DOI: 10.1002/cphc.201200343 Continuous Synthesis of Peralkylated Imidazoles and their Transformation into Ionic Liquids with Improved (Electro)Chemical Stabilities Cedric Maton, [a] Nils De Vos, [a] Bart I. Roman, [a] Evert Vanecht, [b] Neil R. Brooks, [b] Koen Binnemans, [b] Stijn Schaltin, [c] Jan Fransaer, [c] and Christian V. Stevens* [a] 1. Introduction The development of different classes of ionic liquids (ILs) as well as the implementation of microreactor technology in order to facilitate the scale-up of industrially important reac- tions are both booming research fields. Ionic liquids are often considered as green solvents since they have a low volatility, [1] a high flash point and autoignition temperature [2] and are recy- clable. These solvents have tuneable characteristics [3] and have therefore found interest in a great variety of (industrial) appli- cations, [4] for example, solvents in both organic synthesis [5] and biocatalysis, [6] solvents for the dissolution and modification of biomacromolecules, [7] as gas-stripping fluids, [8] as electrolytes [9] and in analytical chromatography. [10] In particular, ionic liquids containing the well-known 1-ethyl-3-methylimidazolium, [C 2 mim] + , and 1-butyl-3-methylimidazolium, [C 4 mim] + cations have been studied intensively because imidazolium ionic liq- uids possess relatively low melting points and viscosities, are readily accessible at a rather low cost and allow for easy modi- fication. [11] In many studies, the imidazolium ion is combined with the weakly coordinating bis(trifluoromethylsulfonyl)imide anion, [NTf 2 ] , leading to hydrophobic ionic liquids. ILs are often considered to have a high thermal stability. However, recent work by Del Sesto et al. shows that the tradi- tional imidazolium ionic liquids such as [C 4 mim][NTf 2 ] start to thermally degrade at temperatures as low as 150 8C. [12] When the anion has a basic or nucleophilic character, its contribution to the stability is believed to be greater than that of the cation. However, the design of more stable cations combined with non-coordinating anions might lead to novel classes of ionic liquids with a high thermal stability. A key point of con- cern in imidazolium-type ionic liquids is the hydrogen atom on the C2 carbon, which is acidic and prone to undergo H/D ex- change in neutral media if the anion is sufficiently basic, such as in 1-butyl-3-methylimidazolium dicyanamide, [C 4 mim] [N(CN) 2 ]. [13] In basic media, deprotonation will occur even more rapidly. [13, 14] Due to this vulnerability towards bases, these typi- cal ILs could not be successfully employed in base-catalysed reactions, such as Baylis–Hillman, [15] Knoevenagel and Claisen– Schmidt condensation reactions, [16] since base depletion and ir- reversible side reactions hinder full conversion. Therefore, com- pletely substituted imidazolium ionic liquids might be conven- ient solvents for employing strong bases such as strong alkalis, Grignard reagents and organo-lithium reagents. For electrochemical applications, a large electrochemical window and a low viscosity are desirable. It was previously re- ported that substitution of the hydrogen atom on the C2 [a] C. Maton, N. De Vos, B. I. Roman, Prof. Dr. C. V. Stevens Department of Sustainable Chemistry and Technology SynBioC Research Group, Ghent University Coupure Links 653, 9000 Ghent (Belgium) Fax: (+ 32) (9) 264 59 57 E-mail : [email protected] [b] E. Vanecht, Dr. N. R. Brooks, Prof. Dr. K. Binnemans Department of Chemistry K.U. Leuven Celestijnenlaan 200F, 3001 Heverlee (Belgium) [c] S. Schaltin, Prof. Dr. J. Fransaer Department of Metallurgy and Materials Engineering (MTM) K.U. Leuven Kasteelpark Arenberg 44, 3001 Heverlee (Belgium) Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cphc.201200343. A versatile and efficient method to synthesize tetrasubstituted imidazoles via a one-pot modified Debus–Radziszewski reac- tion and their subsequent transformation into the correspond- ing imidazolium ionic liquids is reported. The tetrasubstituted imidazoles were also synthesized by means of a continuous flow process. This straightforward synthetic procedure allows for a fast and selective synthesis of tetrasubstituted imidazoles on a large scale. The completely substituted imidazolium di- cyanamide and bis(trifluoromethylsulfonyl)imide salts were ob- tained via a metathesis reaction of the imidazolium iodide salts. The melting points and viscosities are of the same order of magnitude as for their non-substituted analogues. In addi- tion to the superior chemical stability of these novel ionic liq- uids, which allows them to be applied in strong alkaline media, the improved thermal and electrochemical stabilities of these compounds compared with conventional imidazolium ionic liquids is also demonstrated by thermogravimetrical anal- ysis (TGA) and cyclic voltammetry (CV). Although increased substitution of the ionic liquids does not further increase ther- mal stability, a definite increase in cathodic stability is observa- ble. ChemPhysChem 0000, 00, 1 – 13 # 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim &1& These are not the final page numbers! ÞÞ

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

DOI: 10.1002/cphc.201200343
Continuous Synthesis of Peralkylated Imidazoles and theirTransformation into Ionic Liquids with Improved(Electro)Chemical StabilitiesCedric Maton,[a] Nils De Vos,[a] Bart I. Roman,[a] Evert Vanecht,[b] Neil R. Brooks,[b]
Koen Binnemans,[b] Stijn Schaltin,[c] Jan Fransaer,[c] and Christian V. Stevens*[a]
1. Introduction
The development of different classes of ionic liquids (ILs) aswell as the implementation of microreactor technology inorder to facilitate the scale-up of industrially important reac-tions are both booming research fields. Ionic liquids are oftenconsidered as green solvents since they have a low volatility,[1]
a high flash point and autoignition temperature[2] and are recy-clable. These solvents have tuneable characteristics[3] and havetherefore found interest in a great variety of (industrial) appli-cations,[4] for example, solvents in both organic synthesis[5] andbiocatalysis,[6] solvents for the dissolution and modification ofbiomacromolecules,[7] as gas-stripping fluids,[8] as electrolytes[9]
and in analytical chromatography.[10] In particular, ionic liquidscontaining the well-known 1-ethyl-3-methylimidazolium,[C2mim]+ , and 1-butyl-3-methylimidazolium, [C4mim]+ cationshave been studied intensively because imidazolium ionic liq-uids possess relatively low melting points and viscosities, arereadily accessible at a rather low cost and allow for easy modi-fication.[11] In many studies, the imidazolium ion is combinedwith the weakly coordinating bis(trifluoromethylsulfonyl)imideanion, [NTf2]� , leading to hydrophobic ionic liquids.
ILs are often considered to have a high thermal stability.However, recent work by Del Sesto et al. shows that the tradi-tional imidazolium ionic liquids such as [C4mim][NTf2] start tothermally degrade at temperatures as low as 150 8C.[12] Whenthe anion has a basic or nucleophilic character, its contributionto the stability is believed to be greater than that of thecation. However, the design of more stable cations combinedwith non-coordinating anions might lead to novel classes ofionic liquids with a high thermal stability. A key point of con-cern in imidazolium-type ionic liquids is the hydrogen atom on
the C2 carbon, which is acidic and prone to undergo H/D ex-change in neutral media if the anion is sufficiently basic, suchas in 1-butyl-3-methylimidazolium dicyanamide, [C4mim][N(CN)2] .[13] In basic media, deprotonation will occur even morerapidly.[13, 14] Due to this vulnerability towards bases, these typi-cal ILs could not be successfully employed in base-catalysedreactions, such as Baylis–Hillman,[15] Knoevenagel and Claisen–Schmidt condensation reactions,[16] since base depletion and ir-reversible side reactions hinder full conversion. Therefore, com-pletely substituted imidazolium ionic liquids might be conven-ient solvents for employing strong bases such as strong alkalis,Grignard reagents and organo-lithium reagents.
For electrochemical applications, a large electrochemicalwindow and a low viscosity are desirable. It was previously re-ported that substitution of the hydrogen atom on the C2
[a] C. Maton, N. De Vos, B. I. Roman, Prof. Dr. C. V. StevensDepartment of Sustainable Chemistry and TechnologySynBioC Research Group, Ghent UniversityCoupure Links 653, 9000 Ghent (Belgium)Fax: (+ 32) (9) 264 59 57E-mail : [email protected]
[b] E. Vanecht, Dr. N. R. Brooks, Prof. Dr. K. BinnemansDepartment of ChemistryK.U. LeuvenCelestijnenlaan 200F, 3001 Heverlee (Belgium)
[c] S. Schaltin, Prof. Dr. J. FransaerDepartment of Metallurgy and Materials Engineering (MTM)K.U. LeuvenKasteelpark Arenberg 44, 3001 Heverlee (Belgium)
Supporting information for this article is available on the WWW underhttp://dx.doi.org/10.1002/cphc.201200343.
A versatile and efficient method to synthesize tetrasubstitutedimidazoles via a one-pot modified Debus–Radziszewski reac-tion and their subsequent transformation into the correspond-ing imidazolium ionic liquids is reported. The tetrasubstitutedimidazoles were also synthesized by means of a continuousflow process. This straightforward synthetic procedure allowsfor a fast and selective synthesis of tetrasubstituted imidazoleson a large scale. The completely substituted imidazolium di-cyanamide and bis(trifluoromethylsulfonyl)imide salts were ob-tained via a metathesis reaction of the imidazolium iodidesalts. The melting points and viscosities are of the same order
of magnitude as for their non-substituted analogues. In addi-tion to the superior chemical stability of these novel ionic liq-uids, which allows them to be applied in strong alkalinemedia, the improved thermal and electrochemical stabilities ofthese compounds compared with conventional imidazoliumionic liquids is also demonstrated by thermogravimetrical anal-ysis (TGA) and cyclic voltammetry (CV). Although increasedsubstitution of the ionic liquids does not further increase ther-mal stability, a definite increase in cathodic stability is observa-ble.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim &1&
These are not the final page numbers! ��

carbon atom by a methyl group increases the cathodic stabilityof imidazolium ionic liquids.[17] This phenomenon was ex-plained by the increased steric hindrance.[18] The same effect isalso observed upon extension of the alkyl chains.[19] The iden-tification of the degradation products formed upon applyingan electric current to ionic liquids has only been attempted bya few research groups, but it is evident that not only electro-chemical reactions play a role. Further reactions take place be-tween the formed reactive decomposition products as well,[20]
for instance Hoffman elimination[21] and many other secondaryreactions can occur at the electrodes.[22] Density functionaltheory (DFT) calculations have been applied to compare theelectrochemical stabilities of different ionic liquids.[23] For imi-dazolium ionic liquids, it is assumed that electrons are trans-ferred to the lowest unoccupied molecular orbital (LUMO)upon reduction. It was found that the higher the LUMOenergy, the lower the reduction potential.[23a] Gifford et al.stated that methyl substitution at the C2 position leads to aninductive effect, which increases the electron density in the ar-omatic ring.[17b] This stabilizing effect was also reflected in theelectron affinity (EA) values calculated by Ong et al.[23b]
It is evident that the C2 position of an imidazolium core isvery vulnerable to chemical attack and that it influences the(electro)chemical stability of imidazolium ionic liquids. There-fore, it is of importance to investigate the performance of C2-substituted imidazolium ionic liquids. In order to avoid any in-fluence of the hydrogen atoms on the C4 and C5 positionsduring stability testing experiments, it is also necessary to in-troduce substituents in these positions. Herein, a continuousmulticomponent reaction that allows the synthesis of a libraryof polysubstituted imidazoles is reported. These imidazoles allhave methyl substituents on C4 and C5, but different substitu-ents on C2 (H, Me, Et, isoPr). Either an ethyl or hexyl groupwas chosen as the alkyl chain on N1, the latter leading tolower melting points and an increased hydrophobicity of theionic liquids.[4, 5c] Subsequently, the corresponding bis(trifluoro-methylsulfonyl)imide and dicyanamide peralkylated ionic liq-uids were synthesized.
Not only does the applied microreactor technology allow foran easy scale-up (referred to as numbering-up),[24] the technol-ogy is also well suited for multicomponent reactions.[25] Earlierresearch showed the straightforward application of continu-ous-flow synthesis for a modified Radziszewski reaction to-wards N-alkylated imidazoles.[26] Due to small temperature andconcentration gradients in the microreactor channels (approx.100 mm), the synthesis of the imidazoles proceeds in optimalconditions.[24]
2. Results and Discussion
2.1. Synthesis of the Imidazole Core
Peralkylated imidazoles were synthesized via a modifiedDebus–Radziszewski reaction.[27] This condensation reaction ofa 1,2-diketo compound, ammonia, an amine and an aldehydeforms tetrasubstituted imidazoles and water. Earlier researchshowed that the imidazole synthesis is very effective at
120 8C.[26] Since the starting materials are volatile, pressure vialswere used in order to keep these volatile components in thereaction mixture. This also allows for the use of methanol asa solvent. Methanol can easily be removed by means of rotaryevaporation and also takes up some of the reaction water,hence it is the most appropriate solvent. The substituents onthe C2 and N1 positions are built in directly through the corre-sponding aldehydes and amines, eliminating the need forbases and haloalkanes for the N-alkylation or for the nucleo-philic substitution at the C2 positions, which normally suffersfrom a low yield.[28]
The condensation was performed as a two-step multicom-ponent reaction, which allows the selective synthesis of N-alky-lated imidazoles. In a first step, the intermediate imine wasgenerated, which was reacted with the ammonia source andthe 1,2-diketone in a second step (Scheme 1). After reaction,the conversion of starting material was between 32 and 42 %and the product was extracted from the reaction mixture witha 0.5 m HCl solution. The aqueous phase was made alkalineand extracted. After removal of the solvent by rotary evapora-tion, the resulting oil was purified by distillation under reducedpressure.
This reaction sequence yielded pure N-alkylated imidazolesas expected, although in the synthesis of hexyl derivatives,a small amount (up to 12 mol % of the extracted mixture) of1-H-imidazoles was obtained. In order to remove the 1-H-imid-azoles from the reaction mixture, it was sufficient to extractthe alkaline aqueous phase with hexanes, since the 1-H-imida-zoles are not soluble in this solvent. There is, however, an ex-ception to the selectivity. After reaction with propionaldehydeto form 1,2-diethyl-4,5-dimethylimidazole (5 c), 38 mol % of 1-ethyl-2,4,5-trimethylimidazole (5 b) was found in the extractedmixture. This is assumed to be caused by a [1,3]-prototropicshift during the imine formation, as depicted in Scheme 2.
During the synthesis of the N-hexyl derivative 2-ethyl-1-hexyl-4,5-dimethylimidazole (5 g), the same formation of the2-methyl analogue (5 f) was observed, but the latter was never
Scheme 1. One-pot modified Radziszewski reaction with intermediate imineformation. An overview of the corresponding analogues with different Rgroups is given in Table 1.
&2& www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 0000, 00, 1 – 13
�� These are not the final page numbers!
C. V. Stevens et al.

formed with other aldehydes. To circumvent this problem, analternative approach was followed, in which the 1,2-diketoneand the amine were mixed in the first step. In this way, no1-alkyl-2,4,5-trimethylimidazole was formed.
2.2. Continuous-Flow Synthesis of the Imidazole Core
Batch experiments led to the selective synthesis of N-alkylatedimidazoles through a priori imine formation. Earlier research atour department using a CPC college system showed thatimidazoles were optimally synthesized at 120 8C in a continu-ous-flow reactor with a residence time of 118 min and usingsolutions of the starting materials at a 0.5 m concentration. Al-though in this former set-up the amine and the aldehyde werenot mixed before being pumped into the reactor, here thegoal was to synthesize the imine in batches, and subsequentlyto react it with NH4OAc and the 1,2-diketone in the continu-ous-flow reactor (Figure 1).
Continuous-flow synthesis of N-hexyl-imidazoles was per-formed in n-pentanol, allowing the reaction to be carried outat a temperature of 120 8C. In a first attempt, the solubilitylimit of starting materials was examined and 1.0 m was foundto be the maximum concentration for dissolution of NH4OAcin n-pentanol. However, at this concentration, a substantialamount of 1-H-imidazole was formed. Therefore, a lower con-centration was used. At a concentration of 0.5 m of NH4OAc,no 1-H-imidazole was found, and this concentration was usedduring further experiments. In a second step, the residencetime and flow rate were optimized.
The yield and throughput of 1-hexyl-2,4,5-trimethylimidazolewere determined by GC analysis and are presented in Figure 2as a function of the residence time (tres). The yield of the otheranalogues showed a similar trend. The best yields of extractedproduct (36 %) were those obtained with a flow rate of2 � 0.5 mL min�1, that is, a tres of 48 min. Additional prolonga-tion of the residence time would not increase the yield signifi-cantly (Figure 2). To obtain a sufficient throughput combinedwith an acceptable yield, a tres of 20 min was chosen. After col-lection of the reaction mixture, the water formed in the reac-tion and the solvent were distilled off, yielding a dark liquidwhich could be extracted and distilled in the same way as inthe batch procedure. The distilled solvent was successfully re-cycled and used for subsequent microreactor experiments. Thesynthesis of the N-ethyl-imidazole derivatives was not feasiblehowever, due to the formation of ethylamine gas bubbles inthe microreactor channels at the temperature applied. Toavoid the earlier observed prototropic shift during continuoussynthesis of 2-ethyl-1-hexyl-4,5-dimethylimidazole, n-pentanolsolutions of preformed 3-ethylimino-butan-2-one and NH4OAcwith propionaldehyde were used.
2.3. Synthesis of Ionic Liquids
To avoid the presence of starting materials in the end product,iodomethane (MeI) was used for quaternization of the imid-azoles. The use of the reactive MeI allows for a fast and quanti-tative alkylation reaction and a complete removal of the alky-lating reagent, due to the low boiling point in comparisonwith other alkylating agents, for example, dimethylsulfate andbromoalkanes. The corresponding ionic liquids were thus ob-tained by adding MeI dropwise to a solution of the desiredimidazole in dry acetonitrile (Scheme 3).
Scheme 2. [1.3]-prototropic shift of ethylpropylideneamine and consecutiveformation of 1-ethyl-2,4,5-trimethylimidazole in alkaline media.
Figure 1. Microreactor setup. MU: mixing unit ; RTU: residence time unit.
Figure 2. Yield [%] (*) and throughput [g h�1] (~) of 1-hexyl-2,4,5-trimethyl-imidazole (5 f) in the CPC College System microreactor.
Scheme 3. Methylation of the imidazole core with iodomethane.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org &3&
These are not the final page numbers! ��
Peralkylated Imidazoles as Ionic Liquids

This exothermic reaction was conducted at 0 8C, to avoidevaporation of MeI. The reaction was monitored by 1H NMRand after completion, the solvent and excess MeI were re-moved in vacuo. N-ethyl substituted imidazolium iodide saltsare slightly coloured solids. These solids could be recrystallizedin dry acetone which furnished transparent crystals. N-hexylsubstituted imidazolium iodide salts were obtained as trans-parent yellow oils. These oils were dissolved in CH3CN andwashed with hexanes to completely remove any traces of theimidazole precursor. The latter ionic liquids were never thor-oughly dried to avoid thermally induced degradation reactionscaused by the nucleophilic iodide anion, therefore, yieldscannot be reported.
The colouration upon methylation was more pronounced inthe case of the 2-ethyl derivatives, which turned black aftera few hours. The iodide salts were also susceptible to coloura-tion upon storage at ambient temperature, even when shield-ed from light. Therefore, storage at �18 8C under a nitrogen at-mosphere is advised, but immediate use in the metathesis re-action is to be preferred.
The iodide ionic liquids were transformed into the corre-sponding dicyanamide and bis(trifluoromethylsulfonyl)imidesalts according to metathesis reactions described in literature(Scheme 4).[29] Efficient mixing was ensured by mild heating.
Upon metathesis with AgN(CN)2 of the N-hexyl derivatives indemineralized water, precipitation of yellow AgI in an oilymatrix was observed, therefore, a 2:1 mixture of demineralizedwater and CH3CN was used to ensure efficient mixing. ExcessAgN(CN)2 and AgI were filtered off and the solvent was re-moved in vacuo. The metathesis reaction with LiNTf2 was per-formed with only a 3 wt % excess, thereby avoiding the pres-ence of residual LiNTf2, which influences the electrochemistryof the ionic liquids.[30] Excess LiNTf2 and LiI were washed outwith demineralized water. Upon addition of AgNO3 to an aque-ous solution of the [N(CN)2]� salts and to the washing water ofthe [NTf2]� salts, no precipitation was visible.[3]
After metathesis, the ionic liquids were thoroughly driedbefore further experiments were conducted. Drying proceededfor 4 to 8 h at a pressure of 0.5 mbar at temperatures of 80and 120 8C for the [N(CN)2] and [NTf2] salts, respectively. The
water content of the samples was analysed by a coulometricKarl Fischer titration and the [N(CN)2]� salts all contained lessthan 500 ppm of H2O, while the [NTf2]� salts all had water con-tents lower than 200 ppm.
2.4. Melting Point and Viscosity of the Ionic Liquids
Alkylation on C2 prevents the formation of a hydrogen bond.Normally, this hydrogen bond reduces both melting point andviscosity in three ways: 1) the charge transfer established bythis bond decreases the Coulomb forces,[31] 2) the hydrogenbond can hinder a good stacking of the molecules by distor-tion of the crystal lattice[14a] and 3) the C2 proton is an interac-tion site that allows the free movement of the anion throughthe plane of symmetry of the cation, resulting in increased en-tropy.[32] Therefore, alkylation at the C2 position is expected toincrease the melting points, as was shown earlier by several re-search groups.[17a, 32] For the same reasons, the substitution ofthe C4 and C5 protons, which can form weak hydrogenbonds,[32] by methyl groups, might be expected to further in-crease the melting points.
Most of the N-ethylimidazolium salts were solid at roomtemperature. However, all the N-hexylimidazolium salts re-mained liquid at room temperature. In general, the observedmelting points are higher than the reported values for the cor-responding non-peralkylated salts ([C2mim][N(CN)2]: 21 8C,[C2mim][NTf2] �3 8C).[17a, 29a] It is suggested that increased vander Waals attraction forces and the suppression of hydrogenbonding are the main causes for this increase in meltingpoints.
However, as presented in Table 1, melting points do not in-crease upon methylation at the C2 position as expected. Con-trarily, Tm seems to be inversely related to the degree of substi-tution. Only for [C2isoC3m3im][NTf2] (8 d), a higher meltingpoint is observed than for [C2C2m3im][NTf2] (8 c). Therefore, thepossibility of entropy reduction by hindered isopropyl rotationin the [C2isoC3m3im]+ cation was investigated. From NMR spec-tral data and semi-empirical calculations (RM1 model, Hyper-chem 8.0), which attribute a rotational energy barrier of80 kJ mol�1, it is assumed that there is free rotation. As a con-clusion, the increased melting point of [C2isoC3m3im][NTf2]compared to [C2C2m3im][NTf2] cannot be attributed to a de-crease in entropy when going from ethyl to isopropyl C2 sub-stitution.
All N-ethyl derivatives solidify rapidly upon cooling, but donot crystallize. This behaviour was already previously reportedfor the [C2C1mim][NTf2] ionic liquid.[17a] On the other hand, theN-hexyl derivatives did not show any phase transition afterkeeping the samples at �60 8C for several hours. Since theN-hexyl salts are expected to have melting points of about60–70 8C lower than their iodide precursors (as is the case forthe N-ethyl derivatives), this observation suggests that theseionic liquids undercool very well.
As can be seen in Table 2, the viscosity of the [C6m3im][NTf2]salt is about the same as that of the regular [C6mim][NTf2](71 MPa s, 25 8C).[33] Furthermore, methyl substitution at the2-position increases the viscosity by 51 MPa s, which is within
Scheme 4. Aqueous metathesis of iodide salts into their corresponding di-cyanamide and bis(trifluoromethylsulfonyl)imide salts.
&4& www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 0000, 00, 1 – 13
�� These are not the final page numbers!
C. V. Stevens et al.
the same range as the viscosity increase of [C2C1mim][NTf2] asmeasured by Noack et al.[14a] This increase was already reportedby Hunt et al. to be more pro-nounced for the larger and morediffuse anions.[32] On the otherhand, the viscosities of the di-cyanamide salts are substantiallylarger than those of the non-4,5-alkylated imidazolium ionic liq-uids (e.g. [C2mim][N(CN)2]:21 MPa s (20 8C),[29a] [C6mim][N(CN)2]: 47 MPa s (25 8C)[34]).Only small variations in viscosi-ties of the dicyanamide salts areobserved. In general, an incre-ment of the viscosities can be
measured when the degree of substitution increases.In previous research, [N(CN)2]� salts were found to have
lower viscosities than their [NTf2]� analogues.[29a] However, thiswas found to be true only for the 2-isopropyl imidazolium salts([C6isoC3m3im]+). Since molar densities of [N(CN)2]� are higherthan those of [NTf2]� salts,[35] the cations in the [N(CN)2]� saltsare presumed to be more densely packed and thus the vander Waals contribution to the melting points and viscositiesmight be larger.
2.5. Chemical Stability
The base stability of the ionic liquids was first investigated bymeans of deuterium exchange of the ionic liquids in neutraland alkaline media, as demonstrated in Table 3. All ILs weresoluble in the 7:3 CD3OD/D2O mixture. After one week, no deu-terium exchange was observable for the [C6m3im][NTf2] (8 e)ionic liquid (entry 1). After a fast complete deuterium exchangein 0.1 m KOH on C2 (entry 2), which was also reported earlier,[28]
no further reaction took place in the timespan of one week. Ina 0.1 m KOH solution, no deuteration of any of the[C6isoC3m3im][NTf2] (8 h) protons was observed (entry 3).
The stability against Hoffman elimination of both hydrophilic[C6isoC3m3im][N(CN)2] (7 h) and hydrophobic [C6isoC3m3im]-[NTf2] (8 h) was tested by refluxing the ionic liquids in a strongbasic aqueous solution (entries 4 and 5). After 24 h, the reac-tion mixtures were extracted multiple times with CH2Cl2. Inboth cases, the extracted fraction was pure IL, however in theformer case, only 53 % was recuperated. Additional extractionof the organic phase could attribute this loss to solvation ofthe hydrophilic IL in the aqueous phase, furthermore, no otherproducts than IL were found in the aqueous phase by HPLC-MS.
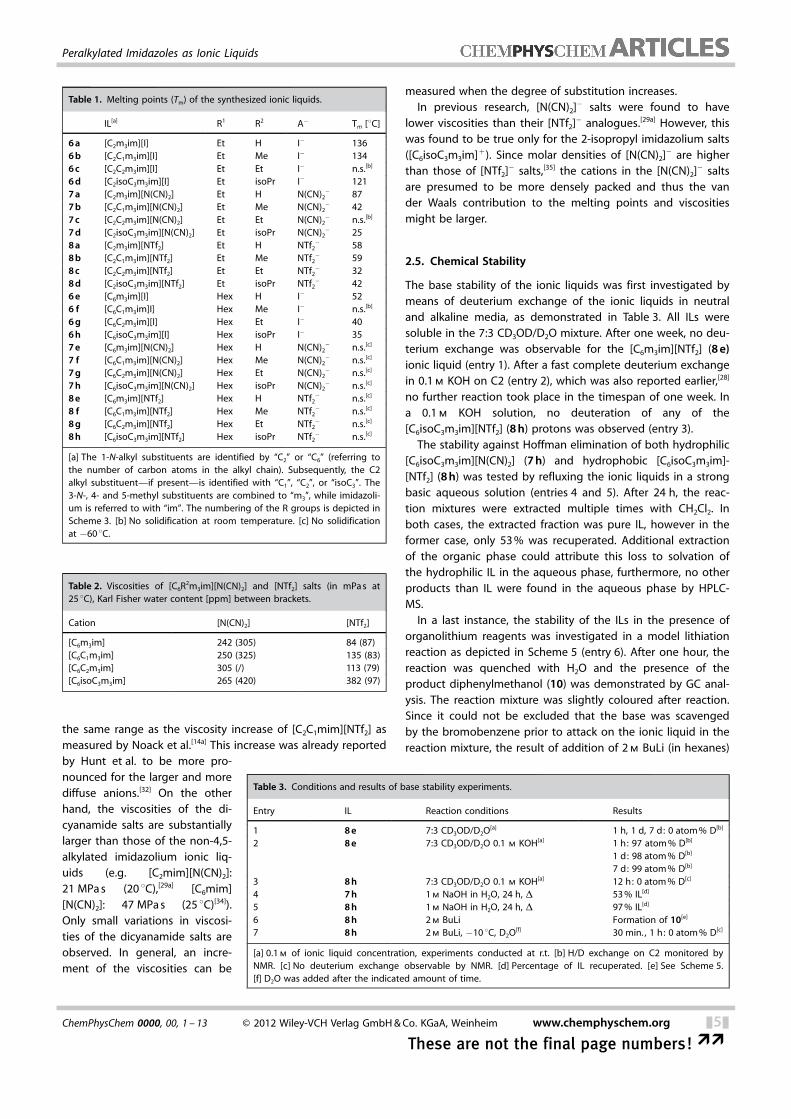
In a last instance, the stability of the ILs in the presence oforganolithium reagents was investigated in a model lithiationreaction as depicted in Scheme 5 (entry 6). After one hour, thereaction was quenched with H2O and the presence of theproduct diphenylmethanol (10) was demonstrated by GC anal-ysis. The reaction mixture was slightly coloured after reaction.Since it could not be excluded that the base was scavengedby the bromobenzene prior to attack on the ionic liquid in thereaction mixture, the result of addition of 2 m BuLi (in hexanes)
Table 1. Melting points (Tm) of the synthesized ionic liquids.
IL[a] R1 R2 A� Tm [8C]
6 a [C2m3im][I] Et H I� 1366 b [C2C1m3im][I] Et Me I� 1346 c [C2C2m3im][I] Et Et I� n.s.[b]
6 d [C2isoC3m3im][I] Et isoPr I� 1217 a [C2m3im][N(CN)2] Et H N(CN)2
� 877 b [C2C1m3im][N(CN)2] Et Me N(CN)2
� 427 c [C2C2m3im][N(CN)2] Et Et N(CN)2
� n.s.[b]
7 d [C2isoC3m3im][N(CN)2] Et isoPr N(CN)2� 25
8 a [C2m3im][NTf2] Et H NTf2� 58
8 b [C2C1m3im][NTf2] Et Me NTf2� 59
8 c [C2C2m3im][NTf2] Et Et NTf2� 32
8 d [C2isoC3m3im][NTf2] Et isoPr NTf2� 42
6 e [C6m3im][I] Hex H I� 526 f [C6C1m3im]I] Hex Me I� n.s.[b]
6 g [C6C2m3im][I] Hex Et I� 406 h [C6isoC3m3im][I] Hex isoPr I� 357 e [C6m3im][N(CN)2] Hex H N(CN)2
� n.s.[c]
7 f [C6C1m3im][N(CN)2] Hex Me N(CN)2� n.s.[c]
7 g [C6C2m3im][N(CN)2] Hex Et N(CN)2� n.s.[c]
7 h [C6isoC3m3im][N(CN)2] Hex isoPr N(CN)2� n.s.[c]
8 e [C6m3im][NTf2] Hex H NTf2� n.s.[c]
8 f [C6C1m3im][NTf2] Hex Me NTf2� n.s.[c]
8 g [C6C2m3im][NTf2] Hex Et NTf2� n.s.[c]
8 h [C6isoC3m3im][NTf2] Hex isoPr NTf2� n.s.[c]
[a] The 1-N-alkyl substituents are identified by “C2” or “C6” (referring tothe number of carbon atoms in the alkyl chain). Subsequently, the C2alkyl substituent—if present—is identified with “C1”, “C2”, or “isoC3”. The3-N-, 4- and 5-methyl substituents are combined to “m3”, while imidazoli-um is referred to with “im”. The numbering of the R groups is depicted inScheme 3. [b] No solidification at room temperature. [c] No solidificationat �60 8C.
Table 2. Viscosities of [C6R2m3im][N(CN)2] and [NTf2] salts (in mPa s at25 8C), Karl Fisher water content [ppm] between brackets.
Cation [N(CN)2] [NTf2]
[C6m3im] 242 (305) 84 (87)[C6C1m3im] 250 (325) 135 (83)[C6C2m3im] 305 (/) 113 (79)[C6isoC3m3im] 265 (420) 382 (97)
Table 3. Conditions and results of base stability experiments.
Entry IL Reaction conditions Results
1 8 e 7:3 CD3OD/D2O[a] 1 h, 1 d, 7 d: 0 atom % D[b]
2 8 e 7:3 CD3OD/D2O 0.1 m KOH[a] 1 h: 97 atom % D[b]
1 d: 98 atom % D[b]
7 d: 99 atom % D[b]
3 8 h 7:3 CD3OD/D2O 0.1 m KOH[a] 12 h: 0 atom % D[c]
4 7 h 1 m NaOH in H2O, 24 h, D 53 % IL[d]
5 8 h 1 m NaOH in H2O, 24 h, D 97 % IL[d]
6 8 h 2 m BuLi Formation of 10[e]
7 8 h 2 m BuLi, �10 8C, D2O[f] 30 min. , 1 h: 0 atom % D[c]
[a] 0.1 m of ionic liquid concentration, experiments conducted at r.t. [b] H/D exchange on C2 monitored byNMR. [c] No deuterium exchange observable by NMR. [d] Percentage of IL recuperated. [e] See Scheme 5.[f] D2O was added after the indicated amount of time.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org &5&
These are not the final page numbers! ��
Peralkylated Imidazoles as Ionic Liquids
to the IL was evaluated by quenching this mixture with D2O(entry 7). Although the mixture turned completely black, nodeuteration of the ionic liquid was observed.
2.6. Thermogravimetrical Analysis
Although dynamic thermogravimetrical analysis does not givea clear view on the long-term stability of ionic liquids at elevat-ed temperatures, it does provide valuable information on theinfluence of variation of the salt composition on the thermalstability. The onset temperature for thermal decomposition(Tonset), that is, the calculated intercept of the line where noweight loss occurs and the tangent upon weight loss, is givenin Table 4. For most of the salts, a lower thermal stability for
the N-hexyl substituted analogues is observed, although litera-ture states that the alkyl chain length should not affect thethermal degradation temperature.[36] A significant reduction ofTonset for the non-C2-substituted imidazolium salts (8 a, 8 e,Table 4) is apparent, this is in agreement with different experi-mental results (Figure 3).[2, 29a, 37]
Two decomposition mechanisms are proposed: on the onehand an E1-type elimination (Scheme 6 a), forming a volatilecarbene and an isopropyl cation at high temperatures,[36] andon the other hand the reverse Menshutkin reaction(Scheme 6 b).[38] Although the reported ionic liquids have onlyweakly coordinating anions, Hofmann elimination cannot beexcluded.
The decomposition of the bis(trifluoromethylsulfonyl)imidesalts is assumed to proceed via the exothermal decompositionof the anion, releasing SO2.[38] At a heating rate of 10 8C min�1,all the [NTf2]� salts were found to be stable up to 400 8C,which was set as the upper limit of measurement.
2.7. Electrochemical Analysis
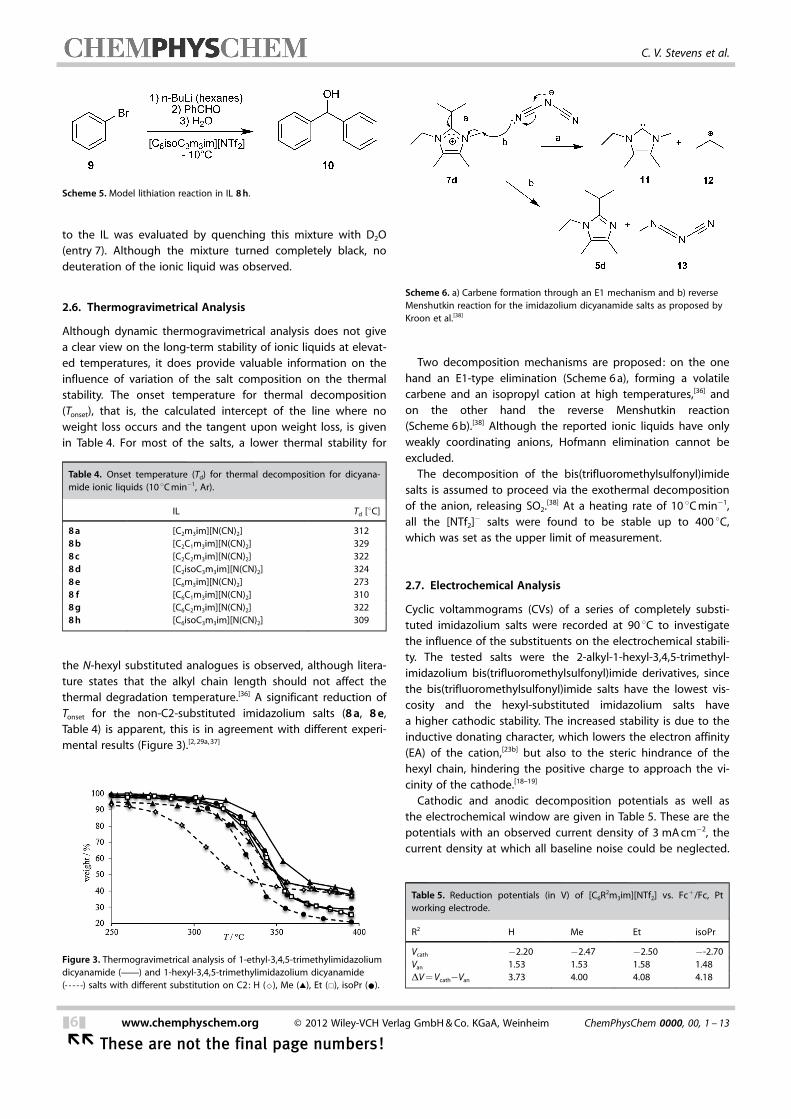
Cyclic voltammograms (CVs) of a series of completely substi-tuted imidazolium salts were recorded at 90 8C to investigatethe influence of the substituents on the electrochemical stabili-ty. The tested salts were the 2-alkyl-1-hexyl-3,4,5-trimethyl-imidazolium bis(trifluoromethylsulfonyl)imide derivatives, sincethe bis(trifluoromethylsulfonyl)imide salts have the lowest vis-cosity and the hexyl-substituted imidazolium salts havea higher cathodic stability. The increased stability is due to theinductive donating character, which lowers the electron affinity(EA) of the cation,[23b] but also to the steric hindrance of thehexyl chain, hindering the positive charge to approach the vi-cinity of the cathode.[18–19]
Cathodic and anodic decomposition potentials as well asthe electrochemical window are given in Table 5. These are thepotentials with an observed current density of 3 mA cm�2, thecurrent density at which all baseline noise could be neglected.
Scheme 5. Model lithiation reaction in IL 8 h.
Table 4. Onset temperature (Td) for thermal decomposition for dicyana-mide ionic liquids (10 8C min�1, Ar).
IL Td [8C]
8 a [C2m3im][N(CN)2] 3128 b [C2C1m3im][N(CN)2] 3298 c [C2C2m3im][N(CN)2] 3228 d [C2isoC3m3im][N(CN)2] 3248 e [C6m3im][N(CN)2] 2738 f [C6C1m3im][N(CN)2] 3108 g [C6C2m3im][N(CN)2] 3228 h [C6isoC3m3im][N(CN)2] 309
Figure 3. Thermogravimetrical analysis of 1-ethyl-3,4,5-trimethylimidazoliumdicyanamide (c) and 1-hexyl-3,4,5-trimethylimidazolium dicyanamide(a) salts with different substitution on C2: H (^), Me (~), Et (&), isoPr (*).
Scheme 6. a) Carbene formation through an E1 mechanism and b) reverseMenshutkin reaction for the imidazolium dicyanamide salts as proposed byKroon et al.[38]
Table 5. Reduction potentials (in V) of [C6R2m3im][NTf2] vs. Fc+/Fc, Ptworking electrode.
R2 H Me Et isoPr
Vcath �2.20 �2.47 �2.50 �-2.70Van 1.53 1.53 1.58 1.48DV = Vcath�Van 3.73 4.00 4.08 4.18
&6& www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 0000, 00, 1 – 13
�� These are not the final page numbers!
C. V. Stevens et al.
Although [NTf2]� anions can influence the cathodic decomposi-tion behaviour,[39] and imidazolium cations can be oxidized atpositive potentials,[40] a clear qualitative effect on the electro-chemical window by the substituents was observed. The stabil-ity against cathodic reduction of the cation increases bychanging the alkyl substituent in the order (H<Me<Et<isoPr), and at positive potentials no significant influence wasapparent. Thus, as for the N-hexyl substitution, the increasedsubstitution on the C2 position is also believed to increase thecathodic stability by increment of both steric bulk and electrondensity in the aromatic system (Figure 4).
3. Conclusions
Tetrasubstituted imidazoles are readily synthesized by meansof a one-pot reaction. Formation of 1-H-imidazole is therebyavoided. It is also possible to synthesize these imidazoles usingcontinuous-flow technology, which can provide the imidazoleson an industrial scale. The imidazole core is easily methylatedwith iodomethane to obtain imidazolium iodides, and consec-utive metathesis reactions give dicyanamide and bis(trifluoro-methylsulfonyl)imide ionic liquids in almost quantitative yields.The N-ethyl-substituted imidazolium iodide salts are solids,which allows for easy purification, while the N-hexyl imidazoli-um iodide analogues are liquid at room temperature. Althoughthese imidazolium ionic liquids are fully substituted, the dicya-namide and bis(trifluoromethylsulfonyl)imide salts all remainliquid below 100 8C and several are liquid at room tempera-ture. The N-hexyl substituted derivatives undercool easily, andthus possess a wide liquid range. Unlike literature values, melt-ing points are found to decrease upon methylation at the C2position. However, viscosities increase upon increasing thealkyl groups, which is in agreement with previous research.Compared to conventional imidazolium ionic liquids, thesefully substituted ionic liquids are applicable in strong alkaline
media. Thermal stability is clearly increased upon alkylation ofthe C2 position, however a distinct effect was not observedwhen changing the alkyl substituent from methyl to isopropyl.On the other hand, the best electrochemical performance wasobserved for the isopropyl substituted derivative. This stabiliz-ing effect was attributed to both steric hindrance and induc-tive neutralization of the positive charge.
Experimental Section
Reactants
Ethylamine (�99.5 %), n-hexylamine (�99 %), diacetyl (�97 %), pro-pionaldehyde (�97 %), isobutyraldehyde (�98 %), ammonium ace-tate (�98 %), silver nitrate (�99.0 %), sodium dicyanamide (96 %),lithium bis(trifluoromethylsulfonyl)imide (�99.0 %), KOH (90 %),NaOH (�97 %), CD3OD (99.96 atom % D), D2O (99.99 atom % D),nBuLi (2.3 m in hexanes), iodomethane (�99.0 %), bromobenzene(99 %), benzaldehyde (�99 %), diphenylmethanol (99 %), methanol,n-pentanol, acetonitrile, dichloromethane and ethyl acetate werepurchased from Sigma–Aldrich, paraformaldehyde and acetalde-hyde from ACROS, acetone from Fisher Chemicals and acetic acidfrom Riedel-de Ha�n. Acetone and acetonitrile were dried over 3 �molecular sieves and CH2Cl2 was refluxed with CaH and distilledprior to use.
Apparatus
Batch reactions were performed in 15 mL and 150 mL glass pres-sure vials with PBT caps (Schott) and PTFE seals for reactions up to20 mmol and 200 mmol respectively. The microreactor used wasa Cytos CPC College System, and the mixing unit and residencetime unit were retained at 120 8C. The internal volume of themixing unit and residence time unit is 48 mL, the applied flow ratewas 0.5 to 2.0 mL min�1. 1H NMR, 13C NMR, 19F NMR and HSQC ex-periments were performed with a JEOL ECP + 300 spectrometer inCDCl3 with TMS as an internal reference. IR spectra were recordedon a PerkinElmer Spectrum FT-IR apparatus (ZnSe crystal ; ATRmode). HPLC-MS spectra were recorded on an Agilent 1100 Series(ES, 4000 V) preceded by a reverse-phase LC column. GC analysiswere performed on a Agilent 6890 GC Plus Series, with an EC5 ca-pillary column (30 m � 0.25 mm; coating thickness 0.25 mm) (injec-tor temp.: 250 8C; carrier gas: He; flow rate: 1.0 mL min�1, temp.profile: 80–200 8C at 10 8C min�1, 200–280 at 30 8C min�1, 5 minhold at 280 8C; detector: FID). Melting points of the iodide saltswere measured with a B�chi B-540 apparatus and are uncorrected.Water contents were measured in Hydranal 34812 (Fluka) witha 719S Titrino coulometric Karl Fischer titrator (Metrohm). DSCmeasurements were done with a Mettler-Toledo 822 DSC appara-tus (heating rate: 10 8C min�1), and all samples were analysedunder a dry helium flow. Viscosities were analysed with a BrookfieldViscometer DV-II Pro at 25 8C and under a nitrogen atmosphere.Cyclic voltammograms were recorded with a Solartron SI 1287Electrochemical Interface (scanning rate: 20 mV s�1; platina workand reference electrodes; counter electrode was platina wire sub-merged in 1-butyl-3-methylpyrrolidinium bis(trifluoromethylsulfo-nyl)imide and calibrated with Fc+/Fc). All CVs were recorded at90 8C under a nitrogen atmosphere, samples were dried prior to re-cording. The thermal stabilities of the ionic liquids were analyzedby thermogravimetric analysis at a heating rate of 10 8C min�1 inalumina pans under an argon atmosphere (100 mL min�1 flow)using a SDT Q600 V8.3 Build 101 apparatus, the sample weightwas between 5 and 15 mg.
Figure 4. Cyclic voltammograms of [C6R2m3im][NTf2] , (R2 : H, Me, Et, isoPr) ; Ptworking electrode at 90 8C.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org &7&
These are not the final page numbers! ��
Peralkylated Imidazoles as Ionic Liquids

Batch Procedure for the Synthesis of 1-Ethyl-2-Isopropyl-4,5-Dimethylimidazole (5 d)
A glass 150 mL pressure vial was filled with MeOH (50 mL) anda PTFE stirring bar. The solvent was allowed to cool to 0 8C and iso-butyraldehyde (100 mmol, 7.21 g) and ethylamine (120 mmol,5.41 g) were added. The vial was closed and submerged in a pre-heated oil bath at 120 8C with temperature controller. After heatingfor 2 h with vigorous stirring, the reaction mixture was carefullycooled to 0 8C, the vial was opened and consecutively diacetyl(100 mmol, 8.61 g) and NH4OAc (100 mmol, 7.71 g) were added.The vessel was closed immediately and heated in a preheated oilbath at 120 8C for 2 h under vigorous stirring. After cooling, MeOHand the reaction water were removed in vacuo. The reaction mix-ture was dissolved in CH2Cl2 and extracted twice with 100 mL of anaqueous 0.5 m HCl solution. The aquatic phase was neutralizedwith 50 mL of 3.0 m NaOH and extracted three times with 40 mL ofEtOAc. The resulting organic phase was washed with 100 mL ofwater and dried over MgSO4. The solvent was removed in vacuoand 6.97 g (41 %) of a black oil was obtained. The resulting oil waspurified by vacuum distillation (52–86 8C/1 mbar), yielding a yellowoil, which was further used for the synthesis of ionic liquids.
Characterisation of Products 5 a–d
1-Ethyl-4,5-Dimethylimidazole (5 a): b.p. 52 8C (1.0 mbar) ; 1H NMR(300 MHz, CDCl3, 25 8C, TMS): d= 1.37 (t, 3J(H,H) = 7.4 Hz, 3 H;CH3CH2), 2.12 (s, 3 H; CCH3), 2.15 (s, 3 H; CCH3), 3.83 (q, 3J(H,H) =
7.4 Hz, 2 H; CH2), 7.33 ppm (s, 1 H; C2H); 13C NMR (75 MHz, CDCl3,25 8C, TMS): d= 8.32 (CH3C), 12.75 (CH3C), 16.21 (CH3CH2), 39.68(CH2), 121.79 (CH3C), 133.79 (CH3C), 134.04 ppm (CCH); IR (ATR): n=1351, 1383, 1415, 2920, 2971 cm�1; MS (ES): m/z [%]: 125.3 (100)[M + H+] ; yield: 40 %, yellow oil.
1-Ethyl-4,5-Dimethylimidazole (5 b): b.p. 58 8C (1.0 mbar) ; 1H NMR(300 MHz, CDCl3, 25 8C, TMS): d= 1.24 (t, 3J(H,H) = 7.2 Hz, 3 H;CH3CH2), 2.10 (s, 6 H; C4/5CH3), 2.15 (s, 3 H; CCH3), 2.34 (s, 3 H;C2CH3), 3.77 ppm (q, 3J(H,H) = 7.2 Hz, 2 H; CH2); 13C NMR (75 MHz,CDCl3, 25 8C, TMS): d= 8.79 (CH3C), 12.49 (CH3), 13.18 (CH3C2), 15.71(CH3CH2), 38.38 (CH2), 121.15 (CH3C), 131.26 (CH3C), 141.72 ppm(CCH); IR (ATR): n= 1311, 1375, 1350, 1413, 2919, 2974 cm�1; MS(ES): m/z [%]: 139.3 (100) [M + H+] ; yield: 39 %, yellow oil.
1,2-Diethyl-4,5-Dimethylimidazole (5 c): b.p. 51 8C (1.0 mbar);1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.25 (t, 3J(H,H) = 7.2 Hz,3 H; CH3CH2), 1.32 (t, 3 H, 3J(H,H) = 7.7 Hz; CCH2CH3), 2.11 (s, 3 H;CCH3), 2.13 (s, 3 H; CCH3), 2.65 (q, 2 H, 3J(H,H) = 7.7 Hz; CCH2),3.78 ppm (q, 3J(H,H) = 7.4 Hz, 2 H; NCH2); 13C NMR (75 MHz, CDCl3,25 8C, TMS): d= 8.70 (CH3C), 12.46 (CH3C), 12.70 (CH3CH2C), 16.02(NCH2CH3), 20.32 (CCH2), 38.08 (NCH2), 121.06 (CH3C), 131.29 (CH3C),146.68 ppm (CCH2); IR (ATR): n= 1068, 1311, 1353, 1376, 1429,2972 cm�1; MS (ES): m/z [%]: 153.3 (100) [M + H+] ; yield: 37 %, clearliquid.
1-Ethyl-2-Isopropyl-4,5-Dimethylimidazole (5 d): b.p. 60 8C(1.0 mbar) ; 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.26 (t,3J(H,H) = 7.2 Hz, 3 H; CH3CH2), 1.32 (d, 3J(H,H) = 6.6 Hz, 6 H;(CH3)2CH), 2.10 (s, 3 H; CCH3), 2.14 (s, 3 H; CCH3), 2.94 (m, 3J(H,H) =6.6 Hz, 1 H; CH), 3.80 ppm (q, 3J(H,H) = 7.2 Hz, 2 H; CH2); 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 8.76 (CH3C), 12.66 (CH3C), 16.38(CH3CH2), 22.25 ((CH3)2CH), 26.02 (CH), 37.80 (CH2), 120.68 (CH3C),131.37 (CH3C), 150.48 ppm (CCH); IR (ATR): n= 1088, 1311, 1321,1438, 2924, 2968 cm�1; MS (ES): m/z [%]: 167.3 (100) [M + H+] ;yield: 42 %, yellow oil.
Optimized Continuous-Flow Procedure for the Synthesis of1-Hexyl-2-Isopropyl-4,5-Dimethylimidazole (5 h)
In an oven-dried 500 mL flask containing dry CH2Cl2 (200 mL) wereadded isobutyraldehyde (0.5 mol, 36.05 g), hexylamine (0.5 mol,50.60 g) and MgSO4 (1 mol, 120.37 g). The mixture was allowed tostir overnight, protected with a drying tube (CaCl2). The reactionmixture was filtered and the solvent was removed in vacuo. Theproduct was diluted with n-pentanol in a 1 L volumetric flask to0.5 m. To a second 1 L volumetric flask containing n-pentanol wereadded diacetyl (0.5 mol, 43.05 g) and NH4OAc (0.5 mol, 38.53 g),the solution was diluted to 0.5 m with n-pentanol. Both volumetricflasks were connected to the pumps on the CYTOS CollegeSystem. The flow rate was set to 1.2 mL min�1 on each pump (tres =20 min) after volumetric calibration. Upon obtaining steady stateconditions (32 min), a red to black solution was collected, contain-ing 0.087 m (35 %) of the product, with a product output of2.55 g. h�1. Vacuum fractional distillation was used to remove thesolvent (85 8C/60 mbar), the residual black oil was dissolved inCH2Cl2, and extracted twice with 500 mL of 0.5 m HCl, the aqueousphase was neutralized with 250 mL of 3.0 m NaOH and extractedwith 3 � 100 mL of hexanes. The organic layer was dried withMgSO4 and the solvent was removed in vacuo, yielding 31.1 g ofa black oil. This oil was fractionally distilled under a high vacuumto afford the product as a yellow oil (83–86 8C/1.0 mbar).
Characterisation of Products 5 e–h
1-Hexyl-4,5-Dimethylimidazole (5 e): b.p. 83 8C (1.0 mbar); 1H NMR(300 MHz, CDCl3, 25 8C, TMS): d= 0.88 (t, 3J(H,H) = 6.6 Hz, 3 H;CH2CH3), 1.27–1.34 (m; 6 H; (CH2)3CH3), 1.63–1.70 (m, 2 H; NCH2CH2),2.11 (s, 3 H; CCH3), 2.15 (s, 3 H; CCH3), 3.76 (t, 3J(H,H) = 7.4 Hz, 2 H;NCH2), 7.31 ppm (s, 1 H; C2H); 13C NMR (75 MHz, CDCl3, 25 8C, TMS):d= 8.44 (CH3C), 12.78 (CH3C), 13.99 (CH3(CH2)5), 22.51 (CH2), 26.29(CH2), 30.78 (NCH2CH2), 31.33 (CH2), 44.96 (NCH2), 121.87 (CH3C),133.70 (CH3C), 134.76 ppm (CCH); IR (ATR): n= 647, 1232, 1450,1500, 2954 cm�1; MS (ES): m/z [%]: 181.3 (100) [M + H+] ; yield:38 %, clear liquid.
1-Hexyl-2,4,5-Trimethylimidazole (5 f): b.p. 84 8C (1.0 mbar); 1H NMR(300 MHz, CDCl3, 25 8C, TMS): d= 0.89 (t, 3J(H,H) = 6.6 Hz, 3 H;CH2CH3), 1.25–1.34 (m; 6 H; (CH2)3CH3), 1.53–1.62 (m, 2 H; NCH2CH2),2.08 (s, 3 H; CCH3), 2.10 (s, 3 H; CCH3), 2.32 (3 H, C2CH3, s), 3.68 ppm(t, 3J(H,H) = 7.5 Hz, 2 H; NCH2); 13C NMR (75 MHz, CDCl3, 25 8C, TMS):d= 8.95 (CH3C), 12.53 (CH3C), 13.36 (CH3C2), 13.98 (CH3(CH2)5), 22.55(CH2), 26.49 (CH2), 30.60 (NCH2CH2), 31.47 (CH2), 43.83 (NCH2),121.40 (CH3C), 131.20 (CH3C), 141.95 ppm (CCH); IR (ATR): n= 726,1128, 1413, 1603, 2925 cm�1; MS (ES): m/z [%]: 195.3 (100) [M + H+
] ; yield: 36 %, yellow oil.
2-Ethyl-1-Hexyl-4,5-Dimethylimidazole (5 g): b.p. 86 8C (1.0 mbar);1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.89 (t, 3J(H,H) = 6.6 Hz,3 H; CH2CH3), 1.26–1.37 (m; 6 H; (CH2)3CH3), 1.53–1.62 (m, 2 H;NCH2CH2), 2.09 (s, 3 H; CCH3), 2.13 (s, 3 H; CCH3), 2.63 (q, 3J(H,H) =7.5 Hz, 2 H; C2CH2CH3), 3.68 ppm (t, 3J(H,H) = 7.7 Hz, 2 H, NCH2);13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.92 (CH3CH2C2), 12.63(CH3C4, CH3C5), 13.97 (CH3CH2C2), 13.97 (CH3(CH2)5), 22.56 (CH2),26.55 (CH2), 30.89 (NCH2CH2), 31.48 (CH2), 43.54 (NCH2), 121.26(CH3C), 131.35 (CH3C), 146.91 ppm (CCH2); IR (ATR): n= 1070, 1310,1372, 1426, 2928, 2957 cm�1; MS (ES): m/z [%]: 209.3 (100) [M + H+
] ; yield: 32 %, yellow oil.
1-Hexyl-2-Isopropyl-4,5-Dimethylimidazole (5 h): b.p. 86 8C(1.0 mbar) ; 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.90 (t,3J(H,H) = 6.6 Hz, 3 H; CH2CH3), 1.24–1.36 (m; 6 H; (CH2)3CH3), 1.31 (d,
&8& www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 0000, 00, 1 – 13
�� These are not the final page numbers!
C. V. Stevens et al.

3J(H,H) = 6.6 Hz, 6 H; (CH3)2CH,), 1.53–1.65 (m, 2 H; NCH2CH2), 2.09(s, 3 H; CCH3), 2.14 (s, 3 H; CCH3), 2.86–2.99 (1 H, CH, m), 3.70 ppm(t, 3J(H,H) = 8.0 Hz, 2 H; NCH2); 13C NMR (75 MHz, CDCl3, 25 8C, TMS):d= 8.92 (CH3C), 12.73 (CH3C), 13.99 (CH3CH2), 22.28 ((CH3)2CH),22.57 (CH2), 26.05 (CH2), 26.57 (NCH2CH2), 31.27 (CH2), 31.48 (CH),43.30 (NCH2), 120.19 (CCH3), 131.32 (CCH3), 150.71 ppm (CCH); IR(ATR): n= 1085, 1312, 1433, 2926, 2960 cm�1; MS (ES): m/z [%]:223.3 (100) [M + H+] ; yield: 40 %, yellow oil.
Batch Procedure for the Synthesis of 1-Ethyl-2-Isopropyl-3,4,5-Trimethylimidazolium Iodide ([C2isoC3m3im][I] , 6 d)
In an oven-dried flask of 100 mL, 1-ethyl-2-isopropyl-4,5-dimethyl-imidazolium (20 mmol, 3.32 g) was dissolved in dry CH3CN (40 mL).The solution was cooled to 0 8C and MeI (60 mmol, 8.47 g) wasadded dropwise at 0 8C. The reaction mixture was allowed to stir atroom temperature for 8 h. When the reaction was completed, thesolvent and residual MeI were removed in vacuo. The resultingyellow solid was recrystallized in dry acetone to furnish transparentto white crystals (1st crop: 75 %, 2nd crop: 11 %). Residual solventwas removed in a high vacuum.
Characterisation of Products 6 a–d
1-Ethyl-3,4,5-Trimethylimidazolium Iodide [C2m3im][I] (6 a): 1H NMR(300 MHz, CDCl3, 25 8C, TMS): d= 1.59 (t, 3J(H,H) = 7.4 Hz, 3 H;NCH2CH3), 2.26 (s, 3 H; CH3C), 2.27 (s, 3 H; CH3C), 3.94 (s, 3 H; NCH3),4.21 (q, 3J(H,H) = 7.4 Hz, 2 H; NCH2CH3), 10.13 ppm (s, 1 H; CH);13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.76 (CH3C), 8.79 (CH3C),15.34 (CH3CH2), 34.41 (NCH3), 42.70 (CH2), 126.30 (CH3C), 127.34(CH3C), 134.92 ppm (CH); IR (ATR): n= 1199, 1241, 1572, 1634,3032 cm�1; MS (ES): m/z [%]: 139.3 (100) [M + H+] ; yield: 87 %,transparent crystals.
1-Ethyl-2,3,4,5-Tetramethylimidazolium Iodide [C2C1m3im][I] (6 b):1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.42 (t, 3J(H,H) = 7.4 Hz,3 H; NCH2CH3), 2.27 (s, 6 H; CH3C4/5), 2.86 (s, 3 H; CH3C2), 3.79 (s, 3 H;NCH3), 4.17 ppm (q, 3J(H,H) = 7.4 Hz, 2 H; NCH2CH3) ; 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 9.04 (CH3C4/5), 9.39 (CH3C4/5), 12.44(CH3C2), 15.33 (CH3CH2), 33.71 (NCH3), 41.52 (CH2), 124.80 (C4/5),126.25 (C4/5), 142.10 ppm (C2) ; IR (ATR): n= 1198, 1456, 1572, 1634,2358, 3032 cm�1; MS (ES): m/z [%]: 153.3 (100) [M + H+] ; yield:89 %, transparent crystals.
1,2-Diethyl-3,4,5-Trimethylimidazolium Iodide [C2C2m3im][I] (6 c):1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.34 (t, 3J(H,H) = 7.7 Hz,3 H; CCH2CH3), 1.46 (t, 3J(H,H) = 7.4 Hz, 3 H; NCH2CH3), 2.28 (s, 6 H;CH3C), 3.22 (q, 3J(H,H) = 7.7 Hz, 2 H; CCH2CH3), 3.81 (s, 3 H; NCH3),4.16 ppm (q, 3J(H,H) = 7.4 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz,CDCl3, 25 8C, TMS): d= 6.99 (CH3C), 7.33 (CH3C), 10.32 (CH3CH2C),13.80 (NCH2CH3), 16.47 (CH2C), 31.33 (NCH3), 39.30 (NCH2), 122.86(CH3C), 124.47 (CH3C), 143.89 ppm (CCH2); IR (ATR): n= 1090, 1259,1448, 1529, 1648, 2975 cm�1; MS (ES): m/z [%]: 167.3 (100) [M + H+
] ; yield: 99 %, reddish oil.
1-Ethyl-2-Isopropyl-3,4,5-Trimethylimidazolium Iodide [C2isoC3m3im][I] (6 d): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.43 (t, 3J(H,H) =7.2 Hz, 3 H; NCH2CH3), 1.55 (d, 3J(H,H) = 7.2 Hz, 6 H; (CH3)2CH), 2.30(s, 6 H; CH3C), 3.66–3.81 (m, 1 H; CH), 3.87 (s, 3 H; NCH3), 4.26 ppm(q, 3J(H,H) = 7.2 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C,TMS): d= 9.26 (CH3C), 9.63 (CH3C), 15.94 (CH3CH2), 19.91 ((CH3)2CH),25.35 (CH), 34.06 (NCH3), 41.65 (CH2), 125.18 (CH3C), 127.17 (CH3C),147.72 ppm (CCH); IR (ATR): n= 1088, 1331, 1448, 1521, 1651,2971 cm�1; MS (ES): m/z [%]: 181.3 (100) [M + H+] ; yield: 86 %,transparent crystals.
Batch Procedure for the Synthesis of 1-Hexyl-2-Isopropyl-3,4,5-Trimethylimidazolium Iodide ([C6 isoC3 m3im][I] , 6 h)
In an oven-dried flask of 100 mL, 1-hexyl-2-isopropyl-4,5-dimethyl-imidazole (20 mmol, 4.44 g) was dissolved in dry CH3CN (40 mL).The solution was cooled to 0 8C and MeI (60 mmol, 8.47 g) wasadded dropwise at 0 8C. The reaction mixture was allowed to stir atroom temperature for 20 h. When the reaction was completed, thesolvent and residual MeI were removed in vacuo. The resultingyellow oil was dissolved in 20 mL CH3CN and washed 3 times with20 mL hexanes. The solvent was removed in vacuo, yielding 6.99 g(96 %) of a yellow oil.
Characterisation of Products 6 e–h
1-Hexyl-3,4,5-Trimethylimidazolium Iodide [C6m3im][I] (6 e): 1H NMR(300 MHz, CDCl3, 25 8C, TMS): d= 0.89 (t, 3J(H,H) = 6.6 Hz, 3 H;CH2CH3), 1.32–1.34 (m; 6 H; (CH2)3CH3), 1.82–1.92 (m, 2 H; NCH2CH2 ;m), 2.27 (s, 3 H; CCH3), 2.29 (s, 3 H; CCH3), 3.95 (s, 3 H; NCH3), 4.15(2 H, t, 3J(H,H) = 7.4 Hz, 2 H; NCH2), 9.96 ppm (s, 1 H; C2H); 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 8.90 (CH3C4, CH3C5), 14.00(CH3(CH2)5), 22.46 (CH2), 26.05 (CH2), 29.93 (NCH2CH2), 31.16 (CH2),34.54 (NCH3), 47.47 (NCH2), 126.36 (CH3C), 127.38 (CH3C),135.21 ppm (CH); IR (ATR): n= 1202, 1452, 1570, 1633, 2928 cm�1;MS (ES): m/z [%]: 195.3 (100) [M + H+] ; yield: 99 %, yellow oil.
1-Hexyl-2,3,4,5-Tetramethylimidazolium Iodide [C6C1m3im][I] (6 f):1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.89 (t, 3J(H,H) = 6.6 Hz,3 H; CH2CH3), 1.27–1.44 (m; 6 H; (CH2)3CH3), 1.67–1.77 (m, 2 H;NCH2CH2 ; m), 2.27 (s, 3 H; CCH3), 2.28 (s, 3 H; CCH3), 2.82 (s, 3 H;C2CH3), 3.81 (s, 3 H; NCH3), 4.06 ppm (2 H, t, 3J(H,H) = 7.4 Hz, 2 H;NCH2); 13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 9.22 (CH3C), 9.47(CH3C), 12.43 (CH3C2), 14.00 (CH3(CH2)5), 22.41 (CH2), 26.26 (CH2),29.79 (NCH2CH2), 31.18 (CH2), 33.77 (NCH3), 46.29 (NCH2), 125.03(CH3C), 126.25 (CH3C), 142.02 ppm (C2CH3); IR (ATR): n= 727, 9201373, 1444, 1533, 1648, 2929 cm�1; MS (ES): m/z [%]: 209.3 (100)[M + H+] ; yield: 99 %, yellow oil.
2-Ethyl-1-Hexyl-3,4,5-Trimethylimidazolium Iodide [C6C2m3im][I](6 g): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.90 (t, 3J(H,H) =6.9 Hz, 3 H; CH2CH3), 1.26–1.44 (m; 6 H; (CH2)3CH3), 1.34 (t, 3J(H,H) =7.7 Hz, 3 H; CCH2CH3), 1.69–1.80 (m, 2 H; NCH2CH2; m), 2.27 (s, 3 H;CCH3), 2.29 (s, 3 H; CCH3), 3.19 (q, 3J(H,H) = 7.7 Hz, 2 H; C2CH2CH3),3.83 (s, 3 H; NCH3), 4.04 ppm (2 H, t, 3J(H,H) = 8.0 Hz, 2 H; NCH2);13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 9.36 (CH3C), 9.56 (CH3C),12.35 (CH3CH2C2), 13.94 (CH3(CH2)5), 18.49 (CH3CH2C2), 22.35 (CH2),26.23 (CH2), 30.38 (NCH2CH2), 31.15 (CH2), 33.64 (NCH3), 46.19(NCH2), 125.14 (CH3C), 126.57 (CH3C), 145.90 ppm (CCH2); IR (ATR):n= 726, 1081, 1452, 1528, 2929 cm�1; MS (ES): m/z [%]: 223.3 (100)[M + H+] ; yield: 99 %, yellow oil.
1-Hexyl-2-Isopropyl-3,4,5-Trimethylimidazolium Iodide[C6isoC3m3im][I] (6 h): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d=0.87–0.89 (m, 3 H; CH2CH3), 1.28–1.48 (m; 6 H; (CH2)3CH3), 1.52–1.77(m, 2 H; NCH2CH2), 1.55 (d, 3J(H,H) = 7.2 Hz, 6 H; (CH3)2CH), 2.28 (s,3 H; CCH3), 2.31 (s, 3 H; CCH3), 3.60–3.72 (m, 1 H; (CH3)2CH), 3.87 (s,3 H; NCH3), 4.10 ppm (2 H, t, 3J(H,H) = 7.4 Hz, 2 H; NCH2); 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 9.50 (CH3C), 9.73 (CH3C), 13.96(CH3(CH2)5), 19.88 ((CH3)2CH), 22.44 (CH2), 25.36 (CH2), 26.28(NCH2CH2), 30.61 (CH2), 31.24 (NCH3), 34.29 (CH), 46.18 (CH2),125.34 (CH3C), 127.24 (CH3C), 147.72 ppm (CCH); IR (ATR): n= 746,1239, 1334, 1454, 1520, 2928 cm�1; MS (ES): m/z [%]: 237.3 (100)[M + H+] ; yield: 99 %, yellow oil.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org &9&
These are not the final page numbers! ��
Peralkylated Imidazoles as Ionic Liquids

Batch Procedure for the Synthesis of 1-Ethyl-2-Isopropyl-3,4,5-Trimethylimidazolium Dicyanamide ([C2isoC3m3im][N(CN)2], 7 d)
To a 50 mL flask charged with the ionic liquid [C2isoC3m3im][I](20 mmol, 6.18 g) was added demineralized water (20 mL) andCH3CN (10 mL). Then silver dicyanamide (24 mmol, 4.15 g) wasadded to the solution. The flask was equipped with a cooler witha cotton plug and the mixture was heated to 50 8C for 8 h. After re-action, the mixture was allowed to cool to 0 8C. The precipitatewas filtered off and the water was removed in vacuo. The resultingyellow oil was dissolved in dry CH2Cl2 and cooled to �18 8C, afterfiltration of a white precipitate. This step was repeated until no fur-ther precipitation was visible, and dichloromethane was removedin vacuo. The ionic liquid was dried for 8 h at 80 8C under reducedpressure (0.5 mbar). An opaque white solid was obtained in quanti-tative yield.
Characterisation of Products 7 a–h
1-Ethyl-3,4,5-Trimethylimidazolium Dicyanamide [C2m3im][N(CN)2](7 a): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.58 (t, 3J(H,H) =7.3 Hz, 3 H; NCH2CH3), 2.30 (s, 3 H; CH3C), 2.31 (s, 3 H; CH3C), 3.87 (s,3 H; NCH3), 4.18 (q, 3J(H,H) = 7.4 Hz, 2 H; NCH2CH3), 9.05 ppm (s, 1 H;CH); 13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.20 (CH3C), 8.31(CH3C), 14.67 (CH3CH2), 33.61 (NCH2), 42.29 (NCH3), 119.50 (N(CN)2),126.86 (CH3C), 127.70 (CH3C), 133.60 ppm (CH); IR (ATR): n= 1313,1571, 2131, 2230, 2362, 2980 cm�1; MS (ES): m/z [%]: 139.3 (100)[M + H+] ; yield: 86 %, yellow wax.
1-Ethyl-2,3,4,5-Tetramethylimidazolium Dicyanamide [C2C1m3im][N(CN)2] (7 b): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.42 (t,3J(H,H) = 7.4 Hz, 3 H; NCH2CH3), 2.29 (s, 3 H; CH3C4/5), 2.31 (s, 3 H;CH3C4/5), 2.71 (s, 3 H; CH3C2), 3.72 (s, 3 H; NCH3), 4.14 ppm (q,3J(H,H) = 7.4 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C,TMS): d= 8.55 (CH3C), 8.76 (CH3C), 10.18 (CH3C2), 14.86 (CH3CH2),32.12 (CH2), 40.83 (NCH3), 119.56 (N(CN)2), 125.03 (CH3C), 126.34(CH3C), 141.67 ppm (C2CH3); IR (ATR): n= 1302, 1447, 1537, 2125,2223 cm�1; MS (ES): m/z [%]: 153.3 (100) [M + H+] ; yield: 83 %,yellow wax.
1,2-Diethyl-3,4,5-Trimethylimidazolium Dicyanamide [C2C2m3im][N(CN)2] (7 c): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 1.36 (t,3J(H,H) = 7.7 Hz, 3 H; CCH2CH3), 1.45 (t, 3J(H,H) = 7.4 Hz, 3 H;NCH2CH3), 2.30 (s, 3 H; CH3C), 2.32 (s, 3 H; CH3C), 3.09 (q, 3J(H,H) =7.7 Hz, 2 H; CCH2CH3), 3.75 (s, 3 H; NCH3), 4.14 ppm (q, 3J(H,H) =7.4 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C, TMS): d=
8.52 (CH3C), 8.69 (CH3C), 11.73 (CH3CH2C), 15.47 (NCH2CH3), 17,36(CH2C), 31.88 (NCH2), 40.67 (NCH3), 119.63 (N(CN)2), 125.09 (CH3C),126.59 (CH3C), 145.55 ppm (CCH2); IR (ATR): n= 736, 919, 1307,1530, 2128, 2227 cm�1; MS (ES): m/z [%]: 167.3 (100) [M + H+] ;yield: 89 %, reddish oil.
1-Ethyl-2-Isopropyl-3,4,5-Trimethylimidazolium Dicyanamide[C2isoC3m3im][N(CN)2] (7 d): 1H NMR (300 MHz, CDCl3, 25 8C, TMS):d= 1.43 (t, 3J(H,H) = 7.4 Hz, 3 H; NCH2CH3), 1.54 (d, 3J(H,H) = 7.2 Hz,6 H; (CH3)2CH), 2.30 (s, 3 H; CH3C), 2.31 (s, 3 H; CH3C), 3.53–3.68 (m,1 H; CH), 3.76 (s, 3 H; NCH3), 4.18 ppm (q, 3J(H,H) = 7.4 Hz, 2 H;NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.64 (CH3C),8.76 (CH3C), 15.59 (CH3CH2), 19.12 (CH3CH), 25.13 (CH), 32.70(NCH3), 40.81 (CH2), 119.56 (N(CN)2), 125.21 (CH3C), 127.05 (CH3C),147.43 ppm (CCH); IR (ATR): n= 1089, 1303, 1523, 2126, 2225 cm�1;MS (ES): m/z [%]: 181.3 (100) [M + H+] ; yield: 82 %, white wax.
1-Hexyl-3,4,5-Trimethylimidazolium Dicyanamide [C6m3im][N(CN)2](7 e): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.90 (t, 3J(H,H) =
6.9 Hz, 3 H; CH2CH3), 1.25–1.41 (m; 6 H; (CH2)3CH3), 1.80–1.90 (m,2 H; NCH2CH2), 2.30 (s, 6 H; CCH3), 3.86 (s, 3 H; NCH3), 4.08 (2 H, t,3J(H,H) = 7.7 Hz, 2 H; NCH2), 8.94 ppm (s, 1 H; C2H); 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 8.44 (CH3C), 8.61 (CH3C), 13.97(CH3(CH2)5), 22.41 (CH2), 25.99 (CH2), 29.70 (NCH2CH2), 31.12 (CH2),33.91 (NCH3), 47.41 (NCH2), 119.79 (N(CN)2), 126.89 (CH3C), 127.79(CH3C), 134.57 ppm (CH); IR (ATR): n= 1303, 1452, 1572, 2126,2224 cm�1; MS (ES): m/z [%]: 195.3 (100) [M + H+] ; yield: 91 %,yellow oil.
1-Hexyl-2,3,4,5-Tetramethylimidazolium Dicyanamide [C6C1m3im][N(CN)2] (7 f): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.90 (t,3J(H,H) = 6.6 Hz, 3 H; CH2CH3), 1.29–1.41 (m; 6 H; (CH2)3CH3), 1.66–1.76 (m, 2 H; NCH2CH2 ; m), 2.29 (s, 6 H; CCH3), 2.69 (s, 3 H; C2CH3),3.73 (s, 3 H; NCH3), 4.03 ppm (2 H, t, 3J(H,H) = 7.7 Hz, 2 H; NCH2);13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.75 (CH3C), 8.81 (CH3C),10.44 (CH3C2), 13.94 (CH3(CH2)5), 22.40 (CH2), 26.17 (CH2), 29.64(NCH2CH2), 31.12 (CH2), 32.19 (NCH3), 45.77 (NCH2), 119.60 (N(CN)2),125.26 (CH3C), 126.36 (CH3C), 141.73 ppm (C2CH3); IR (ATR): n=1310, 1538, 1650, 2132, 2238 cm�1; MS (ES): m/z [%]: 209.3 (100)[M + H+] ; yield: 99 %, yellow oil.
2-Ethyl-1-Hexyl-3,4,5-Trimethylimidazolium Dicyanamide [C6C2m3im][N(CN)2] (7 g): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d= 0.91 (t,3J(H,H) = 6.9 Hz, 3 H; CH2CH3), 1.25–1.44 (m; 6 H; (CH2)3CH3), 1.36 (t,3J(H,H) = 7.7 Hz, 3 H; CCH2CH3), 1.69–1.85 (m, 2 H; NCH2CH2 ; m),2.30 (s, 6 H; CCH3), 3.07 (q, 3J(H,H) = 7.7 Hz, 2 H; C2CH2CH3), 3.75 (s,3 H; NCH3), 4.02 ppm (2 H, t, 3J(H,H) = 8.3 Hz, 2 H; NCH2); 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 8.81 (CH3C), 11.71 (CH3CH2C2),13.94 (CH3(CH2)5), 17.59 (CH3CH2C2), 22.41 (CH2), 26.26 (CH2), 30.31(NCH2CH2), 31.16 (CH2), 32.03 (NCH3), 45.71 (NCH2), 119.73 (N(CN)2),125.35 (CH3C), 126.68 (CH3C), 145.72 ppm (CCH2); IR (ATR): n= 1301,1453, 1531, 2125, 2222 cm�1; MS (ES): m/z [%]: 223.3 (100) [M + H+
] ; yield: 89 %, yellow oil.
1-Hexyl-2-Isopropyl-3,4,5-Trimethylimidazolium Dicyanamide[C6isoC3m3im][N(CN)2] (7 h): 1H NMR (300 MHz, CDCl3, 25 8C, TMS):d= 0.91 (m, 3J(H,H) = 6.9 Hz, 3 H; CH2CH3), 1.30–1.44 (m; 6 H;(CH2)3CH3), 1.53 (d, 3J(H,H) = 7.2 Hz, 6 H; (CH3)2CH),1.66–1.76 (m, 2 H;NCH2CH2 ; m), 2.29 (s, 6 H; CCH3), 3.49–3.63 (m, 1 H; CH), 3.80 (s, 3 H;NCH3), 4.06 ppm (2 H, t, 3J(H,H) = 8.0 Hz, 2 H; NCH2); 13C NMR(75 MHz, CDCl3, 25 8C, TMS): d= 8.84 (CH3C), 8.92 (CH3C), 13.91(CH3(CH2)5), 19.16 ((CH3)2CH), 22.40 (CH2), 25.31 (CH2), 26.15(NCH2CH2), 30.44 (CH2), 31.16 (NCH3), 32.85 (CH), 45.80 (CH2), 119.41(N(CN)2), 125.46 (CH3C), 127.18 (CH3C), 147.53 ppm (CCH); IR (ATR):n= 1300, 1456, 1522, 2124, 2221 cm�1; MS (ES): m/z [%]: 237.3(100) [M + H+] ; yield: 93 %, yellow oil.
Batch Procedure for the Synthesis of 1-Ethyl-2-Isopropyl-3,4,5-Trimethylimidazolium Bis(Trifluoromethylsulfonyl)I-mide ([C2isoC3m3im][NTf2], 8 d)
To a 50 mL flask charged with ionic liquid [C2isoC3m3im][I](20 mmol, 6.18 g) was added demineralized water (20 mL). Thenlithium bis(trifluoromethyl-sulfonyl)imide (20.6 mmol, 5.79 g) wasadded to the solution. The flask was provided with a cooler witha cotton plug and the mixture was heated to 50 8C for 8 h. After re-action, the mixture was allowed to cool to 0 8C and 10 mL ofCH2Cl2 was added. The aqueous layer was decanted, and the or-ganic layer was washed 3 times with 40 mL of demineralizedwater, dichloromethane was removed in vacuo. The ionic liquidwas dried for 8 h at 120 8C under reduced pressure (0.5 mbar). Awhite solid was obtained in 93 % yield.
&10& www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 0000, 00, 1 – 13
�� These are not the final page numbers!
C. V. Stevens et al.

Characterisation of Products 8 a–h
1-Ethyl-3,4,5-Trimethylimidazolium Bis(Trifluoromethyl-Sulfonyl)I-mide [C2m3im]NTf2] (8 a): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d=1.46 (t, 3J(H,H) = 7.2 Hz, 3 H; NCH2CH3), 2.23 (s, 3 H; CH3C), 2.24 (s,3 H; CH3C), 3.72 (s, 3 H; NCH3), 4.07 (q, 3J(H,H) = 7.2 Hz, 2 H;NCH2CH3), 8.47 ppm (s, 1 H; CH); 13C NMR (75 MHz, CDCl3, 25 8C,TMS): d= 8.05 (CH3C), 8.14 (CH3C), 14.60 (CH3CH2), 33.47 (NCH3),42.35 (CH2), 119.87 (q, 1J(C,F) = 320.8 Hz; CF3), 126.85 (CH3C), 127.82(CH3C), 133.44 ppm (CH); 19F NMR (282 MHz, CDCl3, 25 8C): d=�78.95 ppm; IR (ATR): n= 1052, 1134, 1176, 1348, 1575 cm�1; MS(ES): m/z [%]: 139.3 (100) [M + H+] ; yield: 93 %, white solid.
1-Ethyl-2,3,4,5-Tetramethylimidazolium Bis(Trifluoromethyl-Sulfony-l)Imide [C2C1m3im]NTf2] (8 b): 1H NMR (300 MHz, CDCl3, 25 8C, TMS):d= 1.35 (t, 3J(H,H) = 7.4 Hz, 3 H; NCH2CH3), 2.20 (s, 3 H; CH3C), 2.22(s, 3 H; CH3C), 2.56 (s, 3 H; CH3C2), 3.60 (s, 3 H; NCH3), 4.03 ppm (q,3J(H,H) = 7.4 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C,TMS): d= 8.18 (CH3C), 8.38 (CH3C), 9.76 (CH3C2), 14.54 (CH3CH2),31.80 (NCH3), 40.63 (CH2), 119.92 (q, 1J(C,F) = 320.8 Hz; CF3), 124.82(CH3C), 126.27 (CH3C), 141.69 ppm (CH3C2) ; 19F NMR (282 MHz,CDCl3, 25 8C): d M->79.25 ppm; IR (ATR): n= 613, 1049, 1136, 1184,1347 cm�1; MS (ES): m/z [%]: 153.3 (100) [M + H+] ; yield: 95 %,yellow oil.
1,2-Diethyl-3,4,5-Trimethylimidazolium Bis(Trifluoromethyl-Sulfony-l)Imide [C2C2m3im][NTf2] (8 c): 1H NMR (300 MHz, CDCl3, 25 8C, TMS):d= 1.29 (t, 3J(H,H) = 7.7 Hz, 3 H; CCH2CH3), 1.40 (t, 3J(H,H) = 7.4 Hz,3 H; NCH2CH3), 2.23 (s, 3 H; CH3C), 2.25 (s, 3 H; CH3C), 3.00 (q,3J(H,H) = 7.7 Hz, 2 H; CCH2CH3), 3.66 (s, 3 H; NCH3), 4.06 ppm (q,3J(H,H) = 7.4 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C,TMS): d= 8.30 (CH3C), 8.46 (CH3C), 11.48 (CH3CH2C), 15.25(NCH2CH3), 17.24 (CH2C), 31.67 (NCH3), 40.55 (NCH2), 119.82 (qn1J(C,F) = 321.1 Hz; CF3) 124.95 (CH3C), 126.54 (CH3C), 145.54 ppm(CCH2); 19F NMR (282 MHz, CDCl3, 25 8C): d=�78.92 ppm; IR (ATR):n= 314, 788, 1053, 1135, 1176, 1348 cm�1; MS (ES): m/z [%]: 167.3(100) [M + H+] ; yield: 97 %, white solid.
1-Ethyl-2-Isopropyl-3,4,5-Trimethylimidazolium Bis(Trifluoro-Methyl-sulfonyl)Imide [C2isoC3m3im][NTf2] (8 d): 1H NMR (300 MHz, CDCl3,25 8C, TMS): d= 1.37 (t, 3J(H,H) = 7.4 Hz, 3 H; NCH2CH3), 1.47 (d,3J(H,H) = 7.2 Hz, 6 H; (CH3)2CH), 2.21 (s, 3 H; CH3C), 2.23 (s, 3 H;CH3C), 3.44–3.59 (m, 1 H; CH), 3.70 (s, 3 H; NCH3), 4.09 ppm (q,3J(H,H) = 7.4 Hz, 2 H; NCH2CH3) ; 13C NMR (75 MHz, CDCl3, 25 8C,TMS): d= 8.47 (CH3C), 8.58 (CH3C), 15.44 (CH3CH2), 19.01 ((CH3)2CH),25.19 (CH), 32.55 (NCH3), 40.74 (CH2), 119.88 (q, 1J(C,F) = 321.5 Hz;CF3), 125.18 (CH3C), 127.11 (CH3C), 147.40 ppm (CCH); 19F NMR(282 MHz, CDCl3, 25 8C): d=�78.73; IR (ATR): n= 612, 1053, 1137,1180, 1347 cm�1; MS (ES): m/z [%]: 181.3 (100) [M + H+] ; yield:94 %,yellow solid.
1-Hexyl-3,4,5-Trimethylimidazolium Bis(Trifluoromethyl-Sulfonyl)I-mide [C6m3im][NTf2] (8 e): 1H NMR (300 MHz, CDCl3, 25 8C, TMS): d=0.89 (t, 3J(H,H) = 6.6 Hz, 3 H; CH2CH3), 1.27–1.37 (m; 6 H; (CH2)3CH3),1.73–1.84 (m, 2 H; NCH2CH2), 2.24 (s, 6 H; CCH3), 3.75 (s, 3 H; NCH3),4.01 (2 H, t, 3J(H,H) = 7.7 Hz, 2 H; NCH2), 8.55 ppm (s, 1 H; C2H);13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.15 (CH3C), 8.34 (CH3C),13.85 (CH3(CH2)5), 22.32 (CH2), 25.87 (CH2), 29.68 (NCH2CH2), 31.04(CH2), 33.51 (NCH3), 47.29 (NCH2), 119.86 (q, 1J(C,F) = 321.8 Hz; CF3)126.86 (CH3C), 127.78 (CH3C), 134.01 ppm (CH); 19F NMR (282 MHz,CDCl3, 25 8C): d=�74.89; IR (ATR): n= 614, 1053, 1177, 1348, 1574,2934 cm�1; MS (ES): m/z [%]: 195.3 (100) [M + H+] ; yield: 95 %,transparent oil.
1-Hexyl-2,3,4,5-Tetramethylimidazolium Bis(Trifluoromethyl-Sulfony-l)Imide [C6C1m3im][NTf2] (8 f): 1H NMR (300 MHz, CDCl3, 25 8C, TMS):
d= 0.89 (t, 3J(H,H) = 6.6 Hz, 3 H; CH2CH3), 1.26–1.40 (m; 6 H;(CH2)3CH3), 1.60–1.71 (m, 2 H; NCH2CH2; m), 2.20 (s, 6 H; CCH3), 2.55(s, 3 H; C2CH3), 3.59 (s, 3 H; NCH3), 3.92 ppm (2 H, t, 3J(H,H) = 7.7 Hz,2 H; NCH2); 13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.47 (CH3C),10.08 (CH3C2), 13.92 (CH3(CH2)5), 22.41 (CH2), 26.18 (CH2), 29.65(NCH2CH2), 31.16 (CH2), 31.90 (NCH3), 45.64 (NCH2), 119.89 (q,1J(C,F) = 320.8 Hz; CF3) ; 125.05 (CH3C), 126.25 (CH3C), 141.81 ppm(C2CH3); 19F NMR (282 MHz, CDCl3, 25 8C): d=�78.95 ppm; IR (ATR):n= 1053, 1135, 1176, 1331, 1348 cm�1; MS (ES): m/z [%]: 209.3 (100)[M + H+] ; yield: 98 %, yellow oil.
2-Ethyl-1-Hexyl-4,5-Dimethyl-Imidazolium Bis(Trifluoromethyl-Sulfo-nyl)Imide [C6C2m3im][NTf2] (8 g): 1H NMR (300 MHz, CDCl3, 25 8C,TMS): d= 0.91 (t, 3J(H,H) = 6.3 Hz, 3 H; CH2CH3), 1.25–1.42 (m; 6 H;(CH2)3CH3), 1.30 (t, 3J(H,H) = 7.7 Hz, 3 H; CCH2CH3), 1.65–1.73 (m, 2 H;NCH2CH2 ; m), 2.23 (s, 6 H; CCH3), 2.99 (q, 3J(H,H) = 7.7 Hz, 2 H;C2CH2CH3), 3.66 (s, 3 H; NCH3), 3.94 ppm (2 H, t, 3J(H,H) = 8.3 Hz, 2 H;NCH2); 13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.43 (CH3C), 8.47(CH3C), 11.36 (CH3CH2C2), 13.91 (CH3(CH2)5), 17.38 (CH3CH2C2), 22.46(CH2), 26.23 (CH2), 30.24 (NCH2CH2), 31.25 (CH2), 31.74 (NCH3), 45.59(NCH2), 120.02 (q, 1J(C,F) = 321.8 Hz; CF3), 125.29 (CH3C), 126.69(CH3C), 145. ppm(CCH2); 19F NMR (282 MHz, CDCl3, 25 8C): d=
�79.21 ppm; IR (ATR): n= 615, 1053, 1134, 1176 cm�1; MS (ES): m/z[%]: 223.3 (100) [M + H+] ; yield: 98 %, yellow oil.
1-Hexyl-2-Isopropyl-3,4,5-Trimethylimidazolium Bis(Trifluoro-Methyl-sulfonyl)Imide [C6isoC3m3im][NTf2] (8 h): 1H NMR (300 MHz, CDCl3,25 8C, TMS): d= 0.90 (m, 3J(H,H) = 6.6 Hz, 3 H; CH2CH3), 1.24–1.40(m; 6 H; (CH2)3CH3), 1.47 (d, 3J(H,H) = 7.2 Hz, 6 H; (CH3)2CH),1.61–1.69(m, 2 H; NCH2CH2; m), 2.21 (s, 6 H; CCH3), 3.41–3.65 (m, 1 H; CH),3.70 (s, 3 H; NCH3), 3.97 ppm (2 H, t, 3J(H,H) = 8.2 Hz, 2 H; NCH2);13C NMR (75 MHz, CDCl3, 25 8C, TMS): d= 8.53 (CH3C), 8.67 (CH3C),13.88 (CH3CH2), 18.93 ((CH3)2CH), 22.40 (CH2), 25.25 (CH2), 26.12(CH2) 30.40 (CH2) 31.16 (NCH3), 32.63 (CH), 45.68 (NCH2), 119.94 (q,1J(C,F) = 321.1 Hz; CF3), 125.37 (CH3C), 127.18 (CH3C), 147.49 ppm(CCH); 19F NMR (282 MHz, CDCl3, 25 8C): d=�78.96 ppm; IR (ATR):n= 1054, 1135, 1177, 1349, 2360 cm�1; MS (ES): m/z [%]: 237.3 (100)[M + H+] ; yield: 97 %, yellow oil.
Procedure for the Synthesis of Diphenylmethanol in the IL[C6isoC3m3im][NTf2]
A dry 10 mL flask, equipped with septum and dinitrogen flow, wascharged with intensively dried ionic liquid [C6isoC3m3im][NTf2](4.8 mmol, 2.50 g). The ionic liquid was cooled to �10 8C in a NaCl/ice bath and bromobenzene (2 mmol, 314 mg) was added witha syringe. After cooling and controlled stirring for 5 min. , a homo-genous mixture was obtained, to which a 2.3 m nBuli solution(2.1 mmol, 0.91 mL) was added dropwise over 15 min. , while main-taining a constant temperature of �10 8C. After controlled stirringfor 10 min, benzaldehyde (2 mmol, 212 mg) were added dropwiseover 5 min. with a syringe. The mixture was stirred for 60 min. andallowed to obtain room temperature. After reaction, 1 mL of dem-ineralized water was added and the organic fraction was extracted2 times with 5 mL of dichloromethane. The organic solution wasextracted 2 times with 10 mL of diethyl ether, the extracts werecombined and GC samples were prepared from the ethereal solu-tion. The presence of diphenylmethanol was demonstrated bycomparison of the latter with GC analysis of pure commercial di-phenylmethanol.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org &11&
These are not the final page numbers! ��
Peralkylated Imidazoles as Ionic Liquids

Acknowledgements
The authors are grateful for the financial support from theagency for Innovation through Science and Technology (IWT-Vlaanderen) for the funding of the SBO-project Materials Process-ing in Ionic Liquids (MAPIL) and for the funding of a Ph.D grantto S.S. This work was supported by the FWO-Flanders (researchcommunity “Ionic Liquids”). The authors sincerely thank the con-tributors of the project (Peter De Vreese, Kurt Haerens, KaHo SLGhent) for assistance and guidance in the analytical experiments.
Keywords: alkaline stability · aza heterocycles · cyclicvoltammetry · ionic liquids · sustainable chemistry
[1] M. J. Earle, J. Esperanca, M. A. Gilea, J. N. C. Lopes, L. P. N. Rebelo, J. W.Magee, K. R. Seddon, J. A. Widegren, Nature 2006, 439, 831 – 834.
[2] D. M. Fox, W. H. Awad, J. W. Gilman, P. H. Maupin, H. C. De Long, P. C.Trulove, Green Chem. 2003, 5, 724 – 727.
[3] P. Wasserscheid, T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Wein-heim, 2003.
[4] N. V. Plechkova, K. R. Seddon, Chem. Soc. Rev. 2008, 37, 123 – 150.[5] a) T. Welton, Chem. Rev. 1999, 99, 2071 – 2083; b) P. Wasserscheid, W.
Keim, Angew. Chem. 2000, 112, 3926 – 3945; Angew. Chem. Int. Ed. 2000,39, 3772 – 3789; c) M. J. Earle, K. R. Seddon, Pure Appl. Chem. 2000, 72,1391 – 1398.
[6] H. Zhao, J. Chem. Technol. Biotechnol. 2010, 85, 891 – 907.[7] a) O. A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn, T. Heinze, Biomacro-
molecules 2007, 8, 2629 – 2647; b) M. E. Zakrzewska, E. Bogel-Lukasik, R.Bogel-Lukasik, Energy Fuels 2010, 24, 737 – 745; c) A. Pinkert, K. N.Marsh, S. Pang, M. P. Staiger, Chem. Rev. 2009, 109, 6712 – 6728.
[8] a) D. J. Heldebrant, C. R. Yonker, P. G. Jessop, L. Phan, Chem. Eur. J. 2009,15, 7619 – 7627; b) M. Shiflett, D. Drew, R. Cantini, A. Yokozeki, EnergyFuels 2010, 24, 5781 – 5789.
[9] Y. Ito, T. Nohira, Electrochim. Acta 2000, 45, 2611 – 2622.[10] A. Berthod, M. Ruiz-Angel, S. Carda-Broch, J. Chromatogr. A 2008, 1184,
6 – 18.[11] Y. Chauvin, L. Mussmann, H. Olivier, Angew. Chem. 1995, 107, 2941 –
2943; Angew. Chem. Int. Ed. Engl. 1995, 34, 2698 – 2700.[12] R. E. Del Sesto, T. M. McCleskey, C. Macomber, K. C. Ott, A. T. Koppisch,
G. A. Baker, A. K. Burrell, Thermochim. Acta 2009, 491, 118 – 120.[13] S. T. Handy, M. Okello, J. Org. Chem. 2005, 70, 1915 – 1918.[14] a) K. Noack, P. S. Schulz, N. Paape, J. Kiefer, P. Wasserscheid, A. Leipertz,
Phys. Chem. Chem. Phys. 2010, 12, 14153 – 14161; b) R. Giernoth, D.Bankmann, Tetrahedron Lett. 2006, 47, 4293 – 4296.
[15] V. K. Aggarwal, I. Emme, A. Mereu, Chem. Commun. 2002, 1612 – 1613.[16] P. Formentı’n, H. Garcı’a, A. Leyva, J. Mol. Catal. A 2004, 214, 137 – 142.[17] a) P. Bonh�te, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram, M. Gr�t-
zel, Inorg. Chem. 1996, 35, 1168 – 1178; b) P. R. Gifford, J. B. Palmisano, J.Electrochem. Soc. 1987, 134, 610 – 614.
[18] A. B. McEwen, H. L. Ngo, K. LeCompte, J. L. Goldman, J. Electrochem.Soc. 1999, 146, 1687 – 1695.
[19] a) M. Montanino, M. Carewska, F. Alessandrini, S. Passerini, G. B. Appe-tecchi, Electrochim. Acta 2011, 57, 153 – 159; b) B. D. Fitchett, T. N.Knepp, J. C. Conboy, J. Electrochem. Soc. 2004, 151, E219 – E225.
[20] M. C. Kroon, W. Buijs, C. J. Peters, G.-J. Witkamp, Green Chem. 2006, 8,241 – 245.
[21] K. Haerens, E. Matthijs, K. Binnemans, B. Van der Bruggen, Green Chem.2009, 11, 1357 – 1365.
[22] I. A. Shkrob, T. W. Marin, S. D. Chemerisov, J. L. Hatcher, J. F. Wishart, J.Phys. Chem. B 2011, 115, 3889 – 3902.
[23] a) G. C. Tian, X. J. Zhou, J. Li, Y. X. Hua, Trans. Nonferrous Met. Soc. China2009, 19, 1639 – 1644; b) S. P. Ong, G. Ceder, Electrochim. Acta 2010, 55,3804 – 3811.
[24] C. Wiles, P. Watts, Eur. J. Org. Chem. 2008, 1655 – 1671.[25] A. Cukalovic, J.-C. M. R. Monbaliu, C. V. Stevens in Topics in Heterocyclic
Chemistry, Vol. 23 (Eds. : R. V. A. Orru, E. Ruijter), Springer, Heidelberg,2010, pp. 161 – 198.
[26] D. R. J. Acke, R. V. A. Orru, C. V. Stevens, QSAR Comb. Sci. 2006, 25, 474 –483.
[27] a) B. Radziszewski, Ber. Dtsch. Chem. Ges. 1882, 15, 1493 – 1496; b) H.Debus, Justus Liebigs Ann. Chem. 1858, 107, 199 – 208; c) Z. Wang, Com-prehensive Organic Name Reactions and Reagents, Vol. 3, 2293 ed. , Wiley-VCH, Weinheim, 2009.
[28] J.-Y. Cheng, Y.-H. Chu, Tetrahedron Lett. 2006, 47, 1575 – 1579.[29] a) D. R. MacFarlane, S. A. Forsyth, J. Golding, G. B. Deacon, Green Chem.
2002, 4, 444 – 448; b) S. Fang, L. Yang, J. Wang, M. Li, K. Tachibana, K.Kamijima, Electrochim. Acta 2009, 54, 4269 – 4273.
[30] G. Appetecchi, S. Scaccia, C. Tizzani, F. Alessandrini, S. Passerini, J. Elec-trochem. Soc. 2006, 153, A1685 – A1691.
[31] K. Fumino, A. Wulf, R. Ludwig, Angew. Chem. 2008, 120, 8859 – 8862;Angew. Chem. Int. Ed. 2008, 47, 8731 – 8734.
[32] P. A. Hunt, J. Phys. Chem. B 2007, 111, 4844 – 4853.[33] A. Ahosseini, E. Ortega, B. Sensenich, A. M. Scurto, Fluid Phase Equilib.
2009, 286, 72 – 78.[34] Y. Yoshida, O. Baba, G. Saito, J. Phys. Chem. B 2007, 111, 4742 – 4749.[35] M. J. Deng, P. Y. Chen, T. I. Leong, I. W. Sun, J. K. Chang, W. T. Tsai, Electro-
chem. Commun. 2008, 10, 213 – 216.[36] J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker,
R. D. Rogers, Green Chem. 2001, 3, 156 – 164.[37] a) C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki, J. F. Bren-
necke, J. Chem. Eng. Data 2004, 49, 954 – 964; b) W. H. Awad, J. W.Gilman, M. Nyden, R. H. Harris, T. E. Sutto, J. Callahan, P. C. Trulove, H. C.DeLong, D. M. Fox, Thermochim. Acta 2004, 409, 3 – 11; c) H. Tokuda, K.Ishii, M. A. B. H. Susan, S. Tsuzuki, K. Hayamizu, M. Watanabe, J. Phys.Chem. B 2006, 110, 2833 – 2839.
[38] M. C. Kroon, W. Buijs, C. J. Peters, G.-J. Witkamp, Thermochim. Acta2007, 465, 40 – 47.
[39] P. C. Howlett, E. I. Izgorodina, M. Forsyth, D. R. MacFarlane, Z. Phys.Chem. 2006, 220, 1483 – 1498.
[40] H. Sakaebe, H. Matsumoto, Electrochem. Commun. 2003, 5, 594 – 598.
Received: April 19, 2012
Published online on && &&, 2012
&12& www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 0000, 00, 1 – 13
�� These are not the final page numbers!
C. V. Stevens et al.

ARTICLES
C. Maton, N. De Vos, B. I. Roman,E. Vanecht, N. R. Brooks, K. Binnemans,S. Schaltin, J. Fransaer, C. V. Stevens*
&& –&&
Continuous Synthesis of PeralkylatedImidazoles and their Transformationinto Ionic Liquids with Improved(Electro)Chemical Stabilities
Completely alkylated imidazoles aresynthesized via a selective continuousflow process and then transformed intotheir corresponding peralkylated imid-azolium ionic liquids (see picture). Al-though completely substituted, themelting points and viscosities are withinthe workable range. The increased sub-stitution exerts a beneficial influence onthe decomposition temperature andcathodic stability and allows these ionicliquids to be applied in strong alkalinemedia.
ChemPhysChem 0000, 00, 1 – 13 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org &13&
These are not the final page numbers! ��
Related Documents











