Conformational Behavior of a,a-Dialkylated Peptides: Ab Initio and Empirical Results for C ycloprop ylglycine VINCENZO BARONE and FRANCA FRATERNALI, Dipartimnto di Chimica, UniversitcE di Napoli, via Mezzocannone 4, I-80134 Napoli; PIER LUIGI CRISTINZIANO, FRANCESCO L E U , and ANGELA ROSA, Istituto Chimico, UniversitcE &Ua Basilicata, via Nazario Sauro 85, I-851 00 Potenza, Italy Synopsis The preferred conformations of model cyclopropylglycine peptides have been investigated by means of ab initio and empirical methods. Empirical computations performed with fixed bond lengths and valence angles using two well-known force fields show that only values of $ I in the ranges k70" k 20" are sterically allowed, and that the C,-conformation corresponds to the absolute energy minimum irrespective of the terminal groups used. Also, ab initio computations give similar results, but suggest greater stabilities for bridge and, especially, extended structures. These discrepancies can be removed, adding to the empirical force field a twofold torsional potential on \c. and using softer steric repulsive potentials. Complete geometry optimization using both ab initio and empirical methods does not affect the relative stabilities of folded conforma- tions, but leads to a further significant stabilization of the fully extended structure via large modifications of some valence angles. INTRODUCTION In recent years much research has been devoted to the understanding of both ordered and random conformations of polypeptides. Amino acids found in proteins have, of course, attracted prevailing attention, but there are several other naturally occurring amino acids that have interesting conforma- tional properties.'-5 It is known, for example, that the presence of a,a-dialkylated a-amino acids in peptides significantly restricts the available range of backbone conforma- tion~.~-' Studies in this area have also been stimulated by the possibility of using a, a-dialkylated residues as local structure determinants for designing conformationally restricted analogs of biologically active peptides.1° This, in turn, may help in delineating the nature of receptor-bound conformations" and in the rational design of analogs of high biological potency." The simplest and best studied member of this class of amino acids is a-aminoisobutyric acid (Aib). Several studies" 139 l4 have shown that conforma- tions in the 3,,/a-helical regions of the conformational map (#I E [ f6O0, + ZOO]; + E [ f 30°, ZOO]) are significantly more stable in Aib than in other residues. Further worksl5. l6 then showed that the replacement of P-methyl groups by bulkier substituents induces a preference for extended rather than folded Biopolymers, Vol. 27, 1673-1685 (1988) 0 1988 John Wiley & Sons, Inc. CCC oooS-3525/88/101673-13$04.00

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Conformational Behavior of a,a-Dialkylated Peptides: Ab Initio and Empirical Results for
C ycloprop ylgl y cine
VINCENZO BARONE and FRANCA FRATERNALI, Dipartimnto di Chimica, UniversitcE di Napoli, via Mezzocannone 4, I-80134
Napoli; PIER LUIGI CRISTINZIANO, FRANCESCO LEU, and ANGELA ROSA, Istituto Chimico, UniversitcE &Ua Basilicata, via
Nazario Sauro 85, I-851 00 Potenza, Italy
Synopsis
The preferred conformations of model cyclopropylglycine peptides have been investigated by means of ab initio and empirical methods. Empirical computations performed with fixed bond lengths and valence angles using two well-known force fields show that only values of $I in the ranges k70" k 20" are sterically allowed, and that the C,-conformation corresponds to the absolute energy minimum irrespective of the terminal groups used. Also, ab initio computations give similar results, but suggest greater stabilities for bridge and, especially, extended structures. These discrepancies can be removed, adding to the empirical force field a twofold torsional potential on \c. and using softer steric repulsive potentials. Complete geometry optimization using both ab initio and empirical methods does not affect the relative stabilities of folded conforma- tions, but leads to a further significant stabilization of the fully extended structure via large modifications of some valence angles.
INTRODUCTION
In recent years much research has been devoted to the understanding of both ordered and random conformations of polypeptides. Amino acids found in proteins have, of course, attracted prevailing attention, but there are several other naturally occurring amino acids that have interesting conforma- tional properties.'-5
It is known, for example, that the presence of a,a-dialkylated a-amino acids in peptides significantly restricts the available range of backbone conforma- t ion~.~- ' Studies in this area have also been stimulated by the possibility of using a, a-dialkylated residues as local structure determinants for designing conformationally restricted analogs of biologically active peptides.1° This, in turn, may help in delineating the nature of receptor-bound conformations" and in the rational design of analogs of high biological potency."
The simplest and best studied member of this class of amino acids is a-aminoisobutyric acid (Aib). Several studies" 139 l4 have shown that conforma- tions in the 3,,/a-helical regions of the conformational map (#I E [ f6O0, + Z O O ] ; + E [ f 30°, Z O O ] ) are significantly more stable in Aib than in other residues.
Further worksl5. l6 then showed that the replacement of P-methyl groups by bulkier substituents induces a preference for extended rather than folded
Biopolymers, Vol. 27, 1673-1685 (1988) 0 1988 John Wiley & Sons, Inc. CCC oooS-3525/88/101673-13$04.00

1674 BARONE E T AL.
conformations. This modification is accompanied by a strong reduction of the valence angle N-C"-C' (hereafter referred to as T), whereas the other intramo- lecular geometrical parameters remain practically unmodified; so small defor- mations of the valence angles a t the C", requiring little energy, can be very important in fixing the overall conformation of a, a-dialkylated residues.
The 1-a-aminocycloalkanearboxylic acids (abbreviated as Accn, where n is the number of carbon atoms in the cycloalkane ring) have been less exten- sively investigated than other a, a-dialkylated c o m p ~ u n d s . ' ~ - ~ ~ These residues may be expected to impose backbone conformational constraints similar to those observed for Aib, but it will be shown that this is not always the case, especially due to the low values of the CBC"CB' valence angle (hereafter referred to as a), which determine particular steric constraints. In this connection, a comprehensive analysis of 1-aminocyclopropanecarboxylic acid (Acc~), is particularly significant in view of its extremely low value of a (= 60").
Three main points will be addressed in this study, namely, (1) the role played by different terminal groups and modifications of the
local geometry a t the C" in determining the conformational behaviour of Acc3;
(2) a comparison between rigid and flexible geometry calculations; and (3) a comparison between quantum mechanical and empirical methods.
METHODS
Two different systems containing monomeric unities of Acc3 were studied, namely F-Acc3-NH, (F = formyl, NH, = amino), hereafter referred to as model I, and Ac-Acc3-NHMe (Ac = acetyl, NHMe = methylamino), hereafter referred to as model 11.
The standard geometries of Scheraga and co-workers20,21 were used for all the end groups, while the x-ray structure of (E)-m-1-benzamido-1-methoxy- carbonyl-2-chlorocyclopropane (BCP)17 was used for the central part, except for the averaging of the geometrical parameters of the two P-methylene groups (see Fig. 1).
Empirical two-body potential functions were used for describing torsional, steric, electrostatic, and H-bond interactions. Conformational maps were built by the package ICER,22723 which is able to significantly reduce the computa- tional time by means of a preliminary topological analysis. In fact, energy differences between successive points are computed, taking into account only interactions between atoms whose relative positions are modified. Further- more, interactions depending on a single dihedral angle (e.g., torsional terms and 1,4 interactions) are calculated at the beginning of the computation only and stored for successive use. Finally, the program has an user-friendly interface for the input of polypeptide sequences and allows the use of several force fields.
Two different force fields were used in the present study, namely the latest revision21 of the ECEPP potentials proposed by Scheraga and co-workers,20 and the AMBER force field proposed by Kollman and co-workers in its all-atom p a r a m e t r i z a t i ~ n . ~ ~ . ~ ~ Some test computations (vide infra) were per- formed using the Buckingham steric functional of the MM2 force
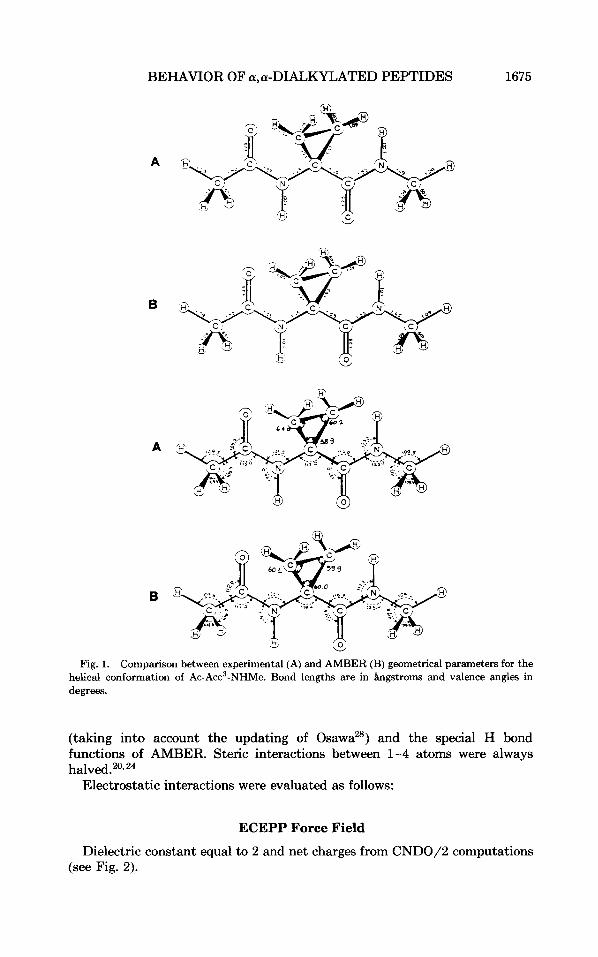
BEHAVIOR OF a,a-DIALKYLATED PEPTIDES 1675
Fig. 1. Comparison between experimental (A) and AMBER (B) geometrical parameters for the helical conformation of Ac-Acc3-NHMe. Bond lengths are in bgstroms and valence angles in degrees.
(taking into account the updating of Osawa28) and the special H bond functions of AMBER. Steric interactions between 1-4 atoms were always
24
Electrostatic interactions were evaluated as follows:
ECEPP Force Field
Dielectric constant equal to 2 and net charges from CND0/2 computations (see Fig. 2).

1676 BARONE ET AL.
MOUt I I
:s ( 3c I
J Y 5 1330)
c . c’ONf O K M A I 1 ON 7
M O U f I 1 1
26
J J 30
454
i i i L
C ’ c , C ’ONf i 3 K M A I I c 3 N
Fig. 2. Atomic charges employed in ECEPP and AMBER (in parentheses) computations are superimposed to schematic drawings of C,-conformation of F-Acc3-NH, and C, confirmation of Ac-Acc3-NHMe.
AMBER and MM2 Force Fields
Dielectric constant equal to ‘2 for 1-4 interactions and equal to 1 for other interactions. The charges (see fig. 2) are generally similar to the original AMBER24 ones, but are derived by empirical rules, which avoid any pre- liminary quantum mechankal omp put at ion.^^ In particular, the atomic charges of the backbone are always taken constant,24 and the total charge on each side chain was set equal to the AMBER value (0.03) for the p-hydrogens of glycine. Next, following the suggestion of Lifson and ~o-workers~~ and some quantum mechanical results,3o electroneutrality was imposed to the CO, NH,,

BEHAVIOR OF a,&-DIALKYLATED PEPTIDES 1677
and NHCH, moieties, and a constant charge (0.03) was assigned to all the aliphatic hydrogen atoms. The charge on oxygen (- 0.5) was the same as in AMBER, but the charges on nitrogen atoms were slightly reduced from the original values in order to enhance the agreement between empirical and ab initio results for glycine and alanine, especially concerning the relative stabil- ity of extended conformation^.^^ Finally, the value of the charge on C" (0.08 compared to the AMBER value of 0.035 in glycine) allows us to always obtain neutral residues. We have, anyway, verified that reasonable modifications of net charges on aliphatic groups have essentially no effect both on structures and relative stabilities of different conformers.
The conformational space was mapped by calculating the conformational energy at 10" intervals for the I#B, J, angles, with w angles fixed at 180" and the terminal methyl groups of model I1 frozen into staggered conformation^.^^ Ab initio computations were performed in a similar way using the GAUSSIAN/82 package32 and the STO-3G basis set,33 but with a grid of 30".
Minimum-energy conformations were then obtained in the low-energy re- gions located in the above search, minimizing the energy with respect to all the dihedral angles by the ICER or GAUSSIAN/82 packages using proce- dures based on analytical gradient^.^^,^^
Complete geometry optimizations (i.e., including variability of bond lengths and bond angles) were performed in the same regions using the AMBER package35 and the complete all-atom force field of Ref. 24, but with net charges evaluated as described above. In view of the negligible effect of bond-length vfuiations, only valence and dihedral angles were instead opti- mized at the STO-3G level.
Conformational energies are expressed as A E = E - E,, where E, is the energy of the most stable conformation for a given compound.
RESULTS AND DISCUSSION
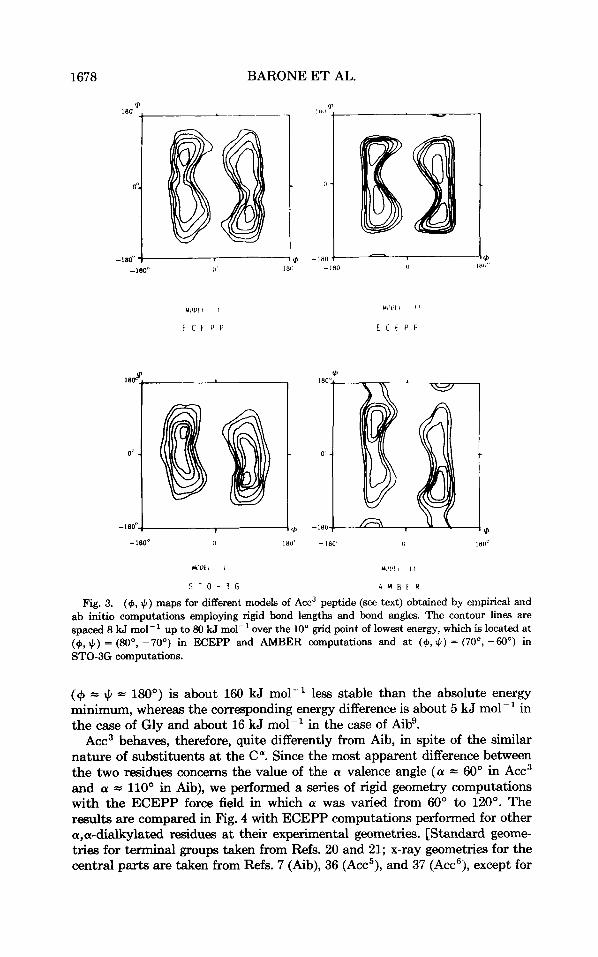
The ($J, J,) maps obtained for both model peptides using the ECEPP force field are shown in Fig. 3. The negligible role played by terminal groups is quite evident in view of the close similarity of the two maps. This result is confirmed by the relative energies of the different conformations obtained upon complete optimization in the torsional angle space (see Table I).
The conformational space available to the backbone for the two models is restricted to a narrow region of centered around f 70", whereas no particu- lar restrictions are found on J,. This region includes the C,-structure (J , = T 70") together with bridge (J , = OO), 3,,-( J, = f 30") and F-( J, = 180") helical conformations.
The energy differences between the absolute energy minimum (C,-struc- ture) and the most stable helix are 6.7 and 6.3 kJ mol-' for model I and model 11, respectively. T h e results are not modified by energy minimization in the complete torsional space, the corresponding energy differences being, in fact, 7.1 and 6.7 kJ mol-'. Table I shows that very similar results are obtained for glycine (hereafter referred to as G ~ Y ) , ~ but in the case of fibg the 3,,-helical structure is more stable than the C, one by about 27 kJ mol-' and the bridge region is not accessible. Furthermore, in the case of Acc3 the C,-conformation
1678 BARONE ET AL.
'I'
7 - - - ~ l
0': 0 -
YOVL 1 I
E C E P P
0 180' - l8OD
-1HO -180
c
oy .
Y ' U t I I
S T O - 3 G
9 - I80 0 IHO"
Fig. 3. (+, 4) maps for different models of Acc3 peptide (see text) obtained by empirical and ab initio computations employing rigid bond lengths and bond angles. The contour lines are spaced 8 kJ mol-' up to 80 kJ mol-' over the 10' grid point of lowest energy, which is located at (g, J.) = (No, -70") in ECEPP and AMBER computations and at (+, 4 ) = (70°, -60") in STO-3G computations.
(+ = # = MOO) is about 160 kJ mol-' less stable than the absolute energy minimum, whereas the corresponding energy difference is about 5 kJ mol-' in the case of Gly and about 16 kJ mol-' in the case of Aib'.
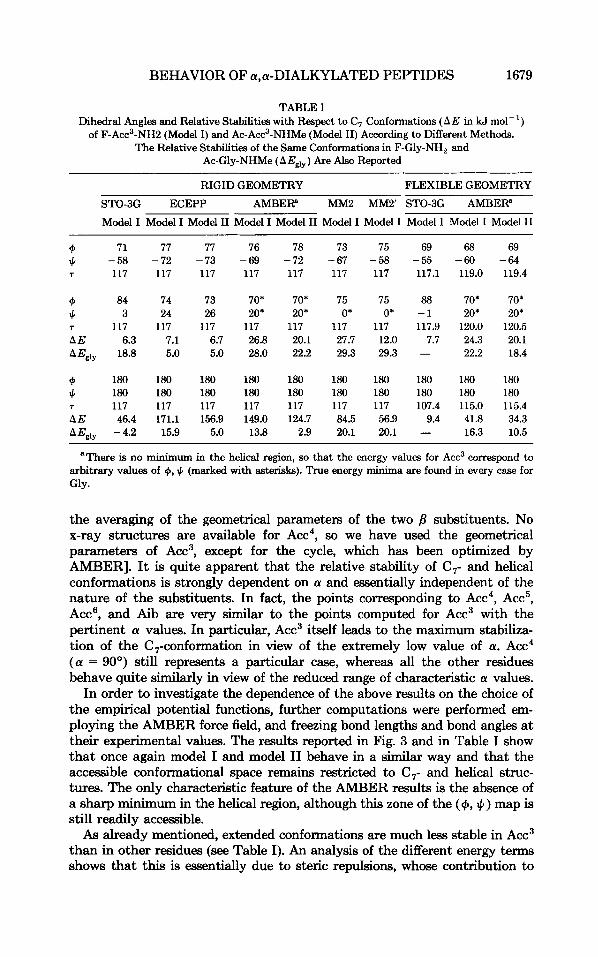
Acc3 behaves, therefore, quite differently from Aib, in spite of the similar nature of substituents at the Ca. Since the most apparent difference between the two residues concerns the value of the a valence angle (a = 60" in Acc3 and a = 110' in Aib), we performed a series of rigid geometry computations with the ECEPP force field in which a was varied from 60' to 120'. The results are compared in Fig. 4 with ECEPP computations performed for other a,a-dialkylated residues at their experimental geometries. [Standard georne- tries for terminal groups taken from Refs. 20 and 21; x-ray geometries for the central parts are taken from Refs. 7 (Aib), 36 (Acc~), and 37 (Acc'), except for
BEHAVIOR OF a,a-DIALKYLATED PEPTIDES 1679
TABLE I Dihedral Angles and Relative Stabilities with Respect to C, Conformations (AE in kJ mol-')
of F-Acc3-NH2 (Model I) and Ac-Acc3-NHMe (Model 11) According to Different Methods. The Relative Stabilities of the Same Conformations in F-Gly-NH, and
Ac-Gly-NHMe ( A EglY) Are Also Reported
RIGID GEOMETRY FLEXIBLE GEOMETRY STO-3G ECEPP AMBEF MM2 MM2' STO-3G AMBERB
Model I Model I Model I1 Model I Model I1 Model I Model I Model I Model I Model I1
9 71 77 4 -58 -72 r 117 117
9 84 74 4 3 24 r 117 117 AE 6.3 7.1 AE,,, 18.8 5.0
9 180 180 4 180 180 7 117 117 AE 46.4 171.1 AE,,, -4.2 15.9
77 - 73 117
73 26
117 6.7 5.0
180 180 117 156.9
5.0
76 - 69 117
70* 20*
26.8 28.0
117
180 180 117 149.0 13.8
78 73 -72 -67 117 117
70* 75 20* O*
117 117 20.1 27.7 22.2 29.3
180 180 180 180 117 117 124.7 84.5
2.9 20.1
75 - 58 117
75
117 O*
12.0 29.3
180 180 117 56.9 20.1
69 68 69 -55 -60 -64 117.1 119.0 119.4
88 70* 70* -1 20* 20* 117.9 120.0 120.5
7.7 24.3 20.1 - 22.2 18.4
180 180 180 180 180 180 107.4 115.0 115.4
9.4 41.8 34.3 - 16.3 10.5
aThere is no minimum in the helical region, so that the energy values for Ace3 correspond to arbitrary values of +, I) (marked with asterisks). True energy minima are found in every case for Gly.
the averaging of the geometrical parametem of the two /3 substituents. No x-ray structures are available for Acc4, so we have used the geometrical parameters of Acc', except for the cycle, which has been optimized by AMBER]. It is quite apparent that the relative stability of C,- and helical conformations is strongly dependent on a and essentially independent of the nature of the substituents. In fact, the points corresponding to Acc', Acc', Acc', and Aib are very similar to the points computed for ACC' with the pertinent a values. In particular, Acc3 itself leads to the maximum stabiliza- tion of the C,-conformation in view of the extremely low value of a. Acc4 (a = 90°) still represents a particular case, whereas all the other residues behave quite similarly in view of the reduced range of characteristic a values.
In order to investigate the dependence of the above results on the choice of the empirical potential functions, further computations were performed em- ploying the AMBER force field, and freezing bond lengths and bond angles at their experimental values. The results reported in Fig. 3 and in Table I show that once again model I and model I1 behave in a similar way and that the accessible conformational space remains restricted to C,- and helical struc- tures. The only characteristic feature of the AMBER results is the absence of a sharp minimum in the helical region, although this zone of the (+, #) map is still readily accessible. As already mentioned, extended conformations are much less stable in ACC'
than in other residues (see Table I). An analysis of the different energy terms shows that this is essentially due to steric repulsions, whose contribution to
1680
J E ( k J I m o 1 ) + * '
+ O .
- 2 .
- 4 .
- 6 . k c " %
A d +
- 8 .
-10 .
BARONE ET AL.
I 1 I I I I I -. 5 0 . 60. 70. 80. 90. 100. 110. 120.
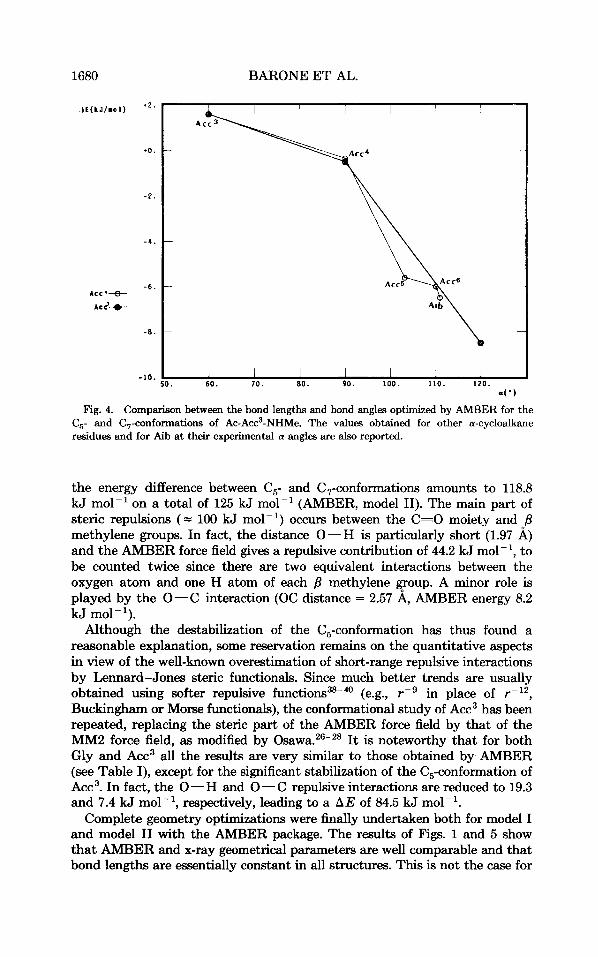
4') Fig. 4. Comparison between the bond lengths and bond angles optimized by AMBER for the
C,- and C7-conformations of Ac-Acc3-NHMe. The values obtained for other a-cycloalkane residues and for Aib at their experimental a angles are also reported.
the energy difference between C,- and C,-conformations amounts to 118.8 kJ mol-' on a total of 125 kJ mol-' (AMBER, model 11). The main part of steric repulsions (= 100 kJ mol-') occurs between the C=O moiety and /3 methylene groups. In fact, the distance 0-H is particularly short (1.97 A) and the AMBER force field gives a repulsive contribution of 44.2 kJ mol-l, to be counted twice since there are two equivalent interactions between the oxygen atom and one H atom of each /3 methylene group. A minor role is played by the 0-C interaction (OC distance = 2.57 A, AMBER energy 8.2 kJ mol-l).
Although the destabilization of the C,-conformation has thus found a reasonable explanation, some reservation remains on the quantitative aspects in view of the well-known overestimation of short-range repulsive interactions by Lennard-Jones steric functionals. Since much better trends are usually obtained using softer repulsive fun~t ions~-~O (e.g., r-' in place of r-12, Buckingham or Morse functionals), the conformational study of Acc3 has been repeated, replacing the steric part of the AMBER force field by that of the MM2 force field, as modified by Osawa.26-28 It is noteworthy that for both Gly and Acc3 all the results are very similar to those obtained by AMBER (see Table I), except for the significant stabilization of the C,-conformation of Acc3. In fact, the 0 - H and 0-C repulsive interactions are reduced to 19.3 and 7.4 kJ mol-l, respectively, leading to a AE of 84.5 kJ mol-l.
Complete geometry optimizations were finally undertaken both for model I and model I1 with the AMBER package. The results of Figs. 1 and 5 show that AMBER and x-ray geometrical parameters are well comparable and that bond lengths are essentially constant in all structures. This is not the case for

BEHAVIOR OF a,a-DIALKYLATED PEPTIDES 1681
Fig. 5. Energy difference ( A E ) between C,- and helical conformations of Ac-Acc3-NHMe as a function of the valence angle C@C"CB'(a). Terminal methyl groups are not included since their geometrical parameters always retain the values shown in Fig. 1 for the helical conformation.
valence angles, whose modification (which requires relatively low energies) is very effective in relaxing strained conformation^.^ It is particularly note- worthy in this connection that the optimized value of the valence angle C"NC' is extremely large in the C,-conformation, thus sensibly increasing the distance between the oxygen atom and the cycle, consequently stabilizing the C 5-conformation (see Table I). Complete geometry optimization has, instead, no effect on the relative stabilities of C,- and helical structures (see Table I) since all the valence angles are very similar in the two conformations (see Fig. 5).
Since the above results indicate that the nature of terminal groups has only a negligible effect on the conformational characteristics of Acc', ab initio computations were performed only for model I. Furthermore bond lengths were never varied in geometry optimizations in view of their constancy both in previous ab initio calculations on different peptides and in AMBER computations on Acc3.
A comparison between rigid geometry empirical and STO-3G (@, #) maps (see Fig. 3) shows a substantial agreement on the accessible conformational space, which is restricted in both cases to the same narrow @ region. The main differences concern (a) the # value for the most stable helix, which is close to 0" in STO-3G computations, but to 20' in empirical computations; and (b) the relative stability of the C,-conformation, which is strongly enhanced by STO-3G computations.
According to all the computations, the C,-conformation corresponds to the absolute energy minimum, the helical structure being less stable by 5-20 kJ mol-', depending on the method of computation. A t first glance there
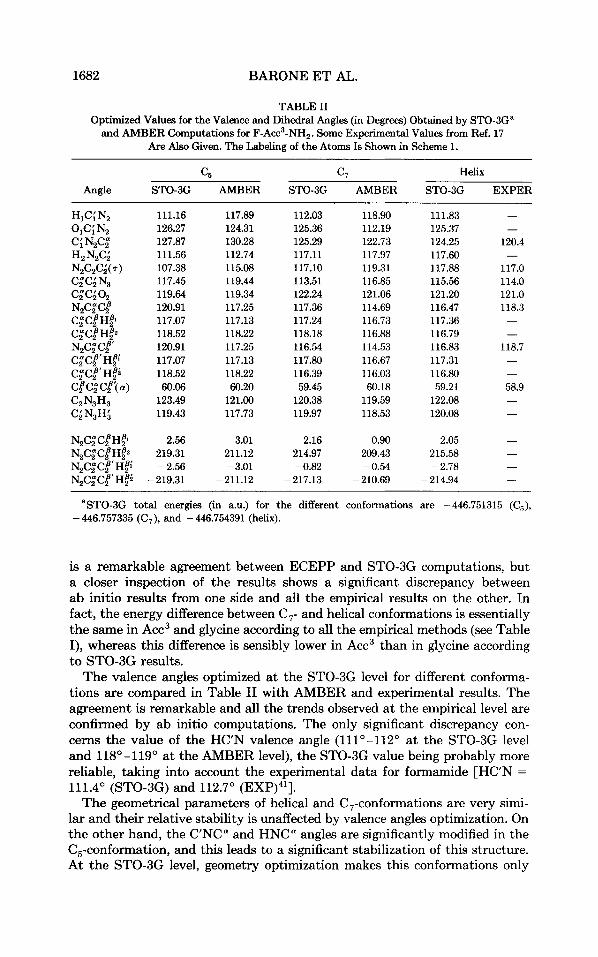
1682 BARONE E T AL.
TABLE I1 Optimized Values for the Valence and Dihedral Angles (in Degrees) Obtained by STO-3G"
and AMBER Computations for F-Acc3-NH,. Some Experimental Values from Ref. 17 Are Also Given. The Labeling of the Atoms Is Shown in Scheme 1.
~ ~~ ~~
c.5 c7 Helix
Angle STO-3G AMBER STO-3G AMBER STO-3G EXPER
111.16 126.27 127.87 111.56 107.38 117.45 119.64 120.91 117.07 118.52 120.91 117.07 118.52 60.06
123.49 119.43
2.56 219.31 - 2.56
-219.31 -
117.89 124.31 130.28 112.74 115.08 119.44 119.34 117.25 117.13 118.22 117.25 117.13 118.22 60.20
121.00 117.73
3.01 211.12 - 3.01 211.12
112.03 118.90 125.36 112.19 125.29 122.73 117.11 117.97 117.10 119.31 113.51 116.85 122.24 121.06 117.36 114.69 117.24 116.73 118.18 116.88 116.54 114.53 117.80 116.67 116.39 116.03 59.45 60.18
120.38 119.59 119.97 118.53
2.16 0.90 214.97 209.43 - 0.82 - 0.54
-217.13 -210.69 -
111.83 - 125.37 - 124.25 120.4 117.60 - 117.88 117.0 115.56 114.0 121.20 121.0 116.47 118.3 117.36 - 116.79 - 116.83 118.7 117.31 - 116.80 -
122.08 - 120.08 -
59.21 58.9
2.05 - 215.58 - - 2.78 -
-214.94 -
'STO-3G total energies (in a.u.) for the different conformations are -446.751315 (C5), - 446.757335 (C7), and - 446.754391 (helix).
is a remarkable agreement between ECEPP and STO-3G computations, but a closer inspection of the results shows a significant discrepancy between ab initio results from one side and all the empirical results on the other. In fact, the energy difference between C,- and helical conformations is essentially the same in Acc3 and glycine according to all the empirical methods (see Table I), whereas this difference is sensibly lower in Acc3 than in glycine according to STO-3G results.
The valence angles optimized a t the STO-3G level for different conforma- tions are compared in Table I1 with AMBER and experimental results. The agreement is remarkable and all the trends observed at the empirical level are confirmed by ab initio computations. The only significant discrepancy con- cerns the value of the H C " valence angle (111°-1120 at the STO-3G level and 118°-1190 at the AMBER level), the STO-3G value being probably more reliable, taking into account the experimental data for formamide [HC'N =
111.4" (STO-3G) and 112.7" (EXP)41]. The geometrical parameters of helical and C,-conformations are very simi-
lar and their relative stability is unaffected by valence angles optimization. On the other hand, the C'NC" and HNCa angles are significantly modified in the C,-conformation, and this leads to a significant stabilization of this structure. At the STO-3G level, geometry optimization makes this conformations only

BEHAVIOR OF a,a-DIALKYLATED PEPTIDES 1683
02 Scheme 1
9.4 kJ mol-l less stable than the C, minimum and 1.7 kJ mol-' less stable than the helical structure, the corresponding AMBER values being 41.8 and 17.5 kJ mol-l. Although no direct evidence can be given about the greater reliability of ab initio results with respect to empirical ones, it is noteworthy that extended conformations have been observed in apolar solvents and even in the solid state for some apdialkylated peptides.15> l6
The differences between empirical and ab initio results are probably related to the peculiar nature of the carbon atoms belonging to the cyclopropane ring, whose behavior cannot be reproduced with the same parameters employed for acyclic alkanes. In particular, it is now well established that the most stable conformations of cyclopropane derivatives containing the COX substituent occur with the CO bond syn or anti to the exocyclic bond of the ring carbon atom that holds the COX g r ~ u p : ~ ~ , ~ The internal rotation around the exocyclic bond of cyclopropane containing the CONH, moiety (Ic, in Acc3) should therefore be g ~ v e r n e d ~ ~ . ~ ~ by a twofold potential with an energy barrier of the order of 20 kJ mol-'.
Taking this effect into account would lead to a stabilization both of C,- and bridge structures (characterized by values of Ic, corresponding to minima of the torsional potential) with respect to the C,-conformation.
In order to verify the above hypothesis we have repeated the MM2 conformational study of model I including for Ic, an intrinsic twofold torsional potential with a barrier of 20 kJ mol-'. The results reported in Table I under the heading MM2' are now in remarkable agreement with STO-3G computa- tions concerning both quantitative aspects and general differences between Acc3 and Gly. Furthermore, the essential differences between cyclic and acyclic a,a-dialkylated peptides (the accessibility of bridge regions only in the former case and of extended regions only in the latter case) are confirmed by these new computations.
In conclusion, the conformational behavior of Acc3 is quite peculiar, and its reliable description requires some modification of standard potential func- tions.
The authors wish to thank Prof. P. A. Kollman for a copy of version 2.0 of the AMBER package.

1684 BARONE ET AL.
References 1. Fox, R. O., Jr. & Richards, F. M. (1982) Nature (London) 300, 325-327. 2. Toniolo, C., Bonora, G. M., Bavmo, A., Benedetti, E., Di Blasio, B., Pavone, V. & Pedone,
3. Goodman, M. (1985) Biopolymers 24, 137-155. 4. Hruby, V. J., Kao, L. F. Himing, L. D. & Burks, J. F. (1985) in Peptides: Structure and
Function, Deber, C. M., Hruby, V. J. & Kopple, K. D., Eds., Pierce Chem. Co., Rockford, IL, pp.
C. (1983) Bwpolymers 22,205-215.
487-490. 5. Toniolo, C., Benedetti, E. & Pedone, C. (1986) Gazz. Chim. Ztal. 116, 355-359. 6. Venkataram Prasad, B. V. & Sasisekharan, V. (1979) Macromolecules 12, 1107-1110. 7. Paterson, Y., Rumsey, S. M., Benedetti, E., Nbmethy, G. & Scheraga, H. A. (1981) J. Am.
8. Peters, D. & Peters, J. (1982) J. Mol. Struct. 86, 341-347. 9. Barone, V., Lelj, F., Bavoso, A., Di Blasio, B., Grimaldi, P., Pavone, V. & Pedone, C. (1985)
10. Marshall, G. R. (1982) in Chemical Regulation of Biological Mechanisms, Cubitt, A. G. &
11. Toniolo, C., Bonora, G. M., Bavosso, A., Benedetti, E., Di Blasio, B., Pavone, V. & Pedone,
12. Komoriya, A. & Chaiken, I. M. (1982) J. Bid . Chem. 257, 2599-2604. 13. Valle, G., Toniolo, C. & Jung, G. (1987) Gazz. Chim. Ztal. 117, 549-553. 14. Jung, G., Briickner, H. & Schmitt, H. (1981) in Structure and Actiuity of Natural Peptzdes,
Voelter, G. & Weitzel, G., Eds., De Gruyter, Berlin, p. 75. 15. Benedetti, E., Toniolo, C., Hardy, P., Barone, V., Bavoso, A., Di Blasio, B., Grimaldi, P.,
Ley, F., Pavone, V., Pedone, C., Bonora, G. M. & Lingham, I. J. (1984) J . Am. Chem. Soc. 106,
16. Benedetti, E., Barone, V., Bavoso, A., Di Blasio, B., Lelj, F., Pavone, V., Pedone, C., Bonora, G. M., Toniolo, C., Laplawy, M. T., Kacmarek, K. & Redlinsky, A. (1988) Bwpolymers
17. Varughese, K. J., Srinivasan, A. R. & Stammer, C. H. (1985) Int. J . Peptide Protein Res. 26, 242-251.
18. Paul, P. K. C., Sukumar, M., Bardi, R., Piazzesi, A. M., Valle, G., Toniolo, C. & Balaram, P. (1986) J. Am. C h m . SOC. 108, 6363-6370.
19. Santini, A., Barone, V., Bavoso, A., Benedetti, E., Di Blasio, B., Fraternali, F. Lelj, F., Pavone, V., Pedone, C., Crisma, M., Bonora, G. M. & Toniolo, C. (1988) Znt. J. Biol. Macromol., in press.
20. Momany, F. A., McGuire, R. F., Burgess, A. W. & Scheraga, H. A. (1975) J. Phys. Chem.
21. Nbmethy, G., Pottle, M. S. & Scheraga, H. A. (1983) J. Phys. Chem. 87, 1883-1887. 22. Fraternali, F. (1987) Thesis, Naples. 23. Barone, V., Cristinziano, P. L. & Lelj, F., submitted for publication. 24. Weiner, S. J., Kollman, P. A., Nguyen, D. T. & Case, D. A. (1986) J. Comput. Chern. 7,
25. Barone, V. & FraternaLi, F., to be published. 26. Allinger, N. L. (1977) J . Am. Chem. SOC. 99, 8127-8133. 27. Allinger, N. L. & Yuh, Y. H. (1981) Quantum Chemistry Program Exchange 13, 395. 28. Jaime, C. & Osawa, E. (1983) Tetrahedron 39, 2769-2778. 29. Hagler, A. T., Huler, E. & Lifson, S. (1974) J. Am. Chem. Soc. 96, 5319-5326. 30. Singh, U. C. & Kollman, P. A. (1984) J. Comput. Chem. 5, 129-145. 31. Zimmerman, S. S., Pottle, M. S., Nbmethy, G. & Scheraga, H. A. (1977) Macromolecules 10,
32. Binkley, J. S., Frisch, M. J., DeFrees, D. J., Raghavachari, K., Whiteside, R. A., Schlegel, H.
33. Hehre, W. J., Stewart, R. F. & Pople, J. A. (1969) J. Chem. Phys. 51, 2657-2664. 34. Schlegel, H. B. (1987) Ado. Chem. Phys. 67, 249-286. 35. Weiner, P. & Kollman, P. (1981) J. Comput. Chem. 2, 287-303. 36. Bardi, R., Piazzesi, A. M., Toniolo, C., Sukumar, M. & Balaram, P. (1986) Biopolymrs 25,
C h m . SOC. 103, 2947-2955.
Bwpolymers 24, 1759-1767.
Creighton, A. M., Eds., The Chemical Society, London.
C. (1983) Bwpolymers 22, 205-215.
8146-8152.
27, 357-371.
79, 2361-2381.
230-252.
1-8.
B., Fluder, E. M. & Pople, J. A. (1982) GAUSSIAN/82, Carnegie-Mellon University.
1635- 1644.

BEHAVIOR OF &,&-DIALKYLATED PEPTIDES 1685
37. Paul, P. K. C., Sukumar, M., Bardi, R., Piazzesi, A. M., Valle, G., Toniolo, C. & Balaram, P.
38. Hagler, A. T., Leiserowitz, L. & Tuval, M. (1976) J . Am. Chem. SOC. 98, 4600-4611. 39. Abraham, R. J. & Stolevik, R. (1978) Chem. Phys. Lett. 58, 622-624. 40. Hillier, I. H. & Robson, B. (1979) J. Theor. Bwl. 76, 83-98. 41. Dory, M, Delhalle, J, Fripiat, J. G. & Andre, J. M. (1987) Znt. J . Quantum Chem., Quantum
42. Volltrauer, H. N. & Schwendeman, R. H. (1971) J . Chem. Phys. 54, 260-267. 43. Volltrauer, H. N. & Schwendeman, R. H. (1971) J . Chem. Phys. 54, 268-273.
(1986) J . Am. C h m . SOC. 108,6363-6370.
BWl. S y v . 14, 85-103.
Received January 1, 1988 Accepted May 5, 1988
Related Documents











