£EA~'R-5928- UJ CO CQ FR0104094 COMMISSARIAT A , L ' É N E B tt I Gestion INIS Doc. Enreg. le ..f.ldiJ..àQû.c. DETERMINATION DES ENTHALPIES LIBRES DE FORMATION DES VERRES BOROSIUCATES APPLICATION À L'ÉTUDE DE L'ALTÉRATION DES VERRES DE CONFINEMENTS DE DÉCHETS RADIOACTIFS o par Yannick LINARD O LU u û DIRECTION DU CYCLE DU COMBUSTIBLE DÉPARTEMENT DE RECHERCHE EN RETRAITEMENT ET EN VITRIFICATION SERVICE DE CONFINEMENT DE DÉCHETS O h- LU Û Centre d'études de la Vallée du Rhône Site de Marcoule DIRECTION DE L'INFORMATION SCIENTIFIQUE ET TECHNIQUE RAPPORT CEA-R-5928

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

£EA~'R-5928-
UJ
CO
CQ
FR0104094C O M M I S S A R I A T A , L ' É N E B tt I
Gestion INISDoc. Enreg. le ..f.ldiJ..àQû.c.
DETERMINATION DES ENTHALPIES LIBRES
DE FORMATION DES VERRES BOROSIUCATES
APPLICATION À L'ÉTUDE DE L'ALTÉRATION
DES VERRES DE CONFINEMENTS DE DÉCHETS RADIOACTIFS
o par
Yannick LINARD
O
LU
u
û
D I R E C T I O N D U C Y C L E D U C O M B U S T I B L E
D É P A R T E M E N T D E R E C H E R C H E E N R E T R A I T E M E N TET E N V I T R I F I C A T I O N
S E R V I C E D E C O N F I N E M E N TD E D É C H E T S
Oh-
LU
Û
Centre d'études de la Vallée du Rhône
Site de Marcoule
D I R E C T I O N DE L ' I N F O R M A T I O N
S C I E N T I F I Q U E E T T E C H N I Q U ERAPPORT
CEA-R-5928

RAPPORT CEA-R-5928 - Yannick LINARD
«DETERMINATION DES ENTHALPIES LIBRES DE FORMATION DES VERRESBOROSILICATES. APPLICATION A L'ETUDE DE L'ALTERATION DES VERRES DECONFINEMENT DE DECHETS RADIO ACTIFS »
Résumé - Ce travail contribue à la caractérisation des propriétés thermochimiques des verres deconfinement de déchets radioactifs. Les résultats sont utilisés pour discuter les mécanismes et lesparamètres intégrés dans les modèles d'altération par l'eau des matrices de confinement dessolutions de produits de fissions.
Le verre est un matériau désordonné, défini thermodynamiquement hors d'équilibre. Enutilisant un paramètre d'ordre qui caractérise l'état de configuration du verre, on peut considérer leverre dans un équilibre métastable et mesurer ses principales propriétés thermodynamiques.
Les techniques calorimétriques ont été utilisées pour mesurer les capacités calorifiques et lesenthalpies de formation de verres borosilicatés (de 3 à 8 oxydes). Les entropies de formation ontégalement été déterminées, en utilisant la théorie entropique des processus de relaxation énoncéepar Adam et Gibbs (1965). Ainsi, les contributions entropiques de configuration ont été déterminéespar des mesures de viscosité.
Cet ensemble de données a permis de calculer les enthalpies libres de dissolution des verresdans l'eau pure. Par comparaison avec des expériences de lixiviation, on montre que la diminutiondes vitesses d'altération observée à fort progrès de la réaction ne peut être associée à l'approche d'unéquilibre réactionnel entre le verre "sain" et la solution altérante. Nous devons envisager la prise encompte de l'évolution de composition de la zone réactionnelle et notamment caractériser l'équilibrethermodynamique d'un verre hydraté désalcalinisé et/ou du gel d'altération avec la solution, pourtraduire le niveau de saturation du lixiviat en produits de corrosion.
2000 — Commissariat à l'Energie Atomique - France
RAPPORT CEA-R-5928 - Yannick LINARD
«DETERMINATION OF THE FREE ENTHALPIES OF FORMATION OFBOROSILICATE GLASSES »
Abstract - This work contributes to the study of the thermochemical properties of nuclear wasteglasses. Results are used to discuss mechanisms and parameters integrated in alteration models ofconditioning materials.
Glass is a disordered material defined thermodynamically as a non-equilibrium state.Taking into account one order parameter to characterise its configurational state, the metastableequilibrium for the glass was considered and the main thermochemical properties were determined.
Calorimetric techniques were used to measure heat capacities and formation enthalpies ofborosilicate glasses ( from 3 to 8 constitutive oxides). Formation Entropies were measured too,using the entropy theory of relaxation processes proposed by Adam and Gibbs (1965). Theconfigurational entropy contribution were determined from viscosity measurements.
This set of data has allowed the calculation of Gibb's free energies of dissolution ofglasses in pure water. By comparison with leaching experiments, It has been demonstrated that thedecreasing of the dissolution rate at high reaction progress cannot be associated to the approach ofan equilibrium between the sound glass and the aqueous solution. The composition changes of thereaction area at the glass surface need to be considered too. To achieve a complete description ofthe thermodynamic stability, the equilibrium between hydrated dealkalinized glass and/or the gellayer with the aqueous solution should also be evaluated.
2000 - Commissariat à l'Energie Atomique - France

PLEASE BE AWARE THATALL OF THE MISSING PAGES IN THIS DOCUMENT
WERE ORIGINALLY BLANK

Université Denis Diderot - Paris VII
U.F.R. des Sciences de la Terre
Thèse présentée à l'Institut de Physique du Globe de Paris
pour l'obtention du titre de Docteur de l'Université Paris VII
spécialité : Géophysique Interne
DETERMINATION
DES ENTHALPIES LIBRES DE FORMATION
DIS VERRES BOROSILICATÉSAPPLICATION A L'ETUDE DE L'ALTERATION DES VERRES DE
CONFINEMENT DE DECHETS RADIOACTIFS
par
LINARD YANNICK
Soutenue le 23 juin 2000 devant la commission d'examen :
M. T. Advocat
Mme Z. Andriambololona
M. G. Calas
M. G. Libourel
M. P. Richet __
M. J. Rogez
Examinateur
Examinateur
Président
Rapporteur
Directeur de thèse
Rapporteur

- Rapport CEA-R-5928-
Centre d'Études de la Vallée du Rhône
Site de Marcoule
Direction du Cycle du Combustible
Département de Recherche en Retraitement et en Vitrification
Service de Confinement des Déchets
DÉTERMINATION DES ENTHALPIES LIBRES
DE FORMATION DES VERRES BOROSILICATES.
APPLICATION À L'ÉTUDE DE L'ALTÉRATION DES VERRES
DE CONFINEMENT DE DÉCHETS RADIOACTIFS.
par
Yannick LINARD
- Août 2000 -

REMERCIEMENTS
Cette thèse a été effectuée d'une part au laboratoire des Géomatériaux de l'Institut dePhysique du Globe de Paris (IPGP) dirigé par Pascal Richet, et d'autre part au Commissariat àl'Energie Atomique (CEA) à Marcoule au sein du Service de Confinement des Déchets (SCD)dirigé successivement par J.P. Moncouyoux et J.P. Nabot et plus précisément dans le laboratoired'Etude de l'Altérabilité des Matériaux dirigé par E. Vernaz.
Je remercie Pascal Richet de m'avoir accepté dans son équipe dès mon stage de DEA endécembre 1995. Il m'a dirigé avec attention. J'ai apprécié sa disponibilité. Ses conseils etencouragements m'ont été d'une aide très précieuses.
Je suis également redevable envers Thierry Advocat qui fut mon contact privilégié auCEA. J'ai profité grandement de ses connaissances sur la gestion des déchets radioactifs et surl'altération des silicates.
Cette étude résulte d'une collaboration entre le CEA, l'Agence Nationale de gestion desDéchets Radioactifs (ANDRA) et l'IPG. Mes remerciements s'adressent donc également à ZoéAndriambololona, chargée du suivi de cette thèse pour l'ANDRA, pour l'intérêt qu'elle a manifestépour ce travail et les discussions que nous avons eues sur les enjeux industriels.
J'exprime ma profonde gratitude à Jacques Rogez qui m'a tout appris sur la calorimétriede dissolution lors de plusieurs semaines passées au Centre de Thermodynamique et deMicrocalorimétrie (CTM) du CNRS à Marseille. Je tiens à remercier également J.C. Mathieuresponsable de l'équipe "physico-chimie des phases condensées" au sein du CTM et tous ceux quim'ont apporté leur aide et leur soutien amical.
Je remercie le professeur Atake qui m'a acceuilli durant 3 semaines dans son laboratoiredans l'Institut de Technologie de Tokyo (Tokyo Institute of Technology) au Japon pourm'apprendre à utiliser un calorimètre adiabatique. Je remercie également les différents membres deson laboratoire et plus particulièrerment Isao Yamashita pour son hospitalité.
Je remercie Daniel Neuville pour sa contribution à mon apprentissage des diversestechniques expérimentales dont dipsose le laboratoire des Géomatériaux. Je le remercie égalementpour son soutien et l'intérêt qu'il a porté sur mon travail.
Christophe Téqui a également largement contribué à l'avancée des mesures encalorimétrie de chute. Il a toujours été présent pour résoudre les petits problèmes techniques. J'aiapprécié son efficacité. Je le remercie pour ses conseils.
Je remercie G. Calas qui a accepté de présider mon jury de thèse ainsi que lesrapporteurs de mon travail, G. Libourel et J. Rogez, pour leur rapidité et efficacité.
Je tiens encore à remercier tous ceux du Laboratoire des Géomatériaux qui m'ont aidépar leurs connaissances techniques, informatiques et scientifiques ou simplement par leur soutienamical : Alain, Ali, Anne, Béatrice, Denis, Dominique, Elrik, Emmanuelle, Fabrice, JB, Joël,Katia, Laurent, Maria, Marie Lyse, Moustafa, Nicola, Pascale, Philipe, Sébastien, Thomas, Vo,Wydia.
J'ai apprécié également la bonne humeur des thésards du SCD :.Christophe,Emmanuelle, Gilles, Isabelle, Mickael, Pierre, Xavier.
Merci à Anne, Max et Omar.
Pour terminer, que toute personne qui m'a donné une part de son temps pour ce travail etque j'aurais honteusement oublié de nommer, m'excuse de cet oubli impardonneble et reçoive mesremerciements d'autant plus sincères.
A Lucas, Sonia et mes parents...

SOMMAIRE
SOMMAIRE 5
INTRODUCTION 7
PREMIÈRE PARTIE LE VERRE (REVUE) 13
CHAPITRE 1 APPROCHE STRUCTURALE 16
1.1 Modèle du réseau continu désordonné, règles de Zachariasen 16
1.2 Les conditions de vitrification 18
1.3 Modèles modernes de la structure des verres 24
CHAPITRE 2 APPROCHE THERMODYNAMIQUE ET CINÉTIQUE 27
2.1 Germination et croissance 27
2.2 Conditions de vitrification 29
2.3 Immiscibilité 31
2.4 La transition vitreuse 33
CHAPITRE 3 LES VERRES DE CONFINEMENT 38
3.1 Les verres « SON » et « AVM » 40
3.2 Choix des compositions étudiées 42
3.3 Connaissances structurales 47
CHAPITRE 4 ALTÉRATION DES SILICATES VITREUX 53
4.1 Mécanismes de dissolution 53
4.2 Diffusion et réactions de surface 55
4.3 L'altération du verre SON 58
4.4 Cinétique de dissolution et modélisation 58
4.5 Un besoin de données thermodynamiques 62
DEUXIÈME PARTIE MÉTHODES EXPÉRIMENTALES 71
CHAPITRE 5 SYNTHÈSES 74
5.7 Protocole de synthèse 74
5.2 La synthèse des verres binaires SiO2-B2Os 78
CHAPITRE 6 CALORIMÉTRIE 80
6.1 Principe 80
6.2 La calorimétrie adiabatique entre 10 et 300 K 81
6.3 La calorimétrie de chute entre 400 et 1600 K (calorimètre à glace) 88
6.4 La calorimétrie différentielle à balayage (DSC) entre 298 et 1100 K 96
6.5 Calorimétrie de dissolution en sels fondus 104
CHAPITRE 7 VISCOSIMÉTRIE 119
7.1 Dispositif de fluage 119
7.2 Viscosimètre rotatif. 124

TROISIEME PARTIE PROPRIETES THERMODYNAMIQUES 133
CHAPITRE 8 CAPACITÉ CALORIFIQUE 135
8.1 Capacité calorifique, vibration et structure 136
8.2 Capacité calorifique basse température 143
8.3 Les mesures de calorimétrie de chute 155
8.4 Les mesures de calorimétrie différentielle à balayage 157
8.5 Capacités calorifiques au-dessus de 273 K 158
8.6 Capacité calorifique de configuration 173
CHAPITRE 9 VISCOSITÉS ET ENTROPIES DE CONFIGURATION 179
9.1 La relaxation structurale et les modèles rhéologiques 779
9.2 Viscosité : effet de la température et de la composition chimique 182
9.3 Entropie de configuration 190
9.4 Entropie deformation 196
CHAPITRE 10 ENTHALPIES DE FORMATION 199
10.1 Les résultats des dissolutions 199
10.2 Calcul des enthalpies deformation 204
QUATRIÈME PARTIE APPORT DE LA THERMODYNAMIQUE À LA
COMPRÉHENSION DES CINÉTIQUES
D'ALTÉRATION DES VERRES 209
CHAPITRE 11 ENTHALPIE LIBRE DE DISSOLUTION DES VERRES SIMPLIFIÉS 212
11.1 Calcul des enthalpies de formation par rapport aux oxydes 212
11.2 Evaluation des erreurs 213
11.3 Influence de la température fictive sur les propriétés deformation 214
11.4 Calcul de l'enthalpie libre de dissolution des verres dans l'eau 216
CHAPITRE 12 COMPARAISON AVEC LES TESTS DE LIXIVIATION 221
12.1 Expériences de lixiviation 221
12.2 Discussion des résultats des essais de lixiviation et des calculs de solubilité
des verres dans l'eau 222
12.3 Quelles modifications apporter ? 228
CONCLUSION 231
BIBLIOGRAPHIE 235
ANNEXE A 257

INTRODUCTION
Les verres silicates, aluminosilicatés et borosilicatés forment la majorité
des compositions de vitrage et d'emballage. Ils ont également un intérêt
géologique car leurs compositions sont celles des assemblages minéraux
terrestres. L'étude des verres naturels, tels que les obsidiennes ou les tectites, est
en soi intéressante, mais n'explique pas le nombre d'études consacrées aux verres
en géologie. Dans ce domaine le réel objet d'étude est le liquide à partir duquel
s'effectue la différenciation des minéraux ; cependant, pour des raisons
expérimentales, les liquides magmatiques sont difficiles à étudier et les verres de
même composition en forment une représentation plus manipulable.
Le bore est un élément mineur voire un élément trace dans les magmas
naturels. Cependant, la présence de bore dans les systèmes magmatiques est
couramment révélée par la cristallisation de tourmaline, un minéral borosilicaté
contenant environ 10 % de B2O3. Les évidences directes de l'implication du bore
dans les processus magmatiques viennent des analyses des verres naturels tels que
les obsidiennes étudiées par Pichavant et al. en 1987 ou des inclusions fluides
observées par London en 1986. A cause de son effet significatif sur les propriétés
physiques et chimiques des liquides silicates, le bore est un constituant
supplémentaire à considérer dans la modélisation expérimentale des systèmes
granitiques.
La chimie unique du bore lui permet d'être concentré dans des formations
géologiques (dépôts de sources chaudes par exemple) sous forme de borax. Ces
formations ont facilité la découverte et l'utilisation du bore dans l'histoire humaine
contrairement à d'autres éléments, tels que le germanium ou le gallium, restés
dissimulés derrière les éléments plus abondants comme le silicium et l'aluminium
pendant très longtemps. Le bore fut en effet utilisé très tôt dans les civilisations
égyptiennes, grecques et chinoises pour faire des verres et des émaux.

Que leur composition relève du domaine industriel ou géologique, la
caractérisation précise de la thermochimie des borosilicates est nécessaire à
l'interprétation de nombreuses propriétés. Ainsi, la connaissance des différences
énergétiques entre le verre et le cristal permet de mieux comprendre la
cristallisation ou la démixtion, que ce soit pour les contrôler (vitro-céramiques) ou
pour prédire ou expliquer des coefficients de partage (différenciation
magmatique). Les phénomènes de transport, avec comme application les verres
conducteurs ioniques, ou l'altération des verres pour confiner des éléments
polluants, ont motivé plusieurs études des propriétés thermodynamiques des
borosilicates. C'est dans le cadre de cette dernière problématique que notre étude a
également été menée.
Le choix d'une matrice vitreuse pour le confinement de déchets
domestiques ou industriels fut dicté par les études qui, dès les années 60,
démontrèrent la capacité du verre à intégrer les divers éléments polluants
(radionucléides de haute activité, éléments lourds, ...) dans le réseau et à assurer
un confinement durable. De nombreux travaux visant à évaluer les effets à long
terme associés aux paramètres susceptibles de modifier les propriétés du verre
(irradiation, altération chimique et dévitrification) ont également été entrepris.
D'un point de vue fondamental, la métastabilité de l'état vitreux a constamment
suscité des interrogations quant à son évolution annoncée vers un état cristallin
thermodynamiquement plus stable. D'autre part, l'état amorphe serait-il plus
résistant à la corrosion aqueuse qu'un état ordonné ? C'est à ce genre
d'interrogations qu'ont dû se heurter nombre de scientifiques s'intéressant au
problème de confinement de déchets.
De manière générale, la cinétique d'altération des verres nucléaires
aluminoborosilicatés peut se décomposer de façon schématique en trois étapes :
Une phase durant laquelle le verre se dissout à une vitesse d'altération
maximale (vitesse initiale Vo),
Une deuxième phase caractérisée par une chute de la vitesse d'altération,
Et enfin une dernière étape au cours de laquelle la vitesse d'altération
reste inférieure de plusieurs ordres de grandeurs à la vitesse initiale Vo.
8

Les deux principaux mécanismes généralement évoqués pour décrire
l'altération des verres nucléaires sont l'interdiffusion et les réactions de surface. Le
premier qui peut être décrit par les lois de Fick est caractéristique d'un échange
d'éléments. Le second est associé aux réactions d'hydrolyse des différentes
liaisons du réseau vitreux. Les effets couplés de ces deux mécanismes conduisent
rapidement à l'établisement d'un état stationnaire. Par ailleurs, l'altération des
verres nucléaires se traduit toujours par la formation à l'interface verre-solution
d'une pellicule d'altération constituée des éléments très peu solubles du verre et
d'une partie de la silice du réseau vitreux.
Grambow (1985) puis Advocat (1991) ont suggéré que, pour les verres
aluminoborosilicatés, la réaction limitant le processus global d'altération est la
seule réaction d'hydrolyse de la silice. Cette hypothèse les a conduit à proposer
une loi cinétique ne dépendant que du pH et de l'activité de l'espèce aqueuse
HUSiC^ à l'interface réactionnelle verre / pellicule d'altération. Dans une telle
approche, l'augmentation de l'activité de l'acide orthosilicique à l'interface
réactionnelle cause donc la chute de vitesse observée expérimentalement.
Cette loi cinétique, intégrée dans les modèles de prédiction du
comportement à long terme des verres nucléaires et dans des codes géochimiques
(Lixiver, Prediver, Glassol et Kindis), s'est avérée insuffisante dans plusieurs cas
expérimentaux : par exemple, certains systèmes complexes verre-eau-argile mais
également dans des systèmes plus simples verre-eau. Or, la méthodologie de
l'étude du comportement à long terme nécessite de développer une modélisation
qui intègre les mécanismes contrôlant la cinétique d'altération des verres
nucléaires.
Dans ce travail, nous avons voulu aborder cette problématique par le
biais d'une description thermodynamique des processus et des matériaux mis en
jeu pour, notamment, déterminer le produit de solubilité du verre, et non plus
seulement de la silice.
Les grandeurs de base que constituent capacités calorifiques, enthalpies
de formation, entropies ou volumes contrôlent la stabilité des phases et les

fonctions d'état nous permettent de décrire des équilibres réactionnels. La courbe
d'équilibre entre deux phases au sein d'un système clos s'exprime par :
àGT p = 0
avec
JAVTtPdP
où AH représente la différence d'enthalpie entre les différents participants à la
réaction considérée, AS la variation d'entropie, ACp celle de chaleur spécifique à
volume constant et A V les données de volume ; les indices associés indiquent les
conditions de pression et de température.
Dans notre problématique, l'équilibre à considérer est celui de la réaction
de dissolution d'un matériau vitreux dans une solution aqueuse. Par conséquent les
différences de propriétés énumérées ci-dessus devront être calculées entre les
verres, les solutions, et les espèces aqueuses résultant de la dissolution.
Notre approche relève de mesures directes de ces propriétés. Les
capacités calorifiques et les enthalpies peuvent être mesurées par des expériences
de calorimétrie (calorimétrie adiabatique, calorimétrie de chute, calorimétrie
différentielle à balayage calorimétrie de dissolution). Le terme entropique est plus
difficile à obtenir. Nous ne ferons pas une mesure réellement directe car celle-ci
n'est possible que lorsque les verres ont un équivalent cristallin fondant de
manière congruente. Depuis une vingtaine d'année, la théorie entropique des
processus de relaxation énoncée par Adam et Gibbs (1965) a été utilisée pour des
systèmes d'intérêt géologique afin de déterminer la partie configurationnelle du
terme entropique. La qualité des résultats obtenus nous a conduit à appliquer cette
approche aux matrices de confinement des déchets. Cette méthode nécessite des
mesures de viscosités des liquides desquelles on déduit l'entropie de configuration
du verre héritée du liquide parental lors du figeage de ce dernier à la transition
vitreuse. L'autre contribution entropique, la partie vibrationnelle, s'obtient à partir
des mesures calorimétriques.
10

Pour bien cerner l'objet de notre étude et pour rappeler les diverses
précautions inhérentes à la détermination des propriétés d'une phase métastable,
nous ferons dans une première partie un descriptif général de l'état vitreux. Nous
verrons notamment ces différentes caractéristiques structurales et
thermodynamiques ainsi que l'état actuel des connaissances sur les mécanismes de
son altération. La seconde partie sera consacrée aux techniques expérimentales
utilisées au cours de notre travail. Les deux dernières parties seront consacrées à
l'exposé des résultats, leur discussion et leur apport à la compréhension des
mécanismes d'altération.
11

PREMIERE PARTIE
LE VERRE
{revue)

Connu et utilisé depuis 5000 ans, le verre a fait l'objet au cours des
cinquante dernières années d'un développement technologique important (semi-
conducteurs amorphes, verres métalliques, etc.) lié à l'effort de recherche de la
science fondamentale. Le retard qu'il pouvait y avoir il y a encore quelques années
tient en grande partie aux difficultés associées à l'étude du matériau vitreux :
- solide désorganisé, le verre ne présente pas de structure périodique se prêtant à
une étude cristallographique fine.
- liquide figé, le verre est un matériau thermodynamiquement métastable qui ne
doit son existence qu'à des problèmes de cinétique.
Et pourtant l'état vitreux, ou amorphe, ou encore non-cristallin, n'est pas
une exception. On le retrouve dans tous les types de matériaux :
solides minéraux : oxydes, phosphates, silicates...
- semi-conducteurs amorphes : silicium, chalcogénures...
métaux vitreux (Metglass) obtenus par hypertrempe
verres organiques : glycérol, sucre de confiserie
- polymères amorphes, verres plastiques.
Pourquoi un matériau se présente-t-il sous forme amorphe plutôt que
cristallisée ? C'est à cette question que nous allons tenter d'apporter quelques
éléments de réponse au travers de cette partie. Nous allons essayer de présenter ici
les différentes idées qui ont été proposées. On trouvera en général deux types
d'approche basée sur :
la structure et la liaison chimique
la thermodynamique et la cinétique.
Ensuite, nous verrons comment l'altération des matériaux vitreux est
abordée et les différentes caractéristiques propres aux verres de confinement des
déchets radioactifs.
15

Chapitre 1
APPROCHE STRUCTURALE
Ce chapitre fera une brève synthèse des modèles structuraux élaborés sur
la base de caractérisations spectroscopiques des matériaux vitreux.
1.1 MODELE DU RESEAU CONTINU DESORDONNE, REGLES
DE ZACHARIASEN
Le modèle du réseau désordonné (random network) proposé par
Zachariasen (1932) et appuyé expérimentalement par Warren (1933) a apporté un
grand nombre d'idées nouvelles par rapport au modèle des microcristallites
proposé en 1835 par Frankenheim et défendu par l'école russe de Lebelev selon
lequel le verre serait composé de petits cristaux appelés les "cristallites".
Prenons l'exemple de la silice. On considère que le motif de base, le
tétraèdre SiÛ4, se retrouve dans le verre comme dans le cristal. On dit que l'ordre
à courte distance est conservé. Dans le cristal (Figure 1.1), ces tétraèdres
s'enchaînent les uns aux autres de façon régulière le long de chaînes (pyroxene),
de plans (mica) ou sous forme d'un réseau tridimensionnel (quartz). Dans le verre
(Figure 1.2), on peut, moyennant quelques modifications des angles et des
longueurs de liaison, imaginer un enchaînement irrégulier tridimensionnel. L'ordre
à grande distance disparaît et on obtient un matériau "amorphe" ne donnant pas de
cliché de diffraction net, mais simplement quelques anneaux plus ou moins flous.
Une telle représentation semble maintenant universellement adoptée. Elle est
confirmée par les études de diffraction qui ont été réalisées depuis et qui montrent
que l'on retrouve dans le verre les distances Si—O, O—O et Si—Si entre atomes
premiers voisins.
16

Figure 1.1: Représentation schématique de la silice cristallisée.
Figure 1.2 : Représentation schématique d'un verre de silice conforme au modèle du réseaualéatoire continu de Waren et Zachariasen.
Ainsi ce modèle décrit les verres comme étant constitués d'unités
tétraédriques ou triangulaires (les formateurs de réseau) liées par leurs sommets et
formant un réseau tridimensionnel. L'enchaînement des polyèdres est tel que le
réseau n'est ni symétrique ni périodique (pas d'ordre structural à plus de 8 Â). Les
modificateurs de réseau s'insèrent aléatoirement dans les "trous" du réseau continu
constitué par les formateurs de réseau. Il s'ensuit, d'après ce modèle, que les
modificateurs de réseau ont une sphère de coordination mal définie.
17

Bien qu'il soit à la base d'un grand nombre d'interprétations des
dépendances compositionnelles des propriétés des verres, ce modèle n'en reste pas
moins qu'une vision schématique qui a été à nombreuses reprises remis en cause
et modifié. Cependant, avant d'exposer les modèles plus récents, regardons dans
quelles conditions un solide prendra cette structure amorphe plutôt qu'une
structure cristalline.
1.2 LES CONDITIONS DE VITRIFICA TION
Zachariasen propose les quatre conditions suivantes applicables aux
verres d'oxydes MxOy :
la coordinence de l'ion métallique doit être faible (3 ou 4),
un atome d'oxygène ne peut être lié à plus de deux atomes métalliques,
les polyèdres d'oxygène ne peuvent être liés que par des sommets, pas par des
arêtes ou des faces,
chaque polyèdre doit être lié à ses voisins par au moins trois sommets.
Les trois premières règles sont essentiellement liées à la condition de
pouvoir désorganiser le réseau cristallin en modifiant très légèrement les angles et
les longueurs de liaison.
Dans la première règle (faible coordinence), intervient aussi la notion de
contre-réaction du réseau. On conçoit que pour créer un désordre, il soit
nécessaire de modifier certaines liaisons M—O. Ceci entrainera une "contre-
réaction," des liaisons voisines. Cette réaction sera évidemment plus importante (
à force de liaison constante) s'il y a beaucoup de liaisons M—O, donc si la
coordinence de l'ion métallique est élevée.
La dernière règle est liée à la nécessité de construire un réseau
tridimensionnel ce qui n'est évidemment plus possible si on a des chaînes dans
lesquelles les tétraèdres SiÛ4 ne sont liés que par deux sommets.
Pour illustrer ce dernier point, regardons ce qui se passe si l'on ajoute du
sodium au réseau de SiÛ2 vitreux. L'ion Na+, trop gros, ne pourra prendre la place
18

du silicium à l'intérieur des tétraèdres. Il se placera donc en insertion comme nous
le montre la Figure 1.3. On voit donc qu'un sodium entraîne la coupure d'une
liaison Si—O—Si. L'oxygène qui servait de pont entre deux tétraèdres n'est plus
lié qu'à un seul silicium. On l'appelle "oxygène non pontant".
oÔ
Figure 1.3 : Représentation schématique d'un silicate alcalin amorphe conforme au modèle duréseau aléatoire continu de Waren et Zachariasen.
Il est évident que si l'on ajoute de plus en plus de sodium, on créera de
plus en plus d'oxygènes non pontants et, lorsque la proportion Na/Si sera égale à
2, les tétraèdres SiÛ2 ne seront plus liés que par deux sommets. Un réseau
tridimensionnel ne pourra plus exister et on n'obtiendra plus un verre.
Effectivement, on constate que la silice vitreuse ne peut accepter qu'une quantité
limitée de sodium qui correspond à peu près au rapport prévu. En revanche, pour
d'autres alcalins, Li et K par exemple, ce seuil existe mais le rapport est différent.
Les règles de Zachariasen font apparaître deux types d'ions métalliques
(pour les systèmes d'oxydes) :
Les ions formateurs, suffisament petits pour former spontanément un réseau
vitreux : As3+, B3+, Si4+, Ge4+, P5+.
Les ions modificateurs, trop gros : Na+, Ca++, Mg++, incapables de donner à
eux seuls des verres et que l'on ne pourra ajouter qu'en quantité limitée.
19

Certains ions, tels Al3+, se trouvent à la frontière séparant ces deux catégories. Ils
pourront, selon les conditions, jouer l'un ou l'autre rôle.
Une autre approche, basée sur des idées différentes, a été avancée par
Dietzel en 1942. Elle consiste à dire qu'un réseau désordonné est impossible si
l'on a :
soit des liaisons totalement orientées (covalentes)
- soit des liaisons totalement désorientées (ionique, métallique, Van der
Waals).
Ce critère, qui implique une diversité au niveau des liaisons chimiques,
se comprend assez bien sur le plan simplement qualitatif.
Des liaisons purement covalentes (carbone diamant) sont fortement
directionnelles. Elles donnent trop peu de degré de liberté. Une légère variation
des angles de liaison nécessite une énergie importante ce qui est défavorable à la
formation d'un verre.
Un solide ionique ou métallique peut se représenter par un modèle de
sphères dures. On conçoit qu'au refroidissement du liquide, au voisinage du point
de fusion, ces sphères puissent glisser facilement les unes sur les autres afin de se
placer dans leur position d'équilibre thermodynamique. On aura alors aisément un
cristal.
On pourra donc plutôt obtenir un verre dans les matériaux présentant
simultanément des liaisons orientées (covalentes) et non orientées :
Covalent + ionique —• verres d'oxydes (SiÛ2)
Covalent + métallique —* metglass (Ni - P)
Covalent + Van der Waals —> verres organiques (glycérol)
Comme pour Zachariasen, on trouvera de nombreuses exceptions (Si
amorphe, métaux vitreux...). Ces règles ne sont évidemment qu'indicatives.
Plusieurs auteurs ont ensuite tenté d'améliorer le modèle en proposant des
critères empiriques plus complexes. L'échelle d'électronégativité de Pauling peut
évidemment être utilisée pour quantifier la notion de liaison mixte. L'ionicité
d'une liaison A—B augmente avec la différence d'électronégativité EA-EB. Pour
20

Si—O, la différence d'électronégativité est égale à 1,7 ce qui donne à la liaison un
caractère à 45 % ionique. Il semble donc qu'un verre pourra se former lorsque les
différences d'électronégativité seront du même ordre.
Il faut toutefois rester prudent car on obtient des verres dans les systèmes
suivants :
- BeF2 : liaison à 70 % ionique,
AS2S3 : liaison à 5 % ionique.
Stanworth en 1948 a regroupé les critères structuraux et de liaisons sous
forme de trois règles (pour les oxydes) :
Cation de coordination faible 3 ou 4,
Cation de rayon ionique inférieur à 0,55 À,
- Electronégativité du cation comprise entre 1,5 et 2,1.
Dietzel (1942) a introduit un paramètre dit "intensité de champ" qui
semble donner une classification bien tranchée entre modificateurs et formateurs.
L'énergie d'interaction entre deux ions s'écrit :
TT Zx.Z2.e2
U = -L-1 , Eq. 1.1
a
Zi et Z2 sont les charges respectives des ions 1 et 2 et a est la distance entre ces
derniers. Dietzel remplace l'énergie t/par l'intensité de champ F,
a
Dans une série homologue, telle que les oxydes, on peut se contenter de :
F = 4- Eq. 1.3a
où Z est la charge du cation et a la distance métal-oxygène.
Les formateurs ont un rayon ionique faible et une coordinence de 3 ou 4
ce qui fait que l'intensité de champ correspondante sera moyenne (entre 1 et 2).
Les modificateurs sont au contraire des ions de faible charge et de fort rayon
ionique. L'intensité de champ correspondante sera faible (inférieure à 0,35).
21

Cole (1966) a suggéré que la liaison doit être assez forte pour s'exercer
dans une direction définie et assez flexible pour accepter des faibles déviations par
rapport à cette direction. Il introduit un paramètre de covalence O défini par :
— Eq. 1.4r
où / est le potentiel d'ionisation du métal. La vitrification intervient pour des
valeurs de O comprises entre 1000 et 3000.
En 1947, Sun introduit une idée nouvelle en considérant qu'au moment
de la solidification d'une fonte, la formation d'un cristal nécessite le réarrangement
d'un certain nombre de liaisons. L'aptitude à la vitrification serait alors liée à la
non-possibilité de casser ces liaisons.
Appelons FL la force de liaison simple M—O d'un oxyde MOX obtenue
en divisant l'énergie de dissociation par la coordinence du cation. Les formateurs
correspondront à des liaisons fortes (FL ~ 100 kcal.mole"1) et les modificateurs à
des liaisons faibles (FL ~ 10 kcal.mole"1).
Cette idée a été reprise et précisée par Rawson (1967) qui prend en
compte l'énergie thermique disponible au point de fusion Tf. Le rapport FL I Tf
donne une mesure de l'énergie nécessaire pour rompre les liaisons par rapport à
l'énergie disponible. Pour un formateur, ce rapport sera grand. S'il est petit, la
solidification conduira facilement à un cristal.
Signalons enfin une idée originale avancée par Garino-Canina (1961). Le
volume d'un cristal peut être décrit comme étant la somme de trois termes :
Vc : volume occupé par les cations
Va : volume occupé par les anions
Vi : volume libre.
On peut considérer que le désordre du verre est obtenu en déplaçant les
ions du cristal. Il sera à priori plus facile de déplacer les cations plus petits. Ce
déplacement sera d'autant plus facile que le volume libre sera grand par rapport à
Vc. Pour les formateurs, VilVc ~ 100 et pour les modificateurs VilVc ~ 10.
22
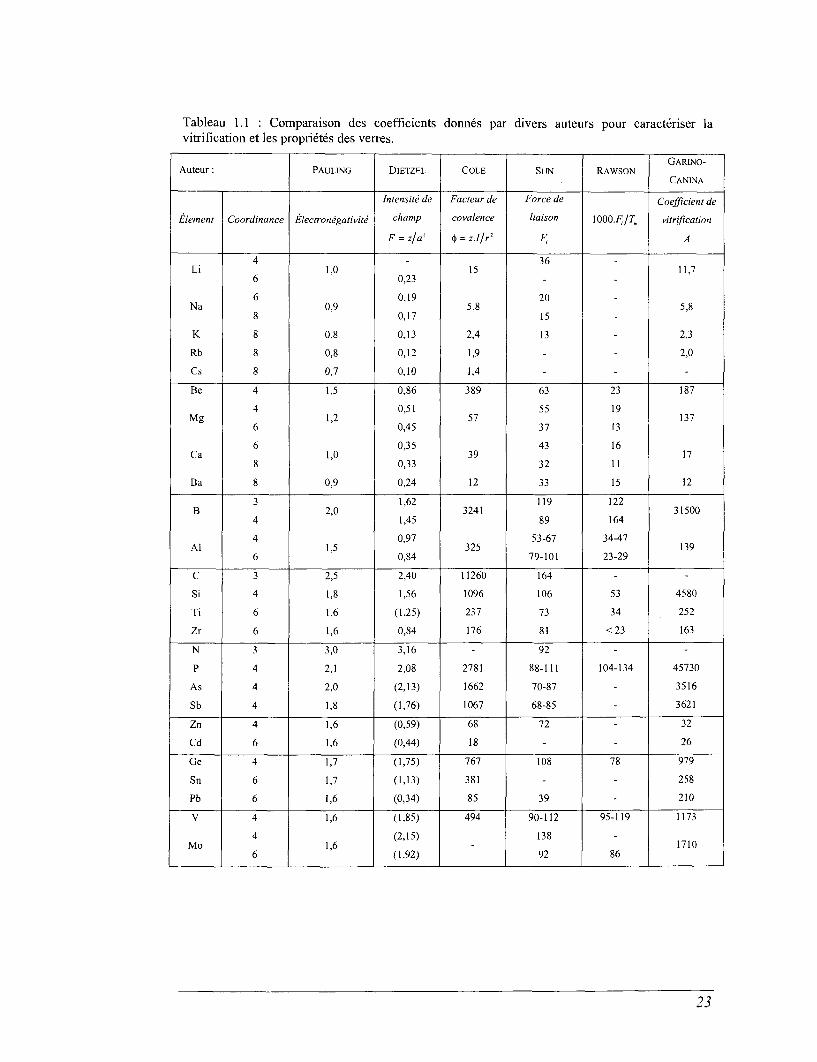
Tableau 1.1 : Comparaison des coefficientsvitrification et les propriétés des verres.
donnés par divers auteurs pour caractériser la
Auteur :
Élément
Li
Na
K
Rb
Cs
Be
Mg
Ca
Ba
B
Al
C
Si
Ti
Zr
N
P
As
Sb
Zn
Cd
Ge
Sn
Pb
V
Mo
Coordinance
4
6
6
8
8
8
8
4
4
6
6
8
8
3
4
4
6
3
4
6
6
3
4
4
4
4
6
4
6
6
4
4
6
PAULING
Electronégativite
1,0
0,9
0,8
0,8
0,7
1,5
1,2
1,0
0,9
2,0
1,5
2,5
1,8
1,6
1,6
3,0
2,1
2,0
1,8
1,6
1,6
1,7
1,7
1,6
1,6
1,6
DlETZEL
Intensité de
champ
F = z/a>
0,23
0,19
0,17
0,13
0,12
0,10
0,86
0,51
0,45
0,35
0,33
0,24
1,62
1,45
0,97
0,84
2,40
1,56
(1,25)
0,84
3,16
2,08
(2,13)
(1,76)
(0,59)
(0,44)
(1,75)
(1,13)
(0,34)
(1,85)
(2,15)
(1,92)
COLE
Facteur de
covalence
4 = z.l/r2
15
5,8
2,4
1,9
1,4
389
57
39
12
3241
325
11260
1096
237
176
-
2781
1662
1067
68
18
767
381
85
494
-
SUN
Force de
liaison
F,
36
20
15
13
-
-
63
55
37
43
32
33
119
89
53-67
79-101
164
106
73
81
92
88-111
70-87
68-85
72
-
108
-
39
90-112
138
92
RAWSON
\OO0.FjTm
;
-
-
-
-
23
19
13
16
11
15
122
164
34-47
23-29
-
53
34
<23
-
104-134
-
-
-
-
78
-
-
95-119
86
GARINO-
CANINA
Coefficient de
vitrification
A
11,7
5,8
2,3
2,0
-
187
137
17
12
31500
139
-
4580
252
163
-
45730
3516
3621
32
26
979
258
210
1173
1710
23

Garino-Canina a ensuite amélioré son modèle en définissant un
coefficient de vitrification A,
A = z2\-L\ , Eq. 1.5
qui fait apparaître une différence importante entre les formateurs et les
modificateurs.
1.3 MODELES MODERNES DE LA STRUCTURE DES VERRES
Au vu des données structurales récentes, les modèles présentés ci-dessus
ont été tantôt remis en cause tantôt affinés.
Le modèle de microcristallite a inspiré plusieurs modèles, dont le plus
récent est celui de l'agrégat mixte contraint présentant le verre comme une
agglomération aléatoire de domaines très ordonnés (clusters ou pseudophases)
entourés par un tissu de connexion continu mais très désordonné (Goodman,
1985). Goodman postule que ces clusters ont jusqu'à 60 voire 80 À de diamètre.
De telles tailles de clusters ont été remises en cause par des mesures récentes de
diffraction des rayons X pour un verre de silice (Wright, 1994) qui montrent que
la taille des clusters ne peut pas dépasser 10 à 12 À
Greaves, en 1985, a proposé le modèle du réseau aléatoire modifié
(Modified Random Network - MRN) qui s'inspire du modèle de Warren-
Zachariasen. Le verre y est représenté comme étant constitué de zones riches en
formateurs de réseau séparées par des domaines riches en modificateurs de réseau
(Figure 1.4). Ce modèle est particulièrement attrayant puisqu'il ne viole pas les
principes de formation des verres établis par Zachariasen et permet d'expliquer la
brutale augmentation de conduction ionique dans les verres d'oxydes.
Pour les verres du système binaire SiO2-Na2Û, Greaves suggère que si la
fraction volumique des alcalins est suffisante (> 16%), les domaines riches en
alcalins se regroupent et forment des chaines de percolation en trois dimensions
par où s'effectue la diffusion ionique. De plus, le regroupement des alcalins et des
24

alcalino-terreux dans les verres silicates, proposé par ce modèle, semble se
confirmer :
Des domaines de percolation riches en Na ont été proposés dans les
titanosilicates (Farges et al., 1996) en s'appuyant sur des considérations de
liaison-valence (Brese and O'Keefe, 1991) qui sont indépendantes du modèle
MRN.
Une répartition non aléatoire des alcalino-terreux a été suggérée par l'analyse
de mesures spectroscopiques récentes. Par diffusion des neutrons, Gaskell a
montré que les octaèdres de CaOô formaient des domaines bidimensionnels
dans le verre de composition CaSiÛ3 mettant ainsi en évidence des zones
riches en modificateurs et riches en formateurs (Gaskell et al., 1991). La
nanoségrégation d'ions Sr dans les binaires SrO-SiÛ2 a été montrée par
diffusion anormale des rayons X (Creux et al., 1995).
La dynamique moléculaire suggère que les alcalins et les alcalino-terreux
soient nanoségrégés dans les verres silicates (Abramo et al., 1992 ; Huang and
Cormack, 1991 ; Vessal et al., 1992) et borosilicatés (Soûles, 1979).
Figure 1.4 : Représentation schématique du modèle de Greaves avec des zones riches enformateurs et des zones riches en modificateurs.
25

A l'heure actuelle, les modèles de structure hétérogène des verres (zones
riches en formateurs et zones riches en modificateurs), comme le MRN ou le
modèle stereochimiquement défini de Gaskell (Gaskell et al., 1995) dans lequel
les éléments s'organisent selon des règles locales et des contraintes à moyenne
distance (rayon, charge, électronégativité...) nous semblent les mieux à même de
décrire la structure des verres.
26

Chapitre 2
APPROCHE THERMODYNAMIQUE ET CINETIQUE
Un verre est en équilibre métastable. Condamné par la thermodynamique,
il ne doit son existence qu'à la cinétique qui empêche sa transformation vers un
état cristallisé plus stable. On connaît des verres naturels dont la formation
géologique remonte à des centaines de millions d'années.
Pour décrire l'approche thermodynamique, nous allons examiner ce qui
se passe au moment de la solidification d'une fonte, lorsque le liquide a le choix
entre donner un solide cristallisé ou donner un verre. Quels sont les facteurs qui
l'orientent dans un sens plutôt que dans l'autre? Nous verrons que la cinétique joue
alors un grand rôle.
La cristalisation d'un liquide met en jeu deux processus élémentaires :
germination (ou nucléation) et croissance que nous allons examiner
successivement.
2.1 GERMINATION ET CROISSANCE
Considérons un liquide au voisinage du point de fusion Tm. Les
fluctuations thermiques amènent à un certain moment un ensemble d'atomes dans
des positions proches de celles qu'ils auraient dans un cristal. Ces amas peuvent
évoluer dans deux sens, soit une dispersion qui redonne un liquide désordonné,
soit au contraire une agglomération qui conduit à un germe cristallin. La
formation d'un germe nécessite une variation d'enthalpie libre AG par rapport au
liquide et fait intervenir les phénomènes de diffusion à travers la zone interfaciale
germe / liquide.
27

La vitesse de nucléation / est proportionnelle à :
T (-W\ f/ oc exp . exp
\RT ) \ RT
Eq. 2.1
AG' est l'énergie d'activation de la diffusion (barrière cinétique de la germination).
AG'
Figure 2.1 : Barrière d'enthalpie libre dans la cinétique de croissance.
/ dépend de deux facteurs (thermodynamique W et cinétique AG) qui
varient en sens inverse. W diminue quand T diminue. AG' augmente quand T
diminue et que la viscosité augmente. La courbe donnant / en fonction de T passe
donc par un maximum.
Au cours de la croissance du germe, on observe deux mouvements de
diffusion, du liquide vers le solide et du solide vers le liquide. La vitesse de
croissance du germe (U) prendra en compte ces deux processus. Elle sera
proportionnelle à :
U oc expRT
r1 - exp
± 1 RT Eq. 2.2
Là encore, on retrouve deux termes. Un terme thermodynamique AG, qui
correspond à la différence d'enthalpie libre entre le liquide et le solide, et un terme
cinétique, qui correspond à la barrière d'activation que doit franchir un atome du
liquide pour se fixer sur le germe à la position qu'il occupera dans le cristal. La
28
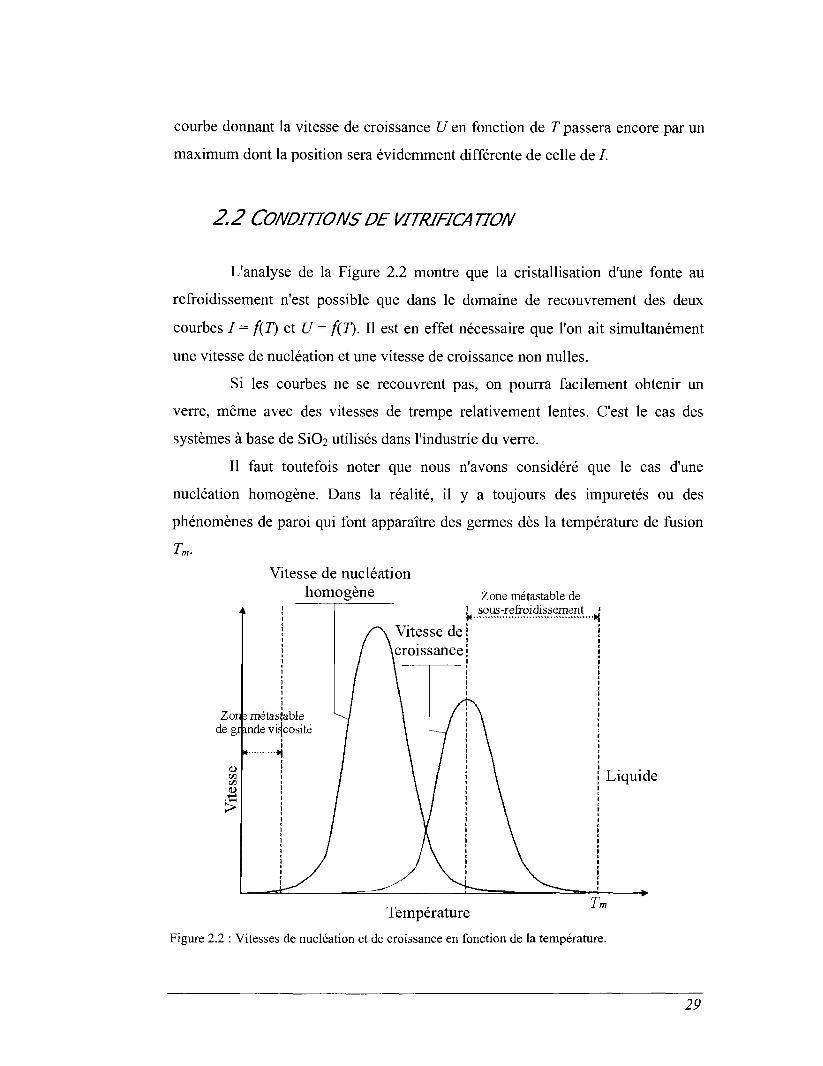
courbe donnant la vitesse de croissance U en fonction de T passera encore par un
maximum dont la position sera évidemment différente de celle de /.
2.2 CONDITIONS DE VITRIFICATION
L'analyse de la Figure 2.2 montre que la cristallisation d'une fonte au
refroidissement n'est possible que dans le domaine de recouvrement des deux
courbes / = f(T) et U = f{T). Il est en effet nécessaire que l'on ait simultanément
une vitesse de nucléation et une vitesse de croissance non nulles.
Si les courbes ne se recouvrent pas, on pourra facilement obtenir un
verre, même avec des vitesses de trempe relativement lentes. C'est le cas des
systèmes à base de S1O2 utilisés dans l'industrie du verre.
Il faut toutefois noter que nous n'avons considéré que le cas d'une
nucléation homogène. Dans la réalité, il y a toujours des impuretés ou des
phénomènes de paroi qui font apparaître des germes dès la température de fusion
Vitesse de nucléationhomogène
Zonde gi mde vi^cositi
Vitesse de\croissance
Zone métastable de• sous-refroidissementF * " ' " " . . . . . . . .
Liquide
Température
Figure 2.2 : Vitesses de nucléation et de croissance en fonction de la température.
29

Une première possibilité d'obtenir un verre consiste à jouer sur la
composition du bain de façon à écarter les courbes I et U (ce point sera développé
dans l'exposé sur la dévitrification).
On peut encore gêner la cristallisation en traversant rapidement la zone
critique ce qui ne laisse pas aux germes le temps de se former ou de se
développer. C'est le principe qu'utilisent les techniques d'hypertrempe dans
lesquelles on réalise des vitesses de trempe dépassant 106 degrés par seconde.
Une autre méthode, pour éviter la formation de germes, consiste à
diminuer le volume de l'échantillon. Le nombre de germes formés est
proportionnel au volume. Les techniques de trempe de brouillard montrent que
plus le volume des gouttelettes est faible et plus la proportion de phase vitreuse est
importante.
La vitrification sera facilitée lorsqu'il y a une grande différence entre la
structure du cristal et celle (ordre à courte distance) du liquide. La réorganisation
des atomes de la fonte nécessite alors une diffusion plus importante. Ceci se
produit en général pour les systèmes complexes. On observe la plupart du temps
que plus les composants sont nombreux et plus la vitrification est facile. C'est une
des raisons pour lesquelles les compositions des verres industriels sont si
complexes.
Un autre facteur important, dans le même ordre d'idée, est lié à la
présence de défauts au sein du cristal. Un cristal contenant de nombreux défauts
se formera plus facilement à partir d'une fonte qu'un cristal ne présentant aucun
défaut. C'est ainsi que SiO2, cristal présentant peu de défauts, vitrifiera facilement
alors que TiO2, cristal susceptible de présenter de nombreux défauts, cristallisera
toujours. La même remarque s'applique à P2O5 et V2O5. Les cristaux d'oxyde de
plomb PbO présentent de nombreux défauts et PbO ne se présente pas sous forme
de verre. On peut diminuer le nombre de défauts du cristal en ajoutant de la silice
mais en contre-partie, on constate que le système binaire vitrifie plus facilement.
C'est ainsi que Pb2SiO4 donne un verre. Pb2GeO4 ne vitrifie pas, du moins si on le
fond dans un creuset de platine. Il vitrifie par contre si on opère dans un creuset de
30

porcelaine ou de silice car une partie des ions aluminium ou silicium se dissolvent
dans le bain.
Un dernier facteur important enfin est lié à la viscosité du liquide au
point de fusion. Une fonte très visqueuse vitrifiera aisément car les mouvements
de diffusion atomique nécessaires à la germination et croissance seront fortement
ralentis. La viscosité est liée à la structure du liquide. La présence de chaînes
(SiC>2, soufre, sélénium) favorise la vitrification. Pour un système donné, elle est
liée à la température de fusion qui aura intérêt à être aussi basse que possible,
d'où, par exemple, l'observation très fréquente selon laquelle des domaines de
vitrification se situent au voisinage des eutectiques profonds.
2.3 IMMISCIBILITE
Dans les domaines de vitrification interviennent parfois des séparations
de phases. Elles conduisent à des verres inhomogènes à des échelles plus ou
moins grandes, qui vont de l'inhomogénéité entraînant l'opalescence du matériau à
une microhétérogénéité détectable uniquement par microscopie électronique. Les
séparations de phases sont utilisées dans les vitro-céramiques lorsqu'elles
précèdent la cristallisation car elles jouent un rôle important dans le contrôle de la
taille des cristallites (Kreidl, 1991). Leur utilisation industrielle est ancienne et
l'on cite souvent l'exemple du Vycor, borosilicate de sodium traité thermiquement
(Paul, 1990). La phase riche en bore est éliminée par traitement à l'acide et le
matériau obtenu est un squelette de silice presque pure qui est densifié en silice
massive à basse température (le procédé permet ainsi d'élaborer à plus basse
température que par fusion de la silice).
L'immiscibilité dans un système binaire se comprend aisément si l'on
considère l'évolution de l'enthalpie libre du système. Lorsque l'enthalpie libre du
mélange binaire homogène est supérieure à l'enthalpie libre de deux phases
séparées il y a séparation de phases thermodynamiques. La connaissance pour
chaque température des compositions répondant à cette condition définit le
31

domaine d'immiscibilité, à l'intérieur duquel on distingue généralement le
domaine dans lequel la séparation de phases ne peut avoir lieu qu'avec un apport
énergétique (c'est la nucleation vue precédement) et un domaine de décomposition
spinodale où la séparation de phases ne rencontre aucune barrière énergétique.
Selon la nature des phases, il existe deux types d'immiscibilités. Le
premier est une immiscibilité à l'état liquide. Elle se rencontre dans les systèmes
silice-oxyde alcalino terreux (Mg, Ca, Sr). La température du liquidus, en-dessous
de laquelle cette séparation en deux liquides n'existe pas, définit le domaine
d'immiscibilité (Doremus, 1994). En pratique, ces séparations à l'état liquide
rendent difficile la formation de verres homogènes. Le second type d'immiscibilité
est qualifié de métastable, il apparaît lorsque la température la plus élevée à
laquelle la séparation de phases se fait est inférieure à la température du liquidus.
Dans ces domaines, un solide cristallisé (souvent un polymorphe de SiCh) est plus
stable ; on pourra pratiquement former des verres mais ces derniers auront
tendance à être inhomogènes.
L'immiscibilité rencontrée dans les binaires à forte teneur en silice peut-
être considérablement réduite par l'ajout d'un troisième oxyde (à condition que son
immiscibilité avec l'un des deux autres ne soit pas plus grande). C'est le cas pour
le sytème CaO-SiO2, où l'ajout de quelques pourcents de Na2Û supprime presque
toute immiscibilité (Dorémus, 1994). D'une façon générale, l'efficacité est plus
grande avec des cations à faible potentiel ionique, les ions à fort potentiel ionique
comme le titane agissant au contraire comme agent nucléant. Le cas de AI2O3,
dont l'ajout en faible quantité est bénéfique (Kreidl, 1991 ; Rawson, 1967) reste à
cet égard mal compris.
La raison structurale des immiscibilités et des séparations de phases dans
les verres n'est pas bien connue. Une relation entre la largeur du domaine
d'immiscibilité et le potentiel ionique ou la force de champ du cation (Doremus,
1994) existe mais elle n'est pas universelle. Une explication qualitative proposée
qui appuie la relation avec le potentiel ionique (Rawson, 1967) est que les cations
entrent en compétition pour se créer un environnement adéquat, les séparations de
phases se produisant lorsque ce but ne peut être atteint dans une seule phase.
32

2.4LA TRANSITION VITREUSE
Plaçons-nous maintenant dans le cas où le bain fondu irait donner
naissance à un verre plutôt qu'à un cristal et regardons comment évoluent
certaines fonctions d'état thermodynamiques, volume V ou enthalpie H par
exemple (Figure 2.3).
2.4.1 Approche thermodynamique
Lorsque le liquide cristallise au cours de la solidification, on observe une
discontinuité brutale du volume (à pression constante) ou de l'enthalpie (chaleur
latente de fusion) au point de fusion. Au-dessus de Tm, le volume augmente assez
vite. En-dessous de Tm, il diminue plus lentement. Le coefficient de dilatation du
liquide est plus grand que celui du solide. On n'observe par contre aucune
discontinuité autour de Tm lorsque le liquide donne un verre. On a un phénomène
de surfusion et la courbe caractéristique du verre est la simple extrapolation de
celle du liquide. Le liquide figé devient un verre.
Imaginons que, grâce à un refroidissement extrêmement lent, on puisse
maintenir cet état métastable jusqu'au zéro absolu. La pente de la courbe
caractérisant le liquide surfondu étant plus grande que celle du cristal, il y aura un
moment où ces deux courbes vont se couper. Au-delà de ce point, on aboutit à une
situation paradoxale dans laquelle le volume V ou l'enthalpie H du verre sont
inférieurs à ceux du cristal, ce qui est contraire à l'expérience (cette possibilité a
été discutée par Kauzmann (1948), qui a donné son nom à ce paradoxe). On sent,
par conséquent, que cette situation de liquide surfondu ne peut
thermodynamiquement pas se prolonger au-delà d'une certaine température To, à
partir de laquelle le verre devra bien se rendre compte qu'il est "devenu" un solide
et non plus un liquide. Son volume ou son enthalpie devront alors varier comme
ceux du cristal. On doit observer à une certaine température une transition entre
un état liquide surfondu et un état "solide". Cette transition, propre aux verres, est
appelée "transition vitreuse".
33
VouH
Liquide
Liquidesurfondt
AmVou AmH
To Tm TempératureFigure 2.3 : Evolution des fonctions d'état thermodynamiques au cours de la
solidification d'un liquide.
Nous avons considéré un cas limite To conduisant à ce que nous appelons
un verre idéal. Dans la réalité, la transition vitreuse se situe autour d'une
température Tg (glass temperature) située avant To. Il faut toutefois remarquer qu'il
ne s'agit pas à proprement parler d'une transition. On observe plutôt un domaine
de transformation, la position de Tg dépendant d'ailleurs de la vitesse de trempe.
Elle est d'autant plus basse (proche de To) que la trempe est lente.
On voit donc que le verre est un état thermodynamiquement métastable.
Il faut d'ailleurs remarquer que pour un système donné, il n'y a pas un "état
vitreux" mais tout un éventail d'états dont la nature est étroitement liée à l'histoire
thermique du matériau (température et vitesse de trempe, recuits...). Par exemple,
Richet et Bottinga (1986) ont montré qu'il y a une différence d'environ 5 kJ.moF1
entre les enthalpies de deux verres de compositions CaMgSi2O6, l'un formé avec
une vitesse de refroidissement rapide caractéristique des expériences de
calorimétrie de chute et l'autre, avec une vitesse plus lente de quelques Kelvin par
34
minutes. Une mesure thermochimique sur un verre n'a donc qu'une valeur
approximative si son histoire thermique n'est pas précisée ; c'est pourquoi il est
nécessaire d'introduire, en plus de la pression et la température, au moins un autre
paramètre pour définir son état : la température fictive, T. Définie par Tool et
Eichlin (1931), la température fictive d'un verre est la température du liquide qui
résulterait de la chauffe instantanée du verre. Ce concept est illustré sur la Figure
2.4 où le volume sert de propriété témoin. Le verre trempé à la vitesse q a sa
température fictive notée T (t=o)- D'après sa définition, la température fictive est
égale à la température de transition vitreuse définie par des méthodes
calorimétriques ou dilatométriques c'est-à-dire le lieu d'intersection des
extrapolations des propriétés de liquide et du verre.
V(t=O)
Zrecuit—T(t=<*>) T(t=Q) Température
Figure 2.4 : Détermination graphique de la température fictive et évolution de celle-ci au coursd'un recuit.

2.4.2 Approche cinétique
L'approche thermodynamique montre qu'à Tg certaines grandeurs varient
de façon brutale (coefficient de dilatation, chaleur spécifique,...) tandis que
d'autres ne montrent aucune discontinuité. D'autre part, on constate que, quelle
que soit la nature du verre (silicate, polymère, verre organique, métal vitreux), la
transition vitreuse se produit toujours lorsque la viscosité dépasse la valeur de 1012
Pa.s. Il semble donc que la transition vitreuse soit liée à un phénomène cinétique
de diffusion et non pas seulement à une nécessité thermodynamique.
Au cours du refroidissement d'un liquide, les mouvements des espèces
moléculaires ou atomiques deviennent de plus en plus lents au fur et à mesure que
la température s'abaisse et que la viscosité augmente. A la transition vitreuse, on
peut imaginer que les mouvements macroscopiques d'ensemble étant gelés, on
assiste au figeage rapide et coopératif des mouvements locaux, et l'on obtient un
verre dont l'état de configuration ne change plus de Tg à 0 K, d'où l'existence d'une
entropie de configuration résiduelle, faisant exception au théorème de Nernst.
Pour préciser cette image, on introduit souvent le concept de nombre de
Deborah*1 comme étant le rapport entre le temps de relaxation structurale ret la
durée d'observation t.
^ArD =-• Eq. 2.3
La transition vitreuse correspondrait au moment où le temps de
relaxation structurale r devient comparable à t (de l'ordre de la minute ou de
l'heure).
Le tableau ci-après, établi pour un verre commercial, indique qu'au-
dessus de Tg, la viscosité et r évoluent de façon parallèle tandis qu'au-dessous, r
se met à croître beaucoup plus vite.
1 Nom d'une prophétesse qui déclara que ce qui apparaissait immobile aux mortels
comme les montagnes ou la taille des océans, ne l'était pas forcément pour une divinité.
36

Tableau 2.1 : Viscosité et nombre de Déborah d'un verre à fibre pour différentes températures.
1 fusion * g •*• ambiante
1500°C 500°C 25°C
Viscosité, rj (Poise) 102 1014 1016
Nombre de Deborah,. \"D 10"11 1 109
II reste à définir ce que l'on entend exactement par temps de relaxation
structurale. Cette notion peut se comprendre au travers de la Figure 2.4.
Considérons par exemple le verre de température fictive T à l'instant t = 0. On
effectue un recuit à une température notée Trecuit et on observe à différents
intervalles de temps (t = 0, t et oo) la variation de température fictive engendrée.
La structure du verre tend à se rapprocher de celle du liquide à la température
Trecuit et passe par des états de configuration qu'auraient pu atteindre des verres
trempés à des vitesses plus lentes (q'<q). Le temps de relaxation correspond alors
au temps caractéristique nécessaire au réarrangement structural nécessaire pour
que le verre adopte la structure du liquide.
Nous avons défini l'état vitreux par ses caractéristiques structurales et
thermodynamiques. Nous allons faire maintenant un bref récapitulatif des
connaissances actuelles sur les verres utilisés pour confiner les déchets radioactifs.
37

Chapitre 3
LES VERRES DE CONFINEMENT
L'idée de solidifier les solutions de produits de fission issues de la chaîne
de retraitement des combustibles irradiés repose sur trois principes :
l'état solide améliore le confinement et facilite les opérations de
manutention,
le passage de la solution aqueuse à l'état solide s'accompagne d'une
réduction de volume des déchets à traiter,
la solidification permet l'obtention d'un matériau répondant aux exigences
de sûreté.
Les études sur le verre ont débuté dès les années 60 et parmi tous les
procédés de solidification envisagés (minéraux, SYNROC), la vitrification est le
seul ayant, pour le moment, abouti sur le plan industriel. En outre, le verre est le
matériau qui répond le mieux au compromis durabilité à long terme / faisabilité
technologique. Le procédé développé et utilisé par le CEA (Jouan et al., 1986)
nécessite plusieurs étapes. La solution de produits de fission est tout d'abord
introduite dans un calcinateur afin d'obtenir un résidu sec qui sera constitué
d'oxyde et de carbonate. Le calcinât est alors mélangé à une fritte de verre inactive
dont la composition est choisie selon certains critères. Le mélange est fondu dans
un creuset à induction, affiné puis coulé dans un conteneur en acier inoxydable.
Le volume de déchets de haute activité, une fois vitrifié, produit par la
France devrait atteindre 6000 m3 en 2020. La responsabilité de la gestion de ces
déchets incombe à l'Agence National de gestion des Déchets Radioactifs
(ANDRA) qui pilote l'ensemble des études de recherche et de développement
dans le domaine. L'option la plus probable pour le stockage de ce type de déchets
est l'enfouissement en site géologique profond. Une loi promulguée le 30
38

décembre 1991 fixe l'étude de la faisabilité et de la sûreté de ce type de stockage
comme axe de recherche prioritaire.
Le stockage des colis, c'est-à-dire le verre lui-même et le conteneur en
acier, doit répondre à certaines exigences. Il doit permettre le confinement des
radioéléments le plus longtemps possible et assurer des conséquences
radiologiques mineures dans le cas d'un retour vers la biosphère. En particulier, un
confinement absolu est nécessaire dans les 500 premières années qui
correspondent à la décroissance des principaux produits de fission. Le concept
multibarrière tente de répondre à ces exigences en isolant les déchets du milieu
extérieur et en empêchant leur dissémination, qui peut se faire principalement par
l'action des eaux souterraines sur les blocs de verre.
La première barrière est le colis, la deuxième est constituée de différents
matériaux, principalement des matériaux argileux qui sont mis en place dans les
puits et qui sont désignés sous le terme "barrière ouvragée". La roche d'accueil
enfin est la troisième barrière.
Ces différentes barrières doivent répondre à certains critères de sûreté et
de fiabilité. Leur rôle peut alors être double si l'on considère qu'elles peuvent
intervenir à la fois sur le passage "aller" de l'eau qui s'effectue depuis les eaux
souterraines vers les blocs de verres et sur le passage "retour" partant des blocs
vers la biosphère. Dans le premier sens, elles doivent retarder voire empêcher
l'action de l'eau sur les colis de verre grâce, en particulier à leur imperméabilité.
Dans l'hypothèse d'une interaction avec l'eau, les colis doivent alors présenter de
bonnes qualités vis-à-vis de l'altération, en confinant le plus longtemps possible
les radioéléments.
Lors du passage "retour", les barrières ouvragées doivent réagir avec les
radioéléments lixiviés du verre en les adsorbant ou en les piégeant, afin de
retarder au maximum leur migration vers les roches d'accueil. Le facteur de
rétention des nucleides dans la formation géologique hôte ainsi que leurs limites
de solubilité dans les eaux du site doivent être également estimés, pour quantifier
le facteur de retard qui peut être apporté par la formation hôte.
39

Ce chapitre présentera brièvement des descriptions de l'état vitreux selon
des approches structurales, thermodynamique et cinétique. Nous verrons en quoi
un verre est dit "métastable", pourquoi ses propriétés sont dépendantes du temps,
comment sa structure intègre les éléments qui composent les déchets radioactifs.
Nous ferons aussi un rapide bilan des connaissances sur l'altération des verres en
mettant en relief les paramètres structuraux, chimiques et thermochimiques qui
influent sur sa cinétique. Nous verrons notamment que la connaissance des
propriétés thermochimiques des verres pourrait être une source de progrès
sensible dans la modélisation des vitesses d'altération. Enfin, nous décrirons les
gammes de compositions chimiques requises pour des solutions industrielles
intéressantes avant de donner les compositions étudiées au cours de cette thèse.
3.1 LES VERRES « SON » ET«AVM»
La France a retenu le verre "SON 681817L1C1A2Z1" comme matériau
de confinement des solutions de produits de fission, issues du retraitement du
combustible des réacteurs "eau-légère". C'est dans les ateliers R7 et T7 de La
Hague que les déchets nucléaires français et étrangers sont respectivement
vitrifiés et c'est pourquoi le verre précédent est habituellement désigné sous le
terme générique de verre "R7T7". Ces ateliers constituent le septième maillon de
la chaîne de retraitement des usines UP2 800 et UP3 qui permettent chacune de
retraiter 800 tonnes/an de combustible irradié. Le domaine AVM correspond à
l'ensemble des verres provenant du traitement des déchets à l'Atelier de
Vitrification de Marcoule, de différents combustibles, principalement de type
graphite-gaz.
Ces verres ont des compositions complexes puisqu'ils contiennent une
trentaine d'éléments. Outre les produits de fission et les transuraniens, les
éléments introduits à partir de la fritte de verre sont choisis selon certaines
qualités requises pour le matériau final :
40

Tableau 3.1 : Composition des verres SON et AVM inactifs exprimée en poids % d'oxydes.
Oxydes
SiO2
B2O3
Na2OAI2O3CaOMgOLi2O
Fe2O3
ZnOZrO2
MOO3Ce2O3
ThO2
La2O3
NiOCr2O3
P2O5
UO2
SrOY2O3
MnO2
CoOAg2OCdOSnO2
TeO2
Cs2OBaO
Pr2O3
Nd2O3
PdGd2O3
FCl
SO4
SON(poids%)
45.4814.029.864.914.04
1.982.912.502.651.700.930.330.900.740.510.280.520.330.200.720.120.030.030.020.231.420.6
0.441.59
AVM(poids%)
39.7216.5516.559.930.805,000.200.95
0.960.750.600.110.560.150.300.500.700.210.120.31
0.100.250.020.14
0.340.280.930.250.600.900.050.05
le silicium, l'aluminium et le bore sont introduits en tant que formateurs du
réseau vitreux,
les alcalins et alcalino-terreux, qui sont des éléments modificateurs de
réseau, permettent d'abaisser le point de fusion et facilitent ainsi son
élaboration; le bore joue aussi le rôle de fondant et permet d'incorporer le
molybdène,
41

le zirconium et l'aluminium confèrent au matériau final de bonnes propriétés
chimiques.
Les compositions données dans le Tableau 3.1 sont celles des verres
SON et AVM inactifs (non radioactifs) simulant les verres actifs pour les études
au laboratoire. Dans ces verres, les actinides sont simulés par le thorium et les
platinoïdes par le manganèse, le cobalt et le nickel.
Toutes les études ont été réalisées jusqu'à présent sur des verres inactifs
pour faciliter la mise en œuvre qui devient très lourde lorsqu'il s'agit d'un matériau
radioactif (travail en cellule blindée, risques d'irradiation). Certaines études ont été
effectuées sur des verres dopés avec un ou plusieurs actinides. Les résultats
obtenus sur le verre inactif ont permis d'établir les lois cinétiques régissant la
dissolution du verre ainsi que les mécanismes d'altération en solution aqueuse.
Cependant, une étape ultérieure de validation sur le verre actif sera nécessaire, car
d'autres phénomènes, tels que la radiolyse de l'eau, peuvent alors intervenir et
modifier les lois préétablies.
3.2 CHOIX DES COMPOSITIONS ÉTUDIÉES
3.2.1 Des verres inactifs simplifiés
L'étude directe des verres SON et AVM radioactifs s'avère délicate. C'est
pourquoi nous avons travaillé sur des verres SON et AVM inactifs. Cependant,
l'étude de ces derniers ne se fait pas non plus aisément. En effet, les résultats
obtenus ne permettent pas une interprétation simple et ceci plus particulièrement à
cause du nombre important de constituants. Les effets observés seront
difficilement attribuables à tel ou tel élément présentant des caractéristiques
voisines.
42

Tableau 3.2 : Compositions nominales et analysées en mol % des verres simplifiés dérivés duverre de référence SON (M : masse molaire, n : nombre d'atome).
SiO2
B2O3
Na2O
A1ACaO
ZrO2
Ce2O3
Li2O
Autres
M
n
SON-3a
nominal
67.74
18.02
14.24
62.066
3.3601
analysé
67.58
18.27
14.12
0.02
0.02
62.117
3.3662
SON-4a
nominal
64.93
17.28
13.65
4.13
0.01
63.714
3.4279
analysé
64.94
17.49
13.39
4.14
0.04
63.779
3.4336
SON-5b
nominal
61.16
16.27
12.86
3.89
5.82
63.275
3.3450
analysé
59.16
17.32
13.12
4.04
6.32
0.01
0.03
63.445
3.3639
SON-6b
nominal
60.11
16.00
12.63
3.83
5.72
1.71
64.310
3.3397
analysé
57.71
17.33
12.91
4.16
6.04
1.80
0.02
0.02
64.680
3.3697
Dosage du bore et analyse par ICP-AES pour les autres constituantsAnalyse par Microsonde
SiO2
B2O3
Na2O
A12O3
CaO
ZrO2
Ce2O3
upAutres
M
n
SON-7b
nominal
59.98
15.96
12.61
3.82
5.70
1.70
0.23
64.901
3.3429
analysé
59.18
16.03
12.15
4.75
5.95
1.70
0.22
0.02
65.251
3.3601
SON-8b
nominal
56.98
15.16
11.98
3.63
5.42
1.62
0.22
4.99
63.165
3.3260
analysé
53.68
15.95
10.66
3.71
7.28
1.57
0.87
6.23
0.04
64.521
3.3377
SON
nominal analysé
52.46
13.96
11.03
3.34
4.99
1.49
0.20
4.59
7.94
69.220
3.3147
*'Dosage du bore et analyse par ICP-AES pour les autres constituantsAnalyse par Microsondeb)
43
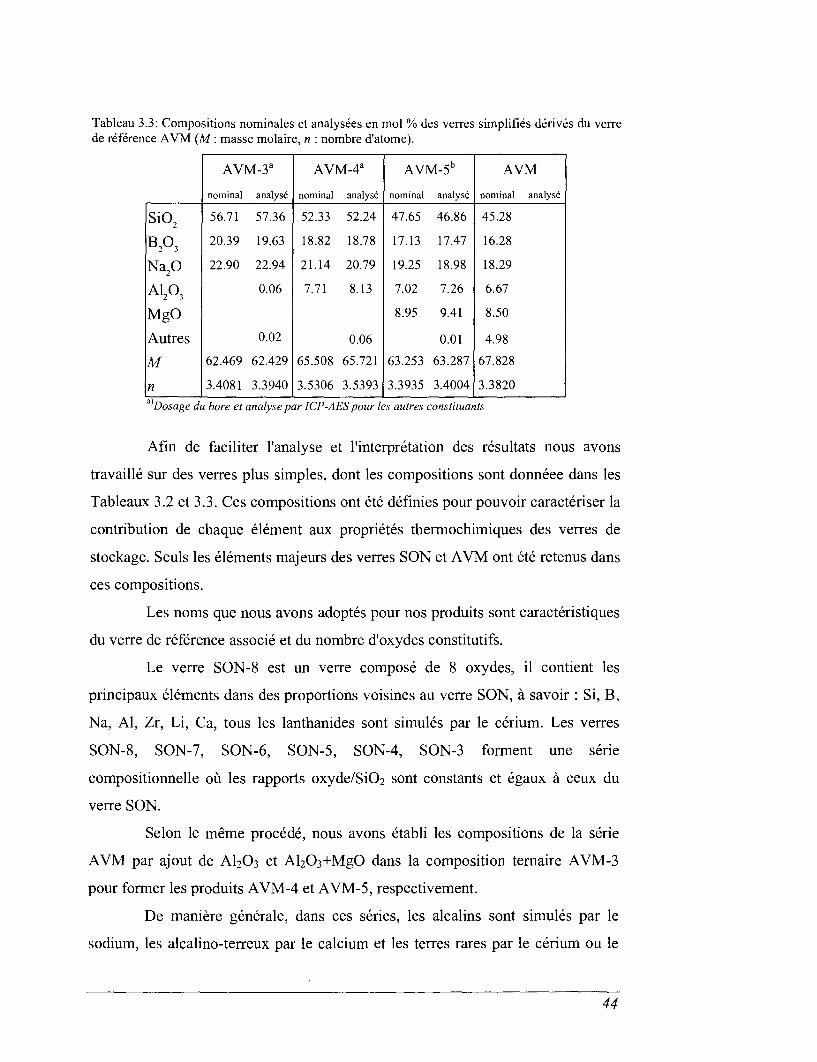
Tableau 3.3: Compositions nominales et analysées en mol % des verres simplifiés dérivés du verrede référence AVM (M : masse molaire, n : nombre d'atome).
SiO2
BANa2O
A12O3
MgO
Autres
M
n
AVM-3a
nominal
56.71
20.39
22.90
62.469
3.4081
analysé
57.36
19.63
22.94
0.06
0.02
62.429
3.3940
AVM-4a
nominal
52.33
18.82
21.14
7.71
65.508
3.5306
analysé
52.24
18.78
20.79
8.13
0.06
65.721
3.5393
AVM-5b
nominal
47.65
17.13
19.25
7.02
8.95
63.253
3.3935
analysé
46.86
17.47
18.98
7.26
9.41
0.01
63.287
3.4004
AVM
nominal analysé
45.28
16.28
18.29
6.67
8.50
4.98
67.828
3.3820
Dosage du bore et analyse par ICP-AES pour les autres constituants
Afin de faciliter l'analyse et l'interprétation des résultats nous avons
travaillé sur des verres plus simples, dont les compositions sont donnéee dans les
Tableaux 3.2 et 3.3. Ces compositions ont été définies pour pouvoir caractériser la
contribution de chaque élément aux propriétés thermochimiques des verres de
stockage. Seuls les éléments majeurs des verres SON et AVM ont été retenus dans
ces compositions.
Les noms que nous avons adoptés pour nos produits sont caractéristiques
du verre de référence associé et du nombre d'oxydes constitutifs.
Le verre SON-8 est un verre composé de 8 oxydes, il contient les
principaux éléments dans des proportions voisines au verre SON, à savoir : Si, B,
Na, Al, Zr, Li, Ca, tous les lanthanides sont simulés par le cérium. Les verres
SON-8, SON-7, SON-6, SON-5, SON-4, SON-3 forment une série
compositionnelle où les rapports oxyde/SiO2 sont constants et égaux à ceux du
verre SON.
Selon le même procédé, nous avons établi les compositions de la série
AVM par ajout de AI2O3 et Al2O3+MgO dans la composition ternaire AVM-3
pour former les produits AVM-4 et AVM-5, respectivement.
De manière générale, dans ces séries, les alcalins sont simulés par le
sodium, les alcalino-terreux par le calcium et les terres rares par le cérium ou le
44

zirconium. Les verres SON-3 et AVM-3 ne sont formés que de 3 éléments mais
ces derniers représentent 77,45 et 79.85 mol% des compositions SON et AVM,
respectivement.
3.2.2 Le system e SiO2-B2OrNa2O
Comme nous venons de le mentionner dans le paragraphe précédent, les
oxydes de silice, de bore et de sodium constituent la plus grande partie des verres
de cette étude. Or, pour de nombreux systèmes vitreux et liquides, les propriétés
physiques et plus particulièrement thermodynamiques se sont révélées être des
fonctions quasi-additives des propriétés des oxydes constitutifs. Sur cette base, il
nous a semblé indispensable d'obtenir une connaissance plus complète du système
SiO2-B2O3-Na2O dont les propriétés thermodynamiques, notamment les capacités
calorifiques et les entropies, n'ont été que très rarement étudiées. D'autre part, la
nature des déchets évoluant, il faudra formuler de nouvelles compositions de
matrice de confinement avec des teneurs en SiO2, B2O3 et Na2O différentes de
l'AVM et du SON, il sera alors plus aisé d'estimer les propriétés de ces nouvelles
matrices à partir des variations observées dans ce système.
Nous avons voulu synthétiser des verres s'alignant le long de joints
passant par les compositions AVM-3 et SON-3. L'un des produits (BSN-8/24) n'a
cependant pas la composition escomptée par suite d'une erreur expérimentale et
d'autre part la synthèse des composés binaires à partir de mélange d'oxyde s'est
avérée très complexe. Nous aborderons dans le chapitre 5 les difficultés de
synthèse.
Les compositions synthétisées sont données dans le Tableau 3.4. Les
noms utilisés rappellent la composition chimique nominale, BSN-x/y indique un
verre ayant x mol% de B2O3, y mol% de Na2O et (100-x-.y) mol% de SiO2. Nous
soulignons que le verre nommé BSN-8/24, ayant été victime d'une erreur lors de
sa synthèse, ne suit pas cette définition et devrait d'après l'analyse chimique se
45

nommer BSN-9/17. D'autre part, les verres SON-3 et AVM-3 pourraient
également s'appeler respectivement BSN-18/14 et BSN-20/23.
SiO;
: SONr3-a -;-@--- ••
o Synthétisé° Non synthétiséA Déjà étudié
Na2O
- p ••<&> - p -
/ AVM-3
B2O3Figure 3.1 : Compositions synthétisées dans le diagramme SiO2-B2O3-Na2O. L'oxyde de bore et lebinaire SiO2-Na2O ont déjà fait l'objet d'un grand nombre de descriptions thermochimiquespubliées.
Tableau 3.4: Compositions nominales et analysées en mol % des verres de la série BSN (Mmasse molaire, n : nombre d'atome).
SiO2
BANa2O
Autres
M
n
BSN-5/163
nominal
78.66
5.34
16.00
60.903
3.1071
analysé
78.45
5.70
15.81
0.04
60.955
3.1147
BSN-8/243
nominal
67.73
8.07
24.20
61.312
3.1614
analysé
74.07
8.80
17.05
0.08
61.296
3.1777
BSN-1
nominal
56.71
10.82
32.47
61.731
3.2164
1/3 2a
analysé
56.60
10.96
32.39
0.05
61.766
3.2198
BSN-29/14a
nominal
56.71
28.86
14.43
63.110
3.5772
analysé
56.62
29.28
13.98
0.13
63.197
3.5871
Dosage du bore et analyse par ICP-AES pour les autres constituants
46
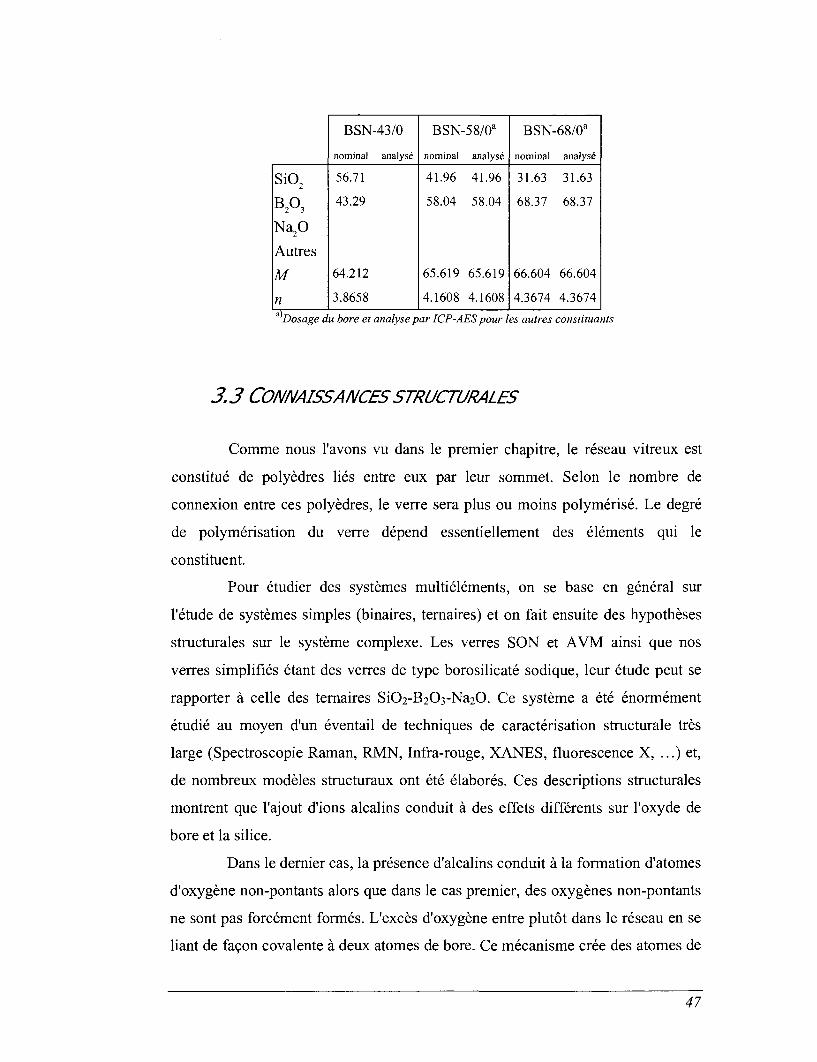
SiO2
BANa2O
Autres
M
n
BSN-43/0
nominal analysé
56.71
43.29
64.212
3.8658
BSN-58/0a
nominal
41.96
58.04
65.619
4.1608
analysé
41.96
58.04
65.619
4.1608
BSN-68/03
nominal
31.63
68.37
66.604
4.3674
analysé
31.63
68.37
66.604
4.3674
Dosage du bore et analyse par ICP-AESpour les autres constituants
3.3 CONNAISSANCES STRUCTURALES
Comme nous l'avons vu dans le premier chapitre, le réseau vitreux est
constitué de polyèdres liés entre eux par leur sommet. Selon le nombre de
connexion entre ces polyèdres, le verre sera plus ou moins polymérisé. Le degré
de polymérisation du verre dépend essentiellement des éléments qui le
constituent.
Pour étudier des systèmes multiéléments, on se base en général sur
l'étude de systèmes simples (binaires, ternaires) et on fait ensuite des hypothèses
structurales sur le système complexe. Les verres SON et AVM ainsi que nos
verres simplifiés étant des verres de type borosilicaté sodique, leur étude peut se
rapporter à celle des ternaires SiO2-B2O3-Na2O. Ce système a été énormément
étudié au moyen d'un éventail de techniques de caractérisation structurale très
large (Spectroscopie Raman, RMN, Infra-rouge, XANES, fluorescence X, ...) et,
de nombreux modèles structuraux ont été élaborés. Ces descriptions structurales
montrent que l'ajout d'ions alcalins conduit à des effets différents sur l'oxyde de
bore et la silice.
Dans le dernier cas, la présence d'alcalins conduit à la formation d'atomes
d'oxygène non-pontants alors que dans le cas premier, des oxygènes non-pontants
ne sont pas forcément formés. L'excès d'oxygène entre plutôt dans le réseau en se
liant de façon covalente à deux atomes de bore. Ce mécanisme crée des atomes de
47

bore liés de manière covalente à quatre oxygènes pontants, l'atome de bore central
et les quatres oxygènes qui l'entourent forment ainsi un oxyanion qui est lié à la
structure vitreuse par chaque oxygène. La charge négative associée à l'oxyanion
n'est pas localisée sur un oxygène particulier mais est compensée par un ion
alcalin dans l'environnement proche. A partir de leurs analyses de spectroscopie
RMN du n B, Bray et ses collaborateurs (Dell et al., 1983 ; Xiao, 1981 ; Yun et
Bray, 1978 ; Silver et Bray, 1958) ont développé un modèle détaillé de la structure
atomique des verres borosilicatés sodiques. Ce modèle donne les fractions
molaires du bore présent dans les sites anioniques tétraèdriques (7V4), symétrique
trigonal (A^s), et assymétrique trigonal (N^a). Dans presque toute la totalité du
domaine vitrifiable de ce système, N4, NiS, et À̂ a dépendent des rapports molaires
Na2O/B2Û3 (R) at SiO2/B2O3 (K). Faisons ici un bref résumé du modèle de Dell et
al. (1983) expliquant la dépendance de la spéciation du bore avec la composition :
R < 0.5. Dans ce domaine de composition à faible teneur en sodium, tout le
bore est soit en site trigonal symétrique soit en site tetragonal. La quantité
de bore tétracoordonné est directement proportionnelle à la teneur en
sodium. Tout le sodium dans le verre compense la charge négative des sites
tétraèdriques du bore contenus dans une matrice purement boratée. La
teneur maximale de sodium que le sous-réseau borate peut intégrer est
supposée correspondre à la composition diborate (Na2O.2B2Û3) pour
laquelle R = 0.5. Les sous-réseaux borate sodique et silicate sont supposés
ne pas se mélanger à une échelle locale ce qui s'accorde avec la tendance de
ces verres à présenter des séparations de phase.
0.5 < R < i?max = 0.5 + K116. Dans ce domaine, la proportion totale de bore
tétracoordonné est encore égale à la teneur en sodium du verre. Cependant,
il est supposé que le sodium en excès par rapport à la composition diborate
est capable d'entrer dans le réseau silicate avec un bore tétracoordonné pour
former des unités Reedmergnerite. Ces unités [BSi40io]~ sont formées d'un
tétraèdre BO4, chaque oxygène participant également à la construction d'un
tétraèdre autour d'un atome de silicium. Tout le sodium supplémentaire (par
rapport à la composition diborate) vient donc compenser la charge négative
_

de ces unités jusqu'à ce que la teneur en bore qui s'intègre au réseau silicate
soit tellement importante que tous les oxygènes pontants formant les sites
tétraèdriques du bore ne puissent plus être liés à des tétraèdres de silice.
•Kmax <R<Rd\ = 0.5+K/4. Dans ce domaine, N4 est constant. Le sodium
en excès par rapport à i?max est supposé s'associer avec des oxygènes non-
pontants sur les tétraèdres de silice. Il est de plus supposé que le nombre
maximum de tétraèdres SiÛ4 à oxygènes non-pontants que le réseau peut
accomoder est de trois pour une paire d'unités Reedmergnerite.
Ru < R < Z?d3 = 2 + K. Une fois que le réseau est saturé en oxygènes non-
pontants dans les unités Reedmergnerite et en bore tétracoordonné, le
sodium supplémentaire est supposé compenser la charge négative crée par la
formation de site trigonal assymétrique du bore contenant un ou deux
oxygènes non-pontants. La formation de ces nouvelles unités résulte de
l'élimination des bores tétracoordonnés jusqu'à ce que la teneur en sodium
excède i?d3, point où tous les sites tétraèdriques ont disparu.
Le modèle structural des verres borosilicatés sodiques ci-dessus est basé
entièrement sur les résultats de résonance magnétique nucléaire du n B . Bien que
le modèle apparaisse qualitativement en accord avec les spectres de vibrations
obtenus par spectroscopie Raman (Furukawa and White, 1981), la plupart des
détails quantitatifs du modèle n'ont pas été vérifiés par d'autres tecnhiques
spectroscopiques. Bunker et al. (1990), Loshagin et Sosnin (1994), Bhasin et al.
(1998), Fleet et Muthupari (1999) avec leurs nouvelles données de spectroscopie
RMN (29Si, 170,23Na et nB), Raman et Xanes au seuil K du bore ont montré que
le mélange des unités boratées et silicatées commençait pour des valeurs de R
inférieures à celle prédite par Dell et al., entre 0.33 et 0.4 au lieu de 0.5. Les
données de Bunker et al. (1990) et Loshagin et Sosnin (1994) ont révélé la
présence d'un groupement structural supplémentaire qu'ils ont nommé groupe
danburite [BSiC>4]\ Celui-ci se compose d'un atome de bore tétracoordonné lié à
un tétraèdre Sidt. Par conséquent sa présence dans les domaines de composition
R>0.5 induit une augmentation de la fraction de bore tétracoordonné par rapport
aux valeurs du modèle de Dell. De plus le réseau silicate serait saturé à R — 3 K
49

/16 (et non Kl 16). En désaccord avec tous ces travaux, l'étude de Wang et
Stebbins (1998) par spectrométrie RMN 17O des verres borosilicatés sodiques a
par ailleurs montré qu'un mélange entre les atomes de bore en coordinence 3 dans
le réseau silicate était possible.
Les compositions de nos verres ayant été choisies pour pouvoir
extrapoler les résultats obtenus aux verres SON et AVM, les valeurs de R de
chaque série sont identiques et égales à celles qui caractérisent les verres de
référence, SON ou AVM, soit 0.79 et 1.12, respectivement. La valeur de R étant
supérieure à 0.5, nous sommes a priori en présence d'un réseau mixte constitué en
particulier d'unités de type Reedmergnerite [BSi40io]~.
Si on peut penser que cette caractérisation structurale est valide pour les
verres SON-3, AVM-3 (ternaires) et la série BSN, l'extrapolation aux verres plus
complexes et notamment au SON et AVM est délicate car l'effet de l'incorporation
d'autres éléments dans le réseau d'oxyde est mal connu. En particulier, la
possibilité d'effets synergiques entre deux cations peut modifier les résultats
obtenus sur les systèmes simples. C'est typiquement le cas des systèmes
quaternaires SiO2-B2O3-Na2O-Al2O3, où la substitution du sodium par
l'aluminium augmente la polymérisation du réseau de silice et diminue la
proportion de bore en coordinence 4 (El-Damrawi et al., 1993). Araujo et Hares
(1981) suggèrent que, dans les cas où la teneur en alumine reste inférieure à celle
des alcalins, l'aluminium soit considéré comme tétracoordonné et qu'un atome
d'aluminium ne puisse être lié via un atome d'oygène à un autre aluminium ou à
un atome de bore tétracoordonné. Cette suggestion permet d'utiliser les
descriptions structurales du système SiO2-B2O3-Na2O en réduisant seulement la
teneur en alcalins potentiellement utilisable pour compenser les charges des
oxyanions de bore de la proportion d'alumine dans la composition du verre.
De nombreuses techniques spectroscopiques (RMN, absorption X,
XANES, Môssbauer) ont été utilisées pour scruter de façon un peu plus précise la
structure du verre SON et de verres simplifiés qui lui sont associés. La RMN Al
a permis de vérifier la coordinence tétraèdrique de l'aluminium qui est donc
complètement formateur de réseau dans le verre (Delorme, 1998 ; Ricol, 1995 ;
50

Tovena, 1995). La spectroscopie d'absorption X au seuil K et L du zirconium
(Galoisy et al., 1999 ; Petit-Maire, 1988 ; Ricol, 1995) a mis en évidence le rôle
de formateur de réseau du zirconium avec une coordinence 6 en degré d'oxydation
+4. La spectroscopie Môssbauer (Ricol, 1995) a attribué une valence 3 au fer dans
des sites tétraèdriques. L'étude EXAFS au seuil K du Fe pour une famille de
verres simplifiés dérivés du SON (Pellegrin, 2000), montre que l'essentiel du fer
est sous forme Fe3+ en coordinence 4 mais aucune liaison entre les tétraèdres FeÛ4
et le réseau vitreux n'a pu être mise en évidence. Le zinc a été étudié par EXAFS
au seuil K du Zn (Le Grand, 1999). Il se présente en sites tétraèdriques liés aux
tétraèdres SiÛ4 par des sommets. Les environnements de l'uranium, du thorium,
du neptunium et, globalement, des éléments présentant un danger important à long
terme ont été étudiés par Petit-Maire (1988). Lorsque ces éléments sont en
valence 6, on observe un mélange de sites de coordinence 6 et de coordinence plus
élevée (7, 8 ou plus). La proportion des sites en coordinence 6 augmente lorsque
la taille du cation diminue.
Tous les cations intermédiaires contenus dans le verre SON sont donc
formateurs du réseau. Leur substitution à Si + provoque un déficit de charge qui
doit être compensé par un cation modificateur de réseau afin d'assurer la neutralité
électrique du matériau. Le bilan des charges négatives créées par les groupements
(A1O4), (ZrO62~), (FeO4) et (BO4~) dans le verre SON donne 5.19 moles pour 100
moles d'éléments (calculé à partir des pourcentages molaires d'éléments). En
contre-partie celui des alcalins et alcalino-terreux est de 9.44 moles. Cette
différence explique le fait que tous les cations intermédiaires puissent être
incorporés dans le verre en tant que formateurs. Le même raisonnement s'applique
aux autres verres que nous avons préparés. Ces considérations montrent que le
réseau est homogène et très bien copolymérisé, du fait de la bonne incorporation
des cations intermédiaires dans le réseau vitreux et qu'il est sans doute constitué
de nombreuses liaisons mixtes Si-O-M. Cette bonne incorporation est due à la
forte présence de cations modificateurs qui permettent un agencement homogène
des autres cations dans le réseau. Ce résultat est en outre en accord avec les
données de la littérature sur des systèmes plus simples quant à l'importance des
51

alcalins et alcalinoterreux sur l'incorporation des cations intermédiaires dans le
réseau en tant que formateurs.
Ce bilan fait apparaître un excédent de cations modificateurs restants
dans le réseau. Ils vont donc s'incorporer au réseau de silice en provoquant la
rupture de liaisons Si-O-Si. Ces ruptures provoquent l'apparition de liaisons Si-O"
compensées par un cation de type Na+. Le réseau silicate du verre SON-8 est donc
a priori faiblement dépolymérisé (4.25 moles d'alcalins restants pour 100 moles
d'éléments).
Nous avons vu que la mesure des propriétés thermochimiques des verres
nécessite la réalisation d'un cycle thermodynamique incluant la phase liquide. Il
est donc utile d'avoir également quelques idées sur la structure des borosilicates
liquides. Très peu d'études structurales se consacrent à la structure des liquides
borosilicates. On mentionnera cependant les travaux de Stebbins et ses
collaborateurs (Stebbins et Ellsworth, 1996 ; Wang et Stebbins, 1998 ; Sen et al.,
1998 ; Stebbins et Sen, 1998) qui se sont intéressés aux rearrangements
structuraux dans la zone de transition vitreuse. Leurs résultats de spectroscopie
RMN UB permettent de suivre les variations de la teneur en oxygène non-pontant
et a posteriori l'évolution des rapports 4B / 3B. Ils ont pu observer des diminutions
significatives de la teneur en bore tétracoordonné quand la température augmente
tout en laissant inchangée la coordinence du silicium.
En résumé, la structure des verres borosilicates a fait l'objet de
nombreuses études. Bien que les interprétations structurales issues de ces
dernières permettent d'expliquer un grand nombre de propriétés, les progrès
d'analyse récents apportent d'importantes modifications permettant de se
rapprocher des détails quantitatifs. Pour les liquides, la situation est plus confuse
car les outils analytiques leur sont moins adaptés. Pourtant, on a clairement
montré que la transition vitreuse marquait le début de changements structuraux
qu'il devrait être possible de sonder par le biais de nos mesures des propriétés
thermochimiques.
Structure et thermodynamique ont été abordées, voyons comment
l'altération des silicates peut être comprise à la lumière de ces définitions..
52

Chapitre 4
ALTERATION DES SILICATES VITREUX
L'étude de la corrosion aqueuse des verres est un élément essentiel pour
estimer la durée de vie des colis de stockage. En effet, l'eau, en cas d'intrusion au
sein d'un site de stockage, constitue le principal facteur d'altération des verres et le
principal vecteur des radionucléides vers la biosphère. La prédiction du
comportement à long terme de la matrice vitreuse repose sur l'étude des
mécanismes et cinétiques de dissolution.
4.1 MECANISMES DE DISSOLUTION
La vitesse d'une réaction est proportionnelle à son affinité réactionnelle,
A (Prigodine, 1968). Celle-ci peut être définie comme une force motrice d'origine
chimique qui détermine le sens de la réaction chimique de la même façon que le
sens du gradient de charge hydraulique détermine le sens d'écoulement d'un
fluide. Sa connaissance n'est cependant pas suffisante pour décrire à quelle vitesse
les transferts de masse entre la solution et le minéral s'opèrent jusqu'à l'équilibre.
Il est nécessaire pour cela de connaître le ou les mécanismes contrôlant les
phénomènes de dissolution et de précipitation afin d'accéder au temps de passage
de l'état initial à l'état final ou intermédiaire.
Lors du processus de dissolution interviennent simultanément des
réactions de surface et des phénomènes de diffusion (transport d'espèces en
solution vers l'interface réactionnelle et transport des produits hors de l'interface).
Il convient d'apprécier à chaque instant la contribution de chacun des mécanismes
à la dissolution. La corrosion des matériaux silicates vitreux et cristallisés
s'explique par deux théories étroitement liées :
53

la théorie de l'état de transition (Lasaga, 1981b>c; Aagaard et Helgeson, 1982) :
la réaction de surface entre le solide et la solution met en jeu des molécules
transitoires appelées complexes activés. Le complexe activé définit l'état des
réactants quand ils passent par un maximum d'énergie libre avant de former
les produits de la réaction d'altération;
la théorie de la chimie de coordination de surface (Sposito, 1986; Schindler et
Stumm., 1987 ; Stumm et al., 1985) : la surface du minéral ou du verre dans
l'eau est comparée à une électrode sur laquelle s'adsorberaient les ions.
Selon ces deux théories, toute réaction chimique hétérogène peut se
décomposer en 5 étapes élémentaires situées au niveau de l'interface
solide/solution :
1. transport des réactifs (H+, H3Û+, H2O) vers la surface du verre,
2. adsorption des réactifs sur les sites réactionnels de la surface,
3. réaction chimique de surface sensus-stricto avec déstabilisation des liaisons
pontantes du réseau autour de la molécule réactive,
4. désorption des produits de la réaction de surface,
5. transport des produits loin de la surface.
L'étape la plus lente contrôle la réaction de dissolution. Quand les étapes
limitantes sont les réactions 2, 3, 4, il s'agit d'un contrôle par une réaction de
surface. Pour les verres et minéraux silicates, à des températures inférieures à
100° C, c'est généralement la réaction 4 de désorption du complexe qui est
cinétiquement la plus lente (Stumm et al., 1985). Quand les étapes limitantes sont
1 ou 5, il s'agit d'un contrôle diffusionnel. Les expériences de Guy (1989) sur des
verres basaltiques illustrent les différents domaines de contrôle des mécanismes
de dissolution en fonction de la température et du pH (Figure 4.1). On constate
que les réactions de surfaces contrôlent la cinétique de dissolution des verres pour
des températures basses (inférieures à 170° C) et pour des pH proches de la
neutralité (3 à 9). Le contrôle diffusionnel se manifeste aux températures les plus
élevées et les pH extrêmes acides ou basiques.
54

o
300 -
200 -
100 -
Contrôle diffusionnel
Contrôle intermédiaire
Contrôle par réactionsde surface
6
pH10 12
Figure 4.1 : Mécanismes de contrôle de la cinétique globale des verres basaltiques (Guy, 1989)
4.2 DIFFUSION ET REACTIONS DE SURFACE
4.2.1 La diffusion des espèces réactantes
Dans le modèle des défauts (Bruce et al., 1986), les cations migrants H+
ou H3Û+ pénétreraient préférentiellement dans le verre grâce aux sites interstitiels
laissant les sites normaux vacants. Pour les modèles plus récents qui postulent
l'existence dans le verre de pseudophases (Goodman, 1986) qui auraient un effet
bloquant de la diffusion ionique, la diffusion de H+, H3Û+ ou H2O se fait alors
plutôt dans le réseau désordonné connecté autour des pseudophases. Ce
phénomène d'échange entre les ions mobiles M+ (principalement les alcalins,
alcalinoterreux) du verre et les ions H+ ou H3Û+ de la solution est beaucoup plus
marqué en milieu acide. Ce processus d'extraction préférentielle encore appelé
lixiviation conduit à une augmentation du pH de la solution altérante et à la
formation d'une couche résiduelle désalcalinisée en surface du verre.
Si—O—Na + H+ => Si—OH + Na+
ou Si—O—Na + H2O=>Si—OH +
55

Dans le cas des verres et minéraux borosilicatés, l'altération s'effectue
également par une extraction préférentielle du bore, alors que cet élément est
formateur du réseau vitreux. Ce comportement analogue à celui du sodium résulte
néanmoins de mécanismes réactionnels différents. La réaction électrophile sur un
oxygène pontant favoriserait d'une part la lixiviation du bore en milieu acide et
d'autre part la conversion des sites de coordinence 4 du bore vers des sites de
coordinence 3 facilement hydrolysables (Bunker et al., 1988). Cet auteur estime
donc que la coordinence 4 du bore et son caractère formateur dans le verre ne rend
pas impossible la dissolution sélective de cet élément. On sait par ailleurs que
l'acide borique et le borate de sodium sont solubles dans l'eau.
r+OHsC B :
H2O
OH
B + H2O
4.2.2 La diffusion des espèces hydratées du verre
Sous l'action de l'ion hydroxyl créé par interaction d'un atome d'oxygène
non pontant avec une molécule d'eau, des liaisons siloxanes peuvent se rompre.
Si—O— Si + OH" => Si—OH + Si—O"
Si—O- + H2O => Si—OH + OH"
En milieu neutre ou basique, la dissolution des verres procède par une
attaque nucléophile des ions hydroxyls sur les liaisons fortes du réseau, donc sur
les liaisons entre oxygènes pontants et cations formateurs. Les éléments
constitutifs du verre passent en solution de manière stoechiométrique : la
dissolution est congruente tant qu'aucune phase secondaire ne se forme.
56

Ainsi la dissolution d'un verre silicate peut être limitée et contrôlée par la
diffusion des ions formateurs de réseau à travers une couche d'altération poreuse
superficielle dans certaines conditions : stade très avancé de la réaction
d'altération ou température d'altération très élevée (Doremus, 1979).
4.2.3 Les réactions de surface
Ces réactions consistent en une succession de réactions élémentaires
d'adsorption du solvant réactif, de formation de molécules réactives appelées
complexes activés et de désorption de ces molécules. Les espèces aqueuses sont
susceptibles de catalyser ou d'inhiber la cinétique de réaction globale. La réaction
globale macroscopique qui résulte de ces réactions élémentaires à l'échelle
moléculaire peut s'écrire sous la forme : aA + bB => cC + dD + eE, où A est le
verre réactif solide, B l'espèce aqueuse éventuellement organique (H+, H3O+,
H2O), C, D, E les produits de la réaction (espèces aqueuses telles que HUSiC ,̂
Al3+, Zn2+) et a, b, c, d et e les nombres de moles de réactifs et de produits qui
participent à la réaction globale de dissolution.
Lee et Clark (1985, 1986) ont étudié la dissolution des verres
borosilicatés et proposent le processus suivant : après immersion dans une
solution aqueuse, une charge de surface négative se développe en surface du
verre. La cause de cette charge négative est la lixiviation des alcalins qui crée en
surface des sites SiO\ Ces groupes réagissent avec les molécules d'eau pour
former en surface des groupes silanols et augmenter le pH de la solution. Lorsque
le pH est suffisament élevé pour hydrolyser les liaisons des cations métalliques,
ces cations métalliques en solutions forment des particules colloïdales chargées.
Elles peuvent rester en solution ou s'adsorber en surface du verre suivant la charge
de surface de celui-ci. Les particules de même charge que le verre sont repoussées
et grossissent en solution en adsorbant d'autres ions (Clark, 1992). Les colloïdes
en solution ainsi formés, consommeraient des éléments provenant de la solution et
diminueraient les effets de saturation. Ils augmenteraient de cette façon la
57

dissolution du verre alors que les colloïdes adsorbés en surface pourraient
constituer une barrière protectrice vis-à-vis d'une altération ultérieure.
4.3L'Ai TERA TION DU VERRE SON
Dans le cas du verre SON, la première étape de l'altération est
l'interdiffusion entre les alcalins (Na, Li), alcalinoterreux (Ca, Sr) et les protons de
la solution et l'extraction préférentielle du bore (cf. § 4.2.1) par rapport au
principal élément formateur (Si). Cette première étape provoque une élévation du
pH (de 6 à 9,5) et conduit à la création d'une fine zone réactionnelle de verre
hydraté plus ou moins désalcalinisé encore appelée front d'attaque.
Dès que le pH devient basique, pour des faibles rapports surface de
verre / volume de solution, il y a une libération par hydrolyse de tous les éléments,
la corrosion se poursuit donc par l'attaque des liaisons Si-O-Si (cf. § 4.2.1) c'est-à-
dire par la dissolution du réseau vitreux. Il se forme alors en surface du verre,
lorsque les conditions de saturation sont atteintes, une pellicule d'altération
principalement amorphe appelée "gel", constituée des éléments non solubles (Fe,
Ni, Zn, Zr) et éventuellement la précipitation de phases minérales de type
smectite, zéolithe ou silicate de calcium hydraté mais en très faible quantité à
basse température (90° C) (Vernaz et Dussossoy, 1992). Les mécanismes
d'hydrolyse par réaction de surface sont prépondérants pour le verre SON; ils sont
inhibés par la présence de SiÛ2 dissoute (Raymond, 1992) en milieu basique et
catalysés par de fortes concentrations en H+ et OH".
4.4 CINETIQUE DE DISSOLUTION ET MODELISATION
Nous avons vu que la cinétique était contrôlée par les réactions de
surface. Ce contrôle peut être décrit à l'aide de la théorie d'état de transition
(Aagard et Hegelson, 1982; Hegelson et al., 1984; Lasaga, 1981a'bc, 1984) ce qui

conduit à une équation générale valable pour les réactions congruentes ou
incongnientes, proche ou loin de l'équilibre. Cette équation peut prendre la forme
suivante (Aagaard et Helgelson, 1982) :
di1-exp - _A_
RTEq. 4.1
, kBTKavec k = j — , Eq. 4.2
hy
où 4 e s t l'avancée du processus globale de dissolution (dans le cas de la
dissolution d'un verre, ^est égal au nombre de moles de verre dissoutes), s est la
surface effective de verre (autrement dit, il s'agit de l'aire des sites actifs exposés à
la phase aqueuse), a\ est l'activité de l'espèce i réactive affectée du coefficient de
réaction «/ défini par /?/ =( dnf / dÇ) où dnf rend compte des variations du nombre
de moles de l'espèce / lors de la réaction, A est l'affinité chimique du processus
globale de dissolution, k désigne une constante de vitesse (mol.cm"2.s"1), K est la
constante d'équilibre de la réaction de formation du complexe activé à la surface
du verre et / est le coefficient d'activité du complexe activé à la surface du verre,
R, ks, h et T sont respectivement la constante des gaz parfaits, les constantes de
Boltzmann, de Planck et la température.
Le dernier terme, entre crochets, de l'équation 4.1 rend compte de
l'approche de l'équilibre thermodynamique, autrement dit de la variation des
potentiels chimiques entre le verre et la solution altérante. A l'équilibre A vaut 0 ;
très loin de l'équilibre, A est très grand et seul le premier terme demeure. On voit
ainsi que la cinétique de dissolution est peu sensible à l'enthalpie libre de la
réaction de dissolution globale dans les premiers stades du processus. Le produit
Y\ ap1' rend compte des effets catalytiques et inhibiteurs sur la réaction globale,
comme, par exemple, la présence des ions H+ en conditions acides et OH" en
conditions basiques qui catalysent la dissolution.
Cette loi a été validée dans la majorité des études relatives aux cinétiques
de réactions hétérogènes (Lasaga, 1981a). Grambow (1985) propose que la
59

dissolution de la matrice vitreuse est contrôlée par la seule activité en acide
orthosilicique et suggère que la solubilité d'un verre à plus de 30 oxydes (tel que
ceux utiliser pour le stockage des déchets radioactifs) se limite à celle de son
constituant majeur, la silice. En accord avec Grambow, Advocat et al.(1991)
justifient la nature purement siliceuse du complexe activé critique par la structure
même du verre. Celui-ci est constitué en majorité de tétraèdres de SiC^ répartis de
façon aléatoire qui passent en solution en entraînant avec eux les autres entités
élémentaires. La loi cinétique de dissolution de ce type de verre complexe pourrait
alors se limiter à l'expression réduite de la loi d'Aagaard et Helgelson qui a été
formulé plus haut avec un complexe activé purement siliceux et pourrait s'écrire
sous la forme :
Eq.4.3-^ = k \ H \ 1 - T T
dt L J [ [H4SiO4]sat
Le terme £+[H+]" caractérise la vitesse initiale VQ. NOUS avons vu que la
première étape dans l'altération du verre est l'extraction préférentielle des alcalins
et alcalinoterreux ainsi que du bore. Lors de cette phase, la vitesse de dissolution
peut être considérée constante pour une composition donnée et un pH donné.
Cette vitesse a donc pu être déterminée expérimentalement en milieu extrêmement
dilué et sur des échelles de temps très courtes (Delage et Dussossoy, 1991). La loi
4.3, souvent appelée loi du premier ordre puisqu'elle ne tient compte que de
l'activité de l'acide silicique comme paramètre limitant, est, en première
approximation, en bon accord avec les résultats expérimentaux (Advocat et al.,
1990; Tovena, 1995; Ricol, 1995). Elle montre par conséquent que l'activité de
l'acide silicique est un paramètre clé de la cinétique de dissolution. Cependant,
cette loi suppose que la vitesse de dissolution est nulle lorsque
[EUSiO/i] = [H4SiO4]sat, c'est-à-dire lorsque la phase aqueuse est saturée en acide
orthosilicique vis-à-vis de la silice du verre. Cet équilibre chimique n'a pas été
observé expérimentalement, au contraire la vitesse est non nulle et peut varier
avec le temps (Advocat et al., 1990; Ricol, 1995). Pour expliquer cet écart au
modèle, Grambow (1985) suggère qu'il existe dans le système un assemblage de
60
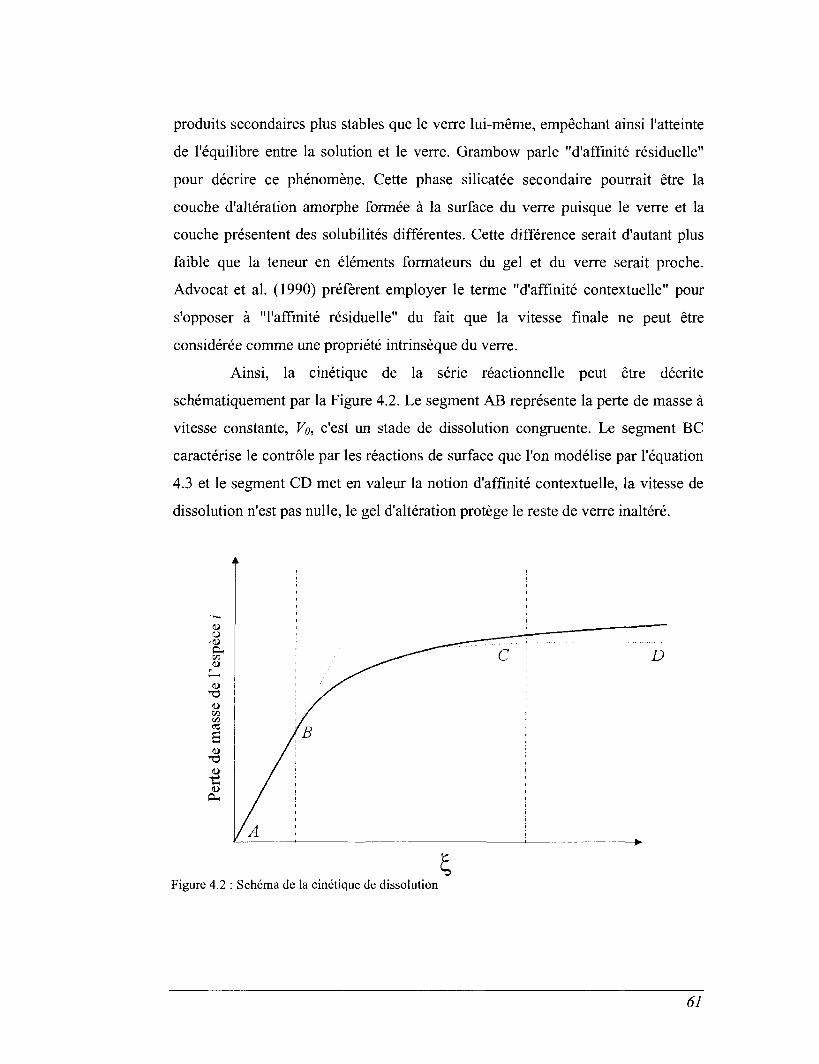
produits secondaires plus stables que le verre lui-même, empêchant ainsi l'atteinte
de l'équilibre entre la solution et le verre. Grambow parle "d'affinité résiduelle"
pour décrire ce phénomène. Cette phase silicatée secondaire pourrait être la
couche d'altération amorphe formée à la surface du verre puisque le verre et la
couche présentent des solubilités différentes. Cette différence serait d'autant plus
faible que la teneur en éléments formateurs du gel et du verre serait proche.
Advocat et al. (1990) préfèrent employer le terme "d'affinité contextuelle" pour
s'opposer à "l'affinité résiduelle" du fait que la vitesse finale ne peut être
considérée comme une propriété intrinsèque du verre.
Ainsi, la cinétique de la série réactionnelle peut être décrite
schématiquement par la Figure 4.2. Le segment AB représente la perte de masse à
vitesse constante, Vo, c'est un stade de dissolution congruente. Le segment BC
caractérise le contrôle par les réactions de surface que l'on modélise par l'équation
4.3 et le segment CD met en valeur la notion d'affinité contextuelle, la vitesse de
dissolution n'est pas nulle, le gel d'altération protège le reste de verre inaltéré.
D
Figure 4.2 : Schéma de la cinétique de dissolution
61

4.5 UN BESOIN DE DONNEES THERMODYNAMIQUES
Les prédictions quantitatives nécessitent des modèles décrivant non
seulement les vitesses auxquels les verres réagissent, de façon congruente ou non,
avec les solutions aqueuses mais aussi les variations de ces vitesses avec la
pression, la température et la composition chimique des verres et des solutions. La
théorie de l'état de transition et la thermodynamique ont permis de comprendre les
principaux mécanismes mis en jeu et de composer des modèles simples décrits
dans la partie précédente. La validation de ces modèles a plus de peine à aboutir.
En effet, ces lois cinétiques font appel à un grand nombre de données
thermochimiques sur les matériaux vitreux. Or ces données sont encore peu
nombreuses actuellement par rapport aux matériaux cristallisés qui ont servi de
base à l'élaboration de ces modèles.
En 1977, Paul suggère que la durabilité chimique d'un verre peut être
estimée à partir de ses propriétés thermodynamiques. Il suppose que les vitesses
de réactions entre l'eau et le verre peuvent être paramétrées par la sommation des
enthalpies libres de dissolution de chaque composant à "structure individualisée";
c'est-à-dire les composants silicates les plus simples et les oxydes qui constituent
le verre. Paul, puis Jantzen et Plodinec ont alors développé une base de données
thermodynamiques pour étudier la dissolution des verres (Paul, 1977; Jantzen et
Plodinec, 1984). Nous rappellerons ici brièvement leur démarche.
Le verre est considéré comme un mélange homogène d'entités telles que
SiÛ2, Na2SiO3, Mg2SiO3, Ca2SiC>3, AI2O3, B2O3... La réaction de ces espèces avec
l'eau crée une variation d'enthalpie libre qui définit thermodynamiquement la
dissolution. L'enthapie libre de dissolution du verre est donc considérée comme la
somme des enthalpies libres de chaque entité (AG°) pondérées par leur fraction
molaire dans le verre,
où Xi représente la fraction molaire de l'entité /. Les enthapies libres les plus
élevées donnent alors les verres les moins altérables.
Discutons maintenant les méthodes d'estimation des données
thermodynamiques concernant les unités de base que les auteurs ont considérées.
Paul calcule les enthalpies libres de dissolution de ces entités de base par
62

différences des enthalpies libres de formation entre réactifs et réactants.
L'enthalpie libre d'un matériau est égale à
AfG(T) = AfH(T)-TAfS(T), Eq. 4.5
où AjH et À/5 sont respectivement les différences d'enthalpie et d'entropie entre le
composé formé et ses constituants. En supposant que le terme TAfS est très
inférieur à AjH dans le cas des systèmes silicates, que les variations d'enthalpie
d'un verre et du cristal de même composition sont très proches, Paul conclue que
A/7overTe~A/7°cristai- Avec l'avancée des connaissances sur les structures et les
propriétés des matériaux vitreux, les hypothèses formulées par Paul sont devenues
caduques.
Si la contribution entropique peut être négligée dans le cas des cristaux, il
n'en va pas de même pour les verres. En effet, on constate, par exemple dans le
cas d'un verre de composition d'anorthite, que la contribution entropique (-18.9
kJ/mol) peut être d'importance semblable à celle de la contribution enthalpique
(-23.6 kJ/mol). L'enthalpie et l'entropie de formation par rapport aux oxydes sont
données par :
= 2>J ASH(TS )' + \CpldT
pT.
-ASH(TS)- \CpdT, Eq.4.6
— . Eq. 4.7T
0 i
où ASH est l'enthalpie de solution dans un solvent approprié (nous reviendrons sur
ce terme par la suite), S est l'entropie, Cp est la capacité calorifique, n exprime une
fraction molaire et T est la température. L'indice i indique une propriété d'un
oxyde constitutif du matériau étudié et l'absence d'indice réfère au matériau lui-
même. Pour les minéraux, comme pour les oxydes constitutifs, S(0) est nulle. De
plus, l'entropie relative d'un silicate peut en général être considérée comme
l'addition des entropies relatives de ces oxydes constitutifs. Par conséquent, on
peut effectivement négliger en première approximation le terme entropique face
au terme enthalpique dans le cas des minéraux. En revanche, dans le cas des
63

verres, l'entropie à 0 Kelvin n'est pas nulle. Cette entropie résiduelle représente
l'entropie de configuration du liquide figée à la température de transition vitreuse,
5(0) = Sfonf(Tg). Eq. 4.8
Cette entropie dépend fortement de la composition chimique du verre. A
température ambiante, elle peut représenter jusqu'à 30 % de l'entropie absolue des
verres de silicates (Richet et Neuville, 1992) et plus de 70% de leur entropie de
formation par rapport aux oxydes.
D'autre part, on peut difficilement assimiler l'enthalpie de formation d'un
verre à celle du cristal de même composition. On peut exprimer l'enthalpie de
formation d'un verre en fonction de l'enthalpie de formation du cristal qui lui est
associé en y ajoutant un terme enthalpique de vitrification,,
AfH(T)verre = AfH(T)crislal + AVH(T). Eq. 4.9
Outre le terme entropique, le modèle de Paul néglige donc également l'énergie
nécessaire à la vitrification. Cette autre approximation est peu propice à une
modélisation efficace puisque l'enthalpie de vitrification de verres silicates peut
être supérieure en valeur absolue à l'enthalpie de formation du cristal par rapport
aux oxydes. Les enthalpies de vitrification du quartz, de la wollastonite et de
l'enstatite sont respectivement 9.12, 33.07 et 42.50 kJ.mor1 alors que leur
enthalpie de formation sont 0, -92.77 et -36.53 kJ.mol"1 (Richet et al., 1982 ;
Richet, 1984 ; Robie et Hemingway, 1995).
La remise en question des hypothèses nécessaires à l'estimation des
enthalpies libres de formation des entités qui composent le verre rend donc
inexploitable les données de Paul pour des modélisations cinétiques quantitatives.
Cependant, si une autre approche permettait d'acquérir ces données, nous
pourrions malgré tout tester le modèle. Nous proposerons donc dans les
paragraphes suivants une nouvelle méthode de détermination basée sur des
mesures calorimétriques et viscosimétriques. C'est cette approche que nous avons
mise en œuvre dans le cadre de cette thèse.
64

Parmi les diverses méthodes utilisables pour déterminer des enthalpies de
formation, celle qui est la mieux adaptée à des composés constitués d'oxydes
consiste à mesurer séparément les parties enthalpiques et entropiques.
La partie enthalpique
L'enthalpie de formation, AjH, est habituellement obtenue à partir de
mesures d'enthalpies de dissolution dans un solvant approprié. Dans le cas de
composés comportant des oxydes réfractaires comme CaO ou MgO, la dissolution
dans des solutions acides (acide fluorhydrique, par exemple, à des températures
proches de l'ambiante) est généralement incomplète. Pour obtenir des résultats
fiables, on doit effectuer les dissolutions dans des oxydes fondus (borate de plomb
par exemple) à des températures de 700° C environ (Navrotsky et al., 1980).
Ayant mesuré les enthalpies de dissolution, ASH, à la température Ts, du verre et
de ces oxydes constitutifs, on obtient l'enthalpie de formation du verre par rapport
aux oxydes avec l'équation 4.6.
La partie entropique
L'entropie de formation est déterminée à partir de mesures de capacités
calorifiques effectuées de 0 K à la température T voulue comme indiqué dans
l'équation 4.7.
Dans le cas des oxydes assimilés à des cristaux parfaits, on a S(0 K)=0;
en revanche, comme nous l'avons vu dans la partie précédente (cf. Eq. 4.8),
l'entropie d'un verre à 0 K est égale à l'entropie de configuration du liquide figée à
la transition vitreuse, Sfon\Tg). Cette entropie de configuration peut être
déterminée par des mesures calorimétriques (Richet, 1984). Cette méthode repose
sur la réalisation du cycle entropique représenté dans la Figure 4.3 en partant
d'une phase cristalline de même composition que le verre considéré et dont le
point de fusion sera noté Tm. La mesure de la capacité calorifique du cristal (Cpc)
de 0 K à Tm permet de déterminer son entropie absolue à Tm:
65
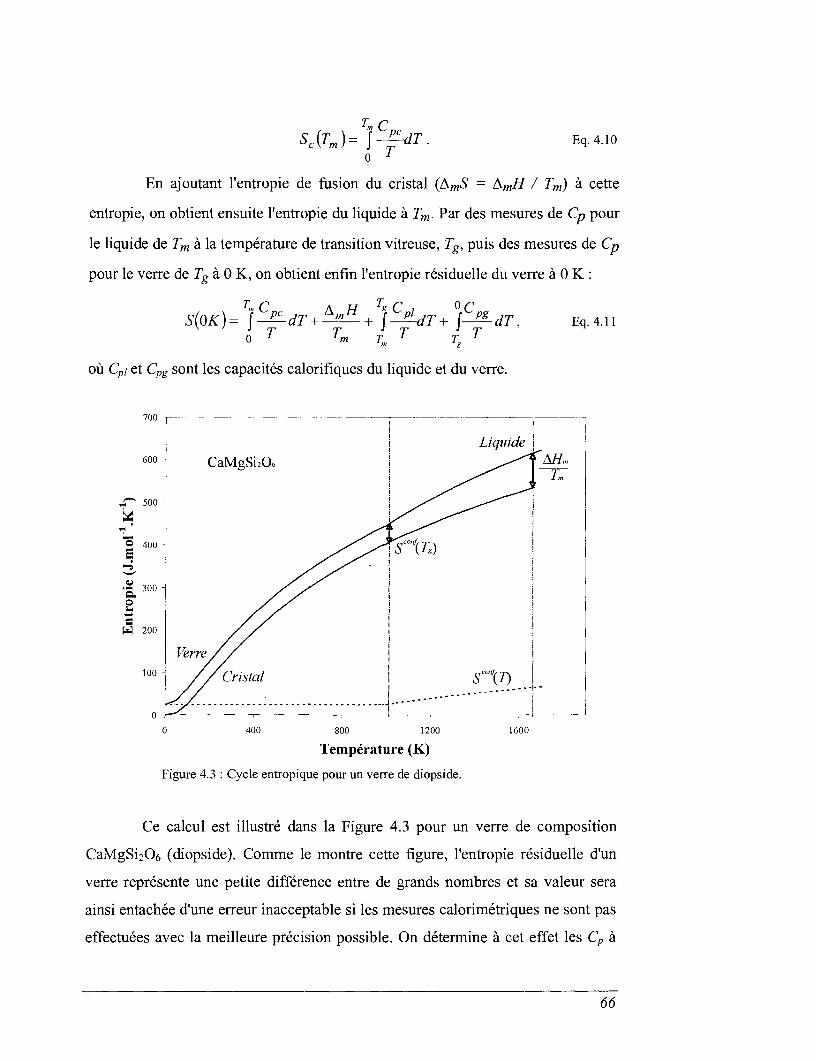
TIT. Eq. 4.10
En ajoutant l'entropie de fusion du cristal (AmS = AmH / Tm) à cette
entropie, on obtient ensuite l'entropie du liquide à Tm. Par des mesures de Cp pour
le liquide de Tm à la température de transition vitreuse, Tg, puis des mesures de Cp
pour le verre de Tg à 0 K, on obtient enfin l'entropie résiduelle du verre à 0 K :
Eq.4.11
où Cpi et Cpg sont les capacités calorifiques du liquide et du verre.
700 -i
400 1600800 1200
Température (K)
Figure 4.3 : Cycle entropique pour un verre de diopside.
Ce calcul est illustré dans la Figure 4.3 pour un verre de composition
CaMgSi2O6 (diopside). Comme le montre cette figure, l'entropie résiduelle d'un
verre représente une petite différence entre de grands nombres et sa valeur sera
ainsi entachée d'une erreur inacceptable si les mesures calorimétriques ne sont pas
effectuées avec la meilleure précision possible. On détermine à cet effet les Cp à
66

mieux que 0.2 % par calorimétrie adiabatique entre 0 K et l'ambiante, et à mieux
que 0.5 % par calorimétrie de chute à haute température.
Cette méthode ne peut être utilisée pour les verres de confinement de
déchets nucléaires car son utilisation est restreinte aux cas particuliers de verre
ayant la composition de cristaux fondant de façon congruente. Il est donc
nécessaire de déterminer l'entropie de configuration à partir d'une autre méthode,
nous avons choisi une méthode viscosimétrique (Richet, 1984). Le fondement
théorique de cette méthode est donné par la théorie entropique des processus de
relaxation (Adam et Gibbs, 1965). Selon cette théorie, un écoulement visqueux
nécessite un changement de configuration d'un liquide. Quand la température
augmente, l'entropie de configuration augmente également, tandis que la viscosité
diminue car ces changements peuvent se produire de manière indépendante dans
des parties de plus en plus petites du liquide. Quantitativement, on obtient :
logr| = ^ e + e—-, Eq.4.12TSœnf{T)
où Ae est un terme pré-exponentiel et Be une constante proportionnelle à la
barrière d'enthalpie libre s'opposant aux réarrangements coopératifs requis pour
l'écoulement visqueux.
En pratique, c'est seulement l'entropie de configuration à une température
donnée, à Tg par exemple, qu'on détermine avec l'équation 4.12 à partir des
mesures de viscosité (en même temps que les paramètres Ae et Be). Entre Tg et une
température quelconque T, l'entropie de configuration augmente suivant la relation
suivante :
y /-iconf
Sconf(T) = Sconf(Tg)+ \-E—dT, Eq.4.13T* T
où Cpconf est la capacité calorifique de configuration. Comme justifié par Richet et
al. (1986), cette dernière peut être prise égale à :
CCp°
nf{T)=Cpl(T)-Cpg{Tg). Eq.4.14
En résumé, des mesures calorimétriques permettent de déterminer avec
l'équation 4.14 la capacité calorifique de configuration en fonction de T. On
67
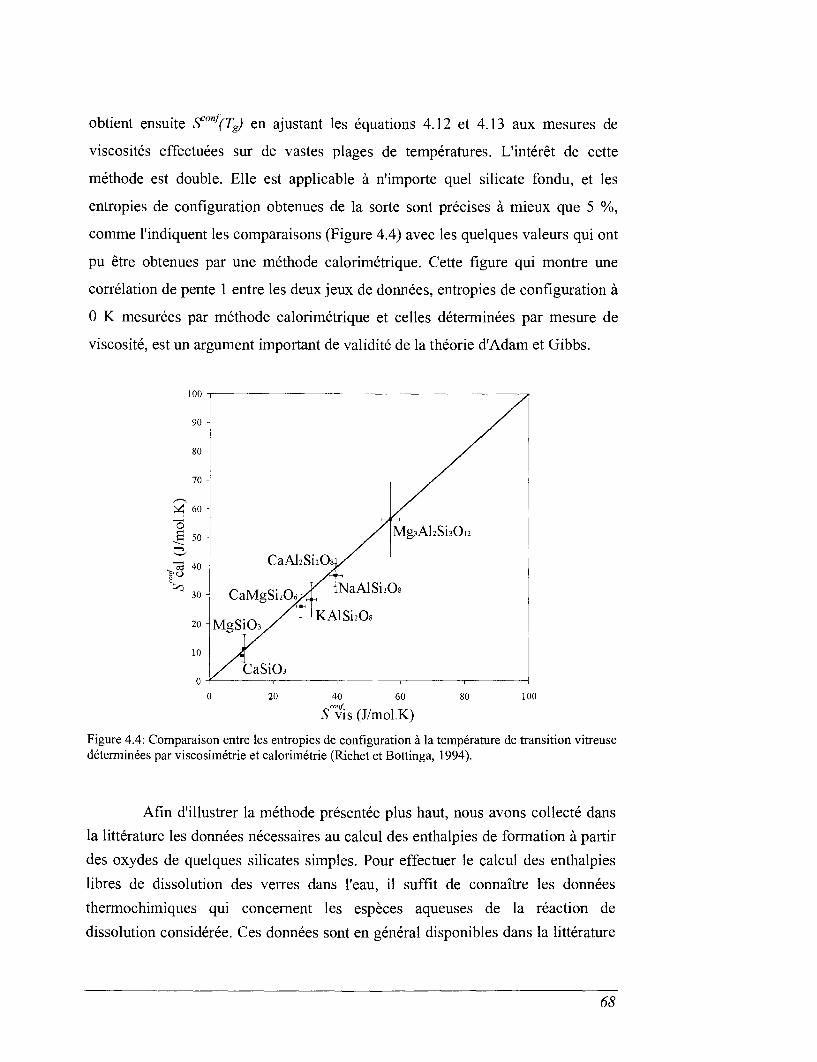
obtient ensuite Sfonf(Tg) en ajustant les équations 4.12 et 4.13 aux mesures de
viscosités effectuées sur de vastes plages de températures. L'intérêt de cette
méthode est double. Elle est applicable à n'importe quel silicate fondu, et les
entropies de configuration obtenues de la sorte sont précises à mieux que 5 %,
comme l'indiquent les comparaisons (Figure 4.4) avec les quelques valeurs qui ont
pu être obtenues par une méthode calorimétrique. Cette figure qui montre une
corrélation de pente 1 entre les deux jeux de données, entropies de configuration à
0 K mesurées par méthode calorimétrique et celles déterminées par mesure de
viscosité, est un argument important de validité de la théorie d'Adam et Gibbs.
100
90
80 -
70 -
40 60
™vis (J/mol.K)80 100
Figure 4.4: Comparaison entre les entropies de configuration à la température de transition vitreusedéterminées par viscosimétrie et calorimétrie (Richet et Bottinga, 1994).
Afin d'illustrer la méthode présentée plus haut, nous avons collecté dans
la littérature les données nécessaires au calcul des enthalpies de formation à partir
des oxydes de quelques silicates simples. Pour effectuer le calcul des enthalpies
libres de dissolution des verres dans l'eau, il suffit de connaître les données
thermochimiques qui concernent les espèces aqueuses de la réaction de
dissolution considérée. Ces données sont en général disponibles dans la littérature
68
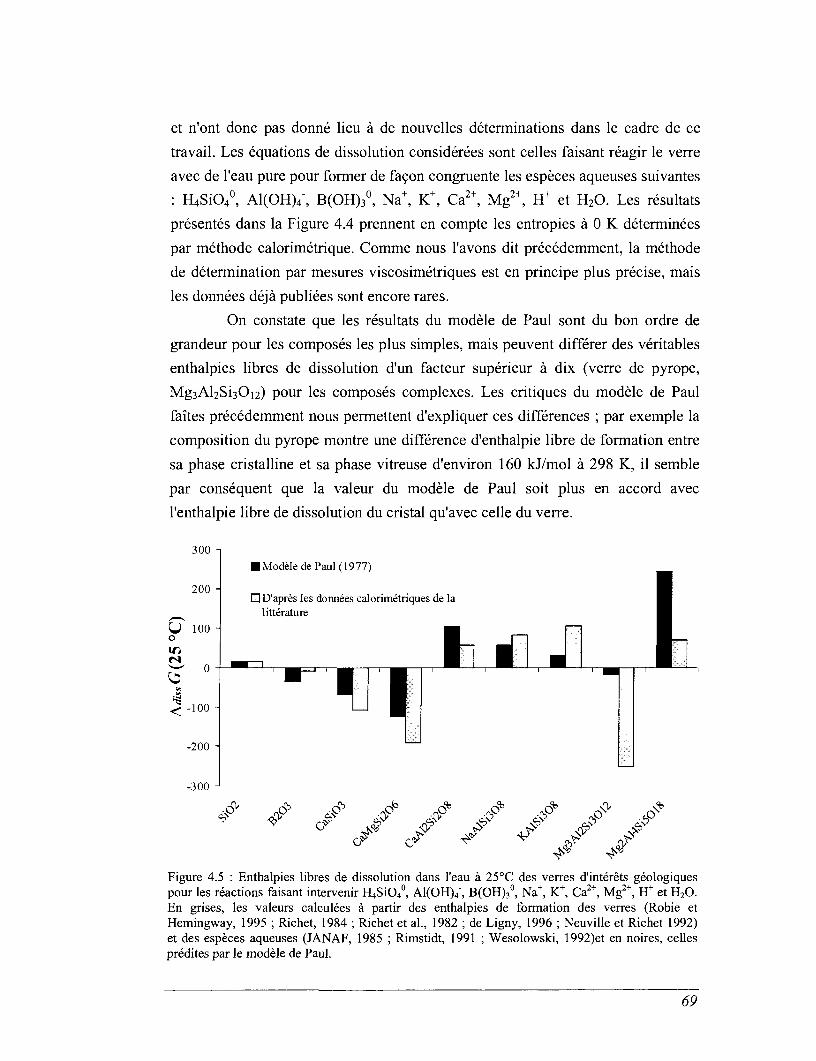
et n'ont donc pas donné lieu à de nouvelles déterminations dans le cadre de ce
travail. Les équations de dissolution considérées sont celles faisant réagir le verre
avec de l'eau pure pour former de façon congruente les espèces aqueuses suivantes
: H4Si04°, Al(OH)4", B(OH)3°, Na+, K+, Ca2+, Mg2+, H+ et H2O. Les résultats
présentés dans la Figure 4.4 prennent en compte les entropies à 0 K déterminées
par méthode calorimétrique. Comme nous l'avons dit précédemment, la méthode
de détermination par mesures viscosimétriques est en principe plus précise, mais
les données déjà publiées sont encore rares.
On constate que les résultats du modèle de Paul sont du bon ordre de
grandeur pour les composés les plus simples, mais peuvent différer des véritables
enthalpies libres de dissolution d'un facteur supérieur à dix (verre de pyrope,
Mg3Al2Si3Oi2) pour les composés complexes. Les critiques du modèle de Paul
faîtes précédemment nous permettent d'expliquer ces différences ; par exemple la
composition du pyrope montre une différence d'enthalpie libre de formation entre
sa phase cristalline et sa phase vitreuse d'environ 160 kJ/mol à 298 K, il semble
par conséquent que la valeur du modèle de Paul soit plus en accord avec
l'enthalpie libre de dissolution du cristal qu'avec celle du verre.
300 i
200 -
!J 100o
«3-100
-200 -
-300 J
• Modèle de Paul (1977)
• D'après les données calorimétriques de lalittérature
°
Figure 4.5 : Enthalpies libres de dissolution dans l'eau à 25°C des verres d'intérêts géologiquespour les réactions faisant intervenir H4S1O40, A1(OH)4", B(OH)3°, Na+, K+, Ca2+, Mg2+, H+ et H2O.En grises, les valeurs calculées à partir des enthalpies de formation des verres (Robie etHemingway, 1995 ; Richet, 1984 ; Richet et al., 1982 ; de Ligny, 1996 ; Neuville et Richet 1992)et des espèces aqueuses (JANAF, 1985 ; Rimstidt, 1991 ; Wesolowski, 1992)et en noires, cellesprédites par le modèle de Paul.
69

Nous avons décrit les traits généraux des verres, de leur structure à leur
altération en passant par leur définition thermodynamique. Les axes de recherche,
que nous avons suivi pour obtenir les valeurs des enthalpies de dissolution, ont été
décrits, nous y reviendrons plus précisément dans les sections suivantes
consacrées à l'exposé des techniques utilisées et des résultats.
70

DEUXIEME PARTIE
Méthodes expérimentales

Les déterminations d'enthalpies libres de formation de verres que nous
avons faites est une approche expérimentale qui permet une mesure de chaque
contribution à l'enthalpie libre du verre (enthalpie de formation, entropie
vibrationnelle, entropie de configuration). Outre la synthèse des produits, cette
partie présentera les techniques suivantes :
calorimétrie adiabatique, qui permet la mesure des entropies relatives entre
l'ambiante et 0 K, S29&-S0,
calorimétrie de chute et calorimétrie différentielle à balayage, qui nous
permettent de mesurer les capacités calorifiques et les propriétés relatives,///-
-#298 et SrS298,
la calorimétrie de dissolution, dont les résultats nous permettent de déterminer
l'enthalpie de formation des verres,
les méthodes de mesures de viscosité qui sont associées aux entropies de
configuration selon la théorie des processus de relaxation (Adam et Gibbs,
1965).
73

Chapitre 5
SYNTHESES
Les échantillons étudiés ont été synthétisés par nos soins à l'exception du
verre SON, qui nous a été fourni par le CEA afin que celui-ci soit en tout point
conforme au verre de référence inactif couramment utilisé, ainsi que les verres de
la série AVM. Les verres de la série SON et les compositions ternaires de la série
BSN ont été préparés suivant le protocole de synthèse décrit par Schairer et
Bowen (1955, 1956) et utilisés au laboratoire depuis des décennies. Les verres
binaire SiO2-B2O3 ont demandé un protocole particulier. Nous décrirons d'abord
les différentes étapes du protocole "classique" puis celles inhérentes à la
particularité des binaires étudiés.
5.1 PROTOCOL E DE SYNTHÈSE
Séchage des poudres
Les poudres d'oxydes et de carbonates utilisées sont légèrement
hygroscopiques. Par conséquent l'eau qu'elles contiennent doit être éliminée pour
ne pas fausser la pesée. Pour cela, les poudres sont mises dans des creusets
d'alumine et séchées dans un four à moufle électrique. Les séchages s'effectuent à
1100°C pour SiO2, A12O3, ZrO2 et ZnO, à 1000°C pour Ce2O3, à 550°C pour
CaCO3) à 500°C pour Na2B4O7 et à 350°C pour Na2CO3 et Li2CO3. Typiquement,
lors de ces séchages, la silice perd environ 0.003 % en poids, alors que ZnO,
A12O3) Ce2O35 Na2B4O7, CaCO3, Na2CO3 perdent quelques pourcents. Ces pertes
sont directement imputables à l'eau.
74

Pesée et broyage
Nous utilisons une balance Sartorius de portée 162 g et de précision 0.1
mg. Nous pesons directement le creuset de platine dans lequel nous effectuerons
la fusion et nous rajoutons les différentes poudres dans les proportions requises.
Après la pesée, le mélange est broyé pendant une heure en présence
d'alcool dans un mortier en agate (RETSCH). A la fin du broyage, nous
transférons le mélange de poudres et d'alcool dans le creuset de platine afin que
l'alcool s'évapore. Pour accélérer la disparition de l'alcool, nous l'enflammons.
Cette combustion peut déposer sur la poudre des résidus carbonés qui disparaient
lors de la fusion.
Décarbonatation et première fusion
Les decarbonatations et les fusions sont effectuées dans un four
"Meeker" composé d'éléments chauffant en Kanthal 33. Les produits qui ont été
synthétisés dans le cadre de cette étude sont pour partie à base de carbonates
(CaCÛ3 et Na2CÛ3). La décarbonatation induit une diminution de la masse de
produit qui permet un premier contrôle des teneurs en CaCÛ3 et Na2CÛ3 présentes
dans le mélange.
La décarbonatation est suivie de la fusion; après le palier au voisinnage
de 900°C pour les produits étudiés ici, qui clot le cycle de décarbonatation, on
augmente la température de 1°C par minute jusqu'à une température légèrement
supérieure au point de fusion du produit.
Première trempe
Cette opération a pour but de figer le liquide pour obtenir un verre.
Généralement les vitesses de trempes sont de l'ordre de la centaine de degrés par
minute. Ces trempes sont effectuées soit en plongeant le fond du creuset dans l'eau
distillée, soit en coulant directement son contenu sur une plaque de cuivre. Lors
de la trempe, les verres se fragmentent sous l'effet des contraintes internes créées
par la différence de vitesse de refroidissement dans le produit, ce qui facilite le
démoulage des creusets.
75

Homogénéisation des produits
Afin d'homogénéiser les produits, à l'issue de la première trempe, on
broyé le verre obtenu puis on le fait fondre en suivant une rampe de l°C/min
jusqu'à une température légèrement supérieure à son liquidus. On fracture alors de
nouveau le produit par une trempe et on recommence le cycle une nouvelle fois.
La dernière trempe doit être extrêmement bien réalisée afin d'obtenir un produit
entièrement vitreux et sans fractures. Pour cela, nous coulons le liquide dans des
cylindres en graphite posés sur une plaque de cuivre afin d'obtenir des "lentilles"
de verre dans lesquelles nous pourrons carotter des éprouvettes aux dimensions
souhaitées. Afin de s'assurer de la non-fracturation de notre produit, nous
transposons la lentille de verre dans un four préchauffé à une temperature
d'environ 500° C. Un relâchement des contraintes internes est obtenu en quelques
heures.
Vérification des synthèses et de la vitrification
En premier lieu, nous effectuons des mesures de densité sur une dizaine
de fragments de verre d'un même échantillon pour nous assurer que le verre est
homogène. La viscosité dépendant fortement de la composition chimique, il faut
que cette dernière soit identique dans tout le verre et surtout dans toutes les
éprouvettes utilisées. Ces mesures sont réalisées selon la méthode d'Archimède,
précise à 0.001, en utilisant du toluène comme fluide d'immersion. Une
vérification de la composition de nos verres sera obtenue, dans un second temps,
par des analyses à la microsonde électronique ou par fluorescence X.
Il faut aussi s'assurer de l'absence de cristaux dans les produits car une
fraction cristalline peut provoquer non seulement une augmentation de viscosité,
mais aussi un comportement non-newtonien (Lejeune et Richet, 1995). Un
premier examen au microscope n'a montré de cristaux ni en lumière naturelle, ni
en lumière polarisée. Cependant, la présence possible de cristaux de symétrie
cubique (spinelle) font que cet examen n'est pas rigoureusement concluant pour
détecter des particules de taille supérieure à quelques microns; c'est pourquoi, des
76
diagrammes de diffraction de rayons X ont également été obtenus pour déterminer
la présence éventuelle de phases cristallines.
Les produits sont également analysés pour s'assurer que la stoechiométrie
souhaitée a été obtenue. Nous avons utilisé deux techniques d'analyse, la
microsonde électronique et la spectrométrie ICP-AES. Ayant observé parfois des
phénomènes caractéristiques d'une volatilisation (B ou Na ?) sous le faisceau de la
microsonde, nous avons voulu nous assurer des compositions par une autre
technique, l'ICP-AES. La comparaison entre les deux types d'analyses montre de
très faibles différences comprises dans les erreurs expérimentales inhérentes aux
techniques. Les compositions analysées sont, à l'exception du verre BSN-8/24, en
bon accord avec les valeurs nominales. Pour le verre BSN-8/24, il est
vraissemblable de penser à une erreur sur la nature des oxydes lors des pesées et
du mélange des poudres.
Les Tableaux 5.1-5.2 font un bref résumé des conditions de synthèse et
indiquent les densités mesurées de tous les produits.
Tableau 5.1 : Récapitulatif des conditions de synthèses des différents verres de la série SON
nombre de cycle
broyage-fusion-trempe
température d'élaboration (°C)
durée d'affinage (heures)
température de recuit (°C)
durée du recuit (heure)
densité des fragments
recuits
non-recuits
SON-3
3
1180
1
480
4
2.448
2.433
SON-4
3
1200
1
480
4
2.405
2.386
SON-5
3
1200
1
500
6
2.467
2.451
SON-6
3
1200
1
500
5
2.490
2.475
SON-7
3
1250
1
500
10
2.514
2.491
SON-8
3
1150
1
450
5
2.611
2.588
77

Tableau 5.2 : Récapitulatif des conditions de synthèses des différents verres de la série BSN
nombre de cycle
broyage-fusion-trempe
température d'élaboration (°C)
durée d'affinage (heures)
température de recuit (°C)
durée du recuit (heure)
densité des fragments
recuits
non-recuits
BSN-5/16
3
1200
1
450
4
2.401
2.392
BSN-8/24
3
1200
1
450
5
2.446
2.438
BSN-11/32
3
1200
1
450
8
2.526
2.511
BSN-29/14
3
1200
1
450
2
2.402
2.388
5.2 LA SYNTHESE DES VERRES BINAIRES SlO2-B2O3
Les verres binaires SiO2-B2O3 ont demandé un protocole de synthèse
particulier du fait des caractéristiques des deux oxydes S1O2 et B2O3. La silice a
un point de fusion très élevé (1999 K) par rapport à B2O3 qui fond à 723 K.
D'autre part, le bore se volatilise très facilement (Asano et al., 1989) dès 1000 K.
Dans les premiers essais de synthèse, suivant le protocole décrit précédemment en
partant de poudre de SiÛ2 et de B2O3 ou H3BO3, on obtenait une suspension de
silice cristallisé dans un bain, liquide, de B2O3 qui s'appauvrissait en bore
lorsqu'on augmentait la température pour s'approcher du point de fusion de la
silice. La volatilisation du bore était clairement mise en évidence par l'apparition
d'une fumée blanche aux environs de 1200 K. Outre un écart important par rapport
à la composition nominale, ces essais n'ont pas abouti puisqu'ils nécessitaient
d'augmenter la température au-delà de 1700 K afin de rendre la silice
suffisamment fluide pour se dissoudre et former un liquide homogène avec B2O3.
Afin de réduire la différence de température de fusion, nous avons choisi
de changer nos produits de départs. Nous avons utilisé de l'acide silicique
(S1O2.MH2O) dont le point de fusion est proche de 1400 K et qui, par sa texture
très floconneuse, permet d'augmenter la "mouillabilité" du bore vis-à-vis de la
78

silice. Après avoir constaté la fonte de tout le mélange d'oxyde vers 1400 K, nous
avons dû porter les mélanges aux environs de 1500 K afin d'homogénéiser la
composition. Ces dernières étapes on fait apparaître des fumées blanches, signes
d'une perte de bore. L'analyse des compositions a confirmé ces pertes qui ont été
de l'ordre de 10 %.
En plus de la difficulté d'obtenir des verres ayant la composition désirée,
cette méthode ne permet de synthétiser que de petites quantités par cycle de pesée-
mélange-fusion car l'acide silicique est beaucoup moins dense que la silice
cristallisée. Pour obtenir la quantité désirée nous avons dû broyer, mélanger et re-
fondre 4 lots de produit ainsi formé. Après l'obtention d'un premier binaire (BSN-
68/0), nous avons enrichi ce produit en silice par ajout d'acide silicique pour
s'approcher par étape successive (BSN-58/0) du verre dont la composition nous
semblait pertinente (BSN-43/0). Ces verres binaires ont un caractère très
hygroscopique qui oblige leur stockage dans un lieu protégé de toute humidité.
79

Chapitre 6
CALORIMETRIE
6.1 PRINCIPE
Le terme de calorimètre désigne un appareil permettant de mesurer les
quantités de chaleur. Il est constitué par un récipient dans lequel on produit les
phénomènes thermiques à étudier. En général ce récipient est placé dans une
cavité dont la paroi est à température constante ou réglable à volonté. Nous
appelons enceinte interne la paroi du récipient calorimétrique et enceinte externe
la paroi de la cavité dans laquelle il est logé.
La chaleur (AQ) dégagée par conduction, rayonnement et convection à
l'intérieur du récipient calorimétrique tend à faire varier la température de
l'enceinte interne et il en résulte des échanges de chaleur entre les deux enceintes
qui tendent à s'équilibrer thermiquement.
Pour un système qui évolue à pression constante, la quantité de chaleur
échangée est la somme des enthalpies des réaction qui l'affectent et d'une variation
d'enthalpie (enthalpie relative ) entre un état initial et un état final. Si on se réfère
à une température finale de 7b, on note l'enthalpie relative HT - HT0. Enthalpie et
capacité calorifique à pression constante sont reliées par les équations suivantes:
Cp=(ÔH/ÔT)p Eq.6.1
T
et HT - HTQ = \CpdT. Eq. 6.2
Expérimentalement, suivant le type de calorimètre, les Cp peuvent être
déterminés "directement", tandis que les enthalpies relatives sont calculées à partir
80

de l'équation 6.2. Ceci n'est possible que si les échanges de chaleur sont réduits au
minimum, auquel cas on peut assimiler AQ/AT à l'équation différentielle 6.1. Les
Cp peuvent aussi être obtenus par dérivation de l'enthalpie relative selon l'équation
6.2.
6.2 LA CALORIMETRIE ADIABATIQUE ENTRE 10 ET 300 K
6.2.1 Le principe
Le flux de chaleur, O, qui passe à chaque instant de l'enceinte interne à
l'enceinte externe est donné par l'équation suivante,
<&=pAT,
où p est la conductibilité du milieu qui sépare les deux enceintes et AT la
difference de température entre celles-ci. Si p est nul, c'est-à-dire si les deux
enceintes sont séparées par un isolant thermique parfait, le calorimètre est dit
adiabatique (imperméable à la chaleur). Dans ces conditions, toute la chaleur AQ
produite au cours de l'expérience est utilisée pour élever la température du
récipient calorimétrique. Celle-ci devient supérieure de AT à la température
initiale et on a
AQ = CPAT.
Il est difficile de réduire à zéro la conductibilité thermique entre les deux
enceintes. Pour répondre aux conditions d'adiabaticité, la température de l'enceinte
externe demeure constamment égale à celle de l'enceinte interne de façon à rendre
toujours nuls les échanges de chaleur entre les deux enceintes (<I> =0). On obtient
ce résultat en modifiant constamment la température de l'enceinte externe grâce à
un système de régulation asservi aux différences de température entre les deux
enceintes.
81

6.2.2 Appareilla ge
Le calorimètre adiabatique que nous avons utilisé se trouve au laboratoire
des Structures et Matériaux de l'institut de Technologie de Tokyo (Japon). Il a été
décrit par Atake et al. (1990).
Ce dispositif est constitué de trois éléments :
• Un calorimètre dans lequel s'effectue la mesure de l'accroissement de
température en réponse à un apport d'énergie,
• Un cryostat permettant de refroidir l'ensemble du dispositif aux basses
températures auxquelles on fait les mesures,
• Une enceinte adiabatique permettant d'isoler thermiquement le
calorimètre.
Une vue en coupe du cryostat est présentée dans la Figure 6.1. La
chambre calorimétrique (F) est entourée par l'enceinte adiabatique (en cuivre
plaqué de chrome, de 1 mm d'épaisseur) formée de trois parties : la partie haute
(Hi), le pourtour (H2) de 70 mm de diamètre et 115 mm de hauteur, et la partie
basse (H3). La chambre calorimétrique est suspendue par des fils de nylon à la
partie haute. Chaque partie de l'enceinte adiabatique est contrôlée
indépendamment par un système p.i.d (d.c. amplificateur, model AM 100IB,
Ohkura Electric Co. et une unité de contrôle p.i.d. modèle 5301, Shinku-Riko
Inc.). La différence de température entre la chambre calorimétrique et le pourtour
de l'enceinte (H2) est détectée par des thermocouples type E et Au / chromel
connectés en série. Pour le contrôle entre le pourtour (H2) et les parties hautes
(Hi) et basse (H3), des thermocouples type E sont utilisés. L'écran thermique E
(bloc de cuivre de 400 g) et le manteau extérieur G (en cuivre plaqué de chrome
de, 1 mm d'épaisseur) sont maintenus à quelques Kelvin sous la température de
l'enceinte adiabatique en utilisant des systèmes p.i.d. (identiques aux précédents).
Sous conditions adiabatiques, la température de la chambre calorimétrique est
gardée constante à des fluctuations de 10"4 K prés.
82

Les tubes L et A sont connectés séparément à un système de pompage
qui permet d'obtenir un vide à mieux que 2 10~4 Pa durant les mesures entre le
réceptacle intérieur I (en cuivre plaqué d'or de 120 mm de diamètre et 1 mm
d'épaisseur) et le receptacle extérieur C d'une part, le manteau G d'autre part.
Le calorimètre est refroidi jusqu'au voisinage de 15 K en cassant le vide
par introduction d'une petite quantité d'hélium. Au-dessus de 20 K environ, le
refroidissement s'effectue uniquement par conduction de chaleur entre la chambre
calorimétrique et le conteneur J (en cuivre plaqué or d'un volume de 1.5 dm3).
M
Figure 6.1 : Vue schématique en coupe du calorimètre adiabatique. (A) Tube d'évacuation duréceptacle extérieur relié à un circuit de pompage. (B) Tube de remplissage du cryostat. (C)Réceptacle extérieur. (D) Obturateur conducteur thermique. (E) Ecran thermique. (F) Chambrecalorimétrique. (G) Manteau extérieur. (H,, H2, H3) Manteau adiabatique. (I) Réceptacle intérieur.(J) Conteneur du liquide refroidissant. (K) Obturateur supérieur. (L) Tube d'évacuation duréceptacle intérieur relié à un circuit de pompage. (M) Caisson Dewar rempli d'azote liquide.(Tanaka et al., 1994)

Celui-ci est utilisé soit vide soit rempli d'hélium liquide ou encore d'azote liquide
et/ou solide selon les températures souhaitées. Pour les mesures aux plus basses
températures, le conteneur I est rempli avec de l'hélium liquide ; avec une seule
charge, l'hélium liquide nous permet des mesures pendant 10 h. L'azote solide et
liquide est utilisé entre 50 et 100 K. A plus haute température, le conteneur est
laissé presque vide, juste rempli d'hélium gazeux sous pression atmosphérique.
Les tubes B servent à remplir ou vider le conteneur. L'ensemble du cryostat est
placé dans le caisson Dewar métallique L qui est toujours rempli d'azote liquide.
Figure 6.2 : Schéma de la chambre calorimétrique. (A) Bouchon. (B) Tube de cuivre. (C) Crochetsd'attache des fils de nylon. (D) Thermocouples. (E) Fils conducteur. (F) Thermomètre à résistancede germanium. (G) Alvéoles. (H) Thermomètre à résistance de platine. (J) Four. (K) Ecran àradiation. (Tanaka et al., 1994)
84

Les détails de la chambre calorimétrique peuvent être observés dans la
Figure 6.2. La chambre est divisée en huits alvéoles (C) pour faciliter l'équilibre
thermique à l'intérieure de la chambre. Le four I est enroulé sur le thermomètre à
résistance de platine F qui est fixé dans la cavité de la chambre calorimétrique. Le
thermomètre à résistance de germanium est placé au contact de l'enveloppe en
cuivre.
L'échelle de température du thermomètre à résistance de platine (type
5187L, Ro=25 Cl, H. Tinsley and Co., Ltd) a été calibré au National Physical
Laboratory en Angleterre sur la base de l'IPTS-68 entre 13.81 et 373.15 K. La
résistance du thermomètre J (platine) est mesurée par un pont a.c. (modèle 5840D,
H. Tinsley and Co., Ltd). Le thermomètre à résistance de germanium (model GR-
200A-500, Lake Shore Cryotonics, Incs.) a été calibré sur la base de l'EPT-76
entre 1.2 et 23 K. Sa résistance est mesurée en utilisant un multimètre digital
haute précision (modèle.TR-7200, Advantest Corp.), un scanner universel
(modèle TR-7200, Advantest Corp.) et un resistor standard (type 2794, 100 Cl,
Yokogawa Hokushin Electric Corp.). Les interruptions du four sont réalisées par
le scanner universel et le temps de chauffage est mesuré par un compteur
universel (modèle TR-5822, Advantest Corp.).
6.2.3 Déroulement des mesures
La mesure des capacités calorifiques s'effectue toujours en chauffant
parce qu'on contrôle beaucoup mieux la température qu'en refroidissant. Après
chaque période de chauffe, l'équilibre thermique de la chambre calorimétrique est
atteint en 4 min à 20 K, 8 min à 50 K, 15 min à 100 K et une vingtaine de minute
au-delà de 150 K. L'apport d'énergie dans le calorimètre s'effectue en débitant un
courant dont on mesure la tension U et l'intensité / pendant un intervalle de temps
A/ variable selon l'incrément de température souhaité. Le temps de l'apport
d'énergie est mesuré par le compteur universel. Lors de cet apport d'énergie, la
température du calorimètre augmente subitement et, à la fin de la période de
chauffe, la température continue à augmenter légèrement avant de se stabiliser à la

température 7}. Ceci est simplement dû à la stabilisation de la température dans
l'échantillon due à la conduction.
La capacité calorifique de l'ensemble échantillon-calorimètre à la
température T se déduit alors facilement par :
P O T T
La mesure à vide du calorimètre permet de déterminer sa capacité
calorifique en fonction de la température. La capacité calorifique est ensuite
soustraite à la valeur donnée par la relation 6.3 pour obtenir la capacité calorifique
de l'échantillon.
De nombreuses données de calorimétrie adiabatique ont été publiées avec
des "corrections de courbure". En effet, la capacité calorifique obtenue n'est
qu'une valeur moyenne correspondant à une élévation de température qui n'est pas
infinitésimale. Une correction de courbure peut être alors apportée car la capacité
calorifique n'est pas une fonction linéaire entre 7/ et Tf. Pour réaliser cette
correction on doit faire l'hypothèse d'une loi locale de variation de la capacité
calorifique avec la température (généralement un polynôme d'ordre trois) et
soustraire la valeur de la dérivée seconde de la capacité calorifique moyenne
lissée par rapport à la température calculée à la température T = (Tt + Tj)l2. Nous
avons choisi de ne pas faire cette correction qui peut générer des erreurs aussi
importantes que celles qu'elle est sensée gommée selon les compositions étudiées
et l'hypothèse de variation locale utilisée en nous astreignant plutôt à des
incréments de température très faibles. Cette démarche a l'inconvénient
d'augmenter considérablement la durée des expériences. Typiquement, les
incréments de température sont de l'ordre de 0.5 à 1 K entre 13 et 20 K, de 1 à 1.5
K entre 20 et 50 K, de 1.5 à 2 K entre 50 et 150 K et entre 2 et 2.5 K entre 150 et
300 K. Ces variations d'incréments sont corrélées aux domaines de plus ou moins
grande linéarité des capacités calorifiques.
Pour tester la qualité des données obtenues, des mesures de capacité
calorifique ont été réalisées sur le matériau standard SRM 720 (un saphir
86

syntéthique, 01-AI2O3) fourni par le U.S. National Institute of Standards and
Technology (U.S. N.I.S.T.). La comparaison de ces mesures avec les valeurs
données par le U.S. N.I.S.T. indique des écarts de 10 % à 10 K, 0.3 % à 50 K et
0.1 % au-dessus de 70 K (Atake et al., 1990).
6.2.4 Lissage des données et extrapolation vers le zéroabsolu
Nous avons choisi de lisser les données expérimentales selon la méthode
des polynômes chevauchants. Pour chaque composition, on a défini des
polynômes de degré 3 à 5 sur des intervalles de températures restreints à des
différences de Cp entre les valeurs expérimentales et celles données par les
polynômes de 0.01 %. Chaque polynôme est "redondant" sur la moitié des
intervalles précédents et suivants. La capacité calorifique à la température T, est
égale à :
()(ff0()(|^)(),où I2 et T\ sont les bornes supérieures et inférieures de l'intervalle de température
commun au domaine de définition des polynômes PA et PB- Cette méthode permet
d'assurrer une parfaite continuité des valeurs lissées tout en s'ajustant au plus prés
aux valeurs expérimentales.
Les deux premiers polynômes étant décisifs pour la qualité de
l'extrapolation des différentes fonctions thermodynamiques à 0 K, on utilise une
fonction de type CP(T) = ai3 , conforme aux approximations du modèle de
vibration de Debye, pour simuler des valeurs inférieures à la plus basse
température mesurée. Ces valeurs et les premières valeurs expérimentales nous
servent à ajuster les coefficients des deux premiers polynômes. Le coefficient a
est pris égal au rapport Cp I T5 de la mesure effectuée à la plus basse température
(entre 13 et 14 K).
87

6.3 LA CALORIMETRIE DE CHUTE ENTRE 400 ET 1600 K(CALORIMETREA GLACE)
6.3.1 Principe
Outre la calorimetrie adiabatique haute température qui n'a pas fait l'objet
d'expérience dans ce travail, la méthode la plus précise au-delà de 1000 K est la
méthode de la chute. On chauffe l'échantillon à la température T, et on le fait
chuter dans un calorimètre, maintenu à la température de la glace fondante, To,
auquel il cède de la chaleur, HTHT0 en s'équilibrant avec lui. Des erreurs
inférieures à 0.1% peuvent être obtenues de 400 K à plus de 2000 K. On peut
ensuite calculer les capacités calorifiques par dérivation de l'enthalpie (Eq. 6.1).
Nous allons présenter le calorimètre à glace que nous avons utilisé au
département des géomatériaux de l'IPG de Paris.
6.3.2 Dispositif expérimental
Pour nos mesures, nous avons utilisé le dispositif comprenant deux
montages en service, l'un dit de basse température (BT) qui permet des mesures
d'enthalpie relative entre 400 et 1150 K et l'autre dit de haute température (HT)
permettant des mesures entre 700 et 1850 K.
Ces calorimètres ont été construits par Deniélou, Fournier, Petitet et
Téqui (Deniélou et al, 1971) et la partie haute température a été réalisée ensuite
par P. Richet (Richet et al., 1982, 1992). Un schéma du dispositif expérimental est
représenté sur la Figure 6.3, dans laquelle nous pouvons voir que le montage
calorimétrique s'articule autour de trois éléments principaux qui sont le four, le
vase calorimétrique et l'enceinte thermostatée.
88

Figure 6.3 : Schéma général du calorimètre à glace. (1) Four. (2) Vase calorimétrique. (3) Mélangeeau-glace fondante. (4) Enceinte thermostatée. (5) Bêcher de mercure. (6) Capillaire calibré (pourmesurer la fuite thermique). (7) Thermocouple de régulation. (8).Thermocouples de mesures. (9)Creuset.
89

Les fours
La température de ces fours (1) est contrôlée par des régulateurs
Eurotherm. Chaque four repose sur un élévateur Fenwick permettant son
positionnement de manière à placer le creuset dans la zone isotherme, pour
laquelle les gradients thermiques résultant de la convection à l'intérieur du four
sont les plus faibles. Les températures sont mesurées à l'intérieur des creusets à
l'aide de deux thermocouples (8) placés à la base de l'échantillon et à son sommet.
Les thermocouples utilisés sont en Pt/Rh 6% - Pt/Rh 30% pour le dispositif haute
température et en Pt - Pt/Rh 10% pour le dispositif basse température. Les f.e.m.,
fonctions de la température, sont directement mesurées avec un galvanomètre par
une méthode d'opposition. Des compensateurs de soudure froide Omega sont
adjoints aux thermocouples Pt - Pt/Rh 10% afin d'ajouter à la f.e.m mesurée de la
température ambiante à la température T, la f.e.m mesurée de 0°C à la température
ambiante. Dans le cas des thermocouples Pt/Rh 6% - Pt/Rh 30%, la f.e.m mesurée
de 0°C à la température ambiante est très faible et constante.
Les porte-échantillons sont constitués par une partie supérieure porteuse
en duralumin prolongée par des tubes verticaux d'alumine permettant l'accrochage
du creuset (9) ainsi que le maintien des thermocouples et d'écrans thermiques qui
permettent de réduire les gradients de température.
Vase calorimétrique
Le vase calorimétrique (2) constitue la partie réceptrice du creuset
chauffé après la chute. La chaleur libérée par l'échantilon est cédée au calorimètre
qui est constitué d'un mélange de deux phases en équilibre, glace et eau à 273 K.
La fusion d'une partie de la glace est accompagnée d'une variation de volume A V,
proportionnelle à l'enthalpie Hj-Hm de l'échantillon.
Le vase est constitué de deux enceintes en verre séparées par un vide
primaire de manière à obtenir une bonne isolation du milieu extérieur. L'intérieur
de la deuxième enceinte contient un mélange de mercure et d'eau bien dégazée par
bidistillation. Un manchon de glace de forme cylindrique qui correspond à
l'expulsion d'environ 450 grammes de mercure hors de l'enceinte interne. Lors de
90

la formation de la glace, le volume augmente en effet, obligeant le mercure à
sortir dans un bêcher extérieur (5), et vice versa lors de la fonte du manchon.
Enceinte thermostatée
L'enceinte thermostatée (4) permet d'isoler le vase calorimétrique du
milieu extérieur. Elle est constituée de glace pilée fondante(3) préparée à partir
d'eau distillée. La glace est préalablement tassée, puis, nous comblons les
interstices avec de l'eau distillée afin d'obtenir une température homogène dans le
thermostat.
Pour éviter une fonte de la glace trop rapide, une circulation d'eau +
glycol, maintenue à une température aux environs de 0.5 °C, a été intégrée entre
l'enceinte en verre et celle en tôle. Cette circulation est gérée par un cryostat
Haake de type F3K. Sur la Figure 6.3, nous pouvons voir les différentes parties
constituant l'enceinte. En partant de l'extérieur, nous avons tout d'abord un
premier container en tôle qui renferme un tube en PVC de 355 mm de diamètre.
Entre les deux se trouve une mousse de polyuréthane expansée constituant un
premier isolant. Entre le tube en PVC et le vase en verre de 300 mm de diamètre,
circule le mélange eau + glycol sur toute la hauteur de l'enceinte.
Pour mesurer les échanges thermiques du vase calorimétrique avec
l'extérieur (fuites thermiques), nous pouvons suivre le niveau de mercure en
fonction du temps dans un capillaire en verre étalonné, à l'aide d'un cathétomètre,
le robinet placé avant le bêcher de mercure étant fermé.
6.3.3 Technique calorimétrique
Un creuset, contenant une masse connue m de produit, est porté à une
température T dans le four. Lors de sa chute dans le calorimètre maintenu à
To=273 K, il apporte une certaine quantité de chaleur (Hr-Hr) au calorimètre, ce
qui entraîne la fusion de glace à l'intérieur du vase calorimétrique. Par voie de
conséquence, le volume de glace diminue et cette perte de volume est compensée
par un apport de mercure venant du bêcher externe. L'enthalpie mesurée (Hm) est
91

donc égale à l'enthalpie apportée par le creuset {Hcr) plus l'enthalpie du produit
Hm = Hpr + Hcr Eq. 6.5
On détermine Hcr par :
2 T 2h(T2-T02)-c(\/T-\/T0) Eq. 6.6
2
où le terme entre crochets est le facteur d'étalonnage du creuset et mcr la masse du
creuset. De plus, 7o=273.15 K et a, b, c sont des paramètres propres à chaque
creuset.
L'enthalpie mesurée est obtenue en faisant la différence de poids de
mercure avant et après la chute. A cette valeur, il faut ajouter une correction de
fuite (quantité de chaleur apportée ou prise au calorimètre par le milieu extérieur)
et une correction de niveau (différence de niveau de mercure dans le capillaire
avant et après la chute lors de la mesure du mercure restant dans le bêcher).
6.3.4 Représentation des mesures calorimétriques
La première représentation possible consiste à exprimer les enthalpies
relatives, HT-H273, en fonction de la température. Un tel graphe a peu d'intérêt car
il y est difficile de différencier le liquide du verre, c'est-à-dire de visualiser la
transition vitreuse. Une seconde représentation, celle que nous utiliserons, nous
permet de mettre en valeur la transition vitreuse par une nette rupture de pente.
Dans cette représentation, les données expérimentales s'expriment sous forme de
capacités calorifiques moyennes, Cm, définies par :
_ HT ~ H*)T>Cm = —£ 221 . Eq. 6.7
T-273.15
En effet, les capacités calorifiques moyennes permettent d'illustrer les
résultats sous une échelle plus dilatée que les enthalpies relatives ne pourraient le
permettre car la capacité calorifique varie peu avec la température.
Ces deux types de représentations sont des représentations brutes des
résultats expérimentaux, elles ne nécessitent aucun calcul long ou fastidieux et
92

nous permet de reconnaître d'éventuels problèmes de cristallisation, mauvaise
chute, etc.. pendant la durée de l'expérience.
Dans un second temps, il est possible de calculer et de représenter les
capacités calorifiques, Cp. Il convient d'abord d'effectuer un lissage des enthalpies
relatives pour obtenir, sur l'intervalle de tempéraure étudié, une représentation
continue des quantités de chaleurs libérées par le produit. Ce lissage s'effectue le
plus fréquemment à partir d'une équation du type Haas et Fisher (1976),
bT2HT-H273 =R273+aT + -bT2 ~ — + 2dyfT+-eT\ Eq. 6.8
où a, b, c, d et e sont des paramètres ajustables ; i?273 est un paramètre ajustable
uniquement pour les liquides, en revanche, pour les verres et les cristaux, il est
fixé par la contrainte HT-H273 = 0 pour T= 273,15 K.
Par dérivation de cette équation, nous obtenons les Cp :
r + -r= + eT2, Eq. 6.92 4Ï
Par cette méthode, des comparaisons avec un étalon calorimétrique
)t nous donnent une précision de 0.2 % pour les enthalpies relatives et
0.5% pour les capacités calorifiques (Richet et al., 1982, 1992).
6.3.5 Etalonnage des creusets
II n'est pas possible d'effectuer des mesures calorimétriques sans avoir
préalablement étalonné le creuset accueillant le produit. Pour ce faire, nous
appliquons au creuset vide le même processus expérimental qu'au creuset plein.
Nous effectuons des mesures dans toute la gamme de température possible (de
400 à 1800 K) de manière à obtenir une courbe régulière. Nous présenterons ici
les étalonnages des creusets 13 et 14 qui ont été fabriqués pour cette étude.
L'étalonnage précis d'un creuset est fondamental car de lui dépend l'équation de
l'enthalpie qui est ensuite incluse dans un programme de dépouillement de
l'enthalpie du produit. Les mesures sur le creuset 13 ont été réalisées
93
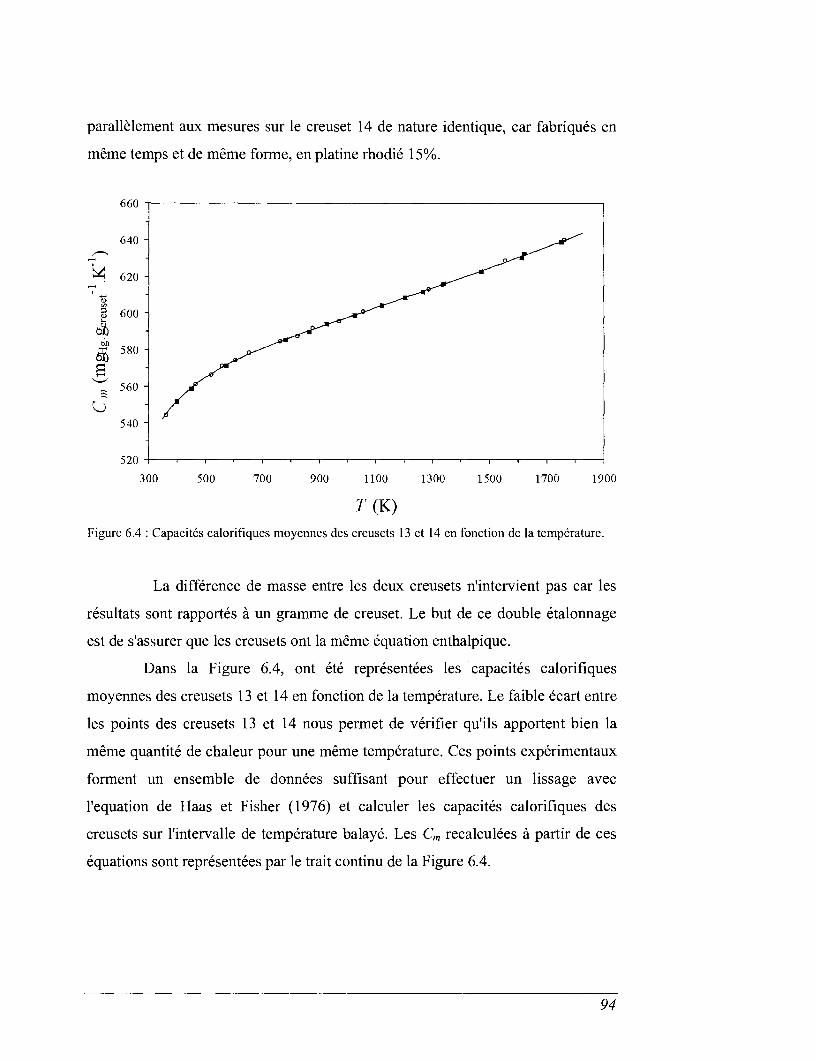
parallèlement aux mesures sur le creuset 14 de nature identique, car fabriqués en
même temps et de même forme, en platine rhodié 15%.
660
520
300 500 700 900 1100 1300 1500 1700 1900
T(K)Figure 6.4 : Capacités calorifiques moyennes des creusets 13 et 14 en fonction de la température.
La différence de masse entre les deux creusets n'intervient pas car les
résultats sont rapportés à un gramme de creuset. Le but de ce double étalonnage
est de s'assurer que les creusets ont la même équation enthalpique.
Dans la Figure 6.4, ont été représentées les capacités calorifiques
moyennes des creusets 13 et 14 en fonction de la température. Le faible écart entre
les points des creusets 13 et 14 nous permet de vérifier qu'ils apportent bien la
même quantité de chaleur pour une même température. Ces points expérimentaux
forment un ensemble de données suffisant pour effectuer un lissage avec
l'équation de Haas et Fisher (1976) et calculer les capacités calorifiques des
creusets sur l'intervalle de température balayé. Les Cm recalculées à partir de ces
équations sont représentées par le trait continu de la Figure 6.4.
94

Tableau 6.1 : Enthalpies relatives des creusets 13 et 14 .
T
(K)
401.15
452.65
573.45
782.15
864.9
926.65
1024.96
1121.59
1204.6
1267.04
1339.87
1472
1614.92
1622.74
1752.24
HT-H273
(mgHg/gcreuset)
70.607
100.317
171.590
297.970
349.030
388.177
450.243
512.408
566.770
607.719
657.382
746.510
845.560
853.477
945.046
T
(K)
362.64
466.52
521.2
558.59
606.5
654.35
762.8
821.85
877.73
971.22
1055.79
1286.2
1335.7
1556.3
1761.38
Hj-H273
(mgHg/gcreuset)
48.719
108.508
140.568
163.042
191.441
220.484
286.223
322.414
357.919
415.731
470.313
621.251
654.190
807.123
952.720
Tableau 6.2 : Coefficients de lissage par l'équation de Haas et Fisher des données descreusets 13 et 14.
creuset
13 et 14
R273
144.112
a
0.3073
\0\b
0.1655
10~5.c
-0.1272 5
d
.574
N"
30
AT{¥L)b
364-1761
AADC
0.05
a N représente le nombre de points pris en compte dans le lissage,b AT (K) représente l'intervalle de température couvert par les points expérimentaux,c AAD est la déviation moyenne absolue entre les valeurs expérimentales et lissées (%).
95

6.4 LA CALORIMETRIE DIFFERENTIELLE A BALAYAGE (DSC)ENTRE298ET1100K
Principe
En accord avec le principe général exposé plus haut (§ 6.1), la
calorimétrie différentielle à balayage consiste à mesurer la quantité de chaleur par
unité de temps qui s'échange entre l'enceinte d'une chambre calorimétrique et une
enceinte extérieure soumise à des variations de température programmées. Le
montage différentiel, qui consiste à associer en opposition deux détecteurs
calorimétriques identiques et à les placer à chaque instant dans les mêmes
conditions de température, permet de supprimer les effets thermiques extérieurs
pour ne mesurer que les flux de chaleurs qui s'opèrent entre la chambre laboratoire
et l'extérieur. Autrement dit, lorsque aucun phénomène énergétique n'est produit
dans la cellule laboratoire, le zéro expérimental est stable et donne lieu à un
enregistrement parallèle à l'axe des temps. Le moyen le plus sûr de soumettre les
deux éléments calorimétriques jumelés à des températures identiques est d'utiliser
un unique thermostat qui entourera les deux chambres de façon équidistante et
symétrique. Pour "balayer" un large domaine de température, le thermostat est un
four qui est régulé selon le phénomène à étudier. Ce principe simple permet
d'étudier les phénomènes énergétiques (fusion, transition de phase, etc..) qui
affectent les matériaux par pointage des températures (ou des intervalles de
température) auxquels se produisent les phénomènes et mesures de l'intensité des
phénomènes par différence entre l'intégration du flux de chaleur sur toute la durée
des phénomènes et l'intégration du flux de chaleur généré par une expérience "à
blanc". Une autre utilisation du DSC (Differential Scanning Calorimeter) est celle
qui nous a permis la détermination des capacités calorifiques à pression constante.
Selon le même principe de mesure, l'intégration de la différence des flux de
chaleur entre l'expérience "chargée" et l'expérience à blanc lors d'un incrément de
température AT dans la chambre laboratoire est égale à la variation d'enthalpie de
96

l'échantillon de T à T+AT. Si l'incrément de température ou la chaleur échangée
sont suffisamment faibles, on peut assimiler cette valeur à la capacité calorifique à
la température T+1/2AT.
6.4.1 Appareilla ge
Nous avons utilisé un calorimètre DSC 121 de la compagnie SETARAM
qui a été acheté par le laboratoire des Géomatériaux (I.P.G.P.) au cours de cette
thèse. Dans la section suivante nous aborderons les étapes de mise en service de
l'appareil (étalonnage, test de reproductibilité, etc.). Le bloc calorimétrique de
DSC 121 est représenté dans la Figure 6.5.
Le bloc calorimétrique (1) est traversé par les deux tubes (2) en alumine
frittée de diamètre intérieur 7 mm. La section intérieure des tubes est entièrement
libre et permet le transit d'un échantillon (3) d'une extrémité à l'autre. Chaque
extrémité des tubes reçoit une embouchure qui permet de connecter un système de
circulation de gaz.
Le cœur du bloc calorimétrique comporte deux cavités dans lesquelles
sont disposés les détecteurs calorimétriques : deux thermopiles consituées de 10
couronnes de 12 thermocouples Au / chromel repartis de façon homogène sur
toute la surface du détecteur (20 mm de longueur). De façon pratique, seuls les 10
mm médians du détecteur sont utilisés pour l'analyse de l'échantillon. Ceci assure
de part et d'autre du creuset une grande longueur de garde thermique. Les
dimensions : diamètre 7 mm, longueur 10 mm définissent ainsi les dimensions
pratiques maximales de l'échantillon étudié. Les détecteurs entourent la partie
médiane des tubes formant ainsi les cellules laboratoire et témoin.
97

U[jUFigure 6.5 : Photo du DSC 121 SETARAM et schéma du bloc calorimétrique (1). (2) Tubes enalumine. (3) Thermopile. (4) Echantillon. (5) Four. (6) Circulation d'eau.
Les deux ensembles, thermopile + tube, sont entourés par un four à effet
Joule qui est placé dans un bloc métallique, lui-même entouré d'une circulation
d'eau. Deux sondes (résistances de platine) sont utilisées pour les mesures de la
température de régulation du four et celle de l'échantillon. La nature des éléments
qui composent l'appareil et leur assemblage ont été conçus pour des utilisations
jussqu'à des températures voisines de 830°C.
Les bornes des thermopiles sont reliées à un amplificateur, lui-même
associé à un convertisseur analogique/numérique. Par ce biais, les variations de
f.e.m. aux bornes des thermopiles sont transmises à un ordinateur qui offre des
possibilités de visualisation graphique des phénomènes en cours. C'est également
à partir de cet ordinateur que l'on contrôle les périodes de chauffe du four ; le four
est asservi à des températures de consigne et des vitesses de chauffe par un
système p.i.d..
La compagnie SETARAM fournit divers types de conteneurs pour placer
les échantillons dans les cellules (barquette et creuset alumine, creuset aluminium,
98

acier inoxydable, platine, etc.) mais ceux-ci n'ont pas été utilisés pour des raisons
évidentes : nos objets d'étude étant des verres et des liquides susceptibles de
s'écouler dans cette géométrie horizontale, il faut des creusets capables de contenir
les échantillons sans risque d'endommager les tubes d'alumine et qui restent inerte
au contact de liquides silicates aux environs de 800°C. Pour répondre à nos
besoins, nous avons fait faire des barquettes de platine (cylindre de longueur 10
mm tronqué au 2/3 selon un plan horizontal parallèle à son axe de révolution) par
les services techniques du Centre de Thermodynamique et de Microcalorimétrie
du CNRS à Marseille. Ces barquettes s'ajustent au tube d'alumine des cellules
pour assurer une bonne conduction thermique et des pertes minimales de chaleur
par les extrémités des tubes.
6.4.2 Mode opératoire
Dans nos expériences, la cellule témoin accueille une barquette vide. La
cellule laboratoire reçoit une barquette contenant entre 150 et 250 mg
d'échantillon ou une barquette vide dans le cas d'une expérience à blanc. En effet,
une expérience typique de détermination de capacité calorifique par DSC consiste
en deux acquisitions : l'une à blanc, l'autre chargée. Les mesures ont été effectuées
dans une atmosphère dynamique d'argon avec un débit de 15 ml/h.
Pour commencer une acquisition, les cellules sont chargées et maintenues
à 25 ou 30°C jusqu'à atteinte de l'équilibre thermique, qui est marqué par une
ligne de base stable et parallèle à l'axe des temps. Un saut de température de 25°C
est alors réalisé, à une vitesse de 3 K/min. La température finale est ensuite
maintenue 600 s pour laisser le flux de chaleur reprendre de nouveau une valeur
stationnaire. Cette température finale est alors utilisée comme nouveau point de
départ pour le prochain saut de température de 25°C. Cette procédure est répétée
de nombreuses fois jusqu'à atteindre la température finale maximale de 820 ou
825°C. Les programmes de chauffe sont consignés dans l'ordinateur avant
l'acquisition, le reste est entièrement automatisé. La durée de chaque acquisition, à
blanc ou non, est de 8 h 30.
99
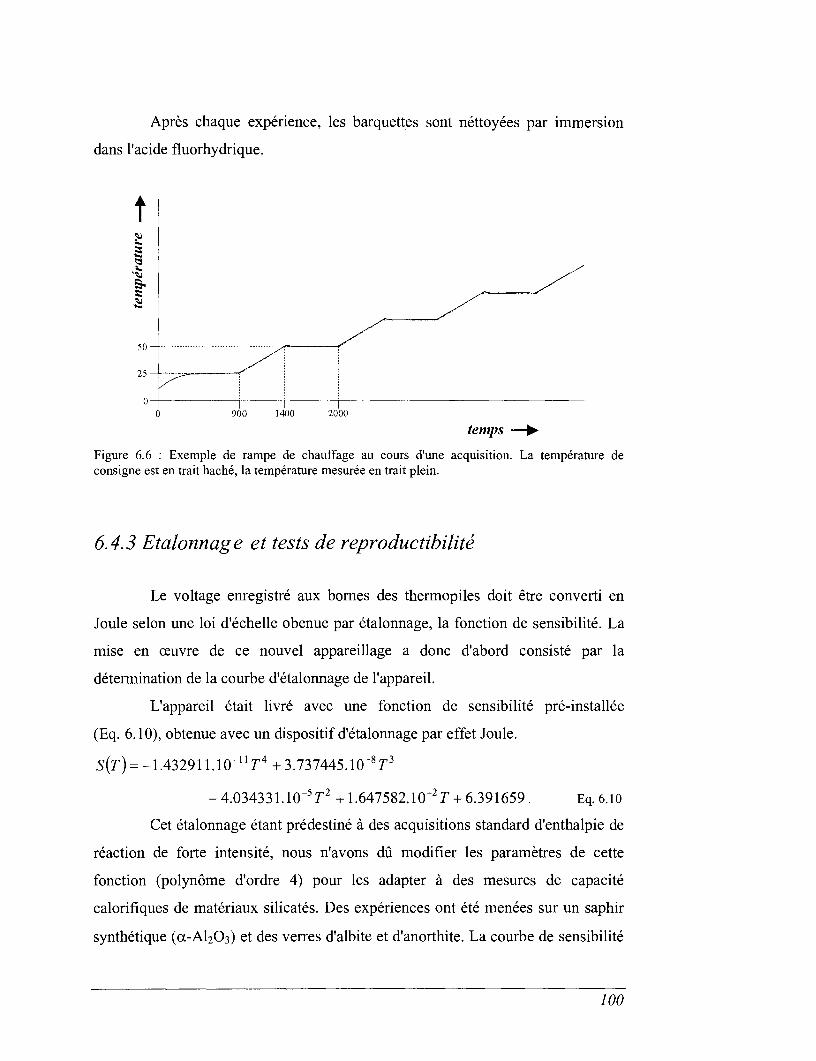
Après chaque expérience, les barquettes sont nettoyées par immersion
dans l'acide fluorhydrique.
900 1400 2000
temps
Figure 6.6 : Exemple de rampe de chauffage au cours d'une acquisition. La température deconsigne est en trait haché, la température mesurée en trait plein.
6.4.3 Etalonnage et tests de reproductibilité
Le voltage enregistré aux bornes des thermopiles doit être converti en
Joule selon une loi d'échelle obenue par étalonnage, la fonction de sensibilité. La
mise en œuvre de ce nouvel appareillage a donc d'abord consisté par la
détermination de la courbe d'étalonnage de l'appareil.
L'appareil était livré avec une fonction de sensibilité pré-installée
(Eq. 6.10), obtenue avec un dispositif d'étalonnage par effet Joule.
= - i .4329i i .Kr 1 1 r 4 + 3.737445. io~8r3
~5 T 2 T2- 4.034331.10~5 T2+1.647582.1(T2 7 + 6.391659. Eq.6.10
Cet étalonnage étant prédestiné à des acquisitions standard d'enthalpie de
réaction de forte intensité, nous n'avons dû modifier les paramètres de cette
fonction (polynôme d'ordre 4) pour les adapter à des mesures de capacité
calorifiques de matériaux silicates. Des expériences ont été menées sur un saphir
synthétique (01-AI2O3) et des verres d'albite et d'anorthite. La courbe de sensibilité
100

a été ajustée par la différence relative entre les mesures obtenues et les valeurs
publiées par Ditmars et Douglas (1971) du National Bureau of Standards, Richet
et al. (1984) et Richet et al. (1984), respectivement. La courbe de sensibilité
obtenue (Eq. 6.10) nous permet de reproduire les valeurs publiées des capacités
calorifiques à mieux que 1% sur tout le domaine de température utilisable.
S(T)= -1.03961. i ( r u : r 4 +2.94555. K r 8 r 3
- 3.40631.10-572+1.3576.10"2r +6.80835. Eq.6.11
Cette courbe de sensibilité est dépendante de la nature des creusets
puisque celle-ci modifie la conductivité thermique entre l'échantillon et le
détecteur calorimétrique. Ceci a été vérifié lors de l'utilisation de barquettes
d'alumine pour lesquels une autre courbe de sensibilité a dû être déterminée.
Une autre calibration doit être effectuée : la calibration en température.
En effet, la mesure de température de l'échantillon est réalisée par une sonde
platine située dans le bloc calorimétrique ; bien que cette mesure se fasse à
proximité de l'échantillon, une petite différence existe avec la température réelle
de l'échantillon due au gradient thermique et au temps de transfert de la chaleur au
travers de la paroi du creuset et du détecteur calorimétrique. Pour effectuer la
correction de température, on utilise divers composés caractérisés par des
transitions (fusion, transition de phase, etc.) bien définies dans le domaine de
température couvert par l'appareil. Les différences entre les températures de
transition réelle et celles pointées sur nos thermogrammes nous permettent
d'obtenir une loi de variation linéaire de la correction a apporté en fonction de la
température. Cet étalonnage ayant déjà été réalisé par la société SETARAM, nous
avons juste vérifié sa validité dans notre configuration de barquette de platine
avec la fusion de l'Indium (429.8 K) et la transition a-P du quartz (847 K).
La correction de température est en théorie sensible à la vitesse de
chauffe programmée, à la nature des creusets employés et à la nature et au débit
du gaz de balayage. Nos tests n'ont pas permis de voir de variations sensibles pour
des vitesses de chauffe entre 0.5 et 3 K/min. De même, les variations des mesures
réalisées en présence ou non d'un balayage d'argon sont restées égales à celles
101
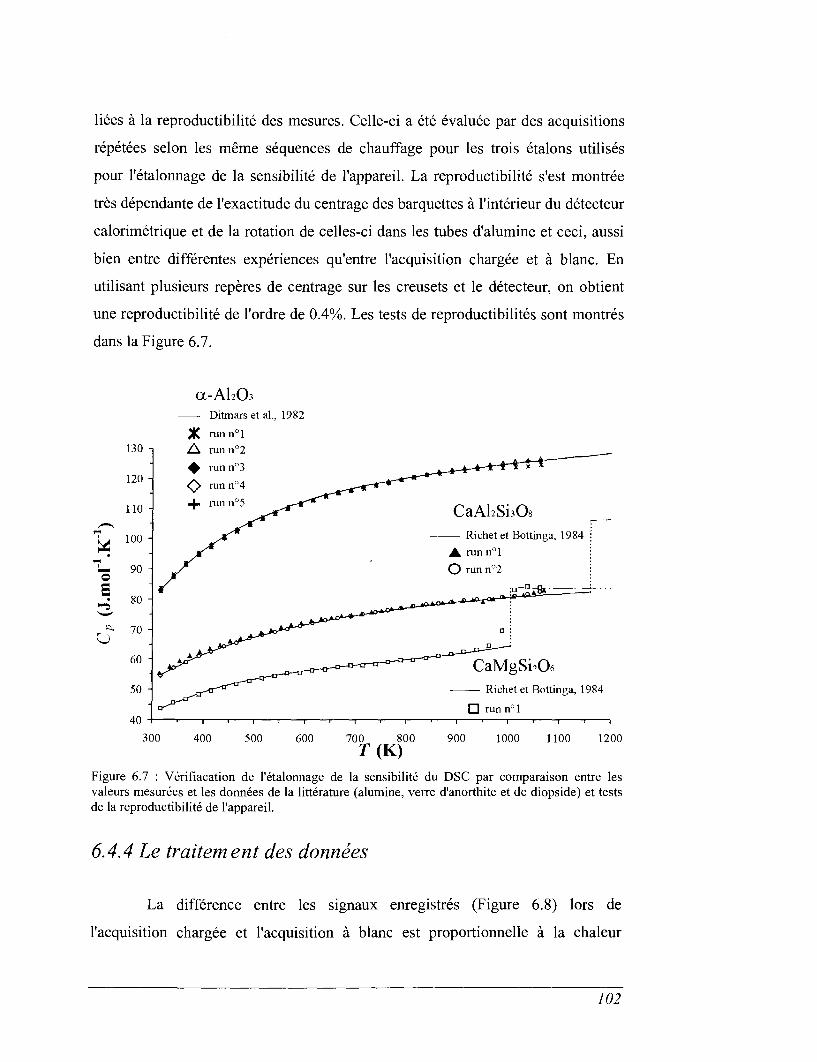
liées à la reproductibilité des mesures. Celle-ci a été évaluée par des acquisitions
répétées selon les même séquences de chauffage pour les trois étalons utilisés
pour l'étalonnage de la sensibilité de l'appareil. La reproductibilité s'est montrée
très dépendante de l'exactitude du centrage des barquettes à l'intérieur du détecteur
calorimétrique et de la rotation de celles-ci dans les tubes d'alumine et ceci, aussi
bien entre différentes expériences qu'entre l'acquisition chargée et à blanc. En
utilisant plusieurs repères de centrage sur les creusets et le détecteur, on obtient
une reproductibilité de l'ordre de 0.4%. Les tests de reproductibilités sont montrés
dans la Figure 6.7.
OC-AI2O3Ditmars et al., 1982
HH
osa.
u
130 -
120 -|
110 -
100 •
90 -
80 -
70 -
60 -
Sf) -J\J
40 -
XA•O+
//
run n° 1run n°2
run n°3
run n°4
run n°5 _^s-^*^
s*
CaAkSisOs __
ivlvllcl Cl -DULLlIltia, 12/oH •
A runn°l :O run n°2 i
n « ;• U iiL.»
• :
n_S——'
Rirliet et Rnttin^n 10ÎM
D runn°l
300 400 500 600 700 800T(K)
900 1000 1100 1200
Figure 6.7 : Vérifiacation de l'étalonnage de la sensibilité du DSC par comparaison entre lesvaleurs mesurées et les données de la littérature (alumine, verre d'anorthite et de diopside) et testsde la reproductibilité de l'appareil.
6.4.4 Le traitement des données
La différence entre les signaux enregistrés (Figure 6.8) lors de
l'acquisition chargée et l'acquisition à blanc est proportionnelle à la chaleur
102
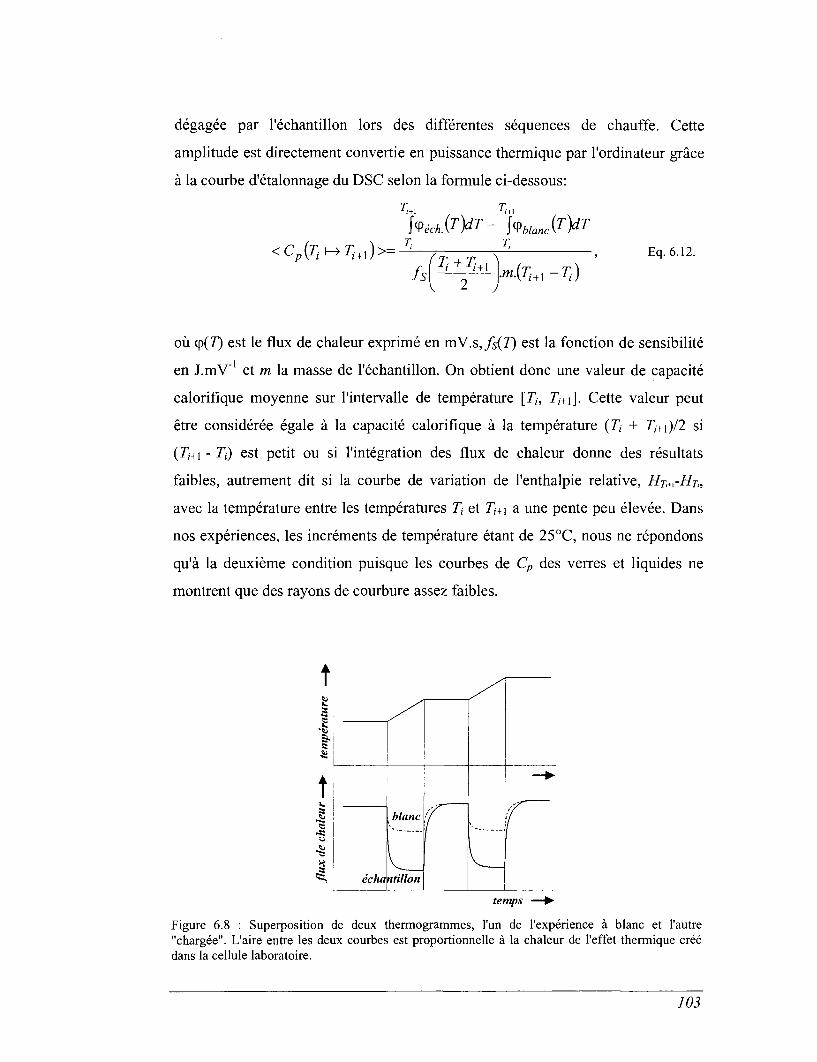
dégagée par l'échantillon lors des différentes séquences de chauffe. Cette
amplitude est directement convertie en puissance thermique par l'ordinateur grâce
à la courbe d'étalonnage du DSC selon la formule ci-dessous:
T
Eq. 6.12.
où cp(7) est le flux de chaleur exprimé en mV.s,fs{T) est la fonction de sensibilité
en J.mV"1 et m la masse de l'échantillon. On obtient donc une valeur de capacité
calorifique moyenne sur l'intervalle de température [T,-, 7/+i]. Cette valeur peut
être considérée égale à la capacité calorifique à la température (Tt + r,+i)/2 si
(Tj+\ - Tt) est petit ou si l'intégration des flux de chaleur donne des résultats
faibles, autrement dit si la courbe de variation de l'enthalpie relative, HTM-HTI,
avec la température entre les températures 7, et Ti+\ a une pente peu élevée. Dans
nos expériences, les incréments de température étant de 25°C, nous ne répondons
qu'à la deuxième condition puisque les courbes de Cp des verres et liquides ne
montrent que des rayons de courbure assez faibles.
temps — •
Figure 6.8 : Superposition de deux thermogrammes, l'un de l'expérience à blanc et l'autre"chargée". L'aire entre les deux courbes est proportionnelle à la chaleur de l'effet thermique créédans la cellule laboratoire.
103

6.5 CALORIMETRIE DE DISSOLUTION EN SELS FONDUS
Les techniques de calorimétrie de dissolution en sels fondus ont été mises
en œuvre avec l'aide de J. Rogez au Centre de Thermodynamique et de
Microcalorimétrie (CTM) du CNRS à Marseille. Celles-ci ont été réalisées dans
un microcalorimètre Tian-Calvet de type haute température. On se référera aux
travaux de Calvet et Prat (1956), à la thèse de C. Brousse (1981) pour la
description détaillée des techniques et de l'appareillage.
La méthode utilisée pour calculer les enthalpies de formation d'un verre
(ou d'un liquide) à partir ses oxydes constitutifs repose sur un cycle
thermodynamique faisant intervenir (n+l) mesures de chaleur de dissolution dans
un même solvant, n étant le nombre d'oxydes constitutifs et la («+l)ieme réaction
étant la dissolution du verre (ou du liquide). De ce fait, l'imprécision affectant
l'enthalpie de formation est importante (de l'ordre du kJ/mole).
6.5.1 La détermination des enthalpies deformation
Pour obtenir l'enthalpie de formation, AjH, des borosilicates vitreux et
liquides par référence à leurs oxides purs constitutifs nous avons dû recourir à la
méthode de dissolution dans un solvant.
La procédure se traduit par la dissolution séparée dans un bain adéquat,
S, du verre et de chacun de ses oxydes constitutifs. Par exemple, pour un verre de
formule molaire xSi02Si02. -^o^Ch. x^p Na2O, les trois réactions suivantes sont
effectuées à la température 7̂ du calorimètre,
verre + xS -> (xsio2 SiO2, xm B2O3, xNaiO Na2O)s, (a)
SiO2+;cS -^(SiO2)s, (b)
B 2 O 3 + J C S ^ ( B 2 O 3 ) S , (c)
Na2O+JcS ^(Na2O) s , (d)
104

cédant chacune au calorimètre une quantité de chaleur proportionnelle à la variation
d'enthalpie de la réaction, ASH(TS).
Pour obtenir l'enthalpie de formation par référence aux constituants purs
solides (les oxides) à la température Ts, il faut que la somme des états finals des
réactions (b), (c) et (d) soit identique à celui de la réaction (a). Ce sera vérifié si les
deux conditions suivantes sont réalisées :
les interactions entre SiCh, Na2Û et AI2O3 dans le bain final résultant de
la réaction (a) sont nulles;
les concentrations en SiC>2, Na2Û et AI2O3 dans ce bain sont les mêmes
que dans les bains finals (b), (c) et (d).
Ces conditions sont remplies si l'on effectue la mesure des enthalpies des
trois réactions précédentes à dilution infinie (AHS°°). L'enthalpie de la réaction à 7̂
xSi0; SiO2 + xBA B2O3 + xN,2o Na2O-^ verre,
s'écrit alors :
AjH(Ts) = xsiOl AsH sio£Ts) + XB^ AJI B2O,(TS) + x^oAJI K,2o(Ts) - AJH vtJJs).
En général, on effectue les mesures d'enthalpie de dissolution en fonction
de la concentration en soluté et on extrapole à concentration nulle. Nous verrons
dans la prochaine section que, avec le solvant choisi, les enthalpies de dissolution
(a), (b), (c) et (d) sont, à forte dilution, indépendantes de la concentration. Il n'est
donc pas nécessaire d'effectuer les mesures en fonction de la concentration et
d'extrapoler à dilution infinie.
6.5.2 Le choix du solvant
Un grand nombre de mélanges d'oxydes fondus a été recherché et étudié
comme solvant. Parmi eux, citons les binaires PbO-V2O5 (Yokokawa and Kleppa,
1964), PbO-B2O3 (Holm and Kleppa, 1967), Na2O-MoO3 (Navrotsky and Kleppa,
1967), PbO-GeO2 (Mûller and Kleppa, 1973), LiBO2-NaBO2 (Amosse and
Mathieu, 1980).
Ces bains d'oxydes doivent satisfaire à plusieurs exigences :
105

ils doivent dissoudre, avec une vitesse suffisante, à la fois les composés acides
(comme SKD2 par exemple) et basiques (comme CaO ou MgO) puisque la
mesure des chaleurs de dissolution est d'autant moins précise que le flux
thermique correspondant est faible et de longue durée ;
leurs températures de liquidus doivent être relativement basses et leurs
viscosités faibles aux températures utilisées ;
les chaleurs de dissolution doivent être reproductibles et indépendantes des
concentrations relatives du soluté et du solvant, et ne doivent pas varier de
façon significative à la suite de légères fluctuations de la composition du bain.
Cette dernière condition est remplie si le solvant choisi présente les
caractéristiques d'une solution tampon.
Nous avons utilisé comme solvant d'oxydes l'une des compositions du
système binaire PbO-B2O3. Dans le domaine situé 0.25 et 0.5 mol% d'anhydrique
borique, les enthalpies molaires partielles des deux constituants varient peu,
autrement dit l'acidité du bain est indépendante de la composition. C'est donc dans
cette zone tampon, due à l'existence d'au moins deux anions borates, qu'il faudra
prendre le solvant. Les auteurs, Holm et Kleppa (1967) conseillent de prendre le
mélange de composition 2PbO.B2Û3. De fait, le métaborate de plomb se prête
bien à la dissolution de composés aluminosilicatés et borosilicatés. Il est
particulièrement efficace entre 900 et 1000 K pour dissoudre les oxydes
réfractaires tels que MgO par rapport aux autres solvants (Navrotsky et al., 1980).
Il est totalement liquide aux alentours de 773 K (Geller et Bunting, 1937) et de
faible viscosité à la température où nous l'utilisons. Il se présente sous forme
vitreuse à température ambiante, et est relativement peu hygroscopique d'où une
certaine simplicité de stockage et de préparation. C'est donc le métaborate de
plomb que nous avons utilisé à une température fixée à 973 K, suivant en cela un
grand nombre d'expérimentateurs. Ce choix permet également de restreindre le
nombre de mesures sur les oxydes constitutifs de nos diverses compositions
puisque les enthalpies de dissolution de la majeure partie des oxydes qui
constituent les verres de référence SON et AVM à plus de 30 oxydes ont déjà été
déterminées dans les mêmes conditions.
106

La température de 973 K étant supérieure à la plupart des températures
de transition vitreuse de nos produits (à l'exception de la série des analogues
naturels), il ne sera pas nécessaire de tenir compte de variation de température
fictive entre les mesures faites avec différentes techniques calorimétriques.
6.5.3 Synthèse du solvant 2PbO.B2O3
Nous nous sommes conformés aux indications fournies par Charlu et al.
(1975) pour sa préparation.
En vue de l'obtention de 1500 g de solvant, nous avons dans un premier
temps synthétisé le metaborate de plomb par fractions de 150g à partir de portions
stoechiométriques d'oxyde de plomb (PbO) et d'acide orthoborique (H3BO3)
(Baker analysed Reagent).
Tableau 6.3 : Compilation des mesures d'enthalpies de dissolution dans 2PbO-B2O3 présentes dansla littérature pour certains oxydes. AHS est l'enthalpie de dissolution de l'oxyde exprimée enkJ/mole à la température Ts (en K) et a est la déviation par rapport à la moyenne des N mesuresréalisées.
Oxyde Ts
SiO2 9651173970970986985975973107010701170970977
Na2O 975
9759751000
AHs(TJ±a
-4.35±0.294.31 ±0.21-5.15±0.29-4.48±0.13-3.18±0.42-3.34±0.12-3.51±0.18-4.23±0.210.84±0.630.79±0.213.77±0.33-4.38±0.13-4.28±0.15
-170.78±0.90
-172.21 ±4.31-170.79±4.89-179.40±3.80
N
558
205617625311536
Références
Shearer et Kleppa, 1973Shearer et Kleppa, 1973Charlu et al., 1975Newton et al., 1980Navrotsky et Coons, 1976Navrotsky et al., 1980Akaogi et Navrotsky, 1984Kiseleva et al., 1979Kiseleva et al., 1979Kiseleva et al., 1979Kiseleva et al., 1979Rogezetal., 1983Ellison et Navrotsky, 1992Kiseleva et al., 1996
Zygan et al., 1978Zyganetal., 1978Hervig et Navrostky, 1985
Produit de départ / Traitement
Quartz nat. de Lisbon, MarylandQuartz nat. de Lisbon, MarylandQuartz nat. de Lisbon, MarylandQuartz nat. de Lisbon, MarylandQuartz naturel brésilien, recuit 48 h. à 1000°CQuartz nat. brésilienQuartz nat. Brésilien, recuit 2 jours à 1073 KQuartz synt. (99.995% SiO2)Monocristal nat. de quartzQuartz synt. (99.995% SiO2)Quartz synt. (99.995% SiO2)Quartz nat. de Lisbon, MarylandQuartz synt.Na2CO3 (produit Aldrich), séché à 473 K,décarbonatationNa2CO3
NaA102
Na2CO3
107
Tableau
Oxyde
B2O3
6.3 (suite)
Ts AH/T^a
1073 -51.46±1.25973 -56.56±0.55
N
34
Références
Holm et Kleppa, 1967Hervig et Navrostky, 1985
HH
3BO33BO3
Produit de départ/ Traitement
AI2O3 965 32.97±1.00 16 Shearer et Kleppa, 1973
1173 31.25±0.54 18 Shearer et Kleppa, 1973
970 32.34±0.33 19 Charlu et al., 1975970 32.55±0.46 16 Newton et al., 1980970 32.92±1.20 9 Rogez et al., 1983
Corindon synt. (Baker Analysed Reagent), recuit 24 hà1400°CCorindon synt. (Baker Analysed Reagent), recuit 24 hà1400°CCorindon synt. (Fisher), recuit 72 h à 1300°CA1(OH)3, recuit 1 semaine à 1350°Alumine a (produit CERAC, 99.7% AI2O3)
CaO 970 -56.36±0.71 6 Newton et al., 1980
986 -54.52±0.92 6 Navrotsky etCoons, 1976970 -55.15±1.21 Charlu et al., 1978973 -55.65±0.29 27 Kiseleva et al., 1979
1070 -52.47±0.96 8 Kiseleva et al., 1979
CaCO3, décarbonatation à 800°C, recuit 1 semaine à1350°CCaCO3, recuit 72 h. à 1350°CCaCO3, recuit 1 semaine à 1450°CCaCO3 (chem. pure) et CaO pureté spéciale,décarbonatation de 72 h. à 1100°C, recuit de 850 à1650°CCaCO3 (chem. pure), décarbonatation de 72 h à1100°C, recuit 168 h. à 1350°C
MgO 1173 8.33±0.46 7 Mûller et Kleppa, 1973
965 3.0U0.12 7 Millier et Kleppa, 1973
1173 8.41±2.26 10 Shearer et Kleppa, 1973
970 4.94±0.33 9 Charlu et al., 1975
986 4.81±0.59 5 Navrotsky et Coons, 1976
1070 7.03±0.96 6 Kiseleva et al., 19791170 8.66±1.72 10 Kiseleva et al., 1979
Monocristaux de periclase, recuit 24 h à 1100°CMonocristaux de MgO (Norton Co.), recuit 24 h à1450°CMonocristaux de MgO (Norton Co.), recuit 24 h à1450°CMonocristaux de MgO (Muscle Shoals Electro-Chemical Corporation)MgO synt. (Baker Analysed Reagent), recuit 18 h. à1250°CMonocristaux synt. de periclase, recuit 5 h à 1000°CMonocristaux synt. de periclase, recuit 5 h à 1000°C
ZnO 9861173
965
18.45±1.0420.33±0.21
17.91±0.84
67
5
Davies et Navrotsky, 1981Mûller et Kleppa, 1973
Navrotsky, 1971
ZnO zinciteZn(NO3)2.4H2O (Merck), décarbonatation, recuit 12 hà1000°CZnO synt. (Mallinckrodt), recuit 24 h à 800°C
NiO 986 36.75±0.73 6 Davies et Navrotsky, 1981986 35.82±1.46 8 Navrotsky et Coons, 1976965 36.23±0.75 7 Mûller et Kleppa, 1973
1173 45.77±0.50 7 Mûller et Kleppa, 1973
965 36.23±0.75 5 Navrotsky, 1971
NiO synt, séché 24 h. à 973KNiO synt. (Clico), recuit 48 h. à 1000°CNi(NO3)2.6H2O (Merck), décarbonatation, recuit 24 hà1200°CNi(NO3)2.6H2O (Merck), décarbonatation, recuit 24 hà1200°CNiO synt. (Mallinckrodt), recuit 24 h à 700°C
MnO
CoO
CuO2
CuO
CdO
Cr2O3
GeO2
K2OZrO2
La2O3
986
986
965
1173
1173
1173
1173
965975977974
5.82±0.71
22.51±1.09
24.81 ±0.46
78.66±0.75
51.92±0.42
12.47±0.25
22.38±0.33
-19.66±0.67-260.98±1.20
15.05±0.52-126.0±4.4
5
10
4
3
6
6
5
6
67
Navrotsky et Coons, 1976
Navrotsky et Coons, 1976
Navrotsky, 1971
Mûller et Kleppa, 1973
Mûller et Kleppa, 1973
Mûller et Kleppa, 1973
Mûller et Kleppa, 1973
Navrotsky, 1971Kiseleva et al., 1996Ellison et Navrotsky, 1992Bolechetal., 1995
Mn2O3 (Baker Analysed Reagent), recuit 5 jours à800°C sous atmosphère tamponné à CO/CO2=3/1CoO synt.(Alfa Inorganics), recuit à l'air 48 h à800°C et réduit à 1000°C sous nitrogèneCo3O4 (Baker Analysed Reagent), décompositionsous N2 à 300-600°C, recuit 24 h. à 1050°Cu(NO3)2.6H2O (Merck), décarbonatation à 1000°C,recuit 12 h. à 1000°Cu(NO3)2.6H2O (Merck), décarbonatation, recuit 48h. à 900°CCd(NO3)2.4H2O (Merck), décarbonatation, recuit 14h. à 1000°CCr(NO3)2.4H2O (Merck), décarbonatation, recuit 12h. à 1000°CGeO2 synt. (Eagle-Picher), recuit 24 h. à 900cCK2CO3 (Aldrich), séché à 413 K, décarbonatationZrO2
La2O3
108

Les produits de départ sont mélangés, puis portés dans un creuset de
platine à la température de 1073 K durant une demi-heure. Le bain résultant est
trempé à l'air sur une plaque métallique.
Les différentes fractions sont broyées puis mélangées. Le tout est refondu
en plusieurs fois (portions d'environ 150 g) à 1323 K, le bain étant maintenu à
cette température durant une heure et demie. Finalement, la totalité du solvant est
rebroyée à une granulométrie inférieure à 50 \xm et mélangée pour produire une
seule quantité homogène.
Ce mode de synthèse a pour but de chasser au maximum l'eau résiduelle
contenue dans le solvant et de réduire la dispersion des mesures pour un produit
donné en disposant d'un stock unique de solvant.
6.5.4 Description du calorimètre
Pour les mesures calorimétriques, nous avons utilisé un microcalorimètre
Tian-Calvet haute température (Figure 6.9). L'appareil se compose de deux piles
thermoélectriques (4) insérées dans une masse isotherme de grande inertie
thermique : le bloc calorimétrique (6). Chaque thermopile est constituée par
l'empilement de couronnes d'alumine frittée autour desquelles s'enroulent les
couples Pt/Pt-Rh 13% (25 couronnes de 18 couples, reliés en série, soit 450
couples au total). Les deux piles sont électriquement reliées en opposition, suivant
en cela le principe des appareils différentiels, ce qui permet de minimiser toutes
les perturbations thermiques d'origine extérieure. Le signal obtenu aux bornes de
la série de thermocouples est amplifié (amplifiacteur Keithley 150B ou
nanovoltmètre EMDM) et enregistré par un potentiomètre (Sensitrace SEFRAM
type BE) et/ou un micro-ordianteur par l'intermédiaire d'un convertisseur
analogique/numérique (Keithley 175). Un jeu d'écrans en alumine isole et fixe le
bloc calorimétrique à l'intérieur d'un four (5) régulé électroniquement. La
température est mesurée par un thermocouple Pt/Pt-Rh 10%, situé au cœur du
bloc calorimétrique et relié à un thermomètre numérique (AOIP). Deux puits
verticaux donnent accès aux chambres calorimétriques dans lesquels on fait passer
109

deux tubes de silice ou "doigts de gant" (3) constituant le laboratoire d'étude. Au
fond de ces tubes, nous déposons un creuset de platine (8) de 0.2 mm d'épaisseur
qui contient le solvant. Par mesure de sécurité et afin de prévenir de toute fuite du
creuset, un tampon de fibres d'alumine (11) est inséré entre le creuset et le fond du
doigt de gant. En cas de fuite, le signal enregistré est augmenté du signal engendré
par la dissolution des fibres d'alumine ce qui nous permet d'être alerté et
d'intervenir sur le calorimètre avant que le solvant ne coule sur les parties
alumineuses du bloc calorimétrique.
Bien que la température de stabilisation du four (973 K) soit supérieure
aux températures de transition vitreuse des compositions étudiées et que les
valeurs obtenues concernent les phases liquides de nos compositions, pour un
grand nombre des produits, la viscosité reste importante. Les échantillons restants
dans leur "domaine de ramollissement", il nous a semblé préférable d'introduire
dans le calorimètre les échantillons sous forme de poudre fine de verre plutôt que
sous forme de fragments millimétriques de manière à augmenter le rapport surface
d'échantillon / volume de solvant et ainsi accroître la cinétique de dissolution. Des
temps d'aquisition important favorisant les risques d'erreurs, une taille de grains
de verre comprise entre 40 et 75 microns augmente la qualité des mesures.
Initialement, l'échantillon en poudre était déposé sur une nacelle en
platine suspendue à trois tiges de platine elles-mêmes reliées à une tige creuse en
alumine frittée. Les premiers résultats sur nos séries borosilicatés témoignant
d'une variation de chaleur très faible durant la dissolution, nous avons triplé la
nacelle (10) afin de multipier par trois la masse d'échantillon dissoute et par
conséquent le signal enregistré.
Il est de plus nécessaire d'agiter la solution afin d'accélérer la dissolution
et d'homogénéiser l'action du solvant sur tout le volume d'échantillon présent.
Suivant l'expérience antérieure acquise par le CTM (Brousse, 1981) nous avons
opté pour une agitation mécanique. Le dispositif (9) utilisé avait pour buts de
stabiliser thermiquement les échantillons au-dessus du bain, de les descendre dans
le bain pour les dissoudre, d'opérer une agitation verticale tout le temps de la
dissolution et, de revenir à l'état initial au-dessus du bain de manière la plus
110

reproductible possible. Le système d'agitation est situé à l'extérieur du
calorimètre. Il est constitué de deux parties. La première se compose d'un moteur
sur l'arbre duquel est fixée une came circulaire portant un roulement à billes
décentré. Sur ce dernier repose la clavette d'une pièce creuse en laiton servant de
guide à la canne porte-échantillon. La rotation de l'arbre du moteur engendre un
mouvement sinusoïdal aux tiges d'alumine qui agite le bain par translation
verticale. La seconde partie se compose d'un moteur, d'une came portant un ergot
décentré d'environ 20 mm sur lequel vient s'appuyer le taquet d'un tube
cylindrique solidaire de la seconde tige d'alumine. Elle permet de maintenir les
nacelles hors du solvant et de les y plonger. Les deux parties peuvent être rendues
solidaires par une pièce en laiton, mais nous verrons par la suite (§ 6.5.6) qu'elles
ne l'ont pas été au cours de nos expériences.
Figure 6.9 : Schéma général d'un calorimètre Tian-Calvet équipé pour la dissolution en selsfondus. (1) Armature métallique. (2) Réfractaire. (3) Doigt de gant. (4) Thermopile. (5) Four. (6)Bloc calorimétrique. (7) Entrée/sortie des thermocouples de mesure, de régulation et desthermopiles. (8) Creuset de platine. (9) Système d'agitation. (10) Nacelle porte-échantillon. (11)Fibres d'alumine.
111

Tout le sytème d'agitation et les cannes porte-échantillon reposent sur des
cales en toile d'alumine posées sur rextrimité des doigts de gant. La hauteur de ces
cales est choisie de manière à ce que les tubes en silice ne soient pas en contact
avec le fond des chambres calorimétriques, ceci afin que les vibrations des cannes
porte-échantillon (engendrées par les moteurs par exemple) ne se transmettent pas
aux thermopiles et n'augmentent pas le bruit du signal enregistré
6.5.5 Principe de la mesure et étalonnage
La grandeur enregistrée est l'écart de température entre les soudures
"internes" et "externes" des thermocouples engendré par la chaleur qui est prise ou
cédée au calorimètre lors de la réaction de dissolution. Les pertes de chaleur
restant assez faibles, on considère que la quantité enregistrée est proportionnelle
au flux thermique, dH/dt, c'est-à-dire à la quantité de chaleur par unité de temps
associée à la dissolution. L'effet calorifique global est alors proportionnel à l'aire
située sous le thermogramme enregistré.
Une fois le facteur d'étalonnage déterminé, l'effet thermique peut être
calculé. Les étalonnages sont réalisés par chute de billes de platine dans le bain de
métaborate de plomb au travers de la tige creuse porte-échantillon. Les capacités
calorifiques de cet étalon sont bien connues. Nous les avons obtenues de la
compilation de Robie et al. (1995).
L'effet endothermique produit dans la cellule calorimétrique par
réchauffement de la bille de platine, de la température ambiante jusqu'à la
température de stabilisation du four, correspond à l'intégration de la capacité
calorifique par rapport à la température entre l'ambiante et Ts = 973 K pondérée
par la masse de la bille de platine. Le facteur d'étalonnage, K, est alors le rapport
de l'enthalpie relative ainsi calculée et de la mesure de l'aire du thermogramme
enregistré. Au cours de nos dépouillements des enregistrements, les mesures
d'aires ont été effectuées à l'aide d'un planimètre OTT ou par des moyens
informatiques.
112
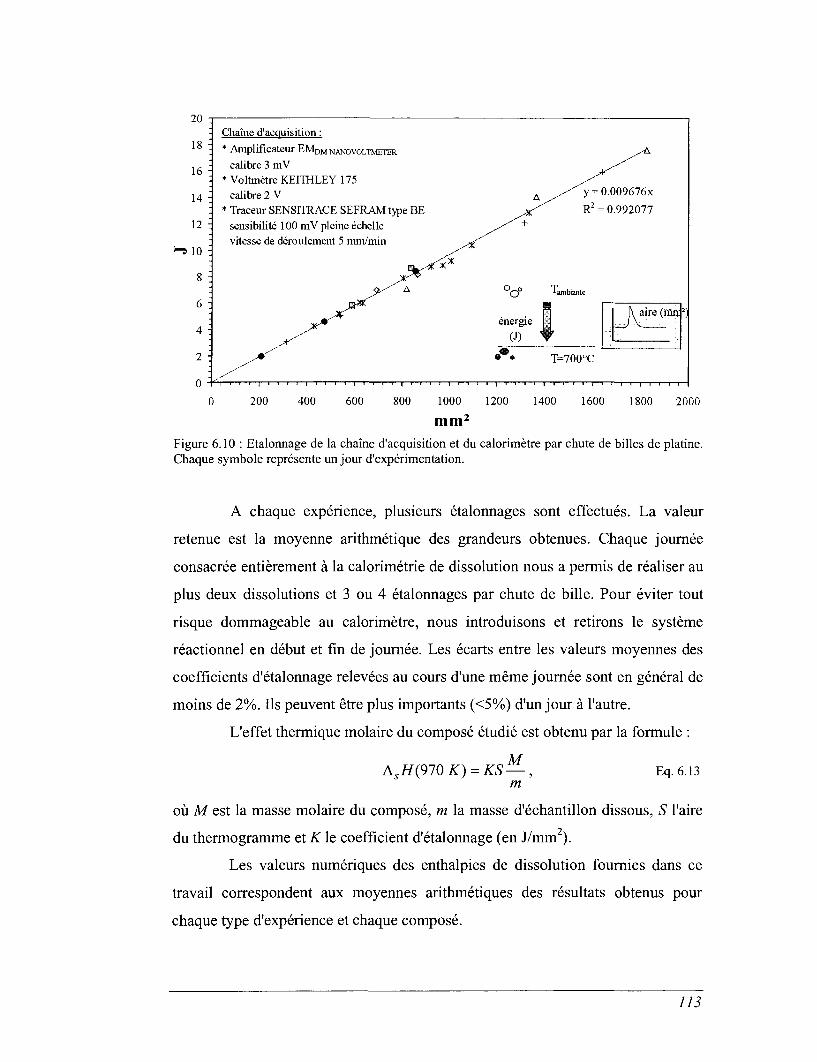
20
18
16
14
12
> 10 :
8 ;
6 :
4 :
2 :
0
Chaîne d'acquisition :
* Amplificateur EMDM NANOVOLTMETER
calibre 3 mV* Voltmètre KEITHLEY 175
calibre 2 V* Traceur SENSITRACE SEFRAM t>pe BE
sensibilité 100 mV pleine échellevitesse de déroulement 5 mm/min
a> • T=700°C
200 400 600 800 1000
mm21200 1400 1600 1800 2000
Figure 6.10 : Etalonnage de la chaîne d'acquisition et du calorimètre par chute de billes de platine.Chaque symbole représente un jour d'expérimentation.
A chaque expérience, plusieurs étalonnages sont effectués. La valeur
retenue est la moyenne arithmétique des grandeurs obtenues. Chaque journée
consacrée entièrement à la calorimétrie de dissolution nous a permis de réaliser au
plus deux dissolutions et 3 ou 4 étalonnages par chute de bille. Pour éviter tout
risque dommageable au calorimètre, nous introduisons et retirons le système
réactionnel en début et fin de journée. Les écarts entre les valeurs moyennes des
coefficients d'étalonnage relevées au cours d'une même journée sont en général de
moins de 2%. Ils peuvent être plus importants (<5%) d'un jour à l'autre.
L'effet thermique molaire du composé étudié est obtenu par la formule :
AsH(970m
Eq. 6.13
où M est la masse molaire du composé, m la masse d'échantillon dissous, S l'aire
du thermogramme et K le coefficient d'étalonnage (en J/mm2).
Les valeurs numériques des enthalpies de dissolution fournies dans ce
travail correspondent aux moyennes arithmétiques des résultats obtenus pour
chaque type d'expérience et chaque composé.
113

L'erreur sur la moyenne est calculée (sauf mention contraire) par la
formule suivante :
G(ASH) = \\^ — Eq. 6.14
6.5.6 Mode opé ratoire
Avant toute série d'expérience, un grand nombre de pièces composant le
calorimètre étant en alumine, le calorimètre doit être stabilisé à 973 K pendant
environ une semaine afin d'obtenir l'équilibre thermique.
On commence une expérience en chargeant la poudre de verre sur les
trois nacelles de la canne dite porte-échantillon. Il est impératif de connaître
précisément la quantité de produit dissoute et par conséquent la charge de chaque
expérience. Pour cela, les pesées sont faites avec une balance Mettler dant la
sensibilité est de 10~5 grammes. La canne porte-échantillon ne pouvant pas de
façon simple être pesée avec son chargement, la masse de soluté est obtenu par
différence de pesée du conteneur de poudre de verre avant et après chargement
des nacelles. L'ordre de grandeur des charges dans le dispositif à trois nacelles est
de 180 mg.
La pesée, la répartition de la poudre sur les nacelles et la descente de la
canne porte-échantillon dans le doigt de gant constituent la partie la plus délicate
de la manipilation car une perte de produit à cet instant entraînerait des erreurs
dans la quantité effectivement dissoute lors de la mesure.
La canne porte-échantillon et son homologue (non chargée) sont
descendues manuellement dans les doigts de gant jusqu'au contact avec la butée
supérieure de l'agitateur (Figure 6.11). Ce niveau que l'on appelera "niveau initial"
correspond à la profondeur nécessaire pour que les trois plateaux soient maintenus
au-dessus du bain, tandis que trois extrémités des tiges de platine qui les
supportent trempant dans le bain. Le fait que ces trois tiges soient en contact avec
le solvant permet de réduire le temps de mise en équilibre thermique du
114

calorimètre. Il faut environ une à deux heures pour atteindre cet équilibre qui se
traduit par une ligne de base stable sur l'enregistreur qui nous servira de référence
lors du dépouillement.
oFigure 6.11 : Schéma explicatif du réglage de l'agitateur et du positionnement des nacelles avant etpendant la dissolution. Etat 1 : les nacelles sont au "niveau initial". Etat 2 : Les deux éléments dusystème d'agitation sont désolidarisés, la canne porte-échantillon subit une agitation par translationverticale autour du "niveau de référence" et les nacelles de la canne témoin sont immobiles au"niveau de référence".
Une fois l'équilibre atteint, la dissolution est amorcée en plongeant les
nacelles dans le solvant à l'aide de l'un des moteurs de la partie haute. Une fois
que la nacelle intermédiaire est positionnée au milieu du bain ("niveau de
référence"), les deux parties de l'agitateur sont désolidarisées. En théorie, et
comme l'avait utilisé initialement Brousse (1981), les deux parties devraient dans
115

l'étape suivante restées solidaires pour reproduire dans les deux doigts de gants le
même phénomèmes oscillatoires et pour assurer un effet thermique similaire.
Cepandant,
comme la solidarité des deux parties ne peut être réalisée sans créer des
vibrations parasites,
comme le viellissement des appareils et leur hisoire expérimentale à créer une
légère différence de sensibilité des deux thermopiles,
et comme le gradient de température le long des axes verticaux n'est pas
parfaitement identique pour les deux piles,
des signaux moins bruités sont enregistrés lorsqu'on désolidarise ces deux parties
mécaniques.
L'agitateur est mis en marche et transmet à la canne porte-échantillon un
mouvement de translation verticale autour du niveau de référence. La dissolution
suit son cours pendant 30 à 60 minutes selon les échantillons que nous avons
étudiés. Un pic positif ou négatif correspondant à un phénomène endothermique
ou exothermique est enregistré, puis une courbe représentative d'une relaxation
thermique apparaît. Au terme de la dissolution, la courbe tend vers une
assymptote témoignant de l'équilibre thermique associé au "niveau de référence".
On peut alors revenir à l'état initial en arrêtant l'agitation, en resolidarisant les
deux parties de l'agitateur et en remontant les deux cannes par activation du
moteur de remontée. Après une heure d'enregistrement, on atteint la ligne de base
initiale correspondant à l'équilibre thermique associé au "niveau initial".
L'effet thermique enregistré est la somme de trois phénomènes : la
translation des plateaux du niveau initial au niveau de référence, l'agitation du
bain et la dissolution de l'échantillon. Afin d'extraire l'information correspondant à
la dissolution du produit, une expérience dite "à blanc" est effectuée après chaque
expérience. Les nacelles, sans charge, sont plongées à nouveau dans le bain
(niveau initial —> niveau de référence) et l'agitation mise en œuvre comme
précédemment. La différence entre les deux effets enregistrés correspond à la
dissolution du verre.
116

Une fois l'expérience terminée la canne porte-échantillon est remontée et
les nacelles sont chauffées afin d'éliminer la partie la plus importante du solvant
résiduel. Enfin les nacelles sont nettoyées par immersion dans une solution
d'acides (HNO3 ou HF) pendant quelques heures avant de pouvoir les réutiliser.
Intervalle d'intégration
Figure 6.12 : Exemple de thermogramme enregisté. (A) Signal de relaxation thermique vers un étatd'équilibre thermique (2) associé à la descente de la canne porte-échantillon jusqu'au niveauintial". (B) Signal d'une expérience chargée. Le signal est la somme des contributions desperturbations thermiques liées à la dissolution, au passage du niveau initial vers le niveau deréférence et l'agitation dans le bain. (C) Signal associé au passage de l'équilibre thermique (1) duniveau de référence vers l'équilibre thermique (2) du niveau initial. (D) Signal d'une expérience àblanc. Le signal correspond à celui des pertubations thermiques de l'agitation et du mouvement descannes du niveau de référence vers le niveau initial. L'aire délimitée par la superposition dessignaux de la dissolution et du blanc est égale à ASH(TS) divisée par le coefficient d'étalonnage K.
6.5.7Intégration des thermogrammes
Pour chaque expérience, deux intégrations sont effectuées: une pour la
dissolution et une autre pour le blanc. La différence entre les deux donne l'aire
proportionnelle à l'enthalpie de dissolution de la masse m de produit dissous
(Figure 6.12).
117

Les décalages entre les lignes de base en début (équilibre thermique
associé à l'état initial) et en fin d'expérience (équilibre thermique associé à l'état de
référence) sont importants ainsi que les différences des amplitudes de ces
décalages entre le blanc et la dissolution. Ces écarts sont dus à des différences de
gradient de température vertical pour les deux états (niveau initial, niveau de
référence) et donc à la différence de fuite thermique.
Lorsque les décalages sont identiques entre l'expérience de dissolution et
l'expérience à blanc, il suffit de soustraire les aires comprises entre la ligne de
base initiale et le signal enregistré sur un intervalle de temps A.t correspondant à
l'atteinte de l'équilibre thermique associé au niveau de référence pour les deux
expériences. Si l'aire résultante est positive la réaction est endothermique et, dans
le cas contraire, la réaction est exothermique pour le soluté étudié.
Lorsque les décalages diffèrent entre l'expérience à blanc et la
dissolution, une intégration rendant compte de la quantité de chaleur dégagée par
la dissolution n'est plus possible sans apporter certaines corrections au signal. La
différence de fuite thermique entre les deux expériences est généralement due à
une mauvaise reproductibilité du positionnement des nacelles entre les deux
expériences. La reproductibilité la meilleure est assurée par l'utilisation du
système mécanique de descente de la canne porte-échantillon. Cependant, il est
parfois arrivé que la reproductibilité voulue ne soit pas atteinte à cause de diverses
sources de frottements (roulement à billes défectueux, usures des pièces,...) entre
les pièces mécaniques. D'autres sources de décalage sont les pertubationss
électriques générales qui peuvent affecter le signal ou encore des perturbations
thermiques extérieures qui n'ont pas été totalement effacées par notre système
différentiel. Dans de tels cas de décalage de ligne de base, nous n'avons effectué
des corrections graphiques directes sur le thermogramme lors de l'intégration
uniquement que pour des deviations brèves et bien marquées des lignes de base.
Pour les autres cas les enregistrements n'ont pas été utilisés.
118

Chapitre 7
VISCOSIMETRIE
La viscosité est une propriété qui varie énormément avec la température
et la composition chimique. Pour observer la variation de viscosité en fonction de
la température, il est donc nécessaire de disposer de plusieurs appareillages.
7.1 DISPOSITIF DEFLUAGE
Les mesures de viscosité comprises entre 10 et 10 Pa.s sont effectuées
avec le dispositif de fluage décrit par Neuville et Richet (1991). Le principe de la
mesure consiste à mesurer la vitesse de déformation d'un échantillon soumis à une
contrainte donnée, à température constante.
7.1.1 Dispositif expérimental
Le viscosimètre est représenté sur la Figure 7.1. Cet appareil permet de
mesurer des viscosités dans une gamme de températures proches de la température
de transition vitreuse, c'est-à-dire entre 108 et 1014 Pa.s.
L'échantillon de verre est placé entre deux pistons (3), l'un fixe (piston
supérieur), l'autre mobile. L'échantillon s'écrase lorsqu'il est soumis à des
contraintes que l'on impose en plaçant des masses sur le plateau (7), la force
produite sur la surface inférieure de l'échantillon étant amplifiée par un bras de
levier. La déformation des éprouvettes est donc due à l'application d'une
contrainte uniaxiale verticale. Deux rondelles d'alumine (8) de 5 mm d'épaisseur
séparent l'échantillon des pistons. Une feuille de platine (9) de 50 microns
intercalée entre l'alumine et le verre évite les réactions chimiques à haute
température.
119
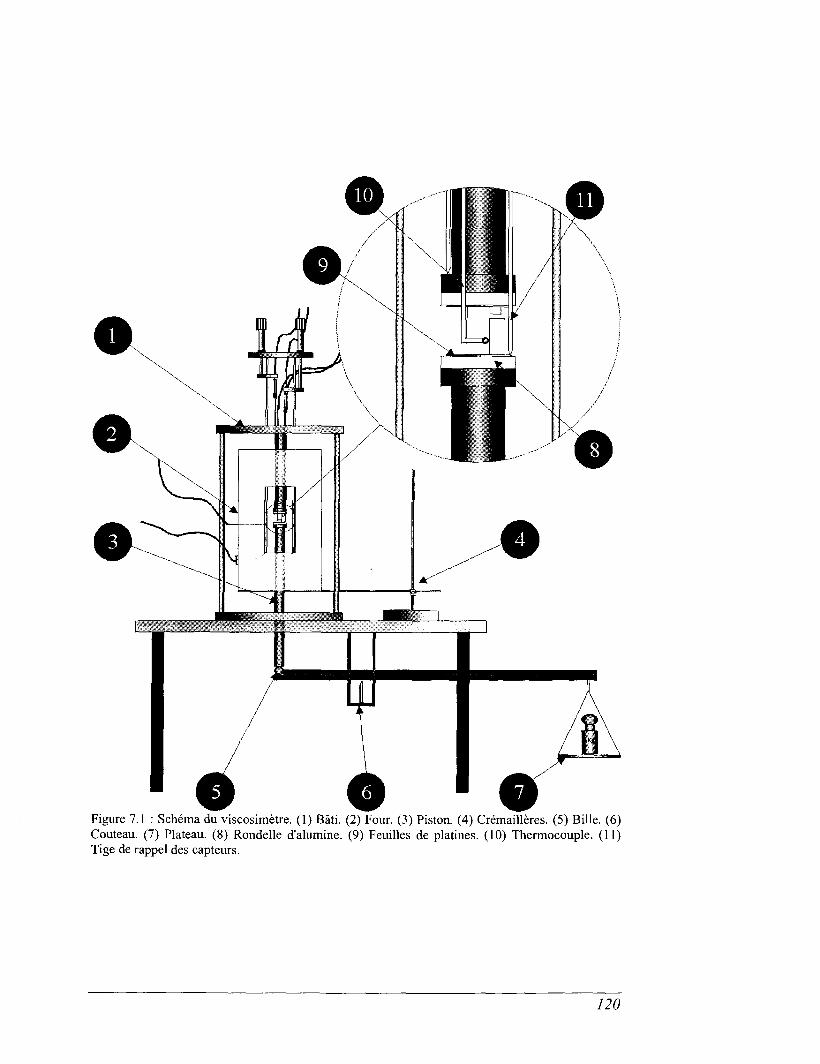
9, i^
Figure 7.1 : Schéma du viscosimètre. (1) Bâti. (2) Four. (3) Piston. (4) Crémaillères. (5) Bille. (6)Couteau. (7) Plateau. (8) Rondelle d'alumine. (9) Feuilles de platines. (10) Thermocouple. (11)Tige de rappel des capteurs.
120

Nous faisons l'hypothèse que les échantillons sont déformés à volume
constant. Ils sont au départ cylindriques ; si l'expérience est prolongée au-delà de
50 % d'écrasement, leur forme sera celle d'un barillet (Neuville, 1992), pour
lequel il faudrait appliquer des corrections de forme. Les mesures seront donc
restreintes à 30 ou 40 % de déformation. La mesure de la longueur de l'éprouvette
est donnée au micron près par deux capteurs linéaires TESATRONIC qui nous
donnent la position de deux tiges d'alumine (11), l'une fixée au piston supérieur
immobile, l'autre étant solidaire du piston inférieur mobile. On mesure, au
dixième de seconde près, le temps nécessaire pour que l'éprouvette se déforme de
5 ou 10 microns à l'aide d'un chronomètre ou de l'horloge interne d'un PC. A partir
de ces mesures il est possible de calculer la vitesse de déformation de
l'échantillon.
La régulation en température du four tubulaire (2) est assurée directement
sur la résistance de celui-ci par un régulateur Eurotherm 720, relié à un
thermocouple Pt/Pt-RhlO % branché sur un compensateur de soudure froide
Omega, qui permet de tenir compte des variations de température de la soudure
froide. Dans ces conditions, nous pouvons effectuer des mesures de déformation
durant plusieurs jours avec des variations de température du four inférieures au
degré. La température au niveau de l'échantillon est connue grâce aux deux
thermocouples Pt/Pt-RhlO % placés aux deux extrémités de l'éprouvette (10). Les
deux thermocouples sont reliés à un compensateur de soudure froide Omega
semblable à celui décrit précédement. Chacun d'entre eux nous donne une
différence de potentiel mesurée par un voltmètre scrutateur Keithley 1992 qui
permet de déterminer précisément la température.
Il existe un gradient de température dans le four. Ce gradient doit être
minimisé au niveau de l'échantillon pour ne pas déformer préferentiellement les
zones les plus chaudes. Un barreau d'argent mis en place autour de l'échantillon
permet de réduire le gradient de température, et celui-ci peut être encore amélioré
en modifiant la position du four: on peut avoir aisément un gradient inférieur à
0,1 °C le long de l'échantillon.
121

7.1.2 Préparation des échantillons
Les échantillons ont été carottés dans les lentilles de verre. Ils sont
ensuite sciés et polis sous eau à l'aide d'une polisseuse rotative de manière à ce
que leurs faces soient planes, parallèles entre elles et perpendiculaires aux
génératrices du cylindre : ces conditions assurent que la déformation est
homogène. On préfère en général faire des mesures de viscosité sur des
éprouvettes de longueur 10 mm, de diamètre 5 mm environ ; il est commode pour
les manipulations avec l'appareil de fluage utilisé que le rapport R=L/D soit
proche de 2. Ces dimensions sont mesurées au micron près avec un palmer
électronique.
7.1.3 Calculs
Dans le cas de l'écoulement isochore d'un fluide newtonien généré par
une contrainte uniaxiale, on a (Lévesque, 1991) :
a = 3r)è, Eq. 7.1
où a est la contrainte cisaillante, n la viscosité et s la vitesse de déformation du
fluide. A haute température, la viscosité des verres devient suffisament faible pour
qu'ils présentent des propriétés analogues à celles des fluides newtoniens. Notre
dispositif expérimental soumet l'échantillon à une contrainte uniaxiale; par
conséquent, la relation précédente nous permet de calculer la viscosité.
La longueur de l'échantillon est calculée à un instant t par L = Lo- dL, où
dL = TtdLj est le raccourcissement depuis le début de la manipulation. L'hypothèse
du volume constant permet de calculer la surface au même instant par :
Eq.7.2L0-dL
La masse réellement appliquée à l'échantillon est connue par la relation
suivante, déterminée par étalonnage du bras de levier,
M = 369.04 + 4.3142m -ma, Eq. 7.3
122

où m est la masse posée sur le plateau de la balance et ma la masse des rondelles
d'alumine (15 g). La masse de l'échantillon vaut approximativement 0,5 g: elle est
négligée puisque les charges appliquées sont supérieures à quelques centaines de
grammes.
Connaissant la masse et la surface, il est possible de calculer la contrainte
que subit l'échantillon:
a - 9 . 8 1 ^ .S
La vitesse de déformation est calculée par :
Eq. 7.4
LdtEq. 7.5
Connaissant cret s on a alors accès à la viscosité par la relation 7.1
14
13
CL»IK
6X
^T 11©
Laboratoire des GéomatériauxIPGP
National Bureauof Standards
• • i
760 780 800 820 840
T(K)860 880 900
Figure 7.2 : Viscosité du verre NBS717. Les ronds correspondent aux valeurs mesurées dans lelaboratoire des Géomatériaux. Les traits continus représentent la fonction donnée par le NationalBureau of Standards.
123

7.1.4 Qualité des mesures
Afin de vérifier la précision de nos mesures de viscosité, un borosilicate
du "National Bureau of Standard", le NBS717, a été étudié dans les mêmes
conditions que nos autres échantillons. Les valeurs mesurées avec la machine de
fluage sont comparées avec celles données par le Bureau des Standards. Dans la
Figure 7.2, nous avons représenté le logarithme décimal de la viscosité en
fonction de l'inverse de la température. Les deux courbes correspondent aux
lissages des valeurs extrêmes données par les équations du Bureau des Standards.
La méthode utilisée pour les obtenir n'a pas été décrite. Les barres d'erreur sur nos
valeurs sont de la dimension des symboles utilisés. On remarque que les valeurs
sont parfaitement encadrées par les valeurs données par le Bureau des Standards.
La reproductibilité est de 0.02 log Pa. s et l'imprécision du même ordre de
grandeur. Ces résultats nous permettent de dire que nous mesurons de façon
absolue les viscosités entre 108 et 1014 Pa.s.
7.2 VISCOSIMETREROTATIF
Contrairement aux mesures de hautes viscosités pour lesquelles il existe
plusieurs méthodes reposant sur des principes différents, on utilise toujours, pour
des faibles viscosités, la méthode de Couette (1988; 1890). Contrairement au
dispositif décrit précédemment, la méthode dite de Couette permet de déterminer
la viscosité dynamique d'un liquide. En effet, nous soumettons un liquide à un
mouvement de rotation qui le cisaille, et nous regardons comment il se comporte.
Le viscosimètre a été décrit par Neuville (1992).
7.2.1 Dispositif expérimental
La mesure de viscosité est effectuée avec un appareil du commerce, le
Rhéomat 115, fabriqué par METTLER et commercialisé par LAMY à Caluire.
Cet appareil permet d'imposer la vitesse de rotation à un plongeur que l'on
124

immerge dans un creuset maintenu fixe qui contient le silicate fondu. Il donne
directement le pourcentage correspondant au couple appliqué. Notre dispositif
peut atteindre des températures de 1700°C en continu et 1750°C
occasionnellement. Dans la Figure 7.3, nous présentons un schéma du
viscosimètre.
Le four
II s'agit d'un four hexagonal dont la coque extérieure est en
duraluminium, et l'intérieur composé de quatre couches de réfractaires fabriqués
par les sociétés KAPYROK et ZIRCAR. Ces réfractaires sont des composés
d'alumine (80 %) et de silice (20 %). Leur résistance à la température vient du fait
que plus ils sont compacts, moins ils résistent à de fortes températures. Les
réfractaires suivants sont utilisés, en partant de la périphérie vers le centre du four
: du KAPYROK 700, du KAPYROK 1200, du KAPYROK 1600 et du Sali
(ZIRCAR). Les températures d'utilisation en continu des trois premières couches
sont respectivement 700°, 1200° et 1600° C. Pour le Sali, elle est de 1700° C.
Dans la partie centrale du four, est disposé un tube laboratoire (2) en
alumine de diamètres extérieur 60 mm et intérieur 50 mm, sa hauteur étant de
1100 mm. Ce tube laboratoire est refroidi à ces deux extrémités par deux
refroidisseurs à eau, le refroidisseur bas étant solidaire du bâti du four. Notre
appareillage est également muni d'un dispositif assurant l'etanchéité à l'air du four
ce qui permet la possibilité de fixer différentes atmosphères.
La régulation
Le système de chauffage est composé de six épingles de super Kanthal
33, de puissance 486 W sous une tension de 11 V, soit une intensité de 45 A. Le
super Kanthal a pour avantage de fonctionner à l'air et de permettre une montée en
température rapide. La régulation est effectuée avec un régulateur 818 S de chez
Eurotherm, qui nous fournit également le bloc de puissance de type 460. Le
montage est relativement simple : le régulateur pilote le bloc de puissance en
fonction de la f.e.m. qu'il reçoit d'un thermocouple de régulation, en PtRh6%-
PtRh30%, placé à 2 cm d'une des épingles de super Khantal. Le bloc de puissance
125

délivre en sortie une tension de 220 V qu'un transformateur (DYNATRA) 220-67
de 12000 Gauss, va baisser à 67 V. Avec cette tension, nous alimentons en série
les épingles.
La montée en température s'effectue de 20°C jusqu'à la première
température voulue pour l'expérience (> 800° C) avec une vitesse de l'ordre de
l°C/min afin de minimiser les chocs thermiques que pourraient subir les tubes
d'alumine. Il faut alors compter une heure pour avoir une température stable.
A 1350°C, le gradient de température dans le four est de ±2° sur 5 cm, le
tube laboratoire étant bouché par une rondelle d'alumine qui sert d'écran et le
tube-porte échantillon (3) étant plein de Sali. Nous respecterons toujours par la
suite ces conditions opératoires de façon à avoir un bon gradient de température.
La tête du viscosimètre
Notre viscosimètre (1) possède une barre de torsion de 50 N/m. Cet
appareil a été choisi car contrairement à d'autres, il permet à lui seul de couvrir la
gamme de 1 à 106 poises. Le Rhéomat possède 30 vitesses comprises entre 0.05 et
780 tr/mn. Le viscosimètre est fixé sur une crémaillère au-dessus du four, ce qui
nous permet de descendre et de remonter le plongeur, relié au Rhéomat par
l'intermédiaire d'une tige en platine irridié 30 %, dans le liquide en fusion.
Le creuset et le plongeur
Le creuset (7) et le plongeur (6) ont été fabriqués par la Compagnie des
Métaux Précieux. Ils sont respectivement en platine irridié 10 % et en platine
irridié 20 %. Ces alliages nous donne une bonne résistance mécanique en
température. Le plongeur est porté par une tige en platine irridié 25 % de 3 mm de
diamètre ce qui lui confère une bonne résistance en torsion.
126
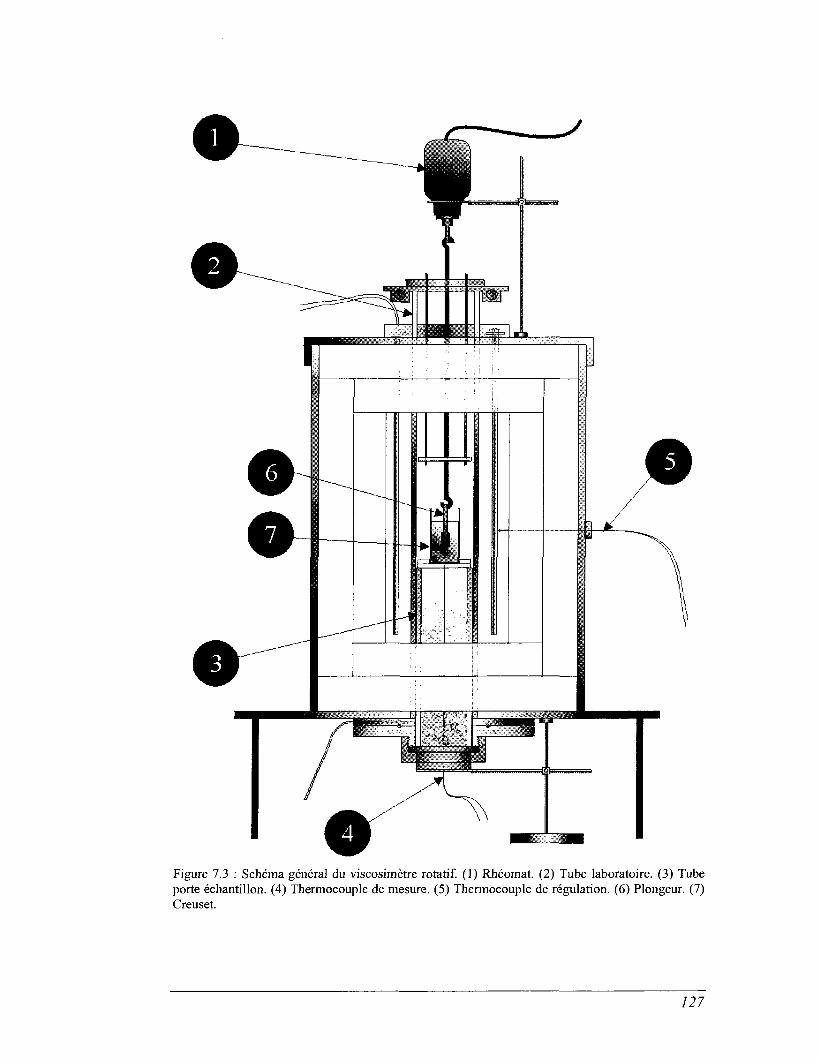
Figure 7.3 : Schéma général du viscosimètre rotatif. (1) Rhéomat. (2) Tube laboratoire. (3) Tubeporte échantillon. (4) Thermocouple de mesure. (5) Thermocouple de régulation. (6) Plongeur. (7)Creuset.
127

'.2.2 Réponse aux conditions de Couette
Avec ce dispositif, il est particulièrement simple de faire des mesures de
viscosité. Une fois que la température est stabilisée, il suffit de fixer la vitesse de
rotation du plongeur et de lire le couple sur l'afficheur du Rhéomat. Il existe
toutefois une difficulté qui résulte des conditions d'immersion du plongeur et de
son centrage. Le dispositif a été conçu pour que le plongeur soit centré. Celui-ci
est relié au Rhéomat par la tige en platine irridié, et par un système de deux
cardans, le premier reliant le plongeur à la tige, le second la tige au viscosimètre.
Le viscosimètre est fixé sur une crémaillère, fixée sur le bâti. L'ensemble bâti
(tube laboratoire), crémaillère et viscosimètre est aligné. Il n'y a qu'un seul
mouvement possible : un déplacement vertical de la crémaillère, donc du réglage
de la profondeur d'immersion du plongeur. Nous avons donc satisfait les deux
premières conditions de Couette. Le creuset est centré dans le tube laboratoire
donc il l'est aussi avec le plongeur (troisième condition satisfaite).
Nous avons cependant essayé volontairement de décentrer le plongeur
par rapport au creuset. On constate dans ces conditions, à vitesse de rotation
constante, une variation anormale et non reproductible de la valeur affichée sur le
Rhéomat. Ce phénomène résulte d'une mauvaise répartition des frottements dans
le cylindre comme l'a montré Couette (1890). Il est possible, si cette exentricité
est faible, de modifier le calcul de viscosité pour en tenir compte (Couette 1890).
Dans la suite, nous pourrons donc attribuer une variation anormale de la valeur de
l'affichage, à température et à vitesse constante pour un liquide newtonien, à une
exentricité du plongeur.
Le creuset fait 50 mm de haut. Nous le remplissons jusqu'à 40 mm avec
le produit à étudier, ce qui représente un volume de 22.9 cm3. Dans ces conditions
d'immersion, le bas du plongeur est à 10 mm du fond du creuset. Nous avons
constaté qu'une variation de hauteur de quelques millimètres n'entraîne pas de
variation de viscosité. Il faudrait se placer dans les conditions limites (plongeur
128

trop près du fond du creuset ou de la surface du liquide) pour voir un effet sur la
viscosité.
7.2.3 Etalonnage
Avec le dispositif de Couette, nous n'effectuons pas de mesure absolue de
viscosité mais une mesure relative. Il faut donc déterminer une constante
d'étalonnage.
Le Rhéomat donne une valeur, qui correspond à un pourcentage de
couple. Il faut multiplier cette valeur par une constante, k, qui est fonction de la
vitesse utilisée, pour connaître la viscosité. Des liquides étalons du "National
Bureau of Standards", le NBS710a et le NBS717 ainsi que le disilicate de sodium
,dont les viscosités ont été mesurées un grand nombre de fois et par des
expérimentateurs différents, ont été utilisés afin d'obtenir ce jeu de coefficients
(Neuville, 1992).
5
£ 4W)O
té
3 -
S 2C/3
2o Na2Si2O5
\
\ -
^ ^ . . NBS717
NBS71Oa ""*-%.
• Etalonnage (Ce travail)
* Etalonnage (Neuville, 1992)
• Etalonnage (Neuville, 1992)
a Etalonnage (Neuville, 1992)
o Fontana and Plummer, 1979
a Meiling and Uhlmann, 1967
» Bockris et al, 1955
Courbe du NBS (Napolitanoand Hawkins, 1964)Courbe du NBS (Napolitanoand Hawkins, 1971)
700 900 1100 1300 1500 1700 1900
T(K)Figure 7.4 : Viscosités des liquides étalons déterminés à l'aide du jeu de coefficient du Tableau 7.1comparées aux données de la littérature.
129
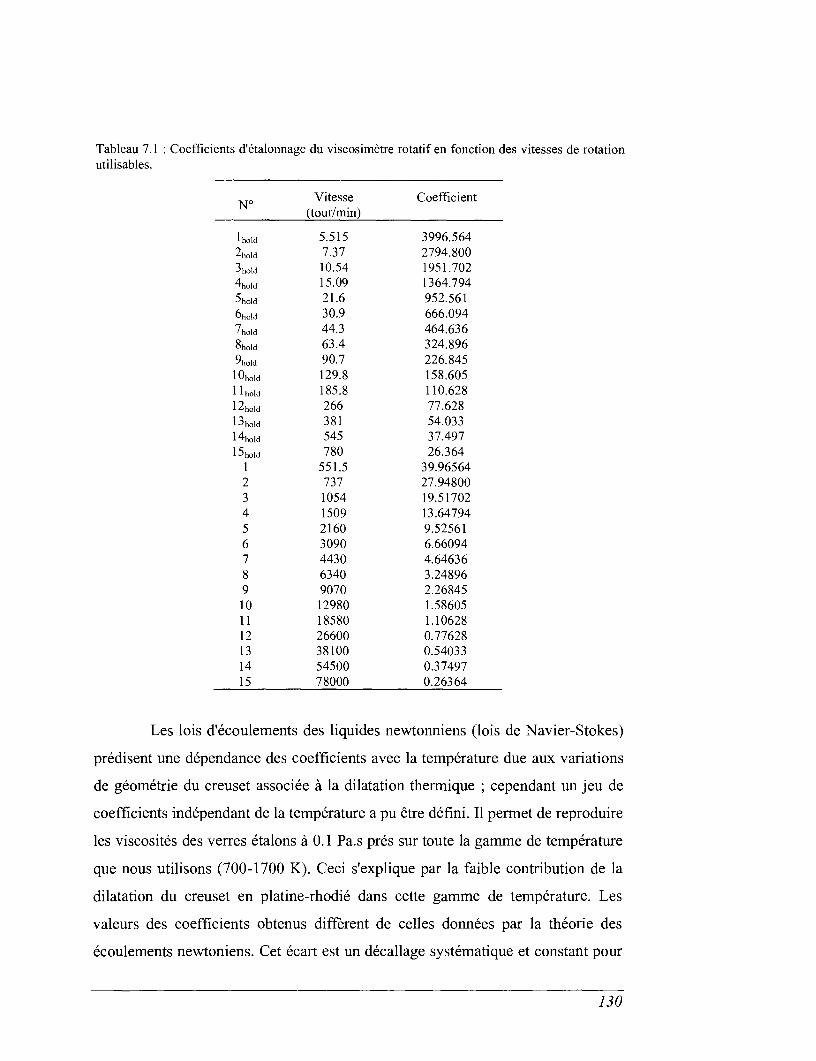
Tableau 7.1 : Coefficients d'étalonnage du viscosimètre rotatif en fonction des vitesses de rotationutilisables.
JN
lhold
2hold
3 hold
4hold
5hold
6hold
7hold
Shold
9hold
1 Ohold
1 lhold
12hold
13hold
14hold
15 hold
123456789101112131415
Vitesse(tour/min)
5.5157.3710.5415.0921.630.944.363.490.7129.8185.8266381545780
551.57371054150921603090443063409070129801858026600381005450078000
Coefficient
3996.5642794.8001951.7021364.794952.561666.094464.636324.896226.845158.605110.62877.62854.03337.49726.364
39.9656427.9480019.5170213.647949.525616.660944.646363.248962.268451.586051.106280.776280.540330.374970.26364
Les lois d'écoulements des liquides newtonniens (lois de Navier-Stokes)
prédisent une dépendance des coefficients avec la température due aux variations
de géométrie du creuset associée à la dilatation thermique ; cependant un jeu de
coefficients indépendant de la température a pu être défini. Il permet de reproduire
les viscosités des verres étalons à 0.1 Pa.s prés sur toute la gamme de température
que nous utilisons (700-1700 K). Ceci s'explique par la faible contribution de la
dilatation du creuset en platine-rhodié dans cette gamme de température. Les
valeurs des coefficients obtenus diffèrent de celles données par la théorie des
écoulements newtoniens. Cet écart est un décallage systématique et constant pour
130

chaque coefficient vers des valeurs plus faibles que nous ne pouvons imputer qu'à
des effets engendrés par l'état de surface de l'intérieur des creusets qu'il nous est
difficile de paramétrer pour en tenir compte dans les modèles théoriques.
Le Tableau 7.1 donne les coefficients que nous avons utilisés pour
chaque vitesse de rotation de la tête du Rhéomat. La Figure 7.4 représente les
viscosités des liquides étalons déterminées avec ce jeu de coefficients comparées
aux valeurs données par le "National Bureau of Standards" et la littérature.
131

TROISIEME PARTIE
Propriétés thermodynamiques

Chapitre 8
CAPACITE CALORIFIQUE
Nous allons décrire dans cette partie les propriétés thermiques des verres.
La capacité calorifique molaire à la température T est la quantité de chaleur
nécessaire pour élever la température d'une mole de matière de T à T+dT.
La contribution majeure aux changements de capacités calorifiques pour l'état
vitreux est vibrationnelle, elle provient de la mobilité des atomes autour de leur
position d'équilibre. Dans un liquide, une autre contribution, attribuée à la
distribution des positions des atomes, apparaît : la contribution configurationnelle.
Dans l'étude des verres borosilicatés à basse température, la contribution
exclusive aux changements d'entropie est vibrationnelle. En effet, bien que
l'entropie configurationnelle ne soit pas nulle, celle-ci est constante et égale à celle
du liquide qui a été figé à la transition vitreuse. Les positions atomiques dans le
verre, solide amorphe, sont déterminées par des minima locaux du potentiel
interatomique séparés par des barrières de potentiel de hauteur et de forme
variable selon les angles et les distances de liaison entre atomes. Lorsqu'on
augmente la température pour se rapprocher de la zone de transition vitreuse,
l'énergie vibrationnelle devient suffisante pour que les atomes franchissent la
barrière de potentiel séparant deux états configurationnels différents. Sur une
courte échelle de temps, seule la partie vibrationnelle de la capacité calorifique
sera alors mesurée mais, si l'échelle de temps devient suffisamment grande pour
laisser place aux changements configurationnels, la capacité calorifique montre un
excès par rapport à la valeur vibrationnelle : c'est la contribution
configurationnelle qui représente la chaleur utilisée pour accroître le potentiel et
non l'énergie vibrationnelle.
Au-delà de Tg, pour les phases liquides, les variations de capacités
calorifiques avec la température sont dues aux changements des vibrations et des
135

états confïgurationnels, la contribution vibrationnelle du liquide étant continue
avec celle du verre.
8.1 CAPACITE CALORIFIQUE, VIBRATION ET STRUCTURE
Les paragraphes suivants décriront quelques aspects théoriques de
l'approche vibrationnelle des capacités calorifiques. La contribution
configurationnelle sera illustrée lors de la discussion des résultats.
8.1.1 Théorie des capacités calorifiques vibrationnelles
Dans un solide (cristallin ou amorphe), les atomes sont attirés entre eux
par les forces de cohésions. Si l'on déplace un atome, il est soumis à une force de
rappel qui, en première approximation, est proportionnelle à l'écart à la position
d'équilibre : c'est l'analogue de la masse suspendue à un ressort.
L'atome vibre dans l'espace : son mouvement peut être considéré comme
la résultante de trois oscillations suivant trois axes perpendiculaires. D'où le
modèle d'Einstein : les mouvements des NA atomes d'une mole de solide sont
schématisés par les mouvements de 3 NA oscillateurs identiques, de même
fréquence, non couplés les uns aux autres et dont l'énergie est quantifiée. Ce sont
ces oscillateurs qui constituent le réservoir d'énergie emmagasinant la chaleur
fournie au solide : quand on chauffe celui-ci, les amplitudes des oscillateurs
augmentent ; elles diminuent quand on le refroidit.
Le modèle d'Einstein soulève de graves objections, pas uniquement à
cause des divergences, somme toute assez mineures, avec l'expérience, mais
surtout parce qu'il repose sur une hypothèse certainement erronée : l'indépendance
des mouvements des atomes voisins. C'est pour tenir compte des interactions entre
les mouvements des atomes dans le solide qu'a été proposé par Debye un autre
modèle de d'agitation thermique dans les solides.
Les interactions dans le solide propagent les vibrations locales selon une
onde progressive. Les modes de vibration du solide forment une suite discrète et
136

finie. La prise en compte des interactions entre atomes conduit à analyser
l'agitation thermique à partir des modes propres de vibration du solide intéressant
tous les atomes de ce solide : les différents modes sont indépendants et tous les
modes se superposent pour décrire l'agitation des atomes dans un solide. Dans les
structures amorphes, l'absence de maille empêche de définir des modes propres de
vibration, mais cela n'implique pas l'inexistence de vibrations. Les vibrations du
verre sont donc interprétables en terme de "pseudo-mode propre de vibration".
A basse température, le comportement de la capacité calorifique prouve
que l'énergie du mode de vibration du solide est quantifiée, comme l'est la
vibration de l'oscillateur d'Einstein. Pour calculer l'énergie d'un mode (ou pseudo-
mode), il faut calculer sa fréquence : c'est là un calcul très difficile qu'on doit
recommencer pour chaque solide et qui fait intervenir les paramètres définissant
les interactions atomiques. Ensuite, il faut faire la somme des contributions de
tous les modes. Pour simplifier, on a recours à une approximation proposée par
Debye.
On suppose que les modes sont répartis avec une symétrie sphérique,
c'est-à-dire que la fréquence d'un mode ne dépend que de la norme de son vecteur
d'onde. Pour les cristaux à haute symétrie, comme les cristaux cubiques,
l'approximation n'est pas mauvaise. Ensuite, on néglige la dispersion ; on admet
que, comme il est vrai pour les petits vecteurs d'onde, la fréquence est
proportionnelle au vecteur d'onde k : co = vk, le facteur v, homogène à une vitesse,
étant choisi intermédiaire entre les vitesses de vibrations longitudinale et
transversales. Pour une structure de volume V, le nombre de modes indépendants
dont la fréquence est comprise entre co et co+dco vaut donc :
( \J 3V 2Jg(COJaCù = r—r- Cù d(ù . Eq. 8.12TCV
La fonction g(co), appelée densité spectrale ou densité d'états vibrationnels, varie
comme co2 à basse fréquence. Debye a fait l'approximation suivante : cette loi en
co2 reste valable jusqu'à une fréquence maximale oc>£>, appelée fréquence de Debye,
calculée de telle façon que le nombre total de modes, c'est-à-dire l'intégrale de
g(co) sur l'intervalle [0, co ]̂ soit égale à 3 TV. On doit donc avoir la relation
137

I 7 Y/3
= v[6n nj , Eq. 8.2
qui relie COD à la vitesse du son, et où n = NA/V est le nombre d'atomes par unité de
volume. En utilisant alors CÙD, on écrit la densité des états, rapportée à la mole,
dans l'approximation de Debye sous la forme :
~ . Eq.8.3
On poursuit alors le calcul de la capacité calorifique en généralisant le calcul
d'Einstein. Dans un intervalle de fréquences compris entre CÙ et co+dco, il y a
g(co)dco modes, on obtient donc l'énergie totale en sommant l'énergie moyenne de
chaque mode sur toute la "bande" de fréquence depuis w = 0 jusqu'à <B = (ÙQ. En
posant x = h(ù I kgT (h est la constante de Planck divisée par 2% et ks la constante
de Boltzmann) et en dérivant l'énergie totale par rapport à la température, on
obtient la capacité calorifique :
avec R = Nks la constante des gaz parfaits (8.3184 J.K'Vmor1). Dans cette
formule, nous avons fait apparaître la température de Debye, ®D = h®D I ks.
Cette fonction a une forme "sigmoïde". On remarque que, lorsque T est
très grand, l'intégrale ne fait intervenir que les petites valeurs de x, ce qui permet
de remplacer ex/(ex-1)2 par \/x2 ; on trouve alors Cv = 3 R qui n'est autre que la loi
de Dulong et Petit. En revanche, lorsque T est au voisinnage de 0 K, on utilise
l'expression de l'intégrale :
yj-xdx = , Eq. 8.5
pour trouver :
Cv=—^M — \. Eq.8.6
Quand T tend vers 0, la capacité calorifique tend vers 0 comme r3, c'est-
à-dire plus lentement que suivant l'exponentielle donnée par le calcul d'Einstein.
138

La formule de Debye, qui interpole entre deux domaines (basses et
hautes températures) est une remarquable approximation pour la dépendance de la
capacité calorifique avec la température pour un grand nombre de substances. En
fait, le calcul entièrement rigoureux consisterait à connaître la forme exacte de la
densité g(co) des modes de vibration dans chacun des cas. Même s'il est vrai que
g(ra) se comporte toujours comme OÙ2 à fréquence suffisamment basse, son
comportement à des fréquences plus élevées dépend des subtilités de la
dynamique des vibrations des solides, spécifiques de chaque solide.
La grandeur thermodynamique mesurable par l'expérience est la capacité
calorifique à pression constante. Les capacités calorifiques à volume constant et
isobare sont dépendantes l'une de l'autre selon la relation :
Cp-Cv=a2VKTT, Eq. 8.7
où a représente le coefficient de dilatation thermique, V le volume molaire, KT le
module d'incompressibilité isotherme et T la température.
Pour les solides, on peut faire l'approximation suivante,
Cp ~ Cv, Eq. 8.8
car la dilatation thermique est extrêmement faible. Cette approximation a été
vérifiée par Richet et Bottinga (1980) pour les silicates vitreux.
8.1.2 Capacités calorifiques et structure des verres
La Figure 8.1 illustre la manière dont la capacité calorifique est sensible à
la densité d'état. Les différentes courbes représentent le produit entre g(a>) et la
fonction d'Einstein prise à des températures différentes. Les capacités calorifiques
sont données par les aires en dessous de chaque courbe. Les capacités calorifiques
ne peuvent informer sur la structure du verre que si la structure recherchée a une
signature vibrationnelle nette.
La densité d'état peut être scinder en différents domaines vibrationnels
attribuables aux modes de vibrations de différentes entités structurales du solide.
Il y a une association étroite entre la fonction des cations et les modes de
vibrations auxquels ils participent (Kieffer, 1979). Les formateurs de réseau sont
139
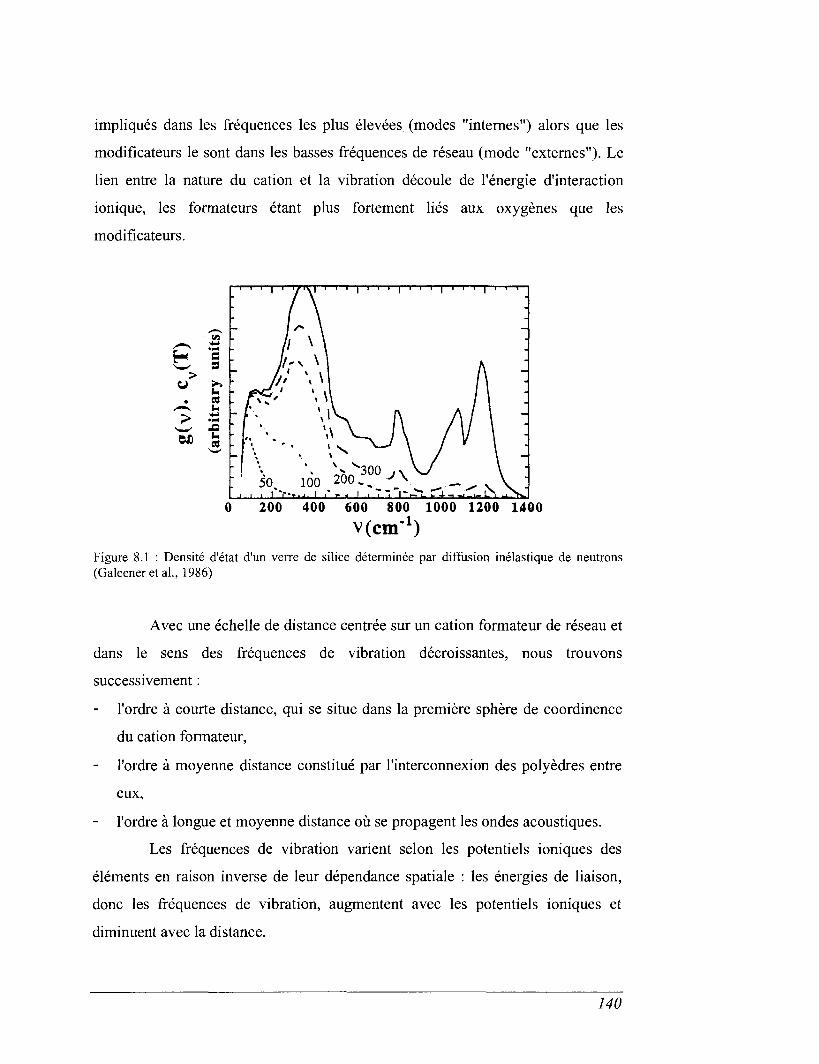
impliqués dans les fréquences les plus élevées (modes "internes") alors que les
modificateurs le sont dans les basses fréquences de réseau (mode "externes"). Le
lien entre la nature du cation et la vibration découle de l'énergie d'interaction
ionique, les formateurs étant plus fortement liés aux oxygènes que les
modificateurs.
e !«
s
: ' /
• J,'!
" • * - .
'. ' 50, , , i v-»
/ • • \ i '
'A
160. J 1 ,
• • 1 •
1
200^°
• 1 | • 1 1 | • 1 1 | . • 1
A A :J \ / \ '-
0 200 400 600 800 1000 1200 1400
Figure 8.1 : Densité d'état d'un verre de silice déterminée par diffusion inélastique de neutrons(Galeener et al., 1986)
Avec une échelle de distance centrée sur un cation formateur de réseau et
dans le sens des fréquences de vibration décroissantes, nous trouvons
successivement :
l'ordre à courte distance, qui se situe dans la première sphère de coordinence
du cation formateur,
l'ordre à moyenne distance constitué par l'interconnexion des polyèdres entre
eux,
l'ordre à longue et moyenne distance où se propagent les ondes acoustiques.
Les fréquences de vibration varient selon les potentiels ioniques des
éléments en raison inverse de leur dépendance spatiale : les énergies de liaison,
donc les fréquences de vibration, augmentent avec les potentiels ioniques et
diminuent avec la distance.
140
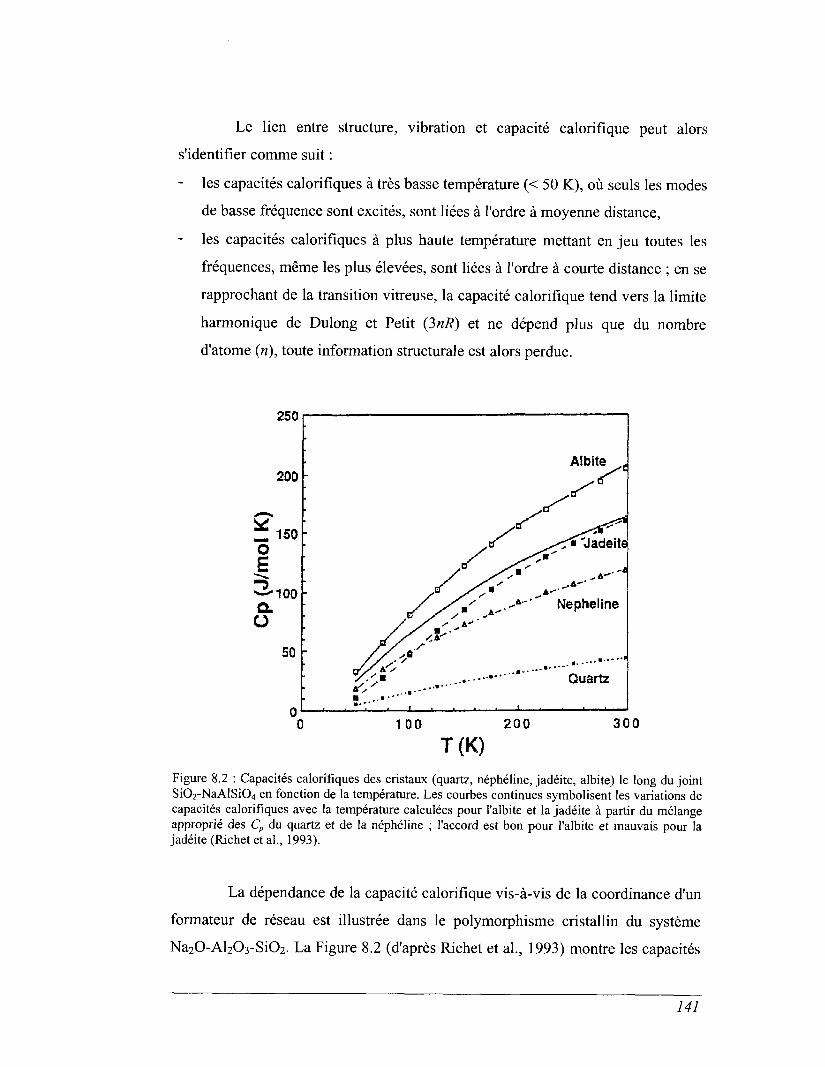
Le lien entre structure, vibration et capacité calorifique peut alors
s'identifier comme suit :
- les capacités calorifiques à très basse température (< 50 K), où seuls les modes
de basse fréquence sont excités, sont liées à l'ordre à moyenne distance,
- les capacités calorifiques à plus haute température mettant en jeu toutes les
fréquences, même les plus élevées, sont liées à l'ordre à courte distance ; en se
rapprochant de la transition vitreuse, la capacité calorifique tend vers la limite
harmonique de Dulong et Petit (3nR) et ne dépend plus que du nombre
d'atome («), toute information structurale est alors perdue.
250
200 •
I I 150O
ao50
;
/ / X*'
> . I .
Aibite\ t
^ A""" "
Néphéline
«Quartz
100 200 300
T(K)Figure 8.2 : Capacités calorifiques des cristaux (quartz, néphéline, jadéite, aibite) le long du jointSiO2-NaAlSiO4 en fonction de la température. Les courbes continues symbolisent les variations decapacités calorifiques avec la température calculées pour l'albite et la jadéite à partir du mélangeapproprié des Cp du quartz et de la néphéline ; l'accord est bon pour l'albite et mauvais pour lajadéite (Richet et al., 1993).
La dépendance de la capacité calorifique vis-à-vis de la coordinance d'un
formateur de réseau est illustrée dans le polymorphisme cristallin du système
Na2O-Al2O3-SiO2. La Figure 8.2 (d'après Richet et al., 1993) montre les capacités
141
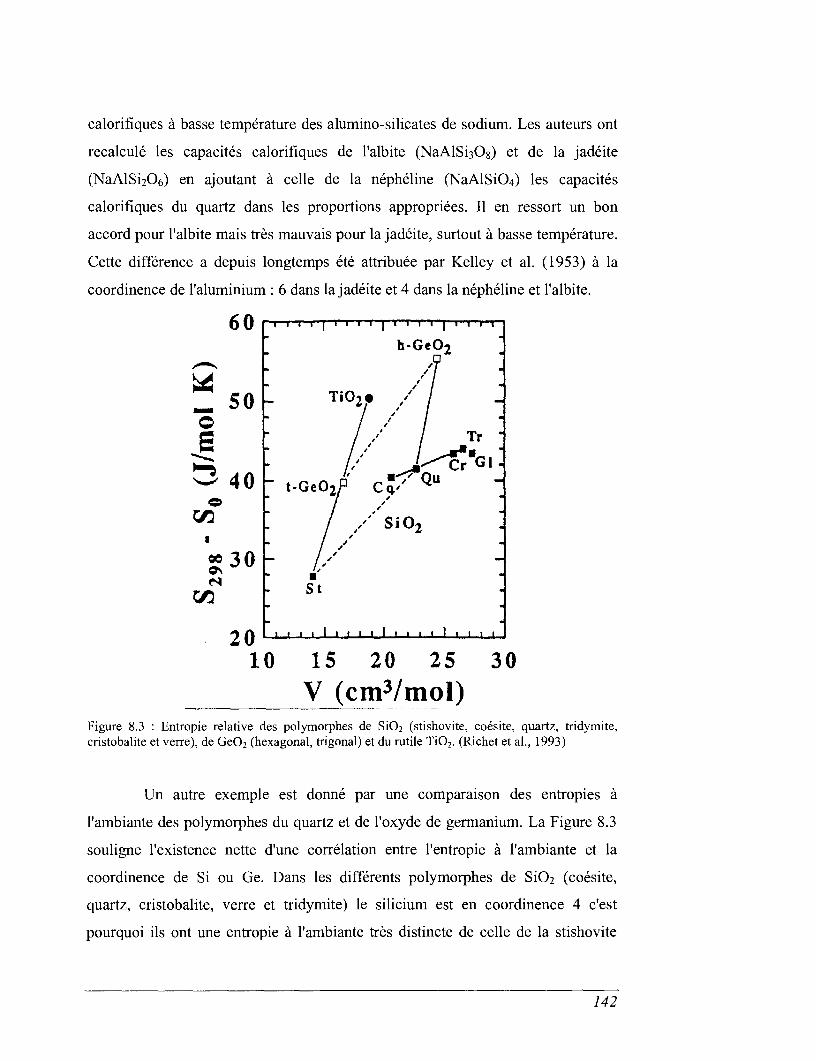
calorifiques à basse température des alumino-silicates de sodium. Les auteurs ont
recalculé les capacités calorifiques de l'albite (NaAISisOg) et de la jadéite
(NaAISiiOô) en ajoutant à celle de la néphéline (NaAlSiC^) les capacités
calorifiques du quartz dans les proportions appropriées. Il en ressort un bon
accord pour l'albite mais très mauvais pour la jadéite, surtout à basse température.
Cette différence a depuis longtemps été attribuée par Kelley et al. (1953) à la
coordinence de l'aluminium : 6 dans la jadéite et 4 dans la néphéline et l'albite.
6 0 i ' • • > i r • • ' i
^ A I i i i i I i i i i I i i i i I i
10 15 20 25 30V (cm3/mol)
Figure 8.3 : Entropie relative des polymorphes de SiC>2 (stishovite, coésite, quartz, tridymite,cristobalite et verre), de GeO2 (hexagonal, trigonal) et du rutile TiO2. (Richet et al., 1993)
Un autre exemple est donné par une comparaison des entropies à
l'ambiante des polymorphes du quartz et de l'oxyde de germanium. La Figure 8.3
souligne l'existence nette d'une corrélation entre l'entropie à l'ambiante et la
coordinence de Si ou Ge. Dans les différents polymorphes de SiÛ2 (coésite,
quartz, cristobalite, verre et tridymite) le silicium est en coordinence 4 c'est
pourquoi ils ont une entropie à l'ambiante très distincte de celle de la stishovite
142

dans laquelle le silicium a une coordinence 6. Le passage d'une coordinence 4 à
une coordinence 6 augmentant la compacité du site polyédrique, on observe une
augmentation de la fréquence des modes de réseaux. Cet effet implique une
importante diminution des capacités calorifiques à basse température.
L'idée de modéliser les propriétés thermiques en fonction de la
coordinence des oxydes constitutifs des matériaux n'est pas nouvelle. Robinson et
Haas (1983) proposent un modèle d'entropie et de capacité calorifique pour les
cristaux basé sur cette idée. Ils considèrent que les propriétés thermiques des
cristaux sont additives par rapport aux propriétés calculées pour des pôles fictifs
d'oxydes de coordinences variées.
8.2 CAPACITE CALORIFIQUE BASSE TEMPERATURE
Les mesures de capacité calorifique à basse température ont été
partiellement réalisées par le professeur T. Atake et son étudiant I. Yamashita du
Laboratoire des Matériaux et Structures de l'Institut de Technologie de Tokyo,
l'autre partie ayant été réalisée par nos soins lors d'un séjour de trois semaines
dans cet institut.
Les masses utilisées pour les mesures sur les verres SON-3, AVM-3,
BSN-5/16, BSN-11/32 et BSN-29/14 étaient respectivement de 15.020, 18.356,
13.925, 15.148 et 14.200 g. Typiquement, les verres contribuent de 60% à la
capacité calorifique mesurée totale à 300 K, 30% à 100 K, 10% à 50 K et 4% à
20 K.
Les capacités calorifiques molaires expérimentales sont données dans le
Tableau 8.1 et représentées dans la Figure 8.4. La dépendance des Cp vis-à-vis de
la température ne montre pas d'évidence de transition ou de discontinuité, elle est
conforme à la forme sigmoïde habituelle observée pour les solides.
Les valeurs de Cp lissées et les fonctions thermodynamiques relatives à
298.15 K sont reportées dans l'Annexe A. Les différences absolues entre les
valeurs expérimentales et les données lissées pour l'ensemble des compositions
143

étudiées par calorimétrie adiabatique n'excèdent pas 0.04 %. Puisque les verres
silicates ne présentent pas de changement configurationnel au-dessous de la
température ambiante, les résultats des Tableaux A.1-A.14 représentent les
entropies vibrationelles uniquement, sans contribution configurationelle.
La Figure 8.5 montre l'excès des capacités calorifiques mesurées par
rapport à la loi de Debye en J3. Cet excès est particulièrement marqué aux plus
basses températures mesurées. Ces déviations ne sont pas surprenantes car elles
sont habituellement observées pour les verres et parfois pour les minéraux
présentant un certain désordre (Pohl, 1981). L'origine de cette déviation est encore
mal comprise mais Leadbetter (1968), grâce à ces expériences de diffusion de
neutron sur un verre de silice, a pu corréler ce phénomène à un excès de densité
des modes vibrationnels de basse fréquence par rapport à la loi de Debye.
Tableau 8.1 : Capacités calorifiques mesurées par calorimétrie adiabatique.
T(K)
13.914.515.115.716.317.118.018.919.820.922.023.224.425.526.627.829.230.531.732.934.135.236.237.338.439.640.841.943.044.145.146.046.947.848.7
cP(J.mol'.K1)
SON-30.410.470.530.590.670.760.870.991.121.281.461.661.872.062.262.502.763.023.273.523.774.014.244.484.724.985.255.515.766.006.236.456.676.887.08
T(K)
14.315.216.116.917.919.020.121.322.523.825.326.728.129.330.531.532.633.734.936.237.538.940.441.843.344.846.548.249.851.553.455.557.358.860.3
cP(J.moï'.K1)
AVM-30.450.540.640.750.871.031.201.401.611.842.112.412.702.973.223.463.713.984.274.584.905.255.615.996.376.767.207.658.078.519.029.5610.0310.4310.82
T(K)
14.315.617.018.319.721.022.323.825.226.627.929.230.732.233.735.236.638.039.540.842.243.745.246.748.249.851.352.954.756.458.259.857.859.461.2
cP(J.mor'.K'1)
BSN-5/160.560.710.901.101.311.521.772.032.302.572.843.133.453.784.114.434.765.095.425.736.066.426.797.147.517.888.248.629.049.469.8710.279.7910.1810.59
T(K)
14.215.116.117.218.419.520.521.422.523.524.425.326.327.428.429.731.232.633.935.236.537.939.440.942.543.945.446.848.249.751.252.754.155.456.3
cP(J.mor'.lC1)
BSN-11/320.500.590.720.871.041.221.381.551.751.942.132.322.552.783.033.333.704.064.394.735.085.465.886.306.757.167.587.998.408.849.299.7210.1410.5310.81
T(K)
13.814.816.017.318.619.921.222.624.025.326.427.528.830.231.532.934.135.436.637.939.240.742.243.645.046.447.749.150.552.053.354.756.157.458.1
cp(J.mor'.K1)
BSN-29/140.470.580.730.911.091.291.511.762.032.282.502.743.013.313.633.934.224.514.805.105.435.796.166.506.847.187.517.858.208.558.899.229.569.8910.07
144

Tableau 8.1 (suite)
T(K)
49.650.551.552.754.055.657.258.960.662.464.265.867.469.170.872.674.476.278.180.081.681.883.483.785.285.587.088.890.592.394.095.897.699.3101.1102.9104.8106.6108.4110.1111.9113.6115.3116.9118.7120.6122.5124.4126.5128.6130.7132.8135.0137.2139.3141.4143.5145.6147.7149.7151.8153.9155.9157.9159.9161.9163.8165.8167.7169.7
cp(J.moï'.K1)
SON-37.287.497.738.008.328.689.079.469.8710.2910.7011.0811.4611.8412.2412.6613.0813.5013.9214.3614.7414.7915.1615.2115.5715.6315.9716.3716.7617.1517.5417.9318.3218.7019.0919.5019.8920.2720.6621.0421.4021.7522.1022.4522.8123.1923.5823.9924.3824.8125.2225.6326.0726.4726.8827.2727.6828.0628.4528.8329.2029.5729.9430.2930.6430.9831.3331.6831.9932.31
T(K)
61.763.264.666.167.568.870.171.472.874.275.677.078.379.781.081.282.382.783.884.385.385.987.689.591.593.595.797.899.9102.2104.7107.1109.4111.7114.0116.2118.4120.6122.7124.8126.9129.0131.0133.0135.0136.9138.9140.8142.7144.6146.4148.3150.1151.9153.7155.5157.3159.1161.1163.1165.3167.4169.5171.7173.9176.0178.1180.3182.4184.6
(J.mol'.K1)
AVM-311.1911.5611.9512.3212.6713.0213.3613.6814.0214.3814.7315.0815.4215.7416.0716.1416.4016.5016.7516.8817.1117.2817.6918.1418.6119.0919.5820.0720.5621.0821.6322.1722.6923.1923.6824.1624.6325.0825.5325.9926.3826.8027.2127.6028.0028.3928.7629.1429.4929.8630.2130.5630.9031.2331.5731.9032.2232.5532.8933.2733.6434.0134.3934.7735.1335.5035.8536.2136.5736.91
T(K)
63.064.967.069.070.972.974.776.778.780.682.684.586.488.490.492.394.496.598.6100.6102.6104.6106.6108.8110.9113.0115.1117.1119.1121.1123.1125.0127.2129.4131.6133.8136.1138.4140.6142.9145.1147.4149.7152.0154.2156.5158.7160.9163.2165.4167.7170.0172.3174.5176.8179.0181.2183.4185.6187.7189.9192.0194.2196.3198.4200.6202.8205.1207.3209.5
(J.mol'.K1)
BSN-5/1611.0211.4911.9712.4412.8913.3513.7914.2414.7015.1515.5916.0316.4716.9117.3517.8018.2618.7219.1719.6120.0420.4620.9121.3521.8022.2322.6623.0723.4723.8624.2624.6525.0625.5025.9326.3326.7727.2027.6128.0128.4228.8229.2429.6330.0330.4230.8031.1731.5631.9332.2932.6633.0333.4033.7534.0934.4434.7835.1035.4435.7536.0836.3936.7037.0137.3437.6537.9738.2738.59
T(K)
cP(J.mol'.K1)
BSN-11/3258.159.861.462.964.566.368.170.071.973.775.577.379.080.882.684.385.987.789.591.593.595.497.399.1101.0102.7104.5106.2107.9109.6111.5113.6115.6117.7119.6121.6123.6125.7127.8129.9132.0134.2136.5138.7140.9143.2145.4147.6149.8152.1154.3156.6158.8161.0163.2165.4167.7169.9172.1174.4176.6178.7180.9183.1185.2187.4189.5191.6193.7195.7
11.3211.8112.2812.7313.1913.6914.2214.7615.2915.8116.3016.7917.2717.7518.2218.6819.1119.5720.0620.5721.0721.5522.0322.4822.9323.3623.7824.2024.6024.9925.4425.9126.3726.8327.2627.6928.1528.5729.0229.4529.9030.3530.8031.2431.6732.1132.5432.9633.3833.7934.2134.6335.0235.4135.8136.2036.5836.9637.3437.7138.0838.4438.7939.1439.4939.8340.1640.4940.8241.15
T(K)
cP(J.mol'.K'')
BSN-29/1458.859.760.161.262.864.466.268.069.871.573.275.076.978.980.882.884.686.588.490.392.294.195.997.799.4101.2103.0104.8106.5108.2109.9111.5113.2114.8116.4118.1119.9121.7123.5125.2127.0128.7130.4132.2134.0135.9137.8139.7141.7143.6145.5147.3149.2151.0152.9154.7156.5158.2160.1161.9163.8165.7167.5169.4171.3173.1175.0176.9178.8180.7
10.2210.4510.5610.8311.2011.6012.0312.4612.8713.2713.6814.1114.5515.0115.4715.9016.3316.7517.1817.6218.0418.4518.8619.2519.6420.0320.4120.7821.1521.5221.8822.2222.5622.8923.2423.6023.9624.3324.7025.0425.3925.7326.0726.4326.7727.1427.5127.8828.2428.6128.9729.3229.6729.9930.3430.6731.0031.3231.6431.9732.3132.6532.9733.3133.6233.9534.2834.6034.9135.23
171.6 32.65 186.7 37.28 211.7 38.90 197.8 41.44 182.5 35.54
145

) — ON f̂ I> CO ON <-3 (
i
t~- r~- r -cooooooooooNONONONON
oooNONONONON
^ D O o r h t i o o û O o o c o û o i j N ^ ^ O v c ^
00
— \ h ; ̂ r s o r - ^ n —> —•^^ooo-^rni/2r^ooo(N^i"^Dt~^ON^-r*^i/~ir^odi—THTI S o\i-« rn </S r-- ON •—•'r*i"/Sr̂ ON
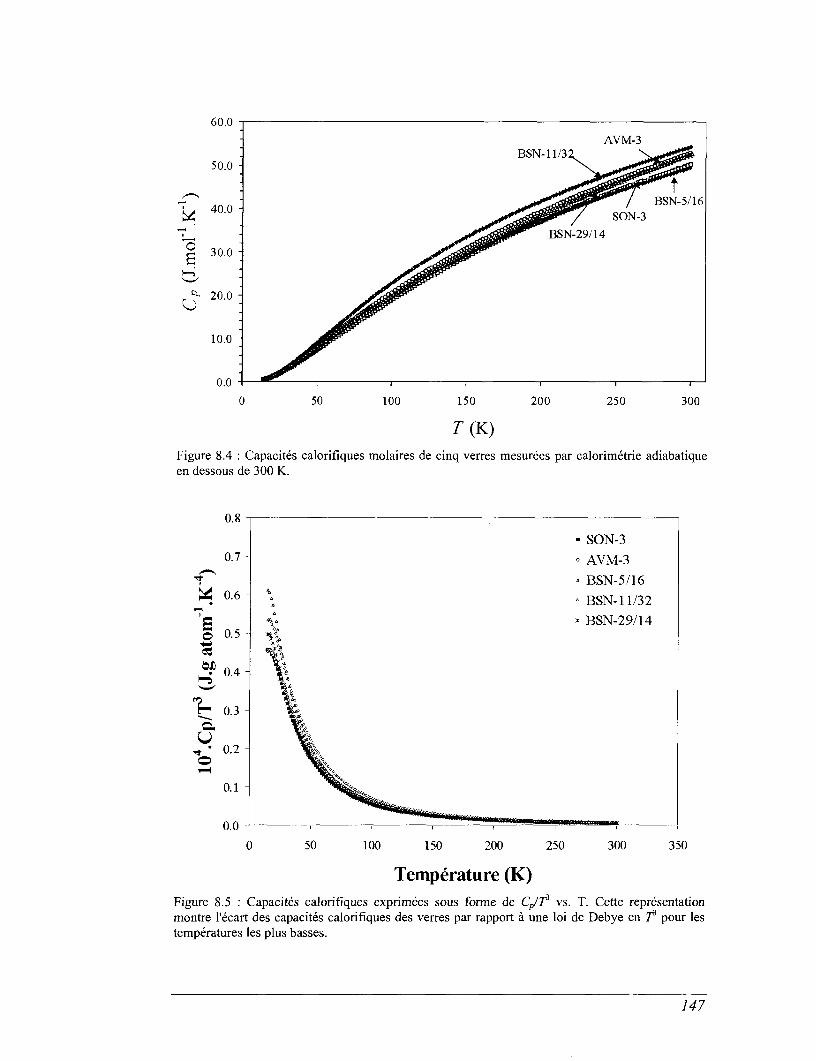
°-
60.0
50.0 -
40.0 -
30.0 -
20.0 -
10.0 -
0.0
AVM-3BSN-11/32
BSN-5/16SON-3
50 100 150 200 250 300
T(K)Figure 8.4 : Capacités calorifiques molaires de cinq verres mesurées par calorimétrie adiabatiqueen dessous de 300 K.
SON-3
AVM-3
BSN-5/16
BSN-11/32
BSN-29/14
50 100 150 200 250 300 350
Température (K)Figure 8.5 : Capacités calorifiques exprimées sous forme de Cplf1 vs. T. Cette représentationmontre l'écart des capacités calorifiques des verres par rapport à une loi de Debye en 7e pour lestempératures les plus basses.
147

On remarque également que ces écarts à la loi de Debye dépendent de la
composition. Les verres BSN-11/32, AVM-3 et BSN-29/14 qui ont des teneurs en
silice semblables mais des rapports R différents montrent des différences
sensibles, le verre s'écartant au moins de la loi de Debye étant le plus proche du
rapport R=l.
8.2.1 Capacité calorifique et entropie vibrationnelle
Pour la plupart des verres complexes, les propriétés thermodynamiques
sont en général estimées à partir d'une loi additive des propriétés de leurs
constituants vitreux les plus simples ou, à défaut de données, de leurs oxydes
constitutifs. Une autre approche, qui s'est révélée fructeuse pour modéliser un
grand nombre de propriétés, est de définir des propriétés partielles des oxydes ou
des entités structurales dans les verres. Celles-ci s'obtiennent en général à partir
des mesures des propriétés sur des systèmes simples (binaire, ternaire) en
considérant que la contribution de chaque entité considérée est proportionnelle à
sa concentration dans le système.
Les capacités calorifiques partielles du sodium à basse température
peuvent être déterminées par les mesures de Cp obtenues dans le système binaire
SiO2-Na2O (SiO2 : Yamashita et al, 2000 ; Na2Si205 et Na2Si307 : Westrum,
1988). Ces valeurs (Tableau 8.2) et les capacités calorifiques de la silice vitreuse
(Yamashita et al., 2000) et de l'oxyde de bore vitreux (de Ligny, 1996) ont été
utilisées pour tester la loi additive (Figure 8.6). Les écarts calculés entre nos
mesures et le modèle additif sont inférieurs à 12% entre 100 et 300 K, 12 à 37%
entre 30 et 100 K et 37 à plus de 100% en dessous de 30 K. Ces écarts nous
indiquent une non-additivité bien nette avec les constituants pris en compte.
D'autre part, les écarts semblent être corrélés à la composition des verres : les
verres BSN-5/16 (5 mol% de B2O3), BSN-11/32(11 mol%) et BSN-29/14(29
mol%) montrent des écarts respectifs à la loi additive de 8, 20 et 35 % à 30 K.
148
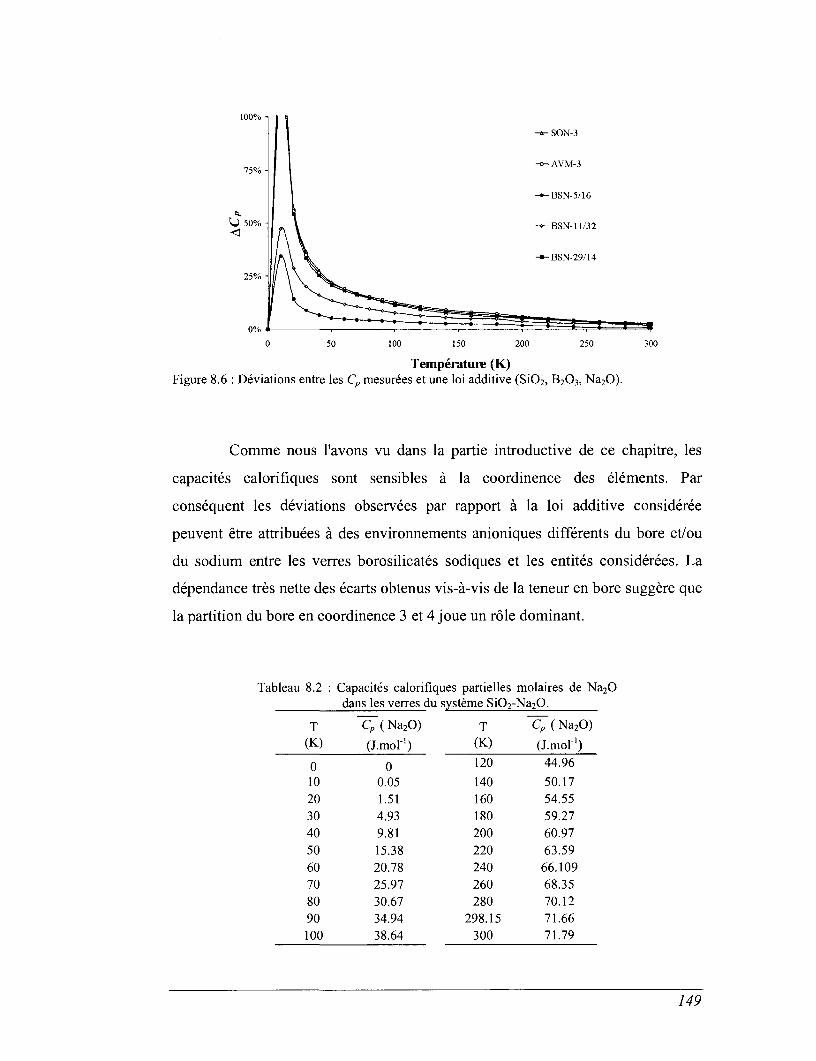
100% -,
0%
Température (K)Figure 8.6 : Déviations entre les Cp mesurées et une loi additive (SiO2, B2O3, Na2O).
Comme nous l'avons vu dans la partie introductive de ce chapitre, les
capacités calorifiques sont sensibles à la coordinence des éléments. Par
conséquent les déviations observées par rapport à la loi additive considérée
peuvent être attribuées à des environnements anioniques différents du bore et/ou
du sodium entre les verres borosilicatés sodiques et les entités considérées. La
dépendance très nette des écarts obtenus vis-à-vis de la teneur en bore suggère que
la partition du bore en coordinence 3 et 4 joue un rôle dominant.
Tableau 8.2 : Capacités calorifiques partielles molaires de Na2Odans les verres du système SiO2-Na2O.
T(K)
0102030405060708090100
C p(Na 20)
(J.mol1)
00.051.514.939.8115.3820.7825.9730.6734.9438.64
T(K)120140160180200220240260280
298.15300
~ÔT(Na2O)(J.mol1)44.9650.1754.5559.2760.9763.5966.10968.3570.1271.6671.79
149

A partir d'hypothèses structurales permettant la détermination des
rapports B/ B, nos mesures de Cp à basse température nous permettent de définir
les propriétés thermodynamiques molaires partielles du bore en coordinence 4 et
celles de Na2Û dans les systèmes borosilicatés (on suppose que les propriétés
partielles de B2O3 avec le bore en coordinence 3 sont égales à celles du verre B2O3
et celles de la silice aux propiétés de la silice vitreuse).
En utilisant le modèle structural de Dell et al. (1983), que nous avons
décrit dans le premier chapitre (§ 3.2.2), nous obtenons les fractions de bore en
coordinence 4 par rapport au nombre total d'atome de bore (notée JV4) que nous
avons dans nos verres. Ces valeurs sont tabulées ci-dessous :
Tableau 8.3 : Estimation de la fraction de boretétracoordonné d'après le modèle de Dell et al. (1983).
Verre
SON-3AVM-3BSN-5/16BSN-11/32BSN-29/14
73%67%100%67%50%
Afin de déterminer les propriétés partielles, nous avons ajouté à notre
base de données les mesures réalisées sur les verres de SiÛ2 (Yamashita et al,
2000), Na2Si205 (Westrum, 1988), Na2Si307 (Westrum, 1988), B2O3 (de Ligny,
1996) et Na2B4O7 (Grenier et Westrum, 1972). Le bore est considéré
complètement trigonal dans le verre B2O3 et réparti à 50% sur les deux
coordinences dans le tétraborate de sodium.
Les capacités calorifiques molaires parielles ( Cp ,) obtenues pour
et Na2Û sont données dans le Tableau 8.4. On remarque que, entre 50 et 300 K,
celles de Na2Û diffèrent à moins de 2% des valeurs obtenues en ne considérant
que le binaire Na2O-SiO2. La Figure 8.7 montre les déviations entre les valeurs
expérimentales lissées et le modèle additif des Cp „
150
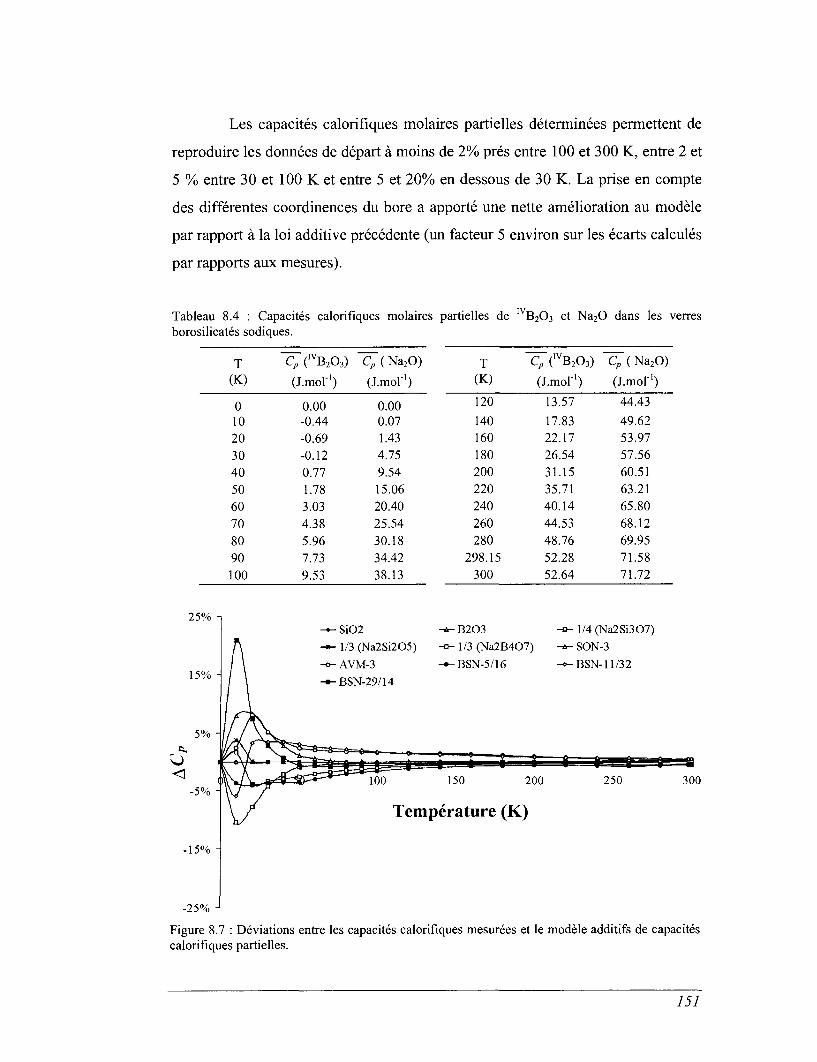
Les capacités calorifiques molaires partielles déterminées permettent de
reproduire les données de départ à moins de 2% prés entre 100 et 300 K, entre 2 et
5 % entre 30 et 100 K et entre 5 et 20% en dessous de 30 K. La prise en compte
des différentes coordinences du bore a apporté une nette amélioration au modèle
par rapport à la loi additive précédente (un facteur 5 environ sur les écarts calculés
par rapports aux mesures).
Tableau 8.4 : Capacités calorifiques molaires partielles de IVB2O3 et Na2O dans les verresborosilicatés sodiques.
T(K)
0102030405060708090100
2O3)
(J.mor1)
0.00-0.44-0.69-0.120.771.783.034.385.967.739.53
C,(Na2O)
(J.mor1)
0.000.071.434.759.5415.0620.4025.5430.1834.4238.13
T(K)
120140160180200220240260280
298.15300
Cp (IVB2O3)
(J.mol" ')
13.5717.8322.1726.5431.1535.7140.1444.5348.7652.2852.64
Cp ( Na2O)
(J.mor1)
44.4349.6253.9757.5660.5163.2165.8068.1269.9571.5871.72
25%
15% -
5% -
-5% -
-15% -
-25% J
•SiO2
• 1/3 (Na2Si2O5)
- AVM-3
•BSN-29/14
B2O3
1/3 (Na2B4O7)
BSN-5/16
•l/4(Na2Si3O7)
- SON-3
-BSN-11/32
200 250
Température (K)
300
Figure 8.7 : Déviations entre les capacités calorifiques mesurées et le modèle additifs de capacitéscalorifiques partielles.
151

Il y a plusieurs interprétations quant aux écarts résiduels. D'une part, les
limites de notre approche sont dictées par l'imprécision relative de l'interprétation
structurale de Dell (discuté dans le chapitre 4). Notons cependant que le bore en
coordinence 3 peut se répartir sur des sites symétriques ou asymétriques et ces
deux symétries pourraient avoir des signatures calorimétriques différentes. Selon
le modèle de Dell, seul le verre BSN-29/14 présente une petite quantité de BO3
asymétriques. Par conséquent, si cette interprétation structurale est correcte, cette
contribution n'est pas suffisante pour expliquer les écarts observés pour toutes les
compositions. D'autre part, les écarts, pour des températures inférieures à 30 K,
peuvent être corrélés aux déviations observées par rapports à la loi de Debye
(Figure 8.5). Cette corrélation ne nous semble pas fortuite : Richet et al. (1993)
ont montré que, entre 40 et 0 K, les capacités calorifiques étaient caractérisées par
une influence marquée de l'histoire thermique des verres et donc par une forte
sensibilité à l'ordre à moyenne distance qui est alors surperposé à la contribution
de l'ordre à courte distance. Par conséquent les imperfections du modèle en
dessous de 30 K reflètent des différences d'ordre à moyenne distance dans nos
verres. Ces différences se traduisent également par des signatures vibrationnelles
différentes (et donc par des écarts à la loi de Debye variables). Ces dépendances
complexes de la capacité calorifique des verres devraient cependant se situer assez
prés de 0 K pour que leurs contributions aux entropies à température ambiante
soient négligeables (Richet et al., 1993).
Nos calculs thermodynamiques nécessitent d'obtenir les entropies
relatives des verres entre 0 K et l'ambiante ; au-delà les variations d'entropie
vibrationnelle pourront être calculées avec les équations Cp obtenues à partir des
mesures de calorimétrie de chute et de DSC.
Les entropies vibrationnelles partielles, déterminées selon la même
méthode que les Cp partielles, sont respectivement 32.40 et 85.08 J.mol'.K"1 pourIVB2Û3 et Na2O. La valeur obtenue pour Na2Û dans les borosilicates est identique
à 1.4% à celle obtenue dans le binaire Na2O-SiO2 (86.26 J.mor'.K"1).
152

Richet et al. (1993) ont calculé les entropies partielles de AI2O3, CaO,
MgO et Li2Û à partir de données publiées pour le système SiO2-Al2O3-CaO-MgO
et le binaire SiO2-Li2O. Pour les compositions de ces systèmes, les capacités
calorifiques sont additives à mieux que 2 % au-dessus de 70 K.
La coordinance de l'aluminium (IV) dans les verres du système SiÛ2-
Al2O3-CaO-MgO étant similaire à celle des borosilicates étudiés (cf. chapitre 1 §
3.3), nous supposerons que l'ajout de bore dans ce système ne modifie pas de
manière importante la contribution de l'aluminium ni celle des unités BO3 et BO4
à la capacité calorifique et l'entropie des verres.
Contrairement à la présence de bore, la présence d'aluminium semble
modifier de façon significative l'entropie partielle des oxydes alcalins. Richet et
al. (1993) donnent des entropies partielles de 85 et 108 J.mor'.K"1 pour Na2Û et
K2O dans les systèmes binaires M2O-SiO2 et des valeurs de 96.7 et 119.1 J.mor
'.K"1 dans les systèmes aluminosilicatés (SiCVJV^AlC^). Ils interprètent ces
variations par des changements de coordinence des alcalins en présence
d'aluminium. Les variations d'entropies des alcalins Na2Û et K2O en présence
d'alumine étant respectivement de 11.7 et 11.1 J.mor1.K'1 nous prendrons pour
l'autre alcalin Li2Û une valeur supérieure de 11.4 J. J.mor'.K"1 à celle déterminée
pour le binaire SiO2-Li2Û soit 60.4 J. J.mor'.K"1.
Nous ne disposons pas de données calorimétriques pour déterminer
précisément les entropies partielles des oxydes de zirconium et de cérium. Nous
utiliserons donc les données calorimétriques des phases cristallines de ces oxydes.
Les capacités calorifiques et entropies vibrationnelles partielles des oxydes dans
les cristaux sont en général assez voisines de celles déterminées pour les verres de
même composition lorsque les environemments anioniques des éléments dans ces
deux phases sont identiques. Par exemple, IVSiÛ2 et IVAl2Û3 ont des entropies
partielles de 43.4 et 69.1 J.mor'.K"1 dans les verres du système SiO2-Al2O3-CaO-
MgO et de 40.3 et 72.1 J.mor'.K"1 dans les cristaux (Richet et al., 1993).
Le Tableau 8.5 donne les entropies partielles molaires que nous
utiliserons pour le calcul des entropies des verres que nous n'avons pu déterminer
expérimentalement.
153

Tableau 8.5 : Valeurs des entropies molaires partielles dans les verres considérés.
SiO2I VB 2O 3m B 2 O 3
A12O3
ZrO 2
Ce 2 O 3
N a 2 O s a n s A , 2 O 3
Na 2O a v e c A 1 2 O3L i 2 O sans A12O3
L i 2 O avec AI2O3
CaOMgO
S "*,.
(J.mol'.K/1)
43.432.467.469.150.4a
151.6a
85.196.749.060.442.830.7
Références
Richetetal. (1982)Ce travaildeLigny(1996)Richetetal. (1993)Robie et Hemingway (1978)Gronvold et Westrum (1960)Ce travailRichetetal. (1993)Richetetal. (1993)estimation (voir texte)Richetetal. (1993)Richetetal. (1993)
a) - Valeur donnée pour la forme cristalline de l'oxyde (voir texte).
Nous avons utilisé le modèle de Dell et al. (1983) et les hypothèses
d'Araujo et Hares (1981) (voir Chapitre 1 § 3.3) étendues à l'ensemble des
formateurs de réseau pour calculer les fractions de bore tetracoordonne :
Tableau 8.6 : Estimation de la fraction de bore tetracoordonne d'après le modèle de Dell et al.(1983) et les approximations d'Araujo et Hares (1981) étendues à tous les cations formateurs deréseau vitreux.
SON-3SON-4SON-5SON-6SON-7SON-8
73%53%71%71%73%66%
AVM-3AVM-4AVM-5
BSN-5/16BSN-8/24BSN-11/32BSN-29/14
68%67%56%100%100%64%48%
En utilisant les valeurs reportées dans les Tableaux 8.5 (entropies
partielles), 8.6 (fraction de bore tetracoordonne) et 3.4 (composition), nous
obtenons les entropies, S^s-Sb, P o u r l'ensemble de nos verres selon le modèle
additif des propriétés partielles. Le Tableau 8.7 donne les résultats obtenus ; à des
fins de comparaison, nous avons également reporté les valeurs mesurées pour les
compositions ternaires. Selon le nombre d'approximations et les incidences de
154

celles-ci sur les entropies, nous avons estimé les incertitudes sur les résultats
obtenus (Tableau 8.7).
Tableau 8.7 : Valeurs des entropies relatives estimées par le modèle additif des entropies partielles.
SON-3SON-4SON-5SON-6SON-7SON-8AVM-3AVM-4AVM-5
BSN-5/16BSN-8/24
BSN-11/32BSN-29/14
rt rr calcOr>->298
(J.mor'.K1)
48.9952.5551.2251.3251.1652.2552.6956.6354.9749.3549.5457.0651.34
r- rt mes
(J.mol"1.K-1)
48.81
52.17
49.70
56.0051.03
Erreurestimée
1%4%5%6%8%12%1%4%5%1%2%2%1%
8.3 LES MESURES DE CALORIMETRIEDE CHUTE
Les enthalpies relatives expérimentales obtenues pour cinq des
compositions borosilicatées étudiées ont été reportées dans le Tableau 8.8. Ces
données sont représentées dans la Figure 8.8 sous forme de capacité calorifique
moyenne (équation 3.27), Cm. Dans cette figure, les points représentent les valeurs
expérimentales tandis que les courbes en trait plein représentent les variations de
Cm avec la température obtenues après un lissage des données que nous
expliciterons dans la section 8.5.
Chaque courbe présente une discontinuité nette qui marque le phénomène
de transition vitreuse réduit à une température : Tg. Ces ruptures peuvent être plus
ou moins accentuées suivant les produits. Ainsi les compositions AVM et AVM-3
présentent les discontinuités (différences de pente) les plus marquées parmi les
produits étudiés en calorimétrie de chute. En outre, cette figure montre que les
températures de transition vitreuse sont échelonnées entre 815 et 895 K pour les 5
755
verres étudiés selon le même protocole (donc le même traitement thermique) et
que, par conséquent, ces températures dépendent de la composition.
Tableau 8.8 : Enthalpies relatives à 273 K des cinq compositions (verres et liquides) mesurées parcalorimétrie de chute.
T(K)
SON-3308.7346.0405.1457.3525.8624.3711.9757.7759.8812.9830.9859.1892.9928.71014.21066.01124.71190.01241.21384.81417.61477.9
H-rHjn(kJ-g1)
0.0280.0600.1150.1670.2400.3530.4580.5160.5190.5870.6100.6470.6910.7530.9000.9901.0911.2051.2941.5431.5971.703
T(K)
SON-6357.5418.8473.3558.9653.2705.9727.5759.0770.0807.1829.7839.6862.7889.6938.2978.81043.81171.01236.31290.81385.91448.3
H-rHjn(kJ.g1)
0.0680.1250.1780.2670.3700.4310.4570.4940.5070.5520.5800.5920.6240.6700.7530.8230.9331.1451.2541.3461.5001.601
T(K)
SON313.9340.9358.5398.2473.2522.2580.8636.1666.5731.5758.6786.1810.7818.7853.1909.2912.6955.41017.21060.81122.71215.11261.11371.51461.1
HrHm(kJ.g1)
0.0300.0520.0660.1000.1700.2180.2770.3360.3690.4420.4730.5050.5310.5450.6020.6970.7020.7730.8740.9451.0461.1941.2671.4431.587
T(K)
AVM-3306.3353.2432.6526.0628.8709.0782.4827.6859.7901.5912.2963.91079.11189.11257.21361.51414.31491.0
HJ-H2TÎ
(kJ.g1)
0.0270.0690.1480.2480.3680.4660.5600.6180.6700.7500.7700.8671.0831.2821.4061.5951.6931.825
T(K)
AVM302.2328.4358.4411.7450.7525.7602.0662.7714.7741.2789.3850.7865.7907.5918.2971.91085.11192.11263.21367.51420.31479.01498.0
HJ-H2JT,
(kJ.g1)
0.0230.0450.0720.1220.1600.2380.3210.3900.4510.4820.5400.6360.6630.7360.7550.8481.0391.2141.3271.4981.5831.6791.702
156
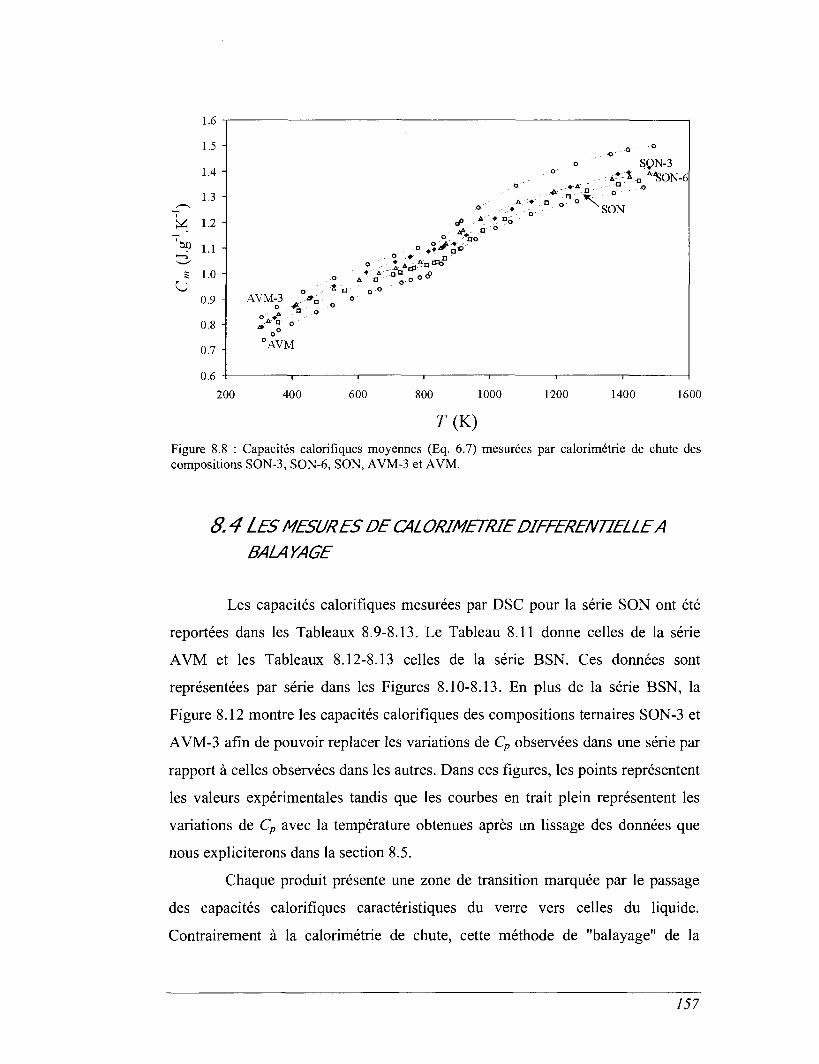
1.6
1.5 •
1.4 -
1.3 -
1.1 -
1.0 -
0.9 -
0.8 -
0.7 -
SON-3
. o - • -
'SON
AVM-3
AVM
o• " - • * • '
A O
D 0°O
200 400 600 800 1000 1200 1400 1600
Figure 8.8 : Capacités calorifiques moyennes (Eq. 6.7) mesurées par calorimétrie de chute descompositions SON-3, SON-6, SON, AVM-3 et AVM.
8.4 LES MESURES DE CALORIMÉTRIE DIFFÉRENTIELLE A
BALAYAGE
Les capacités calorifiques mesurées par DSC pour la série SON ont été
reportées dans les Tableaux 8.9-8.13. Le Tableau 8.11 donne celles de la série
AVM et les Tableaux 8.12-8.13 celles de la série BSN. Ces données sont
représentées par série dans les Figures 8.10-8.13. En plus de la série BSN, la
Figure 8.12 montre les capacités calorifiques des compositions ternaires SON-3 et
AVM-3 afin de pouvoir replacer les variations de Cp observées dans une série par
rapport à celles observées dans les autres. Dans ces figures, les points représentent
les valeurs expérimentales tandis que les courbes en trait plein représentent les
variations de Cp avec la température obtenues après un lissage des données que
nous expliciterons dans la section 8.5.
Chaque produit présente une zone de transition marquée par le passage
des capacités calorifiques caractéristiques du verre vers celles du liquide.
Contrairement à la calorimétrie de chute, cette méthode de "balayage" de la
157

température permet de suivre l'évolution des Cp dans le domaine de transition
vitreuse. En revanche, les courbes lissées, pour des raisons de commodité ne
reproduisent pas cette évolution mais prennent la valeur Tg. En l'absence de toute
autre source de données, la température de transition vitreuse, Tg, déterminée par
ces expériences de calorimétrie différentielle à balayage sera prise égale à
(TA+TB)/2 OÙ les points A et B sont ceux définis sur le schéma explicatif ci-
dessous.
t
Figure 8.9 : Détermination graphique de la température de transition vitreuse à partir des donnéesde DSC. Les points A et B sont définis par l'intersection de l'extrapolation de la courbe de Cp avantet après la transition vitreuse et de la tangente au point d'inflexion (ligne de plus grande pente) dela courbe de Cp dans la zone de transition vitreuse.
8.5 CAPACITES CALORIFIQUES AU-DESSUS DE 273 K
II est possible d'obtenir une représentation continue des capacités
calorifiques et des enthalpies relatives sur l'intervalle de température étudié, en
utilisant diverses fonctions ajustables. Au-dessus de 273 K, l'interpolation est
réalisée avec une équation du type Haas et Fisher (1976) définie dans la section
6.3.4.
Les coefficients de ces relations sont données pour chaque composition,
pour le verre et le liquide, dans le Tableau 8.14. Pour les phases vitreuses, on a
utilisé les trois sources de données : les données adiabatiques au-dessus de 270 K,
les données de DSC et celles de calorimétrie de chute. Pour la phase liquide,
seules les données de DSC et de calorimétrie de chute sont utilisées.
158

Il faut remarquer que les capacités calorifiques de plusieurs des produits
n'ont été mesurées que par DSC, qui est la technique la moins précise et qui, de
plus, se limite à des températures inférieures à 1100 K. Nous utiliserons par la
suite ces coefficients pour extrapoler les Cp à des températures plus hautes, aussi
faudra-t-il rester prudent quant à la précision des interprétations données
concernant ces produits.
40
200 300 400 500 600 700
7(K)
800 900 1000 1100 1200
Figure 8.10 : Capacités calorifiques de la série SON obtenues par calorimétrie différentielle àbalayage.
159
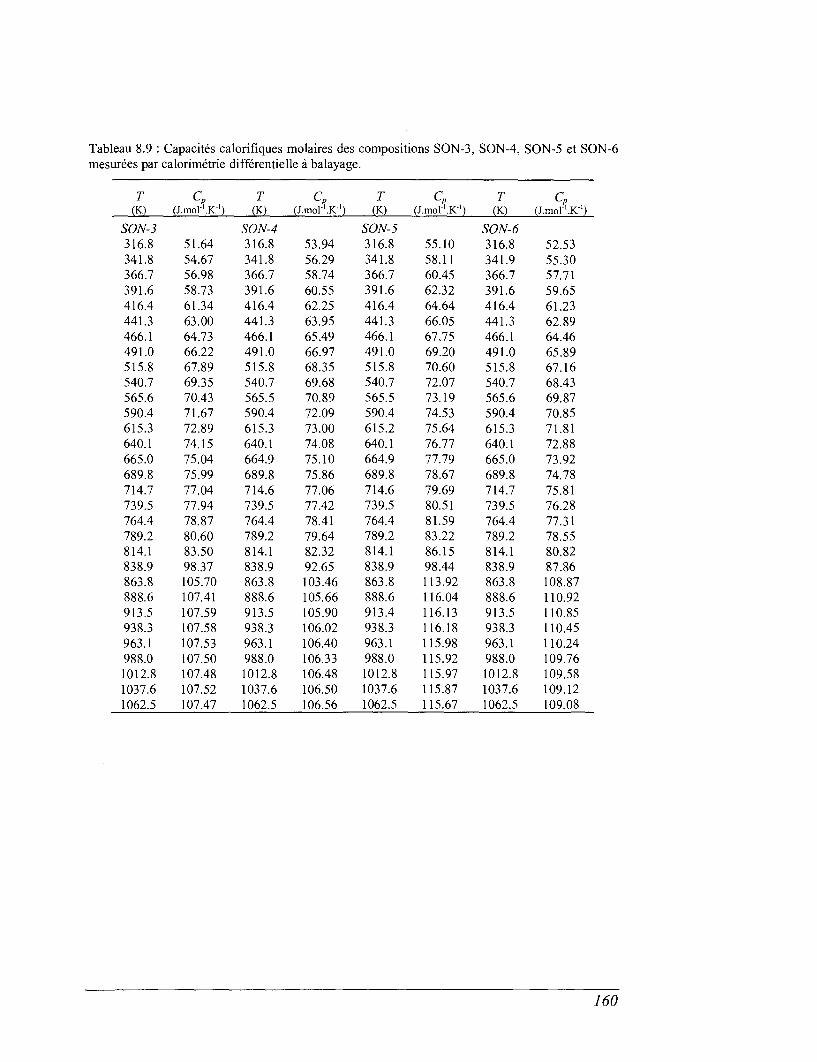
Tableau 8.9 : Capacités calorifiques molaires des compositions SON-3, SON-4, SON-5 et SON-6mesurées par calorimétrie différentielle à balayage.
T(K)
SON-3316.8341.8366.7391.6416.4441.3466.1491.0515.8540.7565.6590.4615.3640.1665.0689.8714.7739.5764.4789.2814.1838.9863.8888.6913.5938.3963.1988.01012.81037.61062.5
cP(J.moï'.K/')
51.6454.6756.9858.7361.3463.0064.7366.2267.8969.3570.4371.6772.8974.1575.0475.9977.0477.9478.8780.6083.5098.37105.70107.41107.59107.58107.53107.50107.48107.52107.47
T(K)
SON-4316.8341.8366.7391.6416.4441.3466.1491.0515.8540.7565.5590.4615.3640.1664.9689.8714.6739.5764.4789.2814.1838.9863.8888.6913.5938.3963.1988.01012.81037.61062.5
cp(J.mol-'.K1)
53.9456.2958.7460.5562.2563.9565.4966.9768.3569.6870.8972.0973.0074.0875.1075.8677.0677.4278.4179.6482.3292.65103.46105.66105.90106.02106.40106.33106.48106.50106.56
T(K)
SON-5316.8341.8366.7391.6416.4441.3466.1491.0515.8540.7565.5590.4615.2640.1664.9689.8714.6739.5764.4789.2814.1838.9863.8888.6913.4938.3963.1988.01012.81037.61062.5
cP(J.mol'.K-1)
55.1058.1160.4562.3264.6466.0567.7569.2070.6072.0773.1974.5375.6476.7777.7978.6779.6980.5181.5983.2286.1598.44113.92116.04116.13116.18115.98115.92115.97115.87115.67
T(K)
SON-6316.8341.9366.7391.6416.4441.3466.1491.0515.8540.7565.6590.4615.3640.1665.0689.8714.7739.5764.4789.2814.1838.9863.8888.6913.5938.3963.1988.01012.81037.61062.5
cP(J.mol'.K')
52.5355.3057.7159.6561.2362.8964.4665.8967.1668.4369.8770.8571.8172.8873.9274.7875.8176.2877.3178.5580.8287.86108.87110.92110.85110.45110.24109.76109.58109.12109.08
160
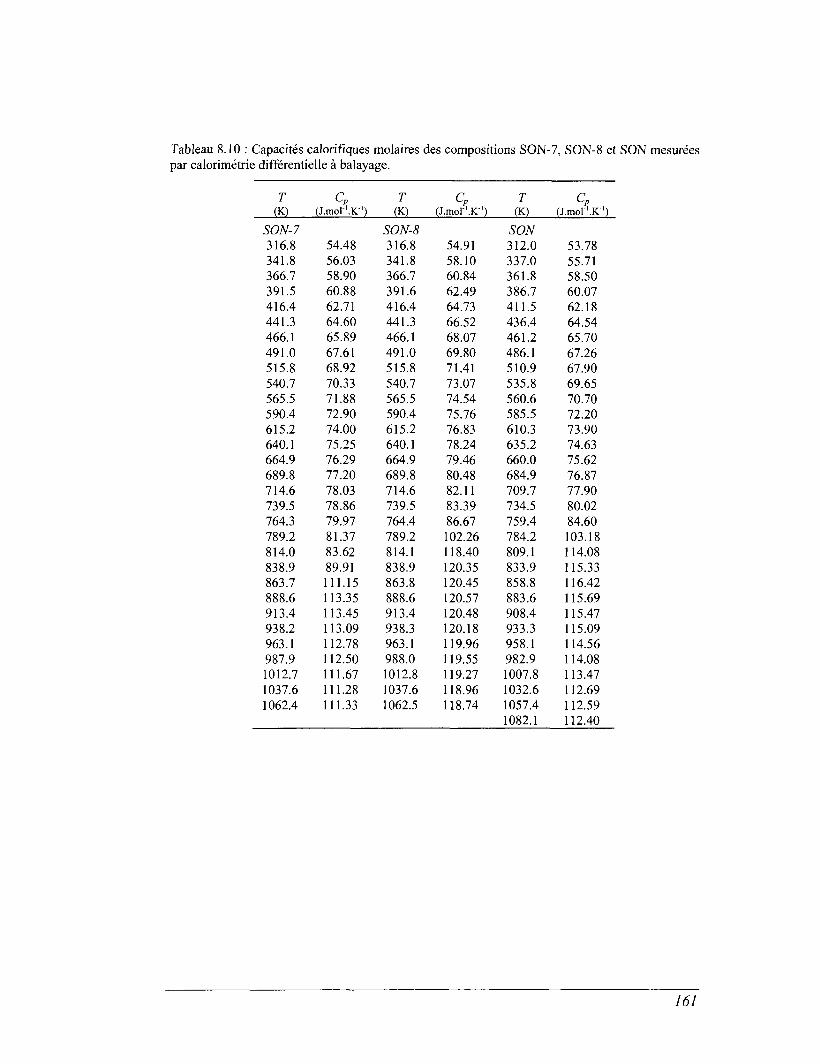
Tableau 8.10 : Capacités calorifiques molaires des compositions SON-7, SON-8 et SON mesuréespar calorimétrie différentielle à balayage.
T(K)
SON-7316.8341.8366.7391.5416.4441.3466.1491.0515.8540.7565.5590.4615.2640.1664.9689.8714.6739.5764.3789.2814.0838.9863.7888.6913.4938.2963.1987.91012.71037.61062.4
(J.mol'.K1)
54.4856.0358.9060.8862.7164.6065.8967.6168.9270.3371.8872.9074.0075.2576.2977.2078.0378.8679.9781.3783.6289.91111.15113.35113.45113.09112.78112.50111.67111.28111.33
T(K)
SON-8316.8341.8366.7391.6416.4441.3466.1491.0515.8540.7565.5590.4615.2640.1664.9689.8714.6739.5764.4789.2814.1838.9863.8888.6913.4938.3963.1988.01012.81037.61062.5
cP(J.mor'.K1)
54.9158.1060.8462.4964.7366.5268.0769.8071.4173.0774.5475.7676.8378.2479.4680.4882.1183.3986.67102.26118.40120.35120.45120.57120.48120.18119.96119.55119.27118.96118.74
T(K)
SON312.0337.0361.8386.7411.5436.4461.2486.1510.9535.8560.6585.5610.3635.2660.0684.9709.7734.5759.4784.2809.1833.9858.8883.6908.4933.3958.1982.91007.81032.61057.41082.1
cP(J.mol'.K1)
53.7855.7158.5060.0762.1864.5465.7067.2667.9069.6570.7072.2073.9074.6375.6276.8777.9080.0284.60103.18114.08115.33116.42115.69115.47115.09114.56114.08113.47112.69112.59112.40
161
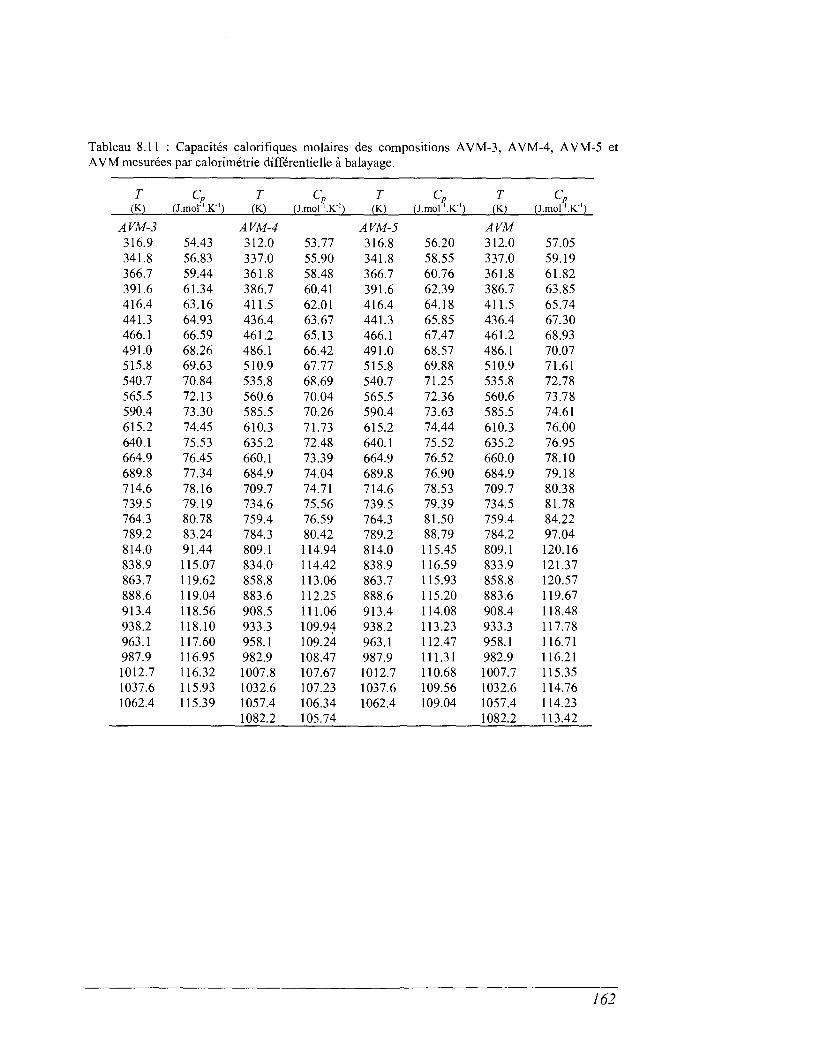
Tableau 8.11 : Capacités calorifiques molaires des compositions AVM-3, AVM-4, AVM-5 etAVM mesurées par calorimetrie différentielle à balayage.
T Cp T Cp T Cp T Cp(K) (J.mol'.K.1) (K) (J.mol'.K1) (K) (J.mol'.K') (K) (J.mol'.K1)
AVM-3316.9341.8366.7391.6416.4441.3466.1491.0515.8540.7565.5590.4615.2640.1664.9689.8714.6739.5764.3789.2814.0838.9863.7888.6913.4938.2963.1987.91012.71037.61062.4
54.4356.8359.4461.3463.1664.9366.5968.2669.6370.8472.1373.3074.4575.5376.4577.3478.1679.1980.7883.2491.44115.07119.62119.04118.56118.10117.60116.95116.32115.93115.39
AVM-4312.0337.0361.8386.7411.5436.4461.2486.1510.9535.8560.6585.5610.3635.2660.1684.9709.7734.6759.4784.3809.1834.0858.8883.6908.5933.3958.1982.91007.81032.61057.41082.2
53.7755.9058.4860.4162.0163.6765.1366.4267.7768.6970.0470.2671.7372.4873.3974.0474.7175.5676.5980.42114.94114.42113.06112.25111.06109.94109.24108.47107.67107.23106.34105.74
AVM-5316.8341.8366.7391.6416.4441.3466.1491.0515.8540.7565.5590.4615.2640.1664.9689.8714.6739.5764.3789.2814.0838.9863.7888.6913.4938.2963.1987.91012.71037.61062.4
56.2058.5560.7662.3964.1865.8567.4768.5769.8871.2572.3673.6374.4475.5276.5276.9078.5379.3981.5088.79115.45116.59115.93115.20114.08113.23112.47111.31110.68109.56109.04
AVM312.0337.0361.8386.7411.5436.4461.2486.1510.9535.8560.6585.5610.3635.2660.0684.9709.7734.5759.4784.2809.1833.9858.8883.6908.4933.3958.1982.91007.71032.61057.41082.2
57.0559.1961.8263.8565.7467.3068.9370.0771.6172.7873.7874.6176.0076.9578.1079.1880.3881.7884.2297.04120.16121.37120.57119.67118.48117.78116.71116.21115.35114.76114.23113.42
162

Tableau 8.12 : Capacités calorifiques molaires des compositions BSN-5/16, BSN-8/24, BSN-11/32et BSN-29/14 mesurées par calorimetrie différentielle à balayage.
T(K)
cp(J.mol'.K1)
T(K)
P
(J.mol'.K')
T(K)
P
(J.mor.K/')
T(K)
cP(J.mol'.K')
BSN-5/16311.9337.0361.8386.7411.5436.4461.2486.1510.9535.8560.6585.5610.3635.2660.0684.9709.7734.6759.4784.2809.1833.9858.8883.6908.4933.3958.1982.91007.81032.61057.41082.2
50.3751.9754.8756.2757.6259.6760.4762.0662.4863.8265.0065.6067.3567.7368.3469.1369.8270.7671.9073.4676.6087.4688.0488.8288.9989.7989.9389.9390.5490.6790.6291.35
BSN-8/24312.0337.0361.8386.7411.5436.4461.2486.1510.9535.8560.6585.5610.3635.2660.0684.9709.7734.5759.4784.2809.1833.9858.8883.6908.4933.3958.1982.91007.81032.61057.41082.2
50.4552.4655.3556.9658.4760.1961.3462.8463.7765.1566.3767.0168.4969.2370.0270.7271.2972.3173.2674.4976.5686.2892.5194.6695.1095.6095.8696.0296.4896.6896.9697.31
BSN-11/32316.9341.9366.7391.6416.5441.3466.2491.0515.9540.7565.6590.4615.3640.2665.0689.8714.7739.6764.4789.3814.1839.0863.9888.7913.6938.4963.2988.11012.91037.81062.6
56.4158.4861.0262.6564.5066.1067.3768.9070.1771.5972.7173.9274.8575.9876.9978.0479.8086.60107.82112.48112.29111.80111.20111.08111.14111.02110.07109.71109.57109.26109.08
BSN-29/14316.8341.8366.7391.6416.4441.3466.1491.0515.8540.7565.5590.4615.2640.1664.9689.8714.6739.5764.3789.2814.0838.9863.7888.6913.4938.3963.1987.91012.81037.61062.4
53.7556.3259.0760.9963.8364.8967.1468.6170.3171.9073.7074.5075.9877.2378.1579.3880.2681.4983.0585.7595.66110.86113.43114.02114.79115.39115.64115.97116.82116.98117.36
163
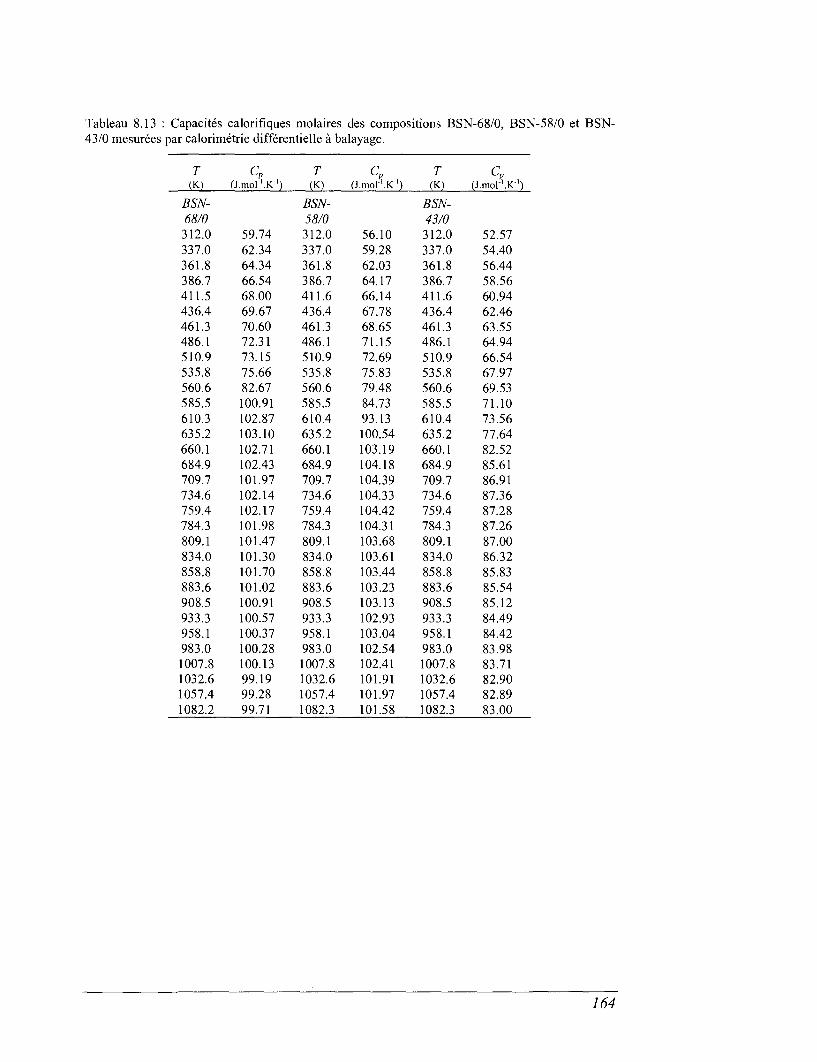
Tableau 8.13 : Capacités calorifiques molaires des compositions BSN-68/0, BSN-58/0 et BSN-)ar calorimétrie différentielle à balayage.
T(K)
BSN-68/0312.0337.0361.8386.7411.5436.4461.3486.1510.9535.8560.6585.5610.3635.2660.1684.9709.7734.6759.4784.3809.1834.0858.8883.6908.5933.3958.1983.01007.81032.61057.41082.2
cP(J.moï'.K1)
59.7462.3464.3466.5468.0069.6770.6072.3173.1575.6682.67100.91102.87103.10102.71102.43101.97102.14102.17101.98101.47101.30101.70101.02100.91100.57100.37100.28100.1399.1999.2899.71
T(K)
BSN-58/0312.0337.0361.8386.7411.6436.4461.3486.1510.9535.8560.6585.5610.4635.2660.1684.9709.7734.6759.4784.3809.1834.0858.8883.6908.5933.3958.1983.01007.81032.61057.41082.3
cP(J.mol-'.K-1)
56.1059.2862.0364.1766.1467.7868.6571.1572.6975.8379.4884.7393.13100.54103.19104.18104.39104.33104.42104.31103.68103.61103.44103.23103.13102.93103.04102.54102.41101.91101.97101.58
T(K)
BSN-43/0312.0337.0361.8386.7411.6436.4461.3486.1510.9535.8560.6585.5610.4635.2660.1684.9709.7734.6759.4784.3809.1834.0858.8883.6908.5933.3958.1983.01007.81032.61057.41082.3
cP(J.mol-'.K'1)
52.5754.4056.4458.5660.9462.4663.5564.9466.5467.9769.5371.1073.5677.6482.5285.6186.9187.3687.2887.2687.0086.3285.8385.5485.1284.4984.4283.9883.7182.9082.8983.00
164
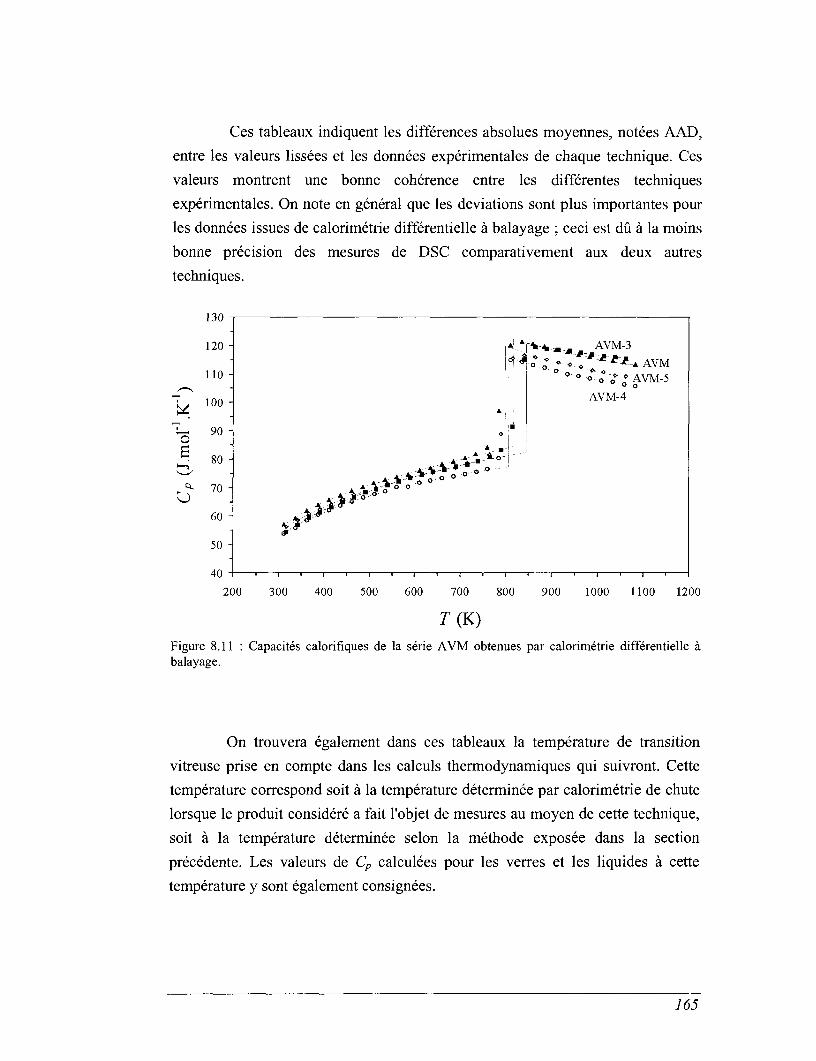
Ces tableaux indiquent les différences absolues moyennes, notées AAD,
entre les valeurs lissées et les données expérimentales de chaque technique. Ces
valeurs montrent une bonne cohérence entre les différentes techniques
expérimentales. On note en général que les deviations sont plus importantes pour
les données issues de calorimétrie différentielle à balayage ; ceci est dû à la moins
bonne précision des mesures de DSC comparativement aux deux autres
techniques.
130
S
120 -
110 -
100 -
90 -
80 -
70 -
60 -
50 -
40
AVM-3
****-* AVM° o <?•« AVM-5
AVM-4
200 300 400 500 600 700 800 900 1000 1100 1200
T(K)Figure 8.11 : Capacités calorifiques de la série AVM obtenues par calorimétrie différentielle àbalayage.
On trouvera également dans ces tableaux la température de transition
vitreuse prise en compte dans les calculs thermodynamiques qui suivront. Cette
température correspond soit à la température déterminée par calorimétrie de chute
lorsque le produit considéré a fait l'objet de mesures au moyen de cette technique,
soit à la température déterminée selon la méthode exposée dans la section
précédente. Les valeurs de Cp calculées pour les verres et les liquides à cette
température y sont également consignées.
165
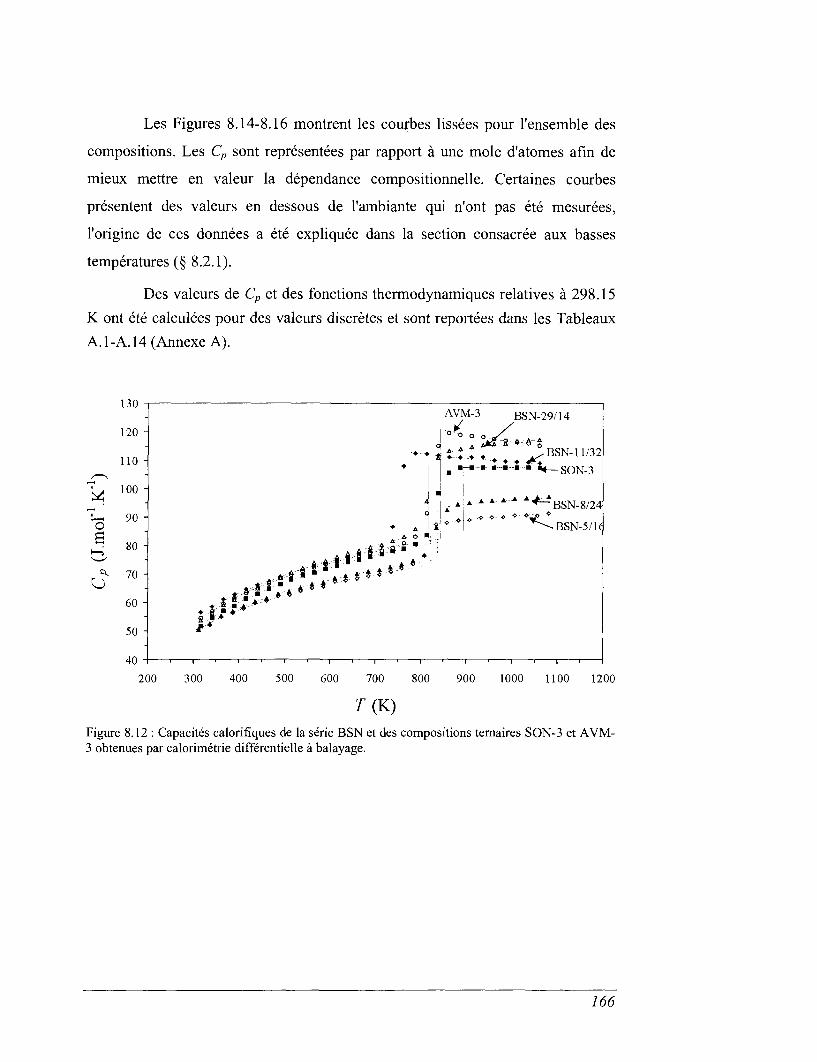
Les Figures 8.14-8.16 montrent les courbes lissées pour l'ensemble des
compositions. Les Cp sont représentées par rapport à une mole d'atomes afin de
mieux mettre en valeur la dépendance compositionnelle. Certaines courbes
présentent des valeurs en dessous de l'ambiante qui n'ont pas été mesurées,
l'origine de ces données a été expliquée dans la section consacrée aux basses
températures (§ 8.2.1).
Des valeurs de Cp et des fonctions thermodynamiques relatives à 298.15
K ont été calculées pour des valeurs discrètes et sont reportées dans les Tableaux
A.1-A.14 (Annexe A).
130
120 -
110 -
/ 100 -
g 9 0 -
^ 80 -
°- 70 -
60 -
50 -
40
AVM-3
\'° ° O -o-
BSN-29/14
* • ? • ./BSN-11/32• • •
•«—SON-3
BSN-8/24
BSN-5/KA O
... o a
A —
200 300 400 500 600 700
T(K)
800 900 1000 1100 1200
Figure 8.12 : Capacités calorifiques de la série BSN et des compositions ternaires SON-3 et AVM-3 obtenues par calorimétrie différentielle à balayage.
166
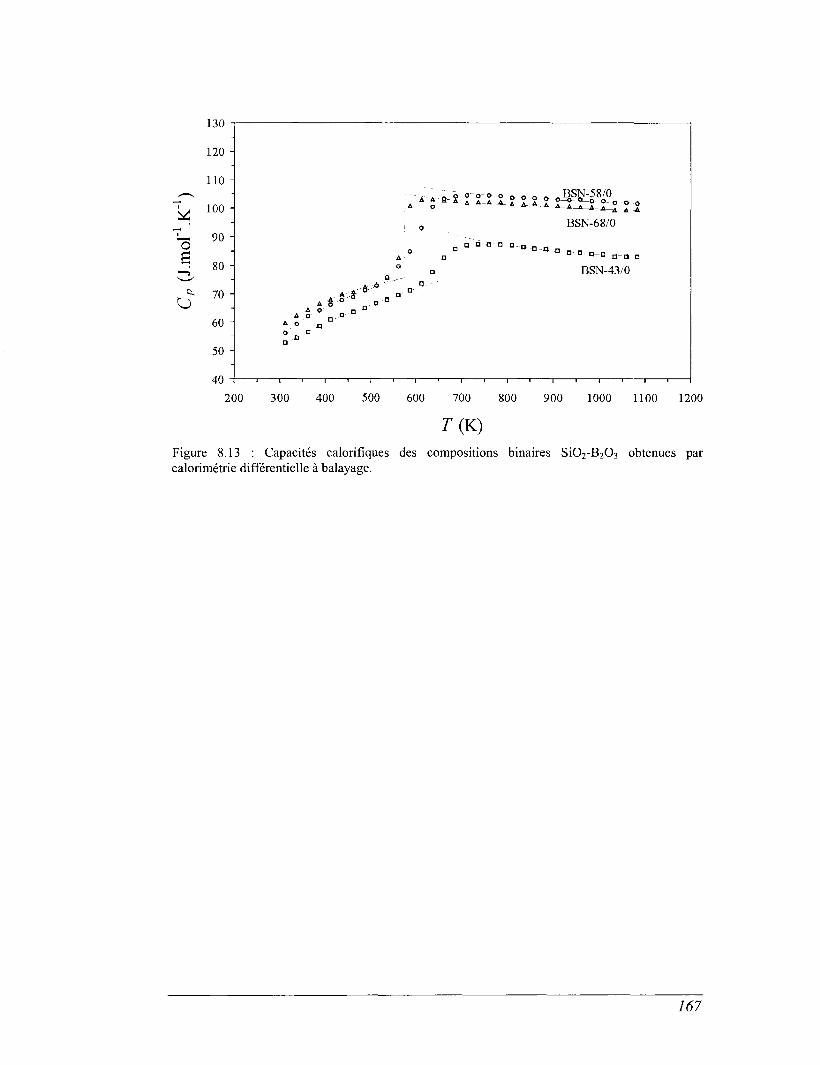
130
-<
*o
Sa.
120 -
110 -
100 -
90 -
80 -
70 -
60 -
50 -
40 -
À A fi
A ; Oo
n
0~O O oA A A A_
a • a a-a
o o oA A-A
n-n-o
BSN-68/0
o a-o a-a ,
BSN-43/0
200 300 400 500 600 700 800 900 1000 1100 1200
Figure 8.13 : Capacités calorifiques des compositions binaires SiO2-B2O3 obtenues parcalorimétrie différentielle à balayage.
167
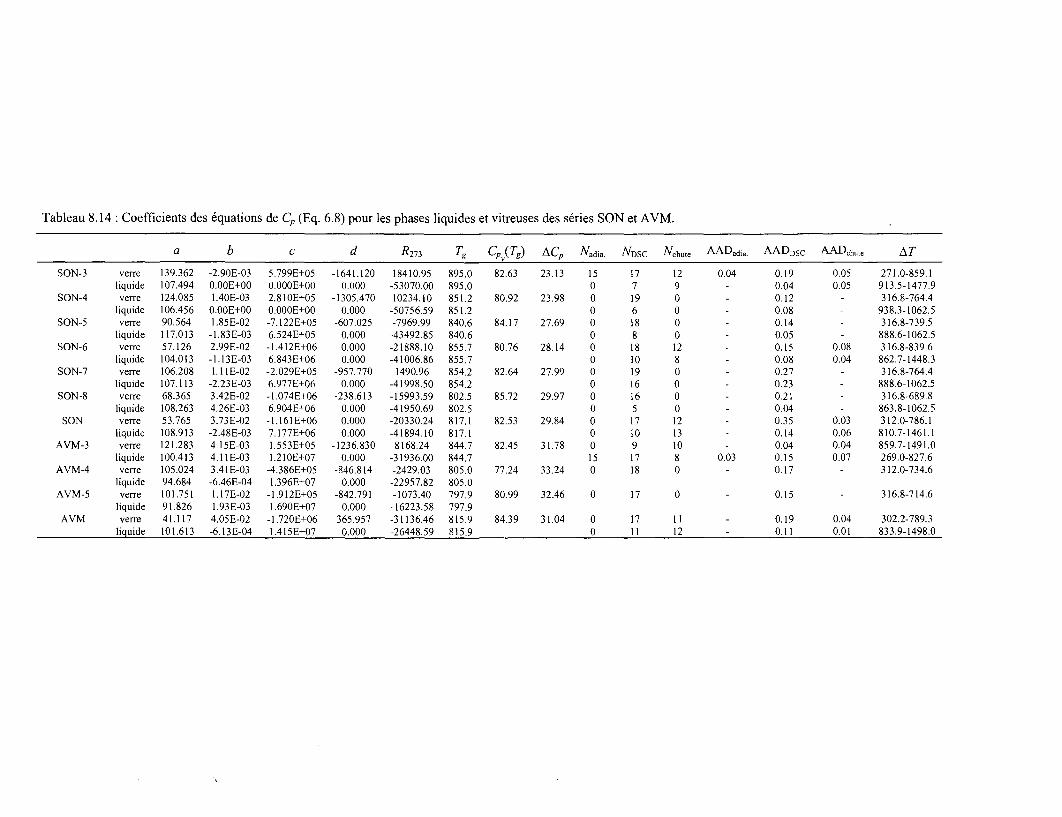
Tableau 8.14 : Coefficients des équations de Cp (Eq. 6.8) pour les phases liquides et vitreuses des séries SON et AVM.
R273 Cp(Tg) AC, Nadia. NDSC
AADadia. AADDSc AADchute AT
SON-3
SON-4
SON-5
SON-6
SON-7
SON-8
SON
AVM-3
AVM-4
AVM-5
AVM
verreliquideverre
liquideverre
liquideverre
liquideverre
liquideverre
liquideverre
liquideverre
liquideverre
liquideverre
liquideverre
liquide
139.362107.494124.085106.45690.564117.01357.126104.013106.208107.11368.365108.26353.765108.913121.283100.413105.02494.684101.75191.82641.117101.613
-2.90E-03O.OOE+001.40E-03
0.00E+001.85E-02
-1.83E-032.99E-02-1.13E-031.11E-02
-2.23E-033.42E-024.26E-033.73E-02-2.48E-034.15E-034.11E-033.41E-03-6.46E-041.17E-021.93E-034.05E-02-6.13E-04
5.799E+050.000E+002.810E+050.000E+00-7.122E+056.524E+05-1.412E+066.843E+06-2.029E+056.977E+06-1.074E+066.904E+06-1.161E+067.177E+061.553E+051.210E+07-4.386E+051.396E+07
-1.912E+051.690E+07
-1.720E+061.415E+07
-1641.1200.000
-1305.4700.000
-607.0250.0000.0000.000
-957.7700.000
-238.6130.0000.0000.000
-1236.8300.000
-846.8140.000
-842.7910.000
365.9570.000
18410.95-53070.0010234.10
-50756.59-7969.99
-43492.85-21888.10-41006.86
1490.96-41998.50-15993.59-41950.69-20330.24-41894.108168.24
-31936.00-2429.03-22957.82-1073.40-16223.58-31136.46-26448.59
895.0895.0851.2851.2840.6840.6855.7855.7854.2854.2802.5802.5817.1817.1844.7844.7805.0805.0797.9797.9815.9815.9
82.63
80.92
84.17
80.76
82.64
85.72
82.53
82.45
77.24
80.99
84.39
23.13
23.98
27.69
28.14
27.99
29.97
29.84
31.78
33.24
32.46
31.04
1500000000000000150
0
00
17719618818101916165171091718
17
1711
1290000128000012131080
0
1112
0.04
0.03
0.190.040.120.080.140.050.150.080.270.230.210.040.350.140.040.150.17
0.15
0.190.11
0.050.05
----
0.080.04
----
0.030.060.040.07
-
-
0.040.01
271.0-859.1913.5-1477.9316.8-764.4
938.3-1062.5316.8-739.5888.6-1062.5316.8-839.6862.7-1448.3316.8-764.4888.6-1062.5316.8-689.8863.8-1062.5312.0-786.1810.7-1461.1859.7-1491.0269.0-827.6312.0-734.6
316.8-714.6
302.2-789.3833.9-1498.0
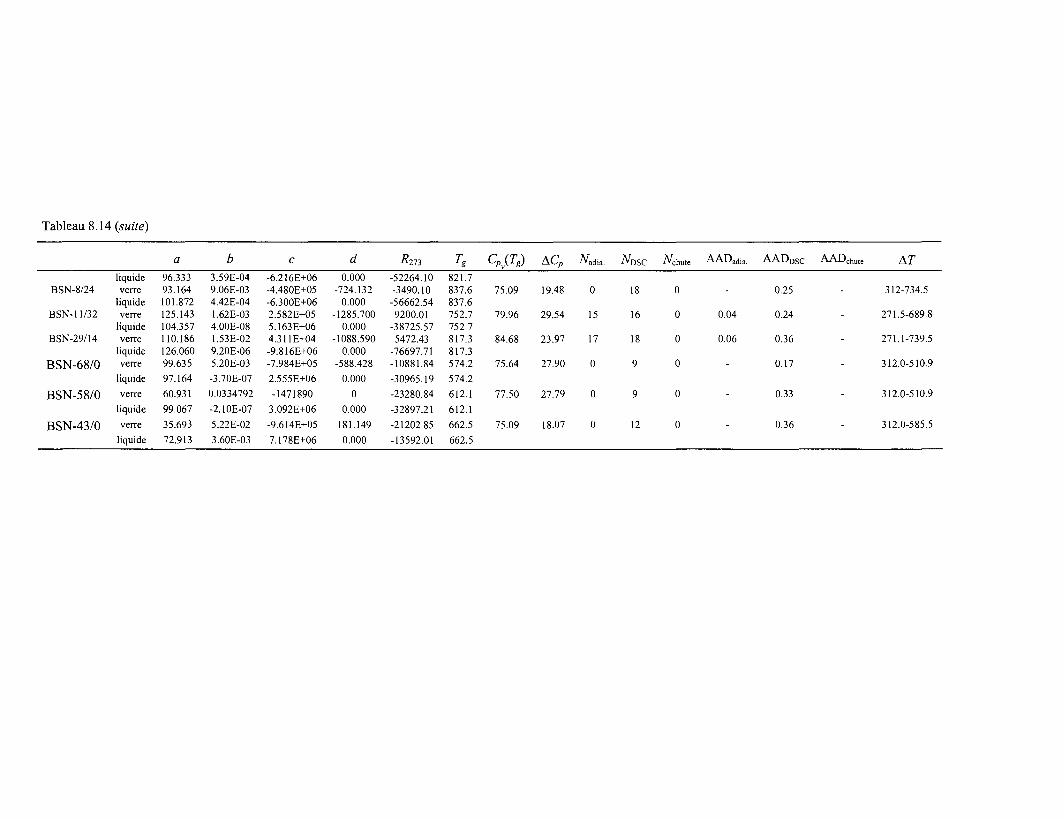
Tableau 8.14 (suite)
R273 CP(Tg) AC AADadia. AAD D S C AADc h u t e AT
BSN-8/24
BSN-11/32
BSN-29/14
BSN-68/0
BSN-58/0
BSN-43/0
liquideverre
liquideverre
liquideverre
liquideverre
liquide
verre
liquide
verre
liquide
96.33393.164101.872125.143104.357110.186126.06099.635
97.164
60.931
99.067
35.693
72.913
3.59E-049.06E-034.42E-041.62E-034.00E-081.53E-029.20E-065.20E-03
-3.70E-07
0.0334792
-2.10E-07
5.22E-02
3.60E-03
-6.216E+06-4.480E+05-6.300E+062.582E+055.163E+064.311E+04-9.816E+06-7.984E+05
2.555E+06
-1471890
3.092E+06
-9.614E+05
7.178E+06
0.000-724.132
0.000-1285.700
0.000-1088.590
0.000-588.428
0.000
0
0.000
181.149
0.000
-52264.10-3490.10-56662.549200.01
-38725.575472.43
-76697.71-10881.84
-30965.19-23280.84
-32897.21
-21202.85
-13592.01
821.7837.6837.6752.7752.7817.3817.3574.2
574.2
612.1
612.1
662.5
662.5
75.09
79.96
75.64
77.50
75.09
19.48
29.54
84.68 23.97
27.90
27.79
18.07
0
15
17
0
0
0
18
16
18
9
9
12
0
0
0
0
0
0
-
0.04
0.06
-
-
0.25
0.24
0.36
0.17
0.33
0.36
312-734.5
271.5-689.8
271.1-739.5
312.0-510.9
312.0-510.9
312.0-585.5
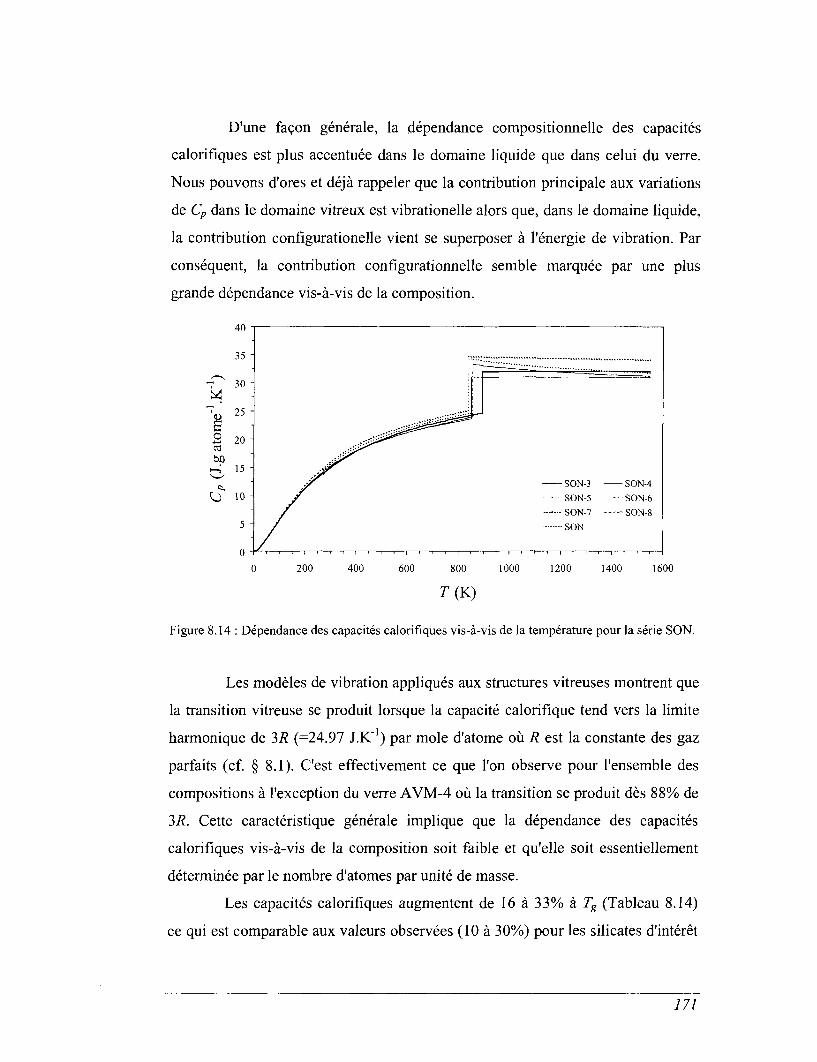
D'une façon générale, la dépendance compositionnelle des capacités
calorifiques est plus accentuée dans le domaine liquide que dans celui du verre.
Nous pouvons d'ores et déjà rappeler que la contribution principale aux variations
de Cp dans le domaine vitreux est vibrationelle alors que, dans le domaine liquide,
la contribution configurationelle vient se superposer à l'énergie de vibration. Par
conséquent, la contribution configurationnelle semble marquée par une plus
grande dépendance vis-à-vis de la composition.
40
35 -
- P 30 -
" C J 25 -
2 2 0 -
ci 15 "«a.
5 -
0 -
SON-3 SON-4
SON-S SON-6
SON-7 SON-8
SON
0 200 400 600 800
T(K)
1000 1200 1400 1600
Figure 8.14 : Dépendance des capacités calorifiques vis-à-vis de la température pour la série SON.
Les modèles de vibration appliqués aux structures vitreuses montrent que
la transition vitreuse se produit lorsque la capacité calorifique tend vers la limite
harmonique de 3R (=24.97 J.K"1) par mole d'atome où R est la constante des gaz
parfaits (cf. § 8.1). C'est effectivement ce que l'on observe pour l'ensemble des
compositions à l'exception du verre AVM-4 où la transition se produit dès 88% de
3R. Cette caractéristique générale implique que la dépendance des capacités
calorifiques vis-à-vis de la composition soit faible et qu'elle soit essentiellement
déterminée par le nombre d'atomes par unité de masse.
Les capacités calorifiques augmentent de 16 à 33% à Tg (Tableau 8.14)
ce qui est comparable aux valeurs observées (10 à 30%) pour les silicates d'intérêt
171
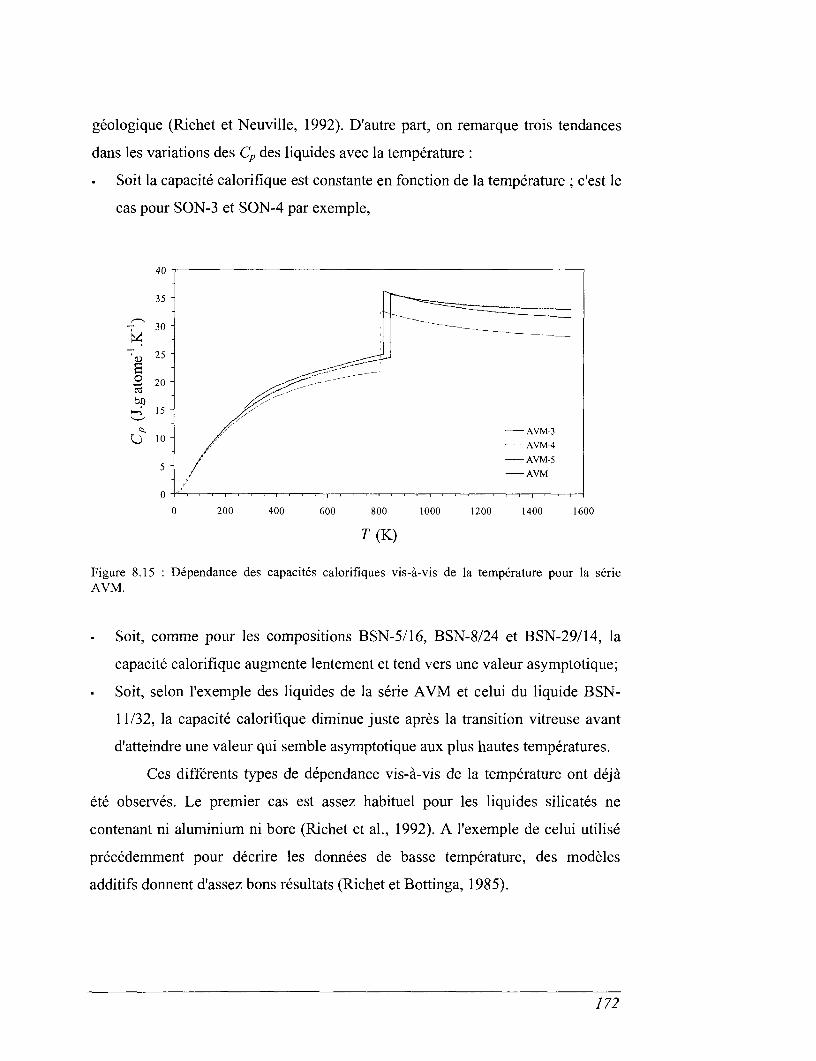
géologique (Richet et Neuville, 1992). D'autre part, on remarque trois tendances
dans les variations des Cp des liquides avec la température :
Soit la capacité calorifique est constante en fonction de la température ; c'est le
cas pour SON-3 et SON-4 par exemple,
40
•ç
o•+-»
H-1
35 "
30 "
25 "
20 -
15 -
10 -
5 "
o -
AVM-3
AVM-4
AVM-5
AVM
200 400 600 800
T(K)
1000 1200 1400 1600
Figure 8.15 : Dépendance des capacités calorifiques vis-à-vis de la température pour la sérieAVM.
• Soit, comme pour les compositions BSN-5/16, BSN-8/24 et BSN-29/14, la
capacité calorifique augmente lentement et tend vers une valeur asymptotique;
Soit, selon l'exemple des liquides de la série AVM et celui du liquide BSN-
11/32, la capacité calorifique diminue juste après la transition vitreuse avant
d'atteindre une valeur qui semble asymptotique aux plus hautes températures.
Ces différents types de dépendance vis-à-vis de la température ont déjà
été observés. Le premier cas est assez habituel pour les liquides silicates ne
contenant ni aluminium ni bore (Richet et al., 1992). A l'exemple de celui utilisé
précédemment pour décrire les données de basse température, des modèles
additifs donnent d'assez bons résultats (Richet et Bottinga, 1985).
172
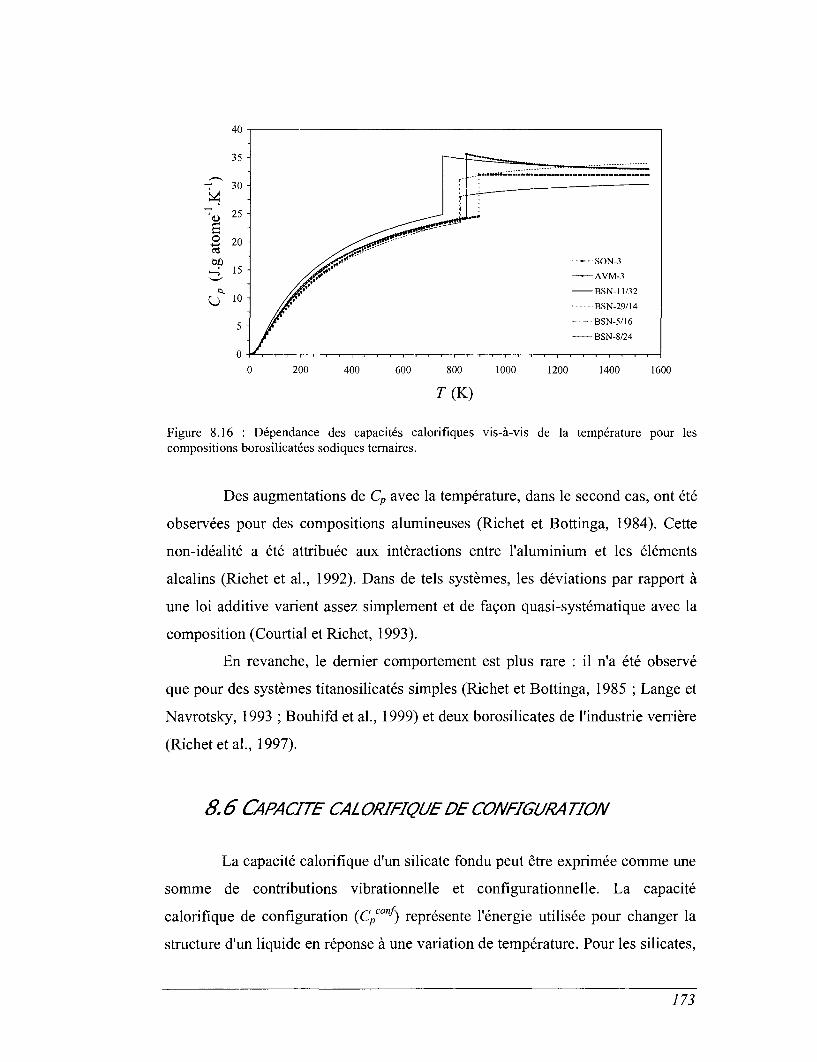
40
35 -
S2 2 0 -
d ! 15"
5 -
0
SON-3
AVM-3
BSN-11/32
BSN-29/14
BSN-5/16
BSN-8/24
200 400 600 800
T(K)
1000 1200 1400 1600
Figure 8.16 : Dépendance des capacités calorifiques vis-à-vis de la température pourcompositions borosilicatées sodiques ternaires.
les
Des augmentations de Cp avec la température, dans le second cas, ont été
observées pour des compositions alumineuses (Richet et Bottinga, 1984). Cette
non-idéalité a été attribuée aux interactions entre l'aluminium et les éléments
alcalins (Richet et al., 1992). Dans de tels systèmes, les déviations par rapport à
une loi additive varient assez simplement et de façon quasi-systématique avec la
composition (Courtial et Richet, 1993).
En revanche, le dernier comportement est plus rare : il n'a été observé
que pour des systèmes titanosilicatés simples (Richet et Bottinga, 1985 ; Lange et
Navrotsky, 1993 ; Bouhifd et al., 1999) et deux borosilicates de l'industrie verrière
(Richet et al., 1997).
8.6 CAPACITE CALORIFIQUE DE CONFIGURATION
La capacité calorifique d'un silicate fondu peut être exprimée comme une
somme de contributions vibrationnelle et configurationnelle. La capacité
calorifique de configuration (Cpconf) représente l'énergie utilisée pour changer la
structure d'un liquide en réponse à une variation de température. Pour les silicates,
173

Richet et al. (1986) suggèrent que la partie vibrationnelle n'est pas marquée de
changements significatifs à la transition vitreuse et que, par conséquent, la
capacité calorifique vibrationnelle d'un liquide peut être obtenue par extrapolation
aux hautes températures de la capacité calorifique du verre. A la transition
vitreuse, l'énergie thermique apportée aux atomes devient suffisamment
importante pour qu'ils puissent franchir les barrières d'énergie qui séparent la
configuration du verre et des configurations caractérisées par des positions
atomiques associées à une valeur du potentiel interatomique plus élevée
(Goldstein, 1969). En augmentant la température, les atomes passent d'un puits de
potentiel à un autre : c'est la conséquence directe de la mobilité des atomes dans le
liquide quelle que soit la complexité du processus dynamique à l'échelle atomique.
Cette description nous conduit à l'expression de la capacité calorifique
configurationnelle suivante (Richet et Bottinga, 1986) :
Ccp°
nf{T)=Cpl{T)-Cpg{Tg). Eq.8.9
Les CpOnf définies par l'équation 8.16 sont représentées dans les Figure
8.17-8.19 pour les liquides étudiés. Puisque les Cp des verres à Tg sont
pratiquement constants (~ 3R I g atom), les capacités calorifiques de configuration
ont les mêmes dépendances vis-à-vis de la température et de la composition que
les Cp des liquides.
174
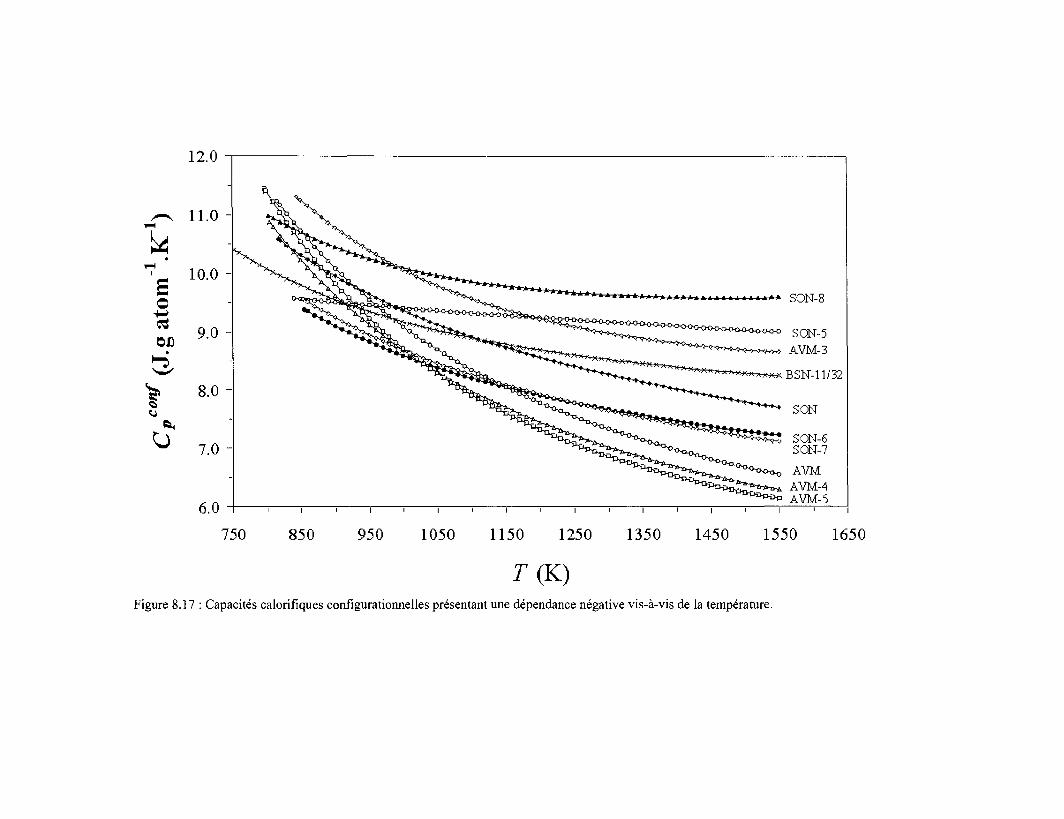
12.0
** SON-8
SON-5AVM-3
; SON-6SON-7
AVMAVM-4AVM-5
750 850 950 1050 1150 1250 1350 1450 1550 1650
BSN-11/32
T(K)Figure 8.17 : Capacités calorifiques configurationnelles présentant une dépendance négative vis-à-vis de la température.
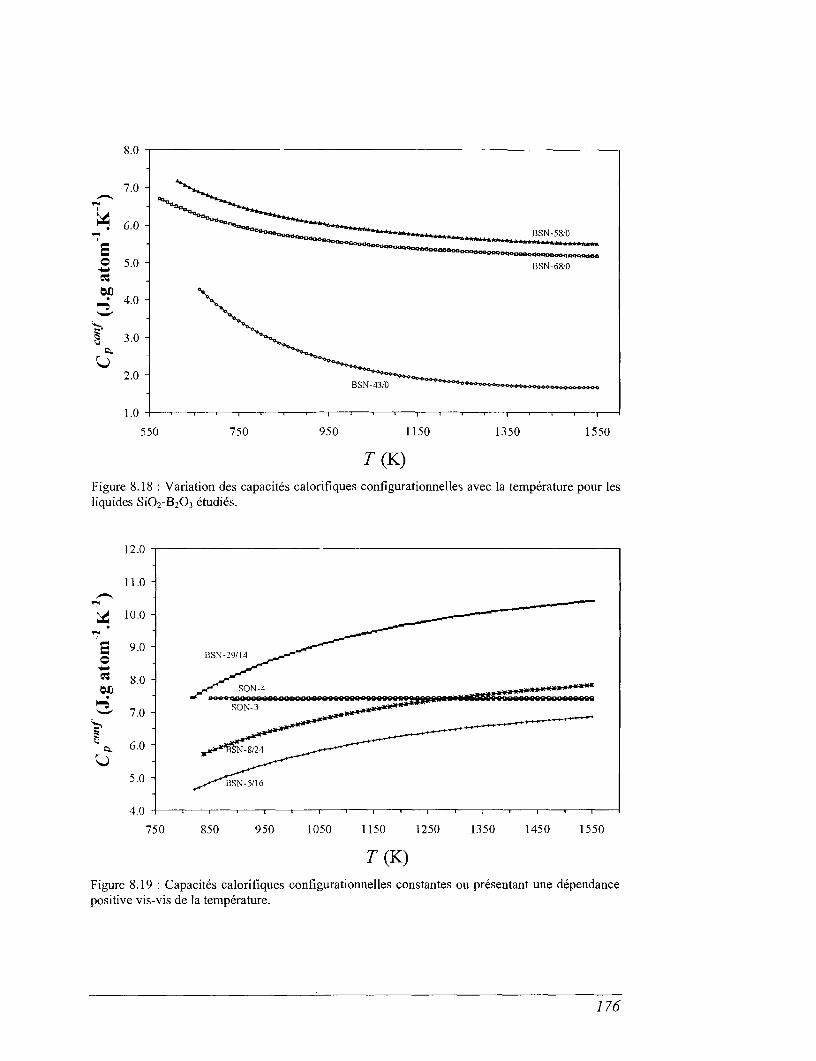
8.0
7.0 -
(J.
t .
a.
O
4.
3.
2.
1
0 -
0 -
o -
o -
BSN-58/0
BSN-43/0
550 750 950 1150 1350 1550
Figure 8.18 : Variation des capacités calorifiques configurationnelles avec la température pour lesliquides SiO2-B2O3 étudiés.
12.0
o
Of
750 850 950 1050 1150 1250 1350 1450 1550
Figure 8.19 : Capacités calorifiques configurationnelles constantes ou présentant une dépendancepositive vis-vis de la température.
176

Il est difficile de rendre compte simplement de la dépendance des Cpconf
avec la composition. Les liquides de la série AVM, caractérisés par des rapports R
(Na2O/B2O3) et K (SiCVE^C^) semblables, présentent tous une forte diminution
de capacité calorifique lorsqu'on augmente la température. En revanche, les
liquides de la série SON, eux aussi caractérisés par des rapports R et K
comparables, présentent les trois types de variations identifiés. Les liquides BSN-
29/14, BSN-11/32 et AVM-3 ont en commun la même teneur en silice ; la Cpconf
du premier augmente avec la température alors que celle des deux autres diminue.
Les teneurs en oxyde de bore des compositions AVM-3 et SON-3 sont semblables
à 2 mol% près : la capacité calorifique de l'un est constante alors que celle de
l'autre montre une forte dépendance négative vis-à-vis de la température. On voit
donc par ces comparaisons que l'existence et les intensités des "anomalies" de Cp
ne sont corrélées ni à la teneur en bore, ni à la teneur en silice, ni même aux
rapports KetR.
Des expériences de spectroscopie RMN (Stebbins, 1991) ont montré que
la proportion d'unité BO4 diminuait quand la température augmente de la
température. Pour une composition de verre borosilicatée de l'industrie verrière
(verre à fibres, E), des variations de 12 à 9% ont été observées pour une variation
de température de 300° (Stebbins et Ellsworth, 1996). De même, un liquide de
composition 20%Na2O-80%B2O3 montre une diminution de sa fraction de bore
tétracoordonné de 75 à 65% entre 580 et 695 K (Stebbins et Ellsworth, 1996).
Bien que les changements de coordinence du bore doivent contribuer aux
variations de capacité calorifique configurationnelle, il n'est pas évident qu'ils
soient à l'origine des anomalies observées. Rappelons d'abord que des
changements de coordinence de Si et Al ont été observés par RMN dans ses
liquides ne présentant pas d'anomalie de Cp (Stebbins, 1991). D'autre part,
l'existence et l'amplitude de ces anomalies ne sont pas corrélées à la teneur en
bore. On pourrait imaginer que le rapport des coordinences du bore soit
indépendant de la teneur en bore totale auquel cas ce dernier argument ne serait
pas justifié. Mais les mesures effectuées par calorimétrie différentielle à balayage
sur les compositions binaires montrent également une diminution de leur capacité
177

calorifique de configuration avec l'augmentation de température. Les verres
binaires sont généralement considérés comme structurés par deux sous-réseaux,
silicate et borate, où le bore ne se trouve qu'en coordinence 3 (Dell et al., 1983 ;
Yun et Bray, 1978). Une étude récente par spectroscopie XANES (Fleet et
Muthupari, 1999) suggère la presence de bore tétracoordonné dans trois
compositions binaires (50, 60, 70 mol% SiCh) mais la fraction de bore en
coordinence 4 n'excède pas 1.2 %. Ainsi, si la présence d'unité BO4 n'est pas
nécessairement à l'origine de ces anomalies, les causes de ces comportements
anormaux peuvent être cherchées dans les interactions entre Si et 3B et/ou 4B.
Nous avons déjà rappelé que les titanosilicates (pour la dépendance
négative vis-à-vis de la température) et les aluminosilicates (pour la dépendance
positive) montraient des comportements semblables. L'aluminium, le titane et le
bore peuvent tous les trois avoir une coordinence semblable à celle du silicium.
Seuls le titane et le bore montrent des diminutions de capacité calorifique quand la
température augmente. Tous deux sont capables de former un réseau vitreux de
leur propre chef. Ces similitudes ne nous semblent pas le fruit du hazard,
cependant nos seules données thermochimiques ne nous permettent pas d'élucider
les mécanismes et la nature des interactions entre ces cations et Si4+ qui pourraient
être la cause des comportements anormaux observés.
178

Chapitre 9
VISCOSITES ET ENTROPIES DE CONFIGURATION
L'écoulement d'un liquide silicate peut être décrit selon plusieurs
approximations de la théorie des déplacements visqueux (la théorie des volumes
libres, la théorie des processus de relaxation, etc.). Bien souvent cependant
quelques hypothèses sur la structure des liquides suffisent à décrire les
dépendances en composition et en température de la viscosité. Dans un bain
silicate par exemple, le silicium forme des liaisons O-Si-O très solides en raison
de sa forte charge et de son rayon ionique très petit. La viscosité sera très élevée
dans les systèmes très riches en silice, fortement polymerises, car la déformation
nécessite la rupture de ces liaisons fortes O-Si-O. Pour un liquide de silice pure,
l'écoulement sous l'action de faibles forces ne pourra être réalisé que pour des
températures très supérieures à 1400 K. La présence de modificateurs de réseau
tels que Na, K, Li, Ca, Mg dans le liquide entrainera une diminution sensible de la
viscosité car les liaisons à caractères ioniques sont plus faibles.
Après avoir rappelé les traits généraux de la théorie des écoulements
visqueux, nous présenterons les résultats qui nous permettront, en accord avec la
théorie des processus de relaxation (Adam et Gibbs, 1965), de déterminer les
entropies de configurations nécessaires pour nos calculs thermochimiques.
9.1 LA RELAXA TION STRUCTURALE ET LES MODÈLES
RHEOLOGIQUES
Les modèles rhéologiques qui tentent d'expliquer le phénomène de
transition vitreuse, la relaxation structurale ou encore les diverses dépendances
des propriétés de transport des verres et des liquides sont basés en général sur
deux sortes d'approximations, l'une consistant à réduire les configurations
179

atomiques à des descriptions utilisables dans des calculs statistiques, l'autre faisant
appel à des descriptions phénoménologiques qui permettent de relier les
paramètres caractéristiques des liquides et des verres. Ces deux types
d'approximation se complètent et, à un certain stade de compréhension, leur
synthèse devient possible.
Nous ne décrirons ici que le modèle entropique des processus de
relaxation énoncé par Adam et Gibbs (1965) qui sera utilisé par la suite pour
décrire les résultats expérimentaux ; les autres approches, le modèle du volume
libre (Turnbull et Cohen, 1961 ; Cohen et Grest, 1979, 1984) notamment pourront
être trouvées dans les publications de Richet et Bottiga (1995, 1992).
Le modèle d'Adam et Gibbs, qui étend les travaux de Gibbs et DiMarzio
(1958) sur les réseaux de polymères organiques linéaires, attribue un rôle majeur à
l'entropie confïgurationnelle. En effet, ce modèle suppose que le temps de
relaxation (T) d'une propriété d'un liquide à la température T est inversement
proportionnel à la probabilité w(T) d'un réarrangement coopératif de sa structure.
S'il existait un liquide d'entropie de configuration nulle, le temps de
relaxation de sa structure serait infini. Si deux configurations seulement étaient
réalisables pour le liquide, son réarrangement structural nécessiterait un
déplacement simultané de toutes les entités. La probabilité d'un tel événement
serait faible mais non nulle et le temps de relaxation serait extrêmement long.
Quand l'entropie de configuration augmente, les réarrangements
coopératifs peuvent se produire indépendamment dans des régions de plus en plus
petites du liquide. Adam et Gibbs utilisèrent un modèle de mécanique statistique
et obtinrent :
f -B \Eq. 9.1
TSconf(T)j
où A est un terme pré-exponentiel et Be est approximativement proportionnelle à
la barrière d'énergie libre empêchant le réarrangement coopératif.
En considérant un ressort parfait et un amortisseur visqueux montés en
série (rhéologie de Maxwell), le temps de relaxation, t, peut s'exprimer par le
180

rapport entre la viscosité n, caractéristique de l'amortisseur, et la constante
élastique du ressort k. Par conséquent, on a :
Eq. 9.2
soit Tl = Ap exp -. avec Ap = kAT . Eq. 9.3
Si Sfonf peut être considérée indépendante de la température, on trouve
alors une loi d'Arrhénius. En dessous de la transition vitreuse, les temps de
relaxation sont importants et, à l'échelle de temps d'une mesure de viscosité
classique, l'entropie de configuration n'est pas modifiée quand on diminue la
température. En accord avec ce modèle, les expériences montrent une dépendance
Arrhénienne des viscosités vis-à-vis de la température.
Ce modèle repose sur deux postulats (Bottinga et al., 1995) :
le liquide est une phase structurale non-homogène qui comprend des sous-
systèmes dont la taille augmente quand la température diminue,
- ces sous-systèmes se réarrangent à cause des mouvements coopératifs des
particules qu'ils contiennent, qui ont lieu indépendamment des sous-systèmes
voisins.
Le premier postulat tend à être confirmé par les analyses structurales aux basses
températures. Le pic de boson, observé en spectroscopie Raman, diffraction X, et
diffusion inélastique des neutrons de basse énergie, résulte d'un excès de densité
d'états vibrationnels à basse fréquence (Lannin, 1977). Cette anomalie reflète des
caractéristiques structurales acquises lors du passage du liquide dans la zone de
transition vitreuse. L'étendue de ces anomalies de densité a été évaluée par
Moynihan et Schroeder (1993) qui ont assimilé des hétérogénéités nanométriques,
observées par diffusion Brillouin dans le liquide prés de la transition vitreuse, à
des fluctuations d'entropie de configuration. Duval et al. (1990) ont quant à eux
interprété des résultats de Raman et de diffusion des neutrons dans la silice
vitreuse et défini des régions "anormales" de l'ordre de 2.1 nm.
181

Le second postulat n'a pas encore été vérifié expérimentalement.
Fredrickson et Brawer (1986) ont cependant montré qu'en introduisant un certain
degré de coopérativité dans leur simulation numérique (modèle d'Ising à 2 spins,
type Monte Carlo), ils pouvaient rendre compte de nombreux aspects
phénoménologiques de la transition vitreuse.
Un autre point important en faveur de ce modèle est fourni par le
paramètre pré-exponentiel Ae. Sa valeur moyenne est de l'ordre de 3.10"3 Pa.s pour
les silicates dont on connaît l'entropie de configuration à 0 K (Richet, 1984 ;
Richet et al., 1986, 1990, 1993). D'après l'équation 9.2, cela conduit à un terme Ax
de l'ordre de 3.1O"13 s (Richet et Bottinga, 1995), une valeur compatible avec les
périodes des vibrations des réseaux silicates vers lesquelles doivent tendre les
temps de relaxation aux plus basses températures.
Comme nous l'avons vu également dans la section 4.5, l'expression de la
viscosité comme une fonction de l'entropie de configuration a aussi été validée
expérimentalement par comparaison entre les entropies déterminées par lissage
des mesures de viscosité et celles mesurées par méthode calorimétrique.
9.2 VISCOSITE : EFFET DE LA TEMPERATURE ET DE LA
COMPOSITION CHIMIQUE
Dans les processus industriels ou naturels, la viscosité diminue de plus de
dix ordres de grandeur entre la transition vitreuse et la température du liquidus. A
température constante, on peut observer les mêmes différences en ce qui concerne
la composition. A 1500 K par exemple, la silice est plus visqueuse d'à peu prés
dix ordres de grandeurs qu'un liquide anorthitique (CaAbSisOg). Nous allons
observer les changements de viscosité dus aux variations de composition des
matrices de confinement et leur dépendance vis-à-vis de la température.
Les viscosités d'équilibre (contraintes relaxées) mesurées avec les
techniques et les procédures expérimentales décrites dans le Chapitre 7 figurent
182

dans les Tableaux 9.1-9.4. L'accord entre les mesures de haute et basse
températures est bon comme nous l'indiquent les Figures 9.1-9.3.
Tableau 9.1: Viscosités des liquides SON-3, SON-4, SON-5 et SON-6.
T(K)
834.6847.8856.2861.9867.6875.7879.4886.7891.1897.7902.1908.0912.4918.6923.7928.11087.91108.11165.91222.11275.11329.21387.31437.31479.51545.11582.61633.7
(Log Pa.s)
SON-313.1012.4312.0211.7011.4211.0010.8210.4710.289.999.819.609.419.158.988.814.804.453.612.972.452.021.631.371.110.880.760.61
T(K)
833.7839.5843.8845.0855.5862.1867.0878.0883.6889.0899.4899.9910.2910.51161.21213.11265.11318.71370.41439.21476.11527.8
il(Log Pa.s)
SON-412.3512.0711.9011.8111.3011.0110.7610.2910.059.859.579.459.129.073.763.152.642.231.901.571.421.23
T(K)
836.0838.9849.5852.7857.2863.0864.2876.6877.7888.0888.4899.1904.2910.5914.71116.61168.11214.51269.21319.01376.61431.41480.91530.6
(Log Pa.s)
SON-512.4412.2311.6211.4511.2310.9410.8910.3310.259.799.819.409.119.038.713.953.322.852.351.951.641.391.100.92
T(K)
839.7846.9853.2855.9860.8864.7871.6875.2882.2886.8893.1897.9903.0908.6914.0920.51162.21211.41267.11315.71371.31428.41477.21528.11574.1
(Log Pa.s)
SON-612.6112.1911.8711.7011.4511.2610.9210.7410.4010.239.939.729.539.299.098.873.643.112.642.221.811.531.221.020.88
183
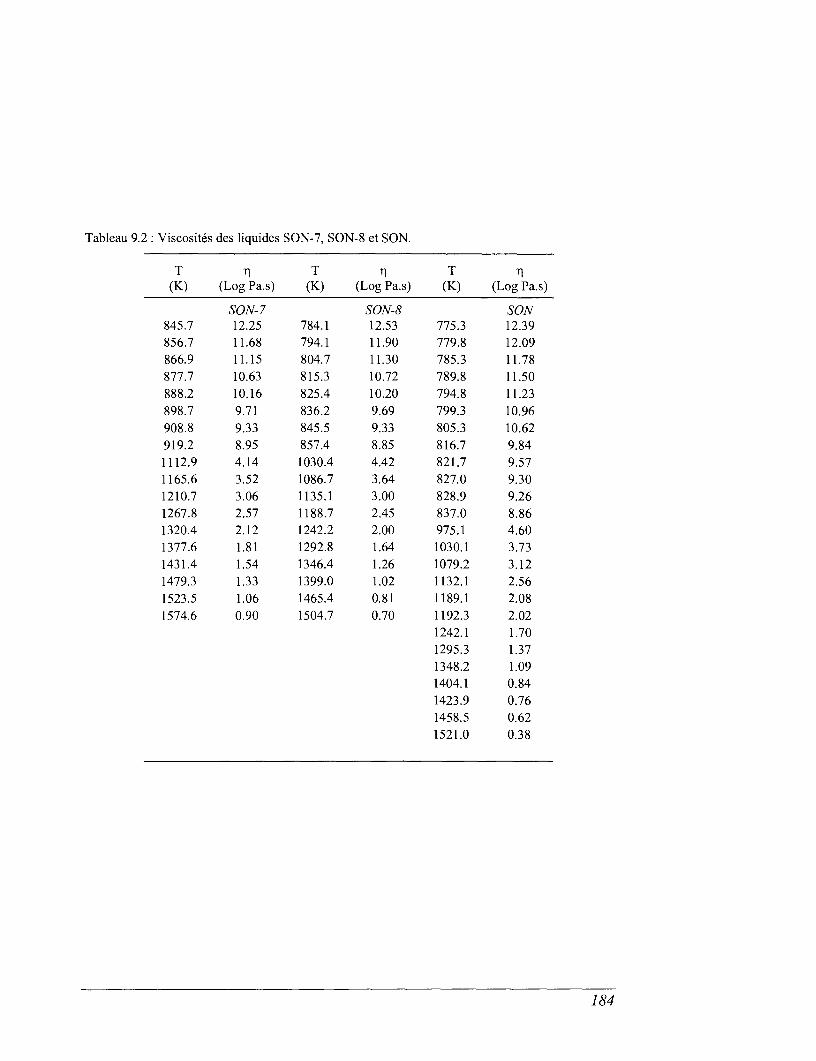
Tableau 9.2 : Viscosités des liquides SON-7, SON-8 et SON.
T(K)
845.7856.7866.9877.7888.2898.7908.8919.21112.91165.61210.71267.81320.41377.61431.41479.31523.51574.6
ri(Log Pa.s)
SON-712.2511.6811.1510.6310.169.719.338.954.143.523.062.572.121.811.541.331.060.90
T(K)
784.1794.1804.7815.3825.4836.2845.5857.41030.41086.71135.11188.71242.21292.81346.41399.01465.41504.7
(Log Pa.s)
SON-812.5311.9011.3010.7210.209.699.338.854.423.643.002.452.001.641.261.020.810.70
T(K)
775.3779.8785.3789.8794.8799.3805.3816.7821.7827.0828.9837.0975.11030.11079.21132.11189.11192.31242.11295.31348.21404.11423.91458.51521.0
(Log Pa.s)
SON12.3912.0911.7811.5011.2310.9610.629.849.579.309.268.864.603.733.122.562.082.021.701.371.090.840.760.620.38
184
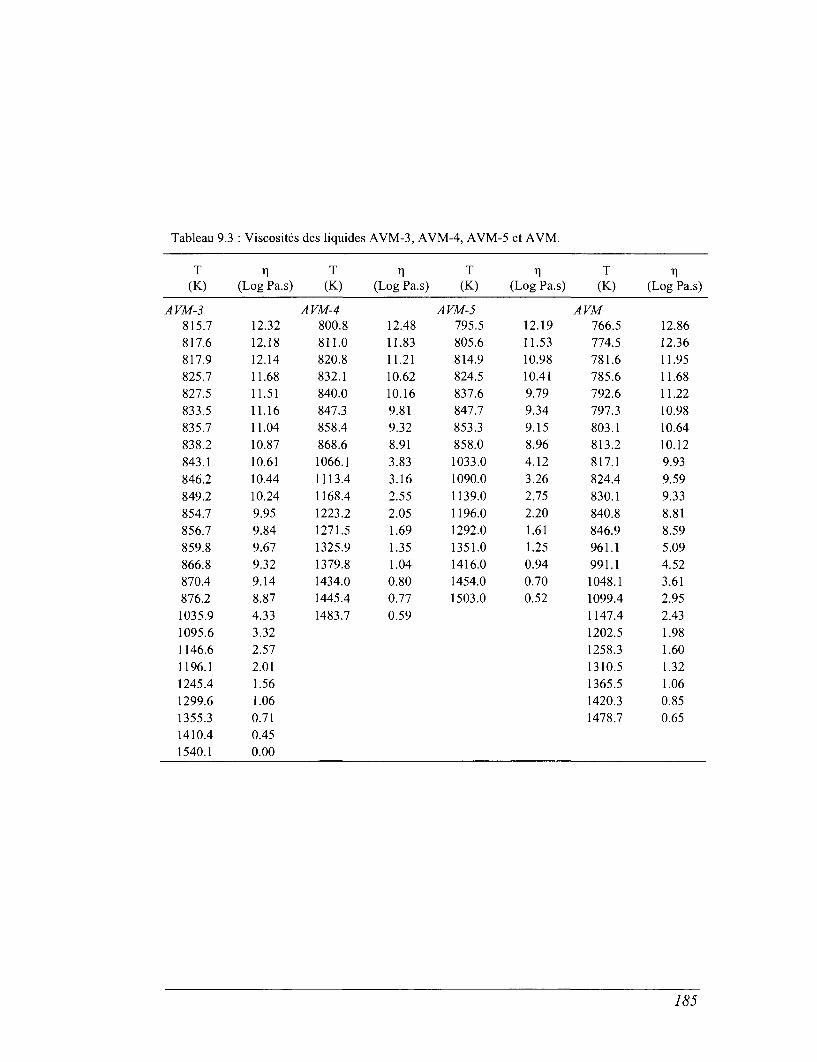
Tableau 9.3 : Viscosités des liquides AVM-3, AVM-4, AVM-5 et AVM.
T(K)
AVM-3815.7817.6817.9825.7827.5833.5835.7838.2843.1846.2849.2854.7856.7859.8866.8870.4876.21035.91095.61146.61196.11245.41299.61355.31410.41540.1
(Log Pa.s)
12.3212.1812.1411.6811.5111.1611.0410.8710.6110.4410.249.959.849.679.329.148.874.333.322.572.011.561.060.710.450.00
T(K)
AVM-4800.8811.0820.8832.1840.0847.3858.4868.61066.11113.41168.41223.21271.51325.91379.81434.01445.41483.7
(Log Pa.s)
12.4811.8311.2110.6210.169.819.328.913.833.162.552.051.691.351.040.800.770.59
T(K)
AVM-5795.5805.6814.9824.5837.6847.7853.3858.01033.01090.01139.01196.01292.01351.01416.01454.01503.0
Tl(Log Pa.s)
12.1911.5310.9810.419.799.349.158.964.123.262.752.201.611.250.940.700.52
T(K)
AVM766.5774.5781.6785.6792.6797.3803.1813.2817.1824.4830.1840.8846.9961.1991.11048.11099.41147.41202.51258.31310.51365.51420.31478.7
n(Log Pa.s)
12.8612.3611.9511.6811.2210.9810.6410.129.939.599.338.818.595.094.523.612.952.431.981.601.321.060.850.65
185
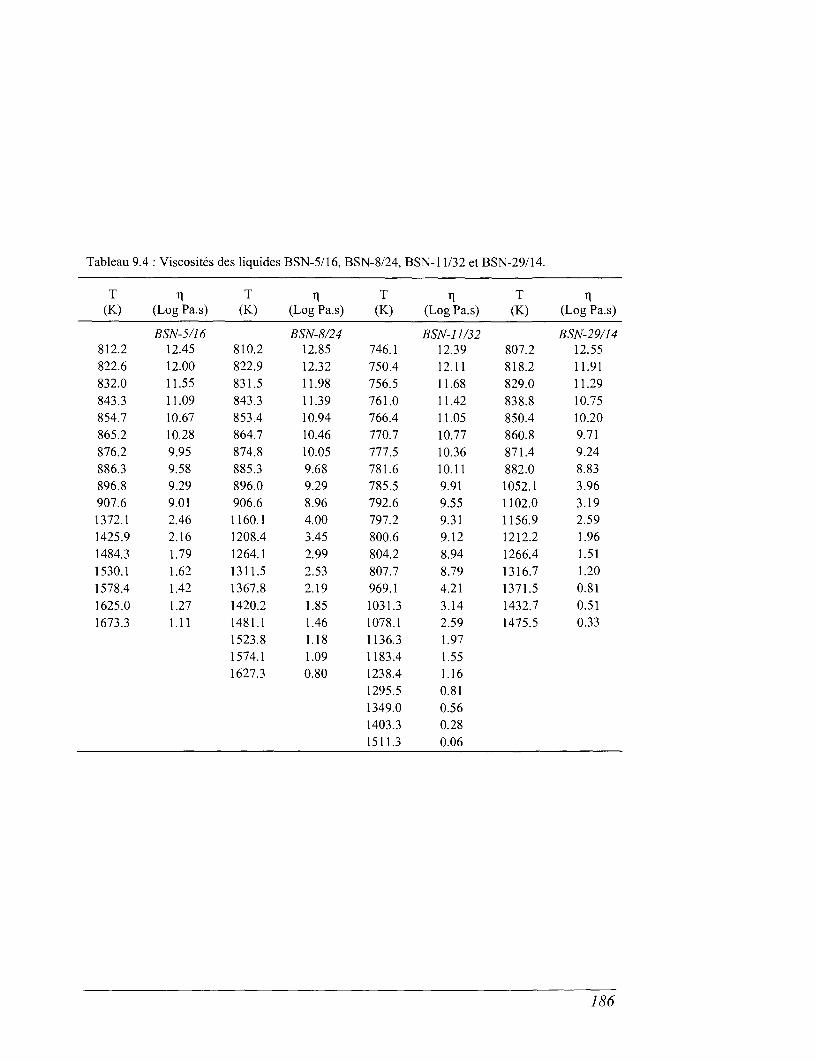
Tableau 9.4 : Viscosités des liquides BSN-5/16, BSN-8/24, BSN-11/32 et BSN-29/14.
T(K)
812.2822.6832.0843.3854.7865.2876.2886.3896.8907.61372.11425.91484.31530.11578.41625.01673.3
(Log Pa.s)
BSN-5/1612.4512.0011.5511.0910.6710.289.959.589.299.012.462.161.791.621.421.271.11
T(K)
810.2822.9831.5843.3853.4864.7874.8885.3896.0906.61160.11208.41264.11311.51367.81420.21481.11523.81574.11627.3
il(Log Pa.s)
BSN-8/2412.8512.3211.9811.3910.9410.4610.059.689.298.964.003.452.992.532.191.851.461.181.090.80
T(K)
746.1750.4756.5761.0766.4770.7777.5781.6785.5792.6797.2800.6804.2807.7969.11031.31078.11136.31183.41238.41295.51349.01403.31511.3
(Log Pa.s)
BSN-11/3212.3912.1111.6811.4211.0510.7710.3610.119.919.559.319.128.948.794.213.142.591.971.551.160.810.560.280.06
T(K)
807.2818.2829.0838.8850.4860.8871.4882.01052.11102.01156.91212.21266.41316.71371.51432.71475.5
(Log Pa.s)
BSN-29/1412.5511.9111.2910.7510.209.719.248.833.963.192.591.961.511.200.810.510.33
186

10
îoVr
11 12
Figure 9.1 : Variation de la viscosité en fonction de la température pour la série SON.
13
14
12 -
OS
a.
Log
SCO
S1
10 -
8 -
6 -
4 -
2 -
0 À
A V M - 3 / ^ - ^
io4/rFigure 9.2 : Variation de la viscosité en fonction de la température pour la série AVM.
13
757

14
12 -
10 11 12 13
io4/rFigure 9.3 : Variation de la viscosité en fonction de la température pour la série BSN.
En général, la viscosité dépend plus fortement de la composition à basse
température. Cette tendance est suivie par les produits des séries SON et AVM
mais ne l'est que de façon approximative pour les compositions ternaires. Par
exemple, la différence de viscosité, qui est de 104 Pa.s à 850 K entre SON-3 et
SON, n'est plus que de 10°6 Pa.s à 1500 K ; en revanche, pour les compositions
ternaires AVM-3 et BSN-5/16, l'écart augmente de 1007 Pa.s à 850 K à 1017 à
1500K. Cette particularité de la série BSN, s'exprime graphiquement par des
écarts à des variations Arrhéniennes moins importants pour la viscosité des
produits BSN-5/16 et BSN-8/24 que pour celle des autres ternaires.
La dépendance de la viscosité vis-à-vis de la température peut être
déterminée par l'application quantitative de la théorie des processus de relaxation
décrite dans la section 9.1. Selon l'équation 9.3, les variations d'entropie de
configuration avec la température déterminent celles de l'énergie d'activation de
l'écoulement visqueux et, par conséquent, gouverne les écarts au modèle
Arrhénien. Entre la température considérée T et la température de transition
vitreuse Tg, cette variation est donnée par :
188

sconf ^ = sconfj çiconf
Eq. 9.4
Les capacités calorifiques de configuration des produits ont été données dans le
chapitre précédent. L'entropie de configuration à Tg peut alors être déduite par
ajustement, avec les constantes Ae et Be, de l'équation 9.4 aux valeurs mesurées.
Les différents coefficients obtenus sont reportés dans le Tableau 9.5 pour
l'ensemble des produits. Pour comparaison, nous avons reporté également les
paramètres de l'équation empirique de Tammann, Vogel et Fulcher (TVF),
Blogr| = A +
T-TiEq. 9.5
déterminés par lissage des mêmes jeux de données (Tableau 9.6).
Comme l'indique le Tableau 9.5, l'équation entropique permet de
reproduire les données mieux ou aussi bien que l'équation TVF (Tableau 9.6).
Tableau 9.5 : Coefficients de l'équation entropique de la viscosité (Eq. 9.3).
Produit
SON-3SON-4SON-5SON-6SON-7SON-8SON
AVM-3AVM-4AVM-5AVM
BSN-5/16BSN-8/24BSN-11/32BSN-29/14
(K)
895.0851.2840.6855.7854.2802.5817.1844.7805.0797.9815.9821.7837.6752.7817.3
S=onf{Tg)(J.mol'.K1)
11.7312.7513.4413.9412.9815.9413.1816.0314.6514.4716.719.1410.4112.5810.67
Ae
-1.5548-1.5121-1.5514-1.8051-1.6009-1.6716-1.3363-2.6666-2.2836-2.1737-2.0168-0.6069-1.0223-1.9237-1.5875
Be
132800152200166260173360160500180470130980191640183420176540176320102170118930140990127560
AAD(%)
0.050.070.060.040.080.050.060.050.060.080.080.030.050.040.06
AT(K)
847.8-1633.7833.6-1527.8836.0-1530.6846.8-1574.1845.6-1574.6784.1-1504.7775.3-1521.0815.6-1540.1800.7-1483.7795.5-1503.0774.4-1478.7812.1-1673.3822.8-1627.3746.1-1511.3818.2-1475.5
N
272224251818252618172317192416
189

Tableau 9.6 : Coefficients de l'équation TVF (Eq. 9.5).
Produit
SON-3SON-4SON-5SON-6SON-7SON-8SON
AVM-3AVM-4AVM-5AVM
BSN-5/16BSN-8/24BSN-11/32BSN-29/14
A
-2.5435-2.6165-2.4787-2.3336-2.0571-2.3655-1.7017-3.1035-2.3625-2.1206-2.0532-2.4410-2.6176-2.4937-3.1261
B
418045744055407537713757287237473520333432555351492032793918
Tx
588.2548.9581.8585.4601.6548.4587.7587.8579.6578.8565.1475.6515.2540.6575.9
AAD(%)
0.060.080.080.040.090.050.060.050.060.080.080.040.030.050.09
AT(K)
847.8-1633.7833.6-1527.8836.0-1530.6846.8-1574.1845.6-1574.6784.1-1504.7775.3-1521.0815.6-1540.1800.7-1483.7795.5-1503.0774.4-1478.7812.1-1673.3822.8-1627.3746.1-1511.3818.2-1475.5
N
272224251818252618172317192416
9.3 ENTROPIE DE CONFIGURATION
Par ajustement aux données viscosimétriques, les entropies de
configuration (Tableau 9.5) à Tg sont déterminées avec une incertitude d'environ
5% (Richet et Neuville, 1992). Il est évident que l'entropie de configuration d'un
verre dépend de sa température fictive. La variation d'entropie engendrée par une
variation de température fictive de Tj à T2 s'écrit
c -Cpl psdT.
TEq. 9.6
Par souci de cohérence, nous avons considéré tout au long de cette étude, des
températures fictives correspondant à des vitesses de refroidissement identiques à
celle imposée par la calorimétrie de chute. Mais des fortes variations de
température fictive pourraient modifier le contenu de la discussion que nous
allons mener.
190

Les variations d'entropie de configuration avec la température sont
représentées dans la Figure 9.4 pour une mole d'atomes du produit considéré. Dès
la transition vitreuse, les entropies de configuration diffèrent d'un facteur 3/2. A
1500 K, la différence maximale est d'environ un facteur 2. Il est difficile de
différencier clairement les produits présentant des anomalies de Cp (Chapitre 8)
d'après ces courbes cependant, comme le montre la comparaison entre les courbes
de BSN-5/16 et celles de la série AVM, on note une linéarité plus importante
lorsque la Cpconf a une dépendance positive vis-à-vis de la température (cas de
BSN-5/16). Au contraire, les courbes d'entropies fléchissent anormalement vite
pour les produits où l'anomalie est particulièrement bien marquée (série AVM).
L'entropie configurationnelle peut être scindée en une entropie de
configuration chimique, qui rend compte de la distribution d'éléments sur des sites
anioniques analogues, et une entropie de configuration déterminée par la
topologie de la structure (distribution des angles et longueurs de liasons par
exemple) (Richet et Neuville, 1991). Pour les liquides silicates simples, les
entropies de configurations à Tg varient de 1.5 à 3 J.g atoirf'.K"1 (Richet et
Neuville, 1992), les plus grandes valeurs étant attribuées aux produits présentant
la plus grande entropie chimique. Les produits de cette étude ont tous des
entropies de configurations supérieures à 3 J.g atom"1.K"1. La plus grande entropie
mesurée est celle du verre AVM (4.9 ± 0.3 J.g atorrf'.K"1). Il n'est pas étonnant de
voir que l'entropie configurationnelle d'une composition aussi complexe que celle
de l'AVM a une valeur parmi les plus élevées recensées à ce jour : les éléments
susceptibles d'occuper des sites de même type sont en nombre important (Si, B,
Al, etc.) d'où une entropie configurationnelle chimique importante. Notons, de
plus, que la majeure partie de l'entropie de configuration chimique est apportée
dès lors qu'il existe des faibles teneurs d'éléments susceptibles de se substituer les
uns aux autres.
191
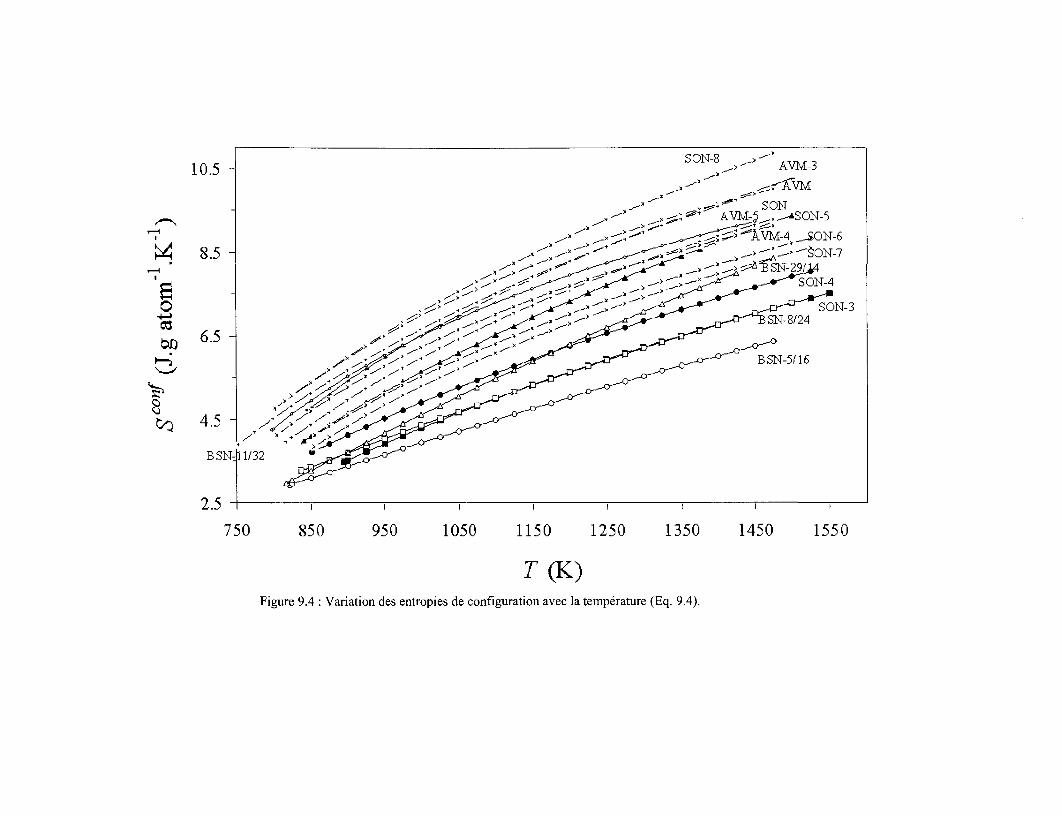
Ibo
10.5 -
-
8.5 -
6.5 -
-
4.5 -
BSN-
2.5 -
A11/32
i i i 1 i
SON-8
v̂ ̂ ̂ ; r^
i
SON
> ̂ CJ^-' ^SON-7
^ _ - ^ ^ S O N - 4
n-^-BSN-8/24
BSN-5/16
i i
750 850 950 1050 1150 1250 1350 1450 1550
Figure 9.4 : Variation des entropies de configuration avec la température (Eq. 9.4).
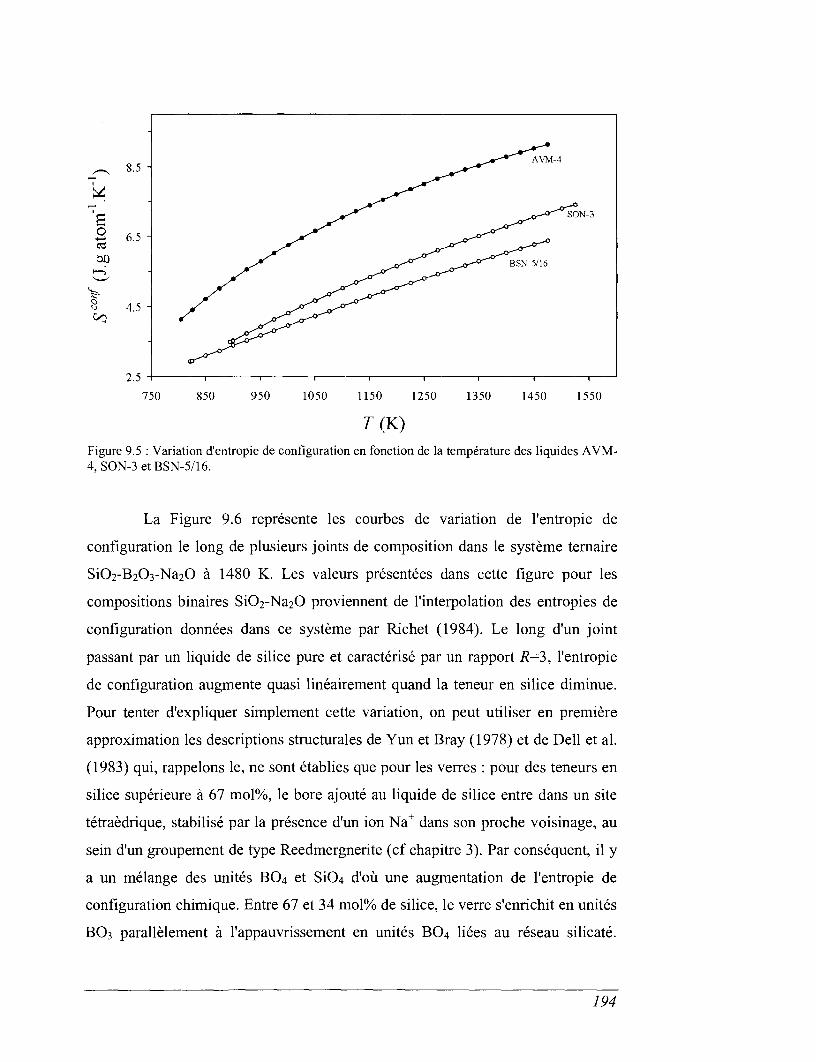
Of)
8.5 -
6.5 -
« 4.5 -
2.5
SON-3
BSN-5/16
750 850 950 1050 1150 1250 1350 1450 1550
T(K)Figure 9.5 : Variation d'entropie de configuration en fonction de la température des liquides AVM-4, SON-3etBSN-5/16.
La Figure 9.6 représente les courbes de variation de l'entropie de
configuration le long de plusieurs joints de composition dans le système ternaire
SiO2-B2O3-Na2Û à 1480 K. Les valeurs présentées dans cette figure pour les
compositions binaires SiO2-Na2O proviennent de l'interpolation des entropies de
configuration données dans ce système par Richet (1984). Le long d'un joint
passant par un liquide de silice pure et caractérisé par un rapport R=3, l'entropie
de configuration augmente quasi linéairement quand la teneur en silice diminue.
Pour tenter d'expliquer simplement cette variation, on peut utiliser en première
approximation les descriptions structurales de Yun et Bray (1978) et de Dell et al.
(1983) qui, rappelons le, ne sont établies que pour les verres : pour des teneurs en
silice supérieure à 67 mol%, le bore ajouté au liquide de silice entre dans un site
tétraèdrique, stabilisé par la présence d'un ion Na+ dans son proche voisinage, au
sein d'un groupement de type Reedmergnerite (cf chapitre 3). Par conséquent, il y
a un mélange des unités BO4 et SiÛ4 d'où une augmentation de l'entropie de
configuration chimique. Entre 67 et 34 mol% de silice, le verre s'enrichit en unités
BO3 parallèlement à l'appauvrissement en unités BO4 liées au réseau silicate.
194
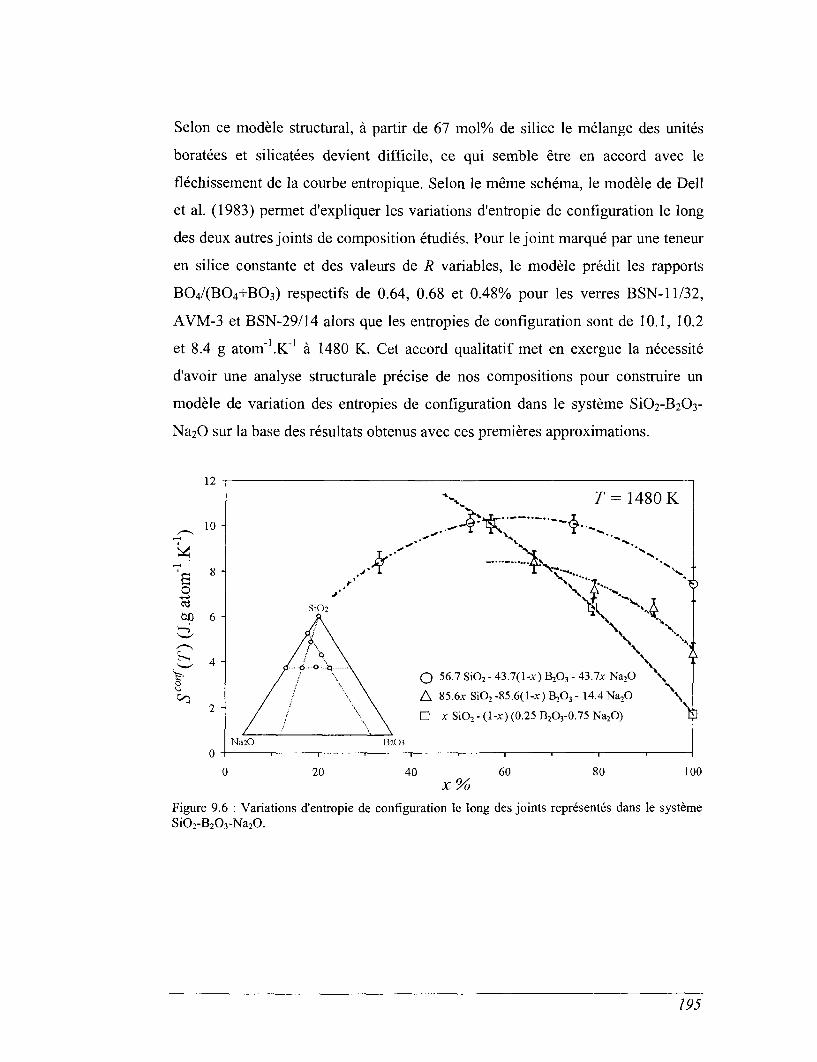
Selon ce modèle structural, à partir de 67 mol% de silice le mélange des unités
boratées et silicatées devient difficile, ce qui semble être en accord avec le
fléchissement de la courbe entropique. Selon le même schéma, le modèle de Dell
et al. (1983) permet d'expliquer les variations d'entropie de configuration le long
des deux autres joints de composition étudiés. Pour le joint marqué par une teneur
en silice constante et des valeurs de R variables, le modèle prédit les rapports
BO4/(BO4+BO3) respectifs de 0.64, 0.68 et 0.48% pour les verres BSN-11/32,
AVM-3 et BSN-29/14 alors que les entropies de configuration sont de 10.1, 10.2
et 8.4 g atom^.K'1 à 1480 K. Cet accord qualitatif met en exergue la nécessité
d'avoir une analyse structurale précise de nos compositions pour construire un
modèle de variation des entropies de configuration dans le système SiO
Na2Û sur la base des résultats obtenus avec ces premières approximations.
12
/—s 1 0 "
o
ÛO 6 -
f4 -
2 -
Na2O
0
S1O2
•*«.. 7 = 1480 K, . . . -1^- - -^ . . .
O 56.7 SiO2 - 43.7(l-x) B,O3 - 43.Ix Na2O
A 85.6x SiO2-85.6(l-x)B2O3- 14.4 Na2O
• xSiO2-(l-x)(0.25B2O3-0.75Na2O)
20 40X%
60 80 100
Figure 9.6 : Variations d'entropie de configuration le long des joints représentés dans le systèmeSiO2-B2O3-Na2O.
195

9.4 ENTROPIE DEFORMATION
Nous disposons maintenant des entropies vibrationnelles et
confïgurationnelles pour chacune de nos compositions. Il est donc possible de
calculer les entropies de formation des verres par rapport à leurs oxydes
constitutifs. Par "oxydes" on entend les composés cristallins y^xOy stables à la
température considérée et à pression ambiante. Ainsi, pour la silice, selon la
température , on fera référence au quartz-a (à 298 K par exemple) ou au quartz-P
(à partir de 847 K) et, dans le cas de B2O3, soit au cristal soit au liquide. Les
autres oxydes ne présentent pas de transition dans l'intervalle de température qui
nous intéresse.
Le calcul est effectué selon la relation suivante à la pression standard :
Eq.9.7
avec 5 y ( r ) = OV(0A )+ — O 7 H . Eq. 9.8A \ / .A. \ / J rri rji *
0 •* -V
A,r/f est la variation d'enthalpie associée à la transition se produisant à Ttr
(comprise entre 0 et T). Pour tous les verres, ce terme est nul. En revanche, pour la
silice à Ttr = 847 K et pour l'oxyde de bore à Ttr = 723 K, cette enthalpie
correspond respectivement à la transition a-p du quartz (Â/r//=0.655 kJ.mol"1,
Richet et al., 1982) et à la fusion du bore (A,r//=24.01 kJ.mol"1, Robie et
Hemingway, 1995). Pour les verres, l'entropie à 0 K est l'entropie de configuration
figée à la température de transition vitreuse ; pour les oxydes, ce terme est nul car
ces phases sont assimilées à des cristaux parfaits. Les données thermodynamiques
relatives aux oxydes proviennent de la base de données thermodynamiques de
Robie et Hemingway (1995) et de la publication de Richet et al. (1982) pour la
silice.
Le Tableau 9.7 donne les entropies de formation calculées à 298.15 K. Le
calcul des incertitudes est réalisé selon les relations suivantes :
196

Eq. 9.9
= ASx{0)+ 4^298 - s o ) X + &(ST ~S29S)x + ̂ Jr£U. Eq. 9.10avec ASX A5X(O)+A(529g 5c), + A(5r 5298)x
Les entropies de configuration à Tg sont connues à 5% prés, les
incertitudes sur les entropies relatives à 0 K sont de moins de 0.1% lorsqu'elles
ont été obtenues par calorimétrie adiabatique, et elles sont données dans le
Tableau 8.7 lorsqu'elles ont été estimées à partir du modèle des Cp partielles. Les
entropies relatives à 298.15 K sont connues à 0.2% près lorsqu'elles sont
déterminées par calorimétrie de chute et de l'ordre de 1 % lorsque seules les
données de DSC sont disponibles. Pour les oxydes, les incertitudes sont données
dans les références citées ci-dessus.
Tableau 9.7 : Entropie de formation desverres par rapport aux oxydes à 298 K.
Produit
SON-3SON-4SON-5SON-6SON-7SON-8AVM-3AVM-4AVM-5BSN-5/16BSN-8/24BSN-11/32BSN-29/14
A/5r(298 K)
(J.mor'.K"1)12.11 ±0.5916.75 ±2.7216.45 ±3.2119.10± 3.7515.93 ±4.7213.08 ±7.0417.08 ±0.8019.70 ±2.9720.07 ± 3.4511.35 ±0.4611.67 ±1.4914.82 ±0.6311.90 ±0.53
197

La contribution aux erreurs calculées la plus importante provient des
incertitudes inhérentes à l'utilisation du modèle de Cp partielles. Nous discuterons
ultérieurement de l'importance de ces erreurs sur le calcul global de l'enthalpie
libre de dissolution des verres.
Par comparaison avec les entropies de configuration (Tableau 9.5), nos
données montrent que l'entropie de configuration est la principale contribution à
l'entropie de formation (de 70 à 100%). Ce résultat n'est pas une surprise
puisqu'on a vu que les entropies vibrationnelles des verres pouvaient s'assimiler en
première approximation à la somme de celles des oxydes.
198

Chapitre 10
ENTHALPIES DE FORMATION
Malgré la faible proportion de travaux consacrés aux mesures
thermochimiques dans les systèmes borosilicatés, les données de calorimétrie de
dissolution sont assez bien représentées. Ostvold et Kleppa (1969), Hervig et
Navrotsky (1985), Julsrud et Kleppa (1986), Geisinger et al. (1988), de Yoreo et
Navrotsky (1990), Borisova et Ushakov (1992) ont utilisé les techniques de
dissolution en sels fondus sur de nombreux verres et liquides de ces systèmes pour
définir leur stabilité intrinsèque (tendance à dévitrifier, immiscibilité, diagramme
de phase, etc.). Suivant leur démarche nous avons mesuré les enthalpies de
dissolution des verres de confinement simplifiés. Après avoir présenté nos
résultats, nous exprimerons les méthodes de calculs des enthalpies de formation.
10.1 LESRESUL TATS DES DISSOLUTIONS
Les enthalpies de dissolution dans 2PbO.B2O3 à 970 K des compositions
de notre étude ont été obtenues selon les méthodes exposées dans le Chapitre 6 (§
6.5). Comme l'ont montré les mesures de capacité calorifique, les températures de
transition vitreuse sont de 100 à 200 K inférieures à la température du calorimètre
(970 K) : les résultats présentés dans le Tableau 10.1 correspondent par
conséquent à la phase liquide de chaque composition. Ce tableau donne l'enthalpie
mesurée à chaque dissolution et la moyenne des valeurs obtenues pour chaque
composition. On remarquera que certaines compositions n'ont fait l'objet que de 2
ou 3 dissolutions ; cette population n'étant pas statistiquement représentative, les
valeurs moyennes indiquées ne le sont qu'à titre indicatif et peuvent ne pas refléter
correctement les quantités de chaleur mises enjeu lors de la dissolution.
199
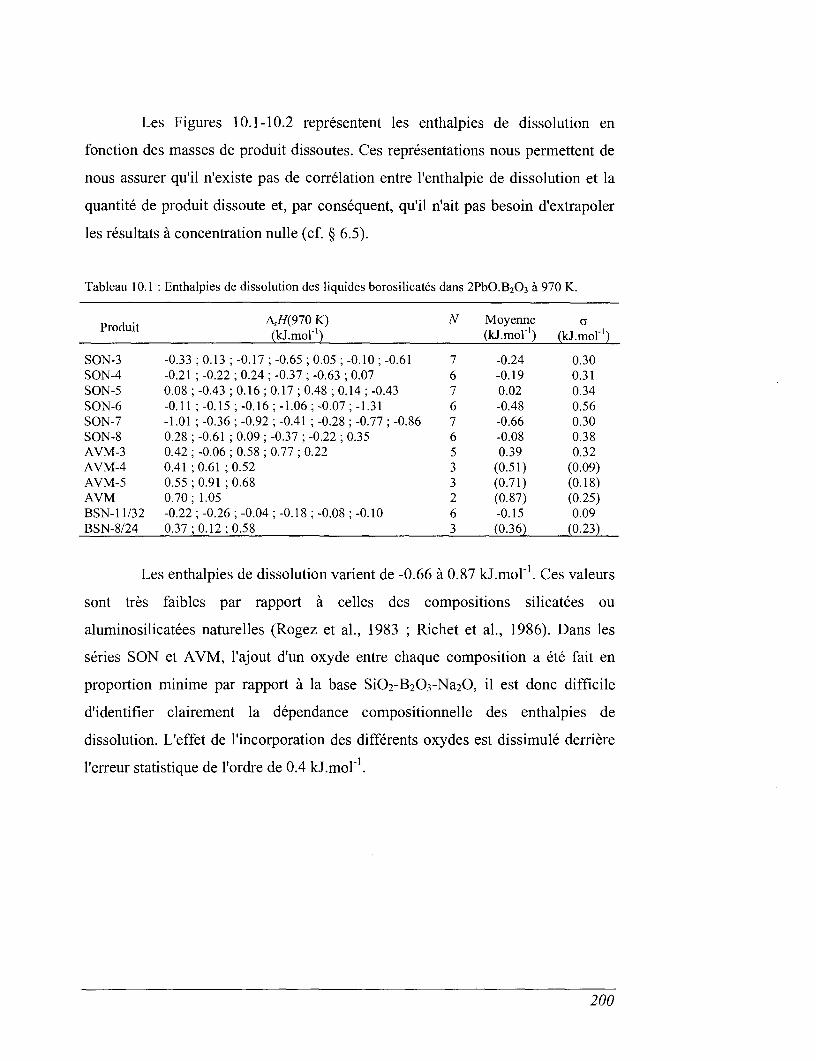
Les Figures 10.1-10.2 représentent les enthalpies de dissolution en
fonction des masses de produit dissoutes. Ces représentations nous permettent de
nous assurer qu'il n'existe pas de corrélation entre l'enthalpie de dissolution et la
quantité de produit dissoute et, par conséquent, qu'il n'ait pas besoin d'extrapoler
les résultats à concentration nulle (cf. § 6.5).
Tableau 10.1 : Enthalpies de dissolution des liquides borosilicatés dans 2PbO.B2O3 à 970 K.
Astf(970K) N Moyenne <j™ U (kJ.mor1) (kJ.mol1) (kJ.mor1)
SON-3 -0.33 ; 0.13 ; -0.17 ; -0.65 ; 0.05 ; -0.10 ; -0.61SON-4 -0.21 ; -0.22 ; 0.24 ; -0.37 ; -0.63 ; 0.07SON-5 0.08 ; -0.43 ; 0.16 ; 0.17 ; 0.48 ; 0.14 ; -0.43SON-6 -0.11 ;-0.15;-0.16;-1.06;-0.07;-1.31SON-7 -1.01 ; -0.36 ; -0.92 ; -0.41 ; -0.28 ; -0.77 ; -0.86SON-8 0.28 ; -0.61 ; 0.09 ; -0.37 ; -0.22 ; 0.35AVM-3 0.42 ; -0.06 ; 0.58 ; 0.77 ; 0.22AVM-4 0.41 ; 0.61 ; 0.52AVM-5 0.55 ; 0.91 ; 0.68AVM 0.70; 1.05BSN-11/32 -0.22 ; -0.26 ; -0.04 ; -0.18 ; -0.08 ; -0.10BSN-8/24 0.37; 0.12; 0.58
Les enthalpies de dissolution varient de -0.66 à 0.87 kJ.mol"1. Ces valeurs
sont très faibles par rapport à celles des compositions silicatées ou
aluminosilicatées naturelles (Rogez et al., 1983 ; Richet et al., 1986). Dans les
séries SON et AVM, l'ajout d'un oxyde entre chaque composition a été fait en
proportion minime par rapport à la base SiO2-B2O3-Na2O, il est donc difficile
d'identifier clairement la dépendance compositionnelle des enthalpies de
dissolution. L'effet de l'incorporation des différents oxydes est dissimulé derrière
l'erreur statistique de l'ordre de 0.4 kJ.mol"!.
767676533263
-0.24-0.190.02-0.48-0.66-0.080.39
(0.51)(0.71)(0.87)-0.15(0.36)
0.300.310.340.560.300.380.32
(0.09)(0.18)(0.25)0.09
(0.23)
200
mol
)/(
970
K)
i on.
050
000
-0 50
-100
-1 50
SON-3
50 100 150 200 250 300 350 400
masse d'échantillon dissoute (mg)
100 150 200 250 300 350 400
masse d'échantillon dissoute (mg)
0 50 100 150 200 250 300 350
masse d'échantillon dissoute (mg)
50 100 150 200 250 300 350 400
masse d'échantillon dissoute (mg)
50 3 150 200 250 303 350
masse d'échantillon dissoute (mg)
50 100 150 200 250 300 350
masse d'échantillon dissoute (mg)
Figure 10.1 : Enthalpies de dissolutions dans 2PbO.B2O3 à 970 K mesurées pour les liquides de lasérie SON en fonction de la masse d'échantillon dissoute.
201
100 200 3OD 400
masse d'échantillon dissoute (mg)
100 200 300
masse d'échantillon dissoute (mg)
(kJ/
mol
)(9
701
i •
05 •
0 •
-0 5 •
-1 .
-1 5 .
•
AVM-5
0 100 200 300 400
masse d'échantillon dissoute (mg)
0 100 200 300 400
masse d'échantillon dissoute (mg)
100 200 300 400
masse d'échantillon dissoute (mg)
0 100 200 300 400
masse d'échantillon dissoute (mg)
Figure 10.2 : Enthalpies de dissolutions dans 2PbO.B2O3 à 970 K mesurées pour les liquides desséries AVM et BSN en fonction de la masse d'échantillon dissoute.
202

La plus grande contribution aux variations d'enthalpies de dissolution le
long d'un joint de composition est due aux équilibres acide-base entre les divers
composants du système considéré (Navrotsky, 1993) : par exemple, les énergies
associées à la rupture des liaisons Si-O-Si et à la formation des liaisons M-O-Si
lorsqu'un oxyde alcalin est ajouté à la silice pure. Les énergies associées au
mélange d'éléments voisins, tel que la substitution d'un oxyde alcalin par un autre
dans un liquide ou un verre (à teneur en silice égale), sont plutôt faibles en
comparaison. Les compositions globales des produits étudiés peuvent être
projetées dans un système simple de composants fictifs qui décrit
schématiquement une information structurale. La Figure 10.3 montre la projection
des produits de cette étude dans le système à trois pôles, S1O2, les composants
susceptibles d'être formateurs de réseau (B2O3, AI2O3, ZrCh) et les modificateurs
de réseau (Na2Û, CaO, MgO). Cette représentation montre que le domaine couvert
par nos compositions est étroit, ce qui peut expliquer la faible amplitude observée
sur l'ensemble des résultats. D'autre part, on distingue les séries AVM et SON
dans ce type de diagramme, en accord avec la tendance endothermique des
dissolutions de la série SON et, au contraire, avec le caractère exothermique des
dissolutions de la série AVM.
SiO2
• Série SONo Série AVMA Série BSN
Na2O + CaO+ MgO
B2O3 + AI2O3+ ZrO2
Figure 10.3 : Projection des produits étudiés dans un diagramme ternaire SiO2, modificateurs deréseau, et éléments pouvant se substituer à la silice dans le réseau vitreux.
203

10.2 CALCUL DES ENTHALPIES DE FORMATION
A 970 K, les enthalpies de formation d'un verre par rapport aux oxydes
sont obtenues par différence de l'enthalpie de dissolution du verre dans
2PbO.B2O3 à 970 K et de la somme des enthalpies de ses oxydes constitutifs (dans
le même solvant et à même température) pondérées par la fraction molaire de ces
derniers.
Le Tableau 10.2 contient les données, concernant les oxydes, nécessaires
au calcul de l'enthalpie de formation de nos verres. Ces données tirées de la
littérature (cf. références dans le Tableau 6.3) sont, en général, des données
expérimentales obtenues par dissolution des oxydes dans un bain de 2PbO-B2Û3
pour des températures voisines de 970 K. Les légères variations de températures
de dissolution sont négligées, car, comme le montre la Figure 10.4, l'enthalpie de
dissolution des oxydes dans 2 PbO.B2O3 ne semble pas dépendre de façon
sensible de la température dans l'intervalle 960-980 K.
D'autre part, les enthalpies de dissolution des oxydes utilisées
proviennent de travaux qui ont tous en commun la détermination de l'enthalpie de
dissolution de la silice amorphe ou de l'alumine. Nos tests préliminaires de
l'appareillage ayant concerné les dissolutions de ces deux matériaux, une
comparaison entre nos mesures sur ces produits et les résultats cités dans les
publications de référence nous permet de vérifier que les compositions des
solvants utilisés sont comparables. Le Tableau 10.3 donne les valeurs
préalablement obtenues ainsi que nos mesures. Le bon accord entre ces résultats
justifie l'utilisation des enthalpies de dissolution des oxydes données dans ces
publications pour le calcul des enthalpies de formation.
204
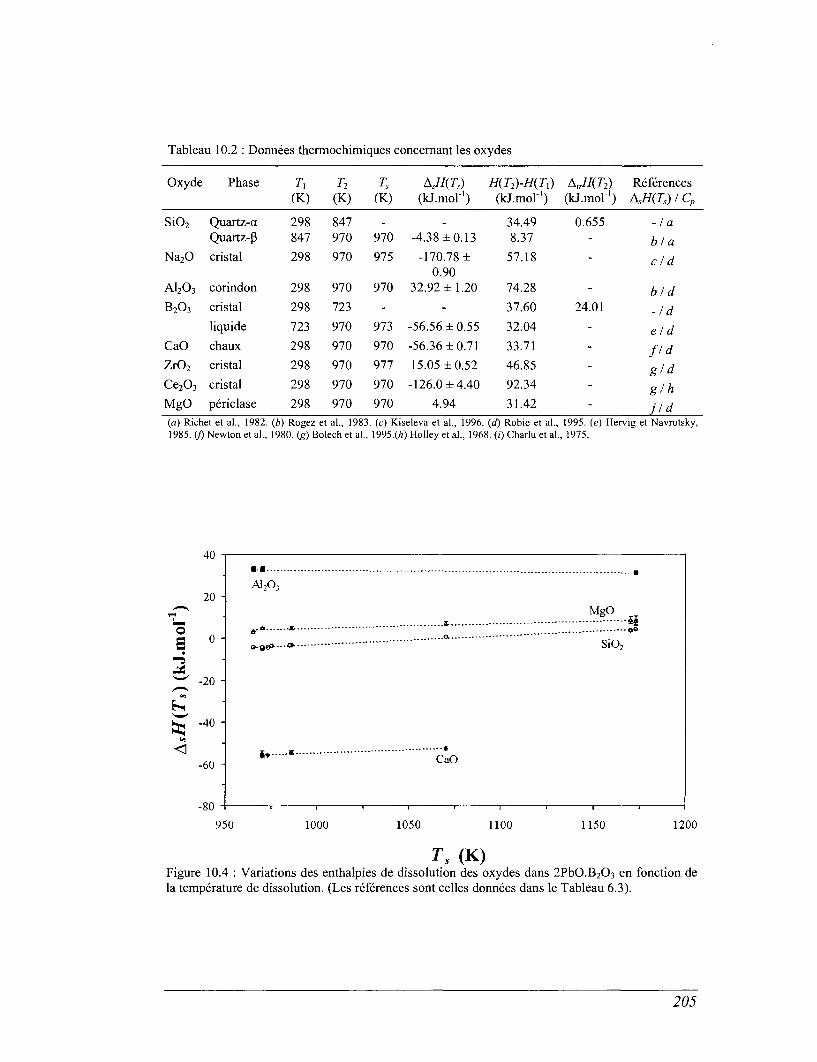
Tableau 10.2 : Données thermochimiques concernant les oxydes
Oxyde
SiO2
Na2O
A12O3
B2O3
CaO
ZrO2
Ce2O3
MgO
Phase
Quartz-aQuartz-p
cristal
corindon
cristal
liquide
chaux
cristal
cristal
périclase(a) Richet et al., 1982.1985. (/) Newton et al.,
Tx(K)
298847
298
298
298
723
298
298
298
298(b) Rogez t
T2
(K)
847970
970
970
723
970
970
970
970
970:t al., 1983
1980. (g)Bolechetal.,
Ts
(K)
_970
975
970
-
973
970
977
970
970
ASH(TS)(kJ.mof1)
-4.38 ±0.13
-170.78 ±0.90
32.92 ±1.20
-
-56.56 ±0.55
-56.36 ±0.71
15.05 ±0.52
-126.0 ±4.40
4.94. (c) Kiseleva et al., 1996.1995.(h) Holley et al., 1968
H(T2)-H(Tx)(kJ.mol"1)
34.498.37
57.18
74.28
37.60
32.04
33.71
46.85
92.34
31.42(d) Robie et al.,:. (0 Charlu et al.
KH{T(kJ.mol
0.655--
-
24.01
-
-
-
-
-1995. (e)
, 1975.
2) Références-1) AsH(Ts)'Cp
-la
bla
cl d
bid
-Id
eld
fidgld
glh
I IdHervig et Navrotsky,
950 1000 1050 1100 1150 1200
(K)Figure 10.4 : Variations des enthalpies de dissolution des oxydes dans 2PbO.B2O3 en fonction dela température de dissolution. (Les références sont celles données dans le Tableau 6.3).
205

Tableau 10.3 : Enthalpies de dissolution de l'alumine et de la silice vitreuse dans 2PbO.B2O3
Références
a-Al2O3
Rogez et al., 1983Kiseleva et al., 1996Hervig et Navrotsky, 1985Newton et al., 1980Charluetal., 1975Ce travailSiO2 (amorphe)Hervig et Navrotsky, 1985Ce travail
T,(K)
970975973970970970
973970
AsH(Ts)(kJ.mol1)
32.92 ±1.2032.8 ±0.3
32.92 ±1.2032.55 ±0.9232.35 ±0.34
32.69 (± 0.58)
-10.92 ±0.23-10.87 (±0.13)
N
99
16193
82
Pour une température T, l'enthalpie de dissolution est obtenue selon la
relation suivante :
T
AfH°(T) = AfH°{970K)+ \AC pdT -J^AtrH, Eq. 10.1970A:
avec ACp =Cpg-^ xt Cpi Eq. 10.2i
où Cpg et Cpi sont les capacités calorifiques du verre considéré et de son oxide
constitutif i, AtrH est la somme des enthalpies de transition pouvant avoir lieu
entre T et 973 K.
Les enthalpies de formation à 970 K et 298 K sont données dans le
Tableau 10.4 pour l'ensemble des verres simplifiés ayant donné lieu à des mesures
d'enthalpie de dissolution.
Les incertitudes sur les enthalpies de formation sont données par la
relation suivante :
A(AfH°{T))= Zxt [à{AsH(Ts ))+ A(HT - HTs )+ A(AtrH)\i
+ [A{ASH{TS))+A{HT -HTJ\g. Eq. 10.3
L'erreur sur les enthalpies de dissolution est assimilée à l'erreur
statistique obtenue sur l'ensemble des dissolutions réalisées pour chaque
composition considérée. L'enthalpie relative est mesurée à 0.5% près par
calorimétrie de chute et à 2% près par calorimétrie différentielle à balayage.
206
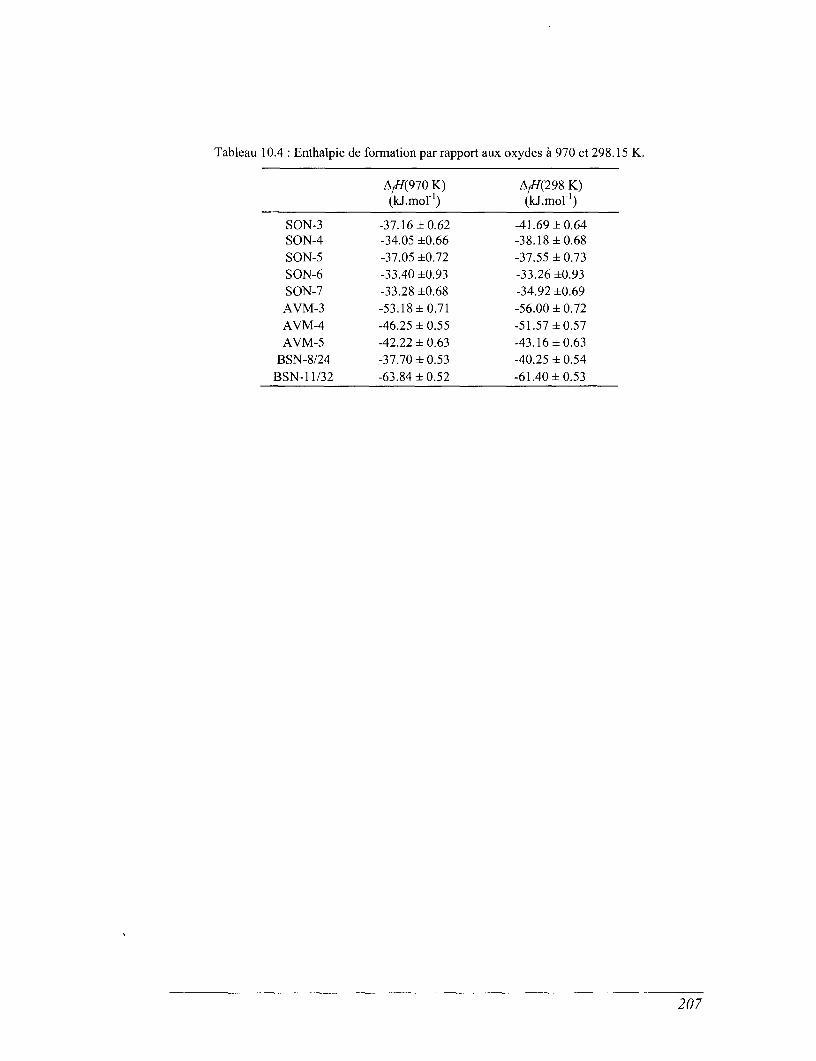
Tableau 10.4 : Enthalpie de formation par rapport aux oxydes à 970 et 298.15 K.
SON-3SON-4SON-5SON-6SON-7AVM-3AVM-4AVM-5
BSN-8/24BSN-11/32
A/f(970 K)(kJ.mol"1)
-37.16 ±0.62-34.05 ±0.66-37.05 ±0.72-33.40 ±0.93-33.28 ±0.68-53.18 ±0.71-46.25 ± 0.55-42.22 ± 0.63-37.70 ± 0.53-63.84 ±0.52
A//(298 K)(kJ.mol1)
-41.69 ±0.64-38.18 ±0.68-37.55 ±0.73-33.26 ±0.93-34.92 ±0.69-56.00 ± 0.72-51.57 ±0.57-43.16 ±0.63-40.25 ± 0.54-61.40 ±0.53
207

QUATRIEME PARTIE
Apport de la thermodynamique
à la compréhension des
cinétiques d'altération des
verres

La vitesse d'altération dans l'eau des verres borosilicatés d'intérêt
nucléaire diminue au cours du temps, au fur et à mesure que la réaction avance.
En effet, au premier instant de l'altération dans de l'eau initialement pure, le verre
s'altère à une vitesse initiale VQ, qui ne dépend que de la composition du solide, de
la température et du pH. Par la suite, la vitesse d'altération peut diminuer de plus
de 4 ordres de grandeur par rapport à VQ. Deux grandes hypothèses sont avancées
pour expliquer la diminution de la vitesse d'altération de ces verres au cours du
temps :
1. la solution altérante se sature en espèces aqueuses provenant de la dissolution
du verre, et tend graduellement vers un équilibre de saturation vis-à-vis du
verre. L'affinité réactionnelle diminue et tend à s'annuler.
2. le produit d'altération appelé gel, qui se forme à l'interface réactionnelle,
devient progressivement protecteur. Il limite l'accès des espèces aqueuses
réactives (OH", H3O+, H2O) vers le verre sain et l'évacuation des espèces
produites tels que H4SiO4° vers la solution altérante. Un tel processus serait
analogue au phénomène de passivation des métaux par formation d'une
couche d'oxyde protectrice.
Grâce à nos mesures des propriétés thermodynamiques, il est possible de
calculer les enthalpies libres de dissolution des verres dans une solution altérante,
et d'évaluer en détail la première hypothèse mécanistique, et la loi cinétique
correspondante.
211

Chapitre 11
ENTHALPIE LIBRE DE DIS SOL UTION DES
VERRES SIMPLIFIES
Les propriétés thermodynamiques des verres de confinement simplifiés
ont été mesurées et permettent le calcul de l'enthalpie libre de formation. Par
différence avec les enthalpies des espèces aqueuses produites lors de la réaction
du verre avec l'eau, nous obtenons les enthalpies libres de dissolution du verre.
Dans ce chapitre nous verrons le détail des calculs en essayant d'identifier les
sources d'erreurs et d'évaluer les incertitudes.
11.1 CALCUL DES ENTHALPIES DE FORMA TION PAR
RAPPORT AUX OXYDES
L'enthapie libre d'une réaction (formation, dissolution, etc.) à la
température T est donnée par :
ArG°(T)=ArH0(T)-TArS°{T). Eq. 11.1
Pour les réactions de formation des verres par rapport aux oxydes stables,
les variations d'enthalpies ont été déterminées dans le Chapitre 10 et celles de
l'entropie dans le Chapitre 9. Le tableau 11.1 donne les valeurs, à 25 et 90°C, des
enthalpies libres de formation par rapport aux oxydes pour les verres simplifiés
pour lesquels toutes les données thermochimiques ont pu être mesurées ou
estimées. Les températures considérées sont typiques des expériences de
lixiviation effectuées en laboratoire.
212

Tableau 11.1: Enthalpie libre de formation des verres par rapport aux oxydes à 25et 90°C.
Produit
SON-3SON-4SON-5SON-6SON-7AVM-3AVM-4AVM-5BSN-8/24BSN-11/32
A/}(25°C)(kJ.mol"1)
-45.30-43.18-42.46-38.95-39.67-61.09-57.44-49.15-43.73-65.82
±0.81±±±±±±±±
1.491.682.052.100.961.461.660.98
±0.72
A/î(90' °C)(kJ.mol"1)
-46.08-44.26-43.53-40.20
±±±±
-40.70±-62.20-58.69-50.45-44.48-66.78
±±±±
0.901.721.952.342.461.071.711.941.13
±0.81
Pour une température donnée, on observe que les variations d'enthalpies
libres de formation des verres dépendent fortement de la composition. Entre les
diverses compositions ternaires, les différences peuvent atteindre 20 kJ.mol"1.
Cette différence maximale est observée entre les compositions marquées des plus
grandes différences en teneur de silice.
11.2 EVALUATION DES ERREURS
L'incertitude est calculée selon la relation suivante :
A ( A / G ° ( 7 ) ) = à(AfH°{T))+TA(AfS°(T)). Eq. 11.2
Regardons le poids respectif de chaque contribution dans le calcul des
erreurs à travers l'exemple du verre AVM-5 :
AjH°(29S K)= -43.16 ± 0.63 kJ.mol"1
A/S°(298 K)= 20.07 ± 3.45 J.mor'.K1
Dans ce cas on constate que l'erreur sur la contribution entropique est
importante car elle représente 62% de l'erreur totale sur l'enthalpie libre. Cette
erreur sur l'entropie est due à l'utilisation du modèle des Cp partielles et ne reflète
213

pas directement une erreur expérimentale. Considérons maintenant un cas où
toutes les propriétés ont été mesurées comme le SON-3 par exemple.
A/#°(298 K)= -41.69 ± 0.62 kJ.mol"1
A/5°(298 K)= 12.11 ± 0.59 mor'.K"1
L'erreur apportée par la contribution entropique n'est ici que de 21%.
Cette valeur est plus représentative de la démarche expérimentale. En effet, les
sources d'erreur sont plus nombreuses dans la détermination des enthalpies de
formation qui nécessite la réalisation d'un cycle de dissolution entre le verre et ses
oxydes constitutifs.
11.3 INFLUENCE DELÀ TEMPERATURE FICTIVE SUR LES
PROPRIETES DEFORMA TION
Le verre étant en équilibre métastable, son enthalpie et son entropie
dépendent de son histoire thermique (cf. Chapitre 2). Pour pouvoir définir les
propriétés thermodynamiques de façon rigoureuse nous avons défini l'état de
configuration de nos verres par une température fictive égale à la température de
transition vitreuse définie lors des mesures de capacité calorifique (cf. Chapitre 8).
Si on effectue un recuit sur l'un des verres étudiés, on modifiera l'état de
configuration qui avait été figé à la transition vitreuse lors de la trempe du liquide
parental et les propriétés du verre devront être reévaluées.
La différence d'enthalpie entre deux verres de même composition mais
avec des températures fictives différentes, Tj et T2 s'écrit:
= j(Cpl-Cpg]lT. Eq.11.3
Une expression analogue est obtenue pour la variation d'entropie
engendrée par un tel changement de configuration (Eq. 9.6).
Prenons l'exemple du verre SON-7 pour lequel les propriétés
thermodynamiques ont été déterminées pour une température fictive de 854 K. Par
analogie avec des expériences de relaxation menées sur d'autres borosilicates

(Sipp et al., 1997), nous estimons qu'une température fictive de l'ordre de 790 K
pourrait être obtenue par un recuit prolongé de plusieurs jours à 829 K,
température qui correspond à une viscosité de 1013-5 Pa.s. A 298 K, l'enthalpie
libre de formation du verre par rapport aux oxydes serait alors de -37.49 kJ/mol
soit une différence de 5.5% par rapport au verre de température fictive égale à 854
K.
Cet exemple montre combien il est imporant de bien caractériser la
température fictive d'un verre pour donner une réelle signification au calcul
d'erreur sur les calculs thermochimiques. Il est cependant rare que, lors des
manipulations de laboratoires, les traitements thermiques que peuvent subir les
échantillons soient de suffisamment longue durée et à des températures permettant
une mobilité atomique suffisante pour modifier la température fictive de l'ordre de
grandeur suggéré dans l'exemple précédent. En revanche, dans les conditions de
stockage souterrain on pourrait s'en approcher. Le scénario thermique subi par un
colis de verre a été déterminé lors d'un essai technologique en disposant plusieurs
thermocouples au cœur et à la surface du conteneur (Maillot, 1989). La
température chute de 1423 K à environ 673 K en moins de 40 heures. Cette phase
correspond à la trempe naturelle de la fonte. Un refroidissement extrêmement lent
s'instaure au-delà de cette première période, limité par la chaleur dégagée par
l'activité résiduelle des radionucléides. Basée sur la décroissance radioactive, une
étude de l'évolution thermique à cœur du conteneur prévoit une température de
500 K après 10 ans, et 475 K après 30 ans (Bernier, 1980). Par extrapolation de la
courbe d'équilibre des viscosités jusqu'à 500 K, nous obtenons pour le verre SON-
7 une viscosité qui suppose l'absence quasi-totale de mobilité atomique. Par
conséquent, pour évaluer l'influence de la relaxation structurale sur les propriétés
thermodynamiques, il faudra évaluer précisément les températures auxquelles sont
soumis les verres au cours de la première année pour pouvoir extrapoler avec
suffisament de fiabilité les données de laboratoire.
215

11.4 CALCUL DE L 'ENTHALPIE LIBRE DE DISSOLUTION DES
VERRES DANS L 'EAU
Selon l'équation 11.1, l'enthalpie libre de dissolution d'un verre dans une
solution altérante s'écrira en fonction des propriétés du verre, de la solution
altérante (l'eau par exemple) et de celles des espèces aqueuses résultant de
l'altération.
Pour calculer les enthalpies de dissolution du verre dans l'eau à partir des
propriétés de formation par rapport aux oxydes, nous effectuons un cycle
réactionnel tel que celui présenté ci-dessous pour le verre hypothétique
SiNa2B2O6 :
SiNa2B2O6 < -^P > SiO2 + Na2O + B2O3
+2H2°\ ^ +2H+ I 2 +3#2O \ 3
HASi0A + 2Na++H2O + 2B(OH)3
La réaction SiNa2B2O6 + AH2O + 2H+ «-• H4Si04 + 2Na+ + 2B(OH\
est équivalente thermodynamiquement à la réaction 5, nous obtenons donc son
enthalpie libre comme suit :
AreacG(T) = AG5 = -AfG + AG{ + AG2 + AG3. Eq. 11.4
Les energies des réactions de dissolution des oxydes dans l'eau (AGi,
AG2, etc.) n'ont pas fait l'objet de mesures dans le cadre de ce travail. Ces données
ont, pour la plupart des oxydes, été reportées pour la température de 25°C dans la
littérature. Cependant, les constantes d'équilibre K de ces réactions sont connues
avec une incertitude variable, allant de quelques pourcent pour les minéraux
simples et bien cristallisés (c'est le cas du quartz) à des facteurs pouvant atteindre
10 pour les oxydes et hydroxydes d'éléments tri-ou tétravalents (Michard, 1989).
Gislason et Arnosson (1993) notent des écarts pouvant atteindre deux ordres de
grandeur entre différentes bases de données thermodynamiques. Les valeurs que
nous avons utilisées (Tableau 11.2) sont des données récentes auxquelles nous
accordons une confiance de 2%.
216

Tableau 11.2: Propriétés thermochimiques d'hydratation des oxydes à 25°C.
Réaction logK25oC AdissG25°c Arf,ss//25.c Arf/MCp25oC Références
(kJ-mol1)
SiO2 + 2 H2O ^> H4Si04° -3.9483 22.54 21.45 -0.027 Rimstidt, 1997
1/2 (B2O3 + 3 H2O) <-> B(OH)3° 3.0064 -17.16 -8.09 -101.291 JANAF, 1985
1/2(A12O3 + 5H2O)^A1(OH)4" + H+ -13.7316 78.38 51.83 -244.630 Wesolowski, 1992
1/2 (Na2O + 2 Ff) <-• Na+ + 1/2 H2O 33.4586 -190.98 -174.26 13.024 JANAF, 1985
CaO + 2 Ff <-+ Ca++ + 2 H2O 32.7003 -186.65 -213.46 -4.165 JANAF, 1985
MgO + 2 Ff <-» Mg++ + H2O 21.5911 -123.24 -151.23 22.108 JANAF, 1985
Pour calculer les valeurs de l'enthalpie libre de dissolution à une
température supérieure à 25°C, AreacG°(25°C) ne suffit pas. Les relations
thermodynamiques montrent que Areac//°(25°C) ou AreacS°(25°C) et à.reacCp(T)
sont également nécessaires. L'expression exacte du logarithme de la constante
d'équilibre à la température T s'écrit en fonction des propriétés connues à une
température de référence To comme suit :
Malheureusement, les variations de capacité calorifique avec la
température des espèces aqueuses n'ont été mesurées que dans très peu de cas. Par
conséquent, afin d'utiliser l'équation 11.4, nous devons faire certaines
approximations. Selon les données disponibles plusieurs approximations sont
possibles :
considérer l'enthalpie de réaction constante,
considérer la variation de capacité calorifique de la réaction constante.
Nous avons receuilli suffisamment de données pour ne faire que la
seconde approximation. Comme cela a été discuté par Puigdomenech et al.
(1996), cette approximation est assez satisfaisante entre 273 et 473 K. En effet, les
capacités calorifiques des espèces aqueuses montrent en général une augmentation
217

avec la température mais ont un maximum entre 325 et 375 K puis commencent à
diminuer. La capacité calorifique à To est donc supposée représenter une valeur
moyenne des capacités calorifiques entre 273 et 473 K.
Les constantes d'équilibre à 90°C des réactions de dissolution calculées
suivant cette approximation, à l'aide de l'équation 11.4 sont reportées dans le
Tableau 11.3.
Tableau 11.3 : Constante d'équilibre et enthalpie libre d'hydratation des oxydes à 90°C
Réaction log K9o°c Arf,MG90»c(kJ.mol"1)
SiO2 + 2 H 2 0 <-<• RtSiCV -3.2758 18.6979
1/2 (B2O3 + 3 H 20) <-> B(OH)3° 2.6563 -15.1620
1/2 (A12O3 + 5 H2O) <-> A1(OH)4" + H+ -12.3394 70.4320
1/2 (Na2O + 2 H+) <-> Na+ + 1/2 H2O 28.0063 -159.8574
CaO + 2 H+ <-• Ca++ + 2 H2O 26.6266 -151.9819
MgO + 2 H+ <-> Mg++ + H2O 16.8699 -96.2916
Nous disposons maintenant de toutes les données pour effectuer le calcul
des enthalpies libres de dissolution des verres simplifiés dans l'eau. Les résultats
sont présentés dans le Tableau 11.4.
Tableau 11.4 : Enthalpies libres et constantes d'équilibre des réactions de dissolution des verresdans l'eau à 25 °C et 90°C (voir texte pour la définition des réactions et les références des donnéesutilisées).
AdissG(25°C) LogK25oC /W?(90oC) LogK90=c
(kJ.mol1) (kJ.mol1)
SON-3 0.28 ±2.32 -0.050 ±0.407 -0.31 ±2.45 0.044 ±0.429SON-4 7.15 ±3.06 -1.253 ±0.535 7.55 ±3.33 -1.086 ±0.584
SON-5AVM-3AVM-4AVM-5BSN-8/24BSN-11/32
-5.73-20.34-3.90
-19.00-7.72
-48.91
± 3 .± 3
± 3 .± 3
±2.
43.1167.9068
±3.52
1.004 ±0.6013.563 ±0.5440.683 ± 0.6423.329 ±0.6831.353 ±0.4708.568 ±0.617
-5.25 ±3 .-21.32 ±3-3.36 ±3 .
-17.82 ±4
74.2698.23
-8.30 ±2.86-50.51 ±3.67
0.755 ±0.6553.067 ± 0.5720.483 ± 0.6982.564 ±0.7411.194 ±0.5027.266 ± 0.644
Les erreurs sur les valeurs à 90°C ont été évaluées en considérant que
l'erreur relative sur les constantes d'équilibre des réactions de dissolution des
218
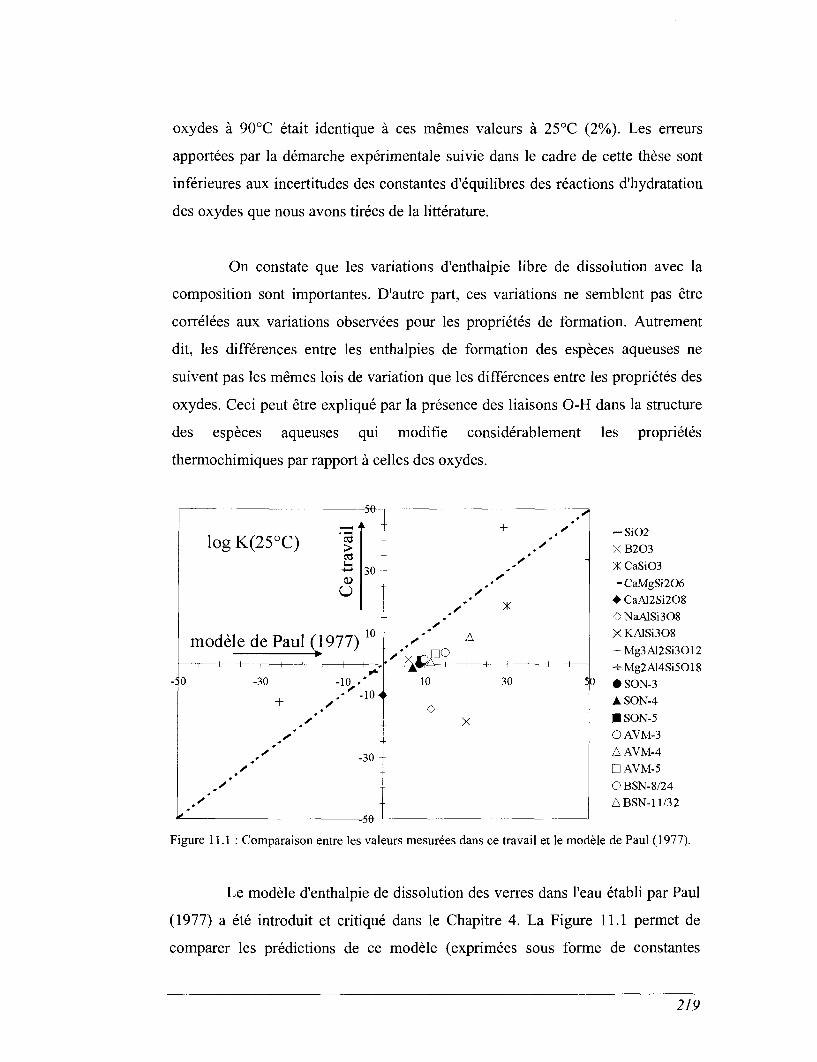
oxydes à 90°C était identique à ces mêmes valeurs à 25°C (2%). Les erreurs
apportées par la démarche expérimentale suivie dans le cadre de cette thèse sont
inférieures aux incertitudes des constantes d'équilibres des réactions d'hydratation
des oxydes que nous avons tirées de la littérature.
On constate que les variations d'enthalpie libre de dissolution avec la
composition sont importantes. D'autre part, ces variations ne semblent pas être
corrélées aux variations observées pour les propriétés de formation. Autrement
dit, les différences entre les enthalpies de formation des espèces aqueuses ne
suivent pas les mêmes lois de variation que les différences entre les propriétés des
oxydes. Ceci peut être expliqué par la présence des liaisons O-H dans la structure
des espèces aqueuses qui modifie considérablement les propriétés
thermochimiques par rapport à celles des oxydes.
— SiO2XB2O3X CaSiO3-CaMgSi2O6
• CaA12Si2O8O NaAlSi3O8X KAlSi3O8+ Mg3A12Si3O12+ Mg2A14Si5O18
0 • SON-3À SON-4• SON-5O AVM-3A AVM-4DAVM-5O BSN-8/24A BSN-11/32
log K(25°C)
modèle de Pau]1 1 1 1 ! f
0 -30
yy
y'
î tra
vail
u
-S6-k
30-
^1977) 1OΗi—i—i—Ï
- îo . ' •*•.- -1(H
yy
-30 -
-56-
y
10
O
*y'
y'A
X
+ y'
yy'
—i 1 1 1
30 5
Figure 11.1: Comparaison entre les valeurs mesurées dans ce travail et le modèle de Paul (1977).
Le modèle d'enthalpie de dissolution des verres dans l'eau établi par Paul
(1977) a été introduit et critiqué dans le Chapitre 4. La Figure 11.1 permet de
comparer les prédictions de ce modèle (exprimées sous forme de constantes
219

d'équilibre des réactions) aux valeurs mesurées dans notre étude et à celles
déterminées grâce aux données d'enthalpie et d'entropie de formation présentes
dans la littérature (cf. référence dans la légende de la Figure 4.5). La droite en
tirets symbolise une corrélation idéale. On constate dans la plupart des cas que les
prédictions du modèle surestiment les enthalpies libres de dissolution. On se
rapportera à la critique du modèle faite dans le Chapitre 4 pour les explications de
ce désaccord.
220

Chapitre 12
COMPARAISON AVEC LES TESTS DE LIXIVIATION
12.1 EXPERIENCES DE LIXIVIATION
Nous avons vu dans le Chapitre 4 que le formalisme cinétique, plus
particulièrement examiné au cours des études d'altération des verres, basé sur les
travaux de Aagard et Helgeson (1982), et Lasaga (1981abc, 1984), était donné par
l'équation 4.1. Nous réécrivons ici de façon simple cette relation, non plus en
terme d'avancement de la réaction et d'affinité mais en terme de vitesse :
l-e RT
où V est la vitesse de dissolution instantanée, Q est le produit d'activité ionique et
K le produit de solubilité pour la réaction de dissolution considérée.
Afin d'évaluer l'aptitude de l'équation 12.1 à décrire la réalité des
processus réactionnels, nous avons utilisé des expériences de lixiviation de fines
poudres de verre SON-3 et d'un verre de composition albitique réalisées au centre
de Marcoule du Commissariat à l'Energie Atomique dans le Laboratoire d'Etude
de l'Altérabilité des Matériaux (LEAM). Le verre SON-3 que nous avons
synthétisé diffère légèrement de la composition utilisée pour ce test de lixiviation
sans excéder pour autant des variations supérieures au pourcent. Ce dernier a été
synthétisé, et son altération caractérisée, dans le cadre de la thèse de C. Jégou
(1998).
Ces tests de lixiviation ont été réalisés dans de l'eau initialement pure à
90°C, avec des rapports S/V (surface de verre divisée par le volume de solution),
221

égaux à 80 cm"1. Ces conditions expérimentales favorisent l'atteinte rapide de
forts progrès de la réaction, avec des états stationnaires de dissolution susceptibles
d'approcher des conditions de saturation. Le protocole opératoire est décrit par
LeTurcqetal(1999).
Les concentrations des éléments Si, B, Na et Al sont dosées en solution
par ICP-AES.
Les vitesses d'altération des verres sont calculées à chaque échéance de
temps à partir du dosage d'un élément chimique considéré comme traceur de
l'altération (B et Na). Les formules suivantes sont employées pour calculer tout
d'abord les pertes de masses élémentaires normalisées NL(i) puis les
vitesses d'altération V(J):
' V
où C(J, t) est la concentration (mg-1"1) de l'élément / dans la solution à l'échéance t,
S/F est le rapport de la surface du verre et du volume de la solution (m"1) et Xt est
la fraction massique de l'élément / dans le verre.
NU (exprimé en g-m"2) est utilisé pour estimer la quantité de verre altéré
quand celle-ci est calculée pour les éléments mobiles (B, Na), ou encore pour
estimer la rétention des éléments moins mobiles (en particulier Si et Al) dans les
produits d'altération.
Eq. 12.3
avec t la durée de lixiviation et / un élément mobile tel que B ou Na.
12.2 DISCUSSION DES RÉSULTATS DES ESSAIS DE
LIXIVIATION ET DES CALCULS DE SOLUBILITE DES
VERRES DANS L 'EAU
Les évolutions en solution, et au cours du temps, des pertes de masses
normalisées NL(i, t) lors de l'altération du verre SON-3 et du verre d'albite sont
222
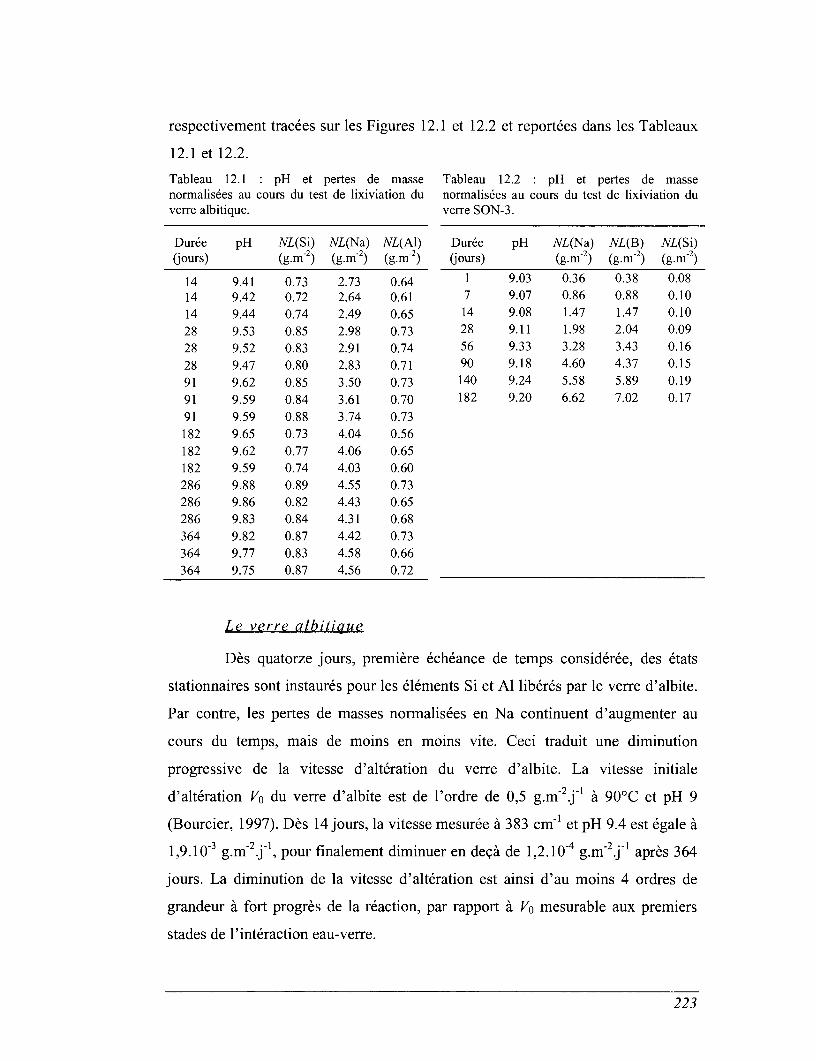
respectivement tracées sur les Figures 12.1 et 12.2 et reportées dans les Tableaux
12.1 et 12.2.
Tableau 12.1 : pH et pertes de massenormalisées au cours du test de lixiviation duverre albitique.
Durée(jours)
pH M-(Si) NLÇNa) M-(Al)(g-m-2) (g-m-2)
Tableau 12.2 : pH et pertes de massenormalisées au cours du test de lixiviation duverre SON-3.
Durée pH M,(Na) M,(B) M,(Si)(jours) (g-m'2) (g.m"2) (g.m"2)
141414282828919191182182182286286286364364364
9.419.429.449.539.529.479.629.599.599.659.629.599.889.869.839.829.779.75
0.730.720.740.850.830.800.850.840.880.730.770.740.890.820.840.870.830.87
2.732.642.492.982.912.833.503.613.744.044.064.034.554.434.314.424.584.56
0.640.610.650.730.740.710.730.700.730.560.650.600.730.650.680.730.660.72
1714285690140182
9.039.079.089.119.339.189.249.20
0.360.861.471.983.284.605.586.62
0.380.881.472.043.434.375.897.02
0.080.100.100.090.160.150.190.17
Le verre albitique
Dès quatorze jours, première échéance de temps considérée, des états
stationnaires sont instaurés pour les éléments Si et Al libérés par le verre d'albite.
Par contre, les pertes de masses normalisées en Na continuent d'augmenter au
cours du temps, mais de moins en moins vite. Ceci traduit une diminution
progressive de la vitesse d'altération du verre d'albite. La vitesse initiale
d'altération Vo du verre d'albite est de l'ordre de 0,5 g.m'lj"1 à 90°C et pH 9
(Bourcier, 1997). Dès 14 jours, la vitesse mesurée à 383 cm"1 et pH 9.4 est égale à-v-3 -2 - 1l,9.10"J g.m~z.j~', pour finalement diminuer en deçà de 1,2.10"4 g.m^.j"1 après 364
jours. La diminution de la vitesse d'altération est ainsi d'au moins 4 ordres de
grandeur à fort progrès de la réaction, par rapport à VQ mesurable aux premiers
stades de l'interaction eau-verre.
223
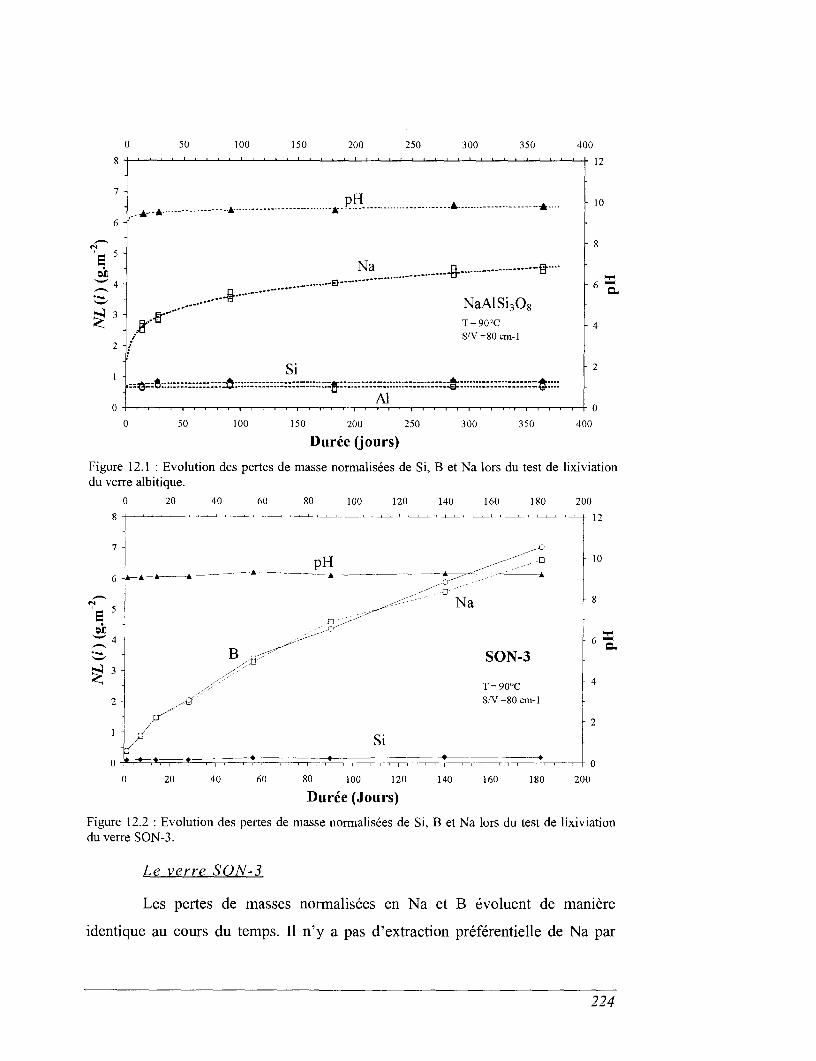
50 100 150 200 250 300 350 400
12
' _.... h 10
6
5 -
I J Na4 -
1 -
0
___ -a-_ „..- NaAlSi3O8
S/V=80cm-l2 H/
Si::::::::::::::g::::::::::::::
Al
- 6 H
0 50 100 150 200 250 300 350 400
Durée (jours)
Figure 12.1 : Evolution des pertes de masse normalisées de Si, B et Na lors du test de lixiviationdu verre albitique.
0 20 40 60 80 100 120 140 160 180 200
12
sSX)
8 -
7 -
6 -
5 -
4 -
3 -
2 -
1 -
n -
* — * A *—• " * ~
B g^-
y"
PH
i i—[—i 1 r—T—i
Si
' " " Na
SON-3
T = 90°CS/V=80cm-l
* »
- 10
0 20 40 60 80 100 120 140 160 180 200
Durée (Jours)
Figure 12.2 : Evolution des pertes de masse normalisées de Si, B et Na lors du test de lixiviationdu verre SON-3.
Le verre SON-3
Les pertes de masses normalisées en Na et B évoluent de manière
identique au cours du temps. Il n'y a pas d'extraction préférentielle de Na par
224

rapport à B. Par contre, ces pertes sont nettement supérieures à celles de Si. Ce
dernier élément est donc retenu à l'interface réactionnelle verre/solution. Il forme
graduellement une pellicule d'altération, clairement identifiée au MEB et TEM
par Jégou (1998). Au cours des 186 jours d'altération, la vitesse d'altération du
verre SON-3 baisse progressivement. La vitesse initiale mesurée par ailleurs à
90°C et pH 9 est ainsi de 5 g.m"2.j"' (Jégou, 1998). Elle est égale, à 3,8.10"2 g.m"2.j"1 après 186 jours, soit une baisse d'environ 1.5 ordres de grandeur.
On met ainsi clairement en évidence, grâce à ces expériences, que les
vitesses d'altération baissent signifïcativement de plusieurs ordres de grandeur. Il
y a donc bien une attaque congruente par l'eau au niveau du réseau vitreux,
jusqu'à ce que des éléments tels que Si et Al soient retenus dans les produits
d'altération secondaires.
Les activités des espèces dissoutes ont été calculées à l'aide du code de
calcul Kindis (Madé et al., 1990), à partir des analyses des concentrations
élémentaires en solution. Les réactions de dissolution de base prises en compte
dans le code de calcul sont les suivantes :
• pour le verre d'albite,
NaAlSi3O% + m2O o Al{OH)4 + Na+ + 3H4Si04
• pour le verre SON-3,
•^4^0.284*0.36802.042 + lJ5*H2° + 02%4H +
O036SB(OH)3 +0.284Na+ +0.674H4SiO4
Les résultats de ces calculs sont reportés respectivement dans les
Tableaux 12.3 et 12.4 pour le verre d'albite et le verre SON-3.
Les produits ioniques d'activité Q ont été calculés à chaque échéance de
temps à partir des activités des espèces aqueuses.
a a a
Q = — - — dans le cas du verre d albite,
0.368 0.284 0.674„ aB(OH\ -aMa+ MH4Si04 , , , „ „,. T -Q = —-—n _ — - — - dans le cas du verre SON-3.
aH;
225

Les rapports QIK ont ensuite été calculés afin d'évaluer le degré de
saturation des lixiviats et l'impact éventuel sur les vitesses d'altération
déterminées expérimentalement. C'est dans ce calcul qu'intervient la valeur que
nous avons déterminée par les techniques calorimétriques et viscosimétriques.
Dans le cas du verre d'albite nous utilisons les données des références citées dans
la Figure 4.5 qui permettent de calculer une constante d'équilibre à 90°C de -
13.25. Les résultats sont reportés dans les Tableaux 12.3 et 12.4.
Tableau 12.3 : Activités des espèces aqueuses présentes en solution en fonction de la durée del'expérience de lixiviation du verre albitique et valeurs du rapport QIK.
DuréeG ours)
141428289191182182182286286286364364364
Al(OH)4-
7.38E-047.74E-048.88E-048.44E-048.40E-048.76E-046.67E-047.77E-047.13E-048.63E-047.67E-048.04E-048.57E-047.79E-048.52E-04
ActivitéNa+
3.77E-033.96E-034.64E-034.31E-034.91E-035.06E-034.79E-034.81E-034.79E-037.01E-036.53E-036.36E-036.44E-035.87E-035.92E-03
H4Si04o
6.71E-046.72E-046.48E-046.87E-045.68E-045.94E-044.42E-044.93E-045.01E-043.33E-043.23E-043.50E-043.72E-043.94E-044.28E-04
H+
3.8OE-1O3.63E-103.02E-103.39E-102.57E-102.57E-102.24E-102.40E-102.57E-101.32E-101.38E-101.48E-101.51E-101.70E-101.78E-10
log(0
-15.076-15.032-14.951-14.927-15.12-15.032-15.559-15.349-15.366-15.651-15.772-15.657-15.546-15.553-15.404
\og{QIK)
-1.826-1.782-1.701-1.677-1.87
-1.782-2.309-2.099-2.116-2.401-2.522-2.407-2.296-2.303-2.154
QIK
1.49E-021.65E-021.99E-022.10E-021.35E-021.65E-024.91E-037.96E-037.66E-033.97E-033.O1E-O33.92E-035.06E-034.98E-037.01E-03
Les résultats des calculs sont les suivants :
1. Le produit ionique d'activité Q (Tableau 12.3) est toujours inférieur au produit
de solubilité du verre d'albite, d'environ 2 unités log. En d'autres termes, le
rapport QIK est de l'ordre de 0.01. D'après l'équation 11.3, la vitesse
d'altération du verre d'albite devrait être proche de VQ. Or, expérimentalement,
la vitesse d'altération du verre d'albite a significativement baissé. Après 14
jours, elle est inférieure de plus de 3 ordres de grandeur à VQ (Figure 11.7).
226

Tableau 12.4 : Activités des espèces aqueuseses présente en solution en fonction de la durée del'expérience de lixiviation du verre SON-3 et valeurs du rapport QIK.
1 7.60E-03 1.25E-02 3.02E-03 1.00E-097 1.54E-02 2.57E-02 3.33E-O3 8.51E-1014 2.43E-02 4.11E-02 3.14E-03 8.32E-1028 3.15E-02 5.41E-02 2.76E-03 7.76E-1056 3.38E-02 9.10E-02 3.07E-03 4.68E-1090 5.12E-02 1.03E-01 3.34E-03 6.61E-10140 5.94E-02 1.26E-01 3.70E-03 5.75E-10182 6.92E-02 1.32E-01 3.26E-03 6.31E-10
-5.57-5.36-5.25-5.22-5.18-5.03-4.97-4.97
-5.6-5.4-5.3-5.3-5.2-5.1-5.0-5.0
2.41E-063.91E-065.04E-065.39E-065.97E-068.41E-069.68E-069.78E-06
2. Les produits ioniques d'activité Q calculés à chaque échéance de temps sont
toujours inférieurs au produit de solubilité du verre SON-3 (Tableau 12.4), de
plus de 5 unités log. Les lixiviats seraient ainsi sous-saturés vis-à-vis du verre
sain. D'après l'équation cinétique 11.3, la vitesse d'altération du verre devrait
toujours être de l'ordre de la vitesse initiale. D'après les données
expérimentales de lixiviation, la vitesse d'altération a baissé de 1.5 ordres de
grandeur par rapport à VQ.
Pour les deux tests de lixiviation décrits ci-dessus, il paraît évident que
l'hypothèse d'un contrôle par l'affinité réactionnelle selon l'équation 11.3 ne peut
être avancée pour expliquer les diminutions de vitesses mesurées
expérimentalement. Il est donc probable que la diminution de la vitesse
d'altération avec le progrès de la réaction soit liée à un autre mécanisme
réactionnel.
D'autres tests de lixiviation ont été effectués sur des verres simplifiés de
type SON et AVM tels que ceux présents dans notre étude. Mais le dépouillement
des données n'est pas encore achevé et les activités des espèces aqueuses n'ont pu
être calculées pour donner lieu à une comparaison dans le cadre de ce mémoire. Il
faudra attendre de confirmer les résultats sur ces autres produits avant de redéfinir
une loi cinétique ne considérant plus que l'équilbre verre/solution pour paramétrer
l'évolution à long terme des vitesses d'altération.
227

12.3 QUELLES MODIFICATIONS APPORTER ?
Pour expliquer ces diminutions de vitesse, nous pouvons envisager par
exemple un processus de passivation de surface, le gel d'altération siliceux à
l'interface réactionnel pouvant jouer éventuellement un rôle protecteur. Cette
hypothèse a souvent été émise (Murakami et al., 1989 ; Bourcier et al., 1990) mais
suppose que la cinétique de l'altération est strictement contrôlée par la diffusion
des espèces réactantes dans le gel d'altération. La vérification d'une telle
hypothèse reste délicate car elle demande d'évaluer les propriétés diffusives d'un
système hydraté, poreux et fracturé qui évolue avec le progrès de la réaction.
Une autre approche consisterait à évaluer non plus la fonction d'affinité
(l-Q/K) du verre sain mais celle du verre hydraté et/ou du gel d'altération
néoformé. Bourcier et Cunnane (1990, 1993) ont tenté d'explorer cette approche
en se fixant uniquement l'interface gel/eau comme système. Le gel est considéré
comme un système polyphasé qui évolue dans le temps jusqu'à montrer un certain
niveau de polymérisation lui faisant contrôler l'altération. La composition du gel
est obtenue par bilan de masse à partir de la composition du verre sain et de
l'évolution de la chimie de la solution. A partir de cette composition, il faut
ensuite évaluer les phases néoformées les plus probables. Enfin, à partir de cette
reconstitution du gel, le produit de solubilité du gel doit être estimé grâce aux
propriétés thermochimiques associées à chaque phase. Une telle approche
demande une bonne caractérisation de la surface réactive pour identifier de façon
exhaustive la nature des phases secondaires précipitées et pour déterminer les
profils de concentration. Cette approche reste délicate car les conditions
d'altération (S/V, mode d'altération,...) fixeront les propriétés du gel (composition,
structure, porosité, fracturation, etc.).
Pour l'étude du système formé par le verre hydraté, l'approche semble
plus proche de celle testée pour le verre sain. On peut considérer qu'une fois que
les étapes conduisant au départ des cations du verre et à l'hydrolyse des liaisons
silicatées de surface (donnant naissance à des groupements silanols) se sont
228

produites, la structure de la zone hydratée se redensifie et tend vers un nouvel
équilibre en réagissant avec l'eau. Les propriétés thermochimiques du verre
hydratée peuvent être évaluées d'une façon proche ou semblable à celle utilisée au
cours de notre travail pour pouvoir ainsi définir une nouvelle fonction d'affinité
chimique. Les mesures de viscosités sur les silicates hydratés (Lejeune et al., 1994
; Dingwell et al., 1996, 1998a'b ; Whittington et al., 2000) montrent que la
viscosité diminue d'autant plus fortement avec la température que la teneur en eau
est importante. Ces diminutions sont interprétées comme des variations d'entropie
de configuration grâce à la théorie entropique des processus de relaxation. De
même, de récentes mesures de capacités calorifiques sur des verres silicates
hydratés (Bouhifd, 2000) indiquent une modification sensible des Cp dès les
faibles teneurs en eau dans le verre par rapport au produit équivalent anhydre. La
tendance générale révélée par ces études est une augmentation des entropies de
configuration, enthalpies et entropies relatives. Sur cette base, il semblerait que la
constante d'équilibre entre le verre hydraté et la solution altérante devrait diminuer
et donc augmenter la valeur de l'affinité chimique. Il me semble nécessaire
d'approfondir cette démarche pour vérifier une amélioration éventuelle d'une
description cinétique telle qu'on peut la formuler sur la base de l'équation 11.3.
229

CONCLUSION
La matrice de confinement des produits de fission est un verre
borosilicaté dont la composition exacte varie selon les types de réacteurs
nucléaires à l'origine de la production du déchet à stocker. Les verres SON
(réacteur "eau légère") et AVM (réacteur "graphite-gaz") en sont deux
représentations, chacun étant formé d'une trentaine d'oxydes. L'évolution des
techniques de vitrification et de la nature des déchets amène à considérer d'autres
compositions de verre qu'il faut également caractériser dans le cadre des études du
comportement à court, moyen et long terme. A ce jour, les modèles géochimiques
qui tendent à reproduire les vitesses de relargages des éléments polluants dans la
biosphère intègrent des lois cinétiques dont la qualité varie selon la composition
de matrice et les conditions d'altération. Cette variabilité à reproduire les données
d'expériences est en partie due à la paramétrisation de ces lois qui souffre d'un
manque de données thermochimiques important.
Le travail que nous avons accompli est une contribution à la
caractérisation des propriétés thermochimiques des matrices de stockage. Le verre
est un matériau désordonné défini thermodynamiquement hors d'équilibre. En
caractérisant chaque verre par au moins un paramètre d'ordre qui rend compte de
son état de configuration, on peut alors considérer que c'est un équilibre
métastable. En utilisant la température fictive de nos verres comme paramètre
d'ordre, nous avons pu déterminer les principales fonctions thermodynamiques de
trois séries de verres simplifiés. Parmi celles-ci, une série formée par des
compositions ternaires du système SiO2-Na2O-B2O3 pourra servir de base à une
modélisation des propriétés thermodynamiques des verres borosilicatés et
notamment à celles des nouvelles compositions de verre de confinement de
déchets.
Les capacités calorifiques à basse température de 5 verres ternaires ont
été mesurées par calorimétrie adiabatique. En utilisant un modèle de structure des
231

borosilicatés, nous avons pu déterminer l'entropie partielle relative à 0 K de l'unité
BO4 dans le système SiO2-Na2O-B2O3. En utilisant les entropies partielles
déterminées dans d'autres systèmes notamment pour les aluminosilicates, nous
avons estimé les variations d'entropie entre 0 et 300 K de l'ensemble de nos
compositions.
Grâce aux techniques de calorimétrie de chute et de calorimétrie
différentielle à balayage, les capacités calorifiques entre l'ambiante et 1500 K de
nos compositions ont été mesurées. Ces mesures rendent compte d'une
dépendance compositionnelle importante des liquides borosilicatés. En effet,
contrairement aux silicates simples, on observe, selon les compositions, des
dépendances de la capacité calorifique des liquides vis-à-vis de la température.
Nous suggérons que ces "anomalies" sont liées à des interactions partagées entre
les cations Si4+ et B3+ mais la nature exacte et la dépendance de ces interactions
vis-à-vis de la composition ne peut être définie facilement sur la seule base de
données thermodynamiques. En revanche, les capacités calorifiques des verres ne
semblent pas marquées de caractère inhabituel ce qui nous laisse envisager une
modélisation simple des Cp des verres basée, comme pour les basses températures,
sur un concept de propriétés molaires partielles rendant compte des coordinences
de chaque élément.
Ainsi, nos mesures pourront conduire à l'élaboration d'un modèle de
capacité calorifique du verre fonction de la température et de la composition pour
des systèmes comprenant le silicium, le bore en coordinence 3 et 4 et, grâce aux
données de la littérature (Richet, 1987), l'aluminium comme formateurs de réseau.
La modélisation des liquides sera plus difficile et demandera notamment une
étude fine de la structure des liquides borosilicatés pour comprendre les causes
des capacités calorifiques anormales observées pour la majeure partie des
compositions de notre étude.
Le troisième volet expérimental fut de mesurer les enthalpies de
dissolution des verres dans un bain de 2PbO.B2C>3 à 970 K. Les résultats
permettent de calculer les enthalpies de formation des verres. Mais l'originalité de
232

notre travail est particulièrement attachée à la détermination des entropies de
configurations des verres étudiés. Les viscosités sont reliées à l'entropie de
configuration du verre à la transition vitreuse par la théorie entropique des
processus de relaxation énoncée par Adam et Gibbs (1965). Les mesures de
viscosités réalisées sur l'ensemble de nos verres nous ont permis de déterminer
leur entropie de configuration et donc d'avoir accès à leur entropie résiduelle à
0 K. Cette méthode est utilisée depuis une vingtaine d'années pour les systèmes
d'intérêt géologique mais, pour la première fois, nous l'avons appliquée aux
compositions des matrices de confinement. Les résultats indiquent des valeurs
particulièrement élevées par rapport aux silicates simples. Encore une fois, cette
observation est à relier au mélange des unités silicatées et boratées qui composent
nos verres.
Cet ensemble de données nous a conduit à la détermination des
enthalpies libres de formations des verres simplifiés. Cette donnée est très riche
d'application puisqu'elle permet de définir les équilibres réactionnels dans lesquels
nos verres peuvent prendre part. Outre les phénomènes d'altération auxquels nous
avons tenté d'apporter un nouvel éclairage, ces données pourront être utiles à
l'élaboration des diagrammes de phases, aux études de démixions ou de
cristallisations qui sont réalisées dans le cadre de la caractérisation des matrices de
stockage.
Grâce à nos mesures des propriétés thermodynamiques, il nous a été
possible de calculer les enthalpies libres de dissolution des verres dans l'eau, et
d'évaluer en détail la loi cinétique qui rend compte de la diminution des vitesses
d'altération des verres par un écart des potentiels chimiques entre le verre sain et
la solution altérante. De nombreux modèles cinétiques décrivent l'évolution des
pertes de masses des éléments traceurs de l'altération (B, Na) par le niveau de
saturation de la solution altérante vis-à-vis des espèces aqueuses provenant de la
dissolution du verre. Nos mesures montrent que dans le cas d'un verre ternaire
(Si0.674Na0.284B0.368O2.042) et d'un verre d'albite (NaAISisOg) les vitesses
d'altération diminuent d'un facteur 2 à 5 alors que les potentiels chimiques du
233

verre et de la solution sont encore très différents. Ces résultats seront à valider sur
les autres tests de lixiviation en cours de dépouillement actuellement mais
suggèrent, au moins pour ces deux compositions, que l'évolution des vitesses
d'altération n'est pas régie par l'écart à l'équilibre thermochimique entre le verre et
la solution.
D'autres mécanismes de contrôle cinétique doivent donc être proposés.
Certaines évidences expérimentales nous montrent deux voies possibles :
Le contrôle par les propriétés diffusives du gel d'altération néoformé,
Le contrôle par l'écart à l'équilibre entre la solution et le verre hydraté
désalcalinisé.
Nos mesures thermochimiques ne permettent pas d'évaluer la pertinence de la
première hypothèse. En revanche, les études menées sur la thermochimie des
silicates hydratées suggèrent des variations des propriétés que nous avons
mesurées selon une tendance générale qui conduirait à une augmentation du
niveau de saturation de la solution altérante par rapport à l'équilibre verre
sain/solution considéré initialement.
L'approche utilisée dans ce travail pourrait être utilisée pour tester de
façon précise cette seconde hypothèse malgré des difficultés expérimentales
supplémentaires liées au risque d'exsolution de l'eau lors des traitements
thermiques effectués au cours des mesures.
234

BIBLIOGRAPHIE
Aagaard P. et Helgeson, 1982, Thermodynamic and kinetic constraints onreaction rates among minerals and aquous solution. I. Theoreticalconsiderations, Am. J. Sci. 282, 237
Abramo M.C., Caccamo C. et Pizzimenti G., 1992, J. Chem. Phys. 96, 9083Adam G. et Gibbs J.H., 1965, On the temperature dependence of cooperative
relaxation properties in glass-forming liquids, J. Chem.Phys. 43, 139Adams, P.B., 1984, Glass Corrosion : A record of the past? A predictor of the
future?, J. Non-Crystal. Solids 67, 193Advocat, T., Crovisier J.L., Fritz B. and Vernaz E., 1990, Thermokinetic model
of borosilicate glass dissolution : contextual affinity, Scientific Basis forNuclear Waste Management XIII. MRS symposium proceeding 176, 241
Akaogi M. et Navrostky A., 1984, Phys. Earth Planet. Inter. 36, 124Arai, T., Yusa Y., Sasaki N., Tsunoda N. et Takano H., 1989, Natural analogue
study of volcanic glass, Scientific Basis for Nuclear Waste ManagementXII. MRS symposium proceeding 127, 73
Araujo R.J. et Hares G.B., 1981, Theoretical determination of the structures ofalkali aluminoborosilicate glasses and their correlation with experimentallydetermined physical properties, Phys. Chem. Glasses 22, 6
Araujo R.J., 1997, Influence of boron-oxygen bonding on glass properties, J.Non-Cryst. Solids 222, 25
Asano M., Kou T. et Mizutani Y., 1989, Vaporization of alkali borosilicateglasses, J. Non-Cryst. Solids 111, 381
Atake T., Kawaji H., Hamano A. et Saito Y., 1990, Construction of an adiabaticcalorimeter for heat capacity measurements from liquid helium temperatureto 330 K, Rep. Res. Lab. Eng. Mater., TIT, 15, 13
Bacon C.R., 1977, High temperature heat content and heat capacity of silicateglasses: Experimental data and a model for calculation, Am. J. Sci. 277, 109
Bashin G., Bhatnagar A., Bhowmik S., Stehle C , Affatigato M., Feller S.,MacKenzie J. et Martin S., 1998, Short range order in sodium borosilicateglasses obtained via deconvolution of 29Si MAS NMR spectra, Phys. Chem.Glasses 39, 269
Bates, J.K. et Gerding T.J., 1988, The performance of actinide-containing SRL165 type glass in unsatured conditions, Scientific Basis for Nuclear WasteManagement XL MRS symposium proceeding 112, 651
Bénard F., Moutou P., Pichavant M., 1985, Phase relations of tourmalineleucogranites and the significance of tourmaline in silicic magmas, J. Geol.93,271
Berman R.G., Engi M., Greenwood H.J. et Brown T.H., 1986, Derivation ofInternally-Consistent Thermodynamic Data by the Technique of
235

Mathematical Programming : a Review with Application to the SystemMgO-SiO2-H2O, J. Petrology 27, 1331
Bernier J.F., 1980, Refroidissement d'un conteneur de produits de fission vitrifiésen phase hors stockage, Rapport DGR 195
Bibler, N.E. et Jurgensen A.R., 1988, Leaching Tc-99 from SRP glass insimulated tuff and salt groundwaters, Scientific Basis for Nuclear WasteManagement XL MRS symposium proceeding 112, 585
Bidoglio, G., Offermann P., De Piano A. et Lazzari G.P., 1988, Influence ofgroundwater composition on glass leaching and actinide speciation,Scientific Basis for Nuclear Waste Management XL MRS symposiumproceeding 112, 621
Borisova N.V. et Ushakov V.M., 1992, Enthalpy of formation of alkali-borosilicate glass,
Bottinga Y., 1994a, Configurational entropy and the non-Newtonian rheology ofhomogeneous silicate liquids, Phys. Rev. B 49,95
Bottinga Y., 1994b, Non-Newtonian rheology of homogeneous silicate melts,Phys. Chem. Min. 20, 454
Bottinga Y. et Weill D.F., 1972, The viscosity of magmatic silicate liquids : amodel for calculation, Am. J. Sci. 272, 438-475
Bottinga Y., Richet P. et Sipp A., 1995, Viscosity regimes of homogeneoussilicate melts, Am. Miner. 80, 305
Bottinga Y., Weill D.F. et Richet P., 1982, Density calculations for silicateliquids : I, Revised method for aluminosilicate compositions, Geochim.Cosmochim. Acta 46, 409-414
Bouhifd M.A., communication personnelle, article en préparationBourcier W.L., 1991, Overview of chemical modeling of nuclear waste glass
dissolution, Scientific Basis for Nuclear Waste Management XIV. MRSsymposium proceeding 212, 3
Bourcier W.L., Peiffer D.W., Knauss K.G., McKeegan K.D. et Smith D.K.,1990, A kinetic model for borosilicate glass dissolution based on thedissolution affinity of surface alteration layer, Scientific Basis for NuclearWaste Management XIII. MRS symposium proceeding 176, 209
Bowen N.L., 1934, Viscosity data for silicate melts, Am. Geophys. Union Trans.,15th Ann. Meeting, EOS Trans. AGU 1, 249
Brawer S., 1985, The Relation between defects and relaxation in viscous liquids,Defects in Glasses. MRS symposium proceeding 61, 21
Bray J., 1985, Structural groupings in borate glasses, Defects in Glasses. MRSsymposium proceeding 61, 121
Bray J. et Dell W.J., 1982, NMR studies of the structure og glass, Physics ofNon-Crystalline Solids. Proceedings of the 5 m International Conference,Montpellier 5-9 July 1982, C9.131
Brousse C , 1981, Contribution à l'étude thermodynamique du sysyèmeNaAlSi3O8-KalSi3O8 (Albite-Orthose), Thèse de doctorat de l'InstitutNational Polytechnique de Grenoble
236

Brousse C , Newton R.C.et Kleppa O.J., 1984, Enthalpy of formation offorsterite, enstatite, akermanite, monticellite and merwinite at 1073 Kdetermined by alkali borate solution calorimetry, Geochim. Cosmochim.Acta 48, 1081
Brousse C , Rogez J., Castanet R. et Mathieu J.C., 1982, Mat. Res. Bull. 7, 125Bruce C, Ingram M.D., MacKenzie M. et Syed R., 1986, Solid state ionics. 410Bruton C.J., 1988, Geochemical simulation of dissolution of West Valley and
DWPF glasses in J-13 water at 90°C, Scientific Basis for Nuclear WasteManagement XL MRS symposium proceeding 112, 607
Bunker B.C., Arnold G.W., Day D.E. et Bray P.J., 1986, The effect ofmolecular structure on borosilicate glass leaching, J. Non-Crystal. Solids 87,226
Bunker, B.C., Arnold G.W. et Wilder J.A., 1984, Phosphate glass dissolution inaqueous solutions, J. Non-Crystal. Solids 64, 291
Bunker B.C., Tallant D.R., Headley T.J., Turner G.L. et Kirkpatrick R.J.,1988, The structure of leached sodium borosilicate glass, Phys. Chem.Glasses 29, 106
Bunker B.C., Tallant D.R., Kirkpatrick R.J. et Turner G.L., 1990,Multinuclear magnetic resonance of sodium borosilicate glass structures,Phys. Chem. Glasses 31, 30
Capobianco C. et Navrotsky A., 1982, Calorimetric evidence for ideal mixing ofsilicon and germanium in glasses and crystals of sodium feldsparcomposition, Am. Miner. 67, 718
Caurel J., Beaufort D. et Vernaz E.Y., 1988, Mineral phase identification alongtwo profiles from the LWR french reference glass : use of an X-ray positionsensitive detector, Scientific Basis for Nuclear Waste Management XI. MRSsymposium proceeding 112, 663
Chai L. et Navrotsky A., 1993, Thermochemistry of carbonate-pyroxeneequilibria, Contrib. Miner. Petrol. 114, 139
Chakraborty I.N., Day D.E., Lapp J.C. et Shelby J.E., 1985, Structure-PropertyRelation in Lanthanide Borate Glasses, J. Am. Ceram. Soc. 68, 368
Chakraborty I.N., Shelby J.E. et Robert A.C.S.R., 1984, Properties andStructure of Lanthanum Borate Glasses, J. Non-Cryst. Solids 67, 782
Charlu T.V., Newton R.C. et Klepa O.J., 1975, Enthalpies of formation at 970K of compounds in the system MgO-Al2O3-SiO2 from high temperaturesolution calorimetry, Geochim. Cosmochim. Acta 39, 1487
Charlu T.V., Newton R.C. et Klepa O.J., 1978, Enthalpies of formation of somelime silicates by high-temperature solution calorimetry, with discussion ofhigh pressur phase equilibria, Geochim. Cosmochim. Acta 42, 367
Charlu T.V., Newton R.C. et Klepa O.J., 1981, Thermochemistry of syntheticCa2Al2SiC>7 (gehlenite)-Ca2MgSi2O7 (akermanite) melilites, Geochim.Cosmochim. Acta 45, 1609
Chatillon-Colinet C, Klepa O.J., Newton R.C. et Perkins III D., 1983,Enthalpy of formation of Fe3Al2Si3Oi2 (almandine) by high temperaturealkali borate solution calorimetry, Geochim. Cosmochim. Acta 47, 439
237

Chatillon-Colinet C, Newton R.C., Perkins III D. et Klepa O.J., 1983,Thermochemistry of (Fe2+,Mg)SiC>3 orthopyroxene, Geochim. Cosmochim.Ada 41, 1597
Chermak J.A. et Rimstidt J.D., 1989, Estimating the thermodynamic properties( A / J ° and AjH°) of silicate minerals at 298 K from the sum of polyhedralcontributions, Am. Miner. 74, 1023
Circone S. et Navrotsky A., 1992, Substitution of [6'4]A1 in phlogopite: high-temperature solution calorimetry, heat capacities, and thermodynamicpropertie of the phlogopite-eastonitejoin, Am. Miner. 77, 1191
Clark D.E., Schultz R.L., Lodding A.R., Tipton A.W. et Wicks G.G., 1992,Effects of metal canister and overpack materials on waste glassperformance. In situ testing of radioactive waste forms and enginneeredbarriers, Scientific Basis for Nuclear Waste Management V
Courtial P. et Richet P., 1993, Heat capacity of magnesium aluminosilicatemelts, Geochim. Cosmochim. Ada 57, 1267
Cowan, R. et Ewing R.C., 1989, Natural analogue study of volcanic glass,Scientific Basis for Nuclear Waste Management XII. MRS symposiumproceeding 127, 49
Creux S., Bouchet-Fabre B. et Gaskell P.H., 1995, Anomalous wide angle X-ray scattering study of strontium silicate and aluminosilicate glasses, J. Non-Cryst. Solids 192, 360
Crovisier J.L., 1985, Dissolution du verre basaltique dans l'eau de mer : approcheexpérimentale et thermodynamique, Thèse de doctorat de l'Université LouisPasteur de Strasbourg
Crovisier J.L., Advocat T., Petit J.C. et Fritz B., 1989, Alteration of basalticglass in Iceland as a natural analogue for nuclear waste glasses :geochemical modelling with dissol, Scientific Basis for Nuclear WasteManagement XII. MRS symposium proceeding 127, 57
Crovisier J.L., Atassi H., Daux V., Honnorez J., Petit J.C. et Eberhart J.P.,1989, A new insight into the nature of the leached layers formed on basalticglasses in relation to the choice of constraints for long term modelling,Scientific Basis for Nuclear Waste Management XII. MRS symposiumproceeding 127, 41
Daux V., Crovisier J.L. et Petit J.C., 1991, Rare Earth elements behaviourduring alteration of basaltic glasses : case of the weathering of icelandichyaloclastites, Scientific Basis for Nuclear Waste Management XIV. MRSsymposium proceeding 212, 107
Davies . et Navrotsky A., (1981)de Ligny D., 1996, Propriétés thermiques de minéraux et de silicates vitreux,
Thèse de doctorat del'Université Paris VIIde Ligny D., Richet P. et Westrum E.F., Jr., 1996, Entropy of calcium and
magnesium aluminosilicate glasses, Chem. Geol. 128, 113De Yoreo J.J., Navrotsky A. et Dingwell D.B., 1990, Energetics of the charge-
coupled substitution Si4+-»Na+ + T3+ in the glasses NaTO2-SiO2 (T=A1, Fe,Ga, B), J. Am. Ceram. Soc. 73, 2068
238

Delage F. et Dussossoy J.L., 1991, R7T7 glass initial dissolution ratemeasurements using a high-temperature SOXHLET device, Scientific Basisfor Nuclear Waste Management XIV. MRS symposium proceeding 212, 41
Dell W.J., Bray P.J. et Xiao S.Z., 1983, n B NMR studies and structuralmodeling of Na2O-B2O3-SiO2 glasses of high soda content, J. Non-Cryst.Solids 58, 1
Delorme L., 1998, Mécanismes de volatilité des verres et des fontesborosilicatées d'intérêt nucléaire. Influence de la structure, Thèse de doctoratde l'université d'Orléans
Deniélou L., Fournier Y., Petitet J. P. et Tequi C, 1971, Etude calorimétriquedes sels entre 273°K et 1533°K. Technique et application aux sulftesalcalins. Rev. Int. Hautes Temp. Refract., 8, 119
Dickenson M.P. et Hess P.C., 1981, Redox equilibria and the structural role ofiron in aluminosilicate melts, Am. Contrib. Miner. Petrol. 78, 352
Dietzel A., 1942, Die Kationenfeldstârken und ihre beziehungen zu entglasungs-vorgângen, zur verbindungsbilbdung und zu den schmelzpunkten vonsilikaten, Z. Electrochemie 48, 9
Dingwell D.B. et Webb S.L., 1990, The onset of non-newtonian rheology ofsilicate melts : A fiber elongation study, Phys. Chem. Minerals 17, 125-132
Ditmars D. A. et Douglas T. B., 1971, Measurements of the relative enthalpy ofpure a-A12O3 (NBS heat capacity and enthalpy standard reference materialN°.72O) from 273 to 1173 K. J. Res. N.B.S., 75A, 401
Doremus R.H., 1973, Glass science, ed. John Wiley and Sons, New-York.Doremus R.H., 1979, Chemical durability of glass, Treat. Mater. Sci. 17, 41Doremus R.H., 1994, Glass Science, ed. John Wiley and SonsDouglas R.W. et El-Shamy T.M.M., 1967, Reactions of Glasses with Aqueous
Solutions, J. Am. Ceram. Soc. 50, 1Duval E., Boukenter A., et Achibat T., 1990, Vibrational dynamics and the
structure of glasses, J. Phys. : Condens. Matter!, 10227Eckert J.O.Jr., Newton R.C. et Klepa O.J., 1991, The àH of reaction and
recalibration of garnet-pyroxene-plagioclase-quartz geobarometers in theCM AS system by solution calorimetry, Am. Miner. 76, 148
El-Damrawi, Mûller-Warmuth W., Doweidar H. et Gohar I.A., 1993, n B , 29Siand 27A1 nuclera magnetic resonance studies of Na2O-Al2O3-B2O3-SiO2glasses, Phys. Chem. Glasses 34, 52
Ellison A.J.G. et Navrotsky A., 1990, Thermochemistry and structure of modelwaste glass compositions, Scientific Basis for Nuclear Waste ManagementXIII. MRS symposium proceeding 176, 193
Elmer T.H., 1985, Role of Acid Concentration in Leaching of Cordierite andAlkali Borosilicate Glass, J. Am. Ceram. Soc. 68, C273
Farges F., Brown G.E.J., Navrotsky A., Gan H. et Rehr J.J., 1996,Coordination Chemistry of Ti(IV) in silicate glasses and melts. III. Glassesand Melts from ambient to high temperatures, Geochim. Cosmochim. Ada60, 3055
239

Farges F., Ponader C.W. et Brown G.E.J, 1991, Structural environments ofincompatible elements in silicate glass/melt systems : part 1. Zr at tracelevels, Geochim. Cosmochim. Acta 55, 1563
Fehrenbach L., Maurette M., Guichard F., Havette A. et Monaco A., 1984,Paleocorrosion studies in deep sea sediments and the geological disposal ofnuclear wastes, J. Non-Crystal. Solids 67, 287
Feng X. et Barkatt A., 1988, Systematic composition studies on the durability ofwaste glass WV205, Scientific Basis for Nuclear Waste Management XLMRS symposium proceeding 112, 673
Feng X., Barkatt A. et Jiang T., 1988, Structural thermodynamic model for thedurability and viscosity of nuclear waste glasses, Scientific Basis forNuclear Waste Management XI. MRS symposium proceeding 112, 543
Fillet S., 1987, Mécanismes de corrosion et comportement des actinides dans leverre nucléaire R7T7, Thèse de doctorat de Sciences des matériaux del'Université des Sciences et Techniques du Languedoc. Montpellier
Fleet M.E., Muthupari S., 1999, Coordination of boron in alkali borosilicatesglasses using XANES, J. Non-Cryst. Solids 255, 233
Fredrickson G.H. et Andersen H.C., 1984, Kinetic Ising model of the glasstransition, Phys. Rev. Lett. 53, 1244
Fredrickson G.H. et Andersen H.C., 1985, Facilitated kinetic ising models andthe glass transition, J. Chem. Phys. 83, 5822
Fredrickson G.H. et Brawer S.A., 1986, Monte Carlo investigation of a kineticising model of the glass transition, J. Chem. Phys. 84, 3351
Fredrickson G.H., 1986, Linear and nonlinear experiments for a spin model withcooperative dynamics, Dynamic aspects of structural changein liquids andglasses. The New York Academy of Sciences 484, 185
Fulcher G.S., 1926, Analysis of recent measurements of the viscosity of glasses,J. Am. Ceram. Soc. 8, 339
Furukawa T. et White W.B., 1981, Raman spectroscopic investigation ofsodium borosilicate glass structure, J. Non-Cryst. Solids 119, 297
Galeener F.L., Leadbetter A.J. et Stringfellow N.W., 1983, Comparaison of theneutron, Raman, and infrared vibrational spectra of vitreous SiC»2, GeCh andBeF2, Phys. Rev. B 27, 1052
Galoisy L. et Calas G., 1993, Structural environment of nickel in silicate glasses /melt systems : part 2. Geochemical implications, Geochim. Cosmochim.Ada 57, 3627
Galoisy L., Calas G., Ramos A., Le Grand M., Allard T., Morin G., Fillet C.et Pacaud F., 1997, Evolution of the structure of simulated nuclear glasseswith the introduction of noble metals (Ru, Pd), Acte des journées sur leverre : Recherche scientifique pour un confinement de haute performance.éd. CEA, 59
Galoisy L., Pélegrin E., Arrio M-A., Ildefonse P., Calas G., Ghaleb D., FilletC. et Pacaud F., 1999, Evidences for 6-coordinated Zr in inactive nuclearwaste glasses,/. Am. Ceram. Soc. 82, 2219
Gaskell P.H., 1995, Structure, glass formation and properties, J. Non-Cryst.Solids 192, 9
240

Gaskell P.H., Eckersley M.C., Barnes A.C. et Chieux P., 1991, Medium rangeorder in the cation distribution of a calcium silicate glass, Nature 350, 675
Geiger C.A., Klepa O.J., Mysen B.O., Lattimer J.M. et Grossman L., 1988,Enthalpy of formation of CaAUOy and CaAli2Oi9 (hibonite) by hightemperature, alkali borate solution calorimetry, Geochim. Cosmochim. Ada52,1729
Geiger, C.A., Newton R.C. et Klepa O.J., 1987, Enthalpy of mixing of syntheticalmandine-grossular and almandine-pyrope garnets from high-temperaturesolution calorimetry,Geochim. Cosmochim. Ada 51, 1755
Geisinger K.L., Gibbs C.V. et Navrotsky A., 1985, Phys. Chem. Miner. 11, 266Geisinger K.L., Oestrike R., Navrotsky A., Turner G.L. et Kirkpatrick R.J.,
1988, Thermochemistry and structure of glasses along the join NaAlSisOs-NaBSi3O8, Geochim. Cosmochim. Ada 52, 2405
Geissberger A.E. et Bray P.J., 1983, J. Non-Cryst. Solids 54, 121Geller R.F. et Bunting E.N., 1937, J. Res. Natl. Bur. Std. 18, 585Gibbs J.H. et Di Marzio E.A., 1958, Nature of the glass transition and the glassy
state, J. Chem. Phys., 28, 373Gin S., 1995, Control of R7T7 nuclear Glass alteration kinetics under saturation
conditions. Scientific Basis for Nuclear Waste Management XIX. MRSsymposium proceeding 412, 189
Goldstein M., 1963, Some thermodynamic aspects of the glass transitions : freevolume, entropy end enthalpy theories, J. Chem. Phys., 39, 3369
Goldstein M., 1973, Viscous liquids and the glass transition. IV. Thermodynamicequations and the transition, J. Phys. Chem. 11, 667
Goldstein M., 1976, Viscous liquids and the glass transition. V. Sources of theexcess specific heat of the liquid, / . Chem. Phys., 64, 4767
Goodman C.H.L., 1985, The structure and properties of glass and the strainedmixed cluster model, Phys. Chem. Glasses 26, 1
Goodman H.L., 1986, The structure of silica glass and its surface, Phys. Chem.Glasses 27, 31
Grambow B., 1985, A general rate equation for nuclear waste glass corrosion,Scientific Basis for Nuclear Waste Management VIII. MRS symposiumproceeding 44, 15
Grambow B. et Lutze W., 1988, Performance assessment of glass as long-termbarrier to the release of radionuclides into the environment, Scientific Basisfor Nuclear Waste Management XI. MRS symposium proceeding 112, 531
Grambow B. et Mûller R., 1990, Chemistry of glass corrosion in high salinebrines, Scientific Basis for Nuclear Waste Management XIII. MRSsymposium proceeding 176, 229
Greaves G.N., 1985, EXAFS and the structure of glass, J. Non-Cryst. Solids 71,203
Greaves G.N., 1992, Glass strucure and ionic transport, The Physics of Non-Crystalline Solids, ed Taylor and Francis
Gupta P.K., Lui M.L. et Bray P.J., 1985, Boron coordination in rapidly cooledand in annealed aluminium borosilicate glass fibers, J.Am. Ceram. Soc. 68,C-82
241

Guy C , 1989, Mécanismes de dissolution des solides dans les solutionshydrothermales déduits du comportement des verres basaltiques et decalcites déformées, Thèse de doctorat de l'Université des SciencesP.Sabatier de Toulouse
Haas J. L. et Fisher J. R., 1976, Simultaneous evaluation and correlation ofthermodynamic data. Am. J. Sci., 276, 525
Haggerty J.S., Cooper A.R. Jr. et Heasley J.H., 1968, Heat capacity of treeinorganic glasses and supercooled liquids, Phys. Chem. Glasses 9, 47
Haselton H.T. et Westrum E.F.Jr., 1980, Low-temperature heat capacities ofsynthetic pyrope, grossular, and pyrope6ogrossular4o, Geochim. Cosmochim.Acta 44, 701
Haselton H.T., Hovis G.L., Hemingway B.S. et Robie R.A., 1983, Calorimetricinvestigation of the excess entropy of mixing in analbite-sanidine solidsolutions : lack of evidence for Na, K short-range order and implications fortwo-feldspar thermometry, Am. Miner. 68, 398
Haselton H.T., Robie R.A. et Hemingway B.S., 1987, Heat capacities ofsynthetic hedenbergite, ferrobustamte, and CaFeSi2Oô glass, Geochim.Cosmochim. Acta 51, 2211
Hayward P.J., 1982, A review and discussion of candidate ceramics forimmobilization of high-level fuel reprocessing wastes. Atomic Energy ofCanada Limited, ed. Whiteshell Nuclear Research Establishement
Helgeson H.C., Murphy W.M. and Aagaard P., 1984, Thermodynamic andkinetic constraints on reaction rates among minerals and aquous solution. II.Rate constants, effective surface area, and the hydrolysis of feldspar,Geochim. Cosmochim. Acta 48, 2405
Hemingway B.S. et Robie R.A., 1977, Enthalpies of formation of low albite(NaAlSi3O8), gibbsite (A1(OH)3), and NaA102; revised values for A//°298and A/G°298 of some aluminosilicate minerals, J. Res. U.S. Geol. Surv. 5,413
Hemingway B.S., Robie R.A., Kittrick J.A., Grew E.S., Nelen J.A. et LondonD., 1984, Amer. Mineral. 69, 701
Hervig R.L. et Navrotsky A., 1985, Thermochemistry of Sodium BorosilicateGlasses, J. Am. Ceram. Soc. 68, 314
Holley CE.Jr., Elmer J.H.Jr. et Floyd B.B., 1968, The Enthalpies, Entropies,and Gibbs Energies of Formation of the Rare Earth Oxides, Progress in theScience and Technology of the Rare Earths, volume 3, chap. 8, 343
Holm J.L. et Kleppa O.J., 1967, Inorg. Chem. 6, 645Holm J.L. et Klepa O.J., 1968, Thermodynamics of the disordering process in
albite, Am. Miner. 53, 123Huang C. et Cormack A.N., 1990, J. Chem. Phys. 93, 8180Hummel W. et Arndt J., 1985, Contrib. Mineral. Petrol. 90, 83JANAF Thermochemical tables, third edition, 1985, J. Phys. Chem. Ref. Data
14 suplement 1Jantzen CM., 1988, Pourbaix diagram for the prediction of waste glass
durability in geologic environments, Scientific Basis for Nuclear WasteManagement XI. MRS symposium proceeding 112, 519
242

Jantzen CM., 1992, Nuclear Waste Glass Durability : I, PredictingEnvironmental Response from Thermodynamic (Pourbaix) Diagrams, J.Am. Ceram. Soc. 75, 2433
Jantzen CM. et Plodinec M.J., 1984, Thermodynamic model of natural,medieval and nuclear waste glass durability, J. Non-Cryst. Solids 67, 207
Jantzen CM. et Ramsey W.G., 1990, Prediction of radioactive waste glassdurability by the hydratation thermodynamic model : application to saturedrepository environments, Scientific Basis for Nuclear Waste ManagementXIII. MRS symposium proceeding 176, 217
Jégou C , 1998, Mise en évidence expérimentale des mécanismes limitantl'altération du verre R7T7 en milieu aqueux. Critique et propositiond'évolution du formalisme cinétique .Thèse de doctorat de l'université deMontpellier IL
Jouan A., Ladirat C. et Moncouyoux J.P., 1986, Present status of the frenchcontinuous fission product vitrification process, Adv. Ceram. 20
Julsrud S. et Kleppa O.J., 1986, A thermochemical study of the liquid systemNaBO2 + SiO2 at 1394 K, Geochim. Cosmochim. Acta 50, 1201
Kaufmann F. et Wiese H., 1987, Vitrification of high level liquid waste,Proceedings of the international conference on Nuclear Fuel Reprocessingand Waste Management "RECOD 87". éd. Société Française d'EnergieNucléaire, volume 2, 279
Kauzmann W., 1948, The nature of the glassy state and the behavior of liquids atlow temperatures, Chem. Rev. 43, 219
Kelley K.K., Todd S.S., Orr L.R., King E.G. et Bonnickson K.R., 1953,Thermodynamic properties of sodium-aluminium and potassium-aluminiumsilicates, U.S. Bureau Mines Rept. Inv. 4955
Kennedy G.C, 1948, Equilibrium between volatiles and iron oxides in igneousrocks, Am. J. Sci. 246, 529
Kieffer S.W., 1979a, Thermodynamics and lattice vibrations of minerals: 1.Mineral heat capacities and their relationships to simple lattice vibrationalmodels, Rev. Geophys. Space Phys. 18, 1
Kieffer S.W., 1979b, Thermodynamics and lattice vibrations of minerals: 2.Vibrational characteristics of silicates, Rev. Geophys. Space Phys. 18, 20
Kieffer S.W., 1979°, Thermodynamics and lattice vibrations of minerals: 3.Lattice dynamics and an approximation for minerals with application tosimple substances and framework silicates, Rev. Geophys. Space Phys. 18,35
Kieffer S.W., 1980, Thermodynamics and lattice vibrations of minerals: 3.Application to chain and sheet silicates and orthosilicates, Rev. Geophys.Space Phys. 18, 862
Kieffer S.W., 1985, Heat capacity and entropy: systematic relations to latticevibrations, Microscopic to macroscopic. Rev. Miner. 14, 65
Kinoshita M., Harada M., Sato Y. et Hariguchi Y., 1991, PercolationPhenomenon for Dissolution of Sodium Borosilicate Glasses in AqueousSolutions, J. Am. Ceram. Soc. 74, 783
243

Kirkpatrick R.J., 1974, Kinetics of crystal growth in the system CaMgSi2O6-CaA\2Si06, Am. J.Sci. 219, 111
Kiseleva L, Navrotsky A., Belitsky LA. et Fursenko B.A., 1996a,Thermochemistry and phase equilibria in calcium zeolites, Am. Miner. 81,658
Kiseleva I., Navrotsky A., Belitsky I. A. et Fursenko B.A., 1996b,Thermochemistry of natural potassium sodium calcium leonhardite and itscation-exchanged forms, Am. Miner. 81, 668
Kiseleva I.A., Ogorodova L.P., Topor N.D. et Chigareva O.G., Geochem.Inter. (1979) p. 122
Kittel C, 1972, Introduction à la physique de l'état solide, éd. DunodKlein, L.C., B.V. Fasano et Jenn Ming Wu, 1983, Viscous flow behavior of
four iron-containing silicates with alumina, effects of composition andoxidation condition, Proceedings of the thirteenth lunar and planetaryscience conference. Part 2, J. Geophys. Res. 88 supplement, A880
Kojitani H. et Akaogi M., 1995, Measurement of heat of fusion of model basaltin the system diopside-forsterite-anorthite, Geophys. Res. Letters 22, 2329
Konijnendijk W.L. et Stevels J.M., 1975, The structure of borate glasses studiedby Raman scattering, J. Non-Cryst. Solids 18, 307
Konijnendijk W.L. et Stevels J.M., 1976, Raman scattering measurements ofsilicate glasses and compounds, J. Non-Cryst. Solids, 447
Konijnendijk W.L. et Stevels J.M., 1978, Structure of borate and borosilicateglasses by Raman spectroscopy, Borate Glasses. MSR 12, 259
Kracek F.C., Neuvonen K., Burley G. et Gordon R.J., 1953, Contributions ofthermochemical and X-ray data to the problem of mineral stability, CarnegieInstitution Washington Yearbook 52, 69
Kreidl E.M., 1991, J. Non-Cryst. Solids 129, 1Krogh-Moe J., 1969, The structure of vitreous and liquid boron oxide, J. Non-
Cryst. Solids 1, 269Kurpka K.M., Hemingway B.S., Robie R.A. et Kerrick D.M., 1985, High-
temperature heat capacities and derived thermodynamic properties ofanthophyllite, diopside, dolomite, enstatite, bronzite, talc, tremolite andwollastonite, Am. Miner. 70, 261
Kurpka K.M., Robie R.A., Hemingway B.S., Robie R.A., Kerrick D.M. et ItoJ., 1985, Low-temperature heat capacities and derived thermodynamicproperties of anthophyllite, diopside, enstatite, and wollastonite, Am. Miner.70, 249
Lange R.A. et Carmichael I.S.E., 1987, Densities of Na2O-K2O-CaO-MgO-FeO-Fe2O3-Al2O3-TiO2-SiO2 liquids: news measurements and derivedpartial molar properties, Geochim. Cosmochim. Acta 51 supplement, 2931
Lange R.A. et Navrotsky A., 1993, Heat capacities of TiO2-bearing silicateliquids : evidence for anomalous changes in configurational entropy withtemperature, Geochim. Cosmochim. Acta 57, 3001
Lannin J.S., 1977, Raman scattering properties of amorphous As and Sb, Phys.Rev. B 15, 3863
244

Lanza F., Manara A., Mammarella L., Blasi P. et Ceccone G., 1988,Borosilicate HLW glass leaching in silica satured solution, Scientific Basisfor Nuclear Waste Management XI. MRS symposium proceeding 112, 685
Lapp J.C. et Shelby J.E., 1985, Structure-Property Relations in LanthanideBorate Glasses, J. Am. Ceram. Soc. 68, 368
Lasaga A.C., 1981a, Rates laws of chemical reactions, Rev. Miner. 8, 1Lasaga A.C., 1981b, Transition State Theory, Rev. Miner. 8, 135Lasaga A.C., 1981 c, The atomistic basis of kinetics : Defects in minerals, Rev.
Miner. 8, 261Lasaga, A.C., 1984, Chemical Kinetics of Water-Rock Interactions, J. Geophys.
Res. 89, 4009Lazarev A.N., 1972, Vibrational Spectra And Structure Of Silicates, ed.
Consultants Bureau, New York-LondonLe Grand M., 1999, Les platinoïdes et le molybdène dans les verres d'intérêt
nucléaire. Etude structurale, Thèse de doctorat de l'Université Paris VIILeadbetter A.J., 1968, The thermal properties of glasses at low temperatures,
Phys. Chem. Glasses 9, 1Lee C.T. et Clark D.E., 1985, Scientific Basis for Nuclear Waste Management
VIII. MRS symposium proceeding 61, 221Lee C.T. et Clark D.E., 1986, Effect of solution cations on waste glass leaching,
Adv. in Ceramics 20, 541Lee J.H. et Byrne R.H., 1992, Examination of comparative rare earth element
complexation behavior using linear free-energy relationships, Geochim.Cosmochimi. Ada 56, 1127
Leedecke C.J. et Bergeron C.G., 1978, Viscous flow in binary borate melts,Borate Glasses. MSR 12..413
Lefèvre J., 1986, Les déchets nucléaires, collection du CEA, ed. Eyrolles, ParisLejeune A.M., 1994, Rhéologie des magmas : influence des cristaux et des bulles
en suspension, Thèse de doctorat de l'Université Paris VIILejeune A.M. et P. Richet, 1995, Rheology of crystal-bearing silicate melts : an
experimental study at high viscosities, J. Geophys. Res., 100, 4215Le Turcq G., Berger G., Advocat T. et Vernaz E., 1999, Initial of long term
dissolution rate of aluminosilicate glasses enreached in Ti, Zr and Nd,Chem. Geol. 160, 39
Lévesque S., 1999, Rhéologie des silicates fondus et de laves partiellementcristallisées, Thèse de doctorat de l'Université Paris VII
London D., 1986, Magmatic-hydrothermal transition in the tanco rare-elementpegmatite : evidence from fluid inclusions and phase-equilibriumexperiments, Am. Miner. 71, 376
Loshagin A.V. et Sosnin E.P., 1994, Boron-11, silicon-29, and sodium-23 NMRin sodium borosilicate glasses, Glass Phys. Chem. 20, 250
Lutze W. et Schubert P., 1988, Chemical corrosion of lead-iron phosphate glass,Scientific Basis for Nuclear Waste Management XI. MRS symposiumproceeding 112, 641
245

Lutze W., Muller R. et Montserrat W., 1988, Chemical corrosion of COGEMAglass R7T7 in high saline brines, Scientific Basis for Nuclear WasteManagement XL MRS symposium proceeding 112, 575
MacGrail B.P., 1988, Modeling the dissolution behavior of defense waste glassin a salt repository environment, Scientific Basis for Nuclear WasteManagement XL MRS symposium proceeding 112, 595
Made B., 1991, Modélisation thermodynamique et cinétique des réactionsgéochimiques dans les interactions eau-roche, Thèse de doctorat del'Université Louis Pasteur. Strasbourg
Madé B., Clément A. et Fritz B., 1989, Modélisation cinétique etthermodynamique de l'altération : le modèlegéochimique KINDIS, C.R.Acad.Sci. 310,31
Madé B., Clément A. et Fritz B., 1994a, Modeling mineral/solution interactions:the thermodynamic and kinetic code KINDIS, Computer. Geosci. 20, 1347
Madé B., Clément A. et Fritz B., 1994b, Modélisation thermodynamique etcinétique des réactions diagénétiques dans les bassins sédimentaires.Présentation du modèle géochimique BONDIS, Rev. Inst. Fr. Petrol. 49, 569
Maillot E., 1989, Scénarios de refroidissement d'un conteneur de verre nominal,Note technique interne du CEA 1207 70 0106, SGN
Malow G., Lutze W. et Ewing R.C., 1984, Alteration effects and leach rates ofbasaltic glasses : implications for the long-term stability of nuclear wasteform borosilicate glasses, J. Non-Crystal. Solids 67, 305
Mao H.K., Virgo D. et Bell P.M., 1973, Analytical study of the orange soilreturned by the Apollo 17 astronauts, Carnegie Inst. Wash. Year Book 72,631
Mark T.D. et Walder G., 1988, Annealing and leaching studies with natural andartificial obsidian glass, Scientific Basis for Nuclear Waste Management XLMRS symposium proceeding 112, 693
Marshall W.L., 1980a, Amorphous silica solubilities I. Behavior in aqueoussodium nitrate solutions; 25-300° C, 0-6 molal, Geochim. Cosmochim. Ada44, 907
Marshall W.L., 1980b, Amorphous silica solubilities III. Activity coefficientrelations and predictions of solubility behavior in salt solution, 0-350° C,Geochim. Cosmochim. Acta 44, 925
Marshall W.L. et Warakomski J.M., 1980, Amorphous silica solubilities II.Effect of aqueous salt solutions at 25° C, Geochim. Cosmochim. Acta 44,915
Marshall W.L. et Chen C.A., 1982a, Amorphous silica solubilities V. Predictionsof solubility behavior in aqueous mixed electrolyte solutions to 300° C,Geochim. Cosmochim. Acta 46, 289
Marshall W.L. et Chen C.A., 1982b, Amorphous silica solubilities VI. Postulatedsulfate-silicic acid solution complex, Geochim. Cosmochim. Acta 46, 367
Martin S.W., Bain D., Budhwani K. et Feller S., 1992, 29Si MAS-NMR Studyof the Short-Range Order in Lithium Borosilicate Glasses, J. Am. Ceram.Soc. 75, 1117
246

Mayer P., Topping J.A. et Lackner J.L., 1977, Effect of Additions ofTransition-Metal Oxides on the Corrosion Kinetics of a K2O-Na2O-BaO-SiÛ2 Glass in Aqueous KOH Solutions, J. Am. Ceram. Soc. 60, 181
Mazurin O.V. et Streltsina M.V., 1972, Determination of TIE-line directions inthe metastable phase-separation regions of ternary systems, J. Non-Crystal.Solids, 199
Michard G., 1983, Recueil des données thermodynamiques concernant leséquilibres eaux-minéraux dans les réservoirs hydrothermaux. Commissiondes Communautés Européennes, EUR 8590 FR
Michard G., 1989, Equilibres chimiques dans les eaux naturelles, éd. PublisudMorey G.W.,1951, J. Soc. Glass Tech. 35, 270Morss L.R. et Williams C.W., 1992, Enthalpies of formation of rare earth and
actinide(III) hydroxydes; their acid-base relationships and estimation oftheir thermodynamic properties, Scientific Basis for Nuclear WasteManagement XV. MRS symposium proceeding, volume 257, 283
Morss L.R., 1986, Thermodynamic Properties, The Chemistry of the ActinideElements, ed. Katz, Seaborg and Morss, volume 2, 1278
Moynihan C.T., 1993, Correlation between the width of the glass transitionregion and the temperature dependence of the viscosity of high-Tg glasses,J. Am. Ceram. Soc. 76, 1081
Moynihan C.T., Bruce A.J., Gavin D.L., Loehr S.R. et Opalka S.M., 1984,Physical aging of heavy metal fluoride glasses - sub_rg enthalpy relaxationin a ZrF4-BaF2-LaF3-AIF3 glass, Polymer Eng. Sci. 24, 1117
Miiller F. et Kleppa O.J., 1973, Thermodynamics of formation of chromitespinels, J. Inorg. Nucl. Chem. 35, 2673
Murakami T., Banba T., Jercinovic M.J. et Ewing R.C., 1989, Formation andevolution of alteration layers on borosilicate and basalt glasses : initialstage, Scientific Basis for Nuclear Waste Management XII. MRSsymposium proceeding 127, 65
Myhra S., Pham D.K., Smart R.St. C. et Turner P.S., 1990, Dissolutionmechanisms of the perovskite and hollandite phases in the synrocassemblage, Scientific Basis for Nuclear Waste Management XIII. MRSsymposium proceedingl76, 249
Mysen B.O. et Virgo D., 1978, Influence of pressure, temperature and bulkcomposition on melt structures in the system NaAlSi2Û6-NaFe3+ Si2Û6, Am.J. Sci. 278, 1307
Mysen B.O., et Virgo D., 1983, Effect of pressure on the structure of iron-bearingsilicate melts, Carnegie Inst. Wash. Year Book 82, 321
Mysen B.O., Seifert F.A. et Virgo D., 1980, Structure and redox equilibria ofiron-bearing silicate melts, Am. Miner. 65, 867
Navrotsky A., 1971, Thermodynamics of formation of the silicates andgermanates of some divalent transition metals and of magnesium, J. Inorg.Nucl. Chem. 33, 4035
Navrotsky A., 1977, Progress and new directions in high temperaturecalorimetry, Phys. Chem. Minerals 2, 89
247

Navrotsky A., 1987, Calorimetric studies of melts, crystals, and glasses,especially in hydrous systems, Magmatic Processes: PhysicochemicalPrinciples , The Geochemical Society, special publication 1,411
Navrotsky A. et Kleppa O.J., 1973, Estimate of Enthalpies of Formation andFusion of Cordierite, J. Am. Ceram. Soc. 56, 198
Navrotsky A. et Coons W.E., 1976, Thermochemistry of some pyroxenes andrelated compounds, Geochim. Cosmochim. Ada 40, 1281
Navrotsky A. et Kasper R.B., 1976, Spinel disproportion at high pressure:calorimetric determination of Mg2SnO4 and Co2SnO4 and someimplications for silicates, Earth Planet. Sci. Lett. 31, 247
Navrotsky A., Hon R., Weill D.F. et Henry D.J., 1980, Thermochemistry ofglasses and liquids in the systems CaMgSi2O6-CaAl2Si2C>8-NaAlSi3O8,SiO2-CaAl2Si2O8-NaAlSi3O8 and SiO2-Al2O3-CaO-Na2O, Geochim.Cosmochim. Ada 44, 1409
Navrotsky A., Rapp R.P., Smelik E., Burnley P., Circone S., Chai L., Bose K.et Westrich H.R., 1994, The behavior of H2O and CO2 in high-temperaturelead borate solution calorimetry of volatile-bearing phases, Am. Miner. 79,1099
Nemilov S.V., 1997, Glass Phys. Chem. 23, 1Neuville R.D. et Richet P., 1991, Viscosity and mixing in molten (Ca, Mg)
pyroxenes and garnets, Geochim. Cosmochim. Ada 55, 1011Neuville R.D., 1992, Etude des propriétés thermodynamiques et rhéologiques des
silicates fondus, Thèse de doctorat de l'Université Paris VIINeuville R.D., Courtial P., Dingwell D. et Richet P., 1993, Thermodynamic and
rhelogical properties of rhyolite and andésite melts, Contrib. Mineral.Petrol. 113, 572
Neuville, R.D.et Richet P., 1990, Viscosité et entropie des silicates fondus,Rivisita délia Staz. Sper Vetro, 6
Newton R.C., Charlu, T.V. et Klepa O.J., 1977, Thermochemistry of highpressure garnets and clinopyroxenes in the system CaO-MgO-Al2O3-SiO2,Geochim. Cosmochim. Ada 41, 369
Newton R.C., Charlu, T.V. et Klepa O.J., 1980, Thermochemistry of the highstructural state plagioclases, Geochim. Cosmochim. Ada 44, 933
Newton R.C., Charlu, T.V., Anderson P.A.M. et Klepa O.J., 1979,Thermochemistry of synthetic clinopyroxenes on the join CaMgSi2O6-Mg2Si2O6, Geochim. Cosmochim. Ada 43, 55
Oh M.S. et Oversby V.M., 1991, The effect of sample preparation methods onglass performance, Scientific Basis for Nuclear Waste Management XIV.MRS symposium proceeding 212, 123
Orlhac X., 1999, Etude de la stabilité thermique du verre nucléaire. Modélisationde son évolution à long terme, Thèse de doctorat de l'Université MontpellierH
Osborn E.F., 1959, Role of oxygen pressure in the crystallisation anddifférenciation of basaltic magma, Am. J. Sci. 257, 609
248

Oestrike R., Navrotsky A., Turner G.L., Montez B. et Kirkpatrick R.J., 1987,Structural environment of Al dissolved in 2PbO.B2O3 glasses used forsolution calorimetry: an 27A1NMR study, Am. Miner. 72, 788
Ostvold T. et Kleppa O.J., 1970, Thermochemistry of liquid borate. II. Partialenthalpies of solution of boric oxide in its liquid mixtures with lithium,sodium, and potassium oxides, Inorg. Chem. 9, 1395
Pacaud F., Fillet C. et Jacquet-Francillon N., 1992, Effect of platinoids onfrench LWR reference glass properties, Scientific Basis for Nuclear WasteManagement XV. MRS symposium proceeding 257, 161
Parks G.S. et Huffman H.M., 1927, Studies on glass. I.The transition betweenthe glassy and liquid states in the case of some simple organic compounds,J.Phys. Chem. 31, 1842
Paul A. et Cooke D., 1978, Chemical Stability of Boron Containing GlassEnamels with Special Reference to Lead Release, Borate Glasses. MSR 12,509
Paul A., 1977, Chemical durability of glasses, Journal Mater. Sci. 12, 2246Paul A., 1990, Chemistry of glasses, ed. Chapman & HallPauling L., 1929, The principles determining the structure of complex ionic
crystals,/. Am. Chem. Soc. 51, 1010Pederson L.R., McGrail B.P., McVay G.L., Petersen-Villalobos D.A. et Settles
N.S., 1993, Kinetics of alkali silicate and aluminosilicate glass reactions inalkali chloride solutions : influence of surface charge, Phys. Chem. Glasses34, 140
Pélegrin E., 2000, Thèse de doctorat de l'Université Paris VIIPetit-Maire D., 1988, Strucure local autour d'actinides et d'éléments nucléants
dans les verres borosilicaté d'intérêt nucléaire : résultats de spectroscopied'absorption des rayons X, Thèse de doctorat de l'Université Pierre et MarieCurie (Paris VI)
Pichavant M., 1978, Etude du sustème SiO2-B2O3 à P=l kbar. Diagramme dephases et interprétation thermodynamique, Bull. Miner. 101, 498
Pichavant M., Herrera J.V., Boulmier S., Briqueu L., Joron J-L., Juteau M.,Marin L., Michard A., Sheppard S.M.F., Treuil M., et Vernet M., 1987,The Macusani glass, SE Peru : evidence of chemical fractionation inperaluminous magmas, Magmatic Processes, physiochemical pronciples.Geochemical Society, Publ. Spec. 1, 359
Pohl R.O., 1981, Low-temperature specific heats of glasses, Topics in currentphysics, ed. Springer Verlag, 24
Rankin W.D.et Wicks G.G., 1983, Chemical Durability of Savannah River PlantWaste Glass as a Function of Waste Loading, J. Am.Ceram. Soc. 66, 417
Rawson H., 1956, The relationship between liquidus temperature, bond strength
and glass formation, Travaux du IVe congrès international du verre. ParisRawson H., 1967, Inorganic glass-forming systems, ed. Academic Press, NYRaymond M., 1992, Mécanismes et vitessses de corrosion du verre R7T7 en
condition de saturation en silice, communication interne CEA
249

Regnard J.R., Chavez F. et Chappert J., 1983, Study of the oxidation states andmagnetic properties of iron in volcanic glasses : Lipari and Teotihuacanobsidians, Bull. Min. 104, 204
Richet P., 1988, Superheating, melting and vitrification through decompression ofhigh-pressure minerals, Nature 331, 56
Richet P., 1991, Propriétés thermodynamiques et rhéologiques des silicatesfondus, Rev. Int. Hautes Temper. Refract. 27, 1
Richet, P., 1984, Viscosity and configurational entropy of silicate melts,Geochim. Cosmochim. Acta 48, 471
Richet, P., 1987, Heat capacity of silicate glasses, Chem. Geol. 62, 111Richet P.et Bottinga Y., 1980, Heat capacity of liquid silicates : new
measurements on NaAlSisOs and K2Si4Û9, Geochim. Cosmochim. Acta 44,1535
Richet, P. et Bottinga Y.,1983, Verres, liquides et transition vitreuse, Bull.Minéral. 106, 147
Richet P. et Bottinga Y.,1984a, Anorthite, andesine, wollastonite, diopside,cordierite and pyrope : thermodynamics of melting, glass transitions andproperties of the amorphous phases, Earth Planet. Sci. Lett. 67, 415
Richet P.et Bottinga Y.,1984b, Glass transitions and thermodynamic properties ofamorphous SiC>2, NaAlSin02n+2 and KAlSisOg, Geochim. Cosmochim. Acta48, 453
Richet P.et Bottinga Y.,1985, Heat capacity of aluminium-free liquid silicates,Geochim. Cosmochim. Acta 49, 471
Richet, P. et Bottinga Y.,1986, Thermochemical Properties of Silicate Glassesand Liquids : A Review, Rev. Geophysics 24, 1
Richet P.et Bottinga Y.,1994, Rheology and configurational entropy of silicatemelts, Rev. Mineral. 32, chap. 3, 67
Richet P. et Fiquet G.,1991, High-Temperature Heat Capacity and Premelting ofMinerals in the System MgO-CaO-Al2O3-SiO2, J. Geophys. Res. 96, 445
Richet P. et Neuville D.R.,1992, Thermodynamics of silicate melts :configurational properties, Adv. Phys. Geochem. 10, 132
Richet P., Bottinga Y. et Téqui C, J.Am. Ceram. Soc. 67 (1984) C-6Richet P., Gillet P. et Fiquet G., 1992, Adv. Phys. Geochem. 10, 98, ed. S.K.
Saxena, Springer VerlagRichet P., Leclerc F. et Benoist L.,1993, Melting of forsterite and spinel, with
implications for the glass transition of Mg2SiO4 liquid, Geophys. Res.Letters 20, 1675
Richet P., Robie R.A. et Hemingway B.S., 1986, Low-temperature heat capacityof diopside glass (CaMgSiaOô) : A calorimetric test of the configurational-entropy theory applied to the viscosity of liquid silicates, Geochim.Cosmochim. Acta 50, 1521
Richet P., Robie R.A. et Hemingway B.S., 1991, Thermodynamic properties ofwollastonite, pseudowollastonite and CaSiC>3 glass and liquid, European J.Mineral. 3, 475
250

Richet P., Robie R.A. et Hemingway B.S.,1992, Entropy and structure of silicateglasses and melts, Geochim. Cosmochim. Ada 57, 2751
Richet, P., Bottinga Y., Denielou L., Petitet J.P. et Téqui C.,1982,Thermodynamic properties of quartz, cristobalite and amorphous SiO2 :drop calorimetry measurements between 1000 and 1800 K and a reviewfrom 0 to 2000 K, Geochim. Cosmochim. Ada 46, 2639
Richet, P., Bouhifd M.A., Courtial P. et Téqui C , 1997, Configurational heatcapacity and entropy of borosilicate melts, J. Non-Cryst. Solids 211, 271
Richet, P., Robie R.A., Rogez J., Hemingway B.S., Courtial P. et TéquiC.,1990, Thermodynamics of Open Networks : Ordering and Entropy inNaAlSiO4 Glass, Liquid and Polymorphs, Phys. Chem. Minerals 17, 385
Ricol, S., 1995, Etude du gel d'altération des verres nucléaires et synthèse de gelsmodèles, Thèse de doctorat de l'Université Paris VI
Riebling, E.F., 1968, Structural similarities between a glass and its melt, J. Am.Ceram. Soc. 51, 143
Robie R.A. et Hemingway B.S., 1992, Calorimeters for heat of solution and low-temperature heat capacity measurements, U.S. Geol. Surv. Prof. Paper 755.
Robie R.A. et Hemingway B.S., 1995, Thermodynamic properties of mineralsand related substances at 298.15 K and 1 bar pressure and highertemperatures, U.S. Geol. Surv. Bull. 2131
Robie R.A. et Hemingway B.S., U.S. Geol. Surv. Prof. Paper 755 (1972)Robie R.A., Bin Z., Hemingway B.S. and Barton D., 1987, Heat capacity and
thermodynamic properties of andradite garnet, Ca3Fe2Si3Oi2, between 10and 1000 K and revised values for AyGo(298.15 K) of hedenbergite andwollastonite, Geochim. Cosmochim. Ada 51, 2219
Robie R.A., Hemingway B.S. and Wilson W.H., 1978, Low-temperature heatcapacities and entropies of feldspar glasses and of anorthite, Am. Miner. 63,109
Robie R.A., Hemingway B.S. et Wilson W.H., 1976, J. Res. U.S. Geol. Surv. 4,631
Robinson G.R. et Haas J.L., 1983, Heat capacity, relative enthalpy, andcalorimetric entropy of silicate minerals: an empirical method of prediction,Am. Miner. 68, 541
Rogers P.S. et Williamson J., 1969, The nucleation of crystalline phases insilicate glasses containig iron oxydes, Glass Technology 10, 128
Rogez J., Chastel R., Bergman C , Brousse C, Castanet R. et Mathieu J-C,1983, Bull. Mineral. 106 , 119
Roy B.N. et Navrotsky A., 1984,Thermochemistry of Charge-CoupledSubstitutions in Silicate Glasses : The Systems Mi/n
n+A102-Si02 (M = Li,Na, K, Rb, Cs, Mg, Ca, Sr, Ba, Pb), J. Am. Ceram. Soc. 67, 606
Sack R.O., Carmichel I.S.E., Rivers M.L. et Ghiorso M.S., 1980, Ferric-ferrousequilibria in natural silicate liquids at 1 bar, Contrib. Mineral. Petrol. 75,369
Sales B.C., White C.W., Begun G.M. et Boatner L.A., 1984, Surface layerformation on corroded nuclear waste glasses, J. Non-Crystal. Solids 67, 245
251

Scarfe CM., Cronin D.J., Wenzel J.T. et Kauffman D.A., 1983, Viscosity-temperature relationships at 1 atm in the system diopside-anorthite, Am.Miner. 68, 1083
Schairer J.F. et Bowen NX., 1955, The system K2O-Al2O3-SiO2, Am. J. Sci.253,681
Schairer J.F. et Bowen N.L., 1956, The system Na2O-Al2O3-SiO2, Am. J. Sci.254,129
Scherer G.W. et Drexhage M.G., 1985, Stress in Leached Phase-SeparetedGlass, J. Am. Ceram. Soc. 68, 419
Schindler P.W. et Stumm W., 1987, The surface chemistry of oxides,hydroxides and oxides minerals, Aquatic surface chemistry, ed. WileyInterscience, New York
Sen S., Xu Z. et Stebbins J.F., 1998, Temperature dependent structural changesin borate, borosilic and boroaluminate liquids: high-resolution UB, 29Si and27Al NMR studies, / . Non-Cryst. Solids 226, 29
Shaw H.R., 1972, Viscosités of magnetic silicate liquids : an empirical method ofprediction, Am. J. Sci. 272, 870
Shearer J.A. et Kleppa O.J., 1973, J. Inorg. Nucl. Chem. 35,1073Sipp A., Neuville D.R. et Richet P., 1997, Viscosity, configurational entropy and
relaxation kinetics of borosilicate melts, J. Non-Cryst. Solids 211, 281Smelik E.A., Jenkins D.M. et Navrotsky A., 1994, A calorimetric study of
synthetic amphiboles along the tremolite-tschermakite join and the heats offormation of magnesiohornblende and tschermakite, Am. Miner. 19, 1110
Smetts B.M.J., Tholen M.G.W. et Lommen T.P.A., 1984, The effect of divalentcations on the leaching kinetics of glass, / . Non-Crystal. Solids 65, 319
Smith CF., 1978, The vibrational analysis of boron in vitreous silica, BorateGlasses. MSR 12, 307
Smith D.K., 1991, Mineralogical, textural and compositional data on thealteration of basaltic glass from Kilauea, Hawaii to 300°C : insights to thecorrosion of a borosilicate glass waste-form, Scientific Basis for NuclearWaste Management XIV. MRS symposium proceeding 212, 115
Sombret C, 1987, Industrial vitrification processes for high level liquid wastes :potential and limitations, Proceedings of the international conference onNuclear Fuel Reprocessing and Waste Management "RECOD 87". volume2,625
Soûles T.F., 1979, A molecular dynamic calculation of the structure of sodiumsilicate glasses, J. Chem. Phys. 71, 4570
Sposito G., 1986, On distinguishing adsorption from surface precipitation,Geochemical Processes at mineral surfaces, volume 323, édité par DavisT.A. et Hayes K.F.
Stebbins J.F., 1988, Effects of temperature and composition on silicate glassstructure and dynamics : 29Si NMR results, J. Non-Cryst. Solids 106, 359
Stebbins J.F., 1991, Nuclear magnetic resonance at high temperature, Chem. Rev.91, 1353
252

Stebbins J.F. et Ellsworth S.E., 1996, Temperature effects on structure anddynamics in borate and borosilicate liquids: high-resolution and high-temperature NMR results, J.Am. Ceram. Soc. 19, 2247
Stebbins J.F. et Sen S., 1998, Microscopic dynamics and viscous flow in aborosilicate glass-forming liquid, J. Non-Cryst. Solids 224, 80
Stebbins J.F., Carmichael I.S.E. et Moret L.K., 1984, Heat capacities andentropies of silicate liquids and glasses, Contrib. Mineral. Petrol. 86, 131
Stebbins J.F., Farnan I. et Xue X., 1992, The structure and dynamics of alkalisilicateliquids : a view from NMR spectroscopy, Chem. Geol. 96, 371
Stebbins J.F., Sen S. et Farnan I., 1995, Silicate species exchange, viscosity,and crystallisation in a low-silica melt : in situ high temperature MAS NMRspectroscopy, Am. Miner. 80, 861
Stumm W. et Furrer G., 1987, The dissolution of oxides and aluminiumsilicates, examples of surface coordination controlled kinetics, Aquaticsurface chemistry, ed. Wiley Interscience, New York
Stumm W., Furrer G., Wieland E. et Zinder B., 1985, The effect of complexforming ligands on the dissolution rates of minerals, Adv. Chem. 52,1969
Sun K.H., 1947, Fundamental condition of glass formation, J. Am. Ceram. Soc.30, 277
Tammann G. et Hesse W., 1926, Die abhângigkeit der viscositàt von dertemperatur bei unterkiihlten fliissigkeiten, Z. anorg. U. allg. Chem. 156, 245
Tang Y., Jiang Z. et Song X., 1989, NMR, IR and Raman spectra study of thestructure of borate and borosilicate glasses, J. Non-Cryst. Solids 112, 13 lq
Tauber, P. et Arndt J., 1987, The relationship between viscosity and temperaturein the sytem anorthite-diopside, Chem. Geol. 62, 71
Taylor M. et Rindone G.E., 1970, J. Amer. Ceram. Soc. 53, 692Téqui C, Robie R.A., Hemingway B.S., Neuville D.R. and Richet P., 1991,
Geochim. Cosmochim. Ada 55, 1005Téqui, C, Grinspan P. et Richet P., 1992, Thermodynamic properties of alkali
silicates : heat capacity of Li2SiÛ3 and lithium-bearing melts, J. Am. Ceram.Soc. 75, 2601
Téqui, C, Robie R.A., Hemingway B.S., Neuville D.R. et Richet P., 1991,Melting and thermodynamic properties of pyrope (Mg3Al2Si3Oi2),Geochimi. Cosmochim. Acta 55, 1005
Terradas R., Neuville D.R., Martinez S. et Richet P., 1997, Viscosity, heatcapacity and configurational entropy of Zn-bearing silicate melts, EUG 97,résumé 72/4P13
Thieblot L., 1996, Etude des propriétés thermodynamiques de l'enstatite et desgrenats à haute température et de la préfusion, Thèse de doctorat del'Université Paris VII
Tool A.Q. et Eichlin C.G., 1931, Variations caused in the heating curves of glassby heat treatment, J. Am. Ceram. Soc. 14, 276
Tovena, I., 1995, Influence de la composition des verres nucléaires sur leuraltérabilité, Thèse de doctorat de l'Université de Montpellier II
253

Trotignon L., 1990, La corrosion aqueuse des verres borosilicatés. Nature etpropriétés des couches d'altération, Thèse de doctorat de l'Université PaulSabatier de Toulouse
Tsukamoto M., Bjorner I.K., Christensen H., Hermansson H.P. et Werme L.,1988, Leaching of AM-241 from a radioactive waste glass corroded in thepresence of stainless steel corrosion products and/or bentonite, ScientificBasis for Nuclear Waste Management XL MRS symposium proceeding112,.565-
Turnbull D. et Cohen M.H., 1961, Free-volume model of the amorphous phase,J. Chem. Phys. 34, 120
Turnbull D., 1969, Under what conditions can a glasss be formed ?, Contemp.Phys. 10, 473
Uhlmann D.R., Kolbeck A.G. et De Witte D.L., 1971, Heat capacities andthermal behavior of alkali borate glasses, J. Non-Crystal. Solids 5, 426
Urbain G., Bottinga Y. et Richet P., 1982, Viscosity of liquid silica, silicates andalumino-silicates, Geochim. Cosmochim. Acta 46, 1061
Van Iseghem P. et Grambow B., 1988, The long-term corrosion and modellingof two simulated belgian reference high-level waste glasses, Scientific Basisfor Nuclear Waste Management XI. MRS symposium proceeding 112, 631
Vernaz E. et Dussossoy J.L., 1992, Current state of knowledge of nuclear wasteglass corrosion mechanism : the case of R7T7 glass, Appl. Geochem.*publication spéciale n°l, 13
Vernaz E., Dussossoy J.L. et Fillet S., 1988, Temperature dependence of R7T7nuclear waste glass alteration mechanisms, Scientific Basis for NuclearWaste Management XL MRS symposium proceeding 112, 555
Vessal B., Greaves G.N., Marten P.T., Chadwick A.V., Mole R. et Houde-Walter S., 1992, Cation macrosegregation and ionic mobility in mixedalkali glasses, Nature 356, 504-506
Virgo D., Mysen B.O., Danckwerth P.A. et Seifert F.A., 1982, Speciation ofFe3+ in 1-atm Na2O-SiO2-FeO melts, Carnegie Inst. Wash. Year Book 81,349
Vogel H., 1921, Das Temperatur-abhângigkeitgesetz der viskositât vonfliissigkeiten, Physik. Zeitschr. 22, 645
Wang S. et Stebbins J.F., 1998, On the structure of borosilicate glasses: a triple-quantum magic-angle spinning 170 nuclear magnetic resonance study, J.Non-Cryst. Solids 231, 286
Wang Y., Wang Y. et Merino E., 1995, Dynamic weathering model : Constraintsrequired by coupled dissolution and pseudomorphic replacement, Geochim.Cosmochim. Acta 59, 1559
Warren B.E., 1933, X-ray diffraction of vitreous silica, Z. Kristallogr. 86, 349Warren B.E., Krutter H. et Morningstar O., 1936, J. Am. Ceram. Soc. 19, 202Westrum E.F. Jr. et Grenier G., 1957, J. Am. Ceram. Soc. 79, 1799Westrum E.F. Jr., 1988, Calorimetric in the range 5-300K, Specific heat of
solids, ed. C.Y. Ho Hemisphere Publishing CorporationWestrum E.F. Jr., Furukawa G.T. et McCullough J.T., 1968, Experimental
Thermodynamics, ed. Butterworths, London
254

Westrum E.FJr., 1964, Thermodynamic and Magnetic Properties of the RareEarth Chalcogenides, Progress in the Science and Technology of the RareEarths, volume 1, 310
Westrum E.F.Jr., 1966, Thermodynamic and Magnetic Properties of the RareEarth Chalcogenides and Pnictides, Progress in the Science and Technologyof the Rare Earths, volume 2, 35
Westrum E.F.Jr., 1968, Thermodynamic and Magnetic Properties of the RareEarth Chalcogenides, Pnictides, Halides and Binary Semi-MetallicCompounds, Progress in the Science and Technology of the Rare Earths.volume 3, 459
White W.B., 1982, J. Non-Cryst. Solids 49, 321Whittington A., Richet P., Linard Y. et Holtz F., 2000, The viscosity of
hydrous phonolites and trachytes, en préparationWieland E., Wehrli B. et Stumm W., 1988, The coordination chemistry of
weathering : III. A generalization on dissolution rates of minerals,Geoc/zz7«.Cosmochim. Acta 52, 1969
Wood B.J. et Klepa O.J., 1981, Thermochemistry of forsterite-fayalite olivinesolutions,Geoc/n>w. Cosmochim. Acta 45, 529
Wood B.J., Holland T.J.B., Newton R.C. et Klepa O.J., 1980,Thermochemistry of jadeite-diopside pyroxenes,Geochim. Cosmochim. Acta44, 1363
Wright A.C., 1994, Neutron scattering from vitreous silica. V. The structure ofvitreous silica: what have we learned from 60 years of diffraction studies ?,/. Non-Cryst. Solids 179, 84
Wright A.C., Hulme R.A., Grimley D.I., Sinclair R.N., Martin S.W., PriceD.L. et Galeener F.L., 1991, The structure of some simple amorphousnetwork solids revisited, J. Non-Cryst. Solids 129, 213
Xiao S., 1985, A reaction series model for the structure of Na2O-B2O3-SiO2
glasses, J. Chin. Silicate Soc. 13, 58Yamanaka S., Akagi J. et Hattori M., 1985, Reaction of Pyrex type Borosilicate
Glass with water in autoclave, J. Non-Crystal. Solids 70, 279Yun Y.H. et Bray P.J., 1978, Nuclear magnetic resonance studies of the glasses
in the system Na2O-B2O3-SiO2, / . Non-Cryst. Solids 27, 363Yun Y.H. et Bray P.J., 1979, Correction and addenum to "NMR studies of the
glasses in the system Na2O-B2O3-SiO2", J. Non-Cryst. Solids 33, 273Zachariasen W.H., 1932, The atomic arrangement in glass, J. Am. Chem. Soc.
54,3841Zarzycki J., 1982, Les verres et l'état vitreux. MassonZhang Z., 1996, Corrosion of 20M2O.80B2O3 (M=Li, Na and K) glasses in water,
Phys. Chem. Glasses 37, 221Zhong J., Wu X., Liu M.L. et Bray P.J., 1988, Structural modeling of lithium
borosilicate glasses via NMR studies, J. Non-Cryst. Solids 107, 81Zhu H., Newton R.C. et Kleppa O.J., 1994, Enthalpy of formation of
wollastonite (CaSiO3) and anorthite (CaAl2Si2Og) by experimental phaseequilibrium measurements and high-temperature solution calorimetry, Am.Miner. 79, 134
255

Zygan V.N., Kesler Y.A., Gordeev, I.V. et Tret'yakov Y.D., 1978, Neorg.Mater. 14, 1087
256

ANNEXE A
257

Tableau A.I : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON-3.
Tableau A.2 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON-4.
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800850
894.96894.96900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
0.000.151.157.3818.8528.8737.1644.1547.0449.9850.1855.3659.7763.5666.8469.7072.2374.4876.4878.2979.9281.4182.63107.49107.49107.49107.49107.49107.49107.49107.49107.49107.49107.49107.49107.49107.49107.49
(kJ/mol)
-7.95-7.95-7.95-7.82-7.17-5.96-4.31-2.27-1.210.000.092.735.628.7011.9615.3818.9322.6026.3730.2434.2038.2341.9241.9242.4647.8453.2158.5963.9669.3374.7180.0885.4690.8396.21101.58106.96112.33
S7S29S(J/mol.K)
-48.81-48.76-48.40-45.11-36.43-26.83-17.35-8.29-4.250.000.318.4516.1323.4030.2736.7742.9548.8254.4259.7664.8669.7573.9873.9874.5880.4085.9191.1596.15100.93105.51109.90114.11118.17122.08125.85129.49133.02
-iGr-HmyT(J/mol.K)
-
843.99445.74201.60108.0966.6038.8917.368.690.00-0.62-16.26-30.17-42.73-54.19-64.73-74.50-83.59-92.09-100.08-107.61-114.73-120.82-120.82-121.76-130.75-139.12-146.95-154.30-161.22-167.77-173.96-179.85-185.45-190.80-195.91-200.80-205.49
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800850
851.20851.20900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
49.2552.0652.2657.0961.1364.5667.5370.1272.4174.4676.3077.9779.4980.8980.92106.46106.46106.46106.46106.46106.46106.46106.46106.46106.46106.46106.46106.46106.46106.46
(kJ/mol)
-1.270.000.102.835.798.9412.2415.6819.2522.9226.6930.5534.4838.5038.5938.5943.7949.1154.4359.7665.0870.4075.7281.0586.3791.6997.02102.34107.66112.98
Sj-S29S(J/mol.K)
-4.440.000.328.7516.6524.0531.0137.5743.7749.6555.2460.5665.6470.5070.6270.6276.5582.3187.7792.9697.91102.65107.18111.52115.70119.72123.59127.32130.93134.42
-(Gj-H2gs)/T
(J/mol.K)
9.070.00-0.64-16.85-31.13-43.91-55.49-66.09-75.85-84.91-93.37-101.29-108.75-115.79-115.96-115.96-125.20-134.00-142.20-149.87-157.08-163.86-170.28-176.36-182.14-187.64-192.88-197.90-202.71-207.32
258

Tableau A.3 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON-5.
Tableau A.4 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON-6.
T(K)
0
10
2050100150200250
273.15
298.15
300350400450500550600650700750800
840.62
840.62
8509009501000
1050
1100
1150
1200
1250
1300
1350
1400
1450
1500
1550
cP(J/mol.K)
49.3452.9153.1558.7863.1666.7569.8172.5074.9077.0979.1181.0082.7884.17116.40116.36116.17116.00115.83115.68115.54115.40115.27115.14115.02114.90114.78114.67114.55114.44
HrH29s(kJ/mol)
-1.280.000.102.905.969.2112.6216.1819.8723.6727.5731.5835.6739.0652.1753.2659.0764.8870.6776.4682.2488.0193.7899.54105.29111.04116.78122.52128.25133.98
S1-S29S(J/mol.K)
-4.480.000.338.9617.1124.7631.9638.7445.1551.2457.0262.5567.8371.9771.9773.2679.9086.1892.1297.77103.15108.28113.19117.89122.41126.75130.92134.95138.83142.59
-(GT-H29S)/T(J/mol.K)
9.160.00-0.66-17.26-32.00-45.22-57.20-68.16-78.26-87.65-96.41-104.65-112.42-118.43-134.02-135.92-145.54-154.47-162.80-170.59-177.91-184.82-191.34-197.53-203.40-209.00-214.34-219.45-224.34-229.03
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800850
855.66855.66900950100010501100115012001250130013501400145015001550
(J/mol.K)
46.3650.1550.4056.0660.2563.6066.4268.8971.1373.2175.1677.0278.8280.5780.76112.39111.44110.52109.72109.03108.42107.89107.41106.98106.59106.24105.92105.63105.36105.11
H1-H29S(kJ/mol)
-1.210.000.092.765.678.7712.0315.4118.9122.5226.2330.0333.9337.9238.3738.3743.3348.8854.3959.8665.2970.7076.0881.4486.7892.1097.41102.69107.97113.23
S7-S298(J/mol.K)
-4.230.000.318.5316.3023.6030.4536.9042.9948.7654.2659.5164.5469.3769.9169.9175.5681.5687.2192.5497.60102.41106.99111.37115.56119.57123.43127.14130.72134.17
(J/mol.K)
8.650.00-0.62-16.42-30.49-43.09-54.50-64.91-74.51-83.41-91.73-99.56-106.95-113.98-114.75-114.75-123.71-133.02-141.60-149.55-156.96-163.89-170.39-176.52-182.31-187.79-193.00-197.96-202.70-207.22
259
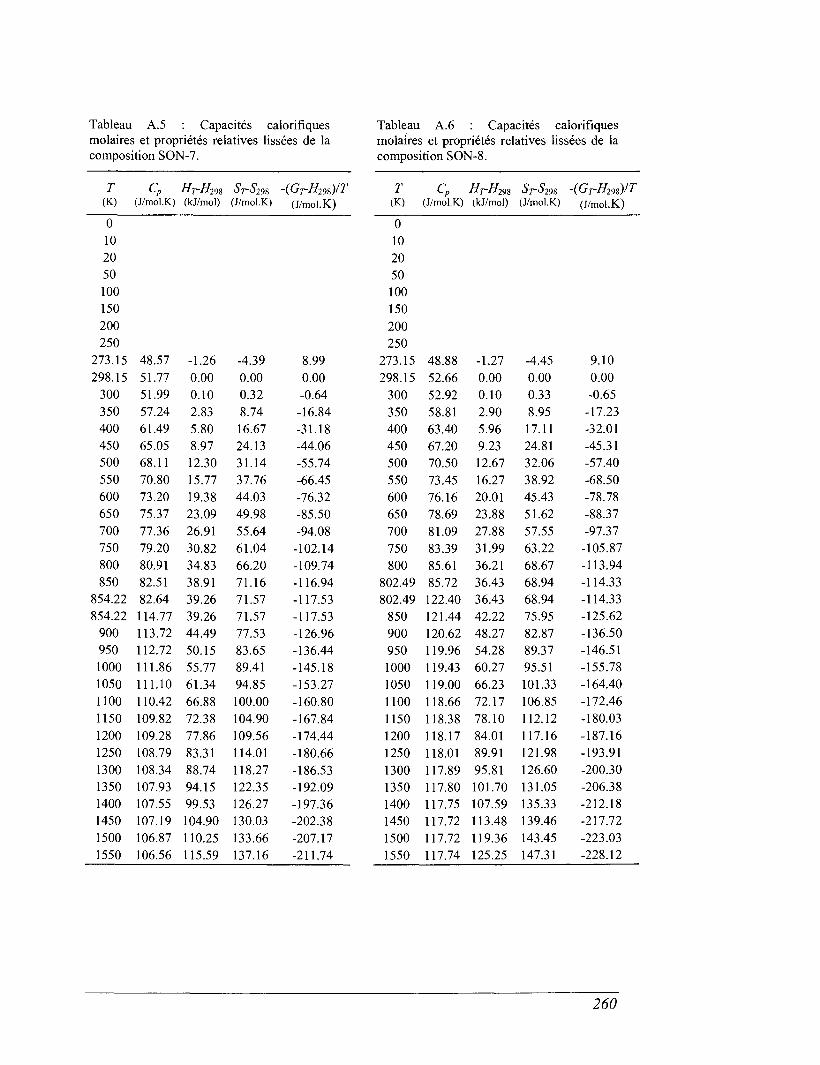
Tableau A.5 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON-7.
Tableau A.6 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON-8.
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800850
854.22854.22900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
48.5751.7751.9957.2461.4965.0568.1170.8073.2075.3777.3679.2080.9182.5182.64114.77113.72112.72111.86111.10110.42109.82109.28108.79108.34107.93107.55107.19106.87106.56
(kj/mol)
-1.260.000.102.835.808.9712.3015.7719.3823.0926.9130.8234.8338.9139.2639.2644.4950.1555.7761.3466.8872.3877.8683.3188.7494.1599.53104.90110.25115.59
SrS29i(J/mol.K)
-4.390.000.328.7416.6724.1331.1437.7644.0349.9855.6461.0466.2071.1671.5771.5777.5383.6589.4194.85100.00104.90109.56114.01118.27122.35126.27130.03133.66137.16
-(Gr//298)/r(J/mol.K)
8.990.00-0.64-16.84-31.18-44.06-55.74-66.45-76.32-85.50-94.08-102.14-109.74-116.94-117.53-117.53-126.96-136.44-145.18-153.27-160.80-167.84-174.44-180.66-186.53-192.09-197.36-202.38-207.17-211.74
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800
802.49802.49850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
48.8852.6652.9258.8163.4067.2070.5073.4576.1678.6981.0983.3985.6185.72122.40121.44120.62119.96119.43119.00118.66118.38118.17118.01117.89117.80117.75117.72117.72117.74
Hj-H29s
(kJ/mol)
-1.270.000.102.905.969.2312.6716.2720.0123.8827.8831.9936.2136.4336.4342.2248.2754.2860.2766.2372.1778.1084.0189.9195.81101.70107.59113.48119.36125.25
SrS29B(J/mol.K)
-4.450.000.338.9517.1124.8132.0638.9245.4351.6257.5563.2268.6768.9468.9475.9582.8789.3795.51101.33106.85112.12117.16121.98126.60131.05135.33139.46143.45147.31
-(Gr//298)/r(J/mol.K)
9.100.00-0.65-17.23-32.01-45.31-57.40-68.50-78.78-88.37-97.37-105.87-113.94-114.33-114.33-125.62-136.50-146.51-155.78-164.40-172.46-180.03-187.16-193.91-200.30-206.38-212.18-217.72-223.03-228.12
260
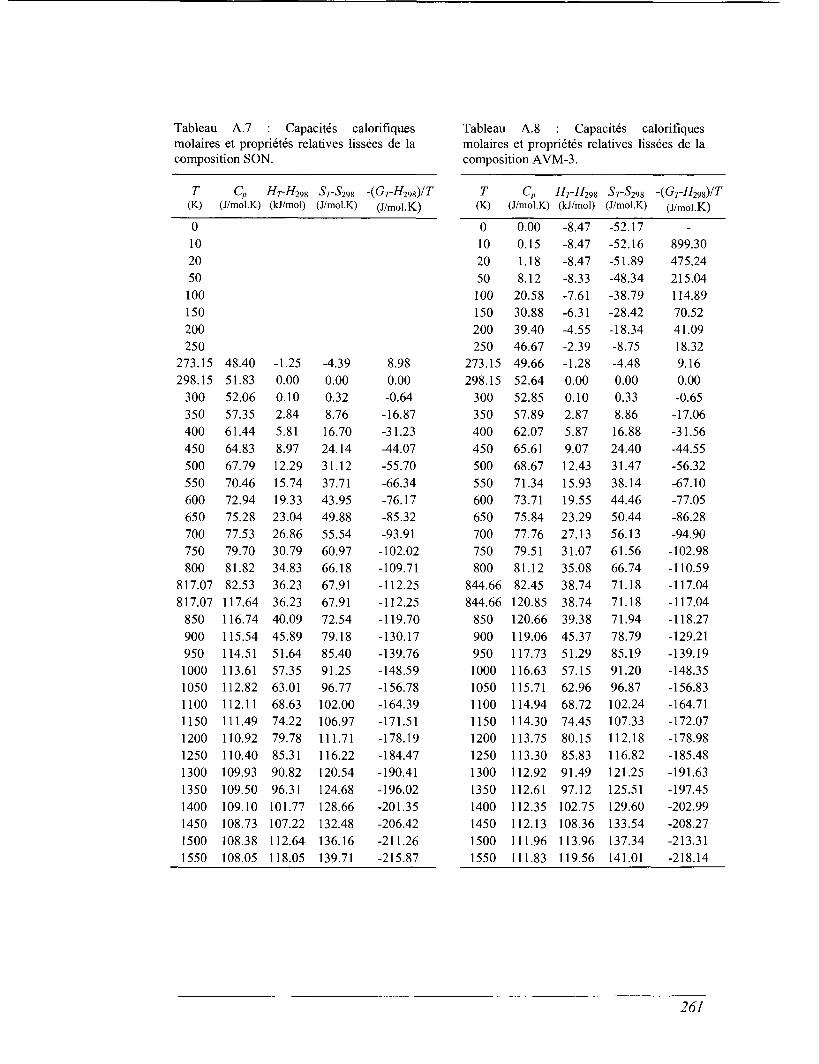
Tableau A.7 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition SON.
Tableau A.8 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition AVM-3.
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800
817.07817.07850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
48.40
51.83
52.06
57.35
61.44
64.83
67.79
70.46
72.94
75.28
77.53
79.70
81.82
82.53
117.64
116.74
115.54
114.51
113.61
112.82
112.11
111.49
110.92
110.40
109.93
109.50
109.10
108.73
108.38
108.05
HrH29i(kJ/mol)
-1.25
0.00
0.10
2.84
5.81
8.97
12.29
15.74
19.33
23.04
26.86
30.79
34.83
36.23
36.23
40.09
45.89
51.64
57.35
63.01
68.63
74.22
79.78
85.31
90.82
96.31
101.77
107.22
112.64
118.05
S7-S298(J/mol.K)
-4.39
0.00
0.32
8.76
16.70
24.14
31.12
37.71
43.95
49.88
55.54
60.97
66.18
67.91
67.91
72.54
79.18
85.40
91.25
96.77
102.00
106.97
111.71
116.22
120.54
124.68
128.66
132.48
136.16
139.71
-(Gr//298yr(J/mol.K)
8.980.00-0.64-16.87-31.23-44.07-55.70-66.34-76.17-85.32-93.91-102.02-109.71-112.25-112.25-119.70-130.17-139.76-148.59-156.78-164.39-171.51-178.19-184.47-190.41-196.02-201.35-206.42-211.26-215.87
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800
844.66844.66850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
0.000.151.188.1220.5830.8839.4046.6749.6652.6452.8557.8962.0765.6168.6771.3473.7175.8477.7679.5181.1282.45120.85120.66119.06117.73116.63115.71114.94114.30113.75113.30112.92112.61112.35112.13111.96111.83
Hr-H29s(kJ/mol)
-8.47
-8.47
-8.47
-8.33
-7.61
-6.31
-4.55
-2.39
-1.28
0.00
0.10
2.87
5.87
9.07
12.43
15.93
19.55
23.29
27.13
31.07
35.08
38.74
38.74
39.38
45.37
51.29
57.15
62.96
68.72
74.45
80.15
85.83
91.49
97.12
102.75
108.36
113.96
119.56
Sj-S29S(J/mol.K)
-52.17
-52.16
-51.89
-48.34
-38.79
-28.42
-18.34
-8.75
-4.48
0.00
0.33
8.86
16.88
24.40
31.47
38.14
44.46
50.44
56.13
61.56
66.74
71.18
71.18
71.94
78.79
85.19
91.20
96.87
102.24
107.33
112.18
116.82
121.25
125.51
129.60
133.54
137.34
141.01
-(Gr//298yr(J/mol.K)
-
899.30
475.24
215.04
114.89
70.52
41.09
18.32
9.16
0.00
-0.65
-17.06
-31.56
-44.55
-56.32
-67.10
-77.05
-86.28
-94.90
-102.98
-110.59
-117.04
-117.04
-118.27
-129.21
-139.19
-148.35
-156.83
-164.71
-172.07
-178.98
-185.48
-191.63
-197.45
-202.99
-208.27
-213.31
-218.14
261

Tableau A. 9 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition AVM-4.
Tableau A. 10 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition AVM-5.
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800
804.96804.96850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
48.8452.0652.2857.3761.3064.4767.1069.3471.2872.9974.5175.8877.1277.24115.71113.46111.34109.54108.00106.67105.51104.50103.60102.81102.10101.47100.90100.3999.9299.49
Hj-H29%(kJ/mol)
-1.260.000.102.845.818.9612.2515.6719.1822.7926.4830.2434.0634.4534.4539.6145.2250.7456.1861.5566.8572.1077.3082.4687.5992.6797.73102.77107.77112.76
(J/mol.K)
-4.420.000.328.7816.7124.1231.0537.5643.6749.4554.9160.1065.0465.5265.5271.7578.1884.1589.7394.9699.90104.56108.99113.21117.22121.07124.74128.28131.67134.94
-(GrH29S)/T(J/mol.K)
9.040.00-0.64-16.91-31.25-44.04-55.56-66.04-75.64-84.51-92.74-100.42-107.62-108.31-108.31-118.35-128.43-137.56-145.91-153.58-160.67-167.26-173.41-179.18-184.60-189.71-194.55-199.15-203.52-207.69
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750
797.88797.88800850900950100010501100115012001250130013501400145015001550
cp(J/mol.K)
51.4054.2954.4959.2563.1266.3669.1771.6473.8675.8877.7379.4580.99119.91119.78116.86114.43112.39110.66109.18107.92106.83105.88105.06104.34103.71103.15102.67102.24101.86
Hj-H29$(kJ/mol)
-1.320.000.102.956.019.2512.6416.1619.8023.5427.3831.3135.1635.1635.4141.3247.1052.7758.3563.8469.2774.6479.9685.2390.4695.66100.83105.98111.10116.20
5*7-5298(J/mol.K)
-4.630.000.349.1117.2824.9132.0538.7645.0951.0856.7762.2067.1667.1667.4874.6581.2687.3993.1198.47103.52108.29112.82117.12121.23125.15128.92132.53136.00139.35
-(Gr//298)/r(J/mol.K)
9.470.00-0.67-17.53-32.31-45.46-57.33-68.14-78.09-87.30-95.89-103.95-111.22-111.22-111.74-123.26-133.60-142.94-151.46-159.27-166.49-173.19-179.45-185.31-190.82-196.02-200.94-205.62-210.07-214.32
262
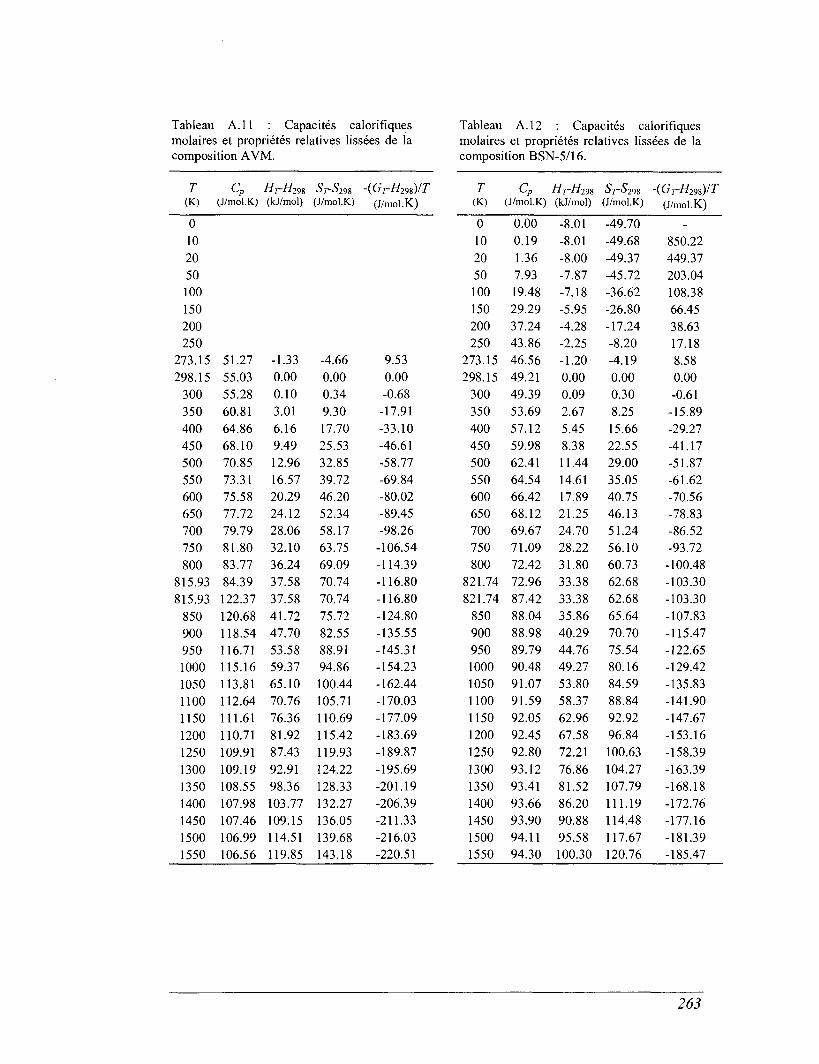
Tableau A. 11 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition AVM.
Tableau A. 12 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition BSN-5/16.
T(K)
0102050100150200250
273.15298.15
300350400450500550600650700750800
815.93815.93
850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
51.2755.0355.2860.8164.8668.1070.8573.3175.5877.7279.7981.8083.7784.39122.37120.68118.54116.71115.16113.81112.64111.61110.71109.91109.19108.55107.98107.46106.99106.56
HrHm(kJ/mol)
-1.330.000.103.016.169.4912.9616.5720.2924.1228.0632.1036.2437.5837.5841.7247.7053.5859.3765.1070.7676.3681.9287.4392.9198.36103.77109.15114.51119.85
SrS29$(J/mol.K)
-4.660.000.349.3017.7025.5332.8539.7246.2052.3458.1763.7569.0970.7470.7475.7282.5588.9194.86100.44105.71110.69115.42119.93124.22128.33132.27136.05139.68143.18
-(Grtf298)/r(J/mol.K)
9.530.00-0.68
-17.91-33.10-46.61-58.77-69.84-80.02-89.45-98.26
-106.54-114.39-116.80-116.80-124.80-135.55-145.31-154.23-162.44-170.03-177.09-183.69-189.87-195.69-201.19-206.39-211.33-216.03-220.51
T(K)
0102050100150200250
273.15298.15
300350400450500550600650700750800
821.74821.74
850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
0.000.191.367.9319.4829.2937.2443.8646.5649.2149.3953.6957.1259.9862.4164.5466.4268.1269.6771.0972.4272.9687.4288.0488.9889.7990.4891.0791.5992.0592.4592.8093.1293.4193.6693.9094.1194.30
HrH29S
(kJ/mol)
-8.01-8.01-8.00-7.87-7.18-5.95-4.28-2.25-1.200.000.092.675.458.3811.4414.6117.8921.2524.7028.2231.8033.3833.3835.8640.2944.7649.2753.8058.3762.9667.5872.2176.8681.5286.2090.8895.58100.30
SrSm(J/mol.K)
-49.70-49.68-49.37-45.72-36.62-26.80-17.24-8.20-4.190.000.308.2515.6622.5529.0035.0540.7546.1351.2456.1060.7362.6862.6865.6470.7075.5480.1684.5988.8492.9296.84100.63104.27107.79111.19114.48117.67120.76
-(Gr//298)/r(J/mol.K)
-
850.22449.37203.04108.3866.4538.6317.188.580.00-0.61
-15.89-29.27-41.17-51.87-61.62-70.56-78.83-86.52-93.72-100.48-103.30-103.30-107.83-115.47-122.65-129.42-135.83-141.90-147.67-153.16-158.39-163.39-168.18-172.76-177.16-181.39-185.47
263
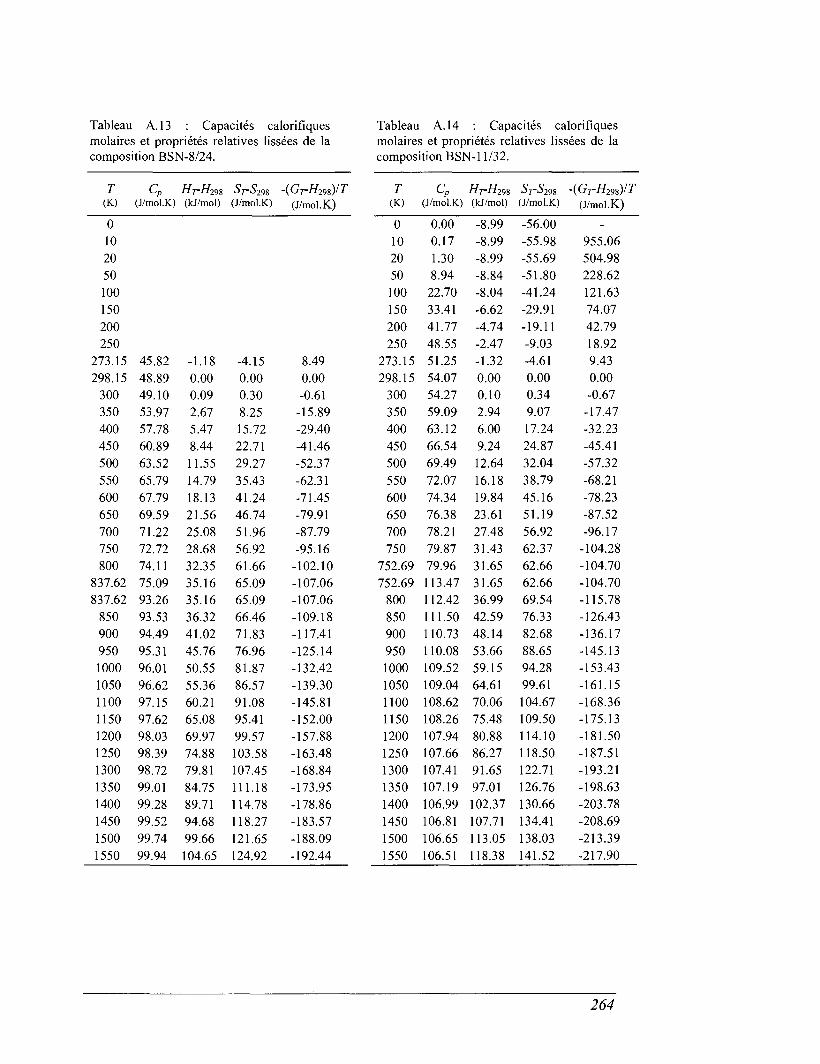
Tableau A. 13 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition BSN-8/24.
Tableau A. 14 : Capacités calorifiquesmolaires et propriétés relatives lissées de lacomposition BSN-11/32.
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800
837.62837.62850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
45.8248.8949.1053.9757.7860.8963.52
65.7967.7969.5971.2272.7274.11
75.0993.2693.5394.4995.3196.0196.6297.1597.6298.0398.3998.7299.0199.2899.5299.7499.94
Hj-H29s(kj/mol)
-1.180.000.092.675.478.4411.5514.7918.1321.5625.0828.6832.3535.1635.1636.3241.0245.7650.5555.3660.2165.0869.9774.8879.8184.7589.7194.6899.66104.65
S7-S29S(J/mol.K)
-4.150.000.308.2515.7222.7129.2735.4341.2446.74
51.9656.9261.6665.0965.0966.4671.8376.9681.8786.5791.0895.4199.57103.58107.45111.18114.78118.27121.65124.92
<GrHm)/T(J/mol.K)
8.490.00-0.61-15.89-29.40-41.46-52.37-62.31-71.45-79.91-87.79-95.16-102.10-107.06-107.06-109.18-117.41-125.14-132.42-139.30-145.81-152.00-157.88-163.48-168.84-173.95-178.86-183.57-188.09-192.44
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750
752.69752.69800850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
0.000.171.308.9422.7033.41
41.7748.5551.2554.0754.2759.0963.1266.5469.4972.0774.3476.3878.2179.8779.96113.47112.42111.50110.73110.08109.52109.04108.62108.26107.94107.66107.41107.19106.99106.81
106.65106.51
Hj-H29S(kJ/mol)
-8.99-8.99-8.99-8.84-8.04-6.62-4.74-2.47-1.320.000.102.946.009.2412.64
16.1819.8423.6127.4831.4331.6531.6536.9942.5948.1453.6659.1564.6170.0675.4880.8886.2791.6597.01102.37107.71113.05118.38
SrS29$(J/mol.K)
-56.00-55.98-55.69
-51.80-41.24-29.91-19.11-9.03-4.610.000.349.0717.2424.8732.0438.7945.1651.1956.9262.3762.6662.6669.5476.3382.6888.6594.2899.61104.67109.50114.10118.50122.71126.76130.66134.41
138.03141.52
-{Gj-H29&yT(J/mol.K)
-
955.06504.98228.62121.6374.0742.7918.929.430.00-0.67-17.47-32.23-45.41-57.32
-68.21-78.23-87.52
-96.17-104.28-104.70-104.70-115.78-126.43-136.17-145.13-153.43-161.15-168.36-175.13-181.50-187.51-193.21-198.63-203.78-208.69-213.39-217.90
264
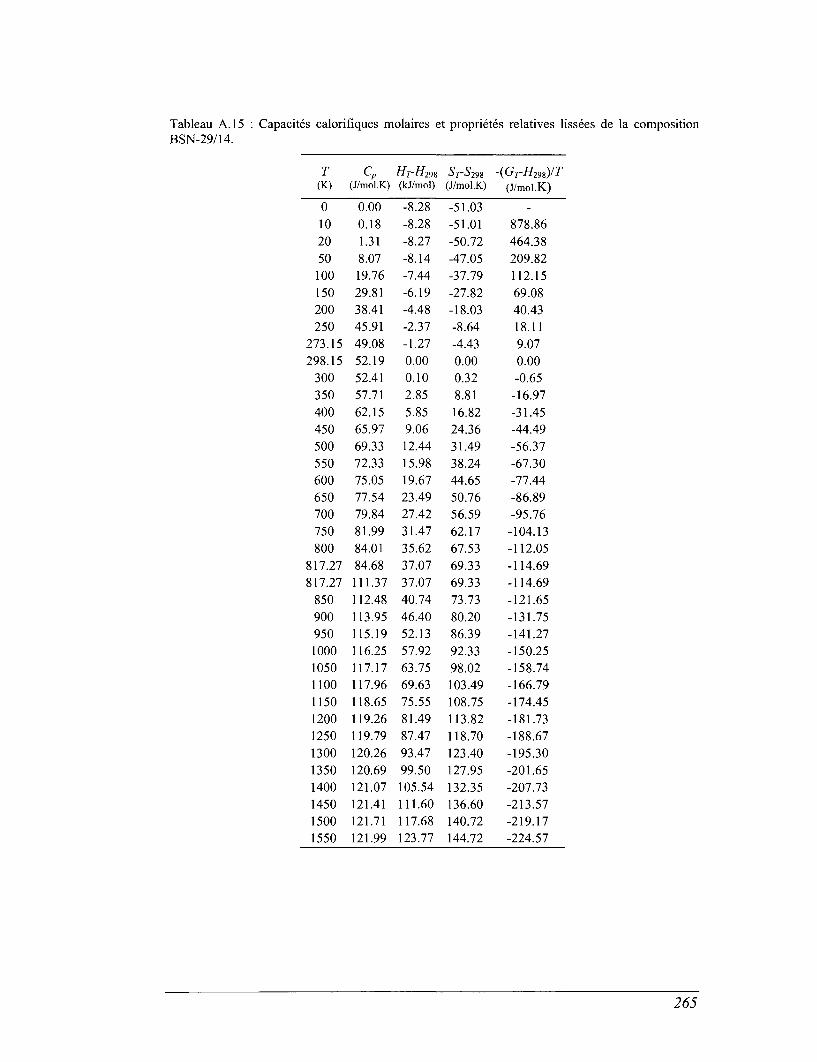
Tableau A. 15 : Capacités calorifiques molaires et propriétés relatives lissées de la compositionBSN-29/14.
T(K)
0102050100150200250
273.15298.15300350400450500550600650700750800
817.27817.27850900950100010501100115012001250130013501400145015001550
cP(J/mol.K)
0.000.181.318.0719.7629.8138.4145.9149.0852.1952.4157.7162.1565.9769.3372.3375.0577.5479.8481.9984.0184.68111.37112.48113.95115.19116.25117.17117.96118.65119.26119.79120.26120.69121.07121.41121.71121.99
HrHm(kj/mol)
-8.28-8.28-8.27-8.14-7.44-6.19-4.48-2.37-1.270.000.102.855.859.0612.4415.9819.6723.4927.4231.4735.6237.0737.0740.7446.4052.1357.9263.7569.6375.5581.4987.4793.4799.50105.54111.60117.68123.77
57-5298(J/mol.K)
-51.03-51.01-50.72-47.05-37.79-27.82-18.03-8.64-4.430.000.328.8116.8224.3631.4938.2444.6550.7656.5962.1767.5369.3369.3373.7380.2086.3992.3398.02103.49108.75113.82118.70123.40127.95132.35136.60140.72144.72
-(GrH29S)/T(J/mol.K)
-
878.86464.38209.82112.1569.0840.4318.119.070.00-0.65-16.97-31.45-44.49-56.37-67.30-77.44-86.89-95.76-104.13-112.05-114.69-114.69-121.65-131.75-141.27-150.25-158.74-166.79-174.45-181.73-188.67-195.30-201.65-207.73-213.57-219.17-224.57
265
Related Documents











