of June 14, 2018. This information is current as Cytomegalovirus Entry into Fibroblast Cells Responses Induced by Human Differential Initiation of Innate Immune Teresa Compton Laura K. Juckem, Karl W. Boehme, Adam L. Feire and http://www.jimmunol.org/content/180/7/4965 doi: 10.4049/jimmunol.180.7.4965 2008; 180:4965-4977; ; J Immunol References http://www.jimmunol.org/content/180/7/4965.full#ref-list-1 , 57 of which you can access for free at: cites 100 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2008 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on June 14, 2018 http://www.jimmunol.org/ Downloaded from by guest on June 14, 2018 http://www.jimmunol.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

of June 14, 2018.This information is current as
Cytomegalovirus Entry into Fibroblast CellsResponses Induced by Human Differential Initiation of Innate Immune
Teresa ComptonLaura K. Juckem, Karl W. Boehme, Adam L. Feire and
http://www.jimmunol.org/content/180/7/4965doi: 10.4049/jimmunol.180.7.4965
2008; 180:4965-4977; ;J Immunol
Referenceshttp://www.jimmunol.org/content/180/7/4965.full#ref-list-1
, 57 of which you can access for free at: cites 100 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 14, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Differential Initiation of Innate Immune Responses Induced byHuman Cytomegalovirus Entry into Fibroblast Cells1
Laura K. Juckem,* Karl W. Boehme,* Adam L. Feire,*† and Teresa Compton2*
Infection of permissive fibroblasts with human CMV (HCMV, AD169) is accompanied by a robust activation of innate immunedefense. In this study, we show that inflammatory cytokine (IC) secretion and activation of the type I IFN pathway (�� IFN) areinitiated through distinct mechanisms. HCMV is recognized by TLR2 leading to the NF-�B activation and IC secretion. However,the IFN response to HCMV is not a TLR2-dependent process, as a dominant negative TLR2 does not affect the antiviral responseto infection. Additionally, bafilomycin, an endosomal acidification inhibitor, has no effect on HCMV-induced IFN responsessuggesting that IFN signaling is independent of endosomal resident TLRs. By contrast, disruption of lipid rafts by depletion ofcellular cholesterol inhibits both HCMV entry as well as IFN responses. Cholesterol depletion had no effect on the induction ofICs by HCMV, illustrating a biological distinction at the cellular level with the initiation of innate immune pathways. Further-more, HCMV entry inhibitors block IFN responses but not IC signaling. In particular, blocking the interaction of HCMV with�1 integrin diminished IFN signaling, suggesting that this virus-cell interaction or subsequent downstream steps in the entrypathway are critical for downstream signal transduction events. These data show that HCMV entry and IFN signaling arecoordinated processes that require cholesterol-rich microdomains, whereas IC signaling is activated through outright sensing viaTLR2. These findings further highlight the complexity and sophistication of innate immune responses at the earliest points inHCMV infection. The Journal of Immunology, 2008, 180: 4965–4977.
H uman CMV (HCMV),3 a member of the Herpesviridaefamily, is a ubiquitous opportunistic pathogen. The out-come of HCMV infection typically correlates with the
immune status of the host. Infection of healthy individuals is oftenasymptomatic; however, infection of immunocompromised hosts,including organ and stem cell transplant recipients and AIDS pa-tients, can be devastating (1, 2). Neonates are also susceptible toHCMV disease, especially when primary infection of the motheroccurs during pregnancy; these infections can result in deafness,mental retardation and mortality (3, 4). In addition, persistentHCMV infection has been associated with chronic diseases such ascoronary artery disease and diabetes (5, 6).
HCMV infection results in the secretion of inflammatory cyto-kines (ICs) and type I IFNs from host cells (7–9). Both classes ofmolecules are hallmarks of innate immunity that contribute signif-icantly to control infections (10, 11). ICs, such as TNF-�, IL-1,IL-6, IL-8, IL-12, and IL-18, have a wide range of biological ef-
fects on tissues and cells and are critical for the recruitment andactivation of phagocytic leukocytes (12). Type I IFNs consist ofIFN-� and multiple forms of IFN-� and are produced in responseto many virus and intracellular bacterial infections (13). IFNs elicitthe expression of IFN-stimulated genes (ISGs), a subset of cellularfactors that inhibit viral replication (7, 8, 11, 14). Together theseresponses serve to limit viral replication and spread at earlytimes following infection and also activate and promote adap-tive immune responses that will ultimately contain or clear theinfection. HCMV does not require virus replication or cellularprotein synthesis for the robust initial induction of innate im-mune responses, suggesting that structural components of thevirus are responsible for the up-regulation of these genes duringvirus entry into cells (7, 9).
The IC branch of innate immunity is defined by activation ofNF-�B, which is responsible for the transcription of genes encod-ing many proinflammatory cytokines and chemokines (15). In re-sponse to stimuli such as cytokines or viruses, activation of thecanonical NF-�B pathway occurs via signal transduction cascadesthat promote the phosphorylation and degradation of I�Bs by ubiq-uitination, thereby releasing the NF-�B heterodimer. Once re-leased, the activated heterodimer comprised of p50 and p65/RelAis able to translocate to the nucleus and drive expression of targetgenes (16). Fibroblasts and monocytes infected with HCMV ex-hibit activated NF-�B as evidenced by its nuclear translocation andincreased DNA binding activity (17, 18).
One means by which viruses elicit ICs and IFNs is throughTLRs that detect and initiate innate immune response to microbialpathogens. TLRs are a class of pattern recognition receptors (19).To date, 12 members of the TLR family have been identified inhumans (20). TLRs are expressed at high levels on phagocyticcells such as dendritic cells and macrophages. However, all celltypes appear to express at least a subset of these receptors (21, 22).TLRs recognize microbial pathogens on the basis of structural mo-tifs, termed pathogen-associated molecular patterns (PAMPs),which differ from those found in host cells (23). Examples of
*McArdle Laboratory for Cancer Research, University of Wisconsin-Madison Med-ical School, Madison, WI 53706; and †Novartis Institute for Biomedical Research,Cambridge, MA 02139
Received for publication January 30, 2008. Accepted for publication February2, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by Grant R01 AI054915 (to T.C.) from the NationalInstitutes of Health and National Institutes of Health Training Grant T32GM07215 (toK.W.B.).2 Address correspondence and reprint requests to Dr. Teresa Compton, Novartis In-stitute for Biomedical Research, 500 Technology Square, Cambridge, MA 02139.E-mail address: [email protected] Abbreviations used in this paper: HCMV, human CMV; IC, inflammatory cytokine;ISG, IFN-stimulated gene; IRF, IFN regulatory factor; NHDF, normal human dermalfibroblast; VSV, vesicular stomatitis virus; M�CD, methyl-�-cyclodextrin; MOI,multiplicity of infection; gB, glycoprotein B; gH, glycoprotein H; PAMP, pathogen-associated molecular pattern.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

PAMPs include dsRNA (TLR3), LPS (TLR4), ssRNA (TLR7/8),and unmethylated CpG DNA (TLR9). A growing body of evidenceindicates that viral envelope glycoproteins can trigger TLR-medi-ated innate immune signaling. For example, the fusion proteinfrom respiratory syncytial virus and the envelope protein of mousemammary tumor virus are sensed by TLR4 (24, 25). Similarly,HCMV envelope glycoprotein B (gB) and glycoprotein H (gH)elicit IC secretion through TLR2 (26, 27).
Induction of type I IFN responses to virus infection can be di-vided into two phases, the activation phase and the amplificationphase. Virus infection activates an initial activation phase througha key regulatory transcription factor, IFN regulatory factor 3(IRF3). Incompletely characterized signal transduction pathwayslead to virus-induced phosphorylation of IRF3 on C-terminalserine residues by related kinases TANK-binding kinase 1 and I�Bkinase � (28–30). This results in the homodimerization and trans-location of IRF3 to the nucleus where it can interact with the co-activators, CREB binding protein and p300, and form a complexthat drives the transcription of IFN-� and a subset of ISGs (31).The nascent IFN is then able to act in an autocrine and paracrinemanner to initiate signaling through the cellular �� IFN receptorleading to activation of a JAK and STAT signal transduction cas-cade. This amplification phase induces the robust expression of abroad panel of ISGs, which further assists the cell in establishingan antiviral state (11).
The mechanisms by which HCMV activates IFN responses re-main largely undefined. HCMV and a soluble version of gB areable to activate IRF3 and the transcription of ISGs (32–34). Recentevidence using several strategies to deplete cellular IRF3 confirmsits requirement and proposes that IRF3 is the primary transcriptionfactor mediating HCMV-induced IFN signaling (35). The abilityof a soluble version of gB to activate IFN signaling suggests thatgB binding to a cell surface receptor during virus entry is sufficientfor antiviral responses. Interestingly, a small molecule HCMV en-try inhibitor that targets gB, as well as neutralizing Abs to both gBand gH, inhibit ISG accumulation (36). These studies suggest thatglycoprotein-cell interactions during the entry process are impor-tant for HCMV-induced IFN activation.
HCMV has evolved many mechanisms to attenuate the host im-mune response to create an environment that is conducive to virusreplication and persistence. The means by which HCMV modu-lates host immune responses is largely attributed to newly synthe-sized viral proteins. The immediate early gene product 1 (IE72)has been shown to prevent the association of STAT1, STAT2 andIRF9 with the promoters of IFN-responsive genes (37). In addi-tion, the immediate early gene product 2 (IE86) has been shown tointerfere with NF-�B binding to the IFN-� promoter (38). HCMValso evades host immune responses through the molecular mimicryof many immune molecules including, but not limited to, the ex-pression of viral chemokines, a MHC class I homolog, chemokinebinding proteins, and virus encoded G protein-coupled receptors(39–43). Due to the ability of live HCMV to modulate the hostinnate immune responses, UV-inactivated virus was used in thecurrent study to focus on the initiation of HCMV-induced innateimmune responses, whereas live virus was used to study virusentry through tegument delivery and the initiation of viral geneexpression.
Understanding the coordination of innate immune activation andvirus entry is a daunting task due in part to the complexity asso-ciated with HCMV entry. Multiple envelope glycoprotein com-plexes decorate HCMV virions and interact with several cellularreceptors to mediate entry into cells. HCMV glycoprotein M andgB interact with cell surface heparan sulfate proteoglycans totether the virion to the cell surface (44). This allows for more
stable docking with additional cellular receptors including �1 and�3 integrins that interact with gB and gH, respectively (45, 46).Epidermal growth factor receptor has also been proposed as anHCMV entry receptor, although its role remains controversial (47–49). HCMV entry may occur in a pH neutral or pH-dependentmanner depending on the cell type (50, 51). The envelope glyco-proteins gB, gH, and glycoprotein L are essential for entry andcritical in mediating fusion (52, 53). It is during this entry processthat the cell gets its first glimpse at the invading pathogen and thefirst opportunity to contain it.
We report that HCMV activates IC and IFN responses throughdiscrete mechanisms during virus entry. HCMV activates IC re-sponses via TLR2 apparently independent of events needed forproductive infection. By contrast, HCMV entry and IFN signalingare intimately linked processes critically dependent on the organi-zation of cholesterol-rich microdomains. These findings enhanceour understanding of the complex interplay between virus entryand host detection.
Materials and MethodsCell lines, reagents, and viruses
Normal human dermal fibroblasts (NHDFs; Clonetics) were grown at 37°Cin 5% CO2 in DMEM (Invitrogen Life Technologies) supplemented with10% FBS (HyClone Laboratories), 1% glutamine, and 1% penicillin-strep-tomycin-amphotericin B (Fungizone; BioWhittaker). The NHDF cells wereserum starved for 24 h before all treatments. The AD169 strain of HCMVwas propagated in NHDF cells and titered by immediate early gene product1 (IE1, IE72) and gene product 2 (IE2, IE86) expression by indirect im-munofluorescence (54). HCMV AD169 IE2-GFP virus was provided byD. H. Spector (University of California, San Diego, CA) (55). Wherenoted, purified AD169 virions isolated by density gradient centrifugationwere used (50). UV inactivation of HCMV was performed as previouslydescribed (26). Replication competent recombinant vesicular stomatitis vi-rus (VSV) expressing the enhanced gene encoding GFP was provided by J.Yin (University of Wisconsin, Madison, WI) and propagated in baby ham-ster kidney BHK cells.
Reagents and Abs
NHDFs were transduced with retroviral vectors containing a TLR2 dom-inant negative construct with Toll/IL-1R domain cytoplasmic deletions aspreviously described (27). IFN treatments were performed using a combi-nation of � and � recombinant human IFNs (BioSource International).Recombinant human IL-1� was obtained from R&D Systems. Pam3CSK4
was obtained from EMC Microcollections and polyinosinic-polycytidylicacid potassium salt (poly(I:C)) was obtained from Sigma-Aldrich. A mAbto pp65 was obtained from the Rumbaugh-Goodwin Institute for CancerResearch (Plantation, FL). A mAb to IRF3 was obtained from Active Mo-tif. Rabbit polyclonal Abs to I�B� and p65 (RelA) were obtained fromSanta Cruz Biotechnology. The anti-P56 Ab was a gift from G. Sen (TheCleveland Clinic Foundation, Cleveland, OH) (56). The second generation�-peptide 19, a potent inhibitor of HCMV entry (57), is designated �-pep-tide (�) in this study, and a control �-peptide that does not block HCMVentry is designated �-peptide (�).
VSV plaque reduction assay
Subconfluent NHDFs were washed with PBS, mock infected, treated with�� IFNs (100 IU/ml), or infected with HCMV as indicated. All treatmentswere performed in serum-free medium. Following a 6-h incubation, thecells were washed once with PBS and infected with 100 PFU/ml VSV(New Jersey strain). VSV was absorbed for 1 h at 37°C, the innoculum wasremoved, and the cells overlaid with 2 ml of a 60/40 mixture of 2� Eagle’sMEM (BioWhittaker) and 1% agarose. The cells were incubated at 37°C,and plaques were visualized by crystal violet staining (0.5� PBS, 0.07%crystal violet, 5.5% formaldehyde) at 48 h of postinfection.
Inhibition of endosome acidification
NHDF cells were pretreated for 30 min with bafilomycin (1 �M). HCMVAD169 IE2-GFP at a multiplicity of infection (MOI) of 1 was directlyadded to the bafilomycin-containing medium and adsorbed for 2 h. Non-penetrated virus was inactivated with a low pH citrate buffer wash (40 mMcitric acid, 10 mM KCl, 135 mM NaCl (pH 3.0)) (58). The medium wasreplaced, and cells were incubated in the presence of bafilomycin at 37°C.
4966 DIFFERENTIAL INITIATION OF INNATE IMMUNE RESPONSES BY HCMV
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Cholesterol depletion and replenishment treatments
NHDFs were incubated with 8 mM methyl-�-cyclodextrin (M�CD; Sigma-Aldrich), as determined by a dose-response curve, and diluted in serum-freemedium for 2 h at 37°C. After two washes with serum-free medium, thecellular cholesterol was replenished by incubating with water-soluble cho-lesterol (2.5–20 �g/ml; Sigma-Aldrich) or serum-free medium alone for anadditional 2 h. Cells were washed an additional two times with serum-freemedium and infected with HCMV (MOI � 1) for 2 h. Cells were washedwith citrate buffer and incubated in serum-free medium.
Flow cytometry
At 18–24 h postinfection, cells were released from the tissue-culture platewith trypsin, resuspended in complete medium, pelleted, and suspended inPBS. Propidium iodide (Molecular Probes) was added and samples wereanalyzed using a FACScan flow cytometer (BD Biosciences) using a stan-dard filter set. Samples were gated for propidium iodide exclusion (livecells) and assayed for GFP expression.
Virus attachment assay
NHDF cells were depleted of membrane cholesterol as described andcooled at 4°C for 30 min. HCMV (MOI � 2) was incubated at 4°C for 2 h.Cells were washed three times in cold serum-free medium and harvested inlysis buffer. DNA was extracted using QIAamp Mini Elute Virus Spin kit(Qiagen) according to the manufacturer’s instructions. DNA was eluted in50 �l of nuclease free water (Ambion). Viral genomes were quantitatedusing the primer pair, pp549s and pp821as, and UL83 FAM-TAMRAprobe as previously described (59). Unknown samples were determinedbased on a standard curve of known UL83 copy numbers using pCGN-HA-pp65, which was a gift from R. Kalejta (University of Wisconsin,Madison, WI) (60). PCR contained 2.5 �l of 50 �l extracted DNA, 50 nMprimers and probe, 12.5 �l of TaqMan Universal PCR Master mix (Ap-plied Biosystems), and nuclease free water to a total volume of 25 �l.Real-time PCR was run on an ABI 7900HT, and data were analyzed usingthe SDS 2.2.1 program.
Immunofluorescence
NHDF cells were seeded on glass coverslips and treated as described. Forthe detection of IRF3 and p65 (RelA), infections were performed in thepresence of cycloheximide (100 �g/ml; Sigma-Aldrich). Cells were fixedin 3% paraformaldehyde for 20 min, permeabilized in 0.1% Triton X-100for 10 min, and blocked in 20% purified goat serum (Pierce). Cells werewashed, incubated with anti-IRF3 Ab, anti-pp65 Ab, or anti-p65 (RelA)Ab, followed by AlexaFluor 594 goat anti-mouse secondary Ab (IRF3 andp65) or AlexaFlour 488 goat anti-rabbit secondary Ab (p65/RelA; Molec-ular Probes). Nuclei were counterstained with DAPI (4�,6�-diamidino-2-phenylindole), washed, and viewed on a Nikon TE-2000S inverted fluo-rescence microscope.
Detection of RNA accumulation of ISGs, ICs, and TLRsby RT-PCR
Total cellular RNA was harvested at 6 h postinfection using RNA-STAT60 (“B”; Tel-Test), according to the manufacturer’s instructions. Briefly,cells were lysed by addition of guanidinium thiocyanate-phenol and chlo-roform extracted, and RNA was isopropanol precipitated. RT-PCR wasperformed on 200 ng of recovered total cellular RNA using RecombinantThermus thermophilus rTth DNA Polymerase (Applied Biosystems). Theprimer pairs used were as follows: IFN-� sense 5�-CACTACAGCTCTTTCCATGA, antisense 5�-AGGATTTCCACTCTGACTCTGACTATGGTCC; ISG56 sense 5�-CAT CAG GTC AAG GAT AGT CTG GAG C,antisense 5�-GGA TTC AGG GTT TTC AGG GTC C; IL-6 sense 5�-TGTGTGAAAGCAGCAAAGAGGC, antisense 5�-TTGGGTCAGGGGTGGTTATTG; and GAPDH sense 5�-GAGCCAAAAGGGTCATC anti-sense 5�-GTGGTCATGAGTCCTTC. Endosomal-localized TLR transcriptswere detected using the Multigene-12 RT-PCR profiling kit (Superarray), ac-cording to the manufacturer’s instructions.
Western blot analysis
NHDF cells were harvested at 3 or 7 h postinfection for immunoblot anal-ysis of I�B� or P56 detection, respectively. Experiments to detect I�B�were conducted in the presence of cycloheximide (100 �g/ml). At the timeof harvest cells, were washed in PBS and lysed in harvest buffer (50 mMTris-HCl (pH 7.5), 250 mM NaCl, 50 mM NaF, 5 mM EDTA, 0.1% Non-idet P-40). Total cellular protein was quantified using a Bradford assay, and10 �g of protein was resolved by 10% or 9% reducing SDS-PAGE forI�B� or P56 detection, respectively. Proteins were transferred to a nitro-
cellulose membrane, and blotted with an anti-I�B� Ab or an anti-P56 Abfollowed by goat anti-rabbit HRP secondary Ab (Pierce). Proteins boundwith Ab were visualized using ECL (PerkinElmer). Densitometry was per-formed using ImageQuant software system (Amersham Biosciences).
Cytokine ELISA
Subconfluent NHDFs were infected with HCMV (MOI � 0.1). At 18 hpostinfection, medium samples were collected, and levels of IL-6 weredetermined by ELISA (OptEIA Set Human IL-6; BD Biosciences) accord-ing to the manufacturer’s instructions.
HCMV fusion inhibitor treatments
Soluble proteins, gB-DLD and gB-651, were incubated with NHDFs at aconcentration of 250 �g/ml for 1 h at 37°C. Cells were washed three timeswith buffer, twice with serum-free medium, and subsequently infected withHCMV-IE2-GFP (MOI � 1). �-peptides (50 �M) were incubated withHCMV (MOI � 1) in serum-free medium for 5 min before the addition ofthe entire complex to cells. Infection was allowed to proceed for 1 h at37°C, after which cells were citrate washed and incubated at 37°C untilharvest.
Expression and purification of gB-DLD and gB-651 solubleproteins
A fragment corresponding to aa 57–146 of gB (gB-DLD) was amplifiedfrom pCAGGS-gB (61) by PCR with the following primer: gB-DLD sense5�-GGA ATT CCA TAT GGT AAC GTC TTC TGA AGC C-3�, antisense5�-CGG GAT CCT TAA ACC TTT TGG TAG ACC CG-3�. The amplifiedfragment was cloned into the NdeI and BamHI sites of the bacterial ex-pression vector pET-28a (Novagen) with an N-terminal His-6 tag fragmentcorresponding to aa 651–718 of gB (gB-651) was also amplified frompCAGGS-gB by PCR with the following primers: OML30 5�-CGG GATCCA TGG ATA TCG ACC CGC TGG AA-3� and OML31 5�-CG AGATCT TCG AAT TAC TAC TAC TAC TAC TAC TGA AGG AGC ACCTTG TTC GTC CGG CGA GTA CTC CAG CAG-3�. The amplified frag-ment was cloned into the NcoI and HindIII sites of the bacterial expressionvector pTriEx-1.1 (Novagen) with a C-terminal His-6 tag. Both vectorswere transformed into Escherichia coli DH5�. To produce recombinantprotein, gB-DLD and gB-651 plasmids were isolated by FastPlasmid Miniper the manufacturer’s instructions (Eppendorf) and transformed intoE. coli BL21 (DE3) strain for protein expression. E. coli containing thepET-28a:gB-DLD was grown at 37°C in Luria-Bertani medium containingkanamycin (50 �g/ml) and pTriEx-1.1:gB-651 in Luria-Bertani mediumcontaining ampicillin (50 �g/ml) to an A600 of 0.6. To induce recombinantprotein, isopropyl �-D-thiogalacto pyranoside (Roche) was added to a finalconcentration of 1 mM and incubated for 4 h at 37°C. The cells werechilled on ice and harvested by centrifugation at 4000 rad/min for 10 minat 4°C. Protein was isolated from inclusion bodies as previously described(62). Protein was then solubilized in 8 M urea/300 mM NaCl/10 mM im-idazole/50 mM Tris (pH 7.9). Affinity purification of the proteins wasaccomplished through Ni-NTA agarose columns (Qiagen) per the manu-facturer’s instructions. Eluate from the Ni-NTA column was placed onto anS-200 sizing column, and 1-ml fractions collected. Fractions containingprotein were determined by measuring the absorbance of each fraction at214 nm using a SpectraMax 190 spectrophotometer (Molecular Devices).Fractions that corresponded to absorption peaks were analyzed by SDS-PAGE to determine the size of the protein. The absorption peak fractionsthat contained a protein of the same size as the gB-DLD or gB-651 werepooled and concentrated using a Ni-NTA column we described. The con-centrated fractions of gB-DLD were dialyzed extensively against 55 mMMES (pH 5.5) and 300 mM NaCl, whereas gB-651 fractions were dialyzedagainst 55 mM Tris (pH 8.3) and 300 mM NaCl.
ResultsHCMV induces IFN responses in a TLR2-independent manner
HCMV activates TLR2 through a physical interaction with enve-lope glycoproteins gB and gH, allowing the cell to mount an ICresponse (26, 27). TLR2 has also been shown to mediate IC sig-naling in response to HSV type 1 (HSV-1) and varicella-zostervirus (63, 64). To date, no link has been established between TLR2and the induction of IFN responses (65). However, this hypothesishas not been tested using viral ligands for TLR2. To determinewhether TLR2 can activate the IFN pathway following HCMVinfection, we used a signaling defective, dominant negative TLR2construct (TLR2�C) that lacks the cytoplasmic Toll/IL-1R domain
4967The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

common to all TLRs. NHDFs were transduced with a bicistronicretrovirus vector encoding TLR2�C and GFP. GFP-positive cellswere collected by FACS. To confirm that TLR2-dependent pro-cesses were inhibited by TLR2�C, IL-8 secretion was measuredby ELISA (Fig. 1A). The IL-8 response to the TLR-independentcytokine IL-1� was unaffected by the expression of TLR2�C.However, the cytokine responses to the TLR2-specific ligandPam3CSK4 and UV-inactivated HCMV (UV-HCMV) were mark-edly reduced in the TLR2�C-expressing cells (Fig. 1A). The in-complete block of IL-8 secretion in response to UV-HCMV sug-gests that TLR2 is not the only mechanism by which UV-HCMVcan activate NF-�B. These data confirm that TLR2�C specificallyinhibits TLR2-dependent IC responses.
To test the hypothesis that TLR2 mediates type I IFN responsesto HCMV, we determined the capacity of UV-HCMV to elicit afunctional antiviral response from the TLR2�C-expressing NHDFcells. Plaque formation by vesicular stomatitis virus (VSV), a virussensitive to the effects of IFNs, was assessed following treatmentwith type I IFNs or infection with UV-HCMV (34). As shown inFig. 1B, both control and TLR2�C-expressing NHDFs were pro-tected from VSV infection by recombinant type I IFNs or HCMVvirions. These data indicate that HCMV can induce a functionalantiviral state in the absence of a signaling-competent TLR2 mol-
ecule. Furthermore, these data suggest that HCMV initiates IFNand IC responses through distinct cellular mechanisms.
Endosomal acidification is not required for HCMV-inducedIFN responses
The observation that TLR2 function does not mediate HCMV-induced IFN signaling does not rule out other members of the TLRfamily from serving in this capacity. TLR3, TLR7, TLR8, andTLR9 are a subset of TLRs known to activate IFN responses andare localized to endosomal compartments (66–68). This subset ofintracellular localized TLRs requires a low pH trigger to initiatesignaling and allow for the rapid recognition of viral ligands dur-ing uncoating or degradation processes (69–71). Interestingly, sev-eral members of the Herpesviridae family are reported to induceIFN responses through TLR9, which senses unmethylated CpG2�-deoxyribo dinucleotides in DNA (72). The CpG rich genome ofHSV-1 and HSV type 2 and mouse CMV are recognized by TLR9in plasmacytoid dendritic cells or dendritic cells in vitro (73–75).NHDF cells express high levels of TLR3 and TLR8, whereas thereis a very low level of TLR7 and no detectable expression of TLR9(Fig. 2C).
FIGURE 1. HCMV-induced antiviral state is not reliant on TLR2. A, NHDF cells were mock transduced or transduced with a TLR2 dominant negativeconstruct (TLR2�C). Cells were treated with IL-1� (25 pg/ml), Pam3CSK4 (100 ng/ml), or UV-HCMV (MOI � 0.1). At 18 h posttreatment, thesupernatants were harvested and IL-8 levels were determined by ELISA. Error bars indicate SD for triplicate samples. B, Control (NHDF) and TLR2�C-expressing cells were infected at the indicated MOIs with UV-HCMV, or treated with recombinant type I IFN (100 U/ml). At 6 h poststimulation, thetreatments were removed and the monolayers were challenged with VSV (�100 PFU/well). Plaque formation was visualized by crystal violet staining at48 h postinfection.
4968 DIFFERENTIAL INITIATION OF INNATE IMMUNE RESPONSES BY HCMV
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
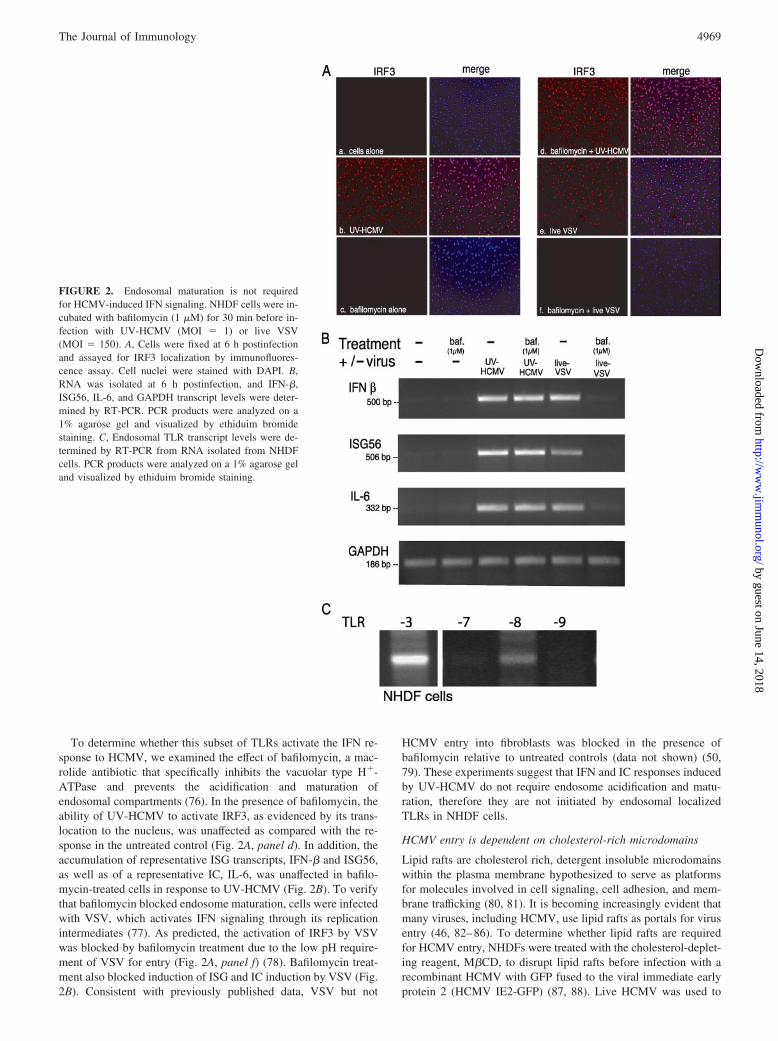
To determine whether this subset of TLRs activate the IFN re-sponse to HCMV, we examined the effect of bafilomycin, a mac-rolide antibiotic that specifically inhibits the vacuolar type H�-ATPase and prevents the acidification and maturation ofendosomal compartments (76). In the presence of bafilomycin, theability of UV-HCMV to activate IRF3, as evidenced by its trans-location to the nucleus, was unaffected as compared with the re-sponse in the untreated control (Fig. 2A, panel d). In addition, theaccumulation of representative ISG transcripts, IFN-� and ISG56,as well as of a representative IC, IL-6, was unaffected in bafilo-mycin-treated cells in response to UV-HCMV (Fig. 2B). To verifythat bafilomycin blocked endosome maturation, cells were infectedwith VSV, which activates IFN signaling through its replicationintermediates (77). As predicted, the activation of IRF3 by VSVwas blocked by bafilomycin treatment due to the low pH require-ment of VSV for entry (Fig. 2A, panel f) (78). Bafilomycin treat-ment also blocked induction of ISG and IC induction by VSV (Fig.2B). Consistent with previously published data, VSV but not
HCMV entry into fibroblasts was blocked in the presence ofbafilomycin relative to untreated controls (data not shown) (50,79). These experiments suggest that IFN and IC responses inducedby UV-HCMV do not require endosome acidification and matu-ration, therefore they are not initiated by endosomal localizedTLRs in NHDF cells.
HCMV entry is dependent on cholesterol-rich microdomains
Lipid rafts are cholesterol rich, detergent insoluble microdomainswithin the plasma membrane hypothesized to serve as platformsfor molecules involved in cell signaling, cell adhesion, and mem-brane trafficking (80, 81). It is becoming increasingly evident thatmany viruses, including HCMV, use lipid rafts as portals for virusentry (46, 82–86). To determine whether lipid rafts are requiredfor HCMV entry, NHDFs were treated with the cholesterol-deplet-ing reagent, M�CD, to disrupt lipid rafts before infection with arecombinant HCMV with GFP fused to the viral immediate earlyprotein 2 (HCMV IE2-GFP) (87, 88). Live HCMV was used to
FIGURE 2. Endosomal maturation is not requiredfor HCMV-induced IFN signaling. NHDF cells were in-cubated with bafilomycin (1 �M) for 30 min before in-fection with UV-HCMV (MOI � 1) or live VSV(MOI � 150). A, Cells were fixed at 6 h postinfectionand assayed for IRF3 localization by immunofluores-cence assay. Cell nuclei were stained with DAPI. B,RNA was isolated at 6 h postinfection, and IFN-�,ISG56, IL-6, and GAPDH transcript levels were deter-mined by RT-PCR. PCR products were analyzed on a1% agarose gel and visualized by ethiduim bromidestaining. C, Endosomal TLR transcript levels were de-termined by RT-PCR from RNA isolated from NHDFcells. PCR products were analyzed on a 1% agarose geland visualized by ethiduim bromide staining.
4969The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

measure virus entry, and the GFP-positive cells were scored at 24 hpostinfection as a surrogate marker of infection (Fig. 3A). Theaverage total percentage of cells infected for the three independent
experiments conducted was 67% at MOI � 1. In cells pretreatedwith M�CD, viral gene expression was reduced by approximately90% compared with expression in untreated control; the effects of
FIGURE 3. HCMV entry is dependent on cholester-ol-rich microdomains. NHDF cells were pretreated with8 mM M�CD to deplete membrane cholesterol, and in-dicated wells were subsequently incubated with water-soluble cholesterol. Cells were washed and infectedwith live HCMV (MOI � 1). A, At 18 h postinfection,GFP expression was assessed by flow cytometry as asurrogate marker of infection. No cellular toxicity wasobserved by propidium iodide staining (data not shown).Data represent the average of three independent exper-iments. B, Delivery of the HCMV tegument proteinpp65 was measured by indirect immunofluorescence at1 h postinfection. At least six random fields werecounted per treatment. Graph is representative of twoindependent experiments. C, Bound HCMV genomeswas measured by quantitative PCR in mock NHDFcells, infected with live HCMV (MOI � 2), pretreatedwith 8 mM M�CD and infected with live HCMV orwith live HCMV treated with soluble heparin (30 �g/ml). Error bars represent SDs.
4970 DIFFERENTIAL INITIATION OF INNATE IMMUNE RESPONSES BY HCMV
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

M�CD could be reversed in a dose-dependent manner with theaddition of water-soluble cholesterol (Fig. 3A).
Due to the important role that lipid rafts play in cell signaling,it is possible that the depletion of lipid rafts disrupts signal trans-duction cascades required for the initiation of viral gene expres-sion. Therefore we assessed virus payload delivery by measuringthe nuclear localization of the tegument protein pp65, which traf-fics to the nucleus almost immediately upon infection with HCMV.Similar to the results obtained with the IE2-GFP reporter virus,M�CD pretreatment resulted in a 70% decrease in the number ofpp65-positive nuclei compared with untreated cells (Fig. 3B). Thisresult suggests that the block to infection is at or before fusion.Notably, pretreatment with M�CD did not alter the binding ofHCMV to cells (Fig. 3C). Together these data suggest that lipidrafts are essential for HCMV entry.
HCMV-induced IFN responses are dependent oncholesterol-rich microdomains
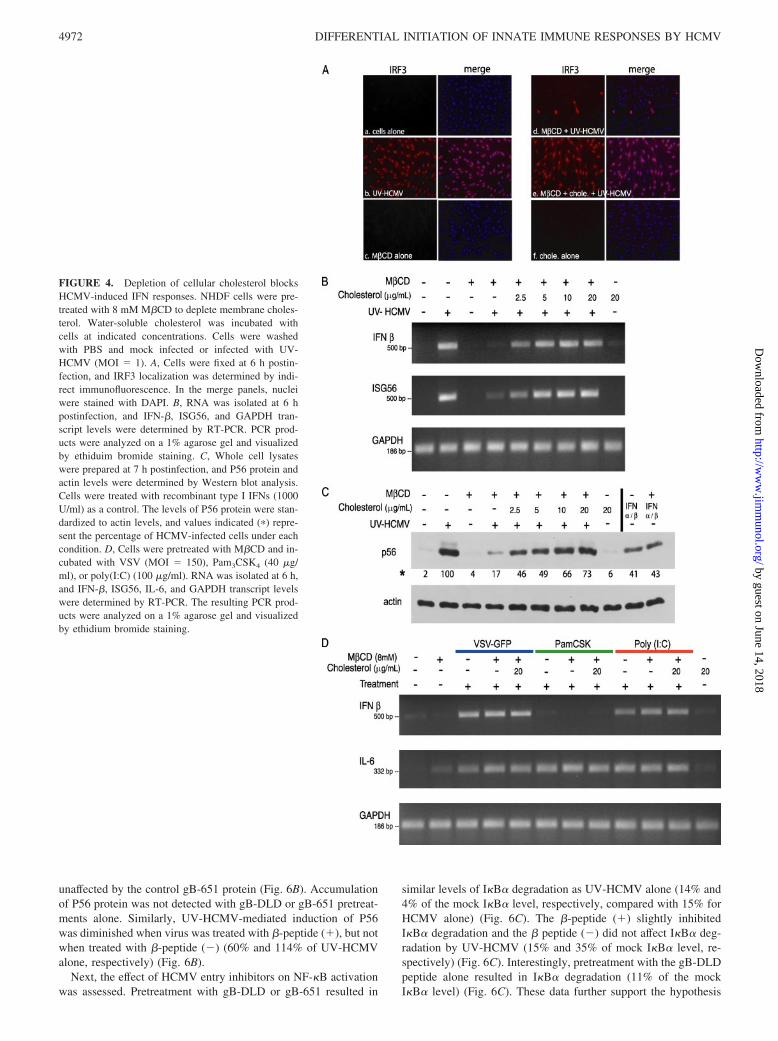
We next assessed whether disruption of lipid rafts by cholesteroldepletion interferes with the induction of the IFN response toHCMV infection. Nuclear translocation of IRF3 induced by UV-HCMV infection was inhibited by pretreatment of cells withM�CD as compared with the infection of untreated cells (Fig. 4A,panels b and d). Activation of IRF3 was restored by the additionof water-soluble cholesterol (Fig. 4A, panel e). Neither M�CD norcholesterol treatment alone activated IRF3 (Fig. 4A, panels c andf). Consistent with the lack of IRF3 nuclear translocation, the ac-cumulation of representative ISG transcripts, IFN-� and ISG56, inresponse to UV-HCMV were inhibited by pretreatment withM�CD (Fig. 4B, lane 4). ISG transcript levels were restored whenmembrane cholesterol was replenished. M�CD and cholesteroltreatments alone did not result in the accumulation of ISG mRNA.Consistent with our transcriptional studies, M�CD pretreatmentdiminished induction of the ISG56 protein (P56) by UV-HCMV.Densitometric analysis revealed a 5- to 6-fold decrease in P56levels with M�CD pretreatment compared with P56 levels inuntreated controls; P56 protein levels were restored by the ad-dition of water-soluble cholesterol (Fig. 4C). Again, treatmentwith M�CD or water-soluble cholesterol alone did not induceP56 accumulation.
To test the specificity of M�CD pretreatment on the HCMV-mediated IFN response, we also measured the accumulation of P56following treatment with recombinant type I IFNs. No differencewas observed upon IFN treatment (Fig. 4C). To further verify thespecificity of M�CD pretreatment, RT-PCR analysis of IFN-� andIL-6 induction was examined in response to additional innate im-mune ligands. VSV, which enters cells through the classical clath-rin-coated pit pathway in a lipid raft-independent manner was un-affected by M�CD pretreatment (Fig. 4D) (78). Similarly,treatment with poly(I:C), a TLR3 specific ligand, induced IFN-� inthe presence and absence of M�CD pretreatment (Fig. 4D). Con-sistent with a previous report (65), the TLR2-specific ligandPamCSK did not cause the accumulation of IFN-� transcripts (Fig.4D). Together these data suggest that virus entry and activation ofthe host IFN responses in fibroblasts are dependent on cholesterol-rich microdomains and are specific to HCMV.
HCMV activates IC secretion independent of the organizationof cholesterol-rich microdomains
We next assessed the role of cholesterol-rich microdomains in theactivation of NF-�B and the induction of ICs by UV-HCMV. NF-�Bactivation was determined by monitoring the degradation of I�B�, aprotein that binds and retains the NF-�B dimer in an inactive state inthe cytoplasm of cells (89). In response to stimuli such as cytokines or
viruses, signal transduction cascades promote the phosphorylation anddegradation of I�B� through ubiquitination, thereby releasing NF-�Bto translocate to the nucleus where it drives target gene expression(16). We measured the degradation of the NF-�B inhibitory subunit,I�B�, as an indirect measure of NF-�B activation. In cells pretreatedwith M�CD and infected with gradient purified UV-HCMV, I�B�was degraded to a similar extent as UV-HCMV alone at both 1 and3 h postinfection (7 and 12% of mock cells, respectively) (Fig. 5A anddata not shown). I�B� degradation following UV-HCMV infectionwas slightly affected by treatment of cells with M�CD or 20 �g/mlwater-soluble cholesterol (90% and 85% of mock cells, respectively)(Fig. 5A). Following I�B� degradation, NF-�B heterodimers com-posed of p50 and p65/RelA rapidly translocate from the cytosol to thenucleus (16). Using indirect immunofluorescence with Abs to RelA aswell as the HCMV tegument protein pp65, we can simultaneouslyvisualize HCMV-induced NF-�B activation and virus entry (Fig. 5B).At 2 h postinfection both RelA and pp65 can be visualized in thenucleus of cells infected with live HCMV. M�CD pretreatmentblocks pp65 uptake but not RelA nuclear localization (Fig. 5B, panelb). Finally, IL-6 induction by UV-HCMV infection was unaffectedfollowing cholesterol depletion or replenishment as measured byELISA (Fig. 5C). Treatment with M�CD or cholesterol alone did notaffect IL-6 secretion. These results suggest that, unlike the IFN re-sponse, activation of NF-�B and subsequent production of ICs is in-dependent of organized cholesterol-rich microdomains. These resultsalso indicate that HCMV-induced IC activation is not dependent onvirus entry into cells, as cholesterol depletion blocks virus entry(Fig. 3).
HCMV entry inhibitors block HCMV-induced IFN signaling butnot IC signaling
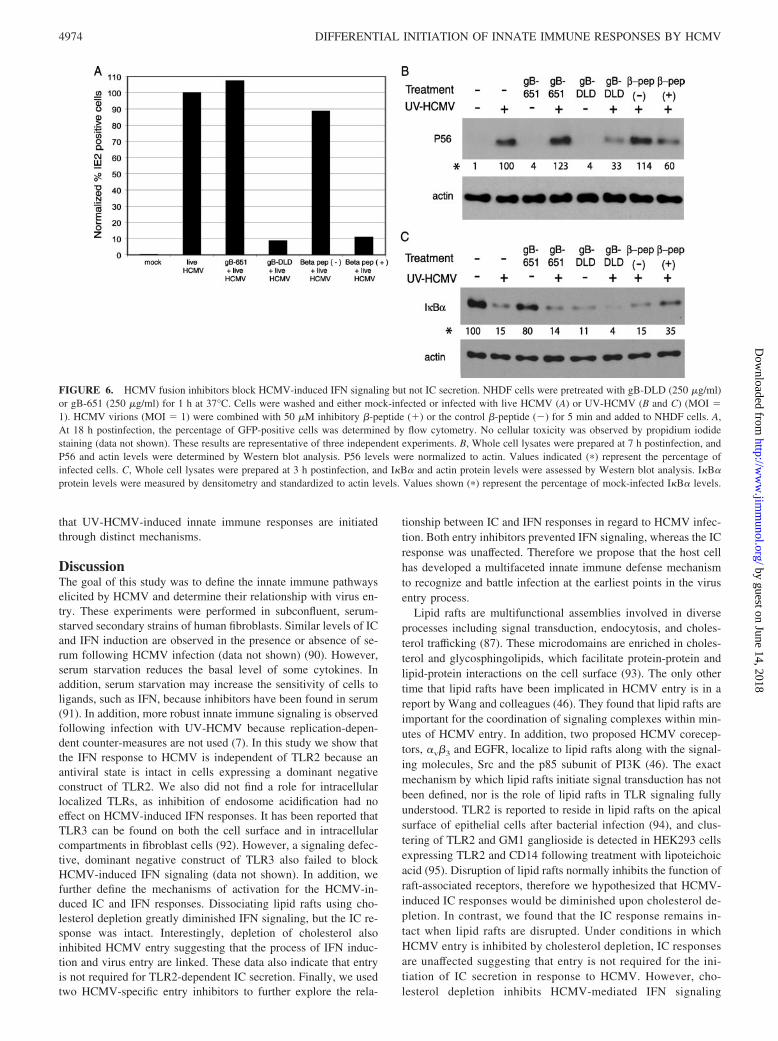
The observation that cholesterol depletion inhibits HCMV entry,as well as the induction of the IFN response (Figs. 3 and 4), sug-gests that 1) lipid rafts serve as a platform on which cellular ma-chinery act to induce the IFN response, or 2) the process of HCMVentry is an important factor for activation of IFN signaling. Tofurther probe the role of HCMV entry in HCMV-induced innateimmune responses, we used two inhibitors of HCMV entry. Theprotein, gB-DLD, contains a highly conserved region of gB en-compassing an integrin binding motif, the disintegrin-like domain.Protein gB-DLD acts as a ligand mimic and interacts with �1 in-tegrin to potently block HCMV entry at a postattachment step (45and A. L. Feire and T. Compton, manuscript in preparation). As acontrol, we used a protein termed gB-651 derived from a region ofgB that contains no recognizable receptor binding motifs. Next, weused �-amino acid oligomers (�-peptide) designed to mimic theheptad repeat region of gB that block HCMV entry at a step beforefusion. A �-peptide that does not block HCMV infection was usedas a control. The inhibitory �-peptide displays a high degree ofspecificity for HCMV, as it does not block entry of mouse CMVor HSV-1 (57). Pretreatment of cells with gB-DLD resulted in a92% decrease in virus infection as assessed by the HCMV-IE2GFP reporter virus (Fig. 6A). The gB-651 protein did not blockHCMV IE2-GFP infection (107% of control infection) (Fig. 6A).The �-peptide inhibitor, �-peptide (�), was preincubated with liveHCMV before infection of NHDFs and caused a 89% decrease inHCMV IE2-GFP infection; whereas the negative control peptide,�-peptide (�), had little effect, resulting in a 12% reduction invirus infection (Fig. 6A).
P56 protein accumulation was used as a measure of IFN acti-vation following treatment with entry inhibitors. Preincubation ofcells with gB-DLD prevented P56 induction after infection withUV-HCMV (33% of UV-HCMV alone). P56 levels were
4971The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
unaffected by the control gB-651 protein (Fig. 6B). Accumulationof P56 protein was not detected with gB-DLD or gB-651 pretreat-ments alone. Similarly, UV-HCMV-mediated induction of P56was diminished when virus was treated with �-peptide (�), but notwhen treated with �-peptide (�) (60% and 114% of UV-HCMValone, respectively) (Fig. 6B).
Next, the effect of HCMV entry inhibitors on NF-�B activationwas assessed. Pretreatment with gB-DLD or gB-651 resulted in
similar levels of I�B� degradation as UV-HCMV alone (14% and4% of the mock I�B� level, respectively, compared with 15% forHCMV alone) (Fig. 6C). The �-peptide (�) slightly inhibitedI�B� degradation and the � peptide (�) did not affect I�B� deg-radation by UV-HCMV (15% and 35% of mock I�B� level, re-spectively) (Fig. 6C). Interestingly, pretreatment with the gB-DLDpeptide alone resulted in I�B� degradation (11% of the mockI�B� level) (Fig. 6C). These data further support the hypothesis
FIGURE 4. Depletion of cellular cholesterol blocksHCMV-induced IFN responses. NHDF cells were pre-treated with 8 mM M�CD to deplete membrane choles-terol. Water-soluble cholesterol was incubated withcells at indicated concentrations. Cells were washedwith PBS and mock infected or infected with UV-HCMV (MOI � 1). A, Cells were fixed at 6 h postin-fection, and IRF3 localization was determined by indi-rect immunofluorescence. In the merge panels, nucleiwere stained with DAPI. B, RNA was isolated at 6 hpostinfection, and IFN-�, ISG56, and GAPDH tran-script levels were determined by RT-PCR. PCR prod-ucts were analyzed on a 1% agarose gel and visualizedby ethiduim bromide staining. C, Whole cell lysateswere prepared at 7 h postinfection, and P56 protein andactin levels were determined by Western blot analysis.Cells were treated with recombinant type I IFNs (1000U/ml) as a control. The levels of P56 protein were stan-dardized to actin levels, and values indicated (�) repre-sent the percentage of HCMV-infected cells under eachcondition. D, Cells were pretreated with M�CD and in-cubated with VSV (MOI � 150), Pam3CSK4 (40 �g/ml), or poly(I:C) (100 �g/ml). RNA was isolated at 6 h,and IFN-�, ISG56, IL-6, and GAPDH transcript levelswere determined by RT-PCR. The resulting PCR prod-ucts were analyzed on a 1% agarose gel and visualizedby ethidium bromide staining.
4972 DIFFERENTIAL INITIATION OF INNATE IMMUNE RESPONSES BY HCMV
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
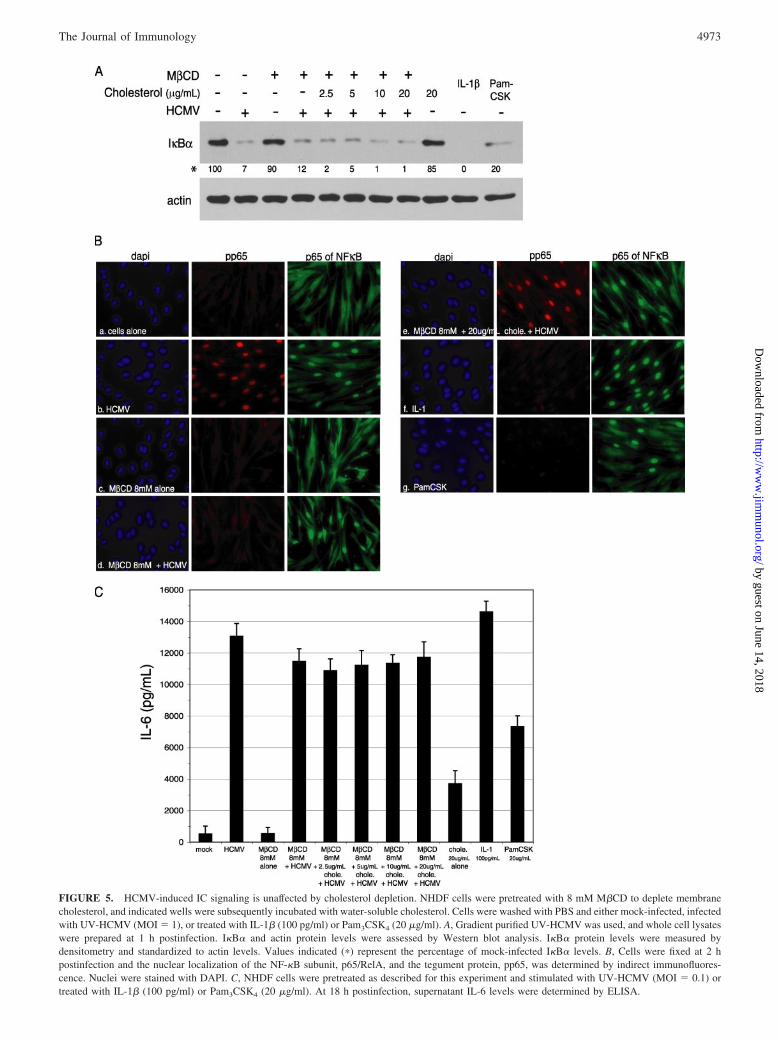
FIGURE 5. HCMV-induced IC signaling is unaffected by cholesterol depletion. NHDF cells were pretreated with 8 mM M�CD to deplete membranecholesterol, and indicated wells were subsequently incubated with water-soluble cholesterol. Cells were washed with PBS and either mock-infected, infectedwith UV-HCMV (MOI � 1), or treated with IL-1� (100 pg/ml) or Pam3CSK4 (20 �g/ml). A, Gradient purified UV-HCMV was used, and whole cell lysateswere prepared at 1 h postinfection. I�B� and actin protein levels were assessed by Western blot analysis. I�B� protein levels were measured bydensitometry and standardized to actin levels. Values indicated (�) represent the percentage of mock-infected I�B� levels. B, Cells were fixed at 2 hpostinfection and the nuclear localization of the NF-�B subunit, p65/RelA, and the tegument protein, pp65, was determined by indirect immunofluores-cence. Nuclei were stained with DAPI. C, NHDF cells were pretreated as described for this experiment and stimulated with UV-HCMV (MOI � 0.1) ortreated with IL-1� (100 pg/ml) or Pam3CSK4 (20 �g/ml). At 18 h postinfection, supernatant IL-6 levels were determined by ELISA.
4973The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
that UV-HCMV-induced innate immune responses are initiatedthrough distinct mechanisms.
DiscussionThe goal of this study was to define the innate immune pathwayselicited by HCMV and determine their relationship with virus en-try. These experiments were performed in subconfluent, serum-starved secondary strains of human fibroblasts. Similar levels of ICand IFN induction are observed in the presence or absence of se-rum following HCMV infection (data not shown) (90). However,serum starvation reduces the basal level of some cytokines. Inaddition, serum starvation may increase the sensitivity of cells toligands, such as IFN, because inhibitors have been found in serum(91). In addition, more robust innate immune signaling is observedfollowing infection with UV-HCMV because replication-depen-dent counter-measures are not used (7). In this study we show thatthe IFN response to HCMV is independent of TLR2 because anantiviral state is intact in cells expressing a dominant negativeconstruct of TLR2. We also did not find a role for intracellularlocalized TLRs, as inhibition of endosome acidification had noeffect on HCMV-induced IFN responses. It has been reported thatTLR3 can be found on both the cell surface and in intracellularcompartments in fibroblast cells (92). However, a signaling defec-tive, dominant negative construct of TLR3 also failed to blockHCMV-induced IFN signaling (data not shown). In addition, wefurther define the mechanisms of activation for the HCMV-in-duced IC and IFN responses. Dissociating lipid rafts using cho-lesterol depletion greatly diminished IFN signaling, but the IC re-sponse was intact. Interestingly, depletion of cholesterol alsoinhibited HCMV entry suggesting that the process of IFN induc-tion and virus entry are linked. These data also indicate that entryis not required for TLR2-dependent IC secretion. Finally, we usedtwo HCMV-specific entry inhibitors to further explore the rela-
tionship between IC and IFN responses in regard to HCMV infec-tion. Both entry inhibitors prevented IFN signaling, whereas the ICresponse was unaffected. Therefore we propose that the host cellhas developed a multifaceted innate immune defense mechanismto recognize and battle infection at the earliest points in the virusentry process.
Lipid rafts are multifunctional assemblies involved in diverseprocesses including signal transduction, endocytosis, and choles-terol trafficking (87). These microdomains are enriched in choles-terol and glycosphingolipids, which facilitate protein-protein andlipid-protein interactions on the cell surface (93). The only othertime that lipid rafts have been implicated in HCMV entry is in areport by Wang and colleagues (46). They found that lipid rafts areimportant for the coordination of signaling complexes within min-utes of HCMV entry. In addition, two proposed HCMV corecep-tors, �v�3 and EGFR, localize to lipid rafts along with the signal-ing molecules, Src and the p85 subunit of PI3K (46). The exactmechanism by which lipid rafts initiate signal transduction has notbeen defined, nor is the role of lipid rafts in TLR signaling fullyunderstood. TLR2 is reported to reside in lipid rafts on the apicalsurface of epithelial cells after bacterial infection (94), and clus-tering of TLR2 and GM1 ganglioside is detected in HEK293 cellsexpressing TLR2 and CD14 following treatment with lipoteichoicacid (95). Disruption of lipid rafts normally inhibits the function ofraft-associated receptors, therefore we hypothesized that HCMV-induced IC responses would be diminished upon cholesterol de-pletion. In contrast, we found that the IC response remains in-tact when lipid rafts are disrupted. Under conditions in whichHCMV entry is inhibited by cholesterol depletion, IC responsesare unaffected suggesting that entry is not required for the ini-tiation of IC secretion in response to HCMV. However, cho-lesterol depletion inhibits HCMV-mediated IFN signaling
FIGURE 6. HCMV fusion inhibitors block HCMV-induced IFN signaling but not IC secretion. NHDF cells were pretreated with gB-DLD (250 �g/ml)or gB-651 (250 �g/ml) for 1 h at 37°C. Cells were washed and either mock-infected or infected with live HCMV (A) or UV-HCMV (B and C) (MOI �1). HCMV virions (MOI � 1) were combined with 50 �M inhibitory �-peptide (�) or the control �-peptide (�) for 5 min and added to NHDF cells. A,At 18 h postinfection, the percentage of GFP-positive cells was determined by flow cytometry. No cellular toxicity was observed by propidium iodidestaining (data not shown). These results are representative of three independent experiments. B, Whole cell lysates were prepared at 7 h postinfection, andP56 and actin levels were determined by Western blot analysis. P56 levels were normalized to actin. Values indicated (�) represent the percentage ofinfected cells. C, Whole cell lysates were prepared at 3 h postinfection, and I�B� and actin protein levels were assessed by Western blot analysis. I�B�protein levels were measured by densitometry and standardized to actin levels. Values shown (�) represent the percentage of mock-infected I�B� levels.
4974 DIFFERENTIAL INITIATION OF INNATE IMMUNE RESPONSES BY HCMV
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

suggesting that it is a process that is independent of IC activa-tion and reliant on the events that occur during the entryprocess.
Next, we tested two inhibitors of HCMV entry. �1 integrin, aHCMV cellular receptor, was blocked using a small protein frag-ment of gB that contains the disintegrin like domain integrin bind-ing motif (A. L. Feire and T. Compton, manuscript in preparation).The gB-DLD effectively inhibited HCMV entry and IFN signalingbut not IC activation. This result suggests that the interaction be-tween �1 integrin and gB or subsequent steps after binding in theHCMV entry process are important determinants for IFN activa-tion. Integrins have been shown to have immunomodulatory rolesin promoting adaptive immune responses but to our knowledgethere has been no link to innate immune activation. In addition, thegB-DLD protein appears to be functioning as a ligand mimic andis able to activate IC signaling alone (Fig. 6C), suggesting thatgB-DLD protein may contain a PAMP from gB that is recognizedby TLR2. The inhibitory �-peptide, designed to mimic the fuso-genic heptad repeat region within gB was also used to impedeHCMV entry (57). It is hypothesized that the �-peptide (�) formsa stable 12-helical conformation that physically interacts with gBof HCMV and blocks a step before fusion. The � peptides aloneare not immunogenic (data not shown) (96, 97), likely due to theirunnatural amino acid composition. The � peptide (�) effectivelyblocked both HCMV entry and IFN signaling. Activation of the ICresponse was observed with both �-peptide treatments and slightvariations in the activation are likely due to alterations in thePAMP recognition of gB by TLR2.
These findings support a model in which virus binding promotesmultiple receptor-ligand interactions between viral envelope gly-coproteins and one or more cellular receptors. The IC responseappears to be activated irrespective of events needed for a produc-tive infection through outright sensing by TLR2. In contrast, theIFN response occurs by a postbinding or fusion-dependent mech-anism. We hypothesize that the congregation of stable dockingreceptors promotes the activation of cellular IFN responsesthrough one or more of the following events: 1) stable binding tocellular receptors; 2) the physical fusion event; or 3) delivery ofvirion contents into the cytoplasm. Our findings illustrate a cleardistinction in the initiation of IFN and IC signal transduction cas-cades by UV-HCMV. RelA translocates to the nucleus in the ab-sence of detectable pp65 localization, a measure of HCMV contentdelivery (Fig. 5B). However, type I IFN signaling is blocked insituations in which HCMV entry is impeded, suggesting a corre-lation between virus entry and IFN activation. The contribution ofNF-�B to HCMV-induced IFN signaling has not been addressed inthese experiments because it is activated before virus entry andIFN signaling. A recent report studying the contribution of NF-�Bto type I IFN signaling following RNA virus infection found thatthe NF-�B subunits p50, RelA, and cRel play a minor role (98).Although the initiation of IFN signaling by RNA viruses and DNAviruses appear to be distinct, the RNA virus VSV was unaffectedby cholesterol depletion (Fig. 4D). It is likely these viruses, at leastpartially, share downstream signaling pathways. An additionalstudy by Wietek et al. (99) found that IRF3-mediated activation ofthe IFN stimulation response element by TLR4, but not TLR3,required the RelA subunit of NF-�B, suggesting that two parallelpathways may exist. Additional studies examining the degree ofcross-talk between these two pathways following DNA virus in-fection are required. Because IL-8 levels are diminished but noteliminated in NHDF cells expressing the TLR2 dominant negativeconstruct (Fig. 1A), we cannot rule out that other mechanisms ofNF-�B activation coincide with later steps in the entry process.This model of innate immune activation is restricted to fibroblasts
because HCMV entry can occur through alternate pathways in dif-ferent cells types.
This report illustrates the existence of multiple mechanismsused by cells to detect HCMV and elicit distinct innate immuneresponses. In this study, we were able to dissect early events in theHCMV entry process and determine an order to the innate immuneinduction. It appears that the IC response is initiated via TLR2before IFN activation and virus entry. The immediate activation ofthe IC response allows the cell to get a jump-start on the beneficialeffects associated with NF-�B activation, such as the infiltration ofprofessional immune cells. Interestingly, HCMV may also benefitfrom rapid IC induction. HCMV contains multiple NF-�B ele-ments in its major immediate early promoter region, although theexact role of these elements in the initiation of viral gene expres-sion remains controversial (100, 101). Postbinding or fusion-de-pendent steps trigger the IFN response, promoting an antiviral stateto protect neighboring cells from the infection. The rapid recog-nition of HCMV by multiple pattern recognition receptors and theactivation of both branches of the innate immune response areadvantageous for the cellular defense mechanism. Increasing ourunderstanding of HCMV-induced innate immune activation allowsfor a more comprehensive view of virus-host interactions and willaid in the identification of new targets for therapeutic intervention.
AcknowledgmentsWe thank Ganes Sen (The Cleveland Clinic Foundation) for the P56 Ab,Deborah Spector (University of California, San Diego) for the HCMVAD169 IE2-GFP virus, John Yin (University of Wisconsin-Madison) forthe VSV, and Rob Kalejta (University of Wisconsin-Madison) for thepCGN-HA-pp65 plasmid. We also thank Sam Gellman and Emily Payne-English (University of Wisconsin-Madison) for synthesizing the � peptidesused in this study and the members of the T. Compton laboratory forcritical review of the manuscript.
DisclosuresThe authors have no financial conflict of interest.
References1. Ljungman, P. 1996. Cytomegalovirus infections in transplant patients. Scand
J. Infect. Dis. Suppl. 100: 59–63.2. Pass, R. F. 2001. Cytomegalovirus. In Fields Virology, Vol. 2.
D. M. Knipe and P. M. Howley, eds. Raven Press, New York, p. 2675.3. Ramsay, M. E., E. Miller, and C. S. Peckham. 1991. Outcome of confirmed
symptomatic congenital cytomegalovirus infection. Arch. Dis. Child. 66:1068–1069.
4. Alford, C. A., and W. J. Britt. 1993. Cytomegalovirus. In The Human Herpes-viruses. B. Roizman, R. J. Whitley, and C. Lopez, eds. Raven Press, New York,p. 227–255.
5. Hendrix, M. G., M. M. Salimans, C. P. van Boven, and C. A. Bruggeman. 1990.High prevalence of latently present cytomegalovirus in arterial walls of patientssuffering from grade III atherosclerosis. Am. J. Pathol. 136: 23–28.
6. Osame, K., Y. Takahashi, H. Takasawa, S. Watanabe, M. Kishimoto,K. Yasuda, Y. Kaburagi, K. Nakanishi, H. Kajio, and M. Noda. 2007. Rapid-onset type 1 diabetes associated with cytomegalovirus infection and islet auto-antibody synthesis. Intern Med. 46: 873–877.
7. Browne, E. P., B. Wing, D. Coleman, and T. Shenk. 2001. Altered cellularmRNA levels in human cytomegalovirus-infected fibroblasts: viral block to theaccumulation of antiviral mRNAs. J. Virol. 75: 12319–12330.
8. Simmen, K. A., J. Singh, B. G. Luukkonen, M. Lopper, A. Bittner, N. E. Miller,M. R. Jackson, T. Compton, and K. Fruh. 2001. Global modulation of cellulartranscription by human cytomegalovirus is initiated by viral glycoprotein B.Proc. Natl. Acad. Sci. USA 98: 7140–7145.
9. Zhu, H., J. P. Cong, and T. Shenk. 1997. Use of differential display analysis toassess the effect of human cytomegalovirus infection on the accumulation ofcellular RNAs: induction of interferon-responsive RNAs. Proc. Natl. Acad. Sci.USA 94: 13985–13990.
10. Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55: 255–281.11. Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber.
1998. How cells respond to interferons. Annu. Rev. Biochem. 67: 227–264.12. Laroux, F. S. 2004. Mechanisms of inflammation: the good, the bad and the
ugly. Front Biosci. 9: 3156–3162.13. Perry, A. K., G. Chen, D. Zheng, H. Tang, and G. Cheng. 2005. The host type
I interferon response to viral and bacterial infections. Cell Res. 15: 407–422.
4975The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

14. Theofilopoulos, A. N., R. Baccala, B. Beutler, and D. H. Kono. 2005. Type Iinterferons (�/�) in immunity and autoimmunity. Annu. Rev. Immunol. 23:307–336.
15. Hayden, M. S., A. P. West, and S. Ghosh. 2006. NF-�B and the immune re-sponse. Oncogene 25: 6758–6780.
16. Baldwin, A. S., Jr. 1996. The NF-�B and I�B proteins: new discoveries andinsights. Annu. Rev. Immunol. 14: 649–683.
17. Yurochko, A. D., and E. S. Huang. 1999. Human cytomegalovirus binding tohuman monocytes induces immunoregulatory gene expression. J. Immunol. 162:4806–4816.
18. Yurochko, A. D., T. F. Kowalik, S. M. Huong, and E. S. Huang. 1995. Humancytomegalovirus upregulates NF-�B activity by transactivating the NF-�Bp105/p50 and p65 promoters. J. Virol. 69: 5391–5400.
19. Takeda, K., and S. Akira. 2003. Toll receptors and pathogen resistance. CellMicrobiol. 5: 143–153.
20. Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innateimmunity. Cell 124: 783–801.
21. Hornung, V., S. Rothenfusser, S. Britsch, A. Krug, B. Jahrsdorfer, T. Giese,S. Endres, and G. Hartmann. 2002. Quantitative expression of toll-like receptor1–10 mRNA in cellular subsets of human peripheral blood mononuclear cellsand sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168: 4531–4537.
22. Zarember, K. A., and P. J. Godowski. 2002. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leu-kocytes in response to microbes, their products, and cytokines. J. Immunol. 168:554–561.
23. Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu.Rev. Immunol. 20: 197–216.
24. Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp,E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, andR. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediateresponse to respiratory syncytial virus. Nat. Immunol. 1: 398–401.
25. Burzyn, D., J. C. Rassa, D. Kim, I. Nepomnaschy, S. R. Ross, and I. Piazzon.2004. Toll-like receptor 4-dependent activation of dendritic cells by a retrovirus.J. Virol. 78: 576–584.
26. Compton, T., E. A. Kurt-Jones, K. W. Boehme, J. Belko, E. Latz,D. T. Golenbock, and R. W. Finberg. 2003. Human cytomegalovirus activatesinflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 77:4588–4596.
27. Boehme, K. W., M. Guerrero, and T. Compton. 2006. Human cytomegalovirusenvelope glycoproteins B and H are necessary for TLR2 activation in permissivecells. J. Immunol. 177: 7094–7102.
28. Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz,D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis. 2003. IKK� andTBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol.4: 491–496.
29. Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott.2003. Triggering the interferon antiviral response through an IKK-related path-way. Science 300: 1148–1151.
30. Servant, M. J., B. tenOever, C. LePage, L. Conti, S. Gessani, I. Julkunen, R. Lin,and J. Hiscott. 2001. Identification of distinct signaling pathways leading to thephosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276: 355–363.
31. Taniguchi, T., and A. Takaoka. 2002. The interferon-�/� system in antiviralresponses: a multimodal machinery of gene regulation by the IRF family oftranscription factors. Curr. Opin. Immunol. 14: 111–116.
32. Navarro, L., K. Mowen, S. Rodems, B. Weaver, N. Reich, D. Spector, andM. David. 1998. Cytomegalovirus activates interferon immediate-early responsegene expression and an interferon regulatory factor 3-containing interferon-stimulated response element-binding complex. Mol. Cell Biol. 18: 3796–3802.
33. Preston, C. M., A. N. Harman, and M. J. Nicholl. 2001. Activation of interferonresponse factor-3 in human cells infected with herpes simplex virus type 1 orhuman cytomegalovirus. J. Virol. 75: 8909–8916.
34. Boehme, K. W., J. Singh, S. T. Perry, and T. Compton. 2004. Human cyto-megalovirus elicits a coordinated cellular antiviral response via envelope gly-coprotein B. J. Virol. 78: 1202–1211.
35. DeFilippis, V. R., B. Robinson, T. M. Keck, S. G. Hansen, J. A. Nelson, andK. J. Fruh. 2006. Interferon regulatory factor 3 is necessary for induction ofantiviral genes during human cytomegalovirus infection. J. Virol. 80:1032–1037.
36. Netterwald, J. R., T. R. Jones, W. J. Britt, S. J. Yang, I. P. McCrone, and H. Zhu.2004. Postattachment events associated with viral entry are necessary for in-duction of interferon-stimulated genes by human cytomegalovirus. J. Virol. 78:6688–6691.
37. Paulus, C., S. Krauss, and M. Nevels. 2006. A human cytomegalovirus antag-onist of type I IFN-dependent signal transducer and activator of transcriptionsignaling. Proc. Natl. Acad. Sci. USA 103: 3840–3845.
38. Taylor, R. T., and W. A. Bresnahan. 2006. Human cytomegalovirus IE86 at-tenuates virus- and tumor necrosis factor �-induced NF�B-dependent gene ex-pression. J. Virol. 80: 10763–10771.
39. Penfold, M. E., D. J. Dairaghi, G. M. Duke, N. Saederup, E. S. Mocarski,G. W. Kemble, and T. J. Schall. 1999. Cytomegalovirus encodes a potent �chemokine. Proc. Natl. Acad. Sci. USA 96: 9839–9844.
40. Wang, D., W. Bresnahan, and T. Shenk. 2004. Human cytomegalovirus encodesa highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. USA 101:16642–16647.
41. Stropes, M. P., and W. E. Miller. 2004. Signaling and regulation of G-proteincoupled receptors encoded by cytomegaloviruses. Biochem. Cell Biol. 82:636–642.
42. Browne, H., G. Smith, S. Beck, and T. Minson. 1990. A complex between theMHC class I homologue encoded by human cytomegalovirus and �2 micro-globulin. Nature 347: 770–772.
43. Leong, C. C., T. L. Chapman, P. J. Bjorkman, D. Formankova, E. S. Mocarski,J. H. Phillips, and L. L. Lanier. 1998. Modulation of natural killer cell cyto-toxicity in human cytomegalovirus infection: the role of endogenous class Imajor histocompatibility complex and a viral class I homolog. J. Exp. Med. 187:1681–1687.
44. Compton, T., D. M. Nowlin, and N. R. Cooper. 1993. Initiation of human cy-tomegalovirus infection requires initial interaction with cell surface heparansulfate. Virology 193: 834–841.
45. Feire, A. L., H. Koss, and T. Compton. 2004. Cellular integrins function as entryreceptors for human cytomegalovirus via a highly conserved disintegrin-likedomain. Proc. Natl. Acad. Sci. USA 101: 15470–15475.
46. Wang, X., D. Y. Huang, S. M. Huong, and E. S. Huang. 2005. Integrin �V�3 isa coreceptor for human cytomegalovirus. Nat. Med. 11: 515–521.
47. Wang, X., S. M. Huong, M. L. Chiu, N. Raab-Traub, and E. S. Huang. 2003.Epidermal growth factor receptor is a cellular receptor for human cytomegalo-virus. Nature 424: 456–461.
48. Isaacson, M. K., A. L. Feire, and T. Compton. 2007. Epidermal growth factorreceptor is not required for human cytomegalovirus entry or signaling. J. Virol.81: 6241–6247.
49. Cobbs, C. S., L. Soroceanu, S. Denham, W. Zhang, W. J. Britt, R. Pieper, andM. H. Kraus. 2007. Human cytomegalovirus induces cellular tyrosine kinasesignaling and promotes glioma cell invasiveness. J. Neurooncol. 85: 271–280.
50. Compton, T., R. R. Nepomuceno, and D. M. Nowlin. 1992. Human cytomeg-alovirus penetrates host cells by pH-independent fusion at the cell surface. Vi-rology 191: 387–395.
51. Ryckman, B. J., M. A. Jarvis, D. D. Drummond, J. A. Nelson, andD. C. Johnson. 2006. Human cytomegalovirus entry into epithelial and endo-thelial cells depends on genes UL128 to UL150 and occurs by endocytosis andlow-pH fusion. J. Virol. 80: 710–722.
52. Keay, S., and B. Baldwin. 1991. Anti-idiotype antibodies that mimic gp86 ofhuman cytomegalovirus inhibit viral fusion but not attachment. J. Virol. 65:5124–5128.
53. Milne, R. S., D. A. Paterson, and J. C. Booth. 1998. Human cytomegalovirusglycoprotein H/glycoprotein L complex modulates fusion-from-without. J. Gen.Virol. 79(Pt. 4): 855–865.
54. Compton, T. 1993. An immortalized human fibroblast cell line is permissive forhuman cytomegalovirus infection. J. Virol. 67: 3644–3648.
55. Sanchez, V., C. L. Clark, J. Y. Yen, R. Dwarakanath, and D. H. Spector. 2002.Viable human cytomegalovirus recombinant virus with an internal deletion ofthe IE2 86 gene affects late stages of viral replication. J. Virol. 76: 2973–2989.
56. Guo, J., K. L. Peters, and G. C. Sen. 2000. Induction of the human protein P56by interferon, double-stranded RNA, or virus infection. Virology 267: 209–219.
57. English, E. P., R. S. Chumanov, S. H. Gellman, and T. Compton. 2006. Rationaldevelopment of �-peptide inhibitors of human cytomegalovirus entry. J. Biol.Chem. 281: 2661–2667.
58. Highlander, S. L., S. L. Sutherland, P. J. Gage, D. C. Johnson, M. Levine, andJ. C. Glorioso. 1987. Neutralizing monoclonal antibodies specific for herpessimplex virus glycoprotein D inhibit virus penetration. J. Virol. 61: 3356–3364.
59. Gault, E., Y. Michel, A. Dehee, C. Belabani, J. C. Nicolas, and A. Garbarg-Chenon. 2001. Quantification of human cytomegalovirus DNA by real-timePCR. J. Clin. Microbiol. 39: 772–775.
60. Baldick, C. J., Jr., A. Marchini, C. E. Patterson, and T. Shenk. 1997. Humancytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity ofviral DNA and accelerates the infectious cycle. J. Virol. 71: 4400–4408.
61. Boyle, K. A., R. L. Pietropaolo, and T. Compton. 1999. Engagement of thecellular receptor for glycoprotein B of human cytomegalovirus activates theinterferon-responsive pathway. Mol. Cell Biol. 19: 3607–3613.
62. Nagai, K., and H. C. Thogersen. 1987. Synthesis and sequence-specific prote-olysis of hybrid proteins produced in Escherichia coli. Methods Enzymol. 153:461–481.
63. Kurt-Jones, E. A., M. Chan, S. Zhou, J. Wang, G. Reed, R. Bronson,M. M. Arnold, D. M. Knipe, and R. W. Finberg. 2004. Herpes simplex virus 1interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc.Natl. Acad. Sci. USA 101: 1315–1320.
64. Wang, J. P., E. A. Kurt-Jones, O. S. Shin, M. D. Manchak, M. J. Levin, andR. W. Finberg. 2005. Varicella-zoster virus activates inflammatory cytokines inhuman monocytes and macrophages via Toll-like receptor 2. J. Virol. 79:12658–12666.
65. Toshchakov, V., B. W. Jones, P. Y. Perera, K. Thomas, M. J. Cody, S. Zhang,B. R. Williams, J. Major, T. A. Hamilton, M. J. Fenton, and S. N. Vogel. 2002.TLR4, but not TLR2, mediates IFN-�-induced STAT1�/�-dependent gene ex-pression in macrophages. Nat. Immunol. 3: 392–398.
66. Ahmad-Nejad, P., H. Hacker, M. Rutz, S. Bauer, R. M. Vabulas, andH. Wagner. 2002. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 32:1958–1968.
67. Latz, E., A. Schoenemeyer, A. Visintin, K. A. Fitzgerald, B. G. Monks,C. F. Knetter, E. Lien, N. J. Nilsen, T. Espevik, and D. T. Golenbock. 2004.TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat.Immunol. 5: 190–198.
68. Nishiya, T., and A. L. DeFranco. 2004. Ligand-regulated chimeric receptorapproach reveals distinctive subcellular localization and signaling properties ofthe toll-like receptors. J. Biol. Chem. 279: 19008–19017.
4976 DIFFERENTIAL INITIATION OF INNATE IMMUNE RESPONSES BY HCMV
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

69. Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, and E. S. C. Reis. 2004. Innateantiviral responses by means of TLR7-mediated recognition of single-strandedRNA. Science 303: 1529–1531.
70. Hacker, H., H. Mischak, T. Miethke, S. Liptay, R. Schmid, T. Sparwasser,K. Heeg, G. B. Lipford, and H. Wagner. 1998. CpG-DNA-specific activation ofantigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 17: 6230–6240.
71. de Bouteiller, O., E. Merck, U. A. Hasan, S. Hubac, B. Benguigui, G. Trinchieri,E. E. Bates, and C. Caux. 2005. Recognition of double-stranded RNA by humantoll-like receptor 3 and downstream receptor signaling requires multimerizationand an acidic pH. J. Biol. Chem. 280: 38133–38145.
72. Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsu-moto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-likereceptor recognizes bacterial DNA. Nature 408: 740–745.
73. Krug, A., G. D. Luker, W. Barchet, D. A. Leib, S. Akira, and M. Colonna. 2004.Herpes simplex virus type 1 activates murine natural interferon-producing cellsthrough toll-like receptor 9. Blood 103: 1433–1437.
74. Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-likereceptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoiddendritic cells. J. Exp. Med. 198: 513–520.
75. Krug, A., A. R. French, W. Barchet, J. A. Fischer, A. Dzionek, J. T. Pingel,M. M. Orihuela, S. Akira, W. M. Yokoyama, and M. Colonna. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cyto-kine responses that activate antiviral NK cell function. Immunity 21: 107–119.
76. Yoshimori, T., A. Yamamoto, Y. Moriyama, M. Futai, and Y. Tashiro. 1991.Bafilomycin A1, a specific inhibitor of vacuolar-type H�-ATPase, inhibits acid-ification and protein degradation in lysosomes of cultured cells. J. Biol. Chem.266: 17707–17712.
77. tenOever, B. R., S. Sharma, W. Zou, Q. Sun, N. Grandvaux, I. Julkunen,H. Hemmi, M. Yamamoto, S. Akira, W. C. Yeh, R. Lin, and J. Hiscott. 2004.Activation of TBK1 and IKKvar� kinases by vesicular stomatitis virus infectionand the role of viral ribonucleoprotein in the development of interferon antiviralimmunity. J. Virol. 78: 10636–10649.
78. Superti, F., L. Seganti, F. M. Ruggeri, A. Tinari, G. Donelli, and N. Orsi. 1987.Entry pathway of vesicular stomatitis virus into different host cells. J. Gen.Virol. 68(Pt. 2): 387–399.
79. Matlin, K. S., H. Reggio, A. Helenius, and K. Simons. 1982. Pathway of ve-sicular stomatitis virus entry leading to infection. J. Mol. Biol. 156: 609–631.
80. Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature387: 569–572.
81. Laude, A. J., and I. A. Prior. 2004. Plasma membrane microdomains: organi-zation, function and trafficking (Review). Mol. Membr. Biol. 21: 193–205.
82. Bender, F. C., J. C. Whitbeck, M. Ponce de Leon, H. Lou, R. J. Eisenberg, andG. H. Cohen. 2003. Specific association of glycoprotein B with lipid rafts duringherpes simplex virus entry. J. Virol. 77: 9542–9552.
83. Chung, C. S., C. Y. Huang, and W. Chang. 2005. Vaccinia virus penetrationrequires cholesterol and results in specific viral envelope proteins associatedwith lipid rafts. J. Virol. 79: 1623–1634.
84. Choi, K. S., H. Aizaki, and M. M. Lai. 2005. Murine coronavirus requires lipidrafts for virus entry and cell-cell fusion but not for virus release. J. Virol. 79:9862–9871.
85. Pietiainen, V., V. Marjomaki, P. Upla, L. Pelkmans, A. Helenius, andT. Hyypia. 2004. Echovirus 1 endocytosis into caveosomes requires lipid rafts,dynamin II, and signaling events. Mol. Biol. Cell 15: 4911–4925.
86. Pelkmans, L., D. Puntener, and A. Helenius. 2002. Local actin polymerizationand dynamin recruitment in SV40-induced internalization of caveolae. Science296: 535–539.
87. Pike, L. J. 2004. Lipid rafts: heterogeneity on the high seas. Biochem. J. 378:281–292.
88. Rodal, S. K., G. Skretting, O. Garred, F. Vilhardt, B. van Deurs, and K. Sandvig.1999. Extraction of cholesterol with methyl-�-cyclodextrin perturbs formationof clathrin-coated endocytic vesicles. Mol. Biol. Cell 10: 961–974.
89. Malek, S., Y. Chen, T. Huxford, and G. Ghosh. 2001. I�B�, but not I�B�,functions as a classical cytoplasmic inhibitor of NF-�B dimers by masking bothNF-�B nuclear localization sequences in resting cells. J. Biol. Chem. 276:45225–45235.
90. Gealy, C., M. Denson, C. Humphreys, B. McSharry, G. Wilkinson, andR. Caswell. 2005. Posttranscriptional suppression of interleukin-6 production byhuman cytomegalovirus. J. Virol. 79: 472–485.
91. Rossman, T. G., and J. Vilcek. 1970. Blocking of interferon action by a com-ponent of normal serum. Arch. Gesamte Virusforsch. 31: 18–27.
92. Matsumoto, M., S. Kikkawa, M. Kohase, K. Miyake, and T. Seya. 2002. Es-tablishment of a monoclonal antibody against human Toll-like receptor 3 thatblocks double-stranded RNA-mediated signaling. Biochem. Biophys. Res. Com-mun. 293: 1364–1369.
93. Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev.Mol. Cell Biol. 1: 31–39.
94. Soong, G., B. Reddy, S. Sokol, R. Adamo, and A. Prince. 2004. TLR2 is mo-bilized into an apical lipid raft receptor complex to signal infection in airwayepithelial cells. J. Clin. Invest. 113: 1482–1489.
95. Triantafilou, M., M. Manukyan, A. Mackie, S. Morath, T. Hartung, H. Heine,and K. Triantafilou. 2004. Lipoteichoic acid and toll-like receptor 2 internal-ization and targeting to the Golgi are lipid raft-dependent. J. Biol. Chem. 279:40882–40889.
96. Arvidsson, P. I., N. S. Ryder, H. M. Weiss, G. Gross, O. Kretz, R. Woessner,and D. Seebach. 2003. Antibiotic and hemolytic activity of a �2/�3 peptidecapable of folding into a 12/10-helical secondary structure. Chembiochem. 4:1345–1347.
97. Patch, J. A., and A. E. Barron. 2002. Mimicry of bioactive peptides via non-natural, sequence-specific peptidomimetic oligomers. Curr. Opin. Chem Biol. 6:872–877.
98. Wang, X., S. Hussain, E. J. Wang, X. Wang, M. O. Li, A. Garcıa-Sastre, andA. A. Beg. 2007. Lack of essential role of NF-�B p50, RelA, and cRel subunitsin virus-induced type 1 IFN expression. J. Immunol. 178: 6770–6776.
99. Wietek, C., S. M. Miggin, C. A. Jefferies, and L. A. O’Neill. 2003. Interferonregulatory factor-3-mediated activation of the interferon-sensitive response el-ement by Toll-like receptor (TLR) 4 but not TLR3 requires the p65 subunit ofNF-�. J. Biol. Chem. 278: 50923–50931.
100. DeMeritt, I. B., L. E. Milford, and A. D. Yurochko. 2004. Activation of theNF-�B pathway in human cytomegalovirus-infected cells is necessary for effi-cient transactivation of the major immediate-early promoter. J. Virol. 78:4498–4507.
101. Gustems, M., E. Borst, C. A. Benedict, C. Perez, M. Messerle, P. Ghazal, andA. Angulo. 2006. Regulation of the transcription and replication cycle of humancytomegalovirus is insensitive to genetic elimination of the cognate NF-�Bbinding sites in the enhancer. J. Virol. 80: 9899–9904.
4977The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Related Documents











