Divergent Synthesis of 1H-Indazoles and 1H-Pyrazoles from Hydrazones via Iodine-Mediated Intramolecular Aryl and sp 3 C–H Amination Wei Wei, a Zhen Wang, a Xikang Yang, a Wenquan Yu, a, * and Junbiao Chang a, * a College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, Henan Province 450001, People)s Republic of China Fax: (+ 86)-(0)371-6778-1588; phone: (+ 86)-(0)371-6778-1788; e-mail: [email protected] or [email protected] Received: June 27, 2017; Revised: July 28, 2017; Published online: September 25, 2017 Supporting information for this article is available under https://doi.org/10.1002/adsc.201700824. Abstract: A divergent intramolecular C–H amination of hydrazones has been developed employing molec- ular iodine (I 2 ) as the sole oxidant. The required hy- drazone substrates were readily obtained by conden- sation of hydrazines with the corresponding ketones. In the presence of potassium iodide, I 2 -mediated oxi- dative cyclization of diaryl and tert-butyl aryl ketone hydrazones produced 1H-indazoles via direct aryl C– H amination. Under similar reaction conditions, pri- mary and secondary alkyl ketone hydrazones were transformed into 1H-pyrazole products in a reaction involving sp 3 C–H amination. This synthetic method- ology does not involve transition metals, and is op- erationally simple, providing a facile access to inda- zole and pyrazole derivatives in an efficient and scal- able fashion. Keywords: C–H amination; hydrazones; 1H-inda- zoles; iodine; 1H-pyrazoles Introduction Nitrogen-containing heterocyclic frameworks such as indazoles and pyrazoles which contain two adjacent nitrogen atoms, are found in many compounds with diverse biological and pharmaceutical properties, [1] in- cluding anti-inflammatory [2] and antimicrobial drugs, [3] kinase inhibitors, [4] and anti-HIV agents. [5] Derivatives of these N-heterocyclic compounds also find wide- spread applications in agricultural chemistry [6] and material science [7] and, consequently, considerable re- search efforts have been devoted to their synthe- sis. [1b–e,8] Recently, several approaches involving intra- molecular C–H amination of hydrazone precursors have been developed for the synthesis of indazoles or pyrazoles through catalyzed aerobic oxidation [9] or using hypervalent iodine reagents. [10] These are ele- gant methods, but they have drawbacks, such as limit- ed substrate scopes or low yields due to the relative instability of the hydrazone substrates. Thus it is im- portant to develop simpler and more efficient ap- proaches, especially those that could lead to either of these heterocyclic frameworks. Carbon-nitrogen (C–N) bond formation is one of the most fundamental and important reactions in or- ganic synthesis. In earlier decades, transition metal- catalyzed coupling reactions, especially Buchwald– Hartwig amination, [11] have become powerful and reli- able tools for C À N bond formation and have been used to synthesize various alkaloids. Built on these achievements, C À N bond formation though direct C– H functionalization [12] has received considerable at- tention in recent years owing to its advantages, such as the rich source of hydrocarbons, high step efficien- cy and atom economy. Previously, we had established several C–H amination reactions employing molecu- lar iodine as the sole oxidant under transition metal- free conditions to produce heterocycles, such as pyra- zoles, [13] triazolopyridines, [14] and benzimidazoles. [15] As a continuation of this work, we describe in this paper the I 2 -mediated cyclization reactions of readily accessible hydrazones for the synthesis of 1H-inda- zoles and 1H-pyrazoles via intramolecular aryl and sp 3 C–H amination reactions, respectively. Results and Discussion The required hydrazone substrates can be readily ob- tained by the condensation of hydrazines with the cor- Adv. Synth. Catal. 2017, 359, 3378 – 3387 # 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 3378 FULL PAPERS DOI: 10.1002/adsc.201700824

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Divergent Synthesis of 1H-Indazoles and 1H-Pyrazoles fromHydrazones via Iodine-Mediated Intramolecular Aryl andsp3 C–H Amination
Wei Wei,a Zhen Wang,a Xikang Yang,a Wenquan Yu,a,* and Junbiao Changa,*a College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, Henan Province 450001, PeopleQs
Republic of ChinaFax: (++86)-(0)371-6778-1588; phone: (++86)-(0)371-6778-1788; e-mail: [email protected] [email protected]
Received: June 27, 2017; Revised: July 28, 2017; Published online: September 25, 2017
Supporting information for this article is available under https://doi.org/10.1002/adsc.201700824.
Abstract: A divergent intramolecular C–H aminationof hydrazones has been developed employing molec-ular iodine (I2) as the sole oxidant. The required hy-drazone substrates were readily obtained by conden-sation of hydrazines with the corresponding ketones.In the presence of potassium iodide, I2-mediated oxi-dative cyclization of diaryl and tert-butyl aryl ketonehydrazones produced 1H-indazoles via direct aryl C–H amination. Under similar reaction conditions, pri-mary and secondary alkyl ketone hydrazones were
transformed into 1H-pyrazole products in a reactioninvolving sp3 C–H amination. This synthetic method-ology does not involve transition metals, and is op-erationally simple, providing a facile access to inda-zole and pyrazole derivatives in an efficient and scal-able fashion.
Keywords: C–H amination; hydrazones; 1H-inda-zoles; iodine; 1H-pyrazoles
Introduction
Nitrogen-containing heterocyclic frameworks such asindazoles and pyrazoles which contain two adjacentnitrogen atoms, are found in many compounds withdiverse biological and pharmaceutical properties,[1] in-cluding anti-inflammatory[2] and antimicrobial drugs,[3]
kinase inhibitors,[4] and anti-HIV agents.[5] Derivativesof these N-heterocyclic compounds also find wide-spread applications in agricultural chemistry[6] andmaterial science[7] and, consequently, considerable re-search efforts have been devoted to their synthe-sis.[1b–e,8] Recently, several approaches involving intra-molecular C–H amination of hydrazone precursorshave been developed for the synthesis of indazoles orpyrazoles through catalyzed aerobic oxidation[9] orusing hypervalent iodine reagents.[10] These are ele-gant methods, but they have drawbacks, such as limit-ed substrate scopes or low yields due to the relativeinstability of the hydrazone substrates. Thus it is im-portant to develop simpler and more efficient ap-proaches, especially those that could lead to either ofthese heterocyclic frameworks.
Carbon-nitrogen (C–N) bond formation is one ofthe most fundamental and important reactions in or-
ganic synthesis. In earlier decades, transition metal-catalyzed coupling reactions, especially Buchwald–Hartwig amination,[11] have become powerful and reli-able tools for C@N bond formation and have beenused to synthesize various alkaloids. Built on theseachievements, C@N bond formation though direct C–H functionalization[12] has received considerable at-tention in recent years owing to its advantages, suchas the rich source of hydrocarbons, high step efficien-cy and atom economy. Previously, we had establishedseveral C–H amination reactions employing molecu-lar iodine as the sole oxidant under transition metal-free conditions to produce heterocycles, such as pyra-zoles,[13] triazolopyridines,[14] and benzimidazoles.[15]
As a continuation of this work, we describe in thispaper the I2-mediated cyclization reactions of readilyaccessible hydrazones for the synthesis of 1H-inda-zoles and 1H-pyrazoles via intramolecular aryl andsp3 C–H amination reactions, respectively.
Results and Discussion
The required hydrazone substrates can be readily ob-tained by the condensation of hydrazines with the cor-
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3378
FULL PAPERS DOI: 10.1002/adsc.201700824
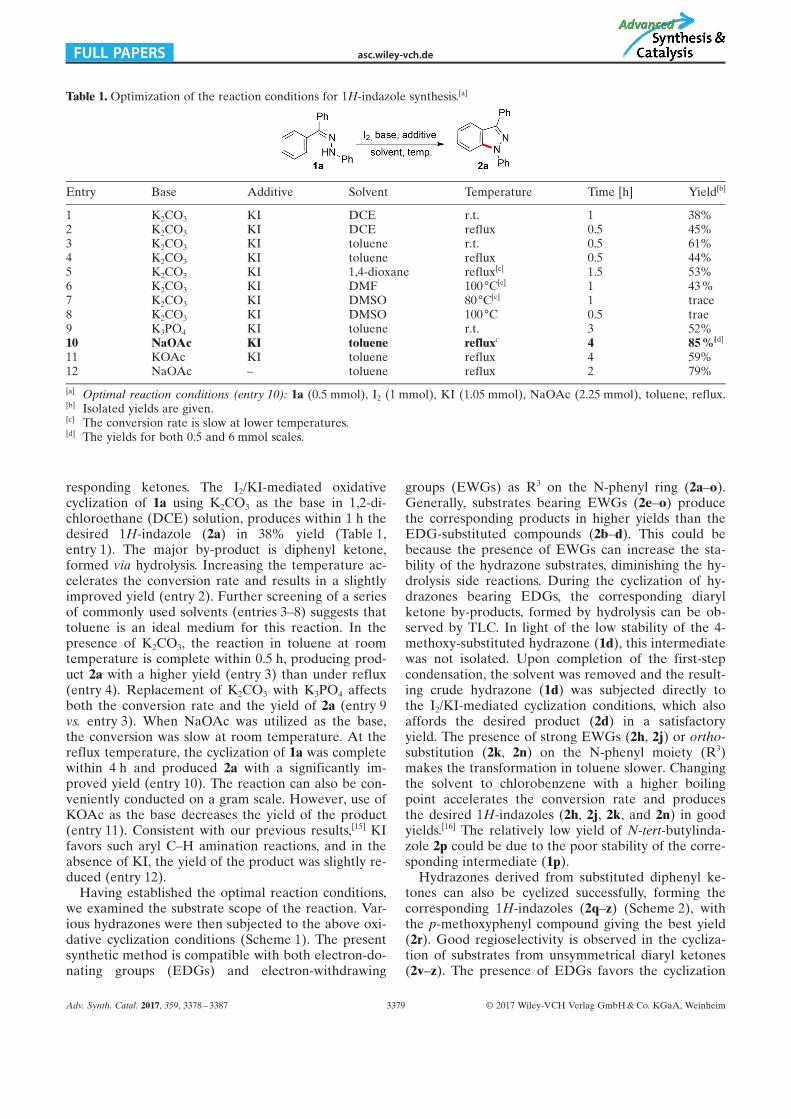
responding ketones. The I2/KI-mediated oxidativecyclization of 1a using K2CO3 as the base in 1,2-di-chloroethane (DCE) solution, produces within 1 h thedesired 1H-indazole (2a) in 38% yield (Table 1,entry 1). The major by-product is diphenyl ketone,formed via hydrolysis. Increasing the temperature ac-celerates the conversion rate and results in a slightlyimproved yield (entry 2). Further screening of a seriesof commonly used solvents (entries 3–8) suggests thattoluene is an ideal medium for this reaction. In thepresence of K2CO3, the reaction in toluene at roomtemperature is complete within 0.5 h, producing prod-uct 2a with a higher yield (entry 3) than under reflux(entry 4). Replacement of K2CO3 with K3PO4 affectsboth the conversion rate and the yield of 2a (entry 9vs. entry 3). When NaOAc was utilized as the base,the conversion was slow at room temperature. At thereflux temperature, the cyclization of 1a was completewithin 4 h and produced 2a with a significantly im-proved yield (entry 10). The reaction can also be con-veniently conducted on a gram scale. However, use ofKOAc as the base decreases the yield of the product(entry 11). Consistent with our previous results,[15] KIfavors such aryl C–H amination reactions, and in theabsence of KI, the yield of the product was slightly re-duced (entry 12).
Having established the optimal reaction conditions,we examined the substrate scope of the reaction. Var-ious hydrazones were then subjected to the above oxi-dative cyclization conditions (Scheme 1). The presentsynthetic method is compatible with both electron-do-nating groups (EDGs) and electron-withdrawing
groups (EWGs) as R3 on the N-phenyl ring (2a–o).Generally, substrates bearing EWGs (2e–o) producethe corresponding products in higher yields than theEDG-substituted compounds (2b–d). This could bebecause the presence of EWGs can increase the sta-bility of the hydrazone substrates, diminishing the hy-drolysis side reactions. During the cyclization of hy-drazones bearing EDGs, the corresponding diarylketone by-products, formed by hydrolysis can be ob-served by TLC. In light of the low stability of the 4-methoxy-substituted hydrazone (1d), this intermediatewas not isolated. Upon completion of the first-stepcondensation, the solvent was removed and the result-ing crude hydrazone (1d) was subjected directly tothe I2/KI-mediated cyclization conditions, which alsoaffords the desired product (2d) in a satisfactoryyield. The presence of strong EWGs (2h, 2j) or ortho-substitution (2k, 2n) on the N-phenyl moiety (R3)makes the transformation in toluene slower. Changingthe solvent to chlorobenzene with a higher boilingpoint accelerates the conversion rate and producesthe desired 1H-indazoles (2h, 2j, 2k, and 2n) in goodyields.[16] The relatively low yield of N-tert-butylinda-zole 2p could be due to the poor stability of the corre-sponding intermediate (1p).
Hydrazones derived from substituted diphenyl ke-tones can also be cyclized successfully, forming thecorresponding 1H-indazoles (2q–z) (Scheme 2), withthe p-methoxyphenyl compound giving the best yield(2r). Good regioselectivity is observed in the cycliza-tion of substrates from unsymmetrical diaryl ketones(2v–z). The presence of EDGs favors the cyclization
Table 1. Optimization of the reaction conditions for 1H-indazole synthesis.[a]
Entry Base Additive Solvent Temperature Time [h] Yield[b]
1 K2CO3 KI DCE r.t. 1 38%2 K2CO3 KI DCE reflux 0.5 45%3 K2CO3 KI toluene r.t. 0.5 61%4 K2CO3 KI toluene reflux 0.5 44%5 K2CO3 KI 1,4-dioxane reflux[c] 1.5 53%6 K2CO3 KI DMF 100 88C[c] 1 43 %7 K2CO3 KI DMSO 80 88C[c] 1 trace8 K2CO3 KI DMSO 100 88C 0.5 trae9 K3PO4 KI toluene r.t. 3 52%10 NaOAc KI toluene refluxc 4 85 %[d]
11 KOAc KI toluene reflux 4 59%12 NaOAc – toluene reflux 2 79%
[a] Optimal reaction conditions (entry 10): 1a (0.5 mmol), I2 (1 mmol), KI (1.05 mmol), NaOAc (2.25 mmol), toluene, reflux.[b] Isolated yields are given.[c] The conversion rate is slow at lower temperatures.[d] The yields for both 0.5 and 6 mmol scales.
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3379
FULL PAPERS asc.wiley-vch.de

process, with methoxy substitution giving the best se-lectivity (2w).
Encouraged by the successful cyclization of diarylketone hydrazones, we continued to explore the sub-strate scope, replacing the aromatic R2 group with ali-phatic groups. Considering that EWGs can increasethe stability of hydrazones and the para-nitrophenyl(PNP) group can be removed[17] for further derivatiza-tion, 4-nitrophenylhydrazine was chosen for substratepreparation. tert-Butyl ketone hydrazone is smoothlyconverted into the expected 1H-indazole (2aa)(Scheme 2) in excellent yield under the optimum cy-clization conditions, but the reaction of methyl ketone
hydrazone 1ab fails to produce the desired product(2ab). This could be due to side reactions caused bythe methyl group of the substrate in the presence ofiodine.[18] For the substrates derived from primary andsecondary alkyl aryl ketones, the corresponding 1H-pyrazole products (3ac–ah) (Table 2) instead wereformed in moderate to good yields in place of the in-dazoles. This reaction involves sp3 C–H amination(see Scheme 3B). The relatively lower yields for pyra-
Scheme 1. Substrate scope of the R3 group for indazole syn-thesis. Optimal reaction conditions: 1 (0.5 mmol), I2
(1 mmol), KI (1.05 mmol), NaOAc (2.25 mmol), toluene,reflux (isolated yields are given).
Table 2. Substrate scope of the R2 group for pyrazole syn-thesis.[a]
[a] Optimal reaction conditions: 1 (0.5 mmol), I2 (1 mmol),NaOAc (2.25 mmol), toluene, reflux.
[b] Isolated yields are given.[c] The reaction resulted in the dihydropyrazole G as the
major product which was further converted into 3ac byDDQ (see the Experimental Section).
[d] Chlorobenzene was used as the solvent.
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3380
FULL PAPERS asc.wiley-vch.de

zole synthesis are due to the formation of the corre-sponding ketones and some other by-products asshown by TLC analysis. Parallel experiments (e.g., forthe synthesis of 3ag and 3ah) indicated that the yieldsare not altered with or without KI. The structure ofpyrazole 3ae was confirmed by X-ray crystallogra-phy.[19] The isobutyl-containing substrate (1ah) wasconverted into a 4,5-dimethylpyrazole product (3ah)via a 1,2-methyl migration process. A plausible mech-anism for this transformation is proposed inScheme 3C.
Tentative mechanisms for these I2-mediated C–Hamination reactions are proposed in Scheme 3. Takingthe cyclization of hydrazone 1a as an example, thebase-promoted iodination of 1a first forms an iodideA. The C@I bond in A is then cleaved with concomi-tant electrocyclic ring closure to give intermediate B.Finally, subsequent deprotonation by base affords the1H-indazole framework 2a. In the case of the hydra-zone 1ac, the resulting iodide intermediate C may un-dergo b-elimination to produce, after tautomerization,a vinylhydrazone E. Intramolecular cycloadditon of Eresults in dihydropyrazoles F and G. The intermediate
G has been isolated and characterized by 1H NMRand mass spectra (see the Experimental Section). Fur-ther oxidative aromatization of the dihydropyra-zole(s) affords the pyrazole product 3ad. For the reac-tion of the isobutyl substrate (1ah), further iodinationof the dihydropyrazole H gives an iodide I. Then,cleavage of the C@I bond in intermediate I triggersthe 1,2-methyl shift to generate compound J. Subse-quent deprotonation of J by base leads to the 4,5-di-methylpyrazole 3ah.
Conclusions
In summary, the intramolecular C–H amination reac-tions of hydrazones employing molecular iodine asthe sole oxidant have been investigated. Under theoptimal reaction conditions, diaryl and tert-butyl arylketone hydrazones are successfully cyclized into 1H-indazoles by direct aryl C–H amination. Oxidativecyclization of primary and secondary alkyl ketonesubstrates affords the corresponding 1H-pyrazoleproducts in a process involving sp3 C–H amination.
Scheme 2. Substrate scope of R1 and R2 groups for indazole synthesis. Optimal reaction conditions: 1 (0.5 mmol), I2
(1 mmol), KI (1.05 mmol), NaOAc (2.25 mmol), toluene, reflux (isolated yields are given).
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3381
FULL PAPERS asc.wiley-vch.de

This transition metal-free synthetic approach to inda-zoles and pyrazoles is operationally simple and broad-ly applicable to a variety of easily accessible hydra-zone substrates. The reaction can be convenientlyconducted on a gram scale.
Experimental Section
General Information1H and 13C NMR spectra were recorded on a 400 MHz(100 MHz for 13C NMR) spectrometer. Chemical shiftvalues are given in ppm (parts per million) with tetramethyl-silane (TMS) as an internal standard. The peak patterns areindicated as follows: s, singlet; d, doublet; t, triplet; q, quar-tet; m, multiplet; dd, doublet of doublets, td, triplet of dou-blets. The coupling constants (J) are reported in Hertz (Hz).Melting points were determined on a micromelting point ap-paratus without correction. High-resolution mass spectra(HR-MS) were obtained on a Q-TOF Mass Spectrometerequipped with an electrospray ion source (ESI), operated inthe positive mode. Flash column chromatography was per-formed over silica gel 200–300 mesh, and the eluent was amixture of CH2Cl2/petroleum ether (PE) or EtOAc/PE.
General Procedure A for the Preparation ofHydrazone Substrates 1
A mixture of a ketone (3 mmol), the corresponding hydra-zine (3.6 mmol, or 4.5 mmol of hydrazine hydrochloride),and AcOH (174 mL, 3 mmol; or 295 mg, 3.6 mmol of
NaOAc) in EtOH (5 mL) was heated to reflux until TLC in-dicated the disappearance of the ketone (12–24 h). Then thesolvent was removed under the reduced pressure, and theresulting residue was purified through silica gel columnchromatography to provide the hydrazone substrate 1.
General Procedure B for the Synthesis of 1H-Indazoles 2
A mixture of I2 (254 mg, 1 mmol) and KI (174 mg,1.05 mmol) in toluene (5 mL; chlorobenzene was used forthe synthesis of 2h, 2j–k, 2n, 2aa) was stirred at room tem-perature for 10 min, then treated with the hydrazone sub-strate 1 (0.5 mmol) and NaOAc (185 mg, 2.25 mmol) in se-quence, and finally heated to reflux. If the reaction was notcomplete within 4 h with more than 1/4 of substrate 1 re-maining (monitored by TLC), another portion of iodine (0.5or 1 mmol, with 1 or 2 mmol of NaOAc) was added. Uponcompletion of the reaction, it was quenched with 5%Na2S2O3 (10 mL), and then extracted with CH2Cl2 (4X10 mL). The combined organic layer was washed with brine(10 mL), dried over anhydrous Na2SO4, and concentrated.The residue was purified by column chromatographythrough silica gel to afford the 1H-indazole 2.
1,3-Diphenyl-1H-indazole (2a): For 0.5 mmol scale: 4 h[1 mmol of I2 was used; yield: 115 mg (85%)]; for 6 mmolscale: 6 h [12 mmol of I2 was used; yield: 1.385 g (85%)];eluent: CH2Cl2/petroleum ether (PE) 15:85; white solid; mp96–98 88C (lit.[20] mp 95–98 88C); 1H NMR (400 MHz, CDCl3):d= 8.08 (d, J= 8.0 Hz, 1 H), 8.04 (d, J= 7.2 Hz, 2 H), 7.79 (t,J=8.0 Hz, 3 H), 7.54 (dd, J=15.2, 7.6 Hz, 4 H), 7.44 (dd, J=15.6, 8.0 Hz, 2 H), 7.36 (t, J=7.6 Hz, 1 H), 7.28 (t, J= 7.6 Hz,1 H); 13C NMR (100 MHz, CDCl3): d =146.1, 140.4, 140.2,
Scheme 3. Proposed mechanisms for the I2-mediated C–H amination reactions.
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3382
FULL PAPERS asc.wiley-vch.de

133.3, 129.5, 128.9, 128.3, 127.8, 127.1, 126.7, 123.2, 123.0,122.0, 121.6, 110.7; HR-MS: m/z= 271.1231 [M++H]++, calcd.for C19H15N2 : 271.1230.
3-Phenyl-1-(p-tolyl)-1H-indazole (2b): 2 h (1 mmol of I2);eluent: CH2Cl2/PE 20:80; yield: 125 mg (88%); white solid;mp 92–93 88C (lit.[9d] mp 88–90 88C); 1H NMR (400 MHz,CDCl3): d =8.07 (d, J=8.4 Hz, 1 H), 8.05–8.03 (m, 2 H), 7.73(d, J=8.4 Hz, 1 H), 7.66 (d, J=8.4 Hz, 2 H), 7.52 (t, J=7.2 Hz, 2 H), 7.45–7.40 (m, 2 H), 7.34 (d, J= 8.4 Hz, 2 H),7.27 (t, J= 7.6 Hz, 1 H), 2.43 (s, 3 H); 13C NMR (100 MHz,CDCl3): d=145.8, 140.4, 137.7, 136.6, 133.4, 130.0, 128.8,128.2, 127.8, 127.0, 123.1, 123.0, 121.8, 121.6, 110.7, 21.1;HR-MS: m/z= 285.1388 [M++H]++, calcd. for C20H17N2 :285.1386.
1-(4-Isopropylphenyl)-3-phenyl-1H-indazole (2c): 2 h(1 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 129 mg(83%); colorless oil ; 1H NMR (400 MHz, CDCl3): d= 8.08(d, J=8.4 Hz, 1 H), 8.04 (d, J= 8.4 Hz, 2 H), 7.75 (d, J=8.4 Hz, 1 H), 7.69 (d, J=8.4 Hz, 2 H), 7.52 (t, J= 7.6 Hz,2 H), 7.46–7.39 (m, 4 H), 7.27 (t, J= 8.0 Hz, 1 H), 3.05–2.95(m, 1 H), 1.31 (d, J=6.8 Hz, 6 H); 13C NMR (100 MHz,CDCl3): d=147.7, 145.7, 140.4, 137.9, 133.4, 128.8, 128.2,127.8, 127.4, 126.9, 123.2, 123.0, 121.8, 121.5, 110.8, 33.9,24.1; HR-MS: m/z= 313.1691 [M++ H]++, calcd. for C22H21N2 :313.1699.
1-(4-Methoxyphenyl)-3-phenyl-1H-indazole (2d): Afterthe condensation of diphenyl ketone (0.5 mmol) and hydra-zine salt (0.9 mmol), prepared according to General Proce-dure A was complete, the reaction mixture was concentratedand the resulting crude hydrazone (1d) was directly subject-ed to the General Procedure B : 2 h (1.5 mmol of I2); eluent:CH2Cl2/PE 35:65; yield: 110 mg (73%); white solid; mp128–129 88C (lit.[9b] mp 129–131 88C); 1H NMR (400 MHz,CDCl3): d= 8.07 (d, J= 8.0 Hz, 1 H), 8.05–8.03 (m, 2 H),7.68–7.65 (m, 3 H), 7.52 (t, J= 7.6 Hz, 2 H), 7.44–7.40 (m,2 H), 7.26 (m, 1 H, overlapped with the peak of chloroform),7.08–7.04 (m, 2 H), 3.87 (s, 3 H); 13C NMR (100 MHz,CDCl3): d=158.5, 145.6, 140.6, 133.4, 133.3, 128.9, 128.2,127.8, 126.9, 124.9, 122.7, 121.7, 121.5, 114.7, 110.5, 55.6;HR-MS: m/z =301.1337 [M++ H]++, calcd. for C20H17N2O:301.1335.
1-(4-Fluorophenyl)-3-phenyl-1H-indazole (2e):[9c] 6 h(1 mmol of I2); eluent: CH2Cl2/PE 25:75; yield: 137 mg(95%); white solid; mp 114–115 88C; 1H NMR (400 MHz,CDCl3): d= 8.08 (d, J= 8.0 Hz, 1 H), 8.02 (d, J=7.2 Hz, 2 H),7.76–7.72 (m, 2 H), 7.68 (d, J= 8.8 Hz, 1 H), 7.53 (t, J=7.2 Hz, 2 H), 7.47–7.41 (m, 2 H), 7.28 (t, J= 7.6 Hz, 1 H),7.26–7.21 (m, 2 H, overlapped with the peak of chloroform);13C NMR (100 MHz, CDCl3): d=161.2 (d, JC,F =245.0 Hz),146.2, 140.5, 136.3 (d, JC,F =2.9 Hz), 133.1, 128.9, 128.4,127.8, 127.3, 124.9 (d, JC,F =8.3 Hz), 123.0, 122.0, 121.7, 116.4(d, JC,F =22.9 Hz), 110.4; HR-MS: m/z =289.1131 [M++H]++,calcd. for C19H14FN2 : 289.1136..
1-(4-Chlorophenyl)-3-phenyl-1H-indazole (2f):[9c] 5 h(1 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 148 mg(97%); white solid; mp 121–122 88C; 1H NMR (400 MHz,CDCl3): d= 8.08 (d, J= 8.0 Hz, 1 H), 8.02 (d, J=7.2 Hz, 2 H),7.76–7.72 (m, 3 H), 7.75–7.42 (m, 6 H), 7.29 (d, J= 7.6 Hz,1 H); 13C NMR (100 MHz, CDCl3): d =146.5, 140.2, 138.7,133.0, 132.1, 129.6, 128.9, 128.5, 127.8, 127.4, 124.0, 123.3,122.2, 121.8, 110.5; HR-MS: m/z =305.0847 [M++ H]++, calcd.for C19H14ClN2 : 305.0840.
1-(4-Bromophenyl)-3-phenyl-1H-indazole (2g):[21] 5 h(1 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 167 mg(96%); white solid; mp 123–124 88C; 1H NMR (400 MHz,CDCl3): d= 8.07 (d, J= 8.4 Hz, 1 H), 8.01 (d, J=7.2 Hz, 2 H),7.73–7.63 (m, 5 H), 7.52 (t, J= 7.6 Hz, 2 H), 7.47–7.41 (m,2 H), 7.28 (t, J=7.6 Hz, 1 H); 13C NMR (100 MHz, CDCl3):d= 146.6, 140.2, 139.3, 133.0, 132.6, 128.9, 128.5, 127.8, 127.4,124.2, 123.4, 122.2, 121.8, 119.8, 110.5; HR-MS: m/z=349.0331 [M++ H]++, calcd. for C19H14BrN2 : 349.0335.
4-(3-Phenyl-1H-indazol-1-yl)benzonitrile (2h):[9c] 8 h(1.5 mmol of I2); eluent: EtOAc/PE 15:85; yield: 130 mg(88%); white solid; mp 161–163 88C; 1H NMR (400 MHz,CDCl3): d= 8.09 (d, J= 8.4 Hz, 1 H), 8.03–7.97 (m, 4 H),7.85–7.81 (m, 3 H), 7.57–7.45 (m, 4 H), 7.35 (t, J= 7.6 Hz,1 H); 13C NMR (100 MHz, CDCl3): d =147.9, 143.8, 140.0,133.6, 132.5, 129.0, 128.9, 128.0, 127.9, 124.1, 122.9, 122.1,122.0, 118.6, 110.7, 109.2; HR-MS: m/z =296.1175 [M++H]++,calcd. for C20H14N3 : 296.1182.
3-Phenyl-1-[4-(trifluoromethyl)phenyl]-1H-indazole(2i):[9b] 10 h (2 mmol of I2); eluent: CH2Cl2/PE 20:80; yield:160 mg (95%); white solid; mp 137–139 88C; 1H NMR(400 MHz, CDCl3): d =8.10 (d, J=8.0 Hz, 1 H), 8.04–8.02(m, 2 H), 7.97 (d, J=8.4 Hz, 2 H), 7.82 (t, J=9.2 Hz, 3 H),7.56–7.44 (m, 4 H), 7.33 (t, J=7.6 Hz, 1 H); 13C NMR(100 MHz, CDCl3): d= 147.3, 143.1, 140.2, 132.8, 128.9,128.7, 128.1 (q, JC,F = 32.6 Hz), 127.9, 127.7, 126.7 (q, JC,F =3.7 Hz), 124.0 (q, JC,F = 270.3 Hz), 123.8, 122.5, 122.2, 122.0,110.6; HR-MS: m/z= 339.1104 [M++H]++, calcd. forC20H14F3N2 : 339.1104.
1-(4-Nitrophenyl)-3-phenyl-1H-indazole (2j): 10 h(2 mmol of I2); eluent: CH2Cl2/PE 25:75; yield: 157 mg(99%); yellow solid; mp 162–163 88C (lit.[9a] mp 160–162 88C);1H NMR (400 MHz, CDCl3): d=8.39 (d, J= 9.2 Hz, 2 H),8.09 (d, J=8.0 Hz, 1 H), 8.04–8.01 (m, 4 H), 7.87 (d, J=8.4 Hz, 1 H), 7.57–7.53 (m, 3 H), 7.50–7.46 (m, 1 H), 7.36 (t,J=7.6 Hz, 1 H); 13C NMR (100 MHz, CDCl3): d= 148.3,145.4, 144.9, 140.1, 132.3, 129.0, 128.2, 127.9, 125.3, 124.3,123.1, 122.2, 121.4, 110.9; HR-MS: m/z =316.1083 [M++H]++,calcd. for C19H14N3O2 : 316.1081.
1-(2-Chlorophenyl)-3-phenyl-1H-indazole (2k):[10b] 10 h(2 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 149 mg(98%); colorless oil ; 1H NMR (400 MHz, CDCl3): d= 8.10(d, J= 8.4 Hz, 1 H), 8.05 (d, J= 7.6 Hz, 2 H), 7.63–7.58 (m,2 H), 7.52 (d, J=7.6 Hz, 2 H), 7.46–7.41 (m, 4 H), 7.31–7.26(m, 2 H); 13C NMR (100 MHz, CDCl3): d=146.5, 141.9,137.1, 133.2, 131.7, 130.8, 129.9, 129.8, 128.9, 128.4, 127.82,127.76, 127.0, 122.2, 121.8, 121.5, 110.9; HR-MS: m/z=305.0850 [M++ H]++, calcd. for C19H14ClN2 : 305.0840.
1-(3-Chlorophenyl)-3-phenyl-1H-indazole (2l):[9c] 6 h(1.5 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 131 mg(86%); colorless oil. ; 1H NMR (400 MHz, CDCl3): d= 8.07(d, J=8.0 Hz, 1 H), 8.02 (d, J=8.0 Hz, 2 H), 7.84 (s, 1 H),7.77 (d, J=8.8 Hz, 1 H), 7.70 (d, J=8.0 Hz, 1 H), 7.53 (t, J=8.0 Hz, 2 H), 7.49–7.42 (m, 3 H), 7.33–7.24 (m, 2 H);13C NMR (100 MHz, CDCl3): d =146.8, 141.3, 140.2, 135.2,132.9, 130.4, 128.9, 128.6, 127.9, 127.5, 126.6, 123.5, 122.9,122.3, 121.8, 120.6, 110.6; HR-MS: m/z =305.0849 [M++H]++,calcd. for C19H14ClN2 : 305.0840.
1-(2,4-Dichlorophenyl)-3-phenyl-1H-indazole (2m):[9b]
10 h (2 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 169 mg(99%); colorless oil ; 1H NMR (400 MHz, CDCl3): d= 8.09(d, J= 8.0 Hz, 1 H), 8.04–8.02 (m, 2 H), 7.63 (d, J= 2.0 Hz,
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3383
FULL PAPERS asc.wiley-vch.de

1 H), 7.54–7.50 (m, 3 H), 7.46–7.41 (m, 3 H), 7.30 (t, J=7.2 Hz, 1 H), 7.25 (d, J=8.4 Hz, 1 H, overlapped with thepeak of chloroform); 13C NMR (100 MHz, CDCl3): d=146.9, 141.9, 135.9, 135.1, 133.0, 132.4, 130.6, 130.5, 128.9,128.5, 128.1, 127.8, 127.2, 122.3, 122.0, 121.6, 110.7; HR-MS:m/z= 361.0270 [M++ Na]++, calcd. for C19H12Cl2N2Na:361.0270.
1-(2,5-Dichlorophenyl)-3-phenyl-1H-indazole (2n): 10 h(2 mmol of I2); eluent: CH2Cl2/PE 15:75; yield: 169 mg(99%); white solid; mp 95–98 88C; 1H NMR (400 MHz,CDCl3): d =8.09 (d, J=8.4 Hz, 1 H), 8.04–8.02 (m, 2 H), 7.62(d, J=2.4 Hz, 1 H), 7.53 (t, J= 8.4 Hz, 3 H), 7.47–7.40 (m,3 H), 7.32–7.27 (m, 2 H); 13C NMR (100 MHz, CDCl3): d =147.1, 141.7, 138.1, 133.3, 132.9, 131.6, 129.9, 129.8, 129.7,128.9, 128.6, 127.8, 127.3, 122.4, 122.1, 121.6, 110.9; HR-MS:m/z= 339.0442 [M++H]++, calcd. for C19H13Cl2N2 : 339.0450.
3-Phenyl-1-(2,4,6-trichlorophenyl)-1H-indazole (2o):[9c]
12 h (2 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 185 mg(99%); colorless oil ; 1H NMR (400 MHz, CDCl3): d= 8.11(d, J= 8.4 Hz, 1 H), 8.03 (d, J= 8.0 Hz, 2 H), 7.55–7.51 (m,4 H), 7.47–7.42 (m, 2 H), 7.31 (t, J=7.6 Hz, 1 H), 7.11 (d, J=8.4 Hz, 1 H); 13C NMR (100 MHz, CDCl3): d =147.2, 141.9,136.5, 136.1, 133.7, 132.9, 129.0, 128.9, 128.6, 127.9, 127.5,122.1, 122.0, 121.7, 109.8; HR-MS: m/z =373.0052 [M++H]++,calcd. for C19H12Cl3N2 : 373.0061.
1-(tert-Butyl)-3-phenyl-1H-indazole (2p): After the con-densation of diphenyl ketone (2 mmol) and hydrazine salt(3.6 mmol) according to General Procedure A was complete,the reaction mixture was concentrated and the resultingcrude hydrazone (1p) was directly subjected to the GeneralProcedure B : 1 h (6 mmol of I2); eluent: CH2Cl2/PE 25:75;yield: 121 mg (24%); white solid; mp 104–106 88C; 1H NMR(400 MHz, CDCl3): d =8.02 (d, J=8.4 Hz, 1 H), 7.98–7.96(m, 2 H), 7.72 (d, J=8.8 Hz, 1 H), 7.48 (t, J=8.0 Hz, 2 H),7.38–7.32 (m, 2 H), 7.19–7.15 (m, 1 H), 1.83 (s, 9 H);13C NMR (100 MHz, CDCl3): d =141.9, 139.6, 134.2, 128.7,127.6, 127.5, 125.2, 123.3, 121.5, 120.4, 112.4, 59.8, 29.8; HR-MS: m/z= 251.1547 [M++ H]++, calcd. for C17H19N2 : 251.1543.
6-Methyl-1-phenyl-3-(p-tolyl)-1H-indazole (2q): 2 h(1 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 117 mg(79%); white solid; mp 90–92 88C (lit.[9b] mp 88–90 88C);1H NMR (400 MHz, CDCl3): d=7.94–7.91 (m, 3 H), 7.78 (d,J=7.6 Hz, 2 H), 7.56–7.52 (m, 3 H), 7.37–7.31 (m, 3 H), 7.10(d, J=8.4 Hz, 1 H), 2.51 (s, 3 H), 2.43 (s, 3 H); 13C NMR(100 MHz, CDCl3): d= 146.0, 140.9, 140.3, 138.1, 137.5,130.5, 129.5, 129.4, 127.6, 126.5, 124.0, 123.1, 121.4, 121.3,110.1, 22.1, 21.4; HR-MS: m/z= 299.1534 [M++H]++, calcd.for C21H19N2 : 299.1543.
6-Methoxy-3-(4-methoxyphenyl)-1-phenyl-1H-indazole(2r): 2 h (1 mmol of I2); eluent: CH2Cl2/PE 40:60; yield:142 mg (86%); white solid; mp 135–136 88C (lit.[9a] mp 132–134 88C); 1H NMR (400 MHz, CDCl3): d=7.94 (d, J= 8.8 Hz,2 H), 7.89 (d, J=8.8 Hz, 1 H), 7.76 (d, J= 8.0 Hz, 2 H), 7.54(t, J=8.0 Hz, 2 H), 7.36 (t, J=7.2 Hz, 1 H), 7.10 (d, J=2.0 Hz, 1 H), 7.04 (d, J= 8.8 Hz, 2 H), 6.91 (dd, J= 8.8,2.0 Hz, 1 H), 3.87 (s, 6 H); 13C NMR (100 MHz, CDCl3): d=159.9, 159.8, 146.0, 141.6, 140.3, 129.5, 128.9, 126.5, 125.9,123.0, 122.4, 117.8, 114.3, 113.3, 91.9, 55.6, 55.4; HR-MS: m/z=331.1441 [M++H]++, calcd. for C21H19N2O2 : 331.1441.
6-Fluoro-3-(4-fluorophenyl)-1-phenyl-1H-indazole (2s):2 h (1 mmol of I2); eluent: CH2Cl2/PE 25:75; yield: 129 mg(84%); white solid; mp 137–138 88C (lit.[10a] mp 135–137 88C);
1H NMR (400 MHz, CDCl3): d=7.98–7.93 (m, 3 H), 7.73 (d,J=7.6 Hz, 2 H), 7.56 (t, J=7.6 Hz, 2 H), 7.42–7.37 (m, 2 H),7.22 (t, J=8.8 Hz, 2 H), 7.05 (td, J=8.8, 2.0 Hz, 1 H);13C NMR (100 MHz, CDCl3): d=163.0 (d, JC,F =246.6 Hz),162.7 (d, JC,F = 244.7 Hz), 145.4, 140.7 (d, JC,F =12.2 Hz),139.7, 129.6, 129.4 (d, JC,F =8.3 Hz), 128.9 (d, JC,F =3.4 Hz),127.1, 122.9, 122.8 (d, JC,F = 11.0 Hz), 119.8, 115.9 (d, JC,F =21.6 Hz), 111.8 (d, JC,F = 25.7 Hz), 96.7 (d, JC,F = 26.9 Hz);HR-MS: m/z =307.1046 [M++H]++, calcd. for C19H13F2N2 :307.1041.
6-Chloro-3-(4-chlorophenyl)-1-phenyl-1H-indazole (2t):7 h (1 mmol of I2); eluent: CH2Cl2/PE 15:85; yield: 124 mg(73%); white solid; mp 153–154 88C (lit.[10a] mp 150–151 88C);1H NMR (400 MHz, CDCl3): d= 7.94–7.90 (m, 3 H), 7.74–7.71 (m, 3 H), 7.56 (t, J= 8.4 Hz, 2 H), 7.49 (d, J= 8.8 Hz,2 H), 7.41 (t, J=7.6 Hz, 1 H), 7.26–7.23 (m, 1 H, overlappedwith the peak of chloroform); 13C NMR (100 MHz, CDCl3):d= 145.0, 140.8, 139.5, 134.5, 133.8, 131.2, 129.7, 129.2, 128.9,127.3, 123.2, 123.1, 122.2, 121.4, 110.6; HR-MS: m/z=339.0455 [M++ H]++, calcd. for C19H13Cl2N2 : 339.0450.
6-Bromo-3-(4-bromophenyl)-1-phenyl-1H-indazole (2u):5 h (1 mmol of I2); eluent: CH2Cl2/PE 20:80; yield: 169 mg(79%); white solid; mp 152–154 88C; 1H NMR (400 MHz,CDCl3): d= 7.92 (d, J= 1.2 Hz, 1 H), 7.87–7.85 (m, 3 H),7.73–7.71 (m, 2 H), 7.66–7.63 (m, 2 H), 7.57 (t, J= 8.0 Hz,2 H), 7.43–7.37 (m, 2 H); 13C NMR (100 MHz, CDCl3): d =145.1, 141.2, 139.4, 132.1, 131.6, 129.7, 129.1, 127.4, 125.7,123.2, 122.7, 122.4, 121.8, 121.7, 113.7; HR-MS: m/z =448.9233 [M++ Na]++, calcd. for C19H12Br2N2Na: 448.9259.
6-Methyl-1,3-diphenyl-1H-indazole (2v) and 1-phenyl-3-(p-tolyl)-1H-indazole (2v’’):[9b] 2 h (1 mmol of I2); eluent:CH2Cl2/PE 25:75; yield: 116 mg (82%; 2v :2v’’=77:23 by1H NMR); white solid; mp 114–116 88C; 1H NMR (400 MHz,CDCl3): d =8.08–8.02 (m, 1.77 H), 7.96–7.93 (m, 1.23 H),7.80–7.76 (m, 2.23 H), 7.56–7.50 (m, 4.31 H), 7.46–7.32 (m,2.46 H), 7.27 (t, J= 8.0 Hz, 0.23 H), 7.11 (d, J= 8.4 Hz,0.77 H), 2.51 (s, 2.31 H), 2.43 (s, 0.69 H); 13C NMR(100 MHz, CDCl3): 2v : d =145.9, 141.0, 140.25, 137.5, 133.4,129.6, 129.5, 128.8, 128.2, 127.72, 126.6, 124.1, 123.1, 121.2,110.2, 22.1; 2v’’: d= 146.2, 140.33, 140.2, 138.2, 130.4, 127.69,127.1, 123.2, 123.0, 121.8, 121.7, 121.3,110.7, 21.4; HR-MS:m/z= 285.1382 [M++H]++, calcd. for C20H17N2 : 285.1386.
6-Methoxy-1,3-diphenyl-1H-indazole (2w). 2 h (1 mmol ofI2); eluent: CH2Cl2/PE 33:67; yield: 120 mg (80%); whitesolid; mp 137–139 88C (lit.[10a] mp 138–140 88C); 1H NMR(400 MHz, CDCl3): d= 8.02–8.00 (m, 2 H), 7.93 (d, J=8.8 Hz, 1 H), 7.78 (d, J=6.8 Hz, 2 H), 7.58–7.50 (m, 4 H),7.44–7.36 (m, 2 H), 7.12 (s, 1 H), 6.94 (d, J=8.8 Hz, 1 H),3.88 (s, 3 H); 13C NMR (100 MHz, CDCl3): d= 160.0, 146.1,141.7, 140.2, 133.3, 129.5, 128.8, 128.3, 127.7, 126.7, 123.1,122.4, 117.8, 113.6, 92.0. 55.6; HR-MS: m/z =301.1344 [M++H]++, calcd. for C20H17N2O: 301.1335.
3-(4-Fluorophenyl)-1-phenyl-1H-indazole (2x) and 6-fluoro-1,3-diphenyl-1H-indazole (2x’’):[9c] 2 h (1 mmol of I2);eluent: CH2Cl2/PE 25:75; yield: 119 mg (83%; 2x :2x’’=80:20by 1H NMR); white solid; mp 96–98 88C; 1H NMR (400 MHz,CDCl3): d =8.02–7.97 (m, 3 H), 7.79–7.73 (m, 2.8 H), 7.56–7.50 (m, 2.4 H), 7.46–7.35 (m, 2.2 H), 7.28 (t, J= 8.0 Hz,0.8 H), 7.23–7.19 (m, 1.6 H, overlapped with the peak ofchloroform), 7.03 (td, J= 8.8, 2.0 Hz, 0.2 H); 13C NMR(100 MHz, CDCl3): 2x : d=162.9 (d, JC,F =246.1 Hz), 145.2,140.4, 140.1, 129.6, 129.5, 129.4, 127.2, 126.8, 123.0, 122.1,
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3384
FULL PAPERS asc.wiley-vch.de

121.4, 116.0, 115.8, 110.8; 2x’’: d =162.7 (d, JC,F =244.4 Hz),146.4, 140.7 (d, JC,F =12.3 Hz), 139.8, 132.8, 128.9, 128.6,127.8, 127.0, 123.1, 123.0, 122.9, 120.0, 111.6 (d, JC,F =25.6 Hz), 96.6 (d, JC,F =27.0 Hz); HR-MS: m/z= 289.1138[M++H]++, calcd. for C19H14FN2 : 289.1136.
3-(4-Chlorophenyl)-1-phenyl-1H-indazole (2y) and 6-chloro-1,3-diphenyl-1H-indazole (2y’’):[9a] 4 h (1 mmol of I2);eluent: CH2Cl2/PE 25:75; yield: 122 mg (80%; 2y :2y’’=85:15by 1H NMR); white solid; mp 114–117 88C; 1H NMR (CDCl3,400 MHz): d= 8.03–7.96 (m, 3 H), 7.78–7.73 (m, 3 H), 7.57–7.44 (m, 5 H), 7.40–7.36 (m, 1 H), 7.29 (d, J=7.2 Hz, 0.85 H),7.25–7.22 (m, 0.15 H, overlapped with the peak of chloro-form); 13C NMR (100 MHz, CDCl3): 2y : d =144.9, 140.4,140.0, 134.2, 131.8, 129.5, 129.1, 128.9, 127.3, 126.9, 123.1,122.9, 122.2, 121.3, 110.8; 2y’’: d= 146.2, 140.8, 139.6, 133.7,132.7, 129.6, 128.6, 127.8, 127.2, 122.6, 121.7, 110.5; HR-MS:m/z= 305.0831 [M++H]++, calcd. for C19H14ClN2 : 305.0840.
3-(4-Bromophenyl)-1-phenyl-1H-indazole (2z)[10a] and 6-bromo-1,3-diphenyl-1H-indazole (2z’’): 4 h (1 mmol of I2);eluent: CH2Cl2/PE 25:75; yield: 138 mg (79%; 2z :2z’’=80:20by 1H NMR); white solid; mp 147–150 88C; 1H NMR (CDCl3,400 MHz): d=8.03 (d, J=8.0 Hz, 0.8 H), 7.99–7.93 (m,0.4 H), 7.93–7.91 (m, 2 H), 7.78–7.73 (m, 2.8 H), 7.65–7.63(m, 1.6 H), 7.57–7.50 (m, 2.4 H), 7.47–7.43 (m, 1 H), 7.39–7.35 (m, 1.2 H), 7.31–7.29 (m, 0.8 H); 13C NMR (100 MHz,CDCl3): 2z : d=144.9, 140.4, 140.0, 132.2, 132.0, 129.5, 129.2,127.3, 126.9, 123.08, 122.9, 122.4, 122.2, 121.3, 110.9; 2z’’: d=146.3, 141.1, 139.6, 132.6, 129.6, 129.0, 128.6, 127.8, 127.2,125.5, 123.14, 122.8, 122.0, 121.7, 113.6; HR-MS: m/z=349.0331 [M++ H]++, calcd. for C19H13BrN2 : 349.0335.
3-(tert-Butyl)-1-(4-nitrophenyl)-1H-indazole (2aa):[22] 10 h(2 mmol of I2); eluent: CH2Cl2/PE 40:60; yield: 147 mg(99%); yellow solid; mp 128–130 88C; 1H NMR (400 MHz,CDCl3): d =8.38 (d, J=9.2 Hz, 2 H), 7.99–7.95 (m, 3 H), 7.84(d, J=8.8 Hz, 1 H), 7.50–7.46 (m, 1 H), 7.29–7.25 (m, 1 H),1.59 (s, 9 H); 13C NMR (100 MHz, CDCl3): d= 157.3, 145.7,144.4, 140.1, 127.5, 125.3, 124.1, 123.0, 121.7, 120.9, 110.8,34.1, 29.8; HR-MS: m/z=296.1389 [M++H]++, calcd. forC17H18N3O2 : 296.1394.
Cleavage of the 4-Nitrophenyl Group for theSynthesis of 1H-Indazole (2j’’)
A solution of indazole 2j (157 mg, 0.5 mmol) in DMSO(5 mL) was treated with NaOEt (102 mg, 1.5 mmol) andstirred at 120 88C under a nitrogen atmosphere for 2 h. Uponcompletion of the reaction, the solution was cooled to roomtemperature, quenched with brine (10 mL), and extractedwith CH2Cl2 (4 X 10 mL). The combined organic layer wasdried over anhydrous Na2SO4, concentrated, and then theresidue was purified by column chromatography throughsilica gel (eluent: EtOAc/PE 16:84) to afford 1H-indazole2j’’;[9c] ; yield: 77 mg (79%); light-brown solid; mp 102–103 88C. 1H NMR (400 MHz, CDCl3): d= 11.64 (br, s, 1 H),8.02 (dd, J= 8.0, 0.8 Hz, 3 H), 7.54 (t, J=7.6 Hz, 2 H), 7.45(t, J=7.6 Hz, 1 H), 7.36–7.33 (m, 1 H), 7.25–7.19 (m, 2 H,overlapped with the peak of chloroform); 13C NMR(100 MHz, CDCl3): d= 145.7, 141.7, 133.5, 129.0, 128.2,127.7, 126.8, 121.3, 121.1, 120.9, 110.3; MS: m/z=195 [M ++H]++, calcd. for C13H11N2 : 195.
General Procedure C for the Synthesis of 1H-Pyrazoles 3
A mixture of the hydrazone substrate 1 (0.5 mmol), I2
(254 mg, 1 mmol) and NaOAc (185 mg, 2.25 mmol) in tolu-ene (5 mL; chlorobenzene was used for the synthesis of 3ag)was heated to reflux. If the reaction was not completewithin 2 h with more than 1/4 of substrate 1 remaining(monitored by TLC), another portion of iodine (0.5 or1 mmol, with 1 or 2 mmol of NaOAc) was added. Uponcompletion of the reaction, it was quenched with 5%Na2S2O3 (10 mL), and then extracted with CH2Cl2 (4X10 mL). The combined organic layer was washed with brine(10 mL), dried over anhydrous Na2SO4, and concentrated.The residue was purified by column chromatographythrough silica gel to afford the 1H-pyrazole 3.
1-(4-Nitrophenyl)-3-phenyl-1H-pyrazole (3ac): Accordingto the General Procedure C for 2 h (1 mmol of I2), the dihy-dropyrazole G was formed as the major product [eluent:CH2Cl2/PE 50:50, yellow solid; 1H NMR (400 MHz, CDCl3):d= 8.15 (d, J= 9.2 Hz, 2 H), 7.75–7.74 (m, 2 H), 7.43–7.38 (m,3 H), 7.03 (d, J=9.2 Hz, 2 H), 3.96 (t, J= 10.4 Hz, 2 H), 3.35(t, J=10.4 Hz, 2 H); MS: m/z =268 [M++H]++, calcd. forC15H14N3O2 : 268.
A solution of the crude intermediate G from above in tol-uene (5 mL) was treated with DDQ (0.25 mmol) and re-fluxed for 1 h to afford pyrazole 3ac. Eluent: CH2Cl2/PE33:67; yield: 66 mg (50%, from the hydrazone); yellowsolid; mp 168–170 88C (lit.[23] mp 170–173 88C); 1H NMR(400 MHz, CDCl3): d =8.33 (d, J= 9.2 Hz, 2 H), 8.04 (d, J=2.8 Hz, 1 H), 7.94–7.91 (m, 4 H), 7.47–7.44 (m, 2 H), 7.40–7.36 (m, 1 H), 6.86 (d, J=2.4 Hz, 1 H); 13C NMR (100 MHz,CDCl3): d 154.5, 145.2, 144.4, 132.2, 128.83, 128.79, 128.3,126.0, 125.4, 118.3, 106.9; HR-MS: m/z =266.0925 [M++H]++,calcd. for C15H12N3O2 : 266.0924.
5-Methyl-1-(4-nitrophenyl)-3-phenyl-1H-pyrazole (3ad):3 h (1.5 mmol of I2); eluent: CH2Cl2/PE 50:50; yield: 106 mg(76%); yellow solid; mp 119–121 88C (lit.[24] mp 124–125 88C);1H NMR (400 MHz, CDCl3): d=8.34 (d, J= 8.8 Hz, 2 H),7.85 (d, J=7.2 Hz, 2 H), 7.77 (d, J= 9.2 Hz, 2 H), 7.44–7.40(m, 2 H), 7.36–7.33 (m, 1 H), 6.59 (s, 1 H), 2.50 (s, 3 H);13C NMR (100 MHz, CDCl3): d =152.9, 145.9, 145.0, 140.6,132.6, 128.7, 128.4, 125.9, 124.8, 123.8, 106.6, 13.4; HR-MS:m/z= 280.1077 [M++H]++, calcd. for C16H14N3O2 : 280.1081.
5-Ethyl-1-(4-nitrophenyl)-3-phenyl-1H-pyrazole (3ae):[25]
3 h (1.5 mmol of I2); eluent: CH2Cl2/PE 50:50; yield: 108 mg(74%); yellow solid; mp 110–111 88C; 1H NMR (400 MHz,CDCl3): d =8.35 (d, J=9.2 Hz, 2 H), 7.88–7.86 (m, 2 H), 7.75(d, J=9.2 Hz, 2 H), 7.44–7.41 (m, 2 H), 7.37–7.33 (m, 1 H),6.63 (s, 1 H), 2.83 (q, J=14.8, 7.2 Hz, 2 H), 1.34 (t, J=7.6 Hz, 3 H); 13C NMR (100 MHz, CDCl3): d =153.0, 147.1,146.0, 145.2, 132.6, 128.7, 128.4, 125.8, 124.7, 124.4, 104.4,20.6, 13.1; HR-MS (m/z) [M ++ H]++ calcd for C17H16N3O2
294.1237, found 294.1239.1-(4-Nitrophenyl)-5-nonyl-3-phenyl-1H-pyrazole (3af): 6 h
(2 mmol of I2); eluent: CH2Cl2/PE 60:40; yield: 165 mg(84%); yellow solid; mp 76–77 88C; 1H NMR (400 MHz,CDCl3): d =8.36 (d, J=8.8 Hz, 2 H), 7.88–7.85 (m, 2 H), 7.74(d, J=9.2 Hz, 2 H), 7.44–7.40 (m, 2 H), 7.36–7.33 (m, 1 H),6.62 (s, 1 H), 2.77 (t, J=7.6 Hz, 2 H), 1.73–1.66 (m, 2 H),1.39–1.26 (m, 12 H), 0.89–0.86 (m, 3 H); 13C NMR (100 MHz,CDCl3): d=152.9, 146.1, 145.8, 145.2, 132.6, 128.7, 128.4,
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3385
FULL PAPERS asc.wiley-vch.de

125.8, 124.7, 124.6, 104.8, 31.9, 29.4, 29.3, 29.2, 28.8, 27.0,22.7, 14.1; HR-MS: m/z=392.2320 [M++H]++, calcd. forC24H30N3O2 : 392.2333.
1-(4-Nitrophenyl)-3-phenyl-4,5,6,7-tetrahydro-1H-indazole(3ag): 6 h (2 mmol of I2); eluent: CH2Cl2/PE 50:50; yield:80 mg (50%); yellow solid; mp 150–152 88C; 1H NMR(400 MHz, CDCl3): d =8.31 (d, J=9.2 Hz, 2 H), 7.83–7.79(m, 4 H), 7.46–7.42 (m, 2 H), 7.38–7.34 (m, 1 H); 2.88–2.86(m, 2 H), 2.81–2.79 (m, 2 H), 1.90–1.85 (m, 4 H); 13C NMR(100 MHz, CDCl3): d= 151.0, 145.1, 140.2, 133.3, 128.6,128.1, 127.1, 124.9, 121.9, 117.8, 24.9, 22.8, 22.7, 22.6; HR-MS: m/z= 320.1393 [M ++H]++, calcd. for C19H18N3O2 :320.1394.
4,5-Dimethyl-1-(4-nitrophenyl)-3-phenyl-1H-pyrazole(3ah): 4 h (1.5 mmol of I2); eluent: CH2Cl2/PE 50:50; yield:81 mg (55%); yellow solid; mp 114–116 88C; 1H NMR(400 MHz, CDCl3): d =8.33 (d, J=9.2 Hz, 2 H), 7.76–7.71(m, 4 H), 7.47–7.31 (m, 2 H), 7.39–7.36 (m, 1 H), 2.42 (s, 3 H),2.22 (s, 3 H); 13C NMR (100 MHz, CDCl3): d= 152.6, 145.7,145.2, 137.4, 133.4, 128.6, 128.0, 127.9, 124.8, 123.9, 115.0,11.8, 9.7; HR-MS: m/z=294.1234 [M++H]++, calcd. forC17H16N3O2 : 294.1237.
Acknowledgements
We thank the Outstanding Young Talent Research Fund ofZhengzhou University (No. 1521316004) and the NationalNatural Science Foundation of China (Nos. 81330075 and81302637) for financial support.
References
[1] a) J. Elguero, in: Comprehensive Heterocyclic Chemis-try, Vol. 5, (Eds.: A. R. Katritzky, C. W. Rees), Perga-mon, New York, 1984, pp 167–303; b) D. D. Gaikwad,A. D. Chapolikar, C. G. Devkate, K. D. Warad, A. P.Tayade, R. P. Pawar, A. J. Domb, Eur. J. Med. Chem.2015, 90, 707–731; c) A. Schmidt, A. Beutler, B. Snovy-dovych, Eur. J. Org. Chem. 2008, 4073–4095; d) A.Schmidt, A. Dreger, Curr. Org. Chem. 2011, 15, 1423–1463; e) A. A. Bekhit, A. Hymete, A. E. A. Bekhit, A.Damtew, H. Y. Aboul-Enein, Mini-Rev. Med. Chem.2010, 10, 1014–1033.
[2] a) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S.Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F.Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J.Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N.Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins,K. Seibert, A. W. Veenhuizen, Y. Y. Zhang, P. C. Isak-son, J. Med. Chem. 1997, 40, 1347–1365; b) A. A.Bekhit, T. Abdel-Aziem, Bioorg. Med. Chem. 2004, 12,1935–1945.
[3] a) X. Li, S. Chu, V. A. Feher, M. Khalili, Z. Nie, S.Margosiak, V. Nikulin, J. Levin, K. G. Sprankle, M. E.Tedder, R. Almassy, K. Appelt, K. M. Yager, J. Med.Chem. 2003, 46, 5663–5673; b) A. Tanitame, Y. Oyama-da, K. Ofuji, M. Fujimoto, N. Iwai, Y. Hiyama, K.Suzuki, H. Ito, H. Terauchi, M. Kawasaki, K. Nagai, M.Wachi, J. Yamagishi, J. Med. Chem. 2004, 47, 3693–3696; c) T. S. Haque, S. Tadesse, J. Marcinkeviciene,
M. J. Rogers, C. Sizemore, L. M. Kopcho, K. Amsler,L. D. Ecret, D. L. Zhan, F. Hobbs, A. Slee, G. L. Train-or, A. M. Stern, R. A. Copel, A. P. Combs, J. Med.Chem. 2002, 45, 4669–4678.
[4] a) K. B. Goodman, H. Cui, S. E. Dowdell, D. E. Gaita-nopoulos, R. L. Ivy, C. A. Sehon, R. A. Stavenger,G. Z. Wang, A. Q. Viet, W. Xu, G. Ye, S. F. Semus, C.Evans, H. E. Fries, L. J. Jolivette, R. B. Kirkpatrick, E.Dul, S. S. Khandekar, T. Yi, D. K. Jung, L. L. Wright,G. K. Smith, D. J. Behm, R. Bentley, C. P. Doe, E. Hu,D. Lee, J. Med. Chem. 2007, 50, 6–9; b) E. Barile, S. K.De, C. B. Carlson, V. Chen, C. Knutzen, M. Riel-Mehan, L. Yang, R. Dahl, G. Chiang, M. Pellecchia, J.Med. Chem. 2010, 53, 8368–8375; c) P. G. Wyatt, A. J.Woodhead, V. Berdini, J. A. Boulstridge, M. G. Carr,D. M. Cross, D. J. Davis, L. A. Devine, T. R. Early,R. E. Feltell, E. J. Lewis, R. L. McMenamin, E. F. Nav-arro, M. A. OQBrien, M. OQReilly, M. Reule, G. Saxty,L. C. A. Seavers, D.-M. Smith, M. S. Squires, G. Trewar-tha, M. T. Walker, A. J.-A. Woolford, J. Med. Chem.2008, 51, 4986–4999; d) J. Regan, A. Capolino, P. F. Ci-rillo, T. Gilmore, A. G. Graham, E. Hickey, R. R. Kroe,J. Madwed, M. Moriak, R. Nelson, C. A. Pargellis, A.Swinamer, C. Torcellini, M. Tsang, N. Moss, J. Med.Chem. 2003, 46, 4676–4686.
[5] L. H. Jones, G. Allan, O. Barba, C. Burt, R. Corbau, T.Dupont, T. Knçchel, S. Irving, D. S. Middleton, C. E.Mowbray, M. Perros, H. Ringrose, N. A. Swain, R.Webster, M. Westby, C. Phillips, J. Med. Chem. 2009,52, 1219–1223.
[6] a) G. P. Lahm, T. M. Stevenson, T. P. Selby, J. H. Freu-denberger, D. Cordova, L. Flexner, C. A. Bellin, C. M.Dubas, B. K. Smith, K. A. Hughes, J. G. Hollingshaus,C. E. Clark, E. A. Benner, Bioorg. Med. Chem. Lett.2007, 17, 6274–6279; b) P. Caboni, R. E. Sammelson,J. E. Casida, J. Agric. Food Chem. 2003, 51, 7055–7061.
[7] a) K. W. Fan, J. J. Roberts, P. J. Martens, M. H. Stenzel,A. M. Granville, J. Mater. Chem. B 2015, 3, 7457–7465;b) P. Yin, L. A. Mitchell, D. A. Parrish, J. M. Shreeve,Chem. Asian J. 2017, 12, 378–384.
[8] a) S. Fustero, M. S#nchez-Roselll, P. Barrio, A. Simln-Fuentes, Chem. Rev. 2011, 111, 6984–7034; b) S. Dadi-boyena, A. Nefzi, Eur. J. Med. Chem. 2011, 46, 5258–5275.
[9] a) T. Zhang, W. Bao, J. Org. Chem. 2013, 78, 1317–1322; b) X. Li, L. He, H. Chen, W. Wu, H. Jiang, J.Org. Chem. 2013, 78, 3636–3646; c) J. Hu, H. Xu, P.Nie, X. Xie, Z. Nie, Y. Rao, Chem. Eur. J. 2014, 20,3932–3938; d) J. Yu, J. W. Lim, S. Y. Kim, J. Kim, J. N.Kim, Tetrahedron Lett. 2015, 56, 1432–1436.
[10] a) M. Kashiwa, M. Sonoda, S. Tanimori, Eur. J. Org.Chem. 2014, 4720–4723; b) Z. Zhang, Y. Huang, G.Huang, G. Zhang, Q. Liu, J. Heterocycl. Chem. 2017,54, 2426–2433.
[11] a) J. P. Wolfe, S. Wagaw, J.-F. Marcoux, S. L. Buchwald,Acc. Chem. Res. 1998, 31, 805–818; b) J. F. Hartwig,Angew. Chem. 1998, 110, 2154–2177; Angew. Chem. Int.Ed. 1998, 37, 2046–2067.
[12] a) Y. Park, Y. Kim, S. Chang, Chem. Rev. 2017, 117,9247–9301; b) P. Subramanian, G. C. Rudolf, K. P. Ka-liappan, Chem. Asian J. 2016, 11, 168–192; c) J. Jiao, K.Murakami, K. Itami, ACS Catal. 2016, 6, 610–633;
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3386
FULL PAPERS asc.wiley-vch.de

d) S. H. Cho, J. Y. Kim, J. Kwak, S. Chang, Chem. Soc.Rev. 2011, 40, 5068–5083; e) F. Collet, R. H. Dodd, P.Dauban, Chem. Commun. 2009, 5061–5074.
[13] X. Zhang, J. Kang, P. Niu, J. Wu, W. Yu, J. Chang, J.Org. Chem. 2014, 79, 10170–10178.
[14] E. Li, Z. Hu, L. Song, W. Yu, J. Chang, Chem. Eur. J.2016, 22, 11022–11027.
[15] Z. Lv, J. Liu, W. Wei, J. Wu, W. Yu, J. Chang, Adv.Synth. Catal. 2016, 358, 2759–2766.
[16] The conversion rate can also be accelerated by using acombination of catalytic iodine, NBS (1.5 equiv.); how-ever, the yields were significantly decreased. For exam-ple, I2/NBS-mediated cyclization of 1j produced prod-uct 2j in only 45 % yield.
[17] Taking indazole 2j as an example, the removal of the p-nitrophenyl group by NaOEt in DMSO at 120 88C af-fords the unprotected 1H-indazole 2j’’ in 79% yield.
[18] Y.-P. Zhu, Z. Fei, M.-C. Liu, F.-C. Jia, A.-X. Wu, Org.Lett. 2013, 15, 378–381.
[19] CCDC 1558444 (3ae) contains the supplementary crys-tallographic data for this paper. These data can be ob-
tained free of charge from The CambridgeCrystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
[20] C. Spiteri, S. Keeling, J. E. Moses, Org. Lett. 2010, 12,3368–3371.
[21] D. Liang, Q. Zhu, Asian J. Org. Chem. 2015, 4, 42–45.[22] M. Barrejln, M. J. Glmez-Escalonilla, J. L. G. Fierro, P.
Prieto, J. R. Carrillo, A. M. Rodr&guez, G. Abell#n,M. C. Llpez-Escalante, M. Gab#s, J. T. Llpez-Navar-rete, F. Langa, Phys. Chem. Chem. Phys. 2016, 18,29582–29590.
[23] D. L. Browne, J. B. Taylor, A. Plant, J. P. A. Harrity, J.Org. Chem. 2010, 75, 984–987.
[24] I. Bouabdallah, R. Touzani, I. Zidane, A. Ramdani, S.Radi, ARKIVOC (Gainesville, FL, U.S.A.) 2006, 138–144.
[25] X.-J. Wang, J. Tan, K. Grozinger, R. Betageri, T. Kir-rane, J. R. Proudfoot, Tetrahedron Lett. 2000, 41, 5321–5324.
Adv. Synth. Catal. 2017, 359, 3378 – 3387 V 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3387
FULL PAPERS asc.wiley-vch.de

本文献由“学霸图书馆-文献云下载”收集自网络,仅供学习交流使用。
学霸图书馆(www.xuebalib.com)是一个“整合众多图书馆数据库资源,
提供一站式文献检索和下载服务”的24 小时在线不限IP
图书馆。
图书馆致力于便利、促进学习与科研,提供最强文献下载服务。
图书馆导航:
图书馆首页 文献云下载 图书馆入口 外文数据库大全 疑难文献辅助工具
Related Documents











