ORIGINAL ARTICLE CTL recognition of a novel HLA-A*0201-binding peptide derived from glioblastoma multiforme tumor cells Cheryl E. Myers • Paul Hanavan • Kwasi Antwi • Daruka Mahadevan • A. Jamal Nadeem • Laurence Cooke • Adrienne C. Scheck • Zachary Laughrey • Douglas F. Lake Received: 24 March 2010 / Accepted: 9 May 2011 / Published online: 29 May 2011 Ó Springer-Verlag 2011 Abstract Genetic instability of tumor cells can result in translation of proteins that are out of frame, resulting in expression of neopeptides. These neopeptides are not self- proteins and therefore should be immunogenic. By eluting peptides from human glioblastoma multiforme (GBM) tumor cell surfaces and subjecting them to tandem mass spectrometry, we identified a novel peptide (KLWGL TPKVTPS) corresponding to a frameshift in the 3 0 beta- hydroxysteroid dehydrogenase type 7 (HSD3B7) gene. HLA-binding algorithms predicted that a 9-amino acid sequence embedded in this peptide would bind to HLA- A*0201. We confirmed this prediction using an HLA- A*0201 refolding assay followed by live cell relative affinity assays, but also showed that the 12-mer binds to HLA-A*0201. Based on the 9-mer sequence, optimized peptide ligands (OPL) were designed and tested for their affinities to HLA-A*0201 and their abilities to elicit anti- peptide and CTL capable of killing GBM in vitro. Wild- type peptides as well as OPL induced anti-peptide CTL as measured by IFN-c ELISPOTS. These CTL also killed GBM tumor cells in chromium-51 release assays. This study reports a new CTL target in GBM and further sub- stantiates the concept that rational design and testing of multiple peptides for the same T-cell epitope elicits a broader response among different individuals than single peptide immunization. Keywords Cytotoxic T lymphocytes Optimized peptide ligand Glioblastoma multiforme Introduction Nearly 11,000 individuals were diagnosed with glioblas- toma multiforme (GBM) in 2009. Statistics suggest that less than 30% of those patients will be alive 5 years after diagnosis. Treatment for GBM includes neurosurgery, chemotherapy, and radiotherapy, but the tumor almost always recurs, likely due to chemo and radio-resistant stem cells and/or the inability to resect the entire tumor due to its location [1, 2]. Obviously, additional studies and approa- ches directed toward cure and therapy are needed for patients with GBM. Although many tumor antigens have been discovered and characterized [3], there remains a need to identify more tumor antigens due to the genetic instability and hetero- geneity of tumors. Over-expressed, tissue-specific, and mutated proteins each have potential to harbor peptide epitopes that could stimulate anti-tumor CTL. Character- ization of peptides that bind to major histocompatibility complex (MHC) class I has revealed that most peptides are presented to CD8 T cells as 8–10 amino acid peptides. However, identification of longer class I T-cell epitopes has been reported [4–6]. HLA-A*0201 (HLA-A2)-binding peptides are typically 8–10 amino acids in length with C. E. Myers P. Hanavan K. Antwi D. F. Lake (&) School of Life Sciences, Arizona State University, Tempe, AZ 85287, USA e-mail: [email protected] Z. Laughrey Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ 85287, USA D. Mahadevan A. J. Nadeem L. Cooke Department of Medicine, Arizona Cancer Center, University of Arizona, Tucson, AZ 85724, USA A. C. Scheck Barrow Neurological Institute of SJHMC, Phoenix, AZ 85013, USA 123 Cancer Immunol Immunother (2011) 60:1319–1332 DOI 10.1007/s00262-011-1032-4

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL ARTICLE
CTL recognition of a novel HLA-A*0201-binding peptide derivedfrom glioblastoma multiforme tumor cells
Cheryl E. Myers • Paul Hanavan • Kwasi Antwi • Daruka Mahadevan •
A. Jamal Nadeem • Laurence Cooke • Adrienne C. Scheck • Zachary Laughrey •
Douglas F. Lake
Received: 24 March 2010 / Accepted: 9 May 2011 / Published online: 29 May 2011
� Springer-Verlag 2011
Abstract Genetic instability of tumor cells can result in
translation of proteins that are out of frame, resulting in
expression of neopeptides. These neopeptides are not self-
proteins and therefore should be immunogenic. By eluting
peptides from human glioblastoma multiforme (GBM)
tumor cell surfaces and subjecting them to tandem mass
spectrometry, we identified a novel peptide (KLWGL
TPKVTPS) corresponding to a frameshift in the 30 beta-
hydroxysteroid dehydrogenase type 7 (HSD3B7) gene.
HLA-binding algorithms predicted that a 9-amino acid
sequence embedded in this peptide would bind to HLA-
A*0201. We confirmed this prediction using an HLA-
A*0201 refolding assay followed by live cell relative
affinity assays, but also showed that the 12-mer binds to
HLA-A*0201. Based on the 9-mer sequence, optimized
peptide ligands (OPL) were designed and tested for their
affinities to HLA-A*0201 and their abilities to elicit anti-
peptide and CTL capable of killing GBM in vitro. Wild-
type peptides as well as OPL induced anti-peptide CTL as
measured by IFN-c ELISPOTS. These CTL also killed
GBM tumor cells in chromium-51 release assays. This
study reports a new CTL target in GBM and further sub-
stantiates the concept that rational design and testing of
multiple peptides for the same T-cell epitope elicits a
broader response among different individuals than single
peptide immunization.
Keywords Cytotoxic T lymphocytes � Optimized peptide
ligand � Glioblastoma multiforme
Introduction
Nearly 11,000 individuals were diagnosed with glioblas-
toma multiforme (GBM) in 2009. Statistics suggest that
less than 30% of those patients will be alive 5 years after
diagnosis. Treatment for GBM includes neurosurgery,
chemotherapy, and radiotherapy, but the tumor almost
always recurs, likely due to chemo and radio-resistant stem
cells and/or the inability to resect the entire tumor due to its
location [1, 2]. Obviously, additional studies and approa-
ches directed toward cure and therapy are needed for
patients with GBM.
Although many tumor antigens have been discovered
and characterized [3], there remains a need to identify more
tumor antigens due to the genetic instability and hetero-
geneity of tumors. Over-expressed, tissue-specific, and
mutated proteins each have potential to harbor peptide
epitopes that could stimulate anti-tumor CTL. Character-
ization of peptides that bind to major histocompatibility
complex (MHC) class I has revealed that most peptides are
presented to CD8 T cells as 8–10 amino acid peptides.
However, identification of longer class I T-cell epitopes has
been reported [4–6]. HLA-A*0201 (HLA-A2)-binding
peptides are typically 8–10 amino acids in length with
C. E. Myers � P. Hanavan � K. Antwi � D. F. Lake (&)
School of Life Sciences, Arizona State University, Tempe, AZ
85287, USA
e-mail: [email protected]
Z. Laughrey
Department of Chemistry and Biochemistry,
Arizona State University, Tempe, AZ 85287, USA
D. Mahadevan � A. J. Nadeem � L. Cooke
Department of Medicine, Arizona Cancer Center,
University of Arizona, Tucson, AZ 85724, USA
A. C. Scheck
Barrow Neurological Institute of SJHMC,
Phoenix, AZ 85013, USA
123
Cancer Immunol Immunother (2011) 60:1319–1332
DOI 10.1007/s00262-011-1032-4

anchor residues at positions P2 and 9 or 10 [7, 8]. Positions
2 and 9 anchor peptides in the HLA-A2-peptide-binding
groove, but auxiliary anchor residues at other positions are
also critical for optimal binding to HLA-A2 [9–14]. Longer
peptides can be accommodated in the MHC class I binding
groove by extending through the carboxy-terminus, bulg-
ing out of the binding cleft or zigzagging across the pep-
tide-binding groove [5, 6]. It is not clear how peptides
longer than 8–10 amino acids are loaded onto MHC class I
molecules. One may speculate that longer peptides loaded
onto class I missed trimming by proteases either before or
after the peptide-MHC-b2 microglobulin (b2 m) complex
has exited from the endoplasmic reticulum. We recently
reported that a large portion of peptides longer than 8-10
amino acids are found on the surfaces of tumor cells,
presumably bound to MHC [4].
Mutations in coding regions may result in translation of
proteins that are out of frame [15–18], but usually truncate
with a stop codon resulting in expression of frameshifted
neopeptides [15–19]. In this study, we identified a frame-
shift (FS) peptide derived from the 3 beta-hydroxysteroid
dehydrogenase type 7 (HSD3B7) gene that binds to the
HLA-A*0201 molecule. HSD3B7 plays a role in the initial
stages of the biosynthesis of bile acids from cholesterol
[20]. It is not known to play any role in tumor development
or suppression. Our results reveal that a new antigenic
12-mer peptide present in brain tumor cell lines is capable
of stimulating CTL. The frameshift and optimized peptide
ligands represent potential tumor-specific antigens that can
be recognized by CTL. Since these frameshift peptides are
expressed in an alternate frame and have nothing to do with
the ‘‘self’’-protein, they should be foreign to the immune
system and elicit an immune response if one were to
immunize with them. Subsequent study of this peptide by
other groups will help to validate its potential for
immunotherapy.
Materials and methods
Acid elution of peptides
1.2 9 108 GBM cells from T98g and CRL2610 were
harvested with Cell Stripper (Mediatech Inc., Manassas,
VA). Cells were centrifuged at 2339g (1,000 rpm) for
5 min and washed in 45 ml phosphate-buffered saline
followed by resuspension in 1 ml cold citric acid buffer,
pH 3.0 [21]. After 5 min at low pH, the acid supernatant
was immediately filtered through a 0.45-lm low protein-
binding filter (Pall Corporation) and then through a 3-kDa
ultrafilter (Millipore, Billerica, MA) at 13000 RPM at 4�C.
The filtered samples were then stored at -20�C in
siliconized tubes until they were analyzed by reverse-phase
high-performance liquid chromatography (RP-HPLC)
coupled to tandem mass spectrometry (MS/MS). LC–MS/
MS was performed as previously described except that the
filtered acid elutions were run on a Thermo-Finnigan LTQ
mass spectrometer [4]. Peptide sequences were identified
by searching spectra against a frameshift database con-
structed by translating the coding sequences from the
NCBI CDS database in ?1 and ?2 frames such that it
contained all possible frameshift peptides longer than 7
amino acids. Spectrum Mill software Rev A.03.03.078
(Agilent Technologies) was used to perform spectra
searches.
Peptides synthesized on TentaGel beads
The following peptides were synthesized on TentaGel S NH2
resin beads (Advanced ChemTech) with a substitution rate of
0.2–0.5 mmol/g and were used in this study: KLWG-12
(KLWGLTPKVTPS) and KLWG-9 (KLWGLTPKV), hep-
atitis B core antigen (FLPSDCFPSV, residues 18–27), and
predicted HLA-B*3901 (NHCQLLKVMV).
HLA-A*0201-binding synthetic peptides
The following HLA-A*0201-binding peptides were used in
this study: KLWG-12 (KLWGLTPKVTPS) and KLWG-9
(KLWGLTPKV); OPL1 (FLWGLTPKV) and OPL2
(FLFWGLTPKV); HIV-1 p17 Gag77-85 epitope (SLYNT-
VATL); and influenza virus matrix peptide (M1: 58–66,
GILGFVFTL) as a positive control for CTL generation
experiments. Peptides were synthesized using standard
FMOC chemistry and purified by HPLC to[90% purity.
Detection of KLWG splice variants from tumor cell
lines and tumor tissue
Total RNA was isolated from 1 9 107 GBM cell lines
CRL-2610, T98g, U-87, and U251 using the RNeasy Mini
Kit (Qiagen, Valencia, CA). An optional on-column DNA
digestion was performed with RNase-Free DNase Set
(Qiagen) for 15 min at room temperature. RNA was eluted
from columns using 50 ll DNase/RNase-free Molecular
Grade Water. RNA from primary brain tumor and normal
brain tissue was graciously provided by Dr. Adrienne
Scheck of Barrow Neurological Institute (BNI), Phoenix,
Arizona.
cDNA synthesis was performed using 1 lg total RNA
that was combined with DEPC-treated water and oligo-dT
primer to a total volume of 25 ll in a 0.5-ml microfuge
tube and incubated at 70�C for 10 min, followed immedi-
ately by 50�C incubation (during which reaction mixtures
1320 Cancer Immunol Immunother (2011) 60:1319–1332
123

were prepared). DEPC-treated water, 10X PCR buffer,
MgCl2, dNTP mix, and Dithiothreitol (DTT) were com-
bined and pre-warmed to 42�C and added to the 50�C RNA
mix. To each cDNA synthesis tube, 200 units of Super-
Script II reverse transcriptase (Invitrogen) was added.
Samples were incubated at 50�C for 50 min, and the
reaction was terminated by 70�C incubation for 15 min.
Samples were immediately chilled followed by addition of
1 ll RNase H and incubation for 20 min at 37�C.
Touchdown was performed as described previously
[22]. Reaction mixes of 50 ll consist of 1X PCR buffer
(Promega), 5% DMSO, 200 lM dNTP mix (Promega), 2
units GoTaq (Promega), 200 nM primers (forward, 50-AG
AAGCTGGTGTACCTGGTC-30; reverse, 50-CTGCTTCG
TATGGGGTGTCT-30) and 40 ng cDNA per reaction; 10
touchdown cycles were performed at -1�C per cycle from
68�C to 58�C, and 25 additional cycles were performed at
58�C annealing temperature and 90 s denaturation/exten-
sion times.
PCR products were electrophoresed on 1% agarose gels,
stained with ethidium bromide, and photographed using a
digital camera.
Cell lines and media
T2 cells (HLA-A*0201?) were used as antigen-presenting
cells (APC) for ELISPOT and chromium-51 release assays.
T2 cells were cultured in RPMI 1640 (Mediatech, Inc.),
supplemented with 10% heat-inactivated fetal bovine
serum (Gibco) with 25,000 U/ml Penicillin–Streptomycin
and 29.2 mg/ml L-glutamine (Lonza, Walkersville, MD).
Glioblastoma cell lines, T98g, CRL2610, and U-87
(molecularly typed, HLA-A*0201) were cultured in
DMEM supplemented with 10% fetal bovine serum with
25,000 U/ml Penicillin–Streptomycin and 29.2 mg/ml
L-glutamine (Lonza, Walkersville, MD) and 250 lg/ml
Amphotericin B (MP Biomedicals, Aurora, CA). Cells
were incubated at 37�C and 5% CO2.
Reconstitution of HLA-A*0201 for peptide screening
Refolding of MHC onto peptide beads was performed as
published previously [14, 23]. Briefly, KLWG-12 and
KLWG-9 peptides were synthesized on resin support
beads. Baculovirus-produced recombinant HLA-A*0201
(kind gift from Dr. Margaret Smith) was denatured in
6 M urea and added to approximately 200 peptide beads
synthesized on resin beads. To allow the peptide-MHC-
b2m complex to assemble on the peptide beads, 100 lg of
purified recombinant b2-microglobulin (kind gift from
Dr. Margaret Smith) was added. The mixture was then put
into a Tube-O-Dialyzer (G-Biosciences, St Louis, MO)
with a 1-kDa MWCO pore size. This was dialyzed against
5 M urea in PBS with a 1 M step-down of the urea
concentration every hour until the peptide-bead-HLA-b2m
refolding buffer was exchanged to PBS containing 0.1%
Tween 20 (PBST). After several washes of PBST, the
resin was then incubated with 1 lg/ml anti-class I mAb
W6/32 for 1 h at room temperature, followed by 2 more
washes with PBST. The peptide beads were then incu-
bated in 1:5000 goat anti-mouse IgG conjugated to
alkaline phosphatase (Jackson Immuno Research) for 1 h
at room temperature. The resin was washed 5 times with
PBST. After a final wash of tris-buffered saline (TBS),
100 uM 5-bromo-4chloro-3-indolyl phosphate (Sigma)
was added to the beads as a colorometric enzyme sub-
strate [24, 25]. Peptide beads that turned blue had HLA-
A*0201 refolded onto them. FLPSDCFPSV peptide is
altered HLA-A*0201 epitope from hepatitis B virus
(HBV) core antigen residues 18–27. This HBV peptide
has been used successfully as a positive control for HLA-
A*0201 binding previously [4, 26, 27]. NHCQLLKVMV
peptide is a predicted HLA-B*3901 binder and has been
used previously as a negative control for HLA-A*0201
binding [4].
HLA-A*0201 binding affinity of frameshift peptides
and optimized peptide ligands
Relative affinity assays were performed to determine
affinities of peptides for HLA-A*0201 molecules as pre-
viously reported [26]. The HBV core antigen
(FLPSDYFPSV) was synthesized with a cysteine residue
substituted for the tyrosine residue at position 6. The cys-
teine residue was conjugated to fluorescein (Fl-peptide) for
flow cytometric detection (FLPSDCFPSV: Fl-peptide). T2
cells (5 9 105 cells/tube) were cultured in serum-free
RPMI with recombinant b2-m (2 lg/ml) and Fl-peptide
(0.1 lg/ml), and incubated for 18–20 h with varying con-
centrations of OPL peptides at 26�C in a 5% CO2 incu-
bator. Mean fluorescence intensity (MFI) values were used
to determine inhibition of the Fl-peptide from binding to
HLA-A*0201 molecules on T cells by optimized peptide
ligands. Percent inhibition was calculated as: [1 - (MFI
T2 ? Fl-peptide ? modified peptide - MFI T2 only)/
((MFI T2 ? Fl-peptide) - (MFI T2 only))] 9 100. The
IC50 values of the OPL and KLWG-9 and 12 peptides were
determined by calculating the concentration of peptide
required to inhibit binding of 50% of the Fl-peptide binding
to T2 cells.
Generation of peptide-specific CTL
PBMC were obtained from normal donors by peripheral
blood draws according to the guidelines set forth by the
Human Subjects Committee at the University of Arizona
Cancer Immunol Immunother (2011) 60:1319–1332 1321
123

and Arizona State University. PBMC were purified using
standard white blood cell separation by density centrifu-
gation with Ficoll Hypaque. Peptide-specific CTL were
generated as follows. Dendritic cells (DC) were generated
by plating HLA-A2? PBMC (1 9 107–1 9 108 cells) in a
T-75 flask in 10 ml of AIM-V medium and incubated at
37�C in a 5% incubator for 2 h. Non-adherent cells were
removed and replaced with 10 ml X-Vivo medium. IL-4
(PeproTech Inc, Rocky Hill, NJ) was added at a concen-
tration of 15 ng/ml along with 1,000 IU/ml Leukine
(Berlex, Seattle, WA) to culture medium. On day 5, IL-4,
GM-CSF, TNF-a (500 IU/ml, R&D Systems, Minneapolis,
MN), and prostaglandin E2 (2 lg/ml, Sigma) were added to
culture medium. On day 7, DC were removed from the
flask and resuspended in Iscove’s modified Dulbecco’s
medium (cIMDM) supplemented with 10% heat-inacti-
vated human AB serum (Gemini Bioproducts, Calabasas,
CA), 25,000 U/ml Penicillin–Streptomycin and 29.2 mg/
ml L-glutamine (Lonza, Walkersville, MD) and 250 lg/ml
Amphotericin B (MP Biomedicals, Aurora, CA) in a
24-well plate. Non-adherent autologous lymphocytes were
added to DC cultures (10:1 ratio of effector cells to stim-
ulator cells). To the DC-PBMC cultures, 10 ng/ml of IL-7,
(R&D Systems, Minneapolis, MN) and 100 IU/ml of IL-2
(PeproTech Inc, Rocky Hill, NJ) were added. KLWG and
OPL were added at 1 lg/ml. Every 3–4 days, 100 IU/ml of
IL-2 was added. Cells were split as needed and prolifera-
tive blasts were frequently dissociated. Weekly peptide
stimulations began seven days after the first stimulation.
Autologous PBMC were irradiated (4,000 rads) and pulsed
with 1 lg/ml peptide. At the end of each stimulation, cells
were analyzed by ELISPOT for peptide recognition.
Detection of IFN-c-producing cells by ELISPOT
Seven days after the third peptide stimulation, PBMC
recognition of KLWG peptides was measured by IFN-cELISPOT. T2 cells (2.5 9 104 cells/well) were loaded with
an HLA-A*0201-binding peptide (10.0 lg/ml) and pan
MHC class I blocking antibody, and W6/32 monoclonal
antibody was added as a control for MHC-dependent CTL
activity prior to incubation with effector cells. Effector
cells (5 9 104 cells/well) and peptide-pulsed T2 cells
(2.5 9 104 cells/well) were added to anti-IFN-c antibody-
coated plates in 200 ll IMDM. Control wells contained un-
stimulated PBMC, PBMC ? PHA (10.0 lg/ml) or
PBMC ? T2 cells. After incubation for 36 h at 37�C in 5%
CO2, cells were removed by washing five times with PBS/
0.05% Tween 20. Captured cytokine was detected by
incubation for 5 h at room temperature with biotinylated
mouse anti-human IFN-c monoclonal antibody (2.5 lg/ml/
well, Pharmingen, San Diego, CA) diluted in PBS/0.05%
Tween 20/0.1% FCS. Plates were washed with PBST
followed by addition of streptavidin-HRP (Pharmingen,
San Diego, CA), diluted 1:1000 in PBST-FCS and incu-
bated for 1 h at room temperature. Staining was performed
with 3-amino-9-ethylcarbazole for 15–20 min at room
temperature. Color development was stopped by thor-
oughly rinsing the plates with distilled water. Spots were
counted with an ELISPOT reader using Immunospot soft-
ware (Cellular Technology Ltd, Cleveland, OH).
Cytolytic activity of PBMC
Standard 51Cr release cytotoxicity assays were performed to
evaluate the ability of CTL generated from PBMC to lyse
target cells pulsed with KLWG-12 and KLWG-9 peptide and
brain tumor target cells. T2 cells (5 9 103 cells/well) were
labeled with 100 lCi of 51Cr (Amersham Pharmacia Biotech
Inc., Piscataway, NJ) for 45 min followed by incubation with
10.0 lg/ml of KLWG-12 or KLWG-9 in cIMDM for 1 h.
Prior to assay, W6/32 mAb was added to T2 cells pulsed with
peptide and tumor cell lines as a control for MHC-dependent
CTL activity. HLA-A2? T98g was a kind gift from
Dr. Walter Storkus. CRL2610 and U-87 were purchased
from American Type Culture Collection, Rockville, MD.
K562 cell line served as an NK cell target. Release of
radioactivity was measured by gamma counting (Top Count,
Packard, Meriden, CT). Maximum release was defined by51Cr released from targets lysed with 10% Triton X-100.
Percent specific lysis was calculated as: ((experimental51Cr release - spontaneous release)/(maximum release
- spontaneous release)) 9 100.
Mode of binding of 9-mer and 12-mer peptides
to HLA-A*0201
MODPROPEP evaluation of the 9-mers was based on a
combination of 26 peptide-HLA-A*0201 crystal structures
which indicated that peptide position 2 (L) and position 9
(V) are invariant and interact in a highly homologous
manner with the peptide-binding site [28]. Further,
MODPROPEP provides binding scores based on peptide-
protein interactions specified by closest neighbor (6 A
cutoff) that encompasses hydrogen bonding, hydrophobic,
aromatic, and van der Waals interactions (data not shown).
Moreover, MODPROPEP also provides a pdb file with
peptide threaded into the MHC class I binding site. The
program MODPROPEP is able to thread 8-mers and
10-mers based on crystal structures; however, no structures
exist for peptides [10 residues bound to HLA class I.
Hence, for the 12-mer, the strategy was to learn from
MODPROPEP to interactively dock utilizing Surflex-Dock
(Sybyl V.8, Tripos) with positions 2 and 9 fixed in their
respective hydrophobic pockets and then optimize binding
based on known protein-peptide interactions.
1322 Cancer Immunol Immunother (2011) 60:1319–1332
123

Results
Glioblastoma acid eluate contains HLA-A*0201
binding peptides
Peptides derived from MHC class I complexes expressed on
the cell surfaces of T98g and CRL2610 were isolated after
cells were treated with pH 3.0 buffer. Acid eluates from tumor
cells were filtered through 3-kDa MW cutoff spin filters and
then subjected to LC–MS/MS on a Thermo-Finnigan Sur-
veyor LC system (Thermo Electron Corp., San Jose, CA)
online with a linear ion trap (LTQ, Thermo Electron Corp.,
San Jose, CA). Peptide sequences were identified by searching
MS/MS spectra against Swissprot and a custom frameshift
protein database. This frameshift database was constructed by
translating the coding sequences from the NCBI CDS data-
base in ?1 and ?2 frames such that it contained all possible
frameshift peptides longer than 7 amino acids.
A total of 90 peptides were eluted and identified using
LC–MS/MS from T98g; 66 peptide sequences matched
proteins in the Swissprot database and 24 matched our cus-
tom frameshift database. However, only 11 peptides from the
Swissprot database (Table 1A), and 7 peptides from our
custom frameshift database (Table 1B) were predicted to
bind HLA-A*0201 by SYFPEITHI (Table 1A) [29]. Fifty-
five peptides were identified by LC–MS/MS from CRL2610
eluate. Thirty-eight peptides matched proteins in the
Swissprot database and 17 peptides corresponded to our
frameshift database. Eight peptides from Swissprot database
(Table 1C) and 6 peptides from the frameshift database
(Table 1D) were predicted to bind HLA-A*0201 by SYF-
PEITHI [29]. Peptides eluted from the cell lines ranged in
size from 9 to 19 amino acids. Peptides larger than 10 amino
acids were predicted to contain internal HLA-A*0201 epi-
topes as shown in Table 1.
One of the frameshift peptides eluted from T98g,
KLWGLTPKVTPS 12-mer, contained an HLA-A*0201
epitope (KLWGLTPKV) and was predicted to bind to
HLA-A*0201 very strongly with a top score of 30. This
sequence was derived from the ?2 frame of the HSD3B7
protein. Neither KLWG-12 nor KLWG-9 was identified in
acid elutions from CRL2610. Because it was possible that
KLWG-12 was misidentified by the mass spectrometer, we
chemically resynthesized KLWG-12 and analyzed it by
LC–MS/MS, demonstrating the same ion fragmentation in
the mass spectra of the synthetic peptide compared with
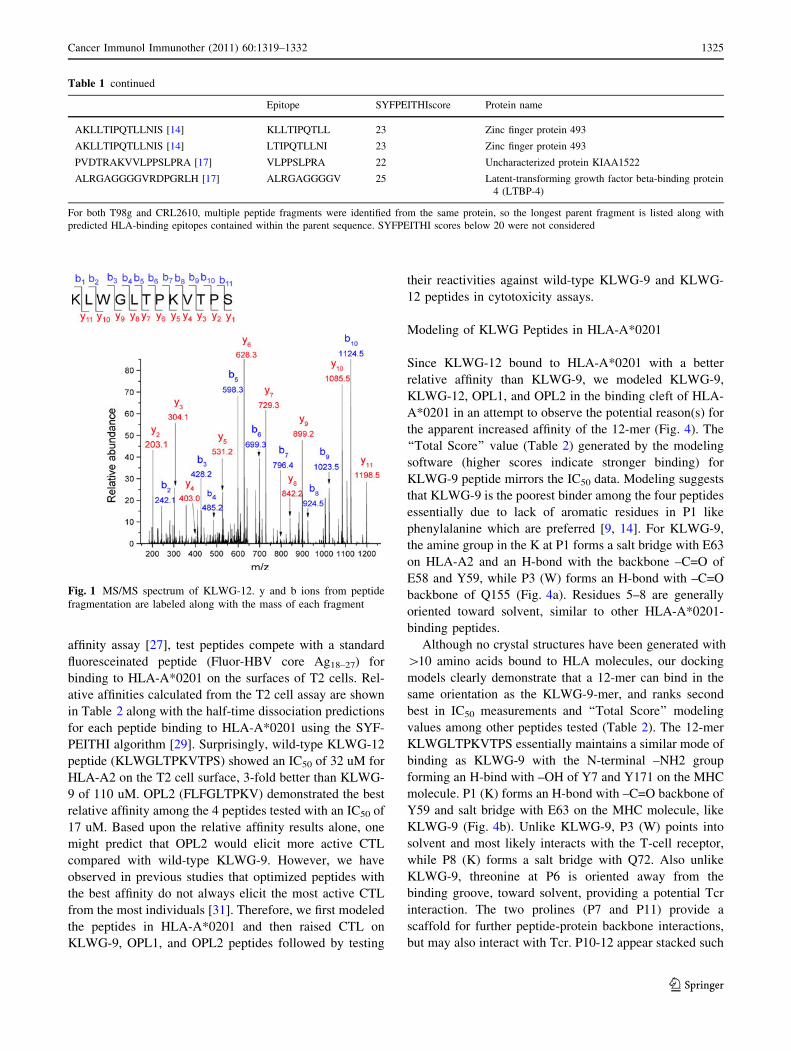
that of the natural, cell-derived peptide (Fig. 1).
Detection of HSD3B7 splice variants in clinical
samples
Primers flanking the KLWG peptide region were used to
amplify cDNA synthesized from polyadenylated RNA
from 4 established GBM cell lines, 5 GBM tumor tissues,
one primary low passage GBM line and one normal brain
tissue. In each PCR amplification, a 438-bp product was
observed, suggesting amplification of exons 2, 3, and 4 as
would be expected from NCBI databases. However, in
established and primary tumor cell lines and GBM tumor
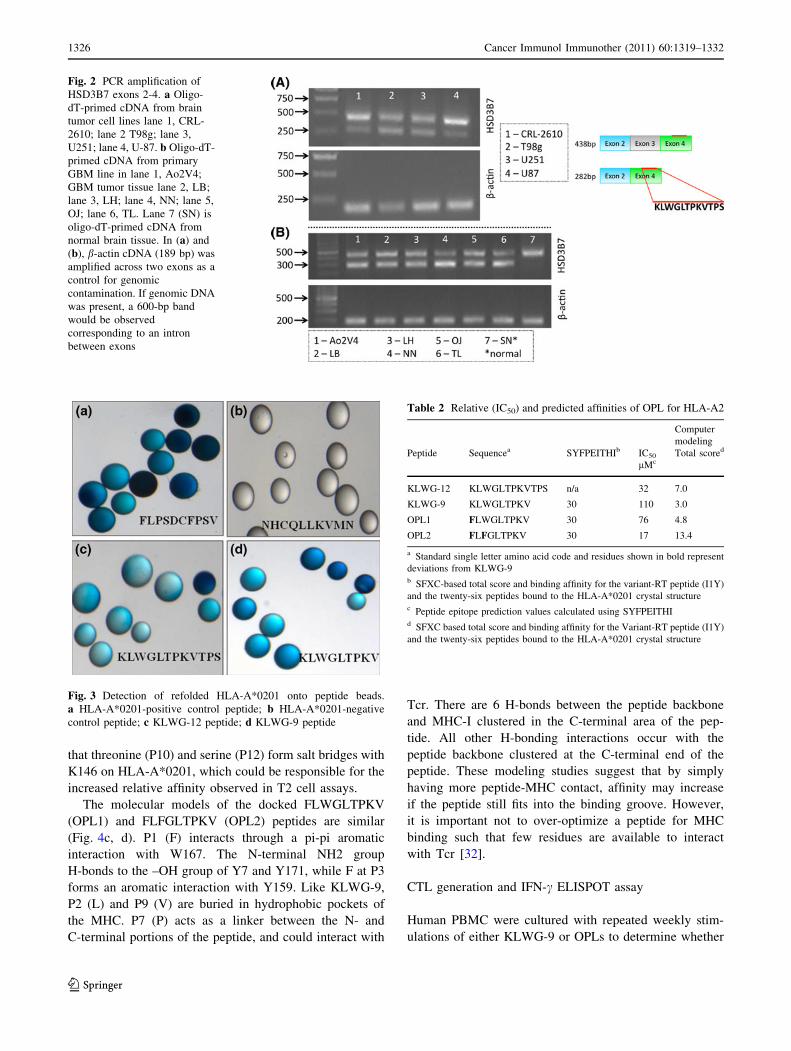
tissues, a second band was observed at 282 bp (Fig. 2).
This 282-bp band was not observed in normal brain tissue
(Fig. 2b, lane 7). Sequence analyses indicated that the
438-bp band contains exons 2, 3, and 4 from HSD3B7,
while the 282-bp band is missing exon 3. Cloning of the
282 bp PCR product and further sequence analysis did not
reveal insertions, deletions, or aberrant splicing between
exons 2 and 4 that would have resulted in translation of the
KLWG peptide. None-the-less, all GBM tumor lines and
tissues tested express mRNA containing an HSD3B7 splice
variant missing exon 3, while normal brain appears not to
express this variant.
HLA-A2 Allele Specific Peptide Binding
To validate the SYFPEITHI algorithm-based HLA-A*0201
binding motif prediction for the HSD3B7 frameshift pep-
tide, baculovirus-produced recombinant HLA-A*0201 was
refolded onto ‘‘KLWG-9’’ and ‘‘KLWG-12’’ peptide beads
as previously described [4, 14, 30]. Since
KLWGLTPKVTPS was acid-eluted from cells, we
hypothesized that the 12-mer would bind to HLA-A*0201
based on our previous findings that it is not uncommon for
peptides longer than 8-10 amino acids to bind class I
molecules [4]. Both KLWG-9 and KLWG-12 peptides
were synthesized onto polyethyleneglycol beads (Tentagel)
and incubated with denatured recombinant HLA-A*0201
and b2m, while 6 M urea was dialyzed out of the peptide-
bead/HLA/b2m mixture to re-nature the trimeric complex.
Monoclonal antibody (mAb), W6/32, was then incubated
with peptide beads followed by the addition of alkaline
phosphatase conjugated to goat anti-mouse IgG antibody
and BCIP substrate. Only properly folded MHC trimeric
complexes recognized by mAb W6/32 turn blue. Both
KLWG-12 and KLWG-9 peptide beads demonstrate their
specificity to the HLA-A*0201 molecule as shown by blue
staining of peptide beads (Fig. 3). A control peptide that is
predicted to bind HLA-B*3901 shows no staining.
Relative affinity of HSD3B7 Frameshift peptide
for HLA-A2
Affinity of peptides for HLA molecules is a function of
their immunogenicity. To assess relative affinity of
KLWG-9, KLWG-12, and two optimized peptide ligands,
FLWGLTPKV (OPL1) and FLFGLTPKV (OPL2), IC50
values of the peptides were obtained. Using the T2 cell
Cancer Immunol Immunother (2011) 60:1319–1332 1323
123

Table 1 Peptide sequences of HLA-A*0201-binding peptides and epitopes eluted from: a T98g Swissprot database, b T98g frameshift database,
c CRL2610 Swissprot database, d CRL2610 frameshift database
Epitope SYFPEITHIscore Protein name
(A) T98g Parent peptide (length) Swissprot database
VLLESEQFL [9] VLLESEQFL 25 Signal recognition particle 14 kDa protein
DFGVSDADIQEL [12] GVSDADIQEL 22 THO complex subunit 4
RVYLGASTPDLQ [12] YLGASTPDL 25 Sulfiredoxin-1
GSLAIKRDPKVND [13] LAIKRDPKV 21 Low-density lipoprotein receptor-related protein 8
precursor
GSLAIKRDPKVND [13] SLAIKRDPKV 27 Low-density lipoprotein receptor-related protein 8
precursor
DIAVDGEPLGRVSF [13] AVDGEPLGRV 22 Peptidyl-prolyl cis–trans isomerase A
MNLGGLAVARDDGL [14] GLAVARDDGL 24 Transgelin-2
LEGKVLPGVDALSNI [15] KVLPGVDAL 26 Phosphoglycerate kinase 1
EQEMATAASSSSLEKS [16] ATAASSSSL 20 ANKRD26-like family C member 1A
IVNTNVPRASVPDGFLSE [18] NTNVPRASV 20 Macrophage migration inhibitory factor
APVSGPVGLLGARRAWDLE [19] LLGARRAWDL 22 G patch domain-containing protein 8
(B) T98g Parent peptide (length) frameshift database
LLCHQPVASRA [11] LLCHQPVASR 20 Prokineticin-1 precursor
KLWGLTPKVTPS [12] KLWGLTPKV 30 3 beta-hydroxysteroid dehydrogenase type 7
IMKTDKLINLVL [12] KTDKLINLV 21 Zinc finger FYVE domain-containing protein 9
IMKTDKLINLVL [12] IMKTDKLINL 23 Zinc finger FYVE domain-containing protein 9
DLRQEAPADLPGDR [14] LRQEAPADL 22 G-protein-signaling modulator 1
AKLLTIPQTLLNIS [14] KLLTIPQTL 25 Zinc finger protein 493
AKLLTIPQTLLNIS [14] LLTIPQTLL 22 Zinc finger protein 493
AKLLTIPQTLLNIS [14] TIPQTLLNI 21 Zinc finger protein 493
AKLLTIPQTLLNIS [14] KLLTIPQTLL 23 Zinc finger protein 493
AKLLTIPQTLLNIS [14] LTIPQTLLNI 23 Zinc finger protein 493
GRTGGEELRKLLGRE [15] RTGGEELRKL 22 TCF7L2 protein
HGWMASLLRVPGYVLS [16] SLLRVPGYV 24 Protein FAM131B
HGWMASLLRVPGYVLS [16] LLRVPGYVL 24 Protein FAM131B
HGWMASLLRVPGYVLS [16] SLLRVPGYVL 25 Protein FAM131B
(C) CRL2610 Parent peptide (length) Swissprot database
VLLESEQFL [9] VLLESEQFL 25 Signal recognition particle 14 kDa protein (SRP14)
(18-kDa Alu RNA-binding protein)
LLRELKHPNI [10] LLRELKHPNI 23 Serine/threonine-protein kinase Nek2 (EC 2.7.11.1)
(NimA-related protein kinase 2) (NimA-like protein
kinase 1) (HSPK 21)
KVNQIGSVTESI [12] QIGSVTESI 20 Enolase 3 (EC 4.2.1.11) (2-phosphoglycerate
dehydratase 3) (2-phospho-D-glycerate hydro-lyase 3)
DIAVDGEPLGRVS [13] AVDGEPLGRV 22 Peptidyl-prolyl cis–trans isomerase A (PPIase A)
(Rotamase A) (EC 5.2.1.8) (Cyclophilin A)
(Cyclosporin A-binding protein)
EGKVLPGVDALSNI [14] KVLPGVDAL 26 Phosphoglycerate kinase 1 (EC 2.7.2.3)
MNLGGLAVARDDGL [14] GLAVARDDGL 24 Transgelin-2 (SM22-alpha homolog)
MSNLDSNRDNEVDF [14] NLDSNRDNEV 23 Protein S100-A4 (S100 calcium-binding protein A4)
(Metastasin)
LEGKVLPGVDALSNI [15] KVLPGVDAL 26 Phosphoglycerate kinase 1 (EC 2.7.2.3)
(D) CRL2610 Parent peptide (length) frameshift database
TQRLPLLQTL [10] QRLPLLQTL 21 Mucin-19
LLCHQPVASRA [11] LLCHQPVASR 20 Prokineticin-1
LPRGAVPAVRGG [11] LPRGAVPAV 20 Smad nuclear-interacting protein 1
AKLLTIPQTLLNIS [14] KLLTIPQTL 25 Zinc finger protein 493
AKLLTIPQTLLNIS [14] LLTIPQTLL 22 Zinc finger protein 493
AKLLTIPQTLLNIS [14] TIPQTLLNI 21 Zinc finger protein 493
1324 Cancer Immunol Immunother (2011) 60:1319–1332
123
affinity assay [27], test peptides compete with a standard
fluoresceinated peptide (Fluor-HBV core Ag18–27) for
binding to HLA-A*0201 on the surfaces of T2 cells. Rel-
ative affinities calculated from the T2 cell assay are shown
in Table 2 along with the half-time dissociation predictions
for each peptide binding to HLA-A*0201 using the SYF-
PEITHI algorithm [29]. Surprisingly, wild-type KLWG-12
peptide (KLWGLTPKVTPS) showed an IC50 of 32 uM for
HLA-A2 on the T2 cell surface, 3-fold better than KLWG-
9 of 110 uM. OPL2 (FLFGLTPKV) demonstrated the best
relative affinity among the 4 peptides tested with an IC50 of
17 uM. Based upon the relative affinity results alone, one
might predict that OPL2 would elicit more active CTL
compared with wild-type KLWG-9. However, we have
observed in previous studies that optimized peptides with
the best affinity do not always elicit the most active CTL
from the most individuals [31]. Therefore, we first modeled
the peptides in HLA-A*0201 and then raised CTL on
KLWG-9, OPL1, and OPL2 peptides followed by testing
their reactivities against wild-type KLWG-9 and KLWG-
12 peptides in cytotoxicity assays.
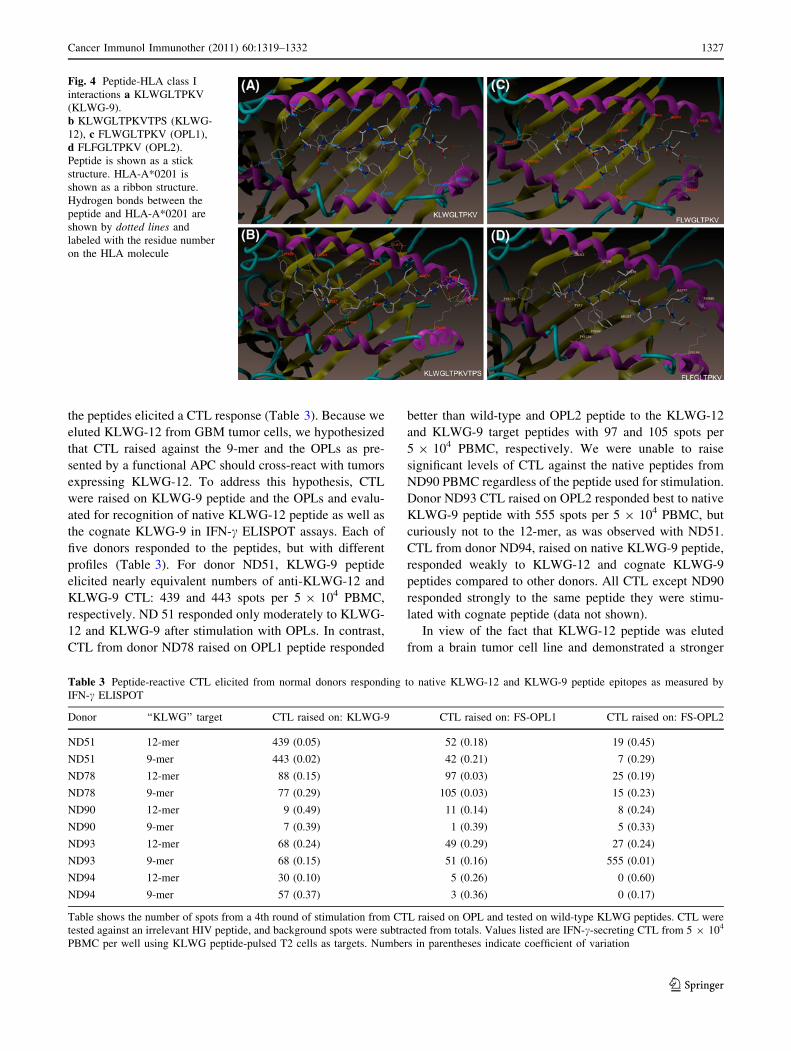
Modeling of KLWG Peptides in HLA-A*0201
Since KLWG-12 bound to HLA-A*0201 with a better
relative affinity than KLWG-9, we modeled KLWG-9,
KLWG-12, OPL1, and OPL2 in the binding cleft of HLA-
A*0201 in an attempt to observe the potential reason(s) for
the apparent increased affinity of the 12-mer (Fig. 4). The
‘‘Total Score’’ value (Table 2) generated by the modeling
software (higher scores indicate stronger binding) for
KLWG-9 peptide mirrors the IC50 data. Modeling suggests
that KLWG-9 is the poorest binder among the four peptides
essentially due to lack of aromatic residues in P1 like
phenylalanine which are preferred [9, 14]. For KLWG-9,
the amine group in the K at P1 forms a salt bridge with E63
on HLA-A2 and an H-bond with the backbone –C=O of
E58 and Y59, while P3 (W) forms an H-bond with –C=O
backbone of Q155 (Fig. 4a). Residues 5–8 are generally
oriented toward solvent, similar to other HLA-A*0201-
binding peptides.
Although no crystal structures have been generated with
[10 amino acids bound to HLA molecules, our docking
models clearly demonstrate that a 12-mer can bind in the
same orientation as the KLWG-9-mer, and ranks second
best in IC50 measurements and ‘‘Total Score’’ modeling
values among other peptides tested (Table 2). The 12-mer
KLWGLTPKVTPS essentially maintains a similar mode of
binding as KLWG-9 with the N-terminal –NH2 group
forming an H-bind with –OH of Y7 and Y171 on the MHC
molecule. P1 (K) forms an H-bond with –C=O backbone of
Y59 and salt bridge with E63 on the MHC molecule, like
KLWG-9 (Fig. 4b). Unlike KLWG-9, P3 (W) points into
solvent and most likely interacts with the T-cell receptor,
while P8 (K) forms a salt bridge with Q72. Also unlike
KLWG-9, threonine at P6 is oriented away from the
binding groove, toward solvent, providing a potential Tcr
interaction. The two prolines (P7 and P11) provide a
scaffold for further peptide-protein backbone interactions,
but may also interact with Tcr. P10-12 appear stacked such
Fig. 1 MS/MS spectrum of KLWG-12. y and b ions from peptide
fragmentation are labeled along with the mass of each fragment
Table 1 continued
Epitope SYFPEITHIscore Protein name
AKLLTIPQTLLNIS [14] KLLTIPQTLL 23 Zinc finger protein 493
AKLLTIPQTLLNIS [14] LTIPQTLLNI 23 Zinc finger protein 493
PVDTRAKVVLPPSLPRA [17] VLPPSLPRA 22 Uncharacterized protein KIAA1522
ALRGAGGGGVRDPGRLH [17] ALRGAGGGGV 25 Latent-transforming growth factor beta-binding protein
4 (LTBP-4)
For both T98g and CRL2610, multiple peptide fragments were identified from the same protein, so the longest parent fragment is listed along with
predicted HLA-binding epitopes contained within the parent sequence. SYFPEITHI scores below 20 were not considered
Cancer Immunol Immunother (2011) 60:1319–1332 1325
123
that threonine (P10) and serine (P12) form salt bridges with
K146 on HLA-A*0201, which could be responsible for the
increased relative affinity observed in T2 cell assays.
The molecular models of the docked FLWGLTPKV
(OPL1) and FLFGLTPKV (OPL2) peptides are similar
(Fig. 4c, d). P1 (F) interacts through a pi-pi aromatic
interaction with W167. The N-terminal NH2 group
H-bonds to the –OH group of Y7 and Y171, while F at P3
forms an aromatic interaction with Y159. Like KLWG-9,
P2 (L) and P9 (V) are buried in hydrophobic pockets of
the MHC. P7 (P) acts as a linker between the N- and
C-terminal portions of the peptide, and could interact with
Tcr. There are 6 H-bonds between the peptide backbone
and MHC-I clustered in the C-terminal area of the pep-
tide. All other H-bonding interactions occur with the
peptide backbone clustered at the C-terminal end of the
peptide. These modeling studies suggest that by simply
having more peptide-MHC contact, affinity may increase
if the peptide still fits into the binding groove. However,
it is important not to over-optimize a peptide for MHC
binding such that few residues are available to interact
with Tcr [32].
CTL generation and IFN-c ELISPOT assay
Human PBMC were cultured with repeated weekly stim-
ulations of either KLWG-9 or OPLs to determine whether
Fig. 2 PCR amplification of
HSD3B7 exons 2-4. a Oligo-
dT-primed cDNA from brain
tumor cell lines lane 1, CRL-
2610; lane 2 T98g; lane 3,
U251; lane 4, U-87. b Oligo-dT-
primed cDNA from primary
GBM line in lane 1, Ao2V4;
GBM tumor tissue lane 2, LB;
lane 3, LH; lane 4, NN; lane 5,
OJ; lane 6, TL. Lane 7 (SN) is
oligo-dT-primed cDNA from
normal brain tissue. In (a) and
(b), b-actin cDNA (189 bp) was
amplified across two exons as a
control for genomic
contamination. If genomic DNA
was present, a 600-bp band
would be observed
corresponding to an intron
between exons
Fig. 3 Detection of refolded HLA-A*0201 onto peptide beads.
a HLA-A*0201-positive control peptide; b HLA-A*0201-negative
control peptide; c KLWG-12 peptide; d KLWG-9 peptide
Table 2 Relative (IC50) and predicted affinities of OPL for HLA-A2
Computer
modeling
Peptide Sequencea SYFPEITHIb IC50
lMcTotal scored
KLWG-12 KLWGLTPKVTPS n/a 32 7.0
KLWG-9 KLWGLTPKV 30 110 3.0
OPL1 FLWGLTPKV 30 76 4.8
OPL2 FLFGLTPKV 30 17 13.4
a Standard single letter amino acid code and residues shown in bold represent
deviations from KLWG-9b SFXC-based total score and binding affinity for the variant-RT peptide (I1Y)
and the twenty-six peptides bound to the HLA-A*0201 crystal structurec Peptide epitope prediction values calculated using SYFPEITHId SFXC based total score and binding affinity for the Variant-RT peptide (I1Y)
and the twenty-six peptides bound to the HLA-A*0201 crystal structure
1326 Cancer Immunol Immunother (2011) 60:1319–1332
123
the peptides elicited a CTL response (Table 3). Because we
eluted KLWG-12 from GBM tumor cells, we hypothesized
that CTL raised against the 9-mer and the OPLs as pre-
sented by a functional APC should cross-react with tumors
expressing KLWG-12. To address this hypothesis, CTL
were raised on KLWG-9 peptide and the OPLs and evalu-
ated for recognition of native KLWG-12 peptide as well as
the cognate KLWG-9 in IFN-c ELISPOT assays. Each of
five donors responded to the peptides, but with different
profiles (Table 3). For donor ND51, KLWG-9 peptide
elicited nearly equivalent numbers of anti-KLWG-12 and
KLWG-9 CTL: 439 and 443 spots per 5 9 104 PBMC,
respectively. ND 51 responded only moderately to KLWG-
12 and KLWG-9 after stimulation with OPLs. In contrast,
CTL from donor ND78 raised on OPL1 peptide responded
better than wild-type and OPL2 peptide to the KLWG-12
and KLWG-9 target peptides with 97 and 105 spots per
5 9 104 PBMC, respectively. We were unable to raise
significant levels of CTL against the native peptides from
ND90 PBMC regardless of the peptide used for stimulation.
Donor ND93 CTL raised on OPL2 responded best to native
KLWG-9 peptide with 555 spots per 5 9 104 PBMC, but
curiously not to the 12-mer, as was observed with ND51.
CTL from donor ND94, raised on native KLWG-9 peptide,
responded weakly to KLWG-12 and cognate KLWG-9
peptides compared to other donors. All CTL except ND90
responded strongly to the same peptide they were stimu-
lated with cognate peptide (data not shown).
In view of the fact that KLWG-12 peptide was eluted
from a brain tumor cell line and demonstrated a stronger
Fig. 4 Peptide-HLA class I
interactions a KLWGLTPKV
(KLWG-9).
b KLWGLTPKVTPS (KLWG-
12), c FLWGLTPKV (OPL1),
d FLFGLTPKV (OPL2).
Peptide is shown as a stick
structure. HLA-A*0201 is
shown as a ribbon structure.
Hydrogen bonds between the
peptide and HLA-A*0201 are
shown by dotted lines and
labeled with the residue number
on the HLA molecule
Table 3 Peptide-reactive CTL elicited from normal donors responding to native KLWG-12 and KLWG-9 peptide epitopes as measured by
IFN-c ELISPOT
Donor ‘‘KLWG’’ target CTL raised on: KLWG-9 CTL raised on: FS-OPL1 CTL raised on: FS-OPL2
ND51 12-mer 439 (0.05) 52 (0.18) 19 (0.45)
ND51 9-mer 443 (0.02) 42 (0.21) 7 (0.29)
ND78 12-mer 88 (0.15) 97 (0.03) 25 (0.19)
ND78 9-mer 77 (0.29) 105 (0.03) 15 (0.23)
ND90 12-mer 9 (0.49) 11 (0.14) 8 (0.24)
ND90 9-mer 7 (0.39) 1 (0.39) 5 (0.33)
ND93 12-mer 68 (0.24) 49 (0.29) 27 (0.24)
ND93 9-mer 68 (0.15) 51 (0.16) 555 (0.01)
ND94 12-mer 30 (0.10) 5 (0.26) 0 (0.60)
ND94 9-mer 57 (0.37) 3 (0.36) 0 (0.17)
Table shows the number of spots from a 4th round of stimulation from CTL raised on OPL and tested on wild-type KLWG peptides. CTL were
tested against an irrelevant HIV peptide, and background spots were subtracted from totals. Values listed are IFN-c-secreting CTL from 5 9 104
PBMC per well using KLWG peptide-pulsed T2 cells as targets. Numbers in parentheses indicate coefficient of variation
Cancer Immunol Immunother (2011) 60:1319–1332 1327
123

affinity for HLA-A2 than the 9-mer, it was essential to
determine whether CTL could be generated against the
KLWG-12 peptide (Table 4). Therefore, normal human
PBMC were stimulated weekly with KLWG-12 peptide as
stated earlier. Donor ND117 CTL raised on KLWG-12
responded *35% more strongly to cognate KLWG-12
peptide than to KLWG-9 epitope with 279 and 181 spots,
respectively. Inhibition of CTL recognition of peptide-
loaded MHC class I molecule with W6/32 mAb demon-
strates the specificity and MHC dependency of the
anti-peptide CTL. ND117 CTL raised on KLWG-9 peptide
responded similarly to KLWG-9 and KLWG-12 peptides
with 204 and 209 spots, respectively. Blocking MHC class
I with W6/32 mAb again showed little response with 35
and 47 spots per 5 9 104 PBMC.
CTL cytotoxicity assay
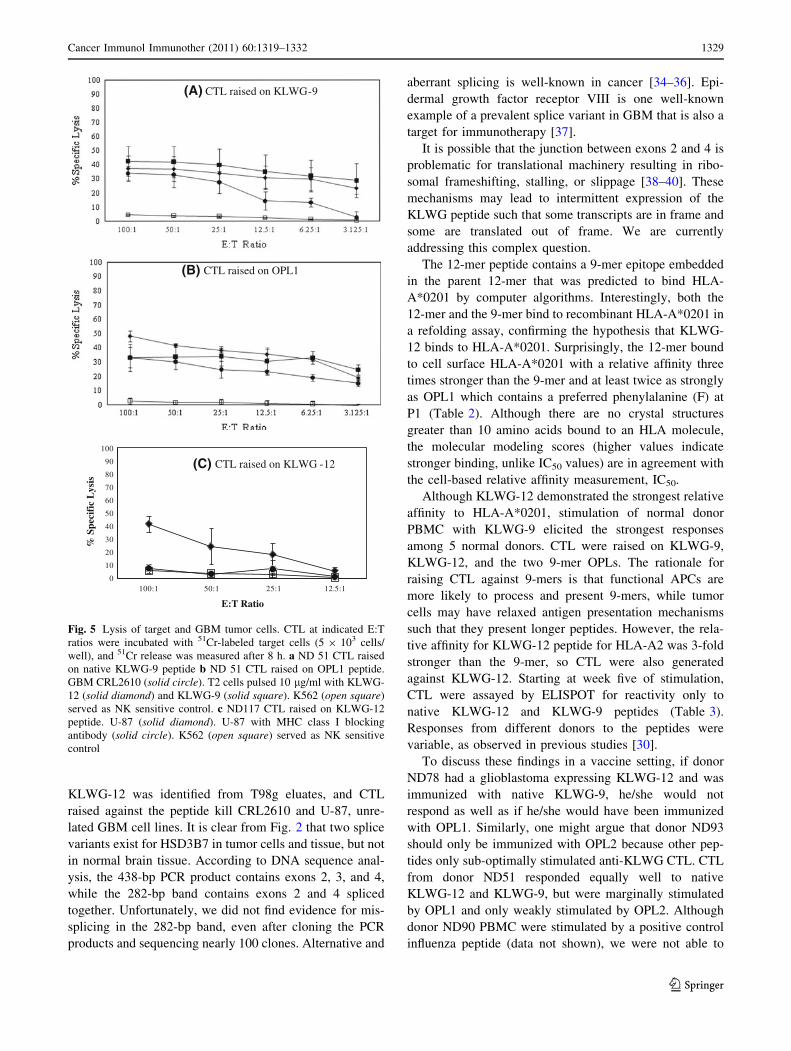
At week 5 of CTL generation, we tested anti-peptide CTL
for the ability to lyse GBM tumor cells in an HLA-A2-
restricted manner in 51Cr release assays. At an effector-to-
target ratio of 100:1, 34% of CRL2610 GBM tumor cells
were lysed by ND 51 anti-KLWG-9 CTL (Fig. 5a). Pep-
tide-pulsed T2 cells were also lysed by ND51 CTL raised
on KLWG-9.
Anti-OPL1 CTL generated from donor ND51 were also
evaluated for the ability to lyse GBM tumor cells. At a
100:1 effector to target ratio, anti-OPL1 CTL lysed 33% of
CRL2610 tumor cells (Fig. 5b). Anti-OPL1 CTL from
ND51 were tested for their ability to lyse T2 cells pulsed
with the 12-mer peptide KLWG-12. At a 100:1 E:T ratio,
anti-OPL1 CTL showed 48% lysis, whereas the anti-
KLWG-9 CTL only showed 37% lysis.
Anti-KLWG-12 CTL generated from ND117 demon-
strated the ability to kill 41% of U-87 glioblastoma tumor
cells at an E:T ratio of 100:1, (Fig. 5c, solid diamonds).
MHC class I–dependent lysis of U-87 was inhibited with
the addition of blocking antibody W6/32 (Fig. 5c, solid
circle). In all cytotoxicity assays, NK activity was between
3 and 5%.
Discussion
Tumors expose their protein contents to CTL by presenting
them as peptides on cell surface MHC class I molecules.
One of the most direct methods for identifying tumor-
associated peptides is to elute them from MHC molecules
on the tumor cell surface and perform tandem mass spec-
trometry on the eluted peptides to determine their
sequences. Because tumor cells are genetically and trans-
criptionally unstable, it is possible that normal transcrip-
tional and translational machinery are dysregulated,
leading to translation of frameshifted and other abnormal
peptides. The abnormal proteins that result should be
ubiquitinylated and targeted to the proteasome where their
subsequent peptides would be sampled by MHC molecules
and presented on the tumor cell surface for T-cell recog-
nition [15–19, 33].
Mass spectrometric analyses of peptides acid-eluted
from GBM tumor cell lines, T98g and CRL2610, resulted
in identification of several potential antigenic peptides. Of
particular interest was a 12-amino acid frameshift peptide
derived from the HSD3B7 gene. It was predicted to have a
very high SYFPEITHI binding score. This peptide was not
found in acid elutions from CRL2610 or PBMC from two
unrelated healthy donors using the same LC–MS/MS
methods. Interestingly, frameshifts from the zinc finger
protein 493 (ZNF493) gene were common to both T98g
and CRL2610, suggesting that this gene may be predis-
posed to transcriptional or translational infidelity. ZNF493
has 3 known variants; isoform 1 lacks 2 exons and utilizes
a downstream start codon, while isoform 2 includes an
exon from a putative 30 untranslated region.
Neither PBMC nor normal brain tissue is available from
the same patient for which the T98g cell line was derived.
However, it is unlikely that this frameshift results from a
unique, patient-specific mutation for 2 reasons. First, PCR
amplification of the RNA region flanking the KLWG
peptide resulted in a splice variant present in 5 GBM
tumors, one primary cell line and all 4 established GBM
cell lines, but not in normal brain tissue (Fig. 2). Second,
Table 4 Peptide-reactive CTL elicited from ND117 responding to native KLWG-12 and KLWG-9 peptide epitopes as measured by IFN-cELISPOT
Donor KLWG ‘‘Target’’ CTL Raised on KLWG-9 CTL Raised on KLWG-12
ND117 12-mer 209 (0.11) 279 (0.09)
ND117 12-mer ? W6/32 35 (0.4) 34 (0.11)
ND117 9-mer 204 (0.11) 181 (0.21)
ND117 9-mer ? W6/32 47 (0.12) 33 (0.12)
Table shows the number of spots from a 4th round of stimulation from CTL generated on KLWG-12, KLWG-9, and tested on wild-type KLWG
peptides with or without MHC class I blocking antibody W6/32. CTL were tested against an irrelevant HIV peptide, and background spots were
subtracted from totals. Values listed are IFN-c-secreting CTL from 5 9 104 PBMC per well using KLWG peptide-pulsed T2 cells as targets.
Numbers in parentheses indicate coefficient of variation
1328 Cancer Immunol Immunother (2011) 60:1319–1332
123
KLWG-12 was identified from T98g eluates, and CTL
raised against the peptide kill CRL2610 and U-87, unre-
lated GBM cell lines. It is clear from Fig. 2 that two splice
variants exist for HSD3B7 in tumor cells and tissue, but not
in normal brain tissue. According to DNA sequence anal-
ysis, the 438-bp PCR product contains exons 2, 3, and 4,
while the 282-bp band contains exons 2 and 4 spliced
together. Unfortunately, we did not find evidence for mis-
splicing in the 282-bp band, even after cloning the PCR
products and sequencing nearly 100 clones. Alternative and
aberrant splicing is well-known in cancer [34–36]. Epi-
dermal growth factor receptor VIII is one well-known
example of a prevalent splice variant in GBM that is also a
target for immunotherapy [37].
It is possible that the junction between exons 2 and 4 is
problematic for translational machinery resulting in ribo-
somal frameshifting, stalling, or slippage [38–40]. These
mechanisms may lead to intermittent expression of the
KLWG peptide such that some transcripts are in frame and
some are translated out of frame. We are currently
addressing this complex question.
The 12-mer peptide contains a 9-mer epitope embedded
in the parent 12-mer that was predicted to bind HLA-
A*0201 by computer algorithms. Interestingly, both the
12-mer and the 9-mer bind to recombinant HLA-A*0201 in
a refolding assay, confirming the hypothesis that KLWG-
12 binds to HLA-A*0201. Surprisingly, the 12-mer bound
to cell surface HLA-A*0201 with a relative affinity three
times stronger than the 9-mer and at least twice as strongly
as OPL1 which contains a preferred phenylalanine (F) at
P1 (Table 2). Although there are no crystal structures
greater than 10 amino acids bound to an HLA molecule,
the molecular modeling scores (higher values indicate
stronger binding, unlike IC50 values) are in agreement with
the cell-based relative affinity measurement, IC50.
Although KLWG-12 demonstrated the strongest relative
affinity to HLA-A*0201, stimulation of normal donor
PBMC with KLWG-9 elicited the strongest responses
among 5 normal donors. CTL were raised on KLWG-9,
KLWG-12, and the two 9-mer OPLs. The rationale for
raising CTL against 9-mers is that functional APCs are
more likely to process and present 9-mers, while tumor
cells may have relaxed antigen presentation mechanisms
such that they present longer peptides. However, the rela-
tive affinity for KLWG-12 peptide for HLA-A2 was 3-fold
stronger than the 9-mer, so CTL were also generated
against KLWG-12. Starting at week five of stimulation,
CTL were assayed by ELISPOT for reactivity only to
native KLWG-12 and KLWG-9 peptides (Table 3).
Responses from different donors to the peptides were
variable, as observed in previous studies [30].
To discuss these findings in a vaccine setting, if donor
ND78 had a glioblastoma expressing KLWG-12 and was
immunized with native KLWG-9, he/she would not
respond as well as if he/she would have been immunized
with OPL1. Similarly, one might argue that donor ND93
should only be immunized with OPL2 because other pep-
tides only sub-optimally stimulated anti-KLWG CTL. CTL
from donor ND51 responded equally well to native
KLWG-12 and KLWG-9, but were marginally stimulated
by OPL1 and only weakly stimulated by OPL2. Although
donor ND90 PBMC were stimulated by a positive control
influenza peptide (data not shown), we were not able to
0
10
20
30
40
50
60
70
80
90
100
100:1 50:1 25:1 12.5:1
% S
peci
fic
Lys
is
E:T Ratio
(A) CTL raised on KLWG-9
(B) CTL raised on OPL1
(C) CTL raised on KLWG -12
Fig. 5 Lysis of target and GBM tumor cells. CTL at indicated E:T
ratios were incubated with 51Cr-labeled target cells (5 9 103 cells/
well), and 51Cr release was measured after 8 h. a ND 51 CTL raised
on native KLWG-9 peptide b ND 51 CTL raised on OPL1 peptide.
GBM CRL2610 (solid circle). T2 cells pulsed 10 lg/ml with KLWG-
12 (solid diamond) and KLWG-9 (solid square). K562 (open square)
served as NK sensitive control. c ND117 CTL raised on KLWG-12
peptide. U-87 (solid diamond). U-87 with MHC class I blocking
antibody (solid circle). K562 (open square) served as NK sensitive
control
Cancer Immunol Immunother (2011) 60:1319–1332 1329
123

elicit CTL from ND90 with any of the KLWG or OPL
peptides. This might suggest that ND90 may have a hole in
his/her T-cell receptor repertoire, or perhaps there is an
OPL not examined in this study that would stimulate CTL
from ND90 to respond to native KLWG peptides. Alter-
natively, donor ND90 may be tolerant to KLWG and
related peptides. Similar to ND90, the response of ND94 to
KLWG-9 was weak, and non-existent to both OPLs.
Overall, ELISPOT data strongly suggest that the KLWG
12-mers and perhaps 9-mers are expressed on the tumor
cell surface for CTL recognition. The ELISPOT data also
parallel to what is observed in patients after vaccination
with tumor-derived peptides; some patients respond, while
some do not. Further studies may indicate whether patients
whose tumors express KLWG peptide possess anti-KLWG
CTL. It is worth speculating that one might increase the
frequency of response among patients if multiple OPL
were used in the vaccine. These findings are also further
evidence that peptides longer than the canonical 8-10 res-
idues can be presented by MHC molecules if they contain a
core HLA-binding epitope [4–6]. To prove that KLWG
peptide recognition was MHC dependent, W6/32 antibody
was used to block ND117 CTL recognition of target cells
in ELISPOT (Table 4).
Cytotoxicity assays were performed using CTL from
five of the six donors raised on each peptide (native
KLWG-9. -12, OPL1 and 2) to determine whether their
CTL would kill HLA-A2? glioblastoma cells. Only CTL
from donor ND51 and ND117 demonstrated the ability to
kill GBM tumor cells. Interestingly, ND51 killed CRL2610
HLA-A2-positive glioblastoma multiforme tumor cells
(Fig. 5), while ND117 killed U-87 tumor cells. Once again
it is not surprising that different donors respond differently
to different tumor cells [30].
We observed similar cytolytic profiles between CTL
raised on native KLWG-9 peptide and CTL raised on
OPL1. Interestingly, KLWG-9 and OPL CTL did not kill
the tumor cell line, T98g, in which the peptide was found
(data not shown). This is not unusual. We have previously
reported that the T98g is more difficult to kill than other
GBM tumor cell lines due to TGF-b secretion [30] and
perhaps other inhibitory cytokines [41, 42]. CTL killing
results indicate that CRL2610 and U-87 tumor cells
express KLWG-9 and/or KLWG-12-mer. This suggests
that CTL are more sensitive to the presence of peptide
than our mass spectrometric detection methods; biology is
more sensitive than technology. Although lysis of GBM
from donor ND51 CTL raised on native peptide or OPL1
were similar, CTL raised on KLWG-9 were most
responsive in ELISPOT (439 spots), but CTL from the
same donor raised on OPL1 peptide showed only 52 spots.
This result is in line with other findings showing a lack of
correlation between cytolytic activity and IFN-c ELISPOT
numbers [43]. This result suggests that the differences in
cytolytic activity may be related more to inherent differ-
ences among donors than to the peptide used for
stimulation.
An additional observation is that the large CTL:target
ratios results in a small variation in killing. It is possible
that some of the CRL2610 and U-87 target cells are het-
erogeneous and do not express high levels of MHC class I.
Alternatively, some of the cells may not express the
KLWG frameshift peptide due to antigenic modulation or
loss of antigen. Either or both of these possibilities would
result in low initial lysis at 100:1 in which most of the cells
expressing peptide-MHC are killed, but those that do not
are not killed.
The results presented here show that by examining the
MHC-binding peptidome of tumor cells, new peptide
antigens can be discovered as a result of genomic, tran-
scriptional, and translational instability. The results also
support rational design and optimization of peptides
derived from tumor-specific antigens for development of
peptides with increased binding affinity for HLA molecules
such that they stimulate a broad T-cell response to an
individual antigenic epitope so that as many anti-epitope
CTL will be stimulated as possible. Relative affinity
measurements of peptides with HLA-A*0201 can be cou-
pled with functional assays to measure peptide stimulation
of CTL. This is especially important given that different
donors respond differentially to native or optimized pep-
tides from the same epitope, suggesting that immunization
with multiple peptides for the same epitope will elicit CTL
from more individuals than immunization with one epitope.
However, it is well established that tumors can down-
modulate individual peptides to escape detection by the
immune system. Our findings do not address this problem,
but instead further substantiate the importance of immu-
nizing individuals not only with multiple tumor antigens
but also with multiple peptides from the same epitope to
elicit the broadest response within the same patient and
among different patients. The results presented here may
explain, in part, the variability in individual patient
responses to immunotherapy.
Acknowledgments We would like to thank HoJoon Lee, M.S. for
the frameshift database.
References
1. Hou LC, Veeravagu A, Hsu AR, Tse VC (2006) Recurrent
glioblastoma multiforme: a review of natural history and man-
agement options. Neurosurg Focus 20:E5
2. Laws ER, Parney IF, Huang W et al (2003) Survival following
surgery and prognostic factors for recently diagnosed malignant
glioma: data from the Glioma Outcomes Project. J Neurosurg
99:467–473
1330 Cancer Immunol Immunother (2011) 60:1319–1332
123

3. Novellino L, Castelli C, Parmiani G (2005) A listing of human
tumor antigens recognized by T cells: March 2004 update. Cancer
Immunol Immunother 54:187–207
4. Antwi K, Hanavan PD, Myers CE, Ruiz YW, Thompson EJ, Lake
DF (2009) Proteomic identification of an MHC-binding pepti-
dome from pancreas and breast cancer cell lines. Mol Immunol
46:2931–2937
5. Stryhn A, Pedersen LO, Holm A, Buus S (2000) Longer peptide
can be accommodated in the MHC class I binding site by a
protrusion mechanism. Eur J Immunol 30:3089–3099
6. Gebreselassie D, Spiegel H, Vukmanovic S (2006) Sampling of
major histocompatibility complex class I-associated peptidome
suggests relatively looser global association of HLA-B*5101
with peptides. Hum Immunol 67:894–906
7. Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG
(1991) Allele-specific motifs revealed by sequencing of self-
peptides eluted from MHC molecules. Nature 351:290–296
8. Stevanovic S, Schild H (1999) Quantitative aspects of T cell
activation–peptide generation and editing by MHC class I mol-
ecules. Semin Immunol 11:375–384
9. Hunt DF, Henderson RA, Shabanowitz J et al (1992) Charac-
terization of peptides bound to the class I MHC molecule HLA-
A2.1 by mass spectrometry. Science 255:1261–1263
10. Kast WM, Brandt RM, Sidney J et al (1994) Role of HLA-A
motifs in identification of potential CTL epitopes in human
papillomavirus type 16 E6 and E7 proteins. J Immunol
152:3904–3912
11. Parker KC, Bednarek MA, Hull LK et al (1992) Sequence motifs
important for peptide binding to the human MHC class I mole-
cule, HLA-A2. J Immunol 149:3580–3587
12. Pogue RR, Eron J, Frelinger JA, Matsui M (1995) Amino-ter-
minal alteration of the HLA-A*0201-restricted human immuno-
deficiency virus pol peptide increases complex stability and in
vitro immunogenicity. Proc Natl Acad Sci USA 92:8166–8170
13. Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A (1993)
Prominent role of secondary anchor residues in peptide binding to
HLA-A2.1 molecules. Cell 74:929–937
14. Smith MH, Nuara AA, Egen JG, Sirjani DB, Lam KS, Grimes WJ
(1998) Baculoviral expressed HLA class I heavy chains used to
screen a synthetic peptide library for allele-specific peptide
binding motifs. Mol Immunol 35:1033–1043
15. Linnebacher M, Gebert J, Rudy W et al (2001) Frameshift pep-
tide-derived T-cell epitopes: a source of novel tumor-specific
antigens. Int J Cancer 93:6–11
16. Saeterdal I, Bjorheim J, Lislerud K et al (2001) Frameshift-
mutation-derived peptides as tumor-specific antigens in inherited
and spontaneous colorectal cancer. Proc Natl Acad Sci USA
98:13255–13260
17. Schwitalle Y, Linnebacher M, Ripberger E, Gebert J, von Knebel
Doeberitz M (2004) Immunogenic peptides generated by frame-
shift mutations in DNA mismatch repair-deficient cancer cells.
Cancer Immun 4:14
18. Townsend A, Ohlen C, Rogers M, Edwards J, Mukherjee S,
Bastin J (1994) Source of unique tumour antigens. Nature
371:662
19. Elliott T, Bodmer H, Townsend A (1996) Recognition of out-of-
frame major histocompatibility complex class I-restricted epi-
topes in vivo. Eur J Immunol 26:1175–1179
20. Cheng JB, Jacquemin E, Gerhardt M et al (2003) Molecular
genetics of 3beta-hydroxy-Delta5–C27-steroid oxidoreductase
deficiency in 16 patients with loss of bile acid synthesis and liver
disease. J Clin Endocrinol Metab 88:1833–1841
21. van der Burg SH, Ras E, Drijfhout JW et al (1995) An HLA class
I peptide-binding assay based on competition for binding to class
I molecules on intact human B cells. Identification of conserved
HIV-1 polymerase peptides binding to HLA-A*0301. Hum
Immunol 44:189–198
22. Korbie DJ, Mattick JS (2008) Touchdown PCR for increased
specificity and sensitivity in PCR amplification. Nat Protocols
3:1452–1456
23. Smith MH, Lam KS, Hersh EM, Lebl M, Grimes WJ (1994)
Peptide sequences binding to MHC class I proteins. Mol Immunol
31:1431–1437
24. Lam KS, Lebl M, Krchnak V (1997) The ‘‘One-Bead-One-Com-
pound’’ combinatorial library method. Chem Rev 97:411–448
25. Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM,
Knapp RJ (1991) A new type of synthetic peptide library for
identifying ligand-binding activity. Nature 354:82–84
26. Dionne SO, Myers CE, Smith MH, Lake DF (2004) Her-2/neu
altered peptide ligand-induced CTL responses: implications for
peptides with increased HLA affinity and T-cell-receptor inter-
action. Cancer Immunol Immunother 53:307–314
27. Parkhurst MR, Salgaller ML, Southwood S et al (1996) Improved
induction of melanoma-reactive CTL with peptides from the
melanoma antigen gp100 modified at HLA-A*0201-binding
residues. J Immunol 157:2539–2548
28. Kumar N, Mohanty D (2007) MODPROPEP: a program for
knowledge-based modeling of protein-peptide complexes.
Nucleic Acids Res 35:W549–W555
29. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Steva-
novic S (1999) SYFPEITHI: database for MHC ligands and
peptide motifs. Immunogenetics 50:213–219
30. Myers CE, Dionne SO, Shakalya K, Mahadevan D, Smith MH,
Lake DF (2008) Variation in cytotoxic T-lymphocyte responses
to peptides derived from tyrosinase-related protein-2. Hum
Immunol 69:24–31
31. Dionne SO, Smith MH, Marincola FM, Lake DF (2003) Func-
tional characterization of CTL against gp100 altered peptide
ligands. Cancer Immunol Immunother 52:199–206
32. Dionne SO, Smith MH, Marincola FM, Lake DF (2001) Antigen
presentation of a modified tumor-derived peptide by tumor
infiltrating lymphocytes. Cell Immunol 214:139–144
33. Ishikawa T, Fujita T, Suzuki Y et al (2003) Tumor-specific
immunological recognition of frameshift-mutated peptides in
colon cancer with microsatellite instability. Cancer Res
63:5564–5572
34. Belicha-Villanueva A, Blickwedehl J, McEvoy S, Golding M,
Gollnick SO, Bangia N (2010) What is the role of alternate
splicing in antigen presentation by major histocompatibility
complex class I molecules? Immunol Res 46:32–44
35. Ward AJ, Cooper TA (2010) The pathobiology of splicing.
J Pathol 220:152–163
36. Kalnina Z, Zayakin P, Silina K, Line A (2005) Alterations of pre-
mRNA splicing in cancer. Genes Chromosomes Cancer
42:342–357
37. Gan HK, Kaye AH, Luwor RB (2009) The EGFRvIII variant in
glioblastoma multiforme. J Clin Neurosci 16:748–754
38. Farabaugh PJ (1996) Programmed translational frameshifting.
Annu Rev Genet 30:507–528
39. Fernandez J, Yaman I, Huang C et al (2005) Ribosome stalling
regulates IRES-mediated translation in eukaryotes, a parallel to
prokaryotic attenuation. Mol Cell 17:405–416
40. Hansen TM, Reihani SN, Oddershede LB, Sorensen MA (2007)
Correlation between mechanical strength of messenger RNA
pseudoknots and ribosomal frameshifting. Proc Natl Acad Sci
USA 104:5830–5835
41. Bodmer S, Strommer K, Frei K et al (1989) Immunosuppression
and transforming growth factor-beta in glioblastoma. Preferential
production of transforming growth factor-beta 2. J Immunol
143:3222–3229
Cancer Immunol Immunother (2011) 60:1319–1332 1331
123

42. Finke J, Ferrone S, Frey A, Mufson A, Ochoa A (1999) Where
have all the T cells gone? Mechanisms of immune evasion by
tumors. Immunol Today 20:158–160
43. van Besouw NM, Zuijderwijk JM, de Kuiper P, Ijzermans JN,
Weimar W, van der Mast BJ (2005) The granzyme B and
interferon-gamma enzyme-linked immunospot assay as alterna-
tives for cytotoxic T-lymphocyte precursor frequency after renal
transplantation. Transplantation 79:1062–1066
1332 Cancer Immunol Immunother (2011) 60:1319–1332
123
Related Documents











