Invited review EEG in Creutzfeldt–Jakob disease Heinz Gregor Wieser a, * , Kaspar Schindler a , Dominik Zumsteg b a Department of Neurology, University Hospital Zu ¨rich, Zu ¨rich, Switzerland b Toronto Western Hospital, University of Toronto, Toronto, Ont., Canada Accepted 5 December 2005 Available online 25 January 2006 Abstract Electroecenphalography (EEG) is an integral part of the diagnostic process in patients with Creutzfeldt–Jakob disease (CJD). The EEG has therefore been included in the World Health Organisation diagnostic classification criteria of CJD. In sporadic CJD (sCJD), the EEG exhibits characteristic changes depending on the stage of the disease, ranging from nonspecific findings such as diffuse slowing and frontal rhythmic delta activity (FIRDA) in early stages to disease-typical periodic sharp wave complexes (PSWC) in middle and late stages to areactive coma traces or even alpha coma in preterminal EEG recordings. PSWC, either lateralized (in earlier stages) or generalized, occur in about two- thirds of patients with sCJD, with a positive predictive value of 95%. PSWC occur in patients with methionine homozygosity and methionine/valine heterozygosity but only rarely in patients with valine homozygosity at codon 129 of the prion protein gene. PSWC tend to disappear during sleep and may be attenuated by sedative medication and external stimulation. Seizures are an uncommon finding, occurring in less than 15% of patients with sCJD. In patients with iatrogenic CJD, PSWC usually present with more regional EEG findings corresponding to the site of inoculation of the transmissible agent. In genetic CJD, PSWC in its typical form are uncommon, occurring in about 10%. No PSWC occur in EEG recordings of patients with variant CJD. q 2006 International Federation of Clinical Neurophysiology. Published by Elsevier Ireland Ltd. All rights reserved. Keywords: Creutzfeldt–Jakob disease; Prion disease; Prion protein; EEG; Periodic patterns; PSWC 1. Introduction Creutzfeldt–Jakob disease (CJD) is a rapidly progressive, uniformly fatal, transmissible spongiform encephalopathy (TSE) characterized by the accumulation of an abnormal isoform (PrP Sc ) of the host encoded cellular prion protein (PrP C ) in the brain (Kretzschmar et al., 1996; Masters et al., 1979). Human prion diseases occur in diverse phenotypes or subtypes and can be familial, sporadic, or acquired. Sporadic CJD (sCJD) is the most common subtype of CJD, occurring worldwide in 84% of cases with an annual mortality rate of 1.39 per million (range 0.8–1.61; Ladogana et al., 2005; nZ4441; sCJDZ3720). Genetic subtypes, including Gerstmann-Stra ¨ussler-Scheinker syndrome (GSS) and fatal familial insomnia (FFI), are less common and occur in about 10%. Iatrogenic CJD (iCJD) caused by the transmission of infection in the course of medical treatment occurs in 3–4%. Variant CJD (vCJD), which has been linked to transmission of the causative agent of bovine spongiform encephalopathy (BSE) to the human population, occurs Clinical Neurophysiology 117 (2006) 935–951 www.elsevier.com/locate/clinph 1388-2457/$30.00 q 2006 International Federation of Clinical Neurophysiology. Published by Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.clinph.2005.12.007 Abbreviations: Asp, aspartic acid; Asn, asparagine; AV, anteroventral nucleus of the thalamus; BSE, bovine spongiform encephalopathy; CJD, Creutzfeldt–Jakob disease; CM, centromedian nucleus of the thalamus; CSF, cerebrospinal fluid; fCJD, familial Creutzfeldt–Jakob disease; FFI, fatal familial insomnia; FIRDA, frontal intermittent rhythmic delta activity; gTSE, genetic transmissible spongiform encephalopathy; GSS, Gerstmann- Stra ¨ussler-Scheinker syndrome; iCJD, iatrogenic Creutzfeldt–Jakob disease; LD, laterodorsal nucleus of the thalamus; ORF, open reading frame; Met, methionine; MM, methionine homozygote; MV, methionine/- valine heterozygote; PLED, periodic lateralized epileptiform discharges; PRNP, prion protein gene; PrP, prion protein; PrP C , cellular prion protein; PrP Sc , disease associated protease resistant prion protein; PSWC, periodic sharp wave complexes; PV, parvalbumine; R, reticular nucleus of the thalamus; sCJD, sporadic Creutzfeldt–Jakob disease; TSE, transmissible spongiform encephalopathy; vCJD, variant Creutzfeldt–Jakob disease; Val, valine; VLa, ventrolateral anterior nucleus; VLp, ventrolateral posterior nucleus; VV, valine homozygote. * Corresponding author. Abteilung fu ¨r Epileptologie & Elektroencepha- lographie, Neurologische Klinik, Universita ¨tsspital Zu ¨rich, Frauenklinik- strasse 26, 8091 Zu ¨rich, Switzerland. Tel.: C41 1 255 5530/31; fax: C41 1 255 4429. E-mail address: [email protected] (H.G. Wieser).

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Invited review
EEG in Creutzfeldt–Jakob disease
Heinz Gregor Wieser a,*, Kaspar Schindler a, Dominik Zumsteg b
a Department of Neurology, University Hospital Zurich, Zurich, Switzerlandb Toronto Western Hospital, University of Toronto, Toronto, Ont., Canada
Accepted 5 December 2005
Available online 25 January 2006
Abstract
Electroecenphalography (EEG) is an integral part of the diagnostic process in patients with Creutzfeldt–Jakob disease (CJD). The EEG has
therefore been included in the World Health Organisation diagnostic classification criteria of CJD. In sporadic CJD (sCJD), the EEG exhibits
characteristic changes depending on the stage of the disease, ranging from nonspecific findings such as diffuse slowing and frontal rhythmic
delta activity (FIRDA) in early stages to disease-typical periodic sharp wave complexes (PSWC) in middle and late stages to areactive coma
traces or even alpha coma in preterminal EEG recordings. PSWC, either lateralized (in earlier stages) or generalized, occur in about two-
thirds of patients with sCJD, with a positive predictive value of 95%. PSWC occur in patients with methionine homozygosity and
methionine/valine heterozygosity but only rarely in patients with valine homozygosity at codon 129 of the prion protein gene. PSWC tend to
disappear during sleep and may be attenuated by sedative medication and external stimulation. Seizures are an uncommon finding, occurring
in less than 15% of patients with sCJD. In patients with iatrogenic CJD, PSWC usually present with more regional EEG findings
corresponding to the site of inoculation of the transmissible agent. In genetic CJD, PSWC in its typical form are uncommon, occurring in
about 10%. No PSWC occur in EEG recordings of patients with variant CJD.
q 2006 International Federation of Clinical Neurophysiology. Published by Elsevier Ireland Ltd. All rights reserved.
Keywords: Creutzfeldt–Jakob disease; Prion disease; Prion protein; EEG; Periodic patterns; PSWC
1388-2457/$30.00 q 2006 International Federation of Clinical Neurophysiology.
doi:10.1016/j.clinph.2005.12.007
Abbreviations: Asp, aspartic acid; Asn, asparagine; AV, anteroventral
nucleus of the thalamus; BSE, bovine spongiform encephalopathy; CJD,
Creutzfeldt–Jakob disease; CM, centromedian nucleus of the thalamus;
CSF, cerebrospinal fluid; fCJD, familial Creutzfeldt–Jakob disease; FFI,
fatal familial insomnia; FIRDA, frontal intermittent rhythmic delta activity;
gTSE, genetic transmissible spongiform encephalopathy; GSS, Gerstmann-
Straussler-Scheinker syndrome; iCJD, iatrogenic Creutzfeldt–Jakob
disease; LD, laterodorsal nucleus of the thalamus; ORF, open reading
frame; Met, methionine; MM, methionine homozygote; MV, methionine/-
valine heterozygote; PLED, periodic lateralized epileptiform discharges;
PRNP, prion protein gene; PrP, prion protein; PrPC, cellular prion protein;
PrPSc, disease associated protease resistant prion protein; PSWC, periodic
sharp wave complexes; PV, parvalbumine; R, reticular nucleus of the
thalamus; sCJD, sporadic Creutzfeldt–Jakob disease; TSE, transmissible
spongiform encephalopathy; vCJD, variant Creutzfeldt–Jakob disease; Val,
valine; VLa, ventrolateral anterior nucleus; VLp, ventrolateral posterior
nucleus; VV, valine homozygote.* Corresponding author. Abteilung fur Epileptologie & Elektroencepha-
lographie, Neurologische Klinik, Universitatsspital Zurich, Frauenklinik-
strasse 26, 8091 Zurich, Switzerland. Tel.: C41 1 255 5530/31; fax: C41 1
255 4429.
E-mail address: [email protected] (H.G. Wieser).
1. Introduction
Creutzfeldt–Jakob disease (CJD) is a rapidly progressive,
uniformly fatal, transmissible spongiform encephalopathy
(TSE) characterized by the accumulation of an abnormal
isoform (PrPSc) of the host encoded cellular prion protein
(PrPC) in the brain (Kretzschmar et al., 1996; Masters et al.,
1979). Human prion diseases occur in diverse phenotypes or
subtypes and can be familial, sporadic, or acquired.
Sporadic CJD (sCJD) is the most common subtype of
CJD, occurring worldwide in 84% of cases with an annual
mortality rate of 1.39 per million (range 0.8–1.61; Ladogana
et al., 2005; nZ4441; sCJDZ3720). Genetic subtypes,
including Gerstmann-Straussler-Scheinker syndrome (GSS)
and fatal familial insomnia (FFI), are less common and
occur in about 10%. Iatrogenic CJD (iCJD) caused by the
transmission of infection in the course of medical treatment
occurs in 3–4%. Variant CJD (vCJD), which has been linked
to transmission of the causative agent of bovine spongiform
encephalopathy (BSE) to the human population, occurs
Clinical Neurophysiology 117 (2006) 935–951
www.elsevier.com/locate/clinph
Published by Elsevier Ireland Ltd. All rights reserved.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951936
in 3% (mainly in the UK). Kuru, finally, reached epidemic
proportions in the mid-1950s in New Guinea, but apparently
disappeared within a generation after the practice of
cannibalism has been eliminated (Goldfarb et al., 2004).
Early and reliable diagnosis of CJD is crucial in
excluding other, potentially treatable, causes of rapidly
progressive encephalopathies and in counseling relatives of
patients suffering from this fatal disease. However, early
diagnosis of CJD is complicated by the marked heterogeneity
of clinical presentation of the disease. For decades,
electroencephalography has been the method of choice to
substantiate the clinical diagnosis of CJD. Periodic sharp
wave complexes (PSWC) were reported to occur in EEG
recordings of about two-thirds of patients with sCJD (Levy
et al., 1986 [nZ28]; Steinhoff et al., 1996 [nZ29, 68
EEGs]; Steinhoff et al., 2004 [nZ150]) and the occurrence
of PSWC was, therefore, included in the World Health
Organisation (1998) diagnostic classification criteria of
sCJD. The current WHO diagnostic criteria allow diagnostic
classification as definite, probable or possible sCJD (World
Health Organisation, 1998). The definite diagnosis of
sporadic CJD still requires neuropathological examination
or detection of scrapie prion protein (PrPSc) by Western blot
analysis of brain bioptic or autoptic specimens
(Kretzschmar et al., 1996; World Health Organisation,
1998). The diagnostic criteria for probable sCJD are: (1)
rapidly evolving dementia (!2 years), (2) typical PSWC
with triphasic morphology in EEG recordings and/or
presence of 14-3-3 protein in cerebrospinal fluid (CSF)
examination, and (3) at least two of the following 4 clinical
signs: (a) myocloni, (b) ataxia and/or visual signs and
symptoms, (c) extrapyramidal and/or pyramidal signs and
symptoms, and (d) akinetic mutism. Patients with the
clinical signs of sCJD but without the EEG and CSF
abnormalities (either not present or investigation not
available) are classified as possible sCJD.
In recent years, several markers of neuronal injury in the
CSF have been monitored in patients with human prion
disease. The most promising of these surrogate markers is
the 14-3-3 protein, for which a specificity of 84% and a
sensitivity of 94% have been reported (Zerr et al., 1998 [nZ484]; Zerr et al., 2000a [nZ805]). Consequently, the 14-3-3
protein has also been included in the diagnostic criteria of
the WHO. Other surrogate markers such as the Tau protein,
neuron-specific enolase (NSE) and the astrocytic protein
S-100-B may also be elevated in the CSF of patients with
CJD (Kropp et al., 1999 [nZ16]; Otto et al., 1997a [nZ53];
Otto et al., 1997b [nZ135]), but these markers are not (yet)
included in the diagnostic criteria of the WHO. Neuroima-
ging studies may show characteristic findings in patients
with CJD. Recent studies revealed increased signal intensity
in caudate nucleus, putamen and parietal or occipital
cortical areas in fluid-attenuated inversion recovery
(FLAIR) and diffusion-weighted (DW) MRI sequences
(Finkenstaedt et al., 1996 [nZ29]; Hirose et al., 1998
[nZ1]; Schroter et al., 2000 [nZ162]; Urbach et al., 1998
[nZ4]; Collie, 2001 [review]; Shiga et al., 2004 [nZ36];
Urbach et al., 2001 [nZ14]), showing an overall sensitivity
of about 60–70% and a specificity of about 80–90%
(Tschampa et al., 2005 [nZ193]; Zeidler et al., 2001 [nZ52]; Zerr et al., 2000b [nZ354]). MRI may be particularly
reliable and helpful in the clinical diagnosis of patients
with certain PrP genotypes (e.g. MV2; Zerr et al., 2000b)
and in patients with vCJD (Zeidler et al., 2000 [nZ56]).
In this article, we review the characteristics and
diagnostic value of EEG findings in patients with sporadic,
acquired and familial CJD. Special emphasis was placed on
the discussion of the neurophysiological concepts under-
lying the typical EEG findings of patients with sCJD.
2. Sporadic Creutzfeldt–Jakob disease (sCJD)
2.1. Clinical presentation, classification and staging
Sporadic CJD commonly develops in the fifth to 7th
decade of life, with a mean age of onset of 62 years (median
65). Mean survival times of about 8.2 months (range 1.5–58
months; Roos et al., 1973 [nZ1435]) or 5.2 months (range
1–15 months; Geiger et al., 2002 [nZ22]) have been
reported. About 5–10% of patients with sCJD, however,
have a clinical course that extends for 2 years or more
(Brown et al., 1984 [nZ357]). The clinical presentation of
sCJD is heterogeneous. As a consequence, the differential
diagnosis of sCJD primarily depends on the predominant
signs and symptoms present at early stages of the disease
and may include Alzheimer’s disease, Pick’s disease,
normal pressure hydrocephalus, Parkinson’s disease, cer-
ebrovascular disease, Huntington’s disease, Hashimoto’s
encephalitis, metabolic and toxic encephalopathies, and
many others. Two rare but well-recognized clinical
presentations of sCJD are the so-called Heidenhain variant
(beginning with progressive visual disturbances and
eventually blindness; Heidenhain (1929) [nZ11, 3 own
cases, 8 from the literature including cases of Creutzfeldt
(1920); Jakob (1921); Kirschbaum (1924)]) and the
Brownell-Oppenheimer variant (beginning with progressive
cerebellar disturbances with unsteadiness and incoordina-
tion; Brownell and Oppenheimer, 1965 [nZ10, 4 own
cases, 6 from the literature]). Other variants are myoclonic
or diffuse cerebral, dyskinetic with or without muscular
atrophy, thalamic, and panencephalopathic forms (Mas-
trianni and Roos, 2000 [review]).
The clinical evolution of sCJD has been arbitrarily
subdivided into 3 sequential stages (Roos et al., 1973). In
Stage 1 (mean duration of 9 weeks, range 1–52), 25% of
patients experienced neurologic or psychiatric symptoms
such as dizziness or vertigo (21%), headache (17%),
depression or anxiety (10%), ‘nervousness’ (7%) and
autonomic disturbances including fatigue, disturbances
of sleep–wakefulness, and loss of appetite or nausea.
Myoclonus occurred in 6% of patients, but otherwise there

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 937
were only minor or ‘subtle’ neurological signs, and patients
were usually not impaired in their day-to-day activities.
Stage 2 (mean duration of 10 weeks, range 1–104) was
defined by the occurrence of a distinct neurological
syndrome with a prominent involvement of higher cortical
functions (but no dementia) and sensorimotor integration
leading to visual disturbances (blurred vision, diplopia,
metamorphopsis or hallucinations) in about 25%, impair-
ment of balance in about 20% and disturbances of stance
and gait in about 33%. More than half of the patients showed
clinical features that could be attributed to involvement of
cerebellar, brain-stem or occipital cortical structures, but
myoclonus was still an uncommon finding during this stage.
The patients were clearly impaired in daily activities. In
stage 3 (mean duration of 14 weeks, range 1–116), moderate
to severe myoclonus and dementia were typical findings.
There was a significant positive correlation between the
duration of stage 1 and the duration of stages 2 and 3
combined. Bernoulli found a significant negative correlation
between the duration of stage 1 and the age of the individual
(Bernoulli et al., unpublished data). More recently, the
Japanese Slow Virus Infection Research Committee (1999;
see also Shiga et al., 2001) suggested a subdivision into 5
clinical stages, with stage 4 representing late stage
disease characterized by reduced movement, and stage 5
representing terminal stage characterized by complete
akinetic mutism.
2.2. Molecular classification
Recent understanding of molecular mechanisms of sCJD
has led to a more rational classification. Most importantly,
the PrP genotype underlying sCJD may have a major
influence on the course of the disease. In the Caucasian
population, codon 129 of the prion protein gene (PRNP;
located on the short arm of chromosome 20) has been shown
to be the site of a common methionine (M)/ valine (V)
polymorphism: 52% of individuals are M homozygous
(MM), 36% are heterozygous (MV) and 12% are V
homozygous (VV) (Palmer et al., 1991 [nZ22, plus nZ23 with suspected sCJD]). Considering that 74% of patients
with sCJD in Europe are methionine homozygotes at codon
129, this genotype has been denoted as a predisposing factor
for sCJD in the Caucasian population. In Japanese
populations, a site of a common polymorphism has been
located at codons 219 (Shibuya et al., 1998 [nZ85, 20 with
definite, 65 with probable sCJD]) and 232 (substitution from
methionine to arginine; Hoque et al., 1996 [nZ3]). In
addition to the PrP genotype, the clinical presentation of
sCJD may also be associated with PrPSc types, which are
responsible for encoding prion strain diversity (Gambetti et al.,
2003 [review]; Tschampa et al., 2002 [nZ21]). Based on the
genotype at codon 129, the size of protease resistant PrPSc
fragments and disease phenotype, sCJD has been subdivided
into several subtypes (Hill et al., 2003 [nZ119]; Parchi et al.,
1999 [nZ300]). Parchi et al. (1999) found that 70% of
individuals showed the classic CJD phenotype (PrPSc type
1) and at least one methionine allele at codon 129, whereas
25% of cases displayed the ataxic and kuru-plaque variants
and were associated with PrPSc type 2 and valine
homozygosity or heterozygosity at codon 129. Valine
homozygotes accounted for approximately 10%. Two
variants, including a thalamic form of CJD and a phenotype
characterized by prominent dementia and cortical pathol-
ogy, were linked to PrPSc type 2 and methionine
homozygosity. Finally, a rare phenotype characterized by
progressive dementia was linked to PrPSc type 1 and valine
homozygosity.
2.3. Typical EEG findings in sCJD
Periodic sharp wave complexes (PSWC) are the
hallmark EEG finding in patients with sCJD (Furlan
et al., 1981 [nZ1]; Hess et al., 2002 [text book article];
Jones and Nevin, 1954 [nZ2]; Levy et al., 1986 [nZ28];
Steinhoff et al., 1996 [nZ29, 68 EEGs]). Morphologically,
typical PSWC consist of either simple sharp waves
(including biphasic and triphasic waves) or complexes
with mixed spikes, polyspikes and slower waves with a
typical duration of 100–600 ms, recurring every 0.5–2 s
(Gloor, 1980). The intervening background usually consists
of generalized low voltage slowing. Fig. 1 shows an
example of typical PSWC in an EEG recording of a patient
with advanced sCJD (stage 3).
There is no satisfactory model to explain the periodicity
of the pattern. It is important to note that PSWC are
generally not related to myoclonic jerks. Traub and Pedley
(1981) suggested that bihemispherically synchronized
discharges such as PSWC do not necessarily imply pacing
from deep midline structures but might be synchronized by
way of the corpus callosum. They further speculated that
fusion of dendritic membranes of affected neurons could
result in increased electrotonic coupling and pathologically
synchronized bursting activity.
Topographically, PSWC commonly reveal a bilateral
voltage distribution with a fronto-precentral midline
maximum. A regional voltage maximum over occipital
areas has been shown in a patient with the Heidenhain
variant of sCJD (Furlan et al., 1981 [nZ1]), but this is a rare
finding. More commonly, PSWC may occur lateralized or
even reveal a strictly unilateral voltage distribution (Au
et al., 1980 [nZ3]; Heye and Cervos-Navarro, 1992
[nZ13]; Heye et al., 1990 [nZ1]; Neufeld and Korczyn,
1992 [nZ4]). Hence, lateralized PSWC may resemble
periodic lateralized epileptiform discharges (PLED). Fig. 2
shows an example of PLED-like PSWC in an EEG
recording of a patient with early stage of sCJD (stage 2).
It has been suggested that the occurrence of lateralized
PSWC (or PLED) in sCJD may reflect an early state of
disease, when the commissural progress has not yet led to
diffuse cortical disease (Heye and Cervos-Navarro, 1992
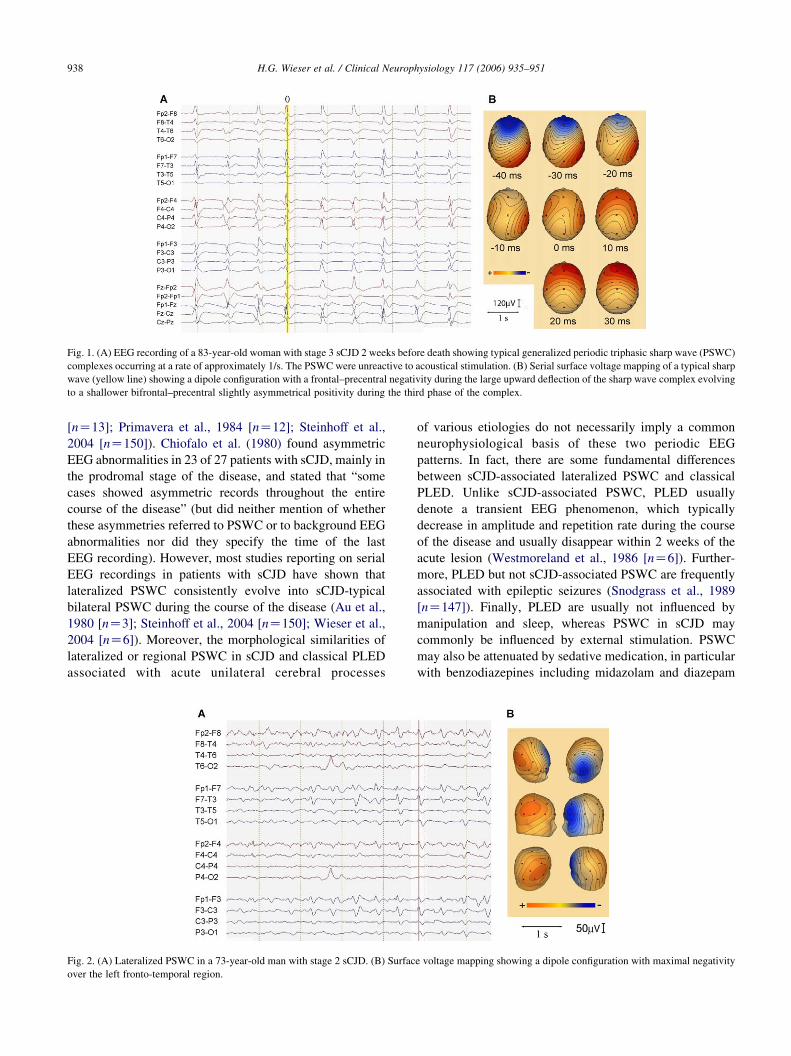
Fig. 1. (A) EEG recording of a 83-year-old woman with stage 3 sCJD 2 weeks before death showing typical generalized periodic triphasic sharp wave (PSWC)
complexes occurring at a rate of approximately 1/s. The PSWC were unreactive to acoustical stimulation. (B) Serial surface voltage mapping of a typical sharp
wave (yellow line) showing a dipole configuration with a frontal–precentral negativity during the large upward deflection of the sharp wave complex evolving
to a shallower bifrontal–precentral slightly asymmetrical positivity during the third phase of the complex.
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951938
[nZ13]; Primavera et al., 1984 [nZ12]; Steinhoff et al.,
2004 [nZ150]). Chiofalo et al. (1980) found asymmetric
EEG abnormalities in 23 of 27 patients with sCJD, mainly in
the prodromal stage of the disease, and stated that “some
cases showed asymmetric records throughout the entire
course of the disease” (but did neither mention of whether
these asymmetries referred to PSWC or to background EEG
abnormalities nor did they specify the time of the last
EEG recording). However, most studies reporting on serial
EEG recordings in patients with sCJD have shown that
lateralized PSWC consistently evolve into sCJD-typical
bilateral PSWC during the course of the disease (Au et al.,
1980 [nZ3]; Steinhoff et al., 2004 [nZ150]; Wieser et al.,
2004 [nZ6]). Moreover, the morphological similarities of
lateralized or regional PSWC in sCJD and classical PLED
associated with acute unilateral cerebral processes
Fig. 2. (A) Lateralized PSWC in a 73-year-old man with stage 2 sCJD. (B) Surfac
over the left fronto-temporal region.
of various etiologies do not necessarily imply a common
neurophysiological basis of these two periodic EEG
patterns. In fact, there are some fundamental differences
between sCJD-associated lateralized PSWC and classical
PLED. Unlike sCJD-associated PSWC, PLED usually
denote a transient EEG phenomenon, which typically
decrease in amplitude and repetition rate during the course
of the disease and usually disappear within 2 weeks of the
acute lesion (Westmoreland et al., 1986 [nZ6]). Further-
more, PLED but not sCJD-associated PSWC are frequently
associated with epileptic seizures (Snodgrass et al., 1989
[nZ147]). Finally, PLED are usually not influenced by
manipulation and sleep, whereas PSWC in sCJD may
commonly be influenced by external stimulation. PSWC
may also be attenuated by sedative medication, in particular
with benzodiazepines including midazolam and diazepam
e voltage mapping showing a dipole configuration with maximal negativity
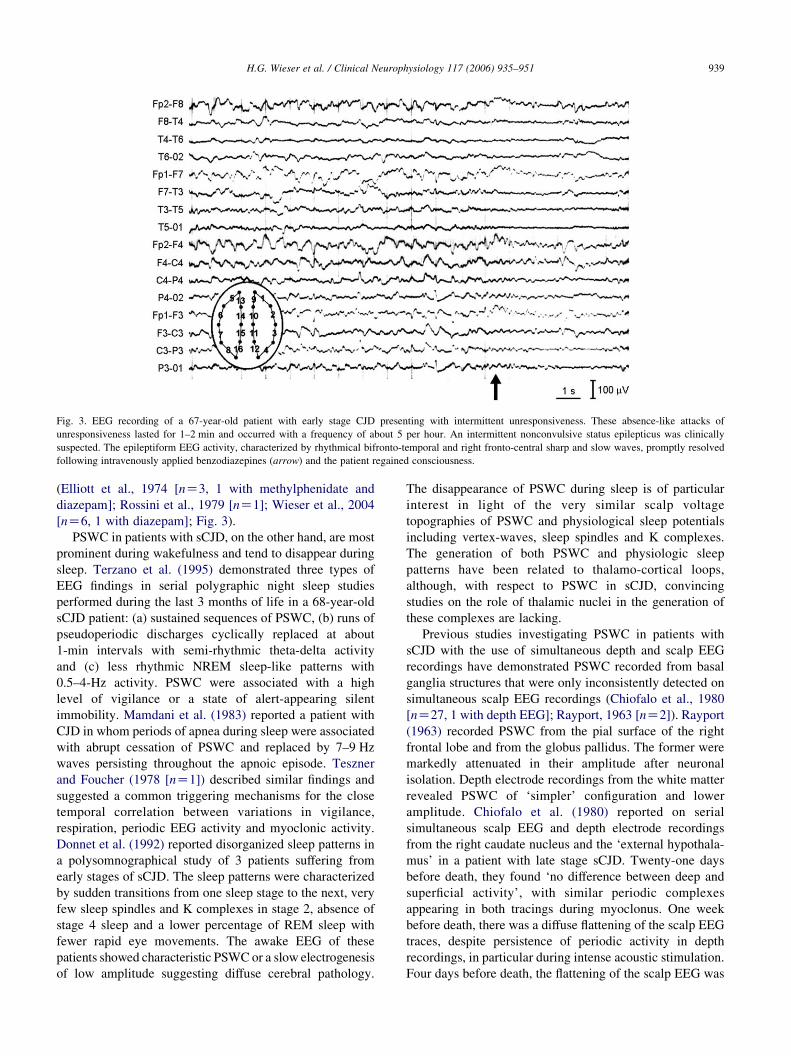
Fig. 3. EEG recording of a 67-year-old patient with early stage CJD presenting with intermittent unresponsiveness. These absence-like attacks of
unresponsiveness lasted for 1–2 min and occurred with a frequency of about 5 per hour. An intermittent nonconvulsive status epilepticus was clinically
suspected. The epileptiform EEG activity, characterized by rhythmical bifronto-temporal and right fronto-central sharp and slow waves, promptly resolved
following intravenously applied benzodiazepines (arrow) and the patient regained consciousness.
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 939
(Elliott et al., 1974 [nZ3, 1 with methylphenidate and
diazepam]; Rossini et al., 1979 [nZ1]; Wieser et al., 2004
[nZ6, 1 with diazepam]; Fig. 3).
PSWC in patients with sCJD, on the other hand, are most
prominent during wakefulness and tend to disappear during
sleep. Terzano et al. (1995) demonstrated three types of
EEG findings in serial polygraphic night sleep studies
performed during the last 3 months of life in a 68-year-old
sCJD patient: (a) sustained sequences of PSWC, (b) runs of
pseudoperiodic discharges cyclically replaced at about
1-min intervals with semi-rhythmic theta-delta activity
and (c) less rhythmic NREM sleep-like patterns with
0.5–4-Hz activity. PSWC were associated with a high
level of vigilance or a state of alert-appearing silent
immobility. Mamdani et al. (1983) reported a patient with
CJD in whom periods of apnea during sleep were associated
with abrupt cessation of PSWC and replaced by 7–9 Hz
waves persisting throughout the apnoic episode. Teszner
and Foucher (1978 [nZ1]) described similar findings and
suggested a common triggering mechanisms for the close
temporal correlation between variations in vigilance,
respiration, periodic EEG activity and myoclonic activity.
Donnet et al. (1992) reported disorganized sleep patterns in
a polysomnographical study of 3 patients suffering from
early stages of sCJD. The sleep patterns were characterized
by sudden transitions from one sleep stage to the next, very
few sleep spindles and K complexes in stage 2, absence of
stage 4 sleep and a lower percentage of REM sleep with
fewer rapid eye movements. The awake EEG of these
patients showed characteristic PSWC or a slow electrogenesis
of low amplitude suggesting diffuse cerebral pathology.
The disappearance of PSWC during sleep is of particular
interest in light of the very similar scalp voltage
topographies of PSWC and physiological sleep potentials
including vertex-waves, sleep spindles and K complexes.
The generation of both PSWC and physiologic sleep
patterns have been related to thalamo-cortical loops,
although, with respect to PSWC in sCJD, convincing
studies on the role of thalamic nuclei in the generation of
these complexes are lacking.
Previous studies investigating PSWC in patients with
sCJD with the use of simultaneous depth and scalp EEG
recordings have demonstrated PSWC recorded from basal
ganglia structures that were only inconsistently detected on
simultaneous scalp EEG recordings (Chiofalo et al., 1980
[nZ27, 1 with depth EEG]; Rayport, 1963 [nZ2]). Rayport
(1963) recorded PSWC from the pial surface of the right
frontal lobe and from the globus pallidus. The former were
markedly attenuated in their amplitude after neuronal
isolation. Depth electrode recordings from the white matter
revealed PSWC of ‘simpler’ configuration and lower
amplitude. Chiofalo et al. (1980) reported on serial
simultaneous scalp EEG and depth electrode recordings
from the right caudate nucleus and the ‘external hypothala-
mus’ in a patient with late stage sCJD. Twenty-one days
before death, they found ‘no difference between deep and
superficial activity’, with similar periodic complexes
appearing in both tracings during myoclonus. One week
before death, there was a diffuse flattening of the scalp EEG
traces, despite persistence of periodic activity in depth
recordings, in particular during intense acoustic stimulation.
Four days before death, the flattening of the scalp EEG was
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951940
complete, but occasional periodic complexes could still be
seen in deeper structures, both occurring spontaneously and
during painful stimulation.
These findings are in good accordance with a recent
study reporting on the correlation between PSWC and
immunoreactivity for the calcium-binding protein parval-
bumine in thalamic nuclei of patients with sCJD (Tschampa
et al., 2002 [nZ21, 5 controls]). The authors found a loss of
parvalbumine positive (PVC) cells in most thalamic nuclei,
including the reticular (R), ventrolateral (VLa and VLp),
laterodorsal (LD) and anteroventral (AV) nucleus. Interest-
ingly, the centromedian nucleus (CM) was the only thalamic
region with an increase of PVC cells. With respect to
PSWC, the most marked differences were found for the AV,
the R and the CM, with a significant increase of PVC cells
in the former and nonsignificant decreases in the latter two
nuclei for patients with PSWC (the decrease in the R was
only significant for patients who had both PSWC and
myoclonus). Based on the generally accepted concept that
the R plays a key role in the generation and maintenance of
thalamic, cortical or thalamocortical spindle rhythms or
synchrony (Avanzini et al., 1993; Fuentealba and Steriade,
2005; Huntsman et al., 1999; Steriade et al., 1993), the
authors hypothesized that the predominant reduction of
PVC cells in the R may determine the generation of PSWC
(associated with myoclonus) in patients with sCJD. The
authors further suggest that the functional integrity (at least
in parts) of the subcortico-cortical or thalamo-cortical
network is a prerequisite for the generation of PSWC.
Ferrer et al. (1993 [nZ3]) also found massive parvalbumin
immunoreactivity abnormalities in cortical neurons.
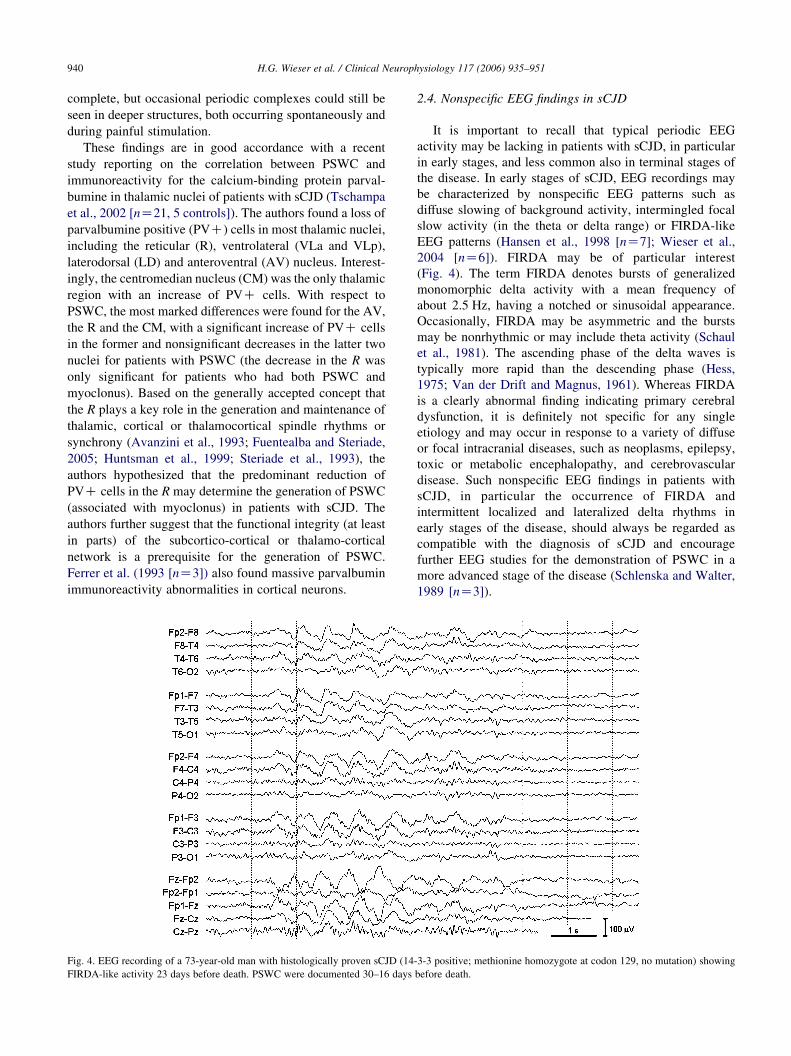
Fig. 4. EEG recording of a 73-year-old man with histologically proven sCJD (14
FIRDA-like activity 23 days before death. PSWC were documented 30–16 days
2.4. Nonspecific EEG findings in sCJD
It is important to recall that typical periodic EEG
activity may be lacking in patients with sCJD, in particular
in early stages, and less common also in terminal stages of
the disease. In early stages of sCJD, EEG recordings may
be characterized by nonspecific EEG patterns such as
diffuse slowing of background activity, intermingled focal
slow activity (in the theta or delta range) or FIRDA-like
EEG patterns (Hansen et al., 1998 [nZ7]; Wieser et al.,
2004 [nZ6]). FIRDA may be of particular interest
(Fig. 4). The term FIRDA denotes bursts of generalized
monomorphic delta activity with a mean frequency of
about 2.5 Hz, having a notched or sinusoidal appearance.
Occasionally, FIRDA may be asymmetric and the bursts
may be nonrhythmic or may include theta activity (Schaul
et al., 1981). The ascending phase of the delta waves is
typically more rapid than the descending phase (Hess,
1975; Van der Drift and Magnus, 1961). Whereas FIRDA
is a clearly abnormal finding indicating primary cerebral
dysfunction, it is definitely not specific for any single
etiology and may occur in response to a variety of diffuse
or focal intracranial diseases, such as neoplasms, epilepsy,
toxic or metabolic encephalopathy, and cerebrovascular
disease. Such nonspecific EEG findings in patients with
sCJD, in particular the occurrence of FIRDA and
intermittent localized and lateralized delta rhythms in
early stages of the disease, should always be regarded as
compatible with the diagnosis of sCJD and encourage
further EEG studies for the demonstration of PSWC in a
more advanced stage of the disease (Schlenska and Walter,
1989 [nZ3]).
-3-3 positive; methionine homozygote at codon 129, no mutation) showing
before death.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 941
In later stages of sCJD, the intensity of the periodic EEG
patterns may gradually decrease. In a longitudinal EEG
study comprising 34 EEG recordings during a 14 months
period in a patient with autopsy confirmed sCJD, Schaffler
et al. (1983) found maximal PSWC occurring at about week
15 after onset of the prodromal stage of the disease. From
the 5th month onwards, the intensity of the periodic EEG
pattern gradually decreased. In the time-span from the 10th
to the 13th month, they found considerable fluctuations of
periodic activity, ranging from pronounced typical PSWC to
complete disappearance of periodic activity. Morphologi-
cally, the PSWC showed a progressive ‘simplification’ of
the complexes during this time, characterized by a broad-
ening of the sharp wave complexes and a slight increase of
the interval duration. The authors discussed a variable
driving by the hypothetical subcortical pacemaker as well as
a decreased capability of cerebral cortex to react to
subcortical stimuli as possible causes for these fluctuations.
Clinically, these EEG changes were associated with a
disappearance of perioral reflexes, deep tendon reflexes,
oral automatisms and myoclonus. No PSWC were found
after month 14, and the last EEG recording performed
3 days before death revealed isoelectric periods, which
alternated with episodes of delta waves or sharp slow wave
complexes. Areactive coma tracings with predominantly
slow waves, low-voltage activity or, rarely, an alpha coma
pattern are a common finding in preterminal EEG
recordings (Lee and Blair, 1973 [nZ3]; Aguglia et al.,
1997 [nZ2]; Asai et al., 2001 [nZ1]).
2.5. Epileptiform and seizure activity in sCJD
Focal motor or generalized seizures have been reported
to occur in 15% of patients with sCJD, typically occurring
late in the disease (Johnson and Gibbs, 1998 [review]; Roos
et al., 1973 [nZ1435]) to 21% (Cokgor et al., 1999
[nZ21]). Seizures as the presenting symptom of sCJD,
however, are an uncommon finding, occurring in only about
3% of cases (Aronyk et al., 1984 [nZ1]). Rarely, EEG and
clinical findings may suggest partial status epilepticus (SE)
including epilepsia partialis continua (Lee et al., 2000
[nZ1]; Parry et al., 2001 [nZ1]), complex partial SE (Rees
et al., 1999 [nZ2]), nonconvulsive SE (Schwinn et al., 2001
[nZ1]; Cohen et al., 2004 [nZ1]; Fernandez-Torre et al.,
2004 [nZ1]; Shapiro et al., 2004 [nZ1]) or generalized SE
(Cokgor et al., 1999 [nZ21]). Fig. 5 shows the EEG
recordings in a patient with sCJD referred to our hospital
with the diagnosis of nonconvulsive frontal SE.
Seizure-like EEG activity in patients with sCJD, in
particular EEG findings suggesting nonconvulsive SE, must
be carefully distinguished from PSWC. At least some cases
of the above-mentioned studies might indeed have revealed
typical PSWC rather than the EEG correlate of clinical
seizures (as noted in the study of Fernandez-Torre et al.,
2004 [nZ1]). PSWC of sCJD are well known to be
attenuated or even abolished by antiepileptic drugs (AEDs)
(Elliott et al., 1974 [nZ3]; Hansen et al., 1998 [nZ7];
Wieser et al., 2004 [nZ6]). Therefore, EEG response to
AEDs alone, in particular to administration of benzo-
diazepines, is definitely not sufficient to conclude that the
periodic discharges represent the electroencephalographic
correlate of seizures. The effects of drugs such as
benzodiazepines on the EEG should always be correlated
to the consequences for mental status: the EEG changes
should be accompanied by a significant clinical improve-
ment (Fountain and Waldman, 2001 [nZ10]). Furthermore,
epileptic discharges are usually time-locked with clinical
motor signs whereas typical sCJD related myocloni are not
and may occur before, during and after the PSWC (Burger
et al., 1972 [nZ8]; Lee and Blair, 1973 [nZ3]).
2.6. Diagnostic yield of EEG findings in sCJD
The diagnostic value of early EEG findings including
slowing of background activity, focal or diffuse slow
activity and FIRDA is limited. As mentioned above, such
EEG findings are compatible, or at best suggestible, but by
no means specific for sCJD. The diagnostic usefulness of
PSWC, on the other hand, is generally accepted and has
been demonstrated in numerous studies. Steinhoff et al.
(2004 [nZ150, 56 controls]) found positive criteria
(Table 1) in the EEG recordings of 96 of 150 (64%)
patients with autopsy confirmed CJD, and falsely positive
criteria in only 5 of 56 (9%) of patients with dementia of
other etiology (4 with Alzheimer’s disease and one with
vascular dementia). These figures correspond to a high
specificity of 91% and a high positive predictive value
(PPV) of 95%, and improved the PPV of clinical symptoms
by no less than 19% to a value of 99%.
Zerr et al. (2000a) reported on a specificity of 74% and a
PPV of 93% for PSWC in a study including 805 patients
with probable or possible CJD (the PPV for detection of the
14-3-3 protein in CSF was 97%, and 99% for the
combination of both tests). Hence, PSWC only rarely
occur in patients with rapidly progressive dementia of other
etiology (e.g. Alzheimer’s disease, vascular dementia and
Lewy body disease; Tschampa et al., 2001 [nZ25 with
definite CJD, nZ56 with probable sCJD; nZ19 with
Alzheimer’s disease, and nZ12 with dementia with Lewy
bodies]; Steinhoff et al., 2004 [nZ150, 56 controls]).
Although these studies unequivocally indicate the high
diagnostic value of PSWC in patients with suspected CJD, it
has to be mentioned that these studies usually included
patients with CJD typical presentation only. Triphasic
waves resembling PSWC, however, may frequently occur
in other neurological or systemic diseases including
metabolic (mostly hepatic) encephalopathy (Karnaze and
Bickford, 1984 [nZ50]), anoxic encephalopathy (usually
associated with fatal outcome; Kuroiwa et al., 1982 [nZ1];
Scollo-Lavizzari and Bassetti, 1987 [nZ26]) and toxic
encephalopathy (in particular baclofen, lithium and the
antidepressant mianserin; Hormes et al., 1988 [nZ1]; Smith
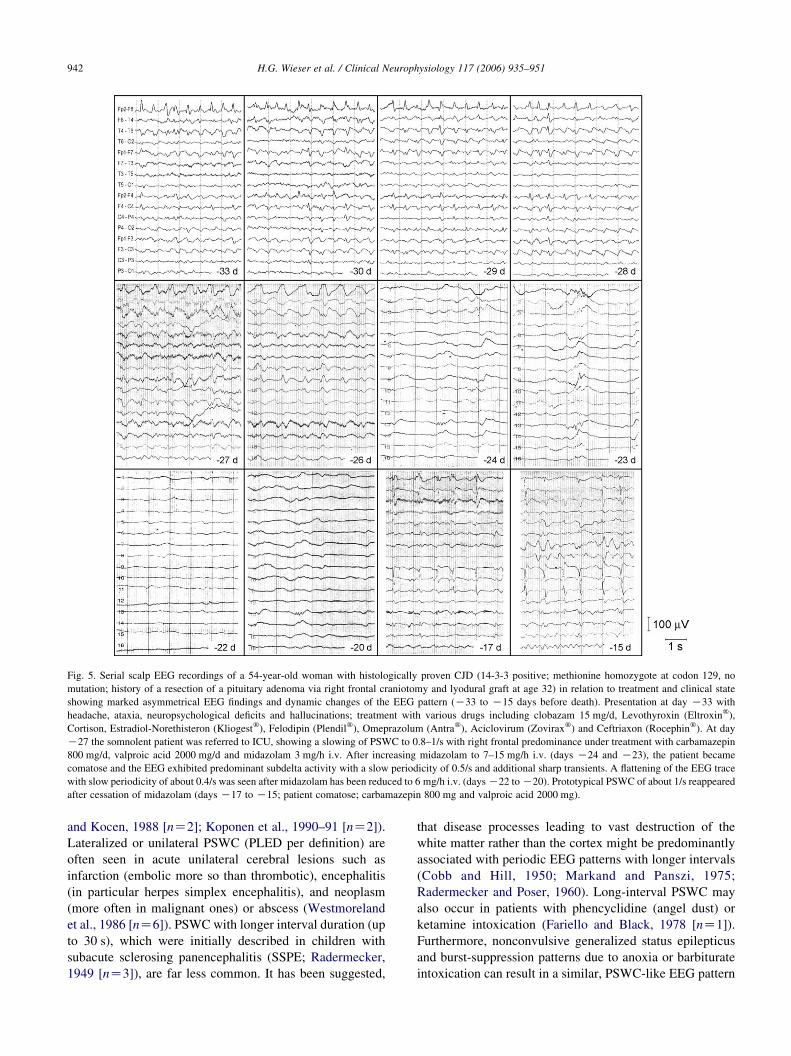
Fig. 5. Serial scalp EEG recordings of a 54-year-old woman with histologically proven CJD (14-3-3 positive; methionine homozygote at codon 129, no
mutation; history of a resection of a pituitary adenoma via right frontal craniotomy and lyodural graft at age 32) in relation to treatment and clinical state
showing marked asymmetrical EEG findings and dynamic changes of the EEG pattern (K33 to K15 days before death). Presentation at day K33 with
headache, ataxia, neuropsychological deficits and hallucinations; treatment with various drugs including clobazam 15 mg/d, Levothyroxin (Eltroxinw),
Cortison, Estradiol-Norethisteron (Kliogestw), Felodipin (Plendilw), Omeprazolum (Antraw), Aciclovirum (Zoviraxw) and Ceftriaxon (Rocephinw). At day
K27 the somnolent patient was referred to ICU, showing a slowing of PSWC to 0.8–1/s with right frontal predominance under treatment with carbamazepin
800 mg/d, valproic acid 2000 mg/d and midazolam 3 mg/h i.v. After increasing midazolam to 7–15 mg/h i.v. (days K24 and K23), the patient became
comatose and the EEG exhibited predominant subdelta activity with a slow periodicity of 0.5/s and additional sharp transients. A flattening of the EEG trace
with slow periodicity of about 0.4/s was seen after midazolam has been reduced to 6 mg/h i.v. (days K22 to K20). Prototypical PSWC of about 1/s reappeared
after cessation of midazolam (days K17 to K15; patient comatose; carbamazepin 800 mg and valproic acid 2000 mg).
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951942
and Kocen, 1988 [nZ2]; Koponen et al., 1990–91 [nZ2]).
Lateralized or unilateral PSWC (PLED per definition) are
often seen in acute unilateral cerebral lesions such as
infarction (embolic more so than thrombotic), encephalitis
(in particular herpes simplex encephalitis), and neoplasm
(more often in malignant ones) or abscess (Westmoreland
et al., 1986 [nZ6]). PSWC with longer interval duration (up
to 30 s), which were initially described in children with
subacute sclerosing panencephalitis (SSPE; Radermecker,
1949 [nZ3]), are far less common. It has been suggested,
that disease processes leading to vast destruction of the
white matter rather than the cortex might be predominantly
associated with periodic EEG patterns with longer intervals
(Cobb and Hill, 1950; Markand and Panszi, 1975;
Radermecker and Poser, 1960). Long-interval PSWC may
also occur in patients with phencyclidine (angel dust) or
ketamine intoxication (Fariello and Black, 1978 [nZ1]).
Furthermore, nonconvulsive generalized status epilepticus
and burst-suppression patterns due to anoxia or barbiturate
intoxication can result in a similar, PSWC-like EEG pattern

Table 1
Objective Diagnostic Criteria of PSWC typical for sCJD (Steinhoff et al.,
1996)
Strictly periodic cerebral potentials, the majority with a duration between
100 and 600 ms and an intercomplex interval between 500 and 2000 ms
Generalized and lateralized complexes accepted
At least 5 repetitive intervals with a duration difference of less than 500 ms
to rule out semiperiodic activity
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 943
(Kuroiwa and Celesia, 1980 [nZ62]; see Fig. 5). Such
conditions, however, usually present with other symptoms
and history than sCJD, and may thus be easily distinguished
on clinical grounds.
The sensitivity and negative predictive value (NPV) of
PSWC in sCJD, in contrast, may be substantially lower.
Steinhoff et al. (2004 [nZ150, 56 controls]) found a
sensitivity of 64% and a NPV of 49% in their cohort. Zerr
et al. (2000b [nZ354]) established a sensitivity of 66%.
Several factors may account for these moderate values.
Most importantly, it has been shown that not all patients
with sCJD develop PSWC during the course of the disease.
With respect to the PrP genotype at codon 129, Zerr et al.
(2000b [nZ354]) found PSWC predominantly in the EEG
recordings of patients with MM1 and MV1 genotypes, but
not in those of valine-homozygous patients. The lack of
PSWC in the EEG recordings of 12 patients with a codon
129-valine homozygote genotype was also reported by the
National CJD Surveillance Unit in Edinburgh (Kovacs et al.,
2000). Hill et al. (2003) reported on PSWC in the EEG
recordings of 5 of 8 MM1 patients, 14 of 18 MM2, 2 of 4
MV2, 1 of 5 MV3, but in none of 4 patients with the VV2
subtype. Hence, PSWC seem to be limited to M
homozygous (MM1 or MM2) and MV heterozygous
(MV1, MV2, and MV3) patients, whereas valine homo-
zygous patients typically do not show PSWC (Table 3). The
reason for this finding is not known. Interestingly, Kovacs
et al. (2000) also reported on the involvement of
hippocampal structures in their PSWC-negative valine
homozygote subgroup. Taking into consideration that the
hippocampal structures are only rarely involved in patients
with sCJD, this finding might be helpful in distinguishing
valine homozygous patients from patients with other
genotypes with the help of MR imaging.
Another explanation for the moderate sensitivity of
PSWC in sCJD might be the timing of the EEG recording in
relation to the disease stage. As mentioned above, the
occurrence of PSWC corresponds to the amount of neuronal
cell loss (Bortone et al., 1994 [nZ15]), the ‘classical’
bilateral diffuse PSWC usually marking the middle and late
stage of sCJD. Hence, the characteristic PSWC may be
missed if the EEG is performed too early in the course of the
disease. Steinhoff et al. (2004) assessed a total number of
443 EEG recordings in 150 patients with CJD and found
typical PSWC after a mean latency of 3.7 months (range
0.2–19.2) after onset of disease. Hansen et al. (1998)
reported on a mean survival time of 8 weeks after onset
of PSWC in a study investigating 7 CJD patients with serial
EEG recordings during the course of the disease. At the
onset of PSWC in the EEG recordings (mean 8.7 weeks
after onset of disease), 5 of 7 patients (71%) had already
progressed to stage 3, showing akinetic mutism and CJD-
typical-movement disorder such as myoclonia, exaggerated
startle reaction or focal dyskinesia. Less commonly, PSWC
may also be missed if the EEG recording is performed too
late in the course of the disease, when PSWC are no longer
present and replaced by prefinal EEG findings in at least
some of the patients with sCJD. In the study of Steinhoff
et al. (2004 [nZ150]), the latest typical EEG was recorded
2.3 months (mean, range day of death to 17.1 months).
3. Iatrogenic Creutzfeldt–Jakob disease (iCJD)
3.1. Clinical presentation and incubation period
Human-to-human transmission of CJD was reported for
the first time in 1974 in a 55-year-old woman who
developed iCJD 18 months after a corneal transplantation
(Duffy et al., 1974). Bernoulli et al. (1977) subsequently
reported on transmission of CJD by EEG depth electrodes in
two young patients (the contaminated electrodes were
reused after sterilization in formaldehyde and alcohol after
cortical recordings in the sCJD patient). Iatrogenic
transmission of CJD by neurosurgical instruments insuffi-
ciently decontaminated by routine sterilization methods was
identified in 6 cases by Will and Matthews (1982 [nZ3,
plus 1 of Jones and Nevin (1954) and 2 of Nevin et al.
(1960)]). Hundred and fourteen cases of putative CJD
transmission caused by cadaveric lyophilized dura trans-
plants from CJD patients were reported by Brown et al.
(2000). Treatment with cadaveric pituitary-derived growth
hormone has resulted in transmission to at least 139 young
people (Brown et al., 2000). Overall, at least 267 cases of
iCJD have been reported up to the end of year 2000 (Brown
et al., 2000).
The incubation period of iCJD may depend on the mode
of transmission. The shortest incubation periods were found
in iCJD caused by contaminated EEG depth electrodes or
neurosurgical instruments, ranging from 16.6 to 28 months
(Bernoulli et al., 1977 [nZ2]; Will and Matthews, 1982
[nZ6]). In iCJD associated with dural grafts and growth
hormone therapy, incubation periods up to 20 years (mean
6–9 years, rarely up to 30 years) have been reported (Brown
et al., 2000 [nZ267]; Croes et al., 2001 [nZ2]; Fushimi et
al., 2002 [nZ1]; Kretzschmar et al., 2003 [nZ1]).
Interestingly, iCJD often presents with cerebellar signs,
whereas sCJD more commonly begins with mental
deterioration. Moreover, the PrP genotype may also
influence the occurrence of iCJD: homozygosity for Met
or Val at the polymorphic codon 129 of the PRNP has been
found to represent a risk factor for iCJD, as for sCJD
(Blattler, 2002 [review]). In hormone cases, homozygosity
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951944
may also promote shorter incubation periods (Huillard
d’Aignaux et al., 1999 [nZ55 out of 1361]).
3.2. Typical EEG findings
The EEG findings in patients with iCJD are similar to
those in patients with sCJD. PSWC with a lateralized or
bilateral voltage distribution as well as nonspecific EEG
patterns such as localized or generalized slowing and
FIRDA have been reported. However, the localized
inoculation of the transmissible agent of CJD (the PrPSc)
may lead to more restricted EEG findings, at least in early
stages. Fushimi et al. (2002) reported on lateralized PSWC
corresponding with the location of the dural graft, which
later evolved into CJD-typical symmetrical PSWC in a
patient who developed iCJD 14 years after surgery. We
observed typical PSWC in a patient who received a lyodura
graft of unknown brand 22 years before disease onset (see
Fig. 5; this patient, however, may also have suffered from
sCJD rather than iCJD). Kretzschmar et al. (2003) described
generalized slowing but no PSWC in a patient with a dura
mater graft following surgery for angioblastoma of the
cerebellum 20 years prior to death.
We have previously reported on the two patients with
iCJD due to the contaminated depth electrode (Bernoulli
et al., 1977; Wieser et al., 2004). These patients with
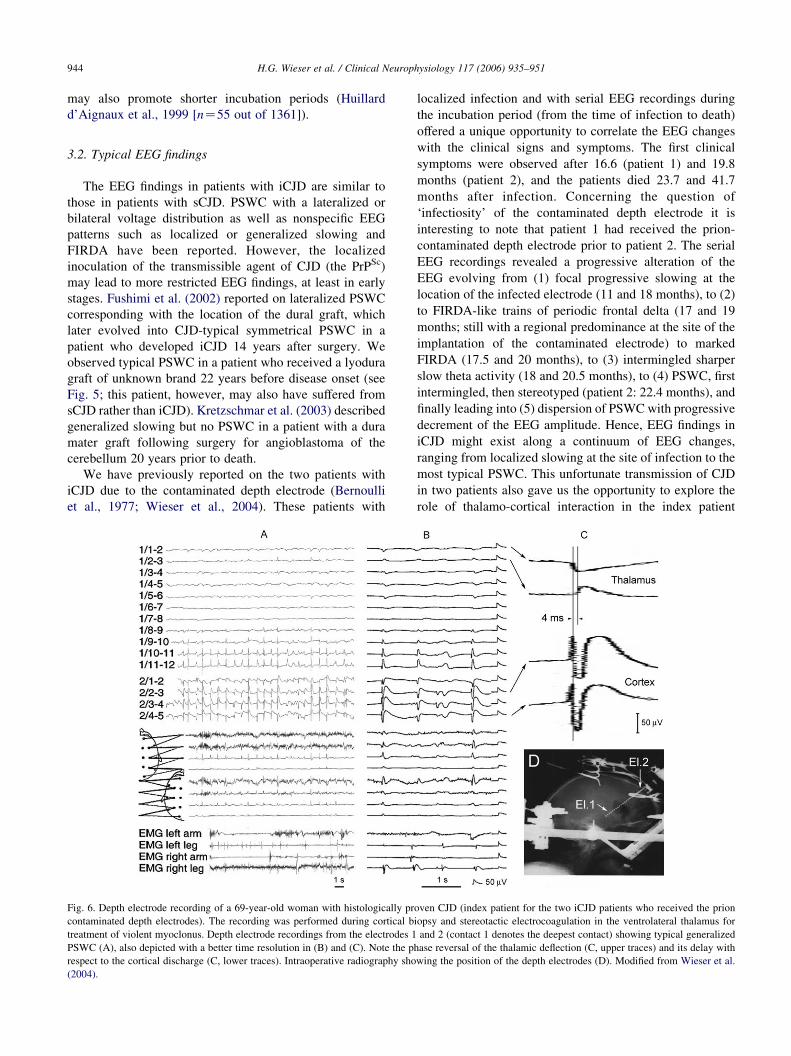
Fig. 6. Depth electrode recording of a 69-year-old woman with histologically pr
contaminated depth electrodes). The recording was performed during cortical b
treatment of violent myoclonus. Depth electrode recordings from the electrodes 1
PSWC (A), also depicted with a better time resolution in (B) and (C). Note the p
respect to the cortical discharge (C, lower traces). Intraoperative radiography sho
(2004).
localized infection and with serial EEG recordings during
the incubation period (from the time of infection to death)
offered a unique opportunity to correlate the EEG changes
with the clinical signs and symptoms. The first clinical
symptoms were observed after 16.6 (patient 1) and 19.8
months (patient 2), and the patients died 23.7 and 41.7
months after infection. Concerning the question of
‘infectiosity’ of the contaminated depth electrode it is
interesting to note that patient 1 had received the prion-
contaminated depth electrode prior to patient 2. The serial
EEG recordings revealed a progressive alteration of the
EEG evolving from (1) focal progressive slowing at the
location of the infected electrode (11 and 18 months), to (2)
to FIRDA-like trains of periodic frontal delta (17 and 19
months; still with a regional predominance at the site of the
implantation of the contaminated electrode) to marked
FIRDA (17.5 and 20 months), to (3) intermingled sharper
slow theta activity (18 and 20.5 months), to (4) PSWC, first
intermingled, then stereotyped (patient 2: 22.4 months), and
finally leading into (5) dispersion of PSWC with progressive
decrement of the EEG amplitude. Hence, EEG findings in
iCJD might exist along a continuum of EEG changes,
ranging from localized slowing at the site of infection to the
most typical PSWC. This unfortunate transmission of CJD
in two patients also gave us the opportunity to explore the
role of thalamo-cortical interaction in the index patient
oven CJD (index patient for the two iCJD patients who received the prion
iopsy and stereotactic electrocoagulation in the ventrolateral thalamus for
and 2 (contact 1 denotes the deepest contact) showing typical generalized
hase reversal of the thalamic deflection (C, upper traces) and its delay with
wing the position of the depth electrodes (D). Modified from Wieser et al.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 945
(Wieser et al., 2004; see Fig. 6). Local-fields attributable to
the PSWC were recorded both from the frontal cortex and
the ventrolateral thalamus, with the discharges recorded
from the frontal cortex generally showing higher amplitudes
and preceding the thalamic complexes by about 4 ms (note
that the thalamic complexes did not show the initial spike;
unfortunately, phase- and coherence analysis was not
carried out at the time of examination, and the available
paper recordings do not allow for such analysis post-hoc
either). In the case of Fushimi et al. (2002), a 61-year-old
man who received a cadaveric dura mater graft and
developed CJD 14 years later, right lateralized PSWC
coinciding with the location of the dural graft were observed
(lateralized PSWC were seen 14 days after admission and
generalized PSWC 1.5 months later).
4. Genetic Creutzfeldt–Jakob disease
4.1. Molecular findings and clinical presentation
Genetic CJD cases are associated with point mutations or
insertions (rarely deletions and silent or influential
polymorphisms) within the open reading frame (ORF) of
the PRNP (the entire ORF resides within a single exon
which encodes for a sequence of 253 amino acids; Basler
et al., 1986). More than 20 mutations have currently been
described, the most common being an amino acid
substitution at codon 200. By definition, familial CJD
(fCJD) is diagnosed when a progressive neuropsychiatric
disorder is encountered in combination with a pathogenic
PRNP mutation (Budka et al., 1995). Mutations within ORF
are also responsible for two rare autosomal inherited
diseases, the Gerstmann-Straussler-Scheinker syndrome
(GSS, several point mutations) and fatal familiar insomnia
(FFI; single point mutation at codon 178, Asp to Asn). It is
the codon 129 genotype that determines whether the patient
segregates with FFI (patients with M at the same allele at the
polymorphic codon 129) or familial CJD (fCJD, patients
with V at the same allele).
Phenotypically, genetic CJD cases are usually classified
on neuropathological grounds: fCJD are characterized by
the classical neuropathological triad of spongiform change,
neuronal loss and gliosis, GSS reveal multicentric amyloid
plaques in the brain, and FFI show predominant thalamic
pathology (Kovacs et al., 2002 [nZ365, review]; Lugaresi
et al., 1998 [review]). The clinical presentation of fCJD
overlaps between mutations and may resemble patients with
sCJD. Nevertheless, some genotypes of fCJD may reveal
characteristic clinical and neuropathological features.
Patient with base pair insertion mutations, for example,
have been shown to exhibit a significant earlier disease
onset and longer disease duration than other fCJD cases
(Kovacs et al., 2002 [nZ365, review]; Vital et al., 1998
[nZ3, review]). GSS usually presents with predominant
cerebellar dysfunction, whereas myoclonus is very rare
and dementia is seen in terminal stages only. FFI is
characterized by progressive and autonomic dysfunction,
ataxia, dementia, and myoclonus. Lugaresi et al. (1998
[review]) have reported on a relationship between selective
atrophy of the anteroventral and mediodorsal limbic
thalamic nuclei and hypovigilance, attention deficit,
inability to generate EEG sleep patterns, sympathetic
hyperactivity and attenuation of vegetative and hormonal
circadian oscillations. Taratuto et al. (2002 [nZ1]) reported
on insomnia associated with thalamic involvement in
E200K 129M haplotype CJD.
4.2. EEG findings
‘Classical’ periodic sharp wave complexes, with or
without triphasic morphology, occur in about 10% of
patients with genetic CJD (Kovacs et al., 2002 [nZ365,
review]; Kovacs et al., 2005 [nZ455, with genetic
transmissible spongiform encephalopathies (gTSE), EEG
available in 82%]. Moreover, a ‘CJD-typical EEG’ was
more frequent in fCJD than in FFI and GSS (only 2 of 26
patients with GGS revealed PSWC in the study of Kovacs
et al., 2005). No PSWC were found in fCJD patients
bearing codon 178, 200 or 210 mutations and in 4 members
of a kindred with CJD in whom myclonus did not develop
(Tietjen and Drury, 1990). Neufeld et al. (2003) reported
on a patient with a codon E200K mutation in the PRNP
gene presenting with status epilepticus (focal motor
seizures with frequent generalization). Seizures are
reported in a few cases of P102L (Hainfellner et al.,
1995 [nZ221 (9 generations)]), A117V (Nochlin et al.,
1989 [nZ24 (5 generations), 2 with seizures]), D178N-
129M (McLean et al., 1997 [nZ6, 2 with seizures]; Julien
et al., 1998 [nZ4, 2 families]), E200K (Seno et al., 2000
[nZ4, 1 with seizures]), 120 BPI (Cochran et al., 1996
[nZ10]), 144 BPI (Collinge et al., 1992 [nZ174]; Poulter
et al., 1992 [same pedigree as in Collinge et al., 1992;
nZ16 with the 144 base pair insertion within the PRNP
ORF]; Oda et al., 1995 [nZ6, one with attacks of
unconsciousness after ‘apoplectic episode’, one with
generalized seizures; and review]; Capellari et al., 1997
[nZ3]), and 192 BPI (Goldfarb et al., 1991 [nZ9, 1 with
seizures]).
5. Variant Creutzfeldt–Jakob disease (vCJD)
5.1. Clinical presentation, classification and staging
In 1996, Will et al. published the first 10 cases with
vCJD. These patients had no family history of CJD, were
unusually young and presented with atypical clinical
features comprising psychiatric symptoms and dysesthesia
at disease onset and ataxia and dementia at later stages.
These patients died within 7.5–22.5 months after onset of
disease. From October 1996 to November 2002, 129 cases
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951946
of vCJD have been reported in the UK, 6 in France and one
each in Canada, Ireland, Italy and the United States of
America. vCJD is strongly linked to exposure to BSE of the
cattle (Bruce et al., 1997 [nZ9, 6 with sCJD and 3 with
vCJD]; Scott et al., 1997 [experimental study (mice)]; Will
et al., 1996). Experimental studies have shown that the prion
protein in BSE-infected cows (and other ruminants and
domestic cats) has an identical glycosylation pattern as the
prion protein of vCJD cases (the PrPvCJD) but not of sCJD
patients (Collinge, 2005; Collinge et al., 1996). By
definition, mutations of PRNP are not found in patients
with vCJD. The incubation time in vCJD may range
between 5 years and more than 40 years, and the mean
disease duration is longer than in patients with sCJD
(median of 14 months as opposed to 4.5 months; Spencer
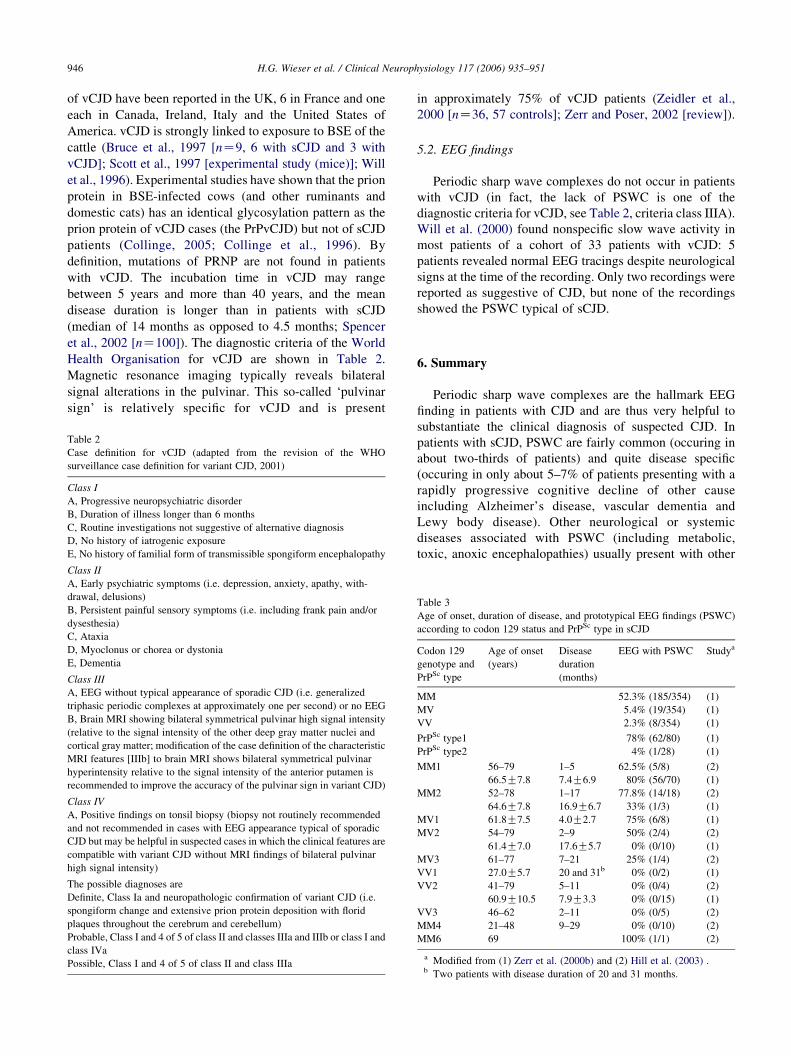
et al., 2002 [nZ100]). The diagnostic criteria of the World
Health Organisation for vCJD are shown in Table 2.
Magnetic resonance imaging typically reveals bilateral
signal alterations in the pulvinar. This so-called ‘pulvinar
sign’ is relatively specific for vCJD and is present
Table 2
Case definition for vCJD (adapted from the revision of the WHO
surveillance case definition for variant CJD, 2001)
Class I
A, Progressive neuropsychiatric disorder
B, Duration of illness longer than 6 months
C, Routine investigations not suggestive of alternative diagnosis
D, No history of iatrogenic exposure
E, No history of familial form of transmissible spongiform encephalopathy
Class II
A, Early psychiatric symptoms (i.e. depression, anxiety, apathy, with-
drawal, delusions)
B, Persistent painful sensory symptoms (i.e. including frank pain and/or
dysesthesia)
C, Ataxia
D, Myoclonus or chorea or dystonia
E, Dementia
Class III
A, EEG without typical appearance of sporadic CJD (i.e. generalized
triphasic periodic complexes at approximately one per second) or no EEG
B, Brain MRI showing bilateral symmetrical pulvinar high signal intensity
(relative to the signal intensity of the other deep gray matter nuclei and
cortical gray matter; modification of the case definition of the characteristic
MRI features [IIIb] to brain MRI shows bilateral symmetrical pulvinar
hyperintensity relative to the signal intensity of the anterior putamen is
recommended to improve the accuracy of the pulvinar sign in variant CJD)
Class IV
A, Positive findings on tonsil biopsy (biopsy not routinely recommended
and not recommended in cases with EEG appearance typical of sporadic
CJD but may be helpful in suspected cases in which the clinical features are
compatible with variant CJD without MRI findings of bilateral pulvinar
high signal intensity)
The possible diagnoses are
Definite, Class Ia and neuropathologic confirmation of variant CJD (i.e.
spongiform change and extensive prion protein deposition with florid
plaques throughout the cerebrum and cerebellum)
Probable, Class I and 4 of 5 of class II and classes IIIa and IIIb or class I and
class IVa
Possible, Class I and 4 of 5 of class II and class IIIa
in approximately 75% of vCJD patients (Zeidler et al.,
2000 [nZ36, 57 controls]; Zerr and Poser, 2002 [review]).
5.2. EEG findings
Periodic sharp wave complexes do not occur in patients
with vCJD (in fact, the lack of PSWC is one of the
diagnostic criteria for vCJD, see Table 2, criteria class IIIA).
Will et al. (2000) found nonspecific slow wave activity in
most patients of a cohort of 33 patients with vCJD: 5
patients revealed normal EEG tracings despite neurological
signs at the time of the recording. Only two recordings were
reported as suggestive of CJD, but none of the recordings
showed the PSWC typical of sCJD.
6. Summary
Periodic sharp wave complexes are the hallmark EEG
finding in patients with CJD and are thus very helpful to
substantiate the clinical diagnosis of suspected CJD. In
patients with sCJD, PSWC are fairly common (occuring in
about two-thirds of patients) and quite disease specific
(occuring in only about 5–7% of patients presenting with a
rapidly progressive cognitive decline of other cause
including Alzheimer’s disease, vascular dementia and
Lewy body disease). Other neurological or systemic
diseases associated with PSWC (including metabolic,
toxic, anoxic encephalopathies) usually present with other
Table 3
Age of onset, duration of disease, and prototypical EEG findings (PSWC)
according to codon 129 status and PrPSc type in sCJD
Codon 129
genotype and
PrPSc type
Age of onset
(years)
Disease
duration
(months)
EEG with PSWC Studya
MM 52.3% (185/354) (1)
MV 5.4% (19/354) (1)
VV 2.3% (8/354) (1)
PrPSc type1 78% (62/80) (1)
PrPSc type2 4% (1/28) (1)
MM1 56–79 1–5 62.5% (5/8) (2)
66.5G7.8 7.4G6.9 80% (56/70) (1)
MM2 52–78 1–17 77.8% (14/18) (2)
64.6G7.8 16.9G6.7 33% (1/3) (1)
MV1 61.8G7.5 4.0G2.7 75% (6/8) (1)
MV2 54–79 2–9 50% (2/4) (2)
61.4G7.0 17.6G5.7 0% (0/10) (1)
MV3 61–77 7–21 25% (1/4) (2)
VV1 27.0G5.7 20 and 31b 0% (0/2) (1)
VV2 41–79 5–11 0% (0/4) (2)
60.9G10.5 7.9G3.3 0% (0/15) (1)
VV3 46–62 2–11 0% (0/5) (2)
MM4 21–48 9–29 0% (0/10) (2)
MM6 69 100% (1/1) (2)
a Modified from (1) Zerr et al. (2000b) and (2) Hill et al. (2003) .b Two patients with disease duration of 20 and 31 months.

Table 4
Human Prion Diseases: Clinical and EEG features (modified from Glatzel et al., 2005)
Human prion
disease
Age of onset
(range)
Disease duration
(range)
Leading clinical signs and symptoms EEG
sCJD 60–70 years 6 months (1–35) Progressive dementia and neurological signs
(myoclonus, cerebellar ataxia, extrapyrami-
dal symptoms, visual disturbances)
PSWC in 60–70% (PPV of 96%); Nonspecific
alterations (slowing, FIRDA) in early stages
fCJD 50–60 years 6 months (2–41) Similar to sCJD PSWC in w10%
GSS 50–60 years 5–6 years (3
months–13 years)
Cerebellar dysfunction (ataxia, nystagmus,
dysarthria)
Nonspecific alterations, PSWC in !10%
FFI 50 years (20–63) 14 months (6–42) Insomnia, autonomic dysfunctions Nonspecific alterations, PSWC uncommon
iCJD 1–30 years
(incubation)
Similar to sCJD Similar to sCJD Similar to sCJD, PSWC often lateralized in early
stages
vCJD 26 years (12–74) 14 months (6–24) Early psychiatric symptoms (depression,
anxiety, social withdrawal) and dysesthesia,
later neurological and cognitive deficits
Nonspecific alterations, no PSWC
H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 947
symptoms and other clinical history than sCJD, and may
thus be distinguished on clinical grounds. Several reasons
may account for the lack of PSWC in patients with CJD.
First, the EEG exhibits characteristic changes during the
course of the sCJD, ranging from nonspecific EEG findings
such as diffuse slowing and FIRDA in early stages to
disease-typical PSWC in middle and late stages to a reactive
coma traces or even alpha coma in preterminal EEG
recordings. Hence, PSWC may be missed if the EEG is
performed too early or, less commonly, too late in the
course of the disease. Nonspecific EEG changes (in
particular the appearance of FIRDA) may help to support
the diagnosis of sCJD and repeated EEG recordings may be
helpful in such situations. Second, EEG findings obviously
depend on the codon 129 status of the PRNP of sCJD
patients. PSWC are likely to occur in patients with
methionine homozygosity and methionine/valine hetero-
zygosity but are only rarely seen in patients with valine
homozygosity at codon 129 of the prion protein gene.
Moreoever, PSWC are more likely to occur in patients with
the PrPSc type 1 than in patients with PrPSc type 2 or 3
(Table 3). Third, PSWC may also occur lateralized or even
reveal a strictly unilateral voltage distribution (in particular
in patients with iCJD) and may thus resemble PLED. Such
lateralized PSWC may reflect an early state of disease and
the pattern usually evolves into sCJD-typical bilateral
PSWC during the course of the disease. Fourth, PSWC are
an uncommon finding in patients with genetic CJD,
occurring in only about 10% of patients with fCJD, even
less often in patients with GSS and rarely in patients with
FFI. No PSWC occur in EEG recordings of patients with
vCJD (Table 4).
Finally, it is pertinent to note that prion diseases and in
particular CJD are characterized by their transmissibility
and the unusual resistance of the infectious agent to
conventional inactivation procedures. Recommendations
to prevent CJD spread in the neurological–neurosurgical
milieu usually differentiate between ‘at risk’ (including
treatment of patients with documented or suspected CJD
and those treated with cadaveric pituitary hormones, dura
mater grafts, fCJD or unexplained neurodegenerative
disease) and ‘at virtual risk’ (including treatment which
involves contact with lymph nodes, blood, intestines, lung,
liver, kidney and placenta). Established decontamination
methods of possibly prion-contaminated materials are: (1)
sodium hypochlorite solution containing 20,000 ppm
available chlorine for 1 h; (2) porous-load autoclaving at
134–138 8C for 18 min; (3) exposure to 1 M sodium
hydroxide for 1 h; and (4) gravity displacement autoclaving
at 132 8C for 1 h (Blattler, 2002).
Acknowledgements
DZ has been supported by the Swiss National Science
Foundation (grant PA00A-101502).
References
Aguglia U, Gambardella A, Le Piane E, Messina D, Farnarie G, Oliveri RL,
Zappia M, Quattrone A. Disappearance of periodic sharp wave
complexes in Creutzfeldt–Jakob disease. Neurophysiol Clin 1997;27:
277–82.
Aronyk K, Petito F, Solomon GE. Partial elementary motor seizures as the
first symptom of Creutzfeldt–Jakob disease. Ann Neurol 1984;15:
210–1.
Asai Y, Shimoda M, Sasaki K, Nakayasu H, Takeshima T, Miyata H,
Ohama E, Nakashima K. Alpha-like activity in terminal stage of
Creutzfeldt–Jakob disease. Acta Neurol Scand 2001;104:118–22.
Au WJ, Gabor AJ, Vijayan N, Markand ON. Periodic lateralized
epileptiform complexes (PLEDs) in Creutzfeldt–Jakob disease. Neu-
rology 1980;30:611–7.
Avanzini G, Vergnes M, Spreafico R, Marescaux C. Calcium-dependent
regulation of genetically determined spike and waves by the reticular
thalamic nucleus of rats. Epilepsia 1993;34:1–7.
Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF,
McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP
isoforms are encoded by the same chromosomal gene. Cell 1986;46:
417–28.
Bernoulli C, Siegfried J, Baumgartner G, Regli F, Rabinowicz T,
Gajdusek DC, Gibbs Jr CJ. Danger of accidental person-to-person
transmission of Creutzfeldt–Jakob disease by surgery. Lancet 1977;26:
478–9.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951948
Blattler T. Implications of prion diseases for neurosurgery. Neurosurg Rev
2002;25:195–203.
Bortone E, Bettoni L, Giorgi C, Terzano MG, Trabattoni GR, Mancia D.
Reliability of EEG in the diagnosis of Creutzfeldt–Jakob disease.
Electroencephalogr Clin Neurophysiol 1994;90:323–30.
Brown P, Rodgers-Johnson P, Cathala F, Gibbs Jr CJ, Gajdusek DC.
Creutzfeldt–Jakob disease of long duration: clinicopathological
characteristics, transmissibility, and differential diagnosis. Ann Neurol
1984;16:295–304.
Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A,
Will RG, Pocchiari M, Cashman NR, d’Aignaux JH, Cervenakova L,
Fradkin J, Schonberger LB, Collins SJ. Iatrogenic Creutzfeldt–Jakob
disease at the millennium. Neurology 2000;55:1075–81.
Brownell B, Oppenheimer DR. An ataxic form of subacute presenile
polioencephalopathy (Creutzfeldt–Jakob disease). J Neurol Neurosurg
Psychiatry 1965;28:350–61.
Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A,
McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H,
Bostock CJ. Transmissions to mice indicate that ‘new variant’ CJD is
caused by the BSE agent. Nature 1997;389:498–501.
Budka H, Aguzzi A, Brown P, Bucher JM, Bugiani O, Colliere J,
Diringer H, Gullotta F, Haltia M, Hanur JJ, Ironside JW,
Kretzschmar HA, Lantos PL, Masullo C, Pocchiari M, Schlote W,
Tateishi J, Will RG. Tissue handling in suspected Creutzfeldt–Jakob
disease and other human spongiform encephalopathies (prion diseases).
Brain Pathol 1995;5:319–22.
Burger LJ, Rowan AJ, Goldensohn ES. Creutzfeldt–Jakob disease: an
electroencephalographic study. Arch Neurol 1972;26:428–33.
Capellari S, Vital C, Parchi P, Petersen RB, Ferrer X, Jarnier D, Pegoraro E,
Gambetti P, Julien J. Familial prion disease with a novel 144-bp
insertion in the prion protein gene in a Basque family. Neurology 1997;
49:133–41.
Chiofalo N, Fuentes A, Galvez S. Serial EEG findings in 27 cases of
Creutzfeldt–Jakob disease. Arch Neurol 1980;37:143–5.
Cobb W, Hill D. Electroencephalogram in subacute progressive encepha-
litis. Brain 1950;73:392–404.
Cochran EJ, Bennett DA, Cervenakova L, Kenney K, Bernard B,
Foster NL, Benson DF, Goldfarb LG, Brown P. Familial Creutzfeldt–
Jakob disease with a five-repeat octapeptide insert mutation. Neurology
1996;47:727–33.
Cohen D, Kutluay E, Edwards J, Peltier A, Beydoun A. Sporadic
Creutzfeldt–Jakob disease presenting with nonconvulsive status
epilepticus. Epilepsy Behav 2004;5:792–6.
Cokgor I, Rozear M, Morgenlander JC. Seizures and Creutzfeldt–Jakob
disease. A case report and series review. N C Med J 1999;2:108–9.
Collie DA. The role of MRI in the diagnosis of sporadic and variant
Creutzfeldt–Jakob disease. JBR-BTR 2001;84:143–6.
Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg
Psychiatry 2005;76:906–19.
Collinge J, Brown J, Hardy J, Mullan M, Rossor MN, Baker H, Crow TJ,
Lofthouse R, Poulter M, Ridley R. Inherited prion disease with 144 base
pair insertion. 2. Clinical and pathological features. Brain 1992;115:
687–710.
Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of
prion strain variation and the aetiology of ‘new variant’ CJD. Nature
1996;383:685–90.
Creutzfeldt H. Ueber eine eigenartige herdformige Erkrankung des
Zentralnervensystems. Z Gesamte Neurol Psychiatr 1920;57:1–18.
Croes EA, Jansen GH, Lemstra AW, Frijns CJ, van Gool WA, van
Duijn CM. The first two patients with dura mater associated
Creutzfeldt–Jakob disease in the Netherlands. J Neurol 2001;248:
877–80.
Donnet A, Famarier G, Gambarelli D, Aguglia U, Regis H. Sleep
electroencephalogram at the early stage of Creutzfeldt–Jakob disease.
Clin Electroencephalogr 1992;23:118–25.
Duffy P, Wolf J, Collins G, Devoe AG, Streen B, Cowen D. Possible
person-to-person transmission of Creutzfeldt–Jakob disease. N Eng
J Med 1974;290:692–3.
Elliott F, Gardner-Thorpe C, Barwick CC, Foster JB. Jakob–Creutzfeldt
disease. Modification of clinical and electroencephalographic activity
with methylphenidate and diazepam. J Neurol Neurosurg Psychiatry
1974;37:879–87.
Fariello RJ, Black JA. Pseudoperiodic bilateral EEG paroxysms in a case of
phencyclidine intoxication. J Clin Psychiatry 1978;39:579–81.
Fernandez-Torre JL, Solar DM, Astudillo A, Cereceda R, Acebes A,
Calatayud MT. Creutzfeldt–Jakob disease and non-convulsive status
epilepticus: a clinical and electroencephalographic follow-up study.
Clin Neurophysiol 2004;115:316–9.
Ferrer I, Casas R, Rivera R. Parvalbumin-immunoreactive cortical neurons
in Creutzfeldt–Jakob disease. Ann Neurol 1993;34:864–6.
Finkenstaedt M, Szudra A, Zerr I, Poser S, Hise JH, Stoebner JM, Weber T.
MR imaging of Creutzfeldt–Jakob disease. Radiology 1996;199:793–8.
Fountain NG, Waldman WA. Effects of benzodiazepines on triphasic
waves: implications for nonconvulsive status epilepticus. J Clin
Neurophysiol 2001;18:345–52.
Fuentealba P, Steriade M. The reticular nucleus revisited: intrinsic and
network properties of a thalamic pacemaker. Prog Neurobiol 2005;75:
125–41.
Furlan AJ, Henry CE, Sweeney PJ, Mitsumoto H. Focal EEG abnormalities
in Heidenhains variant of Jakob–Creutzfeldt disease. Arch Neurol
1981;38:312–4.
Fushimi M, Sato K, Shimizu T, Hadeishi H. PLEDs in Creutzfeldt–Jakob
disease following a cadaveric dural graft. Clin Neurophysiol 2002;113:
1030–5.
Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial
CJD: classification and characterisation. Br Med Bull 2003;66:213–39.
Geiger KD, Brecht U, Schober R, Schlote W. Creutzfeldt–Jakob-Krankheit:
Fehlende Korrelation zwischen zerebraler kortikaler Histologie und
klinischem Verlauf der Erkrankung in 22 Obduktionsfallen [Creutz-
feldt–Jakob disease. Lack or correlation between cerebral cortex
histology and clinical course of the disease in 22 autopsy cases].
Pathologe 2002;23:252–9.
Glatzel M, Stoeck K, Seeger H, Luhrs T, Aguzzi A. Human prion diseases,
molecular and clinical aspects. Arch Neurol 2005;62:545–52.
Gloor P. EEG characteristics in Creutzfeldt–Jakob disease. Ann Neurol
1980;8:341.
Goldfarb LG, Brown P, McCombie WR, Goldgaber D, Swergold GD,
Wills PR, Cervenakova L, Baron H, Gibbs Jr CJ, Gajdusek DC.
Transmissible familial Creutzfeldt–Jakob disease associated with five,
seven and eight extra octapeptide coding repeats in the PRNP gene.
Proc Natl Acad Sci USA 1991;88:10926–30.
Goldfarb LG, Cervenakova L, Gajdusek DC. Genetic studies in relation to
Kuru: an overview. Curr Mol Med 2004;4:375–84.
Hainfellner JA, Brantner-Inthaler S, Cervenakova L, Brown P, Kitamoto T,
Tateishi J, Diringer H, Liberski PP, Regele H, Feucht M, Mayr N,
Wessely P, Summer K, Seitelberger F, Budka H. The original
Gerstmann–Straussler–Scheinker family of Austria: divergent clinico-
pathological phenotypes, but constant PrP genotypes. Brain Pathol
1995;5:201–11.
Hansen HC, Zschocke S, Sturenburg HJ, Kunze K. Clinical changes and
EEG patterns preceding the onset of periodic sharp wave complexes in
Creutzfeldt–Jakob disease. Acta Neurol Scand 1998;97:99–106.
Heidenhain A. Klinische und anatomische Untersuchungen uber eine
eigenartige organische Erkrankung des Zentralnervensystems im
Praesenium. Z Gesamte Neurol Psychiatr 1929;118:49–114.
Hess R. Brain tumours and other space occupying processes. In: Remond A,
editor. Handbook of electroencephalography and clinical neurophysiol-
ogy, vol. 14C. Amsterdam: Elsevier; 1975. p. 11–28.
Hess K, Vingerhoets F, Aguzzi A. Prion-Kankheiten. In: Hess K, Steck J,
editors. Neurologie-Kompendium. Bern: Hans Huber; 2002. p. 150–7.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 949
Heye N, Cervos-Navarro J. Focal involvement and lateralization in
Creutzfeldt–Jakob disease: correlation of clinical, electroencephalo-
graphic and neuropathologic findings. Eur Neurol 1992;32:289–92.
Heye N, Henkes H, Hansen M-L, Gosztonyi G. Focal unilateral
accentuation of changes observed in the early stage of Creutzfeldt–
Jakob disease. J Neurol Sci 1990;95:105–10.
Hill AF, Joiner S, Wadsworth JDF, Sidle KCL, Bell JE, Budka H,
Ironside JW, Collinge J. Molecular classification of sporadic
Creutzfeldt–Jakob disease. Brain 2003;126:1333–46.
Hirose Y, Mokuno K, Abe Y, Sobue G, Matsukawa N. A case of clinically
diagnosed Creutzfeldt–Jakob disease with serial MRI diffusion
weighted images. Rinsho Shinkeigaku 1998;38:779–82.
Hoque MZ, Kitamoto T, Furukawa H, Muramoto T, Tateishi J. Mutation
in the prion protein gene at codon 232 in Japanese patients with
Creutzfeldt–Jakob disease: a clinicopathological, immunohistochem-
ical and transmission study. Acta Neuropathol (Berlin) 1996;92:
441–6.
Hormes JT, Benarroch EE, Rodriguez M, Klass DW. Periodic sharp waves
in baclofen-induced encephalopathy. Arch Neurol 1988;45:814–5.
Huillard d’Aignaux J, Costagliola Maccario J, Billette de Villemeur T,
Brandel JP, Deslys JP, Hauw JJ, Chaussain JL, Agid Y, Dormont D,
Alperovitch A. Incubation period of Creutzfeldt–Jakob disease in
human growth hormone recipients in France. Neurology 1999;53:
1197–201.
Huntsman MM, Porcello DM, Hommanics GE, DeLorey TM,
Huguenard JR. Reciprocal inhibitory connections and network
synchrony in the mammalian thalamus. Science 1999;283:541–3.
Jakob A. Ueber eigenartige Erkrankungen des Zentralnervensystems mit
bemerkenswerten anatomischen Befunden (Spastische Pseudosklerose-
Encephalomyelopathie mit disseminierten Degenerationsherden). Z
Gesamte Neurol Psychiatr 1921;64:147–228.
Johnson RT, Gibbs CJ. Creutzfeldt–Jakob disease and related transmissible
spongiform encephalopathies. N Engl J Med 1998;339:1994–2004.
Jones DP, Nevin S. Rapidly progressive cerebral degeneration with mental
disorder, focal disturbances and myoclonic epilepsy. J Neurol
Neurosurg Psychiatry 1954;17:148–59.
Julien J, Vital C, Delisle MB, Geraud G. The French FFI cases. Brain Pathol
1998;8:555–8.
Karnaze DS, Bickford RG. Triphasic waves: a reassessment of their
significance. Electroencephalogr Clin Neurophysiol 1984;57:193–8.
Kirschbaum WR. Zwei eigenartige Erkrankungen des Zentralnervensys-
tems nach Art der spastischen Pseudosklerose (Jakob). Z Gesamte
Neurol Psychiatr 1924;92:175–220.
Koponen H, Honkonen S, Partanen J, Riekkinen PJ. Epileptic attack,
delirium, and a Creutzfeldt–Jakob-like syndrome during mianserin
treatment. Neuropsychobiology 1990–91;23:164–8.
Kovacs GG, Head MW, Bunn T, Laszlo L, Will RG, Ironside JW.
Clinicopathological phenotype of codon 129 valine homozygote
sporadic Creutzfeldt–Jakob disease. Neuropathol Appl Neurobiol
2000;26:463–72.
Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS,
Budka H. Mutations of the prion protein gene phenotypic spectrum.
J Neurol 2002;249:1567–82.
Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C,
Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N,
Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M,
Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E.
Genetic prion disease: the EUROCJD experience. Hum Genet 2005;
118(2):166–74.
Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic
criteria for sporadic Creutzfeldt–Jakob disease. Arch Neurol 1996;53:
913–20.
Kretzschmar HA, Sethi S, Foldvari Z, Windl O, Querner V, Zerr I, Poser S.
Iatrogenic Creutzfeldt–Jakob disease with florid plaques. Brain Pathol
2003;13:245–9.
Kropp S, Zerr I, Schulz-Schaeffer WJ, Riedemann C, Bodemer M, Laske C,
Kretzschmar HA, Poser S. Increase of neuron-specific enolase in
patients with Creutzfeldt–Jakob disease. Neurosci Lett 1999;261:
124–6.
Kuroiwa Y, Celesia GG. Clinical significance of periodic EEG patterns.
Arch Neurol 1980;37:15–20.
Kuroiwa Y, Celesia GG, Chung HD. Periodic EEG discharges and status
spongiosus of the cerebral cortex in anoxic encephalopathy: a necropsy
case report. J Neurol Neurosurg Psychiatry 1982;45:740–2.
Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S,
Klug GM, Sutcliffe T, Giulivi A, Alperovitch A, Delasnerie-
Laupretre N, Brandel JP, Poser S, Kretzschmar H, Rietveld I,
Mitrova E, Cuesta Jde P, Martinez-Martin P, Glatzel M, Aguzzi A,
Knight R, Ward H, Pocchiari M, van Duijn CM, Will RG, Zerr I.
Mortality from Creutzfeldt–Jakob disease and related disorders in
Europe, Australia, and Canada. Neurology 2005;64:1586–91.
Lee RG, Blair RDG. Evolution of EEG and visual evoked response changes
in Jakob–Creutzfeldt disease. Electroencephalogr Clin Neurophysiol
1973;35:133–42.
Lee K, Haight E, Olejniczak P. Epilepsia partialis continua in Creutzfeldt–
Jakob disease. Acta Neurol Scand 2000;102:398–402.
Levy SR, Chiappa KH, Burke CJ, Young RR. Early evolution of incidence
of electroencephalographic abnormalities in Creutzfeldt–Jakob disease.
J Clin Neurophysiol 1986;3:1–21.
Lugaresi E, Tobler I, Gambetti P, Montagna P. The pathophysiology of fatal
familial insomnia. Brain Pathol 1998;8:521–6.
Mamdani MB, Masdeu J, Ross E, Ohara R. Sleep apnea with unusual EEG
changes in Jakob–Creutzfeldt disease. Electroencephalogr Clin Neuro-
physiol 1983;55:411–6.
Markand ON, Panszi JG. The electroencephalogram in subacute sclerosing
panencephalitis. Arch Neurol 1975;32:719–26.
Masters CL, Harris JO, Gajdusek DC, Gibbs Jr CJ, Bernoulli C, Asher DM.
Creutzfeldt–Jakob disease: patterns of worldwide occurrence and the
significance of familial and sporadic clustering. Ann Neurol 1979;5:
177–88.
Mastrianni JA, Roos RP. The prion diseases. Semin Neurol 2000;20:
337–52.
McLean CA, Storey E, Gardner RJ, Tannenberg AE, Cervenakova L,
Brown P. The D178N (cis-29M) ‘fatal familial insomnia’ mutation
associated with diverse clinicopathologic phenotypes in an Australian
kindred. Neurology 1997;49:552–8.
Neufeld MY, Korczyn AD. Topographic distribution of the periodic
discharges in Creutzfeldt–Jakob disease (CJD). Brain Topogr 1992;4:
201–6.
Neufeld MY, Talianski-Aronov A, Soffer D, Korzyn AD. Generalized
convulsive status epilepticus in Creutzfeldt–Jakob disease. Seizure
2003;12:403–5.
Nevin S, McMenemey WH, Behrman S, Jones DP. Subacute spongiform
encephalopathy—a subacute form of encephalopathy attributable to
vascular dysfunction (spongiform cerebral atrophy). Brain 1960;83:
519–64.
Nochlin D, Sumi SM, Bird TD, Snow AD, Leventhal CM, Beyreuther K,
Masters CL. Familial dementia with PrP positive amyloid plaques: a
variant of Gerstmann–Straussler syndrome. Neurology 1989;39:910–8.
Oda T, Kitamoto T, Tateishi J, Mitsuhashi T, Iwabuchi K, Haga C, Oguni E,
Kato Y, Tominaga I, Yanai K, Kashima H, Kogure T, Hori K, Ogino K.
Prion disease with 144 base pair insertion in a Japanese family line.
Acta Neuropathol (Berlin) 1995;90:80–6.
Otto M, Stein H, Szudra A, Zerr I, Bodemer M, Gefeller O, Poser S,
Kretzschmar HA, Mader M, Weber T. S-100 protein concentration in
the cerebrospinal fluid of patients with Creutzfeldt–Jakob disease.
J Neurol 1997a;244:566–70.
Otto M, Wiltfang J, Tumani H, Zerr I, Lantsch M, Kornhuber J, Weber T,
Kretzschmar HA, Poser S. Elevated levels of tau-protein in
cerebrospinal fluid of patients with Creutzfeldt–Jakob disease. Neurosci
Lett 1997b;225:210–2.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951950
Palmer MS, Dryden AJ, Hughes JT, Colling J. Homozygous prion protein
genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature
1991;31:801–2.
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O,
Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A,
Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P,
Kretzschmar H. Classification of sporadic Creutzfeldt–Jakob disease
based on molecular and phenotypic analysis of 300 subjects. Ann
Neurol 1999;46:224–33.
Parry J, Tuch P, Knezevic W, Fabian V. Creutzfeldt–Jakob syndrome
presenting as epilepsia partialis continua. J Clin Neurosci 2001;3:
266–8.
Poulter M, Baker HF, Frith CD, Leach M, Lofthouse R, Ridley RM, Shah T,
Owen F, Collinge J, Brown J. Inherited Prion disease with 144 base pair
insertion. 1. Genealogical and molecular studies. Brain 1992;115:
675–85.
Primavera A, Tabaton M, Leonardi A. Periodic lateralized discharges in
Creutzfeldt–Jakob disease: serial electroencephalographic studies. Rev
Electroencephalogr Neurophysiol Clin 1984;13:379–82.
Radermecker J. Aspects electroencephalographiques dans trois cas
d’encephalite subaigue. Acta Neurol Psychiatr Belg 1949;49:222–32.
Radermecker J, Poser CM. The significance of repetitive paroxysmal
electroencephalographic patterns. World Neurol 1960;1:422–31.
Rayport M. Electroencephalographic, corticographic and intracerebral
potentials in two anatomically verified cases of Creutzfeldt–Jakob
disease. Electroencephalogr Clin Neurophysiol 1963;15:921.
Rees JH, Smith SJ, Kullmamr DM, Hirsch NP, Howard RS. Creutzfeldt–
Jakob disease presenting as complex partial status epilepticus: a report
of two cases. J Neurol Neurosurg Psychiatry 1999;66:406–7.
Roos R, Gajdusek DC, Gibbs Jr CJ. The clinical characteristics of
transmissible Creutzfeldt–Jakob disease. Brain 1973;96:1–20.
Rossini PM, Caltagirone C, David P, Macchi G. Jakob–Creutzfeldt disease:
analysis of EEG and evoked potentials under basal conditions and
neuroactive drugs. Eur Neurol 1979;18:269–79.
Schaffler L, Karbowski K, Meier C. Langzeitige EEG-Verlaufsuntersu-
chung bei einem Patienten mit Creutzfeldt–Jakobscher Erkrankung. Z
EEG–EMG 1983;14:6–11.
Schaul N, Gloor P, Gotman J. The EEG in deep midline lesions. Neurology
1981;31:157–67.
Schlenska GK, Walter GF. Temporal evolution of electroencephalographic
abnormalities in Creutzfeldt–Jakob disease. J Neurol 1989;236:456–60.
Schroter A, Zerr I, Henkel K, Tschampa HJ, Finkenstadt M, Poser S.
Magnetic resonance imaging in the clinical diagnosis of Creutzfeldt–
Jakob disease. Arch Neurol 2000;57:1751–7.
Schwinn PJ, Krumholz A, Seiden LG. Jakob–Creutzfeldt disease presenting
as non-convulsive status epilepticus. Epilepsia 2001;42(Suppl. 7):
146–7.
Scollo-Lavizzari G, Bassetti C. Prognostic value of EEG in post-anoxic
coma after cardiac arrest. Eur Neurol 1987;26:161–70.
Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M, Koehler R,
Tremblay P, Walther D, Cohen FE, DeArmond SJ, Prusiner SB.
Identification of a prion protein epitope modulating transmission of
bovine spongiform encephalopathy prions to transgenic mice. Proc Natl
Acad Sci USA 1997;94:14279–84.
Seno H, Tashiro H, Ishino H, Inagaki T, Nagasaki M, Morikawa S. New
haplotype of familial Creutzfeldt–Jakob disease with a codon 200
mutation and a codon 219 polymorphism of the prion protein gene in a
Japanese family. Acta Neuropathol 2000;99:125–30.
Shapiro JM, Shujaat A, Wang J, Chen X. Creutzfeldt–Jakob disease
presenting as refractory nonconvulsive status epilepticus. J Intensive
Care Med 2004;19:345–8.
Shibuya S, Higuchi J, Shin R-W, Tateishi J, Kitamoto T. Protective prion
protein polymorphism against sporadic Creutzfeldt–Jakob disease.
Lancet 1998;351:419.
Shiga Y, Seki H, Onuma A, Shimizu H, Itoyama Y. Decrement of N20
amplitude of the median nerve somatosensory evoked potential in
Creutzfeldt–Jakob disease patients. J Clin Neurophysiol 2001;18:576–82.
Shiga Y, Miyazawa K, Sato S, Fukushima R, Shibuya S, Sato Y, Konno H,
Doh-ura K, Mugikura S, Tamura H, Higano S, Takahashi S, Itoyama Y.
Diffusion-weighted MRI abnormalities as an early diagnostic marker
for Creutzfeldt–Jakob disease. Neurology 2004;63:443–9.
Slow Virus Infection Research Committee. Clinical stages of Creutzfeldt–
Jakob disease. In: Kitamoto T, editor. Annual report of the Slow Virus
Infection Research Committee. Tokyo: The Ministry of Health and
Welfare of Japan; 1999. p. 9.
Smith SJM, Kocen RS. A Creutzfeldt–Jakob like syndrome due to lithium
toxicity. J Neurol Neurosurg Psychiatry 1988;51:120–3.
Snodgrass SM, Tsuburaya K, Ajmone-Marsan C. Clinical significance of
periodic lateralized epileptiform discharges: relationship with status
epilepticus. J Clin Neurophysiol 1989;6:159–72.
Spencer MD, Knight RSG, Will RG. First hundred cases of variant
Creutzfeldt–Jakob disease: retrospective case note review of early
psychiatric and neurological features. Br Med J 2002;324:1479–82.
Steinhoff BJ, Racker S, Herrendorf G, Poser S, Grosche S, Zerr I,
Kretzschmar H, Weber T. Accuracy and reliability of periodic sharp
wave complexes (PSWC) in Creutzfeldt–Jakob disease. Arch Neurol
1996;53:162–6.
Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S,
Kretzschmar HA. Diagnostic value of periodic complexes in
Creutzfeldt–Jakob disease. Ann Neurol 2004;56:702–8.
Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in
the sleeping and aroused brain. Science 1993;262:679–85.
Taratuto AL, Piccardo P, Reich EG, Chen SG, Sevlever G, Schultz M,
Luzzi AA, Rugiero M, Abecasis G, Endelman M, Garcia AM,
Capellari S, Xie Z, Lugaresi E, Gambetti P, Dlouhy SR, Ghetti B.
Insomnia associated with thalamic involvement in E200K Creutzfeldt–
Jakob disease. Neurology 2002;58:362–7.
Terzano MG, Parrino L, Pietrini V, Mancia D, Spaggiari MC, Rossi G,
Tagliavini F. Precocious loss of physiological sleep in a case of Creutzfeldt
Jakob disease: a serial polygraphic study. Sleep 1995;18:849–58.
Teszner D, Foucher A. Correlations between variations in vigilance, E.E.G.
activity, respiration, and clonicity in a case of Jakob–Creutzfeldt’s
disease. Rev Electroencephalogr Neurophysiol Clin 1978;8:354–60.
Tietjen GE, Drury I. Familial Creutzfeldt–Jakob disease without periodic
EEG activity. Ann Neurol 1990;28:585–8.
Traub RD, Pedley TA. Virus-induced electrotonic coupling: hypothesis on
the mechanism of periodic EEG discharges in Creutzfeldt–Jakob
disease. Ann Neurol 1981;10:405–10.
Tschampa HJ, Neumann M, Zerr I, Henkel K, Schroter A, Schulz-
Schaeffer WJ, Steinhoff BJ, Kretzschmar HA, Poser S. Patients with
Alzheimer’s disease and dementia with Lewy bodies mistaken for
Creutzfeldt–Jakob disease. J Neurol Neurosurg Psychiatry 2001;71:33–9.
Tschampa HJ, Herms JW, Schulz-Schaeffer WJ, Maruschak B, Windl O,
Jastrow U, Zerr I, Steinhoff BJ, Poser S, Kretzschmar HA. Clinical
findings in sporadic Creutzfeldt–Jakob disease correlate with thalamic
pathology. Brain 2002;125:2558–66.
Tschampa HJ, Kallenberg K, Urbach H, Meissner B, Nicolay C,
Kretzschmar HA, Knauth M, Zerr I. MRI in the diagnosis of sporadic
Creutzfeldt–Jakob disease: a study on inter-observer agreement. Brain
2005;128:2026–33.
Urbach H, Klisch J, Wolf HK, Brechtelsbauer D, Gass S, Solymosi L. MRI
in sporadic Creutzfeldt–Jakob disease: correlation with clinical and
neuropathological data. Neuroradiology 1998;40:65–70.
Urbach H, Paus S, Tschampa HJ, Keller E, Schild HH. Creutzfeldt–Jakob
disease: value of MRI. Rofo 2001;173:509–14 [Article in German].
Van der Drift JH, Magnus O. The value of the EEG in the differential
diagnosis of cases with cerebral lesions. Electroencephalogr Clin
Neurophysiol 1961;(Suppl.19):183–96.
Vital C, Gray F, Vital A, Parchi P, Capellari S, Petersen RB, Ferrer X,
Jarnier D, Julien J, Gambetti P. Prion encephalopathy with insertion of
octapeptide repeats: the number of repeats determines the type of
cerebellar deposits. Neuropathol Appl Neurobiol 1998;24:125–30.
Westmoreland BF, Klass DW, Sharbrough FW. Chronic periodic
lateralized epileptiform discharges. Arch Neurol 1986;43:494–6.

H.G. Wieser et al. / Clinical Neurophysiology 117 (2006) 935–951 951
Wieser HG, Schwarz U, Blattler T, Bernoulli C, Sitzler M, Stoeck K,
Glatzel M. Serial EEG findings in sporadic and iatrogenic Creutzfeldt–
Jakob disease. Clin Neurophysiol 2004;115:2467–78.
Will RG, Matthews WB. Evidence for case-to-case transmission of
Creutzfeldt–Jakob disease. JNeurolNeurosurg Psychiatry1982;45:235–8.
Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A,
Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of
Creutzfeldt–Jakob disease in the UK. Lancet 1996;347:921–5.
Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN,
Mackenzie J, Estibeiro K, Green AJ, Knight RS. Diagnosis of new
variant Creutzfeldt–Jakob disease. Ann Neurol 2000;47:575–82.
World Health Organisation. Consensus on criteria for diagnosis of sporadic
CJD. Wkly Epidemiol Rec 1998;73:361–5.
World Health Organisation. The revision of the surveillance case definition for
variant Creutzfeldt–Jakob Disease (vCJD). Report of a WHO consultation
Edinburgh, United Kingdom; 17 May 2001. http://www.who.int/csr/
resources/publications/bse/WHO_CDS_CSR_EPH_2001_5/en/
Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G, Macleod MA,
Ironside JW, Cousens S, Colchester AC, Hadley DM, Will RG. The
pulvinar sign on magnetic resonance imaging in variant Creutzfeldt–
Jakob disease. Lancet 2000;355(9213):1412–8.
Zeidler M, Collie DA, Macleod MA, Sellar RJ, Knight R. FLAIR MRI in
sporadic Creutzfeldt–Jakob disease. Neurology 2001;56:282.
Zerr I, Poser S. Clinical diagnosis and differential diagnosis of CJD and
vCJD. With special emphasis on laboratory tests. Acta Pathol Microbiol
Immunol Scand 2002;110:88–98.
Zerr I, Bodemer M, Gefeller O, Otto M, Poser S, Wiltfang J, Windl O,
Kretzschmar HA, Weber T. Detection of 14-3-3 protein in the
cerebrospinal fluid supports the diagnosis of Cretzfeldt–Jakob disease.
Ann Neurol 1998;43:32–40.
Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J,
Knight RS, Bernheimer H, Cardone F, Delasnerie-Laupretre N,
Cuadrado Corrales N, Ladogana A, Bodemer M, Fletcher A,
Awan T, Ruiz Bremon A, Budka H, Laplanche JL, Will RG,
Poser S. Analysis of EEG and CSF 14-3-3- protein as aids
to the diagnosis of Creutzfeldt–Jakob disease. Neurology 2000a;
55:811–5.
Zerr I, Schulz-Schaeffer WJ, Giese A, Bodemer M, Schroter A,
Henkel K, Tschampa HJ, Windl O, Pfahlberg A, Steinhoff BJ,
Gefeller O, Kretzschmar HA, Poser S. Current clinical diagnosis in
Creutzfeldt–Jakob disease: identification of uncommon variants.
Ann Neurol 2000b;48:323–9.
Related Documents











