PAPER www.rsc.org/dalton | Dalton Transactions DNA binding and cleavage properties of a newly synthesised Ru(II)-polypyridyl complex† Amrita Ghosh, a Amit Mandoli, a D. Krishna Kumar, a Narendra Singh Yadav, a Tamal Ghosh, b Bhavanath Jha,* a Jim A. Thomas* c and Amitava Das* a Received 3rd April 2009, Accepted 24th August 2009 First published as an Advance Article on the web 9th September 2009 DOI: 10.1039/b906756f The single crystal X-ray structure of the newly synthesized ruthenium (II)-polypyridyl complex, [(bpy) 2 Ru(L)](PF 6 ) 2 (1) (bpy is 2,2¢-bipyridyl and L is 2-methyl-2H-1,3,7,8-tetraaza- cyclopenta[l]phenanthren-2-ol revealed a near planar conformation for the fused imidazole moiety, suggesting the complex may be capable of binding to duplex DNA. Binding-induced changes in spectral properties, along with thermal denaturation and viscosity measurement studies confirmed that the complex binds to CT-DNA with a moderately high binding constant value (4.1 ¥ 10 5 M -1 ) through intercalation. Photocleavage studies with the pBR322 plasmid DNA were performed following excitation of this complex into the Ru(dp) → bpy/L(p*)-based MLCT band and have shown that this complex cleaves the circular pBR322 DNA into linear DNA. Inhibitor studies revealed that the hydroxyl radicals were mainly responsible for the DNA photocleavage reaction. Preliminary studies indicate that the –OH functionality in 1 was also found to hydrolyse the phosphodiester linkage of pBR322 in the dark. Introduction In the pursuit of tools for biomedicine and anticancer drugs, tran- sition metal complexes that bind to DNA have been extensively studied in the last few decades. 1–5 Systems that irreversibly bind to DNA such as cisplatin and its analogues have become some of the most important first-line treatments for solid tumours. 3–9 On the other hand complexes that bind to DNA reversibly have possible application as tools for probing the structure and function of DNA. 7–13 Apart from the possibility of designing systems that bind to specific sites of the DNA strand, one of the attractions of using photoexcitable or redox active metal centres in such systems is that they provide direct or indirect mechanisms for DNA damage and cleavage. Direct reactions include oxidation of deoxyribose residues by hydrogen abstraction or electron transfer from nucleobases to the metal ion, while indirect reactions usually involve the metal centre mediated generation of reactive species such as the hydroxyl radical or singlet oxygen. 14,15 Less often, a Central Salt and Marine Chemicals Research Institute (CSIR), Bhavnagar, 364002, Gujarat, India. E-mail: [email protected], [email protected]; Fax: +91 278 2567562; Tel: +91 278 2567760 b Department of Chemistry, Motilal Nehru National Institute of Technology, Allahabad, 211004, India c Department of Chemistry, University of Sheffield, Sheffield, S3 7H, UK. E-mail: james.thomas@sheffield.ac.uk †Electronic supplementary information (ESI) available: [1] Square wave and cyclic voltammogram of complex 1, [2] crystallographic parameters for 1, [3] packing diagram for 1, [4] plot of I/I o vs. [DNA] for 1, [5] T4 DNA ligase experiment, [6] photocleavage experiment with Ru(phen) 3 Cl 2 as standard, [7] hydrolysis study of phosphodiester of paranitrophenyl phosphate by complex 1. CCDC reference number 671466. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b906756f hydrolytic cleavage has been accomplished using Lewis acid or nucleophile activation. 16 The advantages of this latter pathway is that genetic information is not compromised as there is no chemical damage to individual nucleotides; moreover, systems that have different cleavage preferences to naturally occurring nucleases may be constructed. 17–22 Due to their rich photochemistry polypyridyl complexes of ruthenium(II)-have received considerable attention as DNA bind- ing substrates. They are photochemical stable in their ground and excited state, and have long lived triplet states, rich redox/spectral properties and good water solubility. 23 Furthermore, judicious selection of coordinated ligands can be used to tune the photo- chemistry and DNA binding properties of these systems. The rich optical properties of these complexes also facilitate assessments of their DNA binding capabilities as binding to DNA can be probed through changes in absorption and emission spectra. Although [Ru(bpy) 3 ] 2+ only interacts with DNA through weak external electrostatic binding, many related complexes bind through higher affinity modes such as groove binding or intercalation. 24 For intercalation to occur planar ligands which fit well into the stack between the base pairs of DNA double helix are usually required. 25,26 In previous studies, Ru(II)-polypyridyl complexes with an extended imidazole functionality have received attention as this moiety incorporates a planar conformation and potential hydrogen bonding site. 27 Apart from the possibility of enhancing intercalative interactions, hydrogen bonding can also perturb the Ru(dp) → L¢(p*)-based (L¢ is derivative of 2,2¢-bipyridyl or 1,10-phenanthroline) 3 MLCT excitation used to probe DNA binding. In this article, we report the synthesis, characterization, and X-ray structure of a new Ru(II)-polypyridyl derivative with fused imidazole functionality. We also explore its binding to duplex DNA and cleavage activity. 9312 | Dalton Trans., 2009, 9312–9321 This journal is © The Royal Society of Chemistry 2009

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

PAPER www.rsc.org/dalton | Dalton Transactions
DNA binding and cleavage properties of a newly synthesisedRu(II)-polypyridyl complex†
Amrita Ghosh,a Amit Mandoli,a D. Krishna Kumar,a Narendra Singh Yadav,a Tamal Ghosh,b Bhavanath Jha,*a
Jim A. Thomas*c and Amitava Das*a
Received 3rd April 2009, Accepted 24th August 2009First published as an Advance Article on the web 9th September 2009DOI: 10.1039/b906756f
The single crystal X-ray structure of the newly synthesized ruthenium (II)-polypyridyl complex,[(bpy)2Ru(L)](PF6)2 (1) (bpy is 2,2¢-bipyridyl and L is 2-methyl-2H-1,3,7,8-tetraaza-cyclopenta[l]phenanthren-2-ol revealed a near planar conformation for the fused imidazole moiety,suggesting the complex may be capable of binding to duplex DNA. Binding-induced changes inspectral properties, along with thermal denaturation and viscosity measurement studies confirmed thatthe complex binds to CT-DNA with a moderately high binding constant value (4.1 ¥ 105 M-1) throughintercalation. Photocleavage studies with the pBR322 plasmid DNA were performed followingexcitation of this complex into the Ru(dp) → bpy/L(p*)-based MLCT band and have shown that thiscomplex cleaves the circular pBR322 DNA into linear DNA. Inhibitor studies revealed that thehydroxyl radicals were mainly responsible for the DNA photocleavage reaction. Preliminary studiesindicate that the –OH functionality in 1 was also found to hydrolyse the phosphodiester linkage ofpBR322 in the dark.
Introduction
In the pursuit of tools for biomedicine and anticancer drugs, tran-sition metal complexes that bind to DNA have been extensivelystudied in the last few decades.1–5 Systems that irreversibly bindto DNA such as cisplatin and its analogues have become someof the most important first-line treatments for solid tumours.3–9
On the other hand complexes that bind to DNA reversibly havepossible application as tools for probing the structure and functionof DNA.7–13
Apart from the possibility of designing systems that bindto specific sites of the DNA strand, one of the attractionsof using photoexcitable or redox active metal centres in suchsystems is that they provide direct or indirect mechanisms forDNA damage and cleavage. Direct reactions include oxidation ofdeoxyribose residues by hydrogen abstraction or electron transferfrom nucleobases to the metal ion, while indirect reactions usuallyinvolve the metal centre mediated generation of reactive speciessuch as the hydroxyl radical or singlet oxygen.14,15 Less often,
aCentral Salt and Marine Chemicals Research Institute (CSIR), Bhavnagar,364002, Gujarat, India. E-mail: [email protected], [email protected];Fax: +91 278 2567562; Tel: +91 278 2567760bDepartment of Chemistry, Motilal Nehru National Institute of Technology,Allahabad, 211004, IndiacDepartment of Chemistry, University of Sheffield, Sheffield, S3 7H, UK.E-mail: [email protected]† Electronic supplementary information (ESI) available: [1] Square waveand cyclic voltammogram of complex 1, [2] crystallographic parametersfor 1, [3] packing diagram for 1, [4] plot of I/Io vs. [DNA] for 1, [5] T4DNA ligase experiment, [6] photocleavage experiment with Ru(phen)3Cl2
as standard, [7] hydrolysis study of phosphodiester of paranitrophenylphosphate by complex 1. CCDC reference number 671466. For ESIand crystallographic data in CIF or other electronic format see DOI:10.1039/b906756f
hydrolytic cleavage has been accomplished using Lewis acid ornucleophile activation.16 The advantages of this latter pathwayis that genetic information is not compromised as there is nochemical damage to individual nucleotides; moreover, systems thathave different cleavage preferences to naturally occurring nucleasesmay be constructed.17–22
Due to their rich photochemistry polypyridyl complexes ofruthenium(II)-have received considerable attention as DNA bind-ing substrates. They are photochemical stable in their ground andexcited state, and have long lived triplet states, rich redox/spectralproperties and good water solubility.23 Furthermore, judiciousselection of coordinated ligands can be used to tune the photo-chemistry and DNA binding properties of these systems. The richoptical properties of these complexes also facilitate assessments oftheir DNA binding capabilities as binding to DNA can be probedthrough changes in absorption and emission spectra.
Although [Ru(bpy)3]2+ only interacts with DNA throughweak external electrostatic binding, many related complexesbind through higher affinity modes such as groove binding orintercalation.24 For intercalation to occur planar ligands which fitwell into the stack between the base pairs of DNA double helixare usually required.25,26 In previous studies, Ru(II)-polypyridylcomplexes with an extended imidazole functionality have receivedattention as this moiety incorporates a planar conformation andpotential hydrogen bonding site.27 Apart from the possibilityof enhancing intercalative interactions, hydrogen bonding canalso perturb the Ru(dp) → L¢(p*)-based (L¢ is derivative of2,2¢-bipyridyl or 1,10-phenanthroline) 3MLCT excitation usedto probe DNA binding. In this article, we report the synthesis,characterization, and X-ray structure of a new Ru(II)-polypyridylderivative with fused imidazole functionality. We also explore itsbinding to duplex DNA and cleavage activity.
9312 | Dalton Trans., 2009, 9312–9321 This journal is © The Royal Society of Chemistry 2009

Materials and methods
Analytical measurements
1H NMR spectra were recorded on a Bruker 200 MHz FT NMR(model: Advance-DPX 200) spectrometer at room temperature(RT, 25 ◦C). Tetramethylsilane (TMS) was used as an internalstandard for all 1H NMR studies. ESI MS measurements werecarried out on Waters QTof-Micro instrument. Microanalyses(C, H, N) were performed using a Perkin-Elmer 4100 elementalanalyzer. Infrared spectra were recorded as KBr pellets usinga Perkin Elmer Spectra GX 2000 spectrometer. UV-Vis spectrawere obtained by using either a Shimadzu UV-3101 PC or Cary500 Scan UV-Vis-NIR spectrometer. Room temperature emissionspectra were obtained using a Perkin-Elmer LS 50B luminescencespectrofluorimeter. Electrochemical experiments were performedon a CH-660A (USA) electrochemical instrument with a con-ventional three-electrode cell assembly. The saturated Ag/AgClwas used as a reference electrode and platinum was used forthe working and auxiliary electrodes. [tBu4N]PF6 was used asbackground electrolyte. Ferrocene was added at the end of eachexperiment as an internal standard and all potentials are quotedversus the ferrocenium/ferrocene (Fc+/Fc) couple.
The fluorescence quantum yields, ff were estimated by usingeqn (1) in appropriate solvents (as specified) using the integratedemission intensity of Ru(bpy)3Cl2 (ff = 0.042 in H2O at RT) asreference.28,29
ff = ff¢(Isample/Istd)(Astd/Asample)(n2solvent/n2
std) (1)
where, ff¢ is the absolute quantum yield for the Ru(bpy)3Cl2, usedas reference; Isample and Istd are the integrated emission intensities;Asample and Astd are the absorbances at the excitation wavelength,and n2
solvent and n2std are the respective refractive indices.28,29
Chemicals
[Ru(bpy)2]Cl2·2H2O, [Ru(bpy)3]Cl2, [Ru(phen)3]Cl2 and [Ru(bpy)2-(phen)]Cl2 were prepared following standard literatureprocedures.28,29 RuCl3·xH2O, 2,2¢-bipy, 1,10-phenanthroline,Agarose and Calf thymus DNA (CT-DNA) were purchasedfrom Aldrich Chemical Co (USA) and were used as received.[tBu4N]PF6 was recrystallised from ethanolic solution beforeuse. K4[Fe(CN)6], oleum, nitric acid and acetic acid wereobtained from S.D. Fine Chemicals (India) and were usedwithout any further purification. All solvents were distilledand dried prior to use following standard procedures. pBR322plasmid DNA was obtained from Bangalore Genei (India).The CT-DNA concentration per nucleotide was determinedby absorption spectrum using the molar absorption coefficient(6600 mol-1 dm3 cm-1) at 260 nm.30
Synthesis: synthesis of 5-nitro-1,10-phenanthroline (L1). Syn-thesis of (L1) was achieved by following a known procedure31 withsome modification in the work up: phenanthroline monohydrate(5.0 g, 27.7 mmol), 20% oleum (25mL) and 72% nitric acid (21mL)were used for the reaction. NaHCO3 was used for neutralizationand a pale yellow beige precipitate was collected by filtration.The residue was washed thoroughly with water and dried undervacuum. Yield: 3.75 g (60%); 1H NMR (200 MHz, CDCl3, TMS,d (ppm)): 9.34 (d, 1H, J = 10.4 Hz, aH), 9.31 (d, 1H, J = 10.6 Hz,
dH), 9.023 (d, 1H, J = 8.6 Hz, cH), 8.68 (s, 1H, gH), 8.43 (d, 1H, J =8 Hz, fH), 7.86-7.75 (m, 2H, bH and eH); FT IR(KBr; n/cm-1): 3083(aromatic C-H), 1621, 1590 cm-1 (C=C, C=N), 1518 (asymmetricArNO2 (N=O), 1355 (symmetric ArNO2 (N=O), 832 (Aromatic-NO2, C-N); ESI-MS: m/z 226 (M+ + H) (~65%), 248 (M+ + Na+)(~70 %); Elemental Analysis (Expt. and Calcd. for C12H7N3O2):Expt. C 63.8 %, H 3.0 %, N 18.0 %, Calcd. C 64.0 %, H 3.13 %, N18.66 %.
Synthesis of 5-nitro-6-amino-1,10-phenanthroline (L2). 5-Nitro-1,10-phenanthroline (3.50 g, 15.5 mmol) was taken in a twonecked round bottom flask. To this ~100 ml of a ethanol/dioxan(3:2; v/v) mixture was added and the temperature was raisedto 60 ◦C with stirring to dissolve the solid. The solution wasthen rapidly cooled to give a fine suspension. To this suspension,powdered hydroxylamine hydrochloride (6.83 g, 98.4 mmol) wasadded followed by the slow addition of a methanolic solution ofKOH (7.29 g, 129.4 mmol in 100 mL). The mixture was stirredfor 1 h at 4 ◦C, then at RT for another 1 h. The resultingbrown suspension was poured in ice cold water to give a brightyellow precipitate and was allowed to stand at RT for 18 h. Theprecipitate was filtered and was washed thoroughly with methanol,followed by water. The desired product, obtained as a residue, wasdried under vacuum. Analytical data show that this product wassufficiently pure for further synthesis. Yield: 2.41 g (65 %); 1HNMR (200 MHz, DMSO d6, TMS, d (ppm)): 9.196 (d, 1H, J =4.4 Hz, aH), 9.08 (d, 1H, J = 8.6 Hz, cH), 8.79 (d, 1H, J = 4.4 Hz,dH), 8.72 (d, 1H, J = 8.4 Hz, fH), 8.62 (s, 2H, -NH), 7.86 (dd,1H, J = 4.4 Hz, 3.4 Hz, bH), 7.68 (dd, 1H, J = 4 Hz, 4.3 Hz, eH); FTIR (KBr, n/cm-1): 3145 (N-H), 1631 (N=O), 1596 (C=C/C=N),1263, 737 (br., PF6
-); ESI-MS: m/z 241 (M+ + H+) (~25 %), 263(M+ + Na+) (~100 %); Elemental Analysis (Expt. and Calc. forC12H8N4O2): Expt. C 60.6 %, H 3.3 %, N 23.3 %; Calcd. C 60.0 %,H 3.36 %, N 23.32 %.
Synthesis of 5,6-diamino-1,10-phenanthroline (L3). Synthesisof (L3) was done by a reported methodology with necessarymodification.32,33 To a hot suspension of 5-nitro-6-amino-1,10-phenanthroline (2.0 g, 8.3 mmol) in 300 mL ethanol/dioxan (1:1,v/v), 10 % palladium on activated carbon (1.64 g) was added.To this reaction mixture, a solution of 5.4 mL hydrazine hydratediluted with 20 mL ethanol/dioxan (1:1; v/v) was slowly added ina dropwise manner over a period of 1.5 h. The resulting reactionmixture was refluxed overnight and then filtered hot through acelite bed. The filtrate was collected and allowed to cool to RT andred needles were obtained. This crystalline product was filtered,washed with cold ethanol and dried. Yield: 380 mg (22 %); 1HNMR (200 MHz, DMSO d6, TMS, d (ppm)): 8.78 (d, 2H, J =4.2 Hz, cH), 8.51 (d, 2H, J = 8.4 Hz, aH), 7.62(dd, 2H, J = 4.2 Hz,bH), 3.5(s, 4H,-NH); FT IR(KBr; n/cm-1) 3425 (N-H), 2362, 1619,1555; ESI-MS m/z 210 (M+) (~100 %); Elemental Analysis (Expt.and Calcd. for C12H10N4): Expt. C 67. 9 %, H 4.8 %, N 26.5 %,Calcd. C 68.56 %, H 4.79 %, N 26.65 %.
Synthesis of Ru(bpy)2(L)(PF6)2 (1) (L is 2-methyl-2H-1,3,7,8-tetraaza-cyclopenta[l]phenanthren-2-ol. Ru(bpy)2Cl2.2H2O(0.20 g, 0.38 mmol) and 5,6-diamino-1,10-phenanthroline(0.084 g, 0.4 mmol) was taken into a two neck flask. To this mixture20 mL of ethanol/water (3:1, v/v) was added and allowed to stir for15 min. After the addition of 5 mL of ethanol/acetic acid mixture
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 9312–9321 | 9313

(9:1; v/v) the resulting reaction mixture was refluxed for 4 h afterthis period, the organic solvent was removed under vacuum andthen the acetic acid, that was present in the reaction mixture,was neutralised by triethylamine. To this neutralised solution,an excess of a saturated solution of KPF6 was added. A deeporange precipitate was formed and kept in the refrigerator for16 h. Then this was filtered in cold conditions and the residuewas washed with ice cold water. The crude product was purifiedby recrystallization from acetonitrile–ether mixture following avapor diffusion method. Deep orange crystals were obtained after3 days. Yield: 250 mg (68 %); 1H NMR (200 MHz, CD3CN, TMS,d (ppm)): 8.69 (d, 2H, J = 8 Hz, aH), 8.51 (d, 4H, J = 8.2 Hz, bH),8.07 (t, 4 H, J = 9.6 Hz, cH), 7.93 (d, 2H, J¢= 5.4 Hz, gH), 7.78 (t, 4H, J = 6.6 Hz, dH), 7.59 (t, 2H, J = 6 Hz, fH), 7.4(d, 4H, J = 5.6 Hz,eH), 1.9 (m, 3H, -CH3); FT IR (KBr; n/cm-1): 3369 (O-H), 1604(C=C, C=N), 840 (br, PF6); ESI-MS: m/z 663 (M+-2PF6-H) (~10%) 810 (M+-PF6) (~18 %); Elemental Analysis, (Expt. and Calcd.for C34H26N8ORuP2F12): Expt.: C 42.6 %, H 2.8 %, N 11.9 %.Calc. C 42.82 %, H 2.75 %, N 11.75 %; E1/2 (CH3CN; in V (DE =Ea - Ec/mV) vs. Fc+/Fc couple): 0.78 ((90), Ru(II)/(III) couple);-2.01 ((80), L0/∑- couple); -2.22 ((95), bpy0/∑- couple); -2.4 (fromsquare wave voltammetry, bpy0/∑- couple); -2.52 (from square wavevoltammetry, RuII/I couple) (Supplementary Information†). UV-Vis (CH3CN, l(nm), (log e)): 243 (4.68), 285 (4.86), 441 (4.17);Emission spectra (CH3CN, lem (nm) with lexc = 460 nm): 602, ff =0.0186 using Ru(bpy)3(PF6)2 as standard (ff = 0.062 in CH3CN).34
X-ray diffraction
X-ray single crystal data was collected using MoKa (l = 0.7107 A)radiation on a SMART APEX diffractometer equipped withCCD area detector at 150K. X-ray quality crystals were isolatedfrom the mother liquid and immediately inserted in paratoneoil and then mounted. Data collection, data reduction,35 andstructure solution/refinement36–38 were carried out using thesoftware package of SMART APEX. The structure was solvedby direct method. Crystallographic data for complex 1 are givenin Table 1. The disordered F-atoms of the PF6
- were refined usingFVAR second variable facility provided within SHELXTL. Dueto their higher thermal parameters, nitrogen atom (N25) andtwo carbon atoms (C28 and C30) of the phenanthroline moietyand one of the carbon atoms (C18) of the bpy moiety wererefined isotropically. All other non-hydrogen atoms were refinedanisotropically. All hydrogen atoms were geometrically fixed attheir idealised position. Graphics were generated using Mercury1.4.1.39
DNA binding and cleavage studies
Solutions of CT-DNA were obtained from Sigma-Aldrich Co.(USA). The DNA concentration per nucleotide was determinedby absorption spectrum using the molar absorption coefficient(6600 mol-1 dm3 cm-1) at 260 nm.30 Absorbances at 260 nm (A260)and at 280 nm (A280) for CT-DNA were measured to check forpurity. Tris-buffer (5 mM of tris(hydroxymethyl)amino methane(Tris), pH 7.1, 50 mM NaCl) was used for the absorption andfluorescence titration experiments and phosphate buffer (1 mMphosphate, pH 7.0, 2 mM NaCl) was used for the thermaldenaturation experiments. p-Nitrophenol phosphate disodium
Table 1 Crystallographic parameters for 1
Crystal data 1
Empirical formula C34H26F9N8OP1.5RuFormula weight 881.15Crystal size (mm3) 0.20 ¥ 0.06 ¥ 0.02Crystal system TriclinicSpace group P-1a (A) 8.880(4)b (A) 13.449(6)c (A) 18.076(8)a (◦) 110.642(8)b (◦) 90.305(9)g (◦) 99.427(8)Volume (A3) 1988.2(1)Z 2Dcalc. (g/cm3) 1.472F(000) 883m MoKa (mm-1) 0.535Temperature (K) 150(2)Rint 0.1194q min/max 1.21/25.50Reflections collected/unique/observed 10080/7207/2429Data/restraints/parameters 7207/4/403Goodness of fit on F2 0.914Final R indices [I>2s(I)] R1 = 0.1182
wR2 = 0.2638R indices (all data) R1 = 0.2391
wR2 = 0.3203
salt in Tris-buffer was used as a the model reagent to test thehydrolyzing ability of the complex 1 at the phosphodiester linkageof the DNA molecule.
For the DNA photocleavage experiments, irradiation wascarried out using a 150-Watt lamp, fitted with a monochromaticfilter (Make: Cohu Inc. San Diego, CA) that allows light ofwavelength range of 440–580 nm to pass through, with maximumintensity at 520 nm.
Absorption titrations
A 1.44 ¥ 10-4 M solution of [Ru(bpy)2L]Cl2 was prepared inthoroughly degassed miliQ water and was stored in the dark. Thesesolutions were used for all spectroscopic studies after appropriatedilution with Tris-HCl buffer. All titration experiments were per-formed using 15.0 mM solutions of complex 1 and varying [DNA](0-109 mM) in Tris-HCl buffer at RT. Absorption spectra wererecorded after each successive addition of DNA and equilibrationfor a period of 10 min. The intrinsic binding constant Kb of thecomplex with CT-DNA was evaluated using eqn (2).40
[DNA]/(ea - ef ) = [DNA]/(eb - ef ) + 1/Kb(eb - ef ) (2)
Where ea, ef and eb are the extinction coefficient for apparent,free and bound metal complex, respectively. A best fit plot of[DNA]/(ea - ef ) vs. [DNA] produces a straight line with slope1/(eb - ef ) and intercept 1/Kb(eb - ef ); while Kb is the ratio ofslope to intercept. In order to calculate the binding constant (Kb)in the present study, the change in absorbance values at 430 nmwas monitored.
Luminescence titrations
For fluorescence titration experiments, aqueous Tris-HCl buffersolution of [Ru(bpy)2L]Cl2 was used and concentration was
9314 | Dalton Trans., 2009, 9312–9321 This journal is © The Royal Society of Chemistry 2009

so adjusted that the final concentration of [Ru(bpy)2L]Cl2 was5.0 mM. This was titrated with increasing [DNA], over a range0–3.35 ¥ 10-4 M. Following excitation at 430 nm, the titration pro-cedure was similar to that outlined above for spectrophotometrictitration. For all measurements, a similar slit width (10/10) wasused. Steady state fluorescence intensities at 594 nm for varying[DNA] were used to calculate the binding constant (eqn (3)), asthe observed fluorescence is assumed to be a sum of the weightedcontributions of free (cf ) and bound Ru(II)-complex (cb)
cb = c [(I - I 0)/(Imax - I 0)] (3)
where, c is the total Ru(II)-complex concentration, I and I 0 arethe emission intensities in the presence and absence of DNA,and Imax is the fluorescence of the totally bound complex. Theconcentration of the free complex, cf , is equal to c - cb. A plot ofr/cf vs. r, where r is cb/[DNA], was constructed according to theMcGhee and von Hippel equation (eqn (4)):
2r/cf = Kb(1 - 2nr) [(1 - 2nr)/{1 - 2(n - 1)r}]n-1 (4)
where, Kb represents the intrinsic binding constant of the complexwith DNA and n is the size of a binding site in base pairs.
Thermal denaturation experiments were carried out by monitor-ing the absorption of CT-DNA at 260 nm at various temperatures(25–95 ◦C), in absence and presence of 1. Complex 1 was incubatedwith CT-DNA at [1]/[DNA] = 4.8 ([1] = 25 mM and [DNA] =120 mM). The melting temperature (Tm) and the curve width(sT, the temperature range between which 10% to 90% of theabsorption increase occurred) were calculated following reportedprocedure.41
Steady state emission quenching experiments using varying[Fe(CN)6]4- as the quencher were performed in absence andpresence of DNA. [1] and [DNA] used for this experiment were10 mM and 1.0 ¥ 10-3 M, respectively, while [{Fe (CN)6}4-] wasvaried from 0–2.86 mM. The ratio of the emission intensities inabsence (I 0) and presence of definite [{Fe(CN)6}4-] (I) was plottedagainst the respective [{Fe(CN)6}4-] and the plot was found tofollow the Stern–Volmer equation (eqn (5)).
I 0/I = 1 + K[{Fe(CN)6}4-] (5)
K is linear Stern–Volmer quenching constant and was evaluatedfrom the slope.
Result and discussion
Synthesis
The imidazole derivative (2-methyl-2-hydroxyl-2,3-dihydro-1,10-phenanthrolino-imidazole), was formed by the in situ reactionof acetic acid with 5,6-amino-1,10-phenanthroline, which wasthen reacted with [Ru(bpy)2]Cl2 to yield the desired complex, 1(Scheme 1). All intermediate organic compounds and 1 were char-acterised by a variety of analytical and spectroscopic techniques.Analytical and spectroscopic data matched well with the dataexpected for these complexes. The structure for the complex 1was further confirmed by single crystal X-ray structural analysis.
Recently, a similar one pot synthesis of fluorinated benzimi-dazolines was reported by Olah et. al.42 In the present case, itis presumed that the additional stability gained from extendedconjugation has yielded the final product.
Scheme 1 Reaction scheme followed for synthesis of 1.
X-ray structural studies
Single crystal X-ray diffraction analysis revealed that 1 belongsto the centrosymmetric triclinic space group, P1. The asym-metric unit comprises of one Ru(bpy)2(L)-fragment, one fullyoccupied PF6
- and two PF6- anions whose central P atom sits
on a special position as counter anions. The fluorine atoms of thefully occupied PF6
- anion were found to be disordered. One of thePF6
- anion sitting on the special position was found to be severelydisordered and could not be assigned to any proper model dueto its high thermal parameters (Fig. 1). The metal center, Ru(II)displays a slightly distorted octahedron. The methyl group andthe oxygen atom of the imidazole ring are oriented to the topand bottom of the plane involving the phen moiety. The two bpymoieties are oriented almost perpendicular to the phen plane.
Fig. 1 ORTEP diagram (50% probability) of 1 (anions and disorderedsolvents are not shown).
High thermal parameter and severe disorder, present in thecrystal structure, do not allow us make detailed comment onthe various non-bonded interaction that could be present.43–46
However, the structure was refined enough to reveal the relativeorientation of the coordinated ligands, and a one dimensionaltape like architecture along a-axis involving two molecules ofRu(bpy)2(L) and PF6
- (Supplementary Information†) could be ob-served. This one-dimensional tape architecture was further packedin a two dimensional fashion along b-c plane to from a porousnetwork with channels running down the a-axis (SupplementaryInformation†). The solvent molecules located inside the channelwere also found to be disordered. Smeared electron densitiescorresponding to the solvents as well as one of the disorderedPF6
- anions could not be fully modelled and therefore theoverall contribution of the unaccounted electron densities to thediffraction pattern was subtracted from the observed data using a
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 9312–9321 | 9315
“bypass” technique namely PLATON/SQUEEZE.47 By excludingthe contributions from the solvents and half a molecule of thedisordered PF6
-, the subsequent analysis of relatively ‘noise-free’data enabled a reasonable refinement of the host framework. Theseindicated 190 electrons/unit cell or 95 electrons per molecule,which might be attributed to half of a PF6
- (34.5 electrons) and1.5 molecule of diethyl ether (42 electrons) per molecule. Themolecular formula of the compound was further confirmed bythe elemental analysis performed on the dried crystals of 1.
Spectral analysis
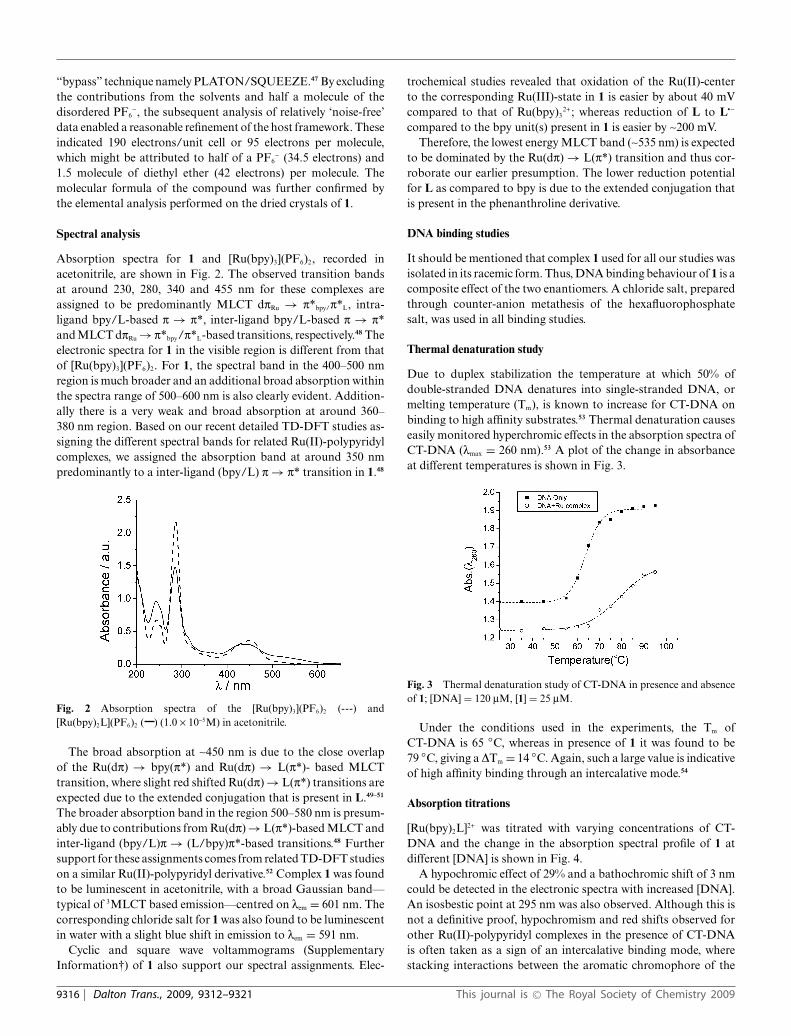
Absorption spectra for 1 and [Ru(bpy)3](PF6)2, recorded inacetonitrile, are shown in Fig. 2. The observed transition bandsat around 230, 280, 340 and 455 nm for these complexes areassigned to be predominantly MLCT dpRu → p*bpy/p*L, intra-ligand bpy/L-based p → p*, inter-ligand bpy/L-based p → p*and MLCT dpRu → p*bpy/p*L-based transitions, respectively.48 Theelectronic spectra for 1 in the visible region is different from thatof [Ru(bpy)3](PF6)2. For 1, the spectral band in the 400–500 nmregion is much broader and an additional broad absorption withinthe spectra range of 500–600 nm is also clearly evident. Addition-ally there is a very weak and broad absorption at around 360–380 nm region. Based on our recent detailed TD-DFT studies as-signing the different spectral bands for related Ru(II)-polypyridylcomplexes, we assigned the absorption band at around 350 nmpredominantly to a inter-ligand (bpy/L) p → p* transition in 1.48
Fig. 2 Absorption spectra of the [Ru(bpy)3](PF6)2 (---) and[Ru(bpy)2L](PF6)2 ( ) (1.0 ¥ 10-5M) in acetonitrile.
The broad absorption at ~450 nm is due to the close overlapof the Ru(dp) → bpy(p*) and Ru(dp) → L(p*)- based MLCTtransition, where slight red shifted Ru(dp) → L(p*) transitions areexpected due to the extended conjugation that is present in L.49–51
The broader absorption band in the region 500–580 nm is presum-ably due to contributions from Ru(dp) → L(p*)-based MLCT andinter-ligand (bpy/L)p → (L/bpy)p*-based transitions.48 Furthersupport for these assignments comes from related TD-DFT studieson a similar Ru(II)-polypyridyl derivative.52 Complex 1 was foundto be luminescent in acetonitrile, with a broad Gaussian band—typical of 3MLCT based emission—centred on lem = 601 nm. Thecorresponding chloride salt for 1 was also found to be luminescentin water with a slight blue shift in emission to lem = 591 nm.
Cyclic and square wave voltammograms (SupplementaryInformation†) of 1 also support our spectral assignments. Elec-
trochemical studies revealed that oxidation of the Ru(II)-centerto the corresponding Ru(III)-state in 1 is easier by about 40 mVcompared to that of Ru(bpy)3
2+; whereas reduction of L to L∑-
compared to the bpy unit(s) present in 1 is easier by ~200 mV.Therefore, the lowest energy MLCT band (~535 nm) is expected
to be dominated by the Ru(dp) → L(p*) transition and thus cor-roborate our earlier presumption. The lower reduction potentialfor L as compared to bpy is due to the extended conjugation thatis present in the phenanthroline derivative.
DNA binding studies
It should be mentioned that complex 1 used for all our studies wasisolated in its racemic form. Thus, DNA binding behaviour of 1 is acomposite effect of the two enantiomers. A chloride salt, preparedthrough counter-anion metathesis of the hexafluorophosphatesalt, was used in all binding studies.
Thermal denaturation study
Due to duplex stabilization the temperature at which 50% ofdouble-stranded DNA denatures into single-stranded DNA, ormelting temperature (Tm), is known to increase for CT-DNA onbinding to high affinity substrates.53 Thermal denaturation causeseasily monitored hyperchromic effects in the absorption spectra ofCT-DNA (lmax = 260 nm).53 A plot of the change in absorbanceat different temperatures is shown in Fig. 3.
Fig. 3 Thermal denaturation study of CT-DNA in presence and absenceof 1; [DNA] = 120 mM, [1] = 25 mM.
Under the conditions used in the experiments, the Tm ofCT-DNA is 65 ◦C, whereas in presence of 1 it was found to be79 ◦C, giving a DTm = 14 ◦C. Again, such a large value is indicativeof high affinity binding through an intercalative mode.54
Absorption titrations
[Ru(bpy)2L]2+ was titrated with varying concentrations of CT-DNA and the change in the absorption spectral profile of 1 atdifferent [DNA] is shown in Fig. 4.
A hypochromic effect of 29% and a bathochromic shift of 3 nmcould be detected in the electronic spectra with increased [DNA].An isosbestic point at 295 nm was also observed. Although this isnot a definitive proof, hypochromism and red shifts observed forother Ru(II)-polypyridyl complexes in the presence of CT-DNAis often taken as a sign of an intercalative binding mode, wherestacking interactions between the aromatic chromophore of the
9316 | Dalton Trans., 2009, 9312–9321 This journal is © The Royal Society of Chemistry 2009
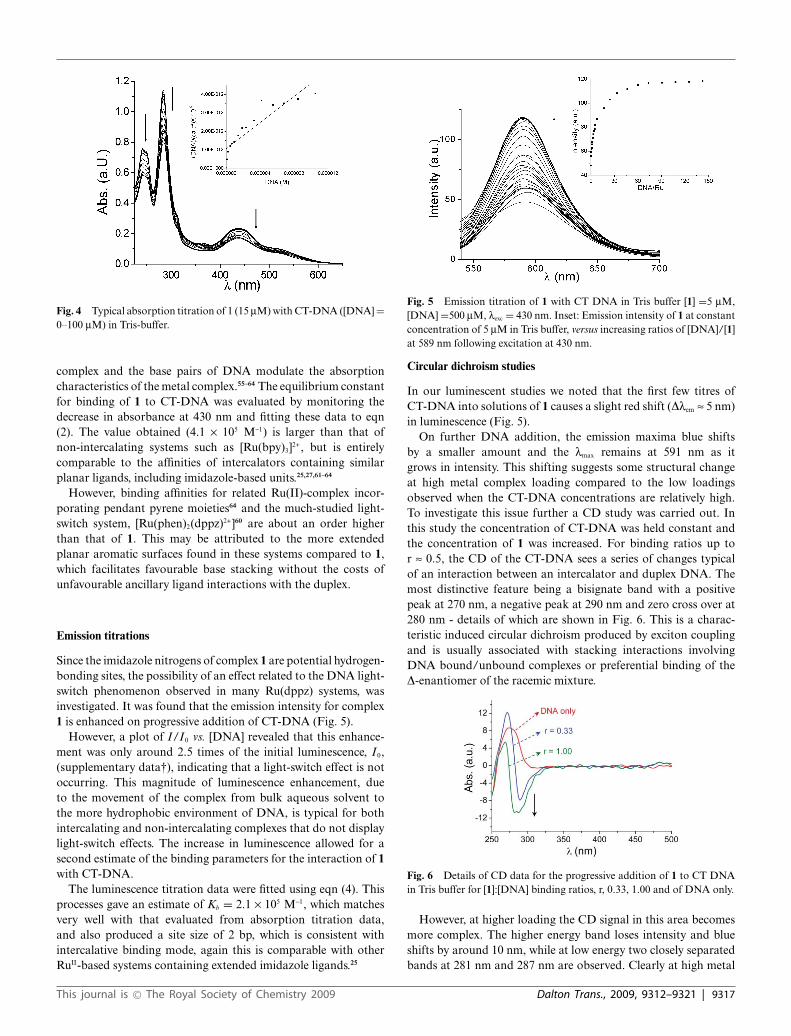
Fig. 4 Typical absorption titration of 1 (15 mM) with CT-DNA ([DNA] =0–100 mM) in Tris-buffer.
complex and the base pairs of DNA modulate the absorptioncharacteristics of the metal complex.55–64 The equilibrium constantfor binding of 1 to CT-DNA was evaluated by monitoring thedecrease in absorbance at 430 nm and fitting these data to eqn(2). The value obtained (4.1 ¥ 105 M-1) is larger than that ofnon-intercalating systems such as [Ru(bpy)3]2+, but is entirelycomparable to the affinities of intercalators containing similarplanar ligands, including imidazole-based units.25,27,61–64
However, binding affinities for related Ru(II)-complex incor-porating pendant pyrene moieties64 and the much-studied light-switch system, [Ru(phen)2(dppz)2+]60 are about an order higherthan that of 1. This may be attributed to the more extendedplanar aromatic surfaces found in these systems compared to 1,which facilitates favourable base stacking without the costs ofunfavourable ancillary ligand interactions with the duplex.
Emission titrations
Since the imidazole nitrogens of complex 1 are potential hydrogen-bonding sites, the possibility of an effect related to the DNA light-switch phenomenon observed in many Ru(dppz) systems, wasinvestigated. It was found that the emission intensity for complex1 is enhanced on progressive addition of CT-DNA (Fig. 5).
However, a plot of I/I 0 vs. [DNA] revealed that this enhance-ment was only around 2.5 times of the initial luminescence, I 0,(supplementary data†), indicating that a light-switch effect is notoccurring. This magnitude of luminescence enhancement, dueto the movement of the complex from bulk aqueous solvent tothe more hydrophobic environment of DNA, is typical for bothintercalating and non-intercalating complexes that do not displaylight-switch effects. The increase in luminescence allowed for asecond estimate of the binding parameters for the interaction of 1with CT-DNA.
The luminescence titration data were fitted using eqn (4). Thisprocesses gave an estimate of Kb = 2.1 ¥ 105 M-1, which matchesvery well with that evaluated from absorption titration data,and also produced a site size of 2 bp, which is consistent withintercalative binding mode, again this is comparable with otherRuII-based systems containing extended imidazole ligands.25
Fig. 5 Emission titration of 1 with CT DNA in Tris buffer [1] =5 mM,[DNA] =500 mM, lexc = 430 nm. Inset: Emission intensity of 1 at constantconcentration of 5 mM in Tris buffer, versus increasing ratios of [DNA]/[1]at 589 nm following excitation at 430 nm.
Circular dichroism studies
In our luminescent studies we noted that the first few titres ofCT-DNA into solutions of 1 causes a slight red shift (Dlem ª 5 nm)in luminescence (Fig. 5).
On further DNA addition, the emission maxima blue shiftsby a smaller amount and the lmax remains at 591 nm as itgrows in intensity. This shifting suggests some structural changeat high metal complex loading compared to the low loadingsobserved when the CT-DNA concentrations are relatively high.To investigate this issue further a CD study was carried out. Inthis study the concentration of CT-DNA was held constant andthe concentration of 1 was increased. For binding ratios up tor ª 0.5, the CD of the CT-DNA sees a series of changes typicalof an interaction between an intercalator and duplex DNA. Themost distinctive feature being a bisignate band with a positivepeak at 270 nm, a negative peak at 290 nm and zero cross over at280 nm - details of which are shown in Fig. 6. This is a charac-teristic induced circular dichroism produced by exciton couplingand is usually associated with stacking interactions involvingDNA bound/unbound complexes or preferential binding of theD-enantiomer of the racemic mixture.
Fig. 6 Details of CD data for the progressive addition of 1 to CT DNAin Tris buffer for [1]:[DNA] binding ratios, r, 0.33, 1.00 and of DNA only.
However, at higher loading the CD signal in this area becomesmore complex. The higher energy band loses intensity and blueshifts by around 10 nm, while at low energy two closely separatedbands at 281 nm and 287 nm are observed. Clearly at high metal
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 9312–9321 | 9317
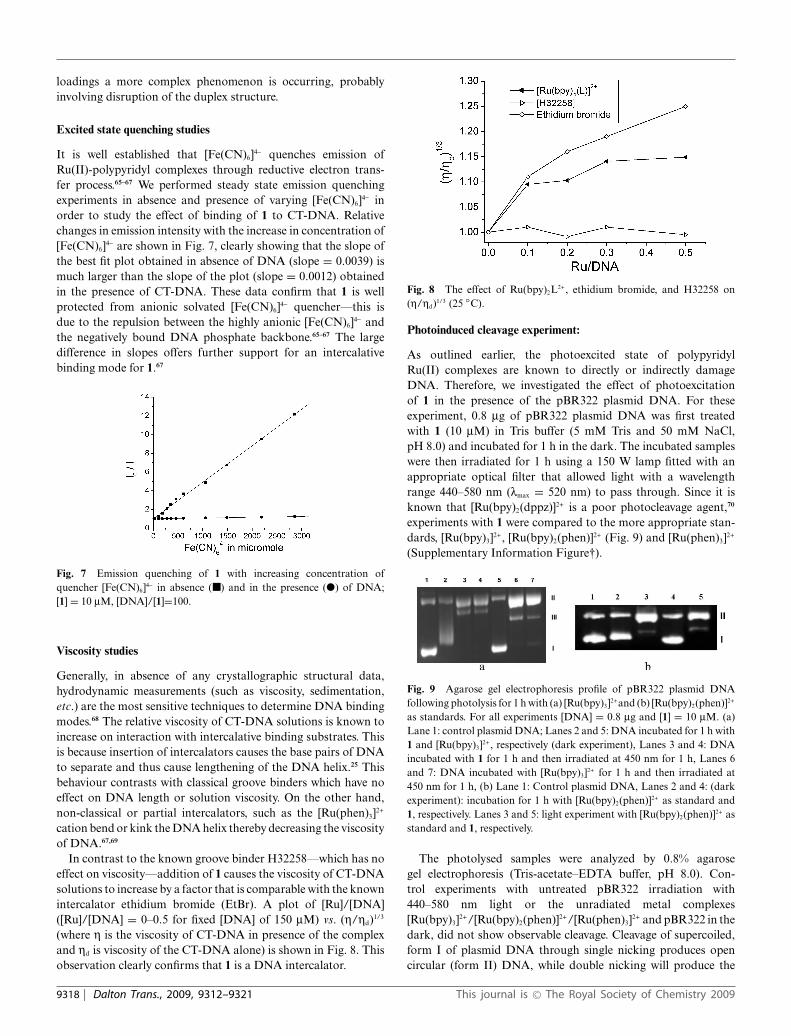
loadings a more complex phenomenon is occurring, probablyinvolving disruption of the duplex structure.
Excited state quenching studies
It is well established that [Fe(CN)6]4- quenches emission ofRu(II)-polypyridyl complexes through reductive electron trans-fer process.65–67 We performed steady state emission quenchingexperiments in absence and presence of varying [Fe(CN)6]4- inorder to study the effect of binding of 1 to CT-DNA. Relativechanges in emission intensity with the increase in concentration of[Fe(CN)6]4- are shown in Fig. 7, clearly showing that the slope ofthe best fit plot obtained in absence of DNA (slope = 0.0039) ismuch larger than the slope of the plot (slope = 0.0012) obtainedin the presence of CT-DNA. These data confirm that 1 is wellprotected from anionic solvated [Fe(CN)6]4- quencher—this isdue to the repulsion between the highly anionic [Fe(CN)6]4- andthe negatively bound DNA phosphate backbone.65–67 The largedifference in slopes offers further support for an intercalativebinding mode for 1.67
Fig. 7 Emission quenching of 1 with increasing concentration ofquencher [Fe(CN)6]4- in absence (�) and in the presence (�) of DNA;[1] = 10 mM, [DNA]/[1]=100.
Viscosity studies
Generally, in absence of any crystallographic structural data,hydrodynamic measurements (such as viscosity, sedimentation,etc.) are the most sensitive techniques to determine DNA bindingmodes.68 The relative viscosity of CT-DNA solutions is known toincrease on interaction with intercalative binding substrates. Thisis because insertion of intercalators causes the base pairs of DNAto separate and thus cause lengthening of the DNA helix.25 Thisbehaviour contrasts with classical groove binders which have noeffect on DNA length or solution viscosity. On the other hand,non-classical or partial intercalators, such as the [Ru(phen)3]2+
cation bend or kink the DNA helix thereby decreasing the viscosityof DNA.67,69
In contrast to the known groove binder H32258—which has noeffect on viscosity—addition of 1 causes the viscosity of CT-DNAsolutions to increase by a factor that is comparable with the knownintercalator ethidium bromide (EtBr). A plot of [Ru]/[DNA]([Ru]/[DNA] = 0–0.5 for fixed [DNA] of 150 mM) vs. (h/hd)1/3
(where h is the viscosity of CT-DNA in presence of the complexand hd is viscosity of the CT-DNA alone) is shown in Fig. 8. Thisobservation clearly confirms that 1 is a DNA intercalator.
Fig. 8 The effect of Ru(bpy)2L2+, ethidium bromide, and H32258 on(h/hd)1/3 (25 ◦C).
Photoinduced cleavage experiment:
As outlined earlier, the photoexcited state of polypyridylRu(II) complexes are known to directly or indirectly damageDNA. Therefore, we investigated the effect of photoexcitationof 1 in the presence of the pBR322 plasmid DNA. For theseexperiment, 0.8 mg of pBR322 plasmid DNA was first treatedwith 1 (10 mM) in Tris buffer (5 mM Tris and 50 mM NaCl,pH 8.0) and incubated for 1 h in the dark. The incubated sampleswere then irradiated for 1 h using a 150 W lamp fitted with anappropriate optical filter that allowed light with a wavelengthrange 440–580 nm (lmax = 520 nm) to pass through. Since it isknown that [Ru(bpy)2(dppz)]2+ is a poor photocleavage agent,70
experiments with 1 were compared to the more appropriate stan-dards, [Ru(bpy)3]2+, [Ru(bpy)2(phen)]2+ (Fig. 9) and [Ru(phen)3]2+
(Supplementary Information Figure†).
Fig. 9 Agarose gel electrophoresis profile of pBR322 plasmid DNAfollowing photolysis for 1 h with (a) [Ru(bpy)3]2+and (b) [Ru(bpy)2(phen)]2+
as standards. For all experiments [DNA] = 0.8 mg and [1] = 10 mM. (a)Lane 1: control plasmid DNA; Lanes 2 and 5: DNA incubated for 1 h with1 and [Ru(bpy)3]2+, respectively (dark experiment), Lanes 3 and 4: DNAincubated with 1 for 1 h and then irradiated at 450 nm for 1 h, Lanes 6and 7: DNA incubated with [Ru(bpy)3]2+ for 1 h and then irradiated at450 nm for 1 h, (b) Lane 1: Control plasmid DNA, Lanes 2 and 4: (darkexperiment): incubation for 1 h with [Ru(bpy)2(phen)]2+ as standard and1, respectively. Lanes 3 and 5: light experiment with [Ru(bpy)2(phen)]2+ asstandard and 1, respectively.
The photolysed samples were analyzed by 0.8% agarosegel electrophoresis (Tris-acetate–EDTA buffer, pH 8.0). Con-trol experiments with untreated pBR322 irradiation with440–580 nm light or the unradiated metal complexes[Ru(bpy)3]2+/[Ru(bpy)2(phen)]2+/[Ru(phen)3]2+ and pBR322 in thedark, did not show observable cleavage. Cleavage of supercoiled,form I of plasmid DNA through single nicking produces opencircular (form II) DNA, while double nicking will produce the
9318 | Dalton Trans., 2009, 9312–9321 This journal is © The Royal Society of Chemistry 2009

linear form (form III). In electrophoresis, form III migrates inbetween form I and form II structures.71 Fig. 9a reveals that1 is more efficient in inducing photo-cleavage of pBR322, than[Ru(bpy)3]2+.
In fact, after irradiation, with 1 the intensity of form IIand III was found to be less than the intensity of formII and III for [Ru(bpy)3]2+. This is due to fragmentation ofpBR322 into small fragments that are not detectable in thegel. This indicates that complex 1 has higher photocleavageactivity than [Ru(bpy)3]2+. The activity of 1 was also comparedto [Ru(bpy)2(phen)]2+ and [Ru(phen)3]2+. In both cases (Fig. 9band supporting information†), complex 1 was found to be moreactive in inflicting cleavage to plasmid DNA. This indicates thatalthough the extended imidazole ligand enhances binding to DNAthrough intercalation, unlike other extended ligand systems suchas dppz, this does not comprise the photocleavage properties ofthe RuII centre.
Since it is well-established that both photogenerated 1O2 and/orOH∑57 can cause DNA cleavage we set out to gain a betterinsight into the cleavage mechanism by probing for evidence ofreactive intermediates being involved in the photocleavage ofpBR322. To detect for the possible presence of these intermediates,cleavage experiments in the presence of respective scavengers werecarried out. DMSO and mannitol are known to scavenge OH∑,whereas sodium azide is an established 1O2 scavenger. Therefore,pBR322 (0.6 mg) was incubated with 1 (10 mM) in the presenceof either DMSO (200 mM)/mannitol (100 mM) or sodium azide(100 mM) for 1 h in the dark, followed by irradiation for 1 h. Theirradiated samples were subjected to gel electrophoresis as shownin Fig. 10. This reveals that photocleavage activity was effectivelysuppressed when experiments were performed in presence ofeither DMSO or mannitol. No such effect was observed whenexperiments were performed in presence of NaN3. These resultsclearly demonstrated that OH∑ has a definite role in the cleavagemechanism involving complex 1. This is a striking result: DNAstrand breakage by polypyridyl ruthenium(II) is usually eitherthrough direct electron transfer processes or indirectly throughsinglet oxygen sensitisation,14,72 although Yam and colleagueshave reported that the [Re(dppn)(CO)3(py)]+ cation cleaves DNAthrough the generation of hydroxyl radicals.73
Fig. 10 Electrophoresis in agarose gel of pBR322 plasmid DNA followingphotolysis for 1 h in presence of different inhibitors. Lane 1: Controlplasmid DNA, lane 2: DNA with 1 in dark, lane 3: DNA with 1 irradiatedfor 1 h, lane 4: DNA with 1 was irradiated for 1 h in presence of mannitol,Lane 5: DNA with 1 was irradiated for 1 h in presence of DMSO, Lane6: DNA with 1 was irradiated for 1 h in presence of NaN3. In all cases[DNA] = 0.8 mg, [1] = 10 mM, [NaN3] = 100 mM, [mannitol] = 100 mM,[DMSO] = 200 mM.
Non-photoinduced cleavage studies
Compared to photocleavage reagents, systems capable of hy-drolysing DNA phosphodiester linkages are much less common.Most DNA hydrolase mimics are based on copper(II) centres74
and there are very few examples of ruthenium(II)-based com-plexes capable of cleavage through hydrolysis.59,75 However, wereasoned that through interaction with DNA, the hydroxyl groupappended to the imidazole based ligand may be brought closeto the phosphodiester linkages and—if it is a sufficiently strongnucleophile—it could facilitate their scission.
To explore its nucleophilic properties, we first treated complex1 with the model system p-nitro phenyl disodium phosphate(p-NPP) salt in Tris buffer (pH = 7.5). We spectrophotometricallymonitored the generation of the p-nitro phenolate ion throughthe increase in its absorbance at 420 nm (lmax for NO2-C6H5-O-
is 420 nm) for 90 minutes. An analysis of the data revealed thatover this period 22% of hydrolyzed product (p-nitro phenolateion) was formed, confirming that complex 1 is capable ofhydrolyzing phosphodiester functionalities. Consequently DNAcleavage studies in the dark using 1 were carried out.
We investigated the effect of treating pBR322 plasmid DNAwith complex 1 in the dark. pBR322 plasmid DNA was incubatedwith complex 1 in the dark for 40 h and was subjected toelectrophoresis in agarose gel along with the control plasmidDNA (untreated pBR322) (Fig. 11a). The result of this experimentclearly reveals conversion of Form I to Form II on incubation withcomplex 1 in the dark. To confirm that cleavage affected by thecomplex 1 in the dark was due to hydrolysis, we carried out furtherstudies involving the enzyme T4 DNA ligase, which is known toexclusively catalyze ligation between 3¢-hydroxy and 5¢-phosphateDNA termini. Open circular plasmid (Form II) obtained from thedark condition cleavage was recovered from agarose gel by phenolchloroform extraction and purified by ethanol precipitation andit was then treated with the T4 DNA ligase. The electrophoresisgel for the product of this process clearly displays evidence of theconversion of Form II to Form I (Fig. 11, Lane 4), confirming that
Fig. 11 (a) Agarose gel electrophoresis profile showing cleavage ofpBR322 plasmid DNA in the dark; Lane 1: 1 Kb ladder, lane 2: Controlplasmid DNA, Lane 3: plasmid DNA incubated in the dark with complex1 (10 mM) for 40 h. In both cases 200 ng of the plasmid DNA was usedfor electrophoresis; (b) Electrophoresis gel profile showing repair of opencircular form (Form II) by T4 DNA ligase; Lane 1: 1 Kb ladder, lane 2:Control plasmid DNA (170 ng)a, Lane 3: Gel purified open circular form(Form II) (45 ng)a obtained from the reaction with complex 1 after anincubation period of 40 h in the dark, Lane 4; Religation of gel purifiedForm II (80 ng)a, produced through reaction with 1, when treated with T4DNA ligase (overnight at 16 ◦C). aActual amount of plasmid loaded forgel electrophoresis.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 9312–9321 | 9319

the hydrolytic cleavage induced by complex 1 in the dark is due tothe hydrolysis of the phosphodiester linkage.
The observed lower activity of 1 towards the hydrolysis of DNAas compared to that of the model system p-NPP is probably dueto the well-known low activity of the phosphodiester functionalityof DNA.
To investigate these initial studies on the possibility of light-freecleavage of DNA, the effect of varying concentration of 1 on thecleavage of pBR322 was then investigated.
Effect of complex loading
A 0.6 mg sample of pBR322 plasmid DNA dissolved in Tris bufferwas incubated in the dark with complex 1 at several differentconcentrations (10, 20, 35, 45 mM) for 1 h at RT. The resultingsamples of pBR322 were then subjected to electrophoresis in 0.8%agarose gel at RT for 4 h at 50 V in TAE buffer. The results areshown in Fig. 12.
Fig. 12 Electrophoresis in agarose gel of pBR322 plasmid DNA incu-bated for 1 h at RT with 1. Lane 5: control plasmid DNA; lanes 1–4: atconcentrations 10, 20, 35 and 45 mM for 1, respectively.
The electrophoretic mobility of plasmid DNA is known tochange in presence of ruthenium(II)-polypyridyl complexes.76–79
Ruthenium(II)-polypyridyl complexes bound to plasmid DNAcause an unwinding of the DNA,77–78 therefore reducing thenumber of supercoils and thus causing a decrease in the rate ofmigration through agarose gel. The control experiment in lane5 shows a major band corresponding to the supercoiled formfor untreated plasmid DNA. However, it is clear that the metalcomplex results in a remarkable decrease in mobility of the formsI and II, so that in lane 2 they almost merge. This indicates that,even in low concentrations, 1 induces unwinding of the DNAsuper helix. More interestingly, when compared to lane 5, it isclear that the ratio of form I: form II has changed, confirmingthe initial study that 1 is capable of DNA hydrolysis. However, ateven higher concentrations ([1] = 35 and 45 mM), it is also clearthat the two bands—form I and form II—again clearly separateas positive supercoiling is induced. At higher complex loadings,there is also a decrease in intensity of both bands. There are twopossible explanations for this observation. It may merely be that atthese higher concentrations, complex 1 competes more efficientlyfor intercalation binding sites compared to the EtBr used to imagethe gel. This seems probable given that 1 is an intercalator andit has a relatively high binding affinity. Alternatively, as indicatedby the previously outlined CD studies, this effect may be due tochanges in the duplex DNA structures at high metal loadings thatinhibits EtBr binding. In all likelihood, a combination of botheffects is occurring.
Conclusions
In summary, we have synthesised and characterised anew [Ru(bpy)2(L)](PF6)2 ((L is 2-methyl-2H-1,3,7,8-tetraaza-cyclopenta[l]phenanthren-2-ol) complex. This luminescent systemcontains a planar coordinated ligand that allows the complexto intercalate into duplex DNA. Binding induced changes inspectral characteristics, melting temperature data, and changesin viscosity confirm an intercalative mode of interaction between1 and DNA. The complex was also found to photocleave theplasmid pBR322 and experimental results confirm that OH∑ isinvolved in the photocleavage reaction. Further studies provideevidence that complex 1 also performs DNA hydrolysis in thedark. Given these properties, analogues of this system could bedeveloped as novel agents in the field of photodynamic therapy,or as probes for nucleic acid structure.
Acknowledgements
AD acknowledges DST and DBT, New Delhi, India for financialsupport. AG thankfully acknowledges CSIR, New Delhi, India fora Sr. Research Fellowship. AD and JAT are grateful for fundingfrom the British Council/DST-UKIERI scheme. The authors alsothank Dr. P. K. Ghosh (Director, CSMCRI) for his keen interestin this work.
Notes and references
1 (a) S. P. Foxon, T. Phillips, M. R. Gill, M. Towrie, A. W. Parker, M.Webb and J. A. Thomas, Angew. Chem., Int. Ed., 2007, 46, 3686; (b) A.Neves, M. Lanznaster, A. J. Bortoluzzi, R. A. Peralta, A. Casellato,E. E. Castellano, P. Herrald, M. J. Riley and G. Schenk, J. Am. Chem.Soc., 2007, 129, 7486; (c) C. Metcalfe and J. A. Thomas, Chem. Soc.Rev., 2003, 32, 215.
2 (a) M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511;(b) B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007,4565.
3 J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611.4 G. Mestroni, E. Alessio, G. Sava, S. Pacor, M. Coluccia and A.
Boccarelli, Met.-Based Drugs, 1994, 1, 41.5 M. Coluccia, G. Sava, F. Loseto, A. Nassi, A. Boccarelli, D. Giordano,
E. Alessio and G. Mestroni, Eur. J. Cancer, 1993, 29, 1873.6 A. Anagnostopoulou, E. Moldrheim, N. Katsaros and E. Sletten,
JBIC, J. Biol. Inorg. Chem., 1999, 4, 199.7 J. D. Aguirre, D. A. Lutterman, A. M. Angeles-Boza, K. R. Dunbar
and C. Turro, Inorg. Chem., 2007, 46, 7494.8 A. K. Patra, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2005,
2798.9 K. E. Erkkila, D. T. Odom and J. K Barton, Chem. Rev., 1999, 99, 2777.
10 R. Tamilarasan and D. R. McMillin, Inorg. Chem., 1990, 29, 2798.11 R. J. Morgan, S. Chatterjee, A. D. Baker and T. C. Strekas, Inorg.
Chem., 1991, 30, 2687.12 B. Norden and F. Tjerneld, FEBS Lett., 1976, 67, 368.13 (a) M. Cusumano, M. Letizia Di Pietro and A. Giannetto, Inorg. Chem.,
2006, 45, 230; (b) Y. An, M. L. Tong, L. N Ji and Z. W. Mao, DaltonTrans., 2006, 2066; (c) T. J. Meade and J. F. Kayyem, Angew. Chem.,1995, 107, 358.
14 B. Armitage, Chem. Rev., 1998, 98, 1171.15 Q. Jiang, N. Xiao, P. Shi, Y. Zhu and Z. Guo, Coord. Chem. Rev., 2007,
251, 1951.16 (a) N. H. Williams, B. Takasaki, M. Wall and J. Chin, Acc. Chem. Res.,
1999, 32, 485; (b) M. W. Grinstaff, Angew. Chem., Int. Ed., 1999, 38,3629.
17 J. A. Cowan, Curr. Opin. Chem. Biol., 2001, 5, 634.18 F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Chem. Commun.,
2005, 2540.19 J. P. Lecomte, A. K. de Mesmaeker and J. M. Kelly, Bull. Soc. Chim.
Belg., 1994, 103, 193.
9320 | Dalton Trans., 2009, 9312–9321 This journal is © The Royal Society of Chemistry 2009

20 L. F. Povirk, S. Neidle and M. J. Waring, Molecular Aspects of Anti-cancer Drug Action, 1983, 157.
21 L. F. Povirk, Mutat. Res., 1991, 257, 127.22 F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Chem. Commun.,
2005, 2540.23 K. Kalyansundaram, Photochemistry of Polypyridine and Porphyrin
Complexes, 1992, Academic Press, London.24 N. J. Turro, J. K. Barton and A. T. Donald, Acc. Chem. Res., 1991, 24,
332.25 L. N. Ji, X. H. Zou and J. G. Liu, Coord. Chem. Rev., 2001, 216–217,
513.26 (a) H. Xu, K. C. Zheng, Y. Chen, Y. Z. Li, L. J. Lin, H. Li, P. X. Zhang
and L. N. Ji, Dalton Trans., 2003, 2260; (b) A. M. Pyle, J. P. Rehmann,R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem.Soc., 1989, 111, 3051; (c) B. M. Goldstein, J. K. Barton and H. M.Berman, Inorg. Chem., 1986, 25, 842.
27 Y. Xiong and L. N. Ji, Coord. Chem. Rev., 1999, 185–186, 711.28 A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von
Zelewsky, Coord. Chem. Rev., 1988, 84, 85.29 V. Balzani, A. Juris and M. Venturi, Chem. Rev., 1996, 96, 759 and
references therein.30 M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem.
Soc., 1954, 76, 3047.31 E. Amouyal, A. Homsi, J. C. Chambron and J. P. Sauvage, J. Chem.
Soc., Dalton Trans., 1990, 1841.32 J. Bolger, A. Gourdon, E. Ishow and J. P. Launay, Inorg. Chem., 1996,
35, 2937.33 R. Nasielski-Hinkens, M. Benedek-Vamos, D. Maetens and
J. Nasielski, J. Organomet. Chem., 1981, 217, 179.34 J. M. Calvert, J. V. Caspar, R. A. Binstead, T. D. Westmoreland and
T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 6620.35 SAINT+, 6.02 ed., Bruker AXS, Madison, WI, 1999.36 G. M. Sheldrick, SHELXTL Reference Manual, version 5.1.37 Bruker AXS: Madison, WI, 1997.38 G. M. Sheldrick, SHELXL-97: Program for Crystal Structure Refine-
ment, University of Gottingen, Gottingen, Germany, 1997.39 Mercury 1.4.1 Supplied with Cambridge Structural Database, Copy-
right CCDC, 2005-2006.40 A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392.41 J. Marmur and P. Doty, J. Mol. Biol., 1962, 5, 109.42 G. K. Suryaprakash, T. Mathew, C. Panja, H. Vaghoo, K. Venkatara-
man and G. A. Olah, Org. Lett., 2007, 9, 179.43 M. Biner, H. B. Burgi, A. Ludi and C. Rohr, J. Am. Chem. Soc., 1992,
114, 5197.44 J. Breu, W. Seidl, D. Huttner and F. Kraus, Chem.–Eur. J., 2002, 8,
4454.45 J. Breu, C. Kratzer and H. Yersin, J. Am. Chem. Soc., 2000, 122,
2548.46 D. P. Rillema, D. S. Jones, C. Woods and H. A. Levy, Inorg. Chem.,
1992, 31, 2935.47 P. VanderSluis and A. L. Spek, Acta Crystallogr., Sect. A: Found.
Crystallogr., 1990, 46, 194.48 A. D. Jose, P. Kar, D. Koley, B. Ganguly, W. Thiel, G. Ramakrishna,
D. K. Palit, H. N. Ghosh and A. Das, Inorg. Chem., 2007, 46, 5576.49 X. H. Zou, B. H. Ye, J. G. Liu, Y. Xiong and L. N. Ji, J. Chem. Soc.,
Dalton Trans., 1999, 19.50 D. P. Rillema, G. Allen, T. J. Meyer and D. C. Conrad, Inorg. Chem.,
1983, 22, 1617.51 X. H. Zou, B. H. Ye, H. Li, Q. L. Zhang, H. Chao, J. G. Liu, L. N. Ji
and X. Y. Li, JBIC, J. Biol. Inorg. Chem., 2001, 6, 143.
52 X. W. Liu, J. Li, H. Li, K. C. Zheng, H. Chao and L. N. Ji, J. Inorg.Biochem., 2005, 99, 2372.
53 D. L. Nelson and M. M. Cox, Lehninger, Principles of Biochemistry,W. H. Freeman, and Company, New York, 2005, Fourth Edition.
54 (a) J. M. Kelly, A. B. Tossi, D. J. McConnell and C. OhUigin, NucleicAcids Res., 1985, 13, 6017; (b) R. B. Lopez, B. L. Loen, T. Boussie andT. J. Meyer, Tetrahedron Lett., 1996, 37, 5437; (c) P. U. Maheswari andM. Palaniandavar, J. Inorg. Biochem., 2004, 98, 219.
55 B. Norden, P. Lincoln, B. Akerman and E. Tuite, Met. Ions Biol. Syst.,1996, 33, 177.
56 C. S. Chow and J. K. Barton, Methods Enzymol., 1992, 212, 219.57 M. Mariappan and B. G. Maiya, Eur. J. Inorg. Chem., 2005, 2164.58 E. Yavin Stemp, L. Weiner, I. Sagi, R. A. Yellin and A. Shanzer, J. Inorg.
Biochem., 2004, 98, 1750.59 M. S. Deshpande, A. A. Kumbhar and A. S. Kumbhar, Inorg. Chem.,
2007, 46, 5450.60 R. M. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919.61 A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K.
Barton, J. Am. Chem. Soc., 1990, 112, 4960.62 J. G. Liu, B. H. Ye, H. Li, L. N. Ji, R. H. Li and J. Y. Zhou, J. Inorg.
Biochem., 1999, 73, 117.63 E. Ruba, J. R. Hart and J. K. Barton, Inorg. Chem., 2004, 43, 4570.64 (a) Q. X. Zhen, B. H. Ye, Q. L. Zhang, J. G. Liu, H. Li, L. N. Ji and
L. Wang, J. Inorg. Biochem., 1999, 76, 47; (b) T. Ghosh, B. G. Maiya,A. Samanta, A. D. Shukla, D. A. Jose, D. Krishna Kumar and A. Das,JBIC, J. Biol. Inorg. Chem., 2005, 10, 496.
65 F. Liu, K. Wang, G. Bai, Y. Zhang and L. Gao, Inorg. Chem., 2004, 43,1799.
66 (a) C. V. Kumar, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1985,107, 5518; (b) A. D. Shukla, H. C. Bajaj and A. Das, Angew. Chem.,Int. Ed., 2001, 40, 446 and references therein.
67 Y. J. Liu, H. Chao, L. F. Tan, Y. X. Yuan, W. Wei and L. N. Ji, J. Inorg.BioChem., 2005, 99.
68 S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry,1992, 31, 9319.
69 H. Deng, H. Xu, Y. Yang, H. Li, H. Zou, L. H. Qu and L. N. Ji, J. Inorg.Biochem., 2003, 97, 207.
70 C. Sentagne, J.-C. Chambron, J.-P. Sauvage and N. Paillous, J. Pho-tochem. Photobiol., B, 1994, 26, 165.
71 J. K. Barton and A. L. Raphael, J. Am. Chem. Soc., 1984, 106, 2466.72 A. Moucheron, K. -De Mesmaeker and J. M. Kelly, J. Photochem.
Photobiol., B, 1997, 40, 91.73 V. W.-W. Yam, K. K.-W. Lo, K.-K. Cheung and R. Y.-C. Kong, J. Chem.
Soc., Dalton Trans., 1997, 2067.74 (a) J. He, P. Hu, Y.-J. Wang, M.-L. Tong, H. Sun, Z.-W. Mao and L.-N.
Ji, Dalton Trans., 2008, 3207; (b) T. Gupta, S. Dhar, M. Nethaji andA. R. Chakravarty, Dalton Trans., 2004, 1896; (c) A. Sreedhara, J. D.Freed and J. A. Cowan, J. Am. Chem. Soc., 2000, 122, 8814.
75 L. A. Basile, A. L. Raphael and J. K. Barton, J. Am. Chem. Soc., 1987,109, 7550.
76 Q. L. Zhang, J. H. Liu, J. Z. Liu, P. X. Zhang, X. Z. Ren, Y. Liu, Y.Huang and L. N. Ji, J. Inorg. Biochem., 2004, 98, 1405.
77 G. L. Cohen, W. R. Bauer, J. K. Barton and S. J. Lippard, Science,1979, 203, 1014.
78 (a) S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153; (b) S. P.Foxon, C. Metcalfe, H. Adams, M. Webb and J. A. Thomas, Inorg.Chem., 2007, 46, 409.
79 (a) J. Sambrook, D. W. Russell, Molecular Cloning, 2001, A laboratorymanual, 3rd edition, Cold spring harbor laboratory press, NY; (b) J. G.Sutcliffe, Cold Spring Harbpr Symp Quant Biol, 1979, 43, 77.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 9312–9321 | 9321
Related Documents











