September 198l 370 TheJournalofPED1ATR1CS Congenital hepatic fibrosis in children Twenty-seven children with congenital hepatic fibrosis were followed for three months to 12 ,Fears. Hepatosplenomegaly, normal liver function tests, and kidney abnormalities were present in most patients, indicating that a correct diagnosis of congenital hepatic fibrosis could be made using simple clinical, biologic', and radiologic criteria. Esophageal endoscopy showed varicesin 21 patients. Sixteen children underwent portal-systemic shunt surgery. Follow-up examinations did not show any impairment of liver function or any sign of hepatic encephalopathy. Cholangitis was present in only three children. F. Alvarez, M.D.,* O. Bernard, M.D., F. Brunelle, M.D., M. Hadchouel, M.D., A. Leblanc, M.D., M. Odi~vre, M.D., and D. Alagille, M.D., Bicbtre, France CONGENITAL HEPATIC FIBROSIS is a recessive auto- somal disease with two major risks: gastrointestinal hemorrhage caused by portal hypertension, and cholangi- tis related to bacterial infection of dilated intrahepatic bile ducts. The liver disease is associated in virtually all cases with kidney abnormalities, consisting either of renal tubular ectasia or polycystic kidney disease ("infantile" type). 1-~ Diagnosis of the disease can be made unequivo- cally on liver biopsy, but it is often necessary to study a large number of portal tracts, which can be provided only by surgical biopsy. ~' We report here our experience with 27 children with this disease. A clear-cut clinical pattern is usually present, allowing a correct diagnosis in most cases before confir- mation by histology. Once problems resulting from portal hypertension are solved, the future of these children is threatened only by the course of the kidney disease or by cholangitis. Neither progressive hepatic failure nor he- patic encephalopathy occurred in our patients, even in those in whom a portal-systemic shunt had to be carried out. From the Unilk de Recherche d'Hkpatologie Infantile INSERM U 56 and Clinique de Pkdiatrie, Hbpital d'Enfants. *Reprint address: Clinique de Pbdiatrie, Hbpital de Biebtre, 78 rue du Gbn~ral Leclerc, 94270 Le Kremlin-Bicetre, France. METHODS AND RESULTS Patients. Between 1960 and 1980 a diagnosis of congen- ital hepatic fibrosis was made in 27 children at Bic~tre Hospital; 13 were males and 14 were females Diagnostic criteria. Histologic features typical of con- genital hepatic fibrosis were observed in 23 patients, either on surgical (18 patients) or needle (5 patients) liver biopsy. The liver was divided by bands of fibrous tissue, with multiple dysmorphic bile ducts of various sizes, often located at the edge of the fibrous strands. Bile ducts often contained mucinous material or inspissated bile in the lumina. In ~ few cases, bile duct-like structures could be found within the lobule. Liver cells appeared to be entirely normal and arranged in regular columns of" normal thickness. Abbreviations used ESR: erythrocyte sedimentation rate IVP: intravenous pyelogram SPG: splenoportography iv: intravenous In the remaining four children, liver changes seen upon needle biopsy examination were compatible with congen- ital hepatic fibrosis: there was portal fibrosis with abun- dant bile duct proliferation, but with no evidence of bile duct dysplasia. Diagnosis was confirmed by the clinical and radiologic findings. Vol. 99, No. 3, pp. 370-375 0022-3476/81/090370+06500.60/0 1981 The C. V. Mosby Co.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

September 198l
370 T h e J o u r n a l o f P E D 1 A T R 1 C S
Congenital hepatic fibrosis in children
Twenty-seven children with congenital hepatic fibrosis were followed for three months to 12 ,Fears.
Hepatosplenomegaly, normal liver function tests, and kidney abnormalities were present in most patients,
indicating that a correct diagnosis of congenital hepatic fibrosis could be made using simple clinical,
biologic', and radiologic criteria. Esophageal endoscopy showed varicesin 21 patients. Sixteen children
underwent portal-systemic shunt surgery. Follow-up examinations did not show any impairment of liver
function or any sign of hepatic encephalopathy. Cholangitis was present in only three children.
F. Alvarez, M.D.,* O. Bernard, M.D., F. Brunelle, M.D.,
M. Hadchouel, M.D., A. Leblanc, M.D., M. Odi~vre, M.D., and
D. Alagille, M.D., Bicb tre , F r a n c e
CONGENITAL HEPATIC FIBROSIS is a recessive auto-
somal disease with two major risks: gastrointestinal hemorrhage caused by portal hypertension, and cholangi-
tis related to bacterial infection of dilated intrahepatic bile ducts. The liver disease is associated in virtually all cases with kidney abnormalities, consisting either of renal tubular ectasia or polycystic kidney disease ("infantile" type). 1-~ Diagnosis of the disease can be made unequivo- cally on liver biopsy, but it is often necessary to study a large number of portal tracts, which can be provided only
by surgical biopsy. ~ ' We report here our experience with 27 children with
this disease. A clear-cut clinical pattern is usually present, allowing a correct diagnosis in most cases before confir- mation by histology. Once problems resulting from portal hypertension are solved, the future of these children is threatened only by the course of the kidney disease or by
cholangitis. Neither progressive hepatic failure nor he- patic encephalopathy occurred in our patients, even in those in whom a portal-systemic shunt had to be carried out.
From the Unilk de Recherche d'Hkpatologie Infantile INSERM U 56 and Clinique de Pkdiatrie, Hbpital d'Enfants. *Reprint address: Clinique de Pbdiatrie, Hbpital de Biebtre, 78 rue du Gbn~ral Leclerc, 94270 Le Kremlin-Bicetre, France.
M E T H O D S AND R E S U L T S
Patients. Between 1960 and 1980 a diagnosis of congen- ital hepatic fibrosis was made in 27 children at Bic~tre
Hospital; 13 were males and 14 were females Diagnostic criteria. Histologic features typical of con-
genital hepatic fibrosis were observed in 23 patients, either on surgical (18 patients) or needle (5 patients) liver biopsy. The liver was divided by bands of fibrous tissue, with multiple dysmorphic bile ducts of various sizes, often
located at the edge of the fibrous strands. Bile ducts often contained mucinous material or inspissated bile in the lumina. In ~ few cases, bile duct-like structures could be
found within the lobule. Liver cells appeared to be entirely normal and arranged in regular columns of" normal thickness.
Abbreviations used ESR: erythrocyte sedimentation rate IVP: intravenous pyelogram SPG: splenoportography iv: intravenous
In the remaining four children, liver changes seen upon needle biopsy examination were compatible with congen- ital hepatic fibrosis: there was portal fibrosis with abun- dant bile duct proliferation, but with no evidence of bile duct dysplasia. Diagnosis was confirmed by the clinical
and radiologic findings.
Vol. 99, No. 3, pp. 370-375 0022-3476/81/090370+06500.60/0 �9 1981 The C. V. Mosby Co.

Volume 99 Congenital hepatic fibrosis 3 7 1 Number 3
Family studies. Data supporting the autosomal reces-
sive transmission of the disease were present in eight of 27 patients. Consanguinity was present in the parents of three patients. Two brothers presented with congenital hepatic fibrosis with the same clinical features; three other patients had a brother or sister not included in this study, but with some features of congenital hepatic fibrosis. In these patients, however, the clinical expression of the disease was somewhat different in the two siblings (Ta-
ble l). Presenting symptoms. The symptoms for which the
patients were referred to us are indicated in Table II. Clinical and laboratory findings. On admission to
Bic~tre hospital, hepatomegaly was present in 25 of the 27 children. In 24 cases, the left lobe was predominant ly enlarged and palpable in the epigastrium. The enlarged liver was considered "firm" or "hard" in all patients. Splenomegaly was present in 24 patients. Liver function tests, including serum bil irubin concentration, transami- nase and alkaline phosphatase activity, cholesterol, and prothrombin time were normal in all children. Excretion of bromsulphalein was delayed in seven of 26 children. Liver function tests were followed for three months to 12 years (mean 5 years 4/,~ months) and remained within normal limits in all patients. No impairment in physical growth was observed in any patient except in those with renal failure.
Portal hypertension: Evaluation and management. Among 24 patients in whom splenomegaly was observed, 18 had thrombocytopenia (isolated or associated with neutropenia) consistent with hypersplenism. Gastrointes- tinal bleeding (hematemesis) was observed in 11 patients and was the first sign of the disease in eight. Esophageal endoscopy was carried out in 22 patients with splenomeg- aly; esophageal varices were present in 21. Splenoportog- raphy was performed in 21 patients; 16 of them had duplication of the intrahepatic branches of the portal vein, as previously described ~ (Fig. 1). Portal thrombosis was present in one patient. Splenic pressure was elevated above 20 cm of water in 19 patients. A natural splenorenal or gastrorenal shunt was observed on splenoportograms in four children. In one of these four children a small varix
seen on esophagoscopy at age 15 years was no longer present one year later; in another a single varix did not
increase in size or number over a 2%-year period. Management of portal hypertension in children with
congenital hepatic fibrosis included medical measures (to prevent variceal bleeding) and portal-systemic surgical shunts. The parents of each child in whom varices were present were given a list of aspirin-containing drugs to be avoided and a Sengstaken-Blakemore tube (the size of
Fig. 1. Duplication of intrahepatic branches of" the portal vein on splenoportogram.
which was adapted to the child's age) to be used in the emergency room in case of massive bleeding. Portal- systemic surgical shunt was carried out in 17 patients. In the six patients who did not bleed from varices, a prophylactic surgical shunt was performed either for social reasons (children coming from countries where no proper medical care could be ensured in case of gastroin- testinal bleeding) or, a few years ago, to avoid repeated blood transfusions when kidney transplantation was fore-
seeable. Portal-systemic shunt was laterolateral portacaval in three patients, lateroterminal mesocaval in three, and
proximal splenorenal in 11. During surgery, the macro- scopic appearance of the liver was found to be quite peculiar, with white stellate spots over a smooth or finely granular liver surface. In one child who had portal thrombosis and hematemesis at the early age of 2 years, the macroscopic lesions were limited to the left lobe of the liver, which looked almost tumoral; biopsies showed fibrosis to be limited to the left lobe, the right lobe being histologically normal. In another child, small cystic spots were visible over the entire surface of the liver. The mean
age at surgery was 9:7,~ years (range: 3~'/~2 to 16~/,~ years). In 16 children anastomosis proved to be effective as judged by disappearance of varices on esophagoscopy, and regression of splenomegaly and hypersplenism. In one patient, who had bled before, thrombosis of the shunt occurred and was followed by a lethal massive hemateme- sis. All patients who were operated on are leading normal fives, with the exception of those presenting with kidney failure. No overt sign of encephalopathy has been observed during follow-up periods ranging from three

3 7 2 A lvarez et al. The Journal of Pediatrics September 1981
Table I. Clinical signs in three children with congenital hepatic fibrosis and in their three siblings
Age at diagnosis"
Patient Sibling
Symptoms Age at
diagnosis Symptoms
3 yr Hepatosplenomegaly; hematemesis; polycystic kidney Birth
3 yr Hepatosplenomegaly; esophageal varices; polycystic kid- Birth ney
9 mo Acute cholangitis with renal tubular ectasia; later on, he- 10 yr patosplenornegaly, portal hypertension, and polycystic kidneys at 8 yr of age
Death from kidney failure with poly- cystic kidney; liver histology: con- genital hepatic fibrosis
Same as above
Tubular ectasia on IVP without hepato. megaly
Table I1. Presenting symptoms and signs in children with congenital hepatic fibrosis.
Clinical No. of Age range findings patients (means)
Polycystic kidneys 4 1 mo-6 yr and hepatomegaly (1 yr 7/12 mo)
Hepatomegaly 10 9 mo*-14 yr (5 yr 3/12 mo)
Hepatosplenomegaly 4 2 yr-13 yr (7 yr)
Splenomegaly 1 10 yr Hematemesis 7* 5 yr-13 yr
(9 yr 6/12 too)
*One patient with portal thrombosis associated with congenital hepatic fibrosis localized to the left lobe of the liver had gastrointestinal bleeding at age 2 years, and is not included in this table because of this additional cause of portal hypertension, possibly responsible for early bleeding.
months to 12 years. Pre- and postprandial blood ammonia concentrations and electroencephalogram were obtained at regular intervals postoperatively in eight Patients, and remained within normal limits in all of them during a follow-up ranging from six months to eight years (mean 28/12 years).
Bile duet abnormalities and related symptoms. Studies of intra- and extrahepatic bile ducts were carried out using intravenous cholangiography, ultrasound and, in one instance, percutaneous transhepatic cho!angiography. Intravenous cholangiography, performed in 17 children,
was normal in five, showed an enlarged gallbladder in eight, and dilatation of intrahepatic bile ducts in 12. Whenever dilatation of intrahepatic bile ducts was Observed, the image appeared as small spots scattered within the liver. Complete visualization of the intrahepat- ic bile tree was very rarely observed and was usually of poor technical quality. In one child, opacification of the entire biliary tree was obtained by transhepatic cholangi- ography; the whole biliary system was abnormal, and
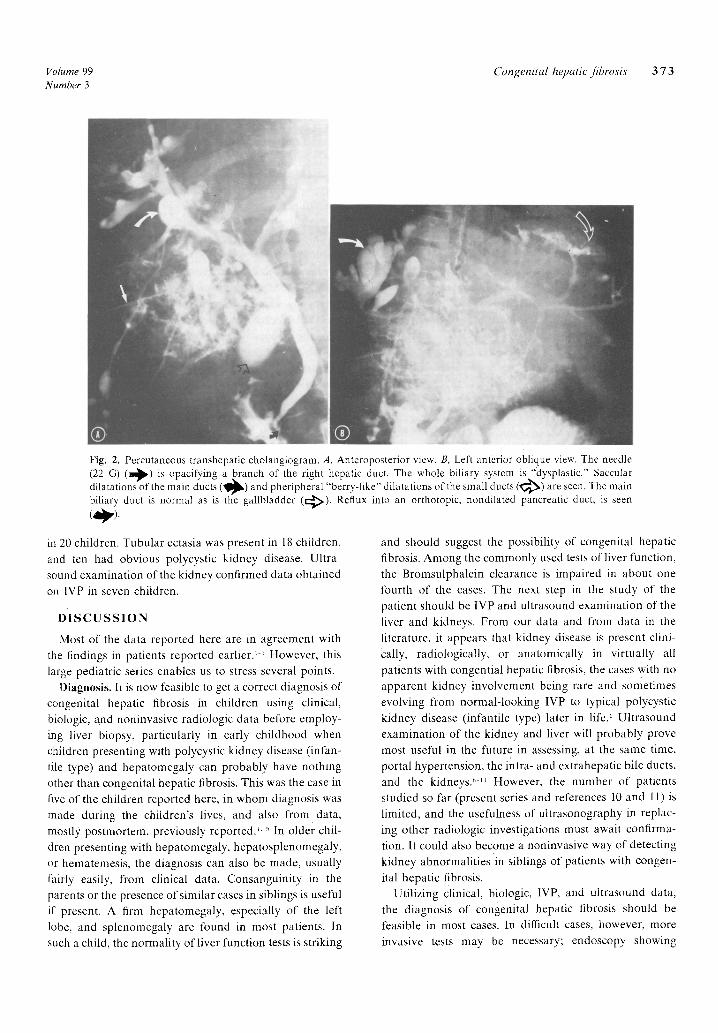
dilatations of the main ducts and peripheral "berry-like" dilatations of the small ducts were seen (Fig. 2). This patient did not have any complication resulting from the procedure. Ultrasound examination of the fiver and biliary tract Was carried out in six patients. It was normal
in one and showed a heterogenous liver in two patients, an enlarged gallbladder in two, and intrahepatic cystic dila- tations in one. None of the children presented with the major dilatation of extrahepatic bile ducts that has been reported to be associated with congenital hepatic fibrosis in a few cases. 7
A diagnosis of cholangitis was made in three children. One 12-year-old child had three recurrent episodes of acu!e cholangitis that could not be controlled by antibiotic therapy. She underwent surgery (cholecystojejunostomy) but this did not improve the course of the biliary infection, and she died nine months after surgery with hepatic coma in spite of antibiotic treatment. In the third patient cholangitis presented as an isolated chronic inflammatory syndrome with increased ESR and raised ~,-globulin values over a three-year period. At age 5, culture of a needle liver biopsy specimen yielded Klebsiella oxytoca.
She was treated with antibiotics for three weeks, and ESR returned to normal within two months. All three patients with cholangitis also presented with symptoms of portal hypertension.
Kidney studies. At the time of admission kidneys were palpable in six of ten patients who were later shown to have polycystic kidney disease. Four of them had arterial hypertension; in two of these patients arterial hyperten- sion was not present initially. One of 27 patients had proteinuria and another had microscopic hematuria. Only one had urinary tract infection. Kidney function tests were carried out in 19 patients; raised blood urea nitrogen concentration was found in four patients. Abnormal maximum concentrating capacity was present in 13. Intravenous pyelography was performed in 27 children, and was normal in three of them. Kidneys were enlarged
Volume 99 Congenital hepatic fibrosis 3 7 3 Number 3
Fig. 2. Percutaneous transhepatic cholangiogram. A, Anteroposterior view. B, Left anterior oblique view. The needle (22 G) ( ~ ) is opacifying a branch of the right hepatic duct. The whole biliary system is "dysplastic." Saccular dilatations of the main ducts (~1~) and pheripheral "berry-like" dilatations of the small ducts ( ~ ) are seen. The main biliary duct is normal as is the gallbladder (c:~) . Reflux into an orthotopic, nondilated pancreatic duct, is seen ( ~ ) .
in 20 children. Tubular ectasia was present in 18 children, and ten had obvious polycystic kidney disease. Ultra- sound examination of the kidney confirmed data obtained on IVP in seven children.
D I S C U S S I O N
Most of the data reported here are in agreement with the findings in patients reported earlier? + However, this large pediatric series enables us to stress several points.
Diagnosis. It is now feasible to get a correct diagnosis of congenital hepatic fibrosis in children using clinical, biologic, and noninvasive radiologic data before employ- ing liver biopsy, particularly in early childhood when children presenting with polycystic kidney disease (infan- tile type) and hepatomegaly can probably have nothing other than congenital hepatic fibrosis. This was the case in five of the children reported here, in whom diagnosis was made during the children's lives, and also from data, mostly postmortem, previously reported." ~ In older chil- dren presenting with hepatomegaly, hepatosplenomegaly, or hematemesis, the diagnosis can also be made, usually fairly easily, from clinical data. Consanguinity in the parents or the presence of similar cases in siblings is useful if" present. A firm hepatomegaly, especially of the left lobe, and splenomegaly are found in most patients. In such a child, the normality of liver function tests is striking
and should suggest the possibility of congenital hepatic fibrosis. Among the commonly used tests of liver function, the Bromsulphalein clearance is impaired in about one fourth of the cases. The next step in the study of the patient should be IVP and ultrasound examination of the liver and kidneys. From our data and from data in the literature, it appears that kidney disease is present clini- cally, radiologically, or anatomically in virtually all patients with congential hepatic fibrosis, the cases with no apparent kidney involvement being rare and sometimes evolving from normal-looking IVP to typical polycystic kidney disease (infantile type) later in life. ~ Ultrasound examination of the kidney and liver will probably prove most useful in the future in assessing, at the same time, portal hypertension, the intra- and extrahepatic bile ducts, and the kidneys? ~ However, the number of patients studied so far (present series and references 10 and 11) is limited, and the usefulness of ultrasonography in replac- ing other radiologic investigations must await confirma- tion. It could also become a noninvasive way of detecting kidney abnormalities ~in siblings of patients with congen-
ital hepatic fibrosis. Utilizing clinical, biologic, 1VP, and ultrasound data,
the diagnosis of congenital hepatic fibrosis should be feasible in most cases. In difficult cases, however, more invasive tests may be necessary; endoscopy showing

3 7 4 A lvarez et al. The Journal of Pediatrics September 1981
varices can prove the presence of portal hypertension in children with isolated hepatosplenomegaly or in patients presenting with acute gastrointestinal bleeding, in whom the spleen can temporarily be nonpalpable because of posthemorrhagic splenic contraction12; intravenous cho- langiography is useful although the pictures obtained are not always of perfect quality. SPG has shown a duplica- tion of the intrahepatic branches of the portal vein in four fifths of the patients with congenital hepatic fibrosis';; except for one of our patients, those in whom SPG did not show duplication either had no portal hypertension or had portal vein thrombosis or a large natural splenorenal shunt that prevented adequate perfusion of the liver by contrast medium.
Finally, it should be stressed that examination of liver needle biopsy specimens can show features typical of congenital hepatic fibrosis but can fail to confirm the diagnosis. We suggest that, since the macroscopic appear- ance of the liver surface is so characteristic in congenital hepatic fibrosis, laparoscopy could be useful in difficult cases, sometimes allowing for a correct diagnosis and thus avoiding surgical wedge fiver biopsy in children who may have to be operated on later in life, either because of portal hypertension or for kidney transplantation.
Follow-up. Although the follow-up of some of our patients has been limited, several points need to be emphasized. Portal hypertension, the most frequent com- plication of congenital hepatic fibrosis, was responsible for gastrointestinal bleeding in 11 of 24 patients with splenomegaly, as proved by esophagoscopy showing vari- ces. Precautions, such as avoiding any aspirin-containing drug in those children, may have limited the number of patients with gastrointestinal bleeding (eight bled from varices before being referred to us). Portal-systemic shunt- ing had to be carried out in children with gastrointestinal bleeding as well as prophylactically in a few others. Shunts were effective in all but one child, as shown by the disappearance of varices on endoscopy six months after surgery. When surgery is decided upon, the type of shunt must be carefully selected so that it will not impair the feasibility of a renal transplantation later on in life, whenever such an operation is foreseeable.
Liver disease did not progress to further impairment of hepatic function. Liver function tests, including coagula- tion studies, always remained within normal limits, and portal-systemic shunts were always very well tolerated; hepatic encephalopathy was not observed. All patients lead normal lives, with a normal diet, unless kidney failure is present. This result is in agreement with another large series of pediatric patients 2 and with the fact that several cases of congenital hepatic fibrosis are reported in the adult literature, suggesting that patients can reach
adulthood without major health problems. However, it is not clear whether congenital hepatic fibrosis can result in liver failure or hepatic encephalopathy in adulthood. Several toxic and infectious factors added to congenital hepatic fibrosis could be responsible for impairment of liver function in adults. In the largest retrospective study, '~ the patients who died were mostly those with acute cholangitis, which was responsibl e for liver failure in some of them.
Although cholangitis is not a major complication of congenital hepatic fibrosis in children,', ~ it was the presenting sign in a 9-month-old child in our series. In most cases, cholangitis must be treated with antibiotics after the responsible organism is cultured from blood or from liver tissue obtained by needle biopsy. TM Cholangitis can either be induced or severely worsened by all kinds of surgical procedures involving the biliary tract. TM 1G We strongly advocate avoiding surgical manipulation of the bile ducts in such patients. However, patients with major dilatation of the extrahepatic bile ducts associated with congenital hepatic fibrosis might benefit from resection of the dilated extrahepatic ducts?, r Subclinical chronic bac- terial cholangitis was demonstrated in one patient, and this possibility should be considered in each patient with congenital hepatic fibrosis and inflammatory symptoms or inflammatory infiltrate of the liver. Our patient clearly benefited from antibiotic treatment adapted to the sensi- tivity of the organism isolated from culture of a liver biopsy specimen. This treatment may prevent evolution toward more severe liver disease.
Disease of the kidney associated with congenital hepat- ic fibrosis is characterized histologically by tubular dilata- tion in the medulla and cortex that can vary in scope from the mere radiologic image of tubular ectasia to a major form of polycystic kidney disease. 1, 5 Four of our patients presenting early in life had renal failure of various degrees. Milder lesions limited to radiologically visible tubular ectasia are usually associated with a good progno- sis in terms of renal function, with isolated alteration in maximum concentrating capacity. However, one of our patients, as well as a few others in the literature, presented with kidney lesions progressing from tubular ectasia toward a major form of polycystic kidney disease. ~. 17 The risk of renal failure in such patients is unknown. Patients with congenital hepatic fibrosis and minimal kidney lesions should therefore be followed regularly with either IVP or ultrasound examination of the kidney, as well as kidney function tests.
Finally, none of our patients had nephronophthisis, is or any other congenital malformations.
Special aspects. Congenital hepatic fibrosis limited to the left lobe of the fiver was found in one of our patients

Volume 99 Congenital hepatic fibrosis 3 7 5 Number 3
presenting at 2 years o f age with portal hypertension and
portal thrombosis. Two similar cases have been
reported. '" All three patients had typical histologic fea-
tures of congenital hepatic fibrosis limited to one lobe of
the liver, with normal liver function tests and normal 1VP.
In our patients, as well as in the literature, clinical
expression of the disease can be different in two siblings.
This finding, as well as the evolution of the kidney lesions
throughout life in a few patients, provides evidence
against a schematic distinction between several types of
polycystic kidney disease associated with congenital
hepatic fibrosis, each of them supposedly linked to an age
group and hypothetically under the control of a different
gene. '
Dilatation of extrahepatic bile ducts, sometimes re-
stricted to an enlarged gallbladder or to some minor
enlargement of the common bile duct, as in our series, can
be very important in a few cases reported as having
"congenital hepatic fibrosis associated with choledochal
cyst". ~~ The intravenous cholangiograms, ultrasound
examinations and transhepatic cholangiogram reported
here suggest that segmental dilatation of intrahepatic bile
ducts is an authentic feature of congenital hepatic fibrosis,
and should not be used as an argument to diagnose such
patients as having Caroli disease. The latter term should
be used only in patients in whom nonobstructive dilata-
tion of intrahepatic bile ducts occurs as an isolated
abnormality without portal fibrosis. TM
We thank Professeur Ch. Bach for permission to include one of his patients in this group, and Ms. B. Chilvers for reviewing the manuscript.
R E F E R E N C E S
I. Kerr DNS, Harrison CV, Sherlock S, and Milnes Walker R: Congenital hepatic fibrosis, Q J Med 117:91, 1961.
2. Auvert J, and Weisgerber G: ka fibroad+nomatose biliaire intrah6patique, M6d Chir Dig 2:363, 1973.
3. Sommerschild HC, Langmark P, and Maurseth K: Congen- ital hepatic fibrosis: Report of two new cases and review of the literature, Surgery 73:53, 1973.
4. Blyth H, and Ockenden BG: Polycystic disease of kidneys and liver presentingin childhood, J Med Genet 8:257, 1971.
5. Lieberman E, Salinas Madrigal L, Gwinn JL, Brennan LP, Fine RN, and Landing BH; Infantile polycystic disease of the kidneys and liver, Medicine 50:277, 1971.
6. Odi6vre M, Chaumont P, Montagne J Ph, and Alagille D: Anomalies of the intrahepatic portal venous system in congenital hepatic fibrosis, Radiology 122:427, 1977.
7. Buts JP, Otte JB, Claus D, Van Craynest MP, and Demeyer R: Kyste du cholddoque: un cas avec dilatation des voies biliaires intrahdpatiques et fibrose h6patique congdnitale. Helv Paediatr Acta 35:289, 1980.
8. Glazer GM, Laing FC, Brown Th W, and Gooding GA: Sonographic demonstration of portal hypertension: the patent umbilical vein, Radiology 136:161, 1980.
9. Weill FS: Ultrasonography of digestive diseases, ed 1, Saint Louis, 1978, The CV Mosby Company, p 152.
10. Rosenfield AT, Siegel N J, Kappelman NB, and Taylor KJW: Gray scale ultrasonography in medullary cystic disease of the kidney and congenital hepatic fibrosis with tubular ectasia. New observations, Am J Roentgenol 129:297, 1977.
I i. Artaud J, Broassin B, Cadier L, and Diard F; Aspects 6chographiques des polykystoses hepato r6nales rdcessives chez l'enfant, J Radiol 61:243, 1980.
12. Alagille D, and Odi~vre M: Liver and biliary tract disease in children, ed 1, New York, 1979, John Wiley & Sons, Inc, pp 265 and 285.
13. Kerr DNS, Okonkwo S, and Choa RG: Congenital hepatic fibrosis: the long-term prognosis, Gut 19:514, 1978.
14. Rogers Ch A, Isenberg JN, Leonard AS, and Sharp HL: Ascending cholangitis diagnosed by percutaneous hepatic aspiration, J Pediatr 88:83, 1976.
15. Dusol M Jr, Levi JU, Glasser K, and Schiff ER: Congenital hepatic fibrosis with dilatation of intrahepatic bile ducts. A therapeutic approach, Gastroenterology 71:839, 1976.
16. Murray-Lyon IM, Shilkin KB, Laws JW, Illing RC, and Williams R: Non-obstructive dilatation of the intrahepatic biliary tree with cholangitis, Q J Med 16,4:477, 1972.
17. Dupond JL, Miguet JP, Carbiller JP, Hillier YS, Perol C, and Leconte des Floris R: Kidney polycystic disease in adult congenital hepatic fibrosis, Ann Intern Med 88:514, 1978.
18. Detaney V, Mullaney J, and Bourke E: Juvenile nephron- ophthisis, congenital hepatic fibrosis and retinal hypoplasia in twins, Q J Med 186:281, 1978.
19. Hausner RJ, and Alexander RW: Localized congenital hepatic fibrosis presenting as an abdominal mass, Hum Pathol 9:473, 1978.
Related Documents











