Autophagy is a cellular mechanism by which neurons recycle cytosolic proteins and organelles, playing a critical role in neuronal adaptation during nutrient deprivation and other forms of neuronal damage. The molecular mechanism of autophagy involves sequestration of these neuronal macro- molecules and organelles in double membrane cytoplasmic vesicles called autophagosomes, which fuse with lysosomes to form autophagolysosomes, being the macromolecules degraded by lysosomal hydrolases (Klionsky and Emr 2000). Furthermore, autophagy also contributes to maintain cellular homeostasis, because it regulates normal turnover of organ- elles to assure quality control of essential neuronal compo- nents (Degenhardt et al. 2006). Autophagy is also involved in other important processes, such as metabolic stress, cell proliferation, growth, and differentiation (Kroemer and Levine 2008). Thus, defective autophagy has been implicated in the pathogenesis of diverse diseases including myopathies, Received August 2, 2011; revised manuscript received October 24, 2011; accepted October 24, 2011. Address correspondence and reprint requests to Prof. Valentı ´n Cen ˜a, Facultad de Medicina, Unidad Asociada Neurodeath, Avda. Almansa, 14, 02006 Albacete, Spain. E-mail: [email protected] Abbreviations used: 3-MA, 3-methyl adenine; Atg, autophagy-related gene; FAM-siRNA, fluorescein-labeled siRNA; LC3, microtubule-asso- ciated light chain 3; LDH, lactate dehydrogenase; MDC, monodansyl- cadaverine; siRNA, small interfering RNA; TGD, TRANSGEDEN. , *NanoDrugs, S.L. Parque Cientı ´fico y Tecnolo ´gico, Albacete, Spain Departamento de Quı ´mica Inorga ´nica, Orga ´nica y Bioquı ´mica, Facultad de Quı ´mica, Universidad de Castilla-La Mancha, Ciudad Real, Spain àDepartamento de Ciencias Me ´dicas, Facultad de Medicina, UCLM, Albacete, Spain §Departamento de Ciencias Me ´dicas, Unidad Asociada Neurodeath, CSIC-Universidad de Castilla-La Mancha, Albacete, Spain ¶CIBERNED, Instituto de Salud Carlos III, Madrid, Spain Abstract Autophagy is an important process which plays a key role in cellular homeostasis by degrading cytoplasmic components in the lysosomes, which facilitates recycling. Alterations to nor- mal autophagy have been linked to excitotoxicity, but the mechanisms governing its signal transduction remain unclear. The aim of this study was to explore the role of autophagy in neuronal excitotoxic death by delivering small interfering RNA (siRNA) to rat cortical neurons, using a dendrimer to silence the autophagy-related gene 6 (beclin 1) and to determine the role of autophagy in excitotoxicity. We have found that the dendrimer is very efficient to deliver siRNA to rat cortical neurons, leading to almost complete removal of the target protein Beclin 1. In addition, NMDA increases autophagy markers, such as the protein levels of Beclin 1, the micro- tubule-associated light chain 3 (LC3) B-II/LC3B-I ratio, and monodansylcadaverine (MDC) labeling in rat cortical neurons. Moreover, NMDA also increases the formation of auto- phagosomes observed under a transmission electron micro- scope. Silencing beclin 1 expression blocked NMDA-induced autophagy. Moreover, Beclin 1 removal potentiated NMDA- induced neuronal death indicating that autophagy plays a protective role during excitotoxicity and suggesting that tar- geting autophagy might be a helpful therapeutic strategy in neurodegenerative diseases. Keywords: Beclin 1, dendrimer, excitotoxicity, siRNA. J. Neurochem. (2012) 120, 259–268. JOURNAL OF NEUROCHEMISTRY | 2012 | 120 | 259–268 doi: 10.1111/j.1471-4159.2011.07556.x ȑ 2011 The Authors Journal of Neurochemistry ȑ 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268 259

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Autophagy is a cellular mechanism by which neurons recyclecytosolic proteins and organelles, playing a critical role inneuronal adaptation during nutrient deprivation and otherforms of neuronal damage. The molecular mechanism ofautophagy involves sequestration of these neuronal macro-molecules and organelles in double membrane cytoplasmicvesicles called autophagosomes, which fuse with lysosomesto form autophagolysosomes, being the macromoleculesdegraded by lysosomal hydrolases (Klionsky and Emr 2000).Furthermore, autophagy also contributes to maintain cellularhomeostasis, because it regulates normal turnover of organ-elles to assure quality control of essential neuronal compo-nents (Degenhardt et al. 2006). Autophagy is also involved
in other important processes, such as metabolic stress, cellproliferation, growth, and differentiation (Kroemer andLevine 2008). Thus, defective autophagy has been implicatedin the pathogenesis of diverse diseases including myopathies,
Received August 2, 2011; revised manuscript received October 24,2011; accepted October 24, 2011.Address correspondence and reprint requests to Prof. Valentın Cena,
Facultad de Medicina, Unidad Asociada Neurodeath, Avda. Almansa,14, 02006 Albacete, Spain. E-mail: [email protected] used: 3-MA, 3-methyl adenine; Atg, autophagy-related
gene; FAM-siRNA, fluorescein-labeled siRNA; LC3, microtubule-asso-ciated light chain 3; LDH, lactate dehydrogenase; MDC, monodansyl-cadaverine; siRNA, small interfering RNA; TGD, TRANSGEDEN.
,
*NanoDrugs, S.L. Parque Cientıfico y Tecnologico, Albacete, Spain
�Departamento de Quımica Inorganica, Organica y Bioquımica, Facultad de Quımica, Universidad de
Castilla-La Mancha, Ciudad Real, Spain
�Departamento de Ciencias Medicas, Facultad de Medicina, UCLM, Albacete, Spain
§Departamento de Ciencias Medicas, Unidad Asociada Neurodeath, CSIC-Universidad de Castilla-La
Mancha, Albacete, Spain
¶CIBERNED, Instituto de Salud Carlos III, Madrid, Spain
Abstract
Autophagy is an important process which plays a key role in
cellular homeostasis by degrading cytoplasmic components in
the lysosomes, which facilitates recycling. Alterations to nor-
mal autophagy have been linked to excitotoxicity, but the
mechanisms governing its signal transduction remain unclear.
The aim of this study was to explore the role of autophagy in
neuronal excitotoxic death by delivering small interfering RNA
(siRNA) to rat cortical neurons, using a dendrimer to silence
the autophagy-related gene 6 (beclin 1) and to determine the
role of autophagy in excitotoxicity. We have found that the
dendrimer is very efficient to deliver siRNA to rat cortical
neurons, leading to almost complete removal of the target
protein Beclin 1. In addition, NMDA increases autophagy
markers, such as the protein levels of Beclin 1, the micro-
tubule-associated light chain 3 (LC3) B-II/LC3B-I ratio, and
monodansylcadaverine (MDC) labeling in rat cortical neurons.
Moreover, NMDA also increases the formation of auto-
phagosomes observed under a transmission electron micro-
scope. Silencing beclin 1 expression blocked NMDA-induced
autophagy. Moreover, Beclin 1 removal potentiated NMDA-
induced neuronal death indicating that autophagy plays a
protective role during excitotoxicity and suggesting that tar-
geting autophagy might be a helpful therapeutic strategy in
neurodegenerative diseases.
Keywords: Beclin 1, dendrimer, excitotoxicity, siRNA.
J. Neurochem. (2012) 120, 259–268.
JOURNAL OF NEUROCHEMISTRY | 2012 | 120 | 259–268 doi: 10.1111/j.1471-4159.2011.07556.x
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268 259

neuronal degeneration, infectious diseases, and cancer(Shintani and Klionsky 2004). Beclin 1, also known asautophagy-related gene (Atg) 6, plays a key role inautophagy. Beclin 1 was initially isolated as a Bcl-2interacting protein (Furuya et al. 2005). Beclin 1 regulatesthe commencement of the autophagic pathway by forming acomplex with type III phosphatidylinositol-3-kinase, respon-sible for autophagic vesicle nucleation. In the mammalianadult brain, Beclin 1 is expressed both in neurons (Diskinet al. 2005) and astrocytes (Rohn et al. 2011) and has beenfound in brain tissues obtained from elderly humans as wellas from patients with Huntington and Alzheimer disease(Shibata et al. 2006; Pickford et al. 2008).
Excitotoxicity mediated by NMDA receptor activity ischaracterized by massive Ca2+ influx into the neurons, whichis accumulated in the mitochondria, triggering apoptosis orprogrammed cell death in different cell types. The mito-chondria play a key role regulating the apoptotic mechanismsand also in some forms of cell death by necrosis. Calciumoverload induces mitochondrial reactive oxygen speciesgeneration, mitochondrial inner membrane permeabilizationthat promotes mitochondrial swelling, outer membranerupture, and release of intermembrane proapoptotic proteins,such as cytochrome c and apoptosis-inducing factor to thecytoplasm (Bossy-Wetzel and Green 2000). These factorsalso activate caspases and, subsequently, caspase-activatedDNase (Bossy-Wetzel and Green 2000) leading to neuronaldeath. Currently, interest in the role of autophagy in thenervous system has increased because it has been revealedthat autophagy is activated in neurons exposed to hypoxic orexcitotoxic stimuli (Zhu et al. 2005). However, it has notbeen established whether the increment in autophagy plays aprotective role or whether, conversely, it contributes toneuronal death by means of autophagic death.
Small interfering RNAs are double-stranded RNA mole-cules specific to mRNA degradation. The efficiency ofsiRNA and its limited side effects have turned this mecha-nism into a powerful tool for silencing target genes and is ofmajor interest for therapeutic applications (Golzio et al.2007). Moreover, siRNAs are easy to deliver, require onlysmall doses to produce their silencing effects, and caninactivate a gene at almost any stage in development (Perez-Martinez et al. 2011). However, one of the major problemsfor the use of siRNA in neurons, so far, has been the lowtransfection efficiency of non-viral vectors to transfectsiRNA into neurons (Posadas et al. 2010a). Recently, wereported a dendrimer [TRANSGEDEN (TGD)] that com-bines a conjugated rigid polyphenylenevinylene core withflexible polyamidoamine branches at the surface that is ableto efficiently transfect cerebellar granular neurons, suggest-ing that it could be potentially useful to deliver siRNA todifferent types of neurons (Rodrigo et al. 2011).
The aim of this work was to determine the role of autophagyin response to NMDA, a paradigm of excitotoxic death. To
achieve this goal, we knocked down Beclin 1 protein using abeclin 1-siRNA delivered into rat cortical neurons using adendrimer as a carrier for the siRNA. We found that Beclin 1removal prevents NMDA-mediated induction of autophagy inrat cortical neurons and that autophagy inhibition potentiatesNMDA-induced neuronal death suggesting that autophagyplays a neuroprotective role during excitotoxicity.
Materials and methods
Cell culturePrimary culture of rat cortical neurons was prepared as previouslyreported (Posadas et al. 2010b). Cortical neurons were seeded onpoly-L-lysine-coated culture plates or on poly-L-lysine-coated glasscoverslips at a density of 7.5 · 104 cells/cm2 and used forexperiments after 7–10 days in vitro. Experimental procedures wereperformed according to the guidelines of the European Communityon Welfare of Research Animals (Directive 2003/65/EC) and theCastilla-La Mancha University Animal Research Ethics Committee.
Cell death paradigmThe conditions for establishing NMDA-induced neuronal death weredetermined by performing a concentration-response curve (Fig. S1a)and once the NMDA concentration was established (150 lM) a time-course curve was performed (Fig. S1b). The data indicated thatexposure to 150 lM NMDA for 24 h caused the death of about 20–25% of the neurons and this concentration and time of treatment wereselected to be used for all the experiments involving excitotoxicity.
Dendrimer preparation and dendriplex formationThe TGD dendrimer was prepared as previously described (Rodrigoet al. 2011). This novel hybrid dendrimer combines a conjugated rigidpolyphenylenevinylene core with flexible polyamidoamine branchesat the surface. It is a first generation dendrimer that contains at pH 7.4(physiologic pH) nine positive charges as has been shown by 1HNMRand 13C NMR (Rodrigo et al. 2011). siRNA-TGD dendrimerdendriplex formation was performed as previously described (Rodr-igo et al. 2011). In short, pre-designed siRNA targeting rat beclin 1(Rn_Becn1_3) and a control scramble RNA targeting a sequence notsharing homology with the rat genome (AllStars Negative Control)were used (Qiagen, Crawley, UK). Briefly, siRNAor scramble siRNAsolutions were prepared 30 min prior to cell transfection. The ratio ofsiRNA to the TGD dendrimer was 100 nM siRNA to 3 lM TGDdendrimer. The siRNA-TGD dendriplexes were incubated for 30 minat room temperature (20–25�C). Cortical neurons culture mediumwasreplaced with fresh medium and the siRNA-TGD dendriplex wasadded to cells and incubated at 37�C in a humidified atmospherecontaining 5% CO2. To explore the effect of Beclin 1 knockdown,cortical neurons were transfected with the siRNA-TGD dendriplex for72h. Then, the culture medium was changed and neurons wereincubated with NMDA (150 lM; Sigma Chemical Co., St. Louis,MO, USA) or vehicle for 24 h. Experiments were performed threetimes, unless specified otherwise.
Dendriplex uptake assaysFor transfection assays, cortical neurons were incubated with100 nM fluorescein-labeled siRNA (FAM-siRNA) alone or
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268� 2011 The Authors
260 | M. D. Perez-Carrion et al.

100 nM FAM-siRNA/3 lM TGD dendriplexes for 48, 72, and 96 h.Subsequently, cortical neurons were washed with phosphate-buffered saline and examined using a Leica DMRXA microscope(Leica, Wetzlar, Germany) with suitable fluorescence filters (exci-tation wavelength of 490 nm and emission wavelength of 520 nm).Photomicroscopy was performed using a Leica DC500 camera andtransfection efficiency was calculated as previously described(Posadas et al. 2009).
Intracellular calcium determinationIntracellular calcium determination was performed as previouslydescribed (Tornero et al. 2011). Neurons were plated on 20 mmdiameter coverslips and incubated as described above. Fura-2obtained from Molecular Probes (Invitrogen, Carlsbad, CA, USA)was used at 5 lM and loaded with 0.005% Pluronic in Krebssolution for 20–30 min at 37�C in the dark. The coverslips werewashed twice with Krebs solution and placed in the fluorescencecamera. Fluorescence was observed on an inverted microscope(Nikon Eclipse TE-2000-S, Birlingam, CA, USA) equipped with a150 W Xenon lamp and 100·, 1.3 numerical aperture, epifluores-cence oil immersion objective. Excitation wavelength was selectedusing a Life Technology monochromator (Omega Optical Inc,Brattleboro, VT, USA). The dye was excited alternately at 340 and380 nm allowing ratiometric measurements of changes in cytosolicCa2+ levels. Emision wavelength was selected using a filter wheel(Sutter ‘lambda 10’, Novato CA, USA). Images were acquired witha digital camera (ORCA II, Hamamatsu, Shizouka, Japan) atintervals of 10 s using an emission filter of 510 nm. Data wereanalyzed using commercial software (Metamorph; Universal Imag-ing Corporation, Silicon Valley, CA, USA).
Real-time RT-PCR analysisReal-time RT-PCR analysis was performed as previously described(Rodrigo et al. 2011). In short, total RNA was extracted using acommercially available reagent (Tripure; Sigma Chemical Co., St.Louis, MO, USA) and cDNA was synthesized from the purified totalRNAusingaHighCapacitycDNAReverseTranscriptionKit (AppliedBiosystems, Foster City, CA, USA) according to the manufacturer’sinstructions. For real-time RT-PCR, cDNA was amplified usingSYBR Green PCR Master mix with the StepOne Real-Time PCRSystem and the StepOne v2.0 software (Applied Biosystems). Theprimers used to amplify the beclin 1 gene were 5¢-GTGCTCCTGTGGAATGGAAT-3¢ (forward) and 5¢-GCTGCACACAGTCCAGAAAA-3¢ (reverse). To normalize the data, b-actin RNA expression levelwas used as an internal control. To ensure the reliability of the resultsobtained, all the samples were processed by triplicates. Quantificationwas performed by the comparative cycle threshold (Ct) method.
Western blot analysisWestern blot analysis was performed as previously described (Jordanet al. 2002). Cortical neurons were lysed in a buffer containing:50 mM Tris-HCl, pH 7.2; 250 mM NaCl; 0.1% NP-40; 2 mMEDTA; 10% glycerol; 0.5 mM phenylmethylsulfonyl fluoride;1 mM dithiothreitol; 1 lg/mL Leupeptine; and 0.1 lg/mL aprotininesimilar to that described by Wong et al. (2008). Protein content wasdetermined by the Bradford method (Pierce, Rockfork, IL, USA).Samples containing the proteins were freshly used without previousfreezing. Protein samples (30–40 lg) were solubilized in sampling
buffer (ABCAM, Cambridge, UK) following the manufacturerinstructions, heated at 95�C for 5 min and loaded on 10–15%polyacrylamide–sodium dodecyl sulfate gels, followed by transfer tonitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA,USA). Membranes were incubated in blocking buffer with 5% non-fat-dried milk in 20 mM Tris-base (pH 7.5), 150 mM NaCl, and0.1% Tween-20 for 1 h at 4�C and subsequently incubated overnightat 4�C with a monoclonal mouse anti-Beclin 1 antibody (1 : 1000;Santa Cruz Biotechnology, Santa Cruz, CA, USA) or a monoclonalmouse anti-LC3B antibody (1 : 500; Enzo Life Sciences, Faiming-dale, NY, USA). Blots were subsequently washed with 20 mM Tris-base (pH 7.5), 150 mM NaCl, and 0.1% Tween-20 buffer andincubated at 23�C for 1 h with peroxidase-conjugated anti-mouseIgG. The immune complexes were visualized using an enhancedchemiluminiscence system (Millipore, Bedford, MA, USA). b-actinprotein expression was used as an internal control. Densitometricanalysis of immunoreactive bands was performed by using QuantityOne Software (Bio-Rad Laboratories).
Cytotoxicity studiesLactate dehydrogenase (LDH) assays were performed as previouslydescribed (Posadas et al. 2007). Supernatants were collected at thetimes indicated and intact cells were lysed using 0.1% (w/v) TritonX-100 in (0.9%) NaCl. LDH released was determined spectropho-tometrically at 490 nm using the CytoTox96� Non-RadioactiveCytotoxicity Assay kit (Promega, Madison, WI, USA). Thepercentage of LDH released was defined as the ratio LDH released/total LDH present in the cells. Samples were run by cuatriplicates.
Monodansylcadaverine stainingCortical neurons were seeded on poly-L-lysine-coated glass cover-slips, cultured for 7 days in vitro, and then treated for differentperiods of time. For labeling of autophagic vacuoles with theautofluorescent agent MDC (Sigma, Barcelona, Spain) – a specificautophagolysosome marker to analyze the autophagic process –cortical neurons were incubated for 10 min with 50 lM MDC at37�C. Cells were later observed under a fluorescence microscope(excitation wavelength 380 nm, emission filter 525 nm). Theautophagic index was determined as the percentage of MDC-labeledcells out of 200 cells from each treatment group, as previouslydescribed (Takeuchi et al. 2005).
Electron microscopyCultured cortical neurons were fixed with 2% paraformaldehyde and2% glutaraldehyde (Sigma, Barcelona, Spain) in 0.1 M phosphatebuffer (pH 7.4), followed by post-fixation with 1% osmium tetraoxidein 0.1 M phosphate buffer (Sigma, Barcelona, Spain). Corticalneurons were then treated with 1% uranyl acetate and graduallydehydrated in a graded series of ethanol (50–100%) and embedded inepoxy (Durcupan) resin. Following resin polymerization, the blockswere cut into 70 nm-thick sections using an ultramicrotome (Reic-hert-Jung Ultracut E; Leica, Wetzlar, Germany). Ultrastructuralanalyses were performed in a JEOL-1010 electron microscope, aspreviously described (Lum et al. 2005).
Statistical analysisAll data are expressed as mean ± SEM of at least three independentexperiments. Non-parametric variance analysis (Kruskal–Wallis)
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268
Autophagy protects from excitotoxicity | 261
followed by a Dunn test was used to assess statistical differencesbetween groups. p < 0.05 was considered statistically significant.Statistical analyses were performed using the software packageSPSS 13.0 (SPSS, Chicago, IL, USA).
Results
3-Methyl adenine (3-MA) blocks NMDA-mediated Ca2+
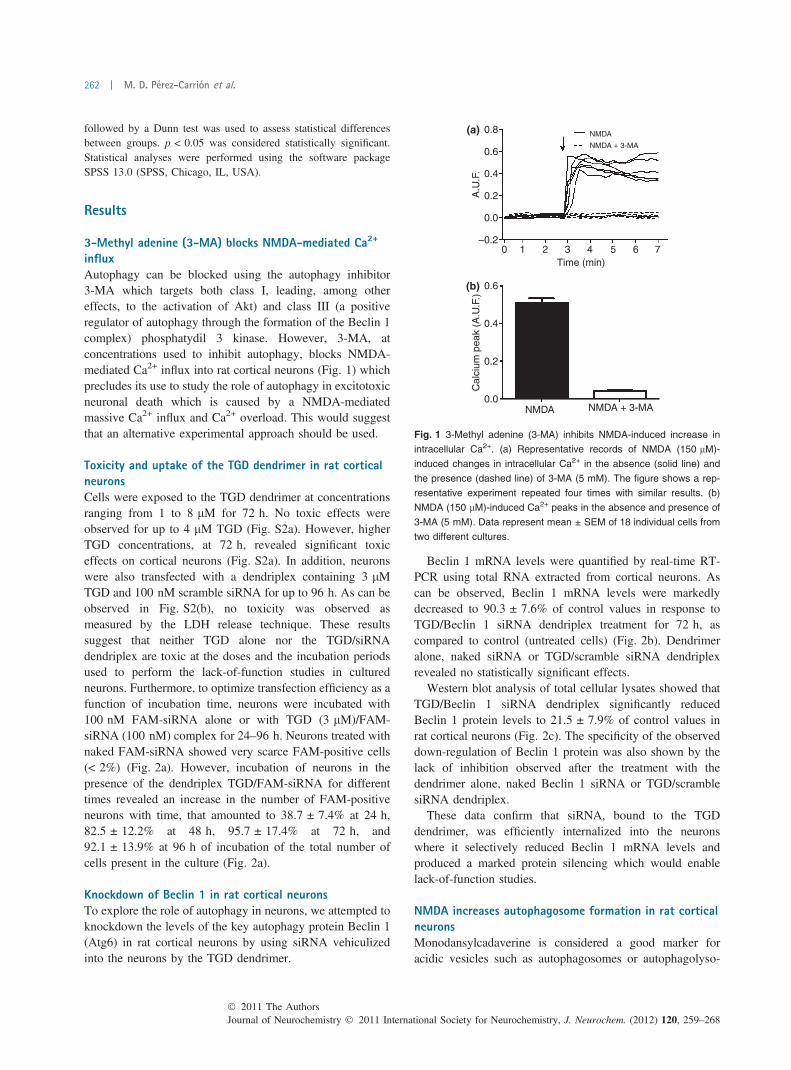
influxAutophagy can be blocked using the autophagy inhibitor3-MA which targets both class I, leading, among othereffects, to the activation of Akt) and class III (a positiveregulator of autophagy through the formation of the Beclin 1complex) phosphatydil 3 kinase. However, 3-MA, atconcentrations used to inhibit autophagy, blocks NMDA-mediated Ca2+ influx into rat cortical neurons (Fig. 1) whichprecludes its use to study the role of autophagy in excitotoxicneuronal death which is caused by a NMDA-mediatedmassive Ca2+ influx and Ca2+ overload. This would suggestthat an alternative experimental approach should be used.
Toxicity and uptake of the TGD dendrimer in rat corticalneuronsCells were exposed to the TGD dendrimer at concentrationsranging from 1 to 8 lM for 72 h. No toxic effects wereobserved for up to 4 lM TGD (Fig. S2a). However, higherTGD concentrations, at 72 h, revealed significant toxiceffects on cortical neurons (Fig. S2a). In addition, neuronswere also transfected with a dendriplex containing 3 lMTGD and 100 nM scramble siRNA for up to 96 h. As can beobserved in Fig. S2(b), no toxicity was observed asmeasured by the LDH release technique. These resultssuggest that neither TGD alone nor the TGD/siRNAdendriplex are toxic at the doses and the incubation periodsused to perform the lack-of-function studies in culturedneurons. Furthermore, to optimize transfection efficiency as afunction of incubation time, neurons were incubated with100 nM FAM-siRNA alone or with TGD (3 lM)/FAM-siRNA (100 nM) complex for 24–96 h. Neurons treated withnaked FAM-siRNA showed very scarce FAM-positive cells(< 2%) (Fig. 2a). However, incubation of neurons in thepresence of the dendriplex TGD/FAM-siRNA for differenttimes revealed an increase in the number of FAM-positiveneurons with time, that amounted to 38.7 ± 7.4% at 24 h,82.5 ± 12.2% at 48 h, 95.7 ± 17.4% at 72 h, and92.1 ± 13.9% at 96 h of incubation of the total number ofcells present in the culture (Fig. 2a).
Knockdown of Beclin 1 in rat cortical neuronsTo explore the role of autophagy in neurons, we attempted toknockdown the levels of the key autophagy protein Beclin 1(Atg6) in rat cortical neurons by using siRNA vehiculizedinto the neurons by the TGD dendrimer.
Beclin 1 mRNA levels were quantified by real-time RT-PCR using total RNA extracted from cortical neurons. Ascan be observed, Beclin 1 mRNA levels were markedlydecreased to 90.3 ± 7.6% of control values in response toTGD/Beclin 1 siRNA dendriplex treatment for 72 h, ascompared to control (untreated cells) (Fig. 2b). Dendrimeralone, naked siRNA or TGD/scramble siRNA dendriplexrevealed no statistically significant effects.
Western blot analysis of total cellular lysates showed thatTGD/Beclin 1 siRNA dendriplex significantly reducedBeclin 1 protein levels to 21.5 ± 7.9% of control values inrat cortical neurons (Fig. 2c). The specificity of the observeddown-regulation of Beclin 1 protein was also shown by thelack of inhibition observed after the treatment with thedendrimer alone, naked Beclin 1 siRNA or TGD/scramblesiRNA dendriplex.
These data confirm that siRNA, bound to the TGDdendrimer, was efficiently internalized into the neuronswhere it selectively reduced Beclin 1 mRNA levels andproduced a marked protein silencing which would enablelack-of-function studies.
NMDA increases autophagosome formation in rat corticalneuronsMonodansylcadaverine is considered a good marker foracidic vesicles such as autophagosomes or autophagolyso-
0.8(a)
(b)
NMDA
NMDA + 3-MA
NMDA NMDA + 3-MA
0.6
0.4
0.2A.U
.F.
–0.20
0.6
0.4
0.2
Cal
cium
pea
k (A
.U.F
.)
0.0
1 2 3Time (min)
4 5 6 7
0.0
Fig. 1 3-Methyl adenine (3-MA) inhibits NMDA-induced increase in
intracellular Ca2+. (a) Representative records of NMDA (150 lM)-
induced changes in intracellular Ca2+ in the absence (solid line) and
the presence (dashed line) of 3-MA (5 mM). The figure shows a rep-
resentative experiment repeated four times with similar results. (b)
NMDA (150 lM)-induced Ca2+ peaks in the absence and presence of
3-MA (5 mM). Data represent mean ± SEM of 18 individual cells from
two different cultures.
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268� 2011 The Authors
262 | M. D. Perez-Carrion et al.

somes (Munafo and Colombo 2001). The mechanism bywhich MDC accumulates in autophagosomes is believed tobe a combination of ionic trapping in acidic compartmentsand interaction with its double membrane lipids. Exposure ofcortical neurons to NMDA (150 lM) increased, in a time-dependent manner, the number of neurons labeled by MDC,indicating that NMDA induced, after 24 h of treatment, theformation of the MDC-labeled vacuoles in virtually all theneurons present in the culture (Fig. 3a). Lower periods ofexposure to NMDA did not induce the formation of theMDC-labeled vacuoles up to 8 h. At this latter time,37.2 ± 4.5% of the cells were MDC-positive. Electronmicroscopy is the gold standard method to visualize doublemembrane structures containing recognizable cellular organ-elles and a high electron-density material characteristic ofautophagosomes (Xu et al. 2007). Autophagosomes wereevident following NMDA treatment as assessed by electronmicroscopy (Fig. 3b).
NMDA increases autophagy in rat cortical neuronsWe explored whether NMDA could increase autophagy incortical neurons. To identify an increase in autophagy, Beclin
1 levels and the ratio LC3B-II/LC3B-I, two known markersfor autophagy, were studied. Enhanced Beclin 1 proteinexpression has been used as a marker of autophagy, aspreviously reported following closed brain injury (Diskinet al. 2005) and focal cerebral ischemia (Rami et al. 2008)while LC3B-I appears as a cytosolic form under normalconditions but during autophagy it is cleaved at theC-terminal portion and converted into an autophagosome-associated form (LC3B-II). An increase in Beclin 1 levelswas observed in response to NMDA from 147.1 ± 9.9% ofcontrol values 8 h after NMDA treatment to 183.4 ± 11% at24 h (Fig. 4a). Moreover, the LC3B-II/LC3B-I ratio in-creased from 0.37 ± 0.03 in untreated neurons to 1.03 ± 0.08after 8 h and up to 1.90 ± 0.06 following 24 h of treatmentwith NMDA, suggesting an up-regulation of autophagy(Fig. 4b).
Beclin 1 knockdown inhibits NMDA-mediated autophagyin rat cortical neuronsBeclin 1 knock down was achieved before and maintainedduring NMDA exposure. Neuronal treatment with Beclin 1siRNA/TGD dendriplex decreased the basal levels of Beclin
Fig. 2 Transfection and Beclin 1 knock down in rat cortical neurons.
(a) Bright-field (Nomarski) and fluorescence images, as well as, the
overlay between both images of neurons treated with either 100 nM
fluorescein-labeled scramble siRNA alone (FAM-siRNA) or 100 nM
FAM-siRNA/3 lM TGD dendriplex for 72 h. In other set of experi-
ments, neurons were incubated with TGD (3 lM), 100 nM naked
Beclin 1 siRNA, 100 nM Beclin 1 siRNA/TGD (3 lM) dendriplex, and
100 nM scramble siRNA/TGD (3 lM) dendriplex for 72 h. Beclin 1
mRNA (b) and protein (c) levels were determined as indicated in
‘Material and methods’. b-actin protein levels were used as reference
for the western blot data. Untreated control neurons were represented
as 100% of the signal. Data represent mean ± SEM of three experi-
ments. ***p < 0.001 as compared with control neurons.
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268
Autophagy protects from excitotoxicity | 263

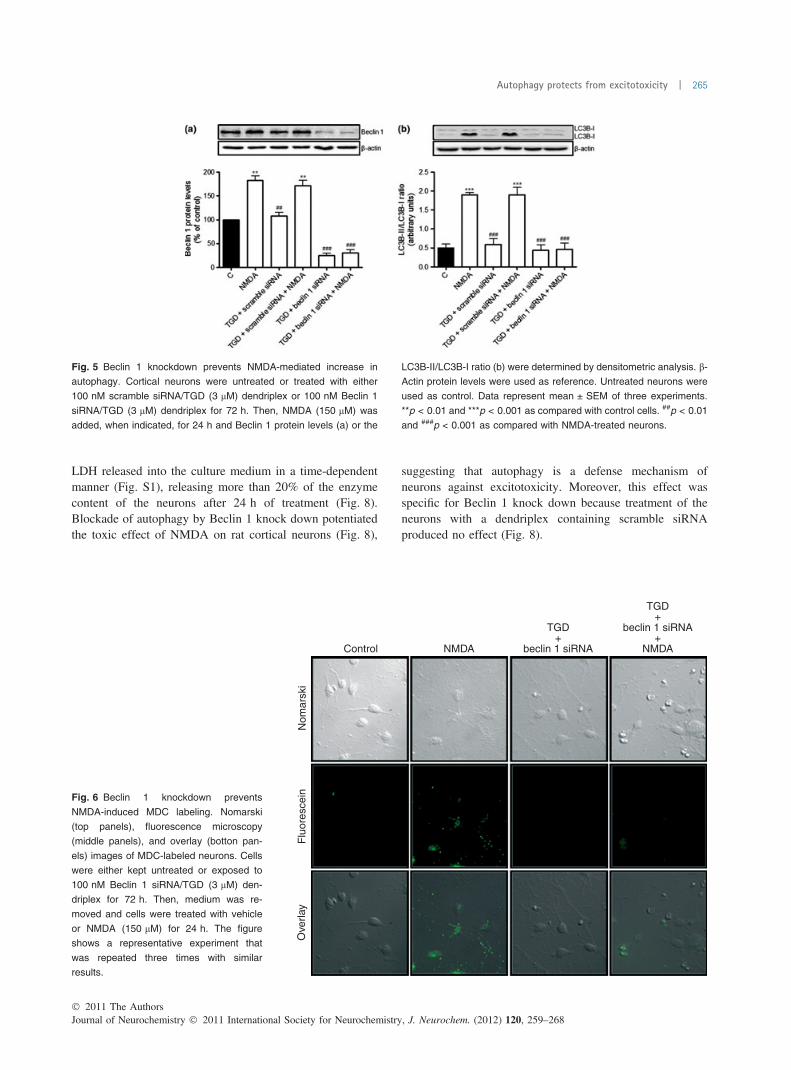
1 and also prevented NMDA-mediated increase in Beclin 1protein levels (Fig. 5a). It also prevented the NMDA-mediated increment in the LC3B-II/LC3B-I ratio (Fig. 5b)suggesting an inhibition of autophagy. Moreover, auto-phagosome labeling with MDC was abolished when Beclin 1was knocked down (Fig. 6). In concordance with this,electron micrographs did not show any evidence of auto-phagosomes after the treatment of the neurons with NMDAin the absence of Beclin 1 while these could be observedafter the treatment with dendriplexes containing TGD and
scramble siRNA (Fig. 7). Taken together, these resultssuggest that knocking down Beclin 1 using siRNA in ratcortical neurons can block the NMDA-mediated increase inautophagic activity.
Autophagy protects neurons against NMDA-mediatedtoxicityNeurotoxic effects of NMDA (150 lM) in rat corticalneurons were assessed by LDH release assays. Treatmentof rat cortical neurons with NMDA (150 lM) increased the
0 h(a)
(b)
Nom
arsk
iF
luor
esce
nce
Ove
rlay
Control NMDA
1 h 3 h 8 h 24 h
Fig. 3 NMDA induces autophagy in rat cortical neurons. (a) Nomarski
and fluorescence images, as well as, the overlay between both images
of neurons exposed to NMDA (150 lM) for 0, 1, 3, 8, and 24 h. The
neurons were labeled with monodansylcadaverine (MDC) as indicated
in ‘Material and methods’. The image shows a representative experi-
ment repeated three times with similar results. (b) NMDA induces
autophagosome formation in rat cortical neurons. Left: Control un-
treated neurons show a low presence of autophagosomes in their
cytoplasm observed by transmission electron microscopy. Right: The
presence of NMDA (150 lM; 24 h) in the culture medium strongly
increased the number of autophagosomes in the neurons. Scale bar:
2 lm.
Fig. 4 Western blot analysis of (a) Beclin 1 protein levels and (b)
LC3B-II/LC3B-I ratio in rat cortical neurons exposed to NMDA
(150 lM) for 1–24 h. b-Actin protein levels were used as reference.
Data represent mean ± SEM of three experiments. *p < 0.05,
**p < 0.01, and ***p < 0.001; as compared with untreated control cells.
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268� 2011 The Authors
264 | M. D. Perez-Carrion et al.
LDH released into the culture medium in a time-dependentmanner (Fig. S1), releasing more than 20% of the enzymecontent of the neurons after 24 h of treatment (Fig. 8).Blockade of autophagy by Beclin 1 knock down potentiatedthe toxic effect of NMDA on rat cortical neurons (Fig. 8),
suggesting that autophagy is a defense mechanism ofneurons against excitotoxicity. Moreover, this effect wasspecific for Beclin 1 knock down because treatment of theneurons with a dendriplex containing scramble siRNAproduced no effect (Fig. 8).
Fig. 5 Beclin 1 knockdown prevents NMDA-mediated increase in
autophagy. Cortical neurons were untreated or treated with either
100 nM scramble siRNA/TGD (3 lM) dendriplex or 100 nM Beclin 1
siRNA/TGD (3 lM) dendriplex for 72 h. Then, NMDA (150 lM) was
added, when indicated, for 24 h and Beclin 1 protein levels (a) or the
LC3B-II/LC3B-I ratio (b) were determined by densitometric analysis. b-
Actin protein levels were used as reference. Untreated neurons were
used as control. Data represent mean ± SEM of three experiments.
**p < 0.01 and ***p < 0.001 as compared with control cells. ##p < 0.01
and ###p < 0.001 as compared with NMDA-treated neurons.
Nom
arsk
iF
luor
esce
inO
verla
y
NMDA
TGD+
beclin 1 siRNA
TGD+
beclin 1 siRNA+
NMDAControl
Fig. 6 Beclin 1 knockdown prevents
NMDA-induced MDC labeling. Nomarski
(top panels), fluorescence microscopy
(middle panels), and overlay (botton pan-
els) images of MDC-labeled neurons. Cells
were either kept untreated or exposed to
100 nM Beclin 1 siRNA/TGD (3 lM) den-
driplex for 72 h. Then, medium was re-
moved and cells were treated with vehicle
or NMDA (150 lM) for 24 h. The figure
shows a representative experiment that
was repeated three times with similar
results.
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268
Autophagy protects from excitotoxicity | 265

Discussion
For this study, we employed a RNA-interference strategy,delivered to rat cortical neurons by a new dendrimer (TGD)to show that autophagy is a neuroprotective mechanismactivated in rat cortical neurons to counteract NMDA-mediated neuronal injury. Moreover, we show here thatNMDA exposure produces a significant accumulation ofautophagosomes in cultured cortical neurons whose inhibi-tion by silencing Beclin 1 leads to an increase in NMDA-mediated neuronal excitotoxic death.
The study of the role of different proteins in pathologyrequires an approach that should include their selectiveknockdown to study a lack-of-function effect. Therefore,siRNA has emerged as an important tool to perform this kindof study to analyze physiologic and pathologic mechanisms.However, the general use of siRNA technology in post-mitotic neuronal cells to study biological problems has beenscarce because the low efficiency of the different non-viralvectors used to deliver siRNA to neurons. Therefore, a 14%transfection efficiency has been reported for polyethyleni-mine in rat hypothalamic neurons (Guerra-Crespo et al.
2003) or 24–27% for Lipofectamine 2000 (Invitrogen,Carlsbad, CA, USA) in rat cortical neurons (Ohki et al.2001). The reported transfection efficiency is higher fordendrimers, approximately 40% for a polyamidoamine-argi-nine dendrimer in rat cortical neurons (Kim et al. 2006), but itis not high enough to enable lack-of-function studies where areduction of about 70–80% of the protein is required. Here, wereport that a dendrimer can decrease Beclin 1 protein levels toabout 20% of the control values, enabling lack-of-functionstudies for this protein. Moreover, this reduction in proteinlevels is very specific because the dendriplex dendrimer/scramble siRNA has no effect. In addition, the dendrimerefficiently delivers siRNA at concentrations that show notoxicity for rat cortical neurons. For this study, we used thedendrimer-delivered siRNA to silence Beclin 1 which isinvolved in the early stage of autophagosome formation and itis essential for the recruitment of other proteins involved inlatter autophagy steps. Therefore, we have demonstrated that adendrimer, coupled to Beclin 1 siRNA, is an effective methodto introduce specific siRNA into rat cortical neurons, enablingknocking down Beclin 1 mRNA and protein levels. Autophagyactivity can be detected by analyzing the LC3B-II/LC3B-Iratio, uptake of the fluorescent dye MDC, and identification ofautophagosomes by ultrastructural microscopy (Klionsky et al.2008). The results presented here indicate that those threemarkers for autophagy are increased strongly suggesting thatexcitoxicity increases autophagy in rat cortical neurons.However, it cannot be completely excluded that autophago-
Fig. 8 Effect of Beclin 1 removal on NMDA-induced neuronal death.
Cortical neurons were untreated or treated with either 100 nM
scramble siRNA/TGD (3 lM) dendriplex, or100 nM Beclin 1 siRNA/
TGD (3 lM) dendriplex for 72 h. Then, NMDA (150 lM) was added,
when indicated, for 24 h and LDH released, taken as index of neuronal
death was determined. Data are expressed as mean ± SEM of 12
experiments. *p < 0.05, **p < 0.01, and ***p < 0.001; as compared
with untreated control (C) cells. #p < 0.05, as compared with NMDA
treatment.
Fig. 7 Beclin 1 knockdown prevents NMDA-induced autophagosome
formation. Top panels: left – ultraestructural analysis of the cytoplasm
of cultured cortical neurons treated with vehicle observed by trans-
mission electron microscopy; middle – NMDA 150 lM, 24 h increased
the number of autophagosomes (arrows) in the cytoplasm of neurons
transfected with scramble siRNA; right – Beclin 1 silencing suppressed
the NMDA effect on the number of autophagosomes. Bottom panels
show a magnified detail of the selected area in top panels, respec-
tively. Scale bar in top panels: 1 lm. Scale bar in bottom panels:
200 nm.
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268� 2011 The Authors
266 | M. D. Perez-Carrion et al.

some accumulation might be because of organelle turnoverinhibition.
Our data reveal that the dendriplex TGD/scramble siRNAdoes not modify the neuron biological responses, because itdoes not affect NMDA-induced autophagy. Conversely, ourdata confirm that NMDA induces an increase in autophagyin rat cortical neurons as previously described (Sadasivanet al. 2010), but they indicate that this induction ofautophagy plays a protective role against NMDA-mediatedexcitotoxic death. It has been previously reported thatautophagy is required for homeostasis in several cell types(Wong and Cuervo 2010). However, autophagy is aparticularly relevant mechanism for quality control innon-dividing post-mitotic cells, such as neurons, in whichthe total content of altered proteins and damaged organellescannot be decreased by redistribution during cell division.In agreement with the results reported here, previousstudies have shown, in vivo, that mice lacking brainexpression of Atg5 or Atg7 which leads to alteredautophagy, quickly develop neurodegenerative phenotypesin selected neuronal populations early on in post-natal life(Hara et al. 2006). In addition, reduced levels of Beclin 1have been found in brain tissues obtained from elderlyhumans as well as from patients with Huntington andAlzheimer disease (Shibata et al. 2006; Pickford et al.2008). It has been also described that genetic down-regulation of autophagy using specific deletion of Atg5 inmouse retinal ganglion cells reduces cell survival after thesection of the optical nerve, whereas pharmacologicalinduction of autophagy in vivo increases the number ofsurviving cells (Rodriguez-Muela et al. 2011). Moreover, arecent study shows that autophagy has a neuroprotectiverole in the ischemic retina and suggests that excitotoxicitynegatively regulates autophagy through calpain-mediatedcleavage of Beclin 1 (Russo et al. 2011). Moreover, otherstudies have postulated that the neuroprotection offered byischemic pre-conditioning is characterized by severalmolecular and cellular mechanisms such as activation ofthe autophagic-lysosomal pathway.
Besides these evidences, it has also been reported that incultured cerebellar granule neurons, the autophagy inhibitor3-methyladenine and Atg7 silencing suppressed NMDA-mediated neuronal death (Sadasivan et al. 2010). Thereasons for this discrepancy might be explained by thedifferent cell type (cerebellar granular cells) used bySadasivan et al. and by the blocking actions of the autophagyinhibitor 3-MA used in that study on NMDA-mediated Ca2+
influx into neurons (Fig. 1). By doing so, 3-MA wouldprevent Ca2+ overload and accordingly the chain of eventsleading to excitotoxic neuronal death. Moreover, thedecrease in Atg7 protein levels reported in that paper wasonly of the order of 30% which makes it difficult to performlack-of-function studies. A better understanding of theprocesses that lead to cell death following NMDA exposure
as well as of the endogenous neuroprotective mechanisms bywhich neurons try to counteract brain aggressions willfacilitate developing new therapeutic approaches for neuro-degenerative diseases. In this study, we show that one ofthese neuroprotective mechanisms, in rat cortical neurons, isautophagy. This would concur with recent studies showingthat a defect in autophagosomal maturation might induceneurodegenerative diseases (Martinez-Vicente and Cuervo2007; Rubinsztein 2007).
In summary, the data reported here reveal that a dendrimercan efficiently deliver siRNA in rat cortical neurons, leadingto a decrease in Beclin 1 protein levels to approximately 20%of control values, enabling lack-of-function studies. Usingthis strategy, we have studied the role of autophagy inNMDA-mediated excitotoxic death and found that ratcortical neurons transfected with beclin 1 siRNA/TGDdendriplexes were defective in autophagy. When these cellswere exposed to NMDA, excitotoxic death increasedsuggesting that rat cortical neurons respond to excitotoxicityby inducing autophagy as a protective mechanism, althoughthis might not be sufficient to maintain cell survival duringexcitotoxic insults. This would suggest that targeting auto-phagy might be a good strategy for therapy in neurodegen-erative diseases.
Acknowledgements
The authors are grateful to Ana Belen Garcıa for her excellenttechnical assistance. FCP-M and MDP-C are recipients of TorresQuevedo contracts from Ministerio de Ciencia e Innovacion (Spain)and NanoDrugs, S.L. This work has been supported, in part, by grantsPI081434 from Fondo de Investigaciones Sanitarias, BFU2011-30161-C02-01 fromMinisterio de Ciencia e Innovacion and PII1I09-0163-4002, and POII10-0274-3182 from Consejerıa de Educacion,JCCM to VC. The authors have no conflict of interest to declare.
Supporting information
Additional supporting information may be found in the onlineversion of this article:
Figure S1. Concentration-response effect and time-course ofNMDA-mediated neuronal death.
Figure S2. Toxicity analysis of TGD and TGD/scramble siRNAcomplex in rat cortical neurons.
As a service to our authors and readers, this journal providessupporting information supplied by the authors. Such materials arepeer-reviewed and may be re-organized for online delivery, but arenot copy-edited or typeset. Technical support issues arising fromsupporting information (other than missing files) should beaddressed to the authors.
References
Bossy-Wetzel E. and Green D. R. (2000) Assays for cytochrome c re-lease from mitochondria during apoptosis. Methods Enzymol. 322,235–242.
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268
Autophagy protects from excitotoxicity | 267

Degenhardt K., Mathew R., Beaudoin B. et al. (2006) Autophagy pro-motes tumor cell survival and restricts necrosis, inflammation, andtumorigenesis. Cancer Cell 10, 51–64.
Diskin T., Tal-Or P., Erlich S., Mizrachy L., Alexandrovich A., ShohamiE. and Pinkas-Kramarski R. (2005) Closed head injury inducesupregulation of Beclin 1 at the cortical site of injur. J. Neuro-trauma 22, 750–762.
Furuya N., Yu J., Byfield M., Pattingre S. and Levine B. (2005) Theevolutionarily conserved domain of Beclin 1 is required for Vps34binding, autophagy and tumor suppressor function. Autophagy 1,46–52.
Golzio M., Mazzolini L., Ledoux A. et al. (2007) In vivo gene silencingin solid tumors by targeted electrically mediated siRNA delivery.Gene Ther. 14, 752–759.
Guerra-Crespo M., Charli J. L., Rosales-Garcia V. H., Pedraza-Alva G.and Perez-Martinez L. (2003) Polyethylenimine improves thetransfection efficiency of primary cultures of post-mitotic rat fetalhypothalamic neurons. J. Neurosci. Methods 127, 179–192.
Hara T., Nakamura K., Matsui M. et al. (2006) Suppression of basalautophagy in neural cells causes neurodegenerative disease inmice. Nature 441, 885–889.
Jordan J., Galindo M. F., Tornero D., Benavides A., Gonzalez C.,Agapito M. T., Gonzalez-Garcia C. and Cena V. (2002) Super-oxide anions mediate veratridine-induced cytochrome c release andcaspase activity in bovine chromaffin cells. Br. J. Pharmacol. 137,993–1000.
Kim J. B., Choi J. S., Nam K., Lee M., Park J. S. and Lee J. K. (2006)Enhanced transfection of primary cortical cultures using arginine-grafted PAMAM dendrimer, PAMAM-Arg. J. Control Release114, 110–117.
Klionsky D. J. and Emr S. D. (2000) Autophagy as a regulated pathwayof cellular degradation. Science 290, 1717–1721.
Klionsky D. J., Abeliovich H., Agostinis P. et al. (2008) Guidelines forthe use and interpretation of assays for monitoring autophagy inhigher eukaryotes. Autophagy 4, 151–175.
Kroemer G. and Levine B. (2008) Autophagic cell death: the story of amisnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010.
Lum J. J., Bauer D. E., Kong M., Harris M. H., Li C., Lindsten T. andThompson C. B. (2005) Growth factor regulation of autophagy andcell survival in the absence of apoptosis. Cell 120, 237–248.
Martinez-Vicente M. and Cuervo A. M. (2007) Autophagy and neu-rodegeneration: when the cleaning crew goes on strike. LancetNeurol. 6, 352–361.
Munafo D. B. and Colombo M. I. (2001) A novel assay to study auto-phagy: regulation of autophagosome vacuole size by amino aciddeprivation. J. Cell Sci. 114, 3619–3629.
Ohki E. C., Tilkins M. L., Ciccarone V. C. and Price P. J. (2001)Improving the transfection efficiency of post-mitotic neurons.J. Neurosci. Methods 112, 95–99.
Perez-Martinez F. C., Guerra J., Posadas I. and Cena V. (2011) Barriersto non-viral vector-mediated gene delivery in the nervous system.Pharm. Res. 28, 1843–1858.
Pickford F., Masliah E., Britschgi M. et al. (2008) The autophagy-re-lated protein beclin 1 shows reduced expression in early Alzheimerdisease and regulates amyloid beta accumulation in mice. J. Clin.Invest. 118, 2190–2199.
Posadas I., Vellecco V., Santos P., Prieto-Lloret J. and Cena V. (2007)Acetaminophen potentiates staurosporine-induced death in a hu-man neuroblastoma cell line. Br. J. Pharmacol. 150, 577–585.
Posadas I., Lopez-Hernandez B., Clemente M. I., Jimenez J. L., OrtegaP., de la M. J., Gomez R., Munoz-Fernandez M. A. and Cena V.(2009) Highly efficient transfection of rat cortical neurons using
carbosilane dendrimers unveils a neuroprotective role for HIF-1alpha in early chemical hypoxia-mediated neurotoxicity. Pharm.Res. 26, 1181–1191.
Posadas I., Guerra F. J. and Cena V. (2010a) Nonviral vectors for thedelivery of small interfering RNAs to the CNS. Nanomedicine(Lond.) 5, 1219–1236.
Posadas I., Santos P., Blanco A., Munoz-Fernandez M. and Cena V.(2010b) Acetaminophen induces apoptosis in rat cortical neurons.PLoS ONE 5, e15360.
Rami A., Langhagen A. and Steiger S. (2008) Focal cerebral ischemiainduces upregulation of Beclin 1 and autophagy-like cell death.Neurobiol. Dis. 29, 132–141.
Rodrigo A. C., Rivilla I., Perez-Martinez F. C. et al. (2011) Efficient,non-toxic hybrid PPV-PAMAM dendrimer as a gene carrier forneuronal cells. Biomacromolecules 12, 1205–1213.
Rodriguez-Muela N., Germain F., Marino G., Fitze P. S. and Boya P.(2011) Autophagy promotes survival of retinal ganglion cells afteroptic nerve axotomy in mice. Cell Death Differ. Published online.doi: 10.1038/cdd.2011.88.
Rohn T. T., Wirawan E., Brown R. J., Harris J. R., Masliah E. andVandenabeele P. (2011) Depletion of Beclin-1 due to proteolyticcleavage by caspases in the Alzheimer’s disease brain. Neurobiol.Dis. 43, 68–78.
Rubinsztein D. C. (2007) Autophagy induction rescues toxicity mediatedby proteasome inhibition. Neuron 54, 854–856.
Russo R., Berliocchi L., Adornetto A., Varano G. P., Cavaliere F., NucciC., Rotiroti D., Morrone L. A., Bagetta G. and Corasaniti M. T.(2011) Calpain-mediated cleavage of Beclin-1 and autophagyderegulation following retinal ischemic injury in vivo. Cell DeathDis. 2, e144.
Sadasivan S., Zhang Z., Larner S. F., Liu M. C., Zheng W., Kobeissy F.H., Hayes R. L. and Wang K. K. (2010) Acute NMDA toxicity incultured rat cerebellar granule neurons is accompanied by auto-phagy induction and late onset autophagic cell death phenotype.BMC Neurosci. 11, 21.
Shibata M., Lu T., Furuya T., Degterev A., Mizushima N., Yoshimori T.,MacDonald M., Yankner B. and Yuan J. (2006) Regulation ofintracellular accumulation of mutant Huntingtin by Beclin 1.J. Biol. Chem. 281, 14474–14485.
Shintani T. and Klionsky D. J. (2004) Autophagy in health and disease: adouble-edged sword. Science 306, 990–995.
Takeuchi H., Kondo Y., Fujiwara K., Kanzawa T., Aoki H., Mills G. B.and Kondo S. (2005) Synergistic augmentation of rapamycin-in-duced autophagy in malignant glioma cells by phosphatidylinositol3-kinase/protein kinase B inhibitors. Cancer Res. 65, 3336–3346.
Tornero D., Posadas I. and Cena V. (2011) Bcl-xL blocks a mitochon-drial inner membrane channel and prevents Ca2+ overload-mediated cell death. PLoSONE 6, e20423. doi:10.1371/journal.pone.0020423.
Wong E. and Cuervo A. M. (2010) Autophagy gone awry in neurode-generative diseases. Nat. Neurosci. 13, 805–811.
Wong J., Zhang J., Si X., Gao G., Mao I., McManus B. M. and Luo H.(2008) Autophagosome supports coxsackievirus B3 replication inhost cells. J. Virol. 82, 9143–9153.
Xu Z. X., Liang J., Haridas V., Gaikwad A., Connolly F. P., Mills G. B.and Gutterman J. U. (2007) A plant triterpenoid, avicin D, inducesautophagy by activation of AMP-activated protein kinase. CellDeath Differ. 14, 1948–1957.
Zhu C., Wang X., Xu F., Bahr B. A., Shibata M., Uchiyama Y., HagbergH. and Blomgren K. (2005) The influence of age on apoptotic andother mechanisms of cell death after cerebral hypoxia-ischemia.Cell Death Differ. 12, 162–176.
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2012) 120, 259–268� 2011 The Authors
268 | M. D. Perez-Carrion et al.
Related Documents











