Computational Molecular Biology Prof. Peng-Yeng Yin Ming Chuan University Taiwan

Computational Molecular Biology Prof. Peng-Yeng Yin Ming Chuan University Taiwan.
Dec 21, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Computational Molecular Biology
Prof. Peng-Yeng Yin
Ming Chuan University
Taiwan

Prof. Peng-Yeng Yin P2
Outlines
• Sequence comparison• Multiple alignment

Prof. Peng-Yeng Yin P3
Sequence Comparison
• Introduction– Mutation
• Natural evolutionary process
• DNA replication errors
• DNA texts ‘editing’
– Similarity between DNA sequences• A clue to common evolutionary origin
• A clue to common function

Prof. Peng-Yeng Yin P4
Sequence Comparison
– Ex: cancer may be caused by a normal gene being switched on at the wrong time
• Oncogenes and PDGF are similar• Oncogenes are genes in viruses that cause a cancer-like transformatio
n of infected cells• PDGF is a protein that stimulates cell growth
– Ex: GenBank database search• 20 yrs ago, it was surprising to find similarities between a cancer-caus
ing gene and a normal gene• Today it would be more surprising not to find any similarity between
a new gene and an old one stored in gene database• Even dynamic programming is too slow to match a gene of length 103
to GenBank of size 109
– Parallel hardware implementation of exact algorithms– Fast heuristics of approximate matching

Prof. Peng-Yeng Yin P5
Sequence Comparison
• Longest common subsequence problem– Common subsequence of V=v1v2…vn and W= w1w2…wm
vit=wjt , 1 <= t <= k
1 <= i1 <…<ik <= n; 1 <= j1 <…<jk <= m
– LCS: the CS with the maximal length
LCS(AGGTCG,ACCTCC)=ATC
– Similarity s(V,W)=k, the length of LCS
Distance d(V,W)=n+m-2s(V,W), minimum number of indels needed to transform V to W

Prof. Peng-Yeng Yin P6
Sequence Comparison
– Dynamic programming approach• For s(V,W), let si,0=s0,j=0
• For d(V,W), let di,0=i, d0,j=j
jiji
jji
iji
ji
wvsWVLCSwsWVLCSvs
s if ,1
),(,),(,
max
1,1
1,
,1
,
jiji
jji
iji
ji
wvdWVLCSwdWVLCSvd
d if ,
),(,1),(,1
min
1,1
1,
,1
,
v1v2…vn
w1w2…wm

Prof. Peng-Yeng Yin P7
Sequence Comparison
– Dynamic programming table

Prof. Peng-Yeng Yin P8
Sequence Comparison
• Sequence alignment
A T - - C G T (X)
- T A G C A T (Y)
- 1 - - 1 - 1 = 3 - -3Similarity score (X,Y)=
indel
match
mismatch

Prof. Peng-Yeng Yin P9
Sequence Comparison
• Dynamic programming
),(
),(
),(
max
1,1
1,
,1
,
jiji
jji
iji
ji
wvs
ws
vs
s
),(),( ji wv
ji
ji
ji
wv
wvwv
if
if1),(
where
deletion
insertion
match or mismatch

Prof. Peng-Yeng Yin P10
Sequence ComparisonEdit graph
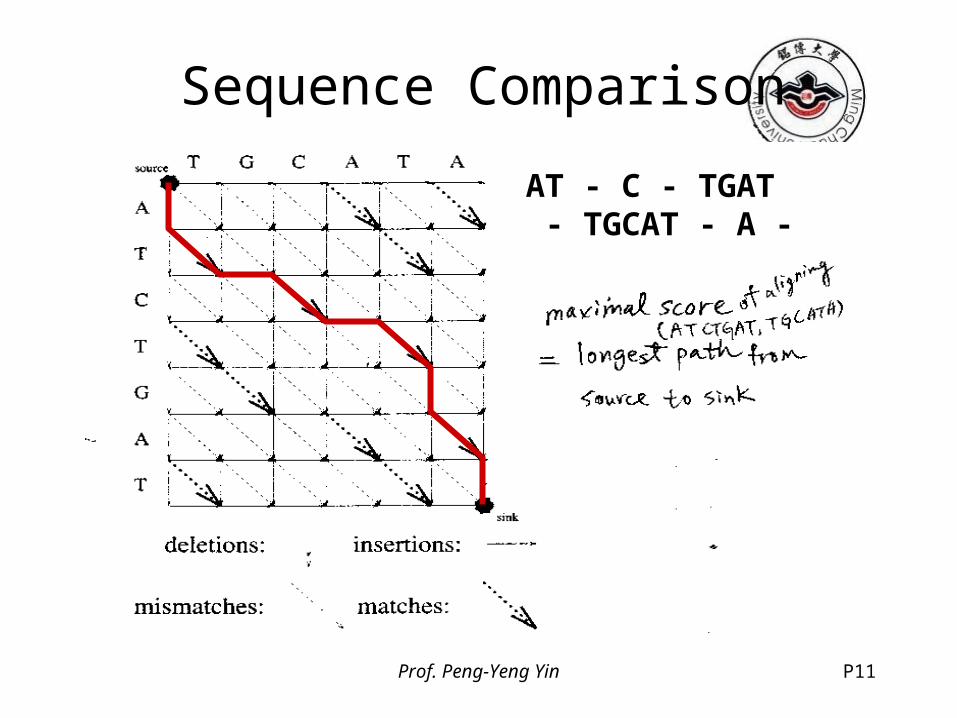
Prof. Peng-Yeng Yin P11
Sequence Comparison
AT - C - TGAT - TGCAT - A -

Prof. Peng-Yeng Yin P12
Sequence Comparison
• Local sequence alignment– Global sequence alignment
s(v1v2…vn, w1w2…wm)
– Local sequence alignment
s(vivi+1…vi’, wjwj+1…wj’)

Prof. Peng-Yeng Yin P13
Sequence Comparison
• Alignment with gap penalties– Gap: consecutive deletions and insertions
– If we define gap penalty as -
A T - - C G T (X)
- T A G C A T (Y)
- 1 - - 1 - 1 = 3 - -3 -2Similarity score (X,Y)=

Prof. Peng-Yeng Yin P14
Sequence Comparison
• Space-efficient sequence alignment– original DP: time complexity O(nm) and space complexi
ty O(nm)
– divide and conquer (D&C): space complexity O(n) but double the running time
• S*,j can be computed by S*,j-1 ( ), hence, Sn,m can be computed column by column with space O(n) but path information is lost
• To obtain the path, use D&C to find consecutive middle vertices
),(max2
,2
,,
reversem
im
iimn sss
This can be solved with time complexity O(nm) and the argument i identifies the middle vertex

Prof. Peng-Yeng Yin P15
Sequence Comparison– Iteratively apply the process, we can find all middle vertices
with time nm+nm/2+nm/4+…<2nm, so this problem can be solved with time complexity O(nm)

Prof. Peng-Yeng Yin P16
Sequence Comparison
• Young tableau– Young diagram of shape
– standard -tableau
9)1,2,2,4(
1 2 3 84 56 79
Each row and column is an increasing sequence

Prof. Peng-Yeng Yin P17
Sequence Comparison
– RSK theorem: =x1x2…xn bitableau(P,Q)RSK

Prof. Peng-Yeng Yin P18
Sequence Comparison– RSK theorem: =x1x2…xn bitableau(P,Q)
– compute (Pk-1,Qk-1) by (Pk,Qk)
• put an empty row above the first row of Pk
• find (i,j) containing k in Qk
• x=Pi,j and erase Qi,j
• R= the (i-1)-th row of Pk
• while R is not the empty row of Pk
– exchange x and Rx-
– R=next row up
• xk=x
– Theorem:longest increasing subsequence (LIS)
LCS(,123…n)=LIS()=length of the first row of Young tableau
RSK

Prof. Peng-Yeng Yin P19
Sequence Comparison
• Average length of LCS– definition
– two interesting problems• Sper(n): LIS in a random permutation
– V:{“12…n”}, W: all permutations of length n (n! strings)
– P(V,W)=1/n!
Hammersley, 1972:Kingman, 1973:Logan, 1977:Baik, 1999:
WwVv
wvpwvsnS,
),(),()(
Spern
nSpern
)(lim
eSper 2
49.259.1 Sper2Sper
...711.1 ),(2)( 61
61
nonnnSper

Prof. Peng-Yeng Yin P20
Sequence Comparison• Sk(n): LCS in a k-letter alphabet
– V, W: all permutations of length n over a k-letter alphabet
– P(V,W)=
Sankoff, 1983:
Steele, 1986:
kk
nS
n
nS
)(lim
2)(lim
kSkk
nnkk
1
kSk
1
2

Prof. Peng-Yeng Yin P21
Sequence Comparison
• Generalized sequence alignment (GSA) and duality– GSA: chains of maximal length in , where is a
partially ordered set (longest -sequence)
– Common subsequence (CS)
– Common inverted forest (CIF)
Assume
),( P
,...21 nvvvV ,...21 mwwwW }:),{( ji wvjiP
Pjipjip ),( ),,( 222111
),(),() ,(Let 121221 jjiipp ji
0,0 21 jipp
0,0 2*
1 jipp

Prof. Peng-Yeng Yin P22
Sequence Comparison• Ex: V = 3 5 7 9 6
W= 5 7 3 6 9)}4,5(),5,4(),2,3(),1,2(),3,1{(P
*
CS
CIF
(1,3) (2,1) (3,2)
(4,5) (5,4)
(1,3) (2,1) (3,2)
(4,5) (5,4)
LCS=longest chain: 5 7 6 , length=3)4,5()2,3()1,2( or 5 7 9 , length=3)5,4()2,3()1,2(
duality
Size of minimal cover
LCS (length ofmaximal chain)

Prof. Peng-Yeng Yin P23
Sequence Comparison
2size )}},5,4()2,3()1,2{()},4,5()3,1{{(
– Theorem: size of minimum cover of P by chains in CS = size of a maximal chain in CIF
• minimal cover of P in CS:
• maximal chain in CIF:
– Theorem: length of LCS = size of minimum cover by CIF• LCS (maximal chain):
• minimal cover by CIF:
2length ),1,2()3,1(
3length ),4,5()2,3()1,2(
3size )}},4,5()5,4{()},2,3()3,1{()},1,2()3,1{{(

Prof. Peng-Yeng Yin P24
Sequence Comparison• Sequence alignment and integer programming
– LCS v.s. IP
LCS(v1v2…vn, w1w2…wn) can be formulated as follows.
Consider the CS graph with the partially ordered set (P, )
e
e PxMAX in chain any for
e
e Px in antichain any for 1 subject to

Prof. Peng-Yeng Yin P25
Sequence Comparison• Approximate string matching
– query matching problemtext: t1t2…tn, query: q1q2…qp, p < n
find all qi…qi+m-1 and tj…tj+m-1 that match with at most k mismatches
– brute force: • k=0, reduced to classical string matching, solvable in O(n)
• k>0, solvable in O(mn)
– lemma: if x1x2…xm and y1y2…ym match with at most k mismatches, then they share an l-substring,
1k
ml

Prof. Peng-Yeng Yin P26
Sequence Comparison– Filtration algorithm
• step 1: [detection] find all common l-substrings for
• step 2: [verification] verify by extending each potential match to the left and to the right
1k
ml
1 2 … k
m
query
text

Prof. Peng-Yeng Yin P27
Sequence Comparison• Comparing a sequence against a database
– protein database search tool• FASTA, 1985
– dot-matrix (v1v2…vn, w1w2…wm) with 1 at entry (i, j) representing there is a common l-tuples (l-substrings) starting at i-th position of V and j-th position of W
– FASTA was a dot-matrix with l=2 in amino acid alphabet
V=DCRNMKFP
W=RNWYCMKFP
DCRNMKFP
RNWYCMKFP0 0 0 0 0 0 0 0 00 0 0 0 0 0 0 0 01 0 0 0 0 0 0 0 00 0 0 0 0 0 0 0 00 0 0 0 0 1 0 0 00 0 0 0 0 0 1 0 00 0 0 0 0 0 0 1 00 0 0 0 0 0 0 0 0
Assemble 1’s on the same diagonal

Prof. Peng-Yeng Yin P28
Sequence Comparison• Comparing a sequence against a database
• BLAST (Basic Local Alignment Search Tool), 1990– segment score for any two l-tuples x1x2…xl , y1y2…yl
– locally maximal segment pairs (LMSP): score can not be improved by extending or shortening both segments
– BLAST finds all LMSPs with scores above a threshold
score similarity:),( ),,(1
yxyxl
iii

Prof. Peng-Yeng Yin P29
Sequence Comparison• Other problems
– parametric sequence alignment• Let alignment score = (# of matches) - (# of mismatches) -
(# of indels)
• what’s the influence of (, ) on the resulting alignment and choose the appropriate ones for biological applications
• polygonal decomposition of (, )-space
Sameopt
alignment 1
Sameopt
alignment 2

Prof. Peng-Yeng Yin P30
Sequence Comparison• Other problems
– alignment statistics and phase transition• local alignment score of two random sequences
),( nH logregion
linearregion
Length of sequences
1})2()log(
),()1{(lim
b
n
HbP n
n
localalignment
score

Prof. Peng-Yeng Yin P31
Sequence Comparison• Other problems
– suboptimal sequence alignment• generate a set of -suboptimal alignment whose deviation fr
om the optimal is at most
– alignment with tandem duplications• tandem duplication is a mutational event in which a stretch
of DNA is duplicated to produce one or more new copies
• Benson, 1997; an efficient algorithm for sequence alignment with tandem duplication
• Landan, 1993; an algorithm for detecting tandem repeats

Prof. Peng-Yeng Yin P32
Sequence Comparison• Other problems
– winnowing database search results• database search programs often report a fixed number of top
matches and truncate lower-scoring but important matches
• define interval I is dominated by interval J if I is contained in J that is with a higher score
• Winnowing database search results is to identify and discard intervals that are dominated by a fixed number of other intervals
– statistical distance between texts (databases)• let X be the set of all l-tuples and x(T) be the number of occ
urrences of x in T
• define the statistical distance between texts V and W as
Xx
WxVxWVd 2))()((),(

Prof. Peng-Yeng Yin P33
Multiple Alignment• Introduction
– similar proteins may not exhibit a strong sequence similarity, one may like to recognize the structural resemblance even when the sequences are very different
– simultaneous comparison of many sequences often allow us to find similarity that are invisible in pairwise comparison
– for comparing k sequences of length n, even DP still costs to find the exact solution
– many heuristics are proposed to find approximate solutions
))2(( knO
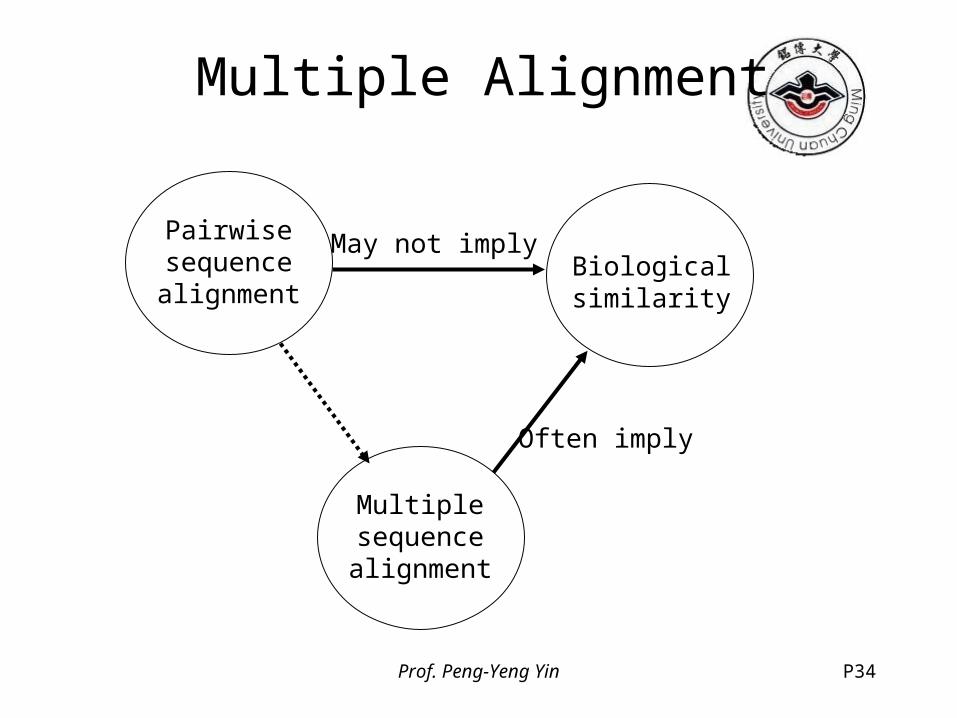
Prof. Peng-Yeng Yin P34
Multiple Alignment
Pairwisesequencealignment
Biologicalsimilarity
Multiplesequencealignment
May not imply
Often imply

Prof. Peng-Yeng Yin P35
Multiple Alignment– Use optimal 2-way alignment to approximate multipl
e k-way alignment• Choose a proper subset of all pairwise alignments and c
ombine then into a multiple alignment
• Feng, 1987; use the star alignment with one center vertex and k-1 leaves, each leaf is optimally aligned to the center vertex following the principle “once a gap, always a gap”. However, there is no performance guarantee
2k
(1) GGAG(2) TGCCAACT(3) GTGTCTAG
(1) ~G ~GA~ ~G(2) TGCCAACT
(2) TGCC~ ~ ~AACT(3) ~ GTGTCTA~ ~G
(1) ~ G~G~ ~ ~A~ ~G(2) TGCC~ ~ ~AACT(3) ~ GTGTCTA~ ~G

Prof. Peng-Yeng Yin P36
Multiple Alignment• Gusfield, 1993; proved that if the star is chosen properly, th
ere is a guaranteed approximation ratio: 2-2/k
– Use optimal l-way alignments to approximate multiple k-way alignment
• Bafna, 1997; with approximation ratio: 2-l/k
– scoring a multiple alignment(1) ~ G~G~ ~ ~A~ ~G(2) TGCC~ ~ ~AACT(3) ~ GTGTCTA~ ~G
Column score
m
iiscoreMIN
1

Prof. Peng-Yeng Yin P37
Multiple Alignment– Definition of column scores
• multiple shortest supersequence problem
• multiple longest common subsequence problem
• biological applications– minimum entropy approach
– distance from consensus
consensus: string of most common character in each column
– sum-of-pair (SP-score)
sum of scores of all pairs of sequences ai and aj
01
Column score = number of different characters in this column
Column score =if all characters in the column are the sameotherwise
x
xx xpp {~}chars all ,logColumn score =
Column score = # of characters that differ from the consensus char
ji
ji aascore,
),(Total score =
Related Documents











![Abstract - arXiv · 2019-11-21 · 1This work was done when Shashanka Venkataramanan was an intern and Kuan-Chuan Peng was a staff scientist at Siemens Corporate Tech- ... [cs.CV]](https://static.cupdf.com/doc/110x72/5f2b287a4b820109cb743eed/abstract-arxiv-2019-11-21-1this-work-was-done-when-shashanka-venkataramanan.jpg)