Contribution of Secretory Antibodies to Intestinal Mucosal Immunity against Helicobacter pylori Rebecca J. Gorrell, a,b * Odilia L. C. Wijburg, a John S. Pedersen, c Anna K. Walduck, a * Terry Kwok, d Richard A. Strugnell, a Roy M. Robins-Browne a,b Department of Microbiology and Immunology, The University of Melbourne, Victoria, Australia a ; Murdoch Childrens Research Institute, Royal Childrens Hospital, Parkville, Victoria, Australia b ; TissuPath Pty. Ltd., Hawthorn, Victoria, Australia c ; Department of Biochemistry and Molecular Biology and Department of Microbiology, Monash University, Victoria, Australia d The natural immune response to Helicobacter pylori neither clears infection nor prevents reinfection. However, the ability of secretory antibodies to influence the course of H. pylori infection has not been determined. We compared the natural progres- sion of H. pylori infection in wild-type C57BL/6 mice with that in mice lacking the polymeric immunoglobulin receptor (pIgR) that is essential for the secretion of polymeric antibody across mucosal surfaces. H. pylori SS1-infected wild-type and pIgR knockout (KO) mice were sampled longitudinally for gastrointestinal bacterial load, antibody response, and histological changes. The gastric bacterial loads of wild-type and pIgR KO mice remained constant and comparable at up to 3 months postin- fection (mpi) despite SS1-reactive secretory IgA in the intestinal contents of wild-type mice at that time. Conversely, abundant duodenal colonization of pIgR KO animals contrasted with the near-total eradication of H. pylori from the intestine of wild-type animals by 3 mpi. H. pylori was cultured only from the duodenum of those animals in which colonization in the distal gastric antrum was of sufficient density for immunohistological detection. By 6 mpi, the gastric load of H. pylori in wild-type mice was significantly lower than in pIgR KO animals. While there was no corresponding difference between the two mouse strains in gas- tric pathology results at 6 mpi, reductions in gastric bacterial load correlated with increased gastric inflammation together with an intestinal secretory antibody response in wild-type mice. Together, these results suggest that naturally produced secretory antibodies can modulate the progress of H. pylori infection, particularly in the duodenum. C arriers of the human gastric bacterial pathogen Helicobacter pylori develop a substantial immune response manifested by cellular infiltration of the gastric mucosa and the development of an antibody response. While this natural immune response is not protective against gastric infection (1) and can even facilitate H. pylori pathogenesis, vaccine-induced immunity may reduce gas- tric loads of H. pylori in animal models of infection when used either prophylactically (2) or therapeutically (3). Investigations of the mechanism(s) of vaccine-induced immunity suggest that cel- lular immunity, perhaps in concert with innate factors, is respon- sible for the observed protection (4). Studies of the contribution of antibodies to vaccine-induced immunity suggest that H. pylori-specific IgA may suppress the development of vaccine-induced protective inflammatory re- sponses (5). Several studies have reported similar or enhanced vaccine-mediated protection in IgA- and B-cell-deficient animals, suggesting that antibodies are dispensable for protective immu- nity against H. pylori (6–9). However, the compensatory contri- bution of enhanced IgM production and significant Th1 skewing in IgA knockout (KO) (7) and MT (10, 11) mouse strains, re- spectively, remains to be examined as a factor contributing to the maintenance of vaccine efficacy in the absence of IgA in these animals. Nevertheless, the lack of any correlation between H. py- lori-specific antibody-mediated immune response and vaccine- induced immunity is in agreement with the apparent inefficacy of antibody-mediated immunity following natural infection. As H. pylori resides within or beneath the mucous-gel layer that protects the gastric mucosa from acid and digestive enzymes, the failure of antibodies to protect against H. pylori is not fully explained by antibody degradation in the gastric lumen. Antibodies destined for mucosal translocation are polymeric structures composed of multiple covalently linked immunoglob- ulin molecules associated with a joining (J)-chain protein (12). In mice, J-chain expression is regulated by interleukin-2 (IL-2)-in- duced downregulation of the negative regulatory element, BSAP (B-cell-specific activator protein) (13). The J-chain facilitates at- tachment of polymeric immunoglobulin to the polymeric immu- noglobulin receptor (pIgR), which is expressed at the basolateral surface of the mucosal epithelium. Epithelial cell expression of pIgR is constitutive in the intestine, although this expression can be upregulated by gamma interferon (IFN-) binding to its recep- tor on the epithelial cell surface (14). Following attachment to pIgR, the polymeric antibody/receptor complex is internalized by endocytosis, translocated through the epithelial cell, and released from the apical mucosal surface following proteolytic cleavage of pIgR, leaving a remnant known as the secretory component at- tached to the secreted antibody. Received 21 January 2013 Returned for modification 20 March 2013 Accepted 21 July 2013 Published ahead of print 5 August 2013 Editor: S. R. Blanke Address correspondence to Rebecca J. Gorrell, [email protected]. * Present address: Rebecca J. Gorrell, Department of Biochemistry and Molecular Biology, Monash University, Clayton, VIC, Australia; Anna K. Walduck, School of Applied Sciences, RMIT University, Bundoora, VIC, Australia. Supplemental material for this article may be found at http://dx.doi.org/10.1128 /IAI.01424-12. Copyright © 2013, American Society for Microbiology. All Rights Reserved. doi:10.1128/IAI.01424-12 3880 iai.asm.org Infection and Immunity p. 3880 –3893 October 2013 Volume 81 Number 10

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Contribution of Secretory Antibodies to Intestinal Mucosal Immunityagainst Helicobacter pylori
Rebecca J. Gorrell,a,b* Odilia L. C. Wijburg,a John S. Pedersen,c Anna K. Walduck,a* Terry Kwok,d Richard A. Strugnell,a
Roy M. Robins-Brownea,b
Department of Microbiology and Immunology, The University of Melbourne, Victoria, Australiaa; Murdoch Childrens Research Institute, Royal Childrens Hospital, Parkville,Victoria, Australiab; TissuPath Pty. Ltd., Hawthorn, Victoria, Australiac; Department of Biochemistry and Molecular Biology and Department of Microbiology, MonashUniversity, Victoria, Australiad
The natural immune response to Helicobacter pylori neither clears infection nor prevents reinfection. However, the ability ofsecretory antibodies to influence the course of H. pylori infection has not been determined. We compared the natural progres-sion of H. pylori infection in wild-type C57BL/6 mice with that in mice lacking the polymeric immunoglobulin receptor (pIgR)that is essential for the secretion of polymeric antibody across mucosal surfaces. H. pylori SS1-infected wild-type and pIgRknockout (KO) mice were sampled longitudinally for gastrointestinal bacterial load, antibody response, and histologicalchanges. The gastric bacterial loads of wild-type and pIgR KO mice remained constant and comparable at up to 3 months postin-fection (mpi) despite SS1-reactive secretory IgA in the intestinal contents of wild-type mice at that time. Conversely, abundantduodenal colonization of pIgR KO animals contrasted with the near-total eradication of H. pylori from the intestine of wild-typeanimals by 3 mpi. H. pylori was cultured only from the duodenum of those animals in which colonization in the distal gastricantrum was of sufficient density for immunohistological detection. By 6 mpi, the gastric load of H. pylori in wild-type mice wassignificantly lower than in pIgR KO animals. While there was no corresponding difference between the two mouse strains in gas-tric pathology results at 6 mpi, reductions in gastric bacterial load correlated with increased gastric inflammation together withan intestinal secretory antibody response in wild-type mice. Together, these results suggest that naturally produced secretoryantibodies can modulate the progress of H. pylori infection, particularly in the duodenum.
Carriers of the human gastric bacterial pathogen Helicobacterpylori develop a substantial immune response manifested by
cellular infiltration of the gastric mucosa and the development ofan antibody response. While this natural immune response is notprotective against gastric infection (1) and can even facilitate H.pylori pathogenesis, vaccine-induced immunity may reduce gas-tric loads of H. pylori in animal models of infection when usedeither prophylactically (2) or therapeutically (3). Investigations ofthe mechanism(s) of vaccine-induced immunity suggest that cel-lular immunity, perhaps in concert with innate factors, is respon-sible for the observed protection (4).
Studies of the contribution of antibodies to vaccine-inducedimmunity suggest that H. pylori-specific IgA may suppress thedevelopment of vaccine-induced protective inflammatory re-sponses (5). Several studies have reported similar or enhancedvaccine-mediated protection in IgA- and B-cell-deficient animals,suggesting that antibodies are dispensable for protective immu-nity against H. pylori (6–9). However, the compensatory contri-bution of enhanced IgM production and significant Th1 skewingin IgA knockout (KO) (7) and �MT (10, 11) mouse strains, re-spectively, remains to be examined as a factor contributing to themaintenance of vaccine efficacy in the absence of IgA in theseanimals. Nevertheless, the lack of any correlation between H. py-lori-specific antibody-mediated immune response and vaccine-induced immunity is in agreement with the apparent inefficacy ofantibody-mediated immunity following natural infection. As H.pylori resides within or beneath the mucous-gel layer that protectsthe gastric mucosa from acid and digestive enzymes, the failure ofantibodies to protect against H. pylori is not fully explained byantibody degradation in the gastric lumen.
Antibodies destined for mucosal translocation are polymeric
structures composed of multiple covalently linked immunoglob-ulin molecules associated with a joining (J)-chain protein (12). Inmice, J-chain expression is regulated by interleukin-2 (IL-2)-in-duced downregulation of the negative regulatory element, BSAP(B-cell-specific activator protein) (13). The J-chain facilitates at-tachment of polymeric immunoglobulin to the polymeric immu-noglobulin receptor (pIgR), which is expressed at the basolateralsurface of the mucosal epithelium. Epithelial cell expression ofpIgR is constitutive in the intestine, although this expression canbe upregulated by gamma interferon (IFN-�) binding to its recep-tor on the epithelial cell surface (14). Following attachment topIgR, the polymeric antibody/receptor complex is internalized byendocytosis, translocated through the epithelial cell, and releasedfrom the apical mucosal surface following proteolytic cleavage ofpIgR, leaving a remnant known as the secretory component at-tached to the secreted antibody.
Received 21 January 2013 Returned for modification 20 March 2013Accepted 21 July 2013
Published ahead of print 5 August 2013
Editor: S. R. Blanke
Address correspondence to Rebecca J. Gorrell, [email protected].
* Present address: Rebecca J. Gorrell, Department of Biochemistry and MolecularBiology, Monash University, Clayton, VIC, Australia; Anna K. Walduck, School ofApplied Sciences, RMIT University, Bundoora, VIC, Australia.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01424-12.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/IAI.01424-12
3880 iai.asm.org Infection and Immunity p. 3880–3893 October 2013 Volume 81 Number 10
In contrast to the lungs, vagina, and most of the gastrointesti-nal tract, the healthy mammalian stomach produces no to littlepIgR (15, 16). In fact, pIgR expression in the gastric mucosa is amarker of intestinal metaplasia (17, 18) and IgA in gastric mucusfrom healthy humans is predominantly nonsecretory (19, 20).Studies in H. pylori-infected humans have shown that baselinepIgR expression by the gastric epithelium can be upregulated inresponse to gastric inflammation (15, 21–23) due to increasedlocal IFN-� production (24). This is accompanied by downregu-lation of J-chain synthesis in local plasma cells (25), probably as aconsequence of reduced IL-2 production in H. pylori-infected gas-tric tissue (24) due to the effects of the VacA cytotoxin on T cells(26). However, despite significantly increased pIgR expressionand IgA plasma cell infiltration in response to H. pylori infection(21, 27), there is no concomitant increase in IgA secretion into thestomach, and it is nonsecretory monomeric IgA which predomi-nates in the stomach of H. pylori-infected individuals (25, 28).This observation is consistent with the leakage of serum antibod-ies across the infected gastric mucosa. Together, these findingssuggest that, although inflammation upregulates pIgR expression,cytokines released in gastric tissue as a consequence of H. pylori-induced gastritis dysregulate the expression of genes encoding ef-fectors required for the efficient translocation of secretory anti-bodies. Antibodies in the stomach may also be sourced fromextragastric sites, namely, from the mouth via saliva and the duo-denum via reflux. Salivary IgA is swallowed continually and maycontribute to vaccine-mediated protection against H. pylori infec-tion (29). In humans, peak IgA levels in the gastric lumen coincidewith gastroduodenal reflux resulting from retrograde peristalsisarising from the interdigestive migrating motor complex (MMC)(30).
Despite substantial evidence that insufficient quantities of an-tibody are translocated across the gastric mucosa to mediate im-munity and that the bulk of gastric antibody is intermittently anddistantly sourced, these factors have not been accounted for in anyprevious studies investigating the contribution of antibodies to H.pylori immunity. In particular, the contribution of intestinal an-tibodies to H. pylori immunity has been poorly examined. In this
study, we compared the progression of gastric H. pylori infectionin naive wild-type C57BL/6 mice with that in pIgR KO C57BL/6mice which lack the ability to secrete antibodies across mucosalsurfaces (31). We also examined duodenal colonization, gastricinflammation, and humoral immune responses in both groups ofmice following infection with H. pylori.
METHODS AND MATERIALSMice and bacterial infections. C57BL/6 wild-type and pIgR knockoutmice (31) were bred and housed in the Department of Microbiology andImmunology Animal Facility at the University of Melbourne as describedpreviously (32). All animal experiments were compliant with the Austra-lian National Health and Medical Research Council guidelines and ap-proved by the University of Melbourne Animal Ethics Committee. A totalof 82 wild-type and 82 pIgR KO female mice were used in this study. Thenumber and age of the animals used were dictated by the availability of thepIgR KO mice from two consecutive breeding cycles. The animals derivedfrom each breeding were handled as independent cohorts in two experi-ments staggered approximately 3 weeks apart for inoculation and subse-quent sample collection (Table 1). H. pylori strain SS1 was used for allexperiments and cultured as described previously (32). Mice were inocu-lated at 6 to 8 weeks of age with 0.1 ml of a suspension of H. pylori SS1 inbrain heart infusion broth (BHI) (108 CFU/ml), or BHI alone, by gavageusing a stainless-steel mouse-feeding needle (Cole-Parmer) (20 gauge; 1.5in. long; curved). Two SS1-inoculated wild-type mice required euthanasiafor reasons unrelated to the experiment and were excluded from the anal-ysis. Mice were maintained on autoclaved potable water for the durationof the experiment and killed by CO2/O2 asphyxiation at 11 days (d), 1month (m; 4 weeks), 3 months (12 weeks), 6 months (30 weeks), or 12months (52 weeks) postinoculation (mpi) (Table 1). Animals were ini-tially caged in groups of 5, and no more than 2 animals were sampled fromany single cage at each time point to eliminate cage effects.
Bacterial culture for assessment of gastric and intestinal bacterialload. During necropsy, the stomach of each animal was dissected alongthe border of the nonglandular region and the glandular stomach wasopened longitudinally along the greater curvature. After partial removalof the gastric contents, the stomach was bisected along the lesser curva-ture. One half was used for culture, and the other half was used for histol-ogy. For recovery of intestinal tissue, approximately 8 cm of proximalintestine was excised from each animal and further dissected to recoverthe first 5 cm. The 5-cm length of intestine was lavaged with 1 ml phos-
TABLE 1 Experimental design with respect to the numbers of animals in each experimental group and sampled at each time pointa
Group
No. of animals sampled at each time point postinoculation Total
Day 11 Mo 1 Mo 3 Mo 6 Mo 12b
WT pIgRWT pIgR WT pIgR WT pIgR WT pIgR WT pIgR
SS1 inoculatedCohort I 4 5 5 5 5 5 5 5c 5 4 24 24Cohort II 5 5 5 5 5 5 5d 5d 4 5 24 25
Total 9 10 10 10 10 10 10 10 9 9 48e 49
BHI inoculatedCohort I 4 5 5 5 5 5 5 5 5 5 24 25Cohort II 0 0 2 2 2 2 2 2 2 2 8 8
Total 4 5 7 7 7 7 7 7 7 7 32 33a See also Fig. S1 in the supplemental material. WT, wild type.b Gastric and intestinal viable bacterial load data unavailable for animals in cohort I sampled at 12 mpi.c Intestinal contents inadvertently not collected from 2 animals in this group.d Intestinal contents inadvertently not collected from 1 animal in each of these groups.e Two additional SS1-inoculated C57BL/6 WT mice required euthanasia due to reasons unrelated to the experiment (malocclusion and eye tumor).
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3881

phate-buffered saline (PBS) to recover the intestinal contents, and therecovered lavage fluid was supplemented immediately thereafter with 40�l 25� Complete protease inhibitor Cocktail (Roche). The intestine wasthen bisected longitudinally, with one half used for culture and the otherused for histology. All tissue recovered for culture was washed twice withsaline solution and blotted before weighing. It was then homogenized in 2ml sterile saline solution and used for quantitative culture on lysed horseblood agar plates supplemented with Dent selective supplement (Oxoid)and 20 �g/ml bacitracin (Sigma-Aldrich) as described previously (32). H.pylori was identified by colony morphology; randomly selected coloniesfrom intestinal tissues were confirmed by measuring urease activity andby colony PCR for species determination as described previously (33).
Preparation and histological analysis of gastric and intestinal tis-sues. Gastric tissue was rinsed in 10% neutral buffered formalin and al-lowed to flatten against the tube wall for several minutes before beingreturned to the formalin. Unrinsed intestinal tissue was gently scrolledaround a cylindrical wooden stick coated with silicone (Sigmacote, Sig-ma-Aldrich) and was pinned in place before immersion in formalin. For-malin-fixed tissues were transferred to 70% ethanol within 24 h of fixa-tion. Following ethanol equilibration, gastric tissues were further bisectedlongitudinally and embedded internal-cut-side down to provide 2 sam-ples per section for analysis. An additional longitudinal strip of tissue (�2mm wide) was recovered from the internal cut side of samples from ani-mals killed at 12 mpi for determination of bacterial load by quantitativePCR (qPCR). Sections (3 �m thick) of gastric and intestinal tissue werestained with hematoxylin and eosin and then assessed by a clinical pathol-ogist who was blinded to details of the study design.
For gastric pathology, the 4-grade scoring scale (0 to 3) of the updatedSydney system (34) was used. Separate scores were assigned to the distalantrum, proximal antrum (corpus-antrum transitional zone), and corpusregions of the stomach for each of the following key pathological features:neutrophil infiltration, mononuclear cell infiltration, atrophy, and intes-tinal metaplasia. The scoring system was modified such that tissues con-taining grade 3 inflammation or atrophy of extensive breadth across aspecific region were assigned a grade of 4. Any additional observations,including lymphoid follicle development and lymphocytic gastritis, werealso recorded. Neutrophil and mononuclear cell infiltration scores werecombined to produce summary inflammation scores for the proximalantrum and corpus (0 to 8). For assessment of intestinal pathology, sec-tions were examined for any architectural changes, lymphoid follicle de-velopment, gastric metaplasia, and cellular infiltration.
qPCR to assess gastric bacterial load. Because quantitative culturedata were not available for the 12-month time point of cohort I due to theinadvertent inclusion of a nonpermissive antibiotic supplement in theculture plates, the bacterial loads of all mice killed at this time point wereassessed by qPCR using previously reported primers specific for H. pylorirRNA (35). Genomic DNA was purified from formalin-fixed (cohorts Iand II) or fresh (cohort II only) gastric tissue, or from broth-grown SS1,using a blood and tissue DNeasy kit (Qiagen) according to the manufac-turer’s protocol for purification of DNA from mammalian tissue. ForDNA extracted from fresh tissue, 300 �l of the same homogenate used forculture was centrifuged (10 min, 20,000 � g, 4°C) and the pelleted tissuewas resuspended in Qiagen ATL buffer and proteinase K solution suppliedin the kit. To extract DNA from formalin-fixed samples, the tissue wassliced finely before incubation in ATL buffer and proteinase K. After over-night incubation, an additional 0.1� volume of ATL buffer and protei-nase K was added to each sample and further incubated with frequentvortexing for 3 h before resuming the manufacturer’s protocol for purifi-cation of genomic DNA from mammalian tissue (Qiagen). To generatetemplate for the qPCR standard curve, genomic DNA was purified fromH. pylori SS1 cells pelleted from a broth culture. The bacterial density ofthis culture, as determined by quantitative culture and by counting in ahemocytometer chamber, was used to confirm the estimation of 1.1 � 106
H. pylori genomes/ng DNA as predicted from the molecular weight of theH. pylori genome.
For qPCR of DNA from fixed tissue, all DNA, including that used toconstruct the standard curve, was preincubated (98°C, 10 min) beforeaddition to the reaction mix to destroy any formalin-induced DNA cross-linking that might have interfered with the early cycles of amplification.qPCR was conducted using Brilliant II master mix (Stratagene) accordingto the manufacturer’s protocol. All reactions were performed in duplicatein a Mx3005P real-time thermal cycler (Stratagene). PCR cycling condi-tions were 10 min at 95°C followed by 40 cycles of 95°C for 30 s, 60°C for1 min (read fluorescence at end of hold), and 72°C for 30 s and 1 cycle of95°C for 1 min and 55°C for 30 s followed by dissociation curve measure-ments. Total gastric DNA was used in reactions at 10 and 1 ng, and thenumber of genomes was estimated against a standard curve ranging from2.0 to 0.00016 ng SS1 genomic DNA. qPCR conducted using DNA ex-tracted from formalin-fixed tissue recovered from animals in cohort IIshowed good agreement with matched fresh tissue qPCR and quantitativeculture results (Pearson r � 0.88 and 0.90 compared with fresh tissue andr � 0.84 and 86 compared with viable count, in duplicate experiments). Asthe DNA purification procedure yielded approximately 500 �g DNA/gram stomach tissue, the number of H. pylori genomes/500 �g DNA wasused as a proxy for CFU/gram stomach to present the qPCR data.
Immunohistochemistry and immunofluorescence. For immunohis-tochemical staining, all antibodies were diluted in 2% (vol/vol) fetal bo-vine serum (FBS)–PBS, and slides were washed between steps with PBS.Dewaxed tissue sections (3 �m thick) were blocked with serum-free pro-tein block (30 min; Dako) before incubation (1 h) with one of the follow-ing primary antibodies: goat anti-mouse pIgR antibody (R&D Systems) (2�g/ml); rabbit anti-H. pylori SS1�ureAB outer membrane vesicle (OMV)polyclonal sera (Sigma) (diluted 1 in 500) produced by hyperimmuniza-tion of a rabbit with 3 doses (125 �g protein per dose in Freund’s incom-plete adjuvant) of OMVs purified from SS1�ureAB spent culture media asdescribed previously (32); or horseradish peroxidase (HRP)-conjugatedgoat anti-mouse IgA (KPL Inc.) (diluted 1 in 200). Following washes(performed once for 5 min and once for 30 min), endogenous peroxidasewas blocked by immersing the slides in 0.05% (vol/vol) peroxide (30 min),and the slides were washed (three times for 5 min each time). Boundanti-pIgR or anti-H. pylori antibodies were revealed by incubation withHRP-conjugated rabbit anti-goat (Sigma-Aldrich) or HRP-conjugatedsheep anti-rabbit (Chemicon) antibodies, respectively, each diluted 1 in200. After washes (performed once for 5 min and once for 30 min), boundantibodies were visualized with DAB/H2O2 substrate prepared from DABtablets (Dako) followed by 3 washes with distilled water and counterstain-ing with hematoyxlin before dehydration and mounting in DePeX(Sigma).
For colocalization of H. pylori and pIgR by two-color immunofluores-cence, antibodies were diluted in 2% FBS–PBS supplemented with 0.02%(vol/vol) Tween 20 (PBS-T0.02; Sigma-Aldrich). All washes were per-formed three times for 5 min each time with PBS-T0.02, unless otherwisestated. Dewaxed sections were blocked with 20% normal donkey serum(Jackson ImmunoResearch Laboratories) before incubation for 1 h withgoat anti-pIgR and rabbit anti-SS1�ureAB OMV polyclonal sera dilutedas described above. Autofluorescence of the formalin-fixed tissue wasquenched by incubation in 0.1% (wt/vol) Sudan Black–70% (vol/vol)ethanol for 15 min followed by washes. Bound primary antibodies weredetected by incubation with Alexa Fluor 594-conjugated donkey anti-goatIg and Alexa Fluor 488-conjugated donkey anti-rabbit Ig (Invitrogen) (1h; both diluted 1 in 500). Following washes, sections were counterstainedwith DAPI (4=,6-diamidino-2-phenylindole) (Invitrogen) (5 min, 400 ng/ml) followed by PBS washes before mounting with fluorescent mountingmedium (Dako).
Quantification of H. pylori-specific IgG and IgA in serum and intes-tinal contents. Serum was assayed for H. pylori SS1-reactive IgG and IgAby endpoint titration enzyme immunoassay (EIA). Intestinal washingswere assayed for total IgA using a mouse IgA-specific EIA (Bethyl Labo-ratories) and for SS1-reactive IgG and IgA using a standardized EIA asdescribed previously (32). For the standardized EIA of intestinal contents,
Gorrell et al.
3882 iai.asm.org Infection and Immunity

EIA units were assigned from a standard curve using pooled serum frommice hyperimmunized with H. pylori SS1. This serum, which had an end-point titer of 1 in 64,000, was assigned an arbitrary value of 1,000 units andused to allocate units of reactivity to the unknown samples. Intestinalcontents were clarified by centrifugation and diluted independently 1 in 4and 1 in 20 for assays. Optical densities (ODs) falling within the linearrange of the curve were assigned units which subsequently were adjustedfor their dilution factor and averaged for each sample. For samples wherethe highest OD was below the lower linear limit of the curve, units wereassigned from the nonlinear region. For comparison of intestinal SS1-reactive antibody levels of individual C57BL/6 wild-type mice, SS1-reac-tive antibody levels were corrected for total IgA content. However, be-cause of the lack of secretory antibody in the intestinal contents of pIgRKO mice, uncorrected SS1-reactive antibody levels were used to comparesamples from wild-type and pIgR KO animals.
Immunoblot analysis of SS1-reactive IgA and IgG in serum and in-testinal contents. Total membrane and soluble fractions of H. pyloriSS1ure� were prepared as described previously (32). Lipopolysaccharide(LPS) was purified from cells of plate-grown SS1 using the method de-scribed by Hitchcock and Brown (36). Antigen preparations were sepa-rated using preparative-well Novex 4% to 12% Bis-Tris gels with Nu-PAGE MOPS (morpholinepropanesulfonic acid) SDS running buffer formembranes and soluble fractions and NuPAGE MES SDS running bufferfor LPS, and separated antigens were transferred onto polyvinylidene flu-oride (PVDF) membranes, all following standard NuPAGE protocols(Life Technologies). Strips of each membrane were blocked with 10%skim milk powder–PBS and incubated with 2.5 ml 5% skim milk powder–PBS-T0.01 containing serum (1 in 500) or intestinal contents (1 in 10)from individual animals at room temperature for 2 h and then overnightat 4°C. Bound mouse IgA and IgG were detected sequentially and visual-ized as described previously (32).
Statistical analysis. Statistical analysis of quantitative data was per-formed using Prism v6.01 (GraphPad Software Inc.) and included thefollowing tests as indicated in the text and/or figure legends: theD’Agostino & Pearson omnibus normality test was used to test individualgroups for normal distribution; graphs illustrating populations that werenormally distributed ions are shown as the mean � 95% confidence in-terval (CI) or standard deviation (SD); data in graphs with one or morenonparametric populations are shown as the median � interquartilerange (IQR); two groups of data were compared by using Student’s t test(parametric) or the Mann-Whitney rank sum test (nonparametric); com-parisons of three or more groups with one variable were done by one-wayanalysis of variance (ANOVA) with the Bonferroni posttest (parametric)or the Kruskal-Wallis test with Dunn’s multiple comparison posttest(nonparametric); comparisons of two or more groups with two variableswere done by two-way ANOVA with the Bonferroni posttest (paramet-ric); contingency tables were analyzed by using Fisher’s exact test; andcorrelations were calculated using Pearson’s correlation coefficient (para-metric) or Spearman’s correlation coefficient (nonparametric). Whereapplicable, tests were unpaired and two tailed. P values of 0.05 wereconsidered significant.
RESULTS
C57BL/6 wild-type and isogenic pIgR KO mice were inoculatedwith H. pylori SS1 or sterile BHI as outlined in Methods and Ma-terials, Table 1, and Fig. S1A in the supplemental material. Exper-imental cohorts I and II represent two independent experimentsthat started approximately 3 weeks apart, and samples from allsubsequent time points were collected and processed with a3-week interval between the two cohorts. Quantitative and quali-tative data sets arising from each cohort were combined for anal-ysis of bacterial load in the stomach and duodenum, serum andintestinal secretory antibody levels, gastrointestinal pathology,and pIgR expression (see Fig. S1B). The contribution of secretory
antibodies to immunity against H. pylori infection in these ani-mals was examined in two ways: (i) comparison of the course ofgastrointestinal infection in C57BL/6 wild-type and pIgR KO miceand (ii) comparison of the course of gastrointestinal H. pyloriinfection in chronically infected responder and nonresponderwild-type and pIgR KO mice (see Fig. S1C).
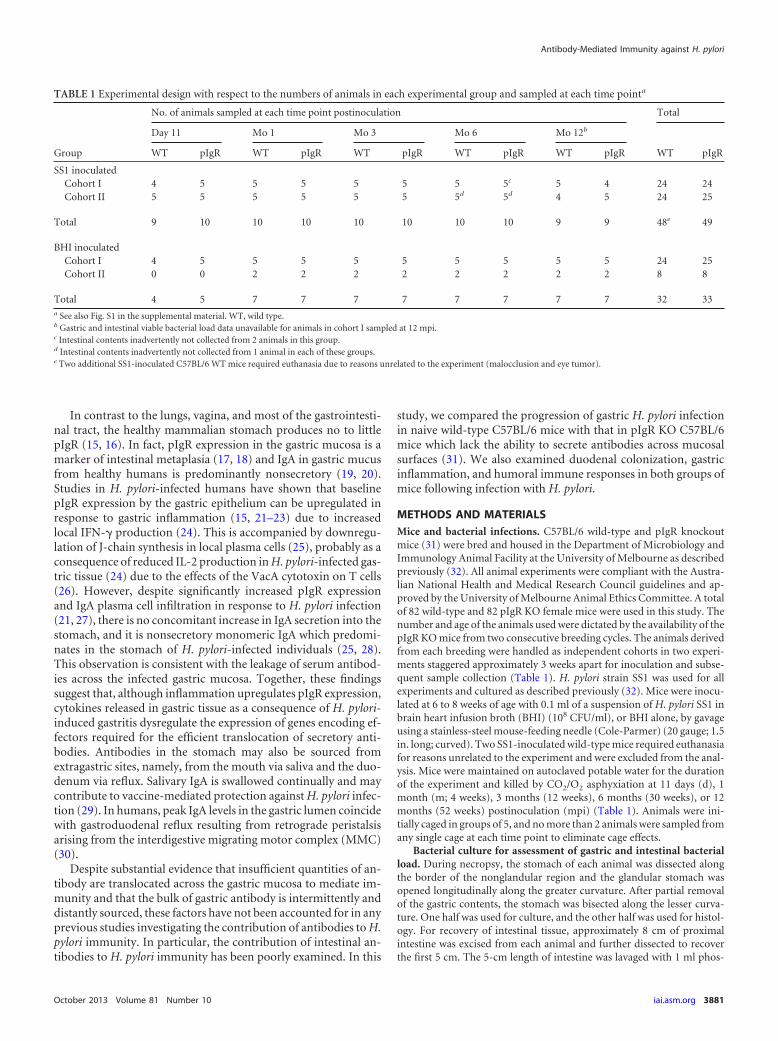
The course of natural H. pylori infection in C57BL/6 wild-type and pIgR KO mice. (i) Quantitative analysis of H. pyloriinfection in the stomach of wild-type and pIgR KO mice. Gastricbacterial loads of wild-type and pIgR KO C57BL/6 mice inocu-lated with H. pylori SS1 were determined 11 days (d) and 1, 3, 6,and 12 months (m) postinoculation (mpi) (Table 1). All SS1-challenged animals of both mouse strains were culture positive ateach of the 5 time points sampled. There was no difference in thegastric bacterial loads of wild-type and pIgR KO mice at up to 3mpi (Fig. 1A; P 0.05; Mann-Whitney test). However, the me-dian gastric bacterial load of wild-type mice determined by quan-titative culture at 6 mpi (Fig. 1A) and by qPCR at 12 mpi (Fig. 1B)was significantly reduced compared to that of matched pIgR KOmice (P � 0.036 and 0.032, respectively; Mann-Whitney test).Moreover, while the median viable gastric bacterial load in pIgRKO mice remained constant for 12 mpi, the median loads of wild-type mice were significantly lower at 6 and 12 mpi, but not at 3mpi, than in acutely infected wild-type mice (both P 0.05;Kruskal-Wallis test, Dunn’s posttest).
(ii) Quantitative culture of H. pylori from the duodenum ofwild-type and pIgR KO mice. We also determined the number ofcultivatable H. pylori bacteria that remained attached to the first 5cm of intestine following lavage of the intestinal lumen with PBSand two PBS washes of the longitudinally bisected tissue. Al-though the duodenal loads of H. pylori were comparable in the twomouse strains at up to 1 mpi (Fig. 1C), duodenal colonization inwild-type mice declined significantly over time (P � 0.0015;Kruskal-Wallis test). Conversely, intestinal colonization persistedin mice lacking secretory antibodies (P 0.05; Kruskal-Wallistest) such that pIgR KO mice had significantly greater intestinalcolonization at 3, 6, and 12 mpi than similarly infected wild-typemice (P � 0.002, 0.002, and 0.02, respectively; Mann-Whitneytest). In addition, the density of H. pylori colonization was higherin the intestine of more pIgR KO mice than wild-type mice (Fig.1D), with more than 1,000 CFU/gram intestine cultured from asignificantly greater number of pIgR KO mice (27/45 pIgR KOmice versus 4/43 wild-type mice; P 0.0001; Fisher’s exact test).Intestinal tissue from all animals gavaged with BHI was culturenegative.
(iii) Distribution of H. pylori in the stomach of infected wild-type and pIgR KO mice. We performed qualitative analysis of thedistribution of H. pylori throughout the stomach of infected ani-mals by using silver staining and immunohistochemical and im-munofluorescent detection of H. pylori in formalin-fixed tissue.The specificity of H. pylori antiserum was confirmed by usinggastric tissue sections from BHI-inoculated mice (see Fig. S2 in thesupplemental material). At 11 dpi, H. pylori was detected along theentire length of duplicate glandular stomach sections of all SS1-inoculated animals. Of the infected mice sampled at 3, 6, and 12mpi, both duodenal culture and gastric immunohistological H.pylori analysis data were available for 19 mice of each strain. Inthese animals, only mice colonized in the distal antrum by H.pylori at a sufficient density for immunohistological visualizationwere duodenum culture positive (see Fig. S3). H. pylori coloniza-
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3883
tion of the distal antrum was visualized in 5/19 wild-type mice and17/19 pIgR KO mice (P � 0.0002; Fisher’s exact test) at these latertime points, and 4 and 16 were culture positive for duodenal in-fection, respectively.
Similar analysis of the corpus region of mice sampled at 3, 6,and 12 mpi showed visible H. pylori colonization in the corpus of14/24 wild-type and 23/24 pIgR KO mice examined (P 0.0001;Fisher’s exact test), including 5 wild-type mice in which the bac-terial density in the distal antrum was below the level required forimmunodetection. All animals lacking visible H. pylori coloniza-tion of the corpus had gastric bacterial loads below 107 CFU/gram.
(iv) Cellular infiltration and atrophic changes in response toH. pylori infection of wild-type and pIgR KO mice. Significantlymore C57BL/6 mice (83%) than pIgR KO mice (48%) developedcorpus inflammation as a consequence of chronic H. pylori infec-
tion (Table 2). This difference was chiefly seen in animals sampledat 3 mpi (see Fig. S4 in the supplemental material) (9 of 10 wild-typeversus 2 of 10 pIgR KO mice with corpus inflammation; P � 0.006;Fisher’s exact test) rather than at the 6 and 12 mpi time points. Incontrast, similar numbers of pIgR KO and C57BL/6 mice showedcellular infiltration in the proximal antrum (Table 2). Apart fromunderlying lymphocytic infiltration of the duodenum in both in-fected and uninfected pIgR KO mice, inflammation was not observedin the distal antrum or duodenum of any animal examined.
Atrophic changes, scored primarily on clear cell metaplasia ofparietal cells, were also observed in the corpus of some animals at3, 6, and 12 mpi (Table 2), but there was no difference in theproportions of affected pIgR KO and wild-type mice.
The many animals with no pathology made the data nonpara-metric and skewed the extent of inflammation toward zero. This
FIG 1 Gastric and intestinal bacterial load in C57BL/6 wild-type and pIgR KO mice after inoculation with H. pylori SS1. (A) Load of H. pylori in the stomachsof mice killed 11 days (d) or 1, 3, 6, or 12 months (m) postinoculation (pi) as determined by spread plate count. Each point represents an individual animal fromindependent experimental cohorts I and II at at up to 6 mpi and cohort II at 12 mpi (Table 1). The median � IQR are shown. The dashed line shows the cutofffor full colonization (107 CFU/g stomach). *, P 0.05 (Mann-Whitney test). (B) Load of H. pylori in the stomachs of mice killed 12 mpi, as determined by H.pylori rRNA gene-specific qPCR of total DNA extracted from formalin-fixed tissue. Each symbol represents individual animals from independent experimentalcohorts I and II as detailed in Table 1 and denotes the mean of the results of two independent qPCR experiments, each performed in duplicate. Filled symbolsdenote gastric tissue samples from individual cohort II animals for which matched viable-count data are shown in Fig. 1A; open symbols denote individual cohortI animals for which viable-count data were unavailable. The median � IQR are shown. *, P 0.05 (Mann-Whitney test). (C) Load of H. pylori in the proximalsmall intestine of mice killed 11 dpi or 1, 3, 6, or 12 mpi, as determined by spread plate count. Each point represents an individual animal from independentexperimental cohorts I and II at up to 6 mpi and from cohort II at 12 mpi (Table 1). The median � IQR are shown. The dashed line indicates the detection limit(68 CFU/g intestine, equivalent to 10 CFU/5 cm intestine). Exact P values, determined by using the Mann-Whitney test (two-tailed), are shown. *, P 0.05; **,P 0.01 (Kruskal-Wallis test with Dunn’s multiple comparison posttest). (D) Frequency of significant intestinal H. pylori colonization in wild-type and pIgR KOmice. The data represent all animals shown in panel C. ****, P 0.0001 (compared to wild-type mice; Fisher’s exact test). Numbers of animals in individualgroups in all graphs are reported in parentheses.
Gorrell et al.
3884 iai.asm.org Infection and Immunity

made statistical analysis of differences in the extent of inflamma-tion or atrophy between the two mouse strains difficult. However,exclusion of unaffected animals from the populations normalizedthe data. Comparison of only affected mice showed no differencebetween C57BL/6 and pIgR KO mice in the degree of gastric in-flammation or atrophy in response to chronic H. pylori infection(Fig. 2). In addition, this comparison showed that although thenumbers of animals with inflammation of the proximal antrumwere similar for infected and uninfected mice of both types (Table2), the extent of inflammation was significantly greater for in-fected mice than uninfected mice (Fig. 2).
Taken together, these data suggest that the inflammatory re-sponse in the gastric corpus of wild-type mice from 3 mpi onwardsmay contribute to the reduction in gastric bacterial load of thesemice at 6 mpi compared to pIgR KO mice. However, comparablecellular infiltration in the proximal antrum, and the absence of H.pylori-related inflammation in the distal antrum and intestine ofall animals, suggests that it is unlikely that the inflammatory re-sponse is responsible for differences between immunocompetentand secretory antibody-deficient mice in the antral and duodenallocalization of H. pylori infection.
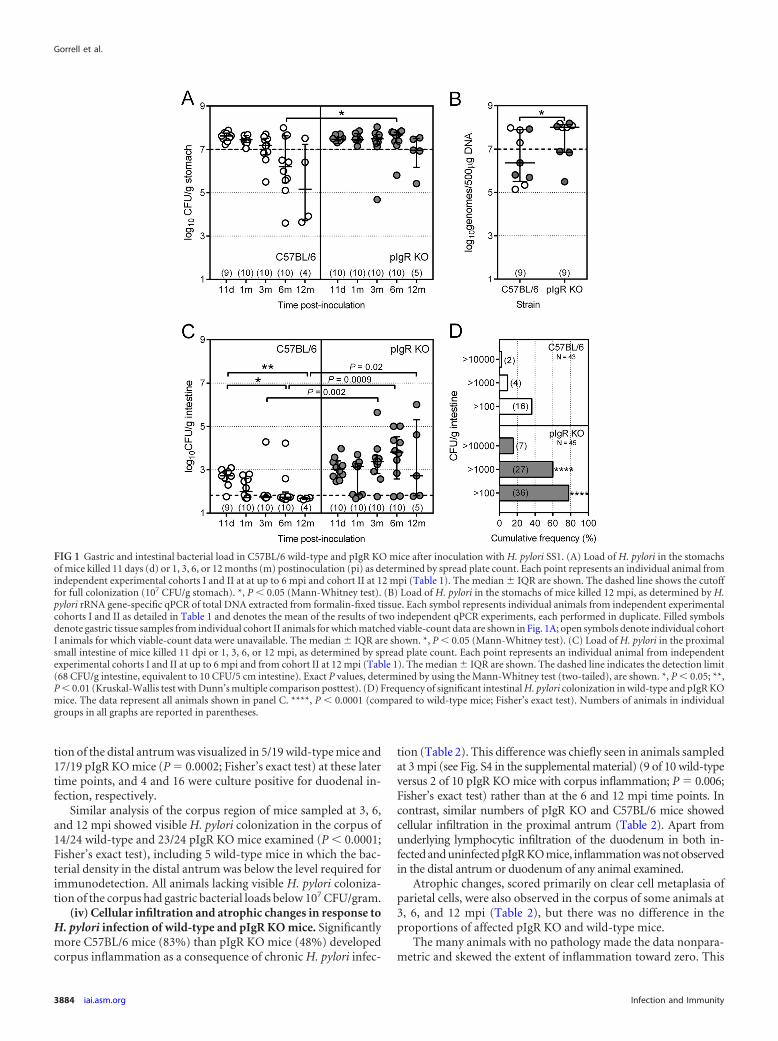
(v) Antibody response of wild-type and pIgR KO mice to in-fection with H. pylori. Serum and intestinal contents from allmice were assayed for H. pylori SS1-reactive IgG and IgA (Fig. 3).In contrast to IgG seroconversion from 1 mpi (Fig. 3A), SS1-reactive IgA was detected in serum samples from both infectedand uninfected pIgR KO mice from 11 dpi (Fig. 3B). This antibodymost likely included the broadly reactive innate secretory IgA thatprotects mucosal surfaces before the development of an adaptiveimmune response (37) and had accumulated in the blood of pIgRKO animals due to continual mucosal IgA production in the ab-sence of active antibody translocation. These preexisting SS1-re-active IgA antibodies continued to accumulate in the serum ofthese animals to the extent that uninfected pIgR KO mice hadSS1-reactive serum IgA titers similar to those of infected wild-typemice by 3 mpi. This increasing IgA baseline for the pIgR mice,together with the wide spread of IgG and IgA titers for mice ofboth strains, precluded accurate statistical analysis of the antibodytiters of the different mouse strains. Instead, we determined thenumbers of animals of each strain that seroconverted �4-fold and�10-fold in response to infection compared to the geometricmean titers of matched uninfected animals sampled at the same
time point after inoculation. IgG and IgA seroconversion rates ofpIgR KO mice were not different from those of matched wild-typemice at any individual time point (see Table S1 in the supplemen-tal material); however, there was a trend toward poorer IgA sero-conversion rates for pIgR mice. Accordingly, the number ofchronically infected (3, 6, and 12 mpi, combined) pIgR KO micewith �10-fold IgA seroconversion was significantly lower thanthat of the wild-type mice (10 of 29 pIgR KO mice compared with20 of 29 wild-type mice; P � 0.02; Fisher’s exact test).
SS1-reactive IgA and IgG were not detected in intestinal con-tents from any animals sampled at 11 dpi and 1 mpi, which wasconsistent with the lack of serum IgA seroconversion in all wild-type animals at those time points. From 3 mpi onwards, measur-able SS1-reactive IgA was detected in the intestine of the majorityof infected wild-type mice (21 of 27 animals) compared to fewinfected pIgR KO mice (7 of 26 animals) and uninfected animalsof both strains (1 of 42 animals) (Fig. 3C). We also observed astrong correlation between the amount of SS1-reactive IgA trans-located into the intestine of wild-type animals and their serumSS1-reactive IgA titer (Fig. 3D; Spearman r � 0.93; P 0.0001).Interestingly, SS1-reactive IgA was detected in the intestinal con-tents of a small number of infected pIgR KO mice at 3, 6, and 12mpi (Fig. 3C). All of these animals had extremely high SS1-reac-tive serum IgA titers (�64,000) compared to other pIgR KO mice,as well as atrophy in the gastric corpus and inflammation in boththe corpus and proximal antrum. These findings agree with theunderlying mucosal leakiness previously reported for pIgR KOmice (38).
Immunoblot analysis of paired serum samples and intestinalcontents from wild-type mice confirmed that IgG was the domi-nant isotype of SS1-reactive serum antibody in these animals andthat IgA was the dominant isotype in the intestinal contents (Fig.4). Some wild-type animals showed restricted profiles of IgA re-
TABLE 2 Histopathological changes in the stomach of C57BL/6 andpIgR KO mice 3 to 12 months after inoculation with H. pylori SS1 orBHIa
InoculumMousestrain
Totalno.
No. (%) of animals with:
Corpusinflammation
Proximalantruminflammation
Corpusatrophy
SS1 C57BL/6a 29 24 (83) 19 (65) 13 (45)pIgR KOb 29 14 (48)* 22 (76) 10 (34)
BHI C57BL/6c 21 2 (9)* 10 (48) 0 (0)*pIgR KOd 21 3 (14)* 12 (57) 2 (9)
a *, statistically significant difference by Fisher’s exact test for corpus inflammation for a
versus b data (P � 0.01), a versus c data (P 0.0001), and b versus d data (P � 0.02) andfor corpus atrophy for a versus c data (P � 0.0002). All other comparisons were notstatistically significant (P 0.05).
FIG 2 Histopathological grading of gastric mucosa for pathology-positive H.pylori-inoculated and control (BHI-inoculated) wild-type (WT) and pIgR KOC57BL/6 mice sampled at 3, 6, and 12 months after inoculation. Pathologyscores (mean � 95% CI) for inflammation of the gastric corpus (SS1-inocu-lated animals only) and gastric proximal antrum (SS1-inoculated and BHI-inoculated animals) represent combined scores for mononuclear and neutro-phil infiltration. Pathology scores for corpus atrophy represent parietal cellclear cell metaplasia. The numbers of animals in each group correspond tothose in Table 2 and are reported in parentheses. All statistical comparisons ofcorpus inflammation and corpus atrophy (Student’s t test) were not significant(P 0.05). Statistical analysis of proximal antrum inflammation by two-wayANOVA with Bonferroni’s posttest showed significant differences betweenmice inoculated with H. pylori and controls. *, P 0.05; **, P 0.01.
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3885
activity to SS1 lipopolysaccharide (see Fig. S5 in the supplementalmaterial; 3 mpi). We did, however, observe some differences inthe reactivity profiles of intestinal IgA compared with serum IgAfrom wild-type mice, suggesting that the intestinal antibody wasproduced locally (Fig. 4 and S5; C57BL/6, 6 mpi). In contrast,intestinal SS1-reactive IgG, where present, reflected the serum IgGprofile (Fig. 4; C57BL/6, 6 mpi). For pIgR KO mice, IgA was thedominant SS1-reactive serum isotype and mainly recognized SS1lipopolysaccharide (see Fig. S5). EIA-positive intestinal contentsfrom pIgR KO animals showed no reactivity to the tested antigensby immunoblot analysis, supporting the reportedly high level ofbroadly reactive, low-specificity IgA in these mice.
Previous studies have suggested that the IgA response to H.pylori infection may be anti-inflammatory (6). However, wefound there was a positive correlation between the serum IgA titerand the corpus inflammation score for both wild-type mice (see
FIG 3 H. pylori SS1-reactive IgG and IgA in serum and intestinal contentsfrom wild-type and pIgR KO C57BL/6 mice inoculated with H. pylori or BHI.(A and B) Serum H. pylori-reactive IgG (A) and IgA (B) titers were determinedby EIA using endpoint titration. Symbols represent individual animals fromcohort I for 11 dpi and 1 mpi and from cohorts I and II for 3, 6, and 12 mpi.Each sample was assayed once in duplicate. Error bars denote median � IQRof the results for each group. The numbers of animals shown above each group
in panel A also apply to panel B. (C) SS1-reactive IgA in intestinal contents.Animal numbers are shown beneath the data for each group. EIA units werederived by using standard curves prepared from pooled H. pylori-immunemouse serum. Symbols represent individual animals, and intestinal SS1-reac-tive antibody levels were averaged from duplicate independent EIAs for 3- and6-mpi animals and from a single EIA for 12-mpi animals (each assay includedindependently diluted duplicates). Error bars denote the median � IQR of theresults for each group. P values were determined by using the Mann-Whitneytest. Significant differences were as follows: *, P 0.05; **, P � 0.01; ***, P �0.001. (D) Correlation analysis of serum and intestinal SS1-reactive IgA. Thelinear regression (solid line) � 95% CI (dotted line) is shown.
FIG 4 Immunoblot analyses of H. pylori-reactive IgG and IgA in serum andintestinal contents from H. pylori SS1-infected C57BL/6 wild-type and pIgRKO mice. NuPAGE-separated H. pylori total membrane fraction antigens weretransferred to PVDF membranes and probed with serum (S) or intestinalcontents (IC) of animals killed at 3 and 6 mpi. Data from one representativeanimal of each strain are shown per time point. All 4 animals represented in thefigure had intestinal contents that were EIA positive for SS1-reactive IgA andhad gastric bacterial loads of 107 CFU/g stomach. Lane M contains Magic-Mark markers (Bio-Rad; estimated sizes are indicated in kDa).
Gorrell et al.
3886 iai.asm.org Infection and Immunity

Fig. S6A in the supplemental material) (Spearman r � 0.72; P 0.0001) and pIgR KO mice (see Fig. S6B) (Spearman r � 0.58; P 0.001). These findings suggest that the smaller number of pIgR KOmice with inflammation in the gastric corpus was not a conse-quence of significantly higher IgA titers in the gastric tissue ofthese animals.
Of additional interest was the small number of animals sam-pled at 3, 6, and 12 mpi that failed to seroconvert following infec-tion with H. pylori. In total, there were 6 mice (4 wild-type and 2pIgR KO mice) with no detectable SS1-reactive serum IgG and 5wild-type mice with no detectable SS1-reactive serum IgA (theseincluded the 4 IgG nonresponders). All of these animals showedno gastric inflammation, despite being fully colonized, as deter-mined by viable count and/or qPCR together with immunostain-ing of gastric tissue.
(vi) Impact of H. pylori infection on body weight of pIgR KOmice. Although body weight was not included as a monitoredparameter in the initial study design, it was evident by 3 monthspostchallenge that pIgR KO mice infected with H. pylori weresmaller than their wild-type counterparts. Consequently, the ani-mals sampled at 6 and 12 mpi were weighed immediately post-mortem. Analysis of the body weights showed that H. pylori infec-tion significantly slowed the growth of the pIgR KO mice but notthat of the wild-type C57BL/6 mice (Fig. 5). Although the differ-ence in body weights of infected and uninfected pIgR KO mice wasonly slight at 6 mpi, by 12 mpi, the mean body weight of H. pylori-infected pIgR KO mice was only 70% of that of mice of the BHI-challenged pIgR KO cohort (P 0.0001; two-way ANOVA).
The course of gastric H. pylori infection in responder andnonresponder mice. As stated above, we observed that somechronically infected wild-type mice failed to seroconvert follow-ing infection with H. pylori. These mice did not mount an inflam-matory response to infection. Some of these animals also had per-sistent duodenal colonization, and all had a gastric bacterial loadsimilar to that in wild-type mice sampled during acute infection(at up to 1 mpi). Interestingly, there were other chronically in-fected wild-type mice with bacterial loads similar to those of
acutely infected animals, despite technically seroconverting totheir infection. These fully colonized (nonresponder) animalscontrasted with a subset of animals (responders) that had reducedbacterial loads together with strong humoral and inflammatoryresponses. These differences provided a unique opportunity toinvestigate immune responses that may contribute to the declinein the gastric bacterial load of wild-type mice compared with pIgRKO mice. More importantly, the responder and nonresponderwild-type mice allowed us to examine a potentially protective im-mune response to natural infection in naive, immunocompetentanimals.
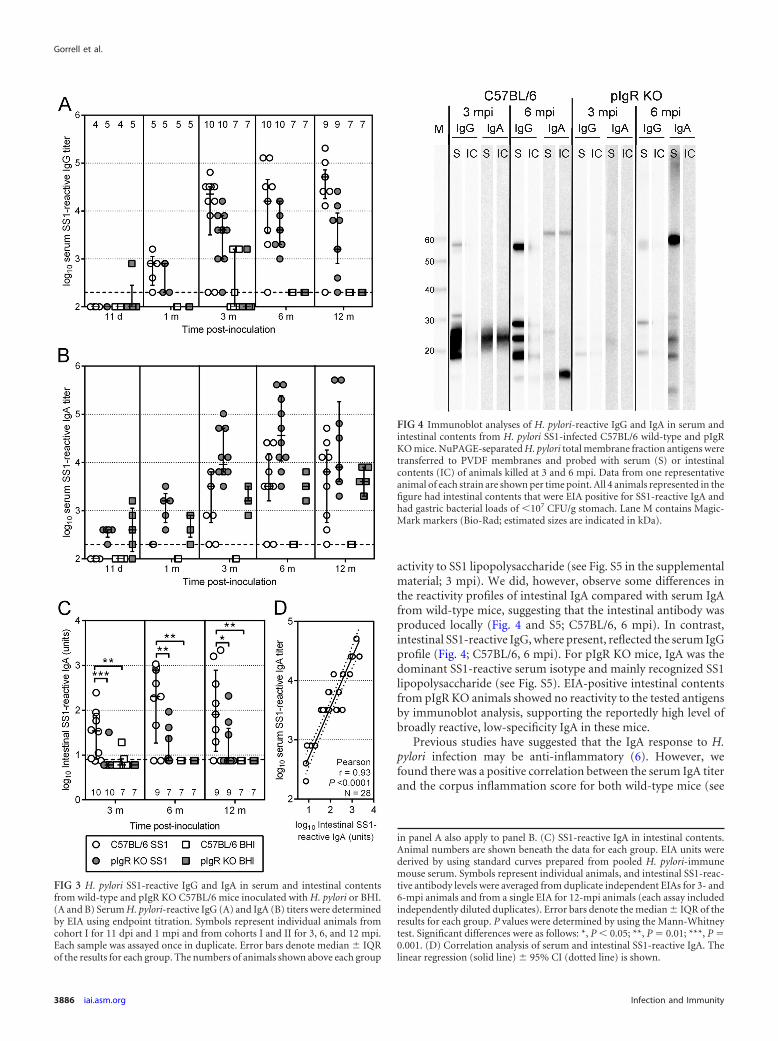
(i) Gastric bacterial load of responder and nonresponderC57BL/6 mice infected with H. pylori. All acutely infected ani-mals (i.e., sampled at 11 dpi and 1 mpi) had bacterial loads ofgreater than 107 CFU/g stomach, and the median bacterial load ofthe wild-type cohort during this period was 107.5 (IQR, 107.4 to107.7) CFU/g stomach. Therefore, we selected 107 CFU/g stomachas an arbitrary threshold for full colonization, and any animal witha gastric bacterial load less than this threshold was considered tohave reduced colonization. Examination of bacterial load datadetermined by using both qPCR (genomes/500 �g DNA) andculture (indicated in Fig. 1A and B) methodologies demon-strated that 107 genomes/500 �g DNA was also a suitablethreshold for categorization of animals whose bacterial loadhad been determined by qPCR. Consequently, bacterial loadsdetermined by qPCR were considered equally with those deter-mined by culture because the relationship between the bacte-rial loads of individual animals was highly reproducible be-tween the two methodologies.
In all, 27 wild-type mice were included in this comparison, ofwhich 13 were fully colonized. Two additional mice from cohort IIwere excluded: intestinal contents were inadvertently not col-lected from one animal; the other animal sampled at 3 mpi had areduced gastric bacterial load by viable count contradicted by verystrong H. pylori-specific labeling throughout the stomach by im-munohistochemistry and immunofluorescence. Comparison ofthe bacterial loads of mice with full and reduced colonizationshowed a greater than 10-fold difference in the median bacterialload between the cohorts (107.6 [IQR 107.4 to 108.0] versus 106.0
[IQR 105.5 to 106.5] CFU/g stomach, respectively; Fig. 6A). Of themice for which duodenal culture data were available, 4 of 10 and 0of 12 with full and reduced colonization, respectively, were culturepositive (P � 0.03; Fisher’s exact test).
(ii) Immune responses correlating with full and reduced gas-tric bacterial load. Animals with a reduced bacterial load hadgreater cellular infiltration into the mucosa of the gastric corpusthan fully colonized animals (Fig. 6B). The negative correlationbetween inflammation and bacterial load was highly significant(Pearson r � �0.77; P 0.0001; Fig. 6C). For comparisons ofintestinal antibody levels from individual wild-type animals, SS1-reactive IgA was standardized against total IgA in the intestinalcontents. Standardized SS1-reactive IgA was 8 times more abun-dant in the intestinal contents of animals with a reduced bacterialload than in fully colonized mice (Fig. 6D) and also showed asignificant negative correlation with bacterial load (Pearson r ��0.69; P 0.0001; Fig. 6E). However, animals with reduced gas-tric infection also had significantly higher levels of total intestinalIgA than fully colonized and uninfected mice (see Fig. S7 in thesupplemental material). This indicates an even greater discor-
FIG 5 Body weights of H. pylori-infected and -uninfected wild-type C57BL/6and pIgR KO mice. Symbols represent individual animals combined fromindependent experimental cohorts I and II as detailed in the text and Table 1;numbers in each group are shown in parentheses. Error bars denote themean � 95% CI of the results for each group. P values were determined byusing two-way ANOVA (Bonferroni’s posttest) with a two-way comparison ofinfection status and mouse strain for each time point. *, P 0.05; ****, P 0.0001.
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3887
dance in the amounts of SS1-reactive IgA in the intestine of re-sponder versus nonresponder mice.
The inflammatory response of individual animals was also as-sessed qualitatively with respect to histological changes. Apartfrom the standard grading of neutrophil and mononuclear cellinfiltration, specific disease-associated histological changes, in-cluding lymphocytic gastritis and gastric lymphoid follicle forma-tion, were identified. These changes, which can also occur in hu-mans infected with H. pylori, were not present in tissue from fullycolonized animals but were identified in 6 of 14 animals withreduced bacterial loads (P � 0.02; Fisher’s exact test), all of whichhad bacterial loads of less than 106 CFU/g stomach.
We also examined the corelationship of both inflammationand antibody with gastric bacterial load. Taking 1 standard devi-ation from the mean of the inflammatory and intestinal IgA re-
sponses of animals with reduced bacterial loads as the minimumprotective threshold (mean minus 1 SD � 3.6 corpus inflamma-tion score and 436 SS1-reactive IgA/�g total IgA, respectively), allanimals with both cellular and secretory antibody responses abovethese thresholds had reduced gastric colonization (Fig. 6F). Con-versely, the majority of fully colonized animals had attenuatedresponses with respect to both corpus inflammation and intestinalIgA.
Although evident in only a few wild-type animals, our datasuggested that having either an inflammatory response or asecretory antibody response alone was insufficient to reducethe gastric bacterial load. To investigate this possibility further,we examined the relationship between inflammation and thegastric bacterial load of pIgR KO mice, which are incapable ofmounting a secretory antibody response. Responder pIgR KO
FIG 6 Analysis of the natural immune responses of C57BL/6 mice with a full (�107 CFU/g stomach) or reduced (107 CFU/g stomach) gastric load of H. pylori.Data included in these analyses are derived from animals in independent experimental cohorts I (n � 15) and II (n � 12) sampled at 3, 6, and 12 mpi, as detailedin the text and Table 1. (A) Median gastric bacterial load in H. pylori-infected C57BL/6 mice with full (n � 13) or reduced (n � 14) bacterial load. Error barsdenote medians � interquartile ranges of the results for each group. P values were determined by using the Mann-Whitney test. (B) Corpus inflammation scoresof H. pylori-infected C57BL/6 mice with a full or reduced bacterial load. Symbols represent individual animals. Inflammation scores were determined fromduplicate sections for each animal. Error bars denote the mean � SD of the results for each group. P values were determined by using Student’s t test. (C)Correlation of gastric bacterial load with corpus inflammation score of individual animals (n � 27). Linear regression (solid line) and 95% CI (dotted lines) areshown. (D) Intestinal secretory IgA levels in H. pylori-infected C57BL/6 mice with a full or reduced bacterial load. Symbols represent individual animals.Intestinal SS1-reactive antibody levels were averaged from duplicate independent assays for 3- and 6-mpi animals and from a single EIA for 12-mpi animals. Allassays were performed using independently diluted duplicates and standardized against total IgA levels determined from a single mouse IgA-specific EIAperformed in duplicate. Error bars denote the mean � SD of the results for each group. P values were determined by using Student’s t test. (E) Correlation ofgastric bacterial load with intestinal IgA titer in individual animals (n � 27); linear regression (solid line) and 95% CI (dotted lines) are shown. (F) Correlationof inflammation and intestinal IgA in individual animals with full (filled circles) and reduced (open circles) H. pylori gastric colonization. Quadrants (brokenlines) represent 1 standard deviation from the mean corpus inflammation score (mean � 1 SD � 3.6) and intestinal SS1-reactive IgA level (mean � 1 SD � 436SS1-reactive EIA units/�g total IgA) measured in samples from animals with full colonization (as shown in panels B and D, respectively). Numbers in the outercorners report the percentages of animals with full/reduced H. pylori colonization represented in each quadrant. The linear regression (solid line) and 95% CI(dotted lines) are shown.
Gorrell et al.
3888 iai.asm.org Infection and Immunity

mice were defined as those animals with corpus inflammationscores greater than 3.6 (i.e., �4), which was the minimumprotective score observed in chronically infected responderwild-type mice. Responder pIgR KO mice with significant cor-pus inflammation had reduced median bacterial loads com-pared to animals with corpus inflammation scores of 3 or less(P � 0.004; Mann-Whitney test; see Fig. S8A in the supplemen-tal material). However, this 4-fold reduction in gastric bacterialload was significantly smaller (P � 0.01; Mann-Whitney test;Fig. S8A) than the 40-fold reduction observed with the sameanalysis of wild-type mice (P � 0.0004; Mann-Whitney test).In agreement with this, there were fewer corpus responderpIgR KO mice with a gastric bacterial load below 107 CFU/gstomach than corpus responder wild-type mice (4 of 9 pIgR KOmice compared with 13 of 14 wild-type mice; P � 0.018; Fish-er’s exact test). These data suggest that corpus inflammation inconjunction with secretory antibodies has a greater impact ongastric bacterial load than inflammation alone.
In contrast to gastric bacterial load, comparative analysis ofcorpus inflammation and duodenal colonization showed no cor-relation. Chronically infected pIgR KO mice with significant cor-pus inflammation and reduced median gastric bacterial load hadduodenal colonization equivalent to that seen with mice with cor-pus inflammation scores 4 and maximal gastric colonization(see Fig. S8B in the supplemental material). Chronically infectedwild-type mice also showed no correlation between inflamma-tion, gastric bacterial load, and duodenal bacterial load, as boththe responder and nonresponder populations (with reduced andfull median gastric bacterial loads, respectively) showed negligibleduodenal colonization.
(iii) Gastric pIgR expression in responder and nonresponderwild-type mice. Gastric tissue from C57BL/6 mice sampled at 3, 6,and 12 mpi was immunostained for pIgR. Colorimetric and fluo-rescent immunohistological detection of pIgR expression in re-sponder and nonresponder animals was compared to that in un-infected control animals and in animals sampled during acuteinfection at 11 dpi.
At 11 dpi, pIgR expression was detected in gastric tissue inonly 1 of 9 infected wild-type animals and was not detected ingastric tissue from any uninfected animal. In contrast, pIgRexpression was readily visible in the intestinal tissue from allanimals sampled at 11 dpi. We observed an increasing gradientof expression along the 5 cm of intestine examined such thatpIgR was barely detectable adjacent to the pyloric sphincter butwas strongly expressed in the small intestine further from thestomach (Fig. 7G to J). Increased intestinal pIgR expression inall animals was associated with increasing age and was possiblya consequence of establishment of the intestinal microbiota. By3 mpi, we also observed infection-associated induction of pIgRin the gastric mucosa insofar as pIgR expression was evidentonly in chronically H. pylori-infected wild-type mice that re-sponded to their gastric infection but not in animals that didnot respond to infection (Fig. 7A to D; see also Fig. S9A and Bin the supplemental material), pIgR KO mice (see Fig. S9C andD), or uninfected animals. Moreover, in contrast to the resultsfrom the gastric corpus and proximal antrum, pIgR expressionwas observed only infrequently and sparsely in the distal an-trum of some responder animals (Fig. 7E and F).
DISCUSSION
In this study, we examined the natural course of H. pylori infectionin naive wild-type and pIgR KO C57BL/6 mice. We observed twomajor effects on the H. pylori load in wild-type mice that were notobserved in pIgR KO mice lacking secretory antibody: (i) clear-ance of transient duodenal colonization by 3 mpi and (ii) a signif-icant reduction in gastric bacterial load by 6 mpi compared to thatmeasured during acute infection. A time-dependent reduction inthe gastric load of H. pylori infection in naive mice has been re-ported previously (2). However, because the present study showedthat the immune responses mounted by pIgR KO mice to H. pyloriwere not protective, the capacity of the immune response to influ-
FIG 7 Immunohistochemical localization of gastrointestinal pIgR expressionin H. pylori nonresponder and responder mice sampled 12 months after inoc-ulation. Formalin-fixed sections were probed with pIgR-specific antibody(brown) and counterstained with hematoxylin (blue nuclei); �20 magnifica-tion. The nonresponder animal (panels A, C, E, G, and I) had a gastric H. pyloribacterial load of 3.2 � 107 CFU/g and no detectable SS1-reactive IgA in theintestine. The responder animal (panels B, D, F, H, and J) had a gastric H. pyloribacterial load of 2.6 � 106 CFU/g. Data representing immunolocalization ofpIgR in the gastric corpus (panels A and B), proximal antrum (transitionalzone; panels C and D) and distal antrum (panels E and F), and the proximal(within 1 cm of the pyloric sphincter) and distal (approximately 3 cm from thepyloric sphincter) duodenum of single nonresponder and responder animalsare shown and are representative of other nonresponder and responder ani-mals, respectively.
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3889

ence H. pylori chronicity in wild-type mice became evident, andmeasurable, for the first time.
The most striking feature of the protective immune response toH. pylori in wild-type mice was the eradication of transient H.pylori colonization from the intestine upon IgA seroconversionand secretion of H. pylori-reactive antibody into the intestinallumen. Conversely, duodenal H. pylori colonization was sustainedin chronically infected pIgR KO mice that inherently lack secre-tory antibody, and the small number of wild-type mice defectivefor duodenal clearance all showed no or minimal seroconversion.The second important feature was the reduction of bacterial loadin the distal antrum of mice with H. pylori-reactive intestinal se-cretory IgA, an event that is evidently linked to the reduction innumber of viable bacteria in the duodenum. However, these twofeatures, which occurred by 3 mpi, were not sufficient to cause ameasurable reduction in the overall gastric bacterial load. By 6mpi, greater inflammation in the gastric mucosa correlated with asignificant reduction in gastric bacterial load in wild-type animals.However, a significantly less pronounced reduction occurred inpIgR KO animals with similar inflammatory changes. Therefore,our observations indicate that effective immunity to H. pylori mayrely on an underlying and fully functional secretory antibody re-sponse, even though this is not sufficient in itself to have an impacton overall gastric H. pylori infection.
These findings suggest that the amount of secretory antibodyavailable to the stomach can contribute to protective immunityaround the gastroduodenal junction and that the altered gastricdistribution of H. pylori colonization in wild-type mice is consis-tent with protective antibody being sourced from the duodenumrather than being translocated locally. This hypothesis is sup-ported by the paucity of pIgR expression in the distal antrum of allanimals, including those mice that had efficiently cleared H. pylorifrom the distal antrum. One should also consider the fact that thisstudy was conducted in C57BL/6 mice which are Th1 skewed (39).As such, our findings illustrate a significant capacity for antibody-mediated immunity even in the context of a dominant cell-medi-ated immune response.
There are several possible explanations for the failure of anti-body-mediated immunity to protect fully against gastric H. pyloriinfection. In the vast majority of the gastric mucosa, antibodylevels are insufficient to influence H. pylori infection. This is be-cause the machinery for translocating antibodies, i.e., pIgR, acrossthe gastric mucosa is not expressed in the normal, healthy gastricmucosa (15). We also observed in this study that when pIgR ex-pression was upregulated during gastric inflammation, it occurredpredominantly in the corpus and proximal antrum, which is likelyto have been a consequence of local IFN-� production. In con-trast, there was no to very little expression of pIgR in the distalantrum, where clearance of H. pylori colonization to below histo-logical detection levels correlated with the presence of H. pylori-specific secretory antibodies in the duodenum of wild-type mice.Given that IgA production is suppressed by IFN-� (40), upregu-lation of gastric pIgR expression does not necessarily lead to in-creased IgA secretion by the gastric mucosa. This is in agreementwith studies showing no increase in gastric secretory IgA levelsduring infection of humans with H. pylori (25, 28) and the obser-vation that H. pylori-infected, inflamed gastric epithelium stainswith similar intensities for secretory components and IgA regard-less of the presence of local IgA-secreting plasma cells (16). Al-though H. pylori-induced IFN-� production may reduce IgA pro-
duction in the stomach despite its contribution to pIgRupregulation, infection with H. pylori did not significantly sup-press intestinal IgA secretion in animals in this study. In fact wemeasured increased total IgA in the intestinal contents of H. pylo-ri-infected mice with reduced gastric bacterial load compared tofully colonized and uninfected animals.
Another important factor contributing to the failure of gastricIgA to convey immunity to H. pylori is the reliance of gastric im-munity on extragastric secreted antibody. As stated earlier, gastricIgA levels peak during transition of the phase III migrating motorcomplex (MMC) from the stomach to the duodenum. The MMC,consisting of progressive phase I, II, or III, occurs between meals,and MMC phase III is responsible for the mechanical and chem-ical cleansing of the empty stomach in preparation for the nextmeal (41). Approximately 50% of MMC activity originates in thestomach (42), and its transition through to the intestine results inthe stomach entering the predominantly idle phase I as the duo-denum enters the highly active phase III (30), during which duo-denal IgA secretion is maximal (43). Retrograde peristalsis is anormal consequence of this transition and facilitates pH restora-tion of the duodenal bulb and the antrum following gastric emp-tying (44). Duodenal retroperistalsis is also a normal consequenceof gastroduodenal motility in mice (45). The resulting reflux de-livers freshly translocated antibody in the duodenal contents tothe gastric lumen, and in humans this is a specific event that occursin the absence of bile reflux (30). Individuals lacking retrogradeduodenal reflux during phase III of the MMC show a higher prev-alence of H. pylori colonization (46). In contrast, IgA-deficienthumans have no increased risk of infection (47). This differencemay be due to immune benefits being conferred by secretory an-tibody of any isotype, and IgM compensation may be sufficient inIgA-deficient individuals.
The evidence in this study for a correlation between duodenalsecretory IgA level and gastric H. pylori distribution is intriguing.In the antrum, H. pylori is mainly found close to the epithelium inthe firmly adherent mucus layer (48), the integrity of which issignificantly compromised during H. pylori infection (49, 50). Se-cretory IgA has a high affinity for mucin glycoproteins and is moresoluble in mucus than other antibody isotypes (51). It has recentlybeen hypothesized that the loosely adherent mucus layer overlyingthe firmly adherent mucus could be sheared from the antrumduring the powerful MMC phase III contractions, thus exposingthe firmly adherent mucus layer in which H. pylori resides tofreshly delivered duodenal fluid containing secretory IgA (52).This hypothesis is readily conceivable given the ease with whichthe loosely adherent mucus layer can be displaced (49).
In this study, we also observed that mice which failed to sero-convert to H. pylori also failed to develop an inflammatory re-sponse or pathological changes. This is in agreement with workshowing that tolerance of H. pylori mediates protection against H.pylori-mediated disease (53). We also found that animals whichdeveloped similar inflammatory and systemic antibody responsesto H. pylori showed a reduced gastric bacterial load only when H.pylori-reactive antibodies were detected in the intestinal lumen.Together with the finding that duodenal colonization persisted inpIgR KO animals, our findings suggest that H. pylori-reactive an-tibodies may be beneficial in controlling H. pylori infection. Morespecifically, our findings suggest that secretory antibody contrib-utes to protective immunity against H. pylori in the distal antrumand duodenum.
Gorrell et al.
3890 iai.asm.org Infection and Immunity

In humans infected with H. pylori, duodenal colonization cor-relates with the development of gastric metaplasia and with theproduction of H. pylori-permissive gastric mucins in the duode-num (54–56). Given that we did not observe gastric metaplasia inthe duodenum of any H. pylori-colonized animals in this study, itis not surprising that duodenal bacterial loads were low comparedto those in the stomach. In contrast to studies of intestinal colo-nization in humans that rely on duodenal biopsy specimens, how-ever, we were able to examine bacterial loads in the entire duode-num of mice. From this analysis we reproducibly detectedduodenal H. pylori colonization in infected mice, but only in theabsence of H. pylori-reactive secretory antibody. This observationwas not due to differences in the ability of H. pylori to colonize theintestine of the mouse strains, as duodenal colonization resultswere indistinguishable between the two strains prior to serocon-version. Moreover, no differences have been observed in overallintestinal normal flora colonization of pIgR KO and wild-typemice (57). In an infected human, the duodenum would be con-tinually exposed to H. pylori from the stomach. Our viable bacte-rial count data from the lavaged and thoroughly washed duodenaltissue of wild-type nonresponder and pIgR KO animals suggestthat in the absence of an effective secretory antibody response, lowlevels of H. pylori may closely associate with the duodenal mucosa.Persistent duodenal colonization correlated with detectable H. py-lori in the distal antrum, suggesting that a continual supply ofbacteria was required. Over a prolonged period, the persistentpresence of H. pylori in the human duodenum may transformareas of the duodenal mucosa, thus facilitating the development ofa gastric metaplasia which harbors the more florid patches of H.pylori colonization in duodenal ulcer patients. Given the limitedcapacity of the gastric mucosa to secrete IgA, the development ofgastric metaplasia in the intestine may suppress the intestinal im-mune response. Indeed, cytokine responses in the duodenal mu-cosa of duodenal ulcer patients have been reported to be sup-pressed compared to those of asymptomatic carriers of H. pylori(58). Our findings suggest that intestinal secretory IgA may exert aprotective effect in two ways: (i) by keeping bacterial numbers inthe distal antrum at a low level, thus reducing the capacity for H.pylori to amass in the normal duodenum, and (ii) by neutralizingany bacteria that reach the duodenum. It would be interesting toexamine the contribution of intestinal secretory antibody to nat-ural and vaccine-mediated H. pylori immunity in the duodenumduring human infection. Such studies might provide a valuableinsight into the protective immunity and/or tolerance associatedwith life-long asymptomatic infection. This area is becoming in-creasingly important as the debate surrounding the protective ca-pacity of H. pylori infection against other diseases, such as chronicinflammatory and atopic disorders (59), esophageal malignancies(60–62) and tuberculosis (63), continues.
An interesting observation in our study was that pIgR KO miceinfected with H. pylori were visibly smaller than their BHI-chal-lenged siblings or H. pylori-infected wild-type mice. Cohortnumbers were too small in this study to ascertain whether thisbody-weight effect on pIgR KO animals was a consequence ofuncontrolled H. pylori infection in general or was specifically as-sociated with intestinal colonization. While weight differences be-tween H. pylori-infected and uninfected experimental animals arenot generally reported, humans display a variety of weight-associ-ated changes in relation to H. pylori infection. Of most importanceis the impact of H. pylori infection on childhood development (64,
65). However, while weight loss is not a factor in adult H. pyloridisease, weight gain in adults following H. pylori eradication hasbeen reported (66). Our findings suggest for the first time that thehumoral immune response may protect against failure-to-thrive-related sequelae resulting from H. pylori infection of children.
In summary, our data suggest that the infection-induced hu-moral immune response to infection with H. pylori can protect theduodenum against long-term colonization by H. pylori. More-over, antibodies to H. pylori may contribute to the maintenance oflow bacterial loads in the gastric antrum, which in turn may facil-itate normal gastric endocrine function for acid production andthus indirectly protect the gastric corpus. While it is highly un-likely that antibodies are sufficiently present or functional in theacid-secreting regions of the stomach to mediate direct immunityin these regions, stimulation of an effective secretory antibodyresponse may be a necessary property of any vaccine destined toprevent H. pylori-related disease or to mediate sterilizing immu-nity.
ACKNOWLEDGMENTS
This work was supported by Australian National Health and Medical Re-search Council (NHMRC) Bacterial Pathogenesis Program grant 284214.O.L.C.W. was supported by an NHMRC Council Career DevelopmentAward.
We are grateful to Laura Del Mastro for coordinating the breeding andage matching of animals used in this study and to Vicki Bennet-Wood forsection preparation and assistance with tissue preparation.
REFERENCES1. Del Giudice G, Covacci A, Telford JL, Montecucco C, Rappuoli R. 2001.
The design of vaccines against Helicobacter pylori and their development.Annu. Rev. Immunol. 19:523–563.
2. Garhart CA, Redline RW, Nedrud JG, Czinn SJ. 2002. Clearance ofHelicobacter pylori infection and resolution of postimmunization gastritisin a kinetic study of prophylactically immunized mice. Infect. Immun.70:3529 –3538.
3. Raghavan S, Hjulstrom M, Holmgren J, Svennerholm AM. 2002. Pro-tection against experimental Helicobacter pylori infection after immuniza-tion with inactivated H. pylori whole-cell vaccines. Infect. Immun. 70:6383– 6388.
4. Aebischer T, Schmitt A, Walduck AK, Meyer TF. 2005. Helicobacterpylori vaccine development: facing the challenge. Int. J. Med. Microbiol.295:343–353.
5. Akhiani AA, Stensson A, Schon K, Lycke NY. 2005. IgA antibodiesimpair resistance against Helicobacter pylori infection: studies on immuneevasion in IL-10-deficient mice. J. Immunol. 174:8144 – 8153.
6. Akhiani AA, Schon K, Franzen LE, Pappo J, Lycke N. 2004. Helicobacterpylori-specific antibodies impair the development of gastritis, facilitatebacterial colonization, and counteract resistance against infection. J. Im-munol. 172:5024 –5033.
7. Blanchard TG, Czinn SJ, Redline RW, Sigmund N, Harriman G, Ne-drud JG. 1999. Antibody-independent protective mucosal immunity togastric helicobacter infection in mice. Cell. Immunol. 191:74 – 80.
8. Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R,Lee CK, Kleanthous H, Monath TP. 1998. Immunization of mice withurease vaccine affords protection against Helicobacter pylori infection inthe absence of antibodies and is mediated by MHC class II-restricted re-sponses. J. Exp. Med. 188:2277–2288.
9. Garhart CA, Nedrud JG, Heinzel FP, Sigmund NE, Czinn SJ. 2003.Vaccine-induced protection against Helicobacter pylori in mice lackingboth antibodies and interleukin-4. Infect. Immun. 71:3628 –3633.
10. Moulin V, Andris F, Thielemans K, Maliszewski C, Urbain J, Moser M.2000. B lymphocytes regulate dendritic cell (DC) function in vivo: in-creased interleukin 12 production by DCs from B cell-deficient mice re-sults in T helper cell type 1 deviation. J. Exp. Med. 192:475– 482.
11. Suto A, Nakajima H, Ikeda K, Kubo S, Nakayama T, Taniguchi M, SaitoY, Iwamoto I. 2002. CD4(�)CD25(�) T-cell development is regulated byat least 2 distinct mechanisms. Blood 99:555–560.
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3891

12. Brandtzaeg P. 1983. Immunohistochemical characterization of intracel-lular J-chain and binding site for secretory component (SC) in humanimmunoglobulin (Ig)-producing cells. Mol. Immunol. 20:941–966.
13. Rinkenberger JL, Wallin JJ, Johnson KW, Koshland ME. 1996. Aninterleukin-2 signal relieves BSAP (Pax5)-mediated repression of the im-munoglobulin J chain gene. Immunity 5:377–386.
14. Piskurich JF, Youngman KR, Phillips KM, Hempen PM, BlanchardMH, France JA, Kaetzel CS. 1997. Transcriptional regulation of thehuman polymeric immunoglobulin receptor gene by interferon-gamma.Mol. Immunol. 34:75–91.
15. Kaneko T, Ota H, Hayama M, Akamatsu T, Katsuyama T. 2000.Helicobacter pylori infection produces expression of a secretory compo-nent in gastric mucous cells. Virchows Arch. 437:514 –520.
16. Isaacson P. 1982. Immunoperoxidase study of the secretory immuno-globulin system and lysozyme in normal and diseased gastric mucosa. Gut23:578 –588.
17. Takemura K, Hirokawa K, Esaki Y, Mishima Y. 1990. Distribution ofimmunoglobulins and secretory component in gastric cancer of the aged.Cancer 66:2168 –2173.
18. Gologan A, Acquafondata M, Dhir R, Sepulveda AR. 2008. Polymericimmunoglobulin receptor-negative tumors represent a more aggressivetype of adenocarcinomas of distal esophagus and gastroesophageal junc-tion. Arch. Pathol. Lab. Med. 132:1295–1301.
19. Spohn M, McColl I. 1979. Studies on human gastric mucosal immuno-globulin A. II. Further evidence for the absence of the secretory compo-nent from the predominant immunoglobulin A of human gastric mucus.Biochim. Biophys. Acta 576:9 –16.
20. Spohn M, McColl I. 1979. Studies on human gastric mucosal immuno-globulin A. Biochim. Biophys. Acta 576:1– 8.
21. Ahlstedt I, Lindholm C, Lonroth H, Hamlet A, Svennerholm AM,Quiding-Jarbrink M. 1999. Role of local cytokines in increased gastricexpression of the secretory component in Helicobacter pylori infection.Infect. Immun. 67:4921– 4925.
22. El Kaissouni J, Bene MC, Faure GC. 1998. Activation of epithelial cells ingastritis. Digestion 59:53–59.
23. Handa Y, Saitoh T, Kawaguchi M, Misaka R, Tsurui M, Ohno H,Morita S, Sanji T, Tani Y, Sakai Y, Umezawa H. 1999. Production ofsecretory component and pathogenesis of gastric cancer in Helicobacterpylori-infected stomach. J. Gastroenterol. 34(Suppl 11):37– 42.
24. Meyer F, Wilson KT, James SP. 2000. Modulation of innate cytokineresponses by products of Helicobacter pylori. Infect. Immun. 68:6265–6272.
25. Berstad AE, Kilian M, Valnes KN, Brandtzaeg P. 1999. Increased mu-cosal production of monomeric IgA1 but no IgA1 protease activity inHelicobacter pylori gastritis. Am. J. Pathol. 155:1097–1104.
26. Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. 2003. Helicobacterpylori vacuolating cytotoxin inhibits T lymphocyte activation. Science301:1099 –1102.
27. Quiding-Järbrink M, Sundström P, Lundgren A, Hansson M, Bäck-ström M, Johansson C, Enarsson K, Hermansson M, Johnsson E,Svennerholm AM. 2009. Decreased IgA antibody production in the stom-ach of gastric adenocarcinoma patients. Clin. Immunol. 131:463– 471.
28. Birkholz S, Schneider T, Knipp U, Stallmach A, Zeitz M. 1998. De-creased Helicobacter pylori-specific gastric secretory IgA antibodies in in-fected patients. Digestion 59:638 – 645.
29. Shirai Y, Wakatsuki Y, Kusumoto T, Nakata M, Yoshida M, Usui T,Iizuka T, Kita T. 2000. Induction and maintenance of immune effectorcells in the gastric tissue of mice orally immunized to Helicobacter pylorirequires salivary glands. Gastroenterology 118:749 –759.
30. Dalenbäck J, Abrahamson H, Björnson E, Fändriks L, Mattsson A, OlbeL, Svennerholm A, Sjövall H. 1998. Human duodenogastric reflux, ret-roperistalsis, and MMC. Am. J. Physiol. 275:R762–R769.
31. Uren TK, Johansen FE, Wijburg OL, Koentgen F, Brandtzaeg P,Strugnell RA. 2003. Role of the polymeric Ig receptor in mucosal B cellhomeostasis. J. Immunol. 170:2531–2539.
32. Gorrell RJ, Robins-Browne RM. 2009. Antibody-mediated protectionagainst infection with Helicobacter pylori in a suckling mouse model ofpassive immunity. Infect. Immun. 77:5116 –5129.
33. Bohr UR, Glasbrenner B, Primus A, Zagoura A, Wex T, MalfertheinerP. 2004. Identification of enterohepatic Helicobacter species in patientssuffering from inflammatory bowel disease. J. Clin. Microbiol. 42:2766 –2768.
34. Dixon MF, Genta RM, Yardley JH, Correa P. 1996. Classification and
grading of gastritis. The updated Sydney System. International Workshopon the Histopathology of Gastritis, Houston 1994. Am. J. Surg. Pathol.20:1161–1181.
35. Tan MP, Kaparakis M, Galic M, Pedersen J, Pearse M, Wijburg OL,Janssen PH, Strugnell RA. 2007. Chronic Helicobacter pylori infectiondoes not significantly alter the microbiota of the murine stomach. Appl.Environ. Microbiol. 73:1010 –1013.
36. Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity amongSalmonella lipopolysaccharide chemotypes in silver-stained polyacryl-amide gels. J. Bacteriol. 154:269 –277.
37. Wijburg OL, Uren TK, Simpfendorfer K, Johansen FE, Brandtzaeg P,Strugnell RA. 2006. Innate secretory antibodies protect against naturalSalmonella typhimurium infection. J. Exp. Med. 203:21–26.
38. Johansen FE, Pekna M, Norderhaug IN, Haneberg B, Hietala MA,Krajci P, Betsholtz C, Brandtzaeg P. 1999. Absence of epithelial immu-noglobulin A transport, with increased mucosal leakiness, in polymericimmunoglobulin receptor/secretory component-deficient mice. J. Exp.Med. 190:915–922.
39. Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. 2004. Innateimmune response in Th1- and Th2-dominant mouse strains. Shock 22:460 – 466.
40. Hodgkin PD, Yamashita LC, Seymour B, Coffman RL, Kehry MR. 1991.Membranes from both Th1 and Th2 T cell clones stimulate B cell prolif-eration and prepare B cells for lymphokine-induced differentiation tosecrete Ig. J. Immunol. 147:3696 –3702.
41. Kelly KA. 1980. Gastric emptying of liquids and solids: roles of proximaland distal stomach. Am. J. Physiol. 239:G71–G76.
42. Luiking YC, van der Reijden AC, van Berge Henegouwen GP, Akker-mans LM. 1998. Migrating motor complex cycle duration is determinedby gastric or duodenal origin of phase III. Am. J. Physiol. 275:G1246 –G1251.
43. Mellander A, Mattsson A, Svennerholm AM, Sjovall H. 1997. Relation-ship between interdigestive motility and secretion of immunoglobulin Ain human proximal small intestine. Dig. Dis. Sci. 42:554 –567.
44. Castedal M, Abrahamsson H. 2001. High-resolution analysis of the du-odenal interdigestive phase III in humans. Neurogastroenterol. Motil. 13:473– 481.
45. Bercik P, Verdu EF, Foster JA, Lu J, Scharringa A, Kean I, Wang L,Blennerhassett P, Collins SM. 2009. Role of gut-brain axis in persistentabnormal feeding behavior in mice following eradication of Helicobacterpylori infection. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296:R587–R594.
46. Testoni PA, Bagnolo F, Bologna P, Colombo E, Bonassi U, Lella F,Buizza M. 1996. Higher prevalence of Helicobacter pylori infection indyspeptic patients who do not have gastric phase III of the migratingmotor complex. Scand. J. Gastroenterol. 31:1063–1068.
47. Bogstedt AK, Nava S, Wadstrom T, Hammarstrom L. 1996. Helicobacterpylori infections in IgA deficiency: lack of role for the secretory immunesystem. Clin. Exp. Immunol. 105:202–204.
48. Shimizu T, Akamatsu T, Sugiyama A, Ota H, Katsuyama T. 1996.Helicobacter pylori and the surface mucous gel layer of the human stom-ach. Helicobacter 1:207–218.
49. Henriksnäs J, Phillipson M, Storm M, Engstrand L, Soleimani M, HolmL. 2006. Impaired mucus-bicarbonate barrier in Helicobacter pylori-infected mice. Am. J. Physiol. Gastrointest. Liver Physiol. 291:G396 –G403.
50. Newton JL, Jordan N, Oliver L, Strugala V, Pearson J, James OF, AllenA. 1998. Helicobacter pylori in vivo causes structural changes in the adher-ent gastric mucus layer but barrier thickness is not compromised. Gut43:470 – 475.
51. McSweegan E, Burr DH, Walker RI. 1987. Intestinal mucus gel andsecretory antibody are barriers to Campylobacter jejuni adherence to INT407 cells. Infect. Immun. 55:1431–1435.
52. Sjovall H. 2011. Meaningful or redundant complexity—mechanisms be-hind cyclic changes in gastroduodenal pH in the fasting state. Acta Physiol.(Oxf) 201:127–131.
53. Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T,Muller A. 2011. Tolerance rather than immunity protects from Helico-bacter pylori-induced gastric preneoplasia. Gastroenterology 140:199 –209.
54. Carrick J, Lee A, Hazell S, Ralston M, Daskalopoulos G. 1989. Campy-lobacter pylori, duodenal ulcer, and gastric metaplasia: possible role offunctional heterotopic tissue in ulcerogenesis. Gut 30:790 –797.
Gorrell et al.
3892 iai.asm.org Infection and Immunity

55. Johan G, Offerhaus A, Molyvas EN, Hoedemaeker PJ. 1990. Helicobacterpylori infection of gastric mucin cell metaplasia: the duodenum revisited.J. Pathol. 162:239 –243.
56. Pietroiusti A, Luzzi I, Gomez MJ, Magrini A, Bergamaschi A, Forlini A,Galante A. 2005. Helicobacter pylori duodenal colonization is a strong riskfactor for the development of duodenal ulcer. Aliment. Pharmacol. Ther.21:909 –915.
57. Sait LC, Galic M, Price JD, Simpfendorfer KR, Diavatopoulos DA, UrenTK, Janssen PH, Wijburg OL, Strugnell RA. 2007. Secretory antibodiesreduce systemic antibody responses against the gastrointestinal commen-sal flora. Int. Immunol. 19:257–265.
58. Strömberg E, Edebo A, Svennerholm AM, Lindholm C. 2003. Decreasedepithelial cytokine responses in the duodenal mucosa of Helicobacter py-lori-infected duodenal ulcer patients. Clin. Diagn. Lab. Immunol. 10:116 –124.
59. Arnold IC, Hitzler I, Müller A. 2012. The immunomodulatory proper-ties of Helicobacter pylori confer protection against allergic and chronicinflammatory disorders. Front. Cell. Infect. Microbiol. 2:10. doi:10.3389/fcimb.2012.00010.
60. Whiteman DC, Parmar P, Fahey P, Moore SP, Stark M, Zhao ZZ,Montgomery GW, Green AC, Hayward NK, Webb PM. 2010. Associa-tion of Helicobacter pylori infection with reduced risk for esophageal can-cer is independent of environmental and genetic modifiers. Gastroenter-ology 139:73– 83.
61. Ye W, Held M, Lagergren J, Engstrand L, Blot WJ, McLaughlin JK,Nyren O. 2004. Helicobacter pylori infection and gastric atrophy: riskof adenocarcinoma and squamous-cell carcinoma of the esophagusand adenocarcinoma of the gastric cardia. J. Natl. Cancer Inst. 96:388 –396.
62. Sonnenberg A, Lash RH, Genta RM. 2010. A national study of Helico-bactor pylori infection in gastric biopsy specimens. Gastroenterology 139:1894 –1901.
63. Perry S, de Jong BC, Solnick JV, de la Luz Sanchez M, Yang S, Lin PL,Hansen LM, Talat N, Hill PC, Hussain R, Adegbola RA, Flynn J,Canfield D, Parsonnet J. 2010. Infection with Helicobacter pylori is asso-ciated with protection against tuberculosis. PLoS One 5:e8804. doi:10.1371/journal.pone.0008804.
64. Mera RM, Correa P, Fontham EE, Reina JC, Pradilla A, Alzate A, BravoLE. 2006. Effects of a new Helicobacter pylori infection on height andweight in Colombian children. Ann. Epidemiol. 16:347–351.
65. Sood MR, Joshi S, Akobeng AK, Mitchell J, Thomas AG. 2005. Growthin children with Helicobacter pylori infection and dyspepsia. Arch. Dis.Child. 90:1025–1028.
66. Lane JA, Murray LJ, Harvey IM, Donovan JL, Nair P, Harvey RF. 2011.Randomised clinical trial: Helicobacter pylori eradication is associated witha significantly increased body mass index in a placebo-controlled study.Aliment. Pharmacol. Ther. 33:922–929.
Antibody-Mediated Immunity against H. pylori
October 2013 Volume 81 Number 10 iai.asm.org 3893
Related Documents










