Decoding asthma: Translating genetic variation in IL33 and IL1RL1 into disease pathophysiology N eomi S. Grotenboer, MSc, a,b,c Maria E. Ketelaar, MSc, a,c Gerard H. Koppelman, MD, PhD, b,c * and Martijn C. Nawijn, PhD a,c * Groningen, The Netherlands Asthma is a complex disease that results from the interaction between genetic predisposition and environmental factors. Recently, genome-wide association studies have identified a number of genes that significantly contribute to asthma. Two of these genes, IL33 and IL-1 receptor–like 1 (IL1RL1), act in one signal transduction pathway. IL33 encodes a cytokine released on damage of cells, whereas IL1RL1 encodes part of the IL-33 receptor complex. Recent progress made in functional studies in human subjects and mouse models of allergic airway disease indicate a central role of IL-33 signaling in driving T H 2 inflammation, which is central to eosinophilic allergic asthma. Here, IL-33 acts on cells of both the adaptive and innate immune systems. Very recently, a novel population of IL-33– responsive innate immune cells, the type 2 innate lymphoid cells, was found to produce hallmark T H 2 cytokines, such as IL-5 and IL-13. The relevance of these cells for asthma is underscored by the identification of retinoic acid–related orphan receptor a (RORA), the gene encoding the transcription factor critical for their differentiation, as another asthma gene in genome-wide association studies. This review describes the mechanisms through which genetic variation at the IL33 and IL1RL1 loci translates into increased susceptibility for asthma. We propose that genetic variation associated with asthma at the IL33 and IL1RL1 loci can be dissected into independent signals with distinct functional consequences for this pathway that is central to asthma pathogenesis. (J Allergy Clin Immunol 2013;131:856-65.) Key words: IL-33, IL-1RL1, ST2, genome-wide association study, nuocytes, innate helper cells, innate type 2 lymphoid cells, expression quantitative trait locus Asthma is a complex, chronic inflammatory disease of the airways currently affecting more than 300 million persons worldwide, with approximately 250,000 annual deaths as a result. 1 It is estimated that by 2025, the number of asthmatic patients will increase by more than 100 million. 2 Asthma is characterized by respiratory symptoms, variable airway obstruction, and airway hyperresponsiveness. The clinical expression of asthma is dependent on the interaction between genetic predisposition and environmental factors. The number of identified asthma susceptibility genes has increased rapidly over the last 5 years, especially with the application of the genome- wide association (GWA) study approach. In a GWA study 300,000 to more than a million DNA polymorphisms covering the genome are investigated for association with asthma in large samples of cases and control subjects. Genetic variation in the IL33 and IL-1 receptor–like 1 (IL1RL1) genes has reproducibly been found to be associated with asthma in GWA studies, identifying IL-33–induced signaling through IL-1RL1 as one of the central pathways in asthmatic patients. Al- though numerous functional studies have revealed a central role for IL-33–induced signaling in T H 2-driven inflammation, which plays a crucial role in allergic asthma, few studies have directly assessed the functional consequences of genetic variation in the IL33 and IL1RL1 genes for the activity of this pathway. Such stud- ies are currently limited by the large number of asthma-associated single nucleotide polymorphisms (SNPs) in these genes and the complex genetic structure of the IL1RL1 locus. Therefore this review aims to offer an interpretation of asthma- associated polymorphisms in IL33 and IL1RL1 as a limited number of discrete genetic signals with distinct functional conse- quences and to discuss these in the context of a newly identified From a the Laboratory of Allergology and Pulmonary Diseases, Department of Pathology and Medical Biology, and b the Department of Pediatric Pulmonology and Pediatric Al- lergology, Beatrix Children’s Hospital, and c GRIAC Research Institute, University of Groningen, University Medical Center Groningen. *These authors contributed equally to this work. Supported by a Netherlands Asthma Foundation grant (no. 3.2.09.081JU). Disclosure of potential conflict of interest: G. H. Koppelman has been supported by one or more grants from or has one or more grants pending with the Netherlands Asthma Foundation, the European Union, and Stichting Astma Bestrijding and has received one or more payments for lecturing from or is on the speakers’ bureau for GlaxoSmithK- line. M. C. Nawijn has received one or more grants from or has one or more grants pending with the Netherlands Asthma Foundation, the European Union, and Stichting Astma Bestrijding, and has received support for travel from the European Respiratory Society. The rest of the authors declare that they have no relevant conflicts of interest. Received for publication July 24, 2012; revised October 30, 2012; accepted for publica- tion November 19, 2012. Available online February 4, 2013. Corresponding author: Martijn C. Nawijn, PhD, Laboratory of Allergology and Pulmo- nary Diseases, Pathology and Medical Biology, IPC EA11, University Medical Center Groningen, GRIAC Research Institute, University of Groningen, Hanzeplein 1, PO Box 30.001, 9700 RB, Groningen, The Netherlands. E-mail: [email protected]. 0091-6749/$36.00 Ó 2013 American Academy of Allergy, Asthma & Immunology http://dx.doi.org/10.1016/j.jaci.2012.11.028 Abbreviations used ASW: African ancestry in Southwest United States CEU: Utah residents with Northern and Western European ancestry from the CEPH collection (referred to as non-Hispanic white) CHB: Han Chinese in Beijing, China GWA: Genome-wide association ILC2: Type 2 innate lymphoid cell IL1RL1: IL-1 receptor–like 1 IL18R1: IL-18 receptor 1 LD: Linkage disequilibrium MEX: Mexican ancestry in Los Angeles, California (referred to as Mexican population) MyD88: Myeloid differentiation primary response gene–88 NF-kB: Nuclear factor kB RORA: Retinoic acid–related orphan receptor a SNP: Single nucleotide polymorphism TIR: Toll-like/IL-1 receptor TLR: Toll-like receptor YRI: Yoruban in Ibadan, Nigeria 856

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Decoding asthma: Translating genetic variation in IL33 andIL1RL1 into disease pathophysiology
N�eomi S. Grotenboer, MSc,a,b,c Maria E. Ketelaar, MSc,a,c Gerard H. Koppelman, MD, PhD,b,c* and
Martijn C. Nawijn, PhDa,c* Groningen, The Netherlands
Abbreviations used
ASW: African ancestry in Southwest United States
CEU: Utah residents with Northern and Western European
ancestry from the CEPH collection (referred to as
non-Hispanic white)
CHB: Han Chinese in Beijing, China
GWA: Genome-wide association
ILC2: Type 2 innate lymphoid cell
IL1RL1: IL-1 receptor–like 1
IL18R1: IL-18 receptor 1
LD: Linkage disequilibrium
MEX: Mexican ancestry in Los Angeles, California (referred
to as Mexican population)
MyD88: Myeloid differentiation primary response gene–88
NF-kB: Nuclear factor kB
RORA: Retinoic acid–related orphan receptor a
SNP: Single nucleotide polymorphism
TIR: Toll-like/IL-1 receptor
TLR: Toll-like receptor
YRI: Yoruban in Ibadan, Nigeria
Asthma is a complex disease that results from the interactionbetween genetic predisposition and environmental factors.Recently, genome-wide association studies have identified anumber of genes that significantly contribute to asthma. Two ofthese genes, IL33 and IL-1 receptor–like 1 (IL1RL1), act in onesignal transduction pathway. IL33 encodes a cytokine releasedon damage of cells, whereas IL1RL1 encodes part of the IL-33receptor complex. Recent progress made in functional studies inhuman subjects and mouse models of allergic airway diseaseindicate a central role of IL-33 signaling in driving TH2inflammation, which is central to eosinophilic allergic asthma.Here, IL-33 acts on cells of both the adaptive and innateimmune systems. Very recently, a novel population of IL-33–responsive innate immune cells, the type 2 innate lymphoid cells,was found to produce hallmark TH2 cytokines, such as IL-5 andIL-13. The relevance of these cells for asthma is underscored bythe identification of retinoic acid–related orphan receptor a(RORA), the gene encoding the transcription factor critical fortheir differentiation, as another asthma gene in genome-wideassociation studies. This review describes the mechanismsthrough which genetic variation at the IL33 and IL1RL1 locitranslates into increased susceptibility for asthma. We proposethat genetic variation associated with asthma at the IL33 andIL1RL1 loci can be dissected into independent signals withdistinct functional consequences for this pathway that iscentral to asthma pathogenesis. (J Allergy Clin Immunol2013;131:856-65.)
Key words: IL-33, IL-1RL1, ST2, genome-wide association study,nuocytes, innate helper cells, innate type 2 lymphoid cells,expression quantitative trait locus
From athe Laboratory of Allergology and Pulmonary Diseases, Department of Pathology
andMedical Biology, and bthe Department of Pediatric Pulmonology and Pediatric Al-
lergology, Beatrix Children’s Hospital, and cGRIAC Research Institute, University of
Groningen, University Medical Center Groningen.
*These authors contributed equally to this work.
Supported by a Netherlands Asthma Foundation grant (no. 3.2.09.081JU).
Disclosure of potential conflict of interest: G. H. Koppelman has been supported by one or
more grants from or has one or more grants pending with the Netherlands Asthma
Foundation, the EuropeanUnion, andStichtingAstmaBestrijding and has received one
or more payments for lecturing from or is on the speakers’ bureau for GlaxoSmithK-
line. M. C. Nawijn has received one or more grants from or has one or more grants
pending with the Netherlands Asthma Foundation, the European Union, and Stichting
Astma Bestrijding, and has received support for travel from the European Respiratory
Society. The rest of the authors declare that they have no relevant conflicts of interest.
Received for publication July 24, 2012; revised October 30, 2012; accepted for publica-
tion November 19, 2012.
Available online February 4, 2013.
Corresponding author: Martijn C. Nawijn, PhD, Laboratory of Allergology and Pulmo-
nary Diseases, Pathology andMedical Biology, IPC EA11, University Medical Center
Groningen, GRIAC Research Institute, University of Groningen, Hanzeplein 1, PO
Box 30.001, 9700 RB, Groningen, The Netherlands. E-mail: [email protected].
0091-6749/$36.00
� 2013 American Academy of Allergy, Asthma & Immunology
http://dx.doi.org/10.1016/j.jaci.2012.11.028
856
Asthma is a complex, chronic inflammatory disease of the
airways currently affecting more than 300 million personsworldwide, with approximately 250,000 annual deaths as aresult.1 It is estimated that by 2025, the number of asthmaticpatients will increase by more than 100 million.2Asthma is characterized by respiratory symptoms, variableairway obstruction, and airway hyperresponsiveness. The clinicalexpression of asthma is dependent on the interaction betweengenetic predisposition and environmental factors. The number ofidentified asthma susceptibility genes has increased rapidly overthe last 5 years, especially with the application of the genome-wide association (GWA) study approach. In a GWA study300,000 to more than a million DNA polymorphisms coveringthe genome are investigated for association with asthma in largesamples of cases and control subjects.Genetic variation in the IL33 and IL-1 receptor–like 1 (IL1RL1)
genes has reproducibly been found to be associated with asthmain GWA studies, identifying IL-33–induced signaling throughIL-1RL1 as one of the central pathways in asthmatic patients. Al-though numerous functional studies have revealed a central rolefor IL-33–induced signaling in TH2-driven inflammation, whichplays a crucial role in allergic asthma, few studies have directlyassessed the functional consequences of genetic variation in theIL33 and IL1RL1 genes for the activity of this pathway. Such stud-ies are currently limited by the large number of asthma-associatedsingle nucleotide polymorphisms (SNPs) in these genes and thecomplex genetic structure of the IL1RL1 locus.
Therefore this review aims to offer an interpretation of asthma-associated polymorphisms in IL33 and IL1RL1 as a limitednumber of discrete genetic signals with distinct functional conse-quences and to discuss these in the context of a newly identified

J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 857
population of innate IL-33–responsive, IL-1RL11 immune cells,the type-2 innate lymphoid cells (ILC2s), in the pathophysiologyof asthma.
GENETIC ASSOCIATION OF THE IL-33/IL-1RL1
PATHWAY WITH ASTHMA SUSCEPTIBILITYIL-33 is an IL-1 family member and the ligand of the IL-1RL1
(ST2) receptor. The IL33 genewas initially found to be associatedwith Cedar pollinosis in a Japanese population.3 One year later, aGWA study in an Icelandic population identified SNPs flankingIL33 to be suggestively associated with blood eosinophils.4
This finding was followed up by a large case-control study ofasthmatic patients and control subjects, indicating significantassociation of IL33 SNPs with asthma.4 Subsequent GWA studymeta-analyses by the European GABRIEL consortium,5 whichwas recently combined with the Analysis in Population-basedCohorts for Asthma Traits consortium meta-analysis,6 and theNorth American EVE consortium7 identified IL33 as one of thetop hits for asthma.Eight IL33 SNPs have been reported to be associated with
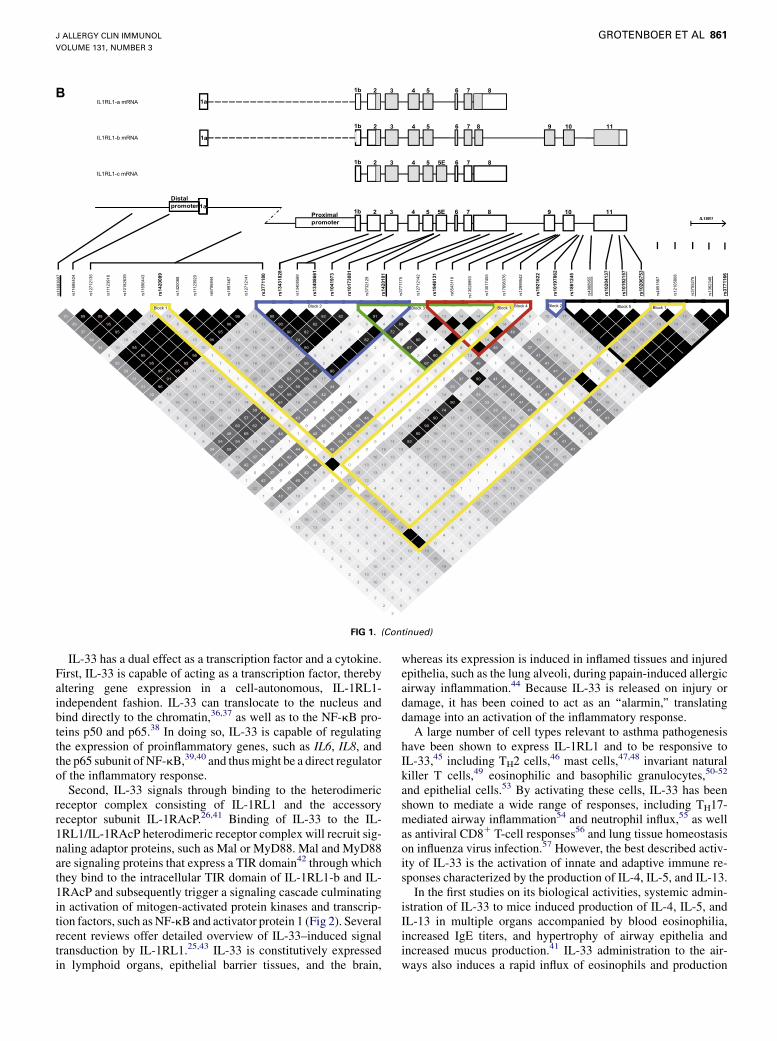
asthma phenotypes (Table I and see the Methods section in thisarticle’s Online Repository at www.jacionlone.org).4-15 Severalof these SNPs are in close proximity to each other and areobserved more frequently together than would be expected bychance based on the allele frequencies in the population. Thisphenomenon is termed linkage disequilibrium (LD) and indicatesthat the SNPs are correlated in populations. In case the SNPs arein full LD, they are always inherited together and therefore repre-sent a single genetic signal. A series of such highly correlatedSNPs form an LD block, and their alleles form a haplotype.Although a single SNP within this LD block might be responsiblefor the functional alteration that contributes to disease, all SNPswithin the LD block will be associated with asthma in a geneticassociation analysis. When the LD structure of IL33 in the Utahresidents with Northern and Western European ancestry fromthe CEPH collection (CEU; referred to as non-Hispanic white)ethnic background, in which most association analyses havebeen performed, is taken into account (Fig 1, A), it becomesapparent that 4 of the asthma-associated SNPs are located in 2LD blocks, whereas the remaining 4 asthma-associated SNPsare not in strong LD with other polymorphisms (Table II [andsee the Methods section in this article’s Online Repository] andFig 1, A).4-21 Interestingly, in the Yoruban in Ibadan, Nigeria(YRI) population and, to some extent in the African ancestry inSouthwest United States (ASW) and Han Chinese in Beijing(CHB) ethnic backgrounds, the LD patterns of IL33 are different(see Fig E1 in this article’s Online Repository at www.jacionline.org), in that the SNPs clustering into LD blocks within the CEUpopulation are independent SNPs in the studied African andAsian populations, indicating that association analyses in patientsof these ethnic backgrounds might reveal the causal SNP retainedwithin the 2 LD blocks.IL1RL1 was first described as a candidate gene for atopic
dermatitis.22 Our group was the first to report IL1RL1 as anasthma susceptibility locus by taking a candidate gene ap-proach.10 In total, 7 candidate gene studies4,9-14 and 4 GWAstudies5,6,7,16 have reported an association of IL1RL1 SNPswith asthma.Thus far 15 different IL1RL1 SNPs have been reported to be
associated with asthma (Table I). These SNPs lie scattered
throughout the IL1RL1 gene (Fig 1, B). Analysis of the LD struc-ture of IL1RL1 and its surrounding genomic region in patients ofthe CEU ethnic background reveals a complex LD pattern inwhich 5 LD blocks containing asthma SNPs and 1 independentasthma SNP can be distinguished, each of which might sepa-rately contribute to asthma susceptibility (Table II). Importantly,several SNPs in IL1RL1 are in LD with SNPs in the genes encod-ing IL-18 receptor 1 (IL18R1) and IL-18 receptor accessory pro-tein, 2 genes juxtaposed to IL1RL1 on chromosome 2q.10,24 Thusgenetic associations in the CEU population cannot conclusivelydetermine which of these genes is implicated in asthma. More-over, analysis of the LD pattern of IL1RL1 in different ethnicbackgrounds reveals that the LD structure of the IL1RL1/IL18R1 locus remains highly complex in all these populations(see Fig E2 in this article’s Online Repository at www.jacionline.org). The IL1RL1 SNP rs1041973 represents an inde-pendent SNP in populations of Africans, Asian, andMexican eth-nicity, whereas it is part of LD block 2 in non-Hispanic whitesubjects (Fig 1, B, and see Fig E2). In the Mexican ancestry inLos Angeles, California, population (MEX; Mexican population)2 SNPs, rs1041973 and rs13431828 (r2 5 0.66 in MEX; both inLD block 2 in CEU) were in fact both found to be significantlyassociated with asthma,11 indicating that there might be 2 inde-pendent asthma signals present within the non-Hispanic whiteLD block 2. Therefore association analyses in other ethnic back-grounds might aid the identification of the causal asthma-associated SNPs, but experimental validation will remain keyto proving the individual contribution of the identified causal var-iants. Potential experimental approaches include in vitro studieson the functional effects of asthma-associated haplotypes in pri-mary cells or in vivo studies using mouse models that have beenspecifically engineered to carry the risk or protective haplotypeof either IL1RL1 or IL18R1.
FUNCTIONAL GENETICS OF IL33 AND IL1RL1The IL33 gene, which is located on chromosome 9, spans
approximately 42.2 kb in length, harboring 8 exons. All IL33asthma-associated SNPs are located 59 of the gene or in the firstintron. Therefore it is tempting to speculate that these SNPs affectIL33 transcription and that asthma susceptibility alleles are asso-ciated with increased IL-33 production. At present, however, nopublished data support this function, warranting further explora-tion of the functional consequences of these SNPs.The IL1RL1 gene, which is located at chromosome 2q12, spans
approximately 40.5 kb in length, harboring 13 exons and a distaland a proximal promoter. The IL1RL1 gene encodes for proteinswith an extracellular region carrying 3 immunoglobulin-likedomains, a transmembrane domain, and an intracellular regionharboring a Toll-like/IL-1 receptor (TIR) domain. Three tran-scripts are expressed through alternative splicing: a short isoformencoding the soluble protein IL1-RL1-a (also called sST2); a longisoform encoding the full transmembrane receptor IL1-RL1-b(ST2L); and a less well-known variant that encodes a truncatedprotein with 2 immunoglobulin-like domains and a hydrophobictail called IL1-RL1-c (ST2V). Functionally, IL1-RL1-b acts totransduce the IL-33 signal to the intracellular compartment,whereas the soluble IL1RL1-a functions as a decoy receptor, cap-turing IL-33 and inhibiting its function.25,26
IL1RL1 asthma-associated SNPs can translate into functionalalterations of the IL-33/IL-1RL1 pathway through several
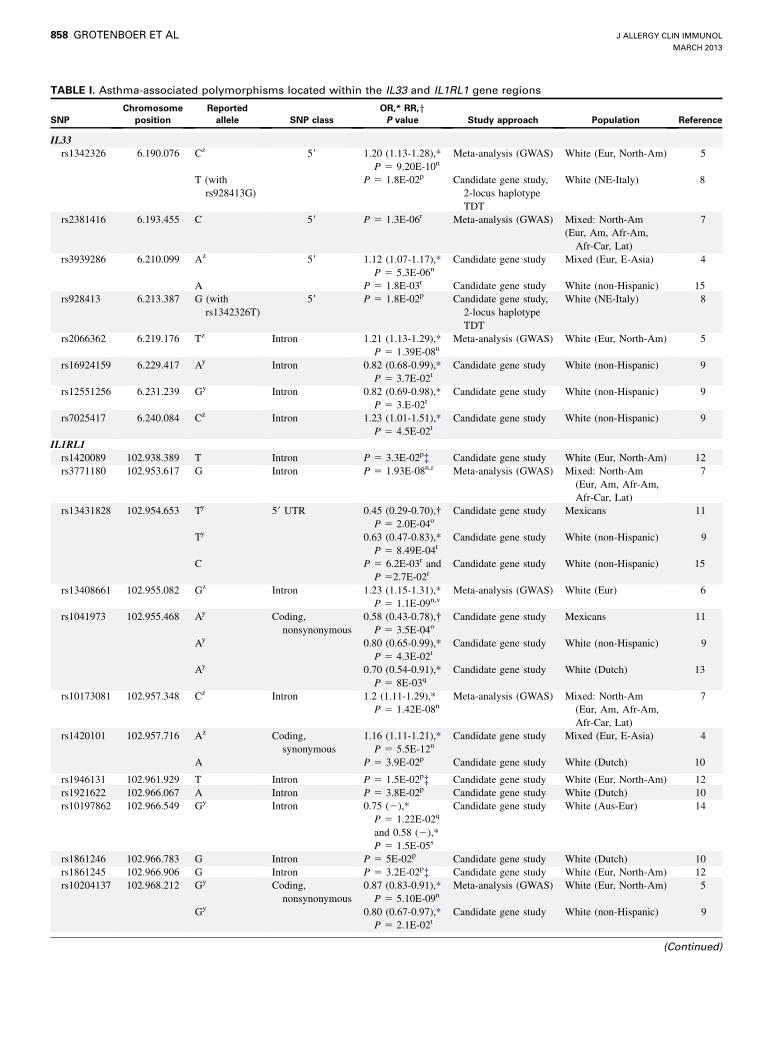
TABLE I. Asthma-associated polymorphisms located within the IL33 and IL1RL1 gene regions
SNP
Chromosome
position
Reported
allele SNP class
OR,* RR,yP value Study approach Population Reference
IL33
rs1342326 6.190.076 Cz
T (with
rs928413G)
59 1.20 (1.13-1.28),*
P 5 9.20E-10n
P 5 1.8E-02p
Meta-analysis (GWAS)
Candidate gene study,
2-locus haplotype
TDT
White (Eur, North-Am)
White (NE-Italy)
5
8
rs2381416 6.193.455 C 59 P 5 1.3E-06r Meta-analysis (GWAS) Mixed: North-Am
(Eur, Am, Afr-Am,
Afr-Car, Lat)
7
rs3939286 6.210.099 Az
A
59 1.12 (1.07-1.17),*
P 5 5.3E-06n
P 5 1.8E-03r
Candidate gene study
Candidate gene study
Mixed (Eur, E-Asia)
White (non-Hispanic)
4
15
rs928413 6.213.387 G (with
rs1342326T)
59 P 5 1.8E-02p Candidate gene study,
2-locus haplotype
TDT
White (NE-Italy) 8
rs2066362 6.219.176 Tz Intron 1.21 (1.13-1.29),*
P 5 1.39E-08nMeta-analysis (GWAS) White (Eur, North-Am) 5
rs16924159 6.229.417 Ay Intron 0.82 (0.68-0.99),*
P 5 3.7E-02tCandidate gene study White (non-Hispanic) 9
rs12551256 6.231.239 Gy Intron 0.82 (0.69-0.98),*
P 5 3.E-02tCandidate gene study White (non-Hispanic) 9
rs7025417 6.240.084 Cz Intron 1.23 (1.01-1.51),*
P 5 4.5E-02tCandidate gene study White (non-Hispanic) 9
IL1RL1
rs1420089 102.938.389 T Intron P 5 3.3E-02p� Candidate gene study White (Eur, North-Am) 12
rs3771180 102.953.617 G Intron P 5 1.93E-08n,r Meta-analysis (GWAS) Mixed: North-Am
(Eur, Am, Afr-Am,
Afr-Car, Lat)
7
rs13431828 102.954.653 Ty
Ty
C
59 UTR 0.45 (0.29-0.70),�P 5 2.0E-04o
0.63 (0.47-0.83),*
P 5 8.49E-04t
P 5 6.2E-03r and
P 52.7E-02r
Candidate gene study
Candidate gene study
Candidate gene study
Mexicans
White (non-Hispanic)
White (non-Hispanic)
11
9
15
rs13408661 102.955.082 Gz Intron 1.23 (1.15-1.31),*
P 5 1.1E-09n,vMeta-analysis (GWAS) White (Eur) 6
rs1041973 102.955.468 Ay
Ay
Ay
Coding,
nonsynonymous
0.58 (0.43-0.78),�P 5 3.5E-04o
0.80 (0.65-0.99),*
P 5 4.3E-02t
0.70 (0.54-0.91),*
P 5 8E-03q
Candidate gene study
Candidate gene study
Candidate gene study
Mexicans
White (non-Hispanic)
White (Dutch)
11
9
13
rs10173081 102.957.348 Cz Intron 1.2 (1.11-1.29),*
P 5 1.42E-08nMeta-analysis (GWAS) Mixed: North-Am
(Eur, Am, Afr-Am,
Afr-Car, Lat)
7
rs1420101 102.957.716 Az
A
Coding,
synonymous
1.16 (1.11-1.21),*
P 5 5.5E-12n
P 5 3.9E-02p
Candidate gene study
Candidate gene study
Mixed (Eur, E-Asia)
White (Dutch)
4
10
rs1946131 102.961.929 T Intron P 5 1.5E-02p� Candidate gene study White (Eur, North-Am) 12
rs1921622 102.966.067 A Intron P 5 3.8E-02p Candidate gene study White (Dutch) 10
rs10197862 102.966.549 Gy Intron 0.75 (2),*
P 5 1.22E-02q
and 0.58 (2),*
P 5 1.5E-05s
Candidate gene study White (Aus-Eur) 14
rs1861246 102.966.783 G Intron P 5 5E-02p Candidate gene study White (Dutch) 10
rs1861245 102.966.906 G Intron P 5 3.2E-02p� Candidate gene study White (Eur, North-Am) 12
rs10204137 102.968.212 Gy
Gy
Coding,
nonsynonymous
0.87 (0.83-0.91),*
P 5 5.10E-09n
0.80 (0.67-0.97),*
P 5 2.1E-02t
Meta-analysis (GWAS)
Candidate gene study
White (Eur, North-Am)
White (non-Hispanic)
5
9
(Continued)
J ALLERGY CLIN IMMUNOL
MARCH 2013
858 GROTENBOER ET AL
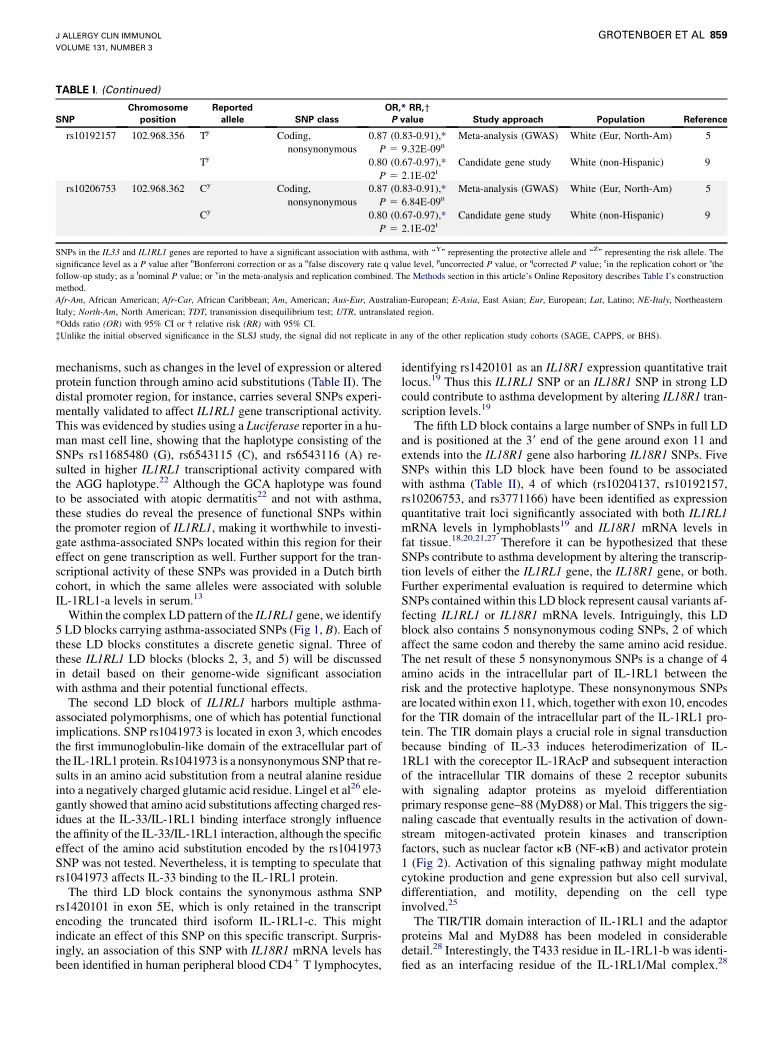
TABLE I. (Continued)
SNP
Chromosome
position
Reported
allele SNP class
OR,* RR,yP value Study approach Population Reference
rs10192157 102.968.356 Ty
Ty
Coding,
nonsynonymous
0.87 (0.83-0.91),*
P 5 9.32E-09n
0.80 (0.67-0.97),*
P 5 2.1E-02t
Meta-analysis (GWAS)
Candidate gene study
White (Eur, North-Am)
White (non-Hispanic)
5
9
rs10206753 102.968.362 Cy
Cy
Coding,
nonsynonymous
0.87 (0.83-0.91),*
P 5 6.84E-09n
0.80 (0.67-0.97),*
P 5 2.1E-02t
Meta-analysis (GWAS)
Candidate gene study
White (Eur, North-Am)
White (non-Hispanic)
5
9
SNPs in the IL33 and IL1RL1 genes are reported to have a significant association with asthma, with ‘‘Y’’ representing the protective allele and ‘‘Z’’ representing the risk allele. The
significance level as a P value after nBonferroni correction or as a ofalse discovery rate q value level, puncorrected P value, or qcorrected P value; rin the replication cohort or sthe
follow-up study; as a tnominal P value; or vin the meta-analysis and replication combined. The Methods section in this article’s Online Repository describes Table I’s construction
method.
Afr-Am, African American; Afr-Car, African Caribbean; Am, American; Aus-Eur, Australian-European; E-Asia, East Asian; Eur, European; Lat, Latino; NE-Italy, Northeastern
Italy; North-Am, North American; TDT, transmission disequilibrium test; UTR, untranslated region.
*Odds ratio (OR) with 95% CI or � relative risk (RR) with 95% CI.
�Unlike the initial observed significance in the SLSJ study, the signal did not replicate in any of the other replication study cohorts (SAGE, CAPPS, or BHS).
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 859
mechanisms, such as changes in the level of expression or alteredprotein function through amino acid substitutions (Table II). Thedistal promoter region, for instance, carries several SNPs experi-mentally validated to affect IL1RL1 gene transcriptional activity.This was evidenced by studies using a Luciferase reporter in a hu-man mast cell line, showing that the haplotype consisting of theSNPs rs11685480 (G), rs6543115 (C), and rs6543116 (A) re-sulted in higher IL1RL1 transcriptional activity compared withthe AGG haplotype.22 Although the GCA haplotype was foundto be associated with atopic dermatitis22 and not with asthma,these studies do reveal the presence of functional SNPs withinthe promoter region of IL1RL1, making it worthwhile to investi-gate asthma-associated SNPs located within this region for theireffect on gene transcription as well. Further support for the tran-scriptional activity of these SNPs was provided in a Dutch birthcohort, in which the same alleles were associated with solubleIL-1RL1-a levels in serum.13
Within the complex LD pattern of the IL1RL1 gene, we identify5 LD blocks carrying asthma-associated SNPs (Fig 1, B). Each ofthese LD blocks constitutes a discrete genetic signal. Three ofthese IL1RL1 LD blocks (blocks 2, 3, and 5) will be discussedin detail based on their genome-wide significant associationwith asthma and their potential functional effects.The second LD block of IL1RL1 harbors multiple asthma-
associated polymorphisms, one of which has potential functionalimplications. SNP rs1041973 is located in exon 3, which encodesthe first immunoglobulin-like domain of the extracellular part ofthe IL-1RL1 protein. Rs1041973 is a nonsynonymous SNP that re-sults in an amino acid substitution from a neutral alanine residueinto a negatively charged glutamic acid residue. Lingel et al26 ele-gantly showed that amino acid substitutions affecting charged res-idues at the IL-33/IL-1RL1 binding interface strongly influencethe affinity of the IL-33/IL-1RL1 interaction, although the specificeffect of the amino acid substitution encoded by the rs1041973SNP was not tested. Nevertheless, it is tempting to speculate thatrs1041973 affects IL-33 binding to the IL-1RL1 protein.The third LD block contains the synonymous asthma SNP
rs1420101 in exon 5E, which is only retained in the transcriptencoding the truncated third isoform IL-1RL1-c. This mightindicate an effect of this SNP on this specific transcript. Surpris-ingly, an association of this SNP with IL18R1 mRNA levels hasbeen identified in human peripheral blood CD41 T lymphocytes,
identifying rs1420101 as an IL18R1 expression quantitative traitlocus.19 Thus this IL1RL1 SNP or an IL18R1 SNP in strong LDcould contribute to asthma development by altering IL18R1 tran-scription levels.19
The fifth LD block contains a large number of SNPs in full LDand is positioned at the 39 end of the gene around exon 11 andextends into the IL18R1 gene also harboring IL18R1 SNPs. FiveSNPs within this LD block have been found to be associatedwith asthma (Table II), 4 of which (rs10204137, rs10192157,rs10206753, and rs3771166) have been identified as expressionquantitative trait loci significantly associated with both IL1RL1mRNA levels in lymphoblasts19 and IL18R1 mRNA levels infat tissue.18,20,21,27 Therefore it can be hypothesized that theseSNPs contribute to asthma development by altering the transcrip-tion levels of either the IL1RL1 gene, the IL18R1 gene, or both.Further experimental evaluation is required to determine whichSNPs contained within this LD block represent causal variants af-fecting IL1RL1 or IL18R1 mRNA levels. Intriguingly, this LDblock also contains 5 nonsynonymous coding SNPs, 2 of whichaffect the same codon and thereby the same amino acid residue.The net result of these 5 nonsynonymous SNPs is a change of 4amino acids in the intracellular part of IL-1RL1 between therisk and the protective haplotype. These nonsynonymous SNPsare located within exon 11, which, together with exon 10, encodesfor the TIR domain of the intracellular part of the IL-1RL1 pro-tein. The TIR domain plays a crucial role in signal transductionbecause binding of IL-33 induces heterodimerization of IL-1RL1 with the coreceptor IL-1RAcP and subsequent interactionof the intracellular TIR domains of these 2 receptor subunitswith signaling adaptor proteins as myeloid differentiationprimary response gene–88 (MyD88) or Mal. This triggers the sig-naling cascade that eventually results in the activation of down-stream mitogen-activated protein kinases and transcriptionfactors, such as nuclear factor kB (NF-kB) and activator protein1 (Fig 2). Activation of this signaling pathway might modulatecytokine production and gene expression but also cell survival,differentiation, and motility, depending on the cell typeinvolved.25
The TIR/TIR domain interaction of IL-1RL1 and the adaptorproteins Mal and MyD88 has been modeled in considerabledetail.28 Interestingly, the T433 residue in IL-1RL1-b was identi-fied as an interfacing residue of the IL-1RL1/Mal complex.28

rs19
2999
6
37
69
71
9
71
71
71
26
18
1
0
0
0
0
23
3
19
2
1
2
0
3
12
13
13
14
12
13
4
13
13
1
1
37
68
71
9
71
71
71
25
19
1
1
0
0
0
23
2
19
3
1
2
0
4
12
13
13
14
12
13
4
13
13
0
1
37
67
71
10
71
71
71
26
18
1
0
0
0
0
23
3
19
2
2
3
0
3
10
13
13
13
11
13
3
13
13
0
1
50
52
2
52
52
52
57
5
1
6
10
10
10
11
4
6
13
14
14
7
14
17
7
7
7
8
7
9
7
7
6
3
95
3
95
95
95
37
12
0
4
0
0
0
22
1
17
0
0
0
2
0
25
7
7
8
9
7
7
7
7
0
1
3
39
15
0
5
0
0
0
20
1
20
0
0
0
2
0
26
9
9
9
10
9
6
9
9
0
0
3
3
3
2
3
0
1
2
2
2
2
1
1
9
9
13
2
14
6
3
3
3
0
3
5
3
3
1
27
39
15
0
5
0
0
0
20
1
20
0
0
0
2
0
26
9
9
9
10
9
6
9
9
0
0
39
15
0
5
0
0
0
20
1
20
0
0
0
2
0
26
9
9
9
10
9
6
9
9
0
0
39
15
0
5
0
0
0
20
1
20
0
0
0
2
0
26
9
9
9
10
9
6
9
9
0
0
12
3
6
2
2
2
11
4
10
6
6
6
7
6
8
4
4
4
4
4
3
4
4
6
0
3
11
17
16
17
74
1
58
17
18
18
12
18
26
18
18
18
16
18
5
18
18
10
5
1
16
19
17
0
1
4
1
4
1
1
1
2
2
2
2
2
2
0
2
2
1
19
64
59
64
8
38
17
48
46
46
88
46
25
6
6
6
6
6
7
6
6
75
2
13
36
26
59
62
58
72
56
17
2
2
3
3
2
5
2
2
64
1
13
32
27
57
60
56
68
54
16
1
1
2
2
1
4
1
1
59
1
13
36
26
59
61
56
72
56
16
2
2
2
3
2
4
2
2
63
1
1
43
17
18
17
9
18
29
24
24
28
26
24
13
24
24
8
4
6
27
27
26
52
26
12
7
7
7
7
7
10
7
7
63
2
35
39
30
19
30
54
41
41
41
44
41
14
41
41
16
8
95
55
95
4
7
7
7
8
7
0
7
7
47
24
95
52
95
6
9
9
9
11
9
0
9
9
46
24
52
3
8
8
8
6
8
0
8
8
47
23
52
28
7
7
7
8
7
9
7
7
88
3
3
8
8
8
5
8
0
8
8
45
23
35
35
36
38
35
47
35
35
24
11
95
71
10
0
95
71
10
0
95
73
10
0
95
68
95
95
11
0
71
10
0
71
71
14
25
10
0
10
0
2
rs47
4216
6
rs14
1242
6
rs
13
42
32
6
rs
23
81
41
6
rs18
8890
9
rs13
2840
60
rs99
2969
rs
39
39
28
6
rs
92
84
13
rs
20
66
36
2
rs16
9241
44
rs11
7944
19
rs22
1046
4
rs10
4358
16
rs10
9754
97
rs47
4084
0
rs
16
92
41
59
rs16
9241
61
rs
12
55
12
56
rs70
3325
8
rs70
3472
0
rs13
75
rs
70
25
41
7
rs47
4217
0
rs70
1957
5
rs10
9755
16
rs13
3038
3
rs19
2999
2
rs11
1357
3
rs10
9755
19
rs14
1242
1
rs70
4792
1
rs10
4827
4
rs10
8153
97
rs20
2699
1
1 2 3 4 5 6 7 8
IL33-variant 3 mRNA
IL33-variant 2 mRNA
IL33-variant 1 mRNA
Block 1 Block 2 Block 2 Block 2
A
FIG 1. LD of the polymorphisms located within the IL33 region (chromosome 9) and the IL1RL1/IL18R1region (chromosome 2). A, LD plot of the IL33 region in the CEU population (see the Methods section in
this article’s Online Repository for more details). B, LD plot of the IL1RL1/IL18R1 region in the CEU popula-
tion (see the Methods section in this article’s Online Repository for more details). Asthma-associated poly-
morphisms are depicted in boldface, and expression quantitative trait loci are underlined.
J ALLERGY CLIN IMMUNOL
MARCH 2013
860 GROTENBOER ET AL
Because the nonsynonymous coding SNP rs4988956 (LD block 5,Table II) results in a change of this polar threonine residue into anonpolar alanine residue,29 it can be hypothesized that this SNPhas a direct effect on the affinity of the interaction between theTIR domains of IL-1RL1-b and Mal. This might directly affectIL-33–induced signaling through IL-1RL1. These data warrantfurther evaluation of the effect of these 4 amino acid substitutionsfor IL-33–induced activation of downstream signaling. We pro-pose that amino acid substitutions contained within the TIR do-main of IL-1RL1 affect coupling of IL-1RL1 to the coreceptorIL-1RAcP and adaptor proteins, such as Mal, and thus affectdownstream signaling.In addition to direct effects on IL-33–induced signaling, the
TIR domain–dependent interaction of IL-1RL1-b with the adap-tor proteins MyD88 and Mal also plays a role in the inhibition ofToll-like receptor (TLR) signaling by IL-1RL1 (Fig 2).30,31 IL-1RL1/Mal orMyD88 complex formationmay prevent the interac-tion betweenMyD88,Mal, or bothwith activated TLRs.28,30 Thusthe presence of multiple amino acid substitutions within the TIRdomain of IL-1RL1-b could result in altered inhibition of TLR ac-tivation. This possibility is especially intriguing given the genetic
interaction we have previously reported for IgE sensitizationbetween polymorphisms in IL1RL1 and TLR4.32
In summary, the IL1RL1 gene carries multiple independent ge-netic signals with possible functional consequences that arehighly divergent, including effects on expression of IL1RL1 andits neighboring gene IL18R1, as well as effects on IL-33/IL-1RL1 binding affinity, TIR domain interactions, and downstreamsignaling.
FROM GENETICS TO BIOLOGY: IL-33/IL-1RL1
SIGNALING IN THE PATHOPHYSIOLOGY OF
ASTHMAGenetic studies strongly implicate the IL-33/IL-1RL1 pathway
in asthmatic patients. This is further corroborated by mechanisticstudies in experimental animal models of asthma and clinicalstudies, which will be reviewed below. Very recently, it has beenshown that the IL-33/IL-1RL1 pathway can act through theactivation of a novel subset of innate immune cells called ILC2sor nuocytes that produce cytokines such as IL-5 and IL-13 in thelungs.33-35
rs11
6854
24
rs12
7121
35
rs11
1239
18
rs10
1826
39
rs11
6904
43
rs
14
20
08
9
rs14
2008
8
rs11
1239
20
rs67
0684
4
rs19
9746
7
rs12
7121
41
rs
37
711
80
rs
13
43
18
28
rs13
4085
69
rs
13
40
86
61
rs
10
41
97
3
rs
10
17
30
81
rs37
3212
9
rs
14
20
10
1
rs37
7117
5
rs12
7121
42
rs
19
46
13
1
rs65
4311
9
rs13
0289
93
rs13
0174
55
rs17
6963
76
rs12
9995
42
rs
19
21
62
2
rs
10
19
78
62
rs
18
61
24
5
rs49
8895
5
rs
10
20
41
37
rs
10
19
21
57
rs
10
20
67
53
rs48
5156
7
rs12
1058
08
rs37
5527
6
rs13
6234
8
Block 1 Block 2 Block 5
42 0153 5E 6 7 8 1191b
1a1a
Distal
promoter
Proximal
promoter
Block 3 Block 4 Block 2
31
35
37
34
34
3
33
33
31
31
32
6
8
8
8
6
8
34
30
8
22
1
22
1
22
3
1
10
6
2
2
2
2
2
3
3
2
2
2
95
10
96
13
16
16
16
21
16
54
59
16
42
0
42
0
42
10
0
16
13
5
5
5
5
5
10
10
5
5
5
95
95
95
10
95
95
95
95
91
14
18
18
18
23
18
48
54
18
37
0
37
0
37
10
0
13
14
3
3
3
3
3
10
10
3
3
3
10
95
9
13
13
13
16
13
60
65
13
45
1
45
0
45
9
0
21
9
4
6
6
6
6
9
9
6
6
6
10
95
11
15
15
15
18
15
57
62
15
42
1
42
0
42
9
0
19
11
4
6
6
6
6
9
9
6
6
6
11
95
10
14
14
14
17
14
59
63
14
44
1
44
0
44
9
0
20
10
5
7
7
7
7
9
10
7
7
7
10
8
10
10
10
1
1
1
1
2
1
5
6
1
8
1
8
1
8
1
17
1
19
19
19
19
19
19
19
19
95
12
15
15
15
20
15
54
61
15
43
0
42
0
42
8
0
15
12
4
6
6
6
6
8
9
6
6
6
95
13
16
16
16
21
16
52
59
16
41
0
40
0
40
7
0
13
13
3
4
4
4
4
7
7
4
4
4
96
13
16
16
16
21
16
53
59
16
42
0
42
0
42
9
0
16
13
5
6
6
6
6
9
9
6
6
6
96
13
16
16
16
21
16
53
59
16
42
0
42
0
42
9
0
16
13
5
6
6
6
6
9
9
6
6
6
13
17
17
17
21
17
56
62
17
44
0
44
0
44
10
0
14
13
6
6
6
6
6
10
10
6
6
6
90
90
90
74
90
5
5
90
7
1
7
1
7
1
1
15
82
17
17
17
17
17
1
1
17
17
17
82
4
5
6
0
6
0
6
1
0
14
90
15
15
15
15
15
1
1
15
15
15
82
4
5
6
0
6
0
6
1
0
14
90
16
15
15
15
15
1
1
15
15
15
82
4
5
6
0
6
0
6
1
0
14
90
16
15
15
15
15
1
1
15
15
15
82
6
1
82
2
0
2
0
2
2
0
7
74
15
15
15
15
15
2
2
15
15
15
4
5
6
0
6
0
6
1
0
14
90
16
15
15
15
15
1
1
15
15
15
91
4
72
2
67
2
67
5
2
33
5
25
30
30
30
30
5
5
30
30
30
5
80
0
80
0
80
6
0
37
5
33
33
33
33
33
6
6
33
33
33
6
0
6
0
6
1
0
14
90
15
15
15
15
15
1
1
15
15
15
13
13
8
13
46
7
41
41
41
41
41
8
8
41
41
41
13
13
1
6
1
5
5
5
5
5
1
1
5
5
5
14
7
14
46
7
37
41
41
41
41
7
7
41
41
41
14
1
6
1
5
5
5
5
5
1
1
5
5
5
7
14
46
7
37
41
41
41
41
7
7
41
41
41
1
17
1
19
19
19
19
19
19
19
19
6
1
5
5
5
5
5
1
1
5
5
5
15
3
5
5
5
5
17
18
5
5
5
17
17
17
17
17
1
1
17
17
17
19
20
19
19
19
19
19
19
19
19 19
19
19
19
19
19
rs11
6936
97
rs
37
711
66
Block 1 Block 1
IL18R1
42 3 5 6 7 81b
IL1RL1-a mRNA
42 0153 6 1187 91b
IL1RL1-b mRNA
42 3 5 5E 6 7 81b
IL1RL1-c mRNA
1a
1a
B
FIG 1. (Continued)
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 861
IL-33 has a dual effect as a transcription factor and a cytokine.First, IL-33 is capable of acting as a transcription factor, therebyaltering gene expression in a cell-autonomous, IL-1RL1-independent fashion. IL-33 can translocate to the nucleus andbind directly to the chromatin,36,37 as well as to the NF-kB pro-teins p50 and p65.38 In doing so, IL-33 is capable of regulatingthe expression of proinflammatory genes, such as IL6, IL8, andthe p65 subunit of NF-kB,39,40 and thusmight be a direct regulatorof the inflammatory response.Second, IL-33 signals through binding to the heterodimeric
receptor complex consisting of IL-1RL1 and the accessoryreceptor subunit IL-1RAcP.26,41 Binding of IL-33 to the IL-1RL1/IL-1RAcP heterodimeric receptor complex will recruit sig-naling adaptor proteins, such as Mal or MyD88. Mal and MyD88are signaling proteins that express a TIR domain42 through whichthey bind to the intracellular TIR domain of IL-1RL1-b and IL-1RAcP and subsequently trigger a signaling cascade culminatingin activation of mitogen-activated protein kinases and transcrip-tion factors, such as NF-kB and activator protein 1 (Fig 2). Severalrecent reviews offer detailed overview of IL-33–induced signaltransduction by IL-1RL1.25,43 IL-33 is constitutively expressedin lymphoid organs, epithelial barrier tissues, and the brain,
whereas its expression is induced in inflamed tissues and injuredepithelia, such as the lung alveoli, during papain-induced allergicairway inflammation.44 Because IL-33 is released on injury ordamage, it has been coined to act as an ‘‘alarmin,’’ translatingdamage into an activation of the inflammatory response.A large number of cell types relevant to asthma pathogenesis
have been shown to express IL-1RL1 and to be responsive toIL-33,45 including TH2 cells,46 mast cells,47,48 invariant naturalkiller T cells,49 eosinophilic and basophilic granulocytes,50-52
and epithelial cells.53 By activating these cells, IL-33 has beenshown to mediate a wide range of responses, including TH17-mediated airway inflammation54 and neutrophil influx,55 as wellas antiviral CD81 T-cell responses56 and lung tissue homeostasison influenza virus infection.57 However, the best described activ-ity of IL-33 is the activation of innate and adaptive immune re-sponses characterized by the production of IL-4, IL-5, and IL-13.In the first studies on its biological activities, systemic admin-
istration of IL-33 to mice induced production of IL-4, IL-5, andIL-13 in multiple organs accompanied by blood eosinophilia,increased IgE titers, and hypertrophy of airway epithelia andincreased mucus production.41 IL-33 administration to the air-ways also induces a rapid influx of eosinophils and production

TABLE II. Discrete genetic signals and predicted function of SNPs associated with asthma in the IL33 and IL1RL1 gene regions
LD block RS no.
Association
reference SNP location
Predicted
function P value Tissue Source database
IL33
2 rs1342326 5, 8 59 of IL33 21 rs2381416
rs3939286
rs928413
7
4, 15
8
59 of IL3359 of IL3359 of IL33
2 rs2066362 5 Intron 1 22 rs16924159 9 Intron 1 22 rs12551256 9 Intron 1 22 rs7025417 9 Intron 1 22 rs2026991 39 of IL33 eQTL* 1.00E-24 Liver GTEx 17
IL1RL1
2 rs11693697 59 of IL1RL1 eQTL� 4.95E-04 Fat tissue GeneVar 18
1 rs1420089 12 Intron 1a Unknown
2 rs3771180
rs13431828
rs13408661
rs1041973
rs10173081
rs10197862
7
9, 11, 15
6
9, 11, 13
7
14
Proximal promoter
Exon 2 (59UTR)Intron 2
Exon 3 (nonsynonymous)
Intron 5
Intron 10
Amino acid
substitution
3 rs1420101
rs12998521
4, 10 Exon 5E (synonymous)
39 of IL1RL1
eQTL�
eQTL�
6.88E-04
1.23E-04
Primary peripheral
blood CD41 lymphocytes
Primary peripheral
blood CD41 lymphocytes
(Murphy 19)
(Murphy 19)
4 rs1946131 12 Intron 8 Unknown
2 rs1921622 10 Intron 10 Unknown
5 rs1861245
rs4988955
rs4988956
rs4988957
rs10192036
rs10204137
rs4988958
rs10192157
rs10206753
rs3755276
rs3771166
12
5, 9
5, 9
5, 9
5, 9, 16
Intron 10
Intron 10
Exon 11 (nonsynonymous)
Exon 11 (synonymous)
Exon 11 (nonsynonymous)
Exon 11 (nonsynonymous)
Exon 11 (synonymous)
Exon 11 (nonsynonymous)
Exon 11 (nonsynonymous)
59 of IL18R1Intron 2 of IL18R1
Amino acid
substitution
and eQTL�
3.05E-04
3.05E-04
3.78E-10
3.05E-04
3.05E-04
3.05E-04
3.05E-04
3.05E-04
3.05E-04
3.05E-04
Fat tissue
Fat tissue
Monocytes
Fat tissue
Fat tissue
Fat tissue
Fat tissue
Fat tissue
Fat tissue
Fat tissue
GeneVar 18
GeneVar 18
SeeQTL 20, 21
GeneVar 18
GeneVar 18
GeneVar 18
GeneVar 18
GeneVar 18
GeneVar 18
GeneVar 18
Polymorphisms with a reference in column 3 have been associated with asthma. eQTLs are depicted in italics, whereas IL1RL1 polymorphisms associated with asthma are depicted
in boldface. The Methods section in this article’s Online Repository describes Table II’s construction method.
eQTL, Expression quantitative trait locus; UTR, untranslated region.
*eQTLs associated with IL33 mRNA levels.
�eQTLs associated with IL1RL1 mRNA levels.
�eQTLs associated with IL18R1 mRNA levels.
J ALLERGY CLIN IMMUNOL
MARCH 2013
862 GROTENBOER ET AL
of IL-5 and IL-13 by innate immune cells independently of IL-4and TH2 lymphocytes.35,58 This innate response characterized byhigh levels of IL-5 and IL-13 is now generally referred to as aninnate type 2 response.Inmurine models IL-33 has been found to be released promptly
after activation or damage of lung resident cells by, for instance,protease-active or glycolipid allergens35,59,60 or influenza infec-tion,61 as well as after allergen challenge in sensitized mice.23
On release, IL-33 activates an innate population of IL-5– andIL-13–producing cells, which were originally identified in mousemodels by multiple groups as ‘‘natural helper cells’’ or ‘‘nuo-cytes’’62,63 and now commonly referred to as ILC2s.64 Also, inthe lung ILC2s have been shown to be present and to respondto IL-33 and to contribute to airway inflammation34 and airwayhyperresponsiveness.33 In fact, ILC2s were found to be required
and sufficient in experimental models of allergic airway diseasefor IL-5 and IL-13 production and mucus hypersecretion on pro-tease treatment.59 Importantly, the induction of airway hyperres-ponsiveness and goblet cell hyperplasia by IL-33 has been foundto be dependent on IL-1RL1 signaling and MyD88-TIR interac-tion.65 In experimental mouse models of allergic airway inflam-mation, ILC2s were found to be a major source of IL-5 andIL-13 but not of IL-4.34 Importantly, the existence of theseIL-33–responsive innate immune cells producing large quantitiesof IL-5 and IL-13 has recently also been described in human sub-jects.57,66 Further support for a central role of the ILC2 cell pop-ulation in the pathogenesis of asthma stems from theidentification of retinoic acid–related orphan receptor a (RORA)as an asthma gene in the GABRIEL GWA study5 and the meta-analysis of the Analysis in Population-based Cohorts for Asthma

TIRTIR
IL-1RAcP
IL1-RL1a
Airway epithelial cells
DendriticCells
Allergen exposure Viral infection
TLRs
IL-1RL1bIL-1RL1b
IL-1RL1b
Naive Th cellsTh2 cells Innate immune cells:
Basophils, Mast cells
Type-2 innatelymphoid cells
Th2 differentationIL-4, IL-5, IL-13 production
enhanced degranulationIL-4, IL-5, IL-13 production
IL-5, IL-13 production
Inhibition of TLR-mediated signaling
TLR2 TLR4
TIR
NF-κB
TIR TIR TIR
IL-1RL1bMALMyD88
IL-33-induced signaling
IL-1RL1a
ERKAP-1
TIR TIR TIR TIR TIR
MALMyD88
IL-1RL1b
NF-κB
IL33IL33 IL33IL33IL33
IL-1RL1b
IL33IL33
TIR
TIR
IL33
TIR TIRTIR
sequestration of
signaling adaptor proteins
inhibition of
TLR signaling
heterotrimeric complex
formation and signalingIL-33 binding
inhibition of
IL-33 binding
IL33
IL33
TIRTIR
IL33IL33
IL33
IL33
IL33
IL33IL33
IL33 IL33 IL33
FIG 2. Biological activity of the IL-33/IL-1RL1 signaling pathway. IL-33–producing and responsive cells in the
lungand the IL-33/IL-1RL1 signaling pathway (in part basedonLingel et al26) are shown.Airway epithelial dam-
agewill induce IL-33 release. IL-33binding toa receptor complexof IL-1RL1-band IL-1RAcP initiates recruitment
of Mal or MyD88 signaling adaptor molecules, leading to activation of downstream pathways (lower insert).IL-1RL1-a canneutralize IL-33. In addition to its role in IL-33 signal transduction, IL-1RL1b can inhibit TLR signal-
ing through TIR domain–dependent sequestration of the adaptor proteins MyD88 andMal (top insert).
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 863
Traits consortium with GABRIEL data7 because RORa was re-cently shown to be critically required for the differentiation ofILC2s in vivo.67 Taken together, a central role in the pathogenesisof asthma for the IL-33/IL-1RL1 pathway, acting in part throughactivation of ILC2s, is supported by the genetic evidence stem-ming from multiple independent GWA studies identifying 3asthma genes operating in this pathway.Next to ILC2s, IL-33 also activates TH2 cells, mast cells, and
basophils, inducing the production of IL-4, as well as IL-5 andIL-13.68 For instance, IL-33 was shown to act as a chemoattrac-tant for TH2 cells,46 and activation of TH2 cells in the presenceof IL-33 greatly enhanced their production of IL-5 and IL-13 inan IL-1RL1- and MyD88-dependent but IL-4–independent fash-ion.49,58 Basophils responded to IL-33 through enhanced basaland IgE cross-linking–induced production of IL-4, IL-5, andIL-13.49,52 Additionally, mast cells responded to IL-33 with en-hanced survival and an increased basal and IgE cross-linking–in-duced production of IL-4, IL-5, and IL-13.47,69-71 Of note, unlikeILC2s, these innate cells are capable of producing IL-4 on IL-33stimulation, a response that has recently been shown to also occurin vivo in a T cell–independent fashion.72
From these in vitro and in vivo studies, a picture is emergingthat higher levels of IL-33, higher activity of IL-1RL1-dependent signal transduction, or both will result in an aggravatedinnate type 2 response mediated by ILC2s, mast cells, and baso-phils, as well as an enhanced adaptive TH2 response. The rele-vance of these observations from experimental mouse models
of asthma for human disease is underscored by studies in asth-matic patients: IL-33 protein was found to be increased in thebronchoalveolar lavage fluid and in airway epithelial cells73 andairway smooth muscle cells of asthmatic patients comparedwith those of healthy control subjects, which positively correlatedwith asthma severity.74,75 Moreover, several cell types derivedfrom allergic subjects, including mast cells, basophils, and eosin-ophils, could be matured and activated to release IL-4, IL-5, and/or IL-13 on IL-33 stimulation, which was increased comparedwith that seen in cells isolated from healthy control subjects.47,49
CONCLUSION AND FUTURE DIRECTIONSSNPswithin the IL33 and IL1RL1 genes are reproducibly found
to associate with asthma in different populations, and evidencefrom experimental models strongly supports a functional rolefor IL-33/IL-1RL1 signaling in asthma pathogenesis. Thereforewe conclude that the IL-33/IL-1RL1 axis plays a critical role inthe susceptibility for this chronic inflammatory disease. On thebasis of currently published data, multiple discrete genetic signalscan be distinguished within the IL33 and IL1RL1 loci, each ofwhich might have an independent contribution to asthma pathol-ogy. However, the complex LD structure at the IL1RL1/IL18R1locus precludes identification of causal asthma polymorphismsby genetic studies. This hurdle could possibly be tackled byperforming association studies in populations characterizedwith less LD, such as the African (American) population,76 or

J ALLERGY CLIN IMMUNOL
MARCH 2013
864 GROTENBOER ET AL
in well-powered meta-analyses with conditional analyses to in-vestigate independence of SNP effects on asthma. Finally,whereas GWA studies typically focus on prevalent variants andare able to explain only a small part of the asthma heritability,it has been shown that asthma susceptibility genes might also har-bor rare variants with potential large effects on gene function.77
Rare variants are therefore thought to explain a part of the ‘‘miss-ing heritability’’ of asthma.78 Hence resequencing of IL33 andIL1RL1 within the context of large-scale sequencing projectsmight answer the question of whether these genes indeed harborrare variants, thereby allowing the analysis of their contribution toasthma susceptibility.In addition to association studies, functional studies focused on
a single SNP or a complete haplotype could also shed light on thebiological relevance of the different genetic signals located withinthe IL-33/IL-1RL1 axis for asthma pathophysiology. One suchstudy performed by Shimizu et al19 revealed that SNPs locatedwithin the distal promoter region of IL1RL1 contribute to alteredIL1RL1 transcription levels. Clearly, more work needs to be donein this area to fully appreciate the relevance of the diversity ofIL33 and IL1RL1 genetic signals in the context of asthmapathogenesis.The recently identified ILC2 is a critical IL-33–responsive
cellular intermediate in the pathogenesis of asthma, whichcontributes to the asthma phenotype through the production ofIL-5 and IL-13. The role of the ILC2s is further supported by theidentification of RORA, encoding the transcription factor criti-cally required for ILC2 differentiation, as another asthma GWAstudy gene.5,7 The relevance of the innate ILC2s, next to the adap-tive and allergen-specific TH2 cells, for the pathogenesis ofasthma will be subject of intense research in the coming years.This research will also need to dissect the relative contributionsof ILC2s and other effector cells of the immune system, such asbasophils and mast cells, to the IL-33/IL-1RL1–driven responses.Such data will likely guide the rational design of novel interven-tions for this chronic inflammatory disease. Moreover, identifica-tion of causal asthma variants and unraveling of the functionalrelevance of these genetic signals in the appropriate cells and tis-sues might lead to a future prospect of personalized therapeuticintervention based on individual genetic risk factors. This mightbe beneficial to understand the clinical variety of asthma pheno-types and offer directions for a rational personalized interventionstrategy.
We thank Dr Mel�en for providing additional details on his studies.
REFERENCES
1. Bousquet J, Clark TJ, Hurd S, Khaltaev N, Lenfant C, O’Byrne P, et al. GINA
guidelines on asthma and beyond. Allergy 2007;62:102-12.
2. Masoli M, Fabian D, Holt S, Beasley R. Global Initiative for Asthma (GINA) Pro-
gram. The global burden of asthma: executive summary of the GINA dissemination
committee report. Allergy 2004;59:469-78.
3. Sakashita M, Yoshimoto T, Hirota T, Harada M, Okubo K, Osawa Y, et al. Asso-
ciation of serum interleukin-33 level and the interleukin-33 genetic variant with
Japanese cedar pollinosis. Clin Exp Allergy 2008;38:1875-81.
4. Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir
GM, et al. Sequence variants affecting eosinophil numbers associate with asthma
and myocardial infarction. Nat Genet 2009;41:342-7.
5. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A
large-scale, consortium-based genomewide association study of asthma. N Engl
J Med 2010;363:1211-21.
6. Ramasamy A, Kuokkanen M, Vedantam S, Gajdos ZK, Couto Alves A, Lyon HN,
et al. Genome-wide association studies of asthma in population-based cohorts
confirm known and suggested loci and identify an additional association near
HLA. PLoS One 2012;7:e44008.
7. Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE,
et al. Meta-analysis of genome-wide association studies of asthma in ethnically di-
verse North American populations. Nat Genet 2011;43:887-92.
8. Belpinati F, Malerba G, Trabetti E, Galavotti R, Xumerle L, Pescollderungg L,
et al. Association of childhood allergic asthma with markers flanking the IL33
gene in Italian families. J Allergy Clin Immunol 2011;128:667-8.
9. Melen E, Himes BE, Brehm JM, Boutaoui N, Klanderman BJ, Sylvia JS, et al.
Analyses of shared genetic factors between asthma and obesity in children.
J Allergy Clin Immunol 2010;126:631-7, e1-8.
10. Reijmerink NE, Postma DS, Bruinenberg M, Nolte IM, Meyers DA, Bleecker ER,
et al. Association of IL1RL1, IL18R1, and IL18RAP gene cluster polymorphisms
with asthma and atopy. J Allergy Clin Immunol 2008;122:651-4.e8.
11. Wu H, Romieu I, Shi M, Hancock DB, Li H, Sienra-Monge JJ, et al. Evaluation of
candidate genes in a genome-wide association study of childhood asthma in Mex-
icans. J Allergy Clin Immunol 2010;125:321-7.e13.
12. Bosse Y, Lemire M, Poon AH, Daley D, He JQ, Sandford A, et al. Asthma and
genes encoding components of the vitamin D pathway. Respir Res 2009;10:98.
13. SavenijeOE,KerkhofM,ReijmerinkNE,BrunekreefB, de Jongste JC,SmitHA, et al.
Interleukin-1 receptor-like 1 polymorphisms are associatedwith serum IL1RL1-a, eo-
sinophils, and asthma in childhood. J Allergy Clin Immunol 2011;127:750-6, e1-5.
14. Ferreira MA, McRae AF, Medland SE, Nyholt DR, Gordon SD, Wright MJ, et al.
Association between ORMDL3, IL1RL1 and a deletion on chromosome 17q21
with asthma risk in Australia. Eur J Hum Genet 2011;19:458-64.
15. Li X, Ampleford EJ, Howard TD, Moore WC, Torgerson DG, Li H, et al. Genome-
wide association studies of asthma indicate opposite immunopathogenesis direc-
tion from autoimmune diseases. J Allergy Clin Immunol 2012;130:861-8.e7.
16. Wan YI, Shrine NR, Soler Artigas M, Wain LV, Blakey JD, Moffatt MF, et al. Ge-
nome-wide association study to identify genetic determinants of severe asthma.
Thorax 2012;67:762-8.
17. Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, et al. Mapping the
genetic architecture of gene expression in human liver. Plos Biol 2008;6:e107.
18. Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, et al. The architecture
of gene regulatory variation across multiple human tissues: the MuTHER study.
PLoS Genet 2011;7:e1002003.
19. Murphy A, Chu JH, Xu M, Carey VJ, Lazarus R, Liu A, et al. Mapping of numer-
ous disease-associated expression polymorphisms in primary peripheral blood
CD41 lymphocytes. Hum Mol Genet 2010;19:4745-57.
20. Xia K, Shabalin AA, Huang S, Madar V, Zhou YH, Wang W, et al. seeQTL: a
searchable database for human eQTLs. Bioinformatics 2012;28:451-2.
21. Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, et al. Genetics
and beyond—the transcriptome of human monocytes and disease susceptibility.
PLoS One 2010;5:e10693.
22. Shimizu M, Matsuda A, Yanagisawa K, Hirota T, Akahoshi M, Inomata N, et al.
Functional SNPs in the distal promoter of the ST2 gene are associated with atopic
dermatitis. Hum Mol Genet 2005;14:2919-27.
23. Kurokawa M, Matsukura S, Kawaguchi M, Ieki K, Suzuki S, Odaka M, et al. Ex-
pression and effects of IL-33 and ST2 in allergic bronchial asthma: IL-33 induces
eotaxin production in lung fibroblasts. Int Arch Allergy Immunol 2011;
155(suppl 1):12-20.
24. Reijmerink NE, Postma DS, Koppelman GH. The candidate gene approach in
asthma: what happens with the neighbours? Eur J Hum Genet 2010;18:17.
25. Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new
kid in the IL-1 family. Nat Rev Immunol 2010;10:103-10.
26. Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, et al. Struc-
ture of IL-33 and its interaction with the ST2 and IL-1RAcP receptors—insight
into heterotrimeric IL-1 signaling complexes. Structure 2009;17:1398-410.
27. Yang TP, Beazley C, Montgomery SB, Dimas AS, Gutierrez-Arcelus M, Stranger
BE, et al. Genevar: a database and java application for the analysis and visualiza-
tion of SNP-gene associations in eQTL studies. Bioinformatics 2010;26:2474-6.
28. Basith S, Manavalan B, Govindaraj RG, Choi S. In silico approach to inhibition
of signaling pathways of toll-like receptors 2 and 4 by ST2L. PLoS One 2011;6:
e23989.
29. Database of single nucleotide polymorphisms (dbSNP). Bethesda (MD): National
Center for Biotechnology Information, National Library of Medicine. dbSNP ac-
cession no. Ss276599472, (dbSNP build ID: 137 for human). Available at: http://
www.ncbi.nlm.nih.gov/SNP/.
30. Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O’Neill LA, et al. ST2 is an in-
hibitor of interleukin 1 receptor and toll-like receptor 4 signaling and maintains en-
dotoxin tolerance. Nat Immunol 2004;5:373-9.
31. Liu J, Buckley JM, Redmond HP, Wang JH. ST2 negatively regulates TLR2 signal-
ing, but is not required for bacterial lipoprotein-induced tolerance. J Immunol
2010;184:5802-8.

J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 865
32. Reijmerink NE, Bottema RW, Kerkhof M, Gerritsen J, Stelma FF, Thijs C, et al.
TLR-related pathway analysis: novel gene-gene interactions in the development
of asthma and atopy. Allergy 2010;65:199-207.
33. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. In-
nate IL-13-producing nuocytes arise during allergic lung inflammation and
contribute to airways hyperreactivity. JAllergy Clin Immunol 2012;129:191-8, e1-4.
34. Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y,
et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in
murine models of allergic asthma. Eur J Immunol 2012;42:1106-16.
35. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-
responsive lineage- CD251 CD44(hi) lymphoid cells mediate innate type 2 immu-
nity and allergic inflammation in the lungs. J Immunol 2012;188:1503-13.
36. Roussel L, Erard M, Cayrol C, Girard JP. Molecular mimicry between IL-33 and
KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO
Rep 2008;9:1006-12.
37. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33,
the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear
factor in vivo. Proc Natl Acad Sci U S A 2007;104:282-7.
38. Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, et al. The dual function
cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-
kappaB-stimulated gene transcription. J Immunol 2011;187:1609-16.
39. Choi YS, Park JA, Kim J, Rho SS, Park H, Kim YM, et al. Nuclear IL-33 is a tran-
scriptional regulator of NF-kB p65 and induces endothelial cell activation. Bio-
chem Biophys Res Commun 2012;421:305-11.
40. Kunisch E, Chakilam S, Gandesiri M, Kinne RW. IL-33 regulates TNF-alpha de-
pendent effects in synovial fibroblasts. Int J Mol Med 2012;29:530-40.
41. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33,
an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2
and induces T helper type 2-associated cytokines. Immunity 2005;23:479-90.
42. Watters TM, Kenny EF, O’Neill LA. Structure, function and regulation of the Toll/
IL-1 receptor adaptor proteins. Immunol Cell Biol 2007;85:411-9.
43. Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel bio-
marker. Nat Rev Drug Discov 2008;7:827-40.
44. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endog-
enous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid
organs, brain, embryos, and inflamed tissues: in situ analysis using a novel il-33-
LacZ gene trap reporter strain. J Immunol 2012;188:3488-95.
45. Mirchandani AS, Salmond RJ, Liew FY. Interleukin-33 and the function of innate
lymphoid cells. Trends Immunol 2012;33:389-96.
46. Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a che-
moattractant for human Th2 cells. Eur J Immunol 2007;37:2779-86.
47. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: the ST2 lig-
and IL-33 potently activates and drives maturation of human mast cells. J Immunol
2007;179:2051-4.
48. Enoksson M, Lyberg K, Moller-Westerberg C, Fallon PG, Nilsson G, Lunderius-
Andersson C. Mast cells as sensors of cell injury through IL-33 recognition.
J Immunol 2011;186:2523-8.
49. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33
amplifies both Th1- and Th2-type responses through its activity on human basophils,
allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol 2008;20:1019-30.
50. Suzukawa M, Iikura M, Koketsu R, Nagase H, Tamura C, Komiya A, et al. An IL-1
cytokine member, IL-33, induces human basophil activation via its ST2 receptor.
J Immunol 2008;181:5981-9.
51. CherryWB,Yoon J, BartemesKR, IijimaK,KitaH.A novel IL-1 family cytokine, IL-
33, potently activates human eosinophils. JAllergy Clin Immunol 2008;121:1484-90.
52. Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human
basophils and eosinophils are the direct target leukocytes of the novel IL-1 family
member IL-33. Blood 2009;113:1526-34.
53. Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsumoto K, et al.
IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol
2010;185:5743-50.
54. Cho KA, Suh JW, Sohn JH, Park JW, Lee H, Kang JL, et al. IL-33 induces Th17-
mediated airway inflammation via mast cells in ovalbumin-challenged mice. Am
J Physiol Lung Cell Mol Physiol 2012;302:L429-40.
55. Hueber AJ, Alves-Filho JC, Asquith DL, Michels C, Millar NL, Reilly JH, et al.
IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur
J Immunol 2011;41:2229-37.
56. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The
alarmin interleukin-33 drives protective antiviral CD81 T cell responses. Science
2012;335:984-9.
57. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA,
et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with
influenza virus. Nat Immunol 2011;12:1045-54.
58. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC,
et al. IL-33 induces antigen-specific IL-51 T cells and promotes allergic-
induced airway inflammation independent of IL-4. J Immunol 2008;181:4780-90.
59. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical
source of Th2 cell-type cytokines in protease allergen-induced airway inflamma-
tion. Immunity 2012;36:451-63.
60. Kim SH, Cho BY, Park CS, Shin ES, Cho EY, Yang EM, et al. Alpha-T-catenin
(CTNNA3) gene was identified as a risk variant for toluene diisocyanate-induced
asthma by genome-wide association analysis. Clin Exp Allergy 2009;39:203-12.
61. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al.
Innate lymphoid cells mediate influenza-induced airway hyper-reactivity indepen-
dently of adaptive immunity. Nat Immunol 2011;12:631-8.
62. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate
production of T(H)2 cytokines by adipose tissue-associated c-kit(1)sca-1(1) lym-
phoid cells. Nature 2010;463:540-4.
63. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes
represent a new innate effector leukocyte that mediates type-2 immunity. Nature
2010;464:1367-70.
64. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators
and effectors of immunity and tissue remodeling. Nat Immunol 2011;12:21-7.
65. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N,
et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell
hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol
2008;20:791-800.
66. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Hu-
man IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by ex-
pression of CRTH2 and CD161. Nat Immunol 2011;12:1055-62.
67. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcrip-
tion factor RORalpha is critical for nuocyte development. Nat Immunol 2012;13:
229-36.
68. Borish L, Steinke JW. Interleukin-33 in asthma: how big of a role does it play?
Curr Allergy Asthma Rep 2011;11:7-11.
69. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can pro-
mote survival, adhesion and cytokine production in human mast cells. Lab Invest
2007;87:971-8.
70. Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, et al. IL-33 induces IL-13
production by mouse mast cells independently of IgE-FcepsilonRI signals.
J Leukoc Biol 2007;82:1481-90.
71. Silver MR, Margulis A, Wood N, Goldman SJ, Kasaian M, Chaudhary D. IL-33
synergizes with IgE-dependent and IgE-independent agents to promote mast cell
and basophil activation. Inflamm Res 2010;59:207-18.
72. Komai-Koma M, Brombacher F, Pushparaj PN, Arendse B, McSharry C, Alexan-
der J, et al. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranu-
lation via interleukin-4 in naive mice. Allergy 2012;67:1118-26.
73. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S,
et al. IL-33 amplifies the polarization of alternatively activated macrophages that
contribute to airway inflammation. J Immunol 2009;183:6469-77.
74. Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko
AJ, et al. Increased expression of IL-33 in severe asthma: Evidence of expression
by airway smooth muscle cells. J Immunol 2009;183:5094-103.
75. Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. In-
creased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Im-
munol 2010;125:752-4.
76. Sleiman PM, Flory J, Imielinski M, Bradfield JP, Annaiah K, Willis-Owen SA,
et al. Variants of DENND1B associated with asthma in children. N Engl J Med
2010;362:36-44.
77. Torgerson DG, Capurso D, Mathias RA, Graves PE, Hernandez RD, Beaty TH,
et al. Resequencing candidate genes implicates rare variants in asthma susceptibil-
ity. Am J Hum Genet 2012;90:273-81.
78. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al.
Finding the missing heritability of complex diseases. Nature 2009;461:747-53.

REFERENCES
E1. The International HapMap Consortium. Integrating common and rare genetic var-
iation in diverse human populations. Nature 2010;467:52-8.
E2. Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, et al. Mapping
the genetic architecture of gene expression in human liver. PLoS Biol 2008;6:
e107.
E3. Yang TP, Beazley C, Montgomery SB, Dimas AS, Gutierrez-Arcelus M, Stranger
BE, et al. Genevar: a database and java application for the analysis and visuali-
zation of SNP-gene associations in eQTL studies. Bioinformatics 2010;26:
2474-6.
E4. Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, et al. Pat-
terns of cis regulatory variation in diverse human populations. PLoS Genet 2012;
8:e1002639.
E5. Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H,
et al. Common regulatory variation impacts gene expression in a cell type-
dependent manner. Science 2009;325:1246-50.
E6. Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, et al. The architec-
ture of gene regulatory variation across multiple human tissues: the MuTHER
study. PLoS Genet 2011;7:e1002003.
E7. GTEX eQTL browser of the National Center for Biotechnology Information, U.S.
National Library of Medicine. Available at: http://www.ncbi.nlm.nih.gov/gtex/
GTEX2/gtex.cgi.
E8. Xia K, Shabalin AA, Huang S, Madar V, Zhou YH, Wang W, et al. seeQTL: a
searchable database for human eQTLs. Bioinformatics 2012;28:451-2.
E9. Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, et al. Genetics
and beyond—the transcriptome of human monocytes and disease susceptibility.
PLoS One 2010;5:e10693.
E10. Wu H, Romieu I, Shi M, Hancock DB, Li H, Sienra-Monge JJ, et al. Evaluation
of candidate genes in a genome-wide association study of childhood asthma in
Mexicans. J Allergy Clin Immunol 2010;125:321-7.
J ALLERGY CLIN IMMUNOL
MARCH 2013
865.e1 GROTENBOER ET AL
METHODS
Methods used for assembling Table IPolymorphisms located in the IL33 and IL1RL1 genes reported to have a
significant association with asthma were included in the table by using the cri-
terion of significance as defined in each study. SNPs retrieved from candidate
studies, genome-wide association studies, and meta-analyses are presented
(see references in the table).
Methods used for assembling Table IITable II includes the reported asthma-associated SNPs from Table I classi-
fied according to LD blocks, as defined and depicted in Fig 1, A and B, based
on the LD structure found in the CEU population (HapMap release 28, phase
I–, II–, and III–merged genotypes and frequencies, based on National Center
for Biotechnology Information B36 assembly).E1 An analysis of publically
available expression quantitative trait locus databases was performed that in-
cluded datasets from the Pritchard Laboratory,E2 GeneVar,E3-E6 and Ge-
Tex,E2,E7 and expression data were retrieved for SNPs from
Caucasians.E2-E9 A search up to 100 kb 59 of the genes IL33, IL1RL1, and
IL18R1 was performed, considering an expression quantitative trait locus
with a P value of less than .001 as significant.
Methods used for defining the LD plots of Fig 1LD of the polymorphisms located within the IL33
region (chromosome 9) and the IL1RL1-IL18R1 region
(chromosome 2). Structural information of the genomic region of
IL33 and IL1RL1, as well as information regarding mRNA transcript variants,
was derived from the National Center for Biotechnology Information Refer-
ence Sequence NC_000009.11 and NC000002.11, respectively (Homo sapi-
ens, GRCh37.p5 Primary Assembly, available at http://www.ncbi.nlm.nih.
gov). LD plots were constructed with HaploView (version 4.2) software,
with SNP genotyping data downloaded from the international HapMap Project
(available at http://hapmap.ncbi.nlm.nih.gov/index.html.en)E1 for the
CEU population. HapMap release 28 (phase I–, II–, and III–merged genotypes
and frequencies) was used and areas {chr9:6170,000..6260,000} and
{chr2:102280,000..102370,000} were downloaded for IL33 and IL1RL1, re-
spectively. For reasons of clarity, a limited number of 40 SNPs was included
within each LD plot. SNP selection was done by prioritizing asthma-
associated SNPs from Table I (main text) while faithfully reporting the under-
lying LD pattern of the loci. Subsequently, LD blocks were defined by using an
r2 value of 0.8 or greater with at least 1 asthma-associated SNP as a cut-off to
include SNPs in a certain LD block.
Methods used for defining the LD plots of Figs E1
and E2We determined the extent to which LD patterns with asthma-associated
SNPs in the IL33 and IL1RL1 genomic regions differ among distinct popu-
lations worldwide. To this end, SNP genotype data were downloaded for
populations of African origin (ASW and YRI), Asian origin (CHB), and
Latin American origin (MEX) from the international HapMap Project,E1 re-
lease 27 (phase I–, II–, and III–merged genotypes and frequencies) was used,
and area {chr9:6170,000..6260,000} was selected for IL33, whereas area
{chr2:102280,000..102370,000} was selected for IL1RL1. SNP genotype
data of IL33 and IL1RL1 were analyzed by using HaploView (version 4.2)
software. LD plots were constructed by including the selected asthma-
associated SNPs shown in Fig 1 (main body text) from the CEU reference
population. SNPs with missing genotypes were excluded from the LD plots.
Subsequently, for each of the analyzed populations, the LD pattern for the
asthma-associated SNPs (see Tables I and II and the main text) was
compared with that reported in the CEU population by using an r2 value
of 0.8 or greater as a cutoff value for each SNP to be considered part of
an LD block.
Interpretation of Figs E1 and E2For Fig E1, the LD pattern of the IL33 genomic region of several popula-
tions was compared with the LD blocks defined in the CEU population. LD
block 1, as observed in the CEU population, could be broken down into inde-
pendent signals in the ASW, YRI, and CHB populations but not in the MEX
population. This resulted in the identification of multiple independent SNPs
in these ethnic backgrounds: SNPs rs3939286 (ASW population), rs928413
(ASW and CHB populations), and rs2381416 (YRI and CHB populations)
constituted independent signals with an LD of less than 0.8 (r2), SNPs that
have been associated with asthma in several white populations (Table II). In
addition, LD block 2, as observed in the CEU ethnic background, are all inde-
pendent SNPs in the YRI population, including asthma-associated SNP
rs7025417. For the asthma-associated SNPs that were independent signals
in the CEU ethnic background, some SNPs (eg, rs16924159 and
rs12551256 in the YRI or rs1342326 and rs2066362 in the MEX) now form
a new LD block, making their individual contributions harder to dissect in
those ethnic backgrounds.
For Fig E2, the LD pattern of asthma-associated SNPs of the IL1RL1/
IL18R1 genomic region of several distinct populations was compared with
the LD blocks defined in the CEU population. For all 4 populations studied,
some differences in LD pattern were apparent with regard to the asthma-
associated SNPs, although overall, (unlike the IL33 gene), the LD pattern
was remarkably similar in all ethnic backgrounds, with a high degree of LD
throughout the locus. However, it is worth noting that the nonsynonymous
SNP rs1041973 (LD block 2 in the CEU, see Tables I and II) represents an in-
dependent SNPwithin all 4 studied populations (ASW, YRI, CHB, andMEX),
an SNP that Wu et alE10 associated with asthma in the Mexican population.
Furthermore, an additional asthma-associated SNP (rs1861246, see also
Table I) that is not present in the HapMap dataset (release 27) of the other pop-
ulations is present in the CHB population and is in LD with block 3 SNPs.
Overall, functional experiments are required to further establish true causal
SNPs in the discussed areas, whereby it should be noted that not only the LD
pattern of SNPs can differ among populations but also the relationship with
asthma can prove distinct.

rs14
1242
6
Ars1342326
rs18
8890
9
rs99
2969
rs3939286
rs928413
rs2066362
rs16
9241
44
rs22
1046
4
rs47
4084
0
rs16
9241
61
rs12551256
rs70
3325
8
rs7025417
rs47
4217
0
rs10
9755
16
rs13
3038
3
rs10
9755
19
rs70
4792
1
rs10
4827
4
rs20
2699
1
19
48
27
42
20
7
1
1
10
10
0
6
0
3
1
6
7
7
7
1
39
0
15
5
9
5
4
5
0
6
6
1
0
4
0
1
1
1
0
45
76
49
0
0
8
13
5
0
11
3
9
0
0
3
3
3
2
63
46
0
1
3
2
3
2
2
4
13
1
0
0
0
0
16
73
0
0
5
14
4
1
9
1
6
2
2
2
2
2
8
1
0
0
2
6
0
1
0
1
0
0
0
0
0
1
3
12
2
3
5
0
6
4
1
12
9
9
9
4
4
13
0
75
17
3
31
2
14
13
13
13
13
32
12
5
13
78
2
22
8
10
10
10
18
4
18
2
25
29
0
13
10
10
10
5
0
3
16
1
5
2
2
2
2
5
23
0
41
0
12
11
11
11
18
19
57
19
20
25
25
25
3
10
22
13
14
14
14
33
8
4
5
5
5
15
42
36
36
36
1
80
80
80
0
0
0
0
FIG E1. LD of the IL33 genomic region for the ASW, YRI, CHB, and MEX populations. A, ASW population.
B, YRI population. C, CHB population. D, MEX population. Boldface, Asthma-associated SNPs; underlined,expression quantitative trait loci.
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 865.e2

rs19
2999
6
B
rs47
4216
6
rs14
1242
6
rs
13
42
32
6
rs
23
81
41
6
rs18
8890
9
rs13
2840
60
rs99
2969
rs
39
39
28
6
rs
92
84
13
rs
20
66
36
2
rs16
9241
44
rs11
7944
19
rs22
1046
4
rs10
4358
16
rs10
9754
97
rs47
4084
0
rs
16
92
41
59
rs16
9241
61
rs
12
55
12
56
rs70
3325
8
rs70
3472
0
rs13
75
rs
70
25
41
7
rs47
4217
0
rs70
1957
5
rs10
9755
16
rs13
3038
3
rs19
2999
2
rs11
1357
3
rs10
9755
19
rs14
1242
1
rs70
4792
1
rs10
4827
4
rs10
8153
97
rs20
2699
1
15
48
37
20
39
21
14
0
2
0
0
0
2
1
11
0
11
11
11
1
6
3
2
6
6
11
12
5
13
13
21
2
15
48
36
19
38
20
13
0
3
0
0
0
2
1
12
0
11
11
11
1
6
3
2
6
6
10
12
4
12
12
18
2
15
45
37
20
39
21
14
0
2
0
0
0
2
1
11
0
11
11
11
1
6
4
2
6
7
11
12
6
13
13
19
2
24
7
0
9
7
6
4
3
0
1
4
0
5
1
2
0
0
0
3
0
0
9
3
2
0
0
0
0
0
4
0
71
37
73
52
0
0
10
0
6
5
2
0
6
0
14
13
13
0
17
0
2
0
0
0
1
2
2
2
9
0
53
51
36
0
0
5
0
1
1
0
0
4
0
23
23
23
0
18
0
2
0
0
2
2
1
3
3
5
0
51
41
7
3
4
4
1
0
0
2
2
2
21
21
21
0
30
0
7
0
0
2
2
0
3
3
2
4
80
1
1
4
0
5
3
1
0
4
1
14
14
14
0
14
1
2
0
0
2
3
2
2
2
5
1
2
3
5
0
13
11
4
1
5
4
12
12
12
1
13
2
0
0
0
1
2
5
1
1
8
1
6
25
15
0
0
0
7
2
7
0
0
0
10
2
4
1
0
0
4
4
5
3
3
6
5
4
6
26
22
19
92
0
92
20
20
20
3
29
1
7
22
22
21
21
3
20
20
1
17
12
19
18
25
5
1
5
2
2
2
6
16
3
13
24
27
10
9
0
8
8
0
3
25
23
34
3
8
7
5
5
5
48
2
25
26
10
11
12
12
28
9
9
6
7
74
22
2
28
16
14
14
12
32
7
1
0
1
0
0
18
0
0
5
0
75
17
2
25
11
8
8
10
31
7
0
1
3
0
0
17
0
0
6
0
16
3
21
7
7
7
16
19
8
6
1
2
0
0
22
0
0
7
0
0
85
16
16
16
1
25
0
4
19
18
17
17
1
16
16
1
19
0
2
2
2
18
2
34
8
2
2
2
2
15
2
2
42
5
22
22
22
3
32
1
7
24
24
22
22
4
22
22
1
19
16
61
8
34
15
14
28
28
20
26
26
7
0
16
61
9
34
15
15
30
28
21
26
26
6
0
16
61
9
34
15
15
30
28
21
26
26
6
0
11
53
39
9
10
10
10
65
6
6
15
24
5
23
6
5
15
15
13
14
14
4
3
24
7
9
9
8
36
4
4
22
11
31
27
28
28
27
24
24
8
2
96
72
72
8
69
69
2
0
76
78
8
75
75
0
0
20
96
96
6
0
19
96
96
6
0
20
20
35
43
7
0
7
0
13
FIG E1. (Continued)
J ALLERGY CLIN IMMUNOL
MARCH 2013
865.e3 GROTENBOER ET AL

rs19
2999
6
C
rs47
4216
6
rs14
1242
6
rs
13
42
32
6
rs
23
81
41
6
rs18
8890
9
rs13
2840
60
rs99
2969
rs
39
39
28
6
rs
92
84
13
rs
20
66
36
2
rs16
9241
44
rs11
7944
19
rs22
1046
4
rs10
4358
16
rs10
9754
97
rs47
4084
0
rs
16
92
41
59
rs16
9241
61
rs
12
55
12
56
rs70
3325
8
rs70
3472
0
rs13
75
rs
70
25
41
7
rs47
4217
0
rs70
1957
5
rs10
9755
16
rs13
3038
3
rs19
2999
2
rs11
1357
3
rs10
9755
19
rs14
1242
1
rs70
4792
1
rs10
4827
4
rs10
8153
97
rs20
2699
1
82
82
23
38
38
38
41
0
0
0
0
0
2
4
0
0
1
0
0
0
0
0
0
0
0
0
2
0
0
0
48
31
31
31
33
3
0
0
0
0
0
5
0
0
1
0
0
0
0
0
0
0
0
0
3
0
0
0
48
31
31
31
33
3
0
0
0
0
0
5
0
0
1
0
0
0
0
0
0
0
0
0
3
0
0
0
65
65
65
30
1
0
0
0
0
0
2
0
0
2
0
0
0
0
4
3
3
2
3
2
3
3
0
48
1
0
0
0
0
1
1
0
0
1
0
0
0
0
2
2
2
2
2
1
2
2
0
48
1
0
0
0
0
1
1
0
0
1
0
0
0
0
2
2
2
2
2
1
2
2
0
48
1
0
0
0
0
1
1
0
0
1
0
0
0
0
2
2
2
2
2
1
2
2
0
2
1
1
1
1
2
2
1
1
0
1
1
1
1
5
5
5
4
4
3
4
4
0
44
44
47
42
61
18
25
27
27
27
27
30
27
21
17
17
14
19
13
14
16
20
95
16
58
73
77
76
77
77
81
77
65
61
61
51
56
51
51
53
54
95
16
58
73
77
77
78
77
81
78
65
61
62
53
56
53
51
53
55
95
19
58
75
80
79
80
80
84
80
66
61
62
52
56
57
51
53
55
19
61
77
73
71
73
73
77
73
69
65
65
55
60
57
55
50
50
14
22
18
17
18
18
20
18
19
14
14
13
16
13
13
14
13
82
78
81
78
78
78
78
75
71
71
56
58
53
56
47
53
95
95
95
95
95
95
91
86
86
68
71
67
68
59
63
86
82
82
63
65
60
63
63
66
85
85
86
66
67
63
64
64
69
86
82
82
65
65
62
63
63
67
86
82
82
63
65
60
63
63
66
86
81
81
63
65
59
63
63
65
86
82
82
64
65
61
63
63
67
95
95
75
78
75
75
66
53
79
82
79
79
69
56
80
82
80
79
69
58
90
77
90
75
88
73
90
77
76
FIG E1. (Continued)
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 865.e4

rs1342326
D
rs18
8890
9
rs99
2969
rs3939286
rs928413
rs2066362
rs16
9241
44
rs22
1046
4
rs47
4084
0
rs16
9241
61
rs12551256
rs70
3325
8
rs7025417
rs47
4217
0
rs10
9755
16
rs13
3038
3
rs10
9755
19
rs70
4792
1
rs10
4827
4
rs20
2699
1
73
54
61
61
86
25
0
0
2
32
0
0
0
14
22
23
22
22
9
80
74
74
62
28
0
0
3
37
0
0
0
19
25
26
25
26
2
93
93
56
27
1
0
3
35
0
1
0
22
23
25
23
24
0
62
28
1
0
3
37
0
1
0
24
25
26
25
26
2
62
28
1
0
3
37
0
1
0
24
25
26
25
26
2
28
0
0
3
37
0
0
0
18
25
26
25
26
14
19
23
16
76
32
19
33
31
38
39
38
40
9
56
82
25
59
59
20
17
16
17
17
0
42
30
70
56
70
13
9
9
10
10
0
21
48
82
48
16
14
13
14
14
0
42
25
43
43
53
54
53
55
12
59
17
18
17
18
18
6
59
20
17
16
17
17
0
16
17
17
17
18
6
84
83
84
83
6
8
9
8
8
FIG E1. (Continued)
J ALLERGY CLIN IMMUNOL
MARCH 2013
865.e5 GROTENBOER ET AL

rs11
6936
97
Ars
1168
5424
rs12
7121
35
rs
14
20
08
9
rs12
7121
41
rs
37
711
80
rs
13
43
18
28
rs
10
41
97
3
rs
14
20
10
1
rs37
7117
5
rs12
7121
42
rs
19
46
13
1
rs
19
21
62
2
rs
10
19
78
62
rs49
8895
5
rs
10
20
41
37
rs
10
19
21
57
rs
10
20
67
53
rs37
5527
6
rs13
6234
8
rs
37
711
66
1
1
0
3
10
10
0
0
10
0
1
3
10
2
2
2
2
1
1
1
75
34
36
13
13
44
13
13
13
7
4
13
3
2
2
2
1
1
1
44
24
12
12
42
25
12
25
7
1
12
0
0
0
0
0
0
0
28
10
10
33
17
10
17
5
9
10
0
0
0
0
1
1
1
37
37
12
0
37
0
1
12
37
3
4
4
4
2
2
2
31
33
33
10
17
23
21
21
21
24
24
24
31
33
33
10
17
23
21
21
21
24
24
24
1
31
1
16
17
31
11
10
10
10
4
4
4
33
31
23
33
28
28
28
28
34
34
34
33
10
17
23
21
21
21
24
24
24
31
23
33
28
28
28
28
34
34
34
8
10
8
7
7
7
5
5
5
17
55
56
56
56
48
48
48
23
21
21
21
24
24
24
87
87
87
87
87
87
87
87
87
87
87
87
FIG E2. LD of the IL1RL1 genomic region for the ASW, YRI, CHB, and MEX populations. A, ASW population.
B, YRI population. C, CHB population. D, MEX population. Boldface, Asthma-associated SNPs; underlined,expression quantitative trait loci.
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 865.e6

rs11
6854
24
B
rs12
7121
35
rs11
1239
18
rs11
6904
43
rs
14
20
08
9
rs14
2008
8
rs67
0684
4
rs19
9746
7
rs12
7121
41
rs
37
711
80
rs
13
43
18
28
rs13
4085
69
rs13
4086
61
rs
10
41
97
3
rs
10
17
30
81
rs37
3212
9
rs
14
20
10
1
rs37
7117
5
rs12
7121
42
rs
19
46
13
1
rs65
4311
9
rs13
0289
93
rs13
0174
55
rs17
6963
76
rs12
9995
42
rs
19
21
62
2
rs
10
19
78
62
rs
18
61
24
5
rs49
8895
5
rs
10
20
41
37
rs
10
19
21
57
rs
10
20
67
53
rs48
5156
7
rs12
1058
08
rs37
5527
6
rs13
6234
8
rs
37
711
66
77
45
51
10
46
66
52
48
23
23
23
23
58
23
4
15
23
15
8
15
13
15
10
13
0
23
0
0
0
0
0
14
13
0
0
0
33
36
12
30
80
40
36
15
15
15
15
42
15
3
21
15
21
6
21
11
21
12
11
0
15
1
1
1
1
1
2
7
0
0
0
96
14
63
43
89
84
50
50
53
53
30
53
6
1
50
1
0
1
2
1
4
3
0
50
0
0
0
0
0
18
14
0
0
0
14
80
47
46
46
46
46
28
46
9
0
46
0
0
0
1
0
5
2
4
46
2
2
2
2
2
18
14
0
0
0
0
31
15
16
8
8
8
8
20
8
1
7
8
7
2
7
3
7
36
3
4
8
2
2
2
2
2
56
81
2
2
2
25
76
81
44
44
45
45
21
45
6
2
44
2
0
1
5
1
4
5
0
44
0
0
0
0
0
0
0
1
1
1
50
46
22
22
21
21
55
21
4
23
22
23
8
24
14
24
11
14
0
22
0
0
0
0
0
12
19
0
0
0
96
49
49
48
48
32
48
7
0
49
0
0
0
0
0
5
1
3
49
1
1
1
1
1
18
14
0
0
0
53
53
53
53
34
53
7
0
53
0
0
0
0
0
6
1
2
53
0
0
0
0
0
20
17
0
0
0
43
4
25
25
8
25
13
25
3
13
17
21
21
21
21
21
10
8
23
23
23
43
4
25
25
8
25
13
25
3
13
17
21
21
21
21
21
10
8
23
23
23
43
3
25
25
8
24
13
24
3
13
17
19
21
21
21
21
10
7
23
23
23
43
3
25
25
8
24
13
24
3
13
17
19
21
21
21
21
10
7
23
23
23
43
10
4
43
4
12
5
19
5
7
19
3
43
6
6
6
6
6
26
21
4
4
4
3
25
25
8
24
13
24
3
13
17
19
21
21
21
21
10
7
23
23
23
16
4
16
1
16
1
16
0
1
25
4
19
20
20
20
20
1
1
18
18
18
25
33
53
2
53
13
25
9
9
9
9
9
1
3
13
13
13
25
8
25
13
25
3
13
17
21
21
21
21
21
10
8
23
23
23
33
53
2
53
13
25
9
9
9
9
9
1
3
13
13
13
33
63
33
0
63
11
8
7
7
7
7
7
3
2
6
6
6
56
2
56
13
25
10
9
9
9
9
2
3
13
13
13
56
1
3
13
3
2
2
2
2
5
4
6
6
6
2
56
13
25
10
9
9
9
9
2
3
13
13
13
1
1
3
2
2
2
2
2
31
38
2
2
2
2
13
2
2
2
2
2
5
4
5
5
5
17
18
23
23
23
23
6
5
19
19
19
21
21
21
21
21
10
8
23
23
23
7
6
91
91
91
7
7
91
91
91
7
7
91
91
91
7
7
91
91
91
7
7
91
91
91
8
8
8
7
7
7
FIG E2. (Continued)
J ALLERGY CLIN IMMUNOL
MARCH 2013
865.e7 GROTENBOER ET AL

rs11
6936
97
C
rs11
6854
24
rs12
7121
35
rs11
1239
18
rs10
1826
39
rs11
6904
43
rs1420089
rs14
2008
8
rs11
1239
20
rs67
0684
4
rs19
9746
7
rs12
7121
41
rs3771180
rs13431828
rs13
4085
69
rs13
4086
61
rs1041973
rs10173081
rs37
3212
9
rs1420101
rs37
7117
5
rs12
7121
42
rs1946131
rs65
4311
9
rs13
0289
93
rs13
0174
55
rs17
6963
76
rs12
9995
42
rs1921622
rs10197862
rs1861246
rs1861245
rs49
8895
5
rs10204137
rs10192157
rs10206753
rs12
1058
08
rs37
5527
6
rs13
6234
8
rs3771166
81
94
3
10
10
10
20
10
50
68
7
68
5
68
5
68
5
57
10
64
2
2
2
2
2
2
2
2
87
77
88
81
81
81
81
81
3
9
9
9
17
9
37
52
6
53
4
53
4
53
4
58
9
73
14
14
14
14
14
14
14
14
3
10
10
10
18
10
54
69
6
69
4
69
4
69
4
57
10
72
4
4
4
4
4
4
4
4
84
84
84
84
88
3
8
9
9
15
9
46
67
5
62
3
59
3
59
3
57
8
69
4
3
3
3
3
3
3
3
3
9
9
9
17
9
56
70
6
70
4
71
4
71
4
59
9
73
4
3
3
3
3
3
3
3
3
10
10
10
19
10
52
68
7
68
4
69
4
69
4
57
10
66
2
2
2
2
2
2
2
2
3
10
10
10
19
10
52
68
7
68
4
69
4
69
4
57
10
66
2
2
2
2
2
2
2
2
3
10
10
10
19
10
52
68
7
68
4
69
4
69
4
57
10
66
2
2
2
2
2
2
2
2
3
10
10
10
19
10
52
68
7
68
4
69
4
69
4
57
10
66
2
2
2
2
2
2
2
2
3
10
10
10
19
10
50
68
7
68
4
68
4
68
4
57
10
65
2
2
2
2
2
2
2
2
87
87
87
43
87
5
8
64
8
0
8
0
8
0
9
87
0
53
53
53
53
53
53
53
53
52
5
7
73
7
0
7
0
7
0
8
0
63
63
63
63
63
63
63
63
53
4
7
73
7
0
6
0
6
0
8
0
63
63
63
63
63
63
63
63
53
4
7
73
7
0
6
0
6
0
8
0
63
63
63
63
63
63
63
63
53
9
0
38
0
25
0
25
0
25
1
52
3
52
52
52
52
52
52
52
52
4
7
73
7
0
6
0
6
0
8
0
63
63
63
63
63
63
63
63
73
3
74
2
75
2
75
2
61
5
52
7
8
8
8
8
8
8
8
5
7
7
7
82
7
68
11
11
11
11
11
11
11
11
5
0
5
0
5
0
6
73
0
46
46
46
46
46
46
46
46
7
7
7
82
7
68
11
11
11
11
11
11
11
11
7
7
5
0
4
3
3
3
3
3
3
3
3
7
7
82
7
69
10
11
11
11
11
11
11
11
7
5
0
4
3
3
3
3
3
3
3
3
7
82
7
69
10
11
11
11
11
11
11
11
5
0
4
3
3
3
3
3
3
3
3
8
83
0
0
0
0
0
0
0
0
0
63
63
63
63
63
63
63
63
1
1
1
1
1
1
1
1
FIG E2. (Continued)
J ALLERGY CLIN IMMUNOL
VOLUME 131, NUMBER 3
GROTENBOER ET AL 865.e8

rs11
6936
97D
rs11
6854
24
rs12
7121
35
rs1420089
rs12
7121
41
rs3771180
rs13431828
rs1041973
rs1420101
rs37
7117
5
rs12
7121
42
rs1946131
rs1921622
rs10197862
rs49
8895
5
rs10204137
rs10192157
rs10206753
rs13
6234
8
rs3771166
25
28
1
26
17
22
12
6
23
6
0
2
20
4
4
4
4
3
4
95
4
21
22
35
56
22
56
9
37
18
2
2
2
2
2
2
4
95
17
18
31
56
18
56
9
37
15
0
0
0
0
0
0
4
1
1
1
2
1
2
0
3
1
19
19
19
19
19
19
22
24
36
54
24
54
9
35
20
2
2
2
2
2
2
83
59
3
83
3
1
4
75
32
32
32
32
32
32
66
5
5
0
6
91
42
42
42
42
42
42
0
66
0
25
0
59
28
28
28
28
27
28
5
17
76
5
12
11
11
11
11
11
5
0
6
91
42
42
42
42
42
42
17
76
5
12
11
11
11
11
11
13
0
2
2
2
2
2
2
7
0
0
0
0
0
0
46
46
46
46
46
46
FIG E2. (Continued)
J ALLERGY CLIN IMMUNOL
MARCH 2013
865.e9 GROTENBOER ET AL
Related Documents











