Development of Defined Media for the Serum-Free Expansion of Primary Keratinocytes and Human Embryonic Stem Cells SEAN RICHARDS, Ph.D., DAVID LEAVESLEY, Ph.D., GEMMA TOPPING, Hons., and ZEE UPTON, Ph.D. ABSTRACT Primary keratinocyte (Kc) cells and human embryonic stem (hES) cells are routinely propagated on a mouse fibroblast feeder layer in media containing fetal bovine serum or other nondefined factors. One disadvantage of using these nondefined factors is that they may inadvertently contaminate the culture system with infectious agents; thus, there remains a need to develop safe culture conditions free from poorly defined and/or animal products. Our laboratory has discovered that growth factors (GFs) and vitronectin (VN) can bind to each other resulting in synergistic short-term functional effects in several cell types. The aim of the current study was to determine whether primary Kc and hES cells can be established and serially propagated serum-free using medium containing VN, insulin-like growth factor-I, and insulin- like growth factor binding protein-3 (VN:GF). Here we demonstrate that primary Kc cells can be isolated, established, serially propagated, and re-form an epidermal layer using the VN:GF combination. Ad- ditionally, cell proliferation studies indicate that the Kcs proliferate using the VN:GF combination at a rate comparable to cells grown using serum. Similarly, we verified that this VN:GF combination could be employed for the serial propagation of hES cells. Importantly, both the Kc and hES cells retain their undifferentiated phenotype when cultured using the VN:GF combinations as a serum-free medium for up to 4 passages for Kc and at least 10 passages for hES cells as indicated by the expression of a range of cell surface markers. This study demonstrates that the novel, fully defined VN:GF medium is a viable alter- native to media containing serum and highlights the potential of this technology for generating ther- apeutically viable cells and tissues. INTRODUCTION T O DATE, MOST IN VITRO mammalian cell culture methods have required the use of nondefined or animal-derived components, such as fibroblast feeder cells, serum, and/or serum-replacement factors. Certainly, this is the case for the successful propagation of primary keratinocytes (Kcs) and human embryonic stem (hES) cells. Rheinwald and Green first reported in 1975 the successful propagation of human Kc using an irradiated mouse fibroblast feeder cell layer and animal serum, 1 and this was later successfully applied to the propagation of hES cells in 1998 using a similar methodol- ogy. 2 The successful in vitro propagation of these cells generated significant interest due to the therapeutic potential that these cells possess. However, the reliance of these cells on undefined foreign components for their propagation, that is, feeder cells and serum, carries the potential risk of exposing the therapeutic cells to animal pathologies such as bovine spongiform encephalopathy 3 or human-derived pathologies. More recently, the addition of these animal components has been demonstrated to introduce immuno- genic agents into hES cells (e.g., N-glycolylneuraminic acid, Neu5Ac). 4,5 Clearly, improved cell culture technologies need to be developed to eliminate the risk of contaminating Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, Australia. TISSUE ENGINEERING: Part C Volume 14, Number 3, 2008 # Mary Ann Liebert, Inc. DOI: 10.1089/ten.tec.2007.0428 1 (page numbers are temporary)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Development of Defined Media for the Serum-Free Expansion
of Primary Keratinocytes and Human Embryonic Stem Cells
SEAN RICHARDS, Ph.D., DAVID LEAVESLEY, Ph.D., GEMMA TOPPING, Hons.,and ZEE UPTON, Ph.D.
ABSTRACT
Primary keratinocyte (Kc) cells and human embryonic stem (hES) cells are routinely propagated on amouse fibroblast feeder layer in media containing fetal bovine serum or other nondefined factors. Onedisadvantage of using these nondefined factors is that they may inadvertently contaminate the culturesystem with infectious agents; thus, there remains a need to develop safe culture conditions free frompoorly defined and/or animal products. Our laboratory has discovered that growth factors (GFs) andvitronectin (VN) can bind to each other resulting in synergistic short-term functional effects in several celltypes. The aim of the current study was to determine whether primary Kc and hES cells can be establishedand serially propagated serum-free usingmedium containing VN, insulin-like growth factor-I, and insulin-like growth factor binding protein-3 (VN:GF). Here we demonstrate that primary Kc cells can be isolated,established, serially propagated, and re-form an epidermal layer using the VN:GF combination. Ad-ditionally, cell proliferation studies indicate that theKcs proliferate using the VN:GF combination at a ratecomparable to cells grown using serum. Similarly, we verified that this VN:GF combination could beemployed for the serial propagation of hES cells. Importantly, both the Kc and hES cells retain theirundifferentiated phenotype when cultured using the VN:GF combinations as a serum-free medium for upto 4 passages for Kc and at least 10 passages for hES cells as indicated by the expression of a range of cellsurface markers. This study demonstrates that the novel, fully defined VN:GF medium is a viable alter-native to media containing serum and highlights the potential of this technology for generating ther-apeutically viable cells and tissues.
INTRODUCTION
TO DATE, MOST IN VITRO mammalian cell culture methods
have required the use of nondefined or animal-derived
components, such as fibroblast feeder cells, serum, and/or
serum-replacement factors. Certainly, this is the case for the
successful propagation of primary keratinocytes (Kcs) and
human embryonic stem (hES) cells. Rheinwald and Green
first reported in 1975 the successful propagation of human
Kc using an irradiated mouse fibroblast feeder cell layer and
animal serum,1 and this was later successfully applied to the
propagation of hES cells in 1998 using a similar methodol-
ogy.2 The successful in vitro propagation of these cells
generated significant interest due to the therapeutic potential
that these cells possess. However, the reliance of these cells
on undefined foreign components for their propagation, that
is, feeder cells and serum, carries the potential risk of
exposing the therapeutic cells to animal pathologies such
as bovine spongiform encephalopathy3 or human-derived
pathologies. More recently, the addition of these animal
components has been demonstrated to introduce immuno-
genic agents into hES cells (e.g., N-glycolylneuraminic acid,
Neu5Ac).4,5 Clearly, improved cell culture technologies
need to be developed to eliminate the risk of contaminating
Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, Australia.
TISSUE ENGINEERING: Part CVolume 14, Number 3, 2008# Mary Ann Liebert, Inc.DOI: 10.1089/ten.tec.2007.0428
1 (page numbers are temporary)

the cultured cells, whilst at the same time providing the
necessary conditions for their in vitro expansion.
Currently there are serum-free alternatives for the growth
of Kc and hES cells, for example, defined keratinocyte
medium (DKM) and knock-out serum replacement (KSR)
(Invitrogen, Mulgrave, VIC, Australia), respectively. These
products have been demonstrated to support the serum-free,
and in the case of DKM, feeder cell–free propagation of cells.
However, these products still require, more often than not,
the inclusion of undefined human and/or animal products,
such as purified human serum albumin (HSA) or bovine pi-
tuitary extract (BPE), for the long-term survival of the
cells.6,7 Further, cells grown using these products have pre-
viously been isolated using serum and required plating at
high cell-seeding densities. The problem with isolating cells
in the presence of serum is that they can still be potentially
contaminated at this stage, thereby, removing any advan-
tage gained through the use of serum alternatives in the
subsequent steps. Moreover, there are also problems asso-
ciated with the requirement for high seeding densities since
this may not be possible or practical in a clinical setting, for
example, with patients who have large surface area burns and
thus limited biopsy sites. Similarly, with hES cells, only
small cell numbers are available in the initial isolation from
the blastocyst. For this reason, the culture conditions for
these cells often contain fibroblast feeder cells to support
their growth. To date, no serum-free and/or feeder cell–free
technology has provided these cell types with a completely
defined, safe, and viable culture system.
More recently, researchers have been looking to the ex-
tracellular environment as a means to remove serum and fi-
broblasts from the culture of these cells. Extracellular matrix
(ECM) proteins, such as laminin, collagen, and fibronectin,
have been demonstrated to provide a favorable, feeder cell–
free environment for the attachment, proliferation, and mi-
gration of Kc and hES cells.8,9 While this approach has
provided a serum-free and feeder cell–free culture environ-
ment, the addition of large amounts of purified HSA, growth
factors (GFs)/mitogens, and certain xeno-derived products,
such as BPE, are required.6,10,11 Thus, while these novel
approachesaddress thepotentialproblemsofpathogentransfer
that exist through the use of serum and feeder cells, they do
not avoid the problems associated with poorly defined and
uncharacterized compounds such as purified BPE and HSA.
Due to the complexity involved in trying to remove the
fibroblast feeder cells from these culture systems, our la-
boratory initially focused its efforts on the development of
growth promoting ECM:GF protein complexes for the
in vitro serum-free expansion of Kcs for skin grafting ap-
plications.12 This new technology is based on the finding that
a synergistic effect occurs between GFs and a specific ECM
protein called vitronectin (VN).13–16 This has led to the
development of novel dimeric, trimeric, and multimeric
growth-promoting complexes incorporating GFs, such as
insulin-like growth factors (IGFs) and insulin-like growth
factor binding proteins (IGFBPs), in conjunction with VN
(VN:GF). Further, the addition of these complexes to de-
fined media has been demonstrated to stimulate short-term
migration and proliferation in a range of cells, including
adult skin- and corneal-derived epithelial cells.13,16 In ad-
dition, this technology has proved successful in removing
serum from the serial expansion of Kc; however, there is a
need for a feeder cell layer in the culture system. Moreover,
the approach reported by Dawson et al. still used serum in
the initial isolation step and involved binding the ECM:GF
complex to the culture dishes.12 Nevertheless, encouraged
by these results and as reported herein, we have pursued
further studies examining whether the VN:GF complex
technology could be improved to create a serum-free med-
ium, rather than VN:GF-bound coatings, that would allow
both the isolation and serial propagation of primary Kc.
Further, we hypothesized that this technology could be
successfully translated to support the long-term cultivation
of hES cells. We report here the isolation, long-term survi-
val, and biological responses of primary Kc cells derived
from adult human skin when isolated and grown in the pre-
sence of the VN:GF serum-free medium. Additionally, we
report the successful long-term cultivation of hES cells ex-
panded in the VN:GF-containing medium.
MATERIALS AND METHODS
Ethics and material collection
Ethics approval to conduct this project was received from
the Queensland University of Technology (QUT) Human
Research Ethics Committee (HREC) for the hES cell ex-
periments (ID: 2943H) and the HRECs of QUT and the St.
Andrews and Wesley Hospitals, Brisbane, Australia, for
aspects related to Kc cells (ID: 3673H). Skin was obtained
from consenting patients undergoing breast reductions and
abdominoplasties. hES cell lines were obtained in strict
adherence to the state, federal, and ‘‘National Health and
Medical Research Council’’ (NHMRC) guidelines regarding
the conduct of research using hES cells. Additionally, in-
formed donor consent was obtained by the investigators who
initially derived the hES cell lines utilized.
Isolation of primary Kcs
Primary Kcs were isolated from split-thickness skin
biopsies obtained from breast reductions and abdomino-
plastiesasdescribedbyGoberdhanetal.17Briefly,thismethod
involved dissecting the skin biopsy into 0.5 cm2 pieces fol-
lowed by a series of antibiotic wash steps. The skin was then
incubated in 0.125% trypsin (Invitrogen) overnight at 48C.The isolation step here differs significantly from that pre-
viously reported, in which all steps were conducted in
serum-free conditions.
2 RICHARDS ET AL.

Kcs cultured in serum-containing medium
The freshly isolated Kcs were then cultured on gamma-
irradiated (two doses of 25 Gy) (Australian Red Cross Blood
Service, Brisbane, QLD, Australia) mouse i3T3 cells
(ATCC# CCL-92) using Dulbecco’s modified Eagle’s me-
dium (DMEM)/HAMS medium (Invitrogen) containing
0.4 mg/mL hydrocortisone, 10 mg/mL epidermal growth
factor (EGF) (Sigma-Aldrich, Castle Hill, NSW, Australia),
0.1 nM cholera toxin, 1.8�10�4M adenine, 2�10�7 M
triiodo-L-thyronine, 5 mg/mL insulin, 5 mg/mL transferring,
2�10�3M glutamine (Invitrogen), 1000 IU/mL penicillin/
1000 mg/mL streptomycin (Invitrogen), and 10% fetal bo-
vine serum (FBS) (Trace Scientific, Noble Park, VIC,
Australia). The cultures were established at a density of
1�106 cells/25 cm2 flask and incubated at 378C in 5% car-
bon dioxide (CO2), with media changes every third day. The
cells were seeded at 2.5�105 cells per 25 cm2 flask for sub-
sequent passages.
Kcs cultured in VN:GF medium
Kcs were cultured on irradiated i3T3 cells in DMEM/
HAMS medium (Invitrogen) containing 0.4 mg/mL hydro-
cortisone, 0.1 nM cholera toxin, 1.8�10�4M adenine, 2�10�7M triiodo-L-thyronine, 5 mg/mL transferrin, 2�10�3M
glutamine (Invitrogen), and 1000 IU/mL penicillin/1000mg/mL
streptomycin (Invitrogen). The serum-replacement component
of the VN:GF culture medium included 0.6mg/mL of highly
purified human VN (Promega, Annandale, NSW, Australia),
0.6mg/mL recombinant mutant human IGFBP-3 (N109D)
(Auspep, Parkville, VIC, Australia), 0.2mg/mL recombinant
human IGF-I (Novozymes, Adelaide, SA, Australia), and
0.2mg/mL recombinant human EGF (Invitrogen). The Kcs
isolated from skin were seeded at an initial density of 1�106
cells per 25 cm2 flask and incubated at 378C in 5% CO2, and
were refed every third day with media containing half the
amount of the VN:GF supplement described above. The cells
were seeded at 2.5�105 cells per 25 cm2 flask for the sub-
sequent passages.
DKM culture
Kcs were also grown in a commercially available serum-
free Kc medium developed for the in vitro propagation of Kc
cells. The DKM (Invitrogen) evaluated includes animal and
human products; however, these are not clearly defined by
the manufacturer. The DKM cultures were set up in both the
presence and absence of irradiated i3T3 feeder cells. Feeder
cell–free cultures were also established, since the manu-
facturer’s protocol indicates that this medium is suitable for
feeder cell–free and serum-free culture of Kc. The Kcs were
seeded at an initial density of 1�106 cells in 25 cm2 flasks
and incubated at 378C in 5% CO2, with media changes every
third day. The cells were seeded at 2.5�105 cells per 25 cm2
flask for the subsequent passages.
Kc proliferation assays
Proliferation was measured using manual cell counting
and involved propagating the Kc in 25 cm2 flasks. The Kcs
were then removed from the flasks using 0.05% trypsin/
EDTA (Invitrogen) and counted using a hemocytometer.
Proliferation assays were conducted in triplicate, and all
experiments were repeated through four passages (P1–P4),
where P1 cells were Kc freshly isolated from patient skin.
Three different patient samples were used to conduct this
study. The Tukey’s and Student’s t-test were used to analyze
the proliferation data. A level of 0.05 was determined to be
statistically significant.
Kc immunohistochemistry
Immunohistochemistry was performed at several differ-
ent passages to ensure that the Kc had maintained an un-
differentiated phenotype. Mouse antibodies to undiluted
keratin 6 (present in hyperproliferative skin), 1:10 dilution
of keratin 14 (present in basal cells), and a 1:20 dilution of
keratin 1/10/11 (present in more differentiated, suprabasal
cells) (Research Diagnostics, Flanders, CA) were used in
this study. Cells grown in the various treatments were in-
cubated in their respective media treatments for 2 days fol-
lowing seeding in 96-well plates. Media were aspirated from
the plates, followed by two washes in phosphate-buffered
saline (PBS). All treatments were incubated in extraction
buffer (0.5% triton X-100, 0.1M pipes buffer, 5mM mag-
nesium chloride, and 1mM ethylene glycol tetra acetic acid
(EGTA) at pH 7.0) for 2min. This was then followed by a
10-min incubation in a fixation buffer (2% paraformalde-
hyde in extraction buffer). Treatments were then blocked for
1 h in PBS/5% normal goat serum (NGS). The primary anti-
bodies were incubated for an hour in PBS/1% NGS. A series
of 3�1min washing steps were carried out, followed by a 1-
h incubation with a 1/100 dilution of alexa-488 goat anti-
mouse IgG antibody (Molecular Probes, Eugene, OR).
Secondary antibody controls were also examined in these
experiments to ensure that no nonspecific binding had
occurred.
Preparation of dermal and skin equivalent
A three-dimensional human skin equivalent (HSE) model
was prepared as previously described.12 Briefly, sterile
stainless steel rings (Aix Scientifics, Aachen, Germany) with
a 7-mm silicone washer base were placed in the culture wells
on top of decellularized dermis (DED) pieces. P4 Kcs, cul-
tured in either serum or VN:GF medium with i3T3, were
placed into the rings at a concentration of 1.9�104 cells/ring.
Subsequently, the rings were removed after 3 days of cul-
ture, and the dermis plus cells (composite) were then placed
onto stainless steel grids at the air–liquid interface for 5 days
in six-well culture plates.
SERUM-FREE CULTURE FOR KERATINOCYTE AND HES CELLS 3

Immunohistochemistry and histology
of skin composites
The skin composites were washed in PBS three times and
then fixed using 4% formalin; these were then subjected to a
series of ethanol washes to dehydrate the samples. The de-
hydrated composites were then embedded using paraffin and
cut into 5-mm sections. A set of sectioned slides were stained
using hematoxylin and eosin (H&E). The remaining com-
posite sections were probed for keratin 1/10/11 (1:400),
keratin 6 (undiluted), keratin 14 (1:20), collagen IV (1:10),
and P63 (1:100) expression using antibodies (Research Dia-
gnostics). Initially, sections were deparafinized and rehy-
drated in 100% xylene for 10min, 100% xylene for 5min,
100% ethanol for 5min, 95% ethanol for 5min, 70% ethanol
for 5min, and ddH2O for 10min. This was followed by two 5-
min washes in PBS. After incubation with the above primary
antibodies, the sections were stained using a Dako Envision
kit (Dako Denmark A/S, Glostrup, Denmark) as per the
manufacturer’s instructions, with the exception that PBS was
used instead of TBS. Following development of the labeled
secondary antibody with the 3,30-diaminobenzidine chro-
mogen solution, all sections were counterstained with he-
matoxylin for 30 s and analyzed using light microscopy.
hES cell culture
Mouse embryonic fibroblast (MEF) cells (ESI, Melbourne,
Australia) were cultured in 80 cm2 culture flasks in 85%
DMEM (Invitrogen), 10% FBS (Gibco, Mulgrave, VIC,
Australia), 1mM L-glutamine, 0.5% penicillin/streptomycin,
and 0.01% gentamycin. Passage 7 MEFs were used as the
feeder layer for the hES cells. Surfaces to be seeded with the
MEFs were coated in 0.1% gelatin (Sigma) for a minimum
of 1 h before addition of the cells. Mitomycin-C was sub-
sequently added to the flasks containing the MEFs, and the
cells were incubated at 378C, 5% CO2 for 2.5 to 3 h to mi-
totically inactivate the MEFs. The MEFs were seeded into
six-well plates (10 cm2) at a density of 2�104 cells/cm2. The
HUES-9 and H1 cells (Harvard University, Boston and
WiCell, Madison,Wisconsin, respectively) were cultured on
passage 7 mitomycin-C–inactivated MEFs in hES medium
containing DMEM (Invitrogen), 20% KSR (Invitrogen),
1000 IU/mL penicillin/1000 mg/mL streptomycin (Invitro-
gen), 1mM glutamax (Invitrogen), 1% nonessential amino
acids, 0.1mM b-mercaptoethanol, 10 ng/mL recombinant
human basic fibroblast growth factor (bFGF) (Chemicon,
VIC, Melbourne, Australia), and 12 ng/mL leukemia in-
hibitory factor (LIF) (Chemicon). Following the hES cell
colony reaching a certain size, the cells were washed in 2mL
PBS per well (Invitrogen) and exposed to 0.05% trypsin/
EDTA (Invitrogen) for a 1- to 2-min incubation (378C, 5%CO2). The cells were then resuspended in hES media and
spun at 500–600 g for 5min prior to being transferred to
freshly inactivated MEFs. The culture medium was then
changed daily 48 h posttransfer.
hES cell VN:GF culture
The serum-free culture of the hES cells involved the use of
the previously mentioned inactivated MEF cells being pre-
plated 24 h before use and then being extensively washed in
serum-free media prior to being serum starved for 4 h prior
to plating the hES cells. The hES cells were plated onto
the serum-starved MEFs in 2.5mL of serum-free medium
containing DMEM (Invitrogen), 1000 IU/mL penicillin/
1000mg/mL streptomycin (Invitrogen), 1mM glutamax
(Invitrogen), 1% nonessential amino acids, 0.1mM b-mercaptoethanol, 0.6mg/mL VN (Promega), 0.6mg/mL
IGFBP-3 (Auspep), 0.2mg/mL IGF-I (GroPep, Adelaide, SA,
Australia), 12 ng/mL LIF, and 0.02 mg/mL bFGF (Chemi-
con). The cultures were then grown at 378C in 5% CO2 and
refed every day 48 h after the initial transfer. The hES cells
were then split, as previously described above, 1:3 to 1:4 de-
pending on their rate of growth and confluence and were re-
suspendedusing1:1volume trypsin:trypsin inhibitor (Sigma).
hES cell immunohistochemistry
Stage-specific embryonic antigen-1 (SSEA-1) expressed
in differentiated hES cells and stage-specific embryonic
antigen-4 (SSEA-4) expressed by undifferentiated hES cells
were used to monitor the differentiation status of the hES
cells.2 The presence of these proteins in the cultures was
monitored using mouse monoclonal antibodies raised against
the proteins (Millipore, North Ryde, NSW, Australia). The
cultures were fixed using 4% paraformaldehyde/PBS for
15min or in 100% methanol for 2min. The fixing agent was
removed, and the cultures were washed twice for 15min per
wash in 20mM Tris-HCl, 0.15 M sodium chloride, and
0.05% Tween-20, pH 7.4 (TBST). The cells were then
permeabilized with 0.1% Triton X-100/PBS for 10min prior
to a further washing step. The cultures were then blocked in
4% goat serum for 30min at room temperature. The block-
ing solution was removed, and primary antibodies against
SSEA-1 and SSEA-4 (Millipore) were diluted to 1:50 in 4%
goat serum and incubated on the cultures for 1 h. The pri-
mary antibodies were removed and the wash steps repeated.
The anti-mouse secondary antibodies (Chemicon) were di-
luted in PBS at 1:100 and incubated for 1 h. The secondary
antibodies were removed, the wash steps were repeated, and
the colonies were viewed with a Nikon TE-2000 fluores-
cence microscope.
hES cell reverse-transcriptase polymerase
chain reaction analysis
Octamer-4 (Oct-4), human telomerase reverse tran-
scriptase (hTERT), and alkaline phosphatase (ALP) have
been shown to be expressed in the undifferentiated hES
cells2,8; hence, reverse-transcriptase polymerase chain re-
action (RT-PCR) was performed to determine whether the
hES cell colonies maintained an undifferentiated phenotype.
RNA was isolated from the hES colony pieces using trir-
4 RICHARDS ET AL.
eagent and its accompanying protocol (Sigma). The RNA
samples were applied to oligo-dT 18mers to create cDNA.
The Oct-4 primers were sense, 50-CTTGCTGCAGAAGTGGGTG-GAGGAA-30; and antisense, 50-CTGCAGTGTGG-GTTTCGGGCA-30. The hTERT primers were sense, 50-CGGAAGAGTGTCTGGAGCAA-30; and antisense, 50-GGATGA-AGCGGAGTCTGGA-30. The alkaline phosphatase primers
were sense, 50-CGTGGCTAAGAATGTCATCATGTT-30;and antisense, 50-TGGTGGAGCTGACCCTTGA-30. The
18sRNA internal standard primers were sense, 50-TTCGGAACTGAGGCCATGA-T-30; and antisense, 50-CGAACCTCC-GACTTTCGTTCT-30. One microgram of cDNA was
added to each of the four primer sets and subjected to an initial
denaturation step of 948C for 5min, followed by 30 cycles of
denaturation at 948C for 30 s, annealing at 558C for 30 s, and
extension at 728C for 30 s, followed by a final extension at
728C for 5min. The ampliconswere then run on a 2%gel with
the positive and negative controls using a 100-bp DNA ladder
(DeeWhy,NewSouthWales, Australia) prior to stainingwith
ethidium bromide and visualized on a UV transilluminator.
RESULTS
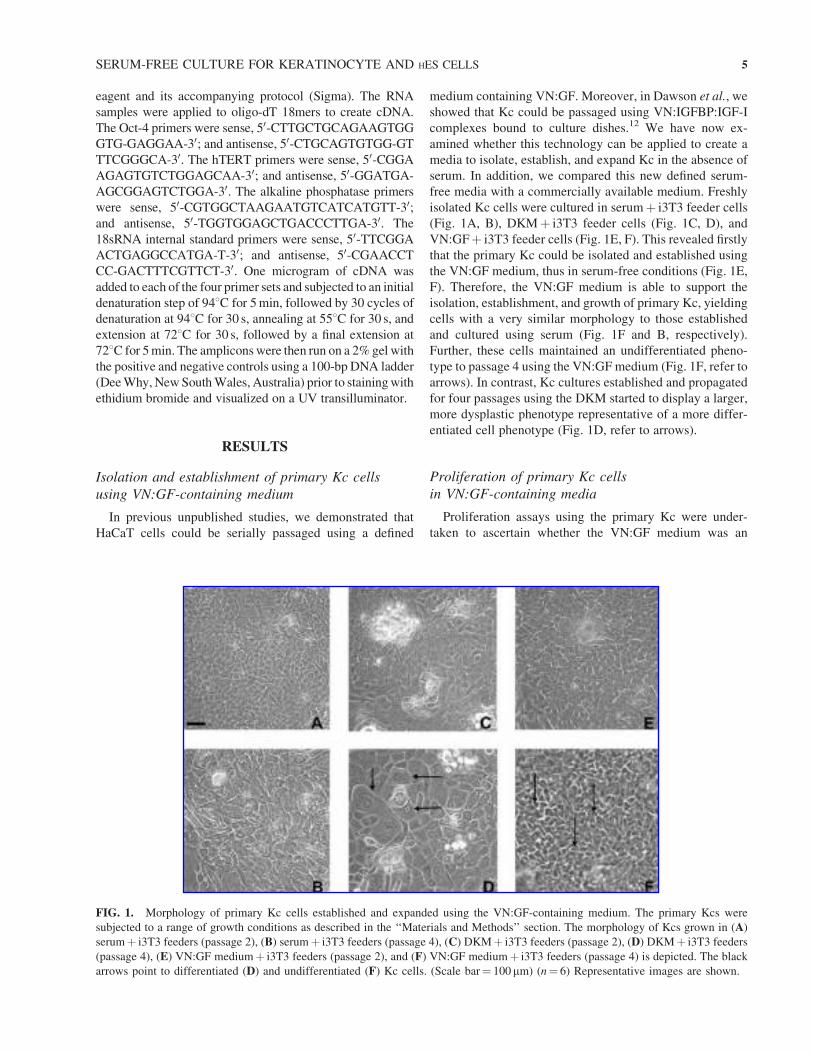
Isolation and establishment of primary Kc cells
using VN:GF-containing medium
In previous unpublished studies, we demonstrated that
HaCaT cells could be serially passaged using a defined
medium containing VN:GF. Moreover, in Dawson et al., we
showed that Kc could be passaged using VN:IGFBP:IGF-I
complexes bound to culture dishes.12 We have now ex-
amined whether this technology can be applied to create a
media to isolate, establish, and expand Kc in the absence of
serum. In addition, we compared this new defined serum-
free media with a commercially available medium. Freshly
isolated Kc cells were cultured in serumþ i3T3 feeder cells
(Fig. 1A, B), DKMþ i3T3 feeder cells (Fig. 1C, D), and
VN:GFþ i3T3 feeder cells (Fig. 1E, F). This revealed firstly
that the primary Kc could be isolated and established using
the VN:GF medium, thus in serum-free conditions (Fig. 1E,
F). Therefore, the VN:GF medium is able to support the
isolation, establishment, and growth of primary Kc, yielding
cells with a very similar morphology to those established
and cultured using serum (Fig. 1F and B, respectively).
Further, these cells maintained an undifferentiated pheno-
type to passage 4 using the VN:GF medium (Fig. 1F, refer to
arrows). In contrast, Kc cultures established and propagated
for four passages using the DKM started to display a larger,
more dysplastic phenotype representative of a more differ-
entiated cell phenotype (Fig. 1D, refer to arrows).
Proliferation of primary Kc cells
in VN:GF-containing media
Proliferation assays using the primary Kc were under-
taken to ascertain whether the VN:GF medium was an
FIG. 1. Morphology of primary Kc cells established and expanded using the VN:GF-containing medium. The primary Kcs were
subjected to a range of growth conditions as described in the ‘‘Materials and Methods’’ section. The morphology of Kcs grown in (A)
serumþ i3T3 feeders (passage 2), (B) serumþ i3T3 feeders (passage 4), (C) DKMþ i3T3 feeders (passage 2), (D) DKMþ i3T3 feeders
(passage 4), (E) VN:GF mediumþ i3T3 feeders (passage 2), and (F) VN:GF mediumþ i3T3 feeders (passage 4) is depicted. The black
arrows point to differentiated (D) and undifferentiated (F) Kc cells. (Scale bar¼ 100mm) (n¼ 6) Representative images are shown.
SERUM-FREE CULTURE FOR KERATINOCYTE AND HES CELLS 5
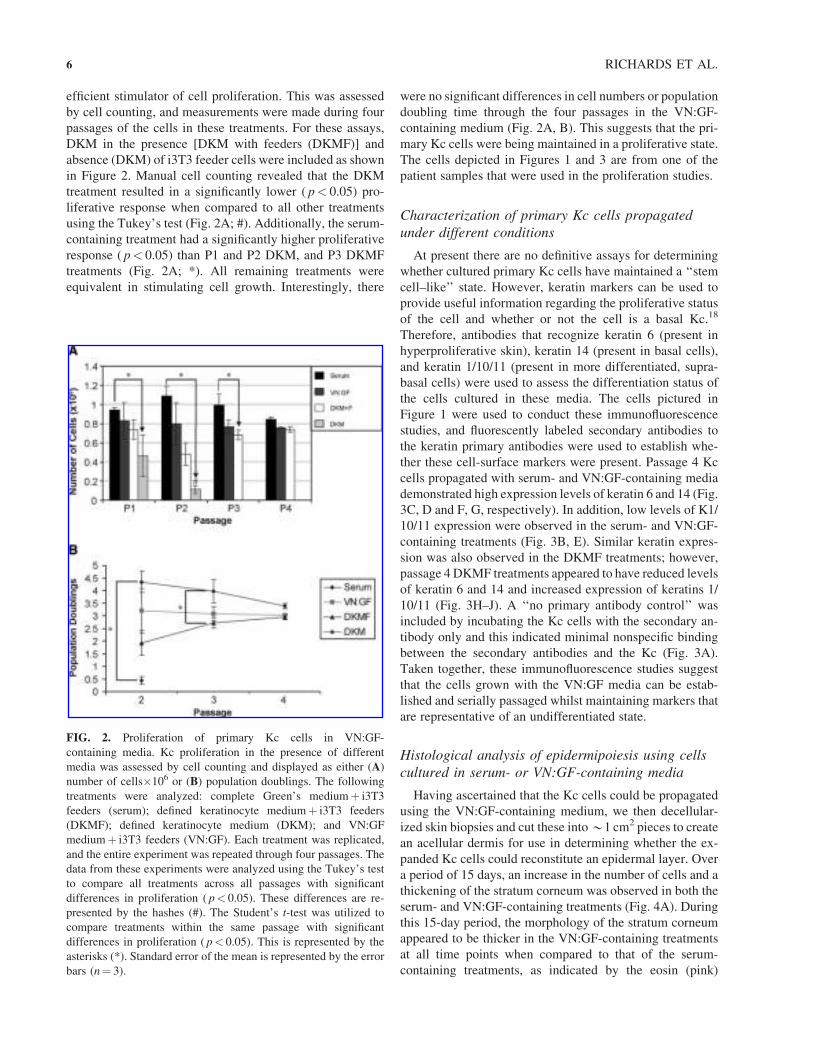
efficient stimulator of cell proliferation. This was assessed
by cell counting, and measurements were made during four
passages of the cells in these treatments. For these assays,
DKM in the presence [DKM with feeders (DKMF)] and
absence (DKM) of i3T3 feeder cells were included as shown
in Figure 2. Manual cell counting revealed that the DKM
treatment resulted in a significantly lower ( p< 0.05) pro-
liferative response when compared to all other treatments
using the Tukey’s test (Fig. 2A; #). Additionally, the serum-
containing treatment had a significantly higher proliferative
response ( p< 0.05) than P1 and P2 DKM, and P3 DKMF
treatments (Fig. 2A; *). All remaining treatments were
equivalent in stimulating cell growth. Interestingly, there
were no significant differences in cell numbers or population
doubling time through the four passages in the VN:GF-
containing medium (Fig. 2A, B). This suggests that the pri-
mary Kc cells were being maintained in a proliferative state.
The cells depicted in Figures 1 and 3 are from one of the
patient samples that were used in the proliferation studies.
Characterization of primary Kc cells propagated
under different conditions
At present there are no definitive assays for determining
whether cultured primary Kc cells have maintained a ‘‘stem
cell–like’’ state. However, keratin markers can be used to
provide useful information regarding the proliferative status
of the cell and whether or not the cell is a basal Kc.18
Therefore, antibodies that recognize keratin 6 (present in
hyperproliferative skin), keratin 14 (present in basal cells),
and keratin 1/10/11 (present in more differentiated, supra-
basal cells) were used to assess the differentiation status of
the cells cultured in these media. The cells pictured in
Figure 1 were used to conduct these immunofluorescence
studies, and fluorescently labeled secondary antibodies to
the keratin primary antibodies were used to establish whe-
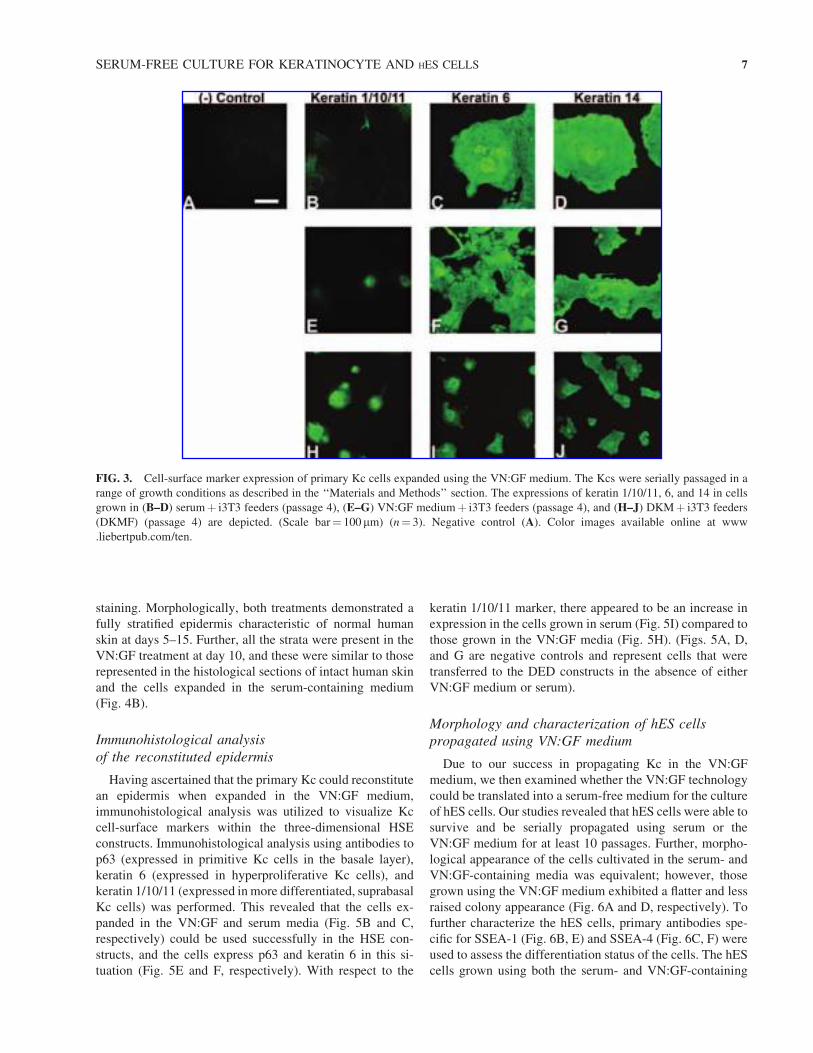
ther these cell-surface markers were present. Passage 4 Kc
cells propagated with serum- and VN:GF-containing media
demonstrated high expression levels of keratin 6 and 14 (Fig.
3C, D and F, G, respectively). In addition, low levels of K1/
10/11 expression were observed in the serum- and VN:GF-
containing treatments (Fig. 3B, E). Similar keratin expres-
sion was also observed in the DKMF treatments; however,
passage 4 DKMF treatments appeared to have reduced levels
of keratin 6 and 14 and increased expression of keratins 1/
10/11 (Fig. 3H–J). A ‘‘no primary antibody control’’ was
included by incubating the Kc cells with the secondary an-
tibody only and this indicated minimal nonspecific binding
between the secondary antibodies and the Kc (Fig. 3A).
Taken together, these immunofluorescence studies suggest
that the cells grown with the VN:GF media can be estab-
lished and serially passaged whilst maintaining markers that
are representative of an undifferentiated state.
Histological analysis of epidermipoiesis using cells
cultured in serum- or VN:GF-containing media
Having ascertained that the Kc cells could be propagated
using the VN:GF-containing medium, we then decellular-
ized skin biopsies and cut these into*1 cm2 pieces to create
an acellular dermis for use in determining whether the ex-
panded Kc cells could reconstitute an epidermal layer. Over
a period of 15 days, an increase in the number of cells and a
thickening of the stratum corneum was observed in both the
serum- and VN:GF-containing treatments (Fig. 4A). During
this 15-day period, the morphology of the stratum corneum
appeared to be thicker in the VN:GF-containing treatments
at all time points when compared to that of the serum-
containing treatments, as indicated by the eosin (pink)
FIG. 2. Proliferation of primary Kc cells in VN:GF-
containing media. Kc proliferation in the presence of different
media was assessed by cell counting and displayed as either (A)
number of cells�106 or (B) population doublings. The following
treatments were analyzed: complete Green’s mediumþ i3T3
feeders (serum); defined keratinocyte mediumþ i3T3 feeders
(DKMF); defined keratinocyte medium (DKM); and VN:GF
mediumþ i3T3 feeders (VN:GF). Each treatment was replicated,
and the entire experiment was repeated through four passages. The
data from these experiments were analyzed using the Tukey’s test
to compare all treatments across all passages with significant
differences in proliferation ( p< 0.05). These differences are re-
presented by the hashes (#). The Student’s t-test was utilized to
compare treatments within the same passage with significant
differences in proliferation ( p< 0.05). This is represented by the
asterisks (*). Standard error of the mean is represented by the error
bars (n¼ 3).
6 RICHARDS ET AL.
staining. Morphologically, both treatments demonstrated a
fully stratified epidermis characteristic of normal human
skin at days 5–15. Further, all the strata were present in the
VN:GF treatment at day 10, and these were similar to those
represented in the histological sections of intact human skin
and the cells expanded in the serum-containing medium
(Fig. 4B).
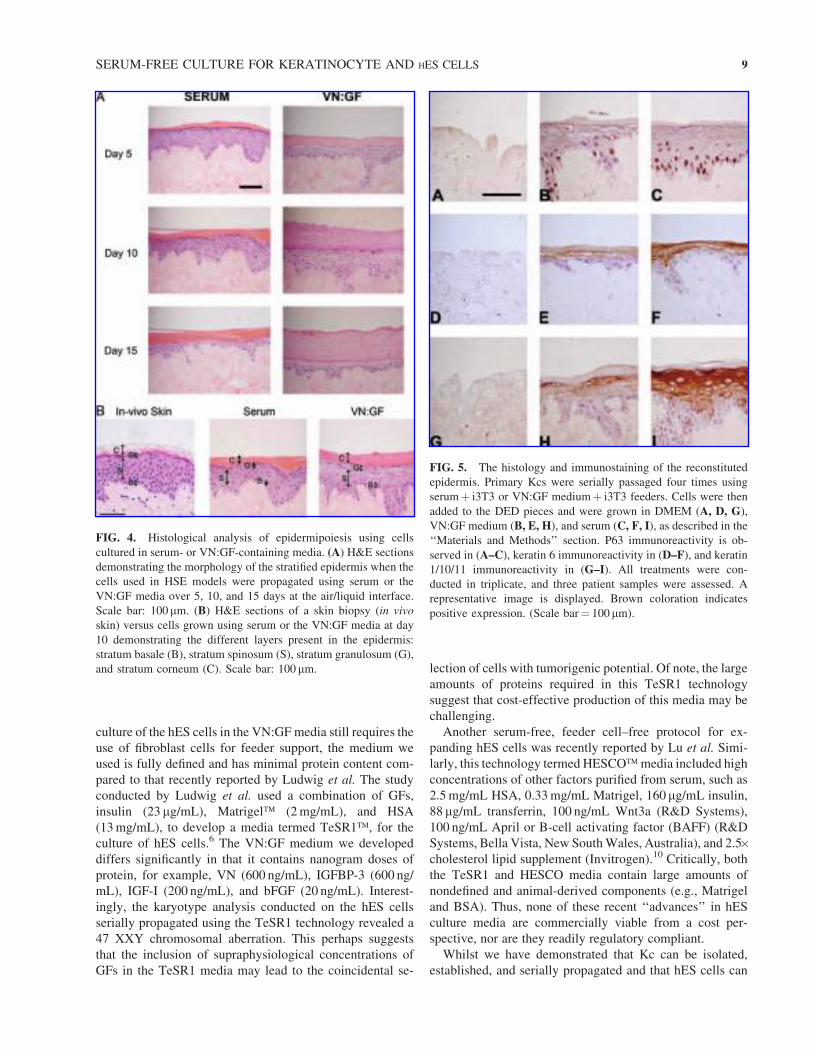
Immunohistological analysis
of the reconstituted epidermis
Having ascertained that the primary Kc could reconstitute
an epidermis when expanded in the VN:GF medium,
immunohistological analysis was utilized to visualize Kc
cell-surface markers within the three-dimensional HSE
constructs. Immunohistological analysis using antibodies to
p63 (expressed in primitive Kc cells in the basale layer),
keratin 6 (expressed in hyperproliferative Kc cells), and
keratin 1/10/11 (expressed inmore differentiated, suprabasal
Kc cells) was performed. This revealed that the cells ex-
panded in the VN:GF and serum media (Fig. 5B and C,
respectively) could be used successfully in the HSE con-
structs, and the cells express p63 and keratin 6 in this si-
tuation (Fig. 5E and F, respectively). With respect to the
keratin 1/10/11 marker, there appeared to be an increase in
expression in the cells grown in serum (Fig. 5I) compared to
those grown in the VN:GF media (Fig. 5H). (Figs. 5A, D,
and G are negative controls and represent cells that were
transferred to the DED constructs in the absence of either
VN:GF medium or serum).
Morphology and characterization of hES cells
propagated using VN:GF medium
Due to our success in propagating Kc in the VN:GF
medium, we then examined whether the VN:GF technology
could be translated into a serum-free medium for the culture
of hES cells. Our studies revealed that hES cells were able to
survive and be serially propagated using serum or the
VN:GF medium for at least 10 passages. Further, morpho-
logical appearance of the cells cultivated in the serum- and
VN:GF-containing media was equivalent; however, those
grown using the VN:GF medium exhibited a flatter and less
raised colony appearance (Fig. 6A and D, respectively). To
further characterize the hES cells, primary antibodies spe-
cific for SSEA-1 (Fig. 6B, E) and SSEA-4 (Fig. 6C, F) were
used to assess the differentiation status of the cells. The hES
cells grown using both the serum- and VN:GF-containing
FIG. 3. Cell-surface marker expression of primary Kc cells expanded using the VN:GF medium. The Kcs were serially passaged in a
range of growth conditions as described in the ‘‘Materials and Methods’’ section. The expressions of keratin 1/10/11, 6, and 14 in cells
grown in (B–D) serumþ i3T3 feeders (passage 4), (E–G) VN:GF mediumþ i3T3 feeders (passage 4), and (H–J) DKMþ i3T3 feeders
(DKMF) (passage 4) are depicted. (Scale bar¼ 100 mm) (n¼ 3). Negative control (A). Color images available online at www
.liebertpub.com/ten.
SERUM-FREE CULTURE FOR KERATINOCYTE AND HES CELLS 7

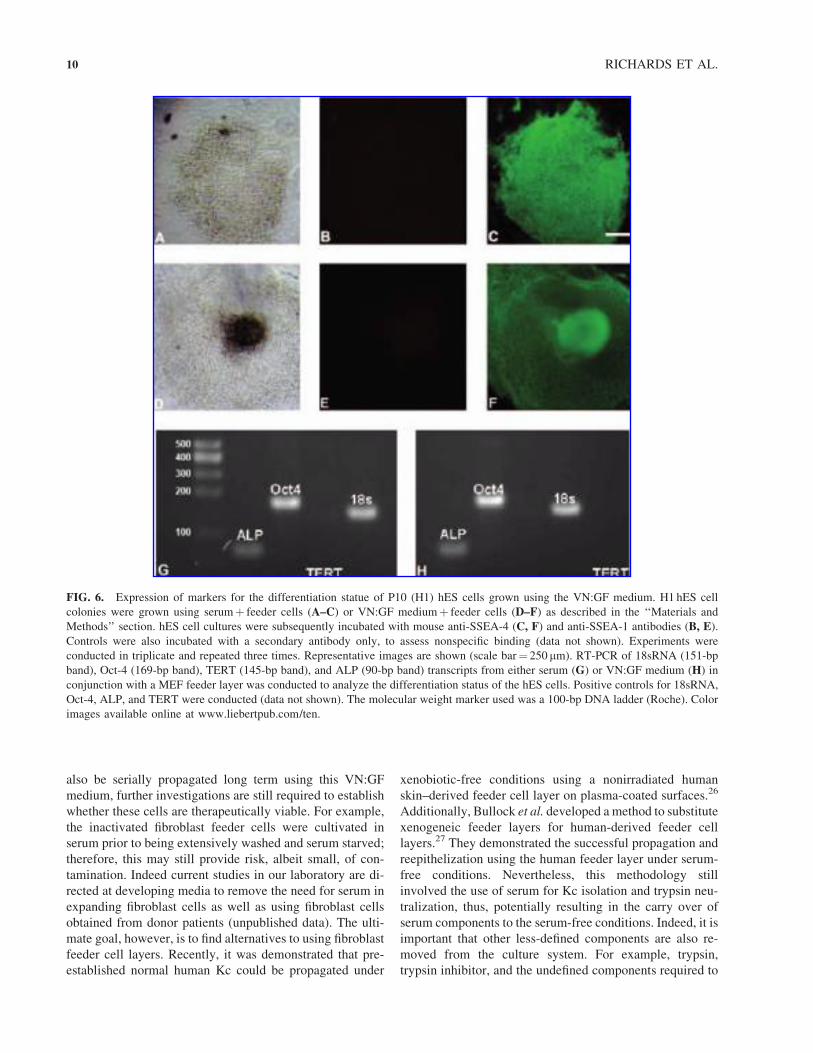
medium (passage 10) demonstrated high expression levels
of SSEA-4 (Fig. 6F). In addition, low levels of SSEA-1
expression were observed in the hES cells grown using
serum- or VN:GF-containing medium (Fig. 6B, F). A no
primary antibody control was included by incubating the
cells with the secondary antibody only, and this indicated
minimal nonspecific binding between the secondary anti-
bodies and the cells (data not shown).
RT-PCR analysis of transcripts collected from the hES
cells grown in either the serum- (Fig. 6G) or the VN:GF-
containing (Fig. 6H) media expressed similar levels of
alkaline phosphatase (ALP) and Oct-4. Unexpectedly, no
levels of hTERT were observed in cells grown in either
treatment. This was an interesting finding as the positive
controls for each of the amplicons produced a band. Taken
together, the immunofluorescence and RT-PCR studies
suggest that the cells grown with the VN:GF combination
can be used as an alternative to serum for the serial propa-
gation of hES cells whilst maintaining markers that are re-
presentative of an undifferentiated state. Negative controls
involved reactions with no addition of the template to ensure
that no contamination occurred in the PCR reaction (data not
shown). The results obtained for the two cell lines evaluated,
H1 and HUES-9, were equivalent. Data for H1 only are
shown. Karyotype analysis also revealed that the hES cells
had maintained their original karyotype when expanded
using the VN:GF medium (data not shown).
DISCUSSION
While the use of Kc and hES cells for cell-based therapies
appears to have great potential to cure a wide variety of
diseases and injuries,19–21 the fact that these culture sys-
tems require xenogeneic and poorly defined components
means that they are unlikely to be readily approved for broad
clinical use. Thus, there is a global move toward serum-free,
animal product–free, and defined media for the culture of
Kc, hES, and indeed other cell types. Many researchers have
investigated ways to minimize the risk of contamination
from animal products such as serum. One such example is
the replacement of FBS with large amounts (10mg/mL) of
purified bovine serum albumin (BSA).22 Whilst this method
did prove successful in the propagation of Kc, it does not
remove the risk of contamination via the BSA present in the
culture system, nor does it result in a fully defined media
since the BSA used was purified from serum rather than
made recombinantly. Further, many of these serum-
replacement systems still require the addition of mouse- or
human-derived fibroblast feeder cells for the successful
culture of the cells of interest.
As irradiated i3T3 feeder cells are known to secrete large
quantities of IGFs and ECM proteins,23 and given that VN
is a major component of serum,24 the hypothesis underlying
the data reported herein was that combinations comprised
of IGFs and VN may trigger matricellular signaling events
that support the serum-free establishment and growth of
human Kc. Indeed, Dawson et al. demonstrated that Kc can
attach and proliferate in response to VN-coated surfaces.25
More recently, Dawson et al. demonstrated that primary Kc
could also be serially propagated using complexes of
VN:IGFBP-3:IGF-I:EGF or VN:IGFBP-5:IGF-I:EGF pre-
bound to culture dishes.12 However, this approach still in-
volved the use of serum during the original isolation of Kc;
thus, potential xenogeneic contamination cannot be elimi-
nated. It was these experimental data that prompted us to
investigate whether the VN:GF complexes could be trans-
lated into a serum-free formulation to support both the
isolation and undifferentiated serial propagation of primary
Kc. We hypothesized that the VN:GF technology could be
improved in two ways: firstly, by examining whether the
complex technology could be used as a serum-free medium
rather than using it as a substrate prebound to the culture
vessel; and secondly, by establishing whether this serum-
free media could be used for steps associated with the
isolation, establishment, and propagation of Kc.
To examine this hypothesis, we examined the potential of
the VN:GF medium to support the isolation and in vitro
expansion of primary human Kc derived from adult skin.
Our results indicate that the VN:GF medium can be suc-
cessfully used for these processes, as the primary cells were
successfully isolated and established using the VN:GF
medium (Figs. 1–4). An interesting observation within these
studies was that the stratum corneum appeared to be thicker
in the VN:GF treatment when compared to the cells grown in
the serum-containing HSE treatment. Whilst we have no
clear explanation for this difference, perhaps the cells in the
VN:GF HSE are proliferating and/or migrating at a greater
rate than those in the serum-containing treatment. Never-
theless, taken together, these results suggest that the VN:GF
medium can maintain the Kc in a proliferative, non-
differentiated state while being expanded in vitro. The next
step in translating this technology for the clinic is to further
validate the biological state of these cells using karyotype
analysis and apoptosis assays. While karyotype analysis of
ex vivo–expanded Kcs is not routine aspect of the production
of clinical epithelial autografts due to shorter cultivation
periods (two to four passages for clinical applications), this
analysis is commonly employed in hES cell biology, where
longer term cultivation (10þ passages) has produced hES
cell chromosomal abberations.6 Nevertheless, karyotyping
of Kcs would be of benefit in ensuring that these cells have
no tumorigenic potential.
Given the similarities in the methods used for the ex-
pansion of hES and Kc cells, and in light of the results above,
we examined whether this technology could also be utilized
for the expansion of hES cells. As reported herein, hES cells
could be serially propagated long term using the VN:GF
medium. Morphological analysis and characterization stu-
dies demonstrated that hES cells grown using VN:GF over
10 passages maintained a similar phenotype to hES cells
grown in serum (Fig. 6). While acknowledging that the
8 RICHARDS ET AL.
culture of the hES cells in the VN:GF media still requires the
use of fibroblast cells for feeder support, the medium we
used is fully defined and has minimal protein content com-
pared to that recently reported by Ludwig et al. The study
conducted by Ludwig et al. used a combination of GFs,
insulin (23 mg/mL), Matrigel� (2mg/mL), and HSA
(13mg/mL), to develop a media termed TeSR1�, for the
culture of hES cells.6 The VN:GF medium we developed
differs significantly in that it contains nanogram doses of
protein, for example, VN (600 ng/mL), IGFBP-3 (600 ng/
mL), IGF-I (200 ng/mL), and bFGF (20 ng/mL). Interest-
ingly, the karyotype analysis conducted on the hES cells
serially propagated using the TeSR1 technology revealed a
47 XXY chromosomal aberration. This perhaps suggests
that the inclusion of supraphysiological concentrations of
GFs in the TeSR1 media may lead to the coincidental se-
lection of cells with tumorigenic potential. Of note, the large
amounts of proteins required in this TeSR1 technology
suggest that cost-effective production of this media may be
challenging.
Another serum-free, feeder cell–free protocol for ex-
panding hES cells was recently reported by Lu et al. Simi-
larly, this technology termed HESCO�media included high
concentrations of other factors purified from serum, such as
2.5mg/mL HSA, 0.33mg/mL Matrigel, 160 mg/mL insulin,
88 mg/mL transferrin, 100 ng/mL Wnt3a (R&D Systems),
100 ng/mL April or B-cell activating factor (BAFF) (R&D
Systems, Bella Vista, New SouthWales, Australia), and 2.5�cholesterol lipid supplement (Invitrogen).10 Critically, both
the TeSR1 and HESCO media contain large amounts of
nondefined and animal-derived components (e.g., Matrigel
and BSA). Thus, none of these recent ‘‘advances’’ in hES
culture media are commercially viable from a cost per-
spective, nor are they readily regulatory compliant.
Whilst we have demonstrated that Kc can be isolated,
established, and serially propagated and that hES cells can
FIG. 4. Histological analysis of epidermipoiesis using cells
cultured in serum- or VN:GF-containing media. (A) H&E sections
demonstrating the morphology of the stratified epidermis when the
cells used in HSE models were propagated using serum or the
VN:GF media over 5, 10, and 15 days at the air/liquid interface.
Scale bar: 100mm. (B) H&E sections of a skin biopsy (in vivo
skin) versus cells grown using serum or the VN:GF media at day
10 demonstrating the different layers present in the epidermis:
stratum basale (B), stratum spinosum (S), stratum granulosum (G),
and stratum corneum (C). Scale bar: 100 mm.
FIG. 5. The histology and immunostaining of the reconstituted
epidermis. Primary Kcs were serially passaged four times using
serumþ i3T3 or VN:GF mediumþ i3T3 feeders. Cells were then
added to the DED pieces and were grown in DMEM (A, D, G),
VN:GF medium (B, E, H), and serum (C, F, I), as described in the
‘‘Materials and Methods’’ section. P63 immunoreactivity is ob-
served in (A–C), keratin 6 immunoreactivity in (D–F), and keratin
1/10/11 immunoreactivity in (G–I). All treatments were con-
ducted in triplicate, and three patient samples were assessed. A
representative image is displayed. Brown coloration indicates
positive expression. (Scale bar¼ 100mm).
SERUM-FREE CULTURE FOR KERATINOCYTE AND HES CELLS 9
also be serially propagated long term using this VN:GF
medium, further investigations are still required to establish
whether these cells are therapeutically viable. For example,
the inactivated fibroblast feeder cells were cultivated in
serum prior to being extensively washed and serum starved;
therefore, this may still provide risk, albeit small, of con-
tamination. Indeed current studies in our laboratory are di-
rected at developing media to remove the need for serum in
expanding fibroblast cells as well as using fibroblast cells
obtained from donor patients (unpublished data). The ulti-
mate goal, however, is to find alternatives to using fibroblast
feeder cell layers. Recently, it was demonstrated that pre-
established normal human Kc could be propagated under
xenobiotic-free conditions using a nonirradiated human
skin–derived feeder cell layer on plasma-coated surfaces.26
Additionally, Bullock et al. developed amethod to substitute
xenogeneic feeder layers for human-derived feeder cell
layers.27 They demonstrated the successful propagation and
reepithelization using the human feeder layer under serum-
free conditions. Nevertheless, this methodology still
involved the use of serum for Kc isolation and trypsin neu-
tralization, thus, potentially resulting in the carry over of
serum components to the serum-free conditions. Indeed, it is
important that other less-defined components are also re-
moved from the culture system. For example, trypsin,
trypsin inhibitor, and the undefined components required to
FIG. 6. Expression of markers for the differentiation statue of P10 (H1) hES cells grown using the VN:GF medium. H1 hES cell
colonies were grown using serumþ feeder cells (A–C) or VN:GF mediumþ feeder cells (D–F) as described in the ‘‘Materials and
Methods’’ section. hES cell cultures were subsequently incubated with mouse anti-SSEA-4 (C, F) and anti-SSEA-1 antibodies (B, E).
Controls were also incubated with a secondary antibody only, to assess nonspecific binding (data not shown). Experiments were
conducted in triplicate and repeated three times. Representative images are shown (scale bar¼ 250 mm). RT-PCR of 18sRNA (151-bp
band), Oct-4 (169-bp band), TERT (145-bp band), and ALP (90-bp band) transcripts from either serum (G) or VN:GF medium (H) in
conjunction with a MEF feeder layer was conducted to analyze the differentiation status of the hES cells. Positive controls for 18sRNA,
Oct-4, ALP, and TERT were conducted (data not shown). The molecular weight marker used was a 100-bp DNA ladder (Roche). Color
images available online at www.liebertpub.com/ten.
10 RICHARDS ET AL.

freeze and store cells also need to be removed to generate
a safe, more-defined culture system for Kcs. Recently,
products such as TrypLE� (Invitrogen), which is a re-
combinant alternative to trypsin, and CryoStor�, a protein-
free freezing medium, have allowed researchers to move
toward establishing a defined culture system.
Taken together, these advances represent an improvement
to the culture system of Kc and hES cells. The ideal scenario,
however, would be the development of culture conditions in
which feeder cells are not required, as the presence of feeder
cells, whether of human or animal origin, adds a potential risk
of contamination and therefore adds to the increased reg-
ulatory burden. In this respect, the VN:GF-containing media
reported here used in combination with a feeder cell alter-
native could represent a significant advance. However, any
culture system developed for widespread use in cell-based
therapies must contain synthetic and/or recombinant com-
ponents. To this end, recombinant VN has recently been
produced in our laboratory and has been shown to be func-
tional when incorporated into the VN:GF combinations (data
not shown); thus, a fully synthetic, defined replacement for
the serum-free culture of Kc and hES cells is a near-term
reality.
ACKNOWLEDGEMENTS
The authors would like to thank Mr. Don Geyer (School
of Life Sciences, QUT) for conducting the H&E staining.
They also would like to acknowledge the Australian Red
Cross Blood Service for the irradiation service they pro-
vided.
REFERENCES
1. Rheinwald, J.G., and H. Green. Serial cultivation of strains of
human epidermal keratinocytes: the formation of keratinizing
colonies from single cells. Cell 6, 331, 1975.
2. Thomson, J.A., J. Itskovitz-Eldor, S.S. Shapiro, M.A.
Waknitz, J.J. Swiergiel, V.S. Marshall, and J.M. Jones.
Embryonic stem cell lines derived from human blastocysts.
Science 282, 1145, 1998.
3. Rolleston, W.B. Bovine serum: reducing the variables
through the use of donor herds. Dev Biol Stand 99, 79, 1999.
4. Martin, M.J., A. Muotri, F. Gage, and A. Varki. Human
embryonic stem cells express an immunogenic nonhuman
sialic acid. Nat Med 11, 228, 2005.
5. Heiskanen, A., T. Satomaa, S. Tiitinen, A. Laitinen, S.
Mannelin, U. Impola, M. Mikkola, C. Olsson, H. Miller-
Podraza, M. Blomqvist, A. Olonen, H. Salo, P. Lehenkari,
T. Tuuri, T. Otonkoski, J. Natunen, J. Saarinen, and J. Laine.
N-glycolylneuraminic acid xenoantigen contamination of
human embryonic and mesenchymal stem cells is sub-
stantially reversible. Stem Cells 25, 197, 2007.
6. Ludwig, T.E., M.E. Levenstein, J.M. Jones, W.T. Berggren,
E.R. Mitchen, J.L. Frane, L.J. Crandall, C.A. Daigh, K.R.
Conard, M.S. Piekarczyk, R.A. Lianas, and J.A. Thomson.
Derivation of human embryonic stem cells in defined condi-
tions. Nat Biotechnol 24, 185, 2006.
7. Wang, H.J., T.M. Chen, L.F. Cheng, T.Y. Cheng, and Y.M.
Tung. Human keratinocyte culture using porcine pituitary
extract in serum-free medium. Burns 21, 503, 1995.
8. Beattie, G.M., A.D. Lopez, N. Bucay, A. Hinton, M.T. Firpo,
C.C. King, and A. Hayek. Activin A maintains pluripotency
of human embryonic stem cells in the absence of feeder
layers. Stem Cells 23, 489, 2005.
9. Pouliot, N., N.A. Saunders and P. Kaur. Laminin 10/11: an
alternative adhesive ligand for epidermal keratinocytes with a
functional role in promoting proliferation and migration. Exp
Dermatol 11, 387, 2002.
10. Lu, J., R. Hou, C.J. Booth, S.H. Yang, and M. Snyder. De-
fined culture conditions of human embryonic stem cells. Proc
Natl Acad Sci USA 103, 5688, 2006.
11. Woodley, D.T., K.C. Wynn, and E.J. O’Keefe. Type IV col-
lagen and fibronectin enhance human keratinocyte thymidine
incorporation and spreading in the absence of soluble growth
factors. J Invest Dermatol 94, 139, 1990.
12. Dawson, R.A., Z. Upton, J. Malda, and D.G. Harkin. Pre-
paration of cultured skin for transplantation using insulin-like
growth factor I in conjunction with insulin-like growth factor
binding protein 5, epidermal growth factor, and vitronectin.
Transplantation 81, 1668, 2006.
13. Hollier, B., D.G. Harkin, D. Leavesley, and Z. Upton. Re-
sponses of keratinocytes to substrate-bound vitronectin:
growth factor complexes. Exp Cell Res 305, 221, 2005.
14. Kricker, J.A., C.L. Towne, S.M. Firth, A.C. Herington, and
Z.Upton. Structural and functional evidence for the interaction of
insulin-like growth factors (IGFs) and IGF binding proteins with
vitronectin. Endocrinology 144, 2807, 2003.
15. Upton, Z., H. Webb, K. Hale, C.A. Yandell, J.P. McMurtry,
G.L. Francis, and F.J. Ballard. Identification of vitronectin
as a novel insulin-like growth factor-II binding protein.
Endocrinology 140, 2928, 1999.
16. Ainscough, S.L., Z. Barnard, Z. Upton, and D.G. Harkin.
Vitronectin supports migratory responses of corneal epithelial
cells to substrate bound IGF-I and HGF, and facilitates serum-
free cultivation. Exp Eye Res 83, 1505, 2006.
17. Goberdhan, N.J., R.A. Dawson, E. Freedlander, and S. Mac
Neil. A calmodulin-like protein as an extracellular mitogen
for the keratinocyte. Br J Dermatol 129, 678, 1993.
18. Watt, F.M. Epidermal stem cells: markers, patterning and the
control of stem cell fate. Philos Trans R Soc Lond B Biol Sci
353, 831, 1998.
19. Meana, A., J. Iglesias, M. Del Rio, F. Larcher, B. Madrigal,
M.F. Fresno, C. Martin, F. San Roman, and F. Tevar. Large
surface of cultured human epithelium obtained on a dermal
matrix based on live fibroblast-containing fibrin gels. Burns
24, 621, 1998.
20. Wright, K.A., K.B. Nadire, P. Busto, R. Tubo, J.M.
McPherson, and B.M. Wentworth. Alternative delivery of
keratinocytes using a polyurethane membrane and the im-
plications for its use in the treatment of full-thickness burn
injury. Burns 24, 7, 1998.
21. Keirstead, H.S., G. Nistor, G. Bernal, M. Totoiu, F. Cloutier,
K. Sharp, and O. Steward. Human embryonic stem cell-
derived oligodendrocyte progenitor cell transplants
SERUM-FREE CULTURE FOR KERATINOCYTE AND HES CELLS 11

remyelinate and restore locomotion after spinal cord injury.
J Neurosci 25, 4694, 2005.
22. Castro-Munozledo, F., M. Hernandez-Quintero, M. Marsch-
Moreno, and W. Kuri-Harcuch. Cultivation, serial transfer,
and differentiation of epidermal keratinocytes in serum-free
medium. Biochem Biophys Res Commun 236, 167, 1997.
23. Barreca, A., M. De Luca, P. Del Monte, S. Bondanza, G.
Damonte, G. Cariola, E. Di Marco, G. Giordano, R. Can-
cedda, and F. Minuto. In vitro paracrine regulation of human
keratinocyte growth by fibroblast-derived insulin-like growth
factors. J Cell Physiol 151, 262, 1992.
24. Schvartz, I., D. Seger, and S. Shaltiel. Vitronectin. Int J
Biochem Cell Biol 31, 539, 1999.
25. Dawson, R.A., N.J. Goberdhan, E. Freedlander, and S. Mac-
Neil. Influence of extracellular matrix proteins on human
keratinocyte attachment, proliferation and transfer to a dermal
wound model. Burns 22, 93, 1996.
26. Sun, T., M. Higham, C. Layton, J. Haycock, R. Short, and S.
MacNeil. Developments in xenobiotic-free culture of human
keratinocytes for clinical use. Wound Repair Regen 12, 626,
2004.
27. Bullock, A.J., M.C. Higham, and S. MacNeil. Use of human
fibroblasts in the development of a xenobiotic-free culture and
delivery system for human keratinocytes. Tissue Eng 12, 245,
2006.
Address reprint requests to:
Sean Richards, Ph.D.
Stem Cell Group, Tissue Repair, and Regeneration
Institute of Health and Biomedical Innovation
Queensland University of Technology
60 Musk Ave., Kelvin Grove
QLD 4059
Australia
E-mail: [email protected]
Received: December 18, 2007
Accepted: April 10, 2008
12 RICHARDS ET AL.
Related Documents











