Utah State University Utah State University DigitalCommons@USU DigitalCommons@USU All Graduate Theses and Dissertations Graduate Studies 12-2011 Effect of Voluntary Exercise and Diet on the Unfolded Protein Effect of Voluntary Exercise and Diet on the Unfolded Protein Response in the Brain of Mice Response in the Brain of Mice Yu Ho Kim Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/etd Part of the Biology Commons Recommended Citation Recommended Citation Kim, Yu Ho, "Effect of Voluntary Exercise and Diet on the Unfolded Protein Response in the Brain of Mice" (2011). All Graduate Theses and Dissertations. 1114. https://digitalcommons.usu.edu/etd/1114 This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Theses and Dissertations by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected].

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Utah State University Utah State University
DigitalCommons@USU DigitalCommons@USU
All Graduate Theses and Dissertations Graduate Studies
12-2011
Effect of Voluntary Exercise and Diet on the Unfolded Protein Effect of Voluntary Exercise and Diet on the Unfolded Protein
Response in the Brain of Mice Response in the Brain of Mice
Yu Ho Kim Utah State University
Follow this and additional works at: https://digitalcommons.usu.edu/etd
Part of the Biology Commons
Recommended Citation Recommended Citation Kim, Yu Ho, "Effect of Voluntary Exercise and Diet on the Unfolded Protein Response in the Brain of Mice" (2011). All Graduate Theses and Dissertations. 1114. https://digitalcommons.usu.edu/etd/1114
This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Theses and Dissertations by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected].

EFFECT OF VOLUNTARY EXERCISE AND DIET ON THE UNFOLDED PROTEIN
RESPONSE IN THE BRAIN OF MICE
by
Yu Ho Kim
A thesis submitted in partial fulfillment
of the requirements for the degree
of
MASTER OF SCIENCE
in
Biology
Approved:
UTAH STATE UNIVERSITY
Logan, Utah
2011
David A. York, Ph.D.
Major Professor
Tim Gilbertson, Ph.D.
Committee Member
Edward M. Heath, Ph.D.
Committee Member
Ilka Nemere, Ph.D.
Committee Member
MieJung Park-York, Ph.D.
Committee Member
Mark R. McLellan, Ph.D.
Vice President for Research and
Dean of the School of Graduate Studies

ii
ABSTRACT
Effect of Voluntary Exercise and Diet on the Unfolded Protein Response
in the Brain of Mice
by
Yu Ho Kim, Master of Science
Utah State University, 2011
Major Professor: Dr. David A. York
Department: Biology
The Endoplasmic Reticulum (ER) is a net-like intracellular organelle where
protein is folded, matures, and is transported. When cellular stressful circumstances affect
the ER, unfolded proteins are stacked in the ER lumen. This cellular stress is called ER
stress. To defeat ER stress, cells have a defensive mechanism called the Unfolded Protein
Response (UPR). Many chronic diseases such as obesity and type 2 diabetes or
neurodegenerative disease such as Alzheimer’s disease have recently been linked to ER
stress. Exercise has a significant effect on ameliorating the development of these chronic
diseases or neurodegenerative diseases. However, no studies have assessed the effect of
exercise on UPR activity in the brain. So this study was mainly focused on identifying
how voluntary running wheel exercise affects the UPR in the brain of C57BL/6 mice
exposed to a variety of dietary conditions of differing levels of dietary fat and different
periods of feeding. As an exercise protocol, access to a voluntary running wheel for 3
weeks was used and running mice were grouped depending on their level of running

iii
activity. Using real-time PCR and western blotting, UPR-related gene/protein expression
(XBP1, ATF6, eIF2α, and GRP78) was assessed in different brain regions. Exercise had a
significant effect on up-regulating UPR activity in the brain of mice fed low fat diet (LFD)
or high fat diet (HFD) for 3 weeks or 3 months. These effects were time and brain region
dependent. However, the effect of exercise on up-regulating UPR disappeared in mice fed
very high fat diet (VHFD) for 4 months. In addition to assessing UPR activity, the
possibility that exercise-induced UPR activation was associated with activation of
apoptosis was investigated. Apoptotic signaling was not affected by exercise. Trophic
factors are activated by exercise and are known to be linked to UPR activity. The
possibility that IGF-1, one such trophic factor, was responsible for exercise-induced UPR
up-regulation without activating apoptosis was studied. The results showed that IGF-1
was not responsible for exercise-related activation of the UPR in the brain. The chemical
chaperone 4-phenylbutyric acid (PBA) was given to mice to reduce ER stress and the
effect of exercise on the UPR of the brain was studied. PBA had a tendency to lower ER
stress in the hypothalamus. In this condition, exercise had a significant effect to decrease
UPR activity. In conclusion, voluntary exercise activates the UPR in several brain regions
of mice exposed to high-fat diet for up-to 3 months without activating apoptotic signaling.
Only long-term exposure to dietary fat increased the brain UPR. It is possible that this
exercise-induced UPR activation without apoptosis may contribute to the protective
effect of exercise on brain health.
(134 pages)

iv
Public Abstract
Beneficial Effect of Exercise on Regulation of Cellular Stress in the Brain
The medical costs for many chronic diseases are increasing dramatically and placing a
major financial burden on nations and individuals in both developed and developing
countries. A number of chronic diseases, such as obesity, type 2 diabetes and some
neurodegenerative disorders are all attenuated by a history of physical activity suggesting
that they may be interconnected in some way. It has been suggested that cellular stress is
a major factor promoting these chronic diseases.
Cellular stress occurs in a specific compartment within the cell, the endoplasmic
reticulum, whose normal function is in the synthesis and folding of proteins into the
correct 3 dimensional structure. Cells have a defensive mechanism to protect against this
cellular stress that is known as the unfolded protein response (UPR). This involves the
activation and/or inhibition of various genes that reduce protein synthesis and increase
folding capacity.
With the support of USTAR (The Utah Science Technology and Research program), Yu
Ho Kim, a Masters student in Dr. York’s research group in the Center for Advanced
Nutrition & the Department of Biology at Utah State University, studied how exercise
affects brain health. The hypothesis was that exercise increased the activity of the UPR to
protect the brain from cellular stress. The experimental model used were mice allowed to
have free access to running wheels for 3 weeks in their cages while fed with either low
fat or high fat diets.
The results of this study confirmed the hypothesis that physical activity increased the
activity of the unfolded protein response in multiple regions of the brain of mice
suggesting that this mechanism may be, in part at least, responsible for the protective
effects of exercise on some neurodegenerative diseases. Future work to identify the
exercise-related signal that enhances the UPR mechanism in the brain may be helpful in
the future treatment of neurodegenerative disorders such as Alzheimer’s disease.

v
ACKNOWLEDGMENTS
I would like to thank Dr. David A. York for helping me to complete my graduate
work and this thesis. I also thank other lab members, Drs. MieJung Park-York and
Stéphane Boghossian, for their experimental support. I would especially like to thank
Hyoung-il Oh, PhD candidate, who was my colleague and a good friend. I would like to
give my thanks to my graduate committee members, Drs. Tim Gilbertson, Edward M.
Heath, and Ilka Nemere, for their sincere assistance from first step to last step.
I always thank my family. Without their support and patience, I could not have
completed my graduate work.
YU HO KIM

vi
CONTENTS
Page
ABSTRACT ...…………………………………………………………….….…………. ii
PUBLIC ABSTRACT ...………………………………………………...……...……..... iv
ACKNOWLEDGMENTS ..………………………………………………..…………..... v
LIST OF TABLES …………..……………………………………………..….…...…..... x
LIST OF FIGURES …………………………..……………………………........…….... xi
CHAPTER
1. INTRODUCTION ………………………………………………………………….. 1
EXERCISE IN CHRONIC DISEASE AND AD …………………………….……. 1
ER STRESS AND UPR MECHANISM ……………….……………………… ….. 3
THE UNFOLDED PROTEIN RESPONSE (UPR) IN OBESITY.…….…………… 6
THE UNFOLDED PROTEIN RESPONSE (UPR) IN ALZHEIMER’S
DISEASE (AD)……………………………………………………….…………8
APOTOSIS …..…………………..………………………………….….…………. 9
ER STRESS-SPECIFIC APOPTOTIC MECHANISM …………….……............... 12
REFERENCES ……………….……………………...….………………………… 14
2. The effect of exercise and diet on the unfolded protein response (UPR) in the
brain of mice …………………………………………..………………………….... 28
INTRODUCTION……………..……………………………………………………. 28
HYPOTHESES……………..………………………………………………………. 33
METHODS……………..……………………………………….…………………. 33
Animals and Diets …………………………………………………………...…. 33
Body Composition ………….………………………….…………………..... 34
Voluntary Running Wheel Exercise ……………...……………………..…….. 34
RNA Isolation and Purification ……………………………..….……………… 36
Quantitative Real-time PCR ……..…..…………………………..………….. 36
Protein Extraction ………………….……..…………………………….…..... 37
Western Blotting ………….……..…..…………………….……..………….. 38
Statistics ………….…………………..…………………………..….……....... 39
RESULTS …………………..……………………………………….……………. 39
The Effect of Very High Fat Diet and Exercise on UPR in the Brain of Mice .. 39
Voluntary running wheel exercise ………………………………….…. 39
The effect of exercise on body weight ….…………….…………….... 40

vii
The effect of exercise on food intake …………...………………….….. 40
The effect of exercise on UPR in the brain ………….………………. 41
The Effect of Exercise on UPR in the Brain of Mice Fed LFD
or HFD for 3 Weeks ……………………………………………………….…. 41
Voluntary running wheel exercise ………..……………………………. 41
The effect of exercise on body weight and body composition ………... 42
The effect of exercise on food intake ………….………………….….. 43
Gene and protein expression in the brain ………………….…………… 44
The effect of exercise in mice fed LFD for 3 weeks ……….... 44
The effect of exercise in mice fed HFD for 3 weeks…………… 45
The effect of exercise on apoptotic signaling in the brain …………… 46
The effect of 3 weeks dietary (LFD/HFD) treatment
on UPR in brain …………………………………………………..…. ...47
The Effect of 3 weeks Exercise on UPR in the Brain of Mice Adapted
to Low or High Fat Diet for 3 Months ………………......….…………………. 47
Voluntary running wheel exercise ………..………………………...…. 47
The effect of exercise on body weight …………………...…………..... 48
The effect of exercise on food intake …………...………………….…. 49
Gene and protein expression in the brain ………………….………….. 49
The effect of exercise in mice fed LFD during 3 months …… 49
The effect of exercise in mice fed HFD during 3 months …… 50
The effect of exercise on apoptotic signaling in the brain …………… 50
The effect of diet on apoptotic signaling in the brain …….....….….….. 50
The effect of prolonged feeding of high fat diet on UPR
in the brain …………………………………….……….................. 51
DISCUSSION …...…………..……………………………………………………. 51
Effect of Diet and Age on Voluntary Running Ability ……………….…. 52
Effect of Exercise on Energy Balance ……...………………………..……. 53
Effect of Diet and Exercise on Food Intake ……………...…………….…. 54
Effect of Exercise on UPR ……….…...………………………...….……… 54
Relationship of Exercise Induced Change in the UPR to Apoptosis .……… 58
Exercise and Neurodegenerative Disease …………………………………. 60
REFERENCES …...……………………………………………………………….. 60
3. The effect of IGF-1 and exercise on UPR activated by voluntary running wheel
exercise mice ………………………………………………………………….... 88
INTRODUCTION …………………..……………………………………………. 88
HYPOTHESES …………………..……………………..…………………………. 91
METHODS ………………..………………………..………………………………. 91

viii
Animals and Diets …………………………………………………………...…. 91
Voluntary Running Wheel Exercise ……………...…………………….……. 92
Osmotic Minipump …………...…………………………………..………….. 92
RNA Isolation and Purification ………...…………………………………….. 93
Quantitative Real-time PCR …………...……..……………………………...... 93
Statistics ………….…………………..……..……………………….……...... 94
RESULTS …………………..…………………………………………………….. 94
Voluntary Running Wheel Exercise ………..………………………………. 94
The Effect of Exercise and Anti-IGF-1 on Change of Body Weight …………. 95
The Effect of Exercise and Anti-IGF-1 on Expression of UPR-related Genes in
the Brain ..…………………………………………………………….……….... 95
The Effect of Exercise and Anti-IGF-1 on Expression of IGF-1 and BDNF in
the Brain ...…………………………………………………………..……….... 96
The Effect of Exercise and Anti-IGF-1 on UPR and IGF-1 Gene Expression
in the Liver ...……………………………...………………………...……….... 96
DISCUSSION ………………..……………………………………………………. 97
REFERENCES ……………..…………………………..……………………….. 101
4. The effect of 4-phenyl butyric acid (PBA) on UPR activated by voluntary
running exercise ………………………………………………………………………. 111
INTRODUCTION …………………..……………………………..……………. 111
HYPOTHESES …………………..………………..…………………………..…. 113
METHODS ……………..……………………………………….…………..……. 113
Animals and Diets …………………………………………………………….. 113
4-Phenylbutyric acid (PBA)………………………………………………….. 113
Voluntary Running Wheel Exercise ………………...…………………..…… 114
RNA Isolation and Purification ………...…………………………..……...…. 114
Quantitative Real-time PCR …………..……..……………………….……..... 114
Statistics ………….…………………..……..……………..………….…….... 115
RESULTS …………………..………………………………………………...…. 115
Voluntary Running Wheel Exercise ………..……………………………….. 115
The Effect of Exercise and PBA on Change of Body Weight ….…….......….. 115
The Effect of Exercise and PBA on Food Intake ……………………..……. 116
The Effect of Exercise and PBA on Drinking ……….………………..……. 116
The Effect of Exercise and PBA on UPR-related Genes Expression
in the Brain ……………………………………………………………..…….. 116
DISCUSSION ……...……………..………………………………………..……. 117
REFERENCES ……...…………..…………….………………..………………. 120
5. OVERALL DISCUSSION AND FUTURE DIRECTIONS …………………….... 127

ix
The regional response …………….……………………………………….. 127
The time-dependent effects of diet and exercise …..……….……………….... 129
Possible implications in neurodegenerative disease ………………………..... 130
Future works ……………………………………………………….….……… 131
References ……………………………………………………...….……………… 132

x
LIST OF TABLES
Table Page
1 Compositions of experimental diet chows ……..………………….…………………. 35

xi
LIST OF FIGURES
Figure Page
1-1 The Unfolded Protein Response (UPR) pathways………….…..…………………… 5
1-2 ER stress activated apoptosis signaling pathway ……………….………………... 12
2-1 The voluntary running wheel ability of mice adapted to 60% HFD for
4 months …………………………………………………………..………………. 68
2-2 The effect of exercise on body weight …………………………………………….. 69
2-3 The effect of exercise on food intake ……………………..……………………… 70
2-4 The effects of three weeks voluntary running wheel exercise on UPR related gene
expressions in the brain of mice adapted to 60% HFD for 4 months …..………. 71
2-5 The effects of three weeks voluntary running wheel exercise on UPR related
protein expressions in the hippocampus of mice adapted to 60% HFD for
4 months …………………………………………………………………..……… 72
2-6 Voluntary running wheel activity of mice fed LFD or HFD for 3 weeks …...….…. 73
2-7 Effect of diet and exercise on body weight and body composition .….……..…….. 74
2-8 The daily caloric intake and cumulated caloric intake of LFD and HFD groups.. 75
2-9 The effect of voluntary running wheel exercise on UPR related gene expressions in
multiple brain regions and liver of mice fed with LFD and HFD for 3 weeks ... 76
2-10 The UPR related protein expression in hippocampus of mice fed LFD or HFD ... 77
2-11 The effect of running exercise on apoptotic protein expression in hippocampus of
mice fed LFD or HFD …………………………….……………………………. 78
2-12 The effect of diet on UPR related gene expressions in multiple brain regions and
liver …………………………………………………………………...………….. 79
2-13 The voluntary running wheel activity of mice adapted to LFD/HFD
for 3 months ……………………………………………………………………..80
2-14 The effect of diet and exercise on body weight ……………………………….. 81
2-15 The effect of diet and exercise on daily and cumulative food intake …..…….. 82

xii
2-16 The effect of exercise on UPR related gene expression in the brain of mice fed LFD
and HFD for 3 months …………………………………………………………… 83
2-17 The effect of exercise on UPR related protein expressions in the hippocampus of
mice fed LFD or HFD during 3 months ………………………….……………... 84
2-18 The effect of exercise on apoptotic related protein expressions in the hippocampus
of mice fed LFD or HFD during 3 months ……………………………………... 85
2-19 The effects of diet on apoptotic related protein expression in the hippocampus . 86
2-20 The effects of 3 months LFD/HFD on UPR-related gene expressions in the brain
…………………………………………………………………………………... 87
3-1 Voluntary running wheel activity in mice treated with IGF-1 antibody ……….. 106
3-2 The effect of exercise and anti IGF-1 on change of body weight ……………… 107
3-3 The effect of exercise and anti IGF-1 on the UPR-related gene expressions in the
brain ……………………………………………………..………………………. 108
3-4 The effect of exercise and anti IGF-1 on IGF-1 and BDNF gene expressions in the
brain ……………………………………………………………………………. 109
3-5 The effect of exercise and anti IGF-1 on UPR-related and IGF-1 gene expressions in
the liver ………………………………………………………………………… 110
4-1 The activity of voluntary running wheel exercise ……………………………… 122
4-2 The effect of exercise and PBA on change of body weight ……………………. 123
4-3 The effect of exercise on the daily food intake in mice treated with PBA ……….. 124
4-4 The effect of exercise and PBA on drinking behavior …………………………. 125
4-5 The effect of PBA and exercise on expression of UPR genes in the hypothalamus and
hippocampus …………………………………………………………………….. 126

CHAPTER 1
INTRODUCTION
EXERCISE IN CHRONIC DISEASES
It has been well established that exercise has significant effects on ameliorating
the development of a variety of chronic diseases such as obesity, type 2 diabetes,
cardiovascular disease as well as in Alzheimer’s disease (AD). In regulating energy
balance, exercise has an important effect by increasing energy expenditure and the resting
metabolic rate (RMR) to maintain energy homeostasis.1-3
This improved metabolic rate,
in turn, ameliorates the onset and/or development of obesity in humans and in rodents. In
addition, many studies have shown that exercise has a positive effect on improving type 2
diabetic symptoms by improving insulin sensitivity.4-10
The risk of developing type 2
diabetes can be inferred by low physical fitness and VO2 max.11
In addition, exercise
activates the expression of the GLUT 4 gene promoting the uptake of glucose into
skeletal muscle.12-13
Habitual physical activity, especially aerobic exercise, improves learning ability
and the plasticity of the brain in humans, especially in the aged.14-17
Attention to the
effects of exercise on cognitive function has recently increased and studies show that
exercise slowed the progress of cognitive decline and decreased the risk of
neurodegenerative disease such as AD.18-20
Though the mechanisms for the effect of
exercise were not well elucidated, there have been a lot of studies showing that exercise
has a strong effect on slowing the progress of neurodegenerative diseases. In recent
studies using an AD transgenic model mouse, exercise not only increased cognitive
function but also ameliorated the characteristics of AD (i.e., reduced expression of Aβ-42)

2
in the brain suggesting that exercise has the important potential of preventing the
development of neurodegenerative disease such as AD.21-24
One interesting result
suggested that exercise has the biggest effect on executive function rather than other tasks
such as controlled, spatial, and speed works.14
Exercise training appeared to improve the
function of learning and memory in rats tested in a water maze or 8-arm radial maze.25-27
It was also shown that physical activity had a significant effect on cognitive function
along with academic achievement in young-people.28-30
Epidemiological studies of physical activity and neurodegenerative disease risk
have been conducted with the purpose of showing the effect of exercise.14, 31-33
One study
showed that physical activity was more effective on AD (i.e., reduction of risk by 45%)
than on dementia (28% reduction) and Parkinson’s disease (no effect).31
Many possible
neuroprotective mechanisms could be induced by physical activity including improved
vascular health, reduced obesity and type 2 diabetes, improved immunological status, and
reduced hypercholesterolemia.34-35
High-technological neuroimaging such as functional
magnetic resonance imaging (fMRI), positron emission tomography,36
and optical
imaging are vigorously being developed and applied to identify the complex linkages
between physical activity and human cognition.19
Rodent models have been used to assess the comprehensive mechanisms of brain
health following exercise. In such studies, it was shown that brain-derived neurotrophic
factor (BDNF) 37-40
and nerve growth factor (NGF) 41
are significantly elevated by
physical activity in multiple brain regions of animals. Animals with high running activity
have a significantly increased expression of BDNF in the hippocampus of brain.42-43
In
the central nervous system, BDNF promotes neuronal plasticity by specific actions on

3
axonal and dendritic remodeling.44-47
BDNF is processed from pro-BDNF and functions
by binding to the trkB receptor, mediating synaptic transmission and cognitive
control.48-49
Taken together, these data show that exercise has a positive effect in that it
improves or decelerates the development of chronic diseases such as obesity and type 2
diabetes and that it has a neuroprotective effect on neurodegenerative diseases including
AD.
ER STRESS AND UPR MECHANISM
The endoplasmic reticulum (ER) is an intra-cellular organelle where many
proteins destined to be secreted or to be membrane components are folded into proper
tertiary structures.50
Once proteins are properly shaped, their hydrophobic amino acid
residues are aggregated inside the entire protein structure. On the other hand, the
presence of misfolded proteins generates the ubiquitin-proteaosome pathway through
which misfolded proteins are covalently bound with the ubiquitin, leading to degradation
in the proteasome.51
Along with folding ability of the ER, cholesterol and many lipids are
synthesized in this cellular organelle.52-53
However, cellular stressful environments such
as glucose deprivation, perturbation of calcium homeostasis and viral infection, can
hinder the role of ER leading to a build-up of unfolded proteins in the ER lumen. This
pathological cellular stress is called ER stress and cells have a defensive mechanism
defined as the unfolded protein response (UPR) in order to counteract to this cellular
stress.54
The overall mechanism of the UPR is depicted in Figure 1-1. There are three
major arms to the UPR mechanism and each is located in the ER lumen; Inositol-
requiring protein 1 (IRE-1), Active Transcription Factor 6 (ATF-6) and PKR-like ER

4
Protein Kinase (PERK).58
In normal cellular status, these three arms are inactivated
through binding with the ER chaperone immunoglobulin binding protein/glucose
response protein 78 (Bip/GRP78). When unfolded proteins are stacked in the ER and ER
stress is aroused, Bip/GRP78 is released from these three regulatory proteins, converting
these UPR molecules into their active forms.59-60
Released Bip/GRP78 binds to and
enhances folding of the unfolded proteins in ER lumen, decreasing the number of
unfolded proteins. Once activated by dissociation of Bip/GRP78, IRE-1 forms a dimer,
leading to autophosphorylation in the cytosolic kinase domain.61
This phosphorylated
kinase acts as an endoribonuclease, a site-specific Ser/Thr protein kinase. Activated IRE-
1 eliminates a 26-nucleotide intron of XBP1 mRNA, releasing spliced XBP1 (X-box
binding protein 1; XBP1s). This frame-shift XBP1 is translocated into the nucleus where
it binds to UPR elements (UPRE) and functions as a transcription factor which up-
regulates genes for glycosylation proteins, disulphide bond proteins,62-65
and genes for
elements of the ER associated degradation 13
by which misfolded proteins are recognized
and transferred to cytosol and degraded by the proteasome.66-67
Upon ER stress, ATF6,
the second arm of UPR, is transported from the ER to the Golgi apparatus by unknown
mechanism where its N-terminal cytoplasmic domain is cleaved by the Site 1 protease
(S1P) and Site 2 protease (S2P). This cleaved ATF6 is translocated into the nucleus and
functions as a transcription factor by binding to the ER stress response elements (ERSE),
increasing the expression of UPR responsive genes including Bip/GRP78,
CCAAT/enhancer-binding protein homologous protein (CHOP), and XBP1.68-71

5
Figure 1-1 The Unfolded Protein Response (UPR) pathways. IRE-1, Inositol-requiring
protein 1; ATF-6, Active transcription factor 6; PERK, PKR-like ER protein kinase;
Bip/GRP78, immunoglobulin binding protein/glucose response protein 78; eIF2α,
eukaryotic translation initiation factor 2α; S1P/S2P, Site 1 protease/Site 2 protease;
UPRE, UPR elements; ERSE, ER stress response element; ERAD, ER-associated
degradation.55-57

6
Along with these two UPR mechanisms, another UPR arm, PERK, affects the
translation rate, reducing the synthesis of proteins and reducing the stress on the ER.
PERK is protein kinase and ER stress activates PERK by phosphorylation. This activated
form of PERK, in turn, phsophorylates eukaryotic translation initiation factor 2α
(eIF2α).72
Because eIF2α is required in the process of binding the initiator methionyl-
tRNA to the small ribosomal subunit, phosphorylated eIF2α no longer binds to the 80S
ribosome, lowering the translational initiation events and eventual protein synthesis. It
was shown that this cellular strategy of translational repression contributes to overall
mRNA stabilization.73
In summary, activated UPR increases the unfolded protein folding capacity along
with decreasing the burden of new protein synthesis on the ER and these mechanisms
eventually contribute to release the ER stress.
THE UNFOLDED PROTEIN RESPONSE (UPR) IN OBESITY
As societies have become westernized, the dietary habits of people all over the
world have changed to the western diet, a high-calorie diet, and this dietary environment
has resulted in an increased prevalence of obesity. Over the last several decades, the
interest in obesity has increased. Many researchers have suggested the possibility that
obesity could be induced by cellular stress signaling and inflammation, but have been
unable to identify what is the exact origin for obesity.74-77
Overnutrition leads to lipid accumulation in nonadipose tissues such as liver,
pancreas, and muscles 78
since fatty acids, triglycerides, and cholesterol are exogenously
taken up.79
The buildup of intracellular lipids results in an increasing level of free fatty
acids (FFA) and other lipids within the tissues which are harmful to intracellular

7
organelles such as ER and mitochondria due to the vulnerability of FFA to oxidative
damage to produce reactive lipid peroxides.80-82
When the ER is overly exposed to FFA
and lipid peroxides, this, in turn, causes structural changes in the ER and unfolded
proteins are eventually accumulated in the ER, leading to the upregulation of UPR.83-84
Hotamisligil’s research group (2004) suggested that ER stress could be a main reason for
obesity and type 2 diabetes. They showed that both obesity-induced by dietary conditions
(High-Fat diet) and genetically obese mice (ob/ob mice) induced the activation of UPR in
liver. In addition, in the UPR related gene depleted model (Xbp1-/-
), UPR responded to
the ER stress inducer tunicamycin and this overexpression led to the impairment of
glucose homeostasis and insulin signaling.85
His group also showed that increased ER
stress in the hypothalamus of obese mice led to leptin resistance.86
They showed that
brain tissue specific UPR gene knock-out led to both obesity and increased leptin levels
after feeding a high-fat diet.
Recently, several researchers have suggested that ER stress could be the link
between obesity and inflammation.87-90
Zhang et al. (2008) suggested that hypothalamic
ER stress induced by high-fat diet was linked to inflammation (IkappaB kinase β/Nuclear
Factor-KappaB [IKKβ/NF-κB]) which led to energy imbalance and obesity.89
IKKβ/NF-
κB is the main switch for the control of intrinsic immune actions 91
. In normal states NF-
κB is inactive through binding with the inhibitory protein IκB, but when activated IKKβ
phosphorylates its substrate IκB to produce an activated form. This activated IκB releases
NF-κB which is translocated into nucleus where it acts as a transcription factor for other
inflammatory actions.89
Viral vector mediated IKKβ deletion in the mediobasal
hypothalamus (MBH), a main region of sensing nutritional status, blocked the effect of

8
IKKβ and reduce the risk of obesity in high-fat diet conditions without increase of ER
stress.89
On the other hand, there have been experimental trials to reduce the ER stress
through applying a chemical chaperon, 4-phenyl butyric acid (PBA).86, 92-93
Oral
treatment with PBA significantly decreased the level of both serum leptin and glucose in
the high-fat dietary condition.86
PBA also reversed the hyperglycemia and improved the
insulin sensitivity in ob/ob mice.93
These results revealed that increased ER stress could
induce obesity as well as type 2 diabetes.
To summarize, overnutrition inducible obesity can be linked with ER stress when
this cellular event leads to activation of the UPR as a defensive mechanism.
THE UNFOLDED PROTEIN RRESPONSE (UPR)
IN ALZHEIMER’S DISEASE (AD)
Alzheimer’s disease (AD) is one of the neurodegenerative diseases and is
characterized by progressive decline of cognitive function. AD is also characterized by
intracellular accumulation of tau protein into neurofibrillary tangles (NFT), and by
extracellular aggregation of amyloid β (Aβ) protein, which forms senile plaques that are
known to be neurotoxic.94
While in health brain β-amyloid precursor protein 92
is
processed by proteases such as α-, β-, and γ-secretases, mutations at the cleavage site of
APP promote the accumulation of Aβ.95-97
Within the last decade, the UPR has been in the spotlight, as it was suggested that
it might be involved in the underlying pathological causes of AD.98-100
One study showed
that Bip/GRP78 chaperone in ER was bound to and enforced APP to be folded correctly,
lowering the production of Aβ.101
In addition, when PERK activity was knock-downed
by application of the PERK siRNA, it was shown that Aβ treatment increased neurotoxity
in vitro 102
due to the destruction of UPR mechanism. Mutations of the presenilin genes

9
(PS1 and PS2) appeared to be the main reason for causing an early onset AD and these
proteins are usually found in ER.103
It was shown that PS1 takes part in UPR activation
and the activation of IRE-1 is controlled by PS1.104
In cells expressing mutant PS1,
mRNA expression level of Bip/GRP78 was significantly decreased.105
In addition, this
mutation of PS1 also decreased the activities of all UPR related arms (PERK, IRE1, and
ATF6).106-107
Loewen and Feany (2010) published their experimental results showing that
upregulated UPR, especially XBP-1, ameliorated the neurotoxicity of tau using
genetically-modified Drosophila.108
Recently, it was shown that calcium homeostasis could be linked to AD.109-111
The
accumulation of Aβ hinders calcium influx at the plasma membrane or ER membrane,
and when calcium homeostasis is impaired, UPR is activated.112-113
In humans, UPR
activity is increased in the brain of AD patients.100, 114
Using immunohistochemistry for
localization of pPERK, peIF2α, and pIRE-1 in the hippocampus of AD patients, it was
shown that these UPR related proteins were expressed in neurodegenerative disease of
human and that the upregulation of PERK was accompanied with phosphorylation of
tau.99-100
To summarize, it is possible that the UPR is linked to the etiology of AD and we
propose that controlling the homeostasis of UPR may lead to the neuroprotective effect of
exercise.
APOPTOSIS
All multicellular animals retain a balance between cell division and cell death,
maintaining the number and size of cells. This tightly controlled normal cell death is

10
called “programmed cell death” or Apoptosis, a term first used by Currie and colleagues
in 1972.115
Overall apoptosis mechanism is depicted in Figure 1-2.
Apoptosis is characterized by its specific morphological changes which are
usually induced by cysteine proteases, one of a protein family known as the caspases
which cleave substrates specifically at Asp-Xxx bonds (i.e., aspartic acid residues).116
Several important substrates for caspase actions have been identified. The DNA nuclease
was firstly identified and shown that it was cleaved and activated by caspase.117
Activated
nuclease cleaves the DNA fragments into shortened DNA fragment of about 180 base
pairs. This DNA ladder is used as a marker of apoptotic cell death.118
It was confirmed
that this DNA ladder nuclease is a caspase-activated DNase, or CAD and it is inactivated
in the normal living cells by binding with an inhibitory subunit (ICAD).119
Activated
caspase-3 cuts the cleavage site of the inhibitory subunit, leading to its release from
nuclease which, in turn, is activated.120-122
Alteration of apoptotic cellular structures is usually followed by caspase action
and it typically occurs at nuclear lamina which have a role as supportive structures for the
nuclear membrane.123-124
Caspases cleave lamina into fragments, resulting in the
destruction of lamina and possible damage to the chromatin structure.125
Caspases are usually activated by three representative mechanisms; caspase
cascade, proximal induction, and holoenzyme formation. In the caspase cascade, an
activation of an initiator caspase delivers a proapoptotic signal which sequentially turns
on effector caspases, causing apoptosis.126
Each initiator caspase has a distinct role for
mediating a proapoptotic signal. For instance, caspase-8 leads to apoptosis related with
death receptors 127
while caspase-9 delivers the signal induced by cytotoxic agents.128-129

11
In addition, it was also shown that specific cofactors are necessary for activation of
initiator caspases. For example, the cofactor Fas-associated protein with death domain
(FADD) is required for the procaspase-8 activation 36, 130
and procaspase-9 is activated by
a complex composed of cofactor called apoptotic protease activating factor-1 (Apaf-1)
with the caspase recruitment domain.8 131
For the case of induced proximity, when CD95
ligand binds to CD95 (the death receptor superfamily), CD95 forms a receptor cluster,
developing the death-inducible signaling complex. This complex, in turn, combines with
procaspase-8 molecules, resulting in the activation of procaspase-8. As mentioned, Apaf-
1 could be not only an activator for caspase-9 but also a necessary subunit for a caspase-9
holoenzyme (also called apoptosome).132
Taken together, initiator caspases are usually
activated by protein-protein interactions while effector caspases are turned on by the
action of an upstream caspase.
Mitochondria are affected by apoptotic death signals.133
It was suggested that the
Bcl-2 family contributes to control the mitochondria homeostasis. Once mitochondria are
damaged by apoptosis, they release cytochrome c, and this combines with Apaf-1 to form
a complex known as apoptosome.134
This complex appears to activate procaspase-9,
which, in turn, activates caspase-3, the main effector caspase.126, 135
It was shown that
Bcl-2 family takes part in this mechanism related to the cytochrome c activation. Bcl-2
proteins seem to be aggregated on the outer mitochondrial membrane where channels
were formed.136
In summary, apoptosis is delicately controlled both catalytically and structurally.
By this complex mechanism, cellular death is controlled and programmed.

12
Figure 1-2 ER stress activated apoptosis signaling pathway. TRAF2, TNF receptor
associated factor-2; CAD, caspase-activated DNase; ICAD, inhibitor of CAD; FADD,
Fas-Associated protein with Death Domain; JNK, Jun N-terminal inhibitory kinase; ASK,
apoptosis signaling kinase; JNK, c-Jun N-terminal kinase; CHOP, CCAAT/enhancer
binding protein (C/EBP); GADD 34, growth arrest and DNA damage gene 34.56, 59, 118, 137-
138
ER STRESS-SPECIFIC APOPTOTIC MECHANISM
If the activated UPR cannot resolve the continued accumulation of unfolded
proteins, the affected cells become toxic and apoptotic signaling is aroused to lead to cell
death. Activation of caspases is also linked to ER stress and caspase-12 is especially
related to this cellular stress. When intracellular calcium homeostasis is impaired, the
perturbed intracellular calcium concentration initiates ER stress and unresolved ER stress
also activates caspase-12 by calpains, a family of Ca2+
-dependent cysteine

13
proteases.139-140
ER stress induced capsapse-12 activity was reduced by treatment with
calpain inhibitors such as E64 and MDL28170.140
Genetic deletion of caspase-12 in vitro
and in vivo inhibited apoptosis in the presence of ER stress inducers thapsigargin and
tunicamycin.141
This ER-stress specific caspase-12 interacts with IRE-1 (one of UPR arm)
and TRAF2, an adaptor protein.142
Upregulated IRE-1 leads to the disassembly of
heterodimers between caspase-12 and TRAF2, inducing the activation of caspase-12.143
Upon ER stress, procaspase-9 is cleaved by caspase-12, resulting in an activated form of
caspase-9 which activates caspase-3, a main effector caspase responsible for the
destruction of cellular substrates.126, 135
Meanwhile, CCAAT/enhancer-binding protein (C/EBP) homologous protein
(CHOP) is known to be linked to the ER stress induced apoptosis by acting as a
transcription factor.144-145
As a transcription factor, CHOP does not affect apoptosis
directly. Instead, CHOP increases the expression of target genes (e.g., GADD34) which
tend to exacerbate the status of ER stress.56
GADD 34 dephosphorylates eIF2α on serine,
deactivating eIF2α and eventually increasing the burden of ER due to the increase of
RNA translation rate.146
Activated CHOP also increases the expression of Ero-1α, a thiol
oxidase, that contributes to disulfide bond formation and protein folding in the ER.
However, Ero-1α also releases a derivative such as reactive oxygen species (ROS) which
can induce apoptosis.147
CHOP is also known as growth arrest- and DNA damage
inducible gene 153 (GADD 153). The GADD 153 gene is one of a group that can be
induced by genotoxic stress and growth arrest signals. It was shown that sustained
exposure to ER stress leads to the upregulation of CHOP expression.148-149
Recently it
was shown that activity of both IRE-1 and PERK was accompanied by the expression of

14
CHOP and that down-regulation of IRE-1 and PERK led to the up-regulation of CHOP,
eventually trigging cell death.102, 150
The c-Jun-N-terminal kinase (JNK) is also known to be involved in apoptotic
signaling and can be induced by uncontrolled UPR activity. Upon ER stress, activated
IRE-1 combines with tumor-necrosis factor-α (TNF-α)-receptor-associated factor 2
(TRAF2) and this complex can interact with apoptosis-signal-regulating kinase
(ASK1).151
Activated ASK1, in turn, may increase the activity of downstream kinase
JNK and lead to cell death.152
Using ASK-/-
of mouse embryonic fibroblasts (MEFs),
these cells were unable to activate JNK and apoptotic signaling after treatment with ER
stress inducers.153
From this result, it was suggested that ASK is an important activator of
JNK and apoptosis. Taken together, it is thought that prolonged ER stress may be linked
to the development of apoptotic signaling by complex mechanisms and cell death can be
induced.
REFERENCES
1. Luis Griera J, Maria Manzanares J, Barbany M, Contreras J, Amigo P, Salas-
Salvado J. Physical activity, energy balance and obesity. Public Health Nutr.
2007;10(10A):1194-9.
2. Colley RC, Hills AP, King NA, Byrne NM. Exercise-induced energy expenditure:
implications for exercise prescription and obesity. Patient Educ Couns. 2010;79(3):327-
32.
3. Hill JO, Wyatt HR. Role of physical activity in preventing and treating obesity. J
Appl Physiol. 2005;99(2):765-70.
4. Ivy JL, Zderic TW, Fogt DL. Prevention and treatment of non-insulin-dependent
diabetes mellitus. Exerc Sport Sci Rev. 1999;27:1-35.

15
5. Amati F, Dube JJ, Coen PM, Stefanovic-Racic M, Toledo FG, Goodpaster BH.
Physical inactivity and obesity underlie the insulin resistance of aging. Diabetes Care.
2009;32(8):1547-9.
6. Bell LM, Watts K, Siafarikas A, et al. Exercise alone reduces insulin resistance in
obese children independently of changes in body composition. J Clin Endocrinol Metab.
2007;92(11):4230-5.
7. Bhaskarabhatla KV, Birrer R. Physical activity and type 2 diabetes: tailoring
exercise to optimize fitness and glycemic control. Phys Sportsmed. 2004;32(1):13-7.
8. Ropelle ER, Pauli JR, Prada PO, et al. Reversal of diet-induced insulin resistance
with a single bout of exercise in the rat: the role of PTP1B and IRS-1 serine
phosphorylation. J Physiol. 2006;577(Pt 3):997-1007.
9. Schrauwen P. Physical activity and diabetes: current considerations. Appl Physiol
Nutr Metab. 2007;32(3):535-6.
10. Solomon TP, Sistrun SN, Krishnan RK, et al. Exercise and diet enhance fat
oxidation and reduce insulin resistance in older obese adults. J Appl Physiol.
2008;104(5):1313-9.
11. Eriksson KF, Lindgarde F. Poor physical fitness, and impaired early insulin
response but late hyperinsulinaemia, as predictors of NIDDM in middle-aged Swedish
men. Diabetologia. 1996;39(5):573-579.
12. Neufer PD, Dohm GL. Exercise induces a transient increase in transcription of the
GLUT-4 gene in skeletal muscle. Am J Physiol. 1993;265(6 Pt 1):C1597-603.
13. Terada S, Yokozeki T, Kawanaka K, et al. Effects of high-intensity swimming
training on GLUT-4 and glucose transport activity in rat skeletal muscle. J Appl Physiol.
2001;90(6):2019-24.
14. Colcombe S, Kramer AF. Fitness effects on the cognitive function of older adults:
a meta-analytic study. Psychol Sci. 2003;14(2):125-30.
15. Kramer AF, Colcombe SJ, McAuley E, et al. Enhancing brain and cognitive
function of older adults through fitness training. J Mol Neurosci. 2003;20(3):213-21.
16. Palleschi L, Vetta F, De Gennaro E, et al. Effect of aerobic training on the
cognitive performance of elderly patients with senile dementia of Alzheimer type. Arch
Gerontol Geriatr. 1996;22 Suppl 1:47-50.

16
17. Powell RR. Psychological effects of exercise therapy upon institutionalized
geriatric mental patients. J Gerontol. 1974;29(2):157-61.
18. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of
Alzheimer disease. JAMA. 2009;302(6):627-37.
19. McAuley E, Kramer AF, Colcombe SJ. Cardiovascular fitness and neurocognitive
function in older adults: a brief review. Brain Behav Immun. 2004;18(3):214-20.
20. Lautenschlager NT, Cox KL, Flicker L, et al. Effect of physical activity on
cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA.
2008;300(9):1027-37.
21. Liu HL, Zhao G, Cai K, Zhao HH, Shi LD. Treadmill exercise prevents decline in
spatial learning and memory in APP/PS1 transgenic mice through improvement of
hippocampal long-term potentiation. Behav Brain Res. 2011;218(2):308-14.
22. Um HS, Kang EB, Koo JH, et al. Treadmill exercise represses neuronal cell death
in an aged transgenic mouse model of Alzheimer's disease. Neurosci Res.
2011;69(2):161-73.
23. Yuede CM, Zimmerman SD, Dong H, et al. Effects of voluntary and forced
exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse
model of Alzheimer's disease. Neurobiol Dis. 2009;35(3):426-32.
24. Hoveida R, Alaei H, Oryan S, Parivar K, Reisi P. Treadmill running improves
spatial memory in an animal model of Alzheimer's disease. Behavioural Brain Res.
2011;216(1):270-274.
25. Berchtold NC, Castello N, Cotman CW. Exercise and time-dependent benefits to
learning and memory. Neuroscience. 2010;167(3):588-97.
26. Kim SE, Ko IG, Kim BK, et al. Treadmill exercise prevents aging-induced failure
of memory through an increase in neurogenesis and suppression of apoptosis in rat
hippocampus. Exper Gerontol. 2010;45(5):357-365.
27. Aguiar AS, Boemer G, Rial D, et al. High-Intensity Physical Exercise Disrupts
Implicit Memory in Mice Involvement of the Striatal Glutathione Antioxidant System
and Intracellular Signaling. Neuroscience. 2010;171(4):1216-1227.
28. Sibley BA, Etnier JL. The relationship between physical activity and cognition in
children: A meta-analysis. Ped. Exercise Sci. 2003;15(3):243-256.

17
29. Davis CL, Tomporowski PD, Boyle CA, et al. Effects of aerobic exercise on
overweight children's cognitive functioning: a randomized controlled trial. Res Q Exerc
Sport. 2007;78(5):510-9.
30. Hillman CH, Pontifex MB, Raine LB, Castelli DM, Hall EE, Kramer AF. The
effect of acute treadmill walking on cognitive control and academic achievement in
preadolescent children. Neuroscience. 2009;159(3):1044-54.
31. Hamer M, Chida Y. Physical activity and risk of neurodegenerative disease: a
systematic review of prospective evidence. Psychol Med. 2009;39(1):3-11.
32. Goodwin VA, Richards SH, Taylor RS, Taylor AH, Campbell JL. The
effectiveness of exercise interventions for people with Parkinson's disease: a systematic
review and meta-analysis. Mov Disord. 2008;23(5):631-40.
33. Coelho FG, Santos-Galduroz RF, Gobbi S, Stella F. Systematized physical
activity and cognitive performance in elderly with Alzheimer's dementia: a systematic
review. Rev Bras Psiquiatr. 2009;31(2):163-70.
34. Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at
midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62(10):1556-
60.
35. Rosendorff C, Beeri MS, Silverman JM. Cardiovascular risk factors for
Alzheimer's disease. Am J Geriatr Cardiol. 2007;16(3):143-9.
36. Muzio M, Chinnaiyan AM, Kischkel FC, et al. FLICE, a novel FADD-
homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--
inducing signaling complex. Cell. 1996;85(6):817-27.
37. Gomez-Pinilla F, Zhuang Y, Feng J, Ying Z, Fan G. Exercise impacts brain-
derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation.
Eur J Neurosci. 2011;33(3):383-90.
38. Griesbach GS, Hovda DA, Gomez-Pinilla F. Exercise-induced improvement in
cognitive performance after traumatic brain injury in rats is dependent on BDNF
activation. Brain Res. 2009;1288:105-15.
39. Gomez-Pinilla F, Vaynman S, Ying Z. Brain-derived neurotrophic factor
functions as a metabotrophin to mediate the effects of exercise on cognition. Eur J
Neurosci. 2008;28(11):2278-87.

18
40. Ying Z, Roy RR, Zhong H, Zdunowski S, Edgerton VR, Gomez-Pinilla F. BDNF-
exercise interactions in the recovery of symmetrical stepping after a cervical hemisection
in rats. Neuroscience. 2008;155(4):1070-8.
41. Chae CH, Jung SL, An SH, et al. Treadmill exercise improves cognitive function
and facilitates nerve growth factor signaling by activating mitogen-activated protein
kinase/extracellular signal-regulated kinase1/2 in the streptozotocin-induced diabetic rat
hippocampus. Neuroscience. 2009;164(4):1665-73.
42. Johnson RA, Rhodes JS, Jeffrey SL, Garland T, Jr., Mitchell GS. Hippocampal
brain-derived neurotrophic factor but not neurotrophin-3 increases more in mice selected
for increased voluntary wheel running. Neuroscience. 2003;121(1):1-7.
43. Johnson RA, Mitchell GS. Exercise-induced changes in hippocampal brain-
derived neurotrophic factor and neurotrophin-3: effects of rat strain. Brain Res.
2003;983(1-2):108-14.
44. Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density
and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J
Neurosci. 1998;18(21):8559-70.
45. Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates
retinal ganglion cell dendritic and axonal arborization in vivo. J Neurosci.
1999;19(22):9928-38.
46. McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu
Rev Neurosci. 1999;22:295-318.
47. Yacoubian TA, Lo DC. Truncated and full-length TrkB receptors regulate distinct
modes of dendritic growth. Nat Neurosci. 2000;3(4):342-9.
48. Soule J, Messaoudi E, Bramham CR. Brain-derived neurotrophic factor and
control of synaptic consolidation in the adult brain. Biochem Soc Trans. 2006;34(Pt
4):600-4.
49. Lu B. Pro-region of neurotrophins: role in synaptic modulation. Neuron.
2003;39(5):735-8.
50. Lee AS. The glucose-regulated proteins: stress induction and clinical applications.
Trends Biochem Sci. 2001;26(8):504-10.
51. Bachmair A, Varshavsky A. The degradation signal in a short-lived protein. Cell.
1989;56(6):1019-32.

19
52. Colgan SM, Tang D, Werstuck GH, Austin RC. Endoplasmic reticulum stress
causes the activation of sterol regulatory element binding protein-2. Int J Biochem Cell
Biol. 2007;39(10):1843-51.
53. Lee JN, Ye J. Proteolytic activation of sterol regulatory element-binding protein
induced by cellular stress through depletion of Insig-1. J Biol Chem.
2004;279(43):45257-65.
54. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded
protein response. Nat Rev Mol Cell Biol. 2007;8(7):519-29.
55. Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress
and apoptosis. Apoptosis. 2009;14(8):996-1007.
56. Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the
unfolded protein response. Physiology (Bethesda). 2007;22:193-201.
57. Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in
immunity and autoimmunity. Nat Rev Immunol. 2008;8(9):663-74.
58. Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res.
2005;569(1-2):29-63.
59. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J
Clin Invest. 2002;110(10):1389-98.
60. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum:
coordination of gene transcriptional and translational controls. Genes Dev.
1999;13(10):1211-33.
61. Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific
endonuclease that initiates mRNA splicing in the unfolded protein response. Cell.
1997;90(6):1031-1039.
62. Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to
secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92-6.
63. Lee K, Tirasophon W, Shen X, et al. IRE1-mediated unconventional mRNA
splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the
unfolded protein response. Genes Dev. 2002;16(4):452-66.

20
64. Shen X, Ellis RE, Lee K, et al. Complementary signaling pathways regulate the
unfolded protein response and are required for C. elegans development. Cell.
2001;107(7):893-903.
65. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced
by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active
transcription factor. Cell. 2001;107(7):881-91.
66. Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the
secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19-57.
67. Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic
reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3(4):246-55.
68. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor
ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response
to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787-3799.
69. Shen JS, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6
localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization
signals. Develop Cell. 2002;3(1):99-111.
70. Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-
bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355-1364.
71. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor
ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response
to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787-99.
72. Harding HP, Zhang YH, Zeng HQ, et al. An integrated stress response regulates
amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619-633.
73. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction
of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol.
2000;2(6):326-32.
74. Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced
insulin resistance with salicylates or targeted disruption of Ikkbeta. Science.
2001;293(5535):1673-7.
75. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from
obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature.
1997;389(6651):610-4.

21
76. Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal
kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54
Suppl 2:S73-8.
77. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and
insulin resistance. Nature. 2002;420(6913):333-6.
78. Schaffer JE. Lipotoxicity: when tissues overeat. Current Opinion in Lipidol.
2003;14(3):281-287.
79. Wu LL, Dunning KR, Yang X, et al. High-fat diet causes lipotoxicity responses in
cumulus-oocyte complexes and decreased fertilization rates. Endocrinology.
2010;151(11):5438-45.
80. Ilieva EV, Ayala V, Jove M, et al. Oxidative and endoplasmic reticulum stress
interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130(Pt 12):3111-23.
81. Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal-
mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology.
2008;47(5):1495-503.
82. Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty
liver disease. Semin Liver Dis. 2008;28(4):360-9.
83. Diakogiannaki E, Welters HJ, Morgan NG. Differential regulation of the
endoplasmic reticulum stress response in pancreatic beta-cells exposed to long-chain
saturated and monounsaturated fatty acids. J Endocrinol. 2008;197(3):553-63.
84. Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of
endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res.
2006;47(12):2726-37.
85. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity,
insulin action, and type 2 diabetes. Science. 2004;306(5695):457-61.
86. Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role
in development of leptin resistance. Cell Metab. 2009;9(1):35-51.
87. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of
metabolic disease. Cell. 2010;140(6):900-17.
88. Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and
diabetes. Int J Obes (Lond). 2008;32 Suppl 7:S52-4.

22
89. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-
kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell.
2008;135(1):61-73.
90. Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and
type 2 diabetes. Novartis Found Symp. 2007;286:86-94; discussion 94-8, 162-3, 196-203.
91. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell.
2008;132(3):344-62.
92. Granell S, Mohammad S, Ramanagoudr-Bhojappa R, Baldini G. Obesity-linked
variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can
be rescued to the cell surface by a chemical chaperone. Mol Endocrinol.
2010;24(9):1805-21.
93. Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and
restore glucose homeostasis in a mouse model of type 2 diabetes. Science.
2006;313(5790):1137-40.
94. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev.
2001;81(2):741-66.
95. Citron M, Oltersdorf T, Haass C, et al. Mutation of the beta-amyloid precursor
protein in familial Alzheimer's disease increases beta-protein production. Nature.
1992;360(6405):672-4.
96. Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a
mutant amyloid beta protein precursor. Science. 1993;259(5094):514-6.
97. Suzuki N, Cheung TT, Cai XD, et al. An increased percentage of long amyloid
beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants.
Science. 1994;264(5163):1336-40.
98. Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in
Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's
pathology. J Neuroinflammation. 2009;6:41.
99. Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P,
Scheper W. The unfolded protein response is activated in pretangle neurons in
Alzheimer's disease hippocampus. Am J Pathol. 2009;174(4):1241-51.
100. Hoozemans JJ, Veerhuis R, Van Haastert ES, et al. The unfolded protein response
is activated in Alzheimer's disease. Acta Neuropathol. 2005;110(2):165-72.

23
101. Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid
precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem.
1998;273(40):25552-5.
102. Lee do Y, Lee KS, Lee HJ, et al. Activation of PERK signaling attenuates Abeta-
mediated ER stress. PLoS One. 2010;5(5):e10489.
103. Walter J, Capell A, Grunberg J, et al. The Alzheimer's disease-associated
presenilins are differentially phosphorylated proteins located predominantly within the
endoplasmic reticulum. Mol Med. 1996;2(6):673-691.
104. Niwa M, Sidrauski C, Kaufman RJ, Walter P. A role for presenilin-1 in nuclear
accumulation of Ire1 fragments and induction of the mammalian unfolded protein
response. Cell. 1999;99(7):691-702.
105. Katayama T, Imaizumi K, Sato N, et al. Presenilin-1 mutations downregulate the
signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1(8):479-85.
106. Katayama T, Imaizumi K, Honda A, et al. Disturbed activation of endoplasmic
reticulum stress transducers by familial Alzheimer's disease-linked presenilin-1 mutations.
Biol Chem. 2001;276(46):43446-43454.
107. Katayama T, Imaizumi K, Sato N, et al. Presenilin-1 mutations downregulate the
signalling pathway of the unfolded-protein response. Nature Cell Biol. 1999;1(8):479-
485.
108. Loewen CA, Feany MB. The unfolded protein response protects from tau
neurotoxicity in vivo. Plos One. 2010;5(9):-.
109. Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid beta
peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys.
Acta. 2004;1742(1-3):81-7.
110. Verkhratsky A, Toescu EC. Endoplasmic reticulum Ca(2+) homeostasis and
neuronal death. J Cell Mol Med. 2003;7(4):351-61.
111. Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD.
Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative
disorders. Trends Neurosci. 2000;23(5):222-9.
112. Pierrot N, Ghisdal P, Caumont AS, Octave JN. Intraneuronal amyloid-beta1-42
production triggered by sustained increase of cytosolic calcium concentration induces
neuronal death. J Neurochem. 2004;88(5):1140-50.

24
113. Sun XD, Mo ZL, Taylor BM, Epps DE. A slowly formed transient conformer of
Abeta(1-40) is toxic to inward channels of dissociated hippocampal and cortical neurons
of rats. Neurobiol Dis. 2003;14(3):567-78.
114. Lee JH, Won SM, Suh J, et al. Induction of the unfolded protein response and cell
death pathway in Alzheimer's disease, but not in aged Tg2576 mice. Exp Mol Med.
2010;42(5):386-94.
115. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with
wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-57.
116. Alnemri ES, Livingston DJ, Nicholson DW, et al. Human ICE/CED-3 protease
nomenclature. Cell. 1996;87(2):171.
117. Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with
endogenous endonuclease activation. Nature. 1980;284(5756):555-6.
118. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770-6.
119. Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256(1):12-8.
120. Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions
downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell.
1997;89(2):175-84.
121. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-
activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature.
1998;391(6662):43-50.
122. Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation
and DNA degradation during apoptosis. Nature. 1998;391(6662):96-9.
123. Takahashi A, Alnemri ES, Lazebnik YA, et al. Cleavage of lamin A by Mch2
alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases
with distinct substrate recognition properties are active in apoptosis. Proc National
Academy of Sciences of the USA. 1996;93(16):8395-8400.
124. Orth K, Chinnaiyan AM, Garg M, Froelich CJ, Dixit VM. The CED-3/ICE-like
protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A.
Biological Chem. 1996;271(28):16443-16446.
125. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science.
1998;281(5381):1312-1316.

25
126. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science.
1998;281(5381):1312-6.
127. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science.
1998;281(5381):1305-8.
128. Hakem R, Hakem A, Duncan GS, et al. Differential requirement for caspase 9 in
apoptotic pathways in vivo. Cell. 1998;94(3):339-52.
129. Kuida K, Haydar TF, Kuan CY, et al. Reduced apoptosis and cytochrome c-
mediated caspase activation in mice lacking caspase 9. Cell. 1998;94(3):325-37.
130. Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a
novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell
death. Cell. 1996;85(6):803-15.
131. Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent
formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell.
1997;91(4):479-89.
132. Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme.
Genes Dev. 1999;13(24):3179-84.
133. Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite
gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727-30.
134. Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein
homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of
caspase-3. Cell. 1997;90(3):405-13.
135. Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12(11):1551-70.
136. Muchmore SW, Sattler M, Liang H, et al. X-ray and NMR structure of human
Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381(6580):335-41.
137. Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated
apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186-94.
138. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular
level. Nat Rev Mol Cell Biol. 2008;9(3):231-41.
139. Nakagawa T, Yuan J. Cross-talk between two cysteine protease families.
Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150(4):887-94.

26
140. Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA. Ubiquitous calpains
promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced
apoptosis. J Biol Chem. 2006;281(23):16016-24.
141. Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-
reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature.
2000;403(6765):98-103.
142. Yoneda T, Imaizumi K, Oono K, et al. Activation of caspase-12, an endoplastic
reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor
2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276(17):13935-
40.
143. Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell
Biol. 2004;14(1):20-8.
144. Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell
death in response to impaired function of the endoplasmic reticulum. Genes Dev.
1998;12(7):982-95.
145. Bruhat A, Jousse C, Wang XZ, Ron D, Ferrara M, Fafournoux P. Amino acid
limitation induces expression of CHOP, a CCAAT/enhancer binding protein-related gene,
at both transcriptional and post-transcriptional levels. J Biol Chem. 1997;272(28):17588-
93.
146. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded
protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol.
2001;153(5):1011-22.
147. Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting
protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev.
2004;18(24):3066-77.
148. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for
translational regulation and cell survival during the unfolded protein response. Mol Cell.
2000;5(5):897-904.
149. Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating
transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like
endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded
protein response. Biochem J. 2002;366(Pt 2):585-94.

27
150. Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the
unfolded protein response. Science. 2007;318(5852):944-9.
151. Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic
reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats.
Genes Dev. 2002;16(11):1345-1355.
152. Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of
JNK protein kinases by transmembrane protein kinase IRE1. Science.
2000;287(5453):664-6.
153. Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic
reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats.
Genes Dev. 2002;16(11):1345-55.

28
CHAPTER 2
THE EFFECT OF EXERCISE AND DIET ON THE UNFOLDED PROTEIN
RESPONSE (UPR) IN THE BRAIN OF MICE
INTRODUCTION
The brain has an important role in regulating energy balance and peripheral
glucose homeostasis and abnormal central regulation can contribute not only to the
development of obesity but also to type 2 diabetes. The brain responds to circulating
leptin which is released from adipose tissue 1-2
and circulating insulin 3-4
both of which
are released relative to the degree of adiposity.
As one way of sensing energy status, the hypothalamus of the brain responds to
leptin and other endocrine and nutrient signals to reduce food intake and increase energy
expenditure, leading to control of body weight.5-7
Friedman’s research group showed that
the obese (ob) gene has a main role in regulating energy balance in the mouse and that
mutation of this gene (lepob
/lepob
) leads to obesity and type 2 diabetes.8 Furthermore, they
showed that the arcuate nucleus of hypothalamus is the central site for regulating leptin
signaling which, in turn, is dependent on the presence of the long form of the leptin
receptor (Ob-Rb) which is known to be absent in db/db mice.9 In high-fat fed mice and
obese humans, circulating leptin levels are chronically increased and their responsiveness
to leptin is severely attenuated.10-12
This down-regulated leptin sensitivity is called “leptin
resistance”. Stimulation of the inhibitory molecule Suppressor of Cytokine Signaling 3
(SOCS3) in the hypothalamus in response to leptin normally modifies its activity and
may have a role in the development of leptin resistance.13-14

29
In addition to leptin actions in the brain, insulin also takes part in the regulation of
energy balance. Although it was initially believed that insulin could not cross the blood-
brain barrier, it is now recognized that insulin can access into the brain across the blood-
brain barrier and that insulin receptors are widely expressed in the brain including the
arcuate nucleus.15
Secretion of insulin is also affected by the amount of stored fat so that
the basal level of circulating insulin is up-regulated according to the individual fat mass.3-
4 Increasing insulin levels with increased adiposity leads to the insulin resistance and the
development of type 2 diabetes. Studies have confirmed that the brain is sensitive to
insulin. Using the technique of intracerebroventricular (icv) infusion, direct insulin
administration into the third ventricle of the brain was shown to decrease food intake
along with body weight.16-18
Numerous studies have also shown that insulin actions in the
brain contribute to glucose homeostasis. Obici et al. (2002) suggested that hypothalamic
insulin receptors are necessary for glucose homeostasis.19
By application of an antisense
oligodeoxynucleotide for the insulin receptor precursor protein, hyperphagia and insulin
resistance are induced in rats indicating that insulin receptor activity is needed in the
hypothalamus for proper regulation of food intake and insulin action.20
Similarly, icv
infusion of antibodies specific to insulin into the hypothalamus increased glucose
production indicating that insulin action in the brain regulates glucose homeostasis.19
Within the last decade, many studies have shown that leptin resistance can result
from ER stress and Ozcan et al. (2004) suggested that uncontrolled ER stress is the
principal cause of obesity as well as type 2 diabetes.21
ER stress inducible chemicals
tunicamycin and dithiothreitol (DTT) induce leptin resistance in vitro by inhibiting leptin
induced tyrosine phosphorylations of both leptin receptor (LepRb) and signal transducer

30
and activator of transcription 3 (STAT3), a down-stream component of the leptin
signaling pathway.22-23
In addition, direct infusion of an ER stress inducer into the brain
of lean mice increased leptin resistance and increased mRNA expression of neuropeptide
Y (NPY) and agouti-related peptide (AgRP), known changes associated with leptin
resistance.22
Furthermore, in neuron specific XBP-1 knock-out mice, not only plasma
leptin level but also fat mass were significantly increased in the presence of high fat
diet.22
Recently, it was shown that ER stress is connected to impaired insulin signaling in
the brain. Insulin activation of phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) in the
mediobasal hypothalamus (MBH), a downstream insulin signaling component, was
severely reduced in neuron specific IkappaB kinase beta (IKKβ) deleted mice in which
ER stress was increased.24
In addition, ER stress triggered by icv injection of the ER
stress inducer thapsigargin down-regulated insulin signaling in the hypothalamus as
detected by western blotting using antibody specific for phosphorylated Akt.25
In the
mHypoE-44 hypothalamic cell line, palmitate induced lipotoxicity up-regulated the
expression of phosphorylated eIF2α, a component of the UPR response, and caspase-3,
an apoptosis marker. This palmitate treatment also decreased the expression of
phosphorylated Akt and prevented insulin signaling.26
Many epidemiological studies have revealed a connection between metabolic
diseases such as obesity and type 2 diabetes, and neuronal diseases such as Alzheimer’s
disease (AD).27-32
Obesity has been recognized as a main risk factor for AD and much
supportive evidence has accumulated. Recent studies in vivo and in vitro have shown that
impaired leptin signaling could also lead to the increased onset of AD. Leptin treatment

31
lowered Aβ levels in a dose- and time-dependent manner in the Neuro2a neuronal cell
line.33
When leptin (daily 20 μg in PBS) was continuously administered into an AD
model mice (Tg2576 mice) using Alzet osmotic minipumps for 8 weeks, brain Aβ levels
were significantly decreased even on high-fat dietary feeding.33
Meanwhile, human
studies have shown that obesity affects the onset of AD. Whitmer et al. (2007) showed
that obesity/overweight, measured by body mass index (BMI), has a strong relationship
with the onset of AD in middle-aged person.34
In another study a significant correlation
between BMI/body fat mass and Aβ levels was identified.35
Although exercise has a significant effect in reversing or slowing the progress of
chronic diseases such as obesity,36-37
type 2 diabetes 38-39
and AD,40-41
there have been
few trials aimed at elucidating the effects of exercise on ER stress which is a possible
main factor for the cause of these diseases. In 2008, Um et al. showed that 16 weeks of
exercise training up-regulated the expression of the ER chaperone ‘GRP78’ proteins and
down-regulated expression of the apoptotic proteins caspase-9 and -3 in the brain of
APPsw transgenic mice, an AD disease model.42
In this study, a water maze test also
showed the effect of exercise on UPR activation in the brain of AD mice along with the
connection to the improvement of behavioral dysfunction.42
Recently, it was shown that 6
weeks of forced running increased UPR related gene expression (Grp78) in liver of rats
fed high-fat diet 43
along with decreasing body fat mass. This implies the effect of
exercise on UPR also occurs in peripheral tissues. In 2011, Wu et al. revealed that one
bout of exercise led to increased expression of UPR related genes in skeletal muscle of
C57BL/6 mice such as quadriceps.44
They also showed that expression of the UPR gene
Xbp1 in skeletal muscle reduced to normal level after longer (4 weeks) exercise

32
training.44
In contrast to these results, some reports have shown opposite results. Ropelle
et al. (2010) showed that physical activity, composed of two 3-hour swimming exercise
sessions along with 45 minutes rest time, lowered the UPR related gene expression
(phosphorylated PERK) in the hypothalamus of rats injected with the ER stress inducer
of thapsigargin.45
It was also shown that chronic exercise of 4 weeks swimming (1 hr/day,
5 day/week) increased phosphorylated PERK protein expression without statistical
significance in hypothalamus of diet-induced obese rats.45
Most recently, it was shown
that 8 weeks of swimming exercise lowered the UPR related phosphorylated PERK and
eIF2α protein expressions in both adipose and liver tissue of rats adapted to high-fat diet
for 2 months.46
Taken together, the effect of exercise on the UPR activity is unclear at
this time. Further experiments are needed to elucidate the effect of exercise on the UPR
mechanism in the brain as well as in other tissues.
In this chapter, we have focused on showing how voluntary running wheel
exercise affects the activities of UPR related genes/proteins in the brain of C57BL/6 mice
and how this response is affected by feeding HFD for differing time periods, 3 weeks
(section 2), 3 months (section 3), and over 3 months (section 1). These experiments have
shown how diet affects the response of the UPR in the brain, information that has
implications for the prevention of chronic diseases.
HYPOTHESES
(1) The UPR mechanism in brain of mice will be up-regulated by voluntary
running wheel exercise and this effect will be enhanced by dietary fat.
(2) Activated UPR in response to exercise will not be connected with apoptotic
signaling up-regulation.

33
(3) Dietary fat will up-regulate the UPR activity and apoptosis will be induced.
METHODS
Animals and Diets
Male C57BL/6 mice (~ 6 weeks) were used in all of the experiments. All animal
studies were approved by the Utah State University Institutional Animal Care and Use
Committee. Once all animals arrived at the animal room, about one week was given for
adaptation to the new environment. Each mouse was then moved to an individual cage
(30.3 x 20.6 x 26 cm, Nalgene® plastic cage) which can be equipped with a running
wheel and the type of diet was also changed as required by each experimental protocol. In
this research, three types of diets were used: Low fat diet (LFD); 10 kcal% Fat (D
12450B, Research Diets, New Brunswick, NJ), High fat diet (HFD); 45 kcal% Fat (D
12451, Research Diets, New Brunswick, NJ) and Very High fat diet (VHFD); 60 kcal%
Fat (TD 03661, Harlan Teklad, Madison, WI). Detailed dietary information is shown in
Table 1. The food was given in feeding cups and both 45% HFD and 60% HFD were
changed every second day due to the possibility of oxidation of the diet fatty acids.
Before starting the 3 weeks running protocol, each diet was given for a different time
(VHFD for 4 months, LFD/HFD for 3 weeks, or LFD/HFD for 3 months). The animal
room was automatically maintained at 22-23 °C with a 12-hour light/dark cycle. All
animals had access to food and water ad libitum.
Body Composition
An Echo nuclear magnetic resonance imaging (MRI)-700TM
system (Echo
Medical Systems, Houston, TX) was used to assay body composition of animals. Using
this system, % fat composition could be obtained along with the results for lean mass and

34
body fluid. After setting up the zero point, each mouse was placed in a transparent plastic
tube which is specifically designed for mice and body compositions were recorded.
Voluntary Running Wheel Exercise
For mice in the running group, mice were individually housed in plastic regular cages
equipped with MiniMitter running wheels (740mm circumference, [MiniMitter, Bend,
OR]). One week was allowed for all mice to become accustomed to the cages containing
the aluminum running wheel and during this period the running wheels were locked with
commercial plastic cables. After the adaptation period, the wheels were released and mice
had free access to the running wheels for the duration of experiments (e.g., 3 weeks).
Revolutions of the running wheel were automatically recorded by the VitalView program
in turns per hour. It was programmed that running activity was recorded and saved every
hour for the duration of the experiment.
RNA Isolation and Purification
Mice were sacrificed by cervical dislocation. The brain was removed and the
hypothalamus, hippocampi and frontal cortex were dissected out on ice. For later analysis,
liver, and white adipose tissues were also taken. All tissues were quickly frozen in liquid
nitrogen and kept at -80 °C until processed. Total RNA from each tissue sample was
isolated using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH). Using a
Teflon pestle, tissues were gently homogenized in 1.5 mL tubes containing TRI Reagent
and 100 µL of 1-bromo-3-chloropropane (BCP, Molecular Research Center, Inc.,
Cincinnati, OH) added for the phase separation. After samples were centrifuged (15 min,
13,000 rpm, 4 °C), the RNA-rich aqueous phase was transferred to a new 1.5 mL tube
and 500 µL isopropanol added to precipitate RNA.

35
Table 1 Compositions of experimental diet chows
10kcal% LFD
(Research Diet, D12450B)
45kcal% HFD
(Research Diet, D12451)
60kcal% HFD
(Harlan Teklad,
TD 03661)
kcal% kcal% kcal%
Protein 20 20 15.3
Carbohydrate 70 35 25.2
Fat 10 45 59.5
Total 100 100 100
kcal/g 3.85 4.73 5.3
Ingredient g/kg g/kg g/kg
Casein 200 200 230
Sucrose 350 172.8 150
Maltodextrin 35 100 170
Cellulose 50 50 26.46
Soybean oil 25 25 20
Lard 20 177.5 330
Mineral Mix 10
(S10026)
10
(S10026)
50
(AIN-93G-MX, 94046)
Calcium
phosphate 13 13 2.5
Vitamin Mix 10
(V10001)
10
(V10001)
14
(AIN-93-VX, 94047)
Choline
Bitartrate 2 2 3.5
Fatty acid % of total fat % of total fat % of total fat
Saturated 25.1 36.3 38
Mono-
unsaturated 34.7 45.3 48
Poly-
unsaturated 40.2 18.5 14

36
After centrifugation at 10,000 rpm at 4 °C for 8 min, the RNA pellet was washed
with 1mL 75 % (v/v) ethanol and then air-dried at room temperature for 5-10 min. The
pellet was resuspended in RNase-free water. Using a TURBO DNA-freeTM kit (Ambion,
Inc., Austin, TX), carry-over DNA was removed. After the DNase treatment, the RNA
concentration was measured by spectrophotometric analysis (A260/A280) and the quality
was visually assessed using 1.5 % agarose gel electrophoresis.
Quantitative Real-Time PCR
Using the Superscript First-Strand Synthesis system for RT-PCR III (Invitrogen,
Carlsbad, CA), 1 µg total RNA from individual tissue samples was used to synthesize
cDNA. The reaction for making cDNA was conducted for 60 min at 50 °C and for 15
min at 70 °C. The expression level of each gene (Xbp1, Grp78, Eif2α and Atf6) was
quantified by the use of SYBR Green (Quanta BioSciences, Inc., Gaithersburg, MD) and
normalized to the expression of cyclophilin B (Ppib) gene. Following the manufacturer’s
protocol, reaction cycles for real-time PCR were set: 1 cycle for 3 min at 95 °C and then
40 cycles with two steps of 10 seconds at 95 °C and 30 seconds at 55 °C. The following
primers were used:
Grp78: Forward, 5 -́CTG GAC TGA ATG TCA TGA GGA TCA-3 ,́ Reverse, 5 -́CTC
TTA TCC AGG CCA TAT GCA ATA G-3 ;́ Xbp1: Forward, 5 -́GGA CTC TGA CAC
TGT TGC CTC TT-3 ,́ Reverse, 5 -́AAC TTG TCC AGA ATG CCC AAA-3 ;́ Atf6:
Forward, 5 -́TGG GCA GGA CTA TGA AGT AAT G-3 ,́ Reverse, 5 -́CAA CGA CTC
AGG GAT GGT GCT G-3 ;́ Eif2α: Forward, 5 -́ATG GAA GCC AAAGCT GAA G-3 ,́
Reverse, 5 -́CTG ACA TGA AGG AGG GCA-3 ;́ Ppib (Cyclophilin B): Forward, 5 -́

37
GCT GGA TGG CAA GCA TGT G-3 ,́ Reverse, 5 -́TGT CTT GGT GCT CTC CAC
CTT-3 ́
Protein Extraction
For the assay of protein expression, the hippocampus was used as its size provides
sufficient protein for the assay and these are also two symmetrical tissues in each brain.
By using a Teflon pestle, hippocampus was homogenized in the 1.5 mL tube containing
whole cell lysis buffer (50 mM KCl, 1 % NP-40, 25 mM 4-(2-hydroxyethyl)-1-
piperazineethanesulfonic acid [HEPES, pH 7.8], 10 µg/ml Leupeptin, 20 µg/ml Aprotinin,
125 µM DTT, 1 mM Phenylmethanesulfonylfluoride [PMSF] and 1 mM sodium
orthovanadate) including one tablet of a protease inhibitor cocktail (Complete Mini®,
Roche, Indianapolis, IN) per 25 ml whole cell lysis buffer. Homogenized tissues were
sonicated with two cycles of 10 strokes. After centrifugation at 13,000 rpm at 4 °C for 15
min, the clear supernatants containing the total protein were transferred to new 1.5 mL
tubes. Using a Pierce Bicinchoninic acid (BCA) protein assay kit (Thermo Scientific Inc.,
Rockford, IL), protein concentration was determined by the amount of reduction of the
Cu+2
to Cu+1
.
Western Blotting
Equal amount of protein from each sample were adjusted to a common volume
with 1x whole cell lysis buffer. 6x Sodium dodecyl sulfate (SDS) loading buffer was
added and samples were heated at 100 °C for 5 min to denature protein structures.
Samples were separated on a 10~12 % SDS-polyacrylamide gel with 1x Tris-Glycine-
SDS electrophoresis running buffer (0.025 M Tris, 0.192 M Glycine and 0.1 % SDS) at
85 ~ 95 V. After confirming that the 10 kD reference band reached the bottom, the

38
protein was transferred to Polyvinylidene fluoride (PVDF) membranes (Thermo
Scientific Inc., Rockford, IL) by western blotting transfer kit of Bio-Rad’s product. Using
transfer buffer (0.5 M Tris base, 3.9 M Glycine and 20 % methanol) which was kept cool
with ice pack, the transfer step was conducted for 3 hours at 80 V. Then, the membrane
was blocked for about 2 hours using 5 % (w/v) nonfat dry milk mixed in Tris Buffer
Saline Tween-20 (TBS-T) buffer (1 M Tris [pH 7.4], 5 M NaCl and 0.1 % Tween 20).
Specific primary antibodies diluted to 1:200-1:1000 with 5 % milk buffer were added to
the buffer and membranes incubated overnight at 4 °C. The antibodies used were:
Rabbit anti-XBP1 (SC-7160), rabbit anti-ATF6 (SC-22799), rabbit anti-phosphorylated
PERK (SC-32577R), rabbit anti-PERK (SC-13073), goat anti-phosphorylated eIF2α (SC-
12412), rabbit anti-CHOP(GADD 153) were all purchased from Santa Cruz
Biotechnology (Santa Cruz, CA), rabbit anti-eIF2α (Cell Signaling, Danvers, MA), rabbit
anti-GRP78 (Stressgen, BC, Canada), β-Actin (Abcam, Cambridge, MA), rabbit anti-
Caspase-12, rabbit anti-Caspase-3 (Cell signaling, Danvers, MA).
After incubation with the primary antibody, each membrane was washed with
TBS-T buffer and incubated with horse radish peroxidase-conjugated secondary antibody
for one hour at room temperature. After washing the membrane with TBS-T buffer, ECL
Western Blotting Substrate (Thermo Scientific Inc., Rockford, IL) was added and protein
expression was detected by exposure to a blue autoradiograph film (Bio Express,
Kaysville, UT). The film was developed by an x-ray film processor (Konica Minolta,
Ramsey, NJ) in the dark room. Image density was assessed on a Bio-Rad Imaging system
using Bio-Rad Quantity One software.

39
Statistics
Data are shown as Means ± SEM and statistically analyzed by the program of
Prism 5 for windows (GraphPad, La Jolla, CA). Two-way ANOVA (running level x diet)
was used to study the interaction between diet and running ability on each physiological
parameter. Individual groups were compared with either an unpaired t-test or a one-way
ANOVA with Tukey’s multiple comparison tests. Values of p<0.05 were considered as
statistically significant.
RESULTS
The Effect of Exercise on the UPR in the Brain
of Mice Fed VHFD for 4 Months
Voluntary running wheel exercise
After feeding mice with 60% HFD for 4 months, mice were given free access to
running wheels for 3 weeks except non-running sedentary (SED) groups. At the time of
beginning running exercise, mice were 25 weeks old. Following their running activity,
running animals were grouped into Low-runner (LR, n=6), Middle-runner (MR, n=8),
and High-runner (HR, n=6). As shown in panel (A) of Figure 2-1, there were variations in
running activity of individual animals. In panel (B), though there was no major difference
in running activity between the running groups in the first weeks of running, MR and HR
mice increased their activity reaching their highest level after 2 or 3 weeks, respectively.
As shown in panel (C), when considering total average running activity during the 3
weeks running period, there were significant differences between running groups. HR
and LR mice made 200.59 ± 20.18 and 36.48 ± 4.94 turns per hour, respectively, and
there was a significant difference between running activity of each running group. In
further experiments, only LR and HR were used for study of the UPR activity.

40
The effect of exercise on body weight
During the 3 weeks running period, both SED (n=5) and LR (n=6) mice slightly
increased their body weights by 1.65 ± 0.43 g and 2.04 ± 0.78 g, respectively. However,
HR mice showed a significantly decreased body weight after three weeks running wheel
exercise relative to LR mice (Figure 2-2, A). As shown in panel (B), the reduction in
body weight (-5.32 ± 0.85 g) of HR mice was significantly different from the change of
body weight in the SED and LR mice.
The effect of exercise on food intake
Although HR mice seemed to ingest fewer daily calories compared to the other
groups during the 3 weeks running period, there was no significant difference on any
specific day between the daily caloric uptake of each group (Figure 2-3, panel A).
However, the cumulative caloric intake during the three weeks running period was
reduced in the HR mice (panel B).
The effect of exercise on UPR in the brain
Three weeks voluntary running exercise decreased the UPR related gene
expression in the brain of mice adapted to very high fat diet (60% HFD) for 4 months
(Figure 2-4). In the hypothalamus, LR mice had significantly decreased gene expression
for Xbp1, Atf6 and Eif2α compared to SED mice. HR mice increased UPR-related gene
expressions relative to the LR. These results in HR mice were still lower than that in the
SED group although there was no significant difference between HR and SED values. In
the hippocampus, UPR related gene expression was not significantly affected by exercise
except for expression of Eif2α which was significantly decreased in the LR group.
Western blotting (Figure 2-5) of hippocampus samples suggested that XBP1 and ATF6

41
protein expression was decreased by running exercise, consistent with the changes in
gene expression. But, contrary to the gene expression patterns, the protein levels of both
eIF2α and GRP78 seemed to be increased by running exercise.
The Effect of Exercise on the UPR in the Brain
of Mice fed LFD or HFD for 3 Weeks
Voluntary running wheel exercise
The 20 mice given LFD or HFD had free access to running wheels during the 3
weeks experimental period after which all running mice were sorted into three groups
relative to their activities; low runner (LR, n=6), middle runner (MR, n=8), and high
runner (HR, n=6). As shown in panel (A) of Figure 2-6, activity levels of mice were
monitored during 3 weeks and there was a big individual variation in running ability
between animals even though mice are of the same genetic background. As shown in
panel (B), there was a significant difference in activity between running groups for 3
weeks running period. While HR mice in LFD and HFD had 573.00 ± 39.90 and 492.90
± 18.80 turns per hour, respectively, LR groups in both diet groups showed less than 200
turns per hour (174.00 ± 55.80 vs. 117.00 ± 24.50, LFD vs. HFD). Comparing the
running activity of each running group between LFD and HFD, HFD fed mice seemed to
be less active than LFD fed mice and a significant difference was observed between MR
mice of LFD and HFD. In panel (C), HR mice eating LFD showed significantly higher
running activity during 3 weeks as compared to LR mice of same diet. In addition, MR
mice showed significantly higher running activity than LR mice after 2 week. As shown
in panel (D), both HR and MR mice fed HFD had significantly more running activity
relative to the activity of LR mice during 3 weeks running period. After 2 weeks, there
was also a significant difference between HR and MR. Interestingly, activity of mice fed

42
the HFD seemed to decrease after 2 weeks of running period. In the subsequent
experimental analysis of the UPR, only tissues from LR and HR mice were used for
comparison.
The effect of exercise on body weight and body composition
As shown in panels (A) and (B) of Figure 2-7, 3 weeks feeding of LFD and HFD
increased the body weight of SED mice by 3.60 ± 0.30 g and 5.80 ± 0.70 g, respectively.
Three weeks of high running activity had a significant effect on lowering the increase of
body weight in mice fed LFD for 3 weeks (panel A). At the end of the running period, the
body weight of HR mice was significantly less than the LR mice. As shown in panel (B),
HR mice fed the HFD had significantly lower body weights during the 3 weeks running
period relative to the SED mice. At 3 weeks, there was a significant difference between
body weights of LR and HR mice.
In this experiment, the Echo MRI system was used for analyzing body
composition change of mice fed with LFD or HFD for 3 weeks (Figure 2-7, C). 3 weeks
HFD feeding significantly increased body fat almost two-fold in comparison to the LFD
group (12.60 ± 0.60 vs. 23.30 ± 1.50). Voluntary running exercise had no effect on body
fat composition of LFD fed mice but decreased the body fat composition in HFD fed
mice. The decrease was significant in HR mice that decreased the body fat composition
to the level of %fat observed in the LFD group. ANOVA also showed that there was a
significant interaction between exercise and diet (F2, 30 = 4.099, p=0.0267). Lean body
mass of animals was also measured by Echo MRI (panel D). Neither diet nor exercise had
any effect on lean body mass of mice.

43
The effect of exercise on food intake
Figure 2-8 shows the daily caloric intake of all groups during the 3 weeks running
period. Both LR and HR mice had a trend of higher caloric intakes compared to SED
group during 3 weeks. However, there was no significant difference between groups on
any specific day except on the first two in mice fed the HFD when LR mice ate
significantly more calories than SED mice. When cumulative caloric intake of each diet
group was compared over the 3-week period (panel C), ANOVA showed that both
exercise (F2, 30 = 12.35, p=0.0001) and diet (F1, 30 = 25.98, p<0.0001) had a significant
effect without interaction. Compared to the equivalent groups of LFD, all groups of HFD
consumed more cumulative calories. In addition, HR mice of each diet group consumed
significantly more calories during the three experimental weeks than did SED mice.
Gene and protein expression in the brain
The effect of exercise in mice fed LFD for 3 weeks. In order to study how
voluntary running wheel exercise affected the three arms (XBP1, ATF6, and PERK) of
the UPR system in multiple brain regions (hypothalamus, hippocampus, and cortex), real-
time PCR and western blotting were used to assay the expression of UPR related genes
(Figure 2-9, panel A) and proteins (Figure 2-10, panel A), respectively.
Gene expressions of Xbp1, Atf6, Eif2α, and Grp78 were normalized to the
expression of a housekeeping gene cyclophilin b. The results of LR and HR mice are
shown as fold changes relative to those of SED mice. The expression of these UPR genes
was generally increased by exercise in each brain region although there was significant
variation in the magnitude of the response and in the level of activity that was required to
induce a significant change. In the hypothalamus, HR mice up-regulated gene expression

44
of two arms, Atf6 and Eif2α (down-stream of PERK signaling), as well as increasing
Grp78 gene expression. Specifically, Atf6 gene expression was the most sensitive to
exercise as this gene was activated even in LR mice. In contrast, LR mice did not show
any change on Eif2α and Grp78 expression although both were significantly increased in
the HR group. However, running activity did not affect the Xbp1 gene expression in this
brain region. In the hippocampus, all genes of the UPR arms in addition to Grp78 were
activated by running exercise and a significant effect for Atf6 gene was observed in LR
mice. In the cortex, although all UPR related genes and Grp78 gene were increased by
exercise, only the changes in Atf6 and Eif2α gene expressions were significant.
When UPR related protein expressions of hippocampus of mice fed LFD were
assessed by western blotting experiments, the results were not consistent with the results
of gene expression (Figure 2-10, panel A). Expression of XBP1 (spliced), phosphorylated
PERK and total PERK were significantly upregulated by voluntary running wheel
exercise. Though other arms and GRP78 had a tendency of being increased by running,
these increases did not reach significance.
In addition to the brain regions, real-time PCR was also conducted on liver
samples from the experimental groups (Figure 2-9, A). Running exercise increased the
expressions of all UPR related genes in the liver, but there was considerable variability in
all groups and only Eif2α gene expression was significantly increased.
The effect of exercise in mice fed HFD for 3 weeks. The same experimental
procedures conducted in LFD animal samples were also applied to mice fed HFD for 3
weeks. UPR related gene expression (Figure 2-9, panel B) and protein expression (Figure
2-10, panel B) were analyzed in the multiple brain regions as well as in the liver.

45
In the hypothalamus the expression of genes in all three UPR arms and Grp78
were significantly increased by voluntary running wheel exercise and these maximal
increases were also seen even in the LR groups. This effect of exercise was also detected
in the hippocampus although it was not as pronounced. HR mice showed a significant
increase in the expression of both Xbp1 and Atf6 genes as well as in Grp78 gene
expression. However, exercise had no significant effect on Eif2α gene expression.
Western blotting of UPR related protein expression in the hippocampus (Figure 2-10,
panel B) identified an increase in XBP1 (spliced) protein expression, consistent with the
increase in gene expression. However, neither ATF6 nor GRP78 protein expression were
affected by exercise in comparison to the results of gene expression. In addition, the
effect of exercise on the pPERK-peIF2α pathway also differed between mRNA level and
protein level. While the Eif2α gene level was not changed by exercise, the protein
expression levels of both pPERK and peIF2α were significantly increased in HR mice. In
cortex of mice fed HFD, exercise only upregulated the Eif2α gene expression
significantly. Although Xbp1 was also shown to be increased by exercise, the increase
was not significant.
All the assessed UPR gene levels were increased in the liver in running mice fed
HFD but only increases in Atf6 and Eif2α gene expression were significant in HR mice.
The effect of exercise on apoptotic signaling
in the brain
In order to see if up-regulation of UPR gene expression induced by 3 weeks
running exercise was associated with an increase in the apoptotic pathway, the protein
expression levels of ER stress specific apoptotic components CHOP, caspase-12, and
caspase-3 were evaluated using western blotting (Figure 2-11).

46
In the brain of mice fed LFD, 3 weeks running exercise had no effect on the level
of apoptotic protein expression (panel A). Exercise had no effect on the level of CHOP,
caspase-12 (full and cleaved) or caspase-3 (full and cleaved). Though caspase-12 (full)
and caspase-12 (cleaved) was decreased by 3 weeks running exercise, these changes were
not significant. In the HFD groups (panel B), exercise also had no effect on apoptotic
signaling in the brain of mice fed HFD. There were trends of decreased expressions in
caspase-12 (full and cleaved) and caspase-3 (full and cleaved) proteins, but these also did
not reach significance.
The effect of three weeks dietary (LFD/HFD) treatment on UPR in brain
To identify the effects of high fat diets alone on UPR activity in the brain and
liver, UPR related gene expressions were compared between sedentary groups of each
diet (LFD or HFD) (Figure 2-12).
Compared to the LFD group, 3 weeks feeding of HFD had no significant effects
on the expression of UPR genes except for Xbp1 gene expression in the hypothalamus
and the Grp78 gene expression in the hippocampus. In the hypothalamus, Xbp1 gene
expression was significantly decreased in mice after 3 weeks of HFD but other UPR
genes were not changed by this dietary treatment. In the hippocampus, HFD feeding
increased Grp78 gene expression significantly. In the cortex, even though no significant
change was observed, 3 weeks HFD treatment showed a tendency to decrease the
expression of UPR related genes.
The effect of diet on UPR in the liver was also studied (panel D). In this case only
Eif2α gene expression was significantly increased. HFD feeding for 3 weeks had no
effect on expressions of other UPR genes as compared to LFD.

47
The Effect of 3 Weeks Exercise on UPR in the Brain
of Mice Adapted to Low or High Fat Diet
for 3 Months
Voluntary running wheel exercise
After feeding mice LFD or HFD for three months, 20 mice of each diet group
were allowed voluntary running wheel exercise for 3 weeks while maintained on these
same diets. As in the previous experiments, running mice of each diet group were divided
into three groups according to their running ability: Low-runner (LR, n=6), Middle-
runner (MR, n=8), and High-runner (HR, n=6). As shown in panel (A) of Figure 2-13,
there was a large variation in the individual exercise level. Diet and running ability had a
significant effect on running activity and both MR and HR of HFD had significantly less
activity than the same groups fed LFD (panel B). There was a significant difference
within running groups of each dietary group. While HR groups of LFD and HFD ran
462.07 ± 13.68 and 339.97 ± 51.39 turns per hour, respectively (p<0.05), LR groups of
LFD and HFD had 129.68 ± 18.55 and 61.63 ± 14.71 turns per hour (p<0.05). Though
LR and HR mice fed LFD maintained their running level during the three weeks of the
running period, running activity of MR mice appeared to decrease with time (panel C).
From the result of mice fed HFD (panel D), MR and HR mice gradually increased their
running activity while LR mice maintained similar low activity during the 3 weeks
running period. There was a significant difference in running activity between running
groups.
The effect of exercise on body weight
Figure 2-14 shows that body weights of mice adapted to LFD/HFD for 3 months
were changed during the 3 weeks running period. At the beginning of the running period

48
(week 0), all LFD mice had a significantly lower body weight than all HFD mice. While
LFD mice weighed 28.30 ± 0.60 g after 3 months feeding of LFD, mice fed HFD for 3
months weighed 36.00 ± 0.85 g (p<0.01). As shown in closed symbols, three weeks
voluntary running activity decreased body weight of LFD fed mice relative to the SED
mice fed LFD. The reduction was significant in the HR group. In mice fed HFD for 3
months (open symbols), the body weights of SED and LR mice increased during the 3
weeks running period while the body weights of HR mice were significantly reduced
compared to both groups SED and LR groups. After 3 weeks of running exercise, HR
mice of LFD and HFD reduced 2.40 ± 0.50 g and 3.40 ± 1.10 g, respectively while SED
mice increased their body weight to 0.50 ± 0.30 g and 4.40 ± 0.90 g in mice fed LFD and
HFD, respectively.
The effect of exercise on food intake
Each group of mice fed LFD had similar daily food intake during the 3 weeks
running exercise period (Figure 2-15, panel A). HR mice of LFD tended to have more
daily calories than other groups and this resulted in a significant cumulative increase over
the 3 weeks of running period (panel C). In mice adapted to HFD (panel B), HR mice
tended to have lower daily food intake and this resulted in a reduction in cumulative
intake over the 3 weeks period (panel C). ANOVA showed (panel C) that there was a
significant interaction between exercise and diet (F2, 30 = 15.31, p<0.0001) but neither
factor had any independent effect on cumulative food intake during the 3 weeks of
running period.

49
Gene and protein expression in the brain
The effect of exercise in mice fed LFD for 3 months. Using real-time PCR, UPR
related gene expressions (Xbp1, Eif2α, and Grp78) were assayed in hypothalamus and
hippocampus (Figure 2-16, panel A). In the hypothalamus, three weeks voluntary running
exercise increased the expression of UPR genes and the increase in Grp78 gene of HR
mice was significant relative to the level in SED mice. In the hippocampus though, HR
mice only increased Eif2α gene expression with significance. There were no significant
effects on either Xbp1 or Grp78. Only XBP1 (spliced) protein level was significantly
increased by 3 weeks running in the hippocampus of mice adapted to LFD for 3 months
(Figure 2-17, panel A). The other proteins levels were not changed by running exercise in
this brain region.
The effect of exercise in mice fed HFD for 3 months. In the brain of mice fed
HFD for 3 months, UPR related gene expressions (Xbp1, Atf6, Eif2α, and Grp78) were
also increased by 3 weeks voluntary running wheel exercise (Figure 2-16, panel B). In the
hypothalamus, it appeared that running exercise increased the expression of UPR genes
but only the increase in Atf6 gene expression reached significance relative to SED mice.
The hippocampus was more responsive to running exercise. Compared to the SED group,
gene expression of Xbp1, Atf6, and Grp78 were all significantly increased in the
hippocampus by 3 weeks running exercise. Western blotting for UPR related proteins
showed that running exercise significantly increased the level of these proteins in the
hippocampus of mice fed HFD for 3 months (Figure 2-17, panel B). The protein
expressions of XBP1 (spliced), ATF6, and peIF2α were all significantly increased by
running exercise related to the results of SED group.

50
The effect of exercise on apoptotic signaling in the brain
Apoptotic related protein expressions (CHOP, caspase-12 and caspase-3) were
assayed by western blotting (Figure 2-18). As shown in panel (A), 3 weeks running
exercise had no effect on apoptotic protein expression in the hippocampus of mice fed
LFD for 3 months. Although HR mice seemed to decrease the protein levels of CHOP
and caspase-12 (full), neither change was statistically significant. Likewise, apoptotic
protein expression in the hippocampus of mice adapted to HFD for 3 months was also not
significantly changed compared to the LFD group (Panel B).
The effect of diet on apoptotic signaling in the brain
To assess the effect of diet, the apoptotic protein expression (CHOP and caspase-
12) between SED mice of 3 weeks HFD and SED mice of 3 months HFD was compared
(Figure 2-19). Long-term (3 months) HFD significantly increased apoptotic signaling in
the hippocampus as compared to short-term (3 weeks) HFD.
The effects of prolonged feeding of high fat diet on UPR in the brain
The effects of long-term (3 months) HFD on UPR related gene expression in the
brain of mice was also compared in the SED groups (Figure 2-20). Compared to the LFD
group, gene expression of Eif2α and Grp78 were considerably increased in the
hypothalamus of mice fed HFD for 3 months. In the hippocampus, 3 months treatment of
HFD increased the expressions of both Eif2α and Grp78 but these changes did not reach
statistical significance.

51
DISCUSSION
Recently, many studies have suggested a linkage between ER stress and chronic
diseases. This was seen in the peripheral tissues such as liver and pancreatic β cells in
obesity and type 2 diabetes.47-49
This possible linkage in the brain regions was also
reported.22, 24, 26
Since exercise has beneficial effects in ameliorating metabolic disease
(obesity and type 2 diabetes),50-51
it was hypothesized that exercise would show a positive
effect on ameliorating the progression of these chronic diseases by up-regulating UPR
and decreasing ER stress. In this study, I focused on the brain because exercise was
shown to have an effect on UPR 45, 52
in the brain and it is known to reduce
neurodegenerative diseases associated with aging.53-54
Since dietary fat is a risk factor for
obesity, type 2 diabetes and neurodegenerative diseases,55-56
the studies reported in this
chapter focused on the interaction between diet and exercise in regulating UPR in the
brain.
Effect of Diet and Age on Voluntary Running Ability
Voluntary running wheel activity of male C57BL/6 mice was decreased in older
mice and in mice eating higher fat diets (45% and 60% HFD). When converting the result
of the running activity (turns per hour) into daily covered distance, HR mice in the 3
weeks diet experiments (LFD and HFD) ran about 10.76 km and 8.74 km a day on
average, respectively. After feeding the diets for 3 months, HR mice ran about 8.20 km
and 6.04 km a day during the 3 weeks of running period in the LFD and HFD groups,
respectively. When mice were adapted to a more dense caloric diet (60% HFD) for a
longer duration (4 months), HR mice on average covered only 3.56 km a day during the 3

52
weeks of running period. It is not clear if the further reduction was principally due to the
older age or prolonged dietary fat intake.
There have been variable reports of the effects of diet on running ability. Animals
adapted to a high-fat diet increased their running endurance capacity as assessed by the
running time to exhaustion.57-58
This was assumed to result from an increased fat
oxidation of animals fed the HFD that could give animals improved exercise ability. In
contrast, another study showed that mice fed HFD ran significantly shorter distances as
compared to mice fed regular chow.59
These different results may reflect the running
protocols. The former study used one-time treadmill exercise to assess exhaustion time,
while others used voluntary running wheel exercise during some period as used in the
experiments in this thesis.
In the current studies, older mice showed less running activity compared to
younger mice with similar dietary treatment. Since mice used in this study were not over
one-year old, the decrease of running activity with age could be primarily induced by
increased body weight, not by biochemical changes such as oxidant stress and
inflammation.
In summary, the type of diet had a significant effect on voluntary running wheel
activity and mice fed HFD showed lower running activity relative to LFD mice. Here,
aging also had an effect on lowering running activity in animals on both diets.
Effect of Exercise on Energy Balance
As expected, this study confirmed that exercise has a significant effect to prevent
weight gain or lower body weight independent of dietary type (LFD or HFD). Although
HR mice did not decrease their body weight during 3 weeks feeding LFD/HFD, the

53
degree of increment was significantly lowered as compared to the SED mice. In the
experiments in which mice were fed 45% HFD and 60% HFD for three months and four
months, respectively, three weeks subsequent voluntary running exercise significantly
decreased their body weight. This effect of exercise on lowering body weight was also
detected in the mice adapted to LFD for three months. This effect of exercise on body
weight has been well described in animals 50, 59
and human studies.60-61
Voluntary running
wheel exercise has a positive effect on maintaining body weight and preventing or
ameliorating obesity.51, 62
The decrease in body weight of exercising mice fed high fat
diet came from a reduced fat mass, confirming a previous report.51
This supports the
suggestion that exercise increased fatty acid oxidation as an important regulatory
response that contributes to the lower body weight.
Taken together, this study confirmed that exercise had a significant effect on
maintaining body weight and contributed to the maintenance of energy balance by
increasing energy expenditure.
Effect of Diet and Exercise on Food Intake
In the three weeks LFD/HFD experiments, HR mice in both diet groups
consumed significantly more energy than LR and SED mice. However, the HR mice fed
either 45% HFD or 60% HFD over three months significantly reduced their cumulative
energy intake during the 3 weeks running period. This suggests that HR mice that
become obese while chronically adapted to HFD get the energy to support activity from
stored fat rather than from the direct energy source of food. It is also possible that three
weeks running affects appetite and changes food intake depending on duration of fat diets.
Other studies using 2~6 week exercise protocols found that exercise animals fed HFD

54
increased their cumulative caloric intake compared to control animals fed chow diet.51, 59
Meanwhile, Patterson et al. (2009) showed that if selectively bred diet-induced obesity
(DIO) rats had access to running wheels, running rats fed HFD had reduced food intake
relative to sedentary rats 3 weeks after starting running but similar food intake after 10
weeks after starting running.50
It is likely that the effects of exercise on food intake are
affected by the length of time on the diet, change in body fat, age, and intensity of
exercise.
Effect of Exercise on UPR
The main focus of my thesis study was to identify how voluntary running wheel
exercise affects the UPR in the brain of mice in a variety of dietary condition. Firstly, it
was assessed how exercise has an effect on UPR in the brain of mice adapted to a highly
calorically dense fat diet (60% HFD) for four months. It was shown that three weeks
running exercise did not up-regulate the UPR in the brain regions. In the hypothalamus,
LR mice had significantly lower UPR related genes expression compared to SED mice
and although HR mice activated UPR, it was still low relative to the SED mice. In the
hippocampus, it appeared that HR mice decreased the UPR gene expression except for
Eif2α. From these results, it was thought that exercise could not increase but decreased
UPR in the brain if mice were totally adapted to HFD and were obese. From these results,
it was supposed that UPR had already reached to the maximum level by the long period
of highly dense fat diet and three weeks voluntary running exercise could not activate the
further UPR. These experiments suggested further studies to show the effect of diet and
the effect of time of exposure to the diet on exercise-regulated UPR.

55
The second experiments showed that, in contrary to previous data of 60% HFD
experiments, mice undertaking voluntary physical activity while feeding with LFD or
HFD for three weeks up-regulated the gene and/or protein expression of a number of
UPR components (XBP1, ATF6, eIF2α, and Bip/GRP78) in multiple brain regions
(hypothalamus, hippocampus, and cortex) though there was frequently discrepancy
between gene and protein expression. The inconsistency between gene and protein levels
could be explained from the possibility that all UPR arms and GRP78 do not respond to
ER stress at the same time. Interestingly, UPR in the hypothalamus of mice fed HFD for
three weeks was more sensitively up-regulated by running exercise than in other brain
regions. This may be important since the hypothalamus is the central brain region that
regulates energy homeostasis and is responsive to physical activity. In this experiment,
UPR activity in the liver was also assessed to know how peripheral tissue responds to the
running activity. The results showed that exercise had similar effects in this peripheral
tissue as in the brain. These results are in line with recently published data. Chapados and
Lavoie (2010) showed that 6 weeks treadmill exercise activated the UPR, specifically
Bip/GRP78, in the liver of rats fed with 45% HFD and this effect of exercise was more
significant in the fatty liver where the expression of more UPR genes (Xbp1, Atf6, Eif2α,
and Grp78) were activated by the exercise protocol.43
The effect of exercise on UPR in the brain of mice which were adapted to HFD
for a longer time period (3 months) was also studied. After feeding mice with LFD or
HFD for three months, the 3 weeks of voluntary running wheel exercise protocol was
applied after which UPR related gene/protein expression were analyzed in different brain
regions. In this experimental paradigm, voluntary running exercise activated the UPR

56
related gene/protein expression in the brain of both diet groups. However, in contrast to
shorter exposure (3 weeks) to HFD, running activity did not significantly affect UPR in
the hypothalamus of mice fed HFD for 3 months. It is possible that the hypothalamus
could be desensitized by longer exposure to dietary fat. Meanwhile, the hippocampus of
mice adapted to three months HFD sensitively responded to voluntary running wheel
exercise and UPR gene/protein expression in this brain region were all activated by
running activity as compared to the result of LFD mice. These results suggest that there is
significant regional variation in the brain in response to exercise. As exercise seemed to
have a significant effect on the hippocampus, which is important for cognitive function,
the activation of UPR by exercise may have a possible effect against neurodegenerative
disease.
The effect of diet itself on UPR activity in the brain was also assessed. Although 3
weeks of exposure to HFD appeared to have no effect on activating UPR as compared to
LFD group, 3 months of HFD treatment was shown to up-regulate the UPR as compared
to LFD mice. Ozcan et al. (2009, 2004) also identified the effect of HFD on activation of
UPR in the hypothalamus22
and the liver,21
respectively. In these studies, they showed
that 25 weeks HFD activated the expression of phosphorylated PERK and
phosphorylated IRE1α (the upstream of XBP1) in the brain and 16 weeks HFD increased
the expression of phosphorylated PERK and phosphorylated eIF2α in the liver. In
addition, they indicated that these upregulated UPR by HFD could be linked to the
insulin resistance in the liver.21
Considering the experimental results of our laboratory in
which Boghossian et al. (2009) showed that 3 days HFD induced central insulin
resistance,63
it is unlikely that UPR activation is responsible for insulin resistance in the

57
brain of mice fed the HFD for a short-term. It could be concluded that the dietary effect
on activation of UPR depends on the length of time animals are exposed to the diet.
This study interestingly indicated that although short-term HFD by itself did not
induce UPR activation, it did enhance the activating effect of exercise on UPR. The effect
of exercise on UPR activity in the brain was greater in the HFD fed group than in LFD
fed group. This result could be explained in a number of ways. Exercise could have an
effect on reversing insulin resistance 64-65
which was known to be induced in the brain by
short-term HFD 63
and that improved insulin signaling, in turn, could activate trophic
factors such as BDNF and IGF-1, contributing to the UPR up-regulation. Many studies
have supported the idea that exercise has a significant effect on reversing insulin
resistance. Specifically, it was recently shown that HFD-induced insulin signaling
impairment in the hypothalamus of rats was restored by exercise.45
In addition, it was
indicated that improved insulin signaling was accompanied with UPR up-regulation 66
and that BDNF action was linked to UPR (i.e., XBP1).67
It could be also assumed that
exercise could reverse any HFD-induced reduction of BDNF, contributing to prompt
UPR up-regulation in the brain. The Gomez-Pinilla research group (2004) showed a
decrease of hippocampal BDNF in animals fed HFD for 2 months that was reversed by
voluntary running wheel exercise.68
A further possibility is that exercise in animals fed
HFD could increase fatty acid oxidation, possibly up-regulating UPR. It has been shown
that increased fat metabolism can activate UPR in adipose tissue 69-70
and in the liver.71
It
is possible that activation of β-oxidation during exercise could produce ketone bodies
which, in turn, could lead to activation of UPR. At this time, there is no supportive
evidence showing a linkage between ketone bodies and UPR. However, if dietary fat is

58
high and exposure time to diet is prolonged, the effect of exercise on up-regulating UPR
disappeared. UPR may have a maximal limit of activation as no additional activation in
response to exercise was possible if UPR was activated chronically by HFD.
In summary, 3 weeks voluntary running wheel exercise up-regulated UPR in
multiple brain regions of mice in various dietary conditions. Short-term HFD increased
UPR in response to exercise. However, short-term HFD treatment alone had no effect on
UPR while HFD feeding for longer duration up-regulated the UPR. This study suggested
that short term HFD induced insulin resistance could not be related to UPR in the brain.
Relationship of Exercise Induced Change
in the UPR to Apoptosis
Many studies have identified that untreated ER stress could sustain UPR up-
regulation and that prolonged UPR activation enhances apoptotic signaling in mice with
obesity or impaired insulin signaling.72-73
Apoptosis in the brain would not be conducive
to the prevention of neurodegenerative diseases. So, in this study, we investigated if the
upregulation of UPR induced by exercise was associated with apoptosis in the brain of
mice fed with LFD or HFD. Apoptotic signaling was assessed using western blotting of
specific ER stress related apoptotic signaling components; CHOP, caspase-12 and
caspase-3. Three weeks voluntary running exercise had no effect on apoptotic signaling
in the brain of mice fed with LFD and HFD for 3 weeks or 3 months. If anything, the
changes that were observed indicated a decrease in the apoptotic response to running
activity in both three weeks and three months experiments although the changes did not
reach statistical significance. Hence, up-regulation of UPR by exercise is not associated
with any increase in apoptosis.

59
The effect of diet on apoptotic signaling in the brain of mice exposed to LFD or
HFD during the short or long term was also studied. In SED mice fed HFD for 3 months,
there was increased expression of apoptotic related proteins (CHOP and cleaved Caspase-
12) in the hippocampus as compared to SED of 3 weeks HFD mice. Hence, long-term
treatment with dietary fat was shown to up-regulate apoptotic signaling in the brain. This
is consistent with results of several other studies which showed that diet could have an
effect on activating apoptosis. 60% HFD for 3 months was shown to increase the CHOP
protein expression in the hypothalamus of rats.45
Another study also showed that 16
weeks HFD activated the expression of phosphorylated JNK in the liver.21
Taken together, it can be concluded that exercise may prevent the normal
activation of apoptotic signaling that is associated with increased UPR. This result, in
turn, supports the idea that UPR up-regulation induced by exercise has beneficial effects
in the brain of mice exposed to high fat diet, inducing the defensive mechanism of
reducing ER stress without the damaging effect of apoptosis.
Exercise and Neurodegenerative Disease
To our knowledge, this study is the first trial to show that voluntary running
exercise increases UPR in the brain of animals independent of the diet composition.
Exercise induced UPR up-regulation of the brain could be helpful to maintain brain
health by clearing malformed proteins associated with neurodegenerative disease. This
effect would be further supported by the absence of any increase in apoptotic signaling.
Exercise is known to have a positive effect on neurodegenerative disease. The data
described in this chapter suggest that the effect of exercise on UPR up-regulation and
apoptotic down-regulation could contribute to the protective effect of exercise on

60
neuronal diseases such as AD. Most recently, Lowen and Feany (2010) suggested that
activated UPR, such as XBP1, was neuroprotective in vivo by decreasing the toxicity of
tau.74
Further, brain-derived neurotrophic factor (BDNF) which is induced by exercise 75-
76 and is needed to activate XBP1,
67 may be responsible for the increase of UPR and
neuronal health. In addition, our laboratory has shown (Park-York, unpublished
observations) that nerve growth factor (NGF) increased XBP1 expression and inhibited
ER stress inducer (thapsigargin) induced CHOP expression in the hippocampal HT-22
cells. Thus we hypothesize that exercise possibly activates UPR by up-regulating BDNF
expression in the brain and that this could have a beneficial effect on brain health.
REFERENCES
1. Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin
concentrations in normal-weight and obese humans. N Engl J Med. 1996;334(5):292-5.
2. Caro JF, Kolaczynski JW, Nyce MR, et al. Decreased cerebrospinal-fluid/serum
leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet.
1996;348(9021):159-61.
3. Polonsky KS, Given BD, Hirsch L, et al. Quantitative study of insulin secretion
and clearance in normal and obese subjects. J Clin Invest. 1988;81(2):435-41.
4. Polonsky KS, Given BD, Van Cauter E. Twenty-four-hour profiles and pulsatile
patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81(2):442-8.
5. Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma
protein encoded by the obese gene. Science. 1995;269(5223):543-6.
6. Leibel RL, Chung WK, Chua SC, Jr. The molecular genetics of rodent single gene
obesities. J Biol Chem. 1997;272(51):31937-40.
7. Seeley RJ, Woods SC. Monitoring of stored and available fuel by the CNS:
implications for obesity. Nat Rev Neurosci .2003;4(11):901-9.

61
8. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional
cloning of the mouse obese gene and its human homologue. Nature.
1994;372(6505):425-32.
9. Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in
diabetic mice. Nature. 1996;379(6566):632-5.
10. Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced
obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab
Disord .2000;24(5):639-46.
11. Schwartz MW, Woods SC, Porte D, Jr., Seeley RJ, Baskin DG. Central nervous
system control of food intake. Nature. 2000;404(6778):661-71.
12. Schwartz MW, Morton GJ. Obesity: keeping hunger at bay. Nature.
2002;418(6898):595-7.
13. Munzberg H, Myers MG, Jr. Molecular and anatomical determinants of central
leptin resistance. Nat Neurosci. 2005;8(5):566-70.
14. Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of
SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1(4):619-25.
15. Baskin DG, Schwartz MW, Sipols AJ, D'Alessio DA, Goldstein BJ, White MF.
Insulin receptor substrate-1 (IRS-1) expression in rat brain. Endocrinology.
1994;134(4):1952-5.
16. Woods SC, Lotter EC, McKay LD, Porte D, Jr. Chronic intracerebroventricular
infusion of insulin reduces food intake and body weight of baboons. Nature.
1979;282(5738):503-5.
17. Air EL, Benoit SC, Blake Smith KA, Clegg DJ, Woods SC. Acute third
ventricular administration of insulin decreases food intake in two paradigms. Pharmacol
Biochem Behav. 2002;72(1-2):423-9.
18. Chavez M, Kaiyala K, Madden LJ, Schwartz MW, Woods SC. Intraventricular
insulin and the level of maintained body weight in rats. Behav Neurosci.
1995;109(3):528-31.
19. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is
required for inhibition of glucose production. Nat Med. 2002;8(12):1376-82.

62
20. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic
insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci.
2002;5(6):566-72.
21. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity,
insulin action, and type 2 diabetes. Science. 2004;306(5695):457-61.
22. Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role
in development of leptin resistance. Cell Metab. 2009;9(1):35-51.
23. Hosoi T, Sasaki M, Miyahara T, et al. Endoplasmic reticulum stress induces leptin
resistance. Mol Pharmacol. 2008;74(6):1610-9.
24. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-
kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell.
2008;135(1):61-73.
25. Won JC, Jang PG, Namkoong C, et al. Central administration of an endoplasmic
reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity.
(Silver Spring) 2009;17(10):1861-5.
26. Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces
endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance
and apoptosis through adenosine 5' monophosphate-activated protein kinase activation.
Endocrinology. 2010;151(2):576-85.
27. Leibson CL, Rocca WA, Hanson VA, et al. Risk of dementia among persons with
diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145(4):301-8.
28. Forette F, Seux ML, Staessen JA, et al. Prevention of dementia in randomised
double-blind placebo-controlled systolic hypertension in Europe (Syst-Eur) trial. Lancet.
1998;352(9137):1347-51.
29. Grant WB. Dietary links to Alzheimer's disease: 1999 update. J Alzheimers Dis.
1999;1(4-5):197-201.
30. Pyorala M, Miettinen H, Halonen P, Laakso M, Pyorala K. Insulin resistance
syndrome predicts the risk of coronary heart disease and stroke in healthy middle-aged
men: the 22-year follow-up results of the Helsinki Policemen Study. Arterioscler Thromb
Vasc Biol. 2000;20(2):538-44.

63
31. Meyer JS, Rauch GM, Rauch RA, Haque A, Crawford K. Cardiovascular and
other risk factors for Alzheimer's disease and vascular dementia. Ann N Y Acad Sci.
2000;903:411-23.
32. Petot GJ, Traore F, Debanne SM, Lerner AJ, Smyth KA, Friedland RP.
Interactions of apolipoprotein E genotype and dietary fat intake of healthy older persons
during mid-adult life. Metabolism. 2003;52(3):279-81.
33. Fewlass DC, Noboa K, Pi-Sunyer FX, Johnston JM, Yan SD, Tezapsidis N.
Obesity-related leptin regulates Alzheimer's Abeta. FASEB J. 2004;18(15):1870-8.
34. Whitmer RA, Gunderson EP, Quesenberry CP, Jr., Zhou J, Yaffe K. Body mass
index in midlife and risk of Alzheimer disease and vascular dementia. Curr Alzheimer
Res. 2007;4(2):103-9.
35. Balakrishnan K, Verdile G, Mehta PD, et al. Plasma Abeta42 correlates positively
with increased body fat in healthy individuals. J Alzheimers Dis. 2005;8(3):269-82.
36. Bruce CR, Thrush AB, Mertz VA, et al. Endurance training in obese humans
improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid
content. Am J Physiol Endocrinol Metab. 2006;291(1):E99-E107.
37. Luis Griera J, Maria Manzanares J, Barbany M, Contreras J, Amigo P, Salas-
Salvado J. Physical activity, energy balance and obesity. Public Health Nutr.
2007;10(10A):1194-9.
38. Balducci S, Zanuso S, Nicolucci A, et al. Anti-inflammatory effect of exercise
training in subjects with type 2 diabetes and the metabolic syndrome is dependent on
exercise modalities and independent of weight loss. Nutr Metab Cardiovasc Dis.
2010;20(8):608-17.
39. Ostergard T, Jessen N, Schmitz O, Mandarino LJ. The effect of exercise, training,
and inactivity on insulin sensitivity in diabetics and their relatives: what is new? Appl
Physiol Nutr Metab. 2007;32(3):541-8.
40. Um HS, Kang EB, Koo JH, et al. Treadmill exercise represses neuronal cell death
in an aged transgenic mouse model of Alzheimer's disease. Neurosci Res.
2011;69(2):161-73.
41. Lautenschlager NT, Cox KL, Flicker L, et al. Effect of physical activity on
cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA.
2008;300(9):1027-37.

64
42. Um HS, Kang EB, Leem YH, et al. Exercise training acts as a therapeutic strategy
for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw-
transgenic model. International Journal of Molecular Medicine. 2008;22(4):529-539.
43. Chapados NA, Lavoie JM. Exercise training increases hepatic endoplasmic
reticulum (er) stress protein expression in MTP-inhibited high-fat fed rats. Cell Biochem
Funct. 2010;28(3):202-10.
44. Wu J, Ruas JL, Estall JL, et al. The unfolded protein response mediates adaptation
to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab.
2011;13(2):160-9.
45. Ropelle ER, Flores MB, Cintra DE, et al. IL-6 and IL-10 anti-inflammatory
activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and
ER stress inhibition. PLoS Biol. 2010;8(8).
46. da Luz G, Frederico MJ, da Silva S, et al. Endurance exercise training ameliorates
insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J
Appl Physiol. 2011.
47. Tsiotra PC, Tsigos C. Stress, the endoplasmic reticulum, and insulin resistance.
Ann N Y Acad Sci. 2006;1083:63-76.
48. Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal
kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54
Suppl 2:S73-8.
49. Kaneto H, Nakatani Y, Kawamori D, et al. Role of oxidative stress, endoplasmic
reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and
insulin resistance. Int J Biochem Cell Biol. 2005;37(8):1595-608.
50. Patterson CM, Bouret SG, Dunn-Meynell AA, Levin BE. Three weeks of
postweaning exercise in DIO rats produces prolonged increases in central leptin
sensitivity and signaling. Am J Physiol Regul Integr Comp Physiol. 2009;296(3):R537-48.
51. Bradley RL, Jeon JY, Liu FF, Maratos-Flier E. Voluntary exercise improves
insulin sensitivity and adipose tissue inflammation in diet-induced obese mice. Am J
Physiol Endocrinol Metab. 2008;295(3):E586-94.
52. Um HS, Kang EB, Leem YH, et al. Exercise training acts as a therapeutic strategy
for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw-
transgenic model. Int J Mol Med. 2008;22(4):529-39.

65
53. Mirochnic S, Wolf S, Staufenbiel M, Kempermann G. Age effects on the
regulation of adult hippocampal neurogenesis by physical activity and environmental
enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus.
2009;19(10):1008-18.
54. Hoveida R, Alaei H, Oryan S, Parivar K, Reisi P. Treadmill running improves
spatial memory in an animal model of Alzheimer's disease. Behavioural Brain Research.
2011;216(1):270-274.
55. McNeilly AD, Williamson R, Sutherland C, Balfour DJ, Stewart CA. High fat
feeding promotes simultaneous decline in insulin sensitivity and cognitive performance in
a delayed matching and non-matching to position task. Behavioural Brain Research.
2011;217(1):134-41.
56. Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin
resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model
of Alzheimer disease. J Biol Chem. 2007;282(50):36275-82.
57. Miller WC, Bryce GR, Conlee RK. Adaptations to a high-fat diet that increase
exercise endurance in male rats. J Appl Physiol. 1984;56(1):78-83.
58. Lee JS, Bruce CR, Spriet LL, Hawley JA. Interaction of diet and training on
endurance performance in rats. Exp Physiol. 2001;86(4):499-508.
59. Murray AJ, Knight NS, Cochlin LE, et al. Deterioration of physical performance
and cognitive function in rats with short-term high-fat feeding. FASEB J.
2009;23(12):4353-60.
60. Sigal RJ, Kenny GP, Wasserman DH, Castaneda-Sceppa C. Physical
activity/exercise and type 2 diabetes. Diabetes Care. 2004;27(10):2518-39.
61. Slentz CA, Houmard JA, Kraus WE. Exercise, abdominal obesity, skeletal muscle,
and metabolic risk: evidence for a dose response. Obesity. (Silver Spring) 2009;17 Suppl
3:S27-33.
62. Haskell-Luevano C, Schaub JW, Andreasen A, et al. Voluntary exercise prevents
the obese and diabetic metabolic syndrome of the melanocortin-4 receptor knockout
mouse. FASEB J. 2009;23(2):642-55.
63. Boghossian S, Lemmon K, Park M, York DA. High-fat diets induce a rapid loss
of the insulin anorectic response in the amygdala. Am J Physiol Regul Integr Comp
Physiol. 2009;297(5):R1302-11.

66
64. Peres SB, de Moraes SM, Costa CE, et al. Endurance exercise training increases
insulin responsiveness in isolated adipocytes through IRS/PI3-kinase/Akt pathway. J
Appl Physiol. 2005;98(3):1037-43.
65. Ropelle ER, Pauli JR, Prada PO, et al. Reversal of diet-induced insulin resistance
with a single bout of exercise in the rat: the role of PTP1B and IRS-1 serine
phosphorylation. J Physiol. 2006;577(Pt 3):997-1007.
66. Ye R, Jung DY, Jun JY, et al. Grp78 heterozygosity promotes adaptive unfolded
protein response and attenuates diet-induced obesity and insulin resistance. Diabetes.
2010;59(1):6-16.
67. Hayashi A, Kasahara T, Iwamoto K, et al. The role of brain-derived neurotrophic
factor (BDNF)-induced XBP1 splicing during brain development. J Biol Chem.
2007;282(47):34525-34.
68. Molteni R, Wu A, Vaynman S, Ying Z, Barnard RJ, Gomez-Pinilla F. Exercise
reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral
plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience.
2004;123(2):429-40.
69. Mollica MP, Lionetti L, Putti R, Cavaliere G, Gaita M, Barletta A. From chronic
overfeeding to hepatic injury: role of endoplasmic reticulum stress and inflammation.
Nutr Metab Cardiovasc Dis. 2011;21(3):222-30.
70. Lionetti L, Mollica MP, Lombardi A, Cavaliere G, Gifuni G, Barletta A. From
chronic overnutrition to insulin resistance: the role of fat-storing capacity and
inflammation. Nutr Metab Cardiovasc Dis. 2009;19(2):146-52.
71. de Roos B, Rungapamestry V, Ross K, et al. Attenuation of inflammation and
cellular stress-related pathways maintains insulin sensitivity in obese type I interleukin-1
receptor knockout mice on a high-fat diet. Proteomics. 2009;9(12):3244-56.
72. Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and
diabetes. Int J Obes. (Lond) 2008;32 Suppl 7:S52-4.
73. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J
Clin Invest. 2002;110(10):1389-98.
74. Loewen CA, Feany MB. The unfolded protein response protects from tau
neurotoxicity in vivo. PLoS One. 2010;5(9).

67
75. Neeper SA, Gomez-Pinilla F, Choi J, Cotman CW. Physical activity increases
mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain
Res. 1996;726(1-2):49-56.
76. Vaynman S, Ying Z, Gomez-Pinilla F. Interplay between brain-derived
neurotrophic factor and signal transduction modulators in the regulation of the effects of
exercise on synaptic-plasticity. Neuroscience. 2003;122(3):647-57.

68
(A)
(B)
(C)
Figure 2-1 Voluntary running wheel ability of mice adapted to 60% HFD for 4 months.
(A) Running activity of individual mice. (B) The change of running activity of each
running group during the 3 weeks running period. High running level group (HR)
increased their activity while low runner group kept their activity low for 3 weeks. (C)
After completing 3 weeks running exercise, running mice could be grouped relative to
average wheel turns per hour; LR (n=6), MR (n=8), and HR (n=6). There were significant
differences between running mice. All data is shown as Mean ± SEM. ++p<0.01 MR vs.
LR; ###p<0.001 HR vs. MR; ***p<0.001, ****p<0.0001 HR vs. LR.

69
(A)
(B)
Figure 2-2 The effect of exercise on body weight. (A) During three weeks of running
exercise, body weights of each group (SED [n=5], LR [n=6], and HR [n=6]) were
measured every week. The results are shown as Mean ± SEM. (B) The change of body
weight after three weeks of running exercise period. ##p<0.01, ###p<0.001 HR vs. LR;
***p<0.001 HR vs. SED.

70
(A)
(B)
Figure 2-3 The effect of exercise on food intake. (A) Daily food intake of each group of
mice adapted to 60% HFD for 4 months during the 3 weeks running period. (B) The
cumulative food intake during the 3 weeks of whole running period. All data is shown as
Mean ± SEM. #p<0.05 HR vs. LR.

71
Figure 2-4 The effects of three weeks voluntary running wheel exercise on UPR related
gene expressions in the brain of mice adapted to 60% HFD for 4 months. The results of
real-time PCR for UPR related gene expressions (Xbp1, Atf6, Eif2α, and Grp78) were
normalized to the expression of the reference gene cyclophilin b (Ppib) and the results of
both LR (n=6) and HR (n=6) was shown relative to the results of SED (n=5). +p<0.05,
++p<0.01 LR vs. SED.

72
(A)
(B)
Figure 2-5 The effects of three weeks voluntary running wheel exercise on UPR related
protein expressions in the hippocampus of mice adapted to 60% HFD for 4 months. Panel
(A) is the image of western blotting for identifying UPR related protein expression in the
hippocampus. Each lane was from pooled samples of three mice of each group. β-Actin
was used as the control for protein expression. Panel (B) shows the result of densitometry
of the western image. The results of LR and HR were shown relative to density of SED
group.

73
(A) (B)
(C) LFD (D) HFD
Figure 2-6 Voluntary running wheel activity of mice fed LFD or HFD for 3 weeks. (A)
The plotted data of running activity of individual mice. (B) Mean of running activity of
mice in each diet for 3 weeks. (C) The result of voluntary running wheel activity in mice
fed LFD for 3 weeks. Activity in each group (LR, n=6; MR, n=8; HR, n=6) is shown as
an average of wheel turns per hour ± SEM during every week. (D) The result of running
activity in mice fed with HFD for 3 weeks. All data is shown as Mean ± SEM. +p<0.05,
++p<0.01, ++++p<0.0001 MR vs. LR, ##p<0.01, ###p<0.001 HR vs. MR ****p<0.0001
HR vs. LR; ap<0.05 LR of HFD vs. LR of LFD.

74
(A) LFD (B) HFD
(C) (D)
Figure 2-7 Effect of diet and exercise on body weight and body composition. (A) The
change of body weight of mice fed 10% LFD. HR mice had a smaller increase of body
weight during 3 weeks. (B) The body weight of mice fed 45% HFD. Though body
weights of all groups (SED, LR, and HR) were increased by 3 weeks HFD, HR had
significantly lower increase of body weight. (C) The effect of 3 weeks diet and exercise
on body fat composition assessed by MRI. Body fat was significantly increased by HFD
as compared to same group of LFD (++p<0.01, +++p<0.001 HFD vs. LFD) and running
exercise had a significant effect on decreasing body fat. (D) Neither exercise nor diet had
any effect on the lean body mass. All data is shown as Mean ± SEM. *p<0.05, **p<0.01
HR vs. SED; #p<0.05 HR vs. LR; +p<0.05 LR vs. SED.

75
(A)
(B)
(C)
Figure 2-8 The daily caloric intake and cumulated caloric intake of LFD (A) and HFD
(B) groups. Except for the first 2 days, there was no significant difference between
groups. (C) The effect of diet and exercise on the cumulative food intake during the 3
weeks running period. During total running period, all of groups in HFD eat significantly
more calories than LFD groups (ap<0.05). HR mice in both diets had significantly more
calories, comparing to SED. +p<0.05 LR vs. SED; *p<0.05, **p<0.01 HR vs. LR.

76
(A) LFD
(B) HFD
Figure 2-9 The effect of voluntary running wheel exercise on UPR related gene
expressions in multiple brain regions and liver of mice fed with LFD and HFD for 3
weeks. All data is shown as Mean ± SEM and the results of running groups (LR and HR)
are shown relative to the results of each SED group. Panel (A) shows the results from the
brain and liver samples of mice fed LFD. Panel (B) shows the results for HFD groups.
All groups had six mice. +p<0.05, ++p<0.01 LR vs. SED; *p<0.05, **p<0.01,
***p<0.001 HR vs. SED.

77
(A) 3 weeks LFD
(B) 3 weeks HFD
Figure 2-10 UPR related protein expression in hippocampus of mice fed LFD or HFD.
(A) The UPR protein expressions in the hippocampus of mice fed LFD. Left panel shows
the results of western blotting of two representative animals in each group. Right bar
graph is the result of the densitometry of UPR protein expressions from four to six mice
in each group. (B) The UPR protein expressions in HFD groups. Left panel shows the
images of western blotting from two representative animals of each group. Right graph
shows the results of densitometry from four to six mice fed with HFD. All data was
shown as Mean ± SEM. +p<0.05, ++p<0.01 LR vs. SED; *p<0.05, **p<0.01,
***p<0.001 HR vs. SED.

78
(A) 3 weeks LFD
(B) 3 weeks HFD
Figure 2-11 The effect of running exercise on apoptotic protein expression in
hippocampus of mice fed LFD or HFD. Exercise affects on expressions of CHOP,
caspase-12, and caspase-3 in the hippocampus of mice fed with LFD (panel A) or HFD
(panel B) for 3 weeks. Left panels show the result of western blotting images for protein
expression from two representative animals. Right panels showed the results of apoptotic
protein expression densitometry for samples from four to six mice per each group. All
data are shown as Mean ± SEM.

79
(A) Hypothalamus (B) Hippocampus
(C) Cortex (D) Liver
Figure 2-12 The effect of diet on UPR related gene expressions in multiple brain regions
and liver. The results of UPR gene expressions from the SED mice of HFD group were
shown in the fold change relative to the results from the SED group of LFD. All data is
shown as Mean ± SEM and came from six mice of each diet group. *p<0.05, **p<0.01
HFD vs. LFD.

80
(A) (B)
(C) LFD (D) HFD
Figure 2-13 Voluntary running wheel activity of mice adapted to LFD/HFD for 3 months.
(A) Running activity of all individual mice in each diet. (B) The effect of diet and
running level or activity. Running mice were grouped into three groups; LR (n=6), MR
(n=8), and HR (n=6). (C) The result of running activity (wheel turns per hour) of LFD
running group during the 3 weeks of running period. (D) The running activity of running
group of HFD during 3 weeks. All data is shown as Mean ± SEM. +p<0.05, +++p<0.001
MR vs. LR, #p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001 HR vs. MR, and
***p<0.001, ****p<0.0001 HR vs. LR.

81
Figure 2-14 The effect of diet and exercise on body weight. These graphs show the
changes of body weight for 3 weeks of running exercise in mice fed LFD (closed
symbols) or HFD (open symbols) for 3 months. On both diets, HR (n=6) significantly
decreased their body weight compared with SED animals (n=6). In HFD, there was
significant difference even between LR and HR. SED and LR group in HFD mice had
significantly increased body weight than same group in LFD mice during three weeks
running periods. **p<0.01, ***p<0.001 HR vs. SED and #p<0.05 HR vs. LR; +p<0.05,
++p<0.01, +++p<0.001, ++++p<0.0001 SED of LFD vs. SED of HFD and ††p<0.01,
†††p<0.001, ††††p<0.0001 LR of LFD vs. LR of HFD.

82
(A)
(B)
(C)
Figure 2-15 The effect of diet and exercise on daily and cumulative food intake. (A)
Daily food intake of mice adapted to 3 months LFD during the 3 weeks running exercise
period. (B) The result of daily food intake of mice fed HFD for 3 months. (C) The effect
of diet and exercise on cumulative food intake during the 3 weeks running exercise. The
data is shown as Mean ± SEM. ##p<0.01 HR vs. MR and *p<0.05, **p<0.01 HR vs.
SED.

83
(A) 3 months LFD
(B) 3 months HFD
Figure 2-16 The effect of exercise on UPR related gene expression in the brain of mice
fed LFD and HFD for 3 months. Panel (A) shows how 3 weeks running exercise affects
UPR related gene expressions in the brain of mice fed LFD for 3 months. Panel (B)
shows the results of UPR related gene expressions of mice fed HFD for 3 months. All
results of both LR (n=6) and HR (n=6) were normalized to the results of SED (n=6) and
were shown as Mean ± SEM. +p<0.05, ++p<0.01 MR vs. SED and *p<0.05, **p<0.01
HR vs. SED.

84
(A) 3 months LFD
(B) 3 months HFD
Figure 2-17 The effect of exercise on UPR related protein expressions in the
hippocampus of mice fed LFD or HFD for 3 months. In the left side panels (A) and (B),
the images show the results of UPR specific western blotting with two individual samples
from group of mice fed LFD or HFD fed 3 months. The expressions of β-Actin were used
for control protein expression. In the right section of both diet (A and B), UPR related
protein expressions are shown in densitometry graph (shown in arbitrary unit) and each
result of both LR and HR was normalized to the result of SED. For constructing
densitometry, protein expressions from four to six mice of each group were used and all
data is shown as Mean ± SEM. +p<0.05 MR vs. SED and *p<0.05 HR vs. SED.

85
(A) 3 months LFD
(B) 3 months HFD
Figure 2-18 The effect of exercise on apoptotic related protein expressions in the
hippocampus of mice fed LFD or HFD for 3 months. The left sides of panel (A) and (B)
show the apoptotic specific western blotting images of two individual animals of each
group fed LFD or HFD for three months. On the right side of each panel (A and B), the
bar graphs show the result of densitometry from four to six mice from each group. The
results of LR and HR were normalized to the results of SED and were shown as Mean ±
SEM.

86
(A)
(B)
Figure 2-19 The effects of diet on apoptotic related protein expression in the
hippocampus. Each lane in the western image (A) comes from individual animals fed
HFD for either 3 weeks or 3 months and the results are shown in densitometry (B). The
protein expression from 3 months HFD mice were normalized to the expression from 3
weeks HFD mice. All data is shown as Mean ± SEM for three mice per group. *p<0.05 3
months vs. 3 weeks.
3 weeks HFD 3 months HFD
CHOP
Caspase-12
(Full)
Caspase-12
(Cleaved)
β - actin

87
(A) (B)
Figure 2-20 The effects of 3 months LFD/HFD on UPR-related gene expressions in the
brain. These bar graphs show the results of comparing the UPR-related gene expression
between SED groups of each diet group (3 months LFD and 3 months HFD). The results
of HFD were normalized to the ones of LFD and were shown as Mean ± SEM. **p<0.01
HFD vs. LFD.

88
CHAPTER 3
THE EFFECT OF IGF-1 AND EXERCISE ON UPR ACTIVATED BY
VOLUNTARY RUNNING EXERCISE
INTRODUCTION
In chapter 2, the preliminary data showed that three weeks voluntary running
wheel exercise up-regulated UPR in multiple brain regions of C57BL/6 mice without
activating any apoptotic signaling pathway such as CHOP, caspase-12, and caspase-3.
Because uncontrolled UPR activity was shown to lead to programmed cell death,1
another question addressed in this thesis is to identify what is the mechanism that leads
to down-regulation of apoptosis despite exercise-induced UPR activation. Exercise is
known to activate many trophic factors such as Insulin-like growth factor-1 (IGF-1),2-3
Brain-derived neurotrophic factor (BDNF),4 Vascular endothelial growth factor (VEGF),
5
and galanin.6 Hence, it is possible that UPR up-regulation or suppression of apoptosis
induced by running wheel exercise could result from activation of these trophic factors.
Specifically, in this thesis chapter, the role of IGF-1 was studied to identify if this trophic
factor has an effect on exercise induced UPR up-regulation.
Recently, it has been shown that trophic factors regulate UPR. BDNF, one of the
neurotrophic factors, is known to control UPR regulation in the brain, contributing to
brain health. Using in situ hybridization, Hayashi et al. (2007) found that the mRNA level
of XBP1 spliced form (XBP1s) was greatly up-regulated in developing mouse brain as
compared to the developed mouse brain.7 Furthermore, they identified that treatment with
BDNF prompted mRNA expression of both Xbp1 and Eif2α in cultured hippocampal
neurons.7 Meanwhile, BDNF had a significant effect on inhibiting apoptotic signaling

89
(caspase-12) in vitro in the presence of an ER stress inducer tunicamycin.8 On the other
hand, exercise appears to have a significant effect on activating Bdnf mRNA expression
in the hippocampus which is the principal site for cognitive function.9-10
BDNF
upregulation followed by exercise was shown to lead to the improvement of learning and
memory.11-12
Gomez-Pinilla et al. (2008) also showed that exercise has a significant
effect on cognitive function by using a strong BDNF receptor inhibitor, a recombinant
TrkB-IgG chimera, to impair BDNF action.11
Wheel running exercise increased Bdnf
mRNA expression in the hippocampus of Alzheimer’s disease mice model (Amyloid
precursor protein [APP]-23 mice) without any changes in spatial learning and
neurogenesis.13
Another trophic factor IGF-1, a polypeptide of 7,500 kDa, is known to have
common downstream signaling with BDNF. After binding to the TrkB receptor, a BDNF
receptor, BDNF activates the phosphatidylinositol 3-kinase (PI3-K) via IRS-1 and -2 14
which are known to be a common component in intracellular signaling pathways of IGF-
1.15
IGF-1 is produced in the liver and secreted into the circulation. It regulates the action
of growth hormone for body development and tissue remodeling in the body.16-17
In high
concentrations (150-400 ng per ml), it is transported in plasma while bound to IGF
binding proteins (IGFBPs) and only a small amount (less than 1% of total) of free IGF-1
is available to bind to their receptors (IGF-1 receptors).18
IGF-1 activity, transport and
half-life is regulated by the presence of IGFBPs.19
Recently, it has been shown that IGF-1
has a critical ability in the central nervous system, contributing to the differentiation and
proliferation of neuronal cells as well as promoting their survival in vitro and in vivo.20

90
Circulating peripheral IGF-1 can cross the blood brain barrier (BBB) and enter into the
brain where it binds to the IGF-1 receptor, a receptor of the tyrosine kinase family.
IGF-1 actions in the brain are important for brain health and this could mediate
some of the effects of exercise.21-22
Carro et al. (2000) showed that 1 hour treadmill
exercise increased the uptake of systemically applied IGF-1 into the rat brain. This was
confirmed by staining digoxigenin-labeled IGF-1 in the brain.21
The role of peripherally
produced IGF-1 in regulating the brain’s response to exercise was shown using anti-IGF-
1 antibody to block the access of IGF-1 into the brain from the circulation.21
The data
suggest that IGF-1 entering into the brain elicits the exercise-related neuronal activation
that can be detected by changes in electrophysiological properties.21
In genetically
modified mice whose liver IGF-1 gene was ablated and circulating IGF-1 level was
significantly lowered, the hippocampal function for spatial learning in the water maze test
was greatly reduced.22
IGF-1, systemically injected, rescued this impaired brain
function.22
IGF-1 acts in the hippocampus. Using autoradiography with [125
I]IGF-1, it was
shown that IGF-1 receptors were located in the dentate gyrus of the hippocampus.23
Aberg et al. (2000) showed that systemically applied IGF-1 had a capacity to induce
neurogenesis even in the adult rat hippocampus.24
They showed that animals treated with
IGF-1 peripherally for either 6 or 20 days up-regulated the proliferation of neural cells in
the adult dentate gyrus and increased neural differentiation selectively in comparison to
astrocytes.
It has been suggested that IGFBP could be a possible mediator of the response to
ER stress. The activity of the bZIP-activating transcription factor 4 (ATF4), which is

91
activated by phosphorylated eIF2α, affects the level of IGFBP which is secreted and
regulates the activity of circulating free IGF-1.25
In this study, ER stress induction with
tunicamycin increased IGFBP-1 mRNA expression. The role of ATF4 was confirmed by
using Atf4 knockout mice. Recent studies have suggested that there is a linkage between
UPR and IGF-1 signaling. There have been some trials showing that IGF-1 signaling
works downstream of an upregulated UPR mechanism in mediating its effects. It was
shown that IGF-1 treatment reversed the ER stress inducible effect of tunicamycin in
vivo and that it also decreased the apoptotic signaling of caspase-3 activity in PC-12
neuronal cells.26
In this chapter, the effect of voluntary running wheel exercise on UPR activity
was studied in the brain of mice whose circulating IGF-1 action was inhibited by anti-
IGF-1 antibody.
HYPOTHESES
(1) IGF-1 will increase UPR activity in the brain.
(2) IGF-1 will increase exercise-activated UPR in the brain.
METHODS
Animals and Diets
Thirty-two male C57BL/6 mice, 6 weeks of age, were used in this experiment. As
in chapter 2, all mice had a 1 week adaptation period once they arrived at the animal
room. During this period, all mice were randomly grouped into four groups;
Saline/Sedentary (Sed), Saline/Run, anti-IGF-1/Sed, and anti-IGF-1/Run. Each group was
composed of eight animals and an osmotic pump filled with saline or anti-IGF-1 antibody
was implanted in each mouse of each treatment group. In this study, all mice were fed

92
with 10% low fat diet (D 12450B, Research Diets, New Brunswick, NJ). Unlike the
experiment of chapter 2 in which food was given in feeding cups, mice had access to
food on the cage lid in this experiment. The animal room was kept at 22-23 °C with a 12-
hour light/dark cycle. This animal study was approved by the Utah State University
Institutional Animal Care and Use Committee (USU-IACUC).
Voluntary Running Wheel Exercise
The detailed exercise protocol was described in the methods of chapter 2. In this
study, only 14 days running period was applied because the working duration of the
osmotic pumps used in this study was only 14 days. Briefly, running mice (n=8) in each
treatment group (Saline or anti-IGF-1) were given free access to running wheels and their
activity (turns per hour) was automatically recorded and stored in the computer
(VitalView program, Mini Mitter, OR) every hour. After completing the 14 days running
protocol, running mice were grouped into two groups depending on their running activity;
Low Runner (LR, n=4) and High Runner (HR, n=4). In this study, only high running
mice were used as running group for the future analysis of mRNA expression:
Saline/RUN or anti-IGF-1/RUN. Saline/SED or anti-IGF-1/SED were used as control
groups.
Osmotic Minipumps
Following the experimental protocol of Carro et al. (2000),21
a rabbit polyclonal
anti-IGF-1 antibody (SC-9013, Santa Cruz Biotechnology, Santa Cruz, CA) was used to
block the entrance of circulating IGF-1 into the brain. Osmotic minipumps (Alzet micro-
osmotic pump, model 1002, Durect Corporation, Cupertino, CA) were used to deliver the
anti-IGF-1 antibody or saline vehicle. The reservoir volume of this pump is 90 µL (±10

93
µL), pumping rate is 0.25 µL per hour, and pumping duration is 14 days. Using a small
syringe (1 mL), pumps were filled with 200 μg of rabbit polyclonal anti-IGF-1 antibody
(20% in saline) or saline under sterile conditions. After inhalational anesthesia with
isoflurane, a small incision was made in the skin in the back neck region of each mouse
and the subcutaneous connective tissue was spread to form a small pocket where each
pump filled with anti-IGF-1 antibody or saline was implanted. After implantation, the
incised skin was closed with an animal wound clip.
RNA Isolation and Purification
The detailed method for RNA isolation and purification was described in chapter
2. In this study, the hypothalamus and hippocampus were used and the liver was also
assessed. Briefly, brain and liver samples were homogenized in TRI reagent (Molecular
Research Center, Cincinnati, OH) using a motor driven Teflon pestle. After adding 100
uL 1-bromo-3-chloropropane (BCP), samples were centrifuged at 13,000 rpm at 4 °C for
15 min. The aqueous upper phase was transferred to a new 1.5 mL tube and 500 uL
isopropanol added. After centrifugation, RNA was precipitated with 75 % (v/v) ethanol
and dried out at room temperature for 5-10 min. The RNA pellets were solubilized in
RNase-free water and heated at 65 °C for 5 min. Extracted RNA samples were cleaned-
up by a TURBO DNA-freeTM kit (Ambion, Inc., Austin, TX). The extracted RNA
quantity was assessed by the A260 absorbance and by 1.5% agarose gel electrophoresis.
Quantitative Real-time PCR
The detailed procedure for real-time PCR was described in chapter 2. In this
experiment, KAPA SYBR green (KAPA Biosystems, Woburn, MA) was used instead of
Quanta SYBR green used in the experiments of chapter 2. Gene expression was

94
normalized to the expression of the reference gene cyclophilin b. PCR reaction cycles
were set as 1 cycle for 20 seconds at 95 °C and then 40 cycles for 1 second at 95 °C and
30 second at 60 °C. Primers additionally used in this chapter were:
Igf1: Forward, 5’-CCTAACACTTGTATTTGTTGAATTTG-3’,
Reverse, 5’-ACAGATGGAGTCAGGTACGTTAAA-3’;
Bdnf: Forward, 5’-AATGTTCCACCAGGTGAGAAGAG-3’,
Reverse, ‘5-TGCAACCGAAGTATGAAATAACCA-3’
Statistics
All data are shown as Mean ± SEM and analyzed using the Prism 5 for windows
(GraphPad, La Jolla, CA). To assess the interaction between exercise and anti-IGF-1 on
each parameter, two-way ANOVA was used. Individual groups were compared with t-
test. Only values of p<0.05 were considered as statistically significant.
RESULTS
Voluntary Running Wheel Exercise
After 14 days of voluntary running wheel exercise, running mice of each
treatment group (Saline or anti IGF-1) were separated into two groups depending on their
running activity; Low Runner (LR, n=4) and High Runner (HR, n=4). For further
analysis, only HR mice of each group were used as running mice. Panel A of Figure 3-1
shows the plotted result of running activity of individual animals for 14 days running
period. As shown in panel B, blocking circulating IGF-1 with the anti-IGF-1 antibody
had no effect on running activity for the 14 days running period and there was no
significant difference between LR and HR of saline or anti-IGF-1 treated mice. As shown
in panel C, HR mice of each treatment group seemed to increase their activity from the

95
second day and there was a significant difference between LR and HR mice treated with
saline at the end of running period.
The Effect of Exercise and Anti-IGF-1
on Change of Body Weight
Figure 3-2 shows the effect of voluntary running exercise and anti-IGF-1
treatment on the change of body weight after the 14 days running period. There was no
effect of anti-IGF-1 antibody on body weight. HR mice (HR, n=4) in both treatment
groups (Saline or anti-IGF-1) had a reduced body weight gain and this decrease was
significant in Saline mice. Here, ANOVA indicated that there was no significant
interaction between anti-IGF-1 and exercise (F1,20 = 0.5489, p=0.4674). However, when
considering all running animals (n=8), the exercise effect on the change of body weight
disappeared.
The Effect of Exercise and Anti-IGF-1
on Expression of UPR-related Genes in the Brain
In the hypothalamus, both exercise and anti-IGF-1 antibody affected the
expression of Xbp1 and Eif2α (Figure 3-3). Compared to saline treated SED mice,
treatment with anti-IGF-1 antibody induced a big increase in the level of both genes in
this brain region of SED mice and the increase in Eif2α gene expression level reached
statistical significance. Running (HR) had no effect on either Eif2α or Xbp1 gene
expression in the control mice. However, comparisons between HR group and SED group
in mice treated with anti-IGF-1 showed that exercise significantly decreased the
expression of both Xbp1 and Eif2α. In the hippocampus, only Xbp1 gene expression was
affected by anti-IGF-1 and exercise. Anti-IGF-1 treatment increased Xbp1 gene
expression compared to SED group of saline. Running exercise significantly lowered this

96
gene expression in the hippocampus of anti IGF-1 treated mice. However, the other UPR
related gene expressions (Atf6, Eif2α, and Grp78) were not changed by anti IGF-1
treatment or exercise in either the hypothalamus or hippocampus.
The Effect of Exercise and Anti-IGF-1 on the Expression
of IGF-1 and BDNF in the Brain
Using real-time PCR, gene expression of both Igf1 and Bdnf were assessed in the
hypothalamus and hippocampus of running and sedentary mice treated with either saline
or anti-IGF-1 antibody (Figure 3-4). Igf1 mRNA expression was reduced by running
wheel exercise in the hypothalamus regardless of blocking circulating IGF-1 although the
effect did not reach statistical significance probably due to the small group sizes. Gene
expression of Igf1 was not changed by exercise in the hippocampus. Neither exercise nor
blocking IGF-1 had any effect on Bdnf mRNA expression in either brain region.
The Effect of Exercise and Anti-IGF-1 on UPR
and IGF-1 Gene Expression in the Liver
In addition to the brain, the effects of exercise and antibody for IGF-1 on UPR
related gene expressions were assessed in the liver (Figure 3-5). Treatment with antibody
for IGF-1 had no effect on expression of Xbp1, Eif2α, or Grp78 in the liver. In the saline
treated mice, exercise appeared to decrease the expression of Xbp1, Grp78, and Eif2α but
none of these changes reached statistical significance probably because of small group
sizes. In addition, it was shown that neither exercise nor anti-IGF-1 antibody had any
effect on Igf1 mRNA expression in the liver.

97
DISCUSSION
In chapter 2, the possibility that exercise had a neuroprotective effect through
activation of UPR was suggested. Hence, we addressed the questions of what might
mediate the effect and what was the signaling pathway linking exercise and UPR
regulation. Exercise was identified to have a neuroprotective effect through increasing the
entrance of circulating IGF-1 into the brain 21
and administration of IGF-1 into sedentary
animals having brain damage improved behavioral abnormalities.27-28
Since the inhibitory
role of IGF-1 on ER stress induced apoptosis has been reported along with the effect of
IGF-1 to up-regulate UPR,25-26, 29
it was hypothesized that IGF-1 takes part in the exercise
regulated UPR. This chapter focused on identifying the effect of exercise on UPR in the
brain of mice while their uptake of circulating IGF-1 into the brain was blocked by
treatment with anti-IGF-1 antibody.
In this study, 14 days of running period was set due to the working duration of the
osmotic minipumps. A previous study already confirmed that implantation of minipump
does not affect exercise activity of animals.21
In addition, it was shown that blockage of
circulating IGF-1 did not affect exercise activity in mice. Indeed increased physical
activity normally has a positive correlation with circulating IGF-1 levels.30-31
Moreover,
in experiments of this chapter, only HR mice in each treatment group were set as running
mice in order to maximize the effect of exercise even though there was no significance
between LR and HR.
In saline treated mice, 14 days wheel running exercise of HR mice significantly
lowered weight gain compared to the sedentary mice. However, when considering all
running animals, this significant effect was diminished. Here, the reason for not reaching

98
significance may be that the food given was low-fat diet and that the running duration
was relatively short compared to the previous exercise protocol applied in chapter 2. It
was indicated that the reduction of body weight in response to exercise was attenuated in
anti-IGF-1 treated mice. Application of a polyclonal antibody against human IGF-1
strengthened the effect of IGF-1 on growth in vivo.32-33
In this study, the whole body
weight of dwarf mice was greatly increased by the treatment of anti-IGF-1 antibody
compared to the mice given solely exogenous IGF-1. They suggested that this growth
promoting effect of anti-IGF-1 could result from the effect of antibody on protecting free
IGF-1 from degradation, sustaining IGF-1 availability to promote anabolic activity.
The results presented in this thesis chapter showed that there was a trend of
increase in Xbp1 signaling by blocking circulating IGF-1 level in both hypothalamus and
hippocampus. The Eif2α mRNA expression was also significantly increased in the
hypothalamus by blocking the circulating IGF-1 comparing between sedentary group of
saline and anti-IGF mice. However, the expression of other UPR related genes were not
changed by treatment of anti-IGF-1 antibody. Hu et al. (2007) showed that XBP-1 has a
capacity to increase the IGF-1 transcription as a transcription regulatory factor.34
Considering their study results, it could be thought that the lowered level of IGF-1 that
followed treatment with anti-IGF-1 antibody reversely activated the Xbp1 gene
expression so as to increase the production of IGF-1 protein and maintain the level of
IGF-1. In another study, addition of IGF-1 promoted the acute upregulation of XBP1
protein expression in MCF-7 cells in the presence of the ER stress inducer thapsigargin
for 3 hours although the effect disappeared with longer incubation time.29
Consistent with
the results reported here in which blocking systemic IGF-1 level increased Eif2α mRNA

99
expression in the hypothalamus, it was shown that IGF-1 treatment lowered the
expression of phosphorylated eIF2α protein in the presence of the same ER stress
inducer.29
Other studies showed that addition of IGF-1 increased the expression of ATF6
protein.29
No change in ATF6 expression was detected in response to IGF-1 antibody in
the current study. This discrepancy could be explained by differences in tissues studied
(brain tissue vs. brain cancer cell line).
Voluntary running wheel exercise for 14 days appeared to be effective on
reducing over expression of Xbp1 and Eif2α mRNA induced by blocking IGF-1 level
back to the level of saline treated running mice. However, running wheel exercise seemed
to have no effect on other UPR related gene expression in the brain of mice whose IGF-1
level was blocked with antibody for IGF-1. In contrast to chapter 2, 14 days running
wheel exercise in this experiment did not increase the UPR related gene expression. This
inconsistent result could be a result of the change of feeding method. Unlike previous
studies where food was provided in cups within the cage, food was given on the cage lid
in this experiment. The physical activity of climbing and holding the lid while eating is
very significant and completely alters energy balance. So, it is likely that all animals had
significant exercise levels regardless of the presence of a running wheel (aerobic exercise
or resistant exercise). One supportive result showed that the feeding method (cup feeding
vs. lid feeding) affected voluntary running wheel activity.35
They showed that animals fed
on the lid had significantly less running activity than ones fed in the cup. In our
experiment, however, exercise seemed to reduce the possibly maximally activated UPR
induced by blocking IGF-1.

100
IGF-1 is expressed in both the liver and brain. Running wheel exercise tended to
decrease the Igf1 mRNA expression in the hypothalamus even in the presence of anti-
IGF-1 antibody. In contrast, in the hippocampus, running exercise had no effect on Igf1
mRNA expression. Only a few mice were involved in these studies. However, variable
effects of exercise in brain IGF-1 expression have been reported. While voluntary
running wheel exercise increased the Igf1 mRNA expression in the cerebral cortex of
mice 36
and hippocampus of rat,11
another study showed that exercise did not induce any
change in Igf1 mRNA expression in the rat hippocampus.21
One possible explanation for
lowered Igf1 mRNA level in hypothalamus in response to exercise in my study is that
running wheel exercise could increase the level of free IGF-1 by exceeding the capacity
of IGF-1 antibody to bind IGF-1 which, in turn, could be sustained by binding with
IGFBP.37-38
This increased circulating IGF-1 level could inhibit Igf1 gene transcription in
the brain. This would explain the decreased Igf1 mRNA expression level in exercising
mice. Alternatively, an exercise-induced increased entrance rate of circulating IGF-1 into
the brain 21
may also lead to decrease the Igf1 mRNA level.
In the small number of mice used, I was unable to show the expected increase in
Bdnf mRNA expression with exercise.4, 9
Again this probably is a result of the feeding lid
protocol that gave the control mice significant activity levels. Likewise in the brain, it
was shown that inhibition of IGF-1 action had no effect on UPR related genes expression.
Although experiments in the previous chapter showed that exercise increased UPR
related gene expression in the liver, this effect of exercise was not found in the current
experiment regardless of the presence of antibody for IGF-1. This difference could be
induced by feeding method, the small number of animals and different duration of

101
exercise. Blocking IGF-1 had no effect on Igf1 mRNA expression in the liver and
exercise also did not give any effect on changing Igf1 mRNA expression in both
treatments of saline and anti-IGF-1. Though one study identified an effect of exercise on
increasing in Igf1 mRNA expression in the liver,39
this different result could be come
from different exercise protocol applied, 4 weeks treadmill exercise using rats.
Taken together, the data suggest that circulating IGF-1 takes part in the regulation
of XBP1 and eIF2α in the brain because blocking IGF-1 led to increase expression of
these mRNAs. Voluntary running wheel exercise appeared to release the increased ER
stress induced by the presence of anti-IGF-1, not up-regulating the UPR. This preliminary
study has a number of limitations that include not assessing whether anti-IGF-1 antibody
affects transport of IGF-1 into the brain or how circulating IGF-1 levels were changed. In
conclusion, increased IGF-1 expression in response to exercise cannot be responsible for
mediating normal increase in UPR in the brain. Instead, it could be speculated that IGF-1
could be responsible for reducing apoptosis.
REFERENCES
1. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J
Clin Invest. 2002;110(10):1389-98.
2. Cappon J, Brasel JA, Mohan S, Cooper DM. Effect of brief exercise on
circulating insulin-like growth factor I. J Appl Physiol. 1994;76(6):2490-6.
3. Schwarz AJ, Brasel JA, Hintz RL, Mohan S, Cooper DM. Acute effect of brief
low- and high-intensity exercise on circulating insulin-like growth factor (IGF) I, II, and
IGF-binding protein-3 and its proteolysis in young healthy men. J Clin Endocrinol Metab.
1996;81(10):3492-7.
4. Gomez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise
induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol.
2002;88(5):2187-95.

102
5. Waters RE, Rotevatn S, Li P, Annex BH, Yan Z. Voluntary running induces fiber
type-specific angiogenesis in mouse skeletal muscle. Am J Physiol Cell Physiol.
2004;287(5):C1342-8.
6. Holmes PV, Yoo HS, Dishman RK. Voluntary exercise and clomipramine
treatment elevate prepro-galanin mRNA levels in the locus coeruleus in rats. Neurosci
Lett. 2006;408(1):1-4.
7. Hayashi A, Kasahara T, Iwamoto K, et al. The role of brain-derived neurotrophic
factor (BDNF)-induced XBP1 splicing during brain development. J Biol Chem.
2007;282(47):34525-34.
8. Shimoke K, Utsumi T, Kishi S, et al. Prevention of endoplasmic reticulum stress-
induced cell death by brain-derived neurotrophic factor in cultured cerebral cortical
neurons. Brain Res. 2004;1028(1):105-11.
9. Neeper SA, Gomez-Pinilla F, Choi J, Cotman CW. Physical activity increases
mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain
Res. 1996;726(1-2):49-56.
10. Vaynman S, Ying Z, Gomez-Pinilla F. Interplay between brain-derived
neurotrophic factor and signal transduction modulators in the regulation of the effects of
exercise on synaptic-plasticity. Neuroscience. 2003;122(3):647-57.
11. Gomez-Pinilla F, Vaynman S, Ying Z. Brain-derived neurotrophic factor
functions as a metabotrophin to mediate the effects of exercise on cognition. Eur J
Neurosci. 2008;28(11):2278-87.
12. Dishman RK, Berthoud HR, Booth FW, et al. Neurobiology of exercise. Obesity.
(Silver Spring) 2006;14(3):345-56.
13. Wolf SA, Kronenberg G, Lehmann K, et al. Cognitive and physical activity
differently modulate disease progression in the amyloid precursor protein (APP)-23
model of Alzheimer's disease. Biol Psychiatry. 2006;60(12):1314-23.
14. Yamada M, Ohnishi H, Sano S, Nakatani A, Ikeuchi T, Hatanaka H. Insulin
receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with
phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured
cerebral cortical neurons. J Biol Chem. 1997;272(48):30334-9.
15. Myers M, Jr, Sun X, Cheatham B, et al. IRS-1 is a common element in insulin and
insulin-like growth factor-I signaling to the phosphatidylinositol 3'-kinase. Endocrinology.
1993;132(4):1421-1430.

103
16. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins:
biological actions. Endocr Rev. 1995;16(1):3-34.
17. Nishijima T, Piriz J, Duflot S, et al. Neuronal activity drives localized blood-
brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron.
2010;67(5):834-46.
18. Clemmons DR. Value of insulin-like growth factor system markers in the
assessment of growth hormone status. Endocrinol Metab Clin North Am. 2007;36(1):109-
29.
19. Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding
proteins. Endocr Rev. 2002;23(6):824-54.
20. Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth
factor system and its pleiotropic functions in brain. Endocr Rev. 2005;26(7):916-43.
21. Carro E, Nunez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth
factor I mediates effects of exercise on the brain. J Neurosci. 2000;20(8):2926-33.
22. Trejo JL, Carro E, Garcia-Galloway E, Torres-Aleman I. Role of insulin-like
growth factor I signaling in neurodegenerative diseases. J Mol Med. 2004;82(3):156-62.
23. Lesniak MA, Hill JM, Kiess W, Rojeski M, Pert CB, Roth J. Receptors for
insulin-like growth factors I and II: autoradiographic localization in rat brain and
comparison to receptors for insulin. Endocrinology. 1988;123(4):2089-99.
24. Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS. Peripheral
infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J
Neurosci. 2000;20(8):2896-903.
25. Marchand A, Tomkiewicz C, Magne L, Barouki R, Garlatti M. Endoplasmic
reticulum stress induction of insulin-like growth factor-binding protein-1 involves ATF4.
J Biol Chem. 2006;281(28):19124-33.
26. Zou CG, Cao XZ, Zhao YS, et al. The molecular mechanism of endoplasmic
reticulum stress-induced apoptosis in PC-12 neuronal cells: the protective effect of
insulin-like growth factor I. Endocrinology. 2009;150(1):277-85.
27. Carro E, Trejo JL, Busiguina S, Torres-Aleman I. Circulating insulin-like growth
factor I mediates the protective effects of physical exercise against brain insults of
different etiology and anatomy. J Neurosci. 2001;21(15):5678-84.

104
28. Fernandez AM, de la Vega AG, Torres-Aleman I. Insulin-like growth factor I
restores motor coordination in a rat model of cerebellar ataxia. Proc Natl Acad Sci U S A.
1998;95(3):1253-8.
29. Novosyadlyy R, Kurshan N, Lann D, Vijayakumar A, Yakar S, LeRoith D.
Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via
enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ.
2008;15(8):1304-17.
30. Eliakim A, Brasel JA, Mohan S, Barstow TJ, Berman N, Cooper DM. Physical
fitness, endurance training, and the growth hormone-insulin-like growth factor I system
in adolescent females. J Clin Endocrinol Metab. 1996;81(11):3986-92.
31. Poehlman ET, Copeland KC. Influence of physical activity on insulin-like growth
factor-I in healthy younger and older men. J Clin Endocrinol Metab. 1990;71(6):1468-73.
32. Stewart CE, Bates PC, Calder TA, Woodall SM, Pell JM. Potentiation of insulin-
like growth factor-I (IGF-I) activity by an antibody: supportive evidence for enhancement
of IGF-I bioavailability in vivo by IGF binding proteins. Endocrinology.
1993;133(3):1462-5.
33. Hill RA, Pell JM. Regulation of insulin-like growth factor I (IGF-I) bioactivity in
vivo: further characterization of an IGF-I-enhancing antibody. Endocrinology.
1998;139(3):1278-87.
34. Hu MC, Gong HY, Lin GH, et al. XBP-1, a key regulator of unfolded protein
response, activates transcription of IGF1 and Akt phosphorylation in zebrafish embryonic
cell line. Biochem Biophys Res Commun. 2007;359(3):778-83.
35. Harri M, Lindblom J, Malinen H, et al. Effect of access to a running wheel on
behavior of C57BL/6J mice. Lab Anim Sci. 1999;49(4):401-5.
36. Nakajima S, Ohsawa I, Ohta S, Ohno M, Mikami T. Regular voluntary exercise
cures stress-induced impairment of cognitive function and cell proliferation accompanied
by increases in cerebral IGF-1 and GST activity in mice. Behavioural Brain Research.
2010;211(2):178-84.
37. Chicharro JL, Lopez-Calderon A, Hoyos J, et al. Effects of an endurance cycling
competition on resting serum insulin-like growth factor I (IGF-I) and its binding proteins
IGFBP-1 and IGFBP-3. Br J Sports Med. 2001;35(5):303-7.

105
38. Koistinen H, Koistinen R, Selenius L, Ylikorkala Q, Seppala M. Effect of
marathon run on serum IGF-I and IGF-binding protein 1 and 3 levels. J Appl Physiol.
1996;80(3):760-4.
39. Zanconato S, Moromisato DY, Moromisato MY, et al. Effect of training and
growth hormone suppression on insulin-like growth factor I mRNA in young rats. J Appl
Physiol. 1994;76(5):2204-9.
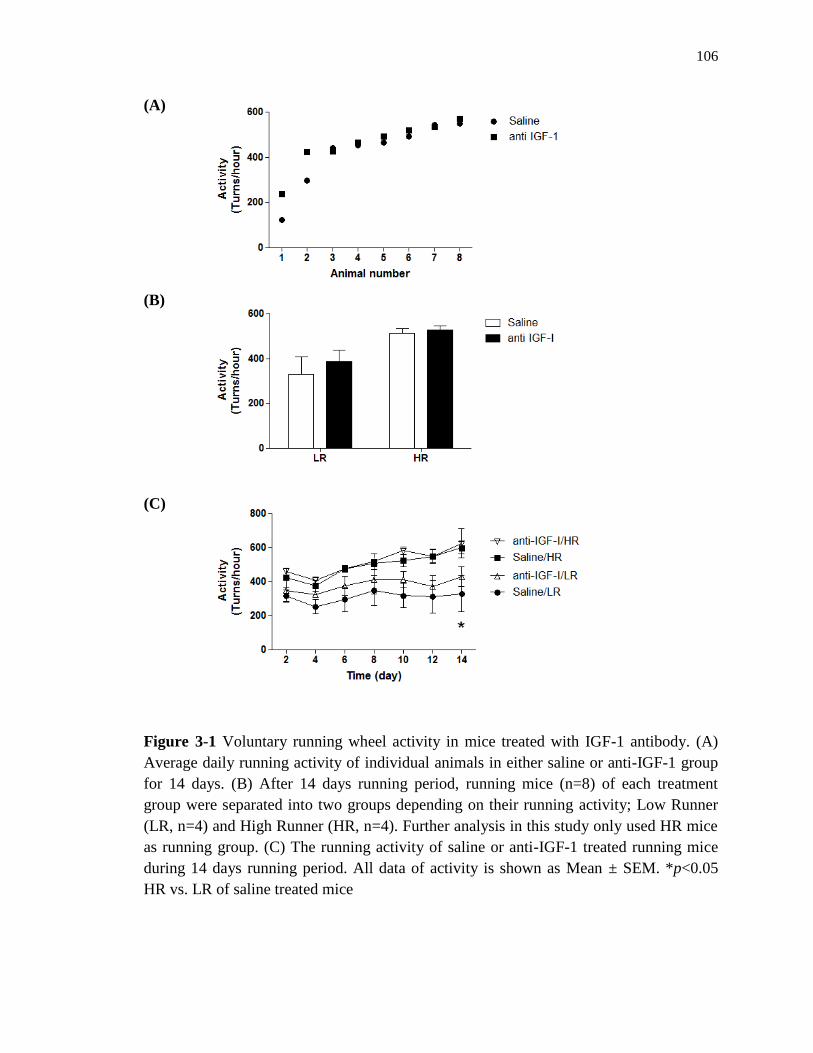
106
(A)
(B)
(C)
Figure 3-1 Voluntary running wheel activity in mice treated with IGF-1 antibody. (A)
Average daily running activity of individual animals in either saline or anti-IGF-1 group
for 14 days. (B) After 14 days running period, running mice (n=8) of each treatment
group were separated into two groups depending on their running activity; Low Runner
(LR, n=4) and High Runner (HR, n=4). Further analysis in this study only used HR mice
as running group. (C) The running activity of saline or anti-IGF-1 treated running mice
during 14 days running period. All data of activity is shown as Mean ± SEM. *p<0.05
HR vs. LR of saline treated mice

107
(A)
(B)
Figure 3-2 The effect of exercise and anti IGF-1 on change of body weight. After 14
days of running period, it was assessed how body weights were changed by both exercise
and antibody for IGF-1. (A) When choosing HR mice (n=4), there was a significant effect
of exercise on the change of body weight. (B) There was no significant effect of exercise
as considering all running animals (n=8). Treatment with antibody for IGF-1 did not
affect the change of body weight comparing SED group of saline and anti IGF-1 mice.

108
(A) Hypothalamus
(B) Hippocampus
Figure 3-3 The effect of exercise and anti IGF-1 on the UPR-related gene expressions in
the brain. Using real-time PCR, UPR-related gene (Xbp1, Atf6, Eif2α, and Grp78)
expressions were assessed in the hypothalamus and hippocampus of mice treated with
saline or anti IGF-1. These groups were again divided into SED (n=4) and HR (n=4)
group. All data is shown as Mean ± SEM. ap<0.05 anti IGF-1 vs. Saline; *p<0.05,
***p<0.001 HR vs. SED.

109
(A) Hypothalamus
(B) Hippocampus
Figure 3-4 The effect of exercise and anti IGF-1 on Igf1 and Bdnf gene expressions in the
brain. The gene levels of Igf1 and Bdnf in the hypothalamus (A) and hippocampus (B)
were also analyzed using real-time PCR. These gene expressions were shown normalized
to the expression of reference gene cyclophilin b. All data is shown as Mean ± SEM.

110
Figure 3-5 The effect of exercise and anti IGF-1 on UPR-related and IGF-1 gene
expressions in the liver. Using liver samples of SED or HR mice treated with saline or
anti IGF-1, the expressions of UPR related genes (Xbp1, Eif2α, and Grp78) and Igf1 gene
were evaluated. Gene expression level is shown normalized to the reference gene
cyclophilin b level. All data is shown as Mean ± SEM.

111
CHAPTER 4
THE EFFECT OF 4-PHENYL BUTYRIC ACID (PBA) ON UPR ACTIVATED BY
VOLUNTARY RUNNING EXERCISE
INTRODUCTION
As mentioned in previous chapters, many pathological events can be linked to ER
stress and cells activate a defensive mechanism called the UPR. However, if cellular
stress is prolonged, the UPR mechanism might be insufficient and cells can be overridden
with the accumulation of unfolded proteins, leading to a cellular death program. To gain
supportive evidence for the role of UPR to promote the folding of unfolded proteins and
release ER stress, the use of chemical chaperones, such as 4-phenyl butyric acid (PBA),
trimethylamine N-oxide dehydrate (TMAO) and dimethyl sulfoxide 1 has been suggested.
PBA was applied to promote the trafficking of cystic fibrosis transmembrane
conductance regulator protein (CFTR) 2-3
and was also approved as a drug for urea cycle
disorder by the FDA.4 This chemical chaperone, a chemical of low molecular weight, was
shown to cross the blood-brain barrier.5 Other studies have confirmed that this chemical
can confer positive effects in the brain through releasing ER stress.6-7
Ozcan et al. (2006) showed that PBA attenuates obesity induced ER stress along
with improving glucose homeostasis and insulin signaling in peripheral tissues (muscle,
liver, and adipose tissues) of ob/ob mice.8 They identified that expression of both
phosphorylated PERK and phosphorylated IRE-1, which are up-regulated in ob/ob mice,
were significantly decreased by PBA treatment. Additionally, tyrosine phosphorylation of
both the insulin receptor (IR) and insulin receptor substrate-1 (IRS-1) were greatly up-
regulated by PBA. Another study identified that adipocyte differentiation induced by

112
over-nutrition was decreased by treatment with PBA.9 In this study, it was shown that the
expression of UPR related proteins GRP78 and phosphorylated eIF2α that is activated
during adipocyte differentiation was significantly reduced by PBA treatment in vitro.
Another study identified that PBA incubation reduced the expression of obesity-linked
MC4R variants in the brain which is known to be linked to up-regulation of the
expression of GRP78 and XBP1.10
Many studies have been conducted to reveal the effect of PBA in the brain and
have suggested the possibility of using PBA for treatment of neurodegenerative diseases
such as Alzheimer’s disease (AD). Administration of PBA appeared to improve memory
function as assessed by a conditioning paradigm in young- and old-AD model mice,
Tg2576.11
Interestingly, this study showed that PBA treatment had an effect on up-
regulating GRP78 level in old AD mice while eIF2α level was decreased by this chemical
chaperone. Wiley et al. (2010) showed that PBA has the capacity to reverse the inhibition
of β-amyloid precursor protein (APP) cleavage which is the pathological characteristic of
AD and is induced by ER stress inducers such as thapsigargin and tunicamycin in
neuronal cells.12
Most recently, they also identified that PBA application decreased the
formation of amyloid plaque in the cortex and hippocampus by up-regulating APP
proteolysis in vivo using AD mice model.6 Along with reduced size of plaques in these
brain regions, cognitive function of AD mice appeared to be improved by this PBA
treatment.
As shown in previous chapters, UPR in both the CNS and liver are affected by
exercise. However, to our knowledge, there has been no trial to study if PBA would
inhibit the exercise induced increase in the UPR. The purpose of the studies reported in

113
this chapter was to confirm the effect of the chemical chaperone PBA on the UPR in the
brain of C57BL/6 mice. Furthermore, the other purpose of this study is to show if PBA
could block the exercise induced increase in UPR in the brain of C57BL/6 mice.
HYPOTHESES
(1) PBA will down-regulate UPR in the brain.
(2) The effect of exercise on up-regulating UPR in the brain will be reduced in the
presence of PBA.
METHODS
Animals and Diets
Total thirty two male C57BL/6 mice (~6 weeks old) were used and this animal
study was approved by the Utah State University Institutional Animal Care and Use
Committee (USU-IACUC). As in other chapters, 1 week was allowed for all animals to
acclimate to their new environment after they arrived at the animal room. All mice were
randomly separated into four groups: Water/Sedentary (Sed) (n=6), Water/Run (n=10),
PBA/Sed (n=6), and PBA/Run (n=10). During the experimental period, a 10 % low fat
diet (D 12450B, Research Diets, New Brunswick, NJ) was given to animals that could
freely access food on cage lid and water. The animal room was automatically controlled,
being maintained at 22-23 °C with a 12-hour light/dark cycle.
4-phenylbutyric acid (PBA)
4-phenylbutyric acid sodium salt (Scandinavian Formulas, Inc., PA) was given to
mice in solubilized in drinking water at a concentration of 2.5 mg/ml to provide a dose of
1g/kg/day. The control group received drinking water without PBA. All mice had access

114
to drinking bottles ad libitum. The amount of water intake was measured every second
day and drinking bottles were replaced after 1 week.
Voluntary Running Wheel Exercise
The detailed exercise protocol was described in the methods of chapter 2. In this
study, mice in the running groups receiving either Water or PBA were allowed to freely
run on wheels for 14 days as in chapter 3. Running activity of mice (n=10) in each group
(Water or PBA) was automatically recorded as turns per hour during 14 days of running
using the VitalView program (Mini Mitter, OR). In addition, 6 mice in each group were
used as control mice (Sedentary mice, SED) and were kept in locked wheel cages during
the running period. Depending on their running activity during running period, running
mice were grouped into Low Runner mice (LR, n=4) and High Runner mice (HR, n=4)
and only HR mice were used for further experiments in this chapter.
RNA Isolation and Purification
The detailed method for RNA isolation and purification was described in the
chapter 2. In this study, the hypothalamus and hippocampus were used for RNA analysis.
Quantitative Real-time PCR
The detailed procedure for real-time PCR was described in chapter 2. As in
chapter 3, KAPA SYBR green (KAPA Biosystems, Woburn, MA) was used for assessing
gene expression which was standardized to the expression of the reference gene
cyclophilin b. PCR reaction cycles were shown in chapter 3.

115
Statistics
For statistical analysis, Prism 5 for windows (GraphPad, La Jolla, CA) was used
and all data is shown as Mean ± SEM. To assess the interaction between exercise and
PBA on each parameter, two-way ANOVA was used. Individual groups were compared
with t-test. Only values of p<0.05 were considered as statistically significant.
RESULTS
Voluntary Running Wheel Exercise
Total 10 mice of each group (Water or PBA) had voluntary running wheel
exercise for 14 days and were grouped into Low Runner (LR, n=4) and High Runner (HR,
n=4) depending on their running activity during the running period (Figure 4-1, A). As
shown in (B) of Figure 4-1, HR mice of both treatment group had significant more
activity as compared to LR mice. As shown in (C), high running activity of HR mice in
both groups appeared to be significantly increased 2 day after the start of running relative
to LR mice and the increase was maintained during the continued running period.
Comparing running activity within either LR or HR group of each group (Water or PBA),
PBA did not affect running activity.
The Effect of Exercise and PBA on Change of Body Weight
As shown in Figure 4-2, 14 days treatment with PBA had no effect on the change
of body weight of SED or RUN mice. HR mice had a tendency toward lower body
weight gains in both water or PBA groups even though there was no statistical
significance. When considering all 10 running animals (B of Figure 4-2), 14 days running
exercise appeared to have no effect on decreasing body weight. The result of ANOVA
indicated that neither PBA nor exercise had any significant effect on the change of body

116
weight and that there was no significant interaction between these two factors (PBA ×
exercise) (F1, 15 = 0.05852, p=0.8121).
The Effect of Exercise and PBA on Food Intake
PBA decreased food intake on day 1 after which food intake increased to levels
similar to those in the water control groups (Figure 4-3, A). As shown in panel A,
running mice (RUN) in each treatment group had higher daily food intake during the 14
days running period as compared to SED mice. The significant differences between RUN
and SED mice were found on day 7 in Water mice and days 11 and 13 in PBA mice,
separately. When comparing the cumulative food intake over the 14-day experimental
period, RUN mice in both treatment groups (Water or PBA) had significantly increased
intake of calories compared to the SED mice.
The Effect of Exercise and PBA on Drinking
As shown in (A) of Figure 4-4, there was no significant difference in fluid intake
between SED and RUN mice in either water or PBA treated groups during 14 days of
running period. However, ANOVA data indicated that PBA had an effect on increasing
the amount of cumulative drinking during the experimental period (p<0.01) (Figure 4-4,
B). This effect of PBA on drinking was shown to be statistically significant in SED mice.
The Effect of Exercise and PBA on UPR-related
Genes Expression in the Brain
Even though the change did not reach statistical significance, PBA had a tendency
to down-regulate the expression of both Eif2α and Grp78 in the hypothalamus (Figure 4-
5, A). In this brain region, exercise also decreased the expression of both Eif2α and
Grp78 genes in the control group (Water group) and the significant decrease was

117
observed in the Eif2α gene level. In the hippocampus (Figure 4-5, B), PBA treatment
itself did not have any effect on the expression of UPR-related genes. However, exercise
had an effect of reducing expression of these genes in the hippocampus of mice treated
with PBA and expression of all UPR related genes (Xbp1, Atf6, Eif2α, and Grp78) were
significantly down-regulated by 14 days running wheel exercise. However, from the
result of two-way ANOVA (exercise × PBA), there was a significant effect of exercise in
the expression of Xbp1 and Atf6 and there was no interaction between exercise and PBA.
DISCUSSION
In chapter 2, it was shown that 3 weeks voluntary running wheel exercise up-
regulated the expression of UPR-related genes in the multiple brain regions of mice
exposed to high-fat diet during a variety of periods. Along with these results, it was
questioned how running wheel exercise affects the UPR in the brain of mice in the
presence of a chemical chaperone such as PBA which has a capacity of reducing the
accumulation of unfolded proteins and decreasing ER stress. So, in this chapter, treatment
with PBA was used to reduce ER stress and we investigated its effects on the exercise
induced UPR-related gene expression in the brain of mice.
PBA treatment for 14 days did not have any effect on running activity of mice.
Neither did PBA treatment have any effect on body weight gain during the 14 days of our
study. This is consistent with previous studies. In a study using ob/ob mice, 20 days PBA
oral treatment did not reduce the body weight as compared to vehicle treated mouse even
though blood glucose level and insulin level were significantly decreased during PBA
treatment.8 In addition, 26 days treatment of PBA did not change the body weight in
ob/ob mice as compared to vehicle treated mice.5

118
In line with the absence of any change of body weight with PBA, PBA treatment
did not affect food intake. However, 14 days running wheel exercise had a significant
effect to increase the cumulative food uptake. Though there are variations in running
periods, other studies have also shown that voluntary running wheel exercise increases
food intake 13-14
and they suggested that this increased food intake followed by running
exercise could be one way to compensate the energy budget increased by high physical
activity.
Contrary to another report,6 14 days PBA treatment significantly increased the
amount of drinking comparing SED groups given PBA with those in the Water group. It
is possible that this reflects the osmolality of the 5 mM PBA, affecting the drinking
behavioral patterns.
PBA treatment tended to lower the expression of both Eif2α and Grp78 in the
hypothalamus. Exercise was shown to decrease these gene levels similar to the levels of
PBA treated SED group. A study also showed that 26 days PBA treatment had no effect
on UPR related phosphorylated PERK in the hypothalamus of lean mice while this
chemical significantly lowered the expression of this gene in this brain region of ob/ob
mice.5 To our knowledge, this thesis study is the first trial to study the effect of exercise
in the brain of mice treated with PBA. Running wheel exercise had no additional effect
on UPR related gene expressions in the hypothalamus of mice over treatment of PBA.
However, the control water treated mice did not show the expected effect of exercise on
the up-regulation of UPR. This different result probably reflected the differences in
feeding way (cup vs. lid). Mice in the previous experiments were provided food in a
feeding cup, but food was given on the cage lid in the current study. Harri et al. (1999)

119
showed that food location could affect behavior of mice that had access to running
wheels.15
They showed that feeding from the cage lid decreased the access time to a
running wheel as compared with feeding on floor. Accessing food on a cage lid appears
to be a major form of exercise for mice. Control SED mice in this study were intact
exercising mice.
When looking at the Figure 4-5 (B), 14 days PBA treatment had no effect on UPR
related gene expressions (Xbp1, Atf6, Eif2α, and Grp78) in the hippocampus of SED mice.
As shown in the hypothalamus, it is possible that 14 days treatment could be too short to
induce the change in UPR and, since mice were not exposed to any stressful circumstance
(i.e., high-fat diet), the effect of the chemical chaperone PBA could be limited. In
contrast, running wheel exercise for 14 days had an effect of lowering all UPR genes
expression in the hippocampus in mice treated with PBA. In this brain region, it is
suggested that exercise may promote the action of PBA.
In summary, the chemical chaperone PBA lowered UPR in the brain. However,
this chapter showed inconsistent results to the ones of previous chapter in that exercise
did not increase the UPR related genes expression. This discrepancy could be induced by
the shorter duration of running or to the altered food location used in this experiment.
This complicates the interpretation of the data such that it is impossible to identify if PBA
prevented the increase in UPR activity that was activated with running in the previous
experiments. Running wheel exercise appeared to reduce the expression of UPR related
genes in the hippocampus of mice treated with PBA for 14 days while it had no effect in
the hypothalamus.

120
REFERENCES
1. Welch WJ, Brown CR. Influence of molecular and chemical chaperones on
protein folding. Cell Stress Chaperones. 1996;1(2):109-15.
2. Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of
CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis
epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100(10):2457-65.
3. Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate
(Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal
epithelial CFTR function. Am J Respir Crit Care Med. 1998;157(2):484-90.
4. Newmark HL, Young CW. Butyrate and phenylacetate as differentiating agents:
practical problems and opportunities. J Cell Biochem Suppl. 1995;22:247-53.
5. Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role
in development of leptin resistance. Cell Metab. 2009;9(1):35-51.
6. Wiley JC, Pettan-Brewer C, Ladiges WC. Phenylbutyric acid reduces amyloid
plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell. 2011.
7. Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J,
Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in
an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34(7):1721-32.
8. Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and
restore glucose homeostasis in a mouse model of type 2 diabetes. Science.
2006;313(5790):1137-40.
9. Basseri S, Lhotak S, Sharma AM, Austin RC. The chemical chaperone 4-
phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J
Lipid Res. 2009;50(12):2486-501.
10. Granell S, Mohammad S, Ramanagoudr-Bhojappa R, Baldini G. Obesity-linked
variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can
be rescued to the cell surface by a chemical chaperone. Mol Endocrinol.
2010;24(9):1805-21.
11. Ricobaraza A, Cuadrado-Tejedor M, Marco S, Perez-Otano I, Garcia-Osta A.
Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse
model of Alzheimer disease. Hippocampus. 2010.

121
12. Wiley JC, Meabon JS, Frankowski H, et al. Phenylbutyric acid rescues
endoplasmic reticulum stress-induced suppression of APP proteolysis and prevents
apoptosis in neuronal cells. PLoS One. 2010;5(2):e9135.
13. Swallow JG, Koteja P, Carter PA, Garland T, Jr. Food consumption and body
composition in mice selected for high wheel-running activity. J Comp Physiol B.
2001;171(8):651-9.
14. Koteja P, Swallow JG, Carter PA, Garland T, Jr. Energy cost of wheel running in
house mice: implications for coadaptation of locomotion and energy budgets. Physiol
Biochem Zool. 1999;72(2):238-49.
15. Harri M, Lindblom J, Malinen H, et al. Effect of access to a running wheel on
behavior of C57BL/6J mice. Lab Anim Sci. 1999;49(4):401-5.

122
(A)
(B)
(C)
Figure 4-1 The activity of voluntary running wheel exercise. (A) Running activity of
individual mice in Water or PBA treated groups. (B) Total 10 mice in each treatment
group (Water or PBA) were separated into Low Runner (LR, n=4) and High Runner (HR,
n=4) relative to their activities for 14 days. (C) The running activity of mice given either
water or PBA during running period of 14 days. All data was shown as Mean ± SEM of
four mice in each group. *p<0.05, **<0.01, ***p<0.001 HR vs. LR of Water mice;
#p<0.05, ##p<0.01, ###p<0.001 HR vs. LR of PBA mice
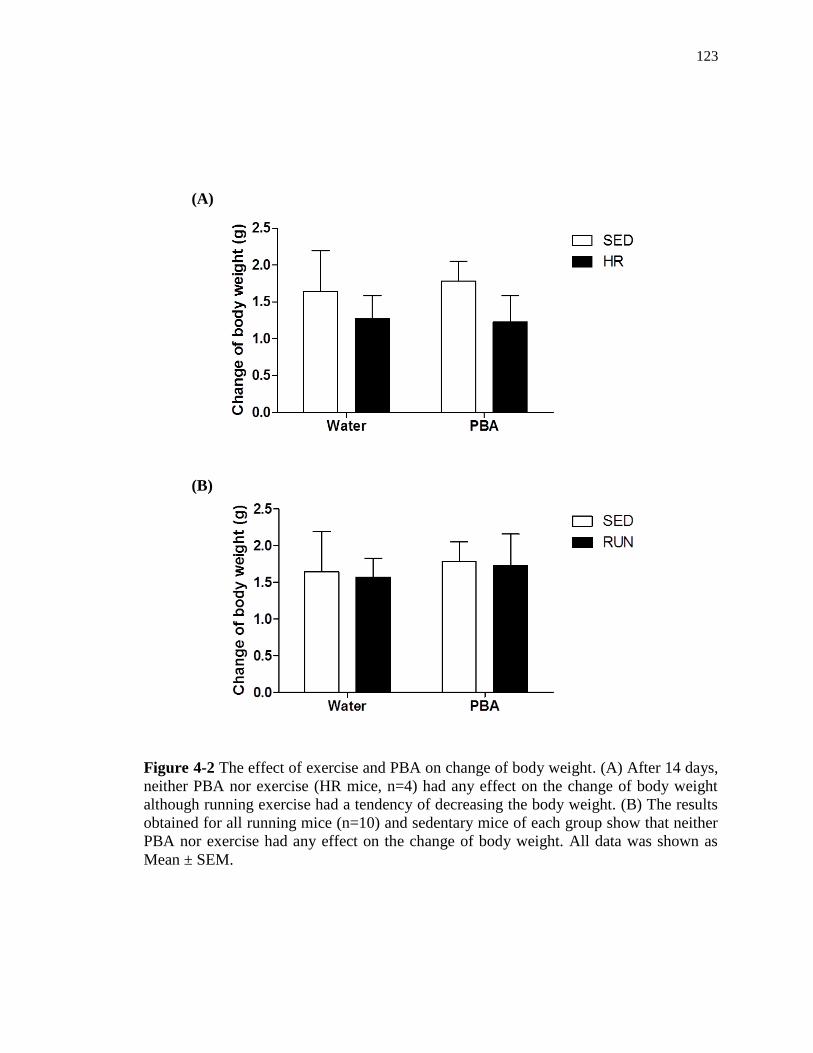
123
(A)
(B)
Figure 4-2 The effect of exercise and PBA on change of body weight. (A) After 14 days,
neither PBA nor exercise (HR mice, n=4) had any effect on the change of body weight
although running exercise had a tendency of decreasing the body weight. (B) The results
obtained for all running mice (n=10) and sedentary mice of each group show that neither
PBA nor exercise had any effect on the change of body weight. All data was shown as
Mean ± SEM.
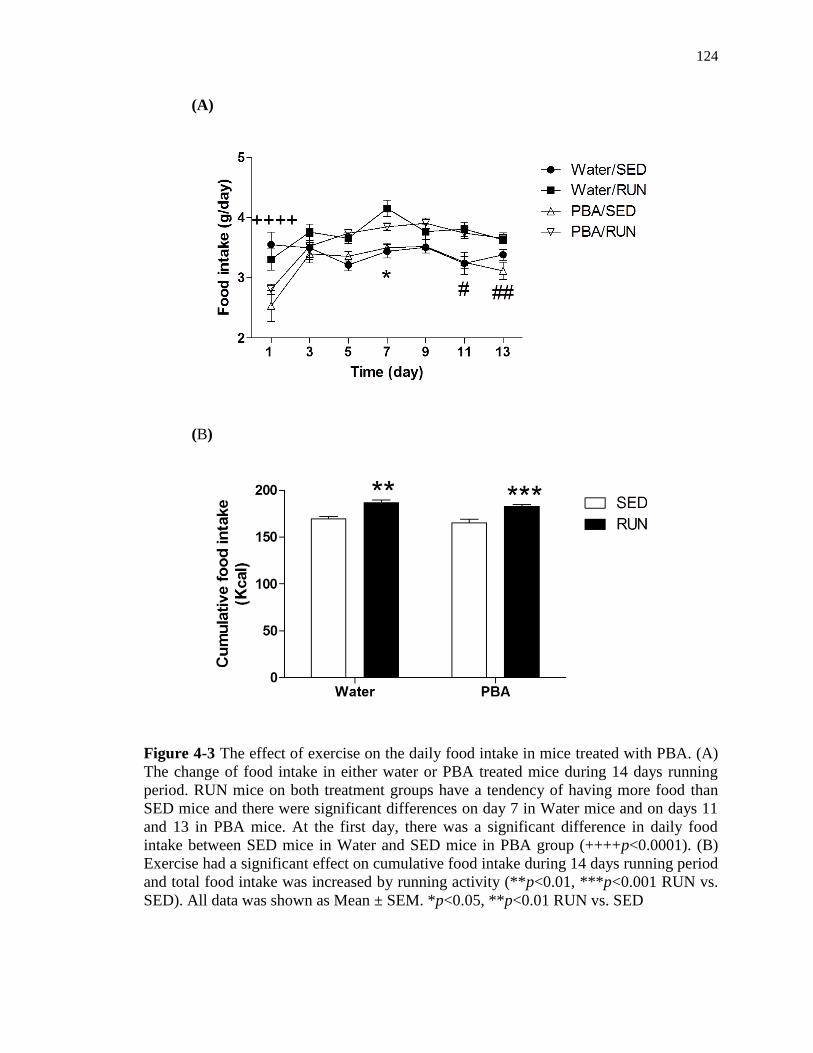
124
(A)
(B)
Figure 4-3 The effect of exercise on the daily food intake in mice treated with PBA. (A)
The change of food intake in either water or PBA treated mice during 14 days running
period. RUN mice on both treatment groups have a tendency of having more food than
SED mice and there were significant differences on day 7 in Water mice and on days 11
and 13 in PBA mice. At the first day, there was a significant difference in daily food
intake between SED mice in Water and SED mice in PBA group (++++p<0.0001). (B)
Exercise had a significant effect on cumulative food intake during 14 days running period
and total food intake was increased by running activity (**p<0.01, ***p<0.001 RUN vs.
SED). All data was shown as Mean ± SEM. *p<0.05, **p<0.01 RUN vs. SED

125
(A)
(B)
Figure 4-4 The effect of exercise and PBA on drinking behavior. (A) The change of daily
water or PBA intake during 14 days. (B) PBA treatment had a significant effect on
cumulative drinking (****p<0.0001 SED PBA vs. SED water). There was no significant
interaction between exercise and PBA.

126
(A) Hypothalamus
(B) Hippocampus
Figure 4-5 The effect of PBA and exercise on expression of UPR genes in the
hypothalamus and hippocampus. In the hypothalamus (A), there was no significant effect
of PBA on the expression of Xbp1 while two genes of both Eif2α and Grp78 had a
tendency to be decreased by PBA treatment as comparing SED mice between Water and
PBA mice. The effect of exercise was observed in expression of both Eif2α and Grp78
genes and Eif2α gene expression was significantly down-regulated by 14 days running
exercise in Water mice (*p<0.05). In the hippocampus (B), PBA treatment had any effect
on all UPR related genes expression (Xbp1, Atf6, Eif2α, and Grp78). In PBA group, the
all UPR related gene expression was significantly decreased by 14 days running wheel
exercise (*p<0.05, **p<0.01 HR vs. SED of PBA mice).

127
CHAPTER 5
OVERALL DISCUSSION AND FUTURE DIRECTIONS
A number of chronic diseases, such as obesity and type 2 diabetes 1-3
and
neurodegenerative diseases such as Alzheimer’s disease (AD),4-6
can be induced with ER
stress. Meanwhile, many studies have shown that exercise has a significant effect on
ameliorating the status/development of obesity 7-8
and type 2 diabetes 9-10
and
neurodegenerative diseases.11-13
In this thesis study, the focus was to understand the
effect of voluntary running wheel exercise on the UPR, a defensive mechanism against
ER stress, in the brain of mice exposed to two dietary circumstances (LFD or HFD) for
short- or long-periods. In addition, the possibility that exercise regulated-UPR activity
could be linked to exercise-responsive IGF-1 signaling in the brain of mice was studied.
Lastly, how the effect of exercise on UPR was changed in the presence of chemical
chaperones which are known to lower ER stress by releasing the accumulation of
unfolded proteins was assessed. So, the overall theme of these studies was to elucidate
the effect of exercise on brain health in terms of regulating UPR.
The regional response
The described results indicated that the UPR to diet and exercise was
differentially regulated in each brain region. Although dietary treatment for 3 weeks did
not activate UPR in the hypothalamus, running wheel exercise activated UPR in this
brain region. One interesting result was that even low running activity up-regulated the
UPR signaling in the hypothalamus of mice fed with HFD. Exercise has a significant
effect on maintaining energy homeostasis by controlling the expression of hypothalamic
neuropeptides such as neuropeptides Y (NPY) and pro-opiomelanocorin (POMC).14
In

128
addition, hypothalamic insulin and leptin signaling is improved by physical activity.
Exercise induced hypothalamic changes were shown to be linked to the reduction of
inflammation (IKKβ/NF-kB) along with reduction of ER stress. So, it is possible that
exercise activated UPR activation in the hypothalamus of mice fed HFD for 3 weeks
could be linked to alterations the expression of neuropeptides as well as to improved
insulin and leptin signaling. When mice were exposed to HFD for 3 months, HFD itself
increased the UPR in the hypothalamus more significantly than in the hippocampus. This
result also suggested that the hypothalamus, the central part for sensing energy status and
keeping energy homeostasis, activated UPR to defeat ER stress induced by a long-term
HFD. However, the effect of exercise on UPR up-regulation was shown more significant
in the hippocampus than in the hypothalamus. This result suggested the possibility that as
the hippocampus is known to take part in cognitive function and memory,15-16
exercise
could have a positive effect on improving cognitive function in the brain of mice exposed
to high-fat diet for a long time.
When circulating IGF-1 was lowered by anti-IGF-1 antibody, UPR signaling in
both the hypothalamus and hippocampus responded similarly. Blocking IGF-1 up-
regulated Xbp1 mRNA expression and appeared to have similar effects in both regions. In
addition, running wheel exercise for 14 days had similar effects in both brain regions,
suggesting that IGF-1 normally down-regulate the UPR in the brain regions. Running
wheel exercise appeared to decrease the UPR that was up-regulated by blocking IGF-1
level in both brain regions. These results suggested that both brain regions respond
similarly to circulating IGF-1.

129
The effect of the chemical chaperone PBA differed depending on the brain region.
While UPR (Eif2α and Grp78) in the hypothalamus seemed to be decreased by PBA,
there was no change in UPR activity of the hippocampus. So, it appeared that the
hypothalamus was more sensitive to PBA treatment than the hippocampus. The effect of
exercise also differed in these two brain regions while in the presence of PBA. In the
hypothalamus, running wheel exercise had no effect on regulating UPR related gene
expressions, while in the hippocampus UPR related genes expression was significantly
reduced by running wheel exercise. It is possible that co-treatment with exercise and PBA
could have an effect on improving cognitive function and memory in the hippocampus by
releasing ER stress.
Taken together, the UPR of each brain region was not responsive simultaneously
to either diet or exercise. Although the responses to IGF-1 and exercise in the
hypothalamus and hippocampus were similar, UPR in these two regions responded
differently to PBA and exercise.
The time-dependent effects of diet and exercise
The effect of diet on the UPR was different depending on how long mice were
exposed to the diet. Although short-term exposure (3 weeks) to HFD itself did not induce
the activation of UPR in the brain, long-term exposure (3 months) to HFD had a
significant effect of up-regulation of UPR. As even HFD feeding for 3 days induced
insulin resistance in the brain,17
it is unlikely that the HFD increase in the UPR is linked
to this change because it is not evident till 3 months. However, voluntary running
exercise activated UPR signaling in the brain of mice fed HFD only for 3 weeks and this
effect of exercise was also shown in the mice fed HFD for 3 months. This exercise

130
induced UPR up-regulation in the brain of mice exposed to short-term HFD could be
linked to exercise-activated insulin sensitivity and this effect could be induced by up-
regulated trophic factors such as BDNF and IGF-1 which, in turn, are activated by
physical activity. However, when exposure time to VHFD was extended over 4 months,
the effect of exercise on activating UPR signaling disappeared. It might be suggested that
the UPR has a maximal limit and if UPR is continuously up-regulated by diet, this could
be linked to cell death.
Although 3 weeks running wheel exercise had an effect on up-regulation of UPR
signaling in the brain of animals fed either LFD or HFD, 14 days running wheel exercise
which was applied in the experiments using anti-IGF-1 antibody and PBA failed to show
an effect on the activation of UPR signaling in the brain. This different result could be
induced by several factors such as the fact that food was placed on the cage lids rather
than in cups in these experiments and that location is associated with a large increase in
physical activity to obtain the food. In summary, UPR in the brain was responsive to diet
and exercise in a time dependent manner.
Possible implications in neurodegenerative disease
This thesis study indicated that exercise induced UPR activation was not linked to
the activation of apoptotic signaling in the brain of mice exposed to either LFD or HFD
for 3 weeks or 3 months. Previous studies have shown that UPR up-regulation was
accompanied with an activation of apoptotic signaling in the brain. Mayer et al. (2010)
showed that treatment of hypothalamic cell with palmitate increased ER stress (eIF2α and
XBP1) along with up-regulation of apoptotic signaling (phosphorylated JNK and cleaved
caspase-3).18
Another study also identified that ER specific apoptosis (caspase-12) was

131
activated in response to disrupted calcium homeostasis in the ER along with ER stress in
vitro and this ER stress induced apoptosis was confirmed using caspase-12 deficient
mice.19
So, it was hypothesized that exercise may prevent UPR associated apoptosis by
activating trophic factors that suppressed apoptotic activation. One such trophic factor,
IGF-1, has an effect on exercise linked UPR activation in the brain. However, it was
revealed that IGF-1 was not responsible for exercise activated UPR signaling, so other
factors (e.g., BDNF) may be involved. On the other hand, it has been shown that UPR
activity is required for preventing or slowing-down the progress of neurodegenerative
disease.20-21
In summary, though the exact mechanisms still remains unknown, exercise
induced UPR activation and suppression of apoptosis might contribute to the known
beneficial effect of exercise on neurodegenerative disease.
Future work
Although this thesis study applied the exercise protocol for only 14 days or 3
weeks, studies of the effects of long-term exercise on UPR activity in the brain of animals
will be needed to provide insight into the possible effects of habitual chronic physical
activity on brain health. Previous studies identified that long term (16 weeks 22
or 3
months 23
) exercise ameliorated the pathological characteristics in neurodegenerative
disease animal models.
An AD animal model could be used to reveal the interactions between running
wheel exercise, UPR signaling, and AD development, identifying the direct effect of
exercise on brain health. Using Alzheimer disease mouse NSE/APPsw-transgenic model,
one research group has already indicated that forced-running has a significant effect on
reducing the pathological phenotype (i.e., the decrease of deposition of Aβ peptides)

132
along with the up-regulation of GRP78.24
Instead of forced-running exercise, the future
study will be able to apply voluntary running wheel exercise to minimize the possible
stress, assessing the change of UPR in the brain of disease model which can be exposed
to HFD for a short-term or a long-term. It will be worth to try to cross UPR gene knock-
out mice with AD mice. It will be likely that the characteristic of AD would be not
induced in this animal model. In addition, it will be possible that exercise protocol will be
also shown to have no effect on up-regulating UPR signaling in this model.
Although IGF-1 in this thesis study was shown not to be responsible for exercise-
induced UPR activation, other trophic factors should be considered to elucidate how
exercise affects the UPR in the brain of mice. Since BDNF has been shown to have an
effect on UPR,25-26
a study of the role of BDNF in exercise-activated UPR without
activating apoptotic signaling is needed. In this future study, BDNF level could be able to
be controlled by the application of the BDNF inhibitor TrkB antibody as used in Gomez-
Pinilla’s research group.27
REFERENCES
1. Tsiotra PC, Tsigos C. Stress, the endoplasmic reticulum, and insulin resistance.
Ann N Y Acad Sci. 2006;1083:63-76.
2. Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal
kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54
Suppl 2:S73-8.
3. Kaneto H, Nakatani Y, Kawamori D, et al. Role of oxidative stress, endoplasmic
reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and
insulin resistance. Int J Biochem Cell Biol. 2005;37(8):1595-608.
4. Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in
Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's
pathology. J Neuroinflammation. 2009;6:41.

133
5. Sekine Y, Takeda K, Ichijo H. The ASK1-MAP kinase signaling in ER stress and
neurodegenerative diseases. Curr Mol Med. 2006;6(1):87-97.
6. Yoshida H. ER stress and diseases. FEBS J. 2007;274(3):630-58.
7. Duggan GE, Hittel DS, Sensen CW, Weljie AM, Vogel HJ, Shearer J.
Metabolomic response to exercise training in lean and diet-induced obese mice. J Appl
Physiol. 2011;110(5):1311-8.
8. Krawczewski Carhuatanta KA, Demuro G, Tschop MH, Pfluger PT, Benoit SC,
Obici S. Voluntary Exercise Improves High-Fat Diet-Induced Leptin Resistance
Independent of Adiposity. Endocrinology. 2011.
9. Haskell-Luevano C, Schaub JW, Andreasen A, et al. Voluntary exercise prevents
the obese and diabetic metabolic syndrome of the melanocortin-4 receptor knockout
mouse. FASEB J. 2009;23(2):642-55.
10. Bradley RL, Jeon JY, Liu FF, Maratos-Flier E. Voluntary exercise improves
insulin sensitivity and adipose tissue inflammation in diet-induced obese mice. Am J
Physiol Endocrinol Metab. 2008;295(3):E586-94.
11. Chen MJ, Russo-Neustadt AA. Running exercise- and antidepressant-induced
increases in growth and survival-associated signaling molecules are IGF-dependent.
Growth Factors. 2007;25(2):118-31.
12. Colcombe SJ, Erickson KI, Raz N, et al. Aerobic fitness reduces brain tissue loss
in aging humans. J Gerontol A Biol Sci Med Sci. 2003;58(2):176-80.
13. Cotman CW, Engesser-Cesar C. Exercise enhances and protects brain function.
Exerc Sport Sci Rev. 2002;30(2):75-9.
14. Ropelle ER, Flores MB, Cintra DE, et al. IL-6 and IL-10 anti-inflammatory
activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and
ER stress inhibition. PLoS Biol. 2010;8(8).
15. Stranahan AM, Zhou Y, Martin B, Maudsley S. Pharmacomimetics of exercise:
novel approaches for hippocampally-targeted neuroprotective agents. Curr Med Chem.
2009;16(35):4668-78.
16. Griffin EW, Bechara RG, Birch AM, Kelly AM. Exercise enhances hippocampal-
dependent learning in the rat: evidence for a BDNF-related mechanism. Hippocampus.
2009;19(10):973-80.

134
17. Boghossian S, Lemmon K, Park M, York DA. High-fat diets induce a rapid loss
of the insulin anorectic response in the amygdala. Am J Physiol Regul Integr Comp
Physiol. 2009;297(5):R1302-11.
18. Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces
endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance
and apoptosis through adenosine 5' monophosphate-activated protein kinase activation.
Endocrinology. 2010;151(2):576-85.
19. Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-
reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature.
2000;403(6765):98-103.
20. Lee do Y, Lee KS, Lee HJ, et al. Activation of PERK signaling attenuates Abeta-
mediated ER stress. PLoS One. 2010;5(5):e10489.
21. Oida Y, Izuta H, Oyagi A, et al. Induction of BiP, an ER-resident protein,
prevents the neuronal death induced by transient forebrain ischemia in gerbil. Brain Res.
2008;1208:217-24.
22. Um HS, Kang EB, Leem YH, et al. Exercise training acts as a therapeutic strategy
for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw-
transgenic model. Int J Mol Med. 2008;22(4):529-39.
23. Leem YH, Lee YI, Son HJ, Lee SH. Chronic exercise ameliorates the
neuroinflammation in mice carrying NSE/htau23. Biochem Biophys Res Commun.
2011;406(3):359-65.
24. Um HS, Kang EB, Leem YH, et al. Exercise training acts as a therapeutic strategy
for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw-
transgenic model. International Journal of Molecular Medicine. 2008;22(4):529-539.
25. Hayashi A, Kasahara T, Iwamoto K, et al. The role of brain-derived neurotrophic
factor (BDNF)-induced XBP1 splicing during brain development. J Biol Chem.
2007;282(47):34525-34.
26. Shimoke K, Utsumi T, Kishi S, et al. Prevention of endoplasmic reticulum stress-
induced cell death by brain-derived neurotrophic factor in cultured cerebral cortical
neurons. Brain Res. 2004;1028(1):105-11.
27. Gomez-Pinilla F, Huie JR, Ying Z, et al. BDNF and learning: Evidence that
instrumental training promotes learning within the spinal cord by up-regulating BDNF
expression. Neuroscience. 2007;148(4):893-906.
Related Documents











