eScholarship provides open access, scholarly publishing services to the University of California and delivers a dynamic research platform to scholars worldwide. Lawrence Berkeley National Laboratory Peer Reviewed Title: Contribution of mobile genetic elements to Desulfovibrio vulgaris genome plasticity Author: Walker, C.B. Publication Date: 03-15-2010 Publication Info: Lawrence Berkeley National Laboratory Permalink: http://escholarship.org/uc/item/6mp2x0nw Local Identifier: LBNL Paper LBNL-2545E Preferred Citation: Environmental Microbiology, 11, 9, 2009

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

eScholarship provides open access, scholarly publishingservices to the University of California and delivers a dynamicresearch platform to scholars worldwide.
Lawrence Berkeley National Laboratory
Peer Reviewed
Title:Contribution of mobile genetic elements to Desulfovibrio vulgaris genome plasticity
Author:Walker, C.B.
Publication Date:03-15-2010
Publication Info:Lawrence Berkeley National Laboratory
Permalink:http://escholarship.org/uc/item/6mp2x0nw
Local Identifier:LBNL Paper LBNL-2545E
Preferred Citation:Environmental Microbiology, 11, 9, 2009

Contribution of mobile genetic elements to Desulfovibrio vulgaris genome plasticity Christopher B. Walker,1,2† Sergey Stolyar,1,2
Dylan Chivian,2,3 Nicolas Pinel,1 Jeffrey A. Gabster,1,2 Paramvir S. Dehal,2,3 Zhili He,2,4 Zamin Koo Yang,2,6 Huei-Che B. Yen,2,5 Jizhong Zhou,2,4
Judy D. Wall,2,5 Terry C. Hazen,2,3 Adam P. Arkin2,3
and David A. Stahl1,2*
1Department of Civil and Environmental Engineering, University of Washington, Seattle, WA, USA.
2Virtual Institute for Microbial Stress and Survival (vimss.lbl.gov).
3Physical Biosciences Division, Lawrence Berkeley National Laboratory , Berkeley, CA, USA.
4Institute for Environmental Genomics, Oklahoma, University, Norman, OK, USA.
5Biochemistry Division, University of Missouri, Columbia, MO, USA.
6Environmental Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, USA.
Summaryemi_1946 2244..2252
The genome of Desulfovibrio vulgaris strain DePue, a sulfate-reducing Deltaproteobacterium isolated from heavy metal-impacted lake sediment, was completely sequenced and compared with the type strain D. vulgaris Hildenborough. The two genomes share a high degree of relatedness and synteny, but harbour distinct prophage and signatures of past phage encounters. In addition to a highly variable phage contribution, the genome of strain DePue contains a cluster of open-reading frames not found in strain Hildenborough coding for the production and export of a capsule exopolysaccharide, possibly ofrelevance to heavy metal resistance. Comparative whole-genome microarray analysis on four additional D. vulgaris strains established greater interstrain variation within regions associated with phage insertion and exopolysaccharide biosynthesis. Introduction Bacteriophage and other mobile elements represent a large reservoir of genetic information affecting the structure and evolution of microbial ecosystems. Complete genome sequences for numerous environmentally relevant microbes have highlighted both a relatively conserved core genome and a remarkable contribution of
bacteriophage to the pan-genome; the pan-genome composed of the sum of all unique genes distributed in a described species (Tettelin et al., 2005; Coleman et al., 2006; Cuadros-Orellana et al., 2007; Lindell et al., 2007). For example, while comparative genomic analysis of only three Prochlorococcus species revealed the core genome, sequencing of 12 isolates suggested a pangenome at least an order of magnitude larger than any single genome (Lindell et al., 2007). Although anaerobes are as yet less well represented by completed genome sequences, the comparative analyses of closely related Desulfovibrio isolates presented here indicates a similar contribution of viruses and other mobile genetic elements to the pan-genome of this representative anaerobe.
Desulfovibrio are sulfate-reducing microorganisms that participate in global sulfur- and carbon-cycling, often forming complex communities with other anaerobes and facilitating the complete decomposition of organic material (for review, see Rabus et al., 2005). The significance of phage to population structure was initially suggested by the isolation and characterization of D. vulgaris strain DePue. This sulfate-reducing microorganism, isolated from a heavy metal-impacted lake sediment, lacks prophages present in strain Hildenborough (Walker et al., 2006). The recently completed genome sequence of strain DePue reported here now provides the basis for a more complete census of phage-associated divergence among different described strains of Desulfovibrio vulgaris and highlights the role of CRISPR (clusters of regularly interspaced palindromic repeats) immunity among closely related strains.
Previous studies have demonstrated that strain DePue was sensitive to lytic infection by viruses carried as prophages in strain Hildenborough (Walker et al., 2006). Complementary whole-genome microarray comparisons with other D. vulgaris strains now shows that among this study set, strain DePue is unique in lacking any of the six prophage annotated in the Hildenborough genome. Thus,
1
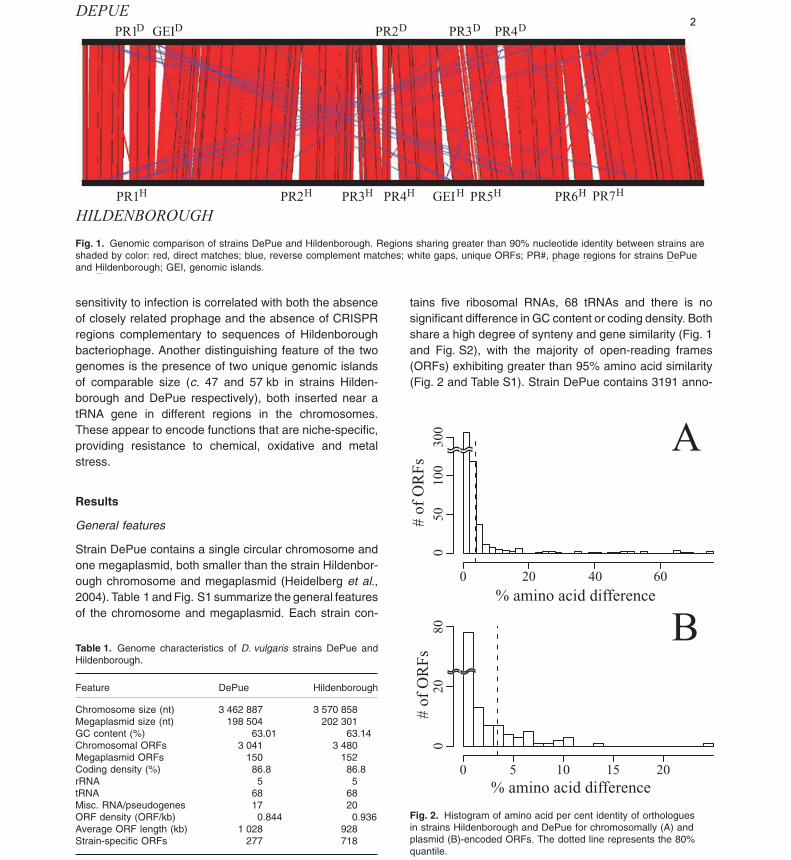
sensitivity to infection is correlated with both the absenceof closely related prophage and the absence of CRISPRregions complementary to sequences of Hildenboroughbacteriophage. Another distinguishing feature of the twogenomes is the presence of two unique genomic islandsof comparable size (c. 47 and 57 kb in strains Hilden-borough and DePue respectively), both inserted near atRNA gene in different regions in the chromosomes.These appear to encode functions that are niche-specific,providing resistance to chemical, oxidative and metalstress.
Results
General features
Strain DePue contains a single circular chromosome andone megaplasmid, both smaller than the strain Hildenbor-ough chromosome and megaplasmid (Heidelberg et al.,2004). Table 1 and Fig. S1 summarize the general featuresof the chromosome and megaplasmid. Each strain con-
tains five ribosomal RNAs, 68 tRNAs and there is nosignificant difference in GC content or coding density. Bothshare a high degree of synteny and gene similarity (Fig. 1and Fig. S2), with the majority of open-reading frames(ORFs) exhibiting greater than 95% amino acid similarity(Fig. 2 and Table S1). Strain DePue contains 3191 anno-
Table 1. Genome characteristics of D. vulgaris strains DePue andHildenborough.
Feature DePue Hildenborough
Chromosome size (nt) 3 462 887 3 570 858Megaplasmid size (nt) 198 504 202 301GC content (%) 63.01 63.14Chromosomal ORFs 3 041 3 480Megaplasmid ORFs 150 152Coding density (%) 86.8 86.8rRNA 5 5tRNA 68 68Misc. RNA/pseudogenes 17 20ORF density (ORF/kb) 0.844 0.936Average ORF length (kb) 1 028 928Strain-specific ORFs 277 718
HILDENBOROUGH
DEPUE
PR1H PR2H PR3H PR4H PR5H PR6H
PR1D PR2D PR4D
PR7H
PR3DGEID
GEIH
Fig. 1. Genomic comparison of strains DePue and Hildenborough. Regions sharing greater than 90% nucleotide identity between strains areshaded by color: red, direct matches; blue, reverse complement matches; white gaps, unique ORFs; PR#, phage regions for strains DePueand Hildenborough; GEI, genomic islands.
# of
OR
Fs
% amino acid difference0 20 40 60
050
100
300 A
# of
OR
Fs
% amino acid difference
020
80
0 5 10 15 20
B
Fig. 2. Histogram of amino acid per cent identity of orthologuesin strains Hildenborough and DePue for chromosomally (A) andplasmid (B)-encoded ORFs. The dotted line represents the 80%quantile.
DePue genome 2245
© 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 2244–2252
2

tated protein-coding sequences, 441 fewer than strainHildenborough. The reduced number of protein-codingsequences appears disproportionate relative to theaccompanying reduction in the genome size of strainDePue. Apart from the use of different ORF-determiningalgorithms, the disproportionately higher number of pre-dicted protein-coding sequences in strain Hildenboroughprimarily reflects of the larger number of small ORFsassigned in regions of presumptive phage insertion (strainHildenborough containing seven insertions as opposed tofour in strain DePue). Their possible biological significancewill likely only be resolved through more extensive com-parative analyses of related Desulfovibrio.
The two genomes share a total of 2914 genes, repre-senting 91% and 80% of the strain DePue and Hilden-borough genomes respectively, with 267 genes uniqueto strain DePue genes and 718 unique to strain Hilden-borough. Assignment of all genes into clusters of orthlo-gous functional groups revealed only minor differencesbetween strains, with the primary divergence in genecontent associated with genomic islands in strain DePue(Fig. S3). Small inversions and rearrangements alsodistinguish the two genomes, most involving tRNAs ormobile genetic elements. Notably, the genome of strainDePue lacks the majority of the insertion sequence ele-ments present in strain Hildenborough.
Genomic islands
The 277 ORFs unique to strain DePue genome assemblein five large genomic islands and four smaller clusters(Fig. 1 and Table S2). Hypothetical, conserved hypotheti-cal and bacteriophage-related ORFs comprise most ofthese unique protein-coding sequences. Four of the largegenomic islands (listed in Fig. 1 as Phage Region 1DePue,PR2D, PR3D and PR4D) contain phage or phage-relatedORFs, with all but PR1D located in different chromosomallocations than the prophage of strain Hildenborough. The
fifth genomic island (GEID, Fig. 1) contains ORFs codingfor exopolysaccharide (EPS) production, modification andtransport. PR1D, PR2D and PR4D contain ORFs exclusivelyfound in strain DePue, while approximately half of theORFs in PR3D are unique (Fig. S4). The rest of the ORFs ofthis region demonstrate homology to protein-codingsequences found in strain Hildenborough at approximatelythe same genomic locus.A tRNA immediately flanks all fivegenomic islands, a general feature among mobile geneticelements and common to bacteriophage insertion sites(Williams, 2002; Campbell, 2003). The four smaller clus-ters unique to strain DePue contain ORFs coding for avariety of functions, with annotation suggesting contribu-tions to outer-membrane protein transport/modification,phage resistance and DNA excision/repair.
Exopolysaccharide genomic island
Unlike the other unique genomic islands in strain DePuecoding for bacteriophage-related functions, the approxi-mately 57 kb region (GEID) located between PR1D andPR2D contains 33 ORFs clustered into two distinct func-tional groups (Fig. 3, Table S3). These ORFs share somehomology with genes in previously sequenced genomes;however, no single organism contains more than a fewhighly similar protein-coding sequences. The first cluster,containing 15 ORFs (Dvul_2564-79) encodes sensor andresponse modules of unknown function. The cluster isflanked by genes for an integrase and transposase andmay be capable of transposition and/or be part of a largergenomic island that includes the adjacent cluster of genes(Dvul_2580-94) encoding for EPS synthesis, export andmodification.
The region containing this EPS biosynthetic gene sharesfeatures with a genomic island present in strain Hildenbor-ough (GEIH in Fig. 1) containing 52 annotated genes(DVU2000–DVU2051). The GEIs in strains DePue andHildenborough are directly inserted in a tRNA-Met gene
76.5%
24.5%
63.1%
kpsMTED kpsCSfkbHexopolysaccharidemodification genes
exopolysaccharidesynthesis genes
5 kb
Dvul_2564 - 96
kpsF
HP HP HP
HP CHP
1 2 3
4
5
76 8 9
HP
HP
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26
Fig. 3. Unique exopolysaccharide biosynthesis region in the genome of strain DePue. The plot illustrates the GC% of ORFs and intergenicregions. ORF numbering refers to descriptions in Table S3, while color-coding indicates ORFs of similar COGs. The red box highlights thesecond ORF cluster coding for capsule exopolysaccharide production, modification and export proteins. The line marking 63.1% correspondsto the average genome GC%.
2246 C. B. Walker et al.
© 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 2244–2252
3
and flanked by identical 49 bp direct repeat sequences.Both GEIs have GC skews well below the genome averageand contain genes for site-specific recombinases andtransposases (Fig. 3) (Johnston et al., 2008). Thissequence-based suggestion of acquisition by lateral trans-fer is now supported by recent studies demonstrating thatthis region in strain Hildenborough is spontaneouslyexcised in a small fraction (c. 3%) of cells maintained understandard laboratory growth conditions (Johnston et al.,2008). Strains lacking this GEI demonstrated faster growthwith lactate and sulfate under anoxic conditions than thosecarrying it (Johnston et al., 2008). However, the presenceof this GEI is strongly implicated in contributing to greaterfitness under environmental conditions of chemical andoxidative stress (Johnston et al., 2008).
The structurally similar GEI in strain DePue likely alsoconfers selective advantage under certain environmentalconditions. For example, key enzymes in the pathway forsialic acid biosynthesis (highlighted in red on Table S3)are among the multiple genes encoding for EPS biosyn-thesis in this GEI. Past studies have shown the capacity ofD. vulgaris to reduce Cr(VI) to the less toxic Cr(III), and to
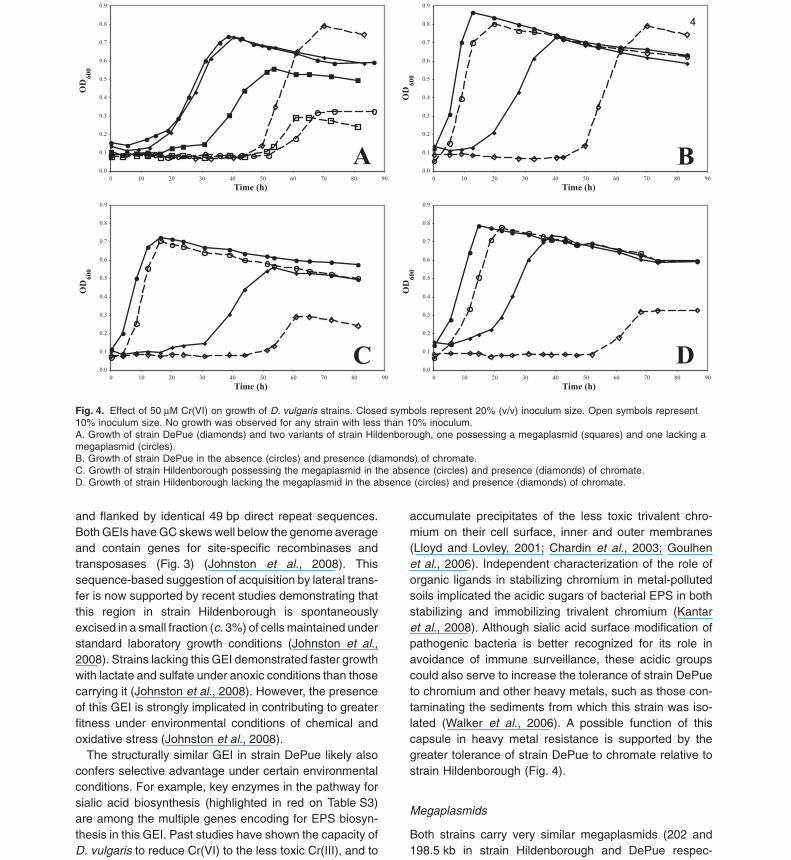
accumulate precipitates of the less toxic trivalent chro-mium on their cell surface, inner and outer membranes(Lloyd and Lovley, 2001; Chardin et al., 2003; Goulhenet al., 2006). Independent characterization of the role oforganic ligands in stabilizing chromium in metal-pollutedsoils implicated the acidic sugars of bacterial EPS in bothstabilizing and immobilizing trivalent chromium (Kantaret al., 2008). Although sialic acid surface modification ofpathogenic bacteria is better recognized for its role inavoidance of immune surveillance, these acidic groupscould also serve to increase the tolerance of strain DePueto chromium and other heavy metals, such as those con-taminating the sediments from which this strain was iso-lated (Walker et al., 2006). A possible function of thiscapsule in heavy metal resistance is supported by thegreater tolerance of strain DePue to chromate relative tostrain Hildenborough (Fig. 4).
Megaplasmids
Both strains carry very similar megaplasmids (202 and198.5 kb in strain Hildenborough and DePue respec-
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0 10 20 30 40 50 60 70 80 90
Time (h)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0 10 20 30 40 50 60 70 80 90
Time (h)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0 10 20 30 40 50 60 70 80 90
Time (h)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0 10 20 30 40 50 60 70 80 90
Time (h)
OD
600
OD
600
OD
600
OD
600
A
C
B
D
Fig. 4. Effect of 50 mM Cr(VI) on growth of D. vulgaris strains. Closed symbols represent 20% (v/v) inoculum size. Open symbols represent10% inoculum size. No growth was observed for any strain with less than 10% inoculum.A. Growth of strain DePue (diamonds) and two variants of strain Hildenborough, one possessing a megaplasmid (squares) and one lacking amegaplasmid (circles).B. Growth of strain DePue in the absence (circles) and presence (diamonds) of chromate.C. Growth of strain Hildenborough possessing the megaplasmid in the absence (circles) and presence (diamonds) of chromate.D. Growth of strain Hildenborough lacking the megaplasmid in the absence (circles) and presence (diamonds) of chromate.
DePue genome 2247
© 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 2244–2252
4

tively). As the plasmid is not required for growth of strainHildenborough under laboratory conditions, being fre-quently lost during routine culture maintenance (J.D. Wall,unpublished), it is notable that strains isolated from differ-ent continents and decades apart carry near identicalplasmids. Differences are relatively minor – the strainDePue megaplasmid lacks 21 ORFs present in strainHildenborough and contains 19 unique genes(Dvul_3099-101, Fig. S5). With the exception of ORFspreviously assigned roles in chromate transport/resistance in strain Hildenborough, most of those absentin DePue are of unknown function (Klonowska et al.,2007). No mobile genetic elements or repetitive signa-tures flank these genes in strain Hildenborough.
The shared presence and conservation of the mega-plasmids in the two isolates are suggestive of strongselective pressure under normal environmental condi-tions. Both megaplasmids code for nitrogen fixation(Dvul_3089-98), EPS biosynthesis (Dvul_3055-72), pos-sible defence against grazers via secretion system III(Dvul_2984-3010), surface lipopolysaccharide modifica-tion proteins (Dvul_3028-39), a protein/peptide trans-port system for potential antibacterial compounds(Dvul_3083-85) and phage immunity systems involvingan abortive infection protein and CRISPR defencemechanisms (Dvul_ 2973-78, Fig. S4 and Table S2). Allcould serve functions in the environment that would haveless adaptive significance in pure culture.
The most striking difference between these closelyrelated strains (sharing greater than 99.8% nucleotideidentity of 16S rRNAgenes) is carriage of distinct prophageand short sequence motifs associated with acquired phageimmunity. The megaplasmids of both strains containunique CRISPR loci and highly similar CRISPR-associated (cas) genes (Fig. 5), generally recognized asproviding bacteriophage immunity (Mojica et al., 2005;Barrangou et al., 2007; Brouns et al., 2008). Both strains
contain a zinc transporter gene (zupT, 97% amino acidsimilarity) following the distal repeat sequence and a highlysimilar leader sequence (87% nucleotide identity) betweenthe cas2 gene (97% amino acid similarity) and the proximalrepeat sequence. Despite the high amino acid similaritiesin flanking genes, each CRISPR region is unique insequence and length (strain DePue contains 45 repeatsversus 28 for strain Hildenborough, Table S4). Both strainscontain nearly identical 32 bp repeat sequences, withstrain-specific variations occurring at common variableregions (Kunin et al., 2007). The same three unique repeatsequences occur in both strains at the distal end of theCRISRP region (Fig. 5, green, black and yellow dia-monds), with strain DePue containing a fourth uniquerepeat sequence (Fig. 5, grey diamond). One of thesedistal repeat sequences (Fig. 5, black diamond) shares100% nucleotide identity with the majority of the strainHildenborough repeat sequences. Small intrastrainvariations in CRISPR repeat sequences are common ingenome sequences available for other microbes; however,the conserved variations in the distal repeat sequencesappear unique for strains DePue and Hildenborough.
Intra- and interstrain comparisons indicate no redun-dant spacer sequences with lengths varying from 29 to 36nucleotides (Table S4). Four strain Hildenborough spacersequences (Fig. 5, open blue boxes) are identical tosequences within PR3D of strain DePue. Our previousobservation that two morphologically distinct bacterioph-age carried by Hildenborough can infect strain DePue(Walker et al., 2006) is consistent with the absence ofspacer sequences in strain DePue having significantnucleotide matches in strain Hildenborough. A search ofthe Global Ocean Sampling metavirome and availableviral genome sequences revealed highly similar, but non-identical matches to strain DePue and Hildenboroughspacer sequences (Rusch et al., 2007; Williamson et al.,2008).
1 kb
cas3 cas1cas5 csd1 csd2 cas4 cas2 zupT
1 2 43
DEPUE
HILDENBOROUGH
GTCGCCCCCCGTGCGGGGGCGTGGATTGAAAC
GTCGCCCCCCGCACGGGGGCGTGGATTGAAAC
GTCGCCCCCCGCGCGGGGGCGTGGATTGAAAC
GTCGCCCCCCACGCGGGGGCGTGGATTGAAAC
GTCGCCCCTCGCGCGGGGGCGTGGATTGAAAC
Fig. 5. CRISPR and cas gene region in megaplasmids of strains DePue and Hildenborough. A zinc transporter (zupT) andCRISPR-associated (cas) genes flank the CRISPR region. Rectangles indicate spacer regions and diamonds indicate repeat structures.Color-coding represents identical repeat sequences. The variable repeat sequence nucleotides are highlighted in red. The numbered openrectangles represent spacer sequences with identical matches in the strain DePue genome: 1, intergenic region between Dvul_1048-9; 2,hypothetical protein (Dvul_1039); 3, NAD-dependent DNA ligase (Dvul_1045); 4, peptidase (Dvul_1049). See Table S4 for spacer sequences.
2248 C. B. Walker et al.
© 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 2244–2252
5

Hybridization array comparisons
Comparative analysis also revealed a correlation betweenthis highly elaborated system of phage immunity and anunusually large variety of prophage harboured by the twostrains of D. vulgaris. Together the two strains contain 11unique phage-specific genomic islands (7 in strain Hilden-borough and 4 in strain DePue, Fig. S4). This density(prophage/Mb of genome, Table S5) is among the highestin available genome sequences and suggests a highimpact of viral infection on the genetic content of D. vul-garis. Based on this observation, we examined the distri-bution of Hildenborough bacteriophage among four otherD. vulgaris strains (Llanelly, Brockhurst Hill, Woolwich andMarburg) by hybridizing genomic DNA from each strain toan array built from the sequence of strain Hildenborough.Each strain hybridized to at least 90% of the genes rep-resented on strain Hildenborough array [genomic DNA(gDNA) sample strain/gDNA strain Hildenborough > 0.70],with the majority of missing ORFs (defined by absence ofhybridization) annotated as phage or phage-related(Fig. 6 and Table S6). Although all strains have highlysimilar (> 99%) 16S rRNA gene nucleotide identity(Table S7), little correlation exists between relatednessand carriage of specific phage. All members of this studyset, except strain DePue, hybridized to array elementstargeting ORFs encoding PR6H, a lambdoid-like phage.Conversely, none of the strains hybridized to probes tar-geting PR7H, a mu-like phage. With the exception of strainLlanely, all did not hybridize with probes for anothermu-like phage located in PR1H. The most divergentstrains (Brockhurst Hill and Woolwich) contain the sameset of Hildenborough viral elements, hybridizing to probesfor PR3H, PR4H and PR6H.
Physiological comparisons
As yet no marked physiological differences have beenobserved between the two strains. Previous characteriza-tion revealed little difference in growth temperature
optimum or substrate utilization (Walker et al., 2006).Given the observation that genes associated with chro-mate resistance are missing from the strain DePue mega-plasmid and in consideration that this strain was isolatedfrom a heavy metal-impacted lake, the response to chro-mate stress was examined. Although both strains exhibitconsiderable chromate tolerance, strain DePue demon-strated greater resistance to chromate at low inoculumtransfer than either the plasmid-plus or plasmid-minusvariant of strain Hildenborough (Fig. 4). Thus, chromateresistance does not appear to be primarily determined byplasmid-encoded traits, including a gene annotated inchromate efflux, but may be associated with the EPSencoded by the strain-specific genomic island.
Discussion
The concept of a microbial species has yet to accom-modate the remarkable diversity revealed by ongoinggenome sequencing projects. Although adaptive featuressuch as those exhibited in Prochlorococcus species likelyexist in anaerobic populations, the reduced number ofsequenced anaerobic organisms limits detailed genomiccomparisons. The characterization described here of twoclosely related D. vulgaris strains provides initial com-parative perspective on the physiological and genomicplasticity within a common species of sulfate reducers.
Both D. vulgaris strains share a high percentage ofgenes (80–91%); however, approximately 1000 arestrain-specific. As is typical for other reported straindivergence, variation occurs primarily within ‘islands’(frequently encoding surface-associated features, mobilegenetic elements and prophage) flanked by more highlyconserved regions. The greatest source of sequencedivergence between these two strains derives from majordifferences in type and number prophage, and in twounique genomic islands. Complementary microarraystudies further documented the presence of a diverse andactive phage population carried among closely related
Fig. 6. Presence/absence distribution mapping of strain Hildenborough bacteriophage among D. vulgaris strains. The presence (blackrectangles) and absence (white rectangles) of bacteriophage are mapped against the genomes of strain DePue and Hildenborough. Thegenomic location of bacteriophage present in strains Llanelly (NCIMB 8446), Brockhurst Hill (NCIMB 8306), Woolwich (NCIMB 8457) andMarburg (ATCC 35115) may differ. Grey arrows indicate ribosomal RNAs.
DePue genome 2249
© 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 2244–2252
6

strains of D. vulgaris. Although variable prophage distri-bution and associated immunity systems are expected toplay important roles in determining population’s structure,the unique GEI elements can be more directly associatedwith niche specialization. Comparison of two variantsof strain Hildenborough, containing or missing the GEIunique to this strain, suggested that this element couldconfer significant adaptive advantage in environmentsexperiencing fluctuations in chemical/oxidative stress(Johnston et al., 2008). Similarly, modification of surfacefeatures attributed to the GEI present in strain DePuewould be expected to also confer selective advantage incertain environments – possibly contributing to greaterchromate tolerance, resistance to protozoal grazing orphage infection by masking receptors. However, the influ-ence of lysogeny on the fitness of Desulfovibrio and thecost of maintaining specific phage resistance mecha-nisms remain questions for future study.
Apart from a major contribution of prophage to thegenomic structure of the two strains, the presence of verysimilar megaplasmids is also notable. As this plasmiddoes not appear to alter the growth of strain Hildenbor-ough under laboratory conditions, it likely confers signifi-cant adaptive advantage in non-laboratory environments(Johnston et al., 2008). As briefly presented in the results,plasmid-encoded functions that could contribute to strongenvironmental selection include systems for nitrogen fixa-tion, and resistance to viral and protozoal attack. In addi-tion to a recognized contribution of the plasmid to biofilmformation (Klonowska et al., 2007), well known to sup-press grazing, both megaplasmids encode a proteinsecretion system III usually associated with plant andanimal pathogenesis (Hueck, 1998).
Experimental procedures
High-molecular-weight genomic DNA preparation
Strain DePue was grown as previously described in 500 ml ofmedia in 1 l pyrex bottles fitted with black butyl rubber stop-pers and screw-top caps (Walker et al., 2006). Cells wereharvested in mid-exponential phase into 500 ml polycarbon-ate wide-mount centrifuge bottle (Nalgene Labware, Roches-ter, NY) previously stored in an anaerobic chamber for 48 h.Cells were centrifuged for 30 min at 6000 r.p.m. (6084 g) and4°C in a Sorvall RC-5B refrigerated centrifuge equipped witha GS-3 rotor. After centrifugation, supernatant was pouredoff and the pellets frozen at -80°C until processing. High-molecular-weight DNA was isolated as previously describedusing phenol : chloroform : isoamyl (Heidelberg et al., 2004).
Genome sequencing
A completely sequenced genome of D. vulgaris strain DePuewas obtained through collaboration with the Joint GenomeInstitute. Whole-genome shotgun sequencing of 3, 8 and40 kb DNA libraries produced at least 8¥ coverage of the
entire genome. Specifics of clone library generation,sequencing and assembly strategies may be found atthe DOE JGI website (http://www.jgi.doe.gov/sequencing/index.html).
Genome analysis
Analysis of the completely sequence D. vulgaris DePuegenome was completed using MicrobesOnline (http://www.microbesonline.org/), the DOE JGI web-browsing soft-ware (http://genome.ornl.gov/microbial/dvul/03oct06/) andthe TIGR Manatee program (http://stahl.ce.washington.edu/genomes).
Direct comparison of the D. vulgaris DePue and Hildenbor-ough genomes was performed using both MicrobesOnlineand the Artemis Comparison Tool (Carver et al., 2005)with a comparison library generated through WebACT(http://www.webact.org/WebACT/home). A list of orthologuesshared by the two genomes was generated using a reciprocalbest-hit approach.
Comparisons between the D. vulgaris DePue and theGlobal Ocean Sampling and ALOHA/HOTS metagenomicdata sets were preformed using CAMERA (http://camera.calit2.net/index.php).
CRISPR analysis
The CRISPR regions were determined using the Java-basedCRISPR recognition tool and FASTA files of both thechromosome and megaplasmid from strains DePue andHildenborough (Bland et al., 2007). The minimum number ofrepeats was set to 3, the minimum repeat length set to 20 andthe maximum repeat length set to 45. Search windows of 6and 9 were used separately and compared, although nodifference was noted. The minimum spacer length was set at15 nt and the maximum spacer length set at 50 nt. All spacerand repeat sequences were compared against the genomesof each strain, the general NCBI database and the CAMERAmetagenomic database using BLAST.
DNA extraction from D. vulgaris strains
Desulfovibrio vulgaris strains DePue, Hildenborough, Brock-hurst Hill (NCIMB 8306), Woolwich (NCIMB 8457), Llanelly(NCIMB 8446) and Marburg (ATCC 35115) were grown andgenomic DNA extracted as previously described (Walkeret al., 2006). In order to verify the identity of the D. vulgarisstrains, PCR amplification and sequencing of most of the 16SrRNA, the complete 16S-23S internal transcribed spacer anda small portion of the 23S rRNA gene were performed aspreviously described (Walker et al., 2006). Phylogenetic rela-tionships were screened using BLAST (Altschul et al., 1997).
Genomic DNA microarray comparison
The gDNA microarray hybridizations and imaging of D. vul-garis strains were performed as previously described (Walkeret al., 2006). Normalized signal intensities for each ORF werecompared manually against the D. vulgaris Hildenboroughcontrol gDNA and signal intensity ratios calculated (sample
2250 C. B. Walker et al.
© 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 2244–2252
7

strain/strain Hildenborough). Signal intensity ratios less than0.5 were considered as gene absent from genomes ofcomparison strains.
Acknowledgements
The authors wish to thank the Marcin Joachimiak (LawrenceBerkeley National Laboratory) for assistance with Microbe-sOnline and David Bruce, Paul Richardson and LynneGoodwin (Department of Energy Joint Genome Institute) formanagement of the genome sequencing. This work was part-ially supported by the U.S. Department of Energy underContract No. DE-AC02-05CH11231.
References
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J.H.,Zhang, Z., Miller, W., and Lipman, D.J. (1997) GappedBLAST and PSI-BLAST: a new generation of protein data-base search programs. Nucleic Acids Res 25: 3389–3402.
Barrangou, R., Fremaux, C., Deveau, H., Richards, M.,Boyaval, P., Moineau, S., et al. (2007) CRISPR providesacquired resistance against viruses in prokaryotes.Science 315: 1709–1712.
Bland, C., Ramsey, T.L., Sabree, F., Lowe, M., Brown, K.,Kyrpides, N.C., and Hugenholtz, P. (2007) CRISPRRecognition Tool (CRT): a tool for automatic detection ofclustered regularly interspaced palindromic repeats. BMCBioinformatics 8: 209.
Brouns, S.J., Jore, M.M., Lundgren, M., Westra, E.R.,Slijkhuis, R.J., Snijders, A.P., et al. (2008) Small CRISPRRNAs guide antiviral defense in prokaryotes. Science 321:960–964.
Campbell, A. (2003) Prophage insertion sites. Res Microbiol154: 277–282.
Carver, T.J., Rutherford, K.M., Berriman, M., Rajandream,M.A., Barrell, B.G., and Parkhill, J. (2005) ACT: the Artemiscomparison tool. Bioinformatics 21: 3422–3423.
Chardin, B., Giudici-Orticoni, M.T., De Luca, G., Guigliarelli,B., and Bruschi, M. (2003) Hydrogenases in sulfate-reducing bacteria function as chromium reductase. ApplMicrobiol Biotechnol 63: 315–321.
Coleman, M.L., Sullivan, M.B., Martiny, A.C., Steglich, C.,Barry, K., DeLong, E.F., and Chisholm, S.W. (2006)Genomic islands and the ecology and evolution of Prochlo-rococcus. Science 311: 1768–1770.
Cuadros-Orellana, S., Martin-Cuadrado, A.B., Legault, B.,D’Auria, G., Zhaxybayeva, O., Papke, R.T., and Rodriguez-Valera, F. (2007) Genomic plasticity in prokaryotes: thecase of the square haloarchaeon. ISME J 1: 235–245.
Goulhen, F., Gloter, A., Guyot, F., and Bruschi, M. (2006)Cr(VI) detoxification by Desulfovibrio vulgaris strain Hilden-borough: microbe–metal interactions studies. Appl Micro-biol Biotechnol 71: 892–897.
Heidelberg, J.F., Seshadri, R., Haveman, S.A., Hemme, C.L.,Paulsen, I.T., Kolonay, J.F., et al. (2004) The genomesequence of the anaerobic, sulfate-reducing bacteriumDesulfovibrio vulgaris Hildenborough. Nat Biotechnol 22:554–559.
Hueck, C.J. (1998) Type III protein secretion systems inbacterial pathogens of animals and plants. Microbiol MolBiol Rev 62: 379–433.
Johnston, S., Lin, S., Lee, P., Caffrey, S.M., Wildschut, J.,Voordouw, J.K., et al. (2008) A genomic island of thesulfate-reducing bacterium Desulfovibrio vulgaris Hilden-borough promotes survival under stress conditions whiledecreasing the efficiency of anaerobic growth. EnvironMicrobiol 11: 981–991.
Kantar, C., Cetin, Z., and Demiray, H. (2008) In situ stabili-zation of chromium(VI) in polluted soils using organicligands: the role of galacturonic, glucuronic and alginicacids. J Hazardous Materials 159: 287–293.
Klonowska, A., He, Z., He, Q., Clark, M.E., Theiman, S.B.,Hazen, T.C., et al. (2007) Global transcriptomic analysis ofchromium(VI) export of Desulfovibrio vulgaris Hilden-borough under sulfate-reducing conditions. In: AmericanSociety for Microbiology 107th General Meeting. Toronto,Canada: American Society for Microbiology Press.
Kunin, V., Sorek, R., and Hugenholtz, P. (2007) Evolutionaryconservation of sequence and secondary structures inCRISPR repeats. Genome Biol 8: R61.
Lindell, D., Jaffe, J.D., Coleman, M.L., Futschik, M.E.,Axmann, I.M., Rector, T., et al. (2007) Genome-wideexpression dynamics of a marine virus and host revealfeatures of co-evolution. Nature 449: 83–86.
Lloyd, J.R., and Lovley, D.R. (2001) Microbial detoxificationof metals and radionuclides. Curr Opin Biotechnol 12: 248–253.
Mojica, F.J.M., Diez-Villasenor, C., Garcia-Martinez, J., andSoria, E. (2005) Intervening sequences of regularly spacedprokaryotic repeats derive from foreign genetic elements.J Mol Evol 60: 174–182.
Rabus, R., Hansen, T., and Widdel, F. (2005) Gram-negativesulfate-reducing bacteria. In The Prokaryotes: An EvolvingElectronic Resource for the Microbiological Community.Ballows, A., Trueper, H.G., Dworkin, M., Harder, W., andSchleifer, K.H. (eds). New York, USA: Springer-Verlag.
Rusch, D.B., Halpern, A.L., Sutton, G., Heidelberg, K.B.,Williamson, S., Yooseph, S., et al. (2007) The Sorcerer IIGlobal Ocean Sampling expedition: Northwest Atlanticthrough Eastern Tropical Pacific. PLoS Biol 5: 398–431.
Tettelin, H., Masignani, V., Cieslewicz, M.J., Donati, C.,Medini, D., Ward, N.L., et al. (2005) Genome analysis ofmultiple pathogenic isolates of Streptococcus agalactiae:implications for the microbial ‘pan-genome’. Proc Natl AcadSci U S A 102: 13950–13955.
Walker, C.B., Stolyar, S.S., Pinel, N., Yen, H.C.B., He, Z.L.,Zhou, J.Z., et al. (2006) Recovery of temperate Des-ulfovibrio vulgaris bacteriophage using a novel host strain.Environ Microbiol 8: 1950–1959.
Williams, K.P. (2002) Integration sites for genetic elements inprokaryotic tRNA and tmRNA genes: sublocation prefer-ence of integrase subfamilies. Nucleic Acids Res 30: 866–875.
Williamson, S.J., Rusch, D.B., Yooseph, S., Halpern, A.L.,Heidelberg, K.B., Glass, J.I., et al. (2008) The Sorcerer IIGlobal Ocean Sampling Expedition: metagenomic charac-terization of viruses within aquatic microbial samples.PLoS ONE 3: e1456.
DePue genome 225
,
8
Related Documents











