Published Ahead of Print 8 June 2007. 2007, 189(16):5816. DOI: 10.1128/JB.00602-07. J. Bacteriol. de Mendoza and Charles O. Rock Luciana Paoletti, Ying-Jie Lu, Gustavo E. Schujman, Diego Bacillus subtilis Synthesis in Coupling of Fatty Acid and Phospholipid http://jb.asm.org/content/189/16/5816 Updated information and services can be found at: These include: REFERENCES http://jb.asm.org/content/189/16/5816#ref-list-1 at: This article cites 35 articles, 26 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on March 1, 2014 by guest http://jb.asm.org/ Downloaded from on March 1, 2014 by guest http://jb.asm.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Published Ahead of Print 8 June 2007. 2007, 189(16):5816. DOI: 10.1128/JB.00602-07. J. Bacteriol.
de Mendoza and Charles O. RockLuciana Paoletti, Ying-Jie Lu, Gustavo E. Schujman, Diego
Bacillus subtilisSynthesis in Coupling of Fatty Acid and Phospholipid
http://jb.asm.org/content/189/16/5816Updated information and services can be found at:
These include:
REFERENCEShttp://jb.asm.org/content/189/16/5816#ref-list-1at:
This article cites 35 articles, 26 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from

JOURNAL OF BACTERIOLOGY, Aug. 2007, p. 5816–5824 Vol. 189, No. 160021-9193/07/$08.00�0 doi:10.1128/JB.00602-07Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Coupling of Fatty Acid and Phospholipid Synthesis in Bacillus subtilis�
Luciana Paoletti,1† Ying-Jie Lu,2†‡ Gustavo E. Schujman,1 Diego de Mendoza,1 and Charles O. Rock2*Instituto de Biologıa Molecular y Celular de Rosario, and Departamento de Microbiologıa, Facultad de Ciencias Bioquımicas y
Farmaceuticas, Universidad Nacional de Rosario, Rosario, Argentina,1 and Department of Infectious Diseases,St. Jude Children’s Research Hospital, 332 N. Lauderdale, Memphis, Tennessee2
Received 18 April 2007/Accepted 29 May 2007
plsX (acyl-acyl carrier protein [ACP]:phosphate acyltransferase), plsY (yneS) (acyl-phosphate:glycerol-phos-phate acyltransferase), and plsC (yhdO) (acyl-ACP:1-acylglycerol-phosphate acyltransferase) function in phos-phatidic acid formation, the precursor to membrane phospholipids. The physiological functions of these geneswas inferred from their in vitro biochemical activities, and this study investigated their roles in gram-positivephospholipid metabolism through the analysis of conditional knockout strains in the Bacillus subtilis modelsystem. The depletion of PlsX led to the cessation of both fatty acid synthesis and phospholipid synthesis. Theinactivation of PlsY also blocked phospholipid synthesis, but fatty acid formation continued due to theappearance of acylphosphate intermediates and fatty acids arising from their hydrolysis. Phospholipid syn-thesis ceased following PlsC depletion, but fatty acid synthesis continued at a high rate, leading to theaccumulation of fatty acids arising from the dephosphorylation of 1-acylglycerol-3-P followed by the deacyla-tion of monoacylglycerol. Analysis of glycerol 3-P acylation in B. subtilis membranes showed that PlsY was anacylphosphate-specific acyltransferase, whereas PlsC used only acyl-ACP as an acyl donor. PlsX was found inthe soluble fraction of disrupted cells but was associated with the cell membrane in intact organisms. Thesedata establish that PlsX is a key enzyme that coordinates the production of fatty acids and membranephospholipids in B. subtilis.
Phosphatidic acid (PtdOH) is synthesized by the successiveacylation of glycerol-P followed by the acylation of 1-acyl-glycerol-P, and there are two enzyme systems that carry out thefirst reaction in bacteria. In the Escherichia coli model system,either acyl-acyl carrier protein (ACP) or acyl-coenzyme A(CoA) thioesters are utilized by the membrane-bound PlsBacyltransferase to acylate the 1 position of glycerol-P (7, 14,25). However, the E. coli paradigm is not universal, and theplsB gene is confined to gram-negative bacteria (primarilygammaproteobacteria) (27). Most gram-positive bacteria uti-lize the recently discovered PlsX/Y pathway for 1-acyl-glyc-erol-P formation (27). PlsX is a soluble protein that catalyzesthe formation of acylphosphate (acyl-PO4) from acyl-ACP.This activated fatty acid is then used by the membrane-boundPlsY acyltransferase to acylate the 1 position of glycerol-P (26).The second acyltransferase in PtdOH formation, PlsC, is uni-versally expressed in bacteria and completes the synthesis ofPtdOH by transferring an acyl chain from acyl-ACP (or acyl-CoA) to the 2 position of 1-acyl-glycerol-P (27). In E. coli, PlsCutilizes either acyl-ACP or acyl-CoA as an acyl donor (5),whereas in Streptococcus pneumoniae, PlsC uses only acyl-ACPas the acyl donor (27).
The PlsX and PlsY reactions were demonstrated in vitro,and their physiological roles were inferred from the biochem-ical analysis and the fact that they are essential genes in bac-
teria that lack plsB (27). Bacillus subtilis is typical of a gram-positive bacterium that lacks a plsB gene. The B. subtilis plsXgene was annotated based on its similarity to the E. coli coun-terpart and is an essential gene in B. subtilis (22). The E. coliplsX gene was discovered as a mutated allele required for aplsB26 mutant strain to exhibit a glycerol-P auxotrophic growthphenotype (23). The E. coli plsB26 mutant produces a PlsBacyltransferase with an elevated Km for glycerol-P (1, 2, 19).Bioinformatic analysis of the B. subtilis genome shows that theyhdO gene corresponds to the plsC gene and that the yneS genecorresponds to the plsY gene. Both of these genes are alsoessential in B. subtilis (22). The goal of this study is to employa genetic approach to investigate the roles of plsX, plsY, andplsC in gram-positive phospholipid metabolism through theanalysis of conditional knockout strains for each of these threegenes in the B. subtilis model system.
MATERIALS AND METHODS
Materials. Sources of supplies were American Radiolabeled Chemicals Inc.([2-14C]acetate [specific activity, 55 Ci/mol] and [U-14C]glycerol-P [specific ac-tivity, 150 Ci/mole]), Avanti Polar Lipids, Inc. (palmitoyl-CoA and phospholip-ids), and Sigma Chemical Co. (glycerol-P and fatty-acid-free bovine serum al-bumin [BSA]); acyl-PO4 was synthesized as described previously (27). Acyl-ACPswere prepared enzymatically by using the acyl-ACP synthase method (32, 33)using E. coli ACP as the starting material. Protein was measured by the Bradfordmethod (3). All other chemicals were reagent grade or better.
Construction of conditional knockout mutant strains. The integrative plasmidpDH88 (30) or pMUTIN4 (35), containing the IPTG (isopropyl-�-D-thiogalac-topyranoside)-inducible Pspac promoter, or plasmid pGES49, a derivative ofpAG58 (34) containing the xylose-inducible Pxyl promoter, was used for condi-tional gene expression in B. subtilis. Plasmid pLP6 (Fig. 1A) was constructedusing a 250-bp DNA fragment, generated by PCR using primers YhdOpU andYhdOpL (Table 1), carrying the ribosome binding site and a 5� portion of yhdO.The amplification product was digested with HindIII and BamHI and cloned intovector pDH88, previously digested with HindIII and BglII. Plasmid pLP35 (Fig.1B) was constructed using a 514-bp DNA fragment, generated by PCR with
* Corresponding author. Mailing address: Department of InfectiousDiseases, St. Jude Children’s Research Hospital, 332 N. Lauderdale,Memphis, TN 38105-2794. Phone: (901) 495-3491. Fax: (901) 495-0399.E-mail: [email protected].
† L.P. and Y.-J.L. contributed equally to the research.‡ Present address: Division of Infectious Diseases, Children’s Hos-
pital, Boston, MA 02115.� Published ahead of print on 8 June 2007.
5816
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from

primers PYneSU and PYneSL (Table 1), containing the ribosome binding siteand the 5� upstream region of the yneS gene. The PCR product was first clonedinto PCR-Blunt II-TOPO. This plasmid was then digested with HindIII andBglII, and the resultant fragment was inserted into the HindIII and BamHI sitesof vector pMUTIN4. Plasmid pLP21 (Fig. 1C) was constructed using a 546-bpDNA fragment, generated by PCR with primers PYlpcU and PYlpcL (Table 1),containing the ribosome binding site and the 5� upstream region of the plsX gene.The product of PCR was digested with HindIII and BamHI and cloned intoplasmid pGES49. Plasmid pGES310 (Fig. 1C) was constructed using a 451-bpDNA fragment containing the ribosome binding site and the 5� upstream frag-ment of the fabD gene generated by PCR using primers FabDmutB and PplsXU(Table 1). The PCR product was digested with BamHI and EcoRI and clonedinto plasmid pMUTIN4. Plasmid pLP36 was constructed using a 389-bp DNAfragment containing the ribosome binding site and the 5� upstream fragment ofthe yneR gene generated by PCR using primers PYneRU and PyneRL (Table 1).The PCR product was digested with BamHI and EcoRI and cloned into plasmidpMUTIN4.
Strains LP15, LP39, GS311, LP61, and LP62 were generated by integration ofthe circular form of the plasmids pLP6, pLP35, pGES310, pLP21, and pLP36,respectively, into the B. subtilis chromosome by a single-crossover event (Fig. 1).This approach results in the conditional inactivation of the target gene whoseexpression can be controlled by either Pspac or Pxyl (Fig. 1). Each of these strains
was checked by PCR to ensure that the plasmid had integrated into the correctsite. B. subtilis strains were cultured in Luria-Bertani (LB) medium supple-mented, where required, with 0.4% xylose and/or 0.2 mM IPTG. B. subtilis strainswere transformed as previously described (34). Transformants were selected onLB agar supplemented, where required, with chloramphenicol (5 �g/ml), eryth-romycin (0.5 �g/ml), lincomycin (12.5 �g/ml), 0.2 mM IPTG, or 0.4% xylose.
The yneR gene is located just downstream of the yneS gene, and although yneRdoes not apparently have a role in lipid metabolism, the two genes may exist inan operon. YneR belongs to a family of proteins involved in iron-sulfur clusterbiosynthesis (Pfam01521) and was not scored as being an essential protein in thescreen for essential Bacillus genes (22). In strain LP61, both genes yneS and yneRwere under the control of the Pspac promoter (Fig. 1B); therefore, we con-structed strain LP62 to rule out an effect of yneR on the yneS phenotype. In thisstrain, only yneR expression depended on the presence of IPTG. Strain LP62grew normally in the presence and absence of the inducer (data not shown),confirming that the yneR gene was not essential and that the growth defect ofstrain LP61 in the absence of IPTG was due to the lack of yneS expression.
Growth and metabolic labeling of mutant strains. LP39 (plsX) was grownovernight in LB medium with 0.2 mM IPTG–0.4% xylose. LP61 (plsY) and LP15(plsC) were grown in LB medium with 0.3 mM IPTG and the correspondingantibiotic. Cells were washed twice and resuspended in LB medium. StrainsLP39, LP61, and LP15 were inoculated at A600 values of 0.06, 0.15, and 0.04,respectively, and grown in the presence or absence of inducers. Each of theconstructs required a slightly different number of cell divisions to dilute out thepreexisting protein in the absence of inducer. The different inoculation densitieswere empirically determined to provide cell cultures making the transition toPlsX-, PlsY-, or PlsC-dependent growth in 3 to 4 h at between 100 and 200 Klettunits.
Cells were labeled with 1 �Ci/ml [14C]acetate for 30 min at the time pointsindicated in the figures. Cells were collected, and total lipids were extracted.Briefly, cells were resuspended in 1.8 ml of chloroform-methanol-acetic acid(1/2/0.02, vol/vol/vol) followed by 0.5 ml water, 0.9 ml of chloroform, and 0.9 mlof 2 M KCl. Samples were vortexed and centrifuged to separate the phases. Thelower phase was transferred into a new tube, and the upper phase was extractedagain with 0.9 ml chloroform and combined with the first extraction. The organicphase was evaporated under nitrogen and redissolved in chloroform. Lipids wereanalyzed using either preadsorbent Silica Gel G layers (Analtech) developedwith hexane-ethyl ether-acetic acid (80/20/1, vol/vol/vol) to separate the neutrallipids or Silica Gel H layers (Analtech) developed with chloroform–methanol–ammonia–water–0.25 M EDTA (45/35/1.5/8.34/0.16, vol/vol/vol/vol/vol) to re-solve the phospholipids. Radioactivity on the plates was visualized using a Ty-phoon 9200 PhosphorImager screen and quantified using ImageQuant software(version 5.2). Lipid species were identified by comigration with standards.
Detection of acyl-PO4 with mass spectroscopy. LP61 cells were grown over-night on an LB plate with 12.5 �g/ml lincomycin, 0.5 �g/ml erythromycin, and 0.3mM IPTG. Cells were scraped off the plate and used to inoculate LB medium toan A600 of 0.03 to initiate growth at 37°C. Cells were collected by centrifugation(6,000 rpm, 4°C, 10 min) when the A600 reached 0.5 and were immediatelyprocessed with total lipid extraction using chloroform-methanol-concentratedHCl (1/2/0.02, vol/vol/vol). The dried sample was dissolved in 50% methanol-water and analyzed by negative-ion electrospray precursor ion-scanning massspectrometry.
Mass spectrometry of acyl-PO4 was performed by the Hartwell Center forBioinformatics and Biotechnology at St. Jude Children’s Research Hospital.Mass spectrometry analysis was performed using a Finnigan TSQ Quantum(Thermo Electron, San Jose, CA) triple-quadrupole mass spectrometerequipped with the nanospray ion source. The instrument was operated in the
TABLE 1. Oligonucleotides used in this study
Oligonucleotide Sequence (5�–3�)
YhdoU .............AAGTGGGATCCGTATAAGTTTTGTGCAAATYhdoL..............CTTACGTCGACAGTTTCAGGACAATCTCTCPYlpcU ............ACCAACAAGCTTTATTACAGATGAAGAACTPYlpcL.............CTACAGGGATCCTCATGCATGCTCCACCTTPYneSU...........ATGGGATCTAGAAAAAGGAGAPYneSL............CCAATAGATCTGGCGAAGATGFabDmutB.......AGTTATGGATCCAACGTTTCATCGGCTTCAPplsX................GTTTGAATTCTGGAAGGCTCTGCGTTGTCPYnerU............GAGAAGCTTTGAAAGGGACACTGGCAACTGPYnerL ............AGTTTCGGATCCTTATAACCATTTTACTTTA
FIG. 1. Diagram of molecular constructs. (A) Initial and finalgenomic organizations of strain LP15 (Pspac-plsC) containing plsC(yhdO) under IPTG control. (B) Initial and final genomic organiza-tions of strain LP61 (Pspac-plsY) containing plsY (yneS) under IPTGcontrol. (C) Initial and final genomic organizations of strain LP39(Pxyl-plsX) containing plsX under the control of the xylose promoter.IPTG was also required for growth of this strain to express the down-stream essential fadD and fabG genes that are located in the plsXoperon as indicated in the diagram.
VOL. 189, 2007 PHOSPHATIDIC ACID FORMATION IN B. SUBTILIS 5817
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from
negative-ion mode using precursor ion scanning to detect the loss of a phosphategroup (27). Ion source parameters were as follows: a spray voltage of 1,600 V, acapillary temperature of 270°C, and a capillary offset of �35 V (tube lens offsetwas set by infusion of the polytyrosine tuning and calibration solution [ThermoElectron, San Jose, CA] in electrospray mode). Mass spectrometry acquisitionparameters for Q1 scanning were as follows: a scan range of 225 to 500 m/z, ascan time zero of 1 s, and a peak width for Q1 of 0.7 FWHM (full width at halfheight). Mass spectrometry acquisition parameters for parent ion scanning wereas follows: a scan range of 225 to 500 m/z, a scan time zero of 1 s, a product massof 79 m/z, a collision energy of 15 V, a peak width for Q1 and Q3 of 0.7 FWHM,and Q2 collision-induced dissociation gas (argon) of 0.5 mTorr. Instrumentcontrol and data acquisition were performed with Finnigan Xcalibur (version 1.4SR1) software (Thermo Electron, San Jose, CA).
PlsC activity assay. The membrane fraction of B. subtilis strain 168 waspurified using sucrose gradient ultracentrifugation according to a method de-scribed previously (27). Reactions were carried out in a solution containing 100mM Tris-HCl (pH 7.4), 150 mM NaCl, 100 �M [14C]glycerol-P, 1 mg/ml BSA,and 5 mM Na3VO4 (to inactive phosphatases) with 5 �g purified membraneprotein. Reactions were started by adding combinations of 200 �M palmitoyl-PO4, 50 �M palmitoyl-ACP, or 50 �M palmitoyl-CoA to the mixtures. Reactionmixtures were extracted by chloroform-methanol-concentrated HCl (1/2/0.02,vol/vol/vol) and spotted onto preadsorbent Silica Gel H layers (Analtech) devel-oped with chloroform–methanol–ammonia–water–0.25 M EDTA (45/35/1.5/8.34/0.16), and product formation was detected with a Typhoon 9200PhosphorImager and quantified using ImageQuant software (version 5.2).
Immunofluorescence microscopy. The strains were grown to the exponentialphase under the conditions described above. Samples were processed for immu-nofluorescence microscopy (16). All cultures were fixed in growth medium witha final concentration of 2.6% paraformaldehyde, 0.006% glutaraldehyde, and 30mM sodium phosphate buffer (pH 7.4) for 15 min at room temperature and 30min on ice. The cells were washed, briefly treated with lysozyme, and affixed topoly-L-lysine-treated multiwell slides. Cells were stained with purified primaryantibodies raised in rabbits against the purified target proteins. The specificity ofthe anti-PlsX, anti-FabF, and anti-PlsC antibodies was analyzed by Westernblotting using protein extracts from wild-type strain 168. These antibodies de-tected a band of the correct molecular weight in the control cells (data notshown). After washing with the fluorescein isothiocyanate (FITC)-conjugatedsecondary antibodies, 1 �g/ml of 4�,6�-diamidino-2-phenylindole (DAPI) wasadded with Slow Fade equilibration buffer (Slow Fade kit from MolecularProbes) for 5 min. Coverslips were mounted with Slow Fade containing glycerol.All photographs were taken using a Nikon Eclipse E800 microscope equippedwith a 100� Plan Apo objective and a Nikon FDX-35 automatic camera system.Two filter sets were used, one for visualizing FITC (FITC-HYQ) and the otherfor visualizing DAPI (UV-2A).
RESULTS
Depletion of PlsX, PlsY, and PlsC. B. subtilis contains threeopen reading frames that have homology with enzymes pre-dicted to be involved in the biosynthesis of PtdOH based ontheir in vitro enzymatic activities (27). Two of these genes, plsXand plsY (yneS), are related to the PlsX/Y pathway for the firstacylation of glycerol-P. The third gene, plsC (yhdO), is pre-dicted to encode an acyltransferase catalyzing the acylation of1-acyl-glycerol-P to give PtdOH. These three genes wereflagged as being essential based on the systematic genome-wide inactivation of B. subtilis genes (22). Therefore, we con-structed strains LP15, LP39, and LP61 that expressed each ofthese genes under the control of a regulated promoter (Fig. 1).Strains LP15 and LP61 contain the plsC or the plsY gene underthe control of the IPTG-inducible Pspac promoter, respec-tively, while in strain LP39, plsX is controlled by the xylose-inducible Pxyl promoter (Fig. 1). In strain LP39, the essentialfabD and fabG genes located downstream of plsX were drivenby Pspac to alleviate the potential polar effect caused by thegeneration of the Pxyl-plsX chromosomal fusion (Fig. 1). StrainLP39 consistently grew to higher densities than the other twostrains due to the supplementation of the medium with xylose
that not only induced plsX expression but also served as acarbon source.
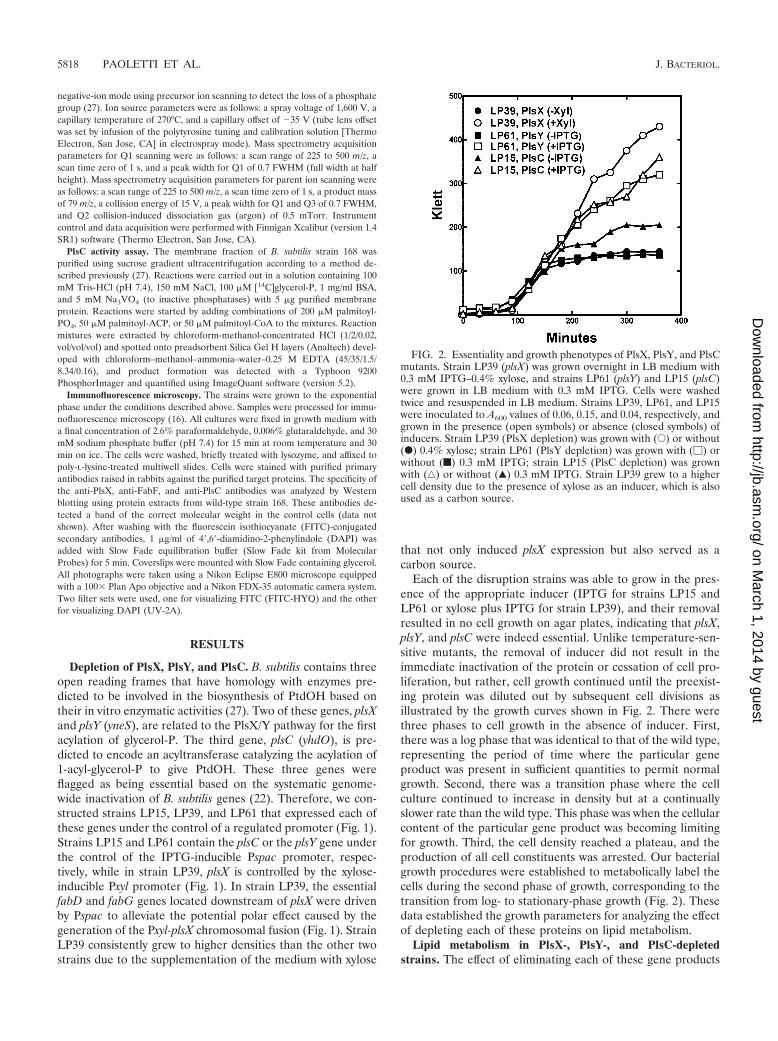
Each of the disruption strains was able to grow in the pres-ence of the appropriate inducer (IPTG for strains LP15 andLP61 or xylose plus IPTG for strain LP39), and their removalresulted in no cell growth on agar plates, indicating that plsX,plsY, and plsC were indeed essential. Unlike temperature-sen-sitive mutants, the removal of inducer did not result in theimmediate inactivation of the protein or cessation of cell pro-liferation, but rather, cell growth continued until the preexist-ing protein was diluted out by subsequent cell divisions asillustrated by the growth curves shown in Fig. 2. There werethree phases to cell growth in the absence of inducer. First,there was a log phase that was identical to that of the wild type,representing the period of time where the particular geneproduct was present in sufficient quantities to permit normalgrowth. Second, there was a transition phase where the cellculture continued to increase in density but at a continuallyslower rate than the wild type. This phase was when the cellularcontent of the particular gene product was becoming limitingfor growth. Third, the cell density reached a plateau, and theproduction of all cell constituents was arrested. Our bacterialgrowth procedures were established to metabolically label thecells during the second phase of growth, corresponding to thetransition from log- to stationary-phase growth (Fig. 2). Thesedata established the growth parameters for analyzing the effectof depleting each of these proteins on lipid metabolism.
Lipid metabolism in PlsX-, PlsY-, and PlsC-depletedstrains. The effect of eliminating each of these gene products
FIG. 2. Essentiality and growth phenotypes of PlsX, PlsY, and PlsCmutants. Strain LP39 (plsX) was grown overnight in LB medium with0.3 mM IPTG–0.4% xylose, and strains LP61 (plsY) and LP15 (plsC)were grown in LB medium with 0.3 mM IPTG. Cells were washedtwice and resuspended in LB medium. Strains LP39, LP61, and LP15were inoculated to A600 values of 0.06, 0.15, and 0.04, respectively, andgrown in the presence (open symbols) or absence (closed symbols) ofinducers. Strain LP39 (PlsX depletion) was grown with (E) or without(F) 0.4% xylose; strain LP61 (PlsY depletion) was grown with (�) orwithout (f) 0.3 mM IPTG; strain LP15 (PlsC depletion) was grownwith (‚) or without (Œ) 0.3 mM IPTG. Strain LP39 grew to a highercell density due to the presence of xylose as an inducer, which is alsoused as a carbon source.
5818 PAOLETTI ET AL. J. BACTERIOL.
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from

on lipid metabolism was assessed by metabolic labeling with[14C]acetate during the time periods when the gene productswere becoming limiting to growth, as indicated in Fig. 2. Ac-etate incorporation into the lipid fraction of strain LP39 (Pxyl-plsX) in the absence of inducer was about 10% of the rate inthe presence of IPTG (Table 2). In strain LP61 (Pspac-plsY),the comparison of acetate incorporation with inducer and thatwithout inducer showed that the acetate incorporation wasapproximately the same. Surprisingly, the result with strainLP15 (Pspac-plsC) showed that the absence of inducer resultedin a 250% increase in acetate labeling (Table 2). The effect ofthe defects in lipid metabolism on general cell metabolic ac-tivity was assessed by the incorporation of [14C]acetate intononlipid (essentially protein) cell components. In all cases, theincorporation of acetate into protein was comparable. In aseparate experiment, we normalized the incorporation of[14C]acetate into cells and normalized the results to total cellprotein. The pattern was essentially the same with the PlsX-depleted strain, incorporating 10.6% of the control, the PlsY-depleted cells, incorporating 98%, and the PlsC-depleted cells,incorporating 202%. These data show that the depletion ofPlsX resulted in the cessation of fatty acid synthesis, while theinactivation of PlsY or PlsC did not.
The next experiments examined the labeled products thatwere found in the depleted cells. The analysis of the lipidproducts is illustrated in Fig. 3, and the distribution of radio-activity is shown in Table 2. Depletion of the plsX gene productresulted in the inhibition of total lipid synthesis without accu-mulation of fatty acids or other intermediates, indicating thatin the absence of PlsX, the syntheses of both fatty acid andphospholipid were severely compromised. There was a trace offatty acid in xylose-supplemented strain LP39 that was notdetected in the PlsX-depleted cells. PlsY depletion alsoblocked phospholipid synthesis, although the synthesis of fattyacid was still active, as evidenced by the accumulation of fattyacid in PlsY-deprived strain LP61 (Fig. 3 and Table 2). Deple-tion of PlsC also compromised phospholipid biosynthesis andled to the incorporation of acetate into lipids at increased
rates. The accumulation of monoacylglycerol and a largeamount of fatty acid was noted following the inactivation ofPlsC. Monoacylglycerol arising from the dephosphorylation ofacyl-glycerol-P was noted previously in acyltransferase assaysusing either E. coli (31) or Rhodopseudomonas spheroids (28)membranes as the in vitro enzyme source. The inactivation ofPlsC in E. coli plsC(Ts) mutants led to a large accumulation ofphosphatidic acid; however, the experiments would not havedetected monoacylglycerol if it was formed (4). In summary,our data indicate that PlsX depletion inhibits total lipid bio-synthesis, while the depletion of either PlsY or PlsC affectsonly phospholipid biosynthesis. Thus, the formation of acyl-PO4 by PlsX is the critical step in the initiation of the phos-pholipid biosynthetic pathway, coupling the biosynthesis offatty acids with phospholipid synthesis.
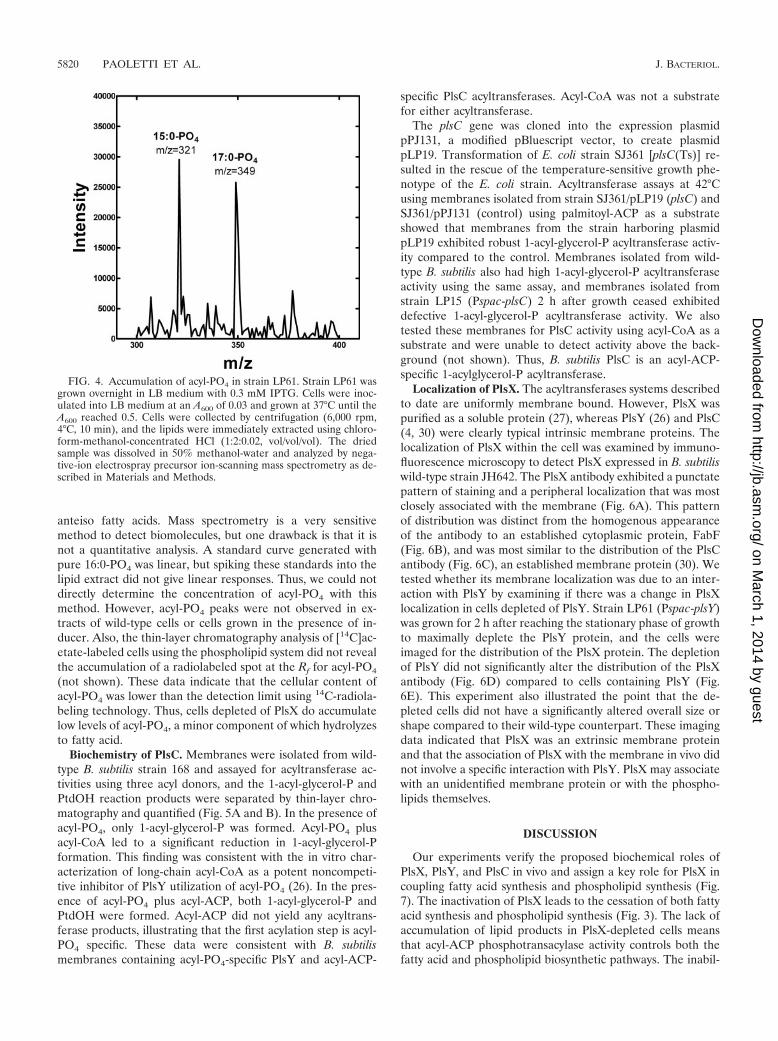
PlsX is responsible for the formation of acyl-PO4. Despiterepeated attempts, we were unable to detect the presence ofacyl-PO4 intermediates in wild-type cells either by metaboliclabeling and thin-layer chromatography or by mass spectrom-etry. These negative experiments were not completely unex-pected, because acyl-PO4 intermediates were not detected in S.pneumoniae wild-type cells either (27). However, we were ableto detect acyl-PO4 peaks in samples of strain LP61 (Pspac-plsY) grown in the absence of IPTG during the transition fromlog-phase growth to growth stasis (Fig. 4). These mass spec-trometry results clearly showed the presence of 15 and 17carbon acyl-PO4s, reflecting the major chain lengths of fattyacids produced by the B. subtilis fatty acid biosynthetic path-way. A small 19-carbon peak was also detected (Fig. 4), indi-cating that some excess elongation of acyl chains occurred inthe absence of utilization. This observation was reminiscent ofa report that the blockade of phospholipid synthesis in E. coliat the acyltransferase step using either plsB or gpsA mutantsgave rise to the formation of abnormally long fatty acid prod-ucts (9). This technology cannot distinguish between iso and
FIG. 3. [14C]Acetate labeling profiles of PlsX-, PlsY-, and PlsC-depleted cells. Cells were labeled with 1 �Ci/ml [14C]acetate for 30 minduring the transition from log- to stationary-phase growth (Fig. 2).Cells were harvested, and total lipids were extracted and analyzedby thin-layer chromatography on preadsorbent Silica Gel G layers(Analtech) developed with hexane-ethyl ether-acetic acid (80/20/1, vol/vol/vol). The radioactive lipid species were visualized using aPhosphorImager screen, and the bands were identified by their comi-gration with standards. FA, fatty acid; DAG, diacylglycerol; MAG,monoacylglycerol; PL, phospholipid. The example shown is typical ofthree experiments performed in both laboratories.
TABLE 2. �14C�Acetate labeling of strains deficient inPlsX, PlsY, or PlsCa
StrainTotal lipid
count/min/108
cells (%)
Distribution (%)
PL MAG DAG FA
LP39(Pxyl-plsX) � xylose 15,782 89.5 — 9.3 0.2LP39(Pxyl-plsX) without
xylose1,679 (10.6) 89.4 — 9.6 —
LP61(Pspac-plsY) � IPTG 9,603 86.2 — 11.8 2.0LP61(Pspac-plsY) without
IPTG8,011 (83.4) 31.8 — 0.5 63.2
LP15(Pspac-plsC) � IPTG 12,938 87.5 — 12.5 —LP15(Pspac-plsC) without
IPTG32,179 (249) 8.0 6.2 0.5 85.3
a Total lipid is given as counts per min per 108 cells. The strains were grown asdescribed in the text with or without IPTG and labeled with �14C�acetate for 30min. The lipids were extracted as described in Materials and Methods. Thedistribution of lipids was determined by thin-layer chromatography using theneutral lipid solvent system shown in Fig. 3, and the percentage of each indicatedlipid class was determined by quantifying the PhosphorImager data. PL, phos-pholipid; MAG, monoacylglycerol; DAG, diacylglycerol; FA, fatty acid; —, notdetected. The data shown are representative of three independent experimentscarried out in both laboratories.
VOL. 189, 2007 PHOSPHATIDIC ACID FORMATION IN B. SUBTILIS 5819
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from
anteiso fatty acids. Mass spectrometry is a very sensitivemethod to detect biomolecules, but one drawback is that it isnot a quantitative analysis. A standard curve generated withpure 16:0-PO4 was linear, but spiking these standards into thelipid extract did not give linear responses. Thus, we could notdirectly determine the concentration of acyl-PO4 with thismethod. However, acyl-PO4 peaks were not observed in ex-tracts of wild-type cells or cells grown in the presence of in-ducer. Also, the thin-layer chromatography analysis of [14C]ac-etate-labeled cells using the phospholipid system did not revealthe accumulation of a radiolabeled spot at the Rf for acyl-PO4
(not shown). These data indicate that the cellular content ofacyl-PO4 was lower than the detection limit using 14C-radiola-beling technology. Thus, cells depleted of PlsX do accumulatelow levels of acyl-PO4, a minor component of which hydrolyzesto fatty acid.
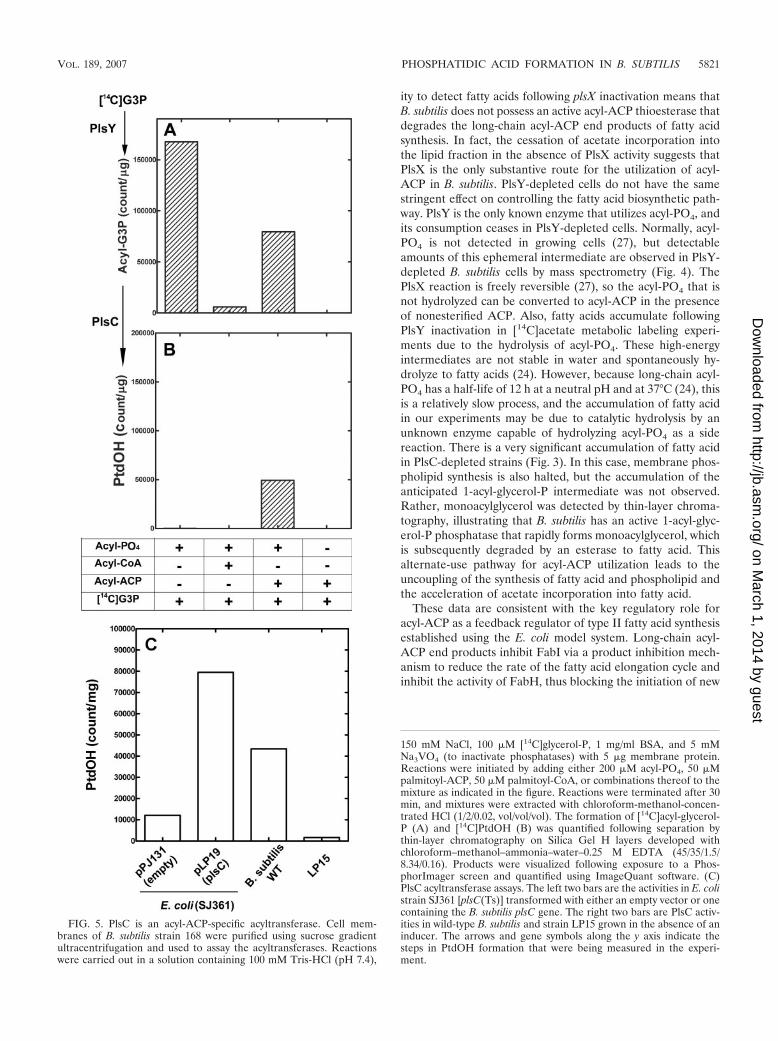
Biochemistry of PlsC. Membranes were isolated from wild-type B. subtilis strain 168 and assayed for acyltransferase ac-tivities using three acyl donors, and the 1-acyl-glycerol-P andPtdOH reaction products were separated by thin-layer chro-matography and quantified (Fig. 5A and B). In the presence ofacyl-PO4, only 1-acyl-glycerol-P was formed. Acyl-PO4 plusacyl-CoA led to a significant reduction in 1-acyl-glycerol-Pformation. This finding was consistent with the in vitro char-acterization of long-chain acyl-CoA as a potent noncompeti-tive inhibitor of PlsY utilization of acyl-PO4 (26). In the pres-ence of acyl-PO4 plus acyl-ACP, both 1-acyl-glycerol-P andPtdOH were formed. Acyl-ACP did not yield any acyltrans-ferase products, illustrating that the first acylation step is acyl-PO4 specific. These data were consistent with B. subtilismembranes containing acyl-PO4-specific PlsY and acyl-ACP-
specific PlsC acyltransferases. Acyl-CoA was not a substratefor either acyltransferase.
The plsC gene was cloned into the expression plasmidpPJ131, a modified pBluescript vector, to create plasmidpLP19. Transformation of E. coli strain SJ361 [plsC(Ts)] re-sulted in the rescue of the temperature-sensitive growth phe-notype of the E. coli strain. Acyltransferase assays at 42°Cusing membranes isolated from strain SJ361/pLP19 (plsC) andSJ361/pPJ131 (control) using palmitoyl-ACP as a substrateshowed that membranes from the strain harboring plasmidpLP19 exhibited robust 1-acyl-glycerol-P acyltransferase activ-ity compared to the control. Membranes isolated from wild-type B. subtilis also had high 1-acyl-glycerol-P acyltransferaseactivity using the same assay, and membranes isolated fromstrain LP15 (Pspac-plsC) 2 h after growth ceased exhibiteddefective 1-acyl-glycerol-P acyltransferase activity. We alsotested these membranes for PlsC activity using acyl-CoA as asubstrate and were unable to detect activity above the back-ground (not shown). Thus, B. subtilis PlsC is an acyl-ACP-specific 1-acylglycerol-P acyltransferase.
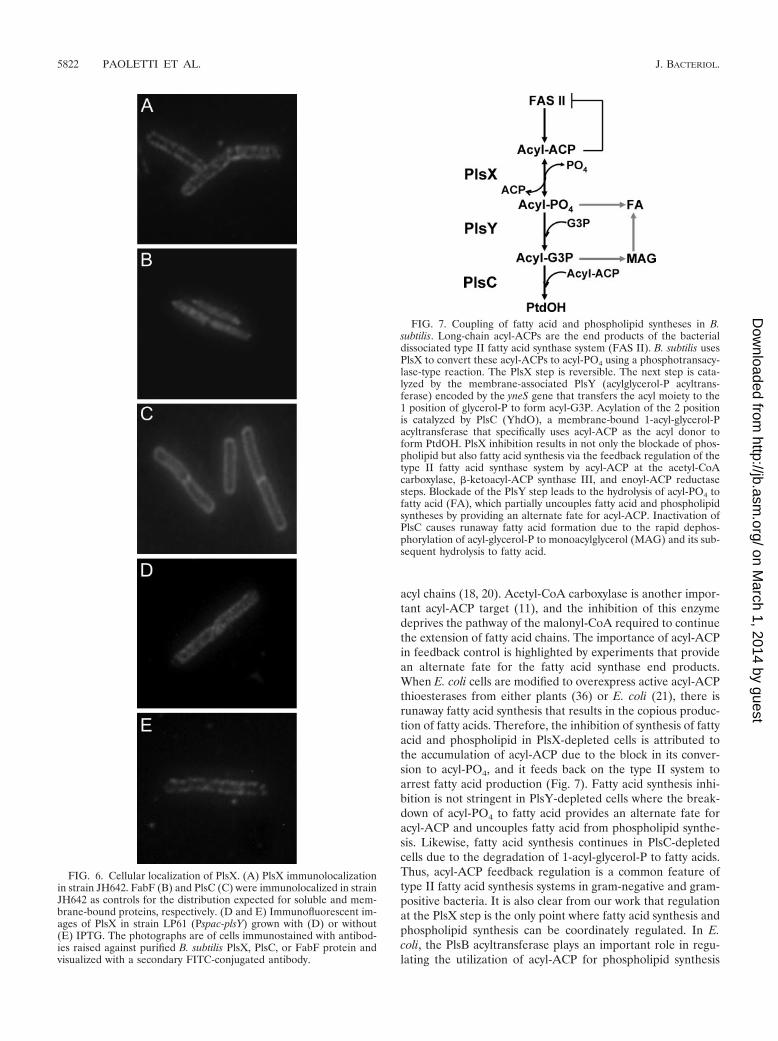
Localization of PlsX. The acyltransferases systems describedto date are uniformly membrane bound. However, PlsX waspurified as a soluble protein (27), whereas PlsY (26) and PlsC(4, 30) were clearly typical intrinsic membrane proteins. Thelocalization of PlsX within the cell was examined by immuno-fluorescence microscopy to detect PlsX expressed in B. subtiliswild-type strain JH642. The PlsX antibody exhibited a punctatepattern of staining and a peripheral localization that was mostclosely associated with the membrane (Fig. 6A). This patternof distribution was distinct from the homogenous appearanceof the antibody to an established cytoplasmic protein, FabF(Fig. 6B), and was most similar to the distribution of the PlsCantibody (Fig. 6C), an established membrane protein (30). Wetested whether its membrane localization was due to an inter-action with PlsY by examining if there was a change in PlsXlocalization in cells depleted of PlsY. Strain LP61 (Pspac-plsY)was grown for 2 h after reaching the stationary phase of growthto maximally deplete the PlsY protein, and the cells wereimaged for the distribution of the PlsX protein. The depletionof PlsY did not significantly alter the distribution of the PlsXantibody (Fig. 6D) compared to cells containing PlsY (Fig.6E). This experiment also illustrated the point that the de-pleted cells did not have a significantly altered overall size orshape compared to their wild-type counterpart. These imagingdata indicated that PlsX was an extrinsic membrane proteinand that the association of PlsX with the membrane in vivo didnot involve a specific interaction with PlsY. PlsX may associatewith an unidentified membrane protein or with the phospho-lipids themselves.
DISCUSSION
Our experiments verify the proposed biochemical roles ofPlsX, PlsY, and PlsC in vivo and assign a key role for PlsX incoupling fatty acid synthesis and phospholipid synthesis (Fig.7). The inactivation of PlsX leads to the cessation of both fattyacid synthesis and phospholipid synthesis (Fig. 3). The lack ofaccumulation of lipid products in PlsX-depleted cells meansthat acyl-ACP phosphotransacylase activity controls both thefatty acid and phospholipid biosynthetic pathways. The inabil-
FIG. 4. Accumulation of acyl-PO4 in strain LP61. Strain LP61 wasgrown overnight in LB medium with 0.3 mM IPTG. Cells were inoc-ulated into LB medium at an A600 of 0.03 and grown at 37°C until theA600 reached 0.5. Cells were collected by centrifugation (6,000 rpm,4°C, 10 min), and the lipids were immediately extracted using chloro-form-methanol-concentrated HCl (1:2:0.02, vol/vol/vol). The driedsample was dissolved in 50% methanol-water and analyzed by nega-tive-ion electrospray precursor ion-scanning mass spectrometry as de-scribed in Materials and Methods.
5820 PAOLETTI ET AL. J. BACTERIOL.
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from
ity to detect fatty acids following plsX inactivation means thatB. subtilis does not possess an active acyl-ACP thioesterase thatdegrades the long-chain acyl-ACP end products of fatty acidsynthesis. In fact, the cessation of acetate incorporation intothe lipid fraction in the absence of PlsX activity suggests thatPlsX is the only substantive route for the utilization of acyl-ACP in B. subtilis. PlsY-depleted cells do not have the samestringent effect on controlling the fatty acid biosynthetic path-way. PlsY is the only known enzyme that utilizes acyl-PO4, andits consumption ceases in PlsY-depleted cells. Normally, acyl-PO4 is not detected in growing cells (27), but detectableamounts of this ephemeral intermediate are observed in PlsY-depleted B. subtilis cells by mass spectrometry (Fig. 4). ThePlsX reaction is freely reversible (27), so the acyl-PO4 that isnot hydrolyzed can be converted to acyl-ACP in the presenceof nonesterified ACP. Also, fatty acids accumulate followingPlsY inactivation in [14C]acetate metabolic labeling experi-ments due to the hydrolysis of acyl-PO4. These high-energyintermediates are not stable in water and spontaneously hy-drolyze to fatty acids (24). However, because long-chain acyl-PO4 has a half-life of 12 h at a neutral pH and at 37°C (24), thisis a relatively slow process, and the accumulation of fatty acidin our experiments may be due to catalytic hydrolysis by anunknown enzyme capable of hydrolyzing acyl-PO4 as a sidereaction. There is a very significant accumulation of fatty acidin PlsC-depleted strains (Fig. 3). In this case, membrane phos-pholipid synthesis is also halted, but the accumulation of theanticipated 1-acyl-glycerol-P intermediate was not observed.Rather, monoacylglycerol was detected by thin-layer chroma-tography, illustrating that B. subtilis has an active 1-acyl-glyc-erol-P phosphatase that rapidly forms monoacylglycerol, whichis subsequently degraded by an esterase to fatty acid. Thisalternate-use pathway for acyl-ACP utilization leads to theuncoupling of the synthesis of fatty acid and phospholipid andthe acceleration of acetate incorporation into fatty acid.
These data are consistent with the key regulatory role foracyl-ACP as a feedback regulator of type II fatty acid synthesisestablished using the E. coli model system. Long-chain acyl-ACP end products inhibit FabI via a product inhibition mech-anism to reduce the rate of the fatty acid elongation cycle andinhibit the activity of FabH, thus blocking the initiation of new
150 mM NaCl, 100 �M [14C]glycerol-P, 1 mg/ml BSA, and 5 mMNa3VO4 (to inactivate phosphatases) with 5 �g membrane protein.Reactions were initiated by adding either 200 �M acyl-PO4, 50 �Mpalmitoyl-ACP, 50 �M palmitoyl-CoA, or combinations thereof to themixture as indicated in the figure. Reactions were terminated after 30min, and mixtures were extracted with chloroform-methanol-concen-trated HCl (1/2/0.02, vol/vol/vol). The formation of [14C]acyl-glycerol-P (A) and [14C]PtdOH (B) was quantified following separation bythin-layer chromatography on Silica Gel H layers developed withchloroform–methanol–ammonia–water–0.25 M EDTA (45/35/1.5/8.34/0.16). Products were visualized following exposure to a Phos-phorImager screen and quantified using ImageQuant software. (C)PlsC acyltransferase assays. The left two bars are the activities in E. colistrain SJ361 [plsC(Ts)] transformed with either an empty vector or onecontaining the B. subtilis plsC gene. The right two bars are PlsC activ-ities in wild-type B. subtilis and strain LP15 grown in the absence of aninducer. The arrows and gene symbols along the y axis indicate thesteps in PtdOH formation that were being measured in the experi-ment.
FIG. 5. PlsC is an acyl-ACP-specific acyltransferase. Cell mem-branes of B. subtilis strain 168 were purified using sucrose gradientultracentrifugation and used to assay the acyltransferases. Reactionswere carried out in a solution containing 100 mM Tris-HCl (pH 7.4),
VOL. 189, 2007 PHOSPHATIDIC ACID FORMATION IN B. SUBTILIS 5821
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from
acyl chains (18, 20). Acetyl-CoA carboxylase is another impor-tant acyl-ACP target (11), and the inhibition of this enzymedeprives the pathway of the malonyl-CoA required to continuethe extension of fatty acid chains. The importance of acyl-ACPin feedback control is highlighted by experiments that providean alternate fate for the fatty acid synthase end products.When E. coli cells are modified to overexpress active acyl-ACPthioesterases from either plants (36) or E. coli (21), there isrunaway fatty acid synthesis that results in the copious produc-tion of fatty acids. Therefore, the inhibition of synthesis of fattyacid and phospholipid in PlsX-depleted cells is attributed tothe accumulation of acyl-ACP due to the block in its conver-sion to acyl-PO4, and it feeds back on the type II system toarrest fatty acid production (Fig. 7). Fatty acid synthesis inhi-bition is not stringent in PlsY-depleted cells where the break-down of acyl-PO4 to fatty acid provides an alternate fate foracyl-ACP and uncouples fatty acid from phospholipid synthe-sis. Likewise, fatty acid synthesis continues in PlsC-depletedcells due to the degradation of 1-acyl-glycerol-P to fatty acids.Thus, acyl-ACP feedback regulation is a common feature oftype II fatty acid synthesis systems in gram-negative and gram-positive bacteria. It is also clear from our work that regulationat the PlsX step is the only point where fatty acid synthesis andphospholipid synthesis can be coordinately regulated. In E.coli, the PlsB acyltransferase plays an important role in regu-lating the utilization of acyl-ACP for phospholipid synthesis
FIG. 6. Cellular localization of PlsX. (A) PlsX immunolocalizationin strain JH642. FabF (B) and PlsC (C) were immunolocalized in strainJH642 as controls for the distribution expected for soluble and mem-brane-bound proteins, respectively. (D and E) Immunofluorescent im-ages of PlsX in strain LP61 (Pspac-plsY) grown with (D) or without(E) IPTG. The photographs are of cells immunostained with antibod-ies raised against purified B. subtilis PlsX, PlsC, or FabF protein andvisualized with a secondary FITC-conjugated antibody.
FIG. 7. Coupling of fatty acid and phospholipid syntheses in B.subtilis. Long-chain acyl-ACPs are the end products of the bacterialdissociated type II fatty acid synthase system (FAS II). B. subtilis usesPlsX to convert these acyl-ACPs to acyl-PO4 using a phosphotransacy-lase-type reaction. The PlsX step is reversible. The next step is cata-lyzed by the membrane-associated PlsY (acylglycerol-P acyltrans-ferase) encoded by the yneS gene that transfers the acyl moiety to the1 position of glycerol-P to form acyl-G3P. Acylation of the 2 positionis catalyzed by PlsC (YhdO), a membrane-bound 1-acyl-glycerol-Pacyltransferase that specifically uses acyl-ACP as the acyl donor toform PtdOH. PlsX inhibition results in not only the blockade of phos-pholipid but also fatty acid synthesis via the feedback regulation of thetype II fatty acid synthase system by acyl-ACP at the acetyl-CoAcarboxylase, �-ketoacyl-ACP synthase III, and enoyl-ACP reductasesteps. Blockade of the PlsY step leads to the hydrolysis of acyl-PO4 tofatty acid (FA), which partially uncouples fatty acid and phospholipidsyntheses by providing an alternate fate for acyl-ACP. Inactivation ofPlsC causes runaway fatty acid formation due to the rapid dephos-phorylation of acyl-glycerol-P to monoacylglycerol (MAG) and its sub-sequent hydrolysis to fatty acid.
5822 PAOLETTI ET AL. J. BACTERIOL.
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from

(17), and our results suggest that PlsX has a similar role inbacteria that lack PlsB and utilize the PlsX/Y/C pathway toPtdOH. However, the mechanism(s) for the regulation of PlsXactivity remains a major unanswered question in bacterial bio-chemistry.
B. subtilis PlsC is the second example of a biochemicallycharacterized gram-positive 1-acyl-glycerol-P acyltransferase.Like S. pneumoniae PlsC (27), B. subtilis PlsC uses only acyl-ACP as the acyl donor. This is in contrast to the E. coli enzyme,which is capable of using acyl-CoA as the acyl donor (4), as dothe plant (15) and mammalian (13) homologs. The S. pneu-moniae genome does not contain homologs of the enzymes forthe synthesis of acyl-CoA or �-oxidation, and therefore, a PlsCcapable of using acyl-CoA is of no practical value for thisorganism. In contrast, B. subtilis has two homologs of acyl-CoAsynthetases (LcfA and YhfL) and a complement of genes pre-dicted to constitute a fatty acid �-oxidation pathway whoseexpression is regulated by YsiA (29). YsiA is an acyl-CoA-responsive transcriptional repressor analogous to FadR (8)that functions in controlling the fatty acid �-oxidation regulonin E. coli. The accumulation of fatty acids, particularly in thePlsC-depleted cells, indicates that this pathway is not rapidlyinduced by the appearance of intracellular fatty acids under thegrowth conditions the we employed. There are reports of ex-ogenous fatty acid incorporation into phospholipids in B. sub-tilis (10) and Bacillus megaterium (12), and our data raise thequestion of the pathway for exogenous fatty acid incorporationinto phospholipids. Acyl-CoAs may be formed, but they couldnot serve as substrates for either the PlsY or PlsC acyltrans-ferases. There are several possible explanations for these ob-servations. There may be fatty acid turnover in membranephospholipids, and the reacylation enzyme may use acyl-CoAas well as acyl-ACP. One such acyltransferase has been bio-chemically characterized in E. coli, but this lysophosphati-dylethanolamine acyltransferase is acyl-ACP specific (6). B.subtilis may possess either an acyl-ACP synthetase or a fattyacid kinase. In this regard, the two putative acyl-CoA syn-thetase homologs of B. subtilis have not been biochemicallycharacterized. This leaves open the possibility that one of themmay be an acyl-ACP synthetase that is a component of theYsiA regulon whose function is to channel exogenous fattyacids into the phospholipid biosynthetic pathway. Fatty acidkinases are unknown to biochemistry, but this postulated ac-tivity would function to introduce exogenous fatty acids intothe phospholipid biosynthetic pathway downstream of PlsX.The accumulation of fatty acids in our experiments suggeststhat neither of these postulated enzymes is operating in ourexperimental setting; however, this may be due to the deple-tion of ACP acceptors for the activated fatty acids when thepathway is arrested.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM34496 (C.O.R.), CancerCenter Support grant CA 21765, the Consejo Nacional de Investiga-ciones Cientıficas y Tecnicas (CONICET, Argentina), the Agencia dePromocion Cientıfica y Tecnologica (FONCYT, Argentina), and theAmerican Lebanese Syrian Associated Charities. L.P. is a fellow andG.E.S. and D.D.M. are Career Investigators at CONICET. D.D.M. isan International Research Scholar of the Howard Hughes MedicalInstitute.
REFERENCES
1. Bell, R. M. 1974. Mutants of Escherichia coli defective in membrane phos-pholipid synthesis: macromolecular synthesis in an sn-glycerol 3-phosphateacyltransferase Km mutant. J. Bacteriol. 117:1065–1076.
2. Bell, R. M. 1975. Mutants of Escherichia coli defective in membrane phos-pholipid synthesis: properties of wild type and Km defective sn-glycerol-3-phosphate acyltransfersae activities. J. Biol. Chem. 250:7147–7152.
3. Bradford, M. M. 1976. A rapid and sensitive method for quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem. 72:248–254.
4. Coleman, J. 1990. Characterization of Escherichia coli cells deficient in1-acyl-sn-glycerol-3-phosphate acyltransferase activity. J. Biol. Chem. 265:17215–17221.
5. Coleman, J. 1992. Characterization of the Escherichia coli gene for 1-acyl-sn-glycerol-3-phosphate acyltransferase (plsC). Mol. Gen. Genet. 232:295–303.
6. Cooper, C. L., L. Hsu, S. Jackowski, and C. O. Rock. 1989. 2-Acylglycerol-phosphoethanolamine acyltransferase/acyl-acyl carrier protein synthetase isa membrane-associated acyl carrier protein binding protein. J. Biol. Chem.264:7384–7389.
7. Cronan, J. E., Jr., and C. O. Rock. 1996. Biosynthesis of membrane lipids, p.612–636. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B.Low, B. Magasanik, W. Reznikoff, M. Riley, M. Schaechter, and H. E.Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecularbiology, 2nd ed. ASM Press, Washington, DC.
8. Cronan, J. E., Jr., and S. Subrahmanyam. 1998. FadR, transcriptional co-ordination of metabolic expediency. Mol. Microbiol. 29:937–943.
9. Cronan, J. E., Jr., L. J. Weisberg, and R. G. Allen. 1975. Regulation ofmembrane lipid synthesis in Escherichia coli. Accumulation of free fatty acidsof abnormal length during inhibition of phospholipid synthesis. J. Biol.Chem. 250:5835–5840.
10. Cybulski, L. E., D. Albanesi, M. C. Mansilla, S. Altabe, P. S. Aguilar, and D.de Mendoza. 2002. Mechanism of membrane fluidity optimization: isother-mal control of the Bacillus subtilis acyl-lipid desaturase. Mol. Microbiol.45:1379–1388.
11. Davis, M. S. and J. E. Cronan, Jr. 2001. Inhibition of Escherichia coli acetylcoenzyme A carboxylase by acyl-acyl carrier protein. J. Bacteriol. 183:1499–1503.
12. Fulco, A. J. 1972. The biosynthesis of unsaturated fatty acids by bacilli. 3.Uptake and utilization of exogenous palmitate. J. Biol. Chem. 247:3503–3510.
13. Ganesh, B. B., P. Wang, J. H. Kim, T. M. Black, T. M. Lewin, F. T. Fiedorek,and R. A. Coleman. 1999. Rat sn-glycerol-3-phosphate acyltransferase: mo-lecular cloning and characterization of the cDNA and expressed protein.Biochim. Biophys. Acta 1439:415–423.
14. Green, P. R., A. H. Merrill, Jr., and R. M. Bell. 1981. Membrane phospho-lipid synthesis in Escherichia coli: purification, reconstitution, and character-ization of sn-glycerol-3-phosphate acyltransferase. J. Biol. Chem. 256:11151–11159.
15. Hanke, C., F. P. Wolter, J. Coleman, G. Peterek, and M. Frentzen. 1995. Aplant acyltransferase involved in triacylglycerol biosynthesis complements anEscherichia coli sn-1-acylglycerol-3-phosphate acyltransferase mutant. Eur.J. Biochem. 232:806–810.
16. Harry, E. J., K. Pogliano, and R. Losick. 1995. Use of immunofluorescenceto visualize cell-specific gene expression during sporulation in Bacillus sub-tilis. J. Bacteriol. 177:3386–3393.
17. Heath, R. J., S. Jackowski, and C. O. Rock. 1994. Guanosine tetraphosphateinhibition of fatty acid and phospholipid synthesis in Escherichia coli isrelieved by overexpression of glycerol-3-phosphate acyltransferase (plsB).J. Biol. Chem. 269:26584–26590.
18. Heath, R. J., and C. O. Rock. 1996. Regulation of fatty acid elongation andinitiation by acyl-acyl carrier protein in Escherichia coli. J. Biol. Chem.271:1833–1836.
19. Heath, R. J., and C. O. Rock. 1999. A missense mutation accounts for thedefect in the glycerol-3-phosphate acyltransferase expressed in the plsB26mutant. J. Bacteriol. 181:1944–1946.
20. Heath, R. J., and C. O. Rock. 1996. Inhibition of �-ketoacyl-acyl carrierprotein synthase III (FabH) by acyl-acyl carrier protein in Escherichia coli.J. Biol. Chem. 271:10996–11000.
21. Jiang, P. and J. E. Cronan, Jr. 1994. Inhibition of fatty acid synthesis inEscherichia coli in the absence of phospholipid synthesis and release ofinhibition by thioesterase action. J. Bacteriol. 176:2814–2821.
22. Kobayashi, K., S. D. Ehrlich, A. Albertini, G. Amati, K. K. Andersen, M.Arnaud, K. Asai, S. Ashikaga, S. Aymerich, P. Bessieres, F. Boland, S. C.Brignell, S. Bron, K. Bunai, J. Chapuis, L. C. Christiansen, A. Danchin, M.Debarbouille, E. Dervyn, E. Deuerling, K. Devine, S. K. Devine, O. Dreesen,J. Errington, S. Fillinger, S. J. Foster, Y. Fujita, A. Galizzi, R. Gardan, C.Eschevins, T. Fukushima, K. Haga, C. R. Harwood, M. Hecker, D. Hosoya,M. F. Hullo, H. Kakeshita, D. Karamata, Y. Kasahara, F. Kawamura, K.Koga, P. Koski, R. Kuwana, D. Imamura, M. Ishimaru, S. Ishikawa, I. Ishio,D. Le Coq, A. Masson, C. Mauel, R. Meima, R. P. Mellado, A. Moir, S.
VOL. 189, 2007 PHOSPHATIDIC ACID FORMATION IN B. SUBTILIS 5823
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from

Moriya, E. Nagakawa, H. Nanamiya, S. Nakai, P. Nygaard, M. Ogura, T.Ohanan, M. O’Reilly, M. O’Rourke, Z. Pragai, H. M. Pooley, G. Rapoport,J. P. Rawlins, L. A. Rivas, C. Rivolta, A. Sadaie, Y. Sadaie, M. Sarvas, T.Sato, H. H. Saxild, E. Scanlan, W. Schumann, J. F. M. L. Seegers, J.Sekiguchi, A. Sekowska, S. J. Seror, M. Simon, P. Stragier, R. Studer, H.Takamatsu, T. Tanaka, M. Takeuchi, H. B. Thomaides, V. Vagner, J. M. vanDijl, K. Watabe, A. Wipat, H. Yamamoto, M. Yamamoto, Y. Yamamoto, K.Yamane, K. Yata, K. Yoshida, H. Yoshikawa, U. Zuber, and N. Ogasawara.2003. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. USA 100:4678–4683.
23. Larson, T. J., D. N. Ludtke, and R. M. Bell. 1984. sn-Glycerol-3-phosphateauxotrophy of plsB strains of Escherichia coli: evidence that a second muta-tion, plsX, is required. J. Bacteriol. 160:711–717.
24. Lehninger, A. L. 1945. Synthesis and properties of the acyl phosphates ofsome higher fatty acids. J. Biol. Chem. 162:333–342.
25. Lightner, V. A., T. J. Larson, P. Tailleur, G. D. Kantor, C. R. H. Raetz, R. M.Bell, and P. Modrich. 1980. Membrane phospholipid synthesis in Escherichiacoli: cloning of a structural gene (plsB) of the sn-glycerol-3-phosphate acyl-transferase. J. Biol. Chem. 255:9413–9420.
26. Lu, Y.-J., F. Zhang, K. D. Grimes, R. E. Lee, and C. O. Rock. 2007. Topologyand active site of PlsY: the bacterial acylphosphate:glycerol-3-phosphateacyltransferase. J. Biol. Chem. 282:11339–11346.
27. Lu, Y.-J., Y.-M. Zhang, K. D. Grimes, J. Qi, R. E. Lee, and C. O. Rock. 2006.Acyl-phosphates initiate membrane phospholipid synthesis in gram-positivepathogens. Mol. Cell 23:765–772.
28. Lueking, D. R., and H. Goldfine. 1975. sn-Glycerol-3-phosphate acyltrans-ferase activity in particulate preparations from anaerobic, light-grown cells of
Rhodopseudomonas spheroides. Involvement of acyl thiolester derivatives ofacyl carrier protein in the synthesis of complex lipids. J. Biol. Chem. 250:8530–8535.
29. Matsuoka, H., K. Hirooka, and Y. Fujita. 2007. Organization and function ofthe YsiA regulon of Bacillus subtilis involved in fatty acid degradation.J. Biol. Chem. 282:5180–5194.
30. Nishibori, A., J. Kusaka, H. Hara, M. Umeda, and K. Matsumoto. 2005.Phosphatidylethanolamine domains and localization of phospholipid syn-thases in Bacillus subtilis membranes. J. Bacteriol. 187:2163–2174.
31. Ray, T. K., J. E. Cronan, Jr., R. D. Mavis, and P. R. Vagelos. 1970. Thespecific acylation of glycerol 3-phosphate to monoacylglycerol 3-phosphatein Escherichia coli. Evidence for a single enzyme conferring this specificity.J. Biol. Chem. 245:6442–6448.
32. Rock, C. O. and J. E. Cronan, Jr. 1981. Acyl-acyl carrier protein synthetasefrom Escherichia coli. Methods Enzymol. 71:163–168.
33. Rock, C. O., and J. L. Garwin. 1979. Preparative enzymatic synthesis andhydrophobic chromatography of acyl-acyl carrier protein. J. Biol. Chem.254:7123–7128.
34. Schujman, G. E., K.-H. Choi, S. Altabe, C. O. Rock, and D. de Mendoza.2001. Response of Bacillus subtilis to cerulenin and the acquisition of resis-tance. J. Bacteriol. 183:3032–3040.
35. Vagner, V., E. Dervyn, and S. D. Ehrlich. 1998. A vector for systematic geneinactivation in Bacillus subtilis. Microbiology 144:3097–3104.
36. Voelker, T. A., and H. M. Davies. 1994. Alteration of the specificity andregulation of fatty acid synthesis of Escherichia coli by expression of a plantmedium-chain acyl-acyl carrier protein thioesterase. J. Bacteriol. 176:7320–7327.
5824 PAOLETTI ET AL. J. BACTERIOL.
on March 1, 2014 by guest
http://jb.asm.org/
Dow
nloaded from
Related Documents











