a primer correspond-ing to residues 751–770 (751CCATCCTCACCATCATCAC770) as the 59-primer together with the primer complimentary to residues 1022–1042as the 39-primer. PCR product was purified with PCR Select-III spincolumns (5 Prime-3 Prime, Boulder, CO) and directly sequenced with theTaq DyeDeoxy Terminator Cycle Sequencing Kit following the manu-facturer’s instructions (Applied Biosystems, Foster City, CA). Two prim-ers were used for sequencing in addition to the above-described primers:a primer corresponding to residues 569–588 (569CCTCCTCAGCATCT-TATCC588) and a primer complimentary to residues 686–706 (686CTG-TACCACCATCCACTACAA706). The reaction products were purifiedwith Centri-Sep spin purification columns (Princeton Separations, Ad-elphia, NJ), electrophoresed on 48-cm 4.75% polyacrylamide/urea gels,and analyzed by an automated DNA sequencing system (model 377A,Applied Biosystems). PCR amplification of the widely expressed isotypeof b-tubulin M40 was performed as previously described (15).
Immunoblot analysis
Cells plated at a density of 4 3 105/well in six-well plates were treatedwith drugs or infected with adenovirus. After incubation for the indi-cated times, cells were lysed and 20 mg protein were separated byelectrophoresis through 12.5 (p21, p53, and tubulin) or 7.5% SDS-PAGEgels (Rb, Mdm-2, and p53) as previously described (15). Immunoblottingwas performed using mouse antihuman WAF-1 (EA10), p53 (PAb 1801),Rb, and Mdm-2 (Oncogene Science, Cambridge, MA) as previouslydescribed (16).
Growth inhibition and cell viability
Cells were plated at a density of 2500–5000/well in 96-well plates in0.1 mL medium in triplicate. Twenty-four hours later, adenovirus in-fection was performed as described above. Chemotherapeutic drugswere added 1 h after adenovirus infections. Three days later, MTTviability assays were performed (14).
Resultsp53 in anaplastic thyroid cancer cell lines is nonfunctional
Under normal circumstances, wt p53 protein accumulatesafter treatment with DNA-damaging agents and induces thetranscription of target genes, including p21WAF1/CIP1 andmdm-2. Induction of p21, in turn, inactivates cyclin-depen-dent kinases that phosphorylate Rb, resulting in its dephos-phorylation. Therefore, accumulation of p53 protein, induc-tion of p21 and Mdm-2, and dephosphorylation of Rb aremarkers of a functional p53.
The levels of p53 protein were highest in KAT-4 cells, ahallmark of mutant p53; intermediate levels were observedin BHT-101 cells, and very low to undetectable levels werefound in SW-1736 cells (Fig. 1). Furthermore, treatment witheither adriamycin or mitomycin C did not increase p53 levelsor result in detectable p21 or Mdm-2 and did not cause Rbdephosphorylation. Therefore, p53 is functionally inactive inall three cell lines. In agreement with this, sequence analysisrevealed mutant p53 status in two of the three cell lines witha substitution in codon 251 in BHT-101 (ATC3ACC;Ile3Thr) and a substitution in codon 273 in KAT-4(CGT3CAT; Arg3His). A mutation could not be identifiedin RNA from SW-1736 cells, which was found on sequenceanalysis to be wt in exons 5–9. However, as shown in Fig. 2,consistent with the low levels of p53 protein, expression ofp53 was markedly reduced, precluding normal p53 protein
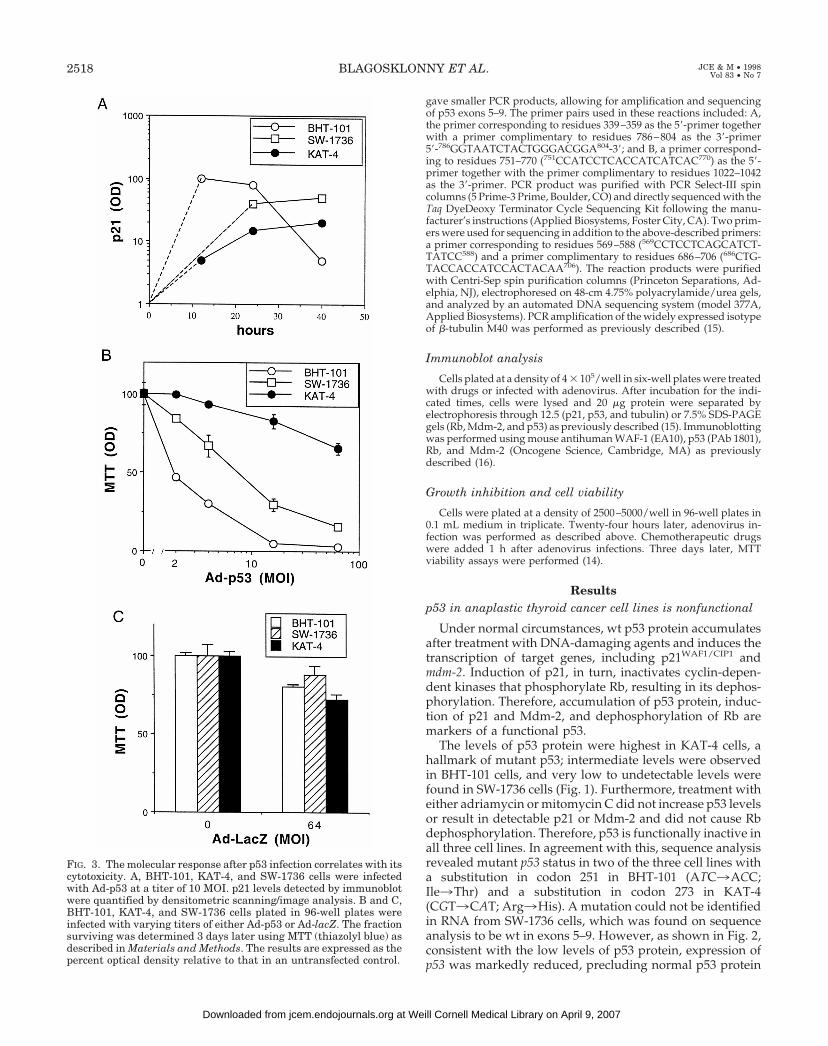
FIG. 3. The molecular response after p53 infection correlates with itscytotoxicity. A, BHT-101, KAT-4, and SW-1736 cells were infectedwith Ad-p53 at a titer of 10 MOI. p21 levels detected by immunoblotwere quantified by densitometric scanning/image analysis. B and C,BHT-101, KAT-4, and SW-1736 cells plated in 96-well plates wereinfected with varying titers of either Ad-p53 or Ad-lacZ. The fractionsurviving was determined 3 days later using MTT (thiazolyl blue) asdescribed in Materials and Methods. The results are expressed as thepercent optical density relative to that in an untransfected control.
2518 BLAGOSKLONNY ET AL. JCE & M • 1998Vol 83 • No 7
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from

expression and function. This pseudo-null p53 status of SW-1736 was functionally equivalent to a p53 null phenotype.
Exogenous wt p53 induces p21 and Mdm-2
Infection of the BHT-101 and SW-1736 cell lines with anadenovirus containing wt p53 (Ad-p53) resulted in rapidaccumulation of p53 protein by 10 h (Fig. 1C). In KAT-4 cells,the increase in p53 protein was not readily apparent becauseof the very high levels of endogenous p53. In all three celllines, introduction of wt p53 resulted in induction of p21 andMdm-2 proteins, indicating that the lack of p21 and Mdm-2induction after DNA-damaging drugs was a consequence ofa nonfunctional p53 and not defective mdm-2 or p21. Induc-tion of p21 reached maximal levels 1 day after infection ofBHT-101 or SW-1736 cells, but not KAT-4 cells (Fig. 3). WithKAT-4 cells it was possible to obtain comparable inductionusing a higher dose of Ad-p53 (40 MOI) or a longer incu-bation time (48 h). Although it has been suggested that de-letion of p21 may be involved in thyroid carcinogenesis (17),in all three cell lines we found defects in p53, but not p21.
Kinetics of p21/Mdm-2 induction determines sensitivityto p53
The differential sensitivity to Ad-p53 cytotoxicity corre-lated with the kinetics of induction of p53-responsive pro-teins. BHT-101 and SW-1736 cells were sensitive to p53. Inthese two cell lines, induction of both p21 and Mdm-2 afterreintroduction of wt p53 occurred within 10 h, and in BHT-101 cells it began to decline by 40 h. In contrast, induction ofp21 in KAT-4 cells was delayed (Fig. 3). Quantitatively, 3days after infection with 2 MOI of Ad p53, only 50% ofBHT-101 cells were alive. SW-1736 cells were also very sen-sitive to Ad p53, with a 50% survival 3 days after infectionwith 8 MOI of Ad p53. The cytotoxicity of Ad-p53 was p53specific, as shown in Fig. 3C, which depicts the lack of asignificant cytotoxic effect of a control Ad-lacZ adenovirus
lacking p53 even at a MOI of 64. In contrast, KAT-4 cells wereas resistant to Ad-p53 as they were to Ad-lacZ.
Resistance of KAT-4 cells to Ad-p53 is associated withlow infectivity
Although the high levels of nonfunctional p53 present inKAT cells could explain the blunted induction of p53-induc-ible proteins, we considered the possibility that, instead, lowinfectivity could explain these observations. The expressionof p53 was not considered a reliable marker because of thevery high levels of endogenous mt p53 in KAT-4 cells. There-fore, we compared the expression of b-galactosidase afterinfection with a b-galactosidase-expressing adenovirus (Ad-lacZ). Expression of Ad-lacZ required a 10-fold higher MOIin KAT-4 cells than in BHT-101 cells (Fig. 4). Nearly allBHT-101 cells were infected at a MOI of 2 (Fig. 4B), withincreased intensity of staining observed with increasingMOI. In contrast, about 10% of KAT-4 cells stained for b-galactosidase at a MOI of 2 (data not shown), whereas themajority of cells were positive only when a MOI of 20 wasused (Fig. 4C). As all KAT-4 cells were infected at a MOIabove 20, we can conclude that KAT-4 cells were refractoryto p53-mediated cytotoxicity.
Expression of wt p53 increases sensitivity to adriamycin
To determine whether wt p53 could enhance drug cyto-toxicity, we infected cells with Ad-p53 and examined drugsensitivity after infection (Fig. 5). Infection with Ad-p53 ren-dered all cells more sensitive to adriamycin; the only differ-ence was the amount of Ad-p53 required for sensitization.The results paralleled the Ad-p53 sensitivity, with enhancedadriamycin cytotoxicity occurring at a MOI of 2 with BHT-101 and SW-1736 cells, but only at a MOI of 64 with KAT-4cells. For example, a MOI of 2 sensitized both BHT-101 andSW-1736 cells, so that the IC50 of adriamycin was approxi-
FIG. 4. The low sensitivity of KAT-4cells to Ad-p53 infection correlates withlow infectivity. BHT-101 and KAT-4cells infected with different MOI of Ad-lacZ were fixed and stained for b-galac-tosidase 30 h later as described in Ma-terials and Methods. A, Photographshowing BHT-101 cells (upper panel)and KAT-4 cells (lower panel) infectedat MOI of 2 and 20, respectively. B andC, Microphotographs comparing b-ga-lactosidase staining of BHT-101 cellsinfected with Ad-lacZ at 2 MOI (B) andof KAT-4 cells infected at 20 MOI (C).
p53 ENHANCES CHEMOSENSITIVITY OF ANAPLASTIC THYROID CELLS 2519
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from
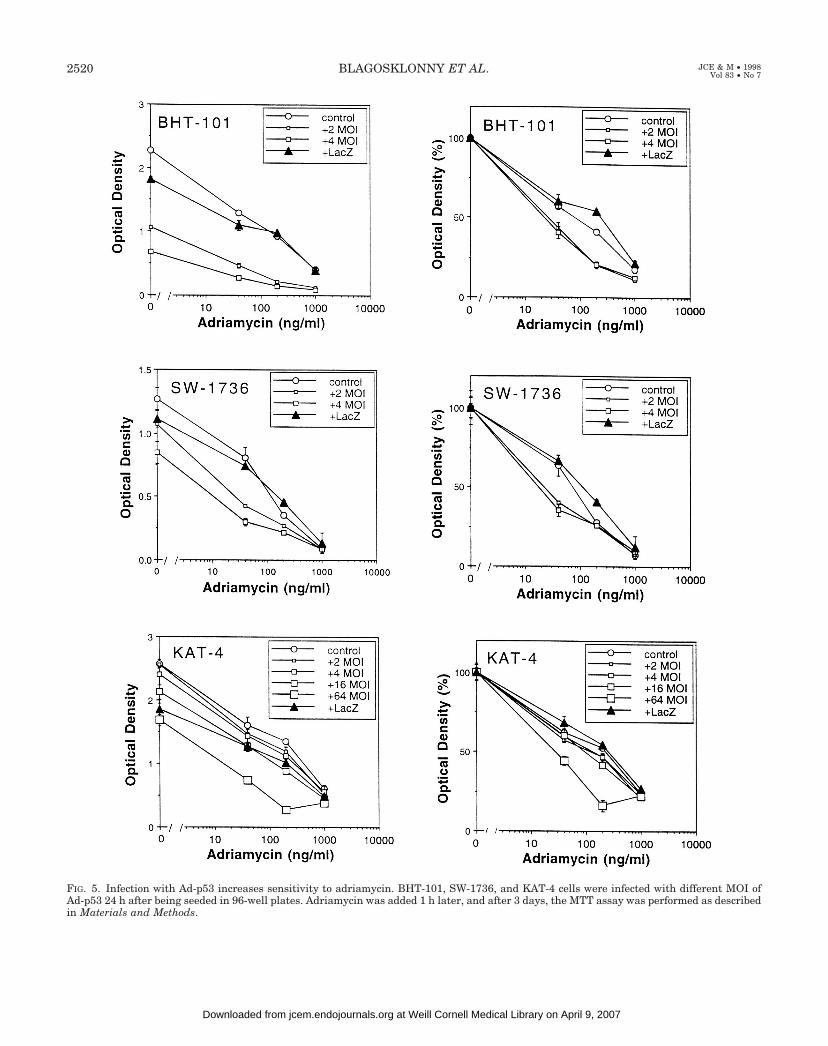
FIG. 5. Infection with Ad-p53 increases sensitivity to adriamycin. BHT-101, SW-1736, and KAT-4 cells were infected with different MOI ofAd-p53 24 h after being seeded in 96-well plates. Adriamycin was added 1 h later, and after 3 days, the MTT assay was performed as describedin Materials and Methods.
2520 BLAGOSKLONNY ET AL. JCE & M • 1998Vol 83 • No 7
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from
mately 20 ng/mL, but a MOI of 64 was required to achievecomparable sensitization with KAT-4 cells.
Morphological alterations of SW-1736 cells by p53and adriamycin
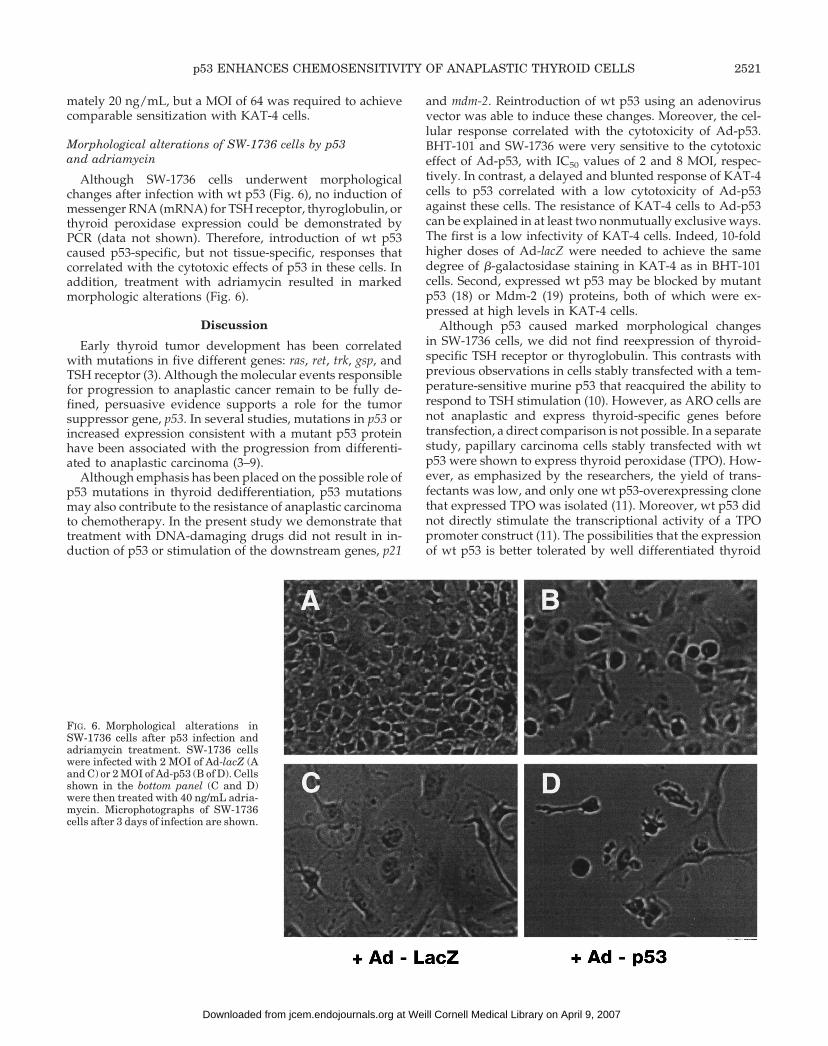
Although SW-1736 cells underwent morphologicalchanges after infection with wt p53 (Fig. 6), no induction ofmessenger RNA (mRNA) for TSH receptor, thyroglobulin, orthyroid peroxidase expression could be demonstrated byPCR (data not shown). Therefore, introduction of wt p53caused p53-specific, but not tissue-specific, responses thatcorrelated with the cytotoxic effects of p53 in these cells. Inaddition, treatment with adriamycin resulted in markedmorphologic alterations (Fig. 6).
Discussion
Early thyroid tumor development has been correlatedwith mutations in five different genes: ras, ret, trk, gsp, andTSH receptor (3). Although the molecular events responsiblefor progression to anaplastic cancer remain to be fully de-fined, persuasive evidence supports a role for the tumorsuppressor gene, p53. In several studies, mutations in p53 orincreased expression consistent with a mutant p53 proteinhave been associated with the progression from differenti-ated to anaplastic carcinoma (3–9).
Although emphasis has been placed on the possible role ofp53 mutations in thyroid dedifferentiation, p53 mutationsmay also contribute to the resistance of anaplastic carcinomato chemotherapy. In the present study we demonstrate thattreatment with DNA-damaging drugs did not result in in-duction of p53 or stimulation of the downstream genes, p21
and mdm-2. Reintroduction of wt p53 using an adenovirusvector was able to induce these changes. Moreover, the cel-lular response correlated with the cytotoxicity of Ad-p53.BHT-101 and SW-1736 were very sensitive to the cytotoxiceffect of Ad-p53, with IC50 values of 2 and 8 MOI, respec-tively. In contrast, a delayed and blunted response of KAT-4cells to p53 correlated with a low cytotoxicity of Ad-p53against these cells. The resistance of KAT-4 cells to Ad-p53can be explained in at least two nonmutually exclusive ways.The first is a low infectivity of KAT-4 cells. Indeed, 10-foldhigher doses of Ad-lacZ were needed to achieve the samedegree of b-galactosidase staining in KAT-4 as in BHT-101cells. Second, expressed wt p53 may be blocked by mutantp53 (18) or Mdm-2 (19) proteins, both of which were ex-pressed at high levels in KAT-4 cells.
Although p53 caused marked morphological changesin SW-1736 cells, we did not find reexpression of thyroid-specific TSH receptor or thyroglobulin. This contrasts withprevious observations in cells stably transfected with a tem-perature-sensitive murine p53 that reacquired the ability torespond to TSH stimulation (10). However, as ARO cells arenot anaplastic and express thyroid-specific genes beforetransfection, a direct comparison is not possible. In a separatestudy, papillary carcinoma cells stably transfected with wtp53 were shown to express thyroid peroxidase (TPO). How-ever, as emphasized by the researchers, the yield of trans-fectants was low, and only one wt p53-overexpressing clonethat expressed TPO was isolated (11). Moreover, wt p53 didnot directly stimulate the transcriptional activity of a TPOpromoter construct (11). The possibilities that the expressionof wt p53 is better tolerated by well differentiated thyroid
FIG. 6. Morphological alterations inSW-1736 cells after p53 infection andadriamycin treatment. SW-1736 cellswere infected with 2 MOI of Ad-lacZ (Aand C) or 2 MOI of Ad-p53 (B of D). Cellsshown in the bottom panel (C and D)were then treated with 40 ng/mL adria-mycin. Microphotographs of SW-1736cells after 3 days of infection are shown.
p53 ENHANCES CHEMOSENSITIVITY OF ANAPLASTIC THYROID CELLS 2521
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from

cells, and that the expression of wt p53 led to the selectionof a well differentiated papillary carcinoma clone cannot beexcluded. Additional studies, consistent with this limitedeffect of wt p53 expression on thyroid differentiation,showed that inactivation of wt p53 by mt p53 did not resultin an anaplastic phenotype, although loss of some tissue-specific markers was reported (3). The results in the presentstudy and the available data suggest that p53 that does notdirectly control the expression of thyroid-specific genes.However, genomic instability secondary to mt p53 may leadto dedifferentiation and thus could explain the clinical ob-servation that a majority of anaplastic thyroid carcinomashave mutant p53. Alternately, loss of wt p53 function couldpermit the growth of rapidly growing anaplastic cells thatwould otherwise be eliminated by wt p53.
Although the problem facing physicians treating anaplas-tic thyroid carcinoma is a systemic dissemination of disease,this tumor often presents as locally advanced disease, re-quiring multimodality therapy (2). Adjuvant chemotherapyand radiotherapy have been used in attempts to enhancelocal control, with only moderate success (2). The presentstudy suggests that although p53 expression may not havea significant impact on the differentiation of anaplastic thy-roid carcinomas, it may have a role in chemosensitivity. Theadvent of therapeutic strategies that target molecular mark-ers such as p53 may provide additional treatment options inpatients with localized anaplastic thyroid carcinoma. Suchapproaches are being actively investigated in head and neckcancers, and have targeted the p53 gene either by attemptingto eradicate cells expressing mutant p53 (20) or by trying torestore normal function (21). Here we showed that expres-sion of wt p53 increased the sensitivity of all three cell linesto adriamycin (10-fold decrease in IC50). Similarly, wt p53increased the sensitivity of KAT-4 cells to cisplatin (notshown). Local administration of Ad-p53 in conjunction withsystemic therapy with DNA-damaging drugs could be usefulin the treatment of locally advanced anaplastic carcinomas.
Finally, we did observe that adriamycin and cisplatin in-duced morphological alterations in SW-1736 cells. A pre-vious study reported conversion of noniodine-concentrat-ing differentiated thyroid carcinoma metastasis into iodine-concentrating foci after anticancer chemotherapy (22). Thepatient presented with metastatic papillary carcinoma andwas treated with cisplatin and doxorubicin. Repeat 131I im-aging after three cycles of chemotherapy showed significant131I uptake in previously noniodine-concentrating lesions.Although these must be regarded as preliminary observa-tions, additional studies are planned to determine whetherthe combination of chemotherapy and wt p53 can causedifferentiation.
References
1. Sweeney PJ, Haraf DJ, Recant W, Kaplan EL, Vokes EE. 1996 Anaplasticcarcinoma of the thyroid. Ann Oncol. 7:739–744.
2. Tan RK, Finley III RK, Driscoll D, Bakamjian V, Hicks Jr WL, Shedd DP.1995 Anaplastic carcinoma of the thyroid: a 24-year experience. Head Neck.17:41–48.
3. Wynford-Thomas D. 1997 Origin and progression of thyroid epithelial tu-mours: cellular and molecular mechanisms. Horm Res. 47:145–157.
4. Pollina L, Pacini F, Fontanini G, Vignati S, Bevilacqua G, Basolo F. 1996 bcl-2,p53 and proliferating cell nuclear antigen expression is related to the degreeof differentiation in thyroid carcinomas. Br J Cancer. 73:139–143.
5. Dobashi Y, Sakamoto A, Sugimura H, et al. 1993 Overexpression of p53 as apossible prognostic factor in human thyroid carcinoma. Am J Surg Pathol.17:375–381.
6. Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA. 1993Gene p53 mutations are restricted to poorly differentiated and undifferentiatedcarcinomas of the thyroid gland. J Clin Invest. 91:1753–1760.
7. Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. 1993 Highprevalence of mutations of the p53 gene in poorly differentiated human thy-roid carcinomas. J Clin Invest. 91:179–184.
8. Ito T, Seyama T, Mizuno T, et al. 1992 Unique association of p53 mutationswith undifferentiated but not with differentiated carcinomas of the thyroidgland. Cancer Res. 52:1369–1371.
9. Nakamura T, Yana I, Kobayashi T, et al. 1992 p53 gene mutations associatedwith anaplastic transformation of human thyroid carcinomas. Jpn J Cancer Res.83:1293–1298.
10. Moretti F, Farsetti A, Soddu S, et al. 1997 p53 re-expression inhibits prolif-eration and restores differentiation of human thyroid anaplastic carcinomacells. Oncogene. 14:729–740.
11. Fagin JA, Tang SH, Zeki K, Di Lauro R, Fusco A, Gonsky R. 1996 Reex-pression of thyroid peroxidase in a derivative of an undifferentiated thyroidcarcinoma cell line by introduction of wild-type p53. Cancer Res. 56:765–771.
12. Lowe SW, Ruley HE, Jacks T, Housman DE. 1993 p53-dependent apoptosismodulates the cytotoxicity of anticancer agents. Cell. 74:957–967.
13. O’Connor PM, Jackman J, Bae I, et al. 1997 Characterization of the p53 tumorsuppressor pathway in cell lines of the National Cancer Institute AnticancerDrug Screen and correlations with growth-inhibitory potency of 123 anticanceragents. Cancer Res. 57:4285–4301.
14. Blagosklonny MV, El-Deiry WS. 1996 In-vitro evaluation of p53-expressingadenovirus as an anti-cancer drug. Int J Cancer. 67:386–392.
15. Giannakakou P, Sackett D, Kang Y-K, et al. 1997 Paclitaxel-resistant humanovarian cancer cells have mutant b-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem. 272:17118–17125.
16. Blagosklonny MV, Toretskey J, Neckers L. 1995 Geldanamycin selectivelydestabilizes and conformationally alters mutated p53. Oncogene. 57:130–135.
17. Shi Y, Zou M, Farid NR, al-Sedairy ST. 1996 Evidence of gene deletion of p21(WAF1/CIP1), a cyclin-dependent protein kinase inhibitor, in thyroid carci-nomas. Br J Cancer. 74:1336–1341.
18. Milner J, Medcalf EA. 1991 Cotranslation of activated mutant p53 with wildtype drives the wild-type p53 protein into the mutant conformation. Cell.65:765–774.
19. Blaydes JP, Gire V, Rowson JM, Wynford-Thomas D. 1997 Tolerance of highlevels of wild-type p53 in transformed epithelial cells dependent on auto-regulation by mdm-2. Oncogene. 14:1859–1868.
20. Kirn DH, Heise G, Mangold G, Von Hoff D. ONYX-015, a selectively rep-licating adenovirus, has antitumoral activity following IV administration aloneand in combination with chemotherapy [Abstract 1564]. Proc of the AmericanSociety of Clinical Oncology. 1997; 437a.
21. Clayman GL, el-Naggar AK, Merritt J, et al. Adenovirus-mediated p53 genetransfer in a phase I trial of patients with advanced recurrent head and necksquamous carcinoma. Proc of the American Society of Clinical Oncology. 1997;383a.
22. Morris JC, Kim CK, Padilla ML, Mechanick JI. 1997 Conversion of non-iodine-concentrating differentiated thyroid carcinoma metastasis into iodine-concentrating foci after anticancer chemotherapy. Thyroid. 7:63–66.
2522 BLAGOSKLONNY ET AL. JCE & M • 1998Vol 83 • No 7
at Weill Cornell Medical Library on April 9, 2007 jcem.endojournals.orgDownloaded from
Related Documents











