Downregulation of cardiac lineage protein-1 confers cardioprotection through the upregulation of redox effectors Narasimman Gurusamy a , Istvan Lekli a , Md. Kaimul Ahsan a , Diptarka Ray a , Subhendu Mukherjee a , Eduardo Mascareno b , M.A.Q. Siddiqui b , Dipak K. Das a, * a Cardiovascular Research Center, University of Connecticut, School of Medicine, Farmington, CT 06030-1110, USA b State University of New York Downstate Medical Center, Brooklyn, NY, USA article info Article history: Received 21 September 2009 Revised 16 October 2009 Accepted 11 November 2009 Available online 21 November 2009 Edited by Stuart Ferguson Keywords: Hexim1 Cardiac lineage protein-1 Redox factor Ischemia–reperfusion Cardiac Ref-1 Nuclear factor erythroid 2-related factor Thioredoxin abstract CLP-1, the mouse homologue of human Hexim1 protein, exerts inhibitory control on transcriptional elongation factor-b of RNA transcript elongation. Previously, we have demonstrated that downreg- ulation of cardiac lineage protein-1 (CLP-1) in CLP-1 +/ heterozygous mice affords cardioprotection against ischemia–reperfusion injury. Our current study results show that the improvement in car- diac function in CLP-1 +/ mice after ischemia–reperfusion injury is achieved through the potentia- tion of redox signaling and their molecular targets including redox effector factor-1, nuclear factor erythroid 2-related factor, and NADPH oxidase 4 and the active usage of thioredoxin-1, thio- redoxin-2, glutaredoxin-1 and glutaredoxin-2. Our results suggest that drugs designed to down reg- ulate CLP-1 could confer cardioprotection through the potentiation of redox cycling. Ó 2009 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved. 1. Introduction Cardiac lineage protein-1, a mouse homologue of human Hexim1 binds with and inhibits the positive transcription elonga- tion factor-b (P-TEFb), which is composed of cyclin-dependent ki- nase 9 (Cdk9) and cyclin T1 [1–3]. P-TEFb phosphorylates the carboxyl-terminal domain (CTD) of RNA polymerase II, a major substrate of P-TEFb, and upon phosphorylation elongates nascent transcripts to form full-length messenger RNAs [2]. Hexim1 forms a protein–RNA complex composed of 7SK small nuclear RNA and P-TEFb, and inhibits the kinase activity of CDK9, leading to the suppression of RNA polymerase II-dependent transcriptional elongation [4]. Previously we have demonstrated that CLP-1 plays an essential role in the regulation of P-TEFb activity, and the dissociation of CLP-1 from P-TEFb complex stimulates cardiac hypertrophic phenotype [1,5,6]. The CTD of human RNA polymer- ase II is composed of 52 repeats of a heptapeptide sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser. Cdk9 specifically phosphorylates Ser2 of the CTD of RNA polymerase II [3], whereas Cdk7 specifi- cally phosphorylates Ser5 [7]. Both Cdk7 and Cdk9 are activated during hypertrophic stimuli, but only Cdk9 is specifically acti- vated during acute overload by aortic banding in the myocar- dium [8]. Activation of Cdk9 causes cardiomyocyte enlargement and defective mitochondrial function, via diminished peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) transcription, and confers a predisposition to heart failure [9]. In our recent study using isolated mouse heart ischemia– reperfusion model, we have demonstrated that downregulation of CLP-1 in heterozygous CLP-1 +/ mice affords cardioprotection against ischemia–reperfusion injury [10], where the CLP-1 remains associated with P-TEFb complex in the CLP-1 +/ mice hearts, and results in a decrease in Cdk7 and Cdk9 activities, and an enhanced level of PGC-1a and hypoxia-inducible factor1a (HIF-1a). Our present study shows that CLP-1 downregulation- mediated cardioprotection against cardiac ischemia–reperfusion injury involved the elicitation of redox active proteins such as redox effector factor-1 (Ref-1), nuclear factor erythroid 2-related factor (Nrf2), NADPH oxidase 4 (NOX4) and thioredoxin super family members. 0014-5793/$36.00 Ó 2009 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved. doi:10.1016/j.febslet.2009.11.054 * Corresponding author. Fax: +1 860 679 4606. E-mail address: [email protected] (D.K. Das). FEBS Letters 584 (2010) 187–193 journal homepage: www.FEBSLetters.org

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

FEBS Letters 584 (2010) 187–193
journal homepage: www.FEBSLetters .org
Downregulation of cardiac lineage protein-1 confers cardioprotection throughthe upregulation of redox effectors
Narasimman Gurusamy a, Istvan Lekli a, Md. Kaimul Ahsan a, Diptarka Ray a, Subhendu Mukherjee a,Eduardo Mascareno b, M.A.Q. Siddiqui b, Dipak K. Das a,*
a Cardiovascular Research Center, University of Connecticut, School of Medicine, Farmington, CT 06030-1110, USAb State University of New York Downstate Medical Center, Brooklyn, NY, USA
a r t i c l e i n f o
Article history:Received 21 September 2009Revised 16 October 2009Accepted 11 November 2009Available online 21 November 2009
Edited by Stuart Ferguson
Keywords:Hexim1Cardiac lineage protein-1Redox factorIschemia–reperfusionCardiacRef-1Nuclear factor erythroid 2-related factorThioredoxin
0014-5793/$36.00 � 2009 Federation of European Biodoi:10.1016/j.febslet.2009.11.054
* Corresponding author. Fax: +1 860 679 4606.E-mail address: [email protected] (D.K. D
a b s t r a c t
CLP-1, the mouse homologue of human Hexim1 protein, exerts inhibitory control on transcriptionalelongation factor-b of RNA transcript elongation. Previously, we have demonstrated that downreg-ulation of cardiac lineage protein-1 (CLP-1) in CLP-1+/� heterozygous mice affords cardioprotectionagainst ischemia–reperfusion injury. Our current study results show that the improvement in car-diac function in CLP-1+/� mice after ischemia–reperfusion injury is achieved through the potentia-tion of redox signaling and their molecular targets including redox effector factor-1, nuclearfactor erythroid 2-related factor, and NADPH oxidase 4 and the active usage of thioredoxin-1, thio-redoxin-2, glutaredoxin-1 and glutaredoxin-2. Our results suggest that drugs designed to down reg-ulate CLP-1 could confer cardioprotection through the potentiation of redox cycling.� 2009 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
1. Introduction
Cardiac lineage protein-1, a mouse homologue of humanHexim1 binds with and inhibits the positive transcription elonga-tion factor-b (P-TEFb), which is composed of cyclin-dependent ki-nase 9 (Cdk9) and cyclin T1 [1–3]. P-TEFb phosphorylates thecarboxyl-terminal domain (CTD) of RNA polymerase II, a majorsubstrate of P-TEFb, and upon phosphorylation elongates nascenttranscripts to form full-length messenger RNAs [2]. Hexim1forms a protein–RNA complex composed of 7SK small nuclearRNA andP-TEFb, and inhibits the kinase activity of CDK9, leading to thesuppression of RNA polymerase II-dependent transcriptionalelongation [4]. Previously we have demonstrated that CLP-1plays an essential role in the regulation of P-TEFb activity, andthe dissociation of CLP-1 from P-TEFb complex stimulates cardiachypertrophic phenotype [1,5,6]. The CTD of human RNA polymer-ase II is composed of 52 repeats of a heptapeptide sequence
chemical Societies. Published by E
as).
Tyr-Ser-Pro-Thr-Ser-Pro-Ser. Cdk9 specifically phosphorylatesSer2 of the CTD of RNA polymerase II [3], whereas Cdk7 specifi-cally phosphorylates Ser5 [7]. Both Cdk7 and Cdk9 are activatedduring hypertrophic stimuli, but only Cdk9 is specifically acti-vated during acute overload by aortic banding in the myocar-dium [8]. Activation of Cdk9 causes cardiomyocyte enlargementand defective mitochondrial function, via diminished peroxisomeproliferator-activated receptor gamma coactivator 1 (PGC-1)transcription, and confers a predisposition to heart failure [9].
In our recent study using isolated mouse heart ischemia–reperfusion model, we have demonstrated that downregulationof CLP-1 in heterozygous CLP-1+/� mice affords cardioprotectionagainst ischemia–reperfusion injury [10], where the CLP-1remains associated with P-TEFb complex in the CLP-1+/� micehearts, and results in a decrease in Cdk7 and Cdk9 activities,and an enhanced level of PGC-1a and hypoxia-inducible factor1a(HIF-1a). Our present study shows that CLP-1 downregulation-mediated cardioprotection against cardiac ischemia–reperfusioninjury involved the elicitation of redox active proteins such asredox effector factor-1 (Ref-1), nuclear factor erythroid 2-relatedfactor (Nrf2), NADPH oxidase 4 (NOX4) and thioredoxin superfamily members.
lsevier B.V. All rights reserved.

188 N. Gurusamy et al. / FEBS Letters 584 (2010) 187–193
2. Materials and methods
2.1. Animals
All animals used in this study received humane care in compli-ance with the regulations relating to animals and experimentsinvolving animals and adheres to principles stated in the Guidefor the Care and Use of Laboratory Animals, NIH Publication,1996 edition, and all the protocols were approved by InstitutionalAnimal Care Committee.
2.2. Isolated working heart preparation and drug treatment
Wild-type CLP-1++ and heterozygous CLP-1+/� mice were anes-thetized with sodium pentobarbital (80 mg/kg, i.p.) (Abbott Labo-ratories, North Chicago, IL, USA), and intraperitonealy heparinsodium (500 IU/kg, i.v.) (Elkins-Sinn Inc., Cherry Hill, NJ, USA)was used as an anticoagulant. After the deep anesthesia was con-formed, hearts were excised, the aorta was canulated, and thehearts were perfused through the aorta in Langendorff mode at aconstant (100 cm of water) perfusion pressure at 37 �C with theKHB for a 5 min washout period as described previously. The per-fusion medium consisted of a modified Krebs–Henseleit bicarbon-ate buffer (millimolar concentration: sodium chloride 118,potassium chloride 4.7, calcium chloride 1.7, sodium bicarbonate25, potassium dihydrogenphosphate 0.36, magnesium sulfate 1.2and glucose 10), and after its oxygenization pH was 7.4 at 37 �C.During the washout period left atria was canulated, and the Lange-ndorff preparation was switched to the working mode for 10 minwith a left atrial filling pressure of 17 cm H2O, aortic afterload pres-sure was set to 100 cm of water. At the end of 10 min, baseline car-diac function like heart rate (HR, beats/min), aortic flow (AF, ml/min), coronary flow (CF, ml/min), left ventricular developed pres-sure (LVDP, mmHg) and first derivative of developed pressure(LVdp/dt, mmHg/s) were recorded. After that 30 min of globalischemia was initiated by clamping the left atrial inflow and aorticoutflow lines at a point close to their origins. At the end of the30 min of ischemia, reperfusion was initiated for 120 min byunclamping the atrial inflow and aortic outflow lines. The first10 min reperfusion was in Langendorff mode to avoid the ventric-ular fibrillations, after the hearts were switched to anterogradeworking mode.
2.3. Cardiac function assessment
Aortic pressure was measured using a Gould P23XL pressuretransducer (Gould Instrument Systems Inc., Valley View, OH,USA) connected to a side arm of the aortic cannula. The signalwas amplified using a Gould 6600 series signal conditioner andmonitored on a CORDAT II real-time data acquisition and analysissystem (Triton Technologies, San Diego, CA, USA). Heart rate, LVDP(defined as the difference of the maximum systolic and diastolicaortic pressures), and dp/dt were all derived or calculated fromthe continuously obtained pressure signal. Aortic flow was mea-sured using a calibrated flow-meter (Gilmont Instrument Inc., Bar-rington, IL, USA) and coronary flow was measured by timedcollection of the coronary effluent dripping from the heart.
2.4. Cytosolic extract preparation
About 100 mg of left ventricular tissue was homogenized in1 ml of buffer containing 25 mM Tris, 25 mM NaCl, 1 mM sodiumorthovanadate, 10 mM sodium fluoride, 10 mM sodium pyro-phosphate, 0.5 mM EDTA, 1 mM PMSF and protease inhibitorscocktail (leupeptin, aprotinin, and pepstatin). Homogenates were
centrifuged at 3000 rpm for 10 min at 4 �C, and the supernatantwas again centrifuged at 10 000 rpm for 20 min at 4 �C. Theresultant supernatant was saved as cytosolic extract. Total pro-tein concentration in the cytosolic extract was determined usingBCA Protein Assay Kit (Pierce, Rockford, IL).
2.5. Western blot analysis
Total cell lysate from mice heart tissue was separated in SDS–PAGE and transferred to nitrocellulose filters. Filters were blockedin 5% non-fat dry milk, and probed with a primary antibody(1:1000 dilution) for overnight. Ref-1 primary antibody was ob-tained from Cell Signaling Technology. Nrf2, heme oxygenase-1and NOX4 primary antibodies were obtained from Santa Cruz Bio-technology. Protein bands were identified with horseradish perox-idase conjugated secondary antibody (1:2000 dilution) andWestern Blotting Luminol Reagent (Santa Cruz Biotechnology).The resulting blots were digitized, subjected to densitometric scan-ning using a standard NIH image program, and normalized againstloading control.
2.6. Immunofluorescence staining
Heart tissue samples collected at the end of experiments werefixed in 4% buffered paraformaldehyde (pH 7.4), embedded in par-affin and sectioned. After deparaffinizing the sections the antigenretrieval treatment was performed using 10 mM sodium citratecontaining 0.05% Tween 20 at 90–95 �C for 30 min. After washingwith PBS, the slides were blocked with Powerblock (BioGenex,San Ramon, CA) for 10 min. Slides were washed with PBS and incu-bated with primary antibodies (1:25 dilution) in PBS containing 1%BSA for 2 h. After washing, the slides were incubated with fluores-cein-conjugated secondary antibodies (anti-rabbit Alexa Fluor 488,green; and anti-goat Alexa Fluor 594, red; both from MolecularProbes and used at 1:1000 dilutions) in the dark for 45 min. For nu-clear staining To-Pro 3 iodide (Molecular Probes; 1:1000 dilution)was used for 45 min in the dark. The slides were washed and cov-ered with mounting medium. Confocal microscopic images wereobtained using a Zeiss LSM 510 (Thornwood, NY) confocal laserscanning microscope with 40 � 1.3 oil immersion objective bysimultaneous recording in the 488k, 560k, and/or 615k channelsas appropriate.
2.7. Semi-quantitative RT-PCR
Total RNA was extracted from liver tissue, using Trizol reagent(Invitrogen, CA, USA). Three micrograms of total RNA was re-verse-transcribed, using a SuperScript-II First-Strand SynthesisSystem (Invitrogen) with oligo dT12-18, according to the manufac-turer’s protocol. The cDNA was amplified by PCR, using PlatinumTaq-DNA polymerase (Clontech, CA, USA) following a standardsemi-quantitative RT-PCR technique in which the amplified prod-ucts were not saturated at the number of cycles performed. Theprimers used for the amplification are enlisted in Table 1. ThePCR products were visualized by electrophoresis in 1% agarosegel (Bio-Rad, CA, USA) with ethidium bromide (0.5 lg/ml; Sigma,MO, USA).
2.8. Statistical analysis
All values are expressed as the means ± standard error of mean(S.E.M.). Student’s t-test or one way analysis of variance test fol-lowed by Bonferoni’s correction was first carried out to test forany differences between the mean values of all groups. The resultswere considered significant if P < 0.05.
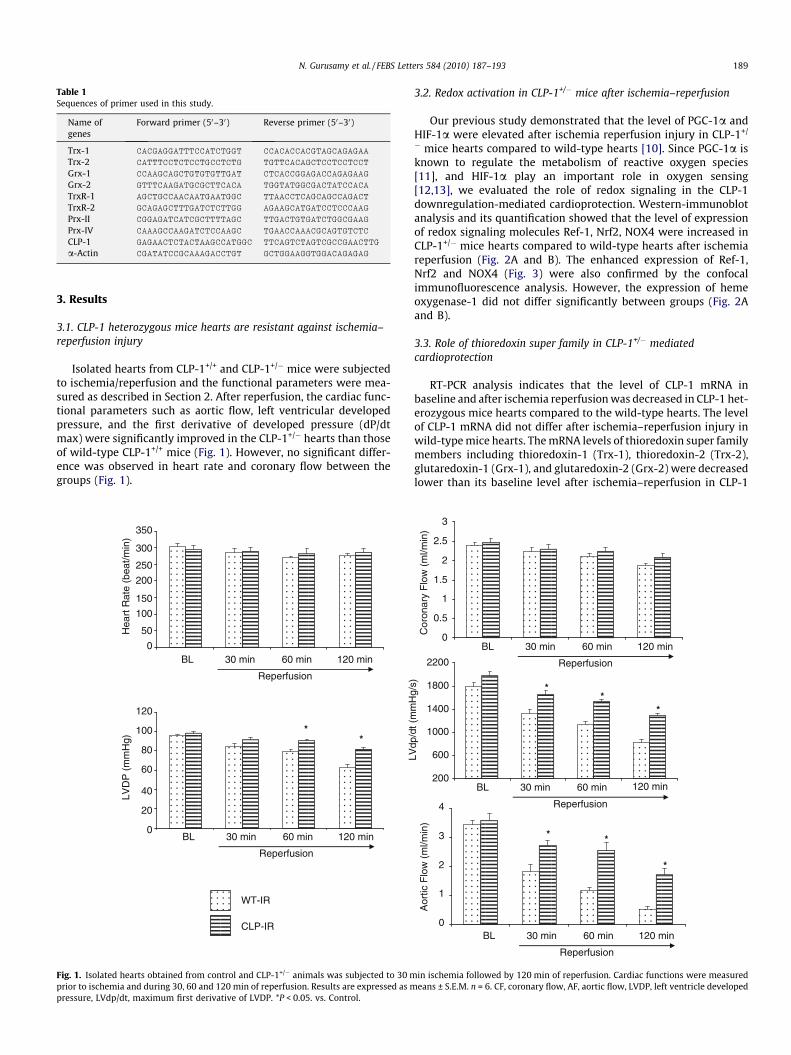
Table 1Sequences of primer used in this study.
Name ofgenes
Forward primer (50–30) Reverse primer (50–30)
Trx-1 CACGAGGATTTCCATCTGGT CCACACCACGTAGCAGAGAA
Trx-2 CATTTCCTCTCCTGCCTCTG TGTTCACAGCTCCTCCTCCT
Grx-1 CCAAGCAGCTGTGTGTTGAT CTCACCGGAGACCAGAGAAG
Grx-2 GTTTCAAGATGCGCTTCACA TGGTATGGCGACTATCCACA
TrxR-1 AGCTGCCAACAATGAATGGC TTAACCTCAGCAGCCAGACT
TrxR-2 GCAGAGCTTTGATCTCTTGG AGAAGCATGATCCTCCCAAG
Prx-II CGGAGATCATCGCTTTTAGC TTGACTGTGATCTGGCGAAG
Prx-IV CAAAGCCAAGATCTCCAAGC TGAACCAAACGCAGTGTCTC
CLP-1 GAGAACTCTACTAAGCCATGGC TTCAGTCTAGTCGCCGAACTTG
a-Actin CGATATCCGCAAAGACCTGT GCTGGAAGGTGGACAGAGAG
N. Gurusamy et al. / FEBS Letters 584 (2010) 187–193 189
3. Results
3.1. CLP-1 heterozygous mice hearts are resistant against ischemia–reperfusion injury
Isolated hearts from CLP-1+/+ and CLP-1+/� mice were subjectedto ischemia/reperfusion and the functional parameters were mea-sured as described in Section 2. After reperfusion, the cardiac func-tional parameters such as aortic flow, left ventricular developedpressure, and the first derivative of developed pressure (dP/dtmax) were significantly improved in the CLP-1+/� hearts than thoseof wild-type CLP-1+/+ mice (Fig. 1). However, no significant differ-ence was observed in heart rate and coronary flow between thegroups (Fig. 1).
WT-IR
CLP-IR
0
50
100
150
200
250
300
350
BL 30 min 60 min 120 min
Hea
rt R
ate
(bea
t/min
)
Reperfusion
0
20
40
60
80
100
120
LVD
P (
mm
Hg) *
*
BL 30 min 60 min 120 min
Reperfusion
Fig. 1. Isolated hearts obtained from control and CLP-1+/� animals was subjected to 30 mprior to ischemia and during 30, 60 and 120 min of reperfusion. Results are expressed as mpressure, LVdp/dt, maximum first derivative of LVDP. *P < 0.05. vs. Control.
3.2. Redox activation in CLP-1+/� mice after ischemia–reperfusion
Our previous study demonstrated that the level of PGC-1a andHIF-1a were elevated after ischemia reperfusion injury in CLP-1+/
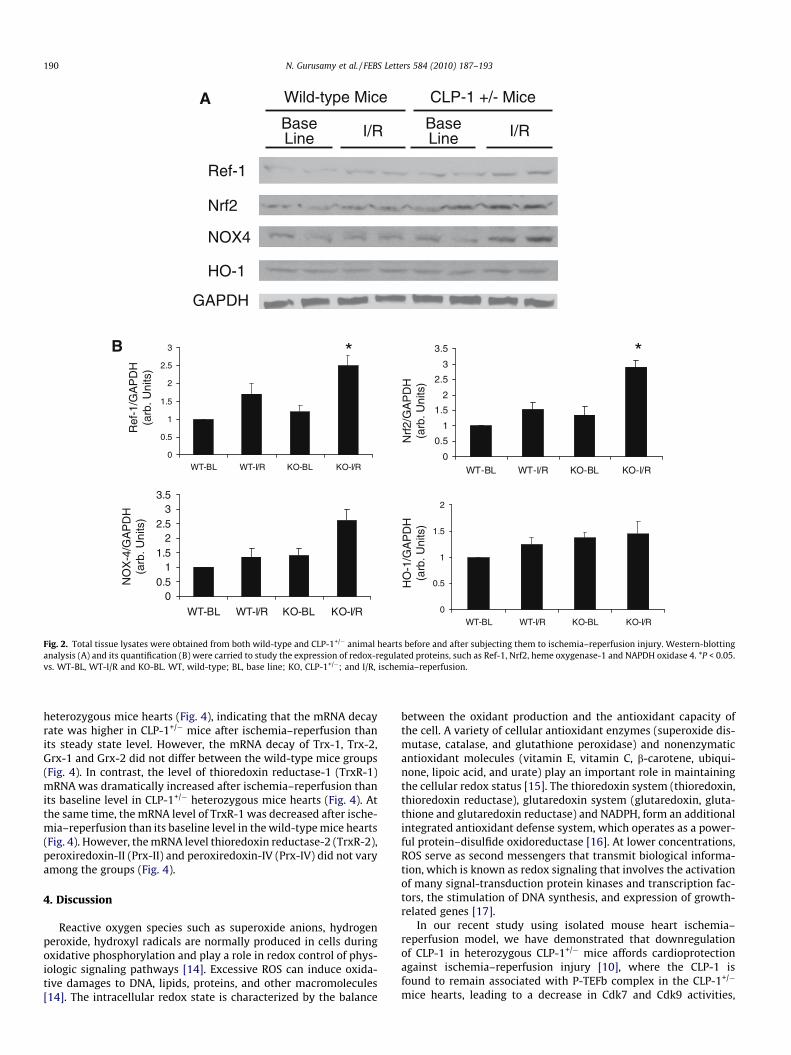
� mice hearts compared to wild-type hearts [10]. Since PGC-1a isknown to regulate the metabolism of reactive oxygen species[11], and HIF-1a play an important role in oxygen sensing[12,13], we evaluated the role of redox signaling in the CLP-1downregulation-mediated cardioprotection. Western-immunoblotanalysis and its quantification showed that the level of expressionof redox signaling molecules Ref-1, Nrf2, NOX4 were increased inCLP-1+/� mice hearts compared to wild-type hearts after ischemiareperfusion (Fig. 2A and B). The enhanced expression of Ref-1,Nrf2 and NOX4 (Fig. 3) were also confirmed by the confocalimmunofluorescence analysis. However, the expression of hemeoxygenase-1 did not differ significantly between groups (Fig. 2Aand B).
3.3. Role of thioredoxin super family in CLP-1+/� mediatedcardioprotection
RT-PCR analysis indicates that the level of CLP-1 mRNA inbaseline and after ischemia reperfusion was decreased in CLP-1 het-erozygous mice hearts compared to the wild-type hearts. The levelof CLP-1 mRNA did not differ after ischemia–reperfusion injury inwild-type mice hearts. The mRNA levels of thioredoxin super familymembers including thioredoxin-1 (Trx-1), thioredoxin-2 (Trx-2),glutaredoxin-1 (Grx-1), and glutaredoxin-2 (Grx-2) were decreasedlower than its baseline level after ischemia–reperfusion in CLP-1
BL 30 min 60 min 120 min0
0.5
1
1.5
2
2.5
3
Cor
onar
y F
low
(m
l/min
)
Reperfusion
120 min200
600
1000
1400
1800
2200
LVdp
/dt(
mm
Hg/
s)
**
*
BL 30 min 60 min
Reperfusion
0
1
2
3
4
Aor
tic F
low
(m
l/min
)
**
*
BL 30 min 60 min 120 min
Reperfusion
in ischemia followed by 120 min of reperfusion. Cardiac functions were measuredeans ± S.E.M. n = 6. CF, coronary flow, AF, aortic flow, LVDP, left ventricle developed
Ref-1
Nrf2
HO-1
NOX4
BaseLine
Wild-type Mice
I/R BaseLine I/R
CLP-1 +/- Mice
GAPDH
0
0.5
1
1.5
2
2.5
3
WT-BL WT-I/R KO-BL KO-I/R0
0.5
1
1.5
2
2.5
3
3.5
WT-BL WT-I/R KO-BL KO-I/R
0
0.5
1
1.5
2
WT-BL WT-I/R KO-BL KO-I/R
00.5
11.5
22.5
33.5
WT-BL WT-I/R KO-BL KO-I/R
Ref
-1/G
AP
DH
(arb
. Uni
ts)
NO
X-4
/GA
PD
H(a
rb. U
nits
)
Nrf
2/G
AP
DH
(arb
. Uni
ts)
HO
-1/G
AP
DH
(arb
. Uni
ts)
* *B
A
Fig. 2. Total tissue lysates were obtained from both wild-type and CLP-1+/� animal hearts before and after subjecting them to ischemia–reperfusion injury. Western-blottinganalysis (A) and its quantification (B) were carried to study the expression of redox-regulated proteins, such as Ref-1, Nrf2, heme oxygenase-1 and NAPDH oxidase 4. *P < 0.05.vs. WT-BL, WT-I/R and KO-BL. WT, wild-type; BL, base line; KO, CLP-1+/�; and I/R, ischemia–reperfusion.
190 N. Gurusamy et al. / FEBS Letters 584 (2010) 187–193
heterozygous mice hearts (Fig. 4), indicating that the mRNA decayrate was higher in CLP-1+/� mice after ischemia–reperfusion thanits steady state level. However, the mRNA decay of Trx-1, Trx-2,Grx-1 and Grx-2 did not differ between the wild-type mice groups(Fig. 4). In contrast, the level of thioredoxin reductase-1 (TrxR-1)mRNA was dramatically increased after ischemia–reperfusion thanits baseline level in CLP-1+/� heterozygous mice hearts (Fig. 4). Atthe same time, the mRNA level of TrxR-1 was decreased after ische-mia–reperfusion than its baseline level in the wild-type mice hearts(Fig. 4). However, the mRNA level thioredoxin reductase-2 (TrxR-2),peroxiredoxin-II (Prx-II) and peroxiredoxin-IV (Prx-IV) did not varyamong the groups (Fig. 4).
4. Discussion
Reactive oxygen species such as superoxide anions, hydrogenperoxide, hydroxyl radicals are normally produced in cells duringoxidative phosphorylation and play a role in redox control of phys-iologic signaling pathways [14]. Excessive ROS can induce oxida-tive damages to DNA, lipids, proteins, and other macromolecules[14]. The intracellular redox state is characterized by the balance
between the oxidant production and the antioxidant capacity ofthe cell. A variety of cellular antioxidant enzymes (superoxide dis-mutase, catalase, and glutathione peroxidase) and nonenzymaticantioxidant molecules (vitamin E, vitamin C, b-carotene, ubiqui-none, lipoic acid, and urate) play an important role in maintainingthe cellular redox status [15]. The thioredoxin system (thioredoxin,thioredoxin reductase), glutaredoxin system (glutaredoxin, gluta-thione and glutaredoxin reductase) and NADPH, form an additionalintegrated antioxidant defense system, which operates as a power-ful protein–disulfide oxidoreductase [16]. At lower concentrations,ROS serve as second messengers that transmit biological informa-tion, which is known as redox signaling that involves the activationof many signal-transduction protein kinases and transcription fac-tors, the stimulation of DNA synthesis, and expression of growth-related genes [17].
In our recent study using isolated mouse heart ischemia–reperfusion model, we have demonstrated that downregulationof CLP-1 in heterozygous CLP-1+/� mice affords cardioprotectionagainst ischemia–reperfusion injury [10], where the CLP-1 isfound to remain associated with P-TEFb complex in the CLP-1+/�
mice hearts, leading to a decrease in Cdk7 and Cdk9 activities,

Fig. 3. Representative confocal immunofluorescence microscopic images of the formaldehyde fixed paraffin embedded heart tissue sections obtained from wild-type andCLP-1+/� mice stained with Ref-1 antibody followed by AlexaFluor 488 (green) conjugated secondary antibody or stained with Nrf2 antibody followed by AlexaFluor 488(green) conjugated secondary antibody or stained with NOX-4 antibody followed by AlexaFluor 594 (red) conjugated secondary antibody. Nuclear staining was performedwith Topro-3-iodide (blue). The nuclear colocalization of Ref-1 and Nrf2 in the CLP-1+/� mice hearts are shown in white (indicated by arrows).
N. Gurusamy et al. / FEBS Letters 584 (2010) 187–193 191
simultaneously causing an enhancement of the expression ofPGC-1a, HIF-1a and pyruvate dehydrogenase kinase-1 (PDK-1)[10]. Our results are in consistent with those reported earlier,where Cdk9 activity was shown to suppress many genes for mito-chondrial proteins including master regulators of mitochondrialfunction PGC-1 [8,9]. Chronic activation of Cdk9 causes not onlycardiomyocyte enlargement but also defective mitochondrialfunction, via diminished PGC-1 transcription, and a resulting sus-ceptibility to apoptotic cardiomyopathy [9]. Since HIF-1a expres-sion during hypoxic conditions is known to induce PDK-1, whichfacilitates cell adaptation to low oxygen pressure by repressingmitochondrial function and by reactive oxygen species production
[18,19]. PDK-1 overexpression in hypoxic HIF-1a null cells in-creases ATP levels, attenuates hypoxic ROS generation, and res-cues them from hypoxia-induced apoptosis [20]. HIF-1stimulates glycolysis, and actively represses mitochondrial func-tion and oxygen consumption by inducing PDK-1 [18,19]. PDK-1phosphorylates and inhibits pyruvate dehydrogenase from usingpyruvate to fuel the mitochondrial TCA cycle [18,19]. This causesa drop in mitochondrial oxygen consumption and results in a rel-ative increase in intracellular oxygen tension [18,19]. Thus, thereduced level of reactive oxygen species production is maintainedin CLP-1+/� mice via enhanced level PDK-1, which reduces theoxidative phosphorylation, and increases the glycolysis. In the

Trx-1
Trx-2
Grx-1
Grx-2
TrxR-1
TrxR-2
αα-Actin
Prx-II
Prx-IV
CLP-1
BaseLine
Wild-type Mice
I/R BaseLine I/R
CLP-1 +/- Mice
Fig. 4. RT-PCR analysis of thioredoxin super family members in CLP-1+/� mediatedcardioprotection. Total RNA was isolated from the isolated heart tissue sections and3 lg of total RNA was reverse-transcribed using First-Strand Superscript-II cDNAsynthesis kit (Invitrogen). PCR was performed using specific primers and conditionsusing clontech Platinum Taq-DNA polymerase, followed by gel electrophoresis.
192 N. Gurusamy et al. / FEBS Letters 584 (2010) 187–193
current study, we found that the oxidative stress generatedthrough ischemia–reperfusion injury in the CLP-1+/� mouse heartsinvolved in the generation of redox signaling as evidenced by theincreased production of NOX4, Nrf2, Ref-1 in CLP-1+/� mousehearts when compared to wild-type mouse hearts. These resultsare in consistent with the previous report that ROS generatedvia UV radiation or H2O2 treatment remarkably induce the levelsof Ref-1, which corresponds with the increase in endonucleaseand redox activities of Ref-1 [21]. Ref-1 facilitates the DNA bind-ing and activation of several transcription factors via reduction ofa cysteine residue [22]. ROS can also affect the functions of Ref-1.For example, oxidative stress severely affects both DNA repair andthe redox regulation activities of Ref-1 [23]. Our results suggestthat oxidative stress induced via ischemia–reperfusion injurycould have significantly affected the redox regulation of Ref-1. Itis tempting to speculate that the redox signaling mediated viaRef-1 and Nrf2 in CLP-1+/� mouse hearts could have attenuatedthe ischemia–reperfusion-induced oxidative stress leading to car-dioprotection in CLP-1+/� mice. As shown in previous studies [24],our results suggest that the enhanced expression of NOX-4 couldplay an important role in the redox signaling in CLP-1+/� mousehearts. Nrf2 is a member of basic leucine zipper transcription fac-tors and is known to induce many genes involved in the stress-responsive and cytoprotective proteins such as glutathione perox-idase, thioredoxin, catalase, and superoxide dismutase [25]. In thecurrent study, we found that the mRNA level of Trx-1, Trx-2, Grx-1 and Grx-2 are reduced. Our results are in consistent with theresults of other studies, where it has been demonstrated thatthe mRNAs that code for proteins produced in response to inter-nal or external stimuli will have short half-lives [26]. In otherwords, the reduced level of Trx-1, Trx-2, Grx-1 and Grx-2 mRNAsindicate that they are actively being used to attenuate the ROSproduced in association with ischemia–reperfusion injury in
CLP-1+/� mice hearts. The mRNA level of TrxR-1, which is re-quired to convert back the oxidized Trx-1 to reduced Trx-1, wasincreased.
Environmental stresses such as heat shock, and UV radiationand myocardial ischemia–reperfusion injury are known to induceoxidative stress. Controlled amount of oxidative stress inducesthe expression of intracellular antioxidants leading to enhancedmyocardial tolerance to ischemia [27], and redox signaling inthe ischemic adaptation induces stress-inducible proteinsthrough the activation of transcription factors [28]. Thioredoxinand glutaredoxin play an essential role in maintaining the intra-cellular environment in a reduced state via redox signaling inthe ischemic myocardium, where redox signaling converts cellu-lar death signal into survival signal [28]. In conclusion, our re-sults suggest that the downregulation of CLP-1 in CLP-1+/�
mice leads to the potentiation of redox signaling thereby gener-ating their molecular targets after ischemia reperfusion injury. Itwould further suggest that drugs designed to down regulate CLP-1 could confer cardioprotection through the potentiation of re-dox cycling.
Acknowledgements
This work was supported by National Heart, Lung and BloodInstitute at the National Institutes of Health [HL 34360, HL22559 and HL 33889].
References
[1] Espinoza-Derout, J., Wagner, M., Shahmiri, K., Mascareno, E., Chaqour, B. andSiddiqui, M.A. (2007) Pivotal role of cardiac lineage protein-1 (CLP-1) intranscriptional elongation factor P-TEFb complex formation in cardiachypertrophy. Cardiovasc. Res. 75, 129–138.
[2] Bres, V., Yoh, S.M. and Jones, K.A. (2008) The multi-tasking P-TEFb complex.Curr. Opin. Cell Biol. 20, 334–340.
[3] Dey, A., Chao, S.H. and Lane, D.P. (2007) HEXIM1 and the control oftranscription elongation: from cancer and inflammation to AIDS and cardiachypertrophy. Cell Cycle 6, 1856–1863.
[4] Shimizu, N. et al. (2005) HEXIM1 forms a transcriptionally abortive complexwith glucocorticoid receptor without involving 7SK RNA and positivetranscription elongation factor b. Proc. Natl. Acad. Sci. USA 102, 8555–8560.
[5] Espinoza-Derout, J., Wagner, M., Salciccioli, L., Lazar, J.M., Bhaduri, S.,Mascareno, E., Chaqour, B. and Siddiqui, M.A. (2009) Positive transcriptionelongation factor b activity in compensatory myocardial hypertrophy isregulated by cardiac lineage protein-1. Circ. Res. 104, 1347–1354.
[6] Huang, F., Wagner, M. and Siddiqui, M.A. (2004) Ablation of the CLP-1 geneleads to down-regulation of the HAND1 gene and abnormality of the leftventricle of the heart and fetal death. Mech. Dev. 121, 559–752.
[7] Cho, E.J., Kobor, M.S., Kim, M., Greenblatt, J. and Buratowski, S. (2001)Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNApolymerase II C-terminal domain. Genes Dev. 15, 3319–3329.
[8] Sano, M. et al. (2002) Activation and function of cyclin T-Cdk9 (positivetranscription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat.Med. 8, 1310–1317.
[9] Sano, M. et al. (2004) Activation of cardiac Cdk9 represses PGC-1 and confers apredisposition to heart failure. EMBO J. 23, 3559–3569.
[10] Mascareno, E., Manukyan, I., Das, D.K. and Siddiqui, M.A. (in press)Downregulation of cardiac lineage protein (CLP-1) expression in CLP-1+/�
mice affords cardioprotection against ischemic stress. J. Cell Mol. Med.[11] Lin, J., Handschin, C. and Spiegelman, B.M. (2005) Metabolic control through
the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370.[12] Ratcliffe, P.J., O’Rourke, J.F., Maxwell, P.H. and Pugh, C.W. (1998) Oxygen
sensing, hypoxia-inducible factor-1 and the regulation of mammalian geneexpression. J. Exp. Biol. 201, 1153–1162.
[13] Millonig, G., Hegedusch, S., Becker, L., Seitz, H.K., Schuppan, D. and Mueller, S.(2009) Hypoxia-inducible factor 1 alpha under rapid enzymatic hypoxia: cellssense decrements of oxygen but not hypoxia per se. Free Radic. Biol. Med. 46,182–191.
[14] Murdoch, C.E., Zhang, M., Cave, A.C. and Shah, A.M. (2006) NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure.Cardiovasc. Res. 71, 208–215.
[15] Gamaley, I.A. and Klyubin, I.V. (1999) Roles of reactive oxygen species:signaling and regulation of cellular functions. Int. Rev. Cytol. 188, 203–255.
[16] Kern, J.C. and Kehrer, J.P. (2005) Free radicals and apoptosis: relationshipswith glutathione, thioredoxin, and the BCL family of proteins. Front. Biosci. 10,1727–1738.

N. Gurusamy et al. / FEBS Letters 584 (2010) 187–193 193
[17] Hensley, K., Robinson, K.A., Gabbita, S.P., Salsman, S. and Floyd, R.A. (2000)Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med.28, 1456–1462.
[18] Papandreou, I., Cairns, R.A., Fontana, L., Lim, A.L. and Denko, N.C. (2006) HIF-1mediates adaptation to hypoxia by actively downregulating mitochondrialoxygen consumption. Cell Metab. 3, 187–197.
[19] Kim, J.W., Tchernyshyov, I., Semenza, G.L. and Dang, C.V. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switchrequired for cellular adaptation to hypoxia. Cell Metab. 3, 177–185.
[20] Simon, M.C. (2006) Coming up for air: HIF-1 and mitochondrial oxygenconsumption. Cell Metab. 3, 150–151.
[21] Tell, G., Damante, G., Caldwell, D. and Kelley, M.R. (2005) The intracellularlocalization of APE1/Ref-1: more than a passive phenomenon? Antioxid.Redox Signal. 7, 367–384.
[22] Gottlieb, T.M., Leal, J.F., Seger, R., Taya, Y. and Oren, M. (2002) Cross-talkbetween Akt, p53 and Mdm2: possible implications for the regulation ofapoptosis. Oncogene 21, 1299–1303.
[23] Walker, L.J., Robson, C.N., Black, E., Gillespie, D. and Hickson, I.D. (1993)Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) thatare essential for redox regulation of Jun DNA binding. Mol. Cell Biol. 13, 5370–5376.
[24] Akki, A., Zhang, M., Murdoch, C., Brewer, A. and Shah, A.M. (2009) NADPHoxidase signaling and cardiac myocyte function. J. Mol. Cell Cardiol. 47, 15–22.
[25] Surh, Y.J., Kundu, J.K. and Na, H.K. (2008) Nrf2 as a master redox switch inturning on the cellular signaling involved in the induction of cytoprotectivegenes by some chemopreventive phytochemicals. Planta. Med. 74, 1526–1539.
[26] Guhaniyogi, J. and Brewer, G. (2001) Regulation of mRNA stability inmammalian cells. Gene 265, 11–23.
[27] Maulik, N., Watanabe, M., Engelman, D.T., Engelman, R.M. and Das, D.K. (1995)Oxidative stress adaptation improves postischemic ventricular recovery. Mol.Cell Biochem. 144, 67–74.
[28] Das, D.K. and Maulik, N. (2004) Conversion of death signal into survival signalby redox signaling. Biochemistry (Moscow) 69, 10–17.
Related Documents











