JOURNAL OF BACTERIOLOGY, Feb. 1996, p. 1162–1171 Vol. 178, No. 4 0021-9193/96/$04.0010 Copyright q 1996, American Society for Microbiology CWH41 Encodes a Novel Endoplasmic Reticulum Membrane N-Glycoprotein Involved in b1,6-Glucan Assembly BO JIANG, 1 JANE SHERATON, 1 ARTHUR F. J. RAM, 2 GERRIT J. P. DIJKGRAAF, 3 FRANS M. KLIS, 2 AND HOWARD BUSSEY 1 * Department of Biology, McGill University, Montreal, Quebec, Canada H3A 1B1, 1 and Institute for Molecular Cell Biology, BioCentrum Amsterdam, 1098 SM Amsterdam, The Netherlands 2 Received 12 September 1995/Accepted 7 December 1995 CWH41 encodes a novel type II integral membrane N-glycoprotein located in the endoplasmic reticulum. Disruption of the CWH41 gene leads to a K1 killer toxin-resistant phenotype and a 50% reduction in the cell wall b1,6-glucan level. CWH41 also displays strong genetic interactions with KRE1 and KRE6, two genes known to be involved in the b1,6-glucan biosynthetic pathway. The cwh41D kre6D double mutant is nonviable; and the cwh41D kre1D double mutation results in strong synergistic defects, with a severely slow-growth phenotype, a 75% reduction in b1,6-glucan level, and the secretion of a cell wall glucomannoprotein, Cwp1p. These results provide strong genetic evidence indicating that Cwh41p plays a functional role, possibly as a new synthetic component, in the assembly of cell wall b1,6-glucan. The yeast cell wall is a complex structure essential for cell growth and viability. Situated between the plasma membrane and the environment, this extracellular matrix protects the cell from external hazards, provides osmotic stability, maintains the cellular shape, and acts as a filter, permitting the passage of some molecules while excluding others (12, 13, 23, 52). The cell wall of Saccharomyces cerevisiae is composed pri- marily of chitin, mannoproteins, and b-glucans. b-Glucans are glucose homopolymers that account for approximately 50% of the cell wall dry weight (12, 13, 23, 52). On the basis of their chemical linkage characteristics, b-glucans can be subdivided into b1,3-glucan and b1,6-glucan (29, 30). The b1,3-glucan is a predominantly 1,3-linked linear molecule with a degree of po- lymerization of approximately 1,500 glucose residues. Bio- chemical studies have indicated that the synthesis of b1,3- glucan is carried out by a plasma membrane enzyme whose activity is regulated by a G protein (7, 33). Recently, a putative b1,3-glucan synthase subunit has been identified indepen- dently by several groups. The gene, FKS1 ETG1 CWH53, en- codes a 215-kDa protein with multiple transmembrane do- mains (10, 42). Disruption of this gene resulted in pleiotropic phenotypes, which include a significant growth defect and a dramatic reduction in b1,3-glucan synthase activity in vitro. b1,6-Glucan is a highly branched molecule composed largely of 1,6-linked residues, with an average size of 140 to 200 glucose residues. In vivo, this polymer also serves as part of the cell wall receptor for K1 killer toxin (5, 19). K1 killer toxin is a secreted protein encoded by a double-stranded RNA virus. The toxin kills sensitive cells by first binding to a b1,6-glucan- based cell wall receptor, possibly a glucomannoprotein, and subsequently forming lethal cation channels in the plasma membrane (31). Thus, mutants with defects in b1,6-glucan synthesis often fail to bind the toxin and become toxin resis- tant. Studies of K1 killer toxin-resistant mutants have led to the identification of several KRE (killer-resistant) genes (4), which include a number of genes required for the synthesis of b1,6- glucan. KRE5 encodes a putative soluble endoplasmic reticulum (ER) protein that has extensive sequence homologies with a Drosophila ER glucosyltransferase (32, 37). The kre5D-null mutant failed to make any detectable amount of the polymer, indicating that Kre5p is essential for b1,6-glucan synthesis. Kre6p and Skn1p are a pair of functional homologs (43–45). They are both type II integral membrane glycoproteins, and the Kre6p protein has been localized in the Golgi apparatus. The C-terminal domains of Kre6p and Skn1p display signifi- cant sequence similarities to two glucan-binding proteins. De- letion of both KRE6 and SKN1 genes caused a major reduction (70 to 80%) in b1,6-glucan levels, consistent with both Kre6p and Skn1p playing direct functional roles in the synthesis of b1,6-glucan. KRE1 codes for a serine- and threonine-rich pro- tein located on the cell surface (1). Disruption of the KRE1 gene leads to a 40% reduction in the level of b1,6-glucan. Polysaccharide structural analysis indicated that the b1,6-glu- can made by the kre1D-null mutant had a reduced polymer size and contained fewer b1,6-linked residues than did the wild- type polymer. These results suggested that Kre1p may play a role to add or extend b1,6-linked side chains at the cell surface. KRE9 is the structural gene for a small serine- and/or threo- nine-rich protein (2, 3). Loss of function of the KRE9 gene led to a dramatic reduction (80%) in b1,6-glucan level, indicating that Kre9p plays an important role in the synthesis of the polymer. The Kre9p protein is O glycosylated, and when over- produced, it can be detected in the extracellular medium. KRE11 codes for a 63-kDa cytosolic protein, and disruption of the gene caused a 50% reduction in b1,6-glucan level (3). Based on the genetic analysis and molecular characterization of these KRE genes, a working model for b1,6-glucan synthesis has been proposed (3, 44). This model suggests that the poly- mer is made within the secretory pathway in a stepwise process which includes a Kre5p-dependent ER step involved in the initiation of the polymer synthesis, a Kre6p- and Skn1p-depen- dent Golgi step for further elaboration of the polymer, and a Kre1p-dependent cell surface step required for side chain ad- dition and maturation of the b1,6-glucan polymer. Because the Kre11p protein is localized in the cytosol, it has been suggested that Kre11p plays a regulatory function in b1,6-glucan assem- bly. We have previously reported the isolation of the cwh41 * Corresponding author. Mailing address: Department of Biology, McGill University, 1205 Dr. Penfield Ave., Montreal, Quebec, Canada H3A 1B1. Phone: (514) 398-6439. Fax: (514) 398-2595. 1162 on August 25, 2015 by guest http://jb.asm.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF BACTERIOLOGY, Feb. 1996, p. 1162–1171 Vol. 178, No. 40021-9193/96/$04.0010Copyright q 1996, American Society for Microbiology
CWH41 Encodes a Novel Endoplasmic Reticulum MembraneN-Glycoprotein Involved in b1,6-Glucan Assembly
BO JIANG,1 JANE SHERATON,1 ARTHUR F. J. RAM,2 GERRIT J. P. DIJKGRAAF,3 FRANS M. KLIS,2
AND HOWARD BUSSEY1*
Department of Biology, McGill University, Montreal, Quebec, Canada H3A 1B1,1 and Institute for Molecular Cell Biology,BioCentrum Amsterdam, 1098 SM Amsterdam, The Netherlands2
Received 12 September 1995/Accepted 7 December 1995
CWH41 encodes a novel type II integral membrane N-glycoprotein located in the endoplasmic reticulum.Disruption of the CWH41 gene leads to a K1 killer toxin-resistant phenotype and a 50% reduction in the cellwall b1,6-glucan level. CWH41 also displays strong genetic interactions with KRE1 and KRE6, two genes knownto be involved in the b1,6-glucan biosynthetic pathway. The cwh41D kre6D double mutant is nonviable; and thecwh41D kre1D double mutation results in strong synergistic defects, with a severely slow-growth phenotype, a75% reduction in b1,6-glucan level, and the secretion of a cell wall glucomannoprotein, Cwp1p. These resultsprovide strong genetic evidence indicating that Cwh41p plays a functional role, possibly as a new syntheticcomponent, in the assembly of cell wall b1,6-glucan.
The yeast cell wall is a complex structure essential for cellgrowth and viability. Situated between the plasma membraneand the environment, this extracellular matrix protects the cellfrom external hazards, provides osmotic stability, maintains thecellular shape, and acts as a filter, permitting the passage ofsome molecules while excluding others (12, 13, 23, 52).The cell wall of Saccharomyces cerevisiae is composed pri-
marily of chitin, mannoproteins, and b-glucans. b-Glucans areglucose homopolymers that account for approximately 50% ofthe cell wall dry weight (12, 13, 23, 52). On the basis of theirchemical linkage characteristics, b-glucans can be subdividedinto b1,3-glucan and b1,6-glucan (29, 30). The b1,3-glucan is apredominantly 1,3-linked linear molecule with a degree of po-lymerization of approximately 1,500 glucose residues. Bio-chemical studies have indicated that the synthesis of b1,3-glucan is carried out by a plasma membrane enzyme whoseactivity is regulated by a G protein (7, 33). Recently, a putativeb1,3-glucan synthase subunit has been identified indepen-dently by several groups. The gene, FKS1 ETG1 CWH53, en-codes a 215-kDa protein with multiple transmembrane do-mains (10, 42). Disruption of this gene resulted in pleiotropicphenotypes, which include a significant growth defect and adramatic reduction in b1,3-glucan synthase activity in vitro.
b1,6-Glucan is a highly branched molecule composed largelyof 1,6-linked residues, with an average size of 140 to 200glucose residues. In vivo, this polymer also serves as part of thecell wall receptor for K1 killer toxin (5, 19). K1 killer toxin is asecreted protein encoded by a double-stranded RNA virus.The toxin kills sensitive cells by first binding to a b1,6-glucan-based cell wall receptor, possibly a glucomannoprotein, andsubsequently forming lethal cation channels in the plasmamembrane (31). Thus, mutants with defects in b1,6-glucansynthesis often fail to bind the toxin and become toxin resis-tant. Studies of K1 killer toxin-resistant mutants have led to theidentification of several KRE (killer-resistant) genes (4), whichinclude a number of genes required for the synthesis of b1,6-glucan.
KRE5 encodes a putative soluble endoplasmic reticulum(ER) protein that has extensive sequence homologies with aDrosophila ER glucosyltransferase (32, 37). The kre5D-nullmutant failed to make any detectable amount of the polymer,indicating that Kre5p is essential for b1,6-glucan synthesis.Kre6p and Skn1p are a pair of functional homologs (43–45).They are both type II integral membrane glycoproteins, andthe Kre6p protein has been localized in the Golgi apparatus.The C-terminal domains of Kre6p and Skn1p display signifi-cant sequence similarities to two glucan-binding proteins. De-letion of both KRE6 and SKN1 genes caused a major reduction(70 to 80%) in b1,6-glucan levels, consistent with both Kre6pand Skn1p playing direct functional roles in the synthesis ofb1,6-glucan. KRE1 codes for a serine- and threonine-rich pro-tein located on the cell surface (1). Disruption of the KRE1gene leads to a 40% reduction in the level of b1,6-glucan.Polysaccharide structural analysis indicated that the b1,6-glu-can made by the kre1D-null mutant had a reduced polymer sizeand contained fewer b1,6-linked residues than did the wild-type polymer. These results suggested that Kre1p may play arole to add or extend b1,6-linked side chains at the cell surface.KRE9 is the structural gene for a small serine- and/or threo-nine-rich protein (2, 3). Loss of function of the KRE9 gene ledto a dramatic reduction (80%) in b1,6-glucan level, indicatingthat Kre9p plays an important role in the synthesis of thepolymer. The Kre9p protein is O glycosylated, and when over-produced, it can be detected in the extracellular medium.KRE11 codes for a 63-kDa cytosolic protein, and disruption ofthe gene caused a 50% reduction in b1,6-glucan level (3).Based on the genetic analysis and molecular characterizationof these KRE genes, a working model for b1,6-glucan synthesishas been proposed (3, 44). This model suggests that the poly-mer is made within the secretory pathway in a stepwise processwhich includes a Kre5p-dependent ER step involved in theinitiation of the polymer synthesis, a Kre6p- and Skn1p-depen-dent Golgi step for further elaboration of the polymer, and aKre1p-dependent cell surface step required for side chain ad-dition and maturation of the b1,6-glucan polymer. Because theKre11p protein is localized in the cytosol, it has been suggestedthat Kre11p plays a regulatory function in b1,6-glucan assem-bly.We have previously reported the isolation of the cwh41
* Corresponding author. Mailing address: Department of Biology,McGill University, 1205 Dr. Penfield Ave., Montreal, Quebec, CanadaH3A 1B1. Phone: (514) 398-6439. Fax: (514) 398-2595.
1162
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

mutant and showed that the cwh41 strain displayed phenotypescharacteristic of b1,6-glucan defects: it was resistant to K1killer toxin and had a reduced glucose/mannose ratio, suggest-ing a lower cell wall glucose content (41). To gain a betterunderstanding of its in vivo function, we cloned the CWH41gene by functional complementation and examined the effectsof the cwh41D-null mutation on cell wall b1,6-glucan assembly.Here we report that the CWH41 gene encodes a novel type IIintegral membrane N-glycoprotein located in the ER. Disrup-tion of the CWH41 gene leads to a K1 killer-resistant pheno-type and a 50% reduction in the cell wall b1,6-glucan level. Wedemonstrate that the cwh41D mutant displayed strong syner-gistic defects with kre1D- or kre6D-null mutations, and we alsoshow that the cwh41D kre1D double mutation resulted in thesecretion of Cwp1p, a glucomannoprotein usually anchoredcovalently within the cell wall matrix. Together, these resultsprovide evidence indicating that CWH41 is involved in theassembly of cell wall b1,6-glucan.
MATERIALS AND METHODS
Yeast and bacterial strains and growth media. The S. cerevisiae strains used inthis study are listed in Table 1. Growth conditions and media for yeast cells wereas described previously (6). Standard procedures were used for genetic crosses,sporulation of diploids, and dissection of tetrads (50). Yeast transformationswere made by the lithium acetate method of Ito et al. (20). Seeded-plate tests forkiller toxin sensitivity were performed as described previously (3). Escherichiacoli XL1-Blue was used for the propagation of all plasmids. LB and 2YT mediawere used for bacterial culture (48).Plasmids. A pRS316-based yeast genomic DNA library (provided by C.
Boone, Simon Fraser University, Burnaby, British Columbia, Canada) was usedto clone the CWH41 gene. The centromeric vector pRS316 was used to subclonethe CWH41 gene. The 2mm-based vector YEp351 was used to overexpress thenative CWH41 gene or the epitope-tagged CWH41-HA gene. pBluescript IIvectors were used for recombinant DNA constructions.DNA purification and recombinant DNA techniques. Yeast DNA was isolated
by the procedure of Hoffman and Winston (18). Plasmid DNA was preparedfrom E. coli as described by Sambrook et al. (48). Restriction endonucleases,Klenow and T4 DNA polymerases, shrimp alkali phosphatase, and T4 DNAligase were purchased from Bethesda Research Laboratories Inc. (Gaithersburg,Md.), Pharmacia LKB Biotechnology (Piscataway, N.J.), Boehringer MannheimBiochemicals (Indianapolis, Ind.), Stratagene Cloning Systems (La Jolla, Calif.),or U.S. Biochemicals (Cleveland, Ohio) and were used according to the instruc-tions of the manufacturers. Radioactive probes for DNA-DNA and RNA-DNAhybridizations were labeled with [a-32P]dCTP (Amersham Canada Limited,Oakville, Ontario) by using the Oligolabelling kit from Pharmacia.DNA sequencing. The CWH41 DNA sequence was determined by a combina-
tion of nested deletion and oligonucleotide primer walking. The 4.4-kb HindIIIsubclone was inserted into the pBluescript II KS1 vector, and nested deletionswere constructed with the pBluescript II Exo/Mung kit from Stratagene CloningSystems. The nucleotide sequence of both DNA strands was determined by thedideoxy-chain termination method of Sanger et al. (49) with the Sequenase kit(U.S. Biochemicals) with [a-35S]dATP as a substrate (Amersham Canada).Gene disruption.Deletion disruption mutants were constructed by the method
of Rothstein (47). An internal 1,152-bp BamHI-XbaI fragment was deleted from
the coding region of the CWH41 gene and replaced with a 1.8-kb BamHIfragment containing the HIS3 gene. After digestion by HindIII, the DNA frag-ment carrying the cwh41D::HIS3 deletion construct was gel purified and used totransform a wild-type diploid strain by selecting for histidine prototrophy. South-ern blot analysis (51) of genomic DNA from the resulting transformants indi-cated that disruptions were at the CWH41 locus (data not shown).Isolation of cell walls. After breaking of the cells with glass beads in ice-cold
lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA, 1 mMphenylmethylsulfonyl fluoride [PMSF], 1.5 mg of leupeptin per ml, and 3.0 mg ofpepstatin A per ml), the cell wall fraction was collected by centrifugation at 1,0003 g for 5 min at 48C. Cell walls were extracted twice with hot sodium dodecylsulfate (SDS) by boiling in lysis buffer containing 2% SDS for 5 min each time,washed three times with cold 1 M NaCl–1 mM PMSF, and washed three timeswith 10 mM Tris-HCl (pH 7.5)–1 mM PMSF.Cell wall analysis. Alkali-insoluble glucans were extracted from isolated cell
walls of stationary-phase cultures. After b1,3-glucanase (Zymolyase; ICN Phar-maceuticals, Inc., Irvine, Calif.) digestion and dialysis, the b1,6-glucan was col-lected and quantified as described by Boone et al. (1). The total alkali-insolubleglucan (b1,3- plus b1,6-glucan) was determined as the hexose content beforedialysis (43), and the b1,3-glucan level was calculated by subtracting the b1,6-glucan content from the total glucan level. The isolated cell wall preparationswere used directly to quantify the cell wall total hexose level.Epitope tagging. Epitope tagging of Cwh41p was performed by inserting a
123-bp BglII fragment, which codes for three tandem copies of the influenza virushemagglutinin (HA) epitope (24), into the unique BamHI site of the CWH41
FIG. 1. Restriction map and cloning of the CWH41 gene. DNA fragmentsisolated from the yeast genomic DNA library are represented as thin lines. Theopen bars indicate various subclones derived from the original genomic DNAfragment. The ability (1) or inability (2) of each fragment to complement thecwh41 mutation is shown on the right. The arrows represent open readingframes. Abbreviations for restriction sites: B, BamHI; E, EcoRI; H, HindIII; S,SpeI; X, XbaI.
TABLE 1. S. cerevisiae strains used in this study
Strain Genotype Source orreference
AR27 MATa ura3-52 41AR49 MATa lys2 41ARC75 MATa cwh41-1 ura3-52 lys2 41SEY6210 MATa leu2-3,112 ura3-52 his3-D200 lys2-801 trp1-D901 suc2-D9 S. D. EmrHAB251-15B SEY6210 autodiploid 42TA405 Autodiploid MATa/MATa his3/his3 leu2/leu2 can1/can1 M. WhitewayHAB635 MATa kre1D::HIS3 in SEY6210 3TR92 MATa kre6D::HIS3 in SEY6210 43TR179 MATa skn1D::LEU2 in SEY6210 44YDK5-3B MATa kre5D::HIS3 in TA405 haploid 32HAB855 MATa cwh41D::HIS3 in SEY6210 This studyHAB856 MATa cwh41D::HIS3 in TA405 haploid This study
VOL. 178, 1996 A NOVEL ER PROTEIN INVOLVED IN b1,6-GLUCAN ASSEMBLY 1163
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

coding region. Subclones carrying the inserted BglII fragment in the correctorientation were confirmed by DNA sequencing.To epitope tag the Cwp1p protein, two complementary oligonucleotides,
oligonucleotide 1 (AATTCATGTACCCATACGACGTCCCAGACTACGCTATGG) and oligonucleotide 2 (AATTCCATAGCGTAGTCTGGGACGTCGTATGGGTACATG), were designed. In addition to containing the sequence en-coding the HA epitope, these oligonucleotides also contain complementaryEcoRI termini at their ends (underlined). The two oligonucleotides were phos-phorylated, annealed, and ligated into the unique EcoRI site located betweencodons 24 and 25 of CWP1. Constructs containing the epitope insertion werescreened by restriction mapping the AatII site (shown in boldface type) presentin each of the oligonucleotides. After sequencing the DNA, we identified positivesubclones carrying one, two, or four copies of the inserted fragment in the correctorientation. Thus, we obtained single-, double-, and quadruple-HA epitope-tagged Cwp1p. All these tagged Cwp1p proteins are correctly targeted andanchored to the cell wall matrix, and we chose to use the quadruple-HA epitope-tagged Cwp1p (Cwp1p-HA) in this study.Preparation of total cell lysates and extraction of membrane proteins. Expo-
nentially growing cells expressing Cwh41p-HA were harvested and broken withglass beads in ice-cold lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5mM EDTA, 1 mM PMSF, 1.5 mg of leupeptin per ml, and 3.0 mg of pepstatin Aper ml). After spinning at 1,000 3 g for 5 min at 48C to remove the cell walls andunbroken cells, the supernatant was collected as the total cell lysate. To deter-mine the nature of Cwh41p-HA’s membrane association, 80 ml of total cell lysatewas mixed with 20 ml of 0.5 M Na2CO3 (pH 11), 3 M NaCl, 8 M urea, 5% TritonX-100, or 2.5% SDS. These mixtures were incubated at 48C for 15 min and thensubjected to a high-speed centrifugation of 150,000 3 g for 15 min at 48C. Theresulting membrane pellets were resuspended in 100 ml of the appropriateextraction buffer, and supernatant and pellet fractions were diluted with SDS-polyacrylamide gel electrophoresis (PAGE) sample loading buffer, heated at958C, and analyzed by Western blotting (immunoblotting).Western blotting analysis. Western blots were performed with a 1:2,500 dilu-
tion of anti-HA antibody (12CA5) and a 1:2,500 dilution of horseradish perox-idase-conjugated goat anti-mouse secondary antibody. The blots were developedwith the ECL chemiluminescence detection kit (Amersham).Immunofluorescence. Log-phase (optical density at 600 nm ' 0.5; approxi-
FIG. 2. DNA and predicted amino acid sequences of CWH41. The nucleotide sequence of a 3.3-kb BclI-BglII fragment is shown. The predicted 833-amino-acidprotein product is shown in the one-letter code below the nucleotide sequence. The stop codon is shown as an asterisk. The potential transmembrane domain isunderlined. The predicted N-glycosylation sites are boxed.
1164 JIANG ET AL. J. BACTERIOL.
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from
mately 2.83 106 cells per ml) homozygous cwh41D diploid cells expressing eitherCwh41p-HA or native Cwh41p protein were fixed with 3.7% formaldehyde.Immunofluorescence microscopy was performed as described by Pringle et al.(40). For cells containing 2mm-based plasmids, anti-HA antibody (12CA5) wasused at a dilution of 1:2,000 and Texas red-conjugated goat anti-mouse second-ary antibody was diluted 1:1,000. For strains carrying centromere-based plas-mids, one additional layer of secondary antibody was used to amplify the immu-nofluorescence signal. Antibody dilutions were 1:2,000 for anti-HA antibody(12CA5), 1:1,000 for Texas red-conjugated goat anti-mouse secondary antibody,and 1:500 for Texas red-conjugated donkey anti-goat secondary antibody. Imageswere recorded on Kodak T-Max 400 black-and-white film with an epifluores-cence microscope (Zeiss Axiophot).Cell labeling and immunoprecipitation. Cells expressing Cwh41p-HA were
grown in 10 ml of YNB (50) medium to log phase (optical density at 600 nm '0.5; approximately 2.8 3 106 cells per ml), harvested, resuspended in 2.5 ml oflow-sulfate minimal medium, and grown for another 30 min in the presence orabsence of tunicamycin (10 mg/ml). Labeling was initiated by adding 100 mCi ofTrans-35S label (ICN Biochemicals) to the cell culture, continued for 20 min, andterminated by adding NaN3 to 10 mM and chilling the cells on ice. Labeled cellswere lysed with glass beads and immunoprecipitated with anti-HA antibody(12CA5) as described by Roemer et al. (45).Cell wall b1,3-glucanase digestion and protein precipitation from growth
media. Washed cell walls were digested with a recombinant b1,3-glucanase,Quantazyme ylg (Quantum Biotechnologies Inc., Montreal, Canada), at a con-centration of 2.5 U/mg of cell wall (wet weight) in 100 ml of a solution containing50 mM Tris-HCl (pH 7.5), 100 mM dithiothreitol, 1 mM PMSF, 1.5 mg ofleupeptin per ml, and 3.0 mg of pepstatin A per ml at 378C for 18 h. After thedigestion, the remaining insoluble material was removed by centrifugation at15,000 3 g for 5 min and the supernatant was analyzed by Western blotting.Proteins secreted into the growth media were recovered by deoxycholate pre-cipitation as described by Ozols (36).Nucleotide sequence accession number. The DNA sequence of the CWH41
gene has been entered in the GenBank database and assigned accession no.U35669.
RESULTS
Cloning and sequencing of the CWH41 gene. The cwh41mutant was originally isolated from a broad cell wall mutantscreen based on the calcofluor white-hypersensitive phenotype.Initial studies indicated that cwh41 displayed a higher cell wallmannose-to-glucose ratio and was resistant to K1 killer toxin,suggesting that this mutant had defects in b1,6-glucan assem-bly (41). To further characterize the gene identified by thismutant allele, we cloned the CWH41 gene by functionalcomplementation. Two overlapping genomic DNA fragmentscomplementing the cwh41-1 calcofluor white-hypersensitivephenotype were isolated from a yeast genomic DNA library.Restriction mapping and subcloning analyses located thecwh41-1 complementing activity to a 4.4-kb HindIII fragment(Fig. 1). To determine if the cloned DNA fragments containedthe CWH41 gene, we crossed the cwh41D::HIS3 deletion mu-tant (see below) with the original cwh41-1 allele. The resultingdiploid strain was sporulated and analyzed by tetrad dissection.Of the 10 tetrads examined, all four spores from each tetradwere calcofluor white hypersensitive, with the HIS3 markersegregating 21:22. The diploid strain showed hypersensitivityto calcofluor white as well. These results demonstrate that thecwh41D::HIS3 deletion not only failed to complement but also
was tightly linked to the original cwh41-1 locus, indicating thatthe cloned DNA fragments contained the CWH41 gene.DNA sequence analysis of the 4.4-kb HindIII fragment re-
vealed a single, 2.5-kb open reading frame encoding a proteinof 833 amino acid residues (Fig. 2). The predicted Cwh41pprotein sequence contains features characteristic of a type IIintegral membrane protein (16, 38): it has a positively chargedN-terminal tail of 10 amino acid residues followed by a stretchof 16 hydrophobic residues that could form a potential mem-brane-spanning domain and a large 807-amino-acid C-terminaldomain containing four potential N-linked glycosylation sites(Asn-X-Ser/Thr). Comparison of the Cwh41p sequence withthose from GenBank, EMBL, PIR, and SwissProt sequencedatabases has not revealed any proteins with significant simi-larities to Cwh41p.A sequence search of the S. cerevisiae GenBank database
revealed that the DNA sequence 59 to the CWH41 codingregion was identical to the DNA sequence 39 to the TRP5 gene(57), thus demonstrating that the CWH41 gene is physicallyadjacent to TRP5 on the left arm of chromosome VII.Phenotypes of the cwh41D::HIS3-null mutant. To study its in
vivo function, a deletion-null mutant of the CWH41 gene wasconstructed and the resulting phenotypes were examined bytetrad analysis. Disruption of the CWH41 gene did not give riseto any detectable growth defects under standard growth con-ditions, indicating that CWH41 is a nonessential gene. How-ever, as found for the original cwh41-1 allele, the cwh41D::HIS3-null mutant displayed cell wall-related defects: the mu-tant was hypersensitive to calcofluor white, more resistant toK1 killer toxin, and showed an approximately 50% reduction incell wall b1,6-glucan levels (Table 2). The levels of the cell walltotal hexose (glucans plus mannans) and b1,3-glucan were notaffected by the cwh41D::HIS3 mutation. These phenotypes in-dicated that the CWH41 gene was involved in b1,6-glucanassembly.Immunodetection of Cwh41p. To facilitate detection and
further characterization of the CWH41 gene product, wetagged the N terminus of CWH41 with a quadruple-HAepitope, which is recognized by the monoclonal antibody12CA5 (see Materials and Methods). The epitope-tagged gene(CWH41-HA) remained fully functional, as judged by its abilityto complement both the killer-resistant phenotype and theb1,6-glucan defect in the cwh41D strain (Table 2).Western blot analysis with 12CA5 antibody detected a sin-
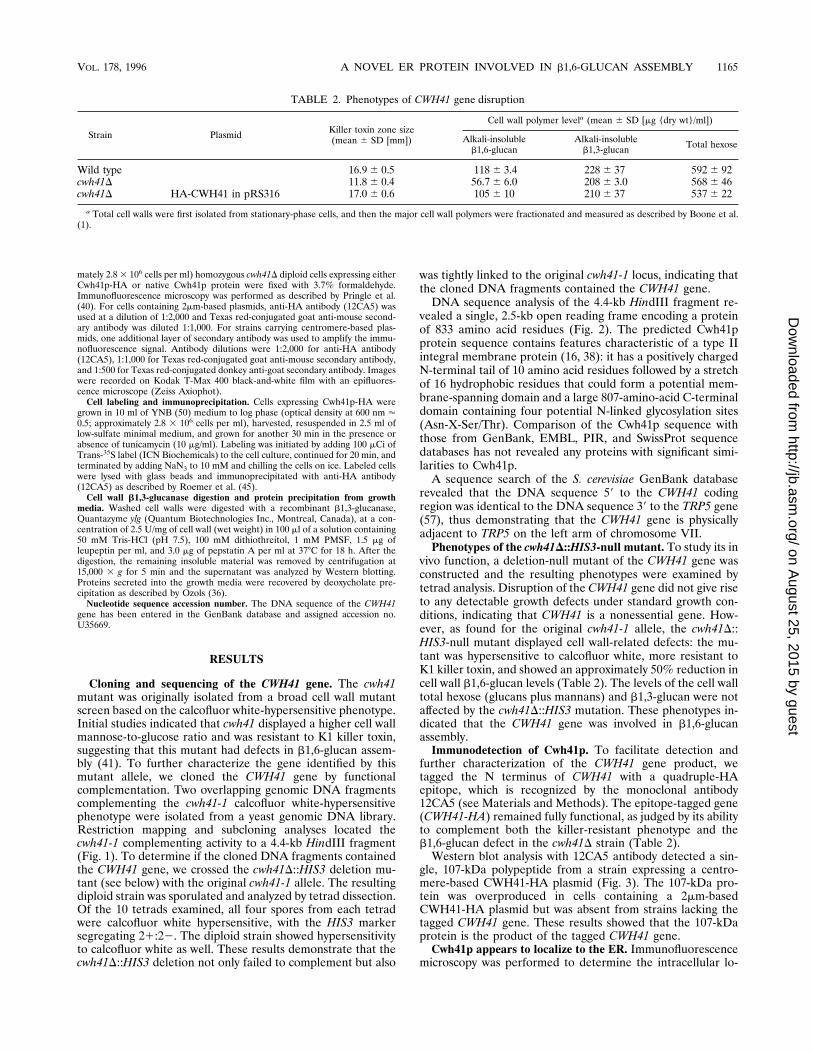
gle, 107-kDa polypeptide from a strain expressing a centro-mere-based CWH41-HA plasmid (Fig. 3). The 107-kDa pro-tein was overproduced in cells containing a 2mm-basedCWH41-HA plasmid but was absent from strains lacking thetagged CWH41 gene. These results showed that the 107-kDaprotein is the product of the tagged CWH41 gene.Cwh41p appears to localize to the ER. Immunofluorescence
microscopy was performed to determine the intracellular lo-
TABLE 2. Phenotypes of CWH41 gene disruption
Strain Plasmid Killer toxin zone size(mean 6 SD [mm])
Cell wall polymer levela (mean 6 SD [mg {dry wt}/ml])
Alkali-insolubleb1,6-glucan
Alkali-insolubleb1,3-glucan Total hexose
Wild type 16.9 6 0.5 118 6 3.4 228 6 37 592 6 92cwh41D 11.8 6 0.4 56.7 6 6.0 208 6 3.0 568 6 46cwh41D HA-CWH41 in pRS316 17.06 0.6 105 6 10 210 6 37 537 6 22
a Total cell walls were first isolated from stationary-phase cells, and then the major cell wall polymers were fractionated and measured as described by Boone et al.(1).
VOL. 178, 1996 A NOVEL ER PROTEIN INVOLVED IN b1,6-GLUCAN ASSEMBLY 1165
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from
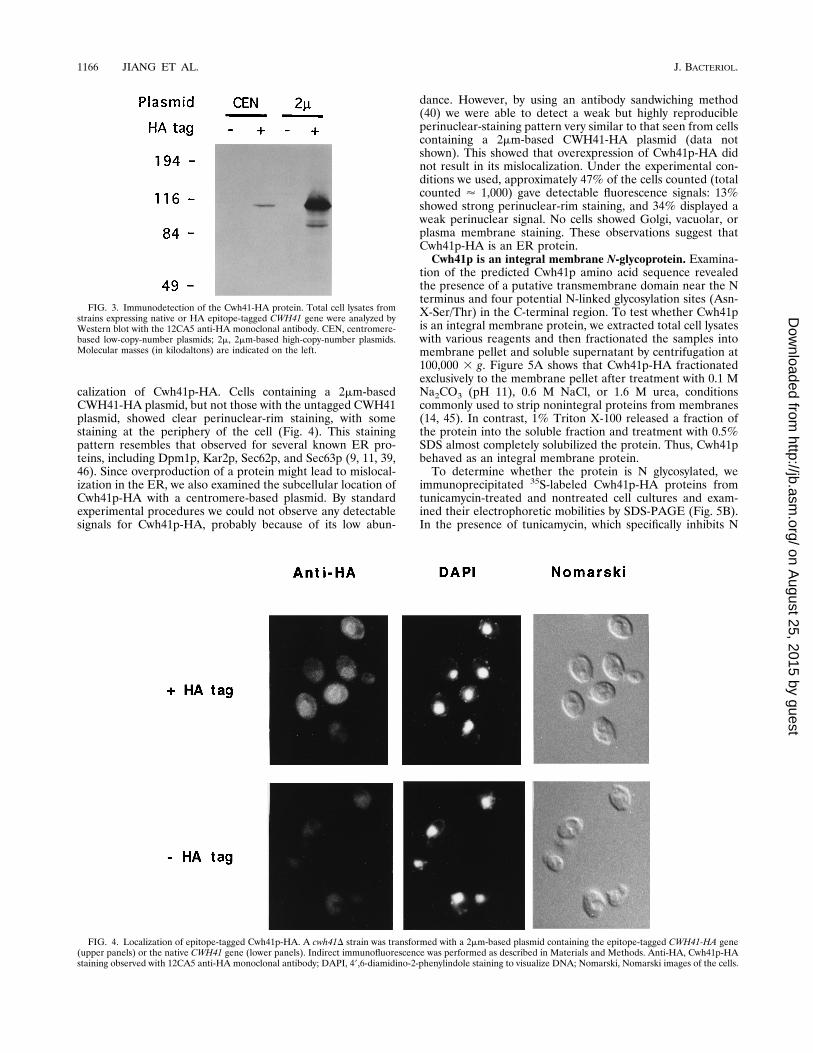
calization of Cwh41p-HA. Cells containing a 2mm-basedCWH41-HA plasmid, but not those with the untagged CWH41plasmid, showed clear perinuclear-rim staining, with somestaining at the periphery of the cell (Fig. 4). This stainingpattern resembles that observed for several known ER pro-teins, including Dpm1p, Kar2p, Sec62p, and Sec63p (9, 11, 39,46). Since overproduction of a protein might lead to mislocal-ization in the ER, we also examined the subcellular location ofCwh41p-HA with a centromere-based plasmid. By standardexperimental procedures we could not observe any detectablesignals for Cwh41p-HA, probably because of its low abun-
dance. However, by using an antibody sandwiching method(40) we were able to detect a weak but highly reproducibleperinuclear-staining pattern very similar to that seen from cellscontaining a 2mm-based CWH41-HA plasmid (data notshown). This showed that overexpression of Cwh41p-HA didnot result in its mislocalization. Under the experimental con-ditions we used, approximately 47% of the cells counted (totalcounted ' 1,000) gave detectable fluorescence signals: 13%showed strong perinuclear-rim staining, and 34% displayed aweak perinuclear signal. No cells showed Golgi, vacuolar, orplasma membrane staining. These observations suggest thatCwh41p-HA is an ER protein.Cwh41p is an integral membrane N-glycoprotein. Examina-
tion of the predicted Cwh41p amino acid sequence revealedthe presence of a putative transmembrane domain near the Nterminus and four potential N-linked glycosylation sites (Asn-X-Ser/Thr) in the C-terminal region. To test whether Cwh41pis an integral membrane protein, we extracted total cell lysateswith various reagents and then fractionated the samples intomembrane pellet and soluble supernatant by centrifugation at100,000 3 g. Figure 5A shows that Cwh41p-HA fractionatedexclusively to the membrane pellet after treatment with 0.1 MNa2CO3 (pH 11), 0.6 M NaCl, or 1.6 M urea, conditionscommonly used to strip nonintegral proteins from membranes(14, 45). In contrast, 1% Triton X-100 released a fraction ofthe protein into the soluble fraction and treatment with 0.5%SDS almost completely solubilized the protein. Thus, Cwh41pbehaved as an integral membrane protein.To determine whether the protein is N glycosylated, we
immunoprecipitated 35S-labeled Cwh41p-HA proteins fromtunicamycin-treated and nontreated cell cultures and exam-ined their electrophoretic mobilities by SDS-PAGE (Fig. 5B).In the presence of tunicamycin, which specifically inhibits N
FIG. 3. Immunodetection of the Cwh41-HA protein. Total cell lysates fromstrains expressing native or HA epitope-tagged CWH41 gene were analyzed byWestern blot with the 12CA5 anti-HA monoclonal antibody. CEN, centromere-based low-copy-number plasmids; 2m, 2mm-based high-copy-number plasmids.Molecular masses (in kilodaltons) are indicated on the left.
FIG. 4. Localization of epitope-tagged Cwh41p-HA. A cwh41D strain was transformed with a 2mm-based plasmid containing the epitope-tagged CWH41-HA gene(upper panels) or the native CWH41 gene (lower panels). Indirect immunofluorescence was performed as described in Materials and Methods. Anti-HA, Cwh41p-HAstaining observed with 12CA5 anti-HA monoclonal antibody; DAPI, 49,6-diamidino-2-phenylindole staining to visualize DNA; Nomarski, Nomarski images of the cells.
1166 JIANG ET AL. J. BACTERIOL.
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

glycosylation (35), the apparent molecular mass of Cwh41p-HAdecreased from 107 to 105 kDa. This demonstrated thatCwh41p was an N-glycoprotein. All N glycoproteins are ini-tially modified by the attachment of a core oligosaccharide(GlcNAc2Man9Glc3) in the ER (17, 26, 53). As each coreoligosaccharide contributes about 2 kDa to the molecular mass(35), the tunicamycin-induced 2-kDa shift suggested that theCwh41p-HA was modified by a core oligosaccharide withoutouter-chain extensions.Genetic interactions between CWH41 and KRE genes. Pre-
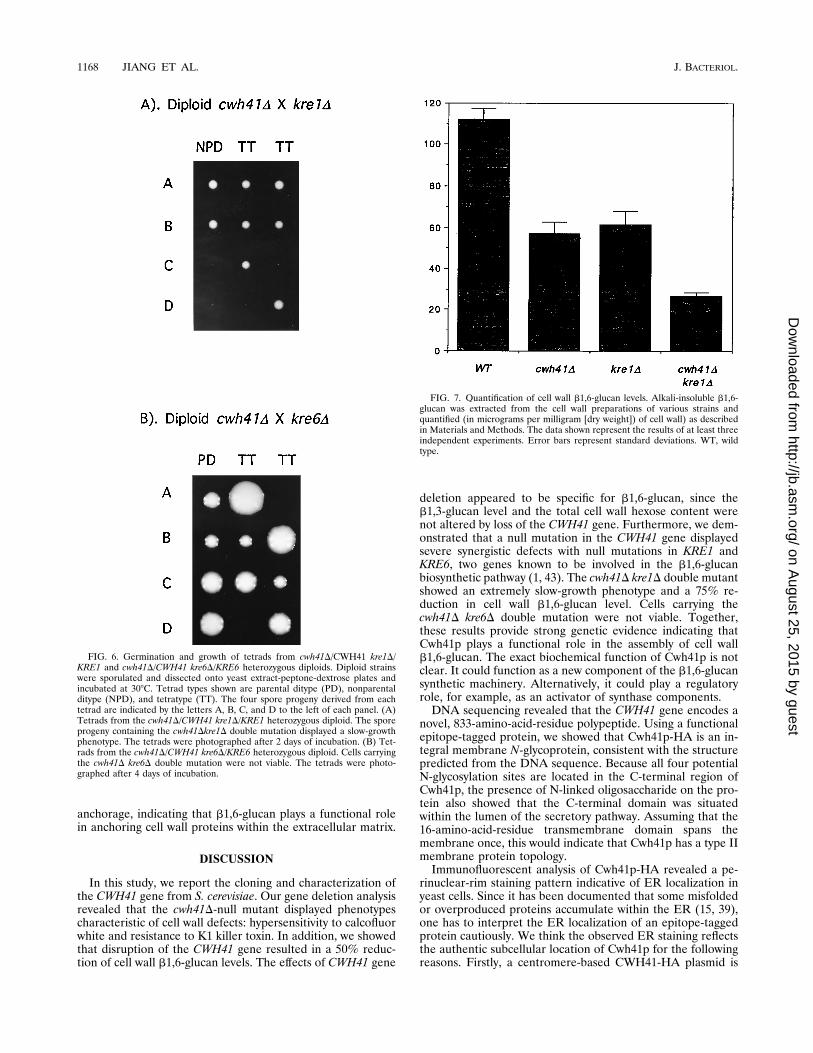
vious studies suggested that b1,6-glucan is synthesized in astepwise manner within the yeast secretory pathway (3, 44). Atleast three distinct steps were identified: a Kre5p-dependentER step involved in the initiation of the polymer synthesis (32),a Kre6p- and Skn1p-dependent Golgi step required for furthermodification of the polymer (45), and a Kre1p-dependent cellsurface step for b1,6-linked side chain addition or elongation(1). Having shown that Cwh41p is an ER membrane proteininvolved in cell wall b1,6-glucan assembly, we searched forpossible genetic interactions between CWH41 and KRE1,KRE6, SKN1, and KRE5. We crossed the cwh41D mutant witheach of these kre-null strains and examined the phenotypes ofthe double mutants by tetrad analysis (Fig. 6). The cwh41Dkre1D double mutant displayed an extremely slow-growth phe-notype and a large reduction (75%) in b1,6-glucan level (Fig.7), and the spores with cwh41D kre6D double mutations werenot viable (Fig. 6). These defects are considerably more severethan those displayed by either single mutant, indicating thatsimultaneous perturbations of the b1,6-glucan synthesis at dif-ferent stages had cumulative effects on the cell growth and thepolymer level. The skn1D mutant, which lacks detectable cellwall phenotypes (44), did not display an exaggerated growthdefect with the cwh41D mutant (data not shown). Since the
kre5D mutation is lethal in the SEY6210 strain background, weinvestigated potential interactions between CWH41 and KRE5in the TA405 strain background. Tetrad analysis indicated thatthe cwh41D kre5D double mutant grew at the same rate as thekre5D single mutant (data not shown), suggesting that thedouble mutations had no more severe effects than did thekre5D single mutation.Overexpressing the KRE5, KRE6, SKN1, or KRE1 genes
from 2mm-based plasmids did not suppress the killer-resistantphenotype of the cwh41D mutation; reciprocally, overproduc-tion of the CWH41 gene failed to suppress the defects associ-ated with the kre5D, kre6D, or kre1Dmutants (data not shown).The cwh41D kre1D double mutant displayed defects in cell
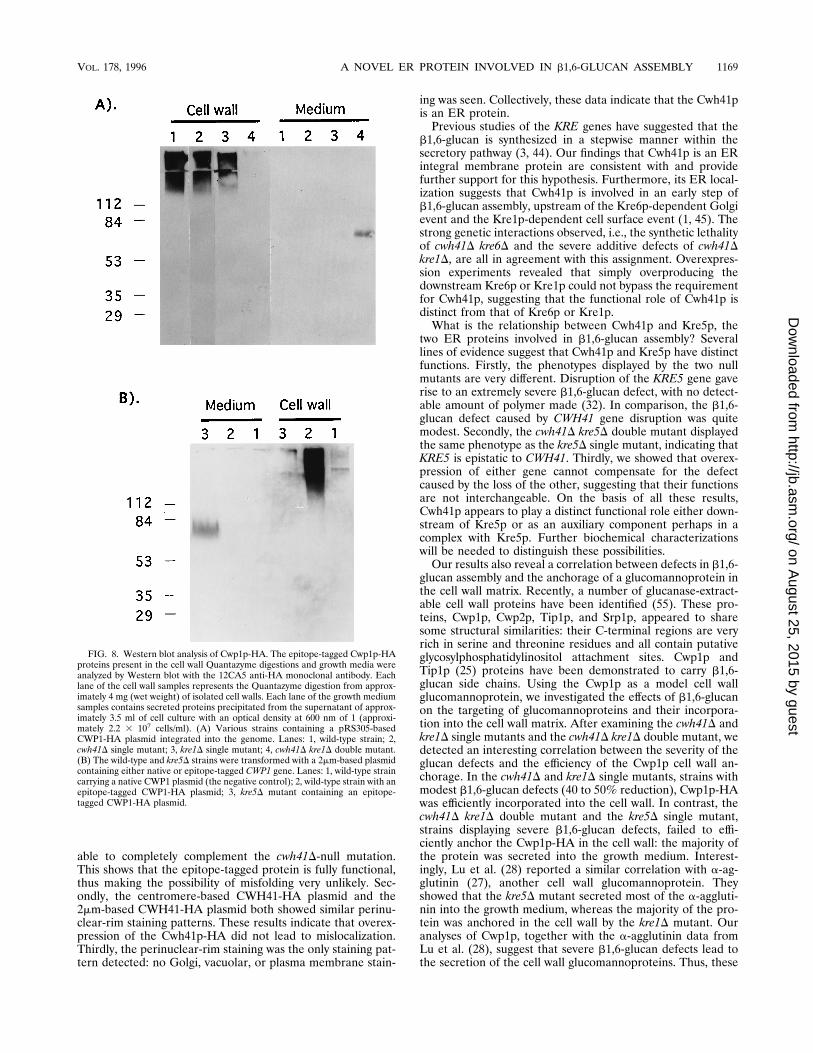
wall anchorage of Cwp1p. It has recently been shown that somecell wall mannoproteins are covalently cross-linked to b1,6-glucans, and these b1,6-glucan side chains have been suggestedto play functional roles in the anchorage of mannoproteins tothe cell wall matrix (28, 34, 56). To obtain additional insightsabout its in vivo function, we examined the effects of CWH41deletion on the cell wall anchorage of Cwp1p, a known b1,6-glucan-modified cell wall protein (55). In wild-type cells, aswell as in the cwh41D and kre1D single mutants, the epitope-tagged Cwp1p-HA protein was correctly anchored into the cellwall matrix (Fig. 8). In the cwh41D kre1D double mutant,however, little if any Cwp1p-HA protein could be detected inthe cell wall fraction. Instead, the protein was secreted into thegrowth medium by the double mutant. To test whether thesecretion of Cwp1p-HA is specific to the cwh41D kre1D mu-tant, we examined the kre5D mutant and found that theCwp1p-HA protein was secreted by the kre5D mutant as well.These results showed that severe b1,6-glucan defects, causedeither by the cwh41D kre1D double mutation or by a kre5Dsingle mutation, resulted in the failure of Cwp1p-HA cell wall
FIG. 5. Membrane association and N glycosylation of Cwh41p-HA. (A) Cell lysates from a strain expressing the 2mm-based CWH41-HA plasmid were treated withH2O, 0.1 M Na2CO3 (pH 11), 0.6 M NaCl, 1.6 M urea, 1% Triton X-100, or 0.5% SDS and separated into supernatant (S) and pellet (P) fractions as described inMaterials and Methods. These fractions were then analyzed by Western blot, with the 12CA5 anti-HA monoclonal antibody. (B) Cells expressing a 2mm-based nativeCWH41 plasmid or a 2mm-based CWH41-HA plasmid were labeled with Trans-35S label in the presence (1) or absence (2) of tunicamycin. The radiolabeledCwh41p-HA protein was immunoprecipitated and analyzed by SDS-PAGE as described in Materials and Methods. Molecular masses (in kilodaltons) are indicated onboth sides of the panel. (C) A schematic diagram of Cwh41p protein. The shaded box indicates the transmembrane domain. The arrow points to the insertion site ofthe HA epitope. The four potential N-glycosylation sites are indicated by asterisks.
VOL. 178, 1996 A NOVEL ER PROTEIN INVOLVED IN b1,6-GLUCAN ASSEMBLY 1167
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from
anchorage, indicating that b1,6-glucan plays a functional rolein anchoring cell wall proteins within the extracellular matrix.
DISCUSSION
In this study, we report the cloning and characterization ofthe CWH41 gene from S. cerevisiae. Our gene deletion analysisrevealed that the cwh41D-null mutant displayed phenotypescharacteristic of cell wall defects: hypersensitivity to calcofluorwhite and resistance to K1 killer toxin. In addition, we showedthat disruption of the CWH41 gene resulted in a 50% reduc-tion of cell wall b1,6-glucan levels. The effects of CWH41 gene
deletion appeared to be specific for b1,6-glucan, since theb1,3-glucan level and the total cell wall hexose content werenot altered by loss of the CWH41 gene. Furthermore, we dem-onstrated that a null mutation in the CWH41 gene displayedsevere synergistic defects with null mutations in KRE1 andKRE6, two genes known to be involved in the b1,6-glucanbiosynthetic pathway (1, 43). The cwh41D kre1D double mutantshowed an extremely slow-growth phenotype and a 75% re-duction in cell wall b1,6-glucan level. Cells carrying thecwh41D kre6D double mutation were not viable. Together,these results provide strong genetic evidence indicating thatCwh41p plays a functional role in the assembly of cell wallb1,6-glucan. The exact biochemical function of Cwh41p is notclear. It could function as a new component of the b1,6-glucansynthetic machinery. Alternatively, it could play a regulatoryrole, for example, as an activator of synthase components.DNA sequencing revealed that the CWH41 gene encodes a
novel, 833-amino-acid-residue polypeptide. Using a functionalepitope-tagged protein, we showed that Cwh41p-HA is an in-tegral membrane N-glycoprotein, consistent with the structurepredicted from the DNA sequence. Because all four potentialN-glycosylation sites are located in the C-terminal region ofCwh41p, the presence of N-linked oligosaccharide on the pro-tein also showed that the C-terminal domain was situatedwithin the lumen of the secretory pathway. Assuming that the16-amino-acid-residue transmembrane domain spans themembrane once, this would indicate that Cwh41p has a type IImembrane protein topology.Immunofluorescent analysis of Cwh41p-HA revealed a pe-
rinuclear-rim staining pattern indicative of ER localization inyeast cells. Since it has been documented that some misfoldedor overproduced proteins accumulate within the ER (15, 39),one has to interpret the ER localization of an epitope-taggedprotein cautiously. We think the observed ER staining reflectsthe authentic subcellular location of Cwh41p for the followingreasons. Firstly, a centromere-based CWH41-HA plasmid is
FIG. 6. Germination and growth of tetrads from cwh41D/CWH41 kre1D/KRE1 and cwh41D/CWH41 kre6D/KRE6 heterozygous diploids. Diploid strainswere sporulated and dissected onto yeast extract-peptone-dextrose plates andincubated at 308C. Tetrad types shown are parental ditype (PD), nonparentalditype (NPD), and tetratype (TT). The four spore progeny derived from eachtetrad are indicated by the letters A, B, C, and D to the left of each panel. (A)Tetrads from the cwh41D/CWH41 kre1D/KRE1 heterozygous diploid. The sporeprogeny containing the cwh41Dkre1D double mutation displayed a slow-growthphenotype. The tetrads were photographed after 2 days of incubation. (B) Tet-rads from the cwh41D/CWH41 kre6D/KRE6 heterozygous diploid. Cells carryingthe cwh41D kre6D double mutation were not viable. The tetrads were photo-graphed after 4 days of incubation.
FIG. 7. Quantification of cell wall b1,6-glucan levels. Alkali-insoluble b1,6-glucan was extracted from the cell wall preparations of various strains andquantified (in micrograms per milligram [dry weight]) of cell wall) as describedin Materials and Methods. The data shown represent the results of at least threeindependent experiments. Error bars represent standard deviations. WT, wildtype.
1168 JIANG ET AL. J. BACTERIOL.
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from
able to completely complement the cwh41D-null mutation.This shows that the epitope-tagged protein is fully functional,thus making the possibility of misfolding very unlikely. Sec-ondly, the centromere-based CWH41-HA plasmid and the2mm-based CWH41-HA plasmid both showed similar perinu-clear-rim staining patterns. These results indicate that overex-pression of the Cwh41p-HA did not lead to mislocalization.Thirdly, the perinuclear-rim staining was the only staining pat-tern detected: no Golgi, vacuolar, or plasma membrane stain-
ing was seen. Collectively, these data indicate that the Cwh41pis an ER protein.Previous studies of the KRE genes have suggested that the
b1,6-glucan is synthesized in a stepwise manner within thesecretory pathway (3, 44). Our findings that Cwh41p is an ERintegral membrane protein are consistent with and providefurther support for this hypothesis. Furthermore, its ER local-ization suggests that Cwh41p is involved in an early step ofb1,6-glucan assembly, upstream of the Kre6p-dependent Golgievent and the Kre1p-dependent cell surface event (1, 45). Thestrong genetic interactions observed, i.e., the synthetic lethalityof cwh41D kre6D and the severe additive defects of cwh41Dkre1D, are all in agreement with this assignment. Overexpres-sion experiments revealed that simply overproducing thedownstream Kre6p or Kre1p could not bypass the requirementfor Cwh41p, suggesting that the functional role of Cwh41p isdistinct from that of Kre6p or Kre1p.What is the relationship between Cwh41p and Kre5p, the
two ER proteins involved in b1,6-glucan assembly? Severallines of evidence suggest that Cwh41p and Kre5p have distinctfunctions. Firstly, the phenotypes displayed by the two nullmutants are very different. Disruption of the KRE5 gene gaverise to an extremely severe b1,6-glucan defect, with no detect-able amount of polymer made (32). In comparison, the b1,6-glucan defect caused by CWH41 gene disruption was quitemodest. Secondly, the cwh41D kre5D double mutant displayedthe same phenotype as the kre5D single mutant, indicating thatKRE5 is epistatic to CWH41. Thirdly, we showed that overex-pression of either gene cannot compensate for the defectcaused by the loss of the other, suggesting that their functionsare not interchangeable. On the basis of all these results,Cwh41p appears to play a distinct functional role either down-stream of Kre5p or as an auxiliary component perhaps in acomplex with Kre5p. Further biochemical characterizationswill be needed to distinguish these possibilities.Our results also reveal a correlation between defects in b1,6-
glucan assembly and the anchorage of a glucomannoprotein inthe cell wall matrix. Recently, a number of glucanase-extract-able cell wall proteins have been identified (55). These pro-teins, Cwp1p, Cwp2p, Tip1p, and Srp1p, appeared to sharesome structural similarities: their C-terminal regions are veryrich in serine and threonine residues and all contain putativeglycosylphosphatidylinositol attachment sites. Cwp1p andTip1p (25) proteins have been demonstrated to carry b1,6-glucan side chains. Using the Cwp1p as a model cell wallglucomannoprotein, we investigated the effects of b1,6-glucanon the targeting of glucomannoproteins and their incorpora-tion into the cell wall matrix. After examining the cwh41D andkre1D single mutants and the cwh41D kre1D double mutant, wedetected an interesting correlation between the severity of theglucan defects and the efficiency of the Cwp1p cell wall an-chorage. In the cwh41D and kre1D single mutants, strains withmodest b1,6-glucan defects (40 to 50% reduction), Cwp1p-HAwas efficiently incorporated into the cell wall. In contrast, thecwh41D kre1D double mutant and the kre5D single mutant,strains displaying severe b1,6-glucan defects, failed to effi-ciently anchor the Cwp1p-HA in the cell wall: the majority ofthe protein was secreted into the growth medium. Interest-ingly, Lu et al. (28) reported a similar correlation with a-ag-glutinin (27), another cell wall glucomannoprotein. Theyshowed that the kre5D mutant secreted most of the a-aggluti-nin into the growth medium, whereas the majority of the pro-tein was anchored in the cell wall by the kre1D mutant. Ouranalyses of Cwp1p, together with the a-agglutinin data fromLu et al. (28), suggest that severe b1,6-glucan defects lead tothe secretion of the cell wall glucomannoproteins. Thus, these
FIG. 8. Western blot analysis of Cwp1p-HA. The epitope-tagged Cwp1p-HAproteins present in the cell wall Quantazyme digestions and growth media wereanalyzed by Western blot with the 12CA5 anti-HA monoclonal antibody. Eachlane of the cell wall samples represents the Quantazyme digestion from approx-imately 4 mg (wet weight) of isolated cell walls. Each lane of the growth mediumsamples contains secreted proteins precipitated from the supernatant of approx-imately 3.5 ml of cell culture with an optical density at 600 nm of 1 (approxi-mately 2.2 3 107 cells/ml). (A) Various strains containing a pRS305-basedCWP1-HA plasmid integrated into the genome. Lanes: 1, wild-type strain; 2,cwh41D single mutant; 3, kre1D single mutant; 4, cwh41D kre1D double mutant.(B) The wild-type and kre5D strains were transformed with a 2mm-based plasmidcontaining either native or epitope-tagged CWP1 gene. Lanes: 1, wild-type straincarrying a native CWP1 plasmid (the negative control); 2, wild-type strain with anepitope-tagged CWP1-HA plasmid; 3, kre5D mutant containing an epitope-tagged CWP1-HA plasmid.
VOL. 178, 1996 A NOVEL ER PROTEIN INVOLVED IN b1,6-GLUCAN ASSEMBLY 1169
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

results provide experimental evidence for the proposal thatb1,6-glucan plays a functional role in anchoring glucomanno-protein in the cell wall, probably by covalently cross-linking theprotein to the extracellular b1,3-glucan matrix (22, 34). Howand where the b1,6-glucan side chain is attached onto theprotein is not clear, although several possibilities, includingthat the b1,6-glucan is linked to the glycosylphosphatidylinosi-tol anchor (8, 21, 28, 54), have been suggested. Detailed anal-yses of the glucosylation of Cwp1p in various kre mutantsshould provide insights on the in vivo functions of these KREgenes and on the molecular mechanisms underlying the pro-cesses of protein glucosylation and cell wall anchorage.
ACKNOWLEDGMENTS
We thank members of the Bussey laboratory for advice and discus-sions, Charlie Boone for his great yeast library, and Diane Oki formanuscript preparation.This work was supported by Operating and Strategic grants from the
Natural Sciences and Engineering Research Council of Canada.
REFERENCES
1. Boone, C., S. S. Sommer, A. Hensel, and H. Bussey. 1990. Yeast KRE genesprovide evidence for a pathway of cell wall b-glucan assembly. J. Cell Biol.110:1833–1843.
2. Brown, J. L., and H. Bussey. 1993. The yeast KRE9 gene encodes an Oglycoprotein involved in cell surface b-glucan assembly. Mol. Cell. Biol.13:6346–6356.
3. Brown, J. L., Z. Kossaczka, B. Jiang, and H. Bussey. 1993. A mutationalanalysis of killer toxin resistance in Saccharomyces cerevisiae identifies newgenes involved in cell wall 1-6-b-glucan synthesis. Genetics 133:837–849.
4. Brown, J. L., T. Roemer, M. Lussier, A.-M. Sdicu, and H. Bussey. 1994. TheK1 killer toxin: molecular and genetic applications to secretion and cellsurface assembly, p. 217–232. In J. R. Johnston (ed.), Molecular genetics ofyeast: a practical approach. IRL Press, Oxford University Press, Oxford.
5. Bussey, H. 1991. K1 killer toxin, a pore-forming protein from yeast. Mol.Microbiol. 5:2339–2343.
6. Bussey, H., W. Sacks, D. Galley, and D. Saville. 1982. Yeast killer plasmidmutations affecting toxin secretion and activity and toxin immunity function.Mol. Cell. Biol. 2:346–354.
7. Cabib, E., and M. S. Kang. 1987. Fungal 1,3-b-glucan synthase. MethodsEnzymol. 138:637–642.
8. De Nobel, H., and P. N. Lipke. 1994. Is there a role for GPIs in yeast cell-wallassembly? Trends Cell Biol. 4:42–45.
9. Deshaies, R. J., and R. Schekman. 1990. Structural and functional dissectionof Sec62p, a membrane-bound component of the yeast endoplasmic reticu-lum protein import machinery. Mol. Cell. Biol. 10:6024–6035.
10. Douglas, C. M., F. Foor, J. A. Marrinan, N. Morin, J. B. Nielsen, A. M. Dahl,P. Mazur, W. Baginsky, W. Li, M. el-Sherbeini, J. A. Clemas, S. M. Mandala,B. R. Frommer, and M. B. Kurtz. 1994. The Saccharomyces cerevisiae FKS1ETG1 gene encodes an integral membrane protein which is a subunit of1,3-b-D-glucan synthase. Proc. Natl. Acad. Sci. USA 91:12907–12911.
11. Feldheim, D., J. Rothblatt, and R. Schekman. 1992. Topology and functionaldomains of Sec63p, an endoplasmic reticulum membrane protein requiredfor secretory protein translocation. Mol. Cell. Biol. 12:3288–3296.
12. Fleet, G. H. 1991. Cell walls, p. 199–277. In A. H. Rose and J. S. Harrison(ed.), The yeasts, 2nd ed., vol. 4. Yeast organelles. Academic Press, London.
13. Fleet, G. H., and H. J. Phaff. 1981. Fungal glucans—structure and metabo-lism, p. 416–440. In W. Tanner and F. A. Loewus (ed.), Encyclopedia ofplant physiology new series, vol. 13B. Plant carbohydrates II. Springer-Verlag, Berlin.
14. Fujiki, Y., A. L. Hubbard, S. Fowler, and P. B. Lazarow. 1982. Isolation ofintracellular membranes by means of sodium carbonate treatment: applica-tion to endoplasmic reticulum. J. Cell Biol. 93:97–102.
15. Gething, M. J., K. McCammon, and J. Sambrook. 1986. Expression ofwild-type and mutant forms of influenza hemagglutinin: the role of folding inintracellular transport. Cell 46:939–950.
16. Hartmann, E., T. A. Rapoport, and H. F. Lodish. 1989. Predicting theorientation of eukaryotic membrane-spanning domains. Proc. Natl. Acad.Sci. USA 86:5786–5790.
17. Herscovics, A., and P. Orlean. 1993. Yeast glycoprotein synthesis. FASEB J.7:540–550.
18. Hoffman, C. S., and F. Winston. 1987. A ten-minute DNA preparation fromyeast efficiently releases autonomous plasmids for transformation of Esche-richia coli. Gene 57:267–272.
19. Hutchins, K., and H. Bussey. 1983. Cell wall receptor for yeast killer toxin:involvement of (136)-b-D-glucan. J. Bacteriol. 154:161–169.
20. Ito, H., Y. Fukuda, M. Murata, and A. Kimura. 1983. Transformations of
intact yeast cells with alkali cations. J. Bacteriol. 153:163–168.21. Kapteyn, J. C., R. C. Montijn, G. J. P. Dijkgraaf, and F. M. Klis. 1994.
Identification of b-1,6-glucosylated cell wall proteins in yeast and hyphalforms of Candida albicans. Eur. J. Cell Biol. 65:402–407.
22. Kapteyn, J. C., R. C. Montijn, G. J. P. Dijkgraaf, H. Van Den Ende, andF. M. Klis. 1995. Covalent association of b-1,3-glucan with b-1,6-glucosy-lated mannoproteins in cell walls of Candida albicans. J. Bacteriol. 177:3788–3792.
23. Klis, F. 1994. Cell wall assembly in yeast. Yeast 10:851–869.24. Kolodziej, P. A., and R. A. Young. 1991. Epitope tagging and protein sur-
veillance. Methods Enzymol. 194:508–519.25. Kondo, K., and M. Inouye. 1991. TIP1, a cold shock-inducible gene of
Saccharomyces cerevisiae. J. Biol. Chem. 266:17537–17544.26. Kukuruzinska, M. A., M. L. E. Bergh, and B. J. Jackson. 1987. Protein
glycosylation in yeast. Annu. Rev. Biochem. 56:915–944.27. Lipke, P., D. Wojciechowicz, and J. Kurjan. 1989. AGa1 is the structural
gene for the Saccharomyces cerevisiae a-agglutinin, a cell surface glycoproteininvolved in cell-cell interactions during mating. Mol. Cell. Biol. 9:3155–3165.
28. Lu, C. F., R. C. Montijn, J. L. Brown, F. Klis, J. Kurjan, H. Bussey, and P. N.Lipke. 1995. Glycosyl phosphatidylinositol-dependent cross-linking of a-ag-glutinin and b 1,6-glucan in the Saccharomyces cerevisiae cell wall. J. CellBiol. 128:333–340.
29. Manners, D. J., A. J. Masson, and J. C. Patterson. 1973. The structure of ab-1-3-D-glucan from yeast cell walls. Biochem. J. 135:19–30.
30. Manners, D. J., A. J. Masson, and J. C. Patterson. 1973. The structure of ab-1-6-D-glucan from yeast cell walls. Biochem. J. 135:31–36.
31. Martinac, B., H. Zhu, A. Kubalski, X. L. Zhou, M. Culbertson, H. Bussey,and C. Kung. 1990. Yeast K1 killer toxin forms ion channels in sensitive yeastspheroplasts and in artificial liposomes. Proc. Natl. Acad. Sci. USA 87:6228–6232.
32. Meaden, P., K. Hill, J. Wagner, D. Slipetz, S. S. Sommer, and H. Bussey.1990. The yeast KRE5 encodes a probable endoplasmic reticulum proteinrequired for (136)-b-D-glucan synthesis and normal cell growth. Mol. Cell.Biol. 10:3013–3019.
33. Mol, P. C., H. M. Park, J. T. Mullins, and E. Cabib. 1994. A GTP-bindingprotein regulates the activity of 133-b-glucan synthase, an enzyme directlyinvolved in yeast cell wall morphogenesis. J. Biol. Chem. 269:31267–31274.
34. Montijn, R. C., J. Van Rinsum, F. A. Van Schagen, and F. M. Klis. 1994.Glucomannoproteins in the cell wall of Saccharomyces cerevisiae contain anovel type of carbohydrate side-chain. J. Biol. Chem. 269:19338–19342.
35. Orlean, P., M. J. Kuranda, and C. F. Albright. 1991. Analysis of glycopro-teins from Saccharomyces cerevisiae. Methods Enzymol. 194:682–697.
36. Ozols, J. 1990. Amino acid analysis. Methods Enzymol. 182:587–601.37. Parker, C. G., L. I. Fessler, R. E. Nelson, and J. H. Fessler. 1995. Drosophila
UDP-glucose:glycoprotein glucosyltransferase: sequence and characteriza-tion of an enzyme that distinguishes between denatured and native proteins.EMBO J. 14:1294–1303.
38. Parks, G. D., and R. A. Lamb. 1991. Topology of eukaryote type II mem-brane proteins: importance of N-terminal positively charged residues flank-ing the hydrophobic domain. Cell 64:777–787.
39. Preuss, D., J. Mulholland, C. A. Kaiser, P. Orlean, C. Albright, M. D. Rose,P. W. Robbins, and D. Botstein. 1991. Structure of the yeast endoplasmicreticulum: localization of ER proteins using immunofluorescence and im-munoelectron microscopy. Yeast 7:891–911.
40. Pringle, J. R., A. E. M. Adams, D. G. Drubin, and B. K. Haarer. 1991.Immunofluorescence methods for yeast. Methods Enzymol. 194:565–601.
41. Ram, A. F. J., A. Wolters, R. ten Hoopen, and F. M. Klis. 1994. A newapproach for isolating cell wall mutants in Saccharomyces cerevisiae byscreening for hypersensitivity to calcofluor white. Yeast 10:1019–1030.
42. Ram, A. F. J., S. S. C. Brekelmans, L. J. W. M. Oehlen, and F. M. Klis. 1995.Identification of two cell cycle regulated genes affecting the b1,3-glucancontent of cell walls in Saccharomyces cerevisiae. FEBS Lett. 358:165–170.
43. Roemer, T., and H. Bussey. 1991. Yeast b-glucan synthesis: KRE6 encodes apredicted type II membrane protein required for glucan synthesis in vivo andfor glucan synthase activity in vitro. Proc. Natl. Acad. Sci. USA 88:11295–11299.
44. Roemer, T., S. Delaney, and H. Bussey. 1993. SKN1 and KRE6 define a pairof functional homologs encoding putative membrane proteins involved inb-glucan synthesis. Mol. Cell. Biol. 13:4039–4048.
45. Roemer, T., G. Paravicini, M. A. Payton, and H. Bussey. 1994. Character-ization of the yeast 1-6-b-glucan biosynthetic components, Kre6p and Skn1p;and genetic interactions between the PKC1 pathway and extracellular matrixassembly. J. Cell Biol. 127:567–579.
46. Rose, M. D., L. M. Misra, and J. P. Vogel. 1989. KAR2, a karyogamy gene,is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57:1211–1221.
47. Rothstein, R. J. 1983. One step gene disruption in yeast. Methods Enzymol.101:202–211.
48. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed., Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
49. Sanger, F. G., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing withchain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463–5467.
1170 JIANG ET AL. J. BACTERIOL.
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

50. Sherman, F., G. R. Fink, and J. B. Hicks. 1982. Methods in yeast genetics.Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
51. Southern, E. M. 1975. Detection of specific sequences among DNA frag-ments separated by gel electrophoresis. J. Mol. Biol. 98:503–517.
52. Stratford, M. 1994. Another brick in the wall? Recent developments con-cerning the yeast cell envelope. Yeast 10:1741–1752.
53. Tanner, W., and L. Lehle. 1987. Protein glycosylation in yeast. Biochim.Biophys. Acta 906:81–99.
54. Van Berkel, M. A. A., L. H. P. Caro, R. C. Montijn, and F. M. Klis. 1994.
Glucosylation of chimeric proteins in the cell wall of Saccharomyces cerevi-siae. FEBS Lett. 349:135–138.
55. Van Der Vaart, J. M., L. H. P. Caro, J. W. Chapman, F. M. Klis, and C. T.Verrips. 1995. Identification of three mannoproteins in the cell wall ofSaccharomyces cerevisiae. J. Bacteriol. 177:3104–3110.
56. Van Rinsum, J., F. M. Klis, and H. Van Den End. 1991. Cell wall glucom-annoproteins of Saccharomyces cerevisiae mnn9. Yeast 7:717–726.
57. Zalkin, H., and C. Yanofsky. 1982. Yeast gene TRP5: structure, function,regulation. J. Biol. Chem. 257:1491–1500.
VOL. 178, 1996 A NOVEL ER PROTEIN INVOLVED IN b1,6-GLUCAN ASSEMBLY 1171
on August 25, 2015 by guest
http://jb.asm.org/
Dow
nloaded from
Related Documents

![Endoplasmic reticulum[1]](https://static.cupdf.com/doc/110x72/58ed5fc71a28aba1678b4611/endoplasmic-reticulum1.jpg)









