Review and recommendations Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations Robert H. Dworkin a,⇑ , Dennis C. Turk b , Sarah Peirce-Sandner c , Laurie B. Burke d , John T. Farrar e , Ian Gilron f , Mark P. Jensen b , Nathaniel P. Katz g,h , Srinivasa N. Raja i , Bob A. Rappaport d , Michael C. Rowbotham j , Misha-Miroslav Backonja k , Ralf Baron l , Nicholas Bellamy m , Zubin Bhagwagar n , Ann Costello d , Penney Cowan o , Weikai Christopher Fang p , Sharon Hertz d , Gary W. Jay q , Roderick Junor r , Robert D. Kerns s,t , Rosemary Kerwin u , Ernest A. Kopecky v , Dmitri Lissin w , Richard Malamut x , John D. Markman c , Michael P. McDermott c , Catherine Munera y , Linda Porter z , Christine Rauschkolb aa , Andrew S.C. Rice ab , Cristina Sampaio ac , Vladimir Skljarevski ad , Kenneth Sommerville ae , Brett R. Stacey af , Ilona Steigerwald ag , Jeffrey Tobias ah , Ann Marie Trentacosti d , Ajay D. Wasan ai , George A. Wells aj , Jim Williams ak , James Witter x , Dan Ziegler al a Departments of Anesthesiology and Neurology and Center for Human Experimental Therapeutics, University of Rochester, Rochester, NY 14642, USA b University of Washington, Seattle, WA, USA c University of Rochester, Rochester, NY, USA d United States Food and Drug Administration, Silver Spring, MD, USA e University of Pennsylvania, Philadelphia, PA, USA f Queen’s University, Kingston, ON, Canada g Analgesic Solutions, Natick, MA, USA h Tufts University, Boston, MA, USA i Johns Hopkins University, Baltimore, MD, USA j California Pacific Medical Center Research Institute, San Francisco, CA, USA k University of Wisconsin, Madison, WI, USA l University of Kiel, Kiel, Germany m University of Queensland, Brisbane, Australia n Bristol-Myers Squibb, Wallingford, CT, USA o American Chronic Pain Association, Rocklin, CA, USA p DePuy Spine, Raynham, MA, USA q Pfizer, New London, CT, USA r Eisai Limited, Mosquito Way, Hatfield, UK s Department of Veterans Affairs, West Haven, CT, USA t Yale University, New Haven, CT, USA u Nuvo Research, West Chester, PA, USA v Endo Pharmaceuticals Inc., Chadds Ford, PA, USA w Durect Corporation, Cupertino, CA, USA x AstraZeneca, Wilmington, DE, USA y Purdue Pharma, Stamford, CT, USA z National Institutes of Health, Bethesda, MD, USA aa Johnson & Johnson Pharmaceutical Research & Development, Titusville, NJ, USA ab Imperial College, London, UK ac Faculdade de Medicina de Lisboa, Lisbon, Portugal ad Eli Lilly & Co., Indianapolis, IN, USA ae King Pharmaceuticals (currently Pfizer), Cary, NC, USA af Oregon Health and Science University, Portland, OR, USA ag Grünenthal GmbH, Aachen, Germany ah NeurogesX, Inc., San Carlos, CA, USA ai Harvard Medical School, Boston, MA, USA aj University of Ottawa, Ottawa, ON, Canada ak Smith & Nephew, Durham, NC, USA al German Diabetes Center, Heinrich Heine University, Düsseldorf, Germany 0304-3959/$36.00 Ó 2012 International Association for the Study of Pain. Published by Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.pain.2012.03.003 ⇑ Corresponding author. Address: Department of Anesthesiology, University of Rochester Medical Center, 601 Elmwood Ave., Box 604, Rochester, NY 14642, USA. Tel.: +1 585 275 8214; fax: +1 585 276 0122. E-mail address: [email protected] (R.H. Dworkin). www.elsevier.com/locate/pain PAIN Ò 153 (2012) 1148–1158

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

w w w . e l s e v i e r . c o m / l o c a t e / p a i n
PAIN�
153 (2012) 1148–1158
Review and recommendations
Considerations for improving assay sensitivity in chronic pain clinical trials:IMMPACT recommendations
Robert H. Dworkin a,⇑, Dennis C. Turk b, Sarah Peirce-Sandner c, Laurie B. Burke d, John T. Farrar e,Ian Gilron f, Mark P. Jensen b, Nathaniel P. Katz g,h, Srinivasa N. Raja i, Bob A. Rappaport d,Michael C. Rowbotham j, Misha-Miroslav Backonja k, Ralf Baron l, Nicholas Bellamy m, Zubin Bhagwagar n,Ann Costello d, Penney Cowan o, Weikai Christopher Fang p, Sharon Hertz d, Gary W. Jay q, Roderick Junor r,Robert D. Kerns s,t, Rosemary Kerwin u, Ernest A. Kopecky v, Dmitri Lissin w, Richard Malamut x,John D. Markman c, Michael P. McDermott c, Catherine Munera y, Linda Porter z, Christine Rauschkolb aa,Andrew S.C. Rice ab, Cristina Sampaio ac, Vladimir Skljarevski ad, Kenneth Sommerville ae, Brett R. Stacey af,Ilona Steigerwald ag, Jeffrey Tobias ah, Ann Marie Trentacosti d, Ajay D. Wasan ai, George A. Wells aj,Jim Williams ak, James Witter x, Dan Ziegler al
a Departments of Anesthesiology and Neurology and Center for Human Experimental Therapeutics, University of Rochester, Rochester, NY 14642, USAb University of Washington, Seattle, WA, USAc University of Rochester, Rochester, NY, USAd United States Food and Drug Administration, Silver Spring, MD, USAe University of Pennsylvania, Philadelphia, PA, USAf Queen’s University, Kingston, ON, Canadag Analgesic Solutions, Natick, MA, USAh Tufts University, Boston, MA, USAi Johns Hopkins University, Baltimore, MD, USAj California Pacific Medical Center Research Institute, San Francisco, CA, USAk University of Wisconsin, Madison, WI, USAl University of Kiel, Kiel, Germanym University of Queensland, Brisbane, Australian Bristol-Myers Squibb, Wallingford, CT, USAo American Chronic Pain Association, Rocklin, CA, USAp DePuy Spine, Raynham, MA, USAq Pfizer, New London, CT, USAr Eisai Limited, Mosquito Way, Hatfield, UKs Department of Veterans Affairs, West Haven, CT, USAt Yale University, New Haven, CT, USAu Nuvo Research, West Chester, PA, USAv Endo Pharmaceuticals Inc., Chadds Ford, PA, USAw Durect Corporation, Cupertino, CA, USAx AstraZeneca, Wilmington, DE, USAy Purdue Pharma, Stamford, CT, USAz National Institutes of Health, Bethesda, MD, USAaa Johnson & Johnson Pharmaceutical Research & Development, Titusville, NJ, USAab Imperial College, London, UKac Faculdade de Medicina de Lisboa, Lisbon, Portugalad Eli Lilly & Co., Indianapolis, IN, USAae King Pharmaceuticals (currently Pfizer), Cary, NC, USAaf Oregon Health and Science University, Portland, OR, USAag Grünenthal GmbH, Aachen, Germanyah NeurogesX, Inc., San Carlos, CA, USAai Harvard Medical School, Boston, MA, USAaj University of Ottawa, Ottawa, ON, Canadaak Smith & Nephew, Durham, NC, USAal German Diabetes Center, Heinrich Heine University, Düsseldorf, Germany
0304-3959/$36.00 � 2012 International Association for the Study of Pain. Published by Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.pain.2012.03.003
⇑ Corresponding author. Address: Department of Anesthesiology, University of Rochester Medical Center, 601 Elmwood Ave., Box 604, Rochester, NY 14642, USA. Tel.: +1585 275 8214; fax: +1 585 276 0122.
E-mail address: [email protected] (R.H. Dworkin).

R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158 1149
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
a r t i c l e i n f o
Article history:Received 27 September 2011Received in revised form 23 February 2012Accepted 5 March 2012
a b s t r a c t
A number of pharmacologic treatments examined in recent randomized clinical trials (RCTs) have failedto show statistically significant superiority to placebo in conditions in which their efficacy had previouslybeen demonstrated. Assuming the validity of previous evidence of efficacy and the comparability of thepatients and outcome measures in these studies, such results may be a consequence of limitations in theability of these RCTs to demonstrate the benefits of efficacious analgesic treatments vs placebo (‘‘assaysensitivity’’). Efforts to improve the assay sensitivity of analgesic trials could reduce the rate of falselynegative trials of efficacious medications and improve the efficiency of analgesic drug development.Therefore, an Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials consensusmeeting was convened in which the assay sensitivity of chronic pain trials was reviewed and discussed.On the basis of this meeting and subsequent discussions, the authors recommend consideration of a num-ber of patient, study design, study site, and outcome measurement factors that have the potential toaffect the assay sensitivity of RCTs of chronic pain treatments. Increased attention to and research onmethodological aspects of clinical trials and their relationships with assay sensitivity have the potentialto provide the foundation for an evidence-based approach to the design of analgesic clinical trials andexpedite the identification of analgesic treatments with improved efficacy and safety.
� 2012 International Association for the Study of Pain. Published by Elsevier B.V. All rights reserved.
1. Introduction
In what appears to be an increasing number of randomized clin-ical trials (RCTs), various analgesic medications have failed to showstatistically significant superiority to placebo in conditions in whichtheir efficacy was previously demonstrated and for which they havebeen approved by regulatory agencies [18,48,71,91,106]. Unfortu-nately, many of these trials remain unpublished, a well-known phe-nomenon that makes it difficult to estimate the incidence of suchresults [79,80]. Because the sets of RCTs submitted to regulatoryagencies are typically comprehensive [58,96], regulatory authori-ties will often be aware of RCTs that have failed to demonstrate effi-cacy and that have not been published; for example, within oneregulatory setting it has been observed that ‘‘We’ve seen over theyears that analgesic clinical trials frequently fail, and often withdrugs that we know work. When opioids that have been known tobe effective for hundreds of years are put in clinical trials, we can’tget a positive result’’ [97].
The interpretation of RCTs in which analgesic treatments thathave previously shown evidence of efficacy do not show statisti-cally significant superiority to placebo can be challenging. One pos-sibility is that changes in study design, patient selection, and/oroutcome measures have improved the validity of these RCTs, andthat treatments previously shown to be efficacious actually lacktrue efficacy. Another possibility is that various characteristics ofclinical trials can compromise their ability to reveal efficacy.Although the demonstration of treatment efficacy can certainly de-pend on the methods and measures used in RCTs, a considerablenumber of trials in which there was a failure to replicate resultshave used similar research designs, patients, and measures (eg,daily pain ratings) [18,48,71,91,106].
In circumstances in which previous demonstrations of analge-sic efficacy can be assumed to be valid and patients and out-come measures are comparable, failures to demonstratestatistically significant evidence of efficacy can be attributed tolimitations in assay sensitivity, which has been defined as theability of a clinical trial ‘‘to distinguish an effective treatmentfrom a less effective or ineffective treatment’’ [39]. Better under-standing of factors—other than chance—that influence the assaysensitivity of analgesic trials to identify truly efficacious treat-ments would expedite the development of new analgesic treat-ments with improved efficacy, safety, or tolerability
[50,104,105]. Such knowledge could also play a critical rolewhen interpreting the results of negative trials in developingevidence-based treatment guidelines. It is important to empha-size that efforts to understand and enhance the assay sensitivityof analgesic RCTs must be clearly distinguished from attempts tosalvage ineffective treatments or weak treatment effects by con-ducting multiple secondary or subgroup analyses and selectivelyreporting the results. To the greatest extent possible, analysesconducted with the objective of identifying factors associatedwith assay sensitivity should be prespecified, comprehensivelydescribed and reported, and followed by the design of prospec-tive studies to confirm the results.
2. Methods
The Initiative on Methods, Measurement, and Pain Assess-ment in Clinical Trials (IMMPACT; www.immpact.org) (eg,[20,22,94,95]) convened a consensus meeting to identify factorsthat appear to influence assay sensitivity and to develop recom-mendations for improvement of assay sensitivity in RCTs ofchronic pain conditions. The meeting included participants fromuniversities, government agencies, industry, and a patient advo-cacy organization selected on the basis of research, clinical, oradministrative expertise relevant to the evaluation of chronicpain treatments. It should be noted that the majority of partici-pants were from North America, with 6 individuals attendingfrom Australia, Germany, Portugal, and the United Kingdom. Asdiscussed below, there may be international differences in pla-cebo group improvement and assay sensitivity in analgesic trials,and the extent to which our recommendations have the poten-tial to improve assay sensitivity in different countries and re-gions is unknown.
Background presentations of literature reviews were deliveredto facilitate discussion; these presentations [37] and a review ofgeneral considerations and research studies relevant to assaysensitivity [21] appear elsewhere. This article presents theauthors’ recommendations for improving assay sensitivity ofchronic pain RCTs based on these background presentations,the deliberations at the consensus meeting, subsequent discus-sions among the participants, and the final development andrevision of the recommendations described in this article.
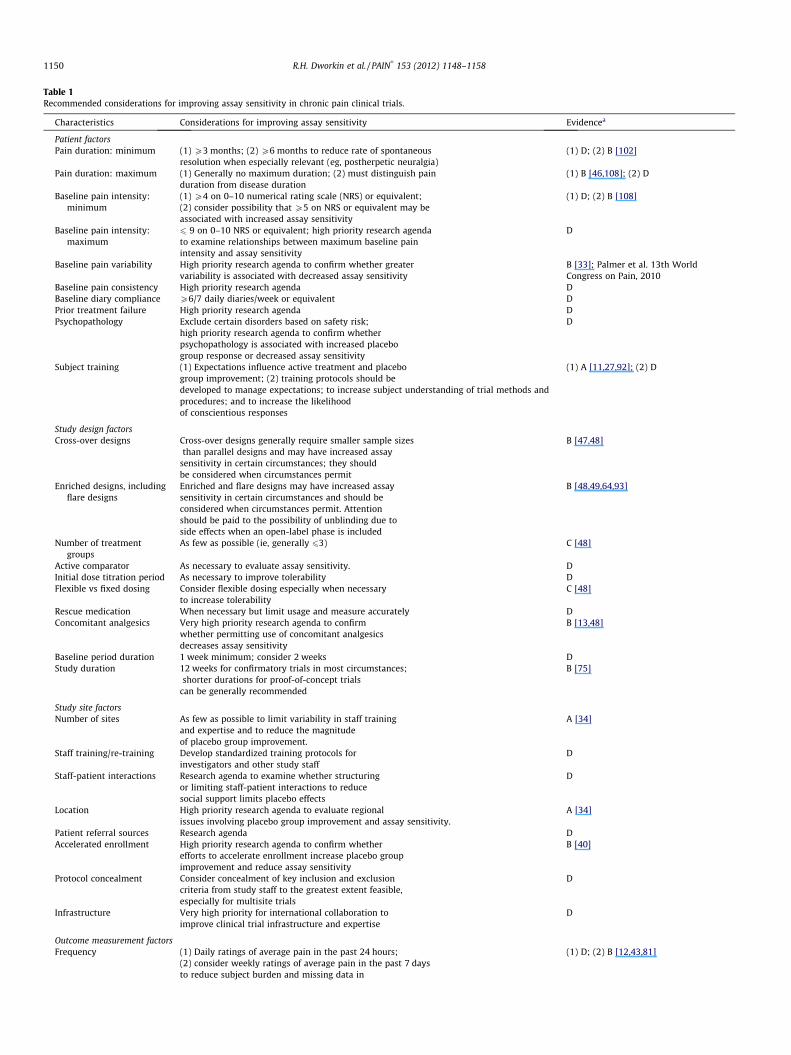
Table 1Recommended considerations for improving assay sensitivity in chronic pain clinical trials.
Characteristics Considerations for improving assay sensitivity Evidencea
Patient factorsPain duration: minimum (1) P3 months; (2) P6 months to reduce rate of spontaneous
resolution when especially relevant (eg, postherpetic neuralgia)(1) D; (2) B [102]
Pain duration: maximum (1) Generally no maximum duration; (2) must distinguish painduration from disease duration
(1) B [46,108]; (2) D
Baseline pain intensity:minimum
(1) P4 on 0–10 numerical rating scale (NRS) or equivalent;(2) consider possibility that P5 on NRS or equivalent may beassociated with increased assay sensitivity
(1) D; (2) B [108]
Baseline pain intensity:maximum
6 9 on 0–10 NRS or equivalent; high priority research agendato examine relationships between maximum baseline painintensity and assay sensitivity
D
Baseline pain variability High priority research agenda to confirm whether greatervariability is associated with decreased assay sensitivity
B [33]; Palmer et al. 13th WorldCongress on Pain, 2010
Baseline pain consistency High priority research agenda DBaseline diary compliance P6/7 daily diaries/week or equivalent DPrior treatment failure High priority research agenda DPsychopathology Exclude certain disorders based on safety risk;
high priority research agenda to confirm whetherpsychopathology is associated with increased placebogroup response or decreased assay sensitivity
D
Subject training (1) Expectations influence active treatment and placebogroup improvement; (2) training protocols should bedeveloped to manage expectations; to increase subject understanding of trial methods andprocedures; and to increase the likelihoodof conscientious responses
(1) A [11,27,92]; (2) D
Study design factorsCross-over designs Cross-over designs generally require smaller sample sizes
than parallel designs and may have increased assaysensitivity in certain circumstances; they shouldbe considered when circumstances permit
B [47,48]
Enriched designs, includingflare designs
Enriched and flare designs may have increased assaysensitivity in certain circumstances and should beconsidered when circumstances permit. Attentionshould be paid to the possibility of unblinding due toside effects when an open-label phase is included
B [48,49,64,93]
Number of treatmentgroups
As few as possible (ie, generally 63) C [48]
Active comparator As necessary to evaluate assay sensitivity. DInitial dose titration period As necessary to improve tolerability DFlexible vs fixed dosing Consider flexible dosing especially when necessary
to increase tolerabilityC [48]
Rescue medication When necessary but limit usage and measure accurately DConcomitant analgesics Very high priority research agenda to confirm
whether permitting use of concomitant analgesicsdecreases assay sensitivity
B [13,48]
Baseline period duration 1 week minimum; consider 2 weeks DStudy duration 12 weeks for confirmatory trials in most circumstances;
shorter durations for proof-of-concept trialscan be generally recommended
B [75]
Study site factorsNumber of sites As few as possible to limit variability in staff training
and expertise and to reduce the magnitudeof placebo group improvement.
A [34]
Staff training/re-training Develop standardized training protocols forinvestigators and other study staff
D
Staff-patient interactions Research agenda to examine whether structuringor limiting staff-patient interactions to reducesocial support limits placebo effects
D
Location High priority research agenda to evaluate regionalissues involving placebo group improvement and assay sensitivity.
A [34]
Patient referral sources Research agenda DAccelerated enrollment High priority research agenda to confirm whether
efforts to accelerate enrollment increase placebo groupimprovement and reduce assay sensitivity
B [40]
Protocol concealment Consider concealment of key inclusion and exclusioncriteria from study staff to the greatest extent feasible,especially for multisite trials
D
Infrastructure Very high priority for international collaboration toimprove clinical trial infrastructure and expertise
D
Outcome measurement factorsFrequency (1) Daily ratings of average pain in the past 24 hours;
(2) consider weekly ratings of average pain in the past 7 daysto reduce subject burden and missing data in
(1) D; (2) B [12,43,81]
1150 R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158

Table 1 (continued)
Characteristics Considerations for improving assay sensitivity Evidencea
circumstances in which information ondaily variability is not needed
Mode of administration High priority research agenda to examine responsivenessof electronic, telephone, and paper capture of painand other outcome data
D
Order of administration Research agenda to examine order effects in theadministration of outcome measures and standardizeorder of administration if warranted
D
a A = meta-regression analysis, systematic review, or prespecified analyses of multiple RCTs; B = retrospective quantitative analysis of RCTs or relevant observationalstudies; C = retrospective qualitative analysis of RCTs or relevant observational studies; D = expert opinion.
R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158 1151
3. Recommendations
3.1. General considerations
Four domains of clinical trial characteristics that potentiallyinfluence assay sensitivity were identified and discussed: (1) pa-tient factors; (2) study design factors; (3) study site factors; and(4) outcome measurement factors (Table 1). Recommendationswere based on 4 levels of evidence that were developed at themeeting specifically to address the limited literature on assay sen-sitivity in analgesic trials: (A) meta-regression analysis, systematicreview, or prespecified analyses of multiple RCTs; (B) retrospectivequantitative analysis of RCTs or relevant observational studies; (C)retrospective qualitative analysis of RCTs or relevant observationalstudies; and (D) expert opinion. However, most existing evidenceis derived from secondary data analyses of clinical trials that werenot specifically designed to evaluate the effects of study methodson assay sensitivity. In the absence of evidence on which to basea recommendation, factors considered sufficiently important towarrant further research were identified and proposed as compo-nents of a research agenda (Table 1). Recommendations were lim-ited to characteristics of chronic pain RCTs in general and notfactors unique to specific conditions, for example, glycemic controlin trials of painful diabetic peripheral neuropathy (DPN). Consider-ations of the likelihood that potential modifications to RCTs mightincrease the rate of false-positive results [57,61] or study costswere generally based on expert opinion, given the lack of dataaddressing these critical issues.
In considering the following recommendations, it must beemphasized that modifications to the research methods of anRCT that are intended to increase its assay sensitivity may havean impact on its generalizability. For example, efforts to increaseassay sensitivity by excluding patients with certain characteristicsmay reduce the generalizability of the study results to clinicalpractice in the community, in which a very heterogeneous patientpopulation will receive the treatment [77]. The extent to which theresults of analgesic clinical trials are relevant to determining theeffectiveness of pain treatments in clinical practice has only re-cently begun to receive attention [68,78]. The emphasis of thepresent article is on the design of RCTs intended to evaluatewhether or not a treatment has an analgesic effect, not the designof studies intended to evaluate the effectiveness of chronic paintreatments in the broader community in which they will be used.It is likely that some of the following recommendations reducethe generalizability of clinical trial results (eg, various types ofenrichment), although it is beyond the scope of this article to ex-pand on this issue in discussing each one of our specific recom-mendations. In addition, the multiple inclusion and exclusioncriteria used in analgesic trials [22,62] already serve to reducethe generalizability of clinical trial results. There are often
challenging trade-offs between research design considerationsand generalizability of results in clinical trials. As recently empha-sized by Rothwell and Watson in an excellent discussion of thegeneralizability of analgesic clinical trials, ‘‘The results of an RCTwill never be relevant to all patients and all settings, but theyshould be designed and reported in a way that allows cliniciansto judge to whom the results can reasonably be applied’’ [78].
A fundamental assumption of placebo-controlled RCTs is thatthe effects of active treatment and placebo are additive and thatthe effects of the active treatment are determined by subtractingthe placebo group response from the active treatment group re-sponse. If this assumption is correct, then modifications to clinicaltrial design intended to decrease placebo group responses will notincrease assay sensitivity because such modifications would be ex-pected to decrease responses in the active treatment group to thesame extent as observed in the placebo group. However, there islimited evidence to support this assumption, and the results ofsome studies suggest that ‘‘sub-additivity’’ of placebo effects inplacebo vs active treatment groups might occur in certain circum-stances [14,51,99,103], potentially reflecting differences in themechanisms of pain relief from placebo effects and from pharma-cologic and other active treatments. In discussing efforts to reduceplacebo group improvement, we assume that responses to activeand placebo treatments are not necessarily additive and that vari-ous methodologic aspects of RCTs could have differential effects onpatients administered placebo and analgesic treatments. For exam-ple, Wise and colleagues [103] found that when treatments werepresented with a neutral message about drug effectiveness, treat-ment efficacy vs placebo was demonstrated, but that when treat-ments were presented with an optimistic bias, ‘‘the placebogroup improved so much that there was no measurable benefit’’of a leukotriene antagonist in patients with asthma. There has beenvery limited attention to the implications of nonadditivity of pla-cebo effects in analgesic clinical trials—for example, in specifyingeffect sizes when determining statistical power—but considerationof these issues is beyond the scope of this article.
As can be seen from the remainder of this article and Table 1,there are major limitations of the evidence base relevant to factorsaffecting the assay sensitivity of chronic pain RCTs. Nevertheless,individuals who are currently designing clinical trials—whetherin academia or industry—must make decisions about the method-ologic aspects of these trials and cannot wait for better evidence tobecome available. In these circumstances, an expert consensusbased on currently available evidence and experience could pro-vide valuable information and improve decisions that are nowbeing made at least partially on an ad hoc or arbitrary basis. A ma-jor objective in presenting the following recommendations is tostimulate discussion and research on important issues that havereceived very limited attention in the literature on evaluatingtreatments for chronic pain [21].

1152 R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158
3.2. Patient factors
3.2.1. Pain durationChronic pain RCTs have typically required a minimum pain
duration ranging from 3 months to 1 year as an inclusion criterion(eg, most neuropathic pain trials require 3 to 6 months pain dura-tion [5,26]). One rationale for requiring a minimum pain durationis to limit the inclusion of subjects more likely to experience spon-taneous pain resolution that could be falsely attributed to thestudy treatment. These considerations provide the basis for recom-mending that a minimum pain duration of 3 months be requiredfor chronic pain RCTs, which is consistent with existing definitionsof chronic pain [19,66]. However, requiring a minimum pain dura-tion of 6 months in some circumstances may reduce the rate ofspontaneous improvement, especially when an appreciable per-centage of patients will experience pain resolution before6 months because of the natural history of the condition (eg, pos-therpetic neuralgia [7,90,102]).
Patients who have had pain for extended periods of time may beless likely to respond to new treatments. Although some RCTs havespecified maximum pain durations, the interaction between painduration and treatment (vs placebo) was not significant in a pooledanalysis of 3 trials of duloxetine for painful DPN [108]. Pain durationwas also not associated with treatment responder status in a chroniclow back pain trial comparing transdermal fentanyl and oral mor-phine, but the relevance of this finding to assay sensitivity is limitedbecause the trial lacked a placebo group [46]. Given these data, spec-ifying a maximum pain duration for chronic pain RCTs does not ap-pear warranted at present. Reports of clinical trials should allinclude information about the use of minimum and maximum painduration as inclusion or exclusion criteria, as well as the mean dura-tion (and range of durations) of pain in the active treatment and pla-cebo groups. In future studies, it will be important to examinewhether poorer assay sensitivity is associated with shorter and long-er pain durations. In all chronic pain clinical trials, careful attentionmust be paid to distinguishing and assessing pain duration separatelyfrom the duration of any underlying diseases and conditions whenpain may not be present at disease onset (eg, diabetes and DPN).
3.2.2. Baseline pain intensity, variability, and adherenceA minimum baseline pain of P4 on a 0–10 numerical rating scale
(or its equivalent) is a standard inclusion criterion for chronic painRCTs, although lower (P3) and higher (P5) values have been used.The results of meta-analyses of RCTs of osteoarthritis (OA) [107],painful DPN [34], and fibromyalgia [34] indicate that higher baselinepain appears to be associated with increased placebo groupimprovement. However, higher baseline pain intensity was associ-ated with increased assay sensitivity in a pooled analysis of 3 trialsin painful DPN [108]. On the basis of the data from these studies, therelationship between baseline pain intensity and assay sensitivitycannot be determined. Nevertheless, a minimum baseline pain ofP4 can be generally recommended to limit the ‘‘floor effect’’ of sub-jects not having sufficient pain for group differences in improve-ment to be detected (cf. [3]). Whether baseline pain P5 can beassociated with increased assay sensitivity should be examined ret-rospectively in existing analgesic trial data and prospectively in fu-ture RCTs.
Maximum levels of baseline pain have been specified in a num-ber of RCTs, including studies of OA [2,53,73] and postherpeticneuralgia [6,7,41]. There are several considerations relevant tothe exclusion of patients with baseline pain that exceeds a pre-specified maximum: (1) patients reporting extreme pain (eg, rat-ings of 9 or 10 on a 0–10 scale) may require more aggressivetreatment than typically can be provided within a placebo-con-trolled trial; (2) reports of extreme pain may reflect psychosocialdistress or psychopathology, which might require treatment that
cannot be provided within a trial; (3) reports of extreme painmay reflect subjects’ lack of comprehension or an inability to accu-rately rate their pain (eg, resulting from subclinical cognitiveimpairment in some cases); and (4) regression to the mean duringan RCT may be greater with extreme levels of pain (which could in-crease variability even if comparable between randomized groups).On the basis of these considerations, it can be recommended thatpatients with baseline pain >9 on a 0–10 scale be excluded fromparticipation in chronic pain RCTs unless doing so is precludedby the specific research objectives or study design. Typically, thisbaseline pain maximum would be based on the mean of multipledaily pain assessments collected in patient diaries, although if asingle pain rating is used as the baseline, similar considerationswould apply to patients who rate their pain over the past 24 hours(or past week) as 10 on a 0–10 scale. Given the absence of dataaddressing this exclusion criterion, examining the relationshipsbetween maximum baseline pain intensity and assay sensitivityin existing clinical trial databases and in future clinical trials is rec-ommended as a high priority research agenda item.
Subjects with greater baseline pain variability (ie, SD of dailypain ratings) have shown greater placebo group improvementand reduced assay sensitivity in RCTs of fibromyalgia ([33], Pal-mer R, Turk D, Hufford M, Wang Y, The impact of pain variabilityon response to milnacipran and placebo in 2 trials of patientswith fibromyalgia. 13th World Congress on Pain, Montreal, Can-ada, August 31-September 2, 2010). Recognizing the episodicnature of some pain conditions and the effects of activity on oth-ers, a recommendation cannot be made to exclude patients withextreme pain variability in the absence of additional researchexamining these issues. However, qualitative research involvingpatient experiences could be conducted on pain variability andother aspects of baseline ratings to increase understanding ofthese rating characteristics and potentially provide a basis formodifying rating instructions and study inclusion criteria to in-crease assay sensitivity.
The possibility that patients who provide unvarying ratings ofbaseline pain intensity (eg, all daily pain ratings for the baselineweek rated 5/10) might compromise assay sensitivity—perhaps be-cause of inability to discriminate changes in their pain—should beconsidered a priority for future research. In this regard, it is inter-esting to note that approximately 25%–30% of patients providedthe same intensity ratings for all specific pain symptoms assessedin recent studies of painful DPN and postherpetic neuralgia [9] andof fibromyalgia [76]. The authors suggested that these patientsmight be ‘‘unable to discriminate between the different sensoryabnormalities and, thus, answer all questions equally [9]’’ and thatthe psychiatric comorbidities present in the subgroup of fibromy-algia patients with very consistent ratings might ‘‘influence theperception of or the ability to discriminate between sensory phe-nomena [76].’’ It is also possible that these patients are accuratelyreporting their pain; therefore, the impact on assay sensitivity ofpatients who provide such ‘‘excessively’’ consistent responses onquestionnaires assessing specific pain symptoms or qualitiesshould be considered a priority for future research.
With respect to specific characteristics of pain, the results of arecent meta-analysis of OA trials found negligible differences be-tween standardized effect sizes for ratings of spontaneous painand pain with movement [17], and replicated relationships be-tween specific pain symptoms and assay sensitivity have not beenreported [4]. Finally, neuropathic pain RCTs frequently require asan inclusion criterion that patients complete a minimum of 4 of7 daily baseline pain ratings because this is thought to provide ameasure of adherence to protocol requirements. However, this rep-resents a relatively low level of adherence, and it is recommendedthat requiring a minimum of 6 of 7 baseline daily pain ratingsshould be considered for chronic pain RCTs (or the equivalent,

R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158 1153
for example, 12 of 14 pain ratings if a 2-week baseline is used). Ifgreater adherence is required, the availability of a larger numberof ratings on which to calculate subjects’ mean baseline pain willalso increase the reliability of these values.
3.2.3. Prior treatment responseIf an investigational analgesic is thought to have the same
mechanism(s) of action as previous treatments received bypatients in whom such treatments were ineffective, it could beargued that their pain is refractory to those specific mechanismsand that such patients could compromise the ability of a trial todemonstrate the efficacy of the new treatment and should there-fore be excluded. However, knowledge of analgesic mechanismsof action is often incomplete, adverse events may also contributeto treatment failure, and obtaining an accurate treatment historyis often challenging. Moreover, patients may respond differentlyto treatments with very similar mechanisms of action [101], anda recent meta-analysis of gabapentin and pregabalin trials foundno evidence that trials that excluded unknown percentages ofprior nonresponders to these medications had greater assay sen-sitivity [89]. Given the increasing number of approved analgesicsfor chronic pain that have become available, the chronic pain pa-tients in the community seeking enrollment in RCTs may bemore likely at present to have a history of being generallyrefractory to treatment and may likewise fail to respond to anew treatment. Although inadequate prior treatment responsecannot be recommended as a basis for exclusion at the presenttime, evaluation of the impact of prior treatment response is rec-ommended as a priority for future research.
3.2.4. Psychiatric comorbidityPatients with current episodes or a recent history of psycho-
sis, major depression, suicidal ideation, and substance abuse aretypically excluded from analgesic clinical trials. This practice isgenerally recommended to promote safety. Psychological distresswas associated with higher placebo group response and loweractive treatment group improvement in a trial of opioids inlow back pain [100], but additional research must be conductedto more fully characterize the relationship between psychopa-thology and assay sensitivity. Given the use of medications thatalso have antidepressant, anxiolytic, and sedative-hypnotic ef-fects in the treatment of chronic pain, careful assessment ofmood and sleep can provide the basis for analyses of the contri-bution of these effects to decreases in pain.
3.2.5. Subject trainingPatients entering analgesic clinical trials often expect their
pain to improve. Study staff and the clinical trial setting mayintentionally or unintentionally reinforce such beliefs and alsoprovide support, both of which could augment placebo effects.It is recommended that easily understood and culturally appro-priate training (and retraining) materials be developed for pa-tients to more consistently address their expectations aboutimprovement and study participation (as discussed below, care-ful training of study staff could also be used to address patientexpectations).
Training and retraining materials should also be developed toensure the correct use of study measures (eg, pain diaries) andthereby maximize the likelihood that subjects will completeassessments in a thoughtful and conscientious manner. Moreelaborate training protocols that seek to improve assay sensitiv-ity by using various approaches to ‘‘calibrating’’ or ‘‘standardiz-ing’’ pain ratings—for example, by matching to standard painstimuli or descriptions of painful events—should also be studied[23,45].
3.3. Study design factors
3.3.1. Research designs and treatment groupsThe efficiencies of cross-over designs are well known, although
carryover effects and, more generally, treatment-by-period inter-action must be carefully considered, as they can have important ef-fects on the results and interpretation of the data [44,83]; the useof washout periods of sufficient duration can be helpful in alleviat-ing these potential problems [54]. Cross-over designs may beassociated with reduced placebo group improvement [47] and pos-sibly greater assay sensitivity. However, there can be importantdifferences between cross-over and parallel design trials in termsof sample size, number of sites needed to obtain the required sam-ple size, investigator experience, patient heterogeneity, and otherfactors, any of which might explain differences between these 2broad types of trial design in their outcomes.
Enriched designs can limit randomization to subjects who haveresponded to a treatment (often on an ‘‘open-label’’ basis) and/ortolerated its side effects, failed to respond to one or more other ac-tive treatments or placebo, or are characterized by various clinicalfeatures. Such designs may provide greater assay sensitivity whenrandomization is limited to subjects who have demonstrated aclinically meaningful treatment response during an open-labelphase [22,46,47,64,74]. However, enriched designs have been crit-icized for limitations in generalizability, specifically, that treat-ment effectiveness is demonstrated in those patients who havealready shown a favorable response to treatment and that the re-sults of the double-blind phase of such trials may therefore not ap-ply to patients in the community [63]. It can be argued, however,that the responses to treatment in the open-label phase are moredirectly applicable to clinical practice. An additional source of con-troversy has involved the possibility that certain enriched designshave potential biases due to unblinding that could result fromexposure to treatment side effects during the open-label phase[57,67,85–87]. Recent instructive examples of trials using enricheddesigns include Phase 3 trials in patients with fibromyalgia [15]and lumbosacral radiculopathy [8] and proof of concept studiesin peripheral neuropathic pain [35,36].
Flare designs have been widely used in OA clinical trials; inthese trials, only patients with continued pain after discontinua-tion of their usual treatment are enrolled. Various approaches todefining ‘‘flare’’ have been used, and more stringent criteria (eg,the combination of a prespecified minimum increase in pain anda minimum pain intensity) appear to be associated with greatertreatment effects vs placebo [93]. Clinical trial research designsthat are intended to increase assay sensitivity—such as cross-over,enrichment, and flare designs—are recommended when circum-stances permit and following careful consideration of their benefitsand limitations.
3.3.2. Treatment groupsEvidence suggests that fewer treatment groups (generally 2 or
3) may be associated with decreased placebo group response andpossibly increased assay sensitivity, presumably by reducingexpectations that subjects are receiving an active treatment[48,70,84]. It is generally recognized that including an active com-parator treatment with well-established efficacy along with a pla-cebo group makes it possible to evaluate assay sensitivity in trialsof new treatments [21]. An informative example is an RCT of agabapentin prodrug in painful DPN that included pregabalin asan active comparator in which neither drug significantly differedfrom placebo, suggesting that the trial lacked assay sensitivity[106]. Including an active comparator to evaluate assay sensitivitycan be generally recommended when circumstances permit,but doing so will add an extra treatment group (with the

1154 R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158
accompanying need for a larger number of subjects), which couldbe especially problematic in RCTs examining multiple dosages.
The use of fewer treatment groups in RCTs is recommended forits potential benefits on reducing placebo group improvement andincreasing assay sensitivity. In addition, the inclusion of an activecomparator should be considered because of the valuable informa-tion that can be provided when interpreting negative trials of newtreatments.
3.3.3. Dosing strategy and concomitant analgesicsFlexible dosing strategies can be expected to decrease the rate
of adverse events leading to withdrawals and may increase assaysensitivity by reducing missing data. Particularly for opioids andother treatments with substantial interindividual differences inefficacious dosages and tolerability, these strategies can reducesubject discontinuations and appear to improve the likelihood ofshowing treatment benefits vs placebo [48]. It is recommendedthat flexible dosing strategies be considered to increase tolerabilitywhen study objectives and existing knowledge of efficacious dos-ages permit.
Providing ‘‘rescue analgesia’’ is generally necessary in placebo-controlled RCTs for ethical reasons and to limit withdrawals. Care-ful assessment of all rescue analgesic use is strongly recom-mended, which makes it possible to examine whether such usediffers between the active treatment and placebo groups.
Permitting continued use of concomitant analgesics has beenthought to reduce assay sensitivity, perhaps because of anassumption that the patients’ pain may have already improvedsubstantially in response to the medication(s) they are takingand that their remaining pain is relatively refractory to treatment[22]. The results of a recent OA trial [13] were consistent with thishypothesis; however, analyses of pregabalin [18] and high-con-centration capsaicin patch [42] trials in postherpetic neuralgiafound no evidence that concomitant analgesics reduced assay sen-sitivity, and excluding use of all concomitant analgesic treatmentshinders enrollment in longer-term trials and might yield refrac-tory or otherwise unrepresentative patient samples [22]. Animportant research priority is clarification of the impact of con-comitant analgesics on assay sensitivity in different clinical condi-tions and research designs. A related priority is furtherconsideration of the assay sensitivity of various types of ‘‘add-on’’ designs, in which all patients in the RCT are maintained forthe duration of the trial on a prespecified analgesic medication,typically at a stable dosage.
3.3.4. Baseline period and study durationBaseline periods serve as the benchmark from which to assess
safety and efficacy and for excluding patients based on pain inten-sity, lack of protocol adherence, and other factors. It is very impor-tant to carefully characterize patients prior to randomization, andlengthening this period from the traditional 1 week might providemore accurate assessments and could reduce regression to themean. However, this could be associated with increased percent-ages of patients who discontinue during the baseline or are ineligi-ble for randomization (both of which might increase assaysensitivity), and might also increase costs.
The placebo group response is highly variable and does not nec-essarily plateau [75], perhaps as a result of an increasing number ofstudy visits [72]. If pain decreases with an active treatment to a le-vel below which further improvement may be unlikely (eg, mildpain) and decreases in pain in the placebo group eventually ‘‘catchup’’ to this ‘‘floor’’ level of pain and do not subsequently dissipate,assay sensitivity will be reduced with studies of longer duration[21].
Trials of 12 weeks in duration are typically required for regula-tory approval and can be generally recommended for confirmatory
trials. However, on the basis of the above considerations and thefact that longer RCTs could be associated with a greater likelihoodof differential discontinuation rates occurring in active treatmentand placebo groups (potentially complicating interpretation ofthe results), shorter durations might maximize assay sensitivityin proof-of-concept studies. Of course, the use of shorter trial dura-tions in proof-of-concept studies may be a limitation when themethods and results provide the basis for designing confirmatorytrials with longer durations. It is important to recognize that tooshort of a study duration might be associated with decreased assaysensitivity if insufficient time has been allotted to measure the fulleffect of the study treatment.
3.4. Study site factors
3.4.1. Number, training, and location of study sitesMultisite trials can make it possible to enroll large numbers of
patients more rapidly, but they are often conducted in multiplecountries including investigators and staff with varying levels ofexperience, which can create substantial challenges in standardiz-ing study procedures. Use of a greater number of study sites wasassociated with a greater magnitude of placebo group improve-ment in RCTs of fibromyalgia but not painful DPN [34]. These con-siderations provide the basis for recommending use of the smallestpossible number of sites when circumstances permit. Although lar-ger sample size is associated with greater placebo group responsesin OA [107], there are multiple possible explanations for this, andmany circumstances preclude sample size reduction.
Training and monitoring of all study staff is strongly recom-mended in order to: (1) maximize staff adherence to the protocol;(2) standardize study procedures; (3) ensure structured provisionof information to subjects (eg, regarding diet and exercise in trialsof painful DPN, which could affect treatment response); and (4) lim-it interactions with subjects, which if extensive and supportivecould augment placebo group responses. Because expectation ofimprovement is a potent source of placebo effects, staff training in-tended to manage patient expectations at enrollment and through-out the trial could be examined as a potential method for decreasingthe component of placebo group improvement due to placebo ef-fects [11,27,92]. Although it would be possible to reduce and stan-dardize staff interactions with subjects by using computer-basedsubject training and administration of all patient-reported outcomemeasures, unintended effects of ‘‘depersonalizing’’ study proce-dures in this way must be evaluated. The impact of such instruc-tional efforts would need to be assessed in RCTs designed for thispurpose, and could be evaluated in tandem with proposals forexamining differences among sites in assay sensitivity [32,cf. 61,65].
Limitations of patient availability often require that confirma-tory RCTs be conducted on a multinational basis [38], which sub-stantially increases cultural and linguistic variability. Differencesamong health care systems and access to clinicians may augmentplacebo group responses in some regions [34,88], perhaps as a re-sult of frequent contact with study staff and access to otherwiseunavailable health care, thereby possibly compromising assay sen-sitivity [91]. It is recommended that existing data be interrogatedto examine such potential effects.
3.4.2. Recruitment methodsMethods of recruitment include clinic and research center dat-
abases, clinician referrals, patient referrals, and advertising. It is pos-sible that patients recruited from the community throughadvertising may compromise assay sensitivity by increasing thenumber of subjects with less severe conditions who have not neces-sarily sought treatment outside the clinical trial setting [38]. Becauseof the increasing use of such recruitment methods, patients enrollingin current analgesic RCTs may have less severe or otherwise different

R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158 1155
conditions than was true in the past, which could be contributing notonly to the increases in placebo group improvement that appear tobe occurring [34], but also to poorer assay sensitivity. However,chronic pain can receive inadequate attention in primary care, andpatients recruited through advertisements have been enrolled in re-cent RCTs that have demonstrated good assay sensitivity [29,30]. Gi-ven these conflicting considerations, retrospective and prospectiveresearch on the relationship between patient referral sources and as-say sensitivity are recommended.
Various strategies can be used to accelerate recruitment, espe-cially when it is not occurring at the anticipated rate. In 4 depres-sion trials, however, late-enrolling patients were more likely to beplacebo responders [59], and in a pooled analysis of 3 painful DPNtrials, placebo group patients from sites with faster recruitmentrates had significantly greater improvement [40]. Although theexplanation for these data is unclear, it is possible that increasedpressure to enroll patients results in the randomization of lessappropriate subjects, perhaps with milder conditions that mightbe more likely to improve after randomization. Such hypothesescould be evaluated in analyses of completed trials in which rela-tionships between ‘‘time of enrollment’’ (eg, early vs late) andinclusion and exclusion criteria and other demographic and clinicalcharacteristics are examined. On the basis of the limited data avail-able, it can be suggested that careful attention must be paid to theuse of any efforts to accelerate recruitment.
3.4.3. Concealment of protocol entry criteriaIf subjects or study staff are aware of specific criteria for pain (or
other variables) required for enrollment, they may knowingly orsubconsciously alter responses to gain participation [55]. Givenevidence that baseline pain intensity, variability, and diary comple-tion could influence assay sensitivity, concealment of key details ofthe eligibility criteria from site study staff could limit these effectsand should be considered to the extent practical [35,61]. An alter-native would be to use less restrictive criteria for randomization,but to clearly prespecify that primary efficacy analyses will be con-ducted on a subgroup of subjects with predefined criteria whosecharacteristics are concealed from site staff. Although this wouldrequire that a larger number of subjects be enrolled, the increasein assay sensitivity could outweigh the additional costs. If suchan approach were to be used, secondary efficacy analyses wouldalso need to be performed for all randomized patients, which couldprovide supportive data with greater generalizability than primaryefficacy analyses conducted on a subgroup of patients. Conceal-ment of certain entry criteria or of the characteristics of patientsubgroups that will provide the basis for primary efficacy analyseshas rarely been a methodologic feature of analgesic clinical trials.Importantly, concealing such aspects of a protocol from investiga-tors should not involve any ethical issues regarding patient safety.It possibly could be argued that concealing protocol features raisesmore general ethical concerns, for example, because it constrainsthe investigators’ knowledge of the RCT and the feedback givento excluded patients about specific reasons for their exclusion.However, any such concerns would be mitigated to the extent thatthese procedures increase the quality of the trial’s data and theintegrity of its methods. Moreover, descriptions of concealed studyfeatures should be prespecified in statistical analysis plans andcould be disclosed to ethics committees in protocol supplementsthat are not made available to sites [61].
The use of single-blind placebo run-ins has not appeared to in-crease assay sensitivity in psychiatry RCTs [25] or in a recent trialin patients with posttraumatic peripheral neuropathic pain [98].However, double-blind variable-duration placebo run-in and run-out periods in depression RCTs [24] have been thought to ‘‘improvedetection of placebo responders . . . and minimize bias in ratings ofefficacy or tolerability associated with the initiation and conclusion
of therapy’’ [31]. Examination of such approaches can be recom-mended for inclusion in methodologically focused studies of assaysensitivity in analgesic RCTs.
3.5. Outcome measurement factors
The reliability, validity, and responsiveness of outcome mea-sures used in primary efficacy analyses have a critical impact onthe assay sensitivity of RCTs. Previous IMMPACT publications haverecommended outcome domains and specific measures for chronicpain trials [20,94], but relatively limited attention has been paid tothe adequacy of each of these domains for specific contexts of usethat may be quite variable even when limited to chronic pain.Limited attention has also been paid to the potential influence ofassessment frequency, mode, and order of administration of thesemeasures. There is little doubt that such practical aspects of clinicaltrial conduct can affect assay sensitivity [25,52]. Moreover, it ispossible that improvements in reliability, validity, and responsive-ness of existing measures could improve assay sensitivity.
Although daily diaries of average pain intensity in the past24 hours are commonly used in chronic pain RCTs, published stud-ies have shown that weekly ratings of average pain in the past7 days are very highly correlated with daily ratings [12,43,81]. Gi-ven the reduction in subject burden, research should be conductedto determine whether reliability, validity, and assay sensitivity aremaintained with use of weekly ratings in circumstances in whichinformation on daily variability is not needed.
Recent research has examined electronic pain diaries of varioustypes [1], including mobile phones [10]. Although there is little evi-dence to date that these approaches show greater responsivenessto treatment effects vs placebo [56], additional research comparingthe reliability, validity, and responsiveness of electronic, telephone,and paper capture of pain ratings and other outcome data is highlyrecommended.
To our knowledge, no attention has been devoted to whetherthe order in which outcome measures are administered affectshow patients respond. There probably is great variability amonganalgesic trials in the order in which assessments are conducted,and because this could influence the reliability, validity, andresponsiveness of patient-reported outcomes, efforts to investigateand standardize assessment order effects are warranted.
3.6. Conclusions
We have recommended consideration of a number of patient,study, site, and outcome measurement factors that have the poten-tial to affect the assay sensitivity of RCTs of chronic pain treat-ments. Space limitations preclude detailed discussion of severalimportant issues. As we have emphasized in Section 3.1, perhapsforemost among these is the effect of efforts to increase the assaysensitivity of RCTs on the generalizability of their results to thetreatment of patients in the community. Because the relevance ofthe results of an analgesic trial to clinical practice depends on itssetting, inclusion and exclusion criteria, treatment protocol, out-come measures, and other factors [78], it is imperative that reportsof RCTs describe this information as comprehensively as possible,especially providing as much information as possible about thecharacteristics of the patients excluded from the trial and the pa-tients included in the data analyses.
We have also not discussed the following important consider-ations and their relationships to improving the assay sensitivityof analgesic trials: (1) the roles of subject retention and of treat-ment of missing data [28,69] and other statistical considerations;(2) the effect of unpublished trials on estimates of (ideally stan-dardized) treatment effect sizes [79,80,96]; (3) patient phenotyp-ing and other approaches to identifying subjects most likely to

1156 R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158
respond to treatment [60,82]; (4) ongoing collaborations and re-search initiatives that seek to increase the assay sensitivity of anal-gesic RCTs [16,21]; and (5) the need for international collaborationto improve clinical trial infrastructure and expertise [38].
Finally, because there is a limited evidence base for consideringapproaches to improving the assay sensitivity of chronic pain RCTs,a major objective of this article is to stimulate further research onimportant methodological issues that have not received adequateattention in the literature. Meta-analyses of publicly available clin-ical trial reports, studies of existing clinical trial raw data, andmethodologically focused prospective studies of clinical trial meth-ods have the potential to provide the foundation for an evidence-based approach to the design of analgesic clinical trials [21].
Conflict of interest statement
The views expressed in this article are those of the authors,none of whom have financial conflicts of interest related to thespecific issues discussed in this manuscript. At the time of the con-sensus meeting on which this article is based, 15 of the authorswere employed by one of the companies that provided unrestrictedgrants to the University of Rochester Office of Continuing Profes-sional Education to support the activities of IMMPACT, includingthe consensus meeting; these companies were AstraZeneca, Bris-tol-Myers Squibb, DePuy Spine, Durect, Eisai, Eli Lilly, Endo, Forest,Grünenthal, Johnson & Johnson, King, NeurogesX, Nuvo, Pfizer, Pur-due, and Smith & Nephew.
Acknowledgements
No official endorsement by the US Department of Veterans Af-fairs, US Food and Drug Administration, US National Institutes ofHealth, or the pharmaceutical companies that provided unre-stricted grants to the University of Rochester Office of ContinuingProfessional Education should be inferred. The authors thank PaulJ. Lambiase and Mary Gleichauf for their invaluable assistance inthe organization of the IMMPACT meeting.
References
[1] Allen KD, Coffman CJ, Golightly YM, Stechuchak KM, Voils CI, Keefe FJ.Comparison of pain measures among patients with osteoarthritis. J Pain2010;11:522–7.
[2] Altman R, Brandt K, Hochberg M, Moskowitz R. Design and conduct of clinicaltrials in patients with osteoarthritis: recommendations from a task force ofthe Osteoarthritis Research Society. Osteoarthritis Cartilage 1996;4:217–43.
[3] Altman RD, Marcussen KC. Effects of a ginger extract on knee pain in patientswith osteoarthritis. Arthritis Rheum 2001;44:2531–8.
[4] Attal N, Bouhassira D, Baron R, Dostrovsky J, Dworkin RH, Finnerup N, GourlayG, Haanpää M, Raja S, Rice ASC, Simpson D, Treede RD, Wells CD. Assessingsymptom profiles in neuropathic pain clinical trials: can it improve outcome?Eur J Pain 2011;15:441–3.
[5] Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, Nurmikko T.EFNS guidelines on the pharmacological treatment of neuropathic pain: 2009revision. Eur J Neurol 2010;17:1113–23.
[6] Backonja MM, Malan TP, Vanhove GF, Tobias JK. NGX-4010, a high-concentration capsaicin patch, for the treatment of postherpetic neuralgia:a randomized, double-blind, controlled study with an open-label extension.Pain Med 2010;11:600–8.
[7] Backonja M, Wallace MS, Blonsky ER, Cutler BJ, Malan Jr P, Rauck R, Tobias J.NGX-4010, a high-concentration capsaicin patch, for the treatment ofpostherpetic neuralgia: a randomised, double-blind study. Lancet Neurol2008;7:1106–12.
[8] Baron R, Freynhagen R, Tölle TR, Cloutier C, Leon T, Murphy TK, Phillips K. Theefficacy and safety of pregabalin in the treatment of neuropathic painassociated with chronic lumbosacral radiculopathy. Pain 2010;150:420–7.
[9] Baron R, Tölle TR, Gockel U, Brosz M, Freynhagen R. A cross sectional cohortsurvey in 2100 patients with painful diabetic neuropathy and postherpeticneuralgia: differences in demographic data and sensory systems. Pain2009;146:34–40.
[10] Bellamy N, Wilson C, Hendrikz J, Whitehouse SL, Patel B, Dennison S, EDCStudy Group. Osteoarthritis Index delivered by mobile phone (m-WOMAC) isvalid, reliable, and responsive. J Clin Epidemiol 2011;64:182–90.
[11] Bingel U, Wanigasekera V, Wiech K, Mhuircheartaigh RN, Lee MC, Ploner M,Tracey I. The effect of treatment expectation on drug efficacy: imaging theanalgesic benefit of the opioid remifentanil. Sci Transl Med 2011;3:70ra14.
[12] Bolton JE. Accuracy of recall of usual pain intensity in back pain patients. Pain1999;83:533–9.
[13] Chappell AS, Ossanna MJ, Liu-Seifert H, Iyengar S, Skljarevski V, Li LC, BennettRM, Collins H. Duloxetine, a centrally acting analgesic, in the treatment ofpatients with osteoarthritis knee pain: a 13-week, randomized, placebo-controlled trial. Pain 2009;146:253–60.
[14] Colagiuri B. Participant expectancies in double-blind randomized placebo-controlled trials: potential limitations to trial validity. Clin Trials 2010;7:246–55.
[15] Crofford LJ, Mease PJ, Simpson SL, Young Jr JP, Martin SA, Haig GM, Sharma U.Fibromyalgia relapse evaluation and efficacy for durability of meaningfulrelief (FREEDOM): a 6-month, double-blind, placebo-controlled trial withpregabalin. Pain 2008;136:419–31.
[16] Department of Health and Human Services, U.S. Food and Drug Administration.Advancing regulatory science for public health. Available from: http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/RegulatoryScience/UCM228444.pdf; 2010 [accessed 30.01.11].
[17] Dworkin RH, Peirce-Sandner S, Turk DC, McDermott MP, Gibofsky A, SimonLS, Farrar JT, Katz NP. Outcome measures in placebo-controlled trials ofosteoarthritis: responsiveness to treatment effects in the REPORT database.Osteoarthritis Cartilage 2011;19:483–92 [Corrigendum. OsteoarthritisCartilage 2011;19:919].
[18] Dworkin RH, Thakur R, Griesing T, Sharma U, Young JP. Randomized clinicaltrials of pregabalin for neuropathic pain: methods, results, and implications.Prog Neurother Neuropsychopharm 2008;3:167–87.
[19] Dworkin RH, Turk DC, Basch E, Berger A, Cleeland C, Farrar JT,Haythornthwaite J, Jensen M, Kerns R, Markman J, Porter L, Raja S, Ross E,Todd K, Wallace M, Woolf CJ. Considerations for extrapolating evidence ofacute and chronic pain analgesic efficacy. Pain 2011;152:1705–8.
[20] Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP,Kerns RD, Stucki G, Allen RR, Bellamy N, Carr DB, Chandler J, Cowan P, DionneR, Galer BS, Hertz S, Jadad AR, Kramer LD, Manning DC, Martin S, McCormickCG, McDermott MP, McGrath P, Quessy S, Rappaport BA, Robbins W, RobinsonJP, Rothman M, Royal MA, Simon L, Stauffer JW, Stein W, Tollett J, Wernicke J,Witter J. Core outcome measures for chronic pain clinical trials: IMMPACTrecommendations. Pain 2005;113:9–19.
[21] Dworkin RH, Turk DC, Katz NP, Rowbotham MC, Peirce-Sandner S, Cerny I,Clingman CS, Eloff BC, Farrar JT, Kamp C, McDermott MP, Rappaport BA,Sanhai WR. Evidence-based clinical trial design for chronic painpharmacotherapy: a blueprint for ACTION. Pain 2011;152:S107–15.
[22] Dworkin RH, Turk DC, Peirce-Sandner S, Baron R, Bellamy N, Burke LB,Chappell A, Chartier K, Cleeland CS, Costello A, Cowan P, Dimitrova R,Ellenberg S, Farrar JT, French JA, Gilron I, Hertz S, Jadad AR, Jay GW,Kalliomaki J, Katz NP, Kerns RD, Manning DC, McDermott MP, McGrath PJ,Narayana A, Porter L, Quessy S, Rappaport BA, Rauschkolb C, Reeve BB, RhodesT, Sampaio C, Simpson DM, Stauffer JW, Stucki G, Tobias J, White RE, Witter J.Research design considerations for confirmatory chronic pain clinical trials:IMMPACT recommendations. Pain 2010;149:177–93.
[23] Eisenach JC, Carpenter R, Curry R. Analgesia from a peripherally active j-opioid receptor agonist in patients with chronic pancreatitis. Pain2003;101:89–95.
[24] Faries DE, Heiligenstein JH, Tollefson GD, Potter WZ. The double-blindvariable placebo lead-in period: results from two antidepressant clinicaltrials. J Clin Psychopharmacol 2001;21:561–8.
[25] Fava M, Evins AE, Dorer DJ, Schoenfeld DA. The problem of the placeboresponse in clinical trials for psychiatric disorders: culprits, possibleremedies, and a novel study design approach. Psychother Psychosom2003;72:115–27.
[26] Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacologicaltreatment of neuropathic pain. Pain 2010;150:573–81.
[27] Finniss DG, Kaptchuk TJ, Miller F, Benedetti F. Biological, clinical, and ethicaladvances of placebo effects. Lancet 2010;375:686–95.
[28] Fleming T. Addressing missing data in clinical trials. Ann Intern Med2011;154:113–7.
[29] Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptylineand gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009;374:1252–61.
[30] Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine,gabapentin, or their combination for neuropathic pain. N Engl J Med2005;352:1324–34.
[31] Goldstein DJ, Lu Y, Detke MJ, Wiltse C, Mallinckrodt C, Demitrack MA.Duloxetine in the treatment of depression: a double-blind placebo-controlledcomparison with paroxetine. J Clin Psychopharmacol 2004;24:389–99.
[32] Hackett D, Haudiquet V, Salinas E. A method for controlling for a high placeboresponse rate in a comparison of venlafaxine XR and diazepam in the short-term treatment of patients with generalised anxiety disorder. Eur Psychiatry2003;18:182–7.
[33] Harris RE, Williams DA, McLean SA, Sen A, Hufford M, Gendreau RM, GracelyRH, Clauw DJ. Characterization and consequences of pain variability inindividuals with fibromyalgia. Arthritis Rheum 2005;52:3670–4.
[34] Häuser W, Bartram-Wunn E, Bartram C, Reinecke H, Tölle T. Systematicreview: placebo response in drug trials of fibromyalgia syndrome and painfulperipheral diabetic neuropathy: magnitude and patient-related factors. Pain2011;152:1709–17.

R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158 1157
[35] Hewitt DJ, Ho TW, Galer B, Backonja M, Markovitz P, Gammaitoni A,Michelson D, Bolognese J, Alon A, Rosenberg E, Herman G, Wang H. Impactof responder definition on the enriched enrollment randomized withdrawaldesign for establishing proof of concept in neuropathic pain. Pain2011;152:514–21.
[36] Ho TW, Backonja M, Ma J, Leibensperger H, Froman S, Polydefkis M. Efficientassessment of neuropathic pain drugs in patients with small fiber sensoryneuropathies. Pain 2009;141:19–24.
[37] Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials.IMMPACT-XII slide presentations. Available from: http://www.immpact.org/meetings/Immpact13/background13.html; 2010 [accessed 31.01.11].
[38] Institute of Medicine. Transforming clinical research in the United States:challenges and opportunities. Washington, DC: National Academies Press.Available from: http://www.nap.edu/catalog.php?record_id=12900; 2010[accessed 30.01.11].
[39] International Conference on Harmonisation. E10: Choice of control groupsand related issues in clinical trials. Available from: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm125912.pdf; 2001[accessed 30.01.11].
[40] Irizarry MC, Webb DJ, Ali Z, Chizh BA, Gold M, Kinrade FJ, Meisner PD, Blum D,Silver MT, Weil JG. Predictors of placebo response in pooled lamotrigineneuropathic pain clinical trials. Clin J Pain 2009;25:469–76.
[41] Irving GA, Backonja MM, Dunteman E, Blonsky ER, Vanhove GF, Lu S-P, TobiasJ. A multicenter, randomized, double-blind, controlled study of NGX-4010, ahigh-concentration capsaicin patch, for the treatment of postherpeticneuralgia. Pain Med 2011;12:99–109.
[42] Irving GA, Backonja M, Rauck R, Webster LR, Tobias JK, Vanhove GF. NGX-4010, a capsaicin 8% dermal patch, administered alone or in combinationwith systemic neuropathic pain medications, reduces pain in patients withpostherpetic neuralgia. Clin J Pain 2012;28:101–7.
[43] Jensen MP, Karoly P. Self-report scales and procedures for assessing pain inadults. In: Turk DC, Melzack R, editors. Handbook of pain assessment. NewYork: Guilford Press; 2011. p. 19–41.
[44] Jones B, Kenward MG. Design and analysis of cross-over trials. 2ndedition. Boca Raton, FL: Chapman & Hall; 2003.
[45] Kane RL, Bershadsky B, Rockwood T, Saleh K, Islam NC. Visual analogue scalepain reporting was standardized. J Clin Epidemiol 2005;58:618–23.
[46] Kalso E, Simpson KH, Slappendel R, Dejonckheere J, Richarz U. Predictinglong-term response to strong opioids in patients with low back pain: findingsfrom a randomized, controlled trial of transdermal fentanyl and morphine.BMC Med 2007;5:39.
[47] Katz J, Finnerup NB, Dworkin RH. Clinical trial outcome in neuropathic pain:relationship to study characteristics. Neurology 2008;70:263–72.
[48] Katz N. Methodological issues in clinical trials of opioids for chronic pain.Neurology 2005;65:S32–49.
[49] Katz N. Enriched enrollment randomized withdrawal trial designs ofanalgesics: focus on methodology. Clin J Pain 2009;25:797–807.
[50] Kissin I. The development of new analgesics over the past 50 years: a lack ofreal breakthrough drugs. Anesthesiology 2010;110:780–9.
[51] Kleijnen J, de Craen AJM, van Everdingen J, Krol L. Placebo effect in double-blind clinical trials: a review of interactions with medications. Lancet1994;344:1347–9.
[52] Kobak KA, Kane JM, Thase ME, Nierenberg AA. Why do clinical trials fail? Theproblem of measurement error in clinical trials: time to test new paradigms?J Clin Psychopharm 2007;27:1–5.
[53] Krüger K, Klasser M, Mössinger J, Becker U. Oxaceprol: a randomised,placebo-controlled clinical study in osteoarthritis with a non-conventionalnon-steroidal anti-inflammatory drug. Clin Exp Rheumatol 2007;25:29–34.
[54] Laird NM, Skinner J, Kenward M. An analysis of two-period crossover designswith carry-over effects. Stat Med 1992;11:1967–79.
[55] Landin R, DeBrota DJ, DeVries TA, Potter WZ, Demitrack MA. The impact ofrestrictive entry criterion during the placebo lead-in period. Biometrics2000;56:271–8.
[56] Lauritsen K, Innocenti AD, Hendel L, Praest J, Lytje MF, Clemmensen-RotneK, Wiklund I. Symptom recording in a randomised clinical trial: paperdiaries vs. electronic or telephone data capture. Control Clin Trials2004;25:585–97.
[57] Leber PD, Davis CS. Threats to the validity of clinical trials employingenrichment strategies for sample selection. Control Clin Trials1998;19:178–87.
[58] Lee K, Bacchetti P, Sim I. Publication of clinical trials supporting successfulnew drug applications: a literature analysis. PLoS Med 2008;5:1348–56.
[59] Liu KS, Snavely DB, Ball WA, Lines CR, Reines SA, Potter WZ. Is bigger betterfor depression trials? J Psychiatr Res 2008;42:622–30.
[60] Maier C, Baron R, Tolle TR, Binder A, Birbaumer N, Birklein F, Gierthmuhlen J,Flor H, Geber C, Huge V, Krumova EK, Landwehrmeyer GB, Magerl W,Maihofner C, Richter H, Rolke R, Scherens A, Schwarz A, Sommer C, TronnierV, Uceyler N, Valet M, Wasner G, Treede RD. Quantitative sensory testing inthe German Research Network on Neuropathic Pain (DFNS): somatosensoryabnormalities in 1236 patients with different neuropathic pain syndromes.Pain 2010;150:439–50.
[61] Mallinckrodt CH, Prucka WR. The enrichment window approach as a meansof dealing with placebo responses in antidepressant clinical trials. ClinPharmacol Ther 2010;88:592–4.
[62] Max MB, Portenoy RK, Laska EM, editors. The design of analgesic clinicaltrials. Philadelphia, PA: Lippincott-Raven; 1991.
[63] McGrath PJ. Randomization within trials does not contribute to externalvalidity. Clin J Pain 2011;27:89.
[64] McQuay HJ, Derry S, Moore RA, Poulain P, Legout V. Enriched enrolment withrandomised withdrawal (EERW): time for a new look at clinical trial design inchronic pain. Pain 2008;135:217–20.
[65] Merlo-Pich E, Alexander RC, Fava M, Gomeni R. A new population-enrichmentstrategy to improve efficiency of placebo-controlled clinical trials ofantidepressant drugs. Clin Pharmacol Ther 2010;88:634–42.
[66] Merskey H, Bogduk N, editors. Classification of chronic pain: descriptions ofchronic pain syndromes and definitions of pain terms. Seattle, WA: IASPPress; 1994.
[67] Moore RA, Derry S, McQuay HJ. Response to: long-term trials of pregabalinand duloxetine for fibromyalgia symptoms: how study designs can affectplacebo factors. Pain 2008;139:477–9.
[68] Moore RA, Derry S, McQuay HJ, Straube S, Aldington D, Wiffen P, Bell RF, KalsoE, Rowbotham MC. Clinical effectiveness: an approach to clinical trial designmore relevant to clinical practice, acknowledging the importance ofindividual differences. Pain 2010;149:173–6.
[69] National Research Council. The prevention and treatment of missing data inclinical trials. Washington, DC: The National Academies Press. Available from:http://www.nap.edu/catalog.php?record_id=12955; 2010 [accessed30.01.11].
[70] Papakostas GI, Fava M. Does the probability of receiving placebo influenceclinical trial outcome? A meta-regression of double-blind, randomizedclinical trials in MDD. Eur Neuropsychopharm 2009;19:34–40.
[71] Pfizer protocol A0081071. A randomized double-blind, placebo-controlled,parallel-group, multi-center trial of pregabalin versus placebo in the treatmentof neuropathic pain associated with diabetic peripheral neuropathy. Availablefrom: http://www.clinicalstudyresults.org/drugdetails/?drug_name_id=203&indication_keyword=neuropathy&sort=c.com pany_name&page=2&drug_id=3972; 2007 [accessed 30.01.11].
[72] Posternak MA, Zimmerman M. Therapeutic effect of follow-up assessmentson antidepressant and placebo response rates in antidepressant efficacytrials: meta-analysis. Br J Psychiatry 2007;190:287–92.
[73] Puopolo A, Boice JA, Fidelholtz JL, Littlejohn TW, Miranda P, Berrocal A, Ko A,Cichanowitz N, Reicin AS. A randomized placebo-controlled trial comparingthe efficacy of etoricoxib 30 mg and ibuprofen 2400 mg for the treatment ofpatients with osteoarthritis. Osteoarthritis Cartilage 2007;15:1348–56.
[74] Quessy SN. Two-stage enriched enrolment pain trials: a brief review ofdesigns and opportunities for broader application. Pain 2010;148:8–13.
[75] Quessy SN, Rowbotham MC. Placebo response in neuropathic pain trials. Pain2008;138:479–83.
[76] Rehm S, Koroschetz J, Gockel U, Bosz M, Freynhagen R, Tölle TR, Baron R. Across-sectional survey of 3035 patients with fibromyalgia: subgroups ofpatients with typical comorbidities and sensory symptom profiles.Rheumatology 2010;49:1146–52.
[77] Rothwell PM. External validity of randomized controlled trials: ‘‘to whom dothe results of this trial apply?’’. Lancet 2005;365:82–93.
[78] Rothwell PM, Watson CPN. External validity of randomized controlled trials:general principles and lessons from trials in neuropathic pain. In: Castro-Lopes J, editor. Current topics in pain: 12th World Congress on Pain. Seattle,WA: IASP Press; 2009. p. 199–220.
[79] Rowbotham MC. The impact of selective publication on clinical research inpain. Pain 2008;140:401–4.
[80] Rowbotham MC. The case for publishing ‘negative’ trials. Pain2009;146:225–6.
[81] Salovey P, Smith AF, Turk DC, Jobe JB, Willis GB. The accuracy of memory forpain: not so bad most of the time. Am Pain Soc J 1993;2:184–91.
[82] Scholz J, Mannion RJ, Hord DE, Griffin RS, Rawal B, Zheng H, Scoffings D,Phillips A, Guo J, Laing RJ, Abdi S, Decosterd I, Woolf CJ. A novel tool for theassessment of pain: validation in low back pain. PLoS Med 2009;6:e1000047.
[83] Senn S. Cross-over trials in clinical research. 2nd edition. West Sussex,England: John Wiley & Sons; 2002.
[84] Sinyor M, Levitt AJ, Cheung AH, Schaffer A, Kiss A, Dowlati Y, Lanctôt KL. Doesinclusion of a placebo arm influence response to active antidepressanttreatment in randomized controlled trials?: results from pooled and meta-analyses. J Clin Psychiatry 2010;71:270–9.
[85] Staud R, Price D. Importance of measuring placebo factors in complex clinicaltrials. Pain 2008;138:474.
[86] Staud R, Price D. Long-term trials of pregabalin and duloxetine forfibromyalgia symptoms: how study designs can affect placebo factors. Pain2008;136:232–4.
[87] Staud R, Price D. Role of placebo factors in clinical trials with special focus onenrichment designs. Pain 2008;139:479–80.
[88] Stein DJ, Baldwin DS, Dolberg OT, Despiegel N, Bandelow B. Which factorspredict placebo response in anxiety disorders and major depression? Ananalysis of placebo-controlled studies of escitalopram. J Clin Psychiatry2006;67:1741–6.
[89] Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrolment: definition andeffects of enrichment and dose in trials of pregabalin and gabapentin inneuropathic pain: a systematic review. Br J Clin Pharmacol 2008;66:266–75.
[90] Thyregod HG, Rowbotham MC, Peters MM, Possehn J, Berro M, Petersen KL.Natural history of pain following herpes zoster. Pain 2007;128:148–56.
[91] Tölle T, Freynhagen R, Versavel M, Trostmann U, Young Jr JP. Pregabalin forrelief of neuropathic pain associated with diabetic neuropathy: a randomized,double-blind study. Eur J Pain 2008;12:203–13.

1158 R.H. Dworkin et al. / PAIN�
153 (2012) 1148–1158
[92] Tracey I. Getting the pain you expect: mechanisms of placebo, nocebo andreappraisal effects in humans. Nat Med 2010;16:1277–83.
[93] Trijau S, Avouac J, Escalas C, Gossec L, Dougados M. Influence of flare designon symptomatic efficacy of non-steroidal anti-inflammatory drugs inosteoarthritis: a meta-analysis of randomized placebo-controlled trials.Osteoarthritis Cartilage 2010;18:1012–8.
[94] Turk DC, Dworkin RH, Allen RR, Bellamy N, Brandenburg N, Carr DB, CleelandC, Dionne R, Farrar JT, Galer BS, Hewitt DJ, Jadad AR, Katz NP, Kramer LD,Manning DC, McCormick CG, McDermott MP, McGrath P, Quessy S, RappaportBA, Robinson JP, Royal MA, Simon L, Stauffer JW, Stein W, Tollett J, Witter J.Core outcome domains for chronic pain clinical trials: IMMPACTrecommendations. Pain 2003;106:337–45.
[95] Turk DC, Dworkin RH, McDermott MP, Bellamy N, Burke LB, Chandler JM,Cleeland CS, Cowan P, Dimitrova R, Farrar JT, Hertz S, Heyse JF, Iyengar S,Jadad AR, Jay GW, Jermano JA, Katz NP, Manning DC, Martin S, Max MB,McGrath P, McQuay HJ, Quessy S, Rappaport BA, Revicki DA, Rothman M,Stauffer JW, Svensson O, White RE, Witter J. Analyzing multiple endpoints inclinical trials of pain treatments: IMMPACT recommendations. Pain2008;139:485–93.
[96] Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selectivepublication of antidepressant trials and its influence on apparent efficacy. NEngl J Med 2008;358:252–60.
[97] Usdin S. Product discovery and development: shaking the cup for pain.BioCentury 2011;19:13–4.
[98] van Seventer R, Bach FW, Toth CC, Serpell M, Temple J, Murphy TK, Nimour M.Pregabalin in the treatment of post-traumatic peripheral neuropathic pain: arandomized double-blind trial. Eur J Neurol 2010;17:1082–9.
[99] Wager TD, Roy M. Separate mechanisms for placebo and opiate analgesia?Pain 2010;150:8–9.
[100] Wasan AD, Davar G, Jamison R. The association between negative affect andopioid analgesia in patients with discogenic low back pain. Pain2005;117:450–61.
[101] Watson CPN, Vernich L, Chipman M, Reed K. Nortriptyline versusamitriptyline in postherpetic neuralgia: a randomized trial. Neurology1998;51:1166–71.
[102] Webster LR, Tark M, Rauck R, Tobias JK, Vanhove GF. Effect of duration ofpostherpetic neuralgia on efficacy analyses in a multicenter, randomized,controlled study of NGX-4010, an 8% capsaicin patch evaluated for thetreatment of postherpetic neuralgia. BMC Neurol 2010;10:92.
[103] Wise RA, Bartlett SJ, Brown ED, Castro M, Cohen R, Holbrook JT, Irvin CG, RandCS, Sockrider MM, Sugar EA. Randomized trial of the effect of drugpresentation on asthma outcomes: the American Lung Association AsthmaClinical Research Centers. J Allergy Clin Immunol 2009;124:436–44.
[104] Woodcock J. A difficult balance. pain management, drug safety, and the FDA.N Engl J Med 2009;361:2105–7.
[105] Woodcock J, Witter J, Dionne RA. Stimulating the development ofmechanism-based individualized pain therapies. Nat Rev Drug Discov2007;6:703–10.
[106] XenoPort, Inc. Phase II results for GSK1838262 (XP13512) reportedfor neuropathic pain associated with diabetic peripheral neuropathy.Available from http://phx.corporate-ir.net/phoenix.zhtml?c=187883&p=irol-newsArticle&ID=1280577&highlight=; 2009 [accessed 30.01.11].
[107] Zhang W, Robertson J, Jones AC, Dieppe PA, Doherty M. The placebo effect andits determinants in osteoarthritis: meta-analysis of randomised controlledtrials. Ann Rheum Dis 2008;67:1716–23.
[108] Ziegler D, Pritchett YL, Wang F, Desaiah D, Robinson MJ, Hall JA, Chappell AS.Impact of disease characteristics on the efficacy of duloxetine in diabeticperipheral neuropathic pain. Diabetes Care 2007;30:664–9.
Related Documents











