Tongue Control for Swallowing in Parkinson’s Disease: Effects of Age, Rate, and Stimulus Consistency Pascal H.H.M. Van Lieshout, PhD, 1,2,3,4 * Catriona M. Steele, PhD, 1,2,4,5,6 and Anthony E. Lang, MD 7 1 Department of Speech-Language Pathology, University of Toronto, Toronto, Ontario, Canada; 2 Institute of Biomaterials & Biomedical Engineering, University of Toronto, Toronto, Ontario, Canada; 3 Department of Psychology, University of Toronto at Mississauga, Mississauga, Ontario, Canada; 4 Toronto Rehabilitation Institute, Toronto, Ontario, Canada; 5 Bloorview Research Institute, Toronto, Ontario, Canada; 6 Graduate Department of Rehabilitation Science, University of Toronto, Toronto, Ontario, Canada; 7 Toronto Western Hospital Research Institute, University Health Network, Toronto, Ontario, Canada ABSTRACT Background: Patients with Parkinson’s disease often suffer from swallowing problems, especially at more advanced stages of the disease. Efficient swallows require well-coordinated tongue movements during bolus flow, but little is known about such movements in Parkinson’s disease. Methods: The current study presents data on tongue movements for patients with mild to moderate Parkin- son’s disease (n 5 10), age-matched adults (n 5 13), and younger healthy adults (n 5 15). Results: Participants with Parkinson’s disease showed smaller and more variable movements in the horizontal movement plane, indicating that tongue movements are affected in early stages of Parkinson’s disease. Conclusions: The small and more variable movements in the horizontal plane of Patients with Parkinson’s dis- ease may pose challenges for swallowing liquids effi- ciently and safely. V C 2011 Movement Disorder Society Key Words: tongue movement; swallowing; Parkin- son’s disease; liquids; articulography; aging Swallowing impairments (dysphagia) are common in patients with Parkinson’s disease (PD) and are gener- ally unresponsive to antiparkinson therapies. 1,2 The most commonly reported features are impaired tongue control, prolonged oral and pharyngeal transit, and reduced strength and amplitude of tongue movement during bolus propulsion. 3–5 These features are claimed to increase the risk of aspiration. 5,6 Those who aspi- rate are at risk for developing pneumonia, which has the highest mortality of all comorbidities in PD. 7–9 In addition to the effects of PD itself on swallowing, medication-related dyskinesias can contribute to swal- lowing difficulties. 10 In clinical settings, texture modi- fications (eg, pureed foods or thickened liquids) and careful control of sip size or swallowing rate (discrete vs sequential swallows) are commonly recommended for the management of dysphagia in PD. 11–13 To date however, there is little objective evidence of the effi- cacy of these strategies, and their influence on tongue movements remains unclear. 13,14 Consequently, the oral control mechanisms that govern safety and effi- ciency of swallowing are not fully understood. PD mainly affects the elderly. Although dysphagia is not an immediate concern for older people, recent studies do report decreased motor flexibility and oral sensitivity with aging. 15–19 It is therefore important that swallowing deficits in PD be dissociated from normal age-related changes. It is of special interest to determine whether age-controlled observations of swallowing in people with early-stage PD (but no clin- ical manifestation of dysphagia) show differences in tongue movement, which might hint at an increased risk for dysphagia with disease progression. The Current Study We present data on tongue movements collected using electromagnetic midsagittal articulography (EMMA) 20,21 in 3 groups of individuals: patients with early-stage PD but no clinical diagnosis of dysphagia, age-matched healthy controls, and younger healthy con- trols. This latter group enabled us to dissociate the effects of aging and PD. In addition, we studied 3 stimuli (water, apple juice, and thickened apple juice) and ------------------------------------------------------------ Additional Supporting Information may be found in the online version of this article. *Correspondence to: Dr. Pascal H.H.M. Van Lieshout, University of Toronto, Department of Speech-Language Pathology, Oral Dynamics Lab (ODL), 500 University Avenue, Toronto, Ontario M5G 1V7, Canada; [email protected] Relevant conflicts of interest/financial disclosures: Nothing to report. This research was supported by funding from the Canadian Institutes of Health Research (Grant 63271) and in part from the Canada Research Chairs Program. Support also came from the Toronto Rehabilitation Institute, which receives funding under the Provincial Rehabilitation Research Program from the Ministry of Health and Long-term Care in Ontario. Full financial disclosures and author roles may be found in the online version of this article. Received: 13 July 2010; Revised: 14 January 2011; Accepted: 31 January 2011 Published online 3 May 2011 in Wiley Online Library (wileyonlinelibrary.com). DOI: 10.1002/mds.23690 BRIEF REPORTS Movement Disorders, Vol. 26, No. 9, 2011 1725

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Tongue Control for Swallowing inParkinson’s Disease: Effects of
Age, Rate, and StimulusConsistency
Pascal H.H.M. Van Lieshout, PhD,1,2,3,4*Catriona M. Steele, PhD,1,2,4,5,6 and Anthony E. Lang, MD7
1Department of Speech-Language Pathology, University of Toronto,
Toronto, Ontario, Canada; 2Institute of Biomaterials & Biomedical
Engineering, University of Toronto, Toronto, Ontario, Canada;3Department of Psychology, University of Toronto at Mississauga,
Mississauga, Ontario, Canada; 4Toronto Rehabilitation Institute,
Toronto, Ontario, Canada; 5Bloorview Research Institute, Toronto,
Ontario, Canada; 6Graduate Department of Rehabilitation Science,
University of Toronto, Toronto, Ontario, Canada; 7Toronto Western
Hospital Research Institute, University Health Network, Toronto,
Ontario, Canada
ABSTRACTBackground: Patients with Parkinson’s disease oftensuffer from swallowing problems, especially at moreadvanced stages of the disease. Efficient swallowsrequire well-coordinated tongue movements duringbolus flow, but little is known about such movementsin Parkinson’s disease.Methods: The current study presents data on tonguemovements for patients with mild to moderate Parkin-son’s disease (n 5 10), age-matched adults (n 5 13),and younger healthy adults (n 5 15).Results: Participants with Parkinson’s disease showedsmaller and more variable movements in the horizontalmovement plane, indicating that tongue movementsare affected in early stages of Parkinson’s disease.
Conclusions: The small and more variable movementsin the horizontal plane of Patients with Parkinson’s dis-ease may pose challenges for swallowing liquids effi-ciently and safely. VC 2011 Movement Disorder Society
Key Words: tongue movement; swallowing; Parkin-son’s disease; liquids; articulography; aging
Swallowing impairments (dysphagia) are common inpatients with Parkinson’s disease (PD) and are gener-ally unresponsive to antiparkinson therapies.1,2 Themost commonly reported features are impaired tonguecontrol, prolonged oral and pharyngeal transit, andreduced strength and amplitude of tongue movementduring bolus propulsion.3–5 These features are claimedto increase the risk of aspiration.5,6 Those who aspi-rate are at risk for developing pneumonia, which hasthe highest mortality of all comorbidities in PD.7–9 Inaddition to the effects of PD itself on swallowing,medication-related dyskinesias can contribute to swal-lowing difficulties.10 In clinical settings, texture modi-fications (eg, pureed foods or thickened liquids) andcareful control of sip size or swallowing rate (discretevs sequential swallows) are commonly recommendedfor the management of dysphagia in PD.11–13 To datehowever, there is little objective evidence of the effi-cacy of these strategies, and their influence on tonguemovements remains unclear.13,14 Consequently, theoral control mechanisms that govern safety and effi-ciency of swallowing are not fully understood.PD mainly affects the elderly. Although dysphagia is
not an immediate concern for older people, recentstudies do report decreased motor flexibility and oralsensitivity with aging.15–19 It is therefore importantthat swallowing deficits in PD be dissociated fromnormal age-related changes. It is of special interest todetermine whether age-controlled observations ofswallowing in people with early-stage PD (but no clin-ical manifestation of dysphagia) show differences intongue movement, which might hint at an increasedrisk for dysphagia with disease progression.
The Current Study
We present data on tongue movements collectedusing electromagnetic midsagittal articulography(EMMA)20,21 in 3 groups of individuals: patients withearly-stage PD but no clinical diagnosis of dysphagia,age-matched healthy controls, and younger healthy con-trols. This latter group enabled us to dissociate the effectsof aging and PD. In addition, we studied 3 stimuli(water, apple juice, and thickened apple juice) and
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Dr. Pascal H.H.M. Van Lieshout, University ofToronto, Department of Speech-Language Pathology, Oral DynamicsLab (ODL), 500 University Avenue, Toronto, Ontario M5G 1V7, Canada;[email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.This research was supported by funding from the Canadian Institutes ofHealth Research (Grant 63271) and in part from the Canada ResearchChairs Program. Support also came from the Toronto RehabilitationInstitute, which receives funding under the Provincial RehabilitationResearch Program from the Ministry of Health and Long-term Care inOntario.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 13 July 2010; Revised: 14 January 2011; Accepted:31 January 2011Published online 3 May 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23690
B R I E F R E P O R T S
Movement Disorders, Vol. 26, No. 9, 2011 1725

swallowing rate (sequential vs discrete). This comprehen-sive design allowed us to determine the specific impact ofearly-stage PD on tongue behaviors in swallowing.
Patients and Methods
Participants
Ten adults with a clinical diagnosis of mild–moder-ate idiopathic PD (�3 on Hoehn and Yahr22) wererecruited from the Morton & Gloria Shulman Move-ment Disorders Centre at Canada’s Toronto WesternHospital (Table 1). They were without prior stroke orother neurological diseases and passed an oral motorand swallowing examination performed by a regis-tered speech-language pathologist. Some were on med-ication during the study, but none presented withdyskinesia at the time of data collection.To differentiate effects of aging from PD, we
recruited 2 groups of healthy adults: a group age-matched to the PD participants (O, n ¼ 13) and agroup of younger adults (Y, n ¼ 15). They all under-went an oral motor and swallowing screening to con-firm eligibility. The study was approved by ResearchEthics Boards at the University Health Network To-ronto and the University of Toronto.
Procedures
The effect of stimulus consistency was tested usingthin (water [H2O], apple juice [APP]) and honey-thick-ened (apple juice [HON]) liquids. As in previousresearch of our laboratory,23,24 EMMA (AG-100,Carstens Medizinelektronik, Bovenden, Germany) was
used to sample movement data at 400 Hz using smalltransducers attached with surgical methacrylate resin(Cyanodent, Ellman International Mfg., Oceanside, NY)in midline to the lips, mandible, tongue blade (1 cmbehind true tongue tip), tongue body (2 cm behind firsttransducer), and tongue dorsum (2 cm behind tonguebody transducer). All data were transformed to a uni-form bite-plane aligned coordinate space.Participants performed 6 discrete swallows (lower-
ing the cup from the lips between swallows) and 6 se-quential swallows (without cup lowering) at acomfortable pace with each stimulus. Data for 144swallows were analyzed (2 sessions � 2 trials of 6 sipseach � 3 stimuli � 2 rates) per participant. Task orderwas randomized. Cup weights were measured beforeand after each trial to calculate average sip volume.25
Signal Processing and Statistical Analysis
Movement signals were band-pass filtered (0.1–6Hz), and tongue movements were corrected for jawinfluence.21 Only tongue body (TB) and tongue dor-sum (TD) data are presented, as these are most rele-vant for swallowing. Two direction-specific kinematicparameters (amplitude and duration) and 1 move-ment-cycle-level parameter (cyclic spatiotemporalindex [cSTI]) were calculated using standard proce-dures developed in our lab.20 Higher cSTI values indi-cate greater variability in movement patterns. Inabsence of session effects, measures were averagedacross 24 swallows prior to statistical analysis.A mixed-design repeated-measures analyses of var-
iance was used with GROUP as between-participant
Table 1. Characteristics of participants
PD patients Older controls Younger controls
Number (M/F) 10 (7/3) 13 (7/6) 15 (7/8)Age (y), mean 6 SD (range) 62.8 6 9.6 (47–75) 66.2 6 4.2 (56–73) 22.6 6 3.4 (19–29)UPDRS-III, median 6 MAD (range) 20.5 6 7 (5–45)Hoehn and Yahr score 2 6 0.5 (1–3)
Individual data on PD patients
Patient code Sex* Age at time of study (y) Disease duration (y) Treatment**
P35 M 74 4 —P42 F 61 2 —P43 F 57 1 PPX 3 mgP44 F 75 5 LD 800 mgP45 M 69 5 —P47 M 70 3 PPX 3 mgP48 M 67 3 LD 450 mgP49 M 54 3 —P50 M 47 3 PPX 1.5 mg (Ama 300)P52 M 54 1 —
For clinical measures, we report median and median absolute deviation (MAD) measures in addition to range.*M, male; F, female.**Five patients on no treatment; 3 only on a dopamine agonist pramipexole (PPX), mean dose 2.5 mg/d (P50 also taking amantadine [Ama] 300 mg/d); 2 onlevodopa/carbidopa (LD), mean dose 625 mg/d.
V A N L I E S H O U T E T A L .
1726 Movement Disorders, Vol. 26, No. 9, 2011

factor and STIMULUS, RATE, and coil LOCATIONas within-subject factors. cSTI analyses were con-ducted separately for the horizontal (X) and vertical(Y) planes. Movement amplitude and duration wereanalyzed separately by direction (up, down, forward,backward). Tukey–Kramer multiple-comparison testsand follow-up ANOVAs were used where appropriate.Effect sizes (Cohen’s F-statistic26) are reported for sig-nificant findings (<0.02, small; 0.15–0.35, moderate;>0.35, strong).
Results
Sip Size
Mean (6 SD) sip size (5.16 6 1.48 mL/sip) did notdiffer significantly between participant groups (F2,35 ¼2.89, P ¼ .069). Main effects were found for STIMU-LUS type (F2,70 ¼ 173.12, P < .001, f ¼ 3.01) andRATE (F1,35 ¼ 16.38, P < .001, f ¼ 0.636). Sip sizeswere smallest for HON (4.2 6 1.31 mL/sip), largestfor H2O (5.8 6 1.28 mL/sip), and intermediate forAPP (5.5 6 1.33 mL/sip). Sequential swallows weresmaller (5.0 6 1.47 mL/sip) than discrete swallows(5.3 6 1.49 mL/sip).
Kinematic Data
Given the large amount of data, we will focus onthe significant group-related findings, followed by ashort description of stimulus, rate, and coil locationeffects. Means (and SDs) for amplitude and durationand an overview of significant main and interactioneffects are provided as supplementary material online.
Group Differences
There were no significant main GROUP effects ontongue movement. Significant GROUP � RATE inter-actions were found for the durations of upward (F2,35¼ 4.39, P ¼ .02, f ¼ 0.422) and forward (F2,35 ¼4.48, P ¼ .019, f ¼ 0.428) movements. In the sequen-tial rate condition, younger controls showed shorterupward movement durations (0.752 6 0.307 seconds)than older controls (0.928 6 0.342 seconds) and PDparticipants (1.001 6 0.441 seconds), who did not dif-fer from each other. For the forward movements,none of the post hoc tests reached significance. Dura-tions of downward movement showed a GROUP �RATE � LOCATION interaction (F2,35 ¼ 4.69, P ¼.016, f ¼ 0.441), which was based on shorter tonguebody durations for PD participants compared withboth control groups. Significant GROUP � STIMU-LUS � RATE interactions were found for the ampli-tudes of upward (F4,70 ¼ 3.07, P ¼ .022, f ¼ 0.467),backward (F4,70 ¼ 3.91, P ¼ .006, f ¼ 0.553), andforward (F4,70 ¼ 5.35, P ¼ .001, f ¼ 0.677) move-ments. Post hoc tests for upward amplitudes revealedsignificantly smaller amplitudes for younger controls
(7.86 6 2.66 mm) for HON, compared with similardata for older controls (9.96 6 2.68 mm) and PD par-ticipants (9.90 6 3.67 mm). In contrast, backwardand forward movement amplitudes were smaller in PDparticipants for both sequential (significantly differentfrom older controls) and discrete (significantly differ-ent from younger controls) swallows of HON. Finally,there was a GROUP � STIMULUS interaction forcSTI in the horizontal movement plane (F4,70 ¼ 2.98,P ¼ .025, f ¼ 0.457). Post hoc tests revealed a signifi-cant group effect for HON only (Fig. 1).
Stimuli, Rate, and Location Differences
In general, HON differed significantly from thinliquids, with larger amplitudes and longer durations.Variability of movement cycles was highest for HON,except for discrete swallows in the X plane. Discreteswallows showed significantly greater variability, largeramplitudes, and longer durations than sequential swal-lows. The tongue dorsum showed larger movementranges than the tongue body, especially in the verticaldirection. Variability for horizontal movements oftongue dorsum was higher than that of tongue body,but the opposite was true for vertical movements.
DiscussionSummarizing the findings, we found rate and liquid-
specific differences between participants with early-stagePD and healthy controls. First, tongue motion in thehorizontal plane showed smaller movement amplitudesfor the PD participants with honey-thickend liquids.Second, PD participants showed consistently shorterdurations of downward movement regardless of stimu-lus and rate conditions. In contrast, upward movementsfor sequential swallows were prolonged with age and inPD compared with younger individuals. Finally, PD par-ticipants showed more variable movements in the hori-zontal plane with honey-thickened liquids. Overall,honey-thickened apple juice elicited larger, slower, andmore variable tongue movements than thin liquids. Dis-crete swallows showed larger, slower, and more vari-able tongue motions than sequential swallows.During the oral phase, impairments in mastication,
oral preparatory tongue movements, and lingual boluscontrol are among the most common symptomsobserved in PD.4,5 In our participants, tongue controlfor the swallowing of both thin and thick liquids wasrelatively preserved. This fits with the notion thatswallowing problems may not manifest themselvesearly in PD. However, the differences that were found(smaller movement ranges and greater variability inhorizontal tongue motions) are of interest, as studiesin speech motor control indicate that smaller move-ments may induce instability because of possiblechanges in kinaesthetic feedback gain.27,28 This might
T O N G U E C O N T R O L F O R S W A L L O W I N G I N P A R K I N S O N ’ S D I S E A S E
Movement Disorders, Vol. 26, No. 9, 2011 1727

impede efficient bolus transport and underlie pro-longed oral transit times in PD.29
The strong stimulus differences highlight the impor-tance of tongue control in handling liquids of differingconsistency.12,23 Greater tongue effort is required totransport thicker liquids through the mouth; associ-ated increases in tongue movement amplitude and du-ration (as shown in the current study) may contributeto longer transit times. Additional time may facilitateimproved pharyngeal coordination and safer swallow-ing, especially with smaller volumes, as shown in thesip-size data. Our results also corroborate earlier find-ings that sequential swallows show shorter durationsand smaller movement amplitudes18,30; this mayimpose higher demands on swallowing motor controland pose additional risks for patients with PD.To conclude, our data indicate that thicker liquids
elicit greater tongue movement amplitudes and dura-tions, perhaps buying more time for subjects to con-trol bolus flow. However, even in the early stages ofPD, smaller and more variable tongue movements mayforecast future difficulties in swallowing, and closemonitoring is warranted.
Acknowledgments: We gratefully acknowledge the assistance ofAravind Namasivayam, Janice Bennett, Lyen Mortensen, Mitsuko Take-uchi, Rebecca Cliffe, Anna Ammoury, and Heidi Diepstra with data col-lection and processing.
References1. Johnston BT, Li Q, Castell JA, Castell DO. Swallowing and
esophageal function in Parkinson’s disease. Am J Gastroenterol1995;90:1741–1746.
2. Coates C, Bakheit AM. Dysphagia in Parkinson’s disease. EurNeurol 1997;38:49–52.
3. Bird MR, Woodward MC, Gibson EM, Phyland DJ, Fonda D.Asymptomatic swallowing disorders in elderly patients with Par-kinson’s disease: a description of findings on clinical examinationand videofluoroscopy in sixteen patients. Age Ageing 1994;23:251–254.
4. Logemann J, Blonsky ER, Boshes B. Lingual control in Parkin-son’s disease. Trans Am Neurol Assoc 1973;98:276–278.
5. Robbins JA, Logemann JA, Kirshner HS. Swallowing and speechproduction in Parkinson’s disease. Ann Neurol 1986;19:283–287.
6. Nakayama Y, Washio M, Mori M. Oral health conditions inpatients with Parkinson’s disease. J Epidemiol 2004;14:143–150.
7. Fernandez H, Lapane K. Predictors of mortality among nursinghome residents with a diagnosis of Parkinson’s disease. Med SciMonit 2002;8:CR241–CR246.
8. Hely M, Morris J, Traficante R, et al. The Sydney multicentrestudy of Parkinson’s disease: progression and mortality at 10years. J Neurol Neurosurg Psychiatry 1999;67:300–307.
9. Marik PE. Aspiration pneumonitis and aspiration pneumonia. NEngl J Med 2001;344(9):665–671.
10. McColl CD, Reardon KA, Shiff M, Kempster PA. Motor responseto levodopa and the evolution of motor fluctuations in the firstdecade of treatment of Parkinson’s disease. Mov Disord 2002;17:1227–1234.
11. Baijens LWJ, Speyer R. Effects of therapy for dysphagia in Par-kinson’s disease: systematic review. Dysphagia 2009;24:91–102.
12. Troche MS, Sapienza CM, Rosenbek JC. Effects of bolus consis-tency on timing and safety of swallow in patients with Parkin-son’s disease. Dysphagia 2008;23:26–32.
13. Deane KH, Whurr R, Clarke CE, Playford ED, Ben-Shlomo Y.Non-pharmacological therapies for dysphagia in Parkinson’s dis-ease. Cochrane Database Syst Rev 2001;(1):CD002816.
14. Logemann JA. Update on clinical trials in Dysphagia. Dysphagia2006;21:116–120.
15. Smith CH, Logemann JA, Burghardt WR, Zecker SG, RademakerAW. Oral and oropharyngeal perceptions of fluid viscosity acrossthe age span. Dysphagia 2006;21:209–217.
16. Logemann JA, Pauloski BR, Rademaker AW, et al. Temporal andbiomechanical characteristics of oropharyngeal swallow in
FIG. 1. Means and SDs for cSTI values in horizontal plane for 3 groups (older controls, Parkinson patients, younger controls) and 3 stimuli (water[H2O], thin apple juice [APP], thick apple juice [HON]).
V A N L I E S H O U T E T A L .
1728 Movement Disorders, Vol. 26, No. 9, 2011

younger and older men. J Speech Lang Hear Res 2000;43:1264–1274.
17. Humbert IA, Fitzgerald ME, McLaren DG, et al. Neurophysiol-ogy of swallowing: effects of age and bolus type. Neuroimage2009;44:982–991.
18. Steele CM, Van Lieshout P. Tongue movements during waterswallowing in healthy young and older adults. J Speech LangHear Res 2009;52:1255–1267.
19. Higashijima M. Influence of age and bolus size on swallowingfunction: basic data and assessment method for care and preven-tive rehabilitation. Am J Occup Ther 2010;64:88–94.
20. van Lieshout PHHM, Moussa W. The assessment of speechmotor behaviors using electromagnetic articulography. Phoneti-cian 2000;81:9–22.
21. Steele CM, Van Lieshout PHHM. Use of electromagnetic midsa-gittal articulography in the study of swallowing. J Speech LangHear Res 2004;47:342–352.
22. Goetz CG, Poewe W, Rascol O, et al;Movement Disorder SocietyTask Force on Rating Scales for Parkinson’s Disease. MovementDisorder Society Task Force report on the Hoehn and Yahr stagingscale: status and recommendations. Mov Disord 2004;19:1020–1028.
23. Steele CM, Van Lieshout PHHM. Influence of bolus consistency onlingual behaviors in sequential swallowing. Dysphagia 2004;19:192–206.
24. Steele CM, Van Lieshout PHHM, Goff HD. The rheology ofliquids: a comparison of clinicians’ subjective impressions andobjective measurement. Dysphagia 2003;18:182–195.
25. Bennett J, Van Lieshout P, Pelletier C, Steele C. Sip-sizing behav-iors in natural drinking conditions compared to instructed experi-mental conditions. Dysphagia 2009;24:152–158.
26. Cohen J. Statistical Power Analysis for the Behavioral Sciences.2nd ed. Hillsdale, NJ: Lawrence Erlbaum Associates;1988.
27. Van Lieshout PHHM, Bose A, Square PA, Steele CM. Speechmotor control in fluent and dysfluent speech production of anindividual with apraxia of speech and Broca’s aphasia. Clin Lin-guist Phon 2007;21:159–188.
28. Van Lieshout PHHM, Rutjens CAW, Spauwen PHM. The dy-namics of interlip coupling in speakers with a repaired unilateralcleft-lip history. J Speech Lang Hear Res 2002;45:5–19.
29. Nagaya M, Kachi T, Yamada T, Igata A. Videofluorographicstudy of swallowing in Parkinson’s disease. Dysphagia 1998;13:95–100.
30. Chi-Fishman G, Sonies BC. Motor strategy in rapid sequentialswallowing: new insights. J Speech Lang Hear Res 2000;43:1481–1492.
Systematic Genetic Analysis of thePITX3 Gene in Patients with
Parkinson Disease
Yi Guo, MS,1,2 Wei-Dong Le, MD, PhD,3
Joseph Jankovic, MD,3 Hua-Rong Yang, MS,1
Hong-Bo Xu, MS,1 Wen-Jie Xie, MD,3 Zhi Song, MD, PhD,1
and Hao Deng, MD, PhD1,3*
1Center for Experimental Medicine and Department of Neurology,
the Third Xiangya Hospital, Central South University, Changsha,
China; 2Department of Physiology, Xiangya Medical School, Central
South University, Changsha, China; 3Department of Neurology,
Baylor College of Medicine, Houston, Texas, USA
ABSTRACTBackground: Paired-like homodomain transcription fac-tor 3 has been found to be important for the differentia-tion and survival of midbrain dopaminergic neurons.Methods: To determine whether genetic variation inthe coding region of the paired-like homodomain tran-scription factor 3 gene plays a role in Parkinson’s dis-ease, genetic analysis was performed in 112 patientswith Parkinson’s disease.Results: We did not identify any mutations exceptrs2281983, but when we extended the analysis ofrs2281983 and 2 intron variants (rs4919621 andrs3758549) in 336 patients with Parkinson’s disease and244 controls, we found that rs2281983 and rs4919621appeared to confer susceptibility to Parkinson’s disease,especially in early-onset Parkinson’s disease and familialParkinson’s disease. VC 2011 Movement Disorder Society
Key Words: Parkinson disease; transcription factor;paired-like homodomain transcription factor 3;susceptibility
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
Yi Guo and Wei-Dong Le contributed equally to this article.
*Correspondence to: Dr. Hao Deng, Professor of Center forExperimental Medicine and Professor of Neurology, Vice Director ofCenter for Experimental Medicine, the Third Xiangya Hospital, CentralSouth University, 138 Tongzipo Road, Changsha, Hunan 410013, China;[email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.This work was supported by Central South University, Changsha, China,and Baylor College of Medicine, Houston, Texas.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 20 June 2010; Revised: 17 January 2011; Accepted:1 February 2011Published online 5 April 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23693
S Y S T E M A T I C G E N E T I C A N A L Y S I S O F T H E P I T X 3 G E N E
Movement Disorders, Vol. 26, No. 9, 2011 1729

Parkinson’s disease (PD) is a common neurodegener-ative disorder characterized by a core phenotype ofmotor abnormalities, particularly tremor, bradykine-sia, rigidity, and postural instability, often coupledwith other motor and nonmotor neurological deficitsduring the progressive course of the disease. PD ispathologically characterized by loss of dopamine neu-rons in the substantia nigra and Lewy body formation.Paired-like homodomain transcription factor 3(PITX3) has been found to be important for the differ-entiation, development, and maintenance of dopami-nergic neurons in the ventral mesencephalon/substantia nigra pars compacta (VM/SNpc).1 PITX3-deficient aphakia mouse has been proposed as a modelof PD because of its selective nigrostriatal DA loss,2
and abnormal features including parkinsonism, micro-phthalmia, and enhanced nociception have beenobserved in spontaneous Pitx3 mutant (416insG)mouse.3 PITX3 induces transcription of miR133B (amicroRNA) that has been found to be deficient inmidbrain tissue from patients with PD,4 although thesignificant association between this variant and PDrisk was queried in a subsequently study.5 To evaluatethe role of the variant(s) in the coding region of thePITX3 gene in whites, we screened PD patients fromNorth America whose DNA had been collected anddeposited in the Genetic Bank at Parkinson DiseaseCenter and Movement Disorders Clinic at Baylor Col-lege of Medicine.
Study Population and MethodsThree hundred and thirty-six unrelated white
patients with PD from North America (mean age,58.9 6 12.3 years; mean age at onset, 53.8 6 12.7years; male/female, 167/169; Table 1) and 244 nor-mal controls (mean age, 58.8 6 14.1 years; male/female, 120/124) were enrolled in this study. Thediagnosis of PD was made according to accepteddiagnostic criteria.6 All subjects signed an informedconsent approved by the Baylor College of MedicineInstitutional Review Board. A detailed history wasobtained from each family member. Among these
patients, 148 had a family history (familial; male/female, 74/74), and 188 had no family history (spo-radic; male/female, 93/95). One hundred and five hadrelatively early-onset PD (EOPD; onset age, �50years), and 231 were late-onset PD (LOPD; onsetage, >50 years). Some of the patients were alsoscreened for and were found to be negative for othergene mutations, such as premutation in the fragile Xmental retardation 1 gene (FMR1),7 the PINK1G309D and W437OPA point mutations,8 the alpha-synuclein gene (SNCA) dosage alternation,9 and theleucine-rich repeat kinase 2 gene (LRRK2) R1441C/G/H and G2019S mutations.10
Genomic DNA was isolated from lymphocytes usingthe standard method. Polymerase chain reactionamplified all coding regions and intron/exon bounda-ries of the PITX3 gene with the primers shown inTable 1. In the first stage, the coding region of thePITX3 gene was analyzed by PCR–single-strand con-formation polymorphism (PCR-SSCP) in 112 PDpatients (age, 58.2 6 9.8 years; onset age, 53.8 610.2 years; male/female, 57/55; 58 familial PD, 54sporadic PD), and PCR products exhibiting the abnor-mally shifted bands were sequenced.11 In the secondstage, 336 PD patients and 244 controls were eval-uated for the distribution of gene frequencies of exonsingle-nucleotide polymorphism (SNP) rs2281983 and2 possibly disease-risk intron SNPs, rs4919621 andrs3758549 (previously evaluated SNP rs2281983and rs4919621 data from 265 patients and 210 con-trols was included), by sequencing.12 Rs3758549forward primer is 50-TCCCCTTGCAAGAGATGATAG-30, and rs3758549 forward primer is 50-CCCAAATAGGCT GGGAATTT-30.A goodness-of-fit v2 test was used to determine
whether the variants were in Hardy–Weinberg equilib-rium. The v2 tests were applied to test for significanceof differences in allele frequencies. All statistical testswere 2-sided. P < .05 was considered significant. Thestatistical analysis was performed with the Stata 8.0Statistical Software Package (Stata Corporation, Col-lege Station, TX) and Haploview 4.2.
Table 1. Primers for detection of the PITX3 gene exons
Fragment Exon Forward primer (50!30) Reverse primer (50!30) Product size (bp)
1 2 CCCATTACCCTGGTCTGTGT TGGGGATGAAGCTGTTATGTC 2152 3 AGCGAGTGGCTTAGGAGGTC GTAGCTGCTGGCTGGTGAAG 1883 3 GACGGTTCGCTGAAAAGAA CGTGCTCATGTCGGGGTAG 1114 3 AGGAGCTAGAGGCGACCTTC AGTCGCGGGTCTGGAGAG 1215 4 GCCACCTCATCTCGTTTAT GGGTACACCTCCTCGTAGGG 2446 4 GAGCTATGCAAAGGCAGCTT AGCTGGGTGGCGAGAAGAC 1997 4 CCTTCAACTCGGTCAACGTG GGCCAGGCTCGAGTTACAC 2788 4 GCCTCTTCCCCCTACGTCTA CCAGTCAAAATGACCCCAGT 240
G U O E T A L .
1730 Movement Disorders, Vol. 26, No. 9, 2011
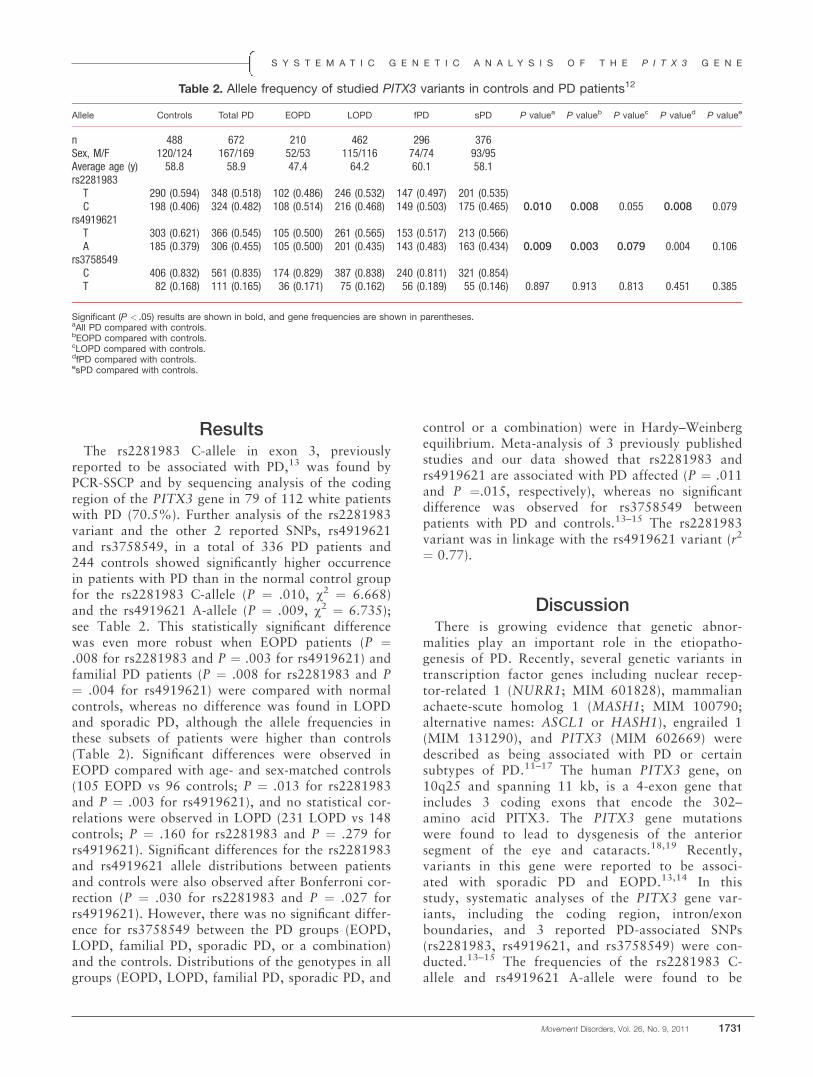
ResultsThe rs2281983 C-allele in exon 3, previously
reported to be associated with PD,13 was found byPCR-SSCP and by sequencing analysis of the codingregion of the PITX3 gene in 79 of 112 white patientswith PD (70.5%). Further analysis of the rs2281983variant and the other 2 reported SNPs, rs4919621and rs3758549, in a total of 336 PD patients and244 controls showed significantly higher occurrencein patients with PD than in the normal control groupfor the rs2281983 C-allele (P ¼ .010, v2 ¼ 6.668)and the rs4919621 A-allele (P ¼ .009, v2 ¼ 6.735);see Table 2. This statistically significant differencewas even more robust when EOPD patients (P ¼.008 for rs2281983 and P ¼ .003 for rs4919621) andfamilial PD patients (P ¼ .008 for rs2281983 and P¼ .004 for rs4919621) were compared with normalcontrols, whereas no difference was found in LOPDand sporadic PD, although the allele frequencies inthese subsets of patients were higher than controls(Table 2). Significant differences were observed inEOPD compared with age- and sex-matched controls(105 EOPD vs 96 controls; P ¼ .013 for rs2281983and P ¼ .003 for rs4919621), and no statistical cor-relations were observed in LOPD (231 LOPD vs 148controls; P ¼ .160 for rs2281983 and P ¼ .279 forrs4919621). Significant differences for the rs2281983and rs4919621 allele distributions between patientsand controls were also observed after Bonferroni cor-rection (P ¼ .030 for rs2281983 and P ¼ .027 forrs4919621). However, there was no significant differ-ence for rs3758549 between the PD groups (EOPD,LOPD, familial PD, sporadic PD, or a combination)and the controls. Distributions of the genotypes in allgroups (EOPD, LOPD, familial PD, sporadic PD, and
control or a combination) were in Hardy–Weinbergequilibrium. Meta-analysis of 3 previously publishedstudies and our data showed that rs2281983 andrs4919621 are associated with PD affected (P ¼ .011and P ¼.015, respectively), whereas no significantdifference was observed for rs3758549 betweenpatients with PD and controls.13–15 The rs2281983variant was in linkage with the rs4919621 variant (r2
¼ 0.77).
DiscussionThere is growing evidence that genetic abnor-
malities play an important role in the etiopatho-genesis of PD. Recently, several genetic variants intranscription factor genes including nuclear recep-tor-related 1 (NURR1; MIM 601828), mammalianachaete-scute homolog 1 (MASH1; MIM 100790;alternative names: ASCL1 or HASH1), engrailed 1(MIM 131290), and PITX3 (MIM 602669) weredescribed as being associated with PD or certainsubtypes of PD.11–17 The human PITX3 gene, on10q25 and spanning 11 kb, is a 4-exon gene thatincludes 3 coding exons that encode the 302–amino acid PITX3. The PITX3 gene mutationswere found to lead to dysgenesis of the anteriorsegment of the eye and cataracts.18,19 Recently,variants in this gene were reported to be associ-ated with sporadic PD and EOPD.13,14 In thisstudy, systematic analyses of the PITX3 gene var-iants, including the coding region, intron/exonboundaries, and 3 reported PD-associated SNPs(rs2281983, rs4919621, and rs3758549) were con-ducted.13–15 The frequencies of the rs2281983 C-allele and rs4919621 A-allele were found to be
Table 2. Allele frequency of studied PITX3 variants in controls and PD patients12
Allele Controls Total PD EOPD LOPD fPD sPD P valuea P valueb P valuec P valued P valuee
n 488 672 210 462 296 376Sex, M/F 120/124 167/169 52/53 115/116 74/74 93/95Average age (y) 58.8 58.9 47.4 64.2 60.1 58.1rs2281983T 290 (0.594) 348 (0.518) 102 (0.486) 246 (0.532) 147 (0.497) 201 (0.535)C 198 (0.406) 324 (0.482) 108 (0.514) 216 (0.468) 149 (0.503) 175 (0.465) 0.010 0.008 0.055 0.008 0.079
rs4919621T 303 (0.621) 366 (0.545) 105 (0.500) 261 (0.565) 153 (0.517) 213 (0.566)A 185 (0.379) 306 (0.455) 105 (0.500) 201 (0.435) 143 (0.483) 163 (0.434) 0.009 0.003 0.079 0.004 0.106
rs3758549C 406 (0.832) 561 (0.835) 174 (0.829) 387 (0.838) 240 (0.811) 321 (0.854)T 82 (0.168) 111 (0.165) 36 (0.171) 75 (0.162) 56 (0.189) 55 (0.146) 0.897 0.913 0.813 0.451 0.385
Significant (P < .05) results are shown in bold, and gene frequencies are shown in parentheses.aAll PD compared with controls.bEOPD compared with controls.cLOPD compared with controls.dfPD compared with controls.esPD compared with controls.
S Y S T E M A T I C G E N E T I C A N A L Y S I S O F T H E P I T X 3 G E N E
Movement Disorders, Vol. 26, No. 9, 2011 1731

significantly increased, especially in patients withEOPD and familial PD, whereas no statisticallysignificant associations were found in LOPD andsporadic PD. Rs2281983 is in exon 3, whereasrs4919621 is in intron 3, and rs3758549 is in thepromoter region of the PITX3 gene. Thers2281983 variant neither changes amino acidsnor affects splicing. These 3 variants are antici-pated to be functional variants because they arethe compositions of domains that may bind toother factors such as AHR-arnt heterodimers andAHR-related factor binding of snRNA-activationprotein complex, EI1-myleiod transforming factor,and GATA binding factor (Genomatix softwareprediction). We and others13 could not replicatefindings from 1 study suggesting a PD-associatedSNP rs3758549 in the putative promoter region ofthe PITX3 gene.15
Our findings, supported by other reports,13–15 sug-gest that certain variants in transcription genes thatcode for proteins involved in the development andmaintenance of the dopaminergic system, such asPITX3, act as susceptibility variants for PD. Althoughthe variants do not represent monogenic causes of PD,they do appear to confer increased risk for the disease.These variants may be involved in the regulation ofgene expression or affect transcription factor binding,native splicing, or other mechanisms that result in adecreased number of dopamine-producing neurons orincreased vulnerability of these neurons to subsequentenvironmental factors.20 Further studies on the PITX3and other transcription genes with linked variants arewarranted.
Acknowledgments: We thank the participating patients and theinvestigators at the Parkinson’s Disease Center and Movement DisordersClinic, Baylor College of Medicine for their cooperation and their effortsin collecting the genetic information and DNA specimens.
References1. Courtois ET, Castillo CG, Seiz EG, et al. In vitro and in
vivo enhanced generation of human A9 dopamine neuronsfrom neural stem cells by Bcl-XL. J Biol Chem 2010;285:9881–9897.
2. Beeler JA, Cao ZF, Kheirbek MA, et al. Dopamine-dependentmotor learning: insight into levodopa’s long-duration response.Ann Neurol 2010;67:639–647.
3. Rosemann M, Ivashkevich A, Favor J, et al. Microphthalmia, par-kinsonism, and enhanced nociception in Pitx3 (416insG) mice.Mamm Genme 2010;21:13–27.
4. Kim J, Inoue K, Ishii J, et al. MicroRNA feedback circuitin midbrain dopamine neurons. Science 2007;317:1220–1224.
5. de Mena L, Coto E, Cardo LF, et al. Analysis of the Micro-RNA-133 and PITX3 genes in Parkinson’s disease. Am J Med Genet BNeuropsychiatr Genet 2010;153B:1235–1239.
6. Jankovic J. Parkinson’s disease: clinical features and diagnosis.J Neurol Neurosurg Psychiatry 2008;79:368–376.
7. Deng H, Le W, Jankovic J. Premutation alleles associated withParkinson disease and essential tremor. JAMA 2004;292:1685–1686.
8. Deng H, Le W, Zhang X, Pan TH, Jankovic J. G309D andW437OPA PINK1 mutations in Caucasian Parkinson’s diseasepatients. Acta Neurol Scand 2005;111:351–352.
9. Deng H, Xie W, Guo Y, Le W, Jankovic J. Gene dosage analysisof alpha-synuclein (SNCA) in patients with Parkinson’s disease.Mov Disord 2006;21:728–729.
10. Deng H, Le WD, Guo Y, Hunter CB, Xie W, Jankovic J.Genetic and clinical identification of Parkinson’s diseasepatients with LRRK2 G2019S mutation. Ann Neurol 2005;57:933–934.
11. Deng H, Yang H, Le W, et al. Examination of the MASH1 genein patients with Parkinson’s disease. Biochem Biophys Res Com-mun 2010;392:548–550.
12. Le W, Nguyen D, Lin XW, et al. Transcription factor PITX3gene in Parkinson’s disease. Neurobiol Aging 2009 [Epub aheadof print].
13. Bergman O, Hakansson A, Westberg L, et al. PITX3 polymor-phism is associated with early onset Parkinson’s disease. Neuro-biol Aging 2010;31:114–117.
14. Fuchs J, Mueller JC, Lichtner P, et al. The transcription factorPITX3 is associated with sporadic Parkinson’s disease. NeurobiolAging 2009;30:731–738.
15. Haubenberger D, Reinthaler E, Mueller JC, et al. Association oftranscription factor polymorphisms PITX3 and EN1 with Parkin-son’s disease. Neurobiol Aging 2011;32:302–307.
16. Ide M, Yamada K, Toyota T, et al. Genetic association analysesof PHOX2B and ASCL1 in neuropsychiatric disorders: evidencefor association of ASCL1 with Parkinson’s disease. Hum Genet2005;117:520–527.
17. Chen CM, Chen IC, Chang KH, et al. Nuclear receptorNR4A2 IVS6 þ18insG and brain derived neurotrophic factor(BDNF) V66M polymorphisms and risk of Taiwanese Parkin-son’s disease. Am J Med Genet B Neuropsychiatr Genet 2007;144B:458–462.
18. Semina EV, Ferrell RE, Mintz-Hittner HA, et al. A novel homeo-box gene PITX3 is mutated in families with autosomal-dominantcataracts and ASMD. Nat Genet 1998;19:167–170.
19. Summers KM, Withers SJ, Gole GA, et al. Anterior segment mes-enchymal dysgenesis in a large Australian family is associatedwith the recurrent 17 bp duplication in PITX3. Mol Vis 2008;14:2010–2015.
20. Le W, Chen S, Jankovic J. Etiopathogenesis of Parkinson’s dis-ease: a new beginning? Neuroscientist 2009;15:28–35.
G U O E T A L .
1732 Movement Disorders, Vol. 26, No. 9, 2011

The LRRK2 R1441C Mutation isMore Frequent Than G2019S in
Parkinson’s Disease Patients fromSouthern Italy
Chiara Criscuolo, MD, PhD,1,2* Anna De Rosa, MD, PhD,2
Anna Guacci, BSc,2 Erik J. Simons,1 Guido J. Breedveld,1
Silvio Peluso, MD,2 Giampiero Volpe, MD,3 Alessandro Filla,MD,2 Ben A. Oostra, PhD,1 Vincenzo Bonifati, MD, PhD,1
and Giuseppe De Michele, MD2
1Department of Clinical Genetics, Erasmus MC, Rotterdam, The
Netherlands; 2Department of Neurological Sciences, Federico II
University, Naples, Italy; 3Department of Neurology, S.Giovanni di
Dio e Ruggi d’Aragona Hospital, Salerno, Italy
ABSTRACTBackground: Mutations in the leucine-rich repeat ki-nase 2 gene are the most frequent cause of familialand sporadic Parkinson’s disease, and G2019S is themost common leucine-rich repeat kinase 2 mutationacross several Mediterranean countries.Methods:One hundred ninety-two patients with Parkin-son’s disease from Campania, a region in southernItaly, were screened for R1441C/H/G and G2019S bydirect sequencing and SfcI digestion.Results: Among 192 patients with Parkinson’s disease(mean age 6 SD, 63.9 6 11.8 years; disease onset,54.0 6 12.5 years; family history for Parkinson’s dis-ease or tremor, 45%), 8 carried a heterozygousR1441C mutation, whereas only 1 had the G2019Smutation. All R1441C patients originate from the prov-ince of Naples and share the same haplotype, suggest-ing a founder effect.Conclusions: G2019S is not ubiquitously the mostcommon leucine-rich repeat kinase 2 mutation; inCampania R1441C is more frequent. Region-specificmutation prevalence data should be taken into accountfor a sensitive and cost-effective molecular diagnosisand counseling of patients with Parkinson’s disease.VC 2011 Movement Disorder Society
Key Words: leucine-rich repeat kinase 2; PARK8;R1441C; G2019S
Mutations in the leucine-rich repeat kinase 2 gene(LRRK2; MIM 609007) are responsible of the mostcommon Mendelian form of Parkinson’s disease (PD),accounting for at least 4% of familial and 1%–2% ofsporadic cases. The clinical phenotype is largely indis-tinguishable from classical PD.1–5
Previous studies indicated G2019S as the most com-mon LRRK2 mutation among populations from
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Dr. Chiara Criscuolo, Dipartimento di ScienzeNeurologiche, Universita degli Studi di Napoli Federico II, Via Pansini 5,80131, Napoli, Italy; [email protected]
Funding agencies: This study was supported by an EFNS fellowship toDr. Chiara Criscuolo and by a research grant from the InternationaalParkinson Fonds (The Netherlands) to Dr. Vincenzo Bonifati.Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 11 December 2010; Revised: 22 February 2011; Accepted:24 February 2011Published online 29 April 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23735
Southern Europe and North Africa.6 We previouslyreported a low G2019S prevalence among PD casesfrom the Campania region, in southern Italy.7 Here,we screened a larger number of patients for G2019Sand for R1441C/H/G, and we found that the R1441Cmutation is more common among PD patients fromthis region than is G2019S. These data have importantimplications for directing future LRRK2 screeningstudies and counseling of PD patients.
Patients and Methods
Patients
One hundred and ninety-two unrelated PD patientswere recruited after informed consent at the Depart-ment of Neurological Sciences of the Federico II Uni-versity in Naples. At the time of screening, mean ageof the patients 6 SD was 63.9 6 11.8 years (range,33–89 years), and PD symptoms onset was 54.0 612.5 years (range, 20–83 years). One hundred andeighteen patients (61%) were men, and 74 patients(39%) were women; 45 (23%) had early disease onset(�45 years) and 147 (77%) late onset (>45 years). Pa-rental consanguinity was reported in 14 cases (1.5%).One hundred and five patients (55%) had sporadicdisease, and 87 (45%) reported at least 1 first-degree(n ¼ 66), second-degree (n ¼ 14), or third-degree (n ¼7) relative affected by PD or tremor. Most patients (n¼ 178) originated from Campania, a region in south-ern Italy that includes 5 provinces (Fig. 1): Naples(131 patients), Salerno (23), Caserta (16), Benevento(5), and Avellino (3). The remaining 14 patients werefrom other regions in central or southern Italy. Allpatients were of white ethnicity and Italian origin.The clinical diagnosis of PD was established accord-
ing to the UK Parkinson’s Disease Society Brain Bankcriteria,8 with the exception that the presence of morethan 1 relative affected by PD was not considered anexclusion criterion.Clinical assessment, including using the Unified Par-
kinson’s Disease Rating Scale (UPDRS) and the modi-fied Hoehn and Yahr scale, was performed by
L R R K 2 R 1 4 4 1 C M U T A T I O N M O R E F R E Q U E N T I N S O U T H E R N I T A L Y
Movement Disorders, Vol. 26, No. 9, 2011 1733

movement disorders specialists (G.D.M., A.D.R.). Afull medical history, including family history, wasobtained from patients and/or caregivers. When avail-able, relatives with suspected movement disorderswere examined.
Genetic Analysis
Genomic DNA was extracted from peripheral bloodcells according to a standard protocol. Primers andpolymerase chain reaction (PCR) protocols for exon41 (p.G2019S) and exon 31 (pR1441C/H/G) havebeen reported previously.7,9 Exon 31 was analyzed bydirect sequencing and G2019S by SfcI digestion.7,9 Forhaplotype analysis, intragenic and flanking markers(short tandem repeats and single-nucleotide polymor-phisms, SNPs) were typed in the R1441C carriers aspreviously described.9 Exonic and intronic LRRK2SNPs were typed by direct sequencing using primersand PCR conditions as previously described.2 Haplo-types were constructed manually.
Results
Prevalence of LRRK2 Mutations
Eight PD index patients with a heterozygousR1441C mutation were detected. A positive family
history was present in 6 (Fig. 2), whereas the remain-ing 2 (families 4 and 190) denied the presence of rela-tives with PD or tremor. The R1441C mutation wasdetected in all 4 secondary PD cases available foranalysis (Fig. 2). The prevalence of the R1441C muta-tion was 4.2% (8 of 192) in the whole sample, 6.9%(6 of 87) in the familial PD patients, and 1.9% (2 of105) in the sporadic cases. R1441H/G mutations werenot found.Only 1 patient carried the G2019S mutation in the
heterozygous state, which has already beenreported.7 The prevalence of the G2019S mutationin our entire sample of PD patients was 0.5% (1/192), which rose to 1.1% (1/87) among those with apositive family history. Therefore, the overall preva-lence of the LRRK2 mutations targeted by this study(G2019S and R1441C) was 4.7% (9 of 192) in thewhole sample, 8.1% (7 of 87) in the familial cases,and 1.9% (2 of 105) in the sporadic cases. All themutation carriers originated from the province ofNaples (Fig. 1).
Clinical Aspects
The main clinical features of the 8 index cases plus4 secondary cases with the R1441C mutation and thecomparison with PD patients without R1441C or
FIG. 1. Geographic origin of the index cases carrying the R1441C (full circle) and G2019S (empty circle) LRRK2 mutation. The insert on the leftshows the origin of the 178 PD cases included in this study and originating from the Campania region: Naples (NA, 3.06 3 1026 inhabitants), Avel-lino (AV, 0.43 3 1026 inhabitants), Benevento (0.29 3 1026 inhabitants), Caserta (0.85 3 1026 inhabitants), and Salerno (1.07 3 1026 inhabitants).The remaining 14 patients originated from other Italian regions.
C R I S C U O L O E T A L .
1734 Movement Disorders, Vol. 26, No. 9, 2011

G2019S are given in Supplementary Table 1. Statisti-cal differences among the 2 groups were not detected.However, the presence of pain was observed in5 R1441C carriers but only in 1 noncarrier (P ¼.155). A positive family history of PD or tremor wasmore frequent among the R1441C carriers, althoughthe difference was not significant (P ¼ .170). Consan-guinity was observed in 2 R1441C families (Fig. 2).
Haplotype Analysis
Haplotype analysis in this study was limited by thelack of DNA from relatives; therefore, phase couldnot be established with certainty for several markers.Nonetheless, the results are compatible with all ourR1441C cases sharing 1 of the major European haplo-types (Supplementary Fig. 1).10,11
DiscussionA previous survey limited to 128 PD patients
showed a low prevalence (0.8%) of the G2019S muta-tion,7 despite the reported high prevalence (33%) ofpositive family history among PD patients from Cam-pania.12 Therefore, we hypothesized that either differ-ent LRRK2 mutations or other, not yet identifiedgenetic factors could be involved in the etiology of PDin our region. We have now extended the study to a
larger sample of patients and to other mutations(R1441C/H/G) of the LRRK2 gene.We report that R1441C, the second most common
LRRK2 mutation worldwide, has a higher prevalence(4.7%) than G2019S (0.5%) in Campania, reaching aprevalence of 6.9% in familial cases. It should benoted that the screened sample is not fully representa-tive of the general population of PD patients becausecases with younger-onset age and/or positive familyhistory were most likely included. However, theR1441C mutation appears to be common in Campa-nia, even if only the sporadic cases are considered(1.9%), and its overall prevalence rises to 6.1% in theprovince of Naples, from where all our R1441C car-riers originated.The R1441C mutation has been reported in 2 white
families from the United States, in Italian, German,Spanish, Belgian, and Irish patients,10,11,13–15 in 1 pro-band from Singapore,16 and in 1 from Iran.17 Its over-all prevalence worldwide is low (0.1%),18 but it was3.4% in 60 Italian PD families with autosomal domi-nant transmission,11 0.6% in Sardinia,19 and 0.2% inan unselected (28% positive family history) large pop-ulation of Italian patients.9 The origin of thesepatients is not detailed, but presumably most of themare from northern Italy.Two main haplotypes for R1441C has been
described in European patients, suggesting 2
FIG. 2. Simplified pedigrees of the 8 families with the LRRK2 R1441C mutation. To protect confidentiality, the order of individuals in sibships wasaltered. Black squares (men) and circles (women) represent individuals affected by Parkinson’s disease; the square with black upper corner indi-cates an individual reported as affected by tremor only. A total of 12 patients (8 index and 4 relatives) were clinically examined, and they all carrythe R1441C mutation. The 8 index cases are indicated by an arrow. The number below symbols indicates age at onset (years).
L R R K 2 R 1 4 4 1 C M U T A T I O N M O R E F R E Q U E N T I N S O U T H E R N I T A L Y
Movement Disorders, Vol. 26, No. 9, 2011 1735

independent founders. A first haplotype is present inso-called family D, whose ancestors probably immi-grated to western Nebraska from England,20 and inseveral Flemish-Belgian families.15 A second haplotypeis common to Italian, German, Spanish, Ameri-can,10,11 and Iranian carriers.17
In our series, all the R1441C carriers are likely toshare the above-mentioned haplotype described inother Italian patients,11 suggesting a common founder.The history of migratory fluxes in Europe does notsuggest that an ancient founder lived in southern Italyand his descendants migrated eastward (Spain), west-ward (Iran), and northward (Germany). Data aremore consistent with the Indo-European migration,which has occurred in several waves since the fourthmillennium BC from the Pontic-Caspian steppe toMediterranean and Central European countries. Fur-ther collaborative studies on the prevalence of theR1441C mutation and the associated haplotypes arenecessary to shed light on the origins and spreading ofthis mutation.Our R1441C patients compared with 33 mutation
carriers collected in an international cooperativestudy10 showed similar Hoehn–Yahr stage, clinicalfeatures, and levodopa response, but slightly lower ageat onset and lower prevalence of family history. Fur-thermore, we found no significant differences compar-ing the phenotypes of R1441C carriers andnoncarriers.In conclusion, our study underlines the importance
of regional surveys of the genetic causes of PD, reveal-ing that in Campania, R1441C is the most commonLRRK2 mutation and should therefore be prioritizedin patients’ screening. Accurate information aboutthe local prevalence of specific mutations can lead tomore cost-effective genetic screening with importantimplications for the molecular diagnosis and geneticcounseling, as well as for future neuroprotective strat-egies.
References1. Gasser T. Molecular pathogenesis of Parkinson disease: insights
from genetic studies. Expert Rev Mol Med. 2009;11:e22.
2. Di Fonzo A, Rohe CF, Ferreira J, et al. A frequent LRRK2 genemutation associated with autosomal dominant Parkinson’s dis-ease. Lancet. 2005;365:412–415.
3. Nichols WC, Pankratz N, Hernandez D, et al. Genetic screeningfor a single common LRRK2 mutation in familial Parkinson’s dis-ease. Lancet. 2005;365:410–412.
4. Gilks WP, Abou-Sleiman PM, Gandhi S, et al. A commonLRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–416.
5. Paisn-Ruiz C. LRRK2 gene variation and its contribution to Par-kinson disease. Hum Mutat. 2009;30:1153–1160.
6. Correia Guedes L, Ferreira JJ, Rosa MM, et al. Worldwide fre-quency of G2019S LRRK2 mutation in Parkinson’s disease: a sys-tematic review. Parkinsonism Relat Disord. 2010;16:237–242.
7. De Rosa A, Criscuolo C, Mancini P, et al. Genetic screening forLRRK2 gene G2019S mutation in Parkinson’s disease patientsfrom Southern Italy. Parkinsonism Relat Disord. 2009;15:242–244.
8. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical di-agnosis of idiopathic Parkinson’s disease: a clinico-pathologicalstudy of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184.
9. Goldwurm S, Di Fonzo A, Simons EJ, et al. The G6055A(G2019S) mutation in LRRK2 is frequent in both early and lateonset Parkinson’s disease and originates from a common ancestor.J Med Genet. 2005;42:e65.
10. Haugarvoll K, Rademakers R, Kachergus JM, et al. Lrrk2R1441C parkinsonism is clinically similar to sporadic Parkinsondisease. Neurology. 2008;70:1456–1460.
11. Di Fonzo A, Tassorelli C, De Mari M, et al. Italian Parkinson’sGenetics Network. Comprehensive analysis of the LRRK2 gene insixty families with Parkinson’s disease. Eur J Hum Genet. 2006;14:322–331.
12. De Michele G, Filla A, Volpe G, et al. Environmental and geneticrisk factors in Parkinson’s disease: a case control study in south-ern Italy. Mov Disord. 1996;11:17–23.
13. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2cause autosomal-dominant parkinsonism with pleomorphic pa-thology. Neuron. 2004;44:601–607.
14. Gosal D, Lynch T, Ross OA, Haugarvoll K, Farrer MJ, GibsonJM. Global distribution and reduced penetrance: Lrrk2 R1441Cin an Irish Parkinson’s disease kindred. Mov Disord. 2007;22:291–292.
15. Nuytemans K, Rademakers R, Theuns J, et al. Founder mutationp.R1441C in the leucine-rich repeat kinase 2 gene in Belgian Par-kinson’s disease patients. Eur J Hum Genet. 2008;16:471–479.
16. Tan EK, Skipper L, Chua E, et al. Analysis of 14 LRRK2 muta-tions in Parkinson’s plus syndromes and late-onset Parkinson’sdisease. Mov Disord. 2006;21:997–1001.
17. Shojaee S, Sina F, Farboodi N, et al. A clinic-based screening ofmutations in exons 31, 34, 35, 41, and 48 of LRRK2 in IranianParkinson’s disease patients. Mov Disord. 2009;24:1023–1027.
18. Healy DG, Falchi M, O’sullivan SS, et al. International LRRK2Consortium. Phenotype, genotype, and worldwide genetic pene-trance of LRRK2-associated Parkinson’s disease: a case-controlstudy. Lancet Neurol. 2008;7:583–590.
19. Floris G, Cannas A, Solla P, et al. Genetic analysis for fiveLRRK2 mutations in a Sardinian parkinsonian population: impor-tance of G2019S and R1441C mutations in sporadic Parkinson’sdisease patients. Parkinsonism Relat Disord. 2009;15:277–280.
20. Wszolek ZK, Pfeiffer B, Fulgham JR, et al. Western Nebraskafamily (family D) with autosomal dominant parkinsonism. Neu-rology. 1995;45:502–505.
C R I S C U O L O E T A L .
1736 Movement Disorders, Vol. 26, No. 9, 2011

Electrogastrographyc Activity inParkinson’s Disease Patients Withand Without Motor Fluctuations
Giovanni Albani, MD,1 Nadia El Assawy,1 Stefania Cattaldo,1
Marilena De Gennaro,1 Francesca Gregorini,1
Luca Pradotto, MD,1* and Alessandro Mauro, MD1,2
1Division of Neurology and Neurorehabilitation, San Giuseppe
Hospital, IRCCS–Istituto Auxologico Italiano, Piancavallo (VB), Italy;2Department of Neuroscience, University of Turin, Turin, Italy
ABSTRACTBackground: Gastroenteric dysfunctions are very com-mon in Parkinson’s disease, but their relationship withdopaminergic response and motor fluctuations is stillunclear. Electrogastrography is a noninvasive methodfor measuring gastric myoelectrical activity.Methods: We evaluated the effects of levodopa intakeon the motility of empty stomachs in Parkinson’s dis-ease patients with and without motor fluctuations.Results: The electrogastrography findings showed anormal pattern not influenced by levodopa intake,unrelated to plasmatic levodopa concentrations and toclinical parameters.Conclusions: Our results suggest that at rest gastricactivity of Parkinson’s disease patients is normal andplasmatic levodopa variability is not influenced by gas-tric motility. VC 2011 Movement Disorder Society
Key Words: Parkinson’s disease; gastric myoelectri-cal activity; motor fluctuations; L-dopa plasmatic levels
Prandial gastric motility is altered in Parkinson’s dis-ease (PD), with a delayed time of gastric emptying in55%–100% of patients1 and associated postprandialbloating, abdominal discomfort, early satiety, and nau-sea.2 Constipation is present in 20% of PD patients,3
frequently years prior to motor signs and correlatedwith autopsy evidence of Lewy bodies in the dorsal
motor nucleus of the vagus,4 in the lower esophagus,5
and in the stomach.6
It has been suggested that altered gastrointestinalfunctions can have pharmacokinetic implicationsbecause delayed gastric emptying and, consequently,delayed levodopa (LD) arrival at intestinal absorptivesites can cause erratic responses to the drug. More-over, retention of LD in the stomach may increase itsexposure to dopa decarboxylase present in the gastricmucosa, which can convert LD to dopamine, makingthe drug unavailable for intestinal absorption.7 Theabsorption troubles may represent one of the mecha-nisms involved in the development of motor fluctua-tions of PD,8 distinct from the reduction of levodopabioavailability due to dietary proteins.LD remains the most effective treatment for PD, but
its effect on visceral smooth muscles is not clear. In par-ticular, the response of dopaminergic terminations ofthe gastroenteric system to LD orally administered isnot well defined, whereas it is known that PD motorsymptoms improve in relation to the achievement of thehighest LD plasma concentrations, generally reachedbetween 30 and 120 minutes after drug intake.9 Indeed,it is still an open question about the behavior of gastricmotility during off periods, with correspondinglydecreased blood concentration of LD, despite sustaineddrug administration.8 Previous reports on gastric motil-ity in PD patients have been performed using radioscin-tigraphy, a technique requiring the administration of aradiolabeled test meal.1,10 Electrogastrography (EGG)is a noninvasive method for the measurement of gastricmyoelectrical activity that uses abdominal surfaceelectrodes to register the dominant frequency of gastro-duodenal contractility.11 Studies have shown a goodcorrelation between cutaneous and serosal recordings,12
so that relative changes in the EGG reflect gastric con-tractility. Very few EGG studies have shown gastricmotility abnormalities, only including after-mealacquisitions, in patients with both early and advancedPD.13,14 To our knowledge, there have been no reportsabout the features of gastric motility in PD patientswhen fasting and after LD intake.The aim of this study was to evaluate gastric motility
when fasting and after LD intake in PD patients withand without motor fluctuations, analyzing the correla-tions between dominant frequency of gastric contra-ctility, LD plasma concentrations, and motor response.
Subjects and MethodsTwenty-three patients with a diagnosis of PD (10
with motor and 13 without motor fluctuations; meanage, 65 years; 12 women, 11 men; mean disease dura-tion, 6.2 years; mean Hoehn & Yahr stage, 2.3) and 20healthy subjects were randomly enrolled. All PDpatients had been treated with antiparkinsonian medi-cations. Clinical features were not significantly different
------------------------------------------------------------*Correspondence to: Luca Pradotto, Divisione di Neurologia eNeuroriabilitazione, Ospedale San Giuseppe, IRCCS–Istituto AuxologicoItaliano, Strada Cadorna n. 90, 28924 Oggebbio (VB), Italy;[email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.This study was partially supported by the Italian Ministry of Health(project RFPS-2007-1-641398).
Received: 7 July 2010; Revised: 23 December 2010; Accepted:6 January 2011Published online 26 May 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23662
E G G F I N D I N G S A F T E R L - D O P A I N T A K E I N P D P A T I E N T S
Movement Disorders, Vol. 26, No. 9, 2011 1737

between the PD group with motor fluctuations and thegroup without motor fluctuations. Patients and controlsubjects (mean age, 64 years; 12 women, 8 men) wereenrolled according to these exclusion criteria: (1) BMI<25 and >30, (2) prior surgery on the esophagus orstomach, (3) a history of peptic ulcer disease, and (4) useof medications that may affect gastrointestinal motorfunction such as prokinetic agents, histamine type 2receptor antagonists, and proton pump inhibitors.After informed consent, the enrolled subjects under-
went overnight fasting without consuming breakfastand a drug withdrawal of 12 hours. The EGG activityhas been studied in all patients at 9 AM (as baseline)and 11 AM, 1 hour after intake of 200 mg of LD. EachEGG registration lasted 45 minutes. In the meantime,in the same patients, LD plasma levels were taken at9 AM (basal), 11 AM, 12 PM, and 1 PM UPDRS part IIImotor scores were obtained by a specialist in move-ment disorders at 9 AM and 11 AM.
EGG Study
Three surface electrodes (1 active, 1 reference, and 1ground electrode) were placed on the subject’s abdo-men after skin preparation, according to the Brownmodel (Brown BH, 1975), as shown in Figure 1. Thesubjects were supine during the recording, with thehead of the bed raised to 30�.The EGG signal was recorded using STEP 32 PC
DemItalia srl (http://www.step32.com) and a specificgastric probe, sampled at 256 Hz; the low-pass filterwas 15 cycles per minute (cpm) and the high-passfilter cutoff was 1.8 cpm, generating a signal ampli-tude in the range of 10–100 lV.The assessed variable was dominant frequency (DF),
defined as the peak frequency of the power spectrumof the overall EGG within the range of 1.0–9.0 cpm.
An overall spectrum analysis was performed using theentire time domain EGG signal (Parkman HP, 1996).
Pharmacokinetic Study
Blood samples, taken at the basal point (12 hours afterlast LD administration) and 1, 2, and 3 hours after theadministration of 200 mg of LD, were stored at �80�C.LD quantification was performed on plasma using aHPLC system equipped with a fluorescence detector.
Statistics
Comparisons were performed using analysis of var-iance, the Student t test, and linear regression.
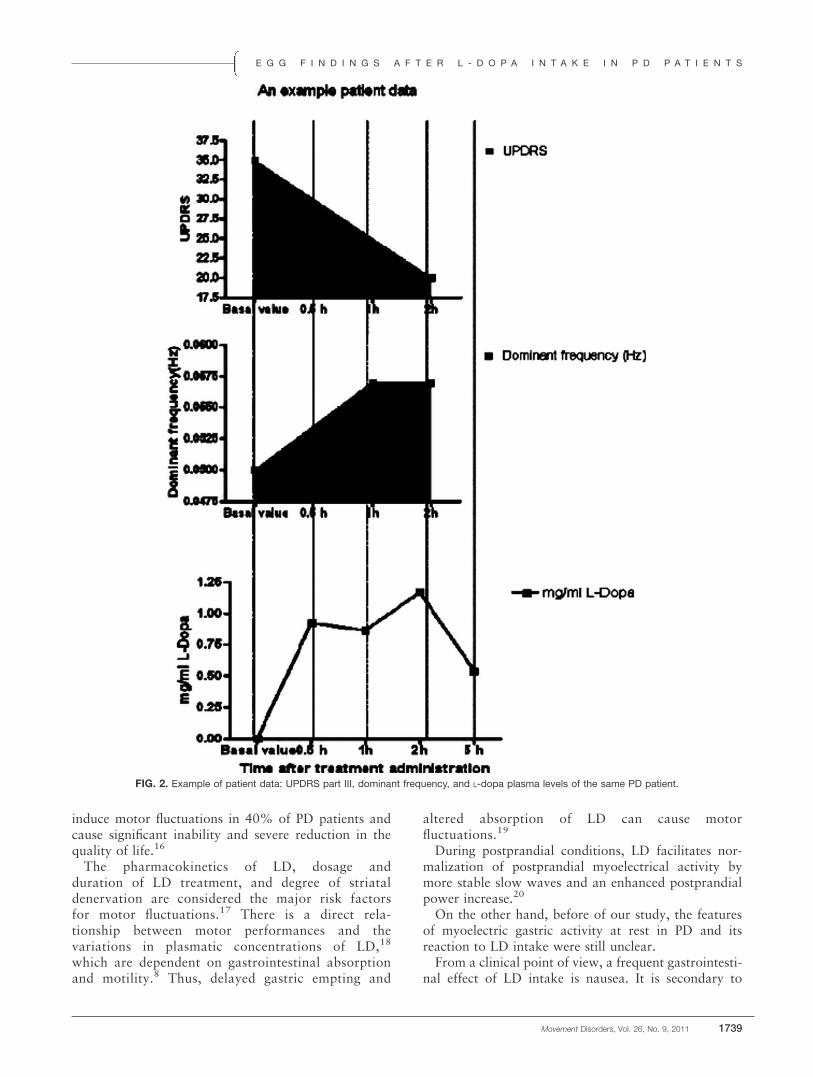
ResultsWe did not find any difference in DF between controls
and PD patients: DF mean in our patient group was 2.766 0.30 cpm, similar to controls (mean, 2.68 6 0.24cpm); no differences were found between the 2 groups ofpatients with motor fluctuations (2.70 cpm) and withoutmotor fluctuations (2.80 cpm). LD intake did not influ-ence DF value (2.74 6 0.34 cpm; Fig. 2). EGG activityin our group of PD patients when fasting was independ-ent of clinical staging and duration of disease.
ConclusionsThe gastrointestinal system has been implicated in the
pathogenesis of PD by the involvement of dopaminergicneurons since the first stages. Initial lesions15 developat 2 specific sites, namely, the dorsal motor nucleus ofthe vagal nerve and the anterior olfactory nucleus,and then progress to other central nervous structures.LD is the most effective treatment of mostly motor
symptoms, but after about 5 years of therapy, it may
FIG. 1. Application of surface electrodes according Brown’s model; representation of dominant frequency of gastric band within 0.035 and 0.050 Hz.[Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
A L B A N I E T A L .
1738 Movement Disorders, Vol. 26, No. 9, 2011
induce motor fluctuations in 40% of PD patients andcause significant inability and severe reduction in thequality of life.16
The pharmacokinetics of LD, dosage andduration of LD treatment, and degree of striataldenervation are considered the major risk factorsfor motor fluctuations.17 There is a direct rela-tionship between motor performances and thevariations in plasmatic concentrations of LD,18
which are dependent on gastrointestinal absorptionand motility.8 Thus, delayed gastric empting and
altered absorption of LD can cause motorfluctuations.19
During postprandial conditions, LD facilitates nor-malization of postprandial myoelectrical activity bymore stable slow waves and an enhanced postprandialpower increase.20
On the other hand, before of our study, the featuresof myoelectric gastric activity at rest in PD and itsreaction to LD intake were still unclear.From a clinical point of view, a frequent gastrointesti-
nal effect of LD intake is nausea. It is secondary to
FIG. 2. Example of patient data: UPDRS part III, dominant frequency, and L-dopa plasma levels of the same PD patient.
E G G F I N D I N G S A F T E R L - D O P A I N T A K E I N P D P A T I E N T S
Movement Disorders, Vol. 26, No. 9, 2011 1739

dopaminergic gastric activation and may be controlledby an antidopaminergic prokinetic agent such asdomperidone.This clinical observation may lead us to expect that
LD intake would induce a reduction in gastric motil-ity, which is already reduced ‘‘per se’’ by the disease.EGG findings obtained in the present study during
fasting are similar to those reported in the literature,where the EGG frequency ranges are defined as 0.5–2.0 cpm for bradygastria, 2.0–4.0 cpm for normalrhythm, and 4.0–9.0 cpm for tachygastria.11
Our results showed when fasting, a normal EGGpattern in PD patients compared with control subjects,with values in agreement with a previous study20
reporting a DF of 2.88 6 0.07 cpm in 13 PD patients.Moreover, our results indicate that the EGG patternin PD patients when fasting is unrelated to clinicaland LD pharmacokinetic parameters. So, differentlythan in scintigraphic after-meal studies, where thedelay in gastric emptying worsened in the advanceddisease stages,19,21 gastric activity is normal at rest inPD and cannot be varied by clinical parameters.This finding of normality at rest emphasizes the im-
portance of features of the diet, suggesting an altera-tion of gastric activity that is meal type–dependent.Our data show that in fasting PD patients, DF is not
influenced by LD intake. This finding would suggest thatgastric motility at rest may be normal. Much more, plas-matic LD levels are time-correlated with motor responsesbut not with the contemporary EGG frequency. Thiswould raise some doubts concerning the role of gastricmotility in the genesis of motor fluctuations.These data are strongly in agreement with recent obser-
vations10 following 13 C-acetate breath test, demonstrat-ing that delayed gastric emptying does not differ betweenPD patients with and without motor fluctuations.In brief, normal gastric activity at rest would indi-
cate that gastroenteric dysfunctions in PD areexpressed as an abnormal response to extrinsic factorssuch as food, emotions, and motor status.
References1. Djaldetti R, Baron J, Ziv I, Melamed E. Gastric emptying in Par-
kinson’s disease: patients with and without response fluctuations.Neurology 1996;46:1051–1054.
2. Edwards LL, Pfeiffer RF, Quigley EMM, Hofman R, Baluff M.Gastrointestinal symptoms in Parkinson’s disease. Mov Dis 1991;6:151–156.
3. Siddiqui MF, Rast S, Lynn MJ, Auchus AP, Pfeiffer RF. Auto-nomic dysfunction in Parkisnon’s disease: a comprehensive symp-tom survey. Parkinsonism Relat Disord 2002;8:277–284.
4. Del Tredici K, Rub U, De Vos RA, Bohl JR, Braak H. Wheredoes Parkinson’s disease pathology begin in the brain?J Neuropa-thology Exp Neurol 2002;61:413–426.
5. Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F. Par-kinson’s disease: the presence of Lewy bodies in Auerbach’s andMeissner plexuses. Acta Neuropathol 1988;76:217–221.
6. Braak H, de Vos RA, Bohl J, Del Tredci K. Gastric alpha-synu-clein immunoreactive inclusions in Meissne’s and Auerbach’splexuses in cases staged for Parkinson’s disease-related brain pa-thology. Neurosci Lett 2006;396:67–72.
7. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease.Lancet Neurol 2003;2:107–116.
8. Kurlan R, Rothfield KP, Woodward WR, et al. Erratic gastricemptying of levodopa may cause ‘‘random’’ fluctuations of par-kinsonian mobility. Neurology 1988;38:419–421.
9. Soykan I, Sarosiek I, Shifflett J, Wooten GF, McCallum RW.Effect of chronic oral domperidone therapy on gastrointestinalsymptoms and gastric emptying in patients with Parkinson’s dis-ease. Mov Disord 1997;12:952–957.
10. Tanaka Y, Kato T, Nishida H, et al. Is there a difference in gas-tric emptying between Parkinson’s disease patients under long-term L-dopa therapy with and without motor fluctuations? Ananalysis using the 13 C-acetate breath test. J Neurol 2009;256:1972–1976.
11. Chen JD, Zou X, Lin X, Ouyang S, Liang J. Detection of gastricslow wave propagation from the cutaneous electrogastrogram.Am J Physiol 1999;277:G424–G430.
12. Hamilton JW, Bellahsene BE, Reicherderfer M, Webster JH, BassP. Human electrogastrograms—comparison of surface and muco-sal recordings. Dig Dis Sci 1986;31:33–39.
13. Krygowska-Wajs A, Lorens K, Thor P, Szczudlik A, Konturek S.Gastric electromechanical dysfunction in Parkinson’s disease.Funct Neurol 2000;15:41–46.
14. Soykan I, Lin Z, Bennett JP, McCallum RW. Gastric myoelectri-cal activity in patients with Parkinson’s disease: evidence of a pri-mary gastric abnormality. Dig Dis Sci 1999;44:927–931.
15. Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sand-mann- Keil, Rub U. Staging of the intracerebral inclusion bodypathology associated with idiopathic Parkinson’s disease (preclini-cal and clinical stages). J Neurol 2002;249:S1–S5.
16. Chapuis S, Ouchchane L, Metz O, Gerbaud L, Durif F. Impact ofthe motor complications of Parkinson’s disease on the quality oflife. Mov Disord 2005;20:224–230.
17. Lloyd KG, Davison L, Hornykiewicz O. The neurochemistry ofParkinson’s disease: effect of L-dopa therapy. J Pharmacol ExpTher 1975;195:453–464.
18. Tolosa ES, Martin WE, Cohen HP, Jacobson RL. Patterns of clin-ical response and plasma dopa levels in Parkinson’s disease. Neu-rology 1975;25:177–183.
19. Hardoff R, Sula M, Tamir A, et al. Gastric emptying time andgastric motility in patients with Parkinson’s disease. Mov Disord2001;16:1041–1047.
20. Lu CL, Shan DE, Chen CY, et al. Impaired gastric myoelectricalactivity in patients with Parkinson’s disease and effect of levo-dopa treatment. Dig Dis Sci 2004;49:744–749.
21. Goetze O, Wieczorek J, Mueller T, Przuntek H, Schmidt WE,Woitalla D. Impaired gastric emptying of a solid test meal inpatients with Parkinson’s disease using 13C–sodium octanatebreath test. Neurosci Lett 2005;375:170–173.
A L B A N I E T A L .
1740 Movement Disorders, Vol. 26, No. 9, 2011

Subthreshold Depression inParkinson’s Disease
Julia Reiff, MD,1 Nele Schmidt, MPsych,2
Bastian Riebe, MD,2 Robert Breternitz,2
Josef Aldenhoff, MD, PhD,1 Gunther Deuschl, MD, PhD,2
and Karsten Witt, MD, PhD2*
1Department of Psychiatry, Zentrum fur Integrative Psychiatrie, Kiel,
Germany; 2Department of Neurology, University Medical Center
Schleswig-Holstein, Campus Kiel, Kiel, Germany
ABSTRACTBackground: Quality of life in Parkinson patients withsubthreshold depression could be improved if the prev-alence and symptom profile were better understood.Methods: Our study used standard DSM-IV and Juddcriteria as well as motor, depression, and quality-of-lifescales to investigate a sample of 110 nondementedParkinson patients. This led to formation of nonde-pressed (48.2%), subthreshold depressed (25.5%), anddepressed (26.4%) groups.Results: Quality of life was seen to be significantlylower in subthreshold depressed patients than in thenondepressed, and there were differences in the fre-quency of depressive symptoms that partially over-lapped with nonmotor symptoms of vegetative origin inParkinson’s disease (appetite, sleep disorders). Keymeasures of depression (diminished interest/pleasure)were more frequent in the depressed group comparedwith the subthreshold depressed, although the motorfunctions of these 2 groups did not differ significantly.Conclusions: The Beck Depression Inventory scoreranging from 9 to 15 points differentiates subthresholddepressed from nondepressed and depressed patientsbest. VC 2011 Movement Disorder Society
Key Words: Parkinson’s disease; subthresholddepression; quality of life
Depressive disorders affect up to 50% of patientswith Parkinson’s disease (PD).1 Most studies havefocused on major depression and its negative impact
on quality of life, motor function, and cognitive func-tion.1,2 Mild depressive symptoms in PD are frequentand are potentially treatable, but are usually difficultto diagnose because motor and nonmotor symptomsof depression and PD overlap.DSM-IV diagnostic criteria of minor depression
include the presence of 2 to 4 symptoms among the 9predefined symptoms of depression, and at least 1 ofthe symptoms must be depressed mood or loss of in-terest or pleasure.3 The 7 remaining items mostlyinclude somatic factors such as weight loss, insomnia,psychomotor alteration, fatigue, and diminished abilityto think or concentrate. The clinical significance crite-rion was introduced to further differentiate the preva-lence of a depressive mental disorder from normalmood reactions to life. Here, depression symptoms aredefined as those that impair social, occupational, orother important areas of functioning.4 However, theclinical significance criterion is not practical for PDpatients because motor, nonmotor, and other symp-toms of depression overlap with symptoms of PD interms of impairment.Patients who do not meet criteria for major depres-
sion, minor depression, or dysthymic disorder, butpresent at least 2 current depressive symptoms dailyfor at least 2 weeks, continuously or for most hours ofthe day, are considered to have subsyndromal or sub-threshold depression.5 Because neither the key DSM-IV criteria nor the clinical significance criterion isobligatory in the diagnosis of subthreshold depressionin PD patients, the subthreshold depression criteriamight be easier to apply. In this study, we investigatedthe prevalence of subthreshold depression and itsimpact on quality of life and motor function in con-secutive PD samples. Furthermore, we evaluated thevalidity of diagnostic screening instruments of depres-sion, such as the Judd criteria,5 for subthresholddepression in PD.
Patients and MethodsThis prospective study included 110 patients en-
rolled for a 5-month period in our outpatient clinic.The inclusion criterion was idiopathic PD based onthe British Parkinson’s Disease Society Brain Bankcriteria.6 Exclusion criteria were dementia (Mattis de-mentia rating scale7 sum score � 130), psychiatric dis-orders other than depression, deep brain stimulation,neurological diseases besides PD, and serious meta-bolic or systemic diseases. All patients gave theirinformed consent to participate, and the study wasapproved by the local ethics committee.The psychiatric and neurological examination was
carried out while the patients were in a stable motorcondition under normal medication. The Mini Interna-tional Neuropsychiatric Inventory8 was conducted toclassify symptoms of depression into the categories‘‘no depression,’’ ‘‘minor depression,’’ ‘‘dysthymia,’’
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Dr. Karsten Witt, Department of Neurology.University Medical Center Schleswig-Holstein, Campus Kiel,Schittenhelmstrasse 10, 24105 Kiel, Germany; [email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 30 December 2009; Revised: 10 January 2011; Accepted:7 February 2011Published online 25 March 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23699
S U B T H R E S H O L D D E P R E S S I O N I N P D
Movement Disorders, Vol. 26, No. 9, 2011 1741
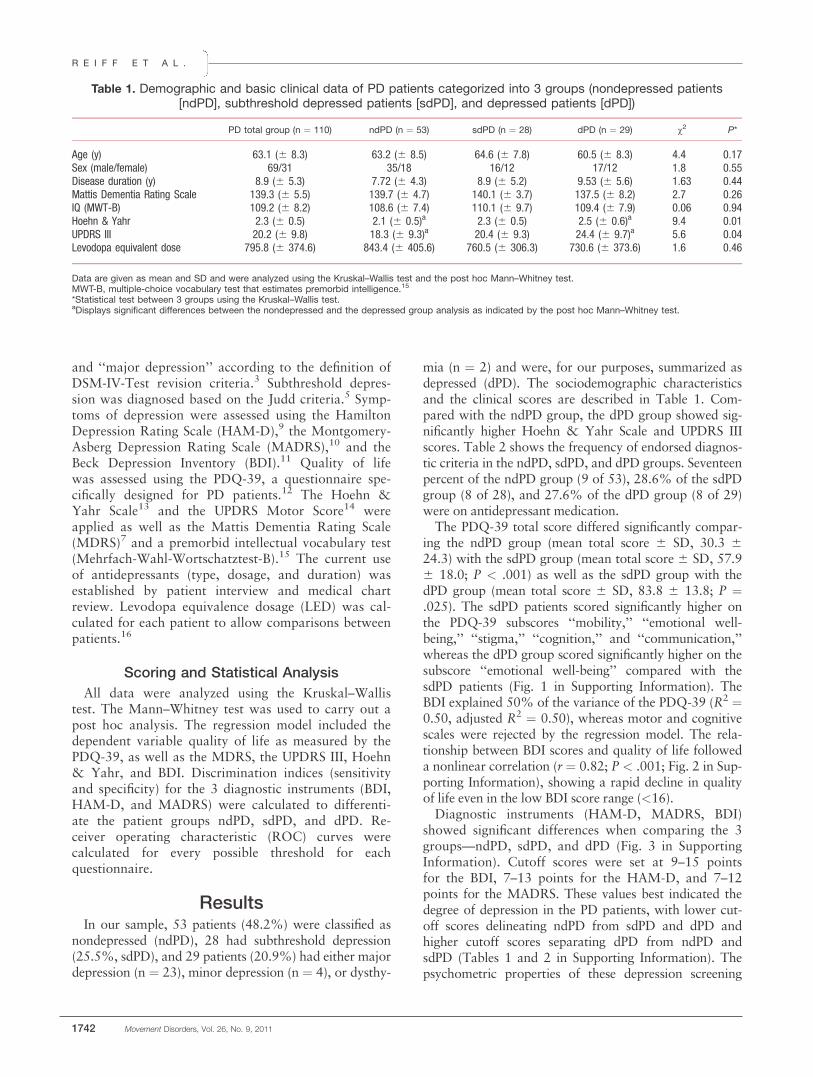
and ‘‘major depression’’ according to the definition ofDSM-IV-Test revision criteria.3 Subthreshold depres-sion was diagnosed based on the Judd criteria.5 Symp-toms of depression were assessed using the HamiltonDepression Rating Scale (HAM-D),9 the Montgomery-Asberg Depression Rating Scale (MADRS),10 and theBeck Depression Inventory (BDI).11 Quality of lifewas assessed using the PDQ-39, a questionnaire spe-cifically designed for PD patients.12 The Hoehn &Yahr Scale13 and the UPDRS Motor Score14 wereapplied as well as the Mattis Dementia Rating Scale(MDRS)7 and a premorbid intellectual vocabulary test(Mehrfach-Wahl-Wortschatztest-B).15 The current useof antidepressants (type, dosage, and duration) wasestablished by patient interview and medical chartreview. Levodopa equivalence dosage (LED) was cal-culated for each patient to allow comparisons betweenpatients.16
Scoring and Statistical Analysis
All data were analyzed using the Kruskal–Wallistest. The Mann–Whitney test was used to carry out apost hoc analysis. The regression model included thedependent variable quality of life as measured by thePDQ-39, as well as the MDRS, the UPDRS III, Hoehn& Yahr, and BDI. Discrimination indices (sensitivityand specificity) for the 3 diagnostic instruments (BDI,HAM-D, and MADRS) were calculated to differenti-ate the patient groups ndPD, sdPD, and dPD. Re-ceiver operating characteristic (ROC) curves werecalculated for every possible threshold for eachquestionnaire.
ResultsIn our sample, 53 patients (48.2%) were classified as
nondepressed (ndPD), 28 had subthreshold depression(25.5%, sdPD), and 29 patients (20.9%) had either majordepression (n ¼ 23), minor depression (n ¼ 4), or dysthy-
mia (n ¼ 2) and were, for our purposes, summarized asdepressed (dPD). The sociodemographic characteristicsand the clinical scores are described in Table 1. Com-pared with the ndPD group, the dPD group showed sig-nificantly higher Hoehn & Yahr Scale and UPDRS IIIscores. Table 2 shows the frequency of endorsed diagnos-tic criteria in the ndPD, sdPD, and dPD groups. Seventeenpercent of the ndPD group (9 of 53), 28.6% of the sdPDgroup (8 of 28), and 27.6% of the dPD group (8 of 29)were on antidepressant medication.The PDQ-39 total score differed significantly compar-
ing the ndPD group (mean total score 6 SD, 30.3 624.3) with the sdPD group (mean total score 6 SD, 57.96 18.0; P < .001) as well as the sdPD group with thedPD group (mean total score 6 SD, 83.8 6 13.8; P ¼.025). The sdPD patients scored significantly higher onthe PDQ-39 subscores ‘‘mobility,’’ ‘‘emotional well-being,’’ ‘‘stigma,’’ ‘‘cognition,’’ and ‘‘communication,’’whereas the dPD group scored significantly higher on thesubscore ‘‘emotional well-being’’ compared with thesdPD patients (Fig. 1 in Supporting Information). TheBDI explained 50% of the variance of the PDQ-39 (R2 ¼0.50, adjusted R2 ¼ 0.50), whereas motor and cognitivescales were rejected by the regression model. The rela-tionship between BDI scores and quality of life followeda nonlinear correlation (r ¼ 0.82; P < .001; Fig. 2 in Sup-porting Information), showing a rapid decline in qualityof life even in the low BDI score range (<16).Diagnostic instruments (HAM-D, MADRS, BDI)
showed significant differences when comparing the 3groups—ndPD, sdPD, and dPD (Fig. 3 in SupportingInformation). Cutoff scores were set at 9–15 pointsfor the BDI, 7–13 points for the HAM-D, and 7–12points for the MADRS. These values best indicated thedegree of depression in the PD patients, with lower cut-off scores delineating ndPD from sdPD and dPD andhigher cutoff scores separating dPD from ndPD andsdPD (Tables 1 and 2 in Supporting Information). Thepsychometric properties of these depression screening
Table 1. Demographic and basic clinical data of PD patients categorized into 3 groups (nondepressed patients[ndPD], subthreshold depressed patients [sdPD], and depressed patients [dPD])
PD total group (n ¼ 110) ndPD (n ¼ 53) sdPD (n ¼ 28) dPD (n ¼ 29) v2 P*
Age (y) 63.1 (6 8.3) 63.2 (6 8.5) 64.6 (6 7.8) 60.5 (6 8.3) 4.4 0.17Sex (male/female) 69/31 35/18 16/12 17/12 1.8 0.55Disease duration (y) 8.9 (6 5.3) 7.72 (6 4.3) 8.9 (6 5.2) 9.53 (6 5.6) 1.63 0.44Mattis Dementia Rating Scale 139.3 (6 5.5) 139.7 (6 4.7) 140.1 (6 3.7) 137.5 (6 8.2) 2.7 0.26IQ (MWT-B) 109.2 (6 8.2) 108.6 (6 7.4) 110.1 (6 9.7) 109.4 (6 7.9) 0.06 0.94Hoehn & Yahr 2.3 (6 0.5) 2.1 (6 0.5)a 2.3 (6 0.5) 2.5 (6 0.6)a 9.4 0.01UPDRS III 20.2 (6 9.8) 18.3 (6 9.3)a 20.4 (6 9.3) 24.4 (6 9.7)a 5.6 0.04Levodopa equivalent dose 795.8 (6 374.6) 843.4 (6 405.6) 760.5 (6 306.3) 730.6 (6 373.6) 1.6 0.46
Data are given as mean and SD and were analyzed using the Kruskal–Wallis test and the post hoc Mann–Whitney test.MWT-B, multiple-choice vocabulary test that estimates premorbid intelligence.15
*Statistical test between 3 groups using the Kruskal–Wallis test.aDisplays significant differences between the nondepressed and the depressed group analysis as indicated by the post hoc Mann–Whitney test.
R E I F F E T A L .
1742 Movement Disorders, Vol. 26, No. 9, 2011

instruments are almost equivalent (Tables 1 and 2 andFig. 4 in Supporting Information).
DiscussionNeuropsychiatric nonmotor symptoms of PD range
from anxiety and mood fluctuations to apathy anddepression. Subthreshold depression criteria focus onneuropsychiatric nonmotor symptoms. Although a dif-ferential diagnosis is difficult to make because of thesimilarity of diagnostic criteria for subthresholddepression and minor depression, we found that sub-threshold depression was about a quarter more fre-quent (28 of 110) than minor depression (4 of 110) inour study sample. The presence of key criteria(depressed mood and diminished interest or pleasure)and the criterion of clinical significance3 ensure thatthe diagnosis of either major or minor depression isfounded on symptoms of depression rather than othernonmotor symptoms of PD. Can the subthresholddepression diagnosis be based simply on somatic fea-tures of PD? Indeed, some symptoms (eg, changes inappetite, sleep disorder) are mixed features of vegeta-tive nonmotor symptoms and depression, and it is notpossible to clarify the etiology (neurovegetative orneuropsychiatric origin) in an individual patient.Other symptoms of depression (eg, depressed mood,suicidal ideation) are not sufficient to differentiatendPD from sdPD. Nevertheless, the profile of depres-sive symptoms in sdPD also shows that typical fea-tures of depression such as diminished pleasure andinterest and feeling of worthlessness are more frequentin sdPD than in ndPD. This suggests that it might bemore practical to consider these features for the sdPDdiagnosis. Subthreshold depression diagnostic factorsmight rely more heavily on signs and symptoms ofdepression and depression severity than on symptometiology. This point and the effects of an antidepres-
sive therapy have to be addressed and clarified in alongitudinal study.Subthreshold depression and its impact on quality of
life have not been studied in PD before. About a quar-ter of our patients had sdPD, and quality of life in thissubgroup was significantly lower than that in thendPD group. Because the results of a cognitive screen-ing test and the motor evaluation did not significantlydiffer between sdPD and ndPD patients, the differencein quality of life factors could be specifically attributedto the severity of depressive symptoms. Regressionanalysis of the relationship between the BDI scoresand the PDQ-39 values showed a significant nonlinearrelationship and demonstrated a rapid decline in qual-ity of life, even in the low BDI score range (<16points).The discrimination values of the 3 rating scales—
BDI, HAM-D, and MADRS—were almost equivalent.A BDI score between 9 and 15 points differentiatedsdPD from ndPD and the dPD group. Scales such asthe BDI are not meant to be used as diagnostic instru-ments, but they can serve as a screening instrumentand facilitate the diagnostic process in clinicalpractice.There were limitations to this study. Our outpatient
clinic population did not reflect the entire clinicalspectrum of Parkinson’s disease. Furthermore, ourstudy was not a longitudinal study. In general, sub-threshold depression may represent a premorbid or aresidual state of major depression.5 The course ofdepressive symptoms and the influence of antidepres-sants on subthreshold depression and quality of life inPD have not been investigated sufficiently.
References1. Reijnders JS, Ehrt U, Weber WE, Aarsland D, Leentjens AF. A
systematic review of prevalence studies of depression in Parkin-son’s disease. Mov Disord. 2008;23:183–189.
Table 2. Diagnostic criteria for depression classification—frequency of positive criteria in nondepressed (ndPD),subthreshold depressed (sdPD), and major depressed (dPD) Parkinson’s disease patients
ndPD (n ¼ 53) sdPD (n ¼ 28) dPD (n ¼ 29)
Depression criteria Positive/negative Positive/negative Positive/negativeDepressed mood 0/53 2/26d 26/3d
Diminished interest or pleasure 0/53a 3/25a,d 27/2d
Decrease or increase in appetite 1/52b 7/21b 13/16Sleep disturbance 12/41b 22/6b 26/3Psychomotor agitation or retardation 20/33b 21/7b 23/6Fatigue or loss of energy 3/50b 13/15b,c 22/7c
Feelings of worthlessness 0/53b 14/14b 18/11Diminished ability to think or concentrate 1/52b 11/17b,d 24/5d
Suicidal ideation 0/53 0/28 3/26
aSignificant differences between ndPD and sdPD group (P ¼ .02, Mann–Whitney test).bSignificant differences between ndPD and sdPD group (P < .001, Mann–Whitney test).cSignificant differences between sdPD and dPD group (P ¼ .02, Mann–Whitney test).dSignificant differences between sdPD and dPD group (P < .001, Mann–Whitney test).
S U B T H R E S H O L D D E P R E S S I O N I N P D
Movement Disorders, Vol. 26, No. 9, 2011 1743

2. Schrag A, Jahanshahi M, Quinn N. What contributes to qualityof life in patients with Parkinson’s disease? J Neurol NeurosurgPsychiatry. 2000;69:308–312.
3. First MB, Gibbon M, Williams JB. Structured clinical interviewfor DSM-IV Axis I disorders (SCID-I). Clinician Version. Arling-ton, VA: American Psychiatric Publishing, Inc.; 2002.
4. Spitzer RL, Wakefield JC. DSM-IV diagnostic criterion for clinicalsignificance: does it help solve the false positives problem? Am JPsychiatry. 1999;156:1856–1864.
5. Judd LL, Rapaport MH, Paulus MP, Brown JL. Subsyndromalsymptomatic depression: a new mood disorder? J Clin Psychiatry.1994;55(Suppl):18–28.
6. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinicaldiagnosis of idiopathic Parkinson’s disease: a clinico-pathologicalstudy of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184.
7. Mattis S. Dementia Rating Scale. Odessa, FL: PsychologicalAssessment Resources Inc.; 1988.
8. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-Interna-tional Neuropsychiatric Interview (M.I.N.I.): the developmentand validation of a structured diagnostic psychiatric interview forDSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl 20):22–33; quiz 34–57.
9. Hamilton M. A rating scale for depression. J Neurol NeurosurgPsychiatry. 1960;23:56–62.
10. Montgomery SA, Asberg M. A new depression scale designed tobe sensitive to change. Br J Psychiatry. 1979;134:382–389.
11. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An in-ventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571.
12. Peto V, Jenkinson C, Fitzpatrick R, Greenhall R. The develop-ment and validation of a short measure of functioning and wellbeing for individuals with Parkinson’s disease. Qual Life Res.1995;4:241–248.
13. Hoehn MM, Yahr MD. Parkinsonism: onset, progression andmortality. Neurology. 1967;17:427–442.
14. Fahn S, Elton RL. Unified Parkinson’s Disease Rating Scale. In:Marsden CD, Calne D, Goldstein M, eds. Recent Developmentsin Parkinson’s Disease. Florham Park, NJ: MacMillian HealthCare Information, 1987:153–163.
15. Lehrl S, Triebig G, Fischer B. Multiple choice vocabulary testMWT as a valid and short test to estimate premorbid intelligence.Acta Neurol Scand. 1995;91:335–345.
16. Deuschl G, Schade-Brittinger C, Krack P, et al. A randomizedtrial of deep-brain stimulation for Parkinson’s disease. N Engl JMed. 2006;355:896–908.
Late-Onset AsymmetricMyoclonus: An Emerging Syndrome
Petra Katschnig, MD,1,2 Joao Massano, MD,1,3
Mark J. Edwards, PhD,1 Petra Schwingenschuh, MD,1,2
Carla Cordivari, MD,4 and Kailash P. Bhatia, MD1*
1Sobell Department of Motor Neuroscience and Movement
Disorders, Institute of Neurology, University College London, Queen
Square, London, United Kingdom; 2Department of Neurology,
Division of Special Neurology, Medical University Graz, Graz, Austria;3Department of Neurology, Hospital de S. Joao, and Faculdade de
Medicina da Universidade do Porto, Porto, Portugal; 4Department of
Clinical Neurophysiology, National Hospital for Neurology and
Neurosurgery, University College London, Queen Square, London,
United Kingdom
ABSTRACTBackground: Asymmetric cortical myoclonus is typi-cally thought to be associated with either contralateralcortical structural lesions or degenerative disorderssuch as corticobasal degeneration when onset is inmiddle-aged or aged adults. This view has been chal-lenged after a recent case series brought to light a syn-drome of senile-onset, asymmetric cortical myoclonusnot associated with any such identifiable disorders,thus, named ‘‘primary progressive myoclonus of aging.’’This is rare and no other reports have been published;hence, further such cases need to be highlighted.Case reports: Here, we describe 3 patients with somesimilarities, namely, adult-onset, asymmetric myoclonusthat is most likely to be cortical, with an unremarkablethorough diagnostic workup, but with younger age atonset and longer follow-up time.Conclusions: This report expands on previous pheno-typical descriptions attempting to further develop andrefine this possible diagnostic entity. VC 2011 MovementDisorder Society
Key Words: myoclonus; cortex; primary; asymmetric;aging
------------------------------------------------------------*Correspondence to: Kailash P. Bhatia, Sobell Department of MotorNeuroscience and Movement Disorders, Institute of Neurology, QueenSquare, London, WC1N 3BG UK, Box 13; [email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.This research was supported by National Institutes of Health Grants R01NS39422, P30 ES09089, P42 ES10349, and R01 CA102484.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 20 March 2010; Revised: 31 December 2010; Accepted:13 January 2011Published online 26 May 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23676
K A T S C H N I G E T A L .
1744 Movement Disorders, Vol. 26, No. 9, 2011

Myoclonus is a brief, sudden, involuntary lightning-like movement, caused by either muscular contraction(positive myoclonus) or inhibition (negative myoclo-nus).1 It can be classified according to its topographicdistribution (focal, segmental, multifocal, generalized),etiology (physiological, essential, epileptic, sympto-matic, psychogenic), and pathophysiology (cortical,subcortical-supraspinal, spinal, peripheral).2,3 Asym-metric cortical myoclonus has been classically associ-ated with an underlying structural lesion or adegenerative condition affecting the contralateral cere-bral cortex, thus being considered secondary.4–9 How-ever, a recent case series has described 7 patients withapparently idiopathic asymmetric cortical myoclonus ofsenile onset for which no causal origin was found; theauthors labeled this syndrome as ‘‘primary progressivemyoclonus of aging.’’10 Briefly, the criteria used by theauthors for patient selection were: (1) asymmetricsymptomatic action myoclonus, (2) age � 65 years, (3)cortical myoclonus confirmed through electrophysiolog-ical testing, (4) no dementia, (5) no other clinical fea-tures suggestive of a neurodegenerative disorder, and(6) no other secondary cause found for the myoclonus.Here, we attempt to further develop and refine thispossible diagnostic entity by describing the cases of 3patients who resemble those described by Alvarez andCaviness,10 including videos to illustrate the phenotype.
Case Reports
Case 1
This 65-year-old woman was in good health untilage 60, when she noticed abnormal movements of her
left foot when attempting to put on her shoes. Overthe following month she developed further difficultiesincluding tripping over the left foot and problemsusing the clutch in her car. Her left arm and left facealso became affected by these movements, and shecomplained of difficulty using her left hand and somestiffness. Five years into the disease her symptoms hadprogressed very little since the first months. Her pastmedical history was unremarkable apart from a headinjury at age 10 and benign abdominal mesentericlipodystrophy. Neurological examination revealedprominent action-induced and stimulus-sensitive myo-clonus of the left leg, some jerks of the left arm, andmyoclonic twitches affecting the left side of her face,which worsened when speaking; occasionally somestimulus-sensitive and action-induced myoclonic jerkscould also be seen on the right lower limb, which madeher gait very difficult. Her reflexes were generally brisk,more so on the left side, but the remainder of the exam-ination was unremarkable. Repeated MRI scans of thebrain showed old damage to the left temporal lobe thatwas thought to be a result of her childhood headtrauma; imaging of the cervical spine, a DaT-SPECTscan, and a whole-body PET were normal. In the EEG,runs of theta/delta activity and spike waves were pres-ent over the right frontocentral region. Surface EMGshowed brief bursts of activity (20–50 ms). Corticalfast activity (C3-Cz/C4-Cz derivations) was found cor-relating with the jerks in the left lower limb (positiveand negative myoclonus), but the high frequency ofthese (around 20 Hz) precluded the correct assessmentof EEG back-averaging of the EMG bursts11; neverthe-less the electrophysiological and clinical findings were
FIG. 1. EEG and EMG tracings in case 1. A: Speed is 1 s/division. There is spiky activity in the vertex, predominantly on the right (obliquearrow), appearing at times in temporal relation with the myoclonic jerks shown in muscle leads, mainly in the left lower limb. B: Magnification(speed is 0.1 s/division) of a segment where fast myoclonic jerks (around 20 Hz) are present with runs of cortical activity seen mainly in the ver-tex, predominantly on the right (vertical arrow). EEG montage was done according to the standard 10-20 system, and derivations are F4-C4, C4-Cz, C3-Cz, F3-C3. EMG leads are (top to bottom, all left side): LBiceps, biceps; LIDIO, first interosseous digitalis; LVM, vastus medialis; LTA, tibi-alis anterior; LGastro, gastrocnemius.
L A T E - O N S E T A S Y M M E T R I C M Y O C L O N U S
Movement Disorders, Vol. 26, No. 9, 2011 1745
suggestive of cortical myoclonus (Fig. 1). SEPs from theleft lower limb did not reach published criteria forgiant SEPs.12 CSF analysis including Whipple’s PCR,14-3-3, and S100 and extensive blood analysis includ-ing routine blood tests, thyroid function and antibod-ies, vitamin B12, ferritin, and serum electrophoresiswere all normal or negative. Screening for antineuro-nal, GluR3, voltage-gated Kþ channel, anti-GAD, anti-amphiphysin, and NMDA-receptor antibodies werenegative. Antigliadin antibodies were positive; how-ever, a subsequent gut biopsy was negative. A gene testfor the epsilon sarcoglycan mutation was also negative.In light of the clinical presentation, the normal investi-gations, and long follow-up (more than 5 years now)with no features suggestive of a neurodegenerativecause, a diagnosis of idiopathic cortical myoclonus wasmade. Treatment with sodium valproate (1200 mg/day) had some effect, whereas clonazepam, levetirace-tam, and topiramate were not tolerated.
Case 2
This 60-year-old woman complained of a 10-year his-tory of clumsiness of the right hand when undertakingdelicate movements, difficulty in coordination, and inabil-ity to write. Apart from migraine with infrequent attacks(1–2/year), her past medical history was unremarkable.On examination, action-induced and stimulus-sensitivemyoclonic jerks affecting her right arm with some postur-ing were present. Tendon jerks were symmetrically brisk,but the remainder of the neurological examinationincluding 2-point discrimination was normal. Investiga-tions carried out early in the disease included a CT brainscan (MRI scan was declined by the patient), carotid an-
giography, EEG, CSF analysis, and routine blood testsincluding thyroid function and antithyroid antibodies,which were all normal or negative. She had been treatedwith clonazepam, diazepam, temazepam, primidone, andphenytoin without major benefit. Nine years into the dis-ease the jerks became worse, and the patient was onlyable to use her hand as a hook but not to grip any more.She complained about paraesthesia and hyperalgesia ofthe fingers without obvious explanation. Additionalinvestigations at that point included plasma electrophore-sis, a muscle biopsy in order to exclude a mitochondrialpathology, and antinuclear antibodies, which were nor-mal or negative. EMG with back-averaging of EEG dem-onstrated cortical spikes related to her myoclonic jerks,and she had giant SEPs (amplitude ¼ 12.1 lV). A diagno-sis of cortical myoclonus was made, and improvement inright hand function was achieved with a combination ofpiracetam (16.8 g/day), phenytoin (340 mg/day), and clo-nazepam (5 mg/day).
Case 3
This 69-year-old man complained of a 2-year his-tory of jerks, which started in his right leg and consid-erably affected his walking. Only a few months laterhe developed jerks of his right arm interfering withmost day-to-day activities. In addition, he noticedspasms of his right face when talking or opening hismouth, causing speech and swallowing problems. In2008 he was diagnosed with bilateral ischemic opticneuropathy and prostate cancer, which responded wellto chemotherapy. On examination there were stimu-lus-sensitive and action-induced positive and negativemyoclonic jerks of his right arm and predominantly
FIG. 2. EEG and EMG tracings in case 3. A: Speed is 1 s/division. There are almost continuous myoclonic jerks shown in all muscle leads. B: Mag-nification (speed is 0.2 s/division) of a segment where fast myoclonic jerks (around 20 Hz) are observed; a rhythmic cortical activity is seen in thevertex. The patient is extending the right wrist and abducting the fingers against resistance, and negative myoclonus is observed (oblique arrow).EEG montage was done according to the standard 10-20 system, and derivations are F4-C4, C4-Cz, C3-Cz, F3-C3. EMG leads are (top to bottom,all right side): Rtrap, trapezius; Rbiceps, biceps; REDC, extensor digitorum communis; RFCR, flexor carpi radialis; RIDIO, first interosseous digitalis.We acknowledge that a referential montage would have given clearer localizations of the EEG activity; however, in this study we were less interestedin precise localization, and therefore we do not believe the montage we used systematically changed the results.
K A T S C H N I G E T A L .
1746 Movement Disorders, Vol. 26, No. 9, 2011

negative myoclonus of his right leg. In addition, hehad jerks in his right face when opening the mouth,with some spread to the left side and concomitantjerks of the tongue; the gait was cautious because ofthe feeling of unsteadiness caused by the existence ofpositive and negative action myoclonus of the rightlower limb. The remainder of the neurological exami-nation was unremarkable. An MRI brain scan, a DaT-SPECT, and a whole-body FDG-PET were normal.Multichannel EEG-EMG recordings revealed no defi-nite epileptiform features. Positive and negative jerksoccurred frequently on posture and action and were ofshort duration (<50 ms), with silent periods between100 and 300 ms (Fig. 2). A correlation between corti-cal and muscle activity was not possible due to thehigh frequency of EMG bursts. However, the durationof the jerks was suggestive of cortical myoclonus.SEPs from the right leg did not fulfill criteria for giantSEPs. Routine blood tests including thyroid functionand a full thrombophilia and antiphospholipid screenwere normal. ANA, ENA, anticardiolipin, antineuro-nal, antitransglutaminase, antiendomysial, antigliadin,anti-GluR3, antiamphiphysin, anti-GAD, and antithy-roid antibodies were all negative. Levetiracetam andsodium valproate made him feel unwell without anyclear improvement, whereas clonazepam 1 mg per dayameliorated the myoclonic jerks to some extent.
DiscussionThe patients described here show some common fea-
tures: (1) markedly asymmetric, almost exclusivelyunilateral action myoclonus of likely cortical origin;(2) absence of other features, like parkinsonism,ataxia, pyramidal signs, or cognitive decline; (3) adultonset, in 2 cases before 65 years; and (4) absence of acause that could account for the symptoms despitecomprehensive laboratory and imaging workup. Theextended follow-up time (mean, 8 years) and compre-hensive workup allow an exclusion of conditions suchas corticobasal degeneration (CBD),4,7,13 paraneoplas-tic syndromes,14 or other disorders5,6,8,9 associatedwith myoclonus. For example, in a large series ofCBD patients, unilateral myoclonus was an isolatedearly feature in 3 of 36, whereas several others pre-sented with myoclonus associated with either limbclumsiness or stiffness or gait difficulty; during the fol-low-up period (mean, 5.2 years), several other patientsdeveloped myoclonus in association with other CBDfeatures, and contralateral spreading of the symptomswas the rule; the total duration of the illness rangedfrom 4 to 8 years.13 We also discounted epilepsia par-tialis continua as a possible differential diagnosisbecause jerks in our patients were seen spontaneouslymuch less than after stimulation or during action andoccurred at intervals of much longer than 10 seconds,inconsistent with the definition by Bien and Elger.15
Unlike the previously reported series,10 our patientswere on average younger at the onset of symptoms(mean, 59.0 years; range, 50–67 years), in contrastwith the patients described by Alvarez and Caviness,where only 1 of 7 had onset before age 70, and 3 of 7had onset at or after age 80. This report brings toattention the finding that patients may be younger andthe symptoms could be present for a longer periodthan previously described. The previous name given tothis syndrome, primary progressive myoclonus ofaging, might be somewhat misleading, as somepatients might develop symptoms well before age 65and ‘‘aging’’ in itself is an indeterminate term, butusually associated with global normal physiologicalprocesses—thus, it should not be associated with aclearly pathological situation. Moreover, at this pointthere are no data implying aging or any neurodegener-ative processes in the pathophysiology of this syn-drome. Our patients (and some of those previouslyreported) do not show a progressive course, but ratherstatic symptoms after an initial period of progression.This syndrome is an evolving one and indeed may
lead to reappraisal of some previously reported casesthat may fit within it.16 With further follow-up (andideally pathological examination) of those casesreported here and previously, we may be able to deter-mine the longer-term clinical outcome and more pre-cisely identify the nature and location of thepathological process behind the myoclonus.Video Case 1. Myoclonus induced by gentle toe
touching is observed on the left but not on the rightsides. Action myoclonus is also observed in the leftlower limb, which hinders ipsilateral voluntarymovements. Bilateral limb ideomotor apraxia testingis normal; when moving the left hand, a fast myoclo-nus (almost resembling a tremor) is triggered on theleft lower limb, which then subsides. The gait is verydifficult because of the constant action-induced myo-clonic jerks.Video Case 2. Movie made approximately 10 years
after the onset of symptoms. At rest no involuntarymovements are observed, but action myoclonus isobserved distally on the right side as soon as thepatient moves the upper limbs. Finger-to-nose testingshows no signs of cerebellar dysfunction, but actionmyoclonus is again seen on the right side. Stimulus-induced myoclonus can be seen, which is triggeredeasily by touching the hand, but not in more proximallocations. Some posturing of the right hand emergesonly during the myoclonic bursts—thus, we are reluc-tant to classify it as dystonic but rather think this is amanifestation of fast runs of myoclonus.Video Case 3. Action myoclonus can be seen in the
right upper limb. The myoclonus is also triggered bysensitive stimulation, which can be observed whentouching either fingers or toes on the right. Finger-to-nose testing shows no signs of cerebellar dysfunction,
L A T E - O N S E T A S Y M M E T R I C M Y O C L O N U S
Movement Disorders, Vol. 26, No. 9, 2011 1747

but action myoclonus is again seen on the right side.On jaw opening, orofacial myoclonus is observed,with some spread to the left side, and concomitanttongue myoclonus is observed on protrusion. The gaitis cautious due to the feeling of unsteadiness causedby the existence of action myoclonus in the rightlower limb.
Acknowledgments: We thank Professor Adam Zeman for helpwith the clinical details in one of the cases.
References1. Marsden CD, Hallett M, Fahn S. The nosology and pathophy-
siology of myoclonus. In: Marsden CD,Fahn S, eds.MovementDisorders. London, UK: Butterworths; 1982:196–248.
2. Caviness JN, Brown P. Myoclonus: current concepts and recentadvances. Lancet Neurol 2004;3:598–607.
3. Chang VC, Frucht SJ. Myoclonus. Curr Treat Options Neurol2008;10:222–229.
4. Bhatia KP, Lee MS, Rinne JO, et al. Corticobasal degenerationlook-alikes. Adv Neurol 2000;82:169–182.
5. Garg RK. Subacute sclerosing panencephalitis. J Neurol 2008;255:1861–1871.
6. Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated po-tassium channel autoimmunity mimicking creutzfeldt-jakob dis-ease. Arch Neurol 2008;65:1341–1346.
7. Mahapatra RK, Edwards MJ, Schott JM, Bhatia KP. Corticobasaldegeneration. Lancet Neurol 2004;3:736–743.
8. Ohnari K, Matsunaga K, Uozumi T, Tamagawa A, Hashimoto T,Tsuji S. Unilateral positive-negative myoclonus in Creutzfeldt-Jakob disease. Mov Disord 2006;21:1963–1966.
9. Takei H, Wilfong A, Malphrus A, et al. Dual pathology in Ras-mussen’s encephalitis: A study of seven cases and review of theliterature. Neuropathology 2010;30:381–391.
10. Alvarez M, Caviness JN. Primary progressive myoclonus of aging.Mov Disord 2008;23:1658–1664.
11. Grosse P, Guerrini R, Parmeggiani L, Bonanni P, PogosyanA, Brown P. Abnormal corticomuscular and intermuscularcoupling in high-frequency rhythmic myoclonus. Brain 2003;126:326–342.
12. Shibasaki H, Yamashita Y, Neshige R, Tobimatsu S, Fukui R.Pathogenesis of giant somatosensory evoked potentials in progres-sive myoclonic epilepsy. Brain 1985;108:225–240.
13. Rinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasaldegeneration. A clinical study of 36 cases. Brain 1994;117:1183–1196.
14. Grant R, Graus F. Paraneoplastic movement disorders. MovDisord 2009;24:1715–1724.
15. Bien CG, Elger CE. Epilepsia partialis continua: semiology anddifferential diagnoses. Epileptic Disord 2008;10:3–7.
16. Obeso JA, Artieda J, Rothwell JC, Day B, Thompson P, MarsdenCD. The treatment of severe action myoclonus. Brain 1989 Jun;112(Pt 3):765–777.
Dystonia Due to Cerebral PalsyResponds to Deep BrainStimulation of the Globus
Pallidus Internus
Warren A. Marks, MD,1,2* John Honeycutt, MD,3
Fernando Acosta Jr., MD,1 MaryAnn Reed, MS, CNRN,1
Laurie Bailey, MS,1,3 Angela Pomykal, PT,2
and Mary Mercer, OTR2
1Department of Neurology, Cook Children’s Medical Center,
Fort Worth, Texas, USA; 2Department of Rehabilitation, Cook
Children’s Medical Center, Fort Worth, Texas, USA; 3Department
of Neurosurgery, Cook Children’s Medical Center, Fort Worth,
Texas, USA
ABSTRACTBackground: Cerebral palsy is the most commoncause of pediatric-onset dystonia. Deep brain stimula-tion is gaining acceptance for treating dystonias in chil-dren. There is minimal reported experience regardingthe efficacy of deep brain stimulation in cerebral palsy.Methods: Fourteen patients, including 8 younger than 16years, received bilateral implants (13 patients) or a unilat-eral implant (1 patient) of the internal globus pallidus andwere observed in a noncontrolled, nonblinded study for atleast 6 months. Motor function was assessed using theBurke-Fahn-Marsden Dystonia Movement and Disabilityscales and the Barry Albright Dystonia Scale.Results: By 6 months, significant improvement wasobserved in the Burke-Fahn-Marsden Dystonia Movementscale (P 5 .004), the Burke-Fahn-Marsden Dystonia Dis-ability scale (P 5 .027), and the Barry Albright DystoniaScale (P 5 .029) for the whole cohort (n 5 14) and in thepatients treated before skeletal maturity (group 1; n 5 8):Burke-Fahn-Marsden Dystonia Movement scale, P 5 .012;Burke-Fahn-Marsden Dystonia Disability scale, P 5 .020;and Barry Albright Dystonia Scale, P5.027.Conclusions: Deep brain stimulation may offer an effec-tive treatment option for cerebral palsy–related dystonia,especially in those treated before skeletal maturity.VC 2011Movement Disorder Society
Key Words: cerebral palsy; deep brain stimulation;dystonia; globus pallidus; pediatric
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Dr. Warren A. Marks, Department of Neurology,Cook Children’s Medical Center, Fort Worth, TX 76104, USA;[email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 31 March 2010; Revised: 8 February 2011; Accepted:24 February 2011Published online 13 April 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23723
M A R K S E T A L .
1748 Movement Disorders, Vol. 26, No. 9, 2011
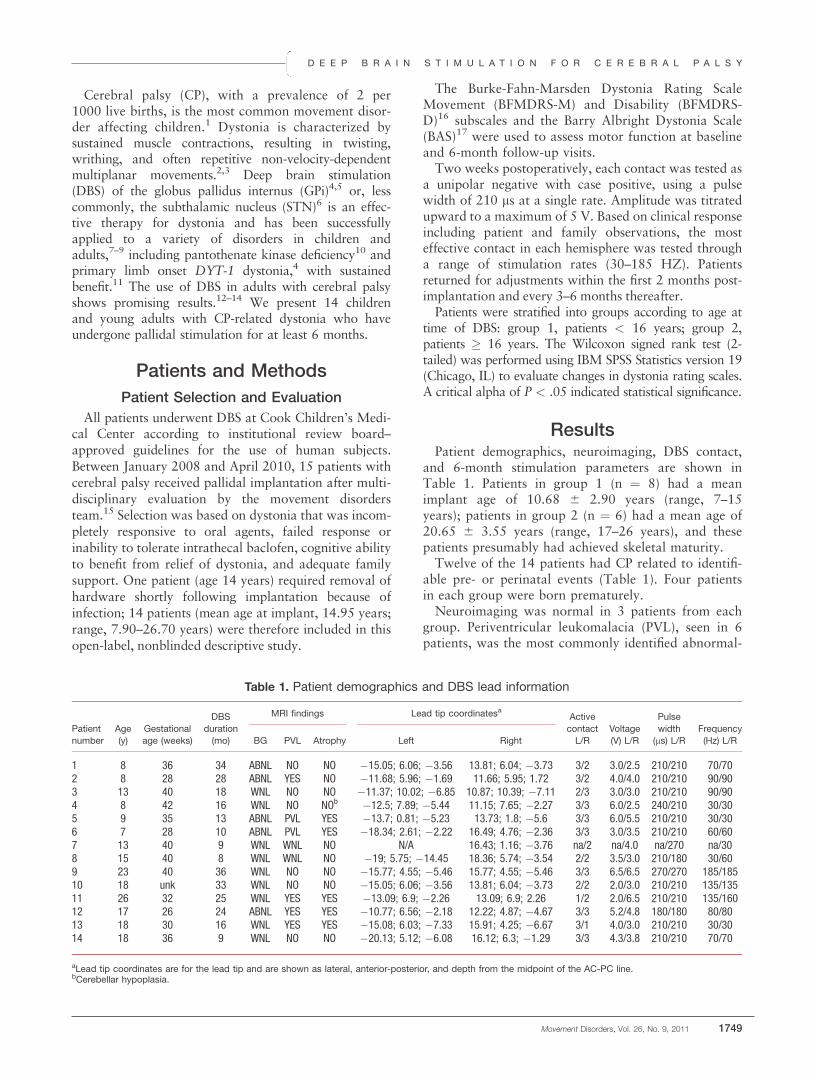
Cerebral palsy (CP), with a prevalence of 2 per1000 live births, is the most common movement disor-der affecting children.1 Dystonia is characterized bysustained muscle contractions, resulting in twisting,writhing, and often repetitive non-velocity-dependentmultiplanar movements.2,3 Deep brain stimulation(DBS) of the globus pallidus internus (GPi)4,5 or, lesscommonly, the subthalamic nucleus (STN)6 is an effec-tive therapy for dystonia and has been successfullyapplied to a variety of disorders in children andadults,7–9 including pantothenate kinase deficiency10 andprimary limb onset DYT-1 dystonia,4 with sustainedbenefit.11 The use of DBS in adults with cerebral palsyshows promising results.12–14 We present 14 childrenand young adults with CP-related dystonia who haveundergone pallidal stimulation for at least 6 months.
Patients and Methods
Patient Selection and Evaluation
All patients underwent DBS at Cook Children’s Medi-cal Center according to institutional review board–approved guidelines for the use of human subjects.Between January 2008 and April 2010, 15 patients withcerebral palsy received pallidal implantation after multi-disciplinary evaluation by the movement disordersteam.15 Selection was based on dystonia that was incom-pletely responsive to oral agents, failed response orinability to tolerate intrathecal baclofen, cognitive abilityto benefit from relief of dystonia, and adequate familysupport. One patient (age 14 years) required removal ofhardware shortly following implantation because ofinfection; 14 patients (mean age at implant, 14.95 years;range, 7.90–26.70 years) were therefore included in thisopen-label, nonblinded descriptive study.
The Burke-Fahn-Marsden Dystonia Rating ScaleMovement (BFMDRS-M) and Disability (BFMDRS-D)16 subscales and the Barry Albright Dystonia Scale(BAS)17 were used to assess motor function at baselineand 6-month follow-up visits.Two weeks postoperatively, each contact was tested as
a unipolar negative with case positive, using a pulsewidth of 210 ls at a single rate. Amplitude was titratedupward to a maximum of 5 V. Based on clinical responseincluding patient and family observations, the mosteffective contact in each hemisphere was tested througha range of stimulation rates (30–185 HZ). Patientsreturned for adjustments within the first 2 months post-implantation and every 3–6 months thereafter.Patients were stratified into groups according to age at
time of DBS: group 1, patients < 16 years; group 2,patients � 16 years. The Wilcoxon signed rank test (2-tailed) was performed using IBM SPSS Statistics version 19(Chicago, IL) to evaluate changes in dystonia rating scales.A critical alpha of P < .05 indicated statistical significance.
ResultsPatient demographics, neuroimaging, DBS contact,
and 6-month stimulation parameters are shown inTable 1. Patients in group 1 (n ¼ 8) had a meanimplant age of 10.68 6 2.90 years (range, 7–15years); patients in group 2 (n ¼ 6) had a mean age of20.65 6 3.55 years (range, 17–26 years), and thesepatients presumably had achieved skeletal maturity.Twelve of the 14 patients had CP related to identifi-
able pre- or perinatal events (Table 1). Four patientsin each group were born prematurely.Neuroimaging was normal in 3 patients from each
group. Periventricular leukomalacia (PVL), seen in 6patients, was the most commonly identified abnormal-
Table 1. Patient demographics and DBS lead information
Patient
number
Age
(y)
Gestational
age (weeks)
DBS
duration
(mo)
MRI findings Lead tip coordinatesa Active
contact
L/R
Voltage
(V) L/R
Pulse
width
(ls) L/RFrequency
(Hz) L/RBG PVL Atrophy Left Right
1 8 36 34 ABNL NO NO �15.05; 6.06; �3.56 13.81; 6.04; �3.73 3/2 3.0/2.5 210/210 70/702 8 28 28 ABNL YES NO �11.68; 5.96; �1.69 11.66; 5.95; 1.72 3/2 4.0/4.0 210/210 90/903 13 40 18 WNL NO NO �11.37; 10.02; �6.85 10.87; 10.39; �7.11 2/3 3.0/3.0 210/210 90/904 8 42 16 WNL NO NOb �12.5; 7.89; �5.44 11.15; 7.65; �2.27 3/3 6.0/2.5 240/210 30/305 9 35 13 ABNL PVL YES �13.7; 0.81; �5.23 13.73; 1.8; �5.6 3/3 6.0/5.5 210/210 30/306 7 28 10 ABNL PVL YES �18.34; 2.61; �2.22 16.49; 4.76; �2.36 3/3 3.0/3.5 210/210 60/607 13 40 9 WNL WNL NO N/A 16.43; 1.16; �3.76 na/2 na/4.0 na/270 na/308 15 40 8 WNL WNL NO �19; 5.75; �14.45 18.36; 5.74; �3.54 2/2 3.5/3.0 210/180 30/609 23 40 36 WNL NO NO �15.77; 4.55; �5.46 15.77; 4.55; �5.46 3/3 6.5/6.5 270/270 185/18510 18 unk 33 WNL NO NO �15.05; 6.06; �3.56 13.81; 6.04; �3.73 2/2 2.0/3.0 210/210 135/13511 26 32 25 WNL YES YES �13.09; 6.9; �2.26 13.09; 6.9; 2.26 1/2 2.0/6.5 210/210 135/16012 17 26 24 ABNL YES YES �10.77; 6.56; �2.18 12.22; 4.87; �4.67 3/3 5.2/4.8 180/180 80/8013 18 30 16 WNL YES YES �15.08; 6.03; �7.33 15.91; 4.25; �6.67 3/1 4.0/3.0 210/210 30/3014 18 36 9 WNL NO NO �20.13; 5.12; �6.08 16.12; 6.3; �1.29 3/3 4.3/3.8 210/210 70/70
aLead tip coordinates are for the lead tip and are shown as lateral, anterior-posterior, and depth from the midpoint of the AC-PC line.bCerebellar hypoplasia.
D E E P B R A I N S T I M U L A T I O N F O R C E R E B R A L P A L S Y
Movement Disorders, Vol. 26, No. 9, 2011 1749
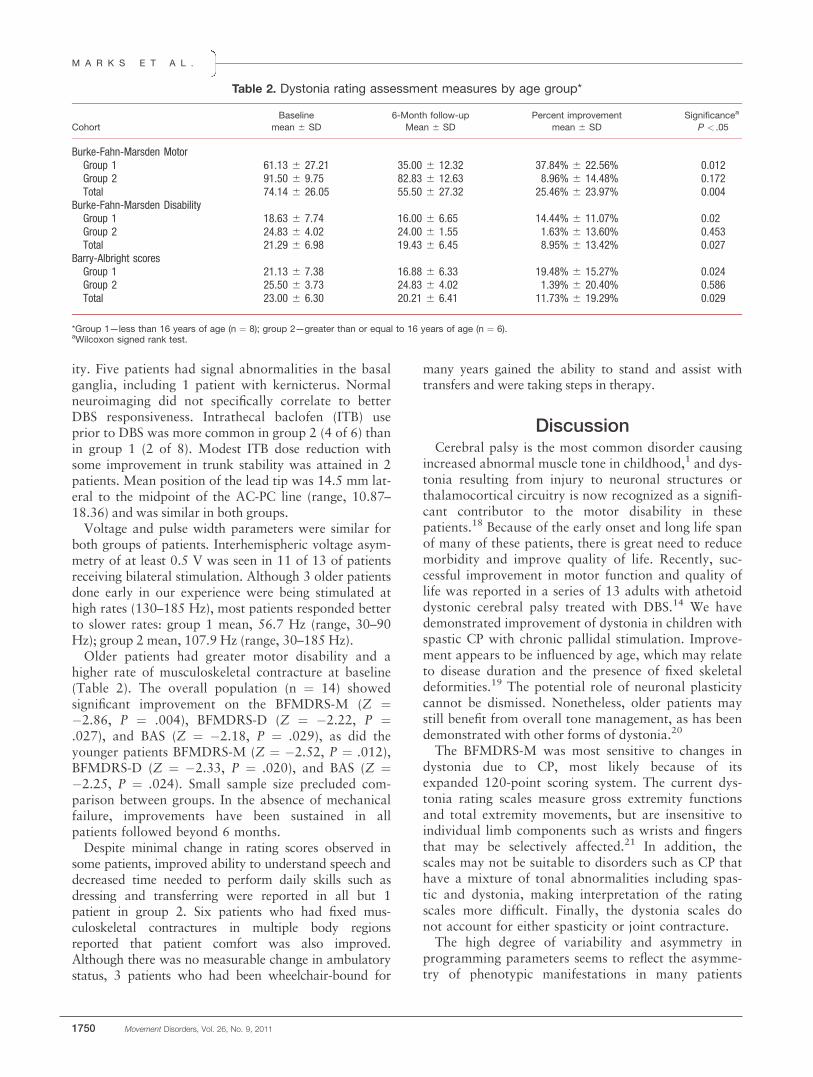
ity. Five patients had signal abnormalities in the basalganglia, including 1 patient with kernicterus. Normalneuroimaging did not specifically correlate to betterDBS responsiveness. Intrathecal baclofen (ITB) useprior to DBS was more common in group 2 (4 of 6) thanin group 1 (2 of 8). Modest ITB dose reduction withsome improvement in trunk stability was attained in 2patients. Mean position of the lead tip was 14.5 mm lat-eral to the midpoint of the AC-PC line (range, 10.87–18.36) and was similar in both groups.Voltage and pulse width parameters were similar for
both groups of patients. Interhemispheric voltage asym-metry of at least 0.5 V was seen in 11 of 13 of patientsreceiving bilateral stimulation. Although 3 older patientsdone early in our experience were being stimulated athigh rates (130–185 Hz), most patients responded betterto slower rates: group 1 mean, 56.7 Hz (range, 30–90Hz); group 2 mean, 107.9 Hz (range, 30–185 Hz).Older patients had greater motor disability and a
higher rate of musculoskeletal contracture at baseline(Table 2). The overall population (n ¼ 14) showedsignificant improvement on the BFMDRS-M (Z ¼�2.86, P ¼ .004), BFMDRS-D (Z ¼ �2.22, P ¼.027), and BAS (Z ¼ �2.18, P ¼ .029), as did theyounger patients BFMDRS-M (Z ¼ �2.52, P ¼ .012),BFMDRS-D (Z ¼ �2.33, P ¼ .020), and BAS (Z ¼�2.25, P ¼ .024). Small sample size precluded com-parison between groups. In the absence of mechanicalfailure, improvements have been sustained in allpatients followed beyond 6 months.Despite minimal change in rating scores observed in
some patients, improved ability to understand speech anddecreased time needed to perform daily skills such asdressing and transferring were reported in all but 1patient in group 2. Six patients who had fixed mus-culoskeletal contractures in multiple body regionsreported that patient comfort was also improved.Although there was no measurable change in ambulatorystatus, 3 patients who had been wheelchair-bound for
many years gained the ability to stand and assist withtransfers and were taking steps in therapy.
DiscussionCerebral palsy is the most common disorder causing
increased abnormal muscle tone in childhood,1 and dys-tonia resulting from injury to neuronal structures orthalamocortical circuitry is now recognized as a signifi-cant contributor to the motor disability in thesepatients.18 Because of the early onset and long life spanof many of these patients, there is great need to reducemorbidity and improve quality of life. Recently, suc-cessful improvement in motor function and quality oflife was reported in a series of 13 adults with athetoiddystonic cerebral palsy treated with DBS.14 We havedemonstrated improvement of dystonia in children withspastic CP with chronic pallidal stimulation. Improve-ment appears to be influenced by age, which may relateto disease duration and the presence of fixed skeletaldeformities.19 The potential role of neuronal plasticitycannot be dismissed. Nonetheless, older patients maystill benefit from overall tone management, as has beendemonstrated with other forms of dystonia.20
The BFMDRS-M was most sensitive to changes indystonia due to CP, most likely because of itsexpanded 120-point scoring system. The current dys-tonia rating scales measure gross extremity functionsand total extremity movements, but are insensitive toindividual limb components such as wrists and fingersthat may be selectively affected.21 In addition, thescales may not be suitable to disorders such as CP thathave a mixture of tonal abnormalities including spas-tic and dystonia, making interpretation of the ratingscales more difficult. Finally, the dystonia scales donot account for either spasticity or joint contracture.The high degree of variability and asymmetry in
programming parameters seems to reflect the asymme-try of phenotypic manifestations in many patients
Table 2. Dystonia rating assessment measures by age group*
Cohort
Baseline
mean 6 SD
6-Month follow-up
Mean 6 SD
Percent improvement
mean 6 SD
Significancea
P < .05
Burke-Fahn-Marsden MotorGroup 1 61.13 6 27.21 35.00 6 12.32 37.84% 6 22.56% 0.012Group 2 91.50 6 9.75 82.83 6 12.63 8.96% 6 14.48% 0.172Total 74.14 6 26.05 55.50 6 27.32 25.46% 6 23.97% 0.004
Burke-Fahn-Marsden DisabilityGroup 1 18.63 6 7.74 16.00 6 6.65 14.44% 6 11.07% 0.02Group 2 24.83 6 4.02 24.00 6 1.55 1.63% 6 13.60% 0.453Total 21.29 6 6.98 19.43 6 6.45 8.95% 6 13.42% 0.027
Barry-Albright scoresGroup 1 21.13 6 7.38 16.88 6 6.33 19.48% 6 15.27% 0.024Group 2 25.50 6 3.73 24.83 6 4.02 1.39% 6 20.40% 0.586Total 23.00 6 6.30 20.21 6 6.41 11.73% 6 19.29% 0.029
*Group 1—less than 16 years of age (n ¼ 8); group 2—greater than or equal to 16 years of age (n ¼ 6).aWilcoxon signed rank test.
M A R K S E T A L .
1750 Movement Disorders, Vol. 26, No. 9, 2011

with CP. This may also be a reflection of disturbedbasal ganglia anatomy in some patients that resulted insome uncertainty about exact lead placement, evenwith the use of microelectrode recording and postopera-tive MR imaging. This is reflected in the variability ofelectrode coordinates, thus making stereotactic place-ment of leads more difficult than in primary dystoniaswith preserved basal ganglia imaging characteristics(Table 1). Lead location in our population did not cor-respond to the reported atlas-based norms for adults.22
Young age, skull shape related to prematurity and atro-phy, and lack of pediatric normative data are all likelycontributing factors to the perceived discrepancies.Younger patients may respond to lower stimulation
rates than older patients. Two patients have had opti-mal response to 30-Hz stimulation, lower than the60–130 Hz more typically used for patients withdystonia. Although our experience suggests that inter-vention before adulthood is more beneficial, moreexperience and longer follow-up are needed with thispopulation to further define optimal stimulationparameters for patients with CP.This is report of a nonrandomized nonblinded study
of a heterogeneous group of patients with cerebralpalsy. Nonetheless, our findings demonstrate the valueof DBS in treating dystonia related to cerebral palsyand suggest that earlier intervention may providemore relief than waiting until after skeletal maturity.The degree of clinical heterogeneity and invasivenessof DBS make it unlikely that randomized blinded trialswill be feasible; thus, alternative strategies to evaluateeffectiveness of this therapy such as delayed, blindedDBS activation will need to be employed.23
ConclusionsDystonia is an often underrecognized contributor to
the motor disability associated with cerebral palsy, themost common pediatric-onset movement disorder.Effective treatment requires thorough identification ofany underlying conditions and tonal abnormalities. Thepresence of both spasticity and dystonia, often with asso-ciated truncal hypotonia, can make treatment particularlychallenging. We have demonstrated that dystonia second-ary to CP can be effectively decreased by pallidal stimula-tion. Results may be better in younger patients and inlimbs without fixed skeletal deformities, indicating thatpatient selection and intervention prior to the develop-ment of contracture are important factors in the successfulapplication of DBS therapy. Although the effects are lessdramatic than those reported in primary dystonias, evensmall gains in movement can provide meaningfulimprovement in quality of life, particularly when consider-ing patient life span as well as caretaker and financial bur-den. Continued experience will further refine theappropriate application of DBS in pediatric patients forameliorating the dystonia associated with CP and othersecondary dystonias.
References1. Winter S, Autry A, Boyle C, Yeargin-Allsopp M. Trends in the
prevalence of cerebral palsy in a population-based study. Pedia-trics 2002;110:1220–1225.
2. Berardelli A, Rothwell JC, Hallett M, Thompson PD, ManfrediM, Marsden CD. The pathophysiology of primary dystonia. Brain1998;121:1195–1212.
3. Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW.Classification and definition of disorders causing hypertonia inchildhood. Pediatrics 2003;111:e89–e97.
4. Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral deep-brainstimulation of the globus pallidus in primary generalized dysto-nia. N Engl J Med 2005;352:459–467.
5. Houeto JL, Yelnik J, Bardinet E, et al. Acute deep-brain stimula-tion of the internal and external globus pallidus in primary dysto-nia: functional mapping of the pallidum. Arch Neurol 2007;64:1281–1286.
6. Sun B, Chen S, Zhan S, Le W, Krahl SE. Subthalamic nucleusstimulation for primary dystonia and tardive dystonia. Acta Neu-rochir Suppl 2007;97:207–214.
7. Coubes P, Cif L, El Fertit H, et al. Electrical stimulation of theglobus pallidus internus in patients with primary generalized dys-tonia: long-term results. J Neurosurg 2004;101:189–194.
8. Zorzi G, Marras C, Nardocci N, et al. Stimulation of the globuspallidus internus for childhood-onset dystonia. Mov Disord 2005;20:1194–1200.
9. Diamond A, Shahed J, Azher S, Dat-Vuong K, Jankovic J. Globus pal-lidus deep brain stimulation in dystonia. Mov Disord 2006;21:692–695.
10. Castelnau P, Cif L, Valente EM, et al. Pallidal stimulationimproves pantothenate kinase-associated neurodegeneration. AnnNeurol 2005;57:738–741.
11. Mehrkens JH, Borggraefe I, Feddersen B, Heinen F, Botzel K.Early globus pallidus internus stimulation in pediatric patientswith generalized primary dystonia: long-term efficacy and safety.J Child Neurol 2010;25:1355–1361.
12. Krauss JK, Loher TJ, Weigel R, Capelle HH, Weber S, BurgunderJM. Chronic stimulation of the globus pallidus internus for treat-ment of non-dYT1 generalized dystonia and choreoathetosis:2-year follow up. J Neurosurg 2003;98:785–792.
13. Zhang JG, Zhang K, Wang ZC, Ge M, Ma Y. Deep brain stimu-lation in the treatment of secondary dystonia. Chin Med J (Engl)2006;119:2069–2074.
14. Vidailhet M, Yelnik J, Lagrange C, et al. Bilateral pallidal deepbrain stimulation for the treatment of patients with dystonia-choreoathetosis cerebral palsy: a prospective pilot study. LancetNeurol 2009;8:709–717.
15. Marks WA, Honeycutt J, Acosta F, Reed M. Deep brain stimulationfor pediatric movement disorders. Semin Pediatr Neurol 2009;16:90–98.
16. Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C,Friedman J. Validity and reliability of a rating scale for the pri-mary torsion dystonias. Neurology 1985;35:73–77.
17. Barry MJ, VanSwearingen JM, Albright AL. Reliability andresponsiveness of the Barry-Albright Dystonia Scale. Dev MedChild Neurol 1999;41:404–411.
18. Volpe JJ. Encephalopathy of prematurity includes neuronalabnormalities. Pediatrics 2005;116:221–225.
19. Isaias IU, Alterman RL, Tagliati M. Outcome predictors of pal-lidal stimulation in patients with primary dystonia: the role ofdisease duration. Brain 2008;131:1895–1902.
20. Alterman RL, Tagliati M. Deep brain stimulation for torsion dys-tonia in children. Childs Nerv Syst 2007;23:1033–1040.
21. Monbaliu E, Ortibus E, Roelens F, et al. Rating scales for dysto-nia in cerebral palsy: reliability and validity. Dev Med Child Neu-rol 2010;52:570–575.
22. Yelnik J, Bardinet E, Dormont D, et al. A three-dimensional,histological and deformable atlas of the human basal ganglia. I.Atlas construction based on immunohistochemical and MRI data.Neuroimage 2007;34:618–638.
23. Kupsch A, Benecke R, MJ, et al. Pallidal deep-brain stimulationin primary generalized or segmental dystonia. N Engl J Med2006;355:1978–1990.
D E E P B R A I N S T I M U L A T I O N F O R C E R E B R A L P A L S Y
Movement Disorders, Vol. 26, No. 9, 2011 1751

Diffusion Tensor MagneticResonance Imaging Tractographyin Progressive Supranuclear Palsy
Elisa Canu, MSc,1 Federica Agosta, MD,1
Francesca Baglio, MD,2 Sebastiano Galantucci, MD,1
Raffaello Nemni, MD,2 and Massimo Filippi, MD1*
1Neuroimaging Research Unit, Institute of Experimental Neurology,
Division of Neuroscience, Scientific Institute and University Hospital
San Raffaele, Milan, Italy; and 2Department of Neurology, Scientific
Institute Fondazione Don Gnocchi, Milan, Italy
ABSTRACTBackground: Diffusion tensor magnetic resonanceimaging tractography allows quantification of in vivowhite matter tract damage.Methods: Using tractography, diffusion tensor magneticresonance imaging metrics were obtained from thesuperior and middle cerebellar peduncles and major cer-ebral white matter tracts in 5 patients with progressivesupranuclear palsy and 13 controls.Results: Patients showed severe intrinsic damage tothe superior cerebellar peduncle, corpus callosum, andcingulum bilaterally. Only decreased axial diffusivitywas found in the left middle cerebellar peduncle.Conclusions: Diffusion tensor magnetic resonanceimaging tractography holds promise for providing accu-rate in vivo cartography of progressive supranuclearpalsy tissue damage. VC 2011 Movement DisorderSociety
Key Words: progressive supranuclear palsy; diffusiontensor magnetic resonance imaging; tractography;superior cerebellar peduncle
Progressive supranuclear palsy (PSP) is a neurodege-nerative disorder classified in the category of tauopa-thies.1 PSP presents clinically with progressive posturalinstability and falls, supranuclear vertical gaze palsy,pseudobulbar palsy, and late frontal cognitive dysfunc-
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Professor Massimo Filippi, NeuroimagingResearch Unit, Institute of Experimental Neurology, Division ofNeuroscience, Scientific Institute and University Vita-Salute San Raffaele,Via Olgettina, 60, 20132 Milan, Italy; [email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 4 January 2011; Revised: 16 February 2011; Accepted:10 March 2011Published online 15 April 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23739
tion.1 Although PSP is easily diagnosed in its laterstages, many patients with PSP are initially thought tohave Parkinson’s disease (PD) or multiple system atro-phy.1 Over the past few years, various neuroimagingtechniques have been proposed to distinguish PSPfrom PD and other atypical parkinsonian syndromes.2
By measuring directional changes in water diffusivity,diffusion tensor (DT) magnetic resonance imaging (MRI)allows quantification of in vivo intrinsic structural whitematter (WM) abnormalities,3 which usually go unde-tected on conventional MRI. Using a region of interest(ROI)–based approach, abnormal diffusivity measures inthe superior cerebellar peduncle (SCP) were consistentlyreported in PSP patients.2 A few voxel-based4,5 studiesshowed that PSP patients also had altered DTMRI meas-ures in the corpus callosum, internal capsulae, pons, anddeep gray matter (GM) structures.Because of the nature of ROI- and voxel-based meth-
ods, however, previous studies could only infer whichspecific tracts are involved by this disease. DT MRI trac-tography allows direct localization and a quantitativeassessment of diffusion metrics in individual neuronalpathways. Although DTMRI tractography has been suc-cessfully applied in several neurodegenerative diseases,only 1 study6 used DT MRI tractography, in 3 PSPpatients, showing abnormal diffusivity in their SCPs.The aim of this study was to explore the ability of
DT MRI tractography to assess the structural damageto the SCP, middle cerebellar peduncle (MCP), andmajor cerebral WM tracts in PSP patients.
Materials and Methods
Subjects
Five patients with probable PSP7 (4 women; meanage, 73 years; mean disease duration, 4.6 years) and 13age- and sex-matched healthy controls (8 women; meanage, 71 years) were studied (Table S1). Subjects wereexcluded if they had: (1) symptoms suggestive of corti-cobasal degeneration syndrome8; (2) significant medicalillnesses or substance abuse that could interfere withcognitive functioning; (3) any other major systemic, psy-chiatric, or neurological illnesses; and (4) other causesof focal or diffuse brain damage including lacunae andextensive cerebrovascular disorders at routine MRI. Atstudy entry, patient disability was scored using the Uni-fied Parkinson’s disease rating scale (UPDRS-III)9; meanUPDRS-III score was 39.6 (standard deviation, 11.0).Cognition was assessed with a short neuropsychologicalbattery (Supplementary Material). The study wasapproved by the local ethical committee. All subjectsprovided written informed consent.
MRI Acquisition
Brain structural and DT MRI scans were obtainedon a 1.5 Tesla Avanto system (Siemens, Enlargen,Germany); see Supplementary Material for details.
C A N U E T A L .
1752 Movement Disorders, Vol. 26, No. 9, 2011
MRI Postprocessing
All MRI analysis was performed by a single experi-enced observer blinded to clinical findings. A detaileddescription of MRI analysis is provided in the Sup-plementary Material. Briefly, WM hyperintensities(WMHs), if any, were identified on dual-echo scansand lesion load measured using the JIM softwarepackage (http://www.xinapse.com). DT analysis wascarried out using in-house software to produce meandiffusivity (MD), fractional anisotropy (FA), andaxial (axD) and radial (radD) diffusivity maps. Anatlas-based automated approach10 was used to obtainDT MRI metrics of the major cerebellar and cerebralWM tracts; these included the SCP, MCP, corpus cal-losum, cingulum, uncinate, inferior fronto-occipitalfasciculus, inferior longitudinal fasciculus, superiorlongitudinal fasciculus, and corticospinal tract. Prob-ability maps of the SCP and MCP are shown in Fig-ure 1. VBM analysis was performed using the
Statistical Parametric Mapping (SPM8) softwarepackage (http://www.fil.ion.ucl.ac.uk/spm) followinga standard procedure.
Statistical Analysis
Differences between groups in demographic, clinical,neuropsychological, and conventional MRI variableswere performed using the Pearson v2 test orthe Mann–Whitney U test. DT MRI indices of WMtracts were compared between groups using a linearmixed-effect model analysis11 and Fisher’s least-signifi-cant-difference test, adjusted for subject age (Supple-mentary Material). Statistical analysis was performedusing SAS release 9.1 (SAS Institute, Cary, NC, USA).Using SPM8, analyses of covariance were performed
to assess GM and WM differences between patientsand controls, adjusting for age and total intracranialvolume (P < .001).
FIG. 1. Top: probability maps of the superior (SCP) and middle cerebellar peduncles (MCP) are shown on the mean fractional anisotropy template.Bottom: regions of gray matter (GM) and white matter (WM) volume loss in PSP patients compared with healthy controls are shown on the T1 Mon-treal Neurological Institute template. Colored bars denote t values.
D T M R I T R A C T O G R A P H Y I N P S P
Movement Disorders, Vol. 26, No. 9, 2011 1753

ResultsThere were no differences in age and sex between PSP
patients and controls (Table S1). Four patients had aneuropsychological score below the cutoff in at least 1test. Two patients had impairment on verbal comprehen-sion, 4 at the phonemic test and 2 at the semantic fluencytest (Table S1). One or more WMHs were seen on theDE scans from all patients and 4 controls. The character-istics of WMHs were always nonspecific. Mean WMHlesion load was 1.27 mL (SD, 1.5 mL) in patients and0.24 mL (SD, 0.4 mL) in controls (P¼ .19).Table 1 shows the estimated means of the DT MRI
metrics of WM tracts of controls and patients. The mul-tivariate tests comparing DT MRI metrics between the2 groups found that WM tract MD, FA, axD, and radDwere significantly different between patients andhealthy controls. Compared with healthy controls,PSP patients showed increased MD of the SCPs(left, P < .001; right, P ¼ .001) and corpus callosum(P ¼ .03), and decreased FA of the SCPs (P < .001,bilaterally), corpus callosum (P ¼ .01), and cingulumbilaterally (left, P ¼ .04; right, P ¼ .002) . PSP patientsrelative to controls also had increased radD of the SCPs(P < .001, bilaterally), corpus callosum (P ¼ .01), andright cingulum (P ¼ .04), and decreased axD of theSCPs (P < .001, bilaterally), cingulum bilaterally (left,P ¼ .03; right, P ¼ .01), and left MCP (P ¼ .03).VBM results showed that PSP patients presented a
pattern of GM volume loss involving the basal ganglia(ie, bilateral putamen, right pallidum, and right cau-
date nucleus) compared with healthy controls (Fig. 1,Table S2). PSP patients relative to healthy controlsalso experienced WM volume loss in a large infraten-torial area (which included the midbrain and ponsbilaterally), the anterior body of the corpus callosumbilaterally, and several areas of the frontal lobe, suchas the right premotor, supplementary motor, inferiororbital frontal, inferior frontal, insular and left middlefrontal regions (Fig. 1, Table S2).
DiscussionThe pattern of brain volume loss observed in our
PSP patients agrees with the observations reported bypast noted studies of pathologically confirmed PSPcases.12 In detail, these patients experienced GM atro-phy of the basal ganglia bilaterally as well as of themidbrain and pons and several motor and extramotorregions in the frontal lobes.Most intriguingly, DT MRI tractography was able
to detect selective damage to the SCP in the brainstem with relative sparing of the MCP, despite thepresence of such a large amount of infratentorial vol-ume loss. In PSP, involvement of the SCP has been pre-viously shown by ROI-based DT MRI studies.2 ROIanalysis, however, is extremely time consuming andpoorly reproducible, especially when applied to patho-logical brains, in which atrophy would increase thelikelihood of partial volume effect from cerebrospinalfluid (CSF) and, hence, possibly result in false-positive
Table 1. DT MRI metrics of WM tracts in PSP patients versus matched healthy controls
FA MD (�10�3 mm2 s�1) RadD (�10�3 mm2 s�1) AxD (�10�3 mm2 s�1)
HC PSP HC PSP HC PSP HC PSP
v2, P a — 217.3, <.0001 — 67.5, <.0001 170.7, <.0001 77.0, <.0001SCP R 0.55 6 0.01 0.42 6 0.02c 0.91 6 0.02 0.97 6 0.03b 0.59 6 0.02 0.73 6 0.03c 1.57 6 0.02 1.44 6 0.03c
L 0.57 6 0.01 0.42 6 0.02c 0.89 6 0.02 0.97 6 0.03c 0.56 6 0.02 0.73 6 0.03c 1.57 6 0.02 1.43 6 0.03c
MCP R 0.54 6 0.01 0.52 6 0.02 0.73 6 0.02 0.72 6 0.03 0.48 6 0.02 0.48 6 0.03 1.22 6 0.02 1.19 6 0.03L 0.54 6 0.01 0.51 6 0.02 0.73 6 0.02 0.71 6 0.03 0.47 6 0.02 0.48 6 0.03 1.23 6 0.02 1.17 6 0.03b
CST R 0.66 6 0.01 0.65 6 0.02 0.71 6 0.02 0.72 6 0.03 0.38 6 0.02 0.40 6 0.03 1.35 6 0.02 1.37 6 0.03L 0.66 6 0.01 0.64 6 0.02 0.71 6 0.02 0.72 6 0.03 0.39 6 0.02 0.41 6 0.03 1.36 6 0.02 1.33 6 0.03
Corpus callosum 0.59 6 0.01 0.54 6 0.02b 0.78 6 0.02 0.82 6 0.03b 0.47 6 0.02 0.54 6 0.03b 1.39 6 0.02 1.38 6 0.03Cingulum R 0.55 6 0.01 0.49 6 0.02b 0.74 6 0.02 0.75 6 0.03 0.49 6 0.02 0.53 6 0.03b 1.26 6 0.02 1.19 6 0.03b
L 0.54 6 0.01 0.51 6 0.02b 0.76 6 0.02 0.76 6 0.03 0.50 6 0.02 0.53 6 0.03 1.29 6 0.02 1.22 6 0.03b
SLF R 0.47 6 0.01 0.44 6 0.02 0.74 6 0.02 0.74 6 0.03 0.53 6 0.02 0.56 6 0.03 1.15 6 0.02 1.12 6 0.03L 0.47 6 0.01 0.45 6 0.02 0.75 6 0.02 0.77 6 0.03 0.54 6 0.02 0.57 6 0.03 1.17 6 0.02 1.17 6 0.03
IFOF R 0.52 6 0.01 0.51 6 0.02 0.81 6 0.02 0.79 6 0.03 0.55 6 0.02 0.54 6 0.03 1.32 6 0.02 1.29 6 0.03L 0.51 6 0.01 0.50 6 0.02 0.82 6 0.02 0.80 6 0.03 0.56 6 0.02 0.56 6 0.03 1.33 6 0.02 1.29 6 0.03
ILF R 0.45 6 0.01 0.43 6 0.02 0.81 6 0.02 0.80 6 0.03 0.60 6 0.02 0.60 6 0.03 1.23 6 0.02 1.20 6 0.03L 0.46 6 0.01 0.44 6 0.02 0.80 6 0.02 0.79 6 0.03 0.58 6 0.02 0.59 6 0.03 1.24 6 0.02 1.20 6 0.03
Uncinate R 0.40 6 0.01 0.38 6 0.02 0.81 6 0.02 0.82 6 0.03 0.62 6 0.02 0.64 6 0.03 1.19 6 0.02 1.17 6 0.03L 0.39 6 0.01 0.39 6 0.02 0.83 6 0.02 0.81 6 0.03 0.64 6 0.02 0.63 6 0.03 1.19 6 0.02 1.18 6 0.03
Numbers are estimated means 6 standard errors. aw2 and P values refer to the multivariate test; bP < .05 and c P < .001 versus healthy controls according toFisher’s least-significant-difference test (see text for further details).AxD, axial diffusivity; CST, corticospinal tract; FA, fractional anisotropy; HC, healthy controls; ILF, inferior longitudinal fasciculus; IFOF, inferior fronto-occipitalfasciculus; L, left; MCP, middle cerebellar peduncle; MD, mean diffusivity; R, right; RadD, radial diffusivity; SCP, superior cerebellar peduncle tract; SLF,superior longitudinal fasciculus.
C A N U E T A L .
1754 Movement Disorders, Vol. 26, No. 9, 2011

findings. Compared to the ROI-based approach, DTMRI tractography is not operator driven, thus avoidingthe variability introduced by the manual drawing ofROIs. Furthermore, our DT MRI tractography methodis atlas based. Such an atlas is highly representative ofthe mean morphology and intensity of the group ana-lyzed, and individuals maps were spatially aligned toit, thus reducing biases associated with the presence ofatrophy. Finally, partial volume effects were minimizedfurther by deriving DT MRI indices from WM tractsafter CSF masking, WM thresholding, and skeletoniz-ing the WM tract probability maps (ie, consideringonly the core of the tracts).In line with previous studies, PSP patients also
showed diffusivity abnormalities in the corpus cal-losum4 and cingulum.13 This may be the result of glo-bose neurofibrillary tangles, tau positive astrocytes,and ballooned argyrophilic neuronal degeneration thatneuropathological studies have shown to involve thefrontal lobes in this condition.1 The disruption ofinterhemispheric and long-range WM tracts in PSPpatients is likely to contribute to the cognitive changesseen in these patients.In addition, in PSP patients compared with controls,
we observed increased radD in the SCPs, corpus cal-losum, and right cingulum and decreased axD in theSCPs, left MCP, and cingulum bilaterally. To date,only 2 studies investigated DT MRI eigenvalues in PSPpatients,4,14 using ROI- or tract-based spatial statisticsapproaches. Decreased axD is thought to be the conse-quence of axonal damage, whereas an increased radDis likely a reflection of myelin breakdown.15 Togetherwith previous findings,4 our results suggest that DTMRI findings in PSP patients are most likely a resultof both axonal damage and demyelination, as occursin secondary degeneration. Although this is the mostintriguing hypothesis, only longitudinal studies havethe potential to clarify the pathological substrates ofthe observed diffusivity abnormalities in PSP patients.This is particularly true when considering our findingof increased radD with normal axD of patients versushealthy controls in the corpus callosum, which sug-gests damage to the myelin only in this region withoutaxonal loss. Conversely, the isolated decreased axD inpatients compared with controls in the left MCP maybe a reflection of the axonal damage or changes in theextracellular space in this region,15 as reported by aprevious study.4 However, the asymmetrical distribu-tion of changes in the MCP is more likely the result ofthe small sample studied.The sample of PSP patients studied was small, and
larger studies are required to confirm our findings. Thismay be the reason why 4 of the 5 PSP patients included
were women. Indeed, previous literature does not sug-gest a sex difference in the PSP population.16 However,given that PSP is a rare condition, this study suggests arole of DT MRI tractography as a tool for detectingtract damage known to be associated with this disease.As a consequence, we believe that DT MRI tractogra-phy holds promise for improving the diagnostic workupon patients suspected of having PSP. Finally, analysis ofthe DT MRI eigenvalues in future larger studies is likelyto offer insight into the possible underlying substratesof WM damage in this disease.
References1. Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopa-
thological concepts and diagnostic challenges. Lancet Neurol2009;8:270–279.
2. Seppi K, Poewe W. Brain magnetic resonance imaging techniquesin the diagnosis of parkinsonian syndromes. Neuroimaging ClinN Am 2010;20:29–55.
3. Basser PJ, Pierpaoli C. Microstructural and physiological featuresof tissues elucidated by quantitative-diffusion-tensor MRI. JMagn Reson B 1996;111:209–219.
4. Knake S, Belke M, Menzler K, et al. In vivo demonstration ofmicrostructural brain pathology in progressive supranuclear palsy:a DTI study using TBSS. Mov Disord 2010;25:1232–1238.
5. Padovani A, Borroni B, Brambati SM, et al. Diffusion tensorimaging and voxel based morphometry study in early progressivesupranuclear palsy. J Neurol Neurosurg Psychiatry 2006;77:457–463.
6. Nilsson C, Markenroth Bloch K, Brockstedt S, Latt J, Widner H,Larsson EM. Tracking the neurodegeneration of parkinsonian dis-orders—a pilot study. Neuroradiology 2007;49:111–119.
7. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for thediagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP internationalworkshop. Neurology 1996;47:1–9.
8. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and itsrelationship to progressive supranuclear palsy and frontotemporaldementia. Ann Neurol 2003;54(Suppl 5):S15–S19.
9. Fahn S, Elton RL, members of the UPDRS Development Commit-tee. Unified Parkinson’s Disease Rating Scale. In: Fahn S, Mars-den CD, Goldstein M, Calne DB, eds. Recent Developments inParkinson’s Disease. New York: MacMillan; 1987:153–163.
10. Pagani E, Filippi M, Rocca MA, Horsfield MA. A method forobtaining tract-specific diffusion tensor MRI measurements in thepresence of disease: application to patients with clinically isolatedsyndromes suggestive of multiple sclerosis. Neuroimage 2005;26:258–265.
11. McCulloch CE, Searle SR, Neuhaus JM. Generalized, Linear, andMixed Models. Hoboken, NJ: Wiley; 2008.
12. Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-basedmorphom-etry in autopsy proven PSP and CBD. Neurobiol Aging 2008;29:280–289.
13. Erbetta A, Mandelli ML, Savoiardo M, et al. Diffusion tensorimaging shows different topographic involvement of the thalamusin progressive supranuclear palsy and corticobasal degeneration.AJNR Am J Neuroradiol 2009;30:1482–1487.
14. Wang J, Wai Y, Lin WY, et al. Microstructural changes inpatients with progressive supranuclear palsy: a diffusion tensorimaging study. J Magn Reson Imaging 2010;32:69–75.
15. Pierpaoli C, Barnett A, Pajevic S, et al. Water diffusion changesin Wallerian degeneration and their dependence on white matterarchitecture. Neuroimage 2001;13:1174–1185.
16. Golbe LI. The epidemiology of progressive supranuclear palsy.Handb Clin Neurol 2008;89:457–459.
D T M R I T R A C T O G R A P H Y I N P S P
Movement Disorders, Vol. 26, No. 9, 2011 1755

Iron-Related MRI Images inPatients with Pantothenate
Kinase–AssociatedNeurodegeneration (PKAN)
Treated with Deferiprone: Resultsof a Phase II Pilot Trial
Giovanna Zorzi, MD,1 Federica Zibordi, MD,1
Luisa Chiapparini, MD,2 Enrico Bertini, MD,3
Lidia Russo, MD,4 Antonio Piga, MD,5
Filomena Longo, MD,5 Barbara Garavaglia, PhD,6
Domenico Aquino, MD,2 Mario Savoiardo, MD,2
Alessandra Solari, MD,7 and Nardo Nardocci, MD1*
1Unit of Child Neurology, Fondazione IRCCS Istituto Neurologico
Carlo Besta, Milan, Italy; 2Neuroradiology, Fondazione IRCCS Istituto
Neurologico Carlo Besta, Milan, Italy; 3Unit of Molecular Medicine for
Neuromuscular and Neurodegenerative Disorders, Ospedale
Bambino Gesu, Roma, Italy; 4Unit of Oncology and Hematology,
Ospedale Bambino Gesu, Roma, Italy; 5Unit of Pediatrics,
Department of Clinical and Biological Sciences, University of Torino,
Torino, Italy; 6Neurogenetics, Fondazione IRCCS Istituto Neurologico
Carlo Besta, Milan, Italy; 7Neuroepidemiology, Fondazione IRCCS
Istituto Neurologico Carlo Besta, Milan, Italy
ABSTRACTBackground: The safety and efficacy of the oral iron-chelating agent deferiprone on magnetic resonancepallida iron concentration and on clinical status wereinvestigated in 10 patients affected by pantothenate ki-nase–associated neurodegeneration.Methods: Nine patients (age range, 7–39 years) com-pleted the study.Results: A significant median reduction in globus pal-lidus iron content as assessed by T2* relaxometry (andcalculated R2* maps; P 5 .008) was observed at theend of the study. None of the patients demonstrated achange in clinical status as assessed by the Burke-Fahn and Marsden Dystonia Rating scales and by ahealth-related quality-of-life scale. Deferiprone was welltolerated, and no serious adverse events occurred.Conclusions: Future trials assessing the clinical effi-cacy of chelating therapy should consider early symp-tomatic patients and a longer treatment period. VC 2011Movement Disorder Society
Key Words: neurodegeneration with brain iron accu-mulation; pantothenate kinase–associated neurodegen-eration; deferiprone; magnetic resonance imaging; iron
Pantothenate kinase–associated neurodegeneration(PKAN) is a rare disorder with progressive dystonia,parkinsonism, spasticity, and brain iron accumula-
------------------------------------------------------------Giovanna Zorzi and Federica Zibordi contributed equally to this work.
*Correspondence to: Dr. Nardo Nardocci, Unit of Child Neurology,Fondazione IRCCS Istituto Neurologico Carlo Besta, Via Celoria 11,20133 Milan, Italy; [email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 18 September 2010; Revised: 1 March 2011; Accepted:15 March 2011Published online 6 May 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23751
tion.1–4 Classical and atypical phenotypes are recog-nized according to the age at onset and diseaseprogression.2 At present, no cure is available. Iron-che-lating treatment has been proposed as a possible treat-ment for PKAN and other neurodegenerative disorderscharacterized by brain iron accumulation.5 Recent datademonstrated radiological and clinical improvementwith iron-chelating treatment in patients affected byFriedreich ataxia and in a case of idiopathic neurode-generation with brain iron accumulation (NBIA).6,7
Deferiprone is an iron-chelating agent effective inpreventing the progression of iron accumulation andin promoting iron elimination.8 We investigated thesafety and efficacy of a 6-month treatment with deferi-prone in reducing iron concentration in the globus pal-lidus on brain MRI in patients with PKAN. We alsoevaluated changes in the Burke-Fahn and MarsdenDystonia Rating Scale (BFMDRS) score and in health-related quality of life, as assessed by the 36-itemshort-form questionnaire (SF-36).
Patients and Methods
Patients
Patients with PKAN followed at 2 centers (IstitutoNeurologico Carlo Besta and Ospedale BambinoGesu) were eligible for inclusion in this phase II pilottrial. Requirements were: radiological and genetic con-firmation and age older than 6 years. Patients wereexcluded if they had any conditions known to be acontraindication to the use of deferiprone, majorcomorbidities (renal, hepatic, or cardiac failure), preg-nancy or breastfeeding, and any condition preventingbrain MRI assessment. Symptomatic treatments andphysiotherapy had to not be changed during the studyunless necessary and were thoroughly recorded. Thestudy was conducted in accordance with the Interna-tional Conference on Harmonisation Guidelines forGood Clinical Practice and the Declaration of Hel-sinki. The protocol was approved by the institutionalreview boards at the 2 institutions. Each patient (orthe patient’s legal representative) gave writteninformed consent.
Z O R Z I E T A L .
1756 Movement Disorders, Vol. 26, No. 9, 2011

Treatment Plan
Patients received deferiprone (Ferriprox) 25 mg/kg/day orally in a liquid formulation in 2 divided dosesfor 6 months.
Clinical Protocol
Patients underwent physical examination and labora-tory testing at screening. Eligible patients returned at base-line visit and received detailed instructions on treatmentadministration and storage. Patients returned to the clinicin months 2, 4, and 6. At every visit, drug compliance, useof concomitant drugs, safety, and laboratory tests wereassessed. The clinical protocol included neurological ex-amination, administration of BFMDRS, and standardizedvideo-recordings.9,10 Health-related quality of life wasassessed at baseline and at the 6-month visit by the SF-36questionnaire.11,12
Safety was assessed on the basis of physical exami-nation, hematological tests, adverse events, and seriousadverse events.Patients had hematological tests performed (cell
blood count, hematocrit, hemoglobin, ferritin, trans-ferrin, transferrin receptor, iron, zinc, and reticulo-cytes) at baseline and every 8 weeks thereafter.Patients were monitored weekly for absolute neutro-phil count and any adverse effect. A confirmed abso-lute neutrophil count (ANC) between 0.5 and 1.5�109/L was defined as neutropenia. A confirmed ANC< 0.5 � 109/L was defined as agranulocytosis.
Neuroradiological Protocol
Patients were imaged on a Siemens Magnetom Avanto1.5 T system with a 4-channel-head coil at baseline andafter 6 months. Basic imaging included axial T2 andproton density–weighted spin echo sequences and axialT2 gradient-recall echo (GRE) sequences. Morphologicalimaging was acquired by volumetric gradient-echo(Flash) and T1 3-D MPRAGE sagittal sequences.To detect iron deposition in the globus pallidus,
T2* Relaxometry was performed as previously
described with a multiecho gradient echo sequencecomposed of 12 echo � 5/55 ms (Dt ¼ 5 ms).13
The physician interpreting the data (L.C.) wasblinded to patient status with regard to treatment.R2* values of the patients were compared with the
normal range R2* values of the controls measured ina previous study.13
Statistical Analysis
Continuous variables were compared betweengroups using the Wilcoxon rank-sum test and withingroups using the Wilcoxon matched-pairs signed-ranktest. Categories were compared using Fisher’s exacttest. All reported P values were based on 2-tailed testsfor significance and unadjusted for multiple testing.The analyses were performed with Stata 10.0.
ResultsTen patients (4 females and 6 males) were recruited
between December 2008 and September 2009. How-ever, only 9 completed the study: 1 patient withdrewher consent after 4 months because she refused toundergo an MRI at the end of the study.The median age of patients was 26 years (range, 7–
39 years). Two sibling patients were from Morocco,the remaining were from 7 Italian families, 2 of whichwere related. Median disease duration was 11 years(range, 4–25 years). Phenotype was defined as classicin 6 patients and as atypical in 3 patients. All patientshad dystonia associated with spasticity and parkinson-ism; obsessive–compulsive disorder was prominent in2. Three patients were able to walk independently, theremaining 6 were wheelchair bound. Seven patientswere under treatment for dystonia at enrollment.Deferiprone was overall well tolerated, and no seri-
ous adverse event occurred. The most frequentadverse symptoms were nausea and gastralgia,reported by 4 patients (44%). No episodes of agranu-locytosis or thrombocytopenia were recorded. Duringa febrile episode, 1 patient experienced an ANC
Table 1. Clinical and genetic data, BFMDRS scores (disability/severity), and R2* values at baseline and at the end ofthe study, percent changes of the 9 PKAN patients, and R2* of controls (values for controls represent the decade
average)
Patient/Age Phenotype
PANK-2
mutations
BMFDRS at
baseline
BFMDRS at
end of study
R2* at
baseline
R2* at end
of study
Change
(%) Controls
1/22 y Classic F228SþF550L 29/116.5 29/116.5 85.83 48.61 �43.4 27.032/39 y Atypical G521RþG259R 12/16 12/21 67.72 47.16 �30.3 28.253/7 y Classic F419fsX472 22/45 25/53 38.31 35.55 �7.20 22.354/10 y Classic F419fsX472 24/57 25/59 33.74 30.66 �9.11 22.355/28 y Classic R264WþS471N 20/62 20/60 61.86 40.0 �33.9 27.036/26 y Atypical R357W 20/44 22/52 80.50 51.62 �35.5 27.037/13 y Classic R264W 13/39 14/39 44.6 31.81 �28.4 25.148/36 y Classic R264W 28/105 28/105 36.75 31.47 �14.3 28.259/27 y Atypical R264WþR286C 10/40 11/40 66.34 25.50 �61.5 27.03Mean — — — — 57.28 38.18 �33.34 26.05
C H E L A T I N G T H E R A P Y I N P K A N
Movement Disorders, Vol. 26, No. 9, 2011 1757
count below the threshold for neutropenia (ANC,1.45 � 109/L), which resolved after deferiprone dis-continuation for 10 days.R2* values, expressed in seconds�1, of patients at base-
line and at the end of the study are summarized in Table1. At baseline, the mean iron content in the pallida of thepatients was 57.28 (range, 33.74–85.83) versus 26.05(range, 22.35–28.25) in the controls (P < .0001). Consid-ering the high dispersion of the normative data, shown inthe column of the controls of Table 1, we decided to usethe average of the measurement obtained for each of thefirst 4 decades rather than the R2* value obtained in eachage-matched control.13 After deferiprone treatment, themean R2* value was 38.18 (range, 25.5–51.92). The me-dian reduction in iron content was 30% (P ¼ .008, Fig.1). Values were reduced in all cases; 3 patients had areduction of less than 15%; in the remaining the reductionranged from 28% to 61% (Table 1). Two of the 3patients showing less reduction were the 2 siblings carry-ing a new homozygous null mutation, whereas all theothers had missense mutations (Table 1).There were no changes in BFMDRS and SF-36 scale
scores (Table 2). None of the patients reported a sub-jective improvement.
DiscussionIn this phase II pilot trial, we found that in PKAN
patients, a 6-month treatment with the chelating agentdeferiprone was well tolerated and produced a significantreduction in iron content in the pallida, as revealed byMRI.Despite the efficacy of treatment on the primary end
point, the clinical status of PKAN patients did notimprove. Several reasons could explain this discrepancy.First, a treatment 6 months in duration might not have
been sufficient to produce clinical amelioration. This dura-tion was sufficient to demonstrate the clinical efficacy ofdeferiprone therapy in Friedreich ataxia patients6; how-ever, in the patient with idiopathic NBIA, clinical statusimproved after 8 months of therapy.7 Second, the damageto neurons in the basal ganglia was far too advanced toallow a rescue of function with removal of iron. In our se-ries, all patients had long disease duration and most ofthem were severely affected. Third, it cannot be excludedthat iron accumulation may be an epiphenomenon andnot the primary cause of the disease.14 There are indeedseveral lines of evidence that iron plays a role in the patho-physiology.14–16 The PANK-2 gene encodes a pantothe-nate kinase that is specifically expressed in the brain andis an essential regulatory enzyme in the biosynthesis ofcoenzyme A.17 The product of pantothenate kinase, phos-phopantothenate, normally condenses with cysteine in thefollowing step of coenzyme A synthesis. High cysteineconcentration, which has iron-chelating properties, hasbeen found in the globus pallida of patients with PKAN.18
This abnormal deposition might account for the regionaliron accumulation, and cysteine-bound iron might be re-sponsible for oxidative damage in these regions.1,19,20
Finally, quantitative imaging of iron could be a not usefulbiomarker of the disease; in fact, in our series, iron con-tent and its reduction after therapy did not correlate withseverity of the phenotype or disease duration.With regard to the safety of deferiprone, the patients
did not experience any serious adverse event or note-worthy hematological effects. These untoward effectshave an incidence of 1%–2% and 5%, respectively.They might not have been observed in our seriesbecause of the low dosage used (25 mg/kg/day) incomparison with the 74 mg/kg/day used in most ofthe published deferiprone studies.21,22
The main limitations of our trial are the study size,the advanced stage of the disease, the short period ofadministration of the chelating agent, and the absenceof a control arm. It is noteworthy that the trial’s pri-mary end point is MRI demonstration of iron re-moval, and results on this point were significant.In conclusion, the good tolerability and the signifi-
cant MRI changes obtained warrant the planning ofinternational placebo-controlled trials of chelatingtreatment in PKAN.
Acknowledgments: We thank Apopharma for supplying the drugdeferiprone for the study.
FIG. 1. Box plot of iron content at baseline and at study end in the 9patients with PKAN (Wilcoxon matched-pairs signed-ranks test, P 50.008). The boxes represent the interquartile range, horizontal linesinside boxes represent medians, and tails represent the 5th–25th and75th–95th percentile ranges.
Table 2. Scores on the BFMDRS and SF-36 at baselineand at the end of the study of the 9 PKAN patients
Baseline
median
(range)
End of study
median (range) P value
BFMDRS Disability Scale 20 (10–29) 22 (11–29) 0.16BFMDRS Severity Scale 45 (11–116.5) 53 (21–116.5) 0.13SF-36 Physical Composite 35.4 (29.2–54.9) 39.5 (30.5–60.9) 0.26SF-36 Mental Composite 44.8 (27.4–58.9) 39.9 (20–58.3) 0.15
BFMDRS, Burke-Fahn-Marsden Dystonia Rating Scales; SF-36, health-related quality-of-life questionnaire.
Z O R Z I E T A L .
1758 Movement Disorders, Vol. 26, No. 9, 2011

References1. Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical,
and radiographic delineation of Hallervorden-Spatz syndrome. NEngl J Med. 2003;348:33–40.
2. Hartig MB, Hortnagel K, Garavaglia B, et al. Genotypic and phe-notypic spectrum of PANK2 mutations in patients with neurode-generation with brain iron accumulation. Ann Neurol. 2006;59:248–256.
3. Sethi KD, Adams RJ, Loring DW, el Gammal T. Hallervorden-Spatz syndrome: clinical and magnetic resonance imaging correla-tions. Ann Neurol. 1988;24:692–694.
4. Angelini L, Nardocci N, Rumi V, Zorzi C, Strada L, SavoiardoM. Hallervorden-Spatz disease: clinical and MRI study of 11cases diagnosed in life. J Neurol. 1992;239:417–425.
5. Whitnall M, Richardson DR. Iron: a new target for pharmacolog-ical intervention in neurodegenerative diseases. Semin PediatrNeurol. 2006;13:186–197.
6. Boddaert N, Le Quan Sang KH, Rotig A, et al. Selective iron che-lation in Friedreich ataxia: Biologic and clinical implications.Blood. 2007;110:401–408.
7. Forni GL, Balocco M, Cremonesi L, Abbruzzese G, Parodi RC,Marchese R. Regression of symptoms after selective iron chela-tion therapy in a case of neurodegeneration with brain iron accu-mulation. Mov Disord. 2008;23:904–907.
8. Kontoghiorghes GJ, Aldouri MA, Sheppard L, Hoffbrand AV.1,2-Dimethyl-3-hydroxypyrid-4-one, an orally active chelator fortreatment of iron overload. Lancet. 1987;1:1294–1295.
9. Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C,Friedman J. Validity and reliability of a rating scale for the pri-mary torsion dystonias. Neurology. 1985;35:73–77.
10. Comella CL, Leurgans S, Wuu J, Stebbins GT, Chmura T, Dysto-nia Study Group. Rating scales for dystonia: A multicenter assess-ment. Mov Disord. 2003;18:303–312.
11. Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form healthsurvey (SF-36). I. conceptual framework and item selection. MedCare. 1992;30:473–483.
12. Apolone G, Mosconi P. The italian SF-36 health survey: Transla-tion, validation and norming. J Clin Epidemiol. 1998;51:1025–1036.
13. Aquino D, Bizzi A, Grisoli M, et al. Age-related iron depositionin the basal ganglia: Quantitative analysis in healthy subjects. Ra-diology. 2009;252:165–172.
14. Schneider SA, Hardy J, Bhatia KP. Iron accumulation in syn-dromes of neurodegeneration with brain iron accumulation 1 and2: causative or consequential? J Neurol Neurosurg Psychiatry.2009;80:589–590.
15. Stankiewicz J, Panter SS, Neema M, Arora A, Batt CE, Bakshi R.Iron in chronic brain disorders: Imaging and neurotherapeuticimplications. Neurotherapeutics. 2007;4:371–386.
16. Benarroch EE. Brain iron homeostasis and neurodegenerative dis-ease. Neurology. 2009;72:1436–1440.
17. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J,Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defec-tive in Hallervorden-Spatz syndrome. Nat Genet 2001;28:345–349.
18. Perry TL, Norman MG, Yong VW, et al. Hallervorden-Spatz dis-ease: cysteine accumulation and cysteine dioxygenase deficiencyin the globus pallidus. Ann Neurol. 1985;18:482–489.
19. Yoon SJ, Koh YH, Floyd RA, Park JW. Copper, zinc superoxidedismutase enhances DNA damage and mutagenicity induced bycysteine/iron. Mutat Res. 2000;448:97–104.
20. Ke Y, Ming Qian Z. Iron misregulation in the brain: a primarycause of neurodegenerative disorders. Lancet Neurol. 2003;2:246–253.
22. Ceci A, Baiardi P, Felisi M, et al. The safety and effectiveness ofdeferiprone in a large-scale, 3-year study in italian patients. Br JHaematol. 2002;118:330–336.
22. Cohen AR, Galanello R, Piga A, De Sanctis V, Tricta F. Safetyand effectiveness of long-term therapy with the oral iron chelatordeferiprone. Blood. 2003;102:1583–1587.
A Reappraisal of Long-LatencyAbdominal Muscle Reflexes inPatients with Propriospinal
Myoclonus
Samar S. Ayache, MD,1,2 Rechdi Ahdab, MD,1,2
Pierre Brugieres, MD,3 Jean-Francois Ejzenbaum, MD,4
Francois-Jerome Authier, MD, PhD,5,6 Gilles Fenelon, MD, PhD,4
and Jean-Pascal Lefaucheur, MD, PhD1,2,5*
1Service de Physiologie—Explorations Fonctionnelles, Hopital Henri
Mondor, APHP, Creteil, France; 2EA 4391, Faculte de Medecine de
Creteil, Universite Paris-Est-Creteil, Creteil, France; 3Service de
Neuroradiologie, Hopital Henri Mondor, APHP, Creteil, France;4Service de Neurologie, Hopital Henri Mondor, APHP, Creteil,
France; 5Centre de reference des maladies rares neuromusculaires,
Hopital Henri Mondor, APHP, Creteil, France; 6INSERM U955-E10,
Universite Paris-Est-Creteil, Creteil, France
ABSTRACTBackground: We report 3 patients with typical clinicaland electrophysiological characteristics of propriospinalmyoclonus propagating from a thoracic spine generator.Methods: In these patients, the pattern of recruitmentof long-latency electromyographic reflexes in abdomi-nal muscles was studied in response to various stimuli.Results: Abdominal reflex latency varied from 60 to140 ms depending on stimulus location. Latencyincreased from magnetic stimulation of the thoracicspine to electrical stimulation of the supraorbital nerve,electrical stimulation of the median nerve, and mag-netic stimulation of the motor cortex.Conclusions: Long-latency abdominal reflex jerks areprobably controlled by the brain stem to propriospinalsystem projections in patients with propriospinal myoclo-nus. The stereotyped pattern of recruitment of thesereflexes could be of clinical utility to differentiate organicpropriospinal myoclonus from psychogenic or mimickedjerks. VC 2011Movement Disorder Society
Key Words: abdominal muscles; electromyography;fiber tracking; propriospinal myoclonus; reflex
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Prof. Jean-Pascal Lefaucheur, ServicePhysiologie, Explorations Fonctionnelles, Hopital Henri Mondor, 51avenue de Lattre de Tassigny, 94010 Creteil cedex, France; [email protected]
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 9 June 2010; Revised: 19 December 2010; Accepted:28 December 2010Published online 26 May 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23645
P R O P R I O S P I N A L M Y O C L O N U S
Movement Disorders, Vol. 26, No. 9, 2011 1759

Propriospinal myoclonus (PSM) is a rare disordercharacterized by repetitive flexor myoclonic jerks ofthe trunk. Myoclonus arises in a myelomere where thegenerator locates, then spreads rostrally and caudallyvia slow-conducting pathways.1,2 Recently, Roze et al3
reported the largest series of patients with PSM todate (10 patients), highlighting the homogenous clini-cal and electrophysiological presentation of this disor-der and the value of diffusion tensor imaging withfiber tracking (DTI-FT) to identify an underlyinglesion of the spinal cord. In the present study, clinicaland electrophysiological data of 3 patients with PSMare reported, including spinal cord DTI-FT examina-tion in 2 cases. Our main goal was to shed light onthe abdominal reflexes that can be elicited by variousstimuli in such patients.3–10
Patients and MethodsSpontaneous electromyographic (EMG) activity was
recorded in multiple muscles to identify the spinallevel of the generator and to measure the velocity ofmuscle jerk propagation, as previously described.1–3
All recordings were performed on the right side (Pha-sis II machine, EB Neuro, Florence, Italy). In addition,reflex EMG activity was recorded in abdominalmuscles in response to magnetic or electrical stimula-tion. Magnetic stimuli were delivered at 80% of maxi-mal stimulator output to the motor cortex or thethoracic spine (double cone or circular coil and Mag-stim 200 stimulator, Magstim, Carmarthenshire, UK).Electrical stimuli were delivered with a handheld bipo-
lar stimulator to the right median nerve at the wristand the supraorbital nerve at the supraorbital notch.Intensity was set at 3 times the sensory threshold(pulse duration, 0.2 ms), which was a low, painless in-tensity of stimulation. Four to 6 responses wererecorded for each stimulation type. In 1 patient(patient 2), the propagation pattern of the reflex jerkswas assessed by recording the responses at multiplesites (including facial and limb muscles) and was com-pared with that of the spontaneous jerks.Additional neurophysiological investigation included
a search for: (1) long-loop reflex (C-reflex) at the handby recording EMG activity in the abductor pollicisbrevis muscle at rest in response to median or superfi-cial radial nerve stimulation, (2) giant somatosensoryevoked potentials to median or tibial nerve stimula-tion, and (3) cortical events preceding myoclonus injerk-locked back-averaged electroencephalography(EEG) recorded at C3, C4, and Cz sites of the Interna-tional 10-20 System. In these EEG recordings, welooked for evidence of a brief cortical potential pre-ceding myoclonus by a short latency of 20–50 ms todemonstrate that myoclonus was of cortical origin11,12
or a slowly rising negativity (Bereitschaftspotential,BP) starting from 0.4 to 2 seconds prior to the EMGto indicate that the jerks were psychogenic.13–15
Neuroradiological investigation included morphologi-cal MRIs of the brain and spinal cord in all patients,and DTI-FT of the spinal cord in 2 patients. For DTI-FTreconstruction of the spinal pathways, a well-establishedstreamline method was used based on the ‘‘fiber assign-ment by continuous tracking’’ algorithm.16 Quantitativeanalysis of DTI-FT data was limited to the dorsal
Table 1. Demographic, clinical, and neurophysiological characteristics
Patient 1 Patient 2 Patient 3
Clinical characteristicsSex M M MAge at onset (y) 46 53 20Disease duration at first exam (y) 4 2 4Triggering factors Lying position, leaning forward,
prolonged standingLying position,sensory stimuli, stress
Lying position,prolonged standing, stress
Medical history Operated cervical disc hernia (C5-C6) Lumbar disc protrusion (L5-S1) NoneNeurological examination Four-limb hyperreflexia
(no other pyramidal sign)No abnormality No abnormality
Treatment Pregabalin Gabapentin, clonazepam ClonazepamEvolution Marked improvement No effect Marked improvement
Spontaneous myoclonic jerksEMG burst duration (ms) 100–800 100–240 200–1000Generator level T8 T8 T10Anatomical limits of propagation T2–T12 T2–L2 C6–L4Spinal cord velocity (m/s) 9–12 5–10 3–4Jerk occurrence/min 0–5 <1 >1
Long-latency reflexes in abdominal wall muscles in response to various stimuli: latency (duration) range, msMagnetic stimulation of motor cortex (ms) 125–155 (80–125) 145–165 (100–130) 120–150 (55–75)Magnetic stimulation of thoracic spine (ms) 55–75 (145–170) 50–60 (145–160) 55–70 (135–175)Electrical stimulation of median nerve (ms) 110–130 (150–175) 85–120 (120–240) Not doneElectrical stimulation of supraorbital nerve (ms) 75–105 (90–120) 75–90 (110–260) Not done
A Y A C H E E T A L .
1760 Movement Disorders, Vol. 26, No. 9, 2011

columns, and comparisons were made of fiber counts ofboth sides of the spinal cord in PSM patients andbetween PSM patients and normal controls.
ResultsDemographic, clinical, and electrophysiological char-
acteristics are summarized in Table 1. A diagnosis ofPSM was established in our 3 patients by well-definedclinical and electrophysiological criteria.3 Clinically, allpatients had repetitive involuntary truncal flexion jerks,more frequently occurring in the lying position. In sur-face EMG recordings, all patients presented spontane-ous myoclonic jerks of long duration (�100 ms),starting at a thoracic myelomere, with slow rostral andcaudal propagation. Surface EMG recordings alsoshowed reflexes in abdominal muscles in response to alltypes of stimuli (Fig. 1). The reflex jerks showed almostthe same slow caudorostral and rostrocaudal spreading,then the spontaneous myoclonic jerks, and were absentin orbicularis oculi and sternocleidomastoid muscles(see Fig. e-1 in the online Additional Supporting Infor-mation). The latency of the reflex jerks increased withthe type of stimulation as follows: magnetic stimulationof the thoracic spine (mean reflex latency, 60 ms), elec-trical stimulation of the supraorbital nerve (mean reflexlatency, 85 ms), electrical stimulation of the mediannerve (mean reflex latency, 110 ms), and magneticstimulation of the motor cortex (mean reflex latency,140 ms). The duration of the reflex jerks was in thesame range as that of the spontaneous myoclonic jerks.Additional neurophysiological investigation showed
no sign of cortical myoclonus, that is, no long-loop C-reflex, no giant somatosensory evoked potentials, and
no cortical potential preceding myoclonus by a shortlatency in jerk-locked back-averaged EEG. There wasalso no BP in back-averaged EEG that could suggestmimicked or psychogenic myoclonus.In neuroradiological investigation, brain MRI was
normal. Conventional spine MRI disclosed sequelae ofan operated-on disc hernia or disc protrusion in 2patients without impact on the spinal cord. Spinal cordDTI-FT revealed bilateral fiber loss in the dorsal col-umns for patient 1, but not for patient 2 (see Fig. e-2in the online Additional Supporting Information).
DiscussionThe 3 patients described in this study had typical,
clinical, and electrophysiological features of PSM,3
with slow rostral and caudal propagation from a gen-erator located at the low thoracic spine level. Thelong duration of the myoclonic jerks, associated withthe absence of the long-loop C-reflex, giant somato-sensory evoked potentials, and time-locked corticalpotential preceding myoclonic jerk in back-averagedEEG, argued against myoclonus of cortical origin.This study focused on the presence of long-latency
abdominal reflexes elicited by various stimuli. To date,such responses have been described only in 16 patientswith PSM,3–10 following electrical nerve stimulationor mechanical tapping with a reflex hammer (seeTable e-1 in the online Additional Supporting Infor-mation). In the literature, abdominal reflex latenciesseemed to be influenced by the type rather than thesite of stimulation. In contrast, we found that stimula-tion site was the main factor of influence. Our resultswere consistent with excitation of a brain stem
FIG. 1. Long-latency reflexes recorded in abdominal wall muscles in response to various stimuli (magnetic stimulation of motor cortex or thoracicspine and electrical stimulation of median or supraorbital nerve). In addition to long-latency reflexes, spinal and cortical magnetic stimulationsevoked ‘‘direct responses’’ (motor evoked potentials) at a latency of 4–8 and 17–22 ms, respectively, in abdominal wall muscles.
P R O P R I O S P I N A L M Y O C L O N U S
Movement Disorders, Vol. 26, No. 9, 2011 1761

structure involved in motor control (possibly the retic-ular formation) and then projection to the propriospi-nal system and the spinal generator of PSM (see Fig.e-3 in the online Additional Supporting Information).The propriospinal system has been described at C3-
C4 spine level in the cat,17 and strong evidence hasbeen provided for the existence of a similar system inhumans.18,19 The reticular formation is known to pro-duce extensive excitation of propriospinal neurons,9
which could in turn activate the thoracic spine genera-tor, where increased excitability exists.6,10 This mayproduce myoclonic jerks in response to the stimuli,with slow rostral and caudal propagation along thepropriospinal pathways, as for spontaneous PSM.Stimulus-evoked abdominal reflexes are rather ster-
eotyped in terms of latency and duration in patientswith PSM. They have been considered a form ofenhanced startle response.20,21 However, the reflexesthat we recorded could not be considered startleresponses in the strict sense for several reasons. First,startle reflexes to electrical stimuli require strongerstimulus intensities (12 times the perception thresholdwith a 0.5-ms pulse duration)22 than those used in ourstudy (3 times the perception threshold with a 0.2-mspulse duration). Second, startle responses are system-atically present in orbicularis oculi and sternoicleido-mastoid muscles, but obtained in fewer than half ofnormal subjects in abdominal wall muscles.23 In con-trast, in our study, median nerve stimulation elicitedlong-latency reflexes in abdominal wall muscles with-out any concomitant response in orbicularis oculi andsternocleidomastoid muscles. Third, as previouslyreported,6,7,10 the long-latency reflexes showed slowrostral and caudal propagation from the thoracic spinegenerator, like the spontaneous myoclonic jerks. Suchspreading is not described for startle responses.23,24
In our experience, abdominal reflex responses can-not be produced in normal subjects with the stimula-tion parameters we used (painless sensory stimuli oflow intensity). It remains to be determined whetherthese reflexes could help to distinguish ‘‘organic PSM’’from psychogenic or mimicked PSM. The value of BPhas recently been emphasized to distinguish betweenmimicked or psychogenic jerk (present BP) and or-ganic myoclonus (absent BP).13–15 The absence of BPin our patients suggests an organic origin of myoclonicjerks. In another study,20 normal subjects wereinstructed to make voluntary generalized flexion jerksin response to median nerve stimulation with stimulusintensity set at 3 times the sensory threshold (as in thepresent study). The voluntary mimicked jerks had amore variable and longer latency (range, 97–189 ms;mean, 139 ms) than our abdominal muscle reflexes(range, 85–130 ms; mean, 110 ms).Recently, DTI-FT analysis was shown to be able to
reveal a subtle spinal cord lesion in PSM.3,25 We have
investigated 2 patients with this technique: the firsthad fiber loss in the dorsal columns, as previouslydescribed,3,25 but the other patient did not show sig-nificant alteration. Thus, the sensitivity of DTI-FTanalysis to support the diagnosis of organic PSMremains to be further evaluated.To conclude, patients with PSM showed a stereo-
typed pattern of recruitment of long-latency abdomi-nal reflexes in response to various stimuli. Thestrategy of using 4 stimulation types and locations, asproposed in this study, is easy to perform and couldbe of clinical utility in the diagnosis of PSM. Suchelectrophysiological testing deserves to be investigatedin larger prospective studies.
References1. Brown P, Thompson PD, Rothwell JC, Day BL, Marsden CD.
Paroxysmal axial spasms of spinal origin. Mov Disord. 1991;6:43–48.
2. Brown P, Thompson PD, Rothwell JC, Day BL, Marsden CD.Axial myoclonus of propriospinal origin. Brain. 1991;114:197–214.
3. Roze E, Bounolleau P, Ducreux D, et al. Propriospinal myoclonusrevisited: Clinical, neurophysiologic, and neuroradiologic findings.Neurology. 2009;72:1301–1309.
4. Kapoor R, Brown P, Thompson PD, Miller DH. Propriospinalmyoclonus in multiple sclerosis. J Neurol Neurosurg Psychiatry.1992;55:1086–1088.
5. Brown P, Rothwell JC, Thompson PD, Marsden CD. Propriospi-nal myoclonus: evidence for spinal ‘‘pattern’’ generators inhumans. Mov Disord. 1994;9:571–576.
6. Kono I, Ueda Y, Araki K, Nakajima K, Shibasaki H. Spinal myo-clonus resembling belly dance. Mov Disord. 1994;9:325–329.
7. Nogues MA, Leiguarda RC, Rivero AD, Salvat F, Manes F. Invol-untary movements and abnormal spontaneous EMG activity insyringomyelia and syringobulbia. Neurology. 1999;52:823–834.
8. Nogues M, Cammarota A, Sola C, Brown P. Propriospinal myo-clonus in ischemic myelopathy secondary to a spinal dural arteri-ovenous fistula. Mov Disord. 2000;15:355–358.
9. Benatru I, Thobois S, Andre-Obadia N, et al. Atypical propriospi-nal myoclonus with possible relationship to alpha interferon ther-apy. Mov Disord. 2003;18:1564–1568.
10. Vetrugno R, Liguori R, D’Alessandro R, D’Angelo R, AlessandriaM, Montagna P. Axial myoclonus in paraproteinemic polyneu-ropathy. Muscle Nerve. 2008;38:1330–1335.
11. Shibasaki H, Kuroiwa Y. Electroencephalographic correlates ofmyoclonus. Electroencephalogr Clin Neurophysiol. 1975;39:455–463.
12. Cassim F, Houdayer E. Neurophysiology of myoclonus. Neuro-physiol Clin. 2006;36:281–291.
13. Terada K, Ikeda A, Van Ness PC, et al. Presence of Bereitschaft-spotential preceding psychogenic myoclonus: clinical applicationof jerk-locked back averaging. J Neurol Neurosurg Psychiatry.1995;58:745–747.
14. Esposito M, Edwards MJ, Bhatia KP, Brown P, Cordivari C. Idio-pathic spinal myoclonus: a clinical and neurophysiological assess-ment of a movement disorder of uncertain origin. Mov Disord.2009;24:2344–2349.
15. Hallett M. Physiology of psychogenic movement disorders. J ClinNeurosci. 2010;17:959–965.
16. Mori S, Crain BJ, Chacko VP, van Zijl PC. Three-dimensionaltracking of axonal projections in the brain by magnetic resonanceimaging. Ann Neurol. 1999;45:265–269.
17. Alstermark B, Lundberg A.The C3-C4 propriospinal system: tar-get-reaching and food-taking. In: Jami L, Pierrot-Deseilligny E,Zytnicki D, eds. Muscle Afferents and Spinal Control of Move-ment. London, UK: Pergamon Press; 1992:327–354.
A Y A C H E E T A L .
1762 Movement Disorders, Vol. 26, No. 9, 2011

18. Pierrot-Deseilligny E. Peripheral and descending control of neuronesmediating non-monosynaptic Ia excitation to motoneurones: a pre-sumed propriospinal system in man. Prog Brain Res. 1989;80:305–314.
19. Pierrot-Deseilligny E. Propriospinal transmission of part of thecorticospinal excitation in humans. Muscle Nerve. 2002;26:155–172.
20. Thompson PD, Colebatch JG, Brown P, et al. Voluntary stimu-lus-sensitive jerks and jumps mimicking myoclonus or pathologi-cal startle syndromes. Mov Disord. 1992;7:257–262.
21. Rothwell JC. The startle reflex, voluntary movement, and thereticulospinal tract. Suppl Clin Neurophysiol. 2006;58:223–231.
22. Alvarez-Blanco S, Leon L, Valls-Sole J. The startle reactionto somatosensory inputs: different response pattern to stimuliof upper and lower limbs. Exp Brain Res. 2009;195:285–292.
23. Vidailhet M, Rothwell JC, Thompson PD, Lees AJ, MarsdenCD. The auditory startle response in the Steele-Richardson-Ols-zewski syndrome and Parkinson’s disease. Brain. 1992;115:1181–1192.
24. Meinck HM. Startle and its disorders. Neurophysiol Clin. 2006;36:357–364.
25. Roze E, Apartis E, Vidailhet M, et al. Propriospinal myoclonus:utility of magnetic resonance diffusion tensor imaging and fibertracking. Mov Disord. 2007;22:1506–1509.
P R O P R I O S P I N A L M Y O C L O N U S
Movement Disorders, Vol. 26, No. 9, 2011 1763
Related Documents











