Consistent methodology for calculating surface and interface energies Krzysztof Rapcewicz, Bin Chen, Boris Yakobson, and J. Bernholc Department of Physics, North Carolina State University, Raleigh, North Carolina 27695-8202 ~Received 20 December 1996; revised manuscript received 1 December 1997! A consistent approach to the calculation of the surface energy valid for all crystal systems is presented. Voronoi polyhedra are introduced and used in conjunction with the energy-density formalism of Chetty and Martin @Phys. Rev. B 45, 6074 ~1992!; 45, 6089 ~1992!# to provide a methodology for the determination of surface energies. The surface energies of the unrelaxed, unreconstructed GaAs ~001! and ~111! surfaces are calculated as a test. As an example of the application of the formalism to a low symmetry system, the energies of selected ~0001! surfaces of the wide-gap semiconductors GaN and SiC are determined. @S0163-1829~98!02012-8# INTRODUCTION There has been tremendous interest and progress in the technology of crystal growth over the past few decades. 1 This has made relevant numerous theoretical questions re- lated to crystal growth and the equilibrium behavior of sur- faces. Indeed, intense efforts have been made to understand the energetics of such surfaces. 2 However, in spite of the substantial progress towards an understanding of the physics of surfaces, the surface energies must be calculated with re- spect to a reference surface, the reference being different for different surfaces except for specific high-symmetry cases. Since the surface energy plays an important role in the de- termination of the mode of growth, namely, layer, island or layer-plus-island growth, the lack of a procedure for the cal- culation of the surface energy is unfortunate. Indeed, if meaningful ab initio predictions about the equilibrium crys- tal shape and preferred growth directions are to be made, it will be necessary to have a database of surface energies of reconstructed surfaces for different directions. This requires that the evaluations of the surface energies be consistent be- tween calculations. The situation can be summarized as follows: On the basis of general considerations, it can be shown that for crystals of sufficiently high symmetry, the surface energy can be calcu- lated unambiguously. For the remaining systems, it can only be calculated consistently, i.e., the surface energy is defined up to a gauge; this gauge is of no consequence in the deter- mination, for instance, of equilibrium crystal shapes. Practi- cally, however, the surface energy, when it is unambiguously defined, can be calculated absolutely with a slab calculation only when the two surfaces are crystallographically identical; most often, only a relative surface energy can be determined. Three cases therefore exist: ~1! the surface energy is un- ambiguously defined and can be determined using a slab calculation; the ~001! surface of a zinc-blende semiconductor is such a case; ~2! the surface energy is unambiguously de- fined, but slab calculations give only relative surface ener- gies; the ~111! surface of a zinc-blende semiconductor serves as a case in point; ~3! the surface energy can only be defined consistently ~the gauge must be set, once and for all, for the calculations!; the ~0001! surface of a wurtzite semiconductor is such an example. Chetty and Martin 3–5 developed an approach that could be used to calculate the surface energies in the second case above. They introduced the concepts of the energy density and symmetry-adapted unit cell to provide a procedure for the calculation of the surface energy and applied their ap- proach to the GaAs ~111! surface and interfaces. However, symmetry-adapted unit cells can be used only if the space group has sufficiently high symmetry, namely, a center of symmetry, two axes of rotation, or an axis of rotation and a mirror plane not through this axis. For the third case, no methodology exists to calculate the surface energy. We have generalized the energy-density approach of Chetty and Martin 3,4 to a consistent methodology for the cal- culation of surface energies for all systems including those of case three above. In this approach, the slab and bulk are considered to be built from blocks whose shape is deter- mined by the symmetry of the bulk crystal. The ‘‘energy cost’’ of each block is evaluated and the ‘‘cost’’ of creating a surface, the surface energy, is obtained by summing up the cost of each block in the slab and subtracting the costs of the equivalent blocks in the bulk. The shape of the blocks is determined according to a geometric rule; thus the approach provides a consistent way of determining the surface energy that can be used for low-symmetry systems. Further, the symmetric nature of the blocks ensures that symmetry- equivalent surfaces will have the same surface energy and hence the methodology will reproduce the results of slab calculations, and those of Chetty and Martin. The blocks can be chosen to be either neutral or charged, the former being our preferred choice because they are more intuitive. The paper is organized as follows: a brief re ´ sume ´ of the surface energy begins the presentation, after which the stan- dard calculations of the surface energy are discussed. In ad- dition, a conceptual approach using only total-energy calcu- lations and scaling arguments, which is applicable to systems of sufficiently high symmetry, is presented. This approach clarifies the issues involved in unambiguously defining the surface energy, but it is not feasible at present. The energy- density approach of Chetty and Martin is then reviewed to- gether with the implementation of the energy-density formal- ism using symmetry-adapted unit cells. A generalization of the energy-density formalism using Voronoi polyhedra ~VP! is introduced and applied to GaAs ~001!, GaAs ~111!, SiC ~0001!, and GaN ~0001!. The latter two cases provide ex- amples of low-symmetry systems in which symmetry- PHYSICAL REVIEW B 15 MARCH 1998-II VOLUME 57, NUMBER 12 57 0163-1829/98/57~12!/7281~11!/$15.00 7281 © 1998 The American Physical Society

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

PHYSICAL REVIEW B 15 MARCH 1998-IIVOLUME 57, NUMBER 12
Consistent methodology for calculating surface and interface energies
Krzysztof Rapcewicz, Bin Chen, Boris Yakobson, and J. BernholcDepartment of Physics, North Carolina State University, Raleigh, North Carolina 27695-8202
~Received 20 December 1996; revised manuscript received 1 December 1997!
A consistent approach to the calculation of the surface energy valid for all crystal systems is presented.Voronoi polyhedra are introduced and used in conjunction with the energy-density formalism of Chetty andMartin @Phys. Rev. B45, 6074 ~1992!; 45, 6089 ~1992!# to provide a methodology for the determination ofsurface energies. The surface energies of the unrelaxed, unreconstructed GaAs~001! and ~111! surfaces arecalculated as a test. As an example of the application of the formalism to a low symmetry system, the energiesof selected ~0001! surfaces of the wide-gap semiconductors GaN and SiC are determined.@S0163-1829~98!02012-8#
s
urta
s
tedoa,-e,s
iret b
asslcunn
etcts
tioa
ne-sloreee
edthr
be
casesityforap-r,aceofa
no
ofl-
reter-ygthetheis
achrgythetry-andlabcaning
tan-ad-
cu-msch
thergy-to-al-of
-y-
INTRODUCTION
There has been tremendous interest and progress intechnology of crystal growth over the past few decade1
This has made relevant numerous theoretical questionslated to crystal growth and the equilibrium behavior of sfaces. Indeed, intense efforts have been made to understhe energetics of such surfaces.2 However, in spite of thesubstantial progress towards an understanding of the phyof surfaces, the surface energies must be calculated withspect to a reference surface, the reference being differendifferent surfaces except for specific high-symmetry casSince the surface energy plays an important role in thetermination of the mode of growth, namely, layer, islandlayer-plus-island growth, the lack of a procedure for the cculation of the surface energy is unfortunate. Indeedmeaningfulab initio predictions about the equilibrium crystal shape and preferred growth directions are to be madwill be necessary to have a database of surface energiereconstructed surfaces for different directions. This requthat the evaluations of the surface energies be consistentween calculations.
The situation can be summarized as follows: On the bof general considerations, it can be shown that for crystalsufficiently high symmetry, the surface energy can be calated unambiguously. For the remaining systems, it can obe calculated consistently, i.e., the surface energy is defiup to a gauge; this gauge is of no consequence in the dmination, for instance, of equilibrium crystal shapes. Pracally, however, the surface energy, when it is unambiguoudefined, can be calculated absolutely with a slab calculaonly when the two surfaces are crystallographically identicmost often, only a relative surface energy can be determi
Three cases therefore exist:~1! the surface energy is unambiguously defined and can be determined using acalculation; the~001! surface of a zinc-blende semiconductis such a case;~2! the surface energy is unambiguously dfined, but slab calculations give only relative surface engies; the~111! surface of a zinc-blende semiconductor servas a case in point;~3! the surface energy can only be definconsistently~the gauge must be set, once and for all, forcalculations!; the ~0001! surface of a wurtzite semiconductois such an example.
Chetty and Martin3–5 developed an approach that could
570163-1829/98/57~12!/7281~11!/$15.00
the.re--nd
icsre-fors.e-rl-if
itofse-
isof-
lyeder-i-lynl;d.
ab
-r-s
e
used to calculate the surface energies in the secondabove. They introduced the concepts of the energy denand symmetry-adapted unit cell to provide a procedurethe calculation of the surface energy and applied theirproach to the GaAs~111! surface and interfaces. Howevesymmetry-adapted unit cells can be used only if the spgroup has sufficiently high symmetry, namely, a centersymmetry, two axes of rotation, or an axis of rotation andmirror plane not through this axis. For the third case,methodology exists to calculate the surface energy.
We have generalized the energy-density approachChetty and Martin3,4 to a consistent methodology for the caculation of surface energies for all systemsincluding those ofcase three above. In this approach, the slab and bulk aconsidered to be built from blocks whose shape is demined by the symmetry of the bulk crystal. The ‘‘energcost’’ of each block is evaluated and the ‘‘cost’’ of creatina surface, the surface energy, is obtained by summing upcost of each block in the slab and subtracting the costs ofequivalent blocks in the bulk. The shape of the blocksdetermined according to a geometric rule; thus the approprovides a consistent way of determining the surface enethat can be used for low-symmetry systems. Further,symmetric nature of the blocks ensures that symmeequivalent surfaces will have the same surface energyhence the methodology will reproduce the results of scalculations, and those of Chetty and Martin. The blocksbe chosen to be either neutral or charged, the former beour preferred choice because they are more intuitive.
The paper is organized as follows: a brief re´sumeof thesurface energy begins the presentation, after which the sdard calculations of the surface energy are discussed. Indition, a conceptual approach using only total-energy callations and scaling arguments, which is applicable to systeof sufficiently high symmetry, is presented. This approaclarifies the issues involved in unambiguously definingsurface energy, but it is not feasible at present. The enedensity approach of Chetty and Martin is then reviewedgether with the implementation of the energy-density formism using symmetry-adapted unit cells. A generalizationthe energy-density formalism using Voronoi polyhedra~VP!is introduced and applied to GaAs~001!, GaAs ~111!, SiC~0001!, and GaN~0001!. The latter two cases provide examples of low-symmetry systems in which symmetr
7281 © 1998 The American Physical Society

w
rgheowtea
nde
ucy
an
t
tis
thre.is
l.
thls
h
f
d-s
ic
or
,th
lly
the
ust
de-
uplabrgyula-ur-thodus-
en-la-is
si-ples of
ofthat-
glethearer,thee-
iscanilib-theof
t bese,t tom
se
7282 57RAPCEWICZ, CHEN, YAKOBSON, AND BERNHOLC
adapted unit cells cannot be used. The paper concludesa summary.
I. SURFACE ENERGY
In this section we review the theory of the surface eneand the role that symmetry plays in the definition of tsurface energy. In particular, for systems that are of lsymmetry the surface energy cannot be defined absolubut only up to a gauge, which must be the same fordirections.
Consider a system containing two phases, 1 and 2, adividing surface. The excess energyE(s) due to the presencof the dividing surface is the~excess! quantity definedthrough the relation
E5E~1!1E~2!1E~s!, ~1!
whereE is the total energy of the system andE(1)(E(2)) isthe energy of the equivalent bulk system of phase 1~2!. Thedefinitions for other surface thermodynamic quantities, sas the entropy,S(s), etc. are similar.6 The surface free energper unit area is defined to be
g5e~s!2Ts~s!2(i
m iG i , ~2!
where small letters denote the corresponding excess quties per unit area with the exception ofG i5Ni
(s)/A, which isthe excess number of particles of thei th species per uniarea, andm i is the chemical potential of thei th species.
The surface free energy defined in this way measureswork required to create a new surface. It is, in general, dtinct from the work needed to deform a surface that issurface stress. In fluids, as is well known, the surface fenergy is isotropic and is the same as the surface stress6 Incontrast, the surface free energy in solids is generally antropic, which is to say that the surface free energy,g(n), isa function of the direction of the outward-pointing normaFurther, the surface free energy and the surface stressdistinct quantities.
The surface free energy per unit area is important indetermination of the equilibrium geometry of small crystaand the equilibrium shape of small particles in contact witsubstrate. Under conditions of constant temperatureT, vol-umeV, and chemical potentialsm i , the excess free energy othe system due to the presence of the surface is
C5E dAg~ n!, ~3!
where dA is the element of surface area with outwarpointing normaln. The equilibrium shape thus minimizethe excess free energy.
For a single crystal at constant temperature and chempotential, the minimization of Eq.~3! subject to the con-straint of fixed volume leads to the Wulff construction fdetermining the equilibrium shape,6 while a modification ofthe Wulff construction, the Winterbottom construction7
gives the shape of a crystal for a given orientation ofsubstrate and, thus, information about the growth mode.
ith
y
lyll
a
h
ti-
he-
ee
o-
are
e
a
al
e
Since the surface energy manifests itself physicathrough the minimization of the integral in Eq.~3!, it is pos-sible to add to the surface energy a gauge termC•n ~whereC is a constant vector! without changing the equilibriumshape.8,9 Symmetry, however, permits a nonzeroC for only10 point groups, namely,C6 , C6v ~which includes wurtzite!,C4 , C4v , C3 , C3v , C2 , C2v , C1h , andC1 . For C1 , C iscompletely arbitrary; for the rest, symmetry constrainschoice ofC. ForC1h , C must lie on the mirror plane; for theremaining eight point groups,C must lie along the uniqueaxis. In other words, one, two, or three arbitrary scalars mbe chosen.9
II. DETERMINATION OF THE SURFACE ENERGYFROM TOTAL ENERGY CALCULATIONS
For the surface energy two situations exist:~i! for highsymmetry systems, the surface energy is unambiguouslyfined ~combining cases one and two of the introduction!; ~ii !for low symmetry systems, the surface energy is definedto a gauge. In this section, we begin by pointing out that scalculations permit a determination of the absolute eneonly in high-symmetry cases. In other cases, slab calctions provide only relative surface energies, even if the sface energy is absolutely defined. We then present a methat permits the calculation of absolute surface energiesing polyhedra and scaling behavior.
The calculations employed to determine the surfaceergy for a given crystal direction are generally slab calcutions. The slab consists of a finite number of layers andmade infinite in the plane of the surface through the impotion of periodic boundary conditions. The need to decouthe two surfaces from one another dictates the thicknesthe slab and also the number of vacuum layers~the calcula-tions are usually carried out using supercells!. We define theenergy of the equivalent bulk as the sum of the numberatoms of each species times the chemical potential ofspecies. Invariably, asingle configuration is used to determine the totalground stateenergy of the slab atT50. In amulticomponent system, the chemical potential for a sinspecies is not defined for a single configuration. In fact,chemical potentials that occur in the slab calculationsexternal parametersand can take arbitrary values; howevephysical arguments are invoked to establish limits uponvariability of the chemical potentials of the individual spcies.
At equilibrium, the chemical potential of a speciesequal in all phases that are in contact. This observationbe exploited to impose constraints on the possible equrium values. In particular, it is generally assumed thatbulk is in equilibrium with the surface, i.e., that the sumthe chemical potentials of the individual species,mA , mB ,etc. equals the bulk chemical potentialmAB¯Z(bulk) ,
mAB¯Z~bulk!5mA1mB1¯1mZ . ~4!
Further, the chemical potential for a given species cannoabove the chemical potential of its elemental bulk phasince the bulk phase would then be unstable with respecprecipitation of the elemental bulk. Thus, the maximuvalue of the chemical potential of a given species, sayA, isequal to the chemical potential of its elemental bulk pha

m
th
ei
dineanb
.esenyyria
u
soicromta
e-
fdriteua
eeg/
o-e-om-orfof
ionthem-
tedallhe
toughdurem-e-
ies,
ems,ingle
ons
l,
dted-thehetral
nythethe
aes
yheue
57 7283CONSISTENT METHODOLOGY FOR CALCULATING . . .
mA(bulk) . The heat of formationDH f , which relates thechemical potential in the elemental bulk system to the checal potential in the compound is defined by
mAB¯Z~bulk!5mA~bulk!1mB~bulk!1¯1mZ~bulk!2DH f .~5!
It can be used to set bounds for the chemical potential ofindividual species in the bulk, viz.
ma~bulk!2DH f<ma<ma~bulk! , ~6!
wherea5A,B, . . . ,Z.10 It is worth emphasizing that thesbounds only delineate the range over which the bulkstable. If the chemical potential falls outside of these bounthe bulk will become metastable and kinetics will determthe subsequent behavior of the system, i.e., whether a chof phase will occur, whether the metastable phase willvery long lived, etc. Thus the bounds Eq.~6! provide area-sonablerange over which to consider the surface energy11
For certain directions of sufficiently high symmetry, thcalculation of the surface energy is simplified. Along thespecial directions, it is possible to find a slab that has idtical surfaces. More explicitly, the surfaces are related breflection about the center of the slab and, possibly, brotation in the plane of the surface. For a slab of mateAB¯Z consisting ofNA atoms of speciesA, NB atoms ofspeciesB, etc., in a sufficiently simple structure@the ~001!surface is an example of such a surface#; the surface energyis
g5 12 ~Eslab2NAmA2NBmB2
¯
NZmZ!, ~7!
where Eslab is the total energy of the slab.12 Along lowersymmetry directions, the slab calculations give only the sface energy relative to some reference, usually taken tothe 131 unreconstructed, unrelaxed surface.
It is possible, at least in principle, to determine the ablute surface energies for the symmetry directions for whslab calculations yield only relative surface energies pvided the crystal has a space group of sufficiently high symetry. Construct a polyhedron with identical faces: the toenergy of a polyhedron,Epoly , that hasnf identical faces,needges, andnv vertices, and containsNA , atoms of speciesA,NB atoms of speciesB, etc., is
Epoly5NAmA1NBmB¯1NzmZ1a fnfEsurfL21aeneEedgeL
1avnvEvertex, ~8!
whereEbulk is the energy per unit volume of the bulk matrial comprising the polyhedron,Esurf is the surface energyper unit area,Eedgeis the edge energy per unit length,Evertexis the energy of a vertex, andL is the characteristic size othe polyhedron. Thea coefficients depend upon the detaileshapes of the surfaces, edges and vertices but the diffescaling behavior of each of the energy terms can be exploto calculate the surface energy. After subtracting out the benergy, the remainder consists of surface terms, which vasL2, edge terms, which vary asL, and vertex terms, whichare independent ofL. For a sufficiently large polyhedron, thcontribution of the edge terms to the remainder will be nligible and the surface energy can be calculated to order 1L,viz.
i-
e
ss,
gee
e-aal
r-be
-h--l
entd
lkry
-
Esurf51
a fnL2 ~Epoly2NAmA2NBmB¯
2NzmZ!1O~1/L !.
~9!
A two-component system with a zinc-blende lattice prvides an example of a crystal with sufficiently high symmtry; a tetrahedron can be constructed that has four faces cprised of~111! surfaces terminated by the same species, AB.13 Clearly this approach is valid only for a limited set osymmetry directions. For the 11 Laue point groups and 11the 21 remaining point groups that have either two rotataxes or one rotation axis with a mirror plane not throughaxis, it is possible to find a polyhedron whose faces are coposed of crystallographically equivalent surfaces.9,13
The surface energies for other directions can be calculaby slicing off one of the vertices; the surface energy forbut one of the faces is known and, in the scaling limit, tunknown surface energy can be determined toO(1/L). Inthis case, too, the polyhedron must be sufficiently largeensure that edge and vertex effects are negligible. Althosuch calculations are not feasible at present, the procedescribed in this section illustrates the existence of unabiguously defined surface energies for crystals with symmtries as described above. For crystals with lower symmetran appropriate convention for defining the vectorC, thesame for all surfaces, must be established. For these systthe surface energies are measured with respect to a sreference surface.
III. ENERGY-DENSITY FORMALISM
The total energy of a charge-neutral system of electrwith densityr~r ! within the density-functional theory is
E5Ts@r#1VH@r#1EXC@r#1Vie@r#1Eii ~$RI%!, ~10!
where Ts@r# is the Kohn-Sham kinetic-energy functionaVH@r# is the Hartree functional,EXC@r# is the exchange-correlation functional,Vie@r# is the electron-ion energy anEii ($RI%) is the electrostatic self-energy of the atoms locaat $RI%. Minimization of this functional subject to the constraint that the number of electrons is fixed leads toKohn-Sham equations, the solution of which gives tground-state energy and density of the charge-neusystem.14,15
The energy density can be defined3,4 through the relation
E5EVd3rE~r !. ~11!
Consistent with this definition of the energy density, afunction f (r ) that integrates to zero can be added toenergy density. In principle, this presents a problem forenergy-density formalism; such an arbitrary function,gauge function, would make the integrals over subvolumarbitrary. In practice, however, such a problemdoes notarise. The energy functional and the energy density arecon-structed with each term having a physical motivationandthus such an arbitrary, unphysical function can beexcludedby construction. Notwithstanding this exclusion of arbitrarunphysical gauge terms, there is an intrinsic variability of tenergy density in any multicomponent system, which is d

thh
enn,o
ar, tTh
gu
e-c-tic
av
-mvrmloiga
roa
cahapo
uao
nersrew
t beuming
elf-sian
heond-
nn anthe
chr
nalt,ce,
s
th
ce.l-
illthere-
7284 57RAPCEWICZ, CHEN, YAKOBSON, AND BERNHOLC
to the nature of the lattice sum for the Coulomb energy ofions. This intrinsic variability can be used to incorporate tvariability of the externally imposed chemical potential.
The forms of many of the components of the energy dsity follow in a straightforward manner from the definitioEq. ~11!, although their concrete realization depends upthe details of the implementation of the calculation. In pticular, the present calculations use a supercell geometrypseudopotential formalism and a plane-wave basis.kinetic-energy density is
T~r !51
2 (n
f n¹cn* ~r !•¹cn~r !, ~12!
where the$cn% are the Kohn-Sham wave functions and$ f n%their occupations. This symmetric form of the kinetic eneris the more basic form that enters into the variational formlation of quantum mechanics.16,17 In systems possessing priodic boundary conditions, the minimization of this funtional gives the standard Laplacian-form of the kineenergy in the Kohn-Sham equations.18 The use of Bloch’stheorem permits the expansion of the Kohn-Sham wfunctions, viz.
T~r !51
2 (i ,k
f ik¹c i ,k* ~r !•¹c i ,k~r !, ~13!
where the sum onk is over the first Brillioun zone. Specializing to a plane-wave basis, it is more efficient, from a coputational point of view, to evaluate the gradient of the wafunction in reciprocal space and then to Fourier transfointo real space, whereT~r ! is a point-wise product. In reaspace, the exchange-correlation energy density is the pwise product of the electron density and the exchancorrelation energy per electron of the homogeneous, intering electron gas taken at the local density, i.e.,
EXC~r !5r~r !«XC~r !. ~14!
The determination of the energy density for the electstatic terms requires a careful treatment. The reasonscomputational and mathematical. The Hartree potentialbe most efficiently calculated for a charge density thatzero net charge, while the lattice sums of the Coulombtentials of the ions must be regularized through the usetechniques related to those introduced by Ewald. The sqof the electric field, which is the negative of the gradientthe Hartree potential, i.e.,
E~r !52¹vh~r !, ~15!
gives the Maxwell energy density, viz.
EM~r !51
8puE~r !u2. ~16!
This term can be most efficiently calculated in a mansimilar to the kinetic-energy density. The electric field is fievaluated in reciprocal space and then transformed intospace, where the energy density is calculated as a pointproduct.
ee
-
n-hee
y-
e
-e
nt-e-ct-
-rens-
ofref
rtal
ise
The pseudoion-pseudoion energy per supercell musmuch more extensively reworked analytically and the srestricted to the supercell by use of the minimum-imagconvention in order to obtain
Eion51
2 (I ,JPV
8ZIZJ
RIJerfcF RIJ
ARc,I2 1Rc,J
2 G21
2 (I PV
2ZI
A2pRc,I
.
~17!
The first term in the pseudoion–pseudoion electrostatic senergy is due to the Coulomb interaction between a Gauspseudoion atRI and one atRJ , while the second term is theelectrostatic self-energy of the charge distribution of tpseudoion atRI . The energy density for this contribution tthe total energy is obtained by taking the energy correspoing to an atom atRI to be the coefficient of ad functioncentered on that atom, viz.
« ion~r !5 (I ,JPV
8ZIZJ
RIJerfcF RIJ
ARc,I2 1Rc,J
2 G3a IJd~r2RI !
21
2 (I PV
2ZI
A2pRc,I
d~r2RI ! ~18!
where the prime on the sum indicates thatIÞJ. The un-known coefficientsa IJ reflect an ambiguity in the resolutioof this term into a density; the electrostatic energy betweepseudoion atRI and one atRJ is a sum evaluated at the iopositions, not an integral evaluated over all space. Whentwo ions in the sum are distinct, there is noa priori way ofassigning a portion of the weights to the contribution of eaion to the sum. Thea IJ may be chosen to be different fodifferent pairs of species subject only to the constraint
a IJ1aJI51 for IÞJ. ~19!
Thus in a two-component system there are two additiodegrees of freedom,a IJ andaJI , and one constraint so thain effect, there is one degree of freedom. For convenienthe $a IJ% were set equal to one-half in the calculations.
The local-pseudopotential energy can be written as
Elps5 (I PV
EV
d3rr~r !Usr,I~r !, ~20!
where
Usr,I~r !5U lps,I~r !2EVd3x
ng,I~x!
ur2xu, ~21!
so that the energy density for the local pseudopotential i
Elps~r !5r~r !Usr~r !. ~22!
In Eq. ~21!, ng,I(x) is a Gaussian charge density wiintegrated chargeZI and widthRc,I centered atRI .
The nonlocal pseudopotential is short range in real spaFollowing Chetty and Martin,3 the nonlocal pseudopotentiaenergy associated with an ion atRI is gathered into adfunction centered on that ion. The smallest volumes that wbe considered are much larger than the region over whichnonlocal pseudopotential is nonzero so that no spurious

dte
rn-n-
acsic
se
ndsvm
ac
el
l
uu
an
u
pcnsu
try
msed.
eces-lcu-ged toeth-be
gfulre-s ofde.
s be
ulkctedali-elab:noice,rgy,e.
nter-ur-eniva-
n avertalmee,s-rgeenak-itz
enofceym-g.anom., isoup
s.s,e
theithill
tral.at, ishe
57 7285CONSISTENT METHODOLOGY FOR CALCULATING . . .
sults are introduced by this treatment of the nonlocal pseupotential. The energy density for the nonlocal pseudopotial is
Enlps~r !5 (I PVEnlps,Id~r2RI !, ~23!
where the coefficient of thed function is
Enlps,I5(i ,k
f ikEV
d3r EV
d3r 8c i ,k* ~r !Unlps,I~r ,r 8!c i ,k~r 8!.
~24!
ThusE~r ! is a sum of densities, viz.
E~r !5Ekin~r !1EXC~r !1EM~r !1Eps~r !1Eion~r !, ~25!
where Ekin(r ) is the kinetic-energy density,EXC(r ) is theexchange-correlation energy density,EM(r ) is the Maxwellenergy density~a particular form of the energy density fothe Hartree term!, Eps(r ) is the pseudopotential energy desity, andEion(r ) is the ion-ion electrostatic self-energy desity.
IV. EVALUATION OF THE SURFACE ENERGY
The present approach to the calculation of the surfenergy necessarily involves two parts: the energy-denmethodology3,4 and an integration procedure. This approaextends the method developed by Chetty and Martin3–5 bygeneralizing the integration method so that it can be ueven in systems of low symmetry.
A. Surface energy using symmetry-adapted cells
The approach of Chetty and Martin3–5 makes use ofsymmetry-adapted unit cells for the integration. The bouaries of a symmetry-adapted unit cell are symmetry planethe crystal. Therefore, the integral of the energy density othis unit cell is gauge independent and the number of atoin the cell can be unambiguously determined. The surfenergy is then
s5EVs
d3rEslab~r !2(i
Nim i , ~26!
whereVs is the volume of the symmetry-adapted unit candNi is the number of atoms of thei th species inside thecell with i 5A,B, . . . ,Z. The externally imposed chemicapotential of thei th species ism i ; their sum is subject to theconstraint, Eq.~4!. This approach can be employed to calclate the surface energy of those directions in which the sface is cut obliquely by these symmetry planes. ChettyMartin4 applied their method to the~100! and~111! surfacesof GaAs. The~111! surfaces of a zinc-blende crystal are cobliquely by the~100! and the~110! planes. However, thisapproach cannot be used for a crystal whose point grouone of the 10 point groups for which the absolute surfaenergy is not defined, i.e., there will be surfaces that arecut obliquely by a sufficient number of symmetry planeWurtzite~0001! is an example of such a surface; as the resof the hexagonal symmetry, the appropriate symmeadapted cell cannot be defined.
o-n-
etyh
d
-oferse
l
-r-d
t
iseot.lt-
B. The Voronoi polyhedron
The integration procedure must permit aconsistentevalu-ation of the surface energy for those low-symmetry systefor which the absolute surface energy cannot be definSuch an approach is needed because, in general, it is nsary to treat the two surfaces of the slab in supercell calations differently. For instance, to minimize the chartransfer between surfaces, one of the surfaces may neepassivated and consequently it is desirable to have a modology that permits the surface energy of each face tocalculated separately. Further, in order to make meaninab initio predictions about equilibrium crystal shape and pferred growth directions, a database of surface energiereconstructed surfaces for different directions must be maThis requires that the evaluations of the surface energieconsistent between calculations.
In the approach that we have developed, we view the band the slab as built of blocks. These blocks are construaccording to a well-defined rule, which is a natural generzation of the definition of the familiar Wigner-Seitz cell. Thsame rule is used to obtain the Voronoi polyhedra in the sdeep in the slab, where the bulk is recovered, the Voropolyhedra are identical to those of the bulk; near the surfathey are deformed. With each block we associate an enenamely, the integral of the energy density over that volumThe ‘‘total cost’’ of constructing a slab and the equivalebulk is determined by simply adding up the respective engies associated with all of the Voronoi polyhedra. The sface energy is then simply the difference in ‘‘cost’’ betwethe portion of the slab containing one surface and the equlent bulk.
In a one-component crystalline system condensed olattice without a basis, the integral of the energy density oany volume that is charge neutral is unique; it is the toenergy per particle. The arbitrariness of the choice of volunotwithstanding, there is a geometrically motivated volumnamely, the Wigner-Seitz cell. The Wigner-Seitz cell posesses the symmetry of the lattice, is space filling and chaneutral. If the crystal has a basis of two identical atoms, ththere is a difference between the positions of the atoms ming up the crystal and the lattice points. The Wigner-Secell for this crystal is the set of all points closer to a givlattice point than to all other lattice points. As for the casea lattice without a basis, the Wigner-Seitz cell for the lattiwith a two-atom basis is charge neutral, possesses the smetry of the point group of the lattice and is space fillinHowever, it contains two atoms. The Wigner-Seitz cell cbe generalized to a Voronoi polyhedron about each atThis polyhedron, which is space filling and charge neutralinvariant under the largest point subgroup of the space grof the lattice.
Consider a lattice that has a basis of two distinct atomAs in the case of a lattice with a basis of two identical atomthe Wigner-Seitz-like volume for a given atom will have thsymmetry of the largest point group that is a subgroup ofspace group of the crystal. The union of such volumes wthe similarly defined cells for the other atom of the basis wbe space filling, but the volume need not be charge neuWe will generally choose to use a Voronoi polyhedron thhas these properties and, because it is most intuitivecharge neutral. This Voronoi polyhedron is obtained if t

nieordrn-m
onti-Ba
ththo
ly
nl.e-
a
a
ma
onrete
el to
rintoea
sux
ta
enoise
h
be-bor.on-
ly-edra
ris-
ionrd
theAlab
yly-
toput
ulk.lk,
hethe
7286 57RAPCEWICZ, CHEN, YAKOBSON, AND BERNHOLC
faces are translated inward or outward along its normal uthe volume is charge neutral. If the normal to a face lalong the vector joining two identical atoms, the face will nbe shifted. Only a face that has its normal pointing towaan atom of a different species than the one at the centethe cell will shift. However, it is also possible to use nocharge-neutral polyhedra. Indeed, in strongly ionic systewhere the charge exchange between the cation and anisignificant or in C60 where the carbon atoms are not idencal, it is necessary to use charged Voronoi polyhedra.cause the role of the Voronoi polyhedra is to provide a wof counting atoms and the energies~chemical potentials! as-sociated with them, it is not necessary to add terms toHamiltonian when using charged Voronoi polyhedra. Maematical aspects of the definition of the Voronoi polyhedrare discussed in Appendix I.
The integral of the energy density over the Voronoi pohedron,Vvp , in bulk is
E* 5EVvp
d3rE~r ! ~27!
and will be referred to as the bulk-atom energy. In a ocomponent system,E* is equal to the chemical potentiaThe integral of the energy density over the Voronoi polyhdron for speciesA will be referred to as the bulk-atom energy of speciesA, etc.
For a multicomponent system, the bulk-atom energy sisfies the constraint of equilibrium
mAB¯Z5EA* 1EB* 1¯1EZ* , ~28!
wheremAB¯Z is the chemical potential. The total energy ofsystem ofNA atoms of speciesA andNB atoms of speciesB,etc., is
Etot5NAEA* 1NBEB* 1¯1NZEZ* . ~29!
The bulk-atom energy plays a role identical to the iposed chemical potential in a slab calculation, namely, ofexternally imposed parameter~cf. Sec. II!. It can assumearbitrary values in the bulk system subject only to the cstraint of Eq.~28!. The same physical constraints that aused to delineate the possible values of the chemical potial also delimit the range of the bulk-atom energy.
For atoms deep inside of a slab where the bulk has brecovered, the Voronoi polyhedra of the slab are identicathe bulk Voronoi polyhedra. The shape of each Voronpolyhedron depends upon the locations of the neighboatoms. Consequently, as the surface is approached and aare displaced from their bulk positions, the volumes will dform. Because the atoms on the surface do not haveneighbors in the vacuum, the volumes for atoms at theface will extend out to infinity, cf. the definition in AppendiI and Fig. 1.
As an example, consider diamond. In a diamond cryswhich has space groupFd3m, the Voronoi polyhedron is asnub tetrahedron19,20 that has sixteen vertices and sixtefaces. The snub-tetrahedron is invariant under the pgroupTd and is pristine, i.e., it is space filling only if it haideal proportions.20 Its four hexagonal faces lie between thnearest neighbors and the atom at the origin, while eac
tilstsof
sis
e-y
e-n
-
e
-
t-
-n
-
n-
enoigms
-nyr-
l,
nt
of
its twelve isosceles triangular faces is a bisecting planetween the atom at the center and a next-nearest neighThe zinc-blende crystal structure for a compound semicductorAB has space groupF43m. In all but the exceptionalcase of equal weights for the two species, the Voronoi pohedra will be deformed snub tetrahedra, i.e., snub tetrahwith nonideal proportions~the union of the two deformedsnub tetrahedra—one forA and the other forB—is spacefilling !. In GaAs, the snub tetrahedra are very close to ptine.
C. Surface energy using Voronoi polyhedra
The use of Voronoi polyhedra permits the determinatof the deviation from bulk behavior in a straightforwamanner. For each atom, there is one polyhedron; deep inslab, the Voronoi polyhedra of the bulk are recovered.section of the slab that extends from a point deep in the sout to a point deep in the vacuum and containsNA atoms ofspeciesA, NB atoms of speciesB, etc., has a total energequal to the sum of the energy of each of the Voronoi pohedra contained in that portion of the slab, i.e.,
Esec5(IE
V I
d3rEslab~r !, ~30!
whereV I is the Voronoi polyhedron for an atom atRI andIruns over all atoms in the section of the slab. In orderobtain the surface energy, the atoms of the slab can beinto a one-to-one correspondence with the atoms of the bThis equivalent bulk is comprised of the appropriate buVoronoi polyhedra. Its total energy is simply
Ebulk5(IE
VVP,I
d3rEbulk~r !5NAEA* 1NBEB* 1¯1NZEZ* ,
~31!
whereVVP,I is the Voronoi polyhedron for an atom atRIand I runs over all atoms in the equivalent bulk@cf. Eq.~29!#. The surface energy is thus
FIG. 1. A two-dimensional slab showing the atomic cells. In tcenter of the slab, the bulk Voronoi polyhedra are recovered;cells at the surface extend to infinity.
e
ro
enossnem
abre9.th
oi
dteve
ofdra
ofe
lent,ing.n ara.int in-of
eal-
mlua-ent
the.s ofting
. ATout
r-hehe
nd
facedsur-
for-hected
ndge-gn,tialnd,
an-al
ari-ace
s
ers
m
it
upis
alb
esst
57 7287CONSISTENT METHODOLOGY FOR CALCULATING . . .
Esurf~EI* !5Esec2Ebulk ~32!
5(I
F EV I
d3rEslab~r !2EI* G ,~33!
where the sum onI runs over all atoms in the section of thslab.
It should be emphasized that the Voronoi polyhedra pvide a way of counting atoms and the energies~bulk-atomenergies! associated with them. In particular, the energy dsity is calculated for the entire cell and not as the superption of energy densities obtained by using the charge denin each Voronoi polyhedron. For instance, the Maxwell eergy density, Eq.~16!, is calculated using the density in thentire cell; Voronoi polyhedra are not used. Thus problerelated to long-range multipole moments do not arise.
Finally, in order to effectively use the formalism, the slmust be sufficiently thick to ensure that the bulk is recovein the center of the slab. In the example shown in Fig. 2,layers were used in order to obtain an accuracy of better0.01 eV.
D. Integrals over the Voronoi polyhedra
Numerous algorithms for the construction of Voronpolyhedra exist.21–26The particular algorithm we followed issimilar to that of Finney.22 A subset of neighbors is selecteand the vertices of the Voronoi polyhedron are then demined. The list of vertices thus generated is sorted and
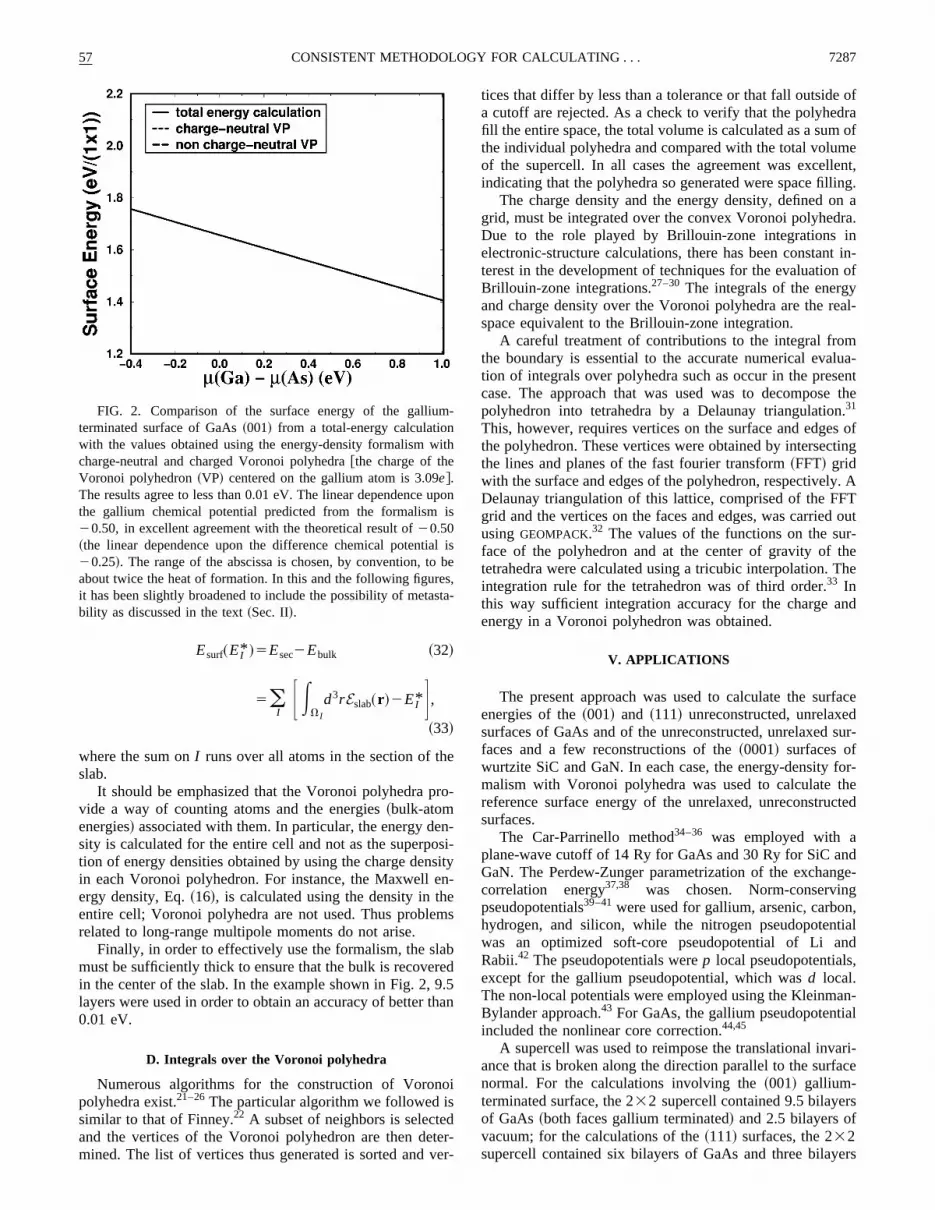
FIG. 2. Comparison of the surface energy of the galliuterminated surface of GaAs~001! from a total-energy calculationwith the values obtained using the energy-density formalism wcharge-neutral and charged Voronoi polyhedra@the charge of theVoronoi polyhedron~VP! centered on the gallium atom is 3.09e#.The results agree to less than 0.01 eV. The linear dependencethe gallium chemical potential predicted from the formalism20.50, in excellent agreement with the theoretical result of20.50~the linear dependence upon the difference chemical potenti20.25!. The range of the abscissa is chosen, by convention, toabout twice the heat of formation. In this and the following figurit has been slightly broadened to include the possibility of metability as discussed in the text~Sec. II!.
-
-i-
ity-
s
d5an
r-r-
tices that differ by less than a tolerance or that fall outsidea cutoff are rejected. As a check to verify that the polyhefill the entire space, the total volume is calculated as a sumthe individual polyhedra and compared with the total volumof the supercell. In all cases the agreement was excelindicating that the polyhedra so generated were space fill
The charge density and the energy density, defined ogrid, must be integrated over the convex Voronoi polyhedDue to the role played by Brillouin-zone integrationselectronic-structure calculations, there has been constanterest in the development of techniques for the evaluationBrillouin-zone integrations.27–30 The integrals of the energyand charge density over the Voronoi polyhedra are the rspace equivalent to the Brillouin-zone integration.
A careful treatment of contributions to the integral frothe boundary is essential to the accurate numerical evation of integrals over polyhedra such as occur in the prescase. The approach that was used was to decomposepolyhedron into tetrahedra by a Delaunay triangulation31
This, however, requires vertices on the surface and edgethe polyhedron. These vertices were obtained by intersecthe lines and planes of the fast fourier transform~FFT! gridwith the surface and edges of the polyhedron, respectivelyDelaunay triangulation of this lattice, comprised of the FFgrid and the vertices on the faces and edges, was carriedusing GEOMPACK.32 The values of the functions on the suface of the polyhedron and at the center of gravity of ttetrahedra were calculated using a tricubic interpolation. Tintegration rule for the tetrahedron was of third order.33 Inthis way sufficient integration accuracy for the charge aenergy in a Voronoi polyhedron was obtained.
V. APPLICATIONS
The present approach was used to calculate the surenergies of the~001! and ~111! unreconstructed, unrelaxesurfaces of GaAs and of the unreconstructed, unrelaxedfaces and a few reconstructions of the~0001! surfaces ofwurtzite SiC and GaN. In each case, the energy-densitymalism with Voronoi polyhedra was used to calculate treference surface energy of the unrelaxed, unreconstrusurfaces.
The Car-Parrinello method34–36 was employed with aplane-wave cutoff of 14 Ry for GaAs and 30 Ry for SiC aGaN. The Perdew-Zunger parametrization of the exchancorrelation energy37,38 was chosen. Norm-conservinpseudopotentials39–41were used for gallium, arsenic, carbohydrogen, and silicon, while the nitrogen pseudopotenwas an optimized soft-core pseudopotential of Li aRabii.42 The pseudopotentials werep local pseudopotentialsexcept for the gallium pseudopotential, which wasd local.The non-local potentials were employed using the KleinmBylander approach.43 For GaAs, the gallium pseudopotentiincluded the nonlinear core correction.44,45
A supercell was used to reimpose the translational invance that is broken along the direction parallel to the surfnormal. For the calculations involving the~001! gallium-terminated surface, the 232 supercell contained 9.5 bilayerof GaAs ~both faces gallium terminated! and 2.5 bilayers ofvacuum; for the calculations of the~111! surfaces, the 232supercell contained six bilayers of GaAs and three bilay
-
h
on
ise
,a-
-thn
eSto
t,enn
tholle
Irmu
f tthorgsis:
ale
in
r tf tseedel
np
thwiethtee
re
th
thethating
ces
ndthe
hantedthetheon-so-
ngan-esonsonallo-ner-asesto
ces
ery-ms
mictern
try
tedism
7288 57RAPCEWICZ, CHEN, YAKOBSON, AND BERNHOLC
of vacuum. A 234 supercell containing five bilayers of material and three bilayers of vacuum was used for SiC;232 GaN supercell contained six bilayers of material afour bilayers of vacuum.
In order to minimize charge transfer between the fachydrogen atoms passivated one face of the GaN andslabs. This was successful as charge transfer was foundsmall.
If the work functions of the two surfaces are differenthen the periodic boundary conditions of the supercellforce a common electrostatic potential in the vacuum aresult in an unphysical change in the electric potential invacuum region equal to the difference between the two wfunctions. The field, induced by this change, was canceby adding a dipole layer in the vacuum region.
A. Surface energy of GaAs„001…
The calculation of the surface energy of the~001! surfaceof GaAs provides a good test of the present formalism.this case a slab in which both surfaces have the same tenation exists. The surface energy can thus be calculateding the total-energy approach and the surface energies otwo surfaces can be independently calculated usingenergy-density approach presented herein. Figure 2 shthe gallium surface energy calculated with the total-enemethod compared to that obtained with the energy-denformalism with Voronoi polyhedra for two different case~1! charge-neutral Voronoi polyhedra and~2! chargedVoronoi polyhedra in which the polyhedra around the glium atoms have charge 3.09e, while those centered on tharsenic atoms have charge 4.91e. Only the calculations ofone face of the slab are shown. Similar results are obtafor the other face.
The surface energies agree to better than 0.01 eV oveentire range. In all of the cases, the linear dependence osurface energy upon the gallium chemical potential haslope of20.50, which is in very good agreement with thexact value of20.50. The value of this slope is not assumin the calculation, but is a result that confirms the correctnof the approach. Thus it is possible to use either neutracharged Voronoi polyhedra.
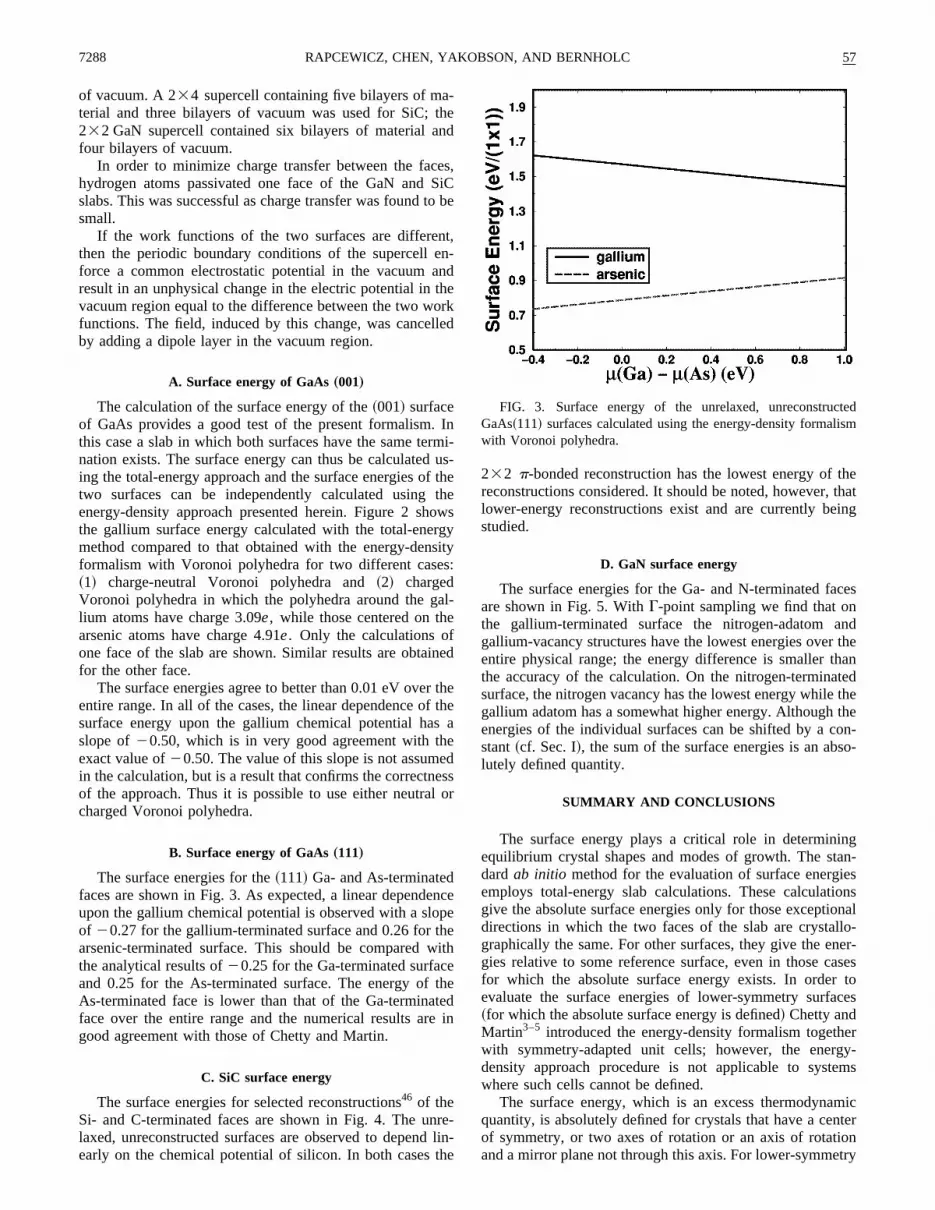
B. Surface energy of GaAs„111…
The surface energies for the~111! Ga- and As-terminatedfaces are shown in Fig. 3. As expected, a linear dependeupon the gallium chemical potential is observed with a sloof 20.27 for the gallium-terminated surface and 0.26 forarsenic-terminated surface. This should be comparedthe analytical results of20.25 for the Ga-terminated surfacand 0.25 for the As-terminated surface. The energy ofAs-terminated face is lower than that of the Ga-terminaface over the entire range and the numerical results argood agreement with those of Chetty and Martin.
C. SiC surface energy
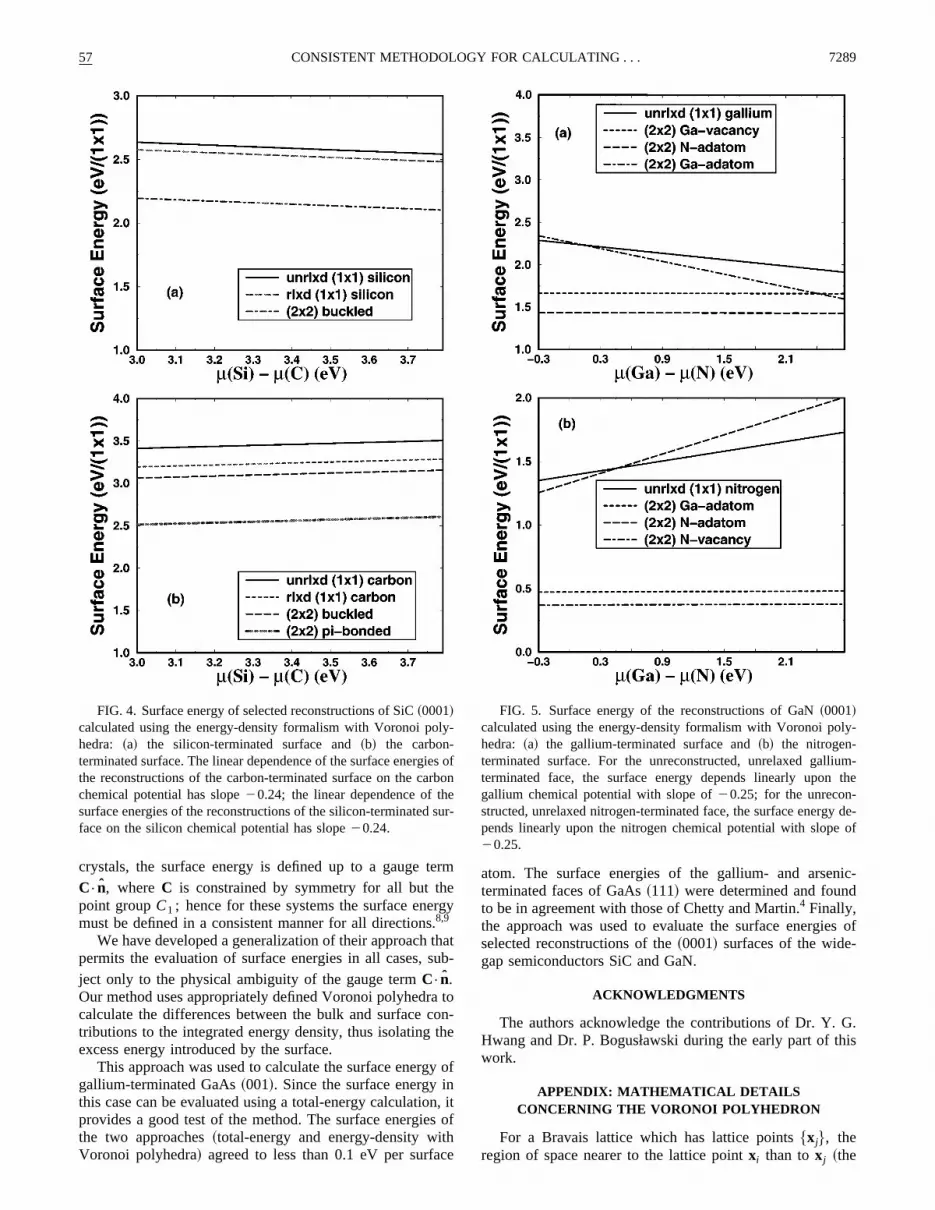
The surface energies for selected reconstructions46 of theSi- and C-terminated faces are shown in Fig. 4. The unlaxed, unreconstructed surfaces are observed to dependearly on the chemical potential of silicon. In both cases
ed
s,iCbe
-derkd
ni-s-
hee
wsyty
-
ed
hehea
ssor
ceeeth
edin
-lin-e
232 p-bonded reconstruction has the lowest energy ofreconstructions considered. It should be noted, however,lower-energy reconstructions exist and are currently bestudied.
D. GaN surface energy
The surface energies for the Ga- and N-terminated faare shown in Fig. 5. WithG-point sampling we find that onthe gallium-terminated surface the nitrogen-adatom agallium-vacancy structures have the lowest energies overentire physical range; the energy difference is smaller tthe accuracy of the calculation. On the nitrogen-terminasurface, the nitrogen vacancy has the lowest energy whilegallium adatom has a somewhat higher energy. Althoughenergies of the individual surfaces can be shifted by a cstant~cf. Sec. I!, the sum of the surface energies is an ablutely defined quantity.
SUMMARY AND CONCLUSIONS
The surface energy plays a critical role in determiniequilibrium crystal shapes and modes of growth. The stdardab initio method for the evaluation of surface energiemploys total-energy slab calculations. These calculatigive the absolute surface energies only for those exceptidirections in which the two faces of the slab are crystalgraphically the same. For other surfaces, they give the egies relative to some reference surface, even in those cfor which the absolute surface energy exists. In orderevaluate the surface energies of lower-symmetry surfa~for which the absolute surface energy is defined! Chetty andMartin3–5 introduced the energy-density formalism togethwith symmetry-adapted unit cells; however, the energdensity approach procedure is not applicable to systewhere such cells cannot be defined.
The surface energy, which is an excess thermodynaquantity, is absolutely defined for crystals that have a cenof symmetry, or two axes of rotation or an axis of rotatioand a mirror plane not through this axis. For lower-symme
FIG. 3. Surface energy of the unrelaxed, unreconstrucGaAs~111! surfaces calculated using the energy-density formalwith Voronoi polyhedra.
eerg.thsu
acoth
ynnshce
nic-d
s of
G.is
ly
ierbesu
ly-
um-the
y de-of
57 7289CONSISTENT METHODOLOGY FOR CALCULATING . . .
crystals, the surface energy is defined up to a gauge tC•n, whereC is constrained by symmetry for all but thpoint groupC1 ; hence for these systems the surface enemust be defined in a consistent manner for all directions8,9
We have developed a generalization of their approachpermits the evaluation of surface energies in all cases,ject only to the physical ambiguity of the gauge termC•n.Our method uses appropriately defined Voronoi polyhedrcalculate the differences between the bulk and surfacetributions to the integrated energy density, thus isolatingexcess energy introduced by the surface.
This approach was used to calculate the surface energgallium-terminated GaAs~001!. Since the surface energy ithis case can be evaluated using a total-energy calculatioprovides a good test of the method. The surface energiethe two approaches~total-energy and energy-density witVoronoi polyhedra! agreed to less than 0.1 eV per surfa
FIG. 4. Surface energy of selected reconstructions of SiC~0001!calculated using the energy-density formalism with Voronoi pohedra: ~a! the silicon-terminated surface and~b! the carbon-terminated surface. The linear dependence of the surface energthe reconstructions of the carbon-terminated surface on the cachemical potential has slope20.24; the linear dependence of thsurface energies of the reconstructions of the silicon-terminatedface on the silicon chemical potential has slope20.24.
rm
y
atb-
ton-e
of
, itof
atom. The surface energies of the gallium- and arseterminated faces of GaAs~111! were determined and founto be in agreement with those of Chetty and Martin.4 Finally,the approach was used to evaluate the surface energieselected reconstructions of the~0001! surfaces of the wide-gap semiconductors SiC and GaN.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Dr. Y.Hwang and Dr. P. Bogusławski during the early part of thwork.
APPENDIX: MATHEMATICAL DETAILSCONCERNING THE VORONOI POLYHEDRON
For a Bravais lattice which has lattice points$xj%, theregion of space nearer to the lattice pointxi than toxj ~the
-
s ofon
r-
FIG. 5. Surface energy of the reconstructions of GaN~0001!calculated using the energy-density formalism with Voronoi pohedra: ~a! the gallium-terminated surface and~b! the nitrogen-terminated surface. For the unreconstructed, unrelaxed galliterminated face, the surface energy depends linearly upongallium chemical potential with slope of20.25; for the unrecon-structed, unrelaxed nitrogen-terminated face, the surface energpends linearly upon the nitrogen chemical potential with slope20.25.

lf
eis
ede
o-th
thareth
ee
thon
tandee
t
is
eilf-
seeram
dep
theuge
inisbe
-
acthealsisof
alf-
the
o-ur-ghee-
ctor
f
bytheif,n be
gedp is
that
7290 57RAPCEWICZ, CHEN, YAKOBSON, AND BERNHOLC
so-called dominance region ofxi over xj ! is the half-space
D~xi ,xj !5$xuux2xi u2,ux2xj u2%; ~A1!
the boundary of this half-space is the plane defined by
ux2xi u25ux2xj u2; ~A2!
and the Wigner-Seitz cell is the intersection of all such haspaces, i.e.,
V~xi !5ù j Þ iD~xi ,xj !. ~A3!
Since the Wigner-Seitz cell is the intersection of convhalf-spaces, it is convex. Because its faces are planes, itpolyhedron.
The isogonal point group of a crystal is the group formfrom all of the point-group operations which occur in thspace group.47 If the space group is symmorphic, the isognal point group will be a subgroup of the space group; ifspace group is nonsymmorphic~it contains either a screwaxis or glide-reflection plane!, it will not be a subgroup. Be-cause the symmetry operations of the point group leavelattice points unchanged, the Wigner-Seitz cell is also invant under the operations of the point group. The WignSeitz cell is also space filling and is charge neutral. Thusthree properties that characterize a Wigner-Seitz cell arfollows: it is a space-filling polyhedron that has the symmtry of the point group of the lattice and is charge neutral.
In the case of diamond, there are two carbon atoms inbasis and the Wigner-Seitz cell as traditionally defined ctains the same number of atoms as there are atoms inbasis, i.e., two carbon atoms. A Wigner-Seitz cell foratom or, more properly, a Voronoi polyhedron, can befined by analogy with the Wigner-Seitz cell for a latticpoint. The region nearer to an atom atRI than to an atom aRJ is the half-space
D~RI ,RJ!5$xuux2RI u2,ux2RJu2%. ~A4!
The intersection of all these half-spaces, i.e.,
V~RI !5ùJÞID~RI ,RJ!, ~A5!
defines the Voronoi polyhedron. Any point inside this cellcloser to the atom atRI than to all other atoms~rather than tothe lattice point as is the case for the traditional Wigner-Scell!. The Voronoi polyhedron, being the intersection of haspaces, is a convex polyhedron.
Defined in this way, the Voronoi polyhedron possesmany properties similar to those of the standard WignSeitz cell. It is a space-filling polyhedron and charge neutHowever, it is no longer necessarily invariant under the symetry operations of the isogonal point group, but only unthe operations of the largest point group that is a subgrou
xy
-
xa
e
ei-r-eas-
e-he
-
tz
sr-l.-rof
the space group of the crystal. Further, the integral ofenergy density over this volume is independent of any gaand is equal to the total energy per atom.
If the basis atoms are not identical, for example asGaAs, the Voronoi polyhedron will no longer be neutral asdesired. In this case, the Voronoi polyhedra is defined toan additively weighted power Voronoi polyhedron.31 The ad-ditively weighted power Voronoi polyhedron or, more simply, the power Voronoi polyhedron extendsnaturally thedefinition of the Voronoi polyhedron from a lattice withbasis of identical atoms to a lattice with a basis of distinatoms. In point of fact, the standard Wigner-Seitz cell, tVoronoi polyhedron for a lattice with a basis of identicatoms and the Voronoi polyhedron for a lattice with a baof distinct atoms are all encompassed within the definitionthe power Voronoi polyhedron.
The additively weighted power-distance of pointx froman atom atRI is
dpw~x,RI ;wI !5ux2RI u22wI ~A6!
wherewI is the weight associated with the atom atRI . Withrespect to the additively weighted power distance, the hspace closer to an atom atRI than to one atRJ is
D~RI ,RJ!5$xuux2RI u22wI,ux2RJu22wJ% ~A7!
and the power Voronoi polyhedron is the intersection ofhalf-spaces, i.e.,
V~RI !5ùJÞID~RI ,RJ!. ~A8!
The use of the additively weighted power distance is mtivated by the observation that the planes defining the sfaces of the traditional Wigner-Seitz cell are defined throuEq. ~A2! and that the additive weights rigidly translate thfaces of the Wigner-Seitz cell, always maintaining a polyhdral shape. WhenwI5w5constant for a lattice without abasis, the standard Wigner-Seitz cell is obtained.
For a tetrahedrally coordinated compound semiconducomprised of elementsA and B, the volume containedwithin the Voronoi polyhedron is a monotonic function othe difference of the weightswA and wB . If charge-neutralpolyhedra are used, the value of this difference is fixedthe requirement that the total electronic charge withinVoronoi polyhedron cell be equal to the ionic charge;instead, charged polyhedra are used, the difference cachosen for convenience. In the case of C60, not all of theatoms are in symmetry-equivalent positions and charVoronoi polyhedra must be used. Further, the space grounonsymmorphic. Consequently, the largest point groupis a subgroup of the space group for solid C60 is notTh
6 , but
3 and the Voronoi polyhedra will have this symmetry.
1J. Tsao,Materials Fundamentals of Molecular Beam Epita~Academic Press, Boston 1993!.
2J. LaFemina, Phys. Rep.16, 133 ~1992!.3N. Chetty and R. M. Martin, Phys. Rev. B45, 6074~1992!.4N. Chetty and R. M. Martin, Phys. Rev. B45, 6089~1992!.5N. Chetty and R. M. Martin, Phys. Rev. B44, 5568~1991!.
6C. Herring, inThe Physics of Powder Metallurgy, edited by W. E.Kingston~American Society of Metals, Cleveland, Ohio, 1951!,pp. 7 and 8.
7W. Winterbottom, Acta Metall.15, 303 ~1967!.8J. Lee, H. Aaronson, and K. Russel, Surf. Sci.51, 302 ~1975!.9E. Arbel and J. Cahn, Surf. Sci.51, 305 ~1975!.

hea
,
,
orh
-
-
ev.
s.
-gs
f
57 7291CONSISTENT METHODOLOGY FOR CALCULATING . . .
10G-X. Qian, R. Martin, and D. Chadi, Phys. Rev. B38, 7649~1988!.
11There may also be other stoichiometries that will further limit tstability range, thus making this choice somewhat arbitrary,though conventional.
12This definition of the surface energy~at T50! corresponds toHerring’s specific surface~free! energy.
13M. Buerger,Elementary Crystallography~MIT Press, Cambridge1978!.
14P. Hohenberg and W. Kohn, Phys. Rev.136, 864B ~1964!.15W. Kohn and L. Sham, Phys. Rev.140, 1133A ~1965!.16J. Slater, Phys. Rev.51, 846 ~1937!.17D. Cook,Schrodinger’s Mechanics~World Scientific, Singapore
1988!.18There is, in principle, no problem in choosing a different form f
the kinetic energy density, provided it is used consistently. Tchoice serves to change the gauge.
19L. Foppl, Phys. Z.15, 191 ~1914!.20K. Critchlow, Order in Space: A Design Source Book~Viking,
New York, 1969!.21W. Brostow, J.-P. Dusrault, and B. Fox, J. Comput. Phys.29, 81
~1978!.22J. Finney, J. Comput. Phys.32, 137 ~1979!.23M. Tanemura, T. Ogawa, and N. Ogifa, J. Comput. Phys.51, 191
~1983!.24N. Medvedev, J. Comput. Phys.67, 223 ~1986!.25R. Riedinger, M. Habar, P. Oelhafen, and H. Gu¨nterodt, J. Com-
put. Phys.74, 61 ~1988!.26M. P. Allen and D. J. Tildesley,Computer Simulation of Liquids
~Clarendon Press, Oxford, 1987!.27O. Jepson and O. K. Anderson, Solid State Commun.9, 1763
~1971!.
l-
e
28G. Lehmann and M. Taut, Phys. Status Solidi B54, 469 ~1972!.29H. Monkhorst and J. Pack, Phys. Rev. B13, 5188~1976!.30P. Blochl, O. Jepsen, and O. K. Anderson, Phys. Rev. B49,
16 223~1994!.31A. Okabe, B. Boots, and K. Sugihara,Spatial Tesselations: Con
cepts and Applications of Voronoi Diagrams~Wiley, Chichester,1992!.
32B. Joe, Adv. Eng. Softw.13, 325 ~1991!.33M. Abramowitz and I. Stegun,Handbook of Mathematical Func
tions ~Dover, N.Y., 1972!, p. 895.34R. Car and M. Parrinello, Phys. Rev. Lett.55, 2471~1985!.35M. Payne, M. Teter, D. Allan, T. Arias, and J. Joannopoulos, R
Mod. Phys.64, 1045~1992!.36D. Remler and P. Madden, Mol. Phys.70, 921 ~1990!.37J. Perdew and A. Zunger, Phys. Rev. B23, 5048~1981!.38D. Ceperley and B. Alder, Phys. Rev. Lett.45, 566 ~1980!.39G. Bachelet, D. Hamann, and M. Schlu¨ter, Phys. Rev. B26, 4199
~1982!.40D. R. Hamann, M. Schlu¨ter, and C. Chiang, Phys. Rev. Lett.43,
1494 ~1979!.41D. R. Hamann, Phys. Rev. B40, 2980~1989!.42G. Li and S. Rabii, 1992~unpublished!.43L. Kleinman and D. Bylander, Phys. Rev. Lett.48, 1425~1982!.44S. Louie, S. Froyen, and M. L. Cohen, Phys. Rev. B26, 1738
~1982!.45M. Buongiorno Nardelli, K. Rapcewicz, and J. Bernholc, Phy
Rev. B55, R7323~1997!.46Y. Hwang, B. Chen, P. Bogusławski, and J. Bernholc, inCovalent
Ceramics II: Non-Oxides, edited by A. R. Barron, G. S. Fischman, M. A. Fury, A. F. Hepp, MRS Symposia ProceedinNo. 327~Materials Research Society, Pittsburgh, 1994!, p. 293.
47C. J. Bradley and A. P. Cracknell,The Mathematical Theory oSymmetry in Solids~Oxford, Clarendon Press, 1972!.
Related Documents











