Deoxygenation of carbon dioxide by electrophilic terminal phosphinidene complexes† Christian Schulten, a Gerd von Frantzius, a Gregor Schnakenburg, a Arturo Espinosa b and Rainer Streubel * a Received 25th July 2012, Accepted 3rd September 2012 DOI: 10.1039/c2sc21081a Deoxygenation of carbon dioxide was achieved using transient terminal phosphinidene chromium and tungsten complexes 2a,b. The overall reaction is exothermic according to DFT calculations on the model terminal P-methyl phosphinidene complex Me-2b; this was also supported by the calculated thermodynamic oxygen-transfer potential. The oxaphosphiran-3-one complex intermediates 3a,b possess an unprecedented bonding situation as some characteristics of a side-on bound carbon dioxide to the (formally) low-coordinated phosphorus centre come to the fore. This is expressed by equidistant P–C and P–O bonds and unusual bond strength relationship, i.e. P–C > P–O, as revealed by the relaxed force constants and other related parameters. The decomposition of 3a,b via CO extrusion yields terminal phosphinidene oxide complexes 4a,b which dimerise to the final products, the 1,3-dioxa-2,4- diphosphetane complexes 5a,b–8a,b. Additional experimental evidence for the transient formation of phosphinidene oxide complexes 4a,b was obtained by a cross dimerisation experiment using transient chromium and tungsten complexes 2a,b. First comparative investigations on the reaction of Li–Cl phosphinidenoid complex 10 and CO 2 at low temperature revealed the formation of the carbamoyl– phosphane complex 11. Introduction The challenge to use carbon dioxide as a synthetic building block has recently received increasing interest in the area of green and sustainable chemistry and catalysis, and the direct conversion of carbon dioxide into urea as an industrially important process provides an extra stimulus. 1,2 In this regard, various addition reactions have been studied involving electrophiles and nucleo- philes 3 and, for example, nucleophilic carbenes add to carbon dioxide to form betaines 4,5 but not to give stable a-lactones (oxiranones) I (Scheme 1, E ¼ C). The latter are obtained only via epoxidation of ketenes having perfluorinated large alkyl substituents; in this case gentle heating also led to ring frag- mentation and thus to carbon monoxide and the corresponding ketone, which represents a net deoxygenation reaction of carbon dioxide. 6 In comparison, transition metal complexes have been widely used to bind and thus activate carbon dioxide. 7 In some cases, deoxygenation of carbon dioxide was initiated by its side- on coordination as in complexes II 8,9 (Scheme 1) and some Mo, 10 Fe, 11 and Ni 12 h 2 –CO 2 complexes were structurally charac- terised. The deoxygenation mechanism based on transient or stable complexes II to give the metal oxo complex and carbon monoxide was corroborated recently by a DFT study 13 for the case of a Ni 0 N-heterocyclic carbene (NHC) complex. 14 Computationally and experimentally described are oxasilir- anone (E ¼ SiH 2 ) 15 and oxaziridinone (E ¼ NH), 16 whereas oxagermiranone (E ¼ GeH 2 ) 17 is known in silico only; oxa- phosphiranones (E ¼ PR) and complexes thereof are unknown, so far. However, with the discovery of metal-free organophos- phorus catalysed fixation of carbon dioxide, 16,18–21 evidence is growing that main group elements may very well enter the clas- sical transition metal domain of small molecule activation 22–24 as recently put forward by Power. 25 Other recent examples are the deoxygenation of CO 2 initiated by coordination to an NHC, 26 and the organocatalytic fixation of CO 2 27 by a frustrated Lewis pair (FLP) system, 28 or by addition Scheme 1 Known deoxygenation processes of CO 2 via transient a-lactones I (oxiranones, E ¼ C) or via side-on carbon dioxide transition metal complexes II. a Institut f € ur Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universit € at Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany. E-mail: [email protected]; Fax: +49-228-73-9616; Tel: +49-228-73-5345 b Departamento de Qu ımica Org anica, Universidad de Murcia, Campus de Espinardo, 30100 Murcia, Spain. E-mail: [email protected]; Fax: +34 868- 88-4149; Tel: +34 868-88-7489 † Electronic supplementary information (ESI) available: Homodesmotic reactions used for computing RSE. CCDC reference number 806428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21081a 3526 | Chem. Sci., 2012, 3, 3526–3533 This journal is ª The Royal Society of Chemistry 2012 Dynamic Article Links C < Chemical Science Cite this: Chem. Sci., 2012, 3, 3526 www.rsc.org/chemicalscience EDGE ARTICLE

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Dynamic Article LinksC<Chemical Science
Cite this: Chem. Sci., 2012, 3, 3526
www.rsc.org/chemicalscience EDGE ARTICLE
Deoxygenation of carbon dioxide by electrophilic terminal phosphinidenecomplexes†
Christian Schulten,a Gerd von Frantzius,a Gregor Schnakenburg,a Arturo Espinosab and Rainer Streubel*a
Received 25th July 2012, Accepted 3rd September 2012
DOI: 10.1039/c2sc21081a
Deoxygenation of carbon dioxide was achieved using transient terminal phosphinidene chromium and
tungsten complexes 2a,b. The overall reaction is exothermic according to DFT calculations on the
model terminal P-methyl phosphinidene complex Me-2b; this was also supported by the calculated
thermodynamic oxygen-transfer potential. The oxaphosphiran-3-one complex intermediates 3a,b
possess an unprecedented bonding situation as some characteristics of a side-on bound carbon dioxide
to the (formally) low-coordinated phosphorus centre come to the fore. This is expressed by equidistant
P–C and P–O bonds and unusual bond strength relationship, i.e. P–C > P–O, as revealed by the relaxed
force constants and other related parameters. The decomposition of 3a,b via CO extrusion yields
terminal phosphinidene oxide complexes 4a,b which dimerise to the final products, the 1,3-dioxa-2,4-
diphosphetane complexes 5a,b–8a,b. Additional experimental evidence for the transient formation of
phosphinidene oxide complexes 4a,b was obtained by a cross dimerisation experiment using transient
chromium and tungsten complexes 2a,b. First comparative investigations on the reaction of Li–Cl
phosphinidenoid complex 10 and CO2 at low temperature revealed the formation of the carbamoyl–
phosphane complex 11.
Introduction
The challenge to use carbon dioxide as a synthetic building block
has recently received increasing interest in the area of green and
sustainable chemistry and catalysis, and the direct conversion of
carbon dioxide into urea as an industrially important process
provides an extra stimulus.1,2 In this regard, various addition
reactions have been studied involving electrophiles and nucleo-
philes3 and, for example, nucleophilic carbenes add to carbon
dioxide to form betaines4,5 but not to give stable a-lactones
(oxiranones) I (Scheme 1, E¼C). The latter are obtained only via
epoxidation of ketenes having perfluorinated large alkyl
substituents; in this case gentle heating also led to ring frag-
mentation and thus to carbon monoxide and the corresponding
ketone, which represents a net deoxygenation reaction of carbon
dioxide.6 In comparison, transition metal complexes have been
widely used to bind and thus activate carbon dioxide.7 In some
cases, deoxygenation of carbon dioxide was initiated by its side-
aInstitut f€ur Anorganische Chemie, RheinischeFriedrich-Wilhelms-Universit€at Bonn, Gerhard-Domagk-Str. 1, 53121Bonn, Germany. E-mail: [email protected]; Fax: +49-228-73-9616;Tel: +49-228-73-5345bDepartamento de Qu�ımica Org�anica, Universidad de Murcia, Campus deEspinardo, 30100 Murcia, Spain. E-mail: [email protected]; Fax: +34 868-88-4149; Tel: +34 868-88-7489
† Electronic supplementary information (ESI) available: Homodesmoticreactions used for computing RSE. CCDC reference number 806428. ForESI and crystallographic data in CIF or other electronic format see DOI:10.1039/c2sc21081a
3526 | Chem. Sci., 2012, 3, 3526–3533
on coordination as in complexes II8,9 (Scheme 1) and some Mo,10
Fe,11 and Ni12 h2–CO2 complexes were structurally charac-
terised. The deoxygenation mechanism based on transient or
stable complexes II to give the metal oxo complex and carbon
monoxide was corroborated recently by a DFT study13 for the
case of a Ni0 N-heterocyclic carbene (NHC) complex.14
Computationally and experimentally described are oxasilir-
anone (E ¼ SiH2)15 and oxaziridinone (E ¼ NH),16 whereas
oxagermiranone (E ¼ GeH2)17 is known in silico only; oxa-
phosphiranones (E ¼ PR) and complexes thereof are unknown,
so far. However, with the discovery of metal-free organophos-
phorus catalysed fixation of carbon dioxide,16,18–21 evidence is
growing that main group elements may very well enter the clas-
sical transition metal domain of small molecule activation22–24 as
recently put forward by Power.25
Other recent examples are the deoxygenation of CO2 initiated
by coordination to an NHC,26 and the organocatalytic fixation of
CO227 by a frustrated Lewis pair (FLP) system,28 or by addition
Scheme 1 Known deoxygenation processes of CO2 via transient
a-lactones I (oxiranones, E ¼ C) or via side-on carbon dioxide transition
metal complexes II.
This journal is ª The Royal Society of Chemistry 2012

to an organic substrate activated by an NHC.29 More recently,
carbon dioxide insertion into a strained P–N bond of a cyclic
amidophosphorane was used for CO2 ‘‘sequestration’’, taking
advantage of ‘‘masked’’ FLP reactivity.30 Regarding this back-
ground, the work of Issleib and others may seem as modern
classics but still is fundamental for metal phosphanides MPHR
(M ¼ Na, K; R ¼ Ph, cyclohexyl) which yield metal phosphi-
nocarboxylates M[HRPCO2] with CO2.31
Here, deoxygenation of carbon dioxide by thermally generated
electrophilic terminal phosphinidene complexes is reported. This
is compared to the reaction of a Li–Cl phosphinidenoid complex
with carbon dioxide that yields a carbamoylphosphane complex,
instead. DFT calculations on the deoxygenation process point to
an oxaphosphiranone complex as the reactive intermediate
which decomposes to yield diastereomeric 1,3-dioxa-2,4-
diphosphetane complexes.
Results and discussion
Transient electrophilic terminal phosphinidene complexes 2a,b,
generated by thermolysis of 2H-azaphosphirene complexes
1a,b,32,33 react at 75 �C with carbon dioxide (50 bar) to yield
selectively cis and trans 1,3-dioxa-2,4-diphosphetane complexes
5a,b and 6a,b which were isolated and unambiguously charac-
terised (Scheme 2).34
The deoxygenation of CO2 by terminal phosphinidene
complexes35 2a,b is assumed to proceed via intermediate oxa-
phosphiranone complexes 3a,b (i), which subsequently extrude
CO to form phosphinidene oxide complexes 4a,b (ii). Subse-
quently, 4a,b dimerise to yield 1,3-dioxa-2,4-diphosphetane
complexes 5a,b and 6a,b36 as final products (iii). Further evidence
for the deoxygenation process and the intermediacy of 4a,b was
obtained by a cross dimerization experiment that furnished the
mixed metal 1,3-dioxa-2,4-diphosphetane complexes cis 7a,b and
trans 8a,b appearing each as pair of diastereomers (Scheme 2). A
related situation was observed recently in thermal reactions of
complexes 1 with isocyanates in which the dinuclear 1,3-dioxa-
2,4-diphosphetane complexes were also obtained as final prod-
ucts (in lower yields).36
A first attempt was made to gain selective access to the
assumed oxaphosphiranone complex 3b using the Li–Cl
phosphinidenoid complex 10,37 which has been successfully
used in the synthesis of oxaphosphirane complexes.37,38
Deprotonation of P-chloro phosphane complex 9 39 by lithium
Scheme 2 Proposed mechanism for the formation of complexes
3a,b–6a,b.
This journal is ª The Royal Society of Chemistry 2012
diisopropylamide in the presence of 12-crown-4 and subsequent
reaction with CO2 at �60 �C yielded complex 11 (Scheme 3) as
the only product to be detected by 31P NMR spectroscopy at
ambient temperature. 31P NMR reaction monitoring (�60
to �30�C) revealed, besides complex 11, exclusively a broad
signal of a transient compound at 112.6 ppm, the 1J(W,P)
coupling of which could not be observed. Two mechanistic
options for the formation of complex 11 seem to be reasonable:
(i) the nucleophilic attack of complex 10 at CO2 and ring
closure to yield 3b with subsequent ring opening by diisopro-
pylamine to give 11 or (ii) reaction of 10 with an initially
formed carbamoylamine to yield 11 (Scheme 3).
The 1,3-dioxa-2,4-diphosphetane complexes 5–8 resonate at
low field (>240 ppm), whereby the 31P NMR shift of the trans
isomers appear downfield of the cis isomers for both metal
complexes. The Dd(31P) value of the tungsten and chromium
complexes is about 65 ppm. The constitution of carbamoyl-
phosphane complex 11 was confirmed by single-crystal X-ray
diffraction analysis (Fig. 1).
DFT calculations
To get further insights into the carbon dioxide deoxygenation
process, the model phosphinidene tungsten complex [(CO)5W-
(PMe)] (Me-3b, Scheme 4) and the phosphinidene oxide model
complex [(CO)5W(OPMe)] (Me-4b) were used (Fig. 2, steps i–ii)
for the DFT calculations; oxaphosphiran-3-one Ia and
complexes thereof (Ib,c,Me-3b, 3bsyn, 3banti) were also calculated
for the first time.
Oxaphosphiranone Ia shows significant differences of the ring
geometry compared with the corresponding parent oxaphos-
phirane (P–C 1.823, P–O 1.724, C–O 1.417 [�A]):40 the phos-
phorus–oxygen bond is shorter in the oxaphosphirane, while the
opposite holds for the endocyclic C–O bond. Upon complexation
with Cr(CO)5 (Ib) the endocyclic P–C, P–O as well as the
exocyclic C–O bond shorten slightly; the characteristic CO
stretch vibration of �1955 cm�1 shifts towards shorter wave-
numbers (Table 1).
The related bond strengths were inspected in order to obtain
insight into the consequences of the geometrical bond changes.
Among several possible bond-strength related descriptors, the
widespread used41 Wiberg’s bond index (WBI)42 was computed.
Additionally, the electron density at bond critical points, r(rc),
Scheme 3 Proposed reaction courses for the formation of complex 11.
Chem. Sci., 2012, 3, 3526–3533 | 3527
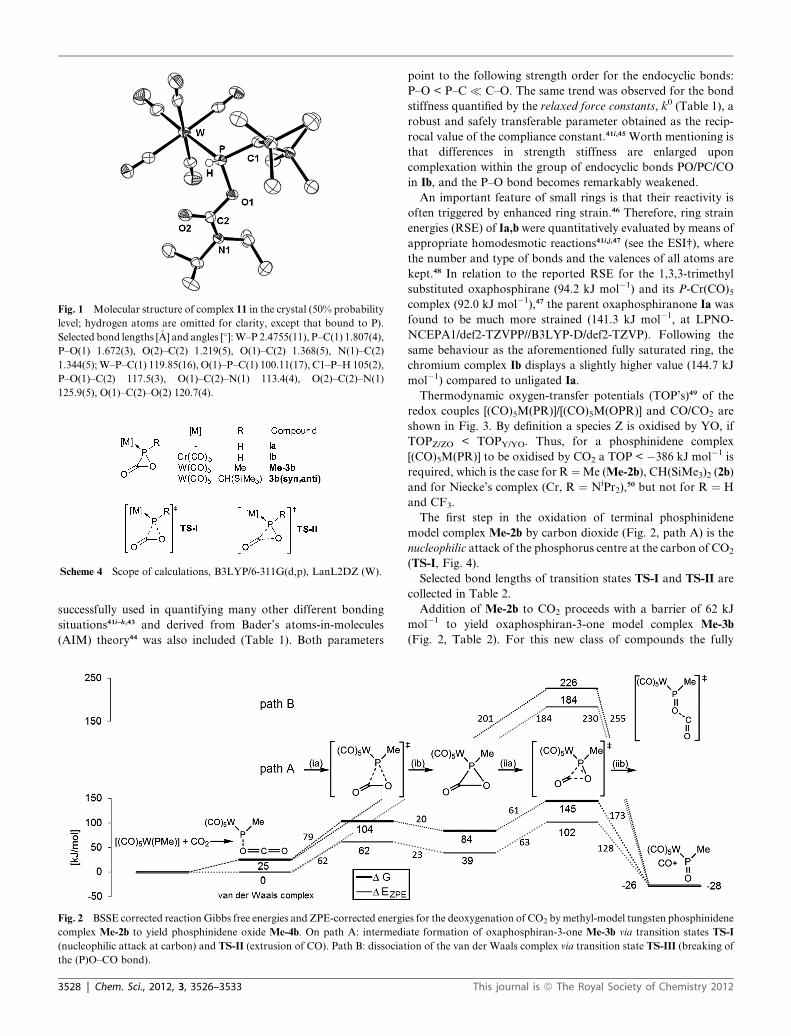
Fig. 1 Molecular structure of complex 11 in the crystal (50% probability
level; hydrogen atoms are omitted for clarity, except that bound to P).
Selected bond lengths [�A] and angles [�]:W–P 2.4755(11), P–C(1) 1.807(4),
P–O(1) 1.672(3), O(2)–C(2) 1.219(5), O(1)–C(2) 1.368(5), N(1)–C(2)
1.344(5);W–P–C(1) 119.85(16), O(1)–P–C(1) 100.11(17), C1–P–H 105(2),
P–O(1)–C(2) 117.5(3), O(1)–C(2)–N(1) 113.4(4), O(2)–C(2)–N(1)
125.9(5), O(1)–C(2)–O(2) 120.7(4).
Scheme 4 Scope of calculations, B3LYP/6-311G(d,p), LanL2DZ (W).
successfully used in quantifying many other different bonding
situations41i–k,43 and derived from Bader’s atoms-in-molecules
(AIM) theory44 was also included (Table 1). Both parameters
Fig. 2 BSSE corrected reaction Gibbs free energies and ZPE-corrected energi
complex Me-2b to yield phosphinidene oxide Me-4b. On path A: intermedi
(nucleophilic attack at carbon) and TS-II (extrusion of CO). Path B: dissociat
the (P)O–CO bond).
3528 | Chem. Sci., 2012, 3, 3526–3533
point to the following strength order for the endocyclic bonds:
P–O < P–C � C–O. The same trend was observed for the bond
stiffness quantified by the relaxed force constants, k0 (Table 1), a
robust and safely transferable parameter obtained as the recip-
rocal value of the compliance constant.41i,45 Worth mentioning is
that differences in strength stiffness are enlarged upon
complexation within the group of endocyclic bonds PO/PC/CO
in Ib, and the P–O bond becomes remarkably weakened.
An important feature of small rings is that their reactivity is
often triggered by enhanced ring strain.46 Therefore, ring strain
energies (RSE) of Ia,b were quantitatively evaluated by means of
appropriate homodesmotic reactions41i,j,47 (see the ESI†), where
the number and type of bonds and the valences of all atoms are
kept.48 In relation to the reported RSE for the 1,3,3-trimethyl
substituted oxaphosphirane (94.2 kJ mol�1) and its P-Cr(CO)5complex (92.0 kJ mol�1),47 the parent oxaphosphiranone Ia was
found to be much more strained (141.3 kJ mol�1, at LPNO-
NCEPA1/def2-TZVPP//B3LYP-D/def2-TZVP). Following the
same behaviour as the aforementioned fully saturated ring, the
chromium complex Ib displays a slightly higher value (144.7 kJ
mol�1) compared to unligated Ia.
Thermodynamic oxygen-transfer potentials (TOP’s)49 of the
redox couples [(CO)5M(PR)]/[(CO)5M(OPR)] and CO/CO2 are
shown in Fig. 3. By definition a species Z is oxidised by YO, if
TOPZ/ZO < TOPY/YO. Thus, for a phosphinidene complex
[(CO)5M(PR)] to be oxidised by CO2 a TOP < �386 kJ mol�1 is
required, which is the case for R¼Me (Me-2b), CH(SiMe3)2 (2b)
and for Niecke’s complex (Cr, R ¼ NiPr2),50 but not for R ¼ H
and CF3.
The first step in the oxidation of terminal phosphinidene
model complex Me-2b by carbon dioxide (Fig. 2, path A) is the
nucleophilic attack of the phosphorus centre at the carbon of CO2
(TS-I, Fig. 4).
Selected bond lengths of transition states TS-I and TS-II are
collected in Table 2.
Addition of Me-2b to CO2 proceeds with a barrier of 62 kJ
mol�1 to yield oxaphosphiran-3-one model complex Me-3b
(Fig. 2, Table 2). For this new class of compounds the fully
es for the deoxygenation of CO2 bymethyl-model tungsten phosphinidene
ate formation of oxaphosphiran-3-one Me-3b via transition states TS-I
ion of the van der Waals complex via transition state TS-III (breaking of
This journal is ª The Royal Society of Chemistry 2012
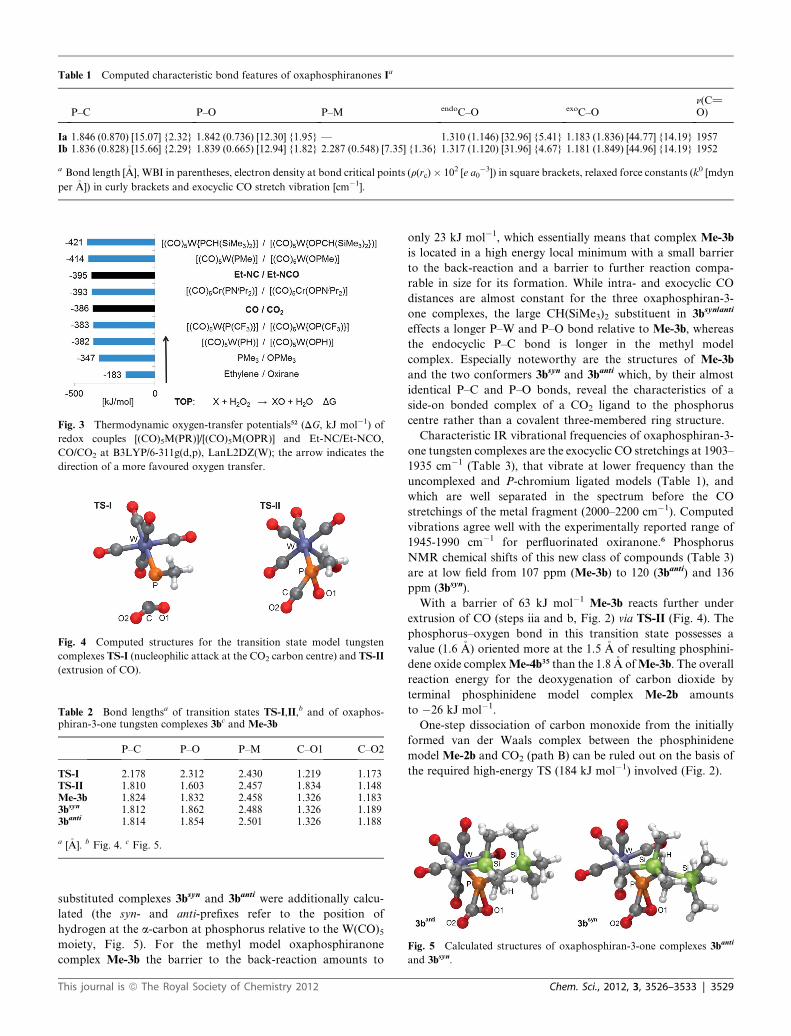
Table 1 Computed characteristic bond features of oxaphosphiranones Ia
P–C P–O P–M endoC–O exoC–On(C]O)
Ia 1.846 (0.870) [15.07] {2.32} 1.842 (0.736) [12.30] {1.95} — 1.310 (1.146) [32.96] {5.41} 1.183 (1.836) [44.77] {14.19} 1957Ib 1.836 (0.828) [15.66] {2.29} 1.839 (0.665) [12.94] {1.82} 2.287 (0.548) [7.35] {1.36} 1.317 (1.120) [31.96] {4.67} 1.181 (1.849) [44.96] {14.19} 1952
a Bond length [�A], WBI in parentheses, electron density at bond critical points (r(rc)� 102 [e a0�3]) in square brackets, relaxed force constants (k0 [mdyn
per �A]) in curly brackets and exocyclic CO stretch vibration [cm�1].
Table 2 Bond lengthsa of transition states TS-I,II,b and of oxaphos-phiran-3-one tungsten complexes 3bc and Me-3b
P–C P–O P–M C–O1 C–O2
TS-I 2.178 2.312 2.430 1.219 1.173TS-II 1.810 1.603 2.457 1.834 1.148Me-3b 1.824 1.832 2.458 1.326 1.1833bsyn 1.812 1.862 2.488 1.326 1.1893banti 1.814 1.854 2.501 1.326 1.188
a [�A]. b Fig. 4. c Fig. 5.
Fig. 3 Thermodynamic oxygen-transfer potentials52 (DG, kJ mol�1) of
redox couples [(CO)5M(PR)]/[(CO)5M(OPR)] and Et-NC/Et-NCO,
CO/CO2 at B3LYP/6-311g(d,p), LanL2DZ(W); the arrow indicates the
direction of a more favoured oxygen transfer.
Fig. 4 Computed structures for the transition state model tungsten
complexes TS-I (nucleophilic attack at the CO2 carbon centre) and TS-II
(extrusion of CO).
Fig. 5 Calculated structures of oxaphosphiran-3-one complexes 3banti
and 3bsyn.
substituted complexes 3bsyn and 3banti were additionally calcu-
lated (the syn- and anti-prefixes refer to the position of
hydrogen at the a-carbon at phosphorus relative to the W(CO)5moiety, Fig. 5). For the methyl model oxaphosphiranone
complex Me-3b the barrier to the back-reaction amounts to
This journal is ª The Royal Society of Chemistry 2012
only 23 kJ mol�1, which essentially means that complex Me-3b
is located in a high energy local minimum with a small barrier
to the back-reaction and a barrier to further reaction compa-
rable in size for its formation. While intra- and exocyclic CO
distances are almost constant for the three oxaphosphiran-3-
one complexes, the large CH(SiMe3)2 substituent in 3bsyn/anti
effects a longer P–W and P–O bond relative to Me-3b, whereas
the endocyclic P–C bond is longer in the methyl model
complex. Especially noteworthy are the structures of Me-3b
and the two conformers 3bsyn and 3banti which, by their almost
identical P–C and P–O bonds, reveal the characteristics of a
side-on bonded complex of a CO2 ligand to the phosphorus
centre rather than a covalent three-membered ring structure.
Characteristic IR vibrational frequencies of oxaphosphiran-3-
one tungsten complexes are the exocyclic CO stretchings at 1903–
1935 cm�1 (Table 3), that vibrate at lower frequency than the
uncomplexed and P-chromium ligated models (Table 1), and
which are well separated in the spectrum before the CO
stretchings of the metal fragment (2000–2200 cm�1). Computed
vibrations agree well with the experimentally reported range of
1945-1990 cm�1 for perfluorinated oxiranone.6 Phosphorus
NMR chemical shifts of this new class of compounds (Table 3)
are at low field from 107 ppm (Me-3b) to 120 (3banti) and 136
ppm (3bsyn).
With a barrier of 63 kJ mol�1 Me-3b reacts further under
extrusion of CO (steps iia and b, Fig. 2) via TS-II (Fig. 4). The
phosphorus–oxygen bond in this transition state possesses a
value (1.6 �A) oriented more at the 1.5 �A of resulting phosphini-
dene oxide complexMe-4b35 than the 1.8�A ofMe-3b. The overall
reaction energy for the deoxygenation of carbon dioxide by
terminal phosphinidene model complex Me-2b amounts
to �26 kJ mol�1.
One-step dissociation of carbon monoxide from the initially
formed van der Waals complex between the phosphinidene
model Me-2b and CO2 (path B) can be ruled out on the basis of
the required high-energy TS (184 kJ mol�1) involved (Fig. 2).
Chem. Sci., 2012, 3, 3526–3533 | 3529
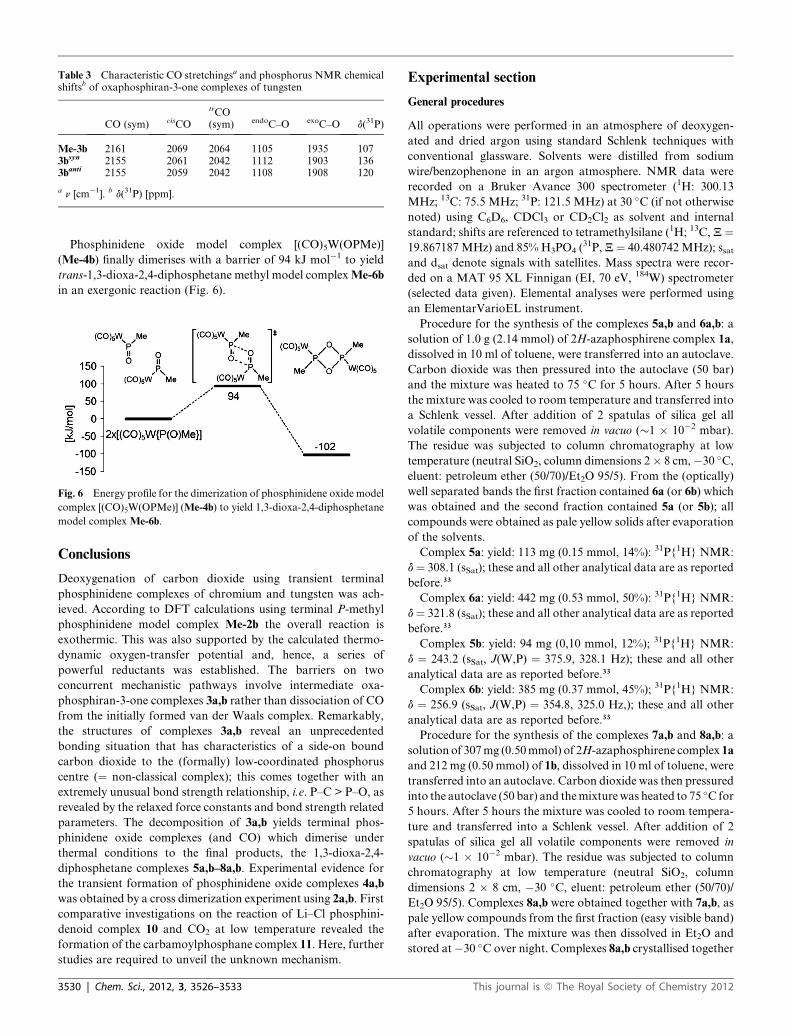
Table 3 Characteristic CO stretchingsa and phosphorus NMR chemicalshiftsb of oxaphosphiran-3-one complexes of tungsten
CO (sym) cisCO
trCO(sym) endoC–O exoC–O d(31P)
Me-3b 2161 2069 2064 1105 1935 1073bsyn 2155 2061 2042 1112 1903 1363banti 2155 2059 2042 1108 1908 120
a n [cm�1]. b d(31P) [ppm].
Phosphinidene oxide model complex [(CO)5W(OPMe)]
(Me-4b) finally dimerises with a barrier of 94 kJ mol�1 to yield
trans-1,3-dioxa-2,4-diphosphetane methyl model complexMe-6b
in an exergonic reaction (Fig. 6).
Fig. 6 Energy profile for the dimerization of phosphinidene oxide model
complex [(CO)5W(OPMe)] (Me-4b) to yield 1,3-dioxa-2,4-diphosphetane
model complex Me-6b.
Conclusions
Deoxygenation of carbon dioxide using transient terminal
phosphinidene complexes of chromium and tungsten was ach-
ieved. According to DFT calculations using terminal P-methyl
phosphinidene model complex Me-2b the overall reaction is
exothermic. This was also supported by the calculated thermo-
dynamic oxygen-transfer potential and, hence, a series of
powerful reductants was established. The barriers on two
concurrent mechanistic pathways involve intermediate oxa-
phosphiran-3-one complexes 3a,b rather than dissociation of CO
from the initially formed van der Waals complex. Remarkably,
the structures of complexes 3a,b reveal an unprecedented
bonding situation that has characteristics of a side-on bound
carbon dioxide to the (formally) low-coordinated phosphorus
centre (¼ non-classical complex); this comes together with an
extremely unusual bond strength relationship, i.e. P–C > P–O, as
revealed by the relaxed force constants and bond strength related
parameters. The decomposition of 3a,b yields terminal phos-
phinidene oxide complexes (and CO) which dimerise under
thermal conditions to the final products, the 1,3-dioxa-2,4-
diphosphetane complexes 5a,b–8a,b. Experimental evidence for
the transient formation of phosphinidene oxide complexes 4a,b
was obtained by a cross dimerization experiment using 2a,b. First
comparative investigations on the reaction of Li–Cl phosphini-
denoid complex 10 and CO2 at low temperature revealed the
formation of the carbamoylphosphane complex 11. Here, further
studies are required to unveil the unknown mechanism.
3530 | Chem. Sci., 2012, 3, 3526–3533
Experimental section
General procedures
All operations were performed in an atmosphere of deoxygen-
ated and dried argon using standard Schlenk techniques with
conventional glassware. Solvents were distilled from sodium
wire/benzophenone in an argon atmosphere. NMR data were
recorded on a Bruker Avance 300 spectrometer (1H: 300.13
MHz; 13C: 75.5 MHz; 31P: 121.5 MHz) at 30 �C (if not otherwise
noted) using C6D6, CDCl3 or CD2Cl2 as solvent and internal
standard; shifts are referenced to tetramethylsilane (1H; 13C, X ¼19.867187 MHz) and 85% H3PO4 (
31P, X¼ 40.480742 MHz); ssatand dsat denote signals with satellites. Mass spectra were recor-
ded on a MAT 95 XL Finnigan (EI, 70 eV, 184W) spectrometer
(selected data given). Elemental analyses were performed using
an ElementarVarioEL instrument.
Procedure for the synthesis of the complexes 5a,b and 6a,b: a
solution of 1.0 g (2.14 mmol) of 2H-azaphosphirene complex 1a,
dissolved in 10 ml of toluene, were transferred into an autoclave.
Carbon dioxide was then pressured into the autoclave (50 bar)
and the mixture was heated to 75 �C for 5 hours. After 5 hours
the mixture was cooled to room temperature and transferred into
a Schlenk vessel. After addition of 2 spatulas of silica gel all
volatile components were removed in vacuo (�1 � 10�2 mbar).
The residue was subjected to column chromatography at low
temperature (neutral SiO2, column dimensions 2� 8 cm,�30 �C,eluent: petroleum ether (50/70)/Et2O 95/5). From the (optically)
well separated bands the first fraction contained 6a (or 6b) which
was obtained and the second fraction contained 5a (or 5b); all
compounds were obtained as pale yellow solids after evaporation
of the solvents.
Complex 5a: yield: 113 mg (0.15 mmol, 14%): 31P{1H} NMR:
d¼ 308.1 (sSat); these and all other analytical data are as reported
before.33
Complex 6a: yield: 442 mg (0.53 mmol, 50%): 31P{1H} NMR:
d¼ 321.8 (sSat); these and all other analytical data are as reported
before.33
Complex 5b: yield: 94 mg (0,10 mmol, 12%); 31P{1H} NMR:
d ¼ 243.2 (sSat, J(W,P) ¼ 375.9, 328.1 Hz); these and all other
analytical data are as reported before.33
Complex 6b: yield: 385 mg (0.37 mmol, 45%); 31P{1H} NMR:
d ¼ 256.9 (sSat, J(W,P) ¼ 354.8, 325.0 Hz,); these and all other
analytical data are as reported before.33
Procedure for the synthesis of the complexes 7a,b and 8a,b: a
solution of 307mg (0.50mmol) of 2H-azaphosphirene complex 1a
and 212 mg (0.50 mmol) of 1b, dissolved in 10 ml of toluene, were
transferred into an autoclave. Carbon dioxide was then pressured
into the autoclave (50 bar) and themixturewas heated to 75 �C for
5 hours. After 5 hours the mixture was cooled to room tempera-
ture and transferred into a Schlenk vessel. After addition of 2
spatulas of silica gel all volatile components were removed in
vacuo (�1 � 10�2 mbar). The residue was subjected to column
chromatography at low temperature (neutral SiO2, column
dimensions 2 � 8 cm, �30 �C, eluent: petroleum ether (50/70)/
Et2O 95/5). Complexes 8a,b were obtained together with 7a,b, as
pale yellow compounds from the first fraction (easy visible band)
after evaporation. The mixture was then dissolved in Et2O and
stored at�30 �C over night. Complexes 8a,b crystallised together
This journal is ª The Royal Society of Chemistry 2012

with 7a,b. Therefore, the following analytical data were obtained
from NMR spectra from solutions of this mixture.
Complex 7a/b: 31P{1H} NMR: d ¼ 306.1 (dSat,2J(P,P) ¼
16.7 Hz), 244.1 (dSat,2J(P,P) ¼ 24.3 Hz; 1J(W,P) could not be
determined); these and all other analytical data are as reported
before.33Complex 8a/b: 31P{1H}NMR: d¼ 319.0 (dSat,2J(P,P)¼
16.7 Hz), 259.3 (dSat,2J(P,P) ¼ 16.7 Hz, 1J(W,P) ¼ 339.8 Hz);
these and all other analytical data are as reported before.33
Procedure for the synthesis of complex 11: a solution of
832 mg (1.45 mmol) of the [bis(trimethylsilyl)methyl]chlor-
ophosphane complex 9 and 256 mg (1.45 mmol) of 12-crown-4,
dissolved in 5 ml of Et2O, was added dropwise to a cooled
(�78 �C) solution of LDA (170 mg (1.59 mmol) in 5 ml Et2O).
The reaction mixture was stirred for 10 min at �78 �C before
warming to�60 �C. CO2 was bubbled through the yellow-orange
solution for about 1 min. An immediate colour change to col-
ourless and precipitation of LiCl was observed. The mixture was
warmed to room temperature (3 hours), filtered and dried in
vacuo (�1� 10�2 mbar). After extraction with n-pentane (10 ml),
the solution was concentrated to 3 ml and stored over night
at �25 �C. Complex 11 crystallised as a pale yellow product.
Complex 11: yield: 750 mg (1.13 mmol 78%); 1H NMR: d ¼0.05 (s, 9H, Si(CH3)3), 0.23 (s, 9H, Si(CH3)3), 0.87 (dd, 6H,3J(H,H) ¼ 6.7 Hz, iPr), 1.15 (d, 1H, 3J(H,H) ¼ 2.3 Hz, PCH),
1.29 (dd, 6H, 3J(H,H) ¼ 6.6 Hz, CHiPr), 2.99 (q, 1H, 3J(H,H) ¼6.6 Hz, CHiPr), 4.29 (q, 1H, 3J(H,H) ¼ 6.6 Hz, CHiPr), 8.40 (dd,
1H, 1J(P,H) ¼ 365.3 Hz, 1J(W,H) ¼ 9.5 Hz, 3J(H,H) ¼ 2.3 Hz,
PH); 31P{1H} NMR: d ¼ 83.59 (1J(W,P) ¼ 277.6); 13C{1H}
NMR: d ¼ 198.7 (d, 2J(P,C) ¼ 27.8 Hz; trans-CO), 196.9 (d,1J(W,C)¼ 125.8 Hz, 2J(P,C)¼ 7.5 Hz, cis-CO), 48.1 (s, iPr), 45.6
(s, iPr), 22.1 (d, 1J(P,C) ¼ 5.8 Hz PCH), 20.3 (d, 1J(P,C) ¼ 5.8
Hz, PCH), 20.2 (s, NCMe2), 1.87 (d, 3J(P,C) ¼ 3.3 Hz, SiMe3),
0.21 (d, 3J(P,C)¼ 3.3 Hz, SiMe3); MS (EI, 70 eV, 184W):m/z (%):
631 ([M]+, 40), 603 ([M � 1CO]+, 100), 575 ([M � 2CO]+, 45),
547 ([M � 3CO]+, 45), 517 ([M � 4CO]+, 63), 502 ([M � 5CO]+,
40); elemental analysis (%) calculated for C19H34NO7PSi2W: C
34.60, H 5.20, N 2.12; found: C 34.64, H 5.20, N 2.00%.
Computational details
Geometry optimizations, frequency and energetic calculations
were performed with the GAUSSIAN03 program suite51 at
B3LYP/6-311g(d,p), with the LanL2DZ basis set at W. Basis set
superposition errors (BSSE) were corrected with the counterpoise
method.All reported energies include correction for the zero point
energy (ZPE) term. Nuclear magnetic shieldings were calculated
using the GIAO method52 with ADF(2009.01)53 at VWNBP86/
TZ2P SO ZORA (LDA: VWN, gradient correction: Becke88/
Perdew86, SO ¼ spin orbit, ZORA ¼ zeroth order regular
approximation).54Minima and transition states of all compounds
were characterised by a number of zero or one imaginary
frequencies. RSEwere computed by single point (SP) calculations
at the LPNO-NCEPA1 level,55 using the def2-TZVPP basis set
and the geometries andZPE correction obtained at theB3LYP-D/
def2-TZVP level of theory,56 using the ORCA electronic structure
program package.57 Bond properties were computed using the
more polarised def2-TZVPP basis set. Electron densities at BCP
were computed with the AIM2000 program58 and WBI were
obtained from the Natural Bond Orbital (NBO) analysis.
This journal is ª The Royal Society of Chemistry 2012
X-Ray crystallographic analysis
Suitable yellow single crystals of 11 were obtained from concen-
trated n-pentane solutions upon decreasing the temperature from
ambient temperature to �25 �C. Data were collected on Nonius
Kappa CCD diffractometer equipped with a low-temperature
device (Cryostream,OxfordCryosystems) at 123Kusing graphite
monochromated Mo-Ka radiation (l ¼ 0.71073 �A). The struc-
tures were solved by Patterson methods (SHELXS-97) and
refined by full-matrix least squares on F2 (SHELXL-97).59
Crystal structure data of complex 11 (C19H34NO7PSi2W):
crystal size 0.24 � 0.24 � 0.22 mm, triclinic, P-1, a ¼10.7620(3), b ¼ 11.0051(3), c ¼ 25.8757(9) �A, a ¼ 85.0791(15)�,b ¼ 82.5425(16)�, g ¼ 64.6070(15)�, V ¼ 2743.59(14) �A3, Z ¼ 4,
rcalc ¼ 1.599 Mg m�3, 2qmax ¼ 29�, collected (independent)
reflections ¼ 30036 (13638), Rint ¼ 0.0391, m ¼ 4.391 mm�1, 589
refined parameters, 1 restraints, R1 (for I > 2s(I)) ¼ 0.0644,
wR2 (for all data) ¼ 0.0727, max./min. residual electron
density ¼ 1.996/�1.525 e �A�3.
The CCDC number 806428 (11) contains the supplementary
crystallographic data for this paper. These data can be obtained
free of charge from the Cambridge Crystallographic Data Centre
via www.ccdc.cam.ac.uk/data_request/cif or in the ESI.†
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft
(STR 411/25-2 and 411/26-1) and the COST action CM0802
‘‘PhoSciNet’’ is gratefully acknowledged. We thank the J€ulich
Supercomputing Centre (JuRoPa@JSC; HBN12) and the
computing centre of the RWTH-Aachen for computing time.
G. S. thanks Prof. A. C. Filippou for support.
Notes and references
1 (a) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH2010; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev.,2007, 107, 2365–2387.
2 C. A. Tsipis and P. A. Karipidis, J. Phys. Chem. A, 2005, 109, 8560–8567.
3 D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780.4 D. E. Milligan and M. E. Jacox, J. Chem. Phys., 1962, 36, 2911–2917.
5 D. Kovacs and J. E. Jackson, J. Phys. Chem. A, 2005, 105, 7579–7587.6 P. L. Coe, A. Sellars, J. Colin Tatlow, G. Whittaker andH. C. Fielding, J. Chem. Soc., Chem. Commun., 1982, 362–363.
7 (a) G. Fachinetti, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Am.Chem. Soc., 1979, 101, 1767–1775; (b) I. Castro-Rodriguez andK. Meyer, Chem. Commun., 2006, 1353–1368, and references citedtherein.
8 U. Jayarathne, P. Chandrasekaran, H. Jacobsen, J. T. Mague andJ. P. Donahue, Dalton Trans., 2010, 39, 9662–9671.
9 S. Gambarotta, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Am.Chem. Soc., 1985, 107, 2985–2986.
10 S. Komiya, M. Akita, N. Kasuga, M. Hirano and A. Fukuoka,J. Chem. Soc., Chem. Commun., 1994, 1115–1116.
11 M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero,J. Chem. Soc., Chem. Commun., 1975, 636–637.
12 J. Li and Z. Lin, Organometallics, 2009, 28, 4231–4234.13 C. H. Lee, D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem.
Soc., 2007, 129, 13802–13803.14 R. Becerra, J. P. Cannady and R. Walsh, J. Phys. Chem. A, 2002, 106,
4922–4927.15 A. Herize, J. R. Mora, J. Lezama, E. Marquez, T. Cordova and
G. Chuchani, J. Phys. Org. Chem., 2009, 22, 170–179.16 S. P. So and W.-K. Li, J. Phys. Chem. A, 2004, 108, 4002–4007.
Chem. Sci., 2012, 3, 3526–3533 | 3531

17 V. Griffiths and J. C. Tebby, J. Chem. Soc., Chem. Commun., 1981,607–608.
18 J. Fournier, C. Bruneau and P. H. Dixneuf, Tetrahedron Lett., 1989,30, 3981–3982.
19 C. Bruneau and P. H. Dixneuf, J. Mol. Catal., 1994, 74, 97–107.20 Y. Kayaki, M. Yamamoto and T. Ikariya, J. Org. Chem., 2007, 72,
647–649.21 G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and
G. Bertrand, Science, 2007, 316, 439–449.22 (a) C. M. Fr€omming, E. Otten, G. Kehr, R. Fr€ohlich, S. Grimme,
D. W. Stephan and G. Erker, Angew. Chem., 2009, 121, 6770–6773;C. M. Fr€omming, E. Otten, G. Kehr, R. Fr€ohlich, S. Grimme,D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48,6643–6646; (b) D. W. Stephan, Dalton Trans., 2009, 3129–3136.
23 R. C. Neu, E. Y. Ouyang, S. J. Geier, X. Zhao, A. Ramos andD. W. Stephan, Dalton Trans., 2010, 39, 4285–4294.
24 (a) D. W. Stephan and G. Erker, Angew. Chem., 2010, 122, 50–81; (b)X. Zhao and D. W. Stephan, Chem. Sci., 2012, 3, 2123–2132.
25 P. P. Power, Nature, 2010, 463, 171–177.26 L. Gu and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 914–915.27 Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., 2009, 121,
4258–4261; Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem.,Int. Ed., 2009, 48, 4194–4197.
28 (a) A. Berkefeld, W. E. Piers andM. Parvez, J. Am. Chem. Soc., 2010,132, 10660–10661; (b) G. M�enard and D. W. Stephan, Angew. Chem.,2011, 123, 8546–8549; G. M�enard and D. W. Stephan, Angew. Chem.,Int. Ed., 2011, 50, 8396–8399; (c) G. M�enard and D. W. Stephan,J. Am. Chem. Soc., 2010, 132, 1796–1797; (d) A. E. Ashley,A. L. Thompson and D. O’Hare, Angew. Chem., 2009, 121, 10023–10027; A. E. Ashley, A. L. Thompson and D. O’Hare, Angew.Chem., Int. Ed., 2009, 48, 9839–9843.
29 V. Nair, V. Varghese, R. R. Paul, A. Jose, C. R. Sinu andR. S. Menon, Org. Lett., 2010, 12, 2653–2655.
30 (a) L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem.,2012, 124, 4792–4795; (b) For reactions of electron-richtrisaminophosphanes with carbon dioxide, see: R. W. Light,L. D. Hutchins, R. T. Paine and C. F. Campana, Inorg. Chem.,1980, 19, 3597–3604; (c) For reactions of acyclicamidophosphoranes with carbon dioxide, see: R. G. Cavell,K. I. The and L. V. Griend, Inorg. Chem., 1981, 20, 3813–3818.
31 (a) K. Issleib and H. Weichmann, Chem. Ber., 1964, 97, 721–724; (b)K. Diemert, T. Hahn and W. Kuchen, J. Organomet. Chem., 1994,476, 173–181; (c) K. Diemert, T. Hahn, W. Kuchen, D. Mootz,W. Poll and P. Tommes, Z. Naturforsch., B: J. Chem. Sci., 1995,50, 209; (d) the parent phosphanyl carboxylic acid was characterisedonly as [(CO)5Cr{H2PCO2H}] (ref. 2 and 3).
32 R. Streubel, F. Ruthe and P. G. Jones, Eur. J. Inorg. Chem., 1998,571–574.
33 R. Streubel, A. Ostrowski, S. Priemer, U. Rohde, J. Jeske andP. G. Jones, Eur. J. Inorg. Chem., 1998, 257–261.
34 Reactions of 1a, b in toluene at 75 �C and atmospheric pressure ofCO2 were unselective; among the products formed are those knownto be formed under argon atmosphere (otherwise unchangedconditions).
35 (a) F. Mathey, N. H. Tran Huy and A. Marinetti, Helv. Chim. Acta,2001, 84, 2938–2957; (b) F. Mathey, Angew. Chem., 2003, 115, 1616–1643; F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604; (c)K. Lammertsma, Top. Curr. Chem., 2003, 229, 95–119; (d)H. Aktas, C. J. Slootweg and K. Lammertsma, Angew. Chem.,2010, 122, 2148–2159; H. Aktas, C. J. Slootweg andK. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 2102–2113.
36 (a) C. Schulten, G. von Frantzius, G. Schnakenburg and R. Streubel,Heteroat. Chem., 2011, 22, 275–286; (b) for 1,3-dioxo-2s4l5,4s4l5-diphosphetanes, see: H. M. Schiffner, PhD thesis, University ofBonn, 1991.
37 A. €Ozbolat, G. von Frantzius, J. M. Perez, M. Nieger and R. Streubel,Angew. Chem., 2007, 119, 9488–9491; A. €Ozbolat, G. von Frantzius,J. M. Perez, M. Nieger and R. Streubel, Angew. Chem., Int. Ed.,2007, 46, 9327–9330.
38 R. Streubel, M. Bode, J. M. P�erez, G. Schnakenburg, J. Daniels andP. G. Jones, Z. Anorg. Allg. Chem., 2009, 635, 1163–1171.
39 R. Streubel, S. Priemer, F. Ruthe and P. G. Jones, Eur. J. Inorg.Chem., 2000, 1253–1259.
40 T. P.M.Goumans,A.W.Ehlers,K.LammertsmaandE.U.W€urthwein,Eur. J. Org. Chem., 2003, 2941–2946, here, the relative stability of
3532 | Chem. Sci., 2012, 3, 3526–3533
phosphiranone was compared to its methylene–oxaphosphirane isomer,but their RSE values were not reported.
41 For recent examples see: (a) C. Bleiholder, F. Rominger andR. Gleiter, Organometallics, 2009, 28, 1014–1017; (b) A. Noor,G. Glatz, R. M€uller, M. Kaupp, S. Demeshko and R. Kempe, Nat.Chem., 2009, 1, 322–325; (c) P. R. Schreiner, H. P. Reisenauer,J. Romanski and G. Mloston, Angew. Chem., Int. Ed., 2009, 48,8133–8136; (d) H. Helten, S. Fankel, O. Feier-Iova, M. Nieger,A. Espinosa Ferao and R. Streubel, Eur. J. Inorg. Chem., 2009,3226–3237; (e) F. Ot�on, I. Ratera, A. Espinosa, K. Wurst,T. Parella, A. T�arraga, J. Veciana and P. Molina, Chem.–Eur. J.,2010, 16, 1532–1542; (f) M. Feller, E. Ben-Ari, M. A. Iron,Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, L. Konstantinovskiand D. Milstein, Inorg. Chem., 2010, 49, 1615–1625; (g)H. Braunschweig, K. Radacki and K. Schwab, Chem. Commun.,2010, 46, 913–915; (h) J. D. Epping, S. Yao, M. Karni, Y. Apeloigand M. Driess, J. Am. Chem. Soc., 2010, 132, 5443–5455; (i)A. Espinosa and R. Streubel, Chem.–Eur. J., 2011, 17, 3166–3178;(j) A. Espinosa, C. G�omez and R. Streubel, Inorg. Chem., 2012, 51,7250–7256; (k) A. Espinosa and R. Streubel, Chem.–Eur. J., 2012,DOI: 10.1002/chem.201201057.
42 K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096.43 For recent examples applied to intramolecular bonding see: (a) J. Ran
and M. W. Wong, Aust. J. Chem., 2009, 62, 1062–1067; (b) H. Wang,I. G. Csizmadia, I. Marsi, G. A. Chasse, D. Fang and B. Viskolcz,J. Chem. Phys., 2009, 131, 035105/1–035105/8; (c) F. Ot�on,A. Espinosa, A. T�arraga, I. Ratera, K. Wurst, J. Veciana andP. Molina, Inorg. Chem., 2009, 48, 1566–1576; (d) H. Raissi,A. F. Jalbout, B. Abbasi, F. Fazli, F. Farzad, E. Nadim and A. deLeon, Int. J. Quantum Chem., 2010, 110, 893–901.
44 R. F. W. Bader, in Atoms in Molecules: A Quantum Theory, OxfordUniversity Press, Oxford, 1990.
45 (a) W. T. Taylor andK. S. Pitzer, J. Res. Natl. Bur. Stand., 1947, 38, 1;(b) P. G. Maslov, Dokl. Akad. Nauk SSSR, 1949, 67, 819; (c)J. C. Decius, J. Chem. Phys., 1963, 38, 241–248; (d) K. Brandhorstand J. Grunenberg, ChemPhysChem, 2007, 8, 1151–1156; (e)K. Brandhorst and J. Grunenberg, Chem. Soc. Rev., 2008, 37,1558–1567.
46 C. J. M. Stirling, Pure Appl. Chem., 1984, 56, 1781–1796.47 O. Krahe, F. Neese and R. Streubel, Chem.–Eur. J., 2009, 15, 2594–
2601. RSE computed at SCS-MP2/def2-TZVPP//BP86/def2-TZVP.48 P. George, M. Trachtman, C. W. Bock and A. M. Brett, Tetrahedron,
1976, 32, 317–323.49 D. V. Deubel, J. Am. Chem. Soc., 2004, 126, 996–997.50 E. Niecke, M. Engelmann, H. Zorn, B. Krebs and G. Henkel, Angew.
Chem., 1980, 92, 738–739; E. Niecke, M. Engelmann, H. Zorn,B. Krebs and G. Henkel, Angew. Chem., Int. Ed. Engl., 1980, 19,710–712.
51 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone,B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li,H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng,J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda,J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao,H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta,F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin,V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari,A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi,N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross,V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann,O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski,R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth,P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels,€O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, andD. J. Fox, Gaussian 03, Revision E.01, Gaussian, Inc., WallingfordCT, 2009.
52 (a) G. Schreckenbach and T. Ziegler, J. Phys. Chem., 1995, 99, 606; (b)G. Schreckenbach and T. Ziegler, Int. J. Quantum Chem., 1997, 61,899; (c) S. K. Wolff and T. Ziegler, J. Chem. Phys., 1998, 109, 895.
53 ADF2009.01, SCM, Theoretical Chemistry, Vrije Universiteit,Amsterdam, The Netherlands, http://www.scm.com.
54 (a) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys.,1993, 99, 4597; (b) E. van Lenthe, E. J. Baerends and J. G. Snijders,J. Chem. Phys., 1994, 101, 9783; (c) E. van Lenthe, J. G. Snijdersand E. J. Baerends, J. Chem. Phys., 1994, 105, 6505–6516; (d)
This journal is ª The Royal Society of Chemistry 2012

E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int.J. Quantum Chem., 1996, 57, 281; (e) C. Fonseca Guerra,J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc.,1998, 99, 391; (f) E. van Lenthe, A. E. Ehlers and E. J. Baerends,J. Chem. Phys., 1999, 110, 8943; (g) G. te Velde, F. M. Bickelhaupt,S. J. A. van Gisbergen, C. Fonseca Guerra, E. J. Baerends,J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931.
55 F. Wennmohs and F. Neese, Chem. Phys., 2008, 343, 217–230.56 Including the Grimme’s damped semiempirical correction accounting
for the major part of the contribution of dispersion forces to theenergy: (a) S. Grimme, J. Comput. Chem., 2004, 25, 1463–1476; (b)S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799.
This journal is ª The Royal Society of Chemistry 2012
57 ORCA – An ab initio, DFT and semiempirical SCF-MO package.Written by F. Neese, Max Planck Institute for BioinorganicChemistry, D-45470 M€ulheim/Ruhr, 2012, Version 2.9.0, http://www.mpibac.mpg.de/bac/logins/neese/description.php; for a recentupdate, see: F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78.
58 AIM2000 v. 2.0, designed by F. Biegler-K€onig, and J. Sch€onbohm,2002, http://www.aim2000.de/(a) F. Biegler-K€onig, J. Sch€onbohmand D. Bayles, J. Comput. Chem., 2001, 22, 545–559; (b) F. Biegler-K€onig and J. Sch€onbohm, J. Comput. Chem., 2002, 23, 1489–1494.
59 (a) SHELXS-97,Acta Crystallogr., Sect. A: Found. Crystallogr., 1990,46, 467; (b) G. M. Sheldrick, SHELXL-97; University of G€ottingen,1997.
Chem. Sci., 2012, 3, 3526–3533 | 3533
Related Documents











