Eur. J. Biochem. 267, 7183–7192 (2000) q FEBS 2000 Dissection of the enzymatic and immunologic functions of macrophage migration inhibitory factor Full immunologic activity of N-terminally truncated mutants Robert Kleemann 1 , Hans Rorsman 2 , Evald Rosengren 3 , Ralf Mischke 1 , Nguyen Tuyet Mai 1 and Ju ¨ rgen Bernhagen 1 1 Laboratory of Biochemistry, Institute for Interfacial Engineering, University of Stuttgart, Germany; 2 Department of Dermatology, and 3 Department of Pharmacology, University Hospital, Lund, Sweden Macrophage migration inhibitory factor (MIF) is a cytokine with broad regulatory functions in innate immunity. MIF belongs to the few cytokines displaying catalytic activities, i.e. MIF has a Pro2-dependent tautomerase and a Cys-Ala-Leu-Cys (CALC) cysteine-based thiol-protein oxidoreductase activity. Previous studies have addressed the roles of the catalytic site residues and the C-terminus. The two activities have not been directly compared. Here we report on the N-terminal mutational analysis and minimization of MIF and on a dissection of the two catalytic activities by comparing mutants P2AMIF, D4MIF, D5MIF, D6MIF, D7MIF, D8MIF, and D10MIF with the cysteine mutants of MIF. As N-terminal deletion was predicted to interfere with protein structure due to disruption of the central b sheet, it was surprising that deletion of up to six N-terminal residues resulted in normally expressed proteins with wild-type conformation. Strikingly, such mutants exhibited full MIF-specific immunologic activity. While mutation of Pro2 eliminated tautomerase activity, the CALC cysteine residues had no influence on this activity. However, mutant C81SMIF, which otherwise has full biologic activity, only had 32% tautomerase activity. Deletion of four N-terminal residues did not interfere with insulin reduction by MIF. By contrast, reduction of 2-hydroxyethyldisulfide (HED) was markedly affected by N-terminal manipulation, with P2AMIF and D2MIF exhibiting 40% activity, and D4MIF completely failing to reduce HED. This study constitutes the first comparison of the two catalytic activities of MIF and should assist in understanding the molecular links between the catalytic and immunologic activities of this cytokine and in providing guidelines for N-terminal protein minimization. Keywords: cytokine; macrophage migration inhibitory factor (MIF); N-terminal mutation; oxidoreductase; tautomerase. Macrophage migration inhibitory factor (MIF) was originally discovered as a T-cell factor acting to inhibit the migration of macrophages [1,2]. More recent research has led to the definition of MIF as a multifunctional, mostly proinflammatory mediator of broad target cell specificity [3–10]. MIF is a secretion product of both endocrine glands and of immune competent cells [11–16], but regulated MIF secretion may also occur from other cell types [17–21]. MIF has been found to be a critical mediator of several inflammatory and immune diseases. Best characterized is the role of MIF as a mediator of endotoxemia and septic shock [11,22–24]. MIF also plays a role in immune- mediated diseases such as delayed-type hypersensitivity reactions [25], chronic crescentic glomerulonephritis [26,27], and rheumatoid arthritis [28]. Disease-promoting MIF- mediated effects could be central to the pathogenesis of lung diseases [8,29]. MIF displays a number of properties that distinguish this factor from cytokines. Of greatest significance, MIF is induced rather than inhibited by glucocorticoids and, once released, can act to counter-regulate the immunosuppressive and anti-inflammatory functions of glucocorticoids [6]. The glucocorticoid-antagonistic effect of MIF could be linked to the suggested roles of MIF in several disease states [6,8,22]. MIF exhibits at least two catalytic activities, i.e. a tautomerase [30–33] and a thiol-protein oxidoreductase [34–36] activity. As a membrane receptor for MIF has not been identified, the catalytic activities are among the prime leads in the search to elucidate the molecular basis of MIF action. Catalytic activities have previously been reported for certain immune mediators. Adult T-cell leukemia-derived factor/thioredoxin (ADF/Trx) is an oxidoreductase with T cell regulatory and chemokine-like functions [37,38]. The cyclos- porin A-binding factor and macrophage-derived factor cyclo- philin [39,40] exhibits prolyl-peptidyl isomerase activity [41]. Other examples exist [42,43]. However, the identified catalytic activities could not generally be linked to the immunologic functions of these molecules. In fact, for some of them, a Correspondence to J. Bernhagen, Laboratory of Biochemistry, Institute for Interfacial Engineering, University of Stuttgart and Fraunhofer Institute FhIGB, Nobelstraße 12, D-70569 Stuttgart, Germany. Fax: 1 49 711 970 4200, Tel.: 1 49 711 970 4020, E-mail: [email protected] Abbreviations: ADF, adult T cell-derived factor; CALC, Cys-Ala-Leu-Cys; CSPD, disodium 3-(4-methoxyspiro(1,2-doxetan-3,2 0 -(5-chloro)tricyclo- decan)-4-yl)phenylphosphate; CXXC, Cys-Xaa-Xaa-Cys; GdnHCl, guanidine hydrochloride; GSH, glutathione; HFIP, 1,1,1,3,3,3-hexafluoroisopropanol; HED, 2-hydroxyethyldisulfide; HPP, p-hydroxyphenylpyruvate; IPTG, isopropyl thio-b-d-galactoside; LPS, lipopolysaccharide; MIF, macrophage migration inhibitory factor; Trx, thioredoxin; wtMIF, renatured recombinant wild-type human MIF. (Received 25 July 2000, accepted 13 October 2000)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Eur. J. Biochem. 267, 7183±7192 (2000) q FEBS 2000
Dissection of the enzymatic and immunologic functions of macrophagemigration inhibitory factorFull immunologic activity of N-terminally truncated mutants
Robert Kleemann1, Hans Rorsman2, Evald Rosengren3, Ralf Mischke1, Nguyen Tuyet Mai1 and JuÈ rgen Bernhagen1
1Laboratory of Biochemistry, Institute for Interfacial Engineering, University of Stuttgart, Germany; 2Department of Dermatology, and3Department of Pharmacology, University Hospital, Lund, Sweden
Macrophage migration inhibitory factor (MIF) is a cytokine with broad regulatory functions in innate immunity.
MIF belongs to the few cytokines displaying catalytic activities, i.e. MIF has a Pro2-dependent tautomerase and a
Cys-Ala-Leu-Cys (CALC) cysteine-based thiol-protein oxidoreductase activity. Previous studies have addressed
the roles of the catalytic site residues and the C-terminus. The two activities have not been directly compared.
Here we report on the N-terminal mutational analysis and minimization of MIF and on a dissection of the two
catalytic activities by comparing mutants P2AMIF, D4MIF, D5MIF, D6MIF, D7MIF, D8MIF, and D10MIF with
the cysteine mutants of MIF. As N-terminal deletion was predicted to interfere with protein structure due to
disruption of the central b sheet, it was surprising that deletion of up to six N-terminal residues resulted in
normally expressed proteins with wild-type conformation. Strikingly, such mutants exhibited full MIF-specific
immunologic activity. While mutation of Pro2 eliminated tautomerase activity, the CALC cysteine residues had
no influence on this activity. However, mutant C81SMIF, which otherwise has full biologic activity, only had
32% tautomerase activity. Deletion of four N-terminal residues did not interfere with insulin reduction by MIF.
By contrast, reduction of 2-hydroxyethyldisulfide (HED) was markedly affected by N-terminal manipulation,
with P2AMIF and D2MIF exhibiting 40% activity, and D4MIF completely failing to reduce HED. This study
constitutes the first comparison of the two catalytic activities of MIF and should assist in understanding the
molecular links between the catalytic and immunologic activities of this cytokine and in providing guidelines for
N-terminal protein minimization.
Keywords: cytokine; macrophage migration inhibitory factor (MIF); N-terminal mutation; oxidoreductase;
tautomerase.
Macrophage migration inhibitory factor (MIF) wasoriginally discovered as a T-cell factor acting to inhibit themigration of macrophages [1,2]. More recent research hasled to the definition of MIF as a multifunctional, mostlyproinflammatory mediator of broad target cell specificity[3±10]. MIF is a secretion product of both endocrineglands and of immune competent cells [11±16], butregulated MIF secretion may also occur from other cell types[17±21].
MIF has been found to be a critical mediator of severalinflammatory and immune diseases. Best characterized isthe role of MIF as a mediator of endotoxemia and septic
shock [11,22±24]. MIF also plays a role in immune-mediated diseases such as delayed-type hypersensitivityreactions [25], chronic crescentic glomerulonephritis [26,27],and rheumatoid arthritis [28]. Disease-promoting MIF-mediated effects could be central to the pathogenesis of lungdiseases [8,29].
MIF displays a number of properties that distinguish thisfactor from cytokines. Of greatest significance, MIF isinduced rather than inhibited by glucocorticoids and, oncereleased, can act to counter-regulate the immunosuppressiveand anti-inflammatory functions of glucocorticoids [6]. Theglucocorticoid-antagonistic effect of MIF could be linked to thesuggested roles of MIF in several disease states [6,8,22].
MIF exhibits at least two catalytic activities, i.e. atautomerase [30±33] and a thiol-protein oxidoreductase[34±36] activity. As a membrane receptor for MIF has notbeen identified, the catalytic activities are among the primeleads in the search to elucidate the molecular basis of MIFaction. Catalytic activities have previously been reported forcertain immune mediators. Adult T-cell leukemia-derivedfactor/thioredoxin (ADF/Trx) is an oxidoreductase with T cellregulatory and chemokine-like functions [37,38]. The cyclos-porin A-binding factor and macrophage-derived factor cyclo-philin [39,40] exhibits prolyl-peptidyl isomerase activity [41].Other examples exist [42,43]. However, the identified catalyticactivities could not generally be linked to the immunologicfunctions of these molecules. In fact, for some of them, a
Correspondence to J. Bernhagen, Laboratory of Biochemistry,
Institute for Interfacial Engineering, University of Stuttgart and Fraunhofer
Institute FhIGB, Nobelstraûe 12, D-70569 Stuttgart, Germany.
Fax: 1 49 711 970 4200, Tel.: 1 49 711 970 4020,
E-mail: [email protected]
Abbreviations: ADF, adult T cell-derived factor; CALC, Cys-Ala-Leu-Cys;
CSPD, disodium 3-(4-methoxyspiro(1,2-doxetan-3,2 0-(5-chloro)tricyclo-
decan)-4-yl)phenylphosphate; CXXC, Cys-Xaa-Xaa-Cys; GdnHCl,
guanidine hydrochloride; GSH, glutathione; HFIP,
1,1,1,3,3,3-hexafluoroisopropanol; HED, 2-hydroxyethyldisulfide; HPP,
p-hydroxyphenylpyruvate; IPTG, isopropyl thio-b-d-galactoside; LPS,
lipopolysaccharide; MIF, macrophage migration inhibitory factor; Trx,
thioredoxin; wtMIF, renatured recombinant wild-type human MIF.
(Received 25 July 2000, accepted 13 October 2000)

physiological relevance of the catalytic activity could not bedemonstrated [43,44].
A number of substrates have been reported to be convertedby the tautomerase function of MIF. These included-dopachrome, 2-hydroxyphenylpyruvate (HPP), and 3,4-dihy-droxyphenylaminechrome [30,31,45]. By its thiol-proteinoxidoreductase activity, MIF catalyzes the glutathione-dependent reduction of insulin and HED [34,35]. Thetautomerase activity is dependent on the presence of the basicPro2 residue [32,46±48] and the oxidoreductase activity ismediated by the central CALC motif. An intramoleculardisulfide bond can form between Cys57 and Cys60, suggestingthat oxidoreductase action of MIF could be similar to that of
thiol-protein oxidoreductases, i.e. be dependent on a CXXCdithiol-disulfide mechanism [34±36].
A link between the catalytic activities of MIF and itsimmunologic functions has not been unequivocally established.The catalytically inactive mutant C60SMIF does not exhibitglucocorticoid-antagonistic activity, nor does it exhibit othermeasurable macrophage-activating activity. Other CALCmutants (C57SMIF and C57S/C60SMIF) show markedlyreduced immune activities, whereas mutant C81SMIF with aremote cysteine substituted behaves in a manner similar towild-type MIF, suggesting that the oxidoreductase activity isinvolved in at least some of the immunologic properties of MIF[34,36]. A physiological role of the tautomerase activity of MIFhas been discussed controversially. Catalytically inactivetautomerase mutants such as P2GMIF, P2SMIF, P2AMIF, andDP2MIF have been found to either be immunologically inactive[33,49] or to exhibit close-to-wild-type-like immunologicactivity [32,50]. C-terminally truncated mutants of MIF exhibitfull oxidoreductase activity [51], but fail to show tautomerase[32] and immunologic activity [32,51]. While the latter dataconfirm the suggested importance of the C-terminus fortautomerase activity [52], they also imply that C-terminaltruncation is unlikely to result in potent minimized MIFanalogs.
N-terminal mutants of MIF, other than those involvingsubstitution or deletion of the Pro2 residue have not beeninvestigated. Moreover, a potential mechanistic interrelation-ship between the two catalytic activities of MIF or theircorresponding catalytic centers has not been addressed. Inparticular, the cysteine mutants have not been examined fortautomerase activity, and, conversely, N-terminal mutants havenot been assayed in the oxidoreductase tests.
Here, we have performed a comprehensive comparison of thetwo catalytic activities by testing various established and novelcatalytic mutants in the enzymatic and immunologic assays. Wehave tested whether N-terminal truncation could be a usefulapproach to generate biologically active minimized MIFs.Although previous studies had indicated that the N-terminalregion was suceptible to structural changes [4], otherconsiderations favoured the argument that the N-terminuscould be a good target structure; i.e. there is no signal sequencenecessary for protein secretion, and, although predicted to berigid, the N-terminal region seemed to be better suited forminimization than inner regions of the molecule, wheremutations had been found to readily lead to protein aggregationand insolubilization [36]. Thus, this study also covers theexpression, purification, and biochemical characterization ofseveral novel N-terminally truncated MIF mutants, i.e. mutantsD4MIF, D5MIF, D6MIF, D7MIF, D8MIF, and D10MIF.
The design of these mutants was based on the followingconsiderations: Pro2 is part of the b1 strand of MIF. This strandis formed by residues Pro2 to Thr8 (Fig. 1A). We anticipatedthat truncation of the b1 strand could lead to an altered overallprotein structure, as b1 is an integral component of the centralfour-stranded b sheet. This b sheet is not only the corecomponent of the MIF monomer, but is also involved insubunit oligomerization by interactions with adjacent subunits[59]. The b1 strand forms an antiparallel b sheet with b4,bringing the residues involved in the two catalytic activities intosomewhat close proximity. Thus, and although only the moreN-terminal portion of b1 has been predicted to be involved insubstrate binding in the N-terminal catalytic pocket, it seemedworthwhile to investigate how b1 truncation would affect thebiological activities of MIF. Mutants D4±D8MIF (Fig. 1B)were generated for this purpose. Mutant D10MIF was examined
Fig. 1. Structure of human MIF. (A) Ribbon structure of the MIF
monomer [74]. (B) Primary sequence and secondary structure of the MIF
monomer. Cylinders represent a-helices and dark arrows show b strands.
Deleted or substituted amino acids are indicated, i.e. N-terminal mutants are
termed according to the number of amino acids deleted. N-termini are
derived from the corresponding cDNA sequences with the start methionine
numbered Met1.
7184 R. Kleemann et al. (Eur. J. Biochem. 267) q FEBS 2000

to address the possibility that b1 truncation would notdrastically interfere with protein structure and activity. In thiscase, the effects of a truncation of the following loop elementcould be assessed. Mutant D4MIF was of particular importance,as the Phe4 residue is close to the tautomerase substrate bindingsite [47,48] and is part of the b1 strand. As for mutant D10MIF,mutants D13MIF and D17MIF were constructed to examine apossible importance of the loop connecting the b1 strand andthe a1 helix. Mutant D26MIF was designed to interrupt thea1 helix structure. Considering that N-terminal deletions couldaffect both mRNA and protein stability, the devised truncationscomprised rather small portions of one or two amino acidsdeleted at a time (Fig. 1B).
M A T E R I A L S A N D M E T H O D S
Materials
Lipopolysaccharide (LPS) of the type O111:B4 was obtainedfrom Sigma-Aldrich Chemicals (Deisenhofen, Germany). Dyeterminator cycle DNA sequencing kits were from Perkin Elmer-Applied Biosystems Inc. (ABI) (Weiterstadt, Germany) andpET expression plasmids were from Novagen (Madison, WI,USA). Oligonucleotide primers were acquired from LifeTechnologies/Gibco BRL (Eggenstein, Germany) and allother molecular biology reagents were from Gibco or NewEngland Biolabs GmbH (Heidelberg, Germany). Enzymes,substrates, and miscellaneous chemicals, which were of thehighest grade commercially available, were bought fromSigma-Aldrich.
Cloning, expression, and purification of the mutants
The cloning, expression, purification, and renaturation of wild-type human MIF (wtMIF) and the cysteine mutants C57SMIF,C60SMIF, and C81SMIF have been described [4,34,36]. TheN-terminal mutants D2MIF, P2AMIF, D4MIF, D5MIF, D6MIF,D7MIF, D8MIF, and D10MIF were generated by the samepolymerase chain reaction-dependent mutagenesis protocol andcloning strategy. The 5 0-primers for these mutants, which allcontained a NdeI restriction site overhang and varied with thedeletion, were: 5 0-GCTAGCGCATATGGCGATGTTCATCG-TAAACAC-3 0 for mutant P2AMIF; 5 0-GCTAGCGCA-TATGTTCATCGTAAACACCAACGTGC-3 0 for mutantD2MIF; 5 0-GGCTAGCGCATATGATCGTAAACACCAACGT-GCCCCGC-3 0 for mutant D4MIF; 5 0-GGCTAGCGCATATGG-TAAACACCAACGTGCCCCGC-3 0 for mutant D5MIF;5 0-GGCTAGCGCATATGAACACCAACGTGCCCCGCGCC-3 0for mutant D6MIF; 5 0-GGCTAGCGCATATGACCAACGTG-CCCCGCGCCTCC-3 0 for mutant D7MIF; 5 0-GGCTAGCGC-ATATGAACGTGCCCCGCGCCTCCGTGCCG-3 0 for mutantD8MIF; and 5 0-GGCTAGCGCATATGCCCCGCGCCTCC-GTGCC-3 0 for mutant D10MIF. The corresponding 3 0-primercontained a BamHI restriction site overhang (5 0-CGGGATCCT-TAGGCGAAGGTGGAGTTGTTCCAGC-3 0). PCR-amplifiedmutant DNA was digested with NdeI and BamHI, and ligatedinto the pET11b expression vector. All generated cDNAsequences were confirmed by bidirectional DNA sequencing.
For expression, purification, and renaturation of N-terminalmutants P2AMIF and D2MIF, the protocol established forwtMIF [4] was followed, except that a French press(1240 p.s.i.) was used for cell disruption. It was noted,however, that mutant D2MIF exhibited a greater tendencythan wtMIF to aggregate during the renaturation procedure(< 30% of the protein precipitated). The other expressible
mutants, i.e. D4MIF, D5MIF, and D6MIF, formed inclusionbodies and were purified by protocols recently devised for theinclusion body-forming catalytic center mutants of MIF [36].Briefly, mutant D4MIF behaved similar to C60SMIF andmarkedly precipitated during the final renaturation step. Thismutant was thus reconstituted at a lower protein concentrationof 40±90 mg´mL21 [36]. Mutants D5MIF and D6MIF were notsoluble in the standard 20 mm sodium phosphate renaturationbuffer. They were refolded by shock dilution from the 100%1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) stock solution (forthe CD measurements) or from the 100% dimethylsulfoxidestock solution (for the glucocorticoid overriding assay) [36]. Allfinal protein preparations were determined to be essentiallyLPS-free as quantified by the Limulus amebocyte lysateQCL-1000 bacterial endotoxin quantitation kit (BioWhittaker,Walkersville MD, USA), containing , 1 pg LPS per mgprotein.
Mutant protein analytics and northern blotting analysis
Identity of the generated mutant proteins was verified by SDS/PAGE/Western blotting analysis [4,5]. SDS/PAGE/Westernblotting also served to estimate the expression levels of themutant proteins in the E. coli expression system. Blots weredeveloped with a donkey anti-(rabbit IgG) horseradishperoxidase-conjugated secondary Ig (Amersham PharmaciaLife Science, Freiburg, Germany); the Super Signal West DuraExtended Duration Substrate (Pierce, St. Augustin, Germany),and the luminescent image analyser LAS-1000Plus (Raytest,Straubenhardt, Germany) were used for detection.
For Northern analysis, a MIF DNA probe correspondingto bases 159±345 of the human MIF cDNA was amplifiedby PCR and was digoxigenin-labelled using the DIG-HighPrime DNA labeling kit (Roche Diagnostics, Mannheim,Germany). Expression of the mutants in the E. coli BL21(DE3) strain was induced with 1 mm isopropyl thio-b-d-galactoside (IPTG) for 90 min. Total cellular RNA wasimmediately isolated with the Trizol Reagent (Gibco), andequal amounts of RNA electrophoresed in a 1% agarose gel andblotted onto positively charged nylon membranes (RocheDiagnostics, Mannheim, Germany) according to standardprocedures. The DNA probe was hybridized with theQuickHyb hybridization solution (Stratagene, Amsterdam,The Netherlands). For luminescent detection, an alkalinephosphatase-conjugated anti-DIG antibody was utilizedtogether with disodium 3-(4-methoxyspiro(1,2-doxetan-3,2'-(5 0chloro)tricyclodecan)-4-yl)phenylphosphate (CSPD) as asubstrate (Roche Diagnostics, Mannheim, Germany).
Circular dichroism spectropolarimetry
Established far-ultraviolet circular dichroism spectroscopy(CD) conditions [4,34] were applied. Briefly, stock solutionsof the renatured proteins dissolved in 20 mm sodium phosphatebuffer (pH 7.2) were diluted to a final concentration of 1 mm inthe same buffer. For mutants D5MIF and D6MIF, the 100%HFIP protein stock solutions (1500 mg´mL21) were diluted to afinal protein concentration of 1 mm, containing a final HFIPconcentration of 0.8%. Spectra were recorded in reference tothe corresponding control spectra and represent net spectra withthe control spectra already subtracted. Secondary structurecontents were calculated as described [34] using the Lincombprogram [53] in combination with the reference protein datasets of Perczel et al. [53], Yang et al. [54], and Brahms &Brahms [55].
q FEBS 2000 Enzymatic and immunologic functions of MIF (Eur. J. Biochem. 267) 7185

Catalytic activity assays
Tautomerase activity was measured by an established tauto-merase assay using HPP as a substrate [31,56]. Tautomeraseactivities of the mutant proteins in comparison to wtMIFwere measured at a final protein concentration of 40±100 nm.Thiol-protein oxidoreductase activity was determined by theHED transhydrogenase and the insulin reduction assay. HEDtranshydrogenase activities were measured as reported pre-viously [34,57]. Final protein concentrations applied were3.4 mm [35,36]. Catalytic activities in the insulin reductionassay were measured using the classical protocol [58] incombination with the previously established MIF-specificmodifications [34,35]. Final protein concentrations in thisassay were 1.8 mm.
Immunologic activity assay
Glucocorticoid counter-regulating activity of MIF was assayedessentially as described [6]. Briefly, MIF-mediated overridingof the anti-inflammatory effect of dexamethasone was deter-mined by measuring LPS-stimulated tumor necrosis factor(TNF) production in peripheral blood monocytes. Peripheralblood monocytes were isolated from an unstressed donor,isolated by Ficoll gradient and adherence, washed, and plated ata concentration of 1 � 106 cells´mL21. Cells were incubatedfor 1 h with 1029 m dexamethasone and 80 pm wtMIF ormutant protein. The corresponding reconstitution buffers alonewere used as controls and showed no measurable macrophage-activating effect. LPS was added at a concentration of1 mg´mL21 together with another 80 pm of test protein andcells were incubated for 14 h. Culture supernatants wereprepared and analyzed for TNF content using a commercialhuman TNF ELISA (R & D Systems GmbH, Wiesbaden,Germany).
R E S U L T S
Expression, purification, and characterization of theN-terminal mutants
PCR-amplified DNA of the N-terminal mutants was cloned intothe pET11b expression vector and mutants overexpressed inE. coli. Bacterial pellets were analyzed for total proteinexpression by SDS/PAGE/silver staining and Western blottinganalysis (Fig. 2). Mutants P2AMIF, D2MIF, D4MIF, andD5MIF were found to be expressed in good amountscomparable to the expression levels seen for wtMIF. Expression
of D6MIF was considerably reduced. Mutants D7MIF, D8MIF,and D10MIF were hardly expressed. Further truncation of theN-terminal region resulted in nonexpressible mutant proteins,i.e. mutants D13MIF, D17MIF, and D26MIF could not beexpressed (not shown).
To clarify whether the observed reduced expression levelswere due to decreased transcription levels, we performedNorthern blotting analysis. Total RNA of the mutants andwtMIF was isolated after IPTG-induced expression andanalyzed with a MIF DNA probe for transcription efficiency(data not shown). All mutants exhibited mRNA levels similar tothose of wtMIF, indicating that the decreased expression levelsof some of the mutants were not due to blocked transcription ordecreased mRNA stability. As inclusion body formation andsolubility (see below) was similar for these mutants, it appearedlikely that the observed differences in protein expression werebased on translational events. Interestingly, in vitro transcrip-tion and translation of two of the badly expressible N-terminalmutants, mutants D7MIF and D10MIF, in a reticulocyte lysatesystem resulted in comparable translation levels as for wtMIF.However, the other poorly expressed mutants failed to expressin the in vitro translation system. While this indicated thatfailure of expression in E. coli could be circumvented for someof the mutants by using another expression system, it also madeit clear that further investigation of the translational effects thatcould be responsible for the observed different expressionlevels of the N-terminal mutants will be necessary.
Mutants P2AMIF and D2MIF could be readily purified tohomogeneity using the method established for wtMIF [4]. Thisresult was in agreement with studies by others who haveexpressed and purified P2AMIF, P2SMIF, P2GMIF, andD2MIF, essentially using the protocols established for thewild-type protein [32,33,46]. Expression of the other expres-sible N-terminal mutants, i.e. mutants D4MIF, D5MIF, andD6MIF led to massive inclusion body formation. These mutantswere exclusively found in the insoluble lysate fraction, and hadto be purified following a recently devised protocol forinsoluble MIF mutants (see Materials and methods) [36].Using these protocols, mutants D4MIF and D5MIF wereobtained in high yields (13.5 mg protein per L bacterialculture); mutant D6MIF was obtained in smaller yields(3.8 mg protein´L21).
Conformational analysis
Far-UV CD was applied to examine the conformation afterreconstitution. Spectra of mutants P2AMIF, D2MIF, andD4MIF were recorded in a standard aqueous buffer and were
Fig. 2. Recombinant expression of the
N-terminal mutants in E. coli BL21(DE3) in
comparison to wtMIF. (A) SDS/PAGE analysis
following IPTG-induced (1) vs. noninduced (±)
bacterial expression. Total bacterial lysates were
electrophoresed in 18% polyacrylamide gels
together with a wtMIF standard (MIF) and
expressed proteins visualized by silver staining.
(B) Western blotting analysis of the indicated
IPTG-induced lysates using a wtMIF-specific
polyclonal antibody and a horse radish
peroxidase-conjugated secondary antibody to
confirm protein identity and to increase
detection sensitivity.
7186 R. Kleemann et al. (Eur. J. Biochem. 267) q FEBS 2000
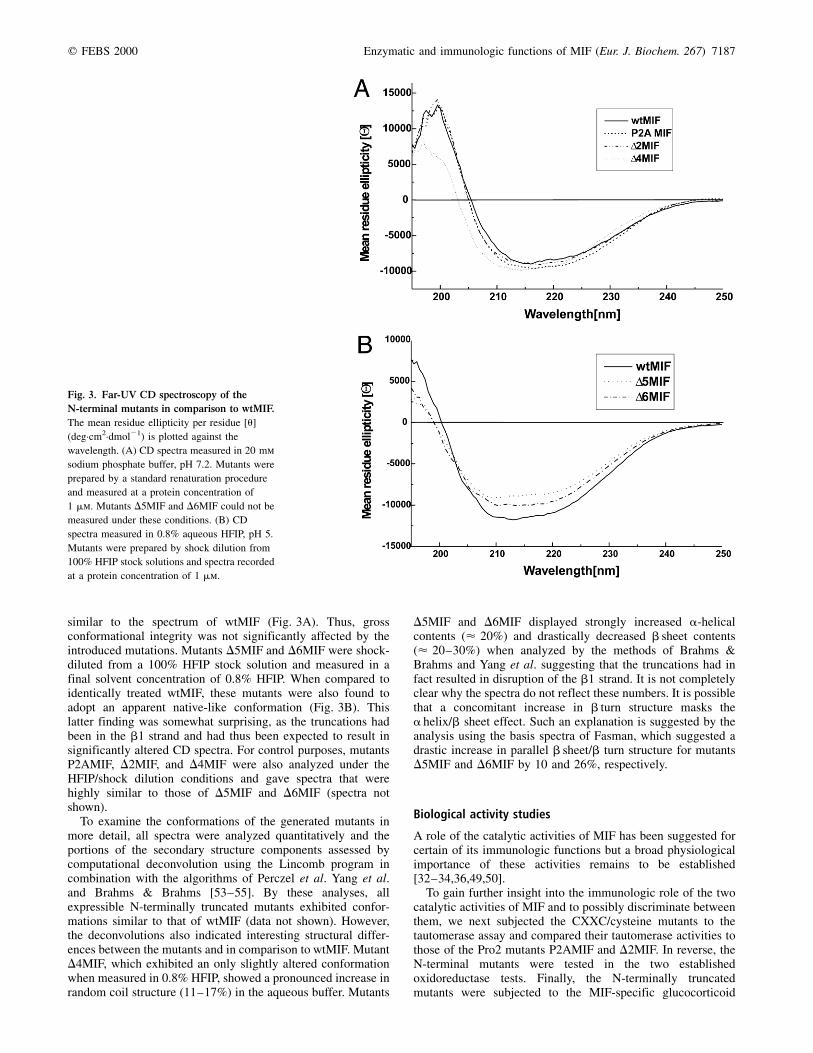
similar to the spectrum of wtMIF (Fig. 3A). Thus, grossconformational integrity was not significantly affected by theintroduced mutations. Mutants D5MIF and D6MIF were shock-diluted from a 100% HFIP stock solution and measured in afinal solvent concentration of 0.8% HFIP. When compared toidentically treated wtMIF, these mutants were also found toadopt an apparent native-like conformation (Fig. 3B). Thislatter finding was somewhat surprising, as the truncations hadbeen in the b1 strand and had thus been expected to result insignificantly altered CD spectra. For control purposes, mutantsP2AMIF, D2MIF, and D4MIF were also analyzed under theHFIP/shock dilution conditions and gave spectra that werehighly similar to those of D5MIF and D6MIF (spectra notshown).
To examine the conformations of the generated mutants inmore detail, all spectra were analyzed quantitatively and theportions of the secondary structure components assessed bycomputational deconvolution using the Lincomb program incombination with the algorithms of Perczel et al. Yang et al.and Brahms & Brahms [53±55]. By these analyses, allexpressible N-terminally truncated mutants exhibited confor-mations similar to that of wtMIF (data not shown). However,the deconvolutions also indicated interesting structural differ-ences between the mutants and in comparison to wtMIF. MutantD4MIF, which exhibited an only slightly altered conformationwhen measured in 0.8% HFIP, showed a pronounced increase inrandom coil structure (11±17%) in the aqueous buffer. Mutants
D5MIF and D6MIF displayed strongly increased a-helicalcontents (< 20%) and drastically decreased b sheet contents(< 20±30%) when analyzed by the methods of Brahms &Brahms and Yang et al. suggesting that the truncations had infact resulted in disruption of the b1 strand. It is not completelyclear why the spectra do not reflect these numbers. It is possiblethat a concomitant increase in b turn structure masks thea helix/b sheet effect. Such an explanation is suggested by theanalysis using the basis spectra of Fasman, which suggested adrastic increase in parallel b sheet/b turn structure for mutantsD5MIF and D6MIF by 10 and 26%, respectively.
Biological activity studies
A role of the catalytic activities of MIF has been suggested forcertain of its immunologic functions but a broad physiologicalimportance of these activities remains to be established[32±34,36,49,50].
To gain further insight into the immunologic role of the twocatalytic activities of MIF and to possibly discriminate betweenthem, we next subjected the CXXC/cysteine mutants to thetautomerase assay and compared their tautomerase activities tothose of the Pro2 mutants P2AMIF and D2MIF. In reverse, theN-terminal mutants were tested in the two establishedoxidoreductase tests. Finally, the N-terminally truncatedmutants were subjected to the MIF-specific glucocorticoid
Fig. 3. Far-UV CD spectroscopy of the
N-terminal mutants in comparison to wtMIF.
The mean residue ellipticity per residue [u]
(deg´cm2´dmol21) is plotted against the
wavelength. (A) CD spectra measured in 20 mm
sodium phosphate buffer, pH 7.2. Mutants were
prepared by a standard renaturation procedure
and measured at a protein concentration of
1 mm. Mutants D5MIF and D6MIF could not be
measured under these conditions. (B) CD
spectra measured in 0.8% aqueous HFIP, pH 5.
Mutants were prepared by shock dilution from
100% HFIP stock solutions and spectra recorded
at a protein concentration of 1 mm.
q FEBS 2000 Enzymatic and immunologic functions of MIF (Eur. J. Biochem. 267) 7187
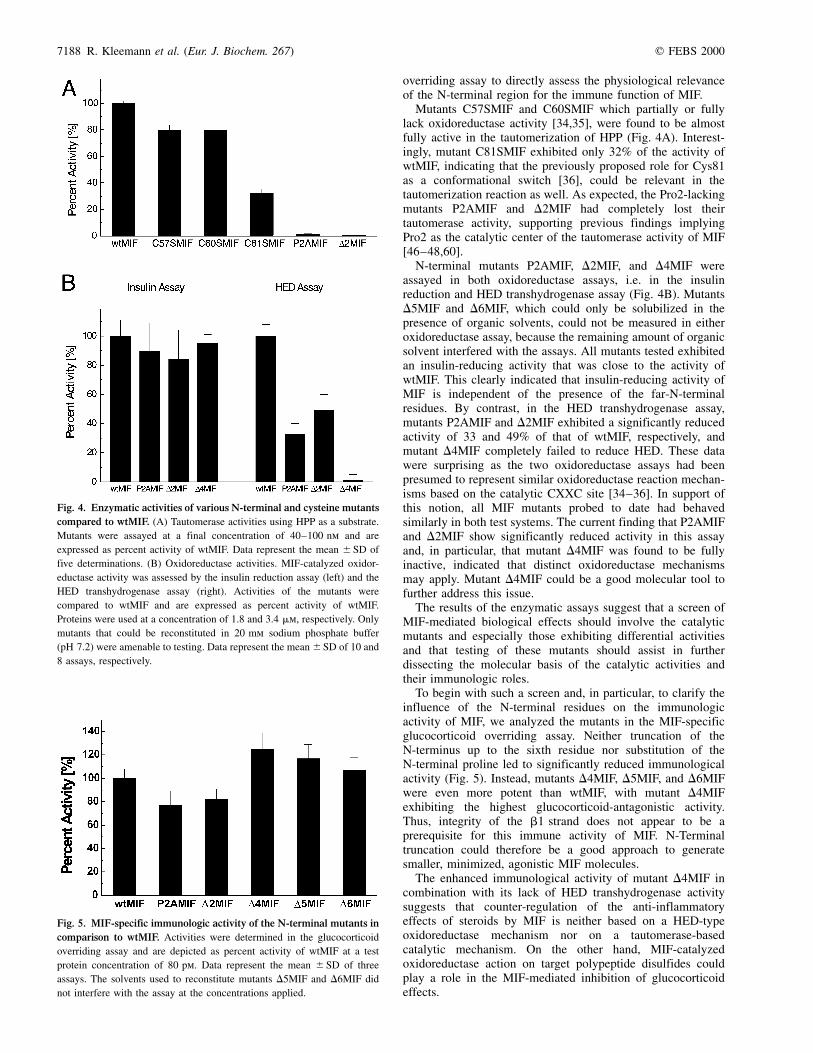
overriding assay to directly assess the physiological relevanceof the N-terminal region for the immune function of MIF.
Mutants C57SMIF and C60SMIF which partially or fullylack oxidoreductase activity [34,35], were found to be almostfully active in the tautomerization of HPP (Fig. 4A). Interest-ingly, mutant C81SMIF exhibited only 32% of the activity ofwtMIF, indicating that the previously proposed role for Cys81as a conformational switch [36], could be relevant in thetautomerization reaction as well. As expected, the Pro2-lackingmutants P2AMIF and D2MIF had completely lost theirtautomerase activity, supporting previous findings implyingPro2 as the catalytic center of the tautomerase activity of MIF[46±48,60].
N-terminal mutants P2AMIF, D2MIF, and D4MIF wereassayed in both oxidoreductase assays, i.e. in the insulinreduction and HED transhydrogenase assay (Fig. 4B). MutantsD5MIF and D6MIF, which could only be solubilized in thepresence of organic solvents, could not be measured in eitheroxidoreductase assay, because the remaining amount of organicsolvent interfered with the assays. All mutants tested exhibitedan insulin-reducing activity that was close to the activity ofwtMIF. This clearly indicated that insulin-reducing activity ofMIF is independent of the presence of the far-N-terminalresidues. By contrast, in the HED transhydrogenase assay,mutants P2AMIF and D2MIF exhibited a significantly reducedactivity of 33 and 49% of that of wtMIF, respectively, andmutant D4MIF completely failed to reduce HED. These datawere surprising as the two oxidoreductase assays had beenpresumed to represent similar oxidoreductase reaction mechan-isms based on the catalytic CXXC site [34±36]. In support ofthis notion, all MIF mutants probed to date had behavedsimilarly in both test systems. The current finding that P2AMIFand D2MIF show significantly reduced activity in this assayand, in particular, that mutant D4MIF was found to be fullyinactive, indicated that distinct oxidoreductase mechanismsmay apply. Mutant D4MIF could be a good molecular tool tofurther address this issue.
The results of the enzymatic assays suggest that a screen ofMIF-mediated biological effects should involve the catalyticmutants and especially those exhibiting differential activitiesand that testing of these mutants should assist in furtherdissecting the molecular basis of the catalytic activities andtheir immunologic roles.
To begin with such a screen and, in particular, to clarify theinfluence of the N-terminal residues on the immunologicactivity of MIF, we analyzed the mutants in the MIF-specificglucocorticoid overriding assay. Neither truncation of theN-terminus up to the sixth residue nor substitution of theN-terminal proline led to significantly reduced immunologicalactivity (Fig. 5). Instead, mutants D4MIF, D5MIF, and D6MIFwere even more potent than wtMIF, with mutant D4MIFexhibiting the highest glucocorticoid-antagonistic activity.Thus, integrity of the b1 strand does not appear to be aprerequisite for this immune activity of MIF. N-Terminaltruncation could therefore be a good approach to generatesmaller, minimized, agonistic MIF molecules.
The enhanced immunological activity of mutant D4MIF incombination with its lack of HED transhydrogenase activitysuggests that counter-regulation of the anti-inflammatoryeffects of steroids by MIF is neither based on a HED-typeoxidoreductase mechanism nor on a tautomerase-basedcatalytic mechanism. On the other hand, MIF-catalyzedoxidoreductase action on target polypeptide disulfides couldplay a role in the MIF-mediated inhibition of glucocorticoideffects.
Fig. 4. Enzymatic activities of various N-terminal and cysteine mutants
compared to wtMIF. (A) Tautomerase activities using HPP as a substrate.
Mutants were assayed at a final concentration of 40±100 nm and are
expressed as percent activity of wtMIF. Data represent the mean ^ SD of
five determinations. (B) Oxidoreductase activities. MIF-catalyzed oxidor-
eductase activity was assessed by the insulin reduction assay (left) and the
HED transhydrogenase assay (right). Activities of the mutants were
compared to wtMIF and are expressed as percent activity of wtMIF.
Proteins were used at a concentration of 1.8 and 3.4 mm, respectively. Only
mutants that could be reconstituted in 20 mm sodium phosphate buffer
(pH 7.2) were amenable to testing. Data represent the mean ^SD of 10 and
8 assays, respectively.
Fig. 5. MIF-specific immunologic activity of the N-terminal mutants in
comparison to wtMIF. Activities were determined in the glucocorticoid
overriding assay and are depicted as percent activity of wtMIF at a test
protein concentration of 80 pm. Data represent the mean ^ SD of three
assays. The solvents used to reconstitute mutants D5MIF and D6MIF did
not interfere with the assay at the concentrations applied.
7188 R. Kleemann et al. (Eur. J. Biochem. 267) q FEBS 2000

D I S C U S S I O N
To date, a comprehensive comparison of the two catalyticactivities of MIF and a systematic assessment of theirphysiological roles has not been attempted. To our knowledge,this report addresses this issue for the first time and investigatesthe catalytic activities of the corresponding mutants in bothenzyme assays, together with testing in a MIF-specificimmunologic assay.
Our data demonstrate that the CXXC motif or thecysteines therein are not necessary for the tautomeraseactivity of MIF. By contrast, the presence of Cys81 isimportant for MIF-mediated tautomerase activity. In reverse,neither the Pro2 residue, which is central to the tautomeraseactivity, nor the other N-terminal residues up to position sixare necessary for the insulin-reducing oxidoreductase activityof MIF.
Our study confirms that the Pro2 residue is not required forthe glucocorticoid overriding activity of MIF [32] and,surprisingly, demonstrates that further truncation of theN-terminus down to the sixth residue does not considerablyinterfere with the conformational integrity of the protein and itsglucocorticoid overriding activity. N-terminal truncation evenappears to result in enhanced immunologic activity.
Intruigingly, the far-N-terminal residues seem to be involvedin the reduction of small molecular mass disulfides such asHED. This finding suggests that the two catalytic activities or,at least, the corresponding underlying structural elements arelinked. For example, as the two oxidoreductase assaysprincipally differ by the substrates converted, i.e. polypeptidedisulfides in the insulin reduction assay and small molecularmass disulfides in the HED test, it is tempting to speculate thatthe N-terminal region of MIF is necessary for the binding/shuttling of small molecular mass disulfides, while polypeptidedisulfides may dock by other mechanisms. Of note, adecapeptide fragment of MIF, spanning the CALC motifcatalyzes insulin reduction, but fails to reduce HED [34],suggesting that polypeptide disulfides such as insulin maydirectly bind to the CXXC region or to residues in closeproximity to this region [61] while small compound substratesmay need more remote docking sites.
The novel N-terminal mutants as examined and characterizedin this study appear to be good molecular tools for thedissection of the catalytic activities. Mutant D4MIF appears tobe particularly suitable for this purpose, as it exhibits a higher-than-wild-type overriding activity, full insulin-reducing activity,but no HED transhydrogenase activity. This mutant shouldtherefore be tested for the established other immunologicactivities of MIF.
Structure activity studies require good amounts of pureprotein. This is usually achieved by overexpression in bacterialsystems such as in the pET11b/E. coli BL21 system, andpurification of the protein investigated from the soluble lysatefraction. We found that mutations within the hydrophobic coreof MIF mostly led to massive inclusion body formation andmutant proteins with delicate solubility characteristics [36].The hydrophobic inner regions of the MIF molecule appear tobecome exposed by the substitutions, which, in turn, favorsprotein±protein association, which competes with the nativefolding process.
Given the solubility characteristics of the N-terminal mutantsas explored in this study, the following considerations seemplausible: N-terminal residues Pro2 to Thr8 form the b1 strand,which is an integral component of the central b sheet of MIF.The adjacent antiparallel b4 strand, which includes parts of the
CALC motif and is formed by amino acids Ala58 to Ser64,interacts with b1. MIF obviously requires an intact four-stranded central b sheet and, in particular, an intact b1/b4 sheetto retain its solubility properties in physiological solvents.N-terminal truncations appear to only be tolerated whenintroduced at the far N-terminal end. The `b1/b4 sheet-model' may also explain why mutant C57SMIF behavesnormally. Although located immediately adjacent to b4,Cys57 is not part of the b1/b4 sheet. While the influence ofthe lateral b strands, b2 and b5, remains to be investigated indetail, work by Bendrat et al. [32] in which a His residue withinb2 was substituted, suggests that a requirement for the integrityof the lateral strands might not be as stringent. From theseconsiderations one might speculate that a `mini-MIF' could beas small as the b1/b4 core or variations thereof.
Our findings concerning the expression and solubilitycharacteristics of the N-terminally truncated MIF variantshave biotechnological implications if minimized MIFanalogs are to be expressed at high level [62]. MIF expressionin E. coli is well established and recombinant MIF proteinfrom E. coli exhibits full immunologic and catalytic activity[4±6,11,31±34,46].
However, expression levels of the N-terminally deletedmutants were significantly decreased in the pET11b/E. coliBL21 system, when more than six amino acids were removedfrom the N-terminus. As mRNAs transcription or stability werenot affected, protein translation or stability is likely to beaffected by the introduced mutations. However, only indirectconclusions may be drawn. The identity of the N-terminalamino acid can be an important determinant of the proteolyticstability of a protein [63±65]. The introduced deletions shouldstabilize rather than destabilize the resulting mutant proteins.The presence of Met (D2MIF), Ile (D4MIF), Val (D5MIF), Asn(D6MIF and D8MIF), Thr (D7MIF), Ser (D13MIF), and Gly(D17MIF) should confer a high stability in E. coli [66]. Thisindicates that ineffective translational events rather than proteinstability are responsible for reduced expression of mutantsD7MIF-D26MIF. The successful expression of two N-terminalmutants in programmed reticulocyte lysates in the presence of apotent proteinase inhibitor cocktail supports this notion.
MIF has been found to catalyze the tautomerization ofseveral compounds including HPP [30,31]. Co-crystallizationand inhibitor experiments have led to the identification of thetautomerase substrate binding site [32,46±48] at the interfacebetween two subunits. None of the residues involved is part ofthe catalytic CALC center or the b4 strand. Our finding that theoxidoreductase mutants exhibit full tautomerase is thus inagreement with the definition of the tautomerase binding site.
Mutant C81SMIF is the only mutant described withsignificantly reduced tautomerase activity, which, at the sametime, exhibits full oxidoreductase and immunologic activity.The observed reduced tautomerase activity of mutant C81SMIFcould be due to conformational changes after the substitution ofCys81 by serine, but could also be due to the low-pH conditionsof the assay itself [33,36,56].
Previous analysis of various MIF mutants in the oxidor-eductase assays had indicated that both assays appeared to becomparable measures of the ability of MIF to reduce disulfidesubstrates [34±36]. We were thus surprised to find that mutantD4MIF behaved drastically different in the two oxidoreductaseassays. Mutants P2AMIF and D2MIF, to a minor degree,showed similar characteristics. Differences in protein structureare unlikely to explain the pronounced differences observedbetween the reduction of insulin and HED. Also, in bothassays, reduced glutathione was used as a reductant. The
q FEBS 2000 Enzymatic and immunologic functions of MIF (Eur. J. Biochem. 267) 7189

most prominent difference thus appears to be the size andnature of the substrate converted. As discussed, our datasuggest that the N-terminus could be a docking site for HED.However, as HED does not appear to share structural propertieswith the known tautomerase substrates, such a site is yet tobe defined. One prominent example for a protein whoseN-terminus is not part of the active site but is important forconformational stabilization and enzymatic functioning, isfirefly luciferase [67].
Protein minimization has been successfully attempted tobetter characterize the mode of action of proteins, to highlightessential determinants of structure and function, and toobtain protein analogs of high potency and stability and lowimmunogenicity for potential therapeutic applications [68±73].Attempts to minimize the 114-amino-acid MIF molecule havebeen disappointing. While predicted to be structurally flexible,deletion of 5±10 residues from the C-terminus of MIF abolishesboth tautomerase and immunologic activity [32,51]. Deletionsof more than one residue at the N-terminus have not beenperformed. Excisions of internal regions such as loops have notbeen attempted and are likely to lead to drasticallyinterfere with protein structure. Our data, although surprisingdue to the predicted structural rigidity of the N-terminus of MIF[52,59], demonstrate that N-terminal deletions may become anexcellent approach for minimization of the MIF molecule.Glucocorticoid overriding is the most important immunologiceffect currently known to be exerted by MIF [6,8] and thetruncated N-terminal mutants investigated exhibited fullactivity in this assay. This finding confirms studies by Bendratet al. [32] and Hermanowski-Vosatka et al. [50] who havefound that Pro2 is not required for immunologic activity ofMIF, but is inconsistent with the findings of Swope et al. [33]and Onodera et al. [49], who report on a link between thepresence of Pro2 and immune function. Closer examination ofthe assays applied suggests that Pro2 and the N-terminus maybe dispensible for MIF action on macrophages ([32,50], andthis study), while it appears to play a role when MIF target cellaction is assessed on neutrophils and stromal cells such asfibroblasts [33,49]. Additional studies including additionalimmune assays and cell types are needed to further resolvethis issue.
As mutants D7±D26MIF were hardly expressible in thebacterial expression system used and thus could not be testedfurther, it is not clear whether truncations beyond Val6 wouldalso lead to biologically active MIF analogs.
We conclude that MIF may be minimized at the N-terminus,at least to some extent, while retaining its most prominentbiological activity, its glucocorticoid antagonistic effect. Thus,N-terminally truncated MIF analogs might be devised to eitheract as MIF agonists or to serve to inhibit or compete withendogenous MIF. The latter application could be of potentialtherapeutic benefit in the various inflammatory conditionswhere MIF has been demonstrated to play a role [8,26,28,29]and which are often treated with glucocorticoids.
This study constitutes the first direct comparison of the twocatalytic activities of MIF and suggests that both activities aredependent on the structural integrity of a joint underlyingstructural element, i.e. the antiparallel b1/b4 sheet. Thesurprising finding that the immunologic activity of MIF isboth independent of the presence of the Pro2 residue and anintact b1 strand indicates that N-terminal truncation offers angood approach for molecule minimization. Our data argue thatdifferential behaviour of the N-terminal mutants in the twooxidoreductase enzyme assays together with the observedreduced tautomerase activity of mutant C81SMIF should allow
to better dissect the molecular links between the catalytic andimmunologic activities of MIF.
A C K N O W L E D G E M E N T S
We are grateful to A. Kapurniotu for assistance with the CD experiments.
We thank N. Greenfield and G. Fasman for kindly providing the CD
software programs and W. Voelter for use of the CD instrument. We are
grateful to A. Beckler for reading of the manuscript. J. B. is supported by
the Deutsche Forschungsgemeinschaft (DFG) grant numbers BE 1977/1-1
and 1-2.
R E F E R E N C E S
1. David, J.R. (1966) Delayed hypersensitivity in vitro: its mediation by
cell-free substances formed by lymphoid cell±antigen interaction.
Proc. Natl Acad. Sci. USA 56, 72±77.
2. Bloom, B. & Bennett, B. (1966) Mechanism of a reaction in vitro
associated with delayed-type hypersensitivity. Science 153, 80±82.
3. Bernhagen, J., Calandra, T. & Bucala, R. (1998) Regulation of the
immune response by macrophage migration inhibitory factor:
biological and structural features. J. Mol. Med. 76, 151±161.
4. Bernhagen, J., Mitchell, R.A., Calandra, T., Voelter, W., Cerami, A. &
Bucala, R. (1994) Purification, bioactivity, and secondary structure
analysis of mouse and human macrophage migration Inhibitory factor
(MIF). Biochemistry 33, 14144±14155.
5. Calandra, T., Bernhagen, J., Mitchell, R.A. & Bucala, R. (1994) The
macrophage is an important and previously unrecognized source of
macrophage migration inhibitory factor. J. Exp. Med. 179,
1985±1902.
6. Calandra, T., Bernhagen, J., Metz, C.N., Spiegel, L.A., Bacher, M.,
Donnelly, T., Cerami, A. & Bucala, R. (1995) MIF as a
glucocorticoid-induced modulator of cytokine production. Nature
377, 68±71.
7. JuÈttner, S., Bernhagen, J., Metz, C., RoÈllinghoff, M., Bucala, R. &
Gessner, A. (1998) Macrophage migration inhibitory factor induces
killing of Leishmania major by macrophages: dependence on reactive
nitrogen intermediates and endogenous TNF-a. J. Immunol. 161,
2383±2390.
8. Donnelly, S.C., Haslett, C., Reid, P.T., Grant, I.S., Wallace, W.A., Metz,
C.N., Bruce, L.J. & Bucala, R. (1997) Regulatory role for
macrophage migration inhibitory factor in acute respiratory distress
syndrome. Nat. Med. 3, 320±323.
9. Bacher, M., Metz, C.N., Calandra, T., Mayer, K., Chesney, J., Lohoff,
M., Gemsa, D., Donnelly, T. & Bucala, R. (1996) An essential
regulatory role for macrophage migration inhibitory factor in T-cell
activation. Proc. Natl Acad. Sci. USA 93, 7849±7854.
10. Mitchell, R.A., Metz, C.N., Peng, T. & Bucala, R. (1999) Sustained
mitogen-activated protein kinase (MAPK) and cytoplasmic phos-
pholipase A2 activation by macrophage migration inhibitory factor
(MIF). Regulatory role in cell proliferation and glucocorticoid action.
J. Biol. Chem. 274, 18100±18106.
11. Bernhagen, J., Calandra, T., Mitchell, R.A., Martin, S.B., Tracey, K.J.,
Voelter, W., Manogue, K.R., Cerami, A. & Bucala, R. (1993) MIF is a
pituitary-derived cytokine that potentiates lethal endotoxaemia.
Nature 365, 756±759.
12. Nishino, T., Bernhagen, J., Shiiki, H., Calandra, T., Dohi, K. & Bucala,
R. (1995) Localization of macrophage migration inhibitory factor
(MIF) to secretory granules within the corticotrophic and thyro-
trophic cells of the pituitary gland. Mol. Med. 1, 781±788.
13. Waeber, G., Calandra, T., Roduit, R., Haefliger, J.A., Bonny, C.,
Thompson, N., Thorens, B., Temler, E., Meinhardt, A., Bacher, M.,
Metz, C.N., Nicod, P. & Bucala, R. (1997) Insulin secretion is
regulated by the glucose-dependent production of islet beta cell
macrophage migration inhibitory factor. Proc. Natl Acad. Sci. USA
94, 4782±4787.
14. Bacher, M., Meinhardt, A., Lan, H.Y., Mu, W., Metz, C.N., Chesney,
J.A., Calandra, T., Gemsa, D., Donnelly, T., Atkins, R. & Bucala, R.
7190 R. Kleemann et al. (Eur. J. Biochem. 267) q FEBS 2000

(1997) Migration inhibitory factor expression in experimentally
induced endotoxemia. Am. J. Pathol. 15, 235±246.
15. Suzuki, H., Kanagawa, H. & Nishihira, J. (1996) Evidence for the
presence of macrophage migration inhibitory factor in murine
reproductive organs and early embryos. Immunol. Lett. 51, 141±147.
16. Meinhardt, A., Bacher, M., McFarlane, J.R., Metz, C.N., Seitz, J.,
Hedger, M.P., deKretser, D.M. & Bucala, R. (1996) Macrophage
migration inhibitory factor production by Leydig cells: evidence for a
role in the regulation of testicular function. Endocrinology 137,
5090±5095.
17. Nishihira, J., Koyama, Y. & Mizue, Y. (1998) Identification of
macrophage migration inhibitory factor (MIF) in human vascular
endothelial cells and its induction by lipopolysaccharide. Cytokine
10, 199±205.
18. Matsuda, A., Tagawa, Y., Matsuda, H. & Nishihira, J. (1997)
Expression of macrophage migration inhibitory factor in corneal
wound healing in rats. Invest. Ophthalmol. Vis. Sci. 38, 1555±1562.
19. Hirokawa, J., Sakaue, S., Furuya, Y., Ishii, J., Hasegawa, A., Tagami,
S., Kawakami, Y., Sakai, M., Nishi, S. & Nishihira, J. (1998)
Tumor necrosis factor-alpha regulates the gene expression of
macrophage migration inhibitory factor through tyrosine kinase-
dependent pathway in 3T3-L1 Adipocytes. J. Biochem. 123,
733±739.
20. Shimizu, T., Abe, R., Ohkawara, A. & Nishihira, J. (1999) Ultraviolet B
radiation upregulates the production of macrophage migration
inhibitory factor (MIF) in human epidermal keratinocytes. J. Invest.
Dermatol. 112, 210±215.
21. Bacher, M., Meinhardt, A., Lan, H.Y., Dhabhar, F.S., Mu, W., Metz,
C.N., Chesney, J.A., Gemsa, D., Donnelly, T., Atkins, R.C. & Bucala,
R. (1998) MIF expression in the rat brain ± Implications for neuronal
function. Mol. Med. 4, 217±230.
22. Calandra, T., Spiegel, L.A., Metz, C.N. & Bucala, R. (1998)
Macrophage migration inhibitory factor is a critical mediator of the
activation of immune cells by exotoxins of Gram-positive bacteria.
Proc. Natl Acad. Sci. USA 95, 11383±11388.
23. Bozza, M., Satoskar, A.R., Lin, G., Lu, B., Humbles, A.A., Gerard, C.
& David, J.R. (1999) Targeted disruption of migration inhibitory
factor gene reveals its critical role in sepsis. J. Exp. Med. 189,
341±346.
24. Calandra, T., Echtenacher, B., Le Roy, D., Pugin, J., Metz, C.N.,
HuÈltner, L., Heumann, D., MaÈnnel, D., Bucala, R. & Glauser, M.
(2000) Protection from septic shock by neutralization of macrophage
migration inhibitory factor (MIF). Nat. Med. 6, 164±169.
25. Bernhagen, J., Bacher, M., Calandra, T., Metz, C.N., Doty, S.B.,
Donnelly, T. & Bucala, R. (1996) An essential role for macrophage
migration inhibitory factor in the tuberculin delayed-type hyper-
sensitivity reaction. J. Exp. Med. 183, 277±282.
26. Lan, H.Y., Mu, W., Yang, N., Meinhardt, A., Nikolic-Paterson, D.J.,
Ng, Y.Y., Bacher, M., Atkins, R.C. & Bucala, R. (1996) De novo
renal expression of macrophage migration inhibitory factor during the
development of rat crescentic glomerulonephritis. Am. J. Pathol. 149,
1119±1127.
27. Lan, H.Y., Bacher, M., Yang, N., Mu, W., Nicolic-Paterson, D.J., Metz,
C., Meinhardt, A., Bucala, R. & Atkins, R.C. (1997) The pathogenic
role of macrophage migration inhibitory factor in immunologically
induced kidney disease in the rat. J. Exp. Med. 185, 1455±1465.
28. Mikulowska, A., Metz, C.N., Bucala, R. & Holmdahl, R. (1997)
Macrophage migration inhibitory factor is involved in the patho-
genesis of collagen type II-induced arthritis in mice. J. Immunol. 158,
5514±5517.
29. Rossi, A.G., Haslett, C., Hirani, N., Greening, A.P., Rahman, I., Metz,
C.N., Bucala, R. & Donnelly, S.C. (1998) Human circulating
eosinophils secrete macrophage migration inhibitory factor (MIF).
Potential role in asthma. J. Clin. Invest. 101, 2869±2874.
30. Rosengren, E., Bucala, R., AÊ man, P., Jacobsson, L., Odh, G., Metz,
C.N. & Rorsman, H. (1996) The immunoregulatory mediator
macrophage migration inhibitory factor (MIF) catalyzes a tauto-
merization reaction. Mol. Med. 2, 143±149.
31. Rosengren, E., Aman, P., Thelin, S., Hansson, C., Ahlfors, S., BjoÈrk, P.,
Jacobsson, L. & Rorsman, H. (1997) The macrophage migration
inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett.
417, 85±88.
32. Bendrat, K., Alabed, Y., Callaway, D.J.E., Peng, T., Calandra, T., Metz,
C.N. & Bucala, R. (1997) Biochemical and mutational investigations
of the enzymatic activity of macrophage migration inhibitory factor.
Biochemistry 36, 15356±15362.
33. Swope, M., Sun, H.-W., Blake, P.R. & Lolis, E. (1998) Direct link
between cytokine activity and a catalytic site for macrophage
migration inhibitory factor. EMBO J. 1, 3534±3541.
34. Kleemann, R., Kapurniotu, A., Frank, R.W., Gessner, A., Mischke, R.,
Flieger, O., JuÈttner, S., Brunner, H. & Bernhagen, J. (1998) Disulfide
analysis reveals a role for macrophage migration inhibitory factor
(MIF) as a thiol-protein oxidoreductase. J. Mol. Biol. 280, 85±102.
35. Kleemann, R., Mischke, R., Kapurniotu, A., Brunner, H. & Berhagen.J.
(1998) Specific reduction of insulin disulfides by macrophage
migration inhibitory factor (MIF) with glutathione and dihydro-
lipoamide: potential role in cellular redox processes. FEBS Lett. 430,
191±196.
36. Kleemann, R., Kapurniotu, A., Mischke, R., Held, J. & Bernhagen, J.
(1999) Characterization of catalytic center mutants of macrophage
migration inhibitory factor (MIF) and comparison with C81S MIF.
Eur. J. Biochem. 261, 753±766.
37. Tagaya, Y., Maeda, Y., Mitsui, A., Kondo, Matsui, H., Hamuro, J.,
Brown, R., Arai, K., Yokota, T., Wakasugi, N. & Yodoi, J. (1989)
ATL-derived factor (ADF), an IL-2 receptor/Tac inducer homologous
to thioredoxin; possible involvement of dithiol-reduction in the IL-2
receptor induction. EMBO J. 8, 757±764.
38. Bertini, R., Howard, O.M.Z., Dong, H.-F., Oppenheim, J.J., Bizzarri, C.,
Caselli, G., Pagliei, S., Romines, B., Wilshire, J.A., Mengozzi, M.,
Nakamura, H., Yodoi, J., Pekkari, K., Gurunath, R., Holmgren, A.,
Herzenberg, L.A., Herzenberg, L.A. & Ghezzi, P. (1999) Thio-
redoxin, a redox enzyme released in infection and inflammation, is a
unique chemoattractant for neutrophils, monocytes, and T cells.
J. Exp. Med. 189, 1783±1789.
39. Sherry, B., Yarlett, N., Strupp, A. & Cerami, A. (1992) Identification of
cyclophilin as a pro-inflammatory secretory product of lipopoly-
saccaride-activated macrophages. Proc. Natl Acad. Sci. USA 89,
3511±3515.
40. Xu, Q., Leiva, M.C., Fischkoff, S.A., Handschumacher, R.E. & Lyttle,
C.R. (1992) Leukocyte chemotactic activity of cyclophilin. J. Biol.
Chem. 267, 11968±11971.
41. Fischer, G. (1994) Peptidyl-prolyl cis/trans isomerases and their
effectors. Angew. Chem. Int 33, 1415±1436.
42. Hoogewerf, A., Leone, J., Reardon, I., Howe, J., Asa, D., Heinrikson,
R. & Ledbetter, S. (1995) CXC chemokines connective tissue
activating peptide-III and neutrophil activating peptide-2 are heparin/
heparan sulfate-degrading enzymes. J. Biol. Chem. 270, 3268±3277.
43. Chaput, M., Claes, V., Portetelle, D., Cludts, I., Cravador, A., Burny, A.,
Gras, H. & Tartar, A. (1988) The neurotrophic factor neuroleukin is
90% homologous with phosphohexose isomerase. Nature 332,
454±455.
44. Fischer, G., Tradler, T. & Zarnt, T. (1998) The mode of action of
peptidyl prolyl cis/trans isomerases in vivo: binding vs. catalysis.
FEBS Lett. 426, 17±20.
45. Matsunaga, J., Sinha, D., Pannell, L., Santis, C., Solano, F., Wistow,
G.J. & Hearing, V.J. (1999) Enzyme activity of macrophage
migration inhibitory factor toward oxidized catecholamines. J. Biol.
Chem. 274, 3268±3271.
46. Stamps, S.L., Fitzgerald, M.C. & Whitman, C.P. (1998) Characteriza-
tion of the role of the amino-terminal proline in the enzymatic
activity catalyzed by macrophage migration inhibitory factor.
Biochemistry 37, 10195±10202.
47. Lubetsky, J.B., Swope, M., Dealwis, C., Blake, P. & Lolis, E. (1999)
Pro-1 of macrophage migration inhibitory factor functions as a
catalytic base in the phenylpyruvate tautomerase activity.
Biochemistry 38, 7346±7354.
48. Taylor, A.B., Johnson, W.H. Jr, Czerwinski, R.M., Li, H.S., Hackert,
M.L. & Whitman, C.P. (1999) Crystal structure of macrophage
q FEBS 2000 Enzymatic and immunologic functions of MIF (Eur. J. Biochem. 267) 7191

migration inhibitory factor complexed with (E)-2-fluoro-p-hydroxy-
cinnamate at 1.8 AÊ resolution: implications for enzymatic catalysis
and inhibition. Biochemistry 38, 7444±7452.
49. Onodera, S., Kaneda, K., Mizue, Y., Koyama, Y., Fuyinaga, M. &
Nishihira, J. (2000) Macrophage migration inhibitory factor
up-regulates expression of matrix metalloproteinases in synovial
fibroblasts of rheumatoid arthritis. J. Biol. Chem. 275, 444±450.
50. Hermanowski-Vosatka, A., Mundt, S.S., Ayala, J.M., Goyal, S.,
Hanlon, W.A., Czerwinski, R.M., Wright, S.D. & Whitman, C.P.
(1999) Enzymatically inactive macrophage migration inhibitory
factor inhibits monocyte chemotaxis and random migration.
Biochemistry 38, 12841±12849.
51. Mischke, R., Gessner, A., Kapurniotu, A., JuÈttner, S., Kleemann, R.,
Brunner, H. & Bernhagen, J. (1997) Structure activity studies of the
cytokine macrophage migration inhibitory factor (MIF) reveal a
critical role for its carboxy terminus. FEBS Lett. 414, 226±232.
52. Suzuki, M., Sugimoto, H., Nakagawa, A., Tanaka, I., Nishihira, J. &
Sakai, M. (1996) Crystal structure of the macrophage migration
inibitory factor from rat liver. Nat. Struct. Biol. 3, 259±266.
53. Perczel, A., Park, K. & Fasman, G.D. (1992) Analysis of the circular
dichroism spectrum of proteins using the convex constraint
algorithm: a practical guide. Anal. Biochem. 203, 83±93.
54. Yang, J.T., Wu, C.S. & Martinez, H.M. (1974) Calculation of protein
conformation from circular dichroism. Methods Enzymol. 130,
208±269.
55. Brahms, S. & Brahms, J. (1980) Determination of protein secondary
structure in solution by vacuum ultraviolet circular dichroism. J. Mol.
Biol. 138, 149±178.
56. Knox, W.E. (1955) p-Hydroxyphenylpyruvate enol-keto tautomerase.
Methods Enzymol. 2, 289±292.
57. Holmgren, A. (1985) Glutaredoxin from Escherichia coli and calf
thymus. Methods Enzymol. 113, 525±528.
58. Holmgren, A. (1979) Thioredoxin catalyses the reduction of insulin
disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254,
9627±9632.
59. Sun, H., Bernhagen, J., Bucala, R. & Lolis, E. (1996) Crystal structure
at 2.6 AÊ resolution of human macrophage migration inhibitory factor.
Proc. Natl Acad. Sci. USA 93, 5191±5196.
60. Bendrat, K., Alabed, Y., Callaway, D.J.E., Peng, T., Calandra, T., Metz,
C.N. & Bucala, R. (1997) Biochemical and nutational investigations
of the enzymatic activity of macrophage migration inhibitory factor.
Biochemistry 36, 15356±15362.
61. Martin, J.L., Bardwell, J.C.A. & Kuriyan, J. (1993) Crystal structure of
the DsbA protein required for disulphide bond formation in vivo.
Nature 365, 464±468.
62. Rudolph, R. (1996) Chapter 10: Successful protein folding on an
industrial scale. In Protein Engeneering: Principles and Practice.
(Cleland, J.L. & Craik, C.S., eds), pp. 283±298. Wiley-Liss, New
York, USA.
63. Varshavsky, A. (1992) The N-end rule. Cell 69, 725±735.
64. Varshavsky, A. (1997) The N-end rule pathway of protein degradation.
Genes Cells 2, 13±28.
65. Tobias, J.W., Shrader, T.E., Rocap, G. & Varshavsky, A. (1991) The
N-end rule in bacteria. Science 254, 1374±1377.
66. Georgiou, G. (1996) Expression of proteins in bacteria. In Protein
Engeneering: Principles and Practice (Cleland, J.L. & Craik, C.S.,
eds), pp. 101±127. Wiley-Liss, New York, USA.
67. Sung, D. & Kang, H. (1998) The N-terminal amino acid sequences of
the firefly luciferase are important for the stability of the enzyme.
Photochem. Photobiol. 68, 749±753.
68. Livnah, O., Stura, E.A., Johnson, D.L., Middleton, S.A., Mulcahy, L.S.,
Wrighton, N.C., Dower, W.J., Jolliffe, L.K. & Wilson, I.A. (1996)
Functional mimicry of a protein hormone by a peptide agonist: the
EPO receptor complex at 2.8 AÊ . Science 273, 464±471.
69. McConnell, S.J., Dinh, T., Le, M.H., Brown, S.J., Becherer, K.,
Blumeyer, K., Kautzer, C., Axelrod, F. & Spinella, D.G. (1998)
Isolation of erythropoietin receptor agonist peptides using evolved
phage libraries. Biol. Chem. 379, 1279±1286.
70. Li, B., Tom, J.Y., Oare, D., Yen, R., Fairbrother, W.J., Wells, J.A. &
Cunningham, B.C. (1995) Minimization of a polypeptide hormone.
Science 270, 1657±1660.
71. McInnes, C., Wang, J.E.A.M.A., Yansouni, C., O'Connor-McCourt, M.
& Sykes, B.D. (1998) Structure-based minimization of transforming
growth factor-alpha (TGF-alpha) through NMR analysis of the
receptor-bound ligand. Design, solution structure, and activity of
TGF-alpha 8±50. J. Biol. Chem. 273, 27357±27363.
72. Hua, Q.X., Hu, S.Q., Jia, W., Chu, Y.C., Burke, G.T., Wang, S.H.,
Wang, R.Y., Katsoyannis, P.G. & Weiss, M.A. (1998) Mini-proinsulin
and mini-IGF-I: homologous protein sequences encoding non-
homologous structures. J. Mol. Biol. 277, 103±118.
73. Domingues, H., Cregut, D., Sebald, W., Oschkinat, H. & Serrano, L.
(1999) Rational design of a GCN-derived mimetic of interleukin-4.
Nat. Struct. Biol. 6, 652±656.
74. Kraulis, P.J. (1991) MOLSCRIPT: a program to produce both detailed
and schematic plots of protein structures. J. Appl. Cryst. 24,
946±950.75.
7192 R. Kleemann et al. (Eur. J. Biochem. 267) q FEBS 2000
Related Documents











