Complementary Vascular and Matrix Regulatory Pathways Underlie the Beneficial Mechanism of Action of Sorafenib in Liver Fibrosis Dominique Thabut, 1,2 * Chittaranjan Routray, 1 * Gwen Lomberk, 1 Uday Shergill, 1 Kevin Glaser, 3 Robert Huebert, 1 Leena Patel, 1 Tetyana Masyuk, 1 Boris Blechacz, 1 Andrew Vercnocke, 3 Erik Ritman, 3 Richard Ehman, 3 Raul Urrutia, 1 and Vijay Shah 1,3 Paracrine signaling between hepatic stellate cells (HSCs) and liver endothelial cells (LECs) modulates fibrogenesis, angiogenesis, and portal hypertension. However, mechanisms regulat- ing these processes are not fully defined. Sorafenib is a receptor tyrosine kinase inhibitor that blocks growth factor signaling in tumor cells but also displays important and not yet fully characterized effects on liver nonparenchymal cells including HSCs and LECs. The aim of this study was to test the hypothesis that sorafenib influences paracrine signaling between HSCs and LECs and thereby regulates matrix and vascular changes associated with chronic liver injury. Complementary magnetic resonance elastography, micro–computed tomography, and histochemical analyses indicate that sorafenib attenuates the changes in both matrix and vascular compartments that occur in response to bile duct ligation–induced liver injury in rats. Cell biology studies demonstrate that sorafenib markedly reduces cell–cell apposition and junctional complexes, thus reducing the proximity typically observed between these sinu- soidal barrier cells. At the molecular level, sorafenib down-regulates angiopoietin-1 and fibro- nectin, both released by HSCs in a manner dependent on the transcription factor Kruppel- like factor 6 , suggesting that this pathway underlies both matrix and vascular changes associ- ated with chronic liver disease. Conclusion: Collectively, the results of this study demonstrate that sorafenib inhibits both matrix restructuring and vascular remodeling that accompany chronic liver diseases and characterize cell and molecular mechanisms underlying this effect. These data may help to refine future therapies for advanced gastrointestinal and liver diseases characterized by abundant fibrosis and neovascularization. (HEPATOLOGY 2011;54:573-585) L iver cirrhosis is associated with variable changes in architecture of both matrix and vasculature within the sinusoidal tree. Matrix changes are characterized by increased deposition of fibronectin, collagen I, and other fibrillar proteins. Concomitant vascular changes primarily include sinusoidal vasocon- striction, angiogenesis, and pathological remodeling of sinusoids typified by increased mural cell coverage and vigorous wrapping by hepatic stellate cells (HSCs) around liver endothelial cells (LECs). 1-3 These vascular Abbreviations: Ang1, angiopoetin-1; BDL, bile duct ligation; CM, conditioned media; HSC, hepatic stellate cell; KLF6, Kruppel-like factor 6; LEC, liver endothelial cell; micro-CT: micro–computed tomography; MRE, magnetic resonance elastography; mRNA, messenger RNA; PCR, polymerase chain reaction; PDGF, platelet-derived growth factor; PDGFR-b, platelet-derived growth factor receptor b; PI3K, phosphoinositide 3-kinase; RT-PCR, reverse-transcription polymerase chain reaction; siRNA, small interfering RNA; TSEC, transformed liver endothelial cell. From the 1 Gastroenterology Research Unit, Mayo Clinic, Rochester, MN; the 2 Department of Hepatology and Gastroenterology, AP-HP, Pierre and Marie Curie University, Paris, France; and the 3 Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, MN. Received February 25, 2011; accepted May 5, 2011. Supported by grants DK59615-06 (to V. S.), P30DK084567 from the National Institute of Diabetes and Digestive and Kidney Diseases, and EB000305 (to E. R.) by National Institute of Biomedical Imaging and Bioengineering. *These authors contributed equally to this work. Address reprint requests to: Vijay H. Shah, M.D., Gastroenterology Research Unit, Mayo Clinic and Foundation, 200 First Street SW, Rochester, MN 55905. E-mail: [email protected]; fax: 507-255-6318. Copyright V C 2011 by the American Association for the Study of Liver Diseases. View this article online at wileyonlinelibrary.com. DOI 10.1002/hep.24427 Potential conflict of interest: Nothing to report. Additional Supporting Information may be found in the online version of this article. 573

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Complementary Vascular and Matrix RegulatoryPathways Underlie the Beneficial Mechanism of
Action of Sorafenib in Liver FibrosisDominique Thabut,1,2* Chittaranjan Routray,1* Gwen Lomberk,1 Uday Shergill,1 Kevin Glaser,3
Robert Huebert,1 Leena Patel,1 Tetyana Masyuk,1 Boris Blechacz,1 Andrew Vercnocke,3 Erik Ritman,3
Richard Ehman,3 Raul Urrutia,1 and Vijay Shah1,3
Paracrine signaling between hepatic stellate cells (HSCs) and liver endothelial cells (LECs)modulates fibrogenesis, angiogenesis, and portal hypertension. However, mechanisms regulat-ing these processes are not fully defined. Sorafenib is a receptor tyrosine kinase inhibitor thatblocks growth factor signaling in tumor cells but also displays important and not yet fullycharacterized effects on liver nonparenchymal cells including HSCs and LECs. The aim ofthis study was to test the hypothesis that sorafenib influences paracrine signaling betweenHSCs and LECs and thereby regulates matrix and vascular changes associated with chronicliver injury. Complementary magnetic resonance elastography, micro–computed tomography,and histochemical analyses indicate that sorafenib attenuates the changes in both matrix andvascular compartments that occur in response to bile duct ligation–induced liver injury inrats. Cell biology studies demonstrate that sorafenib markedly reduces cell–cell appositionand junctional complexes, thus reducing the proximity typically observed between these sinu-soidal barrier cells. At the molecular level, sorafenib down-regulates angiopoietin-1 and fibro-nectin, both released by HSCs in a manner dependent on the transcription factor Kruppel-like factor 6 , suggesting that this pathway underlies both matrix and vascular changes associ-ated with chronic liver disease. Conclusion: Collectively, the results of this study demonstratethat sorafenib inhibits both matrix restructuring and vascular remodeling that accompanychronic liver diseases and characterize cell and molecular mechanisms underlying this effect.These data may help to refine future therapies for advanced gastrointestinal and liver diseasescharacterized by abundant fibrosis and neovascularization. (HEPATOLOGY 2011;54:573-585)
Liver cirrhosis is associated with variable changesin architecture of both matrix and vasculaturewithin the sinusoidal tree. Matrix changes are
characterized by increased deposition of fibronectin,collagen I, and other fibrillar proteins. Concomitant
vascular changes primarily include sinusoidal vasocon-striction, angiogenesis, and pathological remodeling ofsinusoids typified by increased mural cell coverage andvigorous wrapping by hepatic stellate cells (HSCs)around liver endothelial cells (LECs).1-3 These vascular
Abbreviations: Ang1, angiopoetin-1; BDL, bile duct ligation; CM, conditioned media; HSC, hepatic stellate cell; KLF6, Kruppel-like factor 6; LEC, liver endothelialcell; micro-CT: micro–computed tomography; MRE, magnetic resonance elastography; mRNA, messenger RNA; PCR, polymerase chain reaction; PDGF, platelet-derivedgrowth factor; PDGFR-b, platelet-derived growth factor receptor b; PI3K, phosphoinositide 3-kinase; RT-PCR, reverse-transcription polymerase chain reaction; siRNA, smallinterfering RNA; TSEC, transformed liver endothelial cell.From the 1Gastroenterology Research Unit, Mayo Clinic, Rochester, MN; the 2Department of Hepatology and Gastroenterology, AP-HP, Pierre and Marie Curie
University, Paris, France; and the 3Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, MN.Received February 25, 2011; accepted May 5, 2011.Supported by grants DK59615-06 (to V. S.), P30DK084567 from the National Institute of Diabetes and Digestive and Kidney Diseases, and EB000305 (to
E. R.) by National Institute of Biomedical Imaging and Bioengineering.*These authors contributed equally to this work.Address reprint requests to: Vijay H. Shah, M.D., Gastroenterology Research Unit, Mayo Clinic and Foundation, 200 First Street SW, Rochester, MN 55905.
E-mail: [email protected]; fax: 507-255-6318.CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24427Potential conflict of interest: Nothing to report.Additional Supporting Information may be found in the online version of this article.
573

changes disrupt integrity and homeostasis of sinusoidalfunction and, in concert with matrix changes, lead toportal hypertension and its clinical complications.Sorafenib is a multikinase inhibitor compound
recently approved for use in humans with liver cancer.4
Its recent introduction to the clinic has fueled a pleth-ora of studies aimed at understanding not only itstherapeutic potential, but also possible mechanismsunderlying beneficial roles of this drug. In addition toits better-known effects on epithelial cancer cell prolif-eration,5 sorafenib also regulates receptor tyrosine ki-nase pathways in adjacent stromal cells, includingmyofibroblasts and endothelial cells.6 Although the in-hibitory effects of sorafenib on these nonparenchymalcell types are less characterized, they are nonethelesslikely to significantly contribute to antitumoral efficacyof this drug. Furthermore, because HSCs and LECsare integral to the development of matrix and vascularchanges during liver fibrosis, characterizing effects andmechanisms of action of sorafenib in this disease pro-cess is of notable medical importance.Consequently, in the current study, we demonstrate
that sorafenib improves liver fibrosis by acting, at leastin part, through a novel mechanism that is triggeredwithin HSCs and LECs. Our results report a pathwaywhereby angiopoietin-1 (Ang1) cooperates with fibro-nectin to regulate remodeling of sinusoids that accom-panies liver fibrosis. We found that both Ang1 and fi-bronectin are regulated by platelet-derived growthfactor (PDGF) signaling and are functionally linked bya shared transcription factor; the zinc finger protein,Kruppel-like factor 6 (KLF6) . However, these cooper-ative Ang1 and fibronectin pathways are readily inhib-ited by sorafenib through distinct downstream molecu-lar signals that are independent and dependent on Raf,respectively. Complementary in vivo studies revealed arole for these pathways in the process of increased liverstiffness and provide evidence that sorafenib restores si-nusoidal homeostasis by limiting injury-induced matrixand angiogenic changes. Collectively, these findings areof significant importance for building the theoreticalframework necessary to design new therapies to treatfibrosis in the liver and in other gastrointestinal organssusceptible to exuberant fibrogenic responses.
Materials and Methods
Detailed Materials and Methods are provided in theSupporting Information.
Cell Culture and Transfection. Primary murineLECs, human LECs (ScienCell), or a cell line derivedfrom transformed mouse liver endothelial cells
(TSECs)7 were grown with endothelial culture mediawith 10% serum and 1% endothelial growth supple-ment. Human HSCs (ScienCell) were grown in Dul-becco’s modified Eagle’s medium with 10% serum.Isolation of Murine LECs. LECs were isolated from
whole rat liver by way of repeated mincing followed byenzymatic digestion and CD-31–based immunomagneticseparation as described8 with modifications.Preparation of HSC-Derived Conditioned
Media. Human HSCs were serum-starved and treatedwith either vehicle or sorafenib in serum-free Dulbec-co’s modified Eagle’s medium, and conditioned media(CM) was harvested over 12-24 hours.In Vitro LEC Tubulogenesis-Coculture Assays and
Electron Microscopy. Human LECs and HSCs wereplated on Matrigel-coated four-well glass slides, andtubulogenesis was visualized to study angiogenic inter-actions between LECs and HSCs in vitro asdescribed.3 Transmission electron microscopy was per-formed to visualize vascular connections betweenhuman LECs and HSCs cultured in Matrigel.Chemotaxis Assay. Chemotaxis of human LECs
was measured by way of Boyden assay in response toCM with additional compounds added to media asindicated in individual experiments.Confocal Immunofluorescence Microscopy. Immu-
nofluorescence was performed on murine LECs orTSECs as described.9 Murine LECs and TSECs weregrown to monolayer on collagen-coated glass slidesand stained for ZO-1. Images were captured using aconfocal laser scanning microscope.Quantitative and Semiquantitative Polymerase
Chain Reaction. RNA was isolated from human HSC(RNeasy/Qiagen), reverse-transcribed (Superscript/Invi-trogen) and real-time polymerase chain reaction (PCR)was performed (Applied Biosystems 7500).Chromatin Immunoprecipitation Assay. Human
HSCs were transfected with Flag-tagged KLF6 or con-trol vector. After 36 hours, cells were serum-starved for12 hours, stimulated with or without PDGF for 12hours, and chromatin immunoprecipitation was per-formed (EZ-ChIP kit) as described.10
Bile Duct Ligation and Sorafenib AdministrationIn Vivo. Sprague-Dawley rats were subjected to bileduct ligation (BDL) to induce fibrosis as described.11
Rats were injected with vehicle or sorafenib6 (1.5mg/kg body weight) for in vivo experiments. Proce-dures were performed per Mayo Clinic InstitutionalAnimal Care and Use Committee guidelines.Three-dimensional Reconstruction of Hepatic Vas-
culature Using Micro–Computed Tomography. Ani-mals were injected with a radio-opaque liquid-silicone
574 THABUT, ROUTRAY, ET AL. HEPATOLOGY, August 2011

compound (Microfil, MV-122; Flow Tech., Inc., Carver,MA) through the portal vein (infusion rate, 8-10 mL/mi-nute; pressure, 10-12 mm Hg). Intact animals were placedunder refrigeration at 4�C after perfusion to allow polymer-ization. Livers were scanned and reconstructed asdescribed.12
Magnetic Resonance Elastography. Animals weresubjected to magnetic resonance elastography (MRE)at 4 weeks after sham operation or BDL. Liver stiffnesswas measured by generating longitudinal shear wavesthrough the abdominal wall using a pneumatic driverfollowed by detection of propagating shear wave dis-placement pattern using a phase-contrast magnetic res-onance imaging system as described.13-15
Immunostaining of Liver Tissue. Immunohisto-chemistry was performed on paraffin-embedded andfrozen rat liver tissue sections as described.16
Statistical Analysis. Data are expressed as the mean6 SEM of at least three independent experimentsunless indicated otherwise. Groups were comparedusing a two-tailed Student t test. P < 0.05 was consid-ered statistically significant.
ResultsSorafenib Attenuates Injury-Induced Increases in
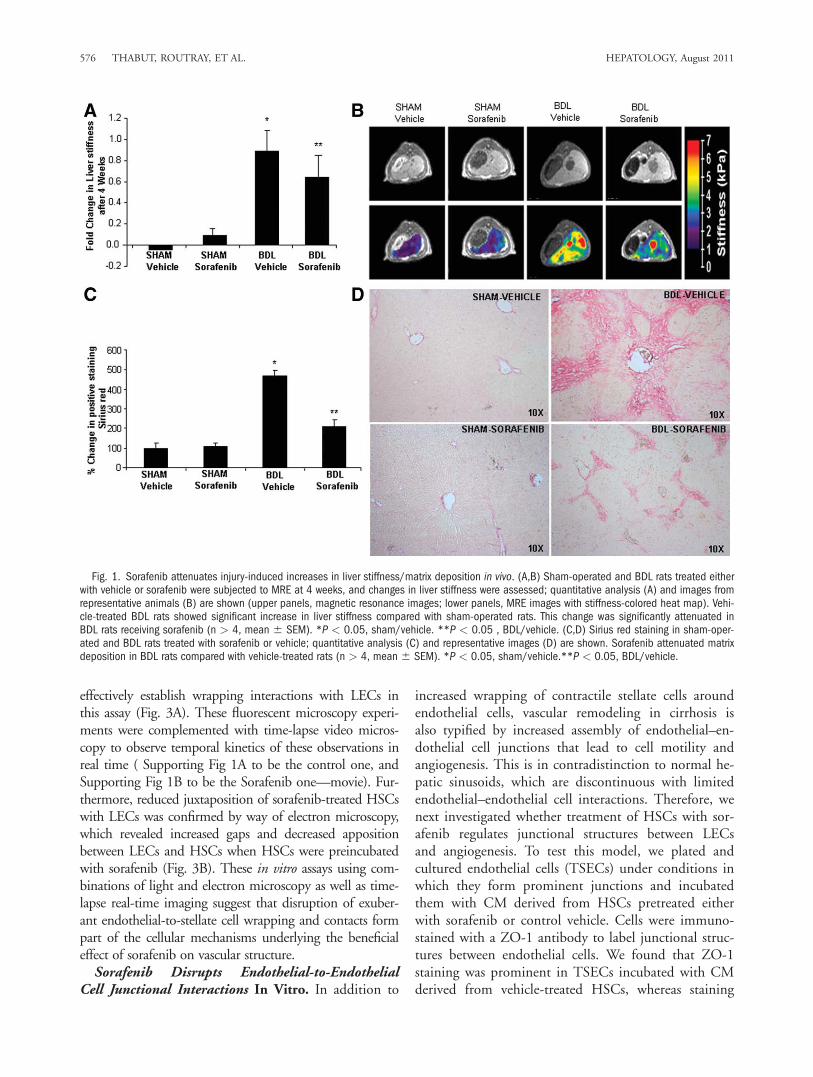
Liver Stiffness, Matrix Deposition, and VascularRemodeling In Vivo. MRE provides one of the best invivo estimations of liver stiffness, a variable that correlateswith both matrix and vascular changes that occur inresponse to liver injury.17 Furthermore, this techniqueprovides an assessment of stiffness throughout the liver, isnoninvasive, and is ideal for performing sequential imag-ing studies in individual animals. In this study, we usedthis technique to evaluate for changes in liver stiffness byway of sorafenib administration. We administered sorafe-nib for 4 weeks to sham-operated or BDL rats beginningimmediately after surgery. Sorafenib was well toleratedwithout overt adverse effects (reduced body weight, diar-rhea, hemorrhage, or mortality). In BDL rats treated withvehicle, MRE revealed a time-dependent increase in liverstiffness compared with sham-operated rats (Fig. 1Ashows composite data, Fig. 1B; top panel shows represen-tative magnetic resonance images, lower panel shows cor-responding MRE). Conversely, in BDL rats receiving sor-afenib, increase in liver stiffness was attenuated (Fig. 1A).Notably, sorafenib also influenced matrix deposition thatoccurs in response to BDL. This attenuated fibrosis insorafenib-treated BDL rats was depicted at 4 weeks byreduced Sirius red staining of liver sections comparedwith BDL rats receiving vehicle (Fig. 1C,D). BecauseMRE reflects angio-architectural changes in addition to
matrix changes, we subsequently ascertained the vascularcomponent that could contribute to liver stiffness and itsmodulation by sorafenib.To investigate the vascular changes in response to sora-
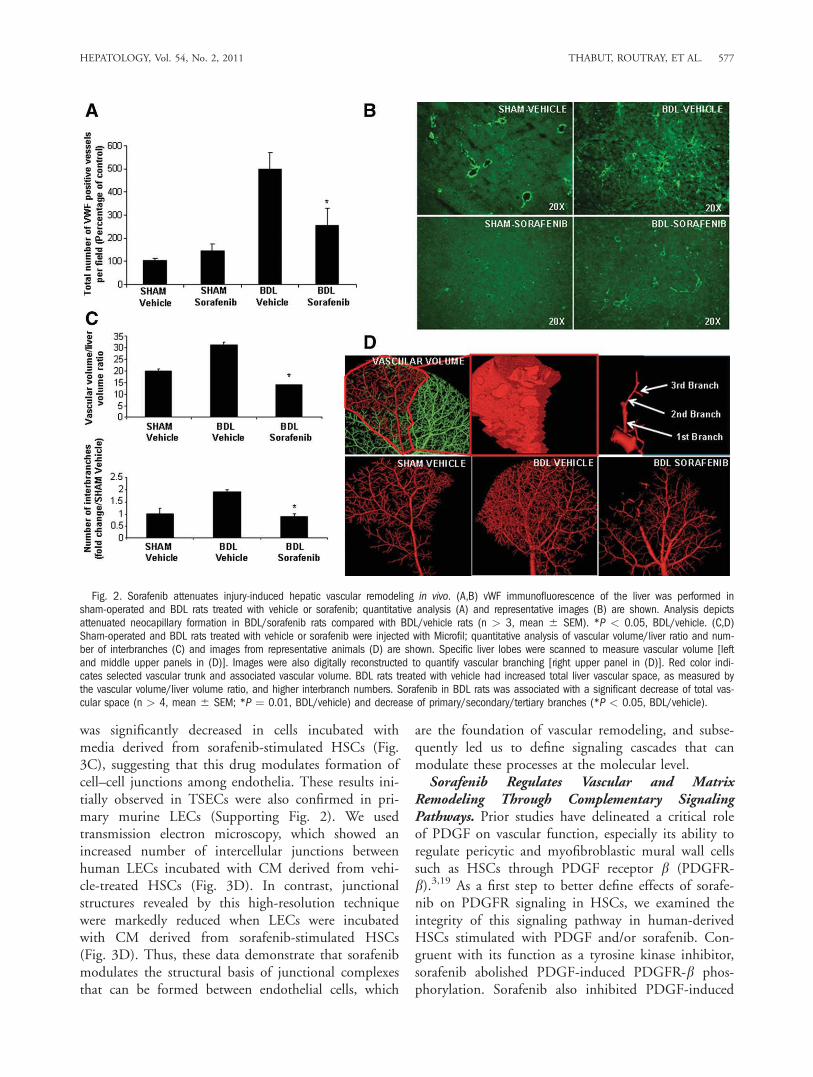
fenib, we initially stained liver tissue sections from treatedanimals with an antibody against vWF, a marker of vascu-lar endothelium that is especially prominent in activelyremodeling vessels in fibrosis.18 Indeed, vWF wasincreased in vehicle-treated BDL rats compared withsham-operated rats as shown by us and others.16 How-ever, when BDL rats were treated with sorafenib, vWFstaining was markedly attenuated (Fig. 2A,B), indicatingthat this drug attenuates vascular changes that occur dur-ing liver wound healing response. To complement thesestudies, we used a micro–computed tomography (micro-CT)–based approach to assess vascular features in sham-operated and BDL rats in greater detail. This techniqueprovides measurement of total vascular volume (Fig. 2D,top left and middle panels) and precise analysis ofbranching of medium and large hepatic vessels (Fig. 2D,right panel), thereby both complementing and corrobo-rating the aforementioned histochemical analyses. Ratioof vascular volume to total liver volume as well as num-ber of branches (primary/secondary/tertiary total) wasincreased in livers of BDL rats (Fig. 2C,D). However,BDL rats receiving sorafenib exhibited attenuation inboth of these micro-CT vascular parameters, consistentwith the vWF histochemical analysis. Thus, sorafenibeffectively attenuates pathobiological vascular changesthat occur in response to BDL.Regulation of Stellate and Endothelial Cell Inter-
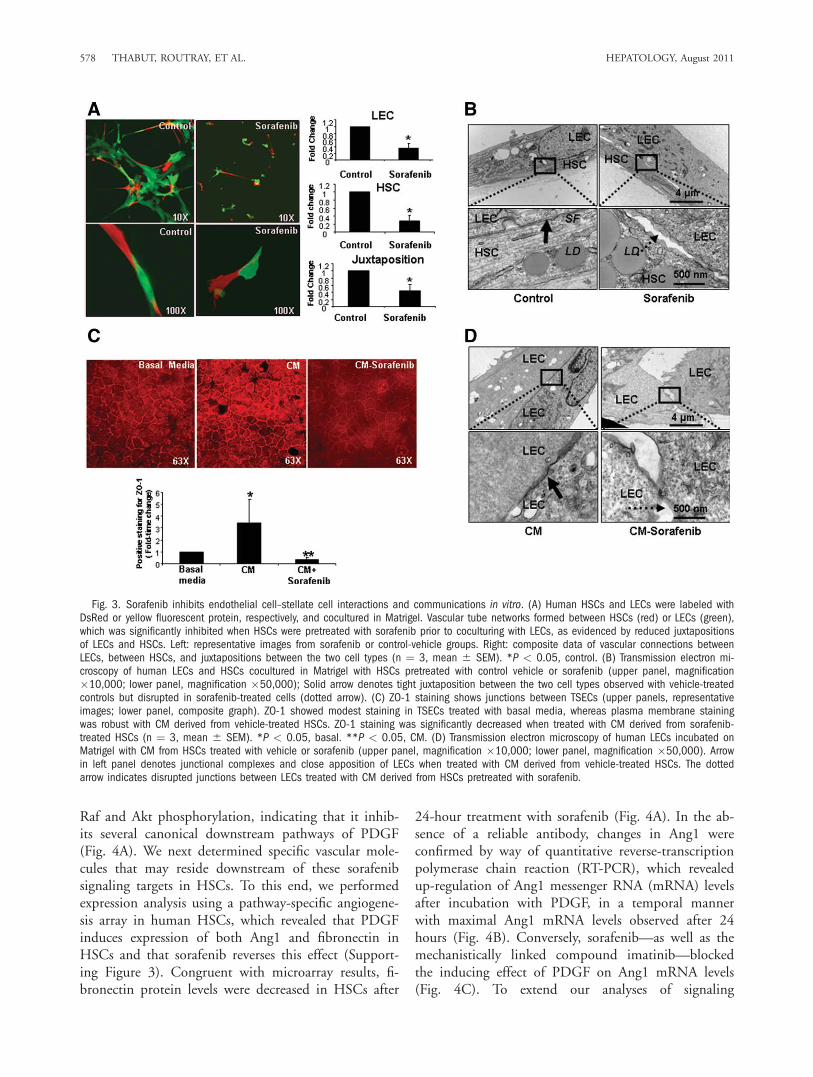
actions Constitutes a Key Cellular MechanismUnderlying the Beneficial Effect of Sorafenib on Vas-cular Restitution. Because sorafenib reverses vasculardefects that are induced during liver injury in vivo, wenext investigated cellular mechanisms underlying theseeffects using a reconstituted cell system composed ofhuman-derived HSCs and LECs, cell populations thatare responsible for matrix and vessel changes in responseto injury. Because fibrotic sinusoids are associated withincreased HSC wrapping of LEC-lined sinusoids,2 wespecifically examined whether sorafenib reverses this pro-cess. We incubated human HSCs with sorafenib or vehi-cle for 24 hours and transferred these cells into coculturein Matrigel with human LECs. Each cell type was labeledwith either yellow fluorescent protein or DsRed plasmidsto distinguish them and facilitate monitoring of theirbehavior. Using this optimized cell culture system, wefound that HSCs and LECs formed robust vascular net-works where both cells juxtapose and form wrapping con-tacts; however, HSCs pretreated with sorafenib showed astark contrast with compromised ability of HSCs to
HEPATOLOGY, Vol. 54, No. 2, 2011 THABUT, ROUTRAY, ET AL. 575
effectively establish wrapping interactions with LECs inthis assay (Fig. 3A). These fluorescent microscopy experi-ments were complemented with time-lapse video micros-copy to observe temporal kinetics of these observations inreal time ( Supporting Fig 1A to be the control one, andSupporting Fig 1B to be the Sorafenib one—movie). Fur-thermore, reduced juxtaposition of sorafenib-treated HSCswith LECs was confirmed by way of electron microscopy,which revealed increased gaps and decreased appositionbetween LECs and HSCs when HSCs were preincubatedwith sorafenib (Fig. 3B). These in vitro assays using com-binations of light and electron microscopy as well as time-lapse real-time imaging suggest that disruption of exuber-ant endothelial-to-stellate cell wrapping and contacts formpart of the cellular mechanisms underlying the beneficialeffect of sorafenib on vascular structure.Sorafenib Disrupts Endothelial-to-Endothelial
Cell Junctional Interactions In Vitro. In addition to
increased wrapping of contractile stellate cells aroundendothelial cells, vascular remodeling in cirrhosis isalso typified by increased assembly of endothelial–en-dothelial cell junctions that lead to cell motility andangiogenesis. This is in contradistinction to normal he-patic sinusoids, which are discontinuous with limitedendothelial–endothelial cell interactions. Therefore, wenext investigated whether treatment of HSCs with sor-afenib regulates junctional structures between LECsand angiogenesis. To test this model, we plated andcultured endothelial cells (TSECs) under conditions inwhich they form prominent junctions and incubatedthem with CM derived from HSCs pretreated eitherwith sorafenib or control vehicle. Cells were immuno-stained with a ZO-1 antibody to label junctional struc-tures between endothelial cells. We found that ZO-1staining was prominent in TSECs incubated with CMderived from vehicle-treated HSCs, whereas staining
Fig. 1. Sorafenib attenuates injury-induced increases in liver stiffness/matrix deposition in vivo. (A,B) Sham-operated and BDL rats treated eitherwith vehicle or sorafenib were subjected to MRE at 4 weeks, and changes in liver stiffness were assessed; quantitative analysis (A) and images fromrepresentative animals (B) are shown (upper panels, magnetic resonance images; lower panels, MRE images with stiffness-colored heat map). Vehi-cle-treated BDL rats showed significant increase in liver stiffness compared with sham-operated rats. This change was significantly attenuated inBDL rats receiving sorafenib (n > 4, mean 6 SEM). *P < 0.05, sham/vehicle. **P < 0.05 , BDL/vehicle. (C,D) Sirius red staining in sham-oper-ated and BDL rats treated with sorafenib or vehicle; quantitative analysis (C) and representative images (D) are shown. Sorafenib attenuated matrixdeposition in BDL rats compared with vehicle-treated rats (n > 4, mean 6 SEM). *P < 0.05, sham/vehicle.**P < 0.05, BDL/vehicle.
576 THABUT, ROUTRAY, ET AL. HEPATOLOGY, August 2011
was significantly decreased in cells incubated withmedia derived from sorafenib-stimulated HSCs (Fig.3C), suggesting that this drug modulates formation ofcell–cell junctions among endothelia. These results ini-tially observed in TSECs were also confirmed in pri-mary murine LECs (Supporting Fig. 2). We usedtransmission electron microscopy, which showed anincreased number of intercellular junctions betweenhuman LECs incubated with CM derived from vehi-cle-treated HSCs (Fig. 3D). In contrast, junctionalstructures revealed by this high-resolution techniquewere markedly reduced when LECs were incubatedwith CM derived from sorafenib-stimulated HSCs(Fig. 3D). Thus, these data demonstrate that sorafenibmodulates the structural basis of junctional complexesthat can be formed between endothelial cells, which
are the foundation of vascular remodeling, and subse-quently led us to define signaling cascades that canmodulate these processes at the molecular level.Sorafenib Regulates Vascular and Matrix
Remodeling Through Complementary SignalingPathways. Prior studies have delineated a critical roleof PDGF on vascular function, especially its ability toregulate pericytic and myofibroblastic mural wall cellssuch as HSCs through PDGF receptor b (PDGFR-b).3,19 As a first step to better define effects of sorafe-nib on PDGFR signaling in HSCs, we examined theintegrity of this signaling pathway in human-derivedHSCs stimulated with PDGF and/or sorafenib. Con-gruent with its function as a tyrosine kinase inhibitor,sorafenib abolished PDGF-induced PDGFR-b phos-phorylation. Sorafenib also inhibited PDGF-induced
Fig. 2. Sorafenib attenuates injury-induced hepatic vascular remodeling in vivo. (A,B) vWF immunofluorescence of the liver was performed insham-operated and BDL rats treated with vehicle or sorafenib; quantitative analysis (A) and representative images (B) are shown. Analysis depictsattenuated neocapillary formation in BDL/sorafenib rats compared with BDL/vehicle rats (n > 3, mean 6 SEM). *P < 0.05, BDL/vehicle. (C,D)Sham-operated and BDL rats treated with vehicle or sorafenib were injected with Microfil; quantitative analysis of vascular volume/liver ratio and num-ber of interbranches (C) and images from representative animals (D) are shown. Specific liver lobes were scanned to measure vascular volume [leftand middle upper panels in (D)]. Images were also digitally reconstructed to quantify vascular branching [right upper panel in (D)]. Red color indi-cates selected vascular trunk and associated vascular volume. BDL rats treated with vehicle had increased total liver vascular space, as measured bythe vascular volume/liver volume ratio, and higher interbranch numbers. Sorafenib in BDL rats was associated with a significant decrease of total vas-cular space (n > 4, mean 6 SEM; *P ¼ 0.01, BDL/vehicle) and decrease of primary/secondary/tertiary branches (*P < 0.05, BDL/vehicle).
HEPATOLOGY, Vol. 54, No. 2, 2011 THABUT, ROUTRAY, ET AL. 577
Raf and Akt phosphorylation, indicating that it inhib-its several canonical downstream pathways of PDGF(Fig. 4A). We next determined specific vascular mole-cules that may reside downstream of these sorafenibsignaling targets in HSCs. To this end, we performedexpression analysis using a pathway-specific angiogene-sis array in human HSCs, which revealed that PDGFinduces expression of both Ang1 and fibronectin inHSCs and that sorafenib reverses this effect (Support-ing Figure 3). Congruent with microarray results, fi-bronectin protein levels were decreased in HSCs after
24-hour treatment with sorafenib (Fig. 4A). In the ab-sence of a reliable antibody, changes in Ang1 wereconfirmed by way of quantitative reverse-transcriptionpolymerase chain reaction (RT-PCR), which revealedup-regulation of Ang1 messenger RNA (mRNA) levelsafter incubation with PDGF, in a temporal mannerwith maximal Ang1 mRNA levels observed after 24hours (Fig. 4B). Conversely, sorafenib—as well as themechanistically linked compound imatinib—blockedthe inducing effect of PDGF on Ang1 mRNA levels(Fig. 4C). To extend our analyses of signaling
Fig. 3. Sorafenib inhibits endothelial cell–stellate cell interactions and communications in vitro. (A) Human HSCs and LECs were labeled withDsRed or yellow fluorescent protein, respectively, and cocultured in Matrigel. Vascular tube networks formed between HSCs (red) or LECs (green),which was significantly inhibited when HSCs were pretreated with sorafenib prior to coculturing with LECs, as evidenced by reduced juxtapositionsof LECs and HSCs. Left: representative images from sorafenib or control-vehicle groups. Right: composite data of vascular connections betweenLECs, between HSCs, and juxtapositions between the two cell types (n ¼ 3, mean 6 SEM). *P < 0.05, control. (B) Transmission electron mi-croscopy of human LECs and HSCs cocultured in Matrigel with HSCs pretreated with control vehicle or sorafenib (upper panel, magnification�10,000; lower panel, magnification �50,000); Solid arrow denotes tight juxtaposition between the two cell types observed with vehicle-treatedcontrols but disrupted in sorafenib-treated cells (dotted arrow). (C) ZO-1 staining shows junctions between TSECs (upper panels, representativeimages; lower panel, composite graph). ZO-1 showed modest staining in TSECs treated with basal media, whereas plasma membrane stainingwas robust with CM derived from vehicle-treated HSCs. ZO-1 staining was significantly decreased when treated with CM derived from sorafenib-treated HSCs (n ¼ 3, mean 6 SEM). *P < 0.05, basal. **P < 0.05, CM. (D) Transmission electron microscopy of human LECs incubated onMatrigel with CM from HSCs treated with vehicle or sorafenib (upper panel, magnification �10,000; lower panel, magnification �50,000). Arrowin left panel denotes junctional complexes and close apposition of LECs when treated with CM derived from vehicle-treated HSCs. The dottedarrow indicates disrupted junctions between LECs treated with CM derived from HSCs pretreated with sorafenib.
578 THABUT, ROUTRAY, ET AL. HEPATOLOGY, August 2011
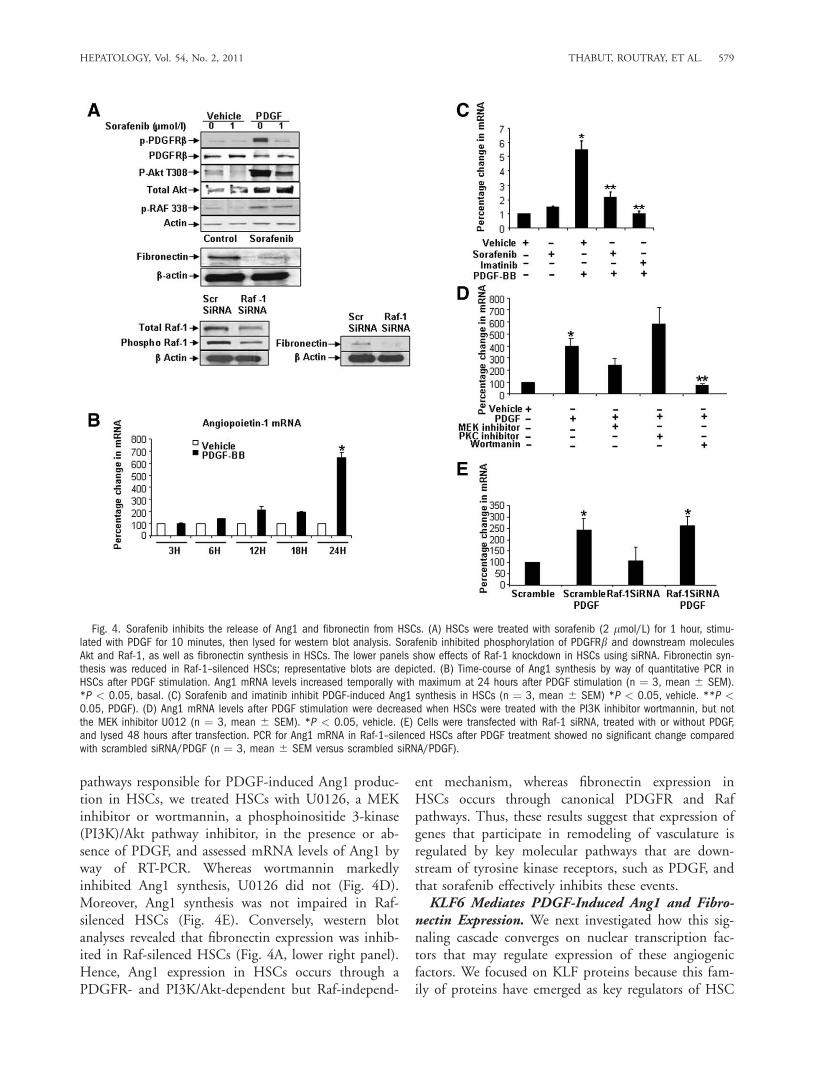
pathways responsible for PDGF-induced Ang1 produc-tion in HSCs, we treated HSCs with U0126, a MEKinhibitor or wortmannin, a phosphoinositide 3-kinase(PI3K)/Akt pathway inhibitor, in the presence or ab-sence of PDGF, and assessed mRNA levels of Ang1 byway of RT-PCR. Whereas wortmannin markedlyinhibited Ang1 synthesis, U0126 did not (Fig. 4D).Moreover, Ang1 synthesis was not impaired in Raf-silenced HSCs (Fig. 4E). Conversely, western blotanalyses revealed that fibronectin expression was inhib-ited in Raf-silenced HSCs (Fig. 4A, lower right panel).Hence, Ang1 expression in HSCs occurs through aPDGFR- and PI3K/Akt-dependent but Raf-independ-
ent mechanism, whereas fibronectin expression inHSCs occurs through canonical PDGFR and Rafpathways. Thus, these results suggest that expression ofgenes that participate in remodeling of vasculature isregulated by key molecular pathways that are down-stream of tyrosine kinase receptors, such as PDGF, andthat sorafenib effectively inhibits these events.KLF6 Mediates PDGF-Induced Ang1 and Fibro-
nectin Expression. We next investigated how this sig-naling cascade converges on nuclear transcription fac-tors that may regulate expression of these angiogenicfactors. We focused on KLF proteins because this fam-ily of proteins have emerged as key regulators of HSC
Fig. 4. Sorafenib inhibits the release of Ang1 and fibronectin from HSCs. (A) HSCs were treated with sorafenib (2 lmol/L) for 1 hour, stimu-lated with PDGF for 10 minutes, then lysed for western blot analysis. Sorafenib inhibited phosphorylation of PDGFRb and downstream moleculesAkt and Raf-1, as well as fibronectin synthesis in HSCs. The lower panels show effects of Raf-1 knockdown in HSCs using siRNA. Fibronectin syn-thesis was reduced in Raf-1–silenced HSCs; representative blots are depicted. (B) Time-course of Ang1 synthesis by way of quantitative PCR inHSCs after PDGF stimulation. Ang1 mRNA levels increased temporally with maximum at 24 hours after PDGF stimulation (n ¼ 3, mean 6 SEM).*P < 0.05, basal. (C) Sorafenib and imatinib inhibit PDGF-induced Ang1 synthesis in HSCs (n ¼ 3, mean 6 SEM) *P < 0.05, vehicle. **P <0.05, PDGF). (D) Ang1 mRNA levels after PDGF stimulation were decreased when HSCs were treated with the PI3K inhibitor wortmannin, but notthe MEK inhibitor U012 (n ¼ 3, mean 6 SEM). *P < 0.05, vehicle. (E) Cells were transfected with Raf-1 siRNA, treated with or without PDGF,and lysed 48 hours after transfection. PCR for Ang1 mRNA in Raf-1–silenced HSCs after PDGF treatment showed no significant change comparedwith scrambled siRNA/PDGF (n ¼ 3, mean 6 SEM versus scrambled siRNA/PDGF).
HEPATOLOGY, Vol. 54, No. 2, 2011 THABUT, ROUTRAY, ET AL. 579
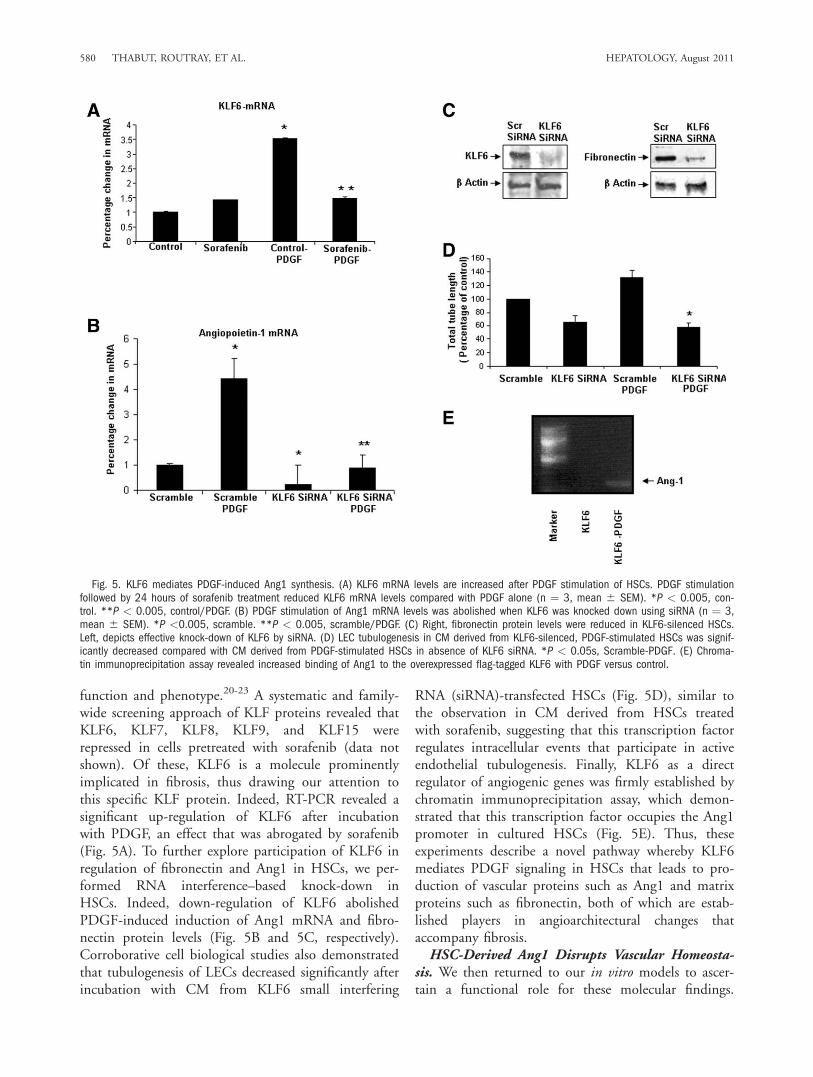
function and phenotype.20-23 A systematic and family-wide screening approach of KLF proteins revealed thatKLF6, KLF7, KLF8, KLF9, and KLF15 wererepressed in cells pretreated with sorafenib (data notshown). Of these, KLF6 is a molecule prominentlyimplicated in fibrosis, thus drawing our attention tothis specific KLF protein. Indeed, RT-PCR revealed asignificant up-regulation of KLF6 after incubationwith PDGF, an effect that was abrogated by sorafenib(Fig. 5A). To further explore participation of KLF6 inregulation of fibronectin and Ang1 in HSCs, we per-formed RNA interference–based knock-down inHSCs. Indeed, down-regulation of KLF6 abolishedPDGF-induced induction of Ang1 mRNA and fibro-nectin protein levels (Fig. 5B and 5C, respectively).Corroborative cell biological studies also demonstratedthat tubulogenesis of LECs decreased significantly afterincubation with CM from KLF6 small interfering
RNA (siRNA)-transfected HSCs (Fig. 5D), similar tothe observation in CM derived from HSCs treatedwith sorafenib, suggesting that this transcription factorregulates intracellular events that participate in activeendothelial tubulogenesis. Finally, KLF6 as a directregulator of angiogenic genes was firmly established bychromatin immunoprecipitation assay, which demon-strated that this transcription factor occupies the Ang1promoter in cultured HSCs (Fig. 5E). Thus, theseexperiments describe a novel pathway whereby KLF6mediates PDGF signaling in HSCs that leads to pro-duction of vascular proteins such as Ang1 and matrixproteins such as fibronectin, both of which are estab-lished players in angioarchitectural changes thataccompany fibrosis.HSC-Derived Ang1 Disrupts Vascular Homeosta-
sis. We then returned to our in vitro models to ascer-tain a functional role for these molecular findings.
Fig. 5. KLF6 mediates PDGF-induced Ang1 synthesis. (A) KLF6 mRNA levels are increased after PDGF stimulation of HSCs. PDGF stimulationfollowed by 24 hours of sorafenib treatment reduced KLF6 mRNA levels compared with PDGF alone (n ¼ 3, mean 6 SEM). *P < 0.005, con-trol. **P < 0.005, control/PDGF. (B) PDGF stimulation of Ang1 mRNA levels was abolished when KLF6 was knocked down using siRNA (n ¼ 3,mean 6 SEM). *P <0.005, scramble. **P < 0.005, scramble/PDGF. (C) Right, fibronectin protein levels were reduced in KLF6-silenced HSCs.Left, depicts effective knock-down of KLF6 by siRNA. (D) LEC tubulogenesis in CM derived from KLF6-silenced, PDGF-stimulated HSCs was signif-icantly decreased compared with CM derived from PDGF-stimulated HSCs in absence of KLF6 siRNA. *P < 0.05s, Scramble-PDGF. (E) Chroma-tin immunoprecipitation assay revealed increased binding of Ang1 to the overexpressed flag-tagged KLF6 with PDGF versus control.
580 THABUT, ROUTRAY, ET AL. HEPATOLOGY, August 2011

Ang1 contributes importantly to vessel maturation.24
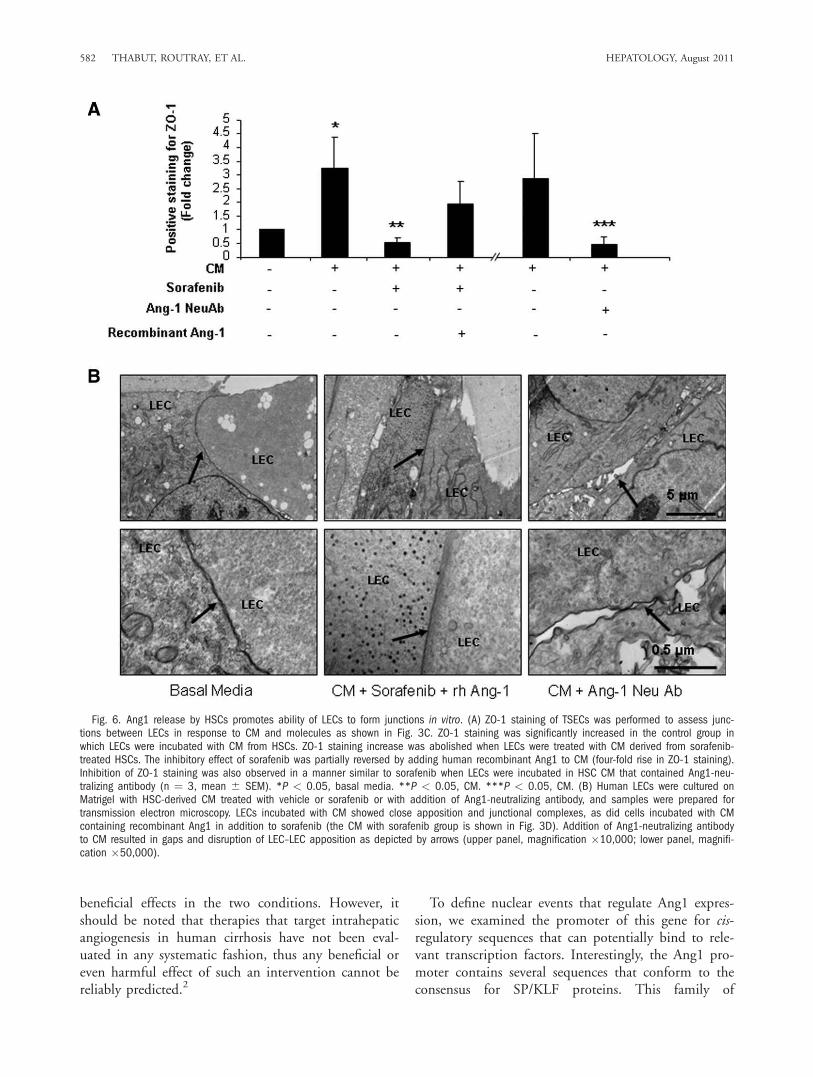
However, excessive Ang1 may disrupt normal vesselsand lead to vascular restructuring and angiogenesis,which characterizes cirrhosis. Therefore, we first inves-tigated whether Ang1 may increase junctional struc-tures between LECs. We plated TSECs, an LEC cellline that forms exuberant junctions at confluence, andimmunostained cells with ZO-1Ab to identify junc-tional structures. To specifically implicate Ang1 in thisprocess, in some experimental groups we examinedZO-1 staining after incubating TSECs with HSC CMcontaining Ang1-neutralizing antibody or supplemen-tal recombinant Ang1. As shown in Fig. 3C, ZO-1staining was significantly increased in the CM-treatedgroup; this effect was abolished when treated with CMderived from sorafenib-treated HSCs (Fig. 6A). Addi-tionally, sorafenib-induced inhibition of junction for-mation between cells was reversed upon addition ofrecombinant Ang1 in HSC-derived media (Fig. 6A). Asimilar pattern to sorafenib was observed when TSECswere incubated with CM pretreated with Ang1-neu-tralizing antibody (Fig. 6A). Those findings were cor-roborated by transmission electron microscopy whichalso revealed a reduction in junctional complexes inLEC upon addition of Ang1-neutralizing antibody toHSC-derived CM (Fig. 6B). These morphologicalanalyses also revealed a reversal of sorafenib-inducedinhibition of junctional complexes between cells byaddition of recombinant Ang1 to HSC CM (Fig. 6B).Thus, these results demonstrate that HSC-derivedAng1 promotes intercellular junctions in LECs, eventsthat could contribute to sinusoidal remodeling andangiogenesis that characterizes fibrotic vasculature.Finally, we extended these cell morphological obser-
vations using functional assays of vascular maturationthat require LEC junctional complexes. Congruentwith the morphological studies, Ang1-neutralizingantibody attenuated tubulogenesis of LECs that occursin response to CM from HSC-stimulated with PDGF(Fig. 7A). Similarly, LEC tubulogenesis was restoredby adding recombinant Ang1 to CM derived from sor-afenib-stimulated HSCs, highlighting the decisive roleof Ang1 and its regulation by sorafenib in this three-dimensional tubulogenic process (Fig. 7B). Theseexperiments were complemented by chemotactic assaysthat require cellular guidance cues and cell motilitymachinery. In this regard, CM from sorafenib-stimu-lated HSCs or those treated with Ang1-neutralizingantibody significantly reduced the ability of LECs tomigrate compared with relevant control groups (Fig.7C). Also, similar to the three-dimensional tubulationstudies, addition of recombinant Ang1 to CM derived
from sorafenib-treated HSCs rescued LEC migration(Fig. 7C). These findings provide a functional hierar-chy to the pathways described here by indicating thatAng1 is both required and sufficient for effects on vas-cular remodeling.
Discussion
The development of cirrhosis requires changes inmatrix composition and turnover as well as conspicu-ous changes in intrahepatic vasculature that requireorchestrated interaction between nonparenchymal livercells, especially endothelial cells and stellate cells.These vascular changes significantly contribute to themorbid complication of portal hypertension thataccompanies advanced fibrosis. In this study, wefocused on (1) identifying novel cellular and molecularpathways underlying angio-matrix changes that occurduring liver fibrosis and (2) defining how sorafenib, acompound that shows promising clinical use inpatients with cirrhosis and liver cancer, affects thesepathways. In this regard, the present study reveals sev-eral novel cellular and molecular phenomena that shedfurther light on angioarchitectural changes that accom-pany fibrosis (Fig. 8). First, we demonstrate that HSCssecrete Ang1, which behaves as a key contributor to fi-brosis-associated vascular changes. We show that exces-sive HSC-derived Ang1 disrupts sinusoidal homeostasisby promoting increased wrapping interactions betweenHSCs and LECs as well as increased junctional con-nections among LECs. These phenomena culminate ina sinusoidal remodeling process that enhances HSCcontraction around sinusoids as well as increasedangiogenesis. Surprisingly, Ang1 production requiresPI3K/Akt activation, though it is independent of Raf,which is the classical target of sorafenib in hepatomacells.4,25 This finding demonstrates that sorafenib usesdistinct pathways to exert its changes in epithelial ver-sus mesenchymal cells. These vascular changes are alsocoordinated with matrix remodeling as shown by anincrease in Raf-dependent fibronectin production,which like Ang1 production relies on integrity of theKLF6 transcriptional pathway, thus revealing a remark-able coordination of vascular and matrix changes thatcontribute to cirrhosis. Finally, we provide clear evi-dence that the multikinase inhibitor sorafenib inhibitsthe KLF6–Ang1–fibronectin molecular triad, therebyattenuating angioarchitectural changes that typify cir-rhosis. These observations also suggest that the func-tion of sorafenib in cancer and cirrhosis might havedistinct differences that can be exploited for tailoringdifferent concentration responses that can achieve
HEPATOLOGY, Vol. 54, No. 2, 2011 THABUT, ROUTRAY, ET AL. 581
beneficial effects in the two conditions. However, itshould be noted that therapies that target intrahepaticangiogenesis in human cirrhosis have not been eval-uated in any systematic fashion, thus any beneficial oreven harmful effect of such an intervention cannot bereliably predicted.2
To define nuclear events that regulate Ang1 expres-sion, we examined the promoter of this gene for cis-regulatory sequences that can potentially bind to rele-vant transcription factors. Interestingly, the Ang1 pro-moter contains several sequences that conform to theconsensus for SP/KLF proteins. This family of
Fig. 6. Ang1 release by HSCs promotes ability of LECs to form junctions in vitro. (A) ZO-1 staining of TSECs was performed to assess junc-tions between LECs in response to CM and molecules as shown in Fig. 3C. ZO-1 staining was significantly increased in the control group inwhich LECs were incubated with CM from HSCs. ZO-1 staining increase was abolished when LECs were treated with CM derived from sorafenib-treated HSCs. The inhibitory effect of sorafenib was partially reversed by adding human recombinant Ang1 to CM (four-fold rise in ZO-1 staining).Inhibition of ZO-1 staining was also observed in a manner similar to sorafenib when LECs were incubated in HSC CM that contained Ang1-neu-tralizing antibody (n ¼ 3, mean 6 SEM). *P < 0.05, basal media. **P < 0.05, CM. ***P < 0.05, CM. (B) Human LECs were cultured onMatrigel with HSC-derived CM treated with vehicle or sorafenib or with addition of Ang1-neutralizing antibody, and samples were prepared fortransmission electron microscopy. LECs incubated with CM showed close apposition and junctional complexes, as did cells incubated with CMcontaining recombinant Ang1 in addition to sorafenib (the CM with sorafenib group is shown in Fig. 3D). Addition of Ang1-neutralizing antibodyto CM resulted in gaps and disruption of LEC–LEC apposition as depicted by arrows (upper panel, magnification �10,000; lower panel, magnifi-cation �50,000).
582 THABUT, ROUTRAY, ET AL. HEPATOLOGY, August 2011
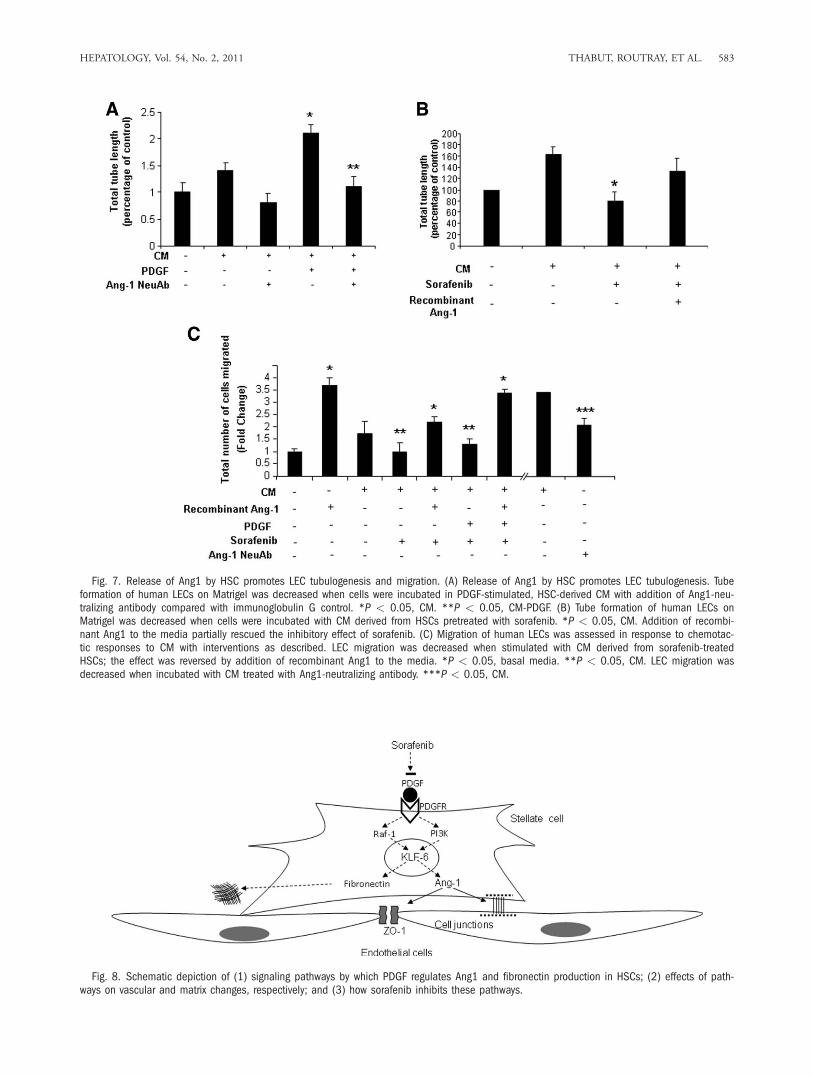
Fig. 7. Release of Ang1 by HSC promotes LEC tubulogenesis and migration. (A) Release of Ang1 by HSC promotes LEC tubulogenesis. Tubeformation of human LECs on Matrigel was decreased when cells were incubated in PDGF-stimulated, HSC-derived CM with addition of Ang1-neu-tralizing antibody compared with immunoglobulin G control. *P < 0.05, CM. **P < 0.05, CM-PDGF. (B) Tube formation of human LECs onMatrigel was decreased when cells were incubated with CM derived from HSCs pretreated with sorafenib. *P < 0.05, CM. Addition of recombi-nant Ang1 to the media partially rescued the inhibitory effect of sorafenib. (C) Migration of human LECs was assessed in response to chemotac-tic responses to CM with interventions as described. LEC migration was decreased when stimulated with CM derived from sorafenib-treatedHSCs; the effect was reversed by addition of recombinant Ang1 to the media. *P < 0.05, basal media. **P < 0.05, CM. LEC migration wasdecreased when incubated with CM treated with Ang1-neutralizing antibody. ***P < 0.05, CM.
Fig. 8. Schematic depiction of (1) signaling pathways by which PDGF regulates Ang1 and fibronectin production in HSCs; (2) effects of path-ways on vascular and matrix changes, respectively; and (3) how sorafenib inhibits these pathways.
HEPATOLOGY, Vol. 54, No. 2, 2011 THABUT, ROUTRAY, ET AL. 583

transcription factors is composed of 24 different pro-teins, with many emerging as key regulators of gastro-intestinal and hepatic cell biology and pathobiology.26
Indeed, we performed a family-wide screen to definewhich KLF protein regulates Ang1 expression. Usingthis approach, we found that KLF6 occupies the pro-moter of Ang1 and that Ang1 expression is inhibitedby siRNA knock-down of KLF6. This is significantbecause KLF6 is the only member of this family whosefunction in liver fibrosis has been well established. Forinstance, KLF6 has been associated to liver woundhealing.22,27 Therefore, these findings, when takenwithin the context of prior studies, indicate that KLF6may be an operator of angio-architectural changes thataccompany fibrosis by virtue of its ability to drive amembrane-to-nucleus pathway in HSCs that beginswith activation of PDGFR and culminates in bindingof KLF6 to the Ang1 promoter.Our study employed state-of-the-art imaging techni-
ques such as MRE and micro-CT for monitoringchanges that occur microscopically but affect the organas a whole. In this regard, our study can also be con-sidered as a preclinical assessment of these innovativemethodologies, the importance of which is under-scored by the fact that MRE is gaining increasingattention as a potentially useful diagnostic modality innoninvasive assessment of liver fibrosis. Interestingly,our MRE assessment of rat liver during BDL-inducedfibrosis revealed increased stiffness. Previous studieshave indicated that such an increased stiffness reflectschanges in matrix remodeling.28 However, increasingevidence (including the results reported in this study)indicates that stiffness also reflects other processes thatfrequently accompany matrix deposition, such asinflammation, edema, and even vascular structure andportal pressure changes.15 Indeed, complementary highresolution micro-CT allowed us to determine thatabnormal MRE signals were accompanied by promi-nent vascular changes. These observations were com-plimented by histological examinations that corrobo-rated increases in vascular density after BDL. Thus,this powerful combination of MRE and micro-CTallowed us to resolve two different important compo-nents of liver cirrhosis, namely matrix changes and vas-cular remodeling.Some (though not all) recent studies suggest that
therapeutic approaches that target aberrant vasculaturestructure in cirrhosis could have a beneficial effect onportal hypertension.18,29-31 Congruent with this idea,it has been recently proposed that multikinase receptortyrosine kinase inhibitors such as sorafenib decreaseportal hypertension in animal models of cirrho-
sis,18,29,30 although detailed molecular mechanisms re-sponsible for this effect have warranted further investi-gation. We here extend our current understanding ofhow this targeted therapy drug may have a potentialbeneficial role in treatment of liver fibrosis and portalhypertension by virtue of its effects on sinusoidal cellcross-talk centered on Ang1.32-34 Indeed, HSCs secreteAng1 to promote formation of junctional complexesbetween LECs, a key step for angiogenesis and vascularrestructuring within a mechanically stressed fibroticmicroenvironment.1,18,35 Importantly, the capillaryresponse regulated by Ang1 in diseased liver in vivoappears to be congruent with molecular mechanismsdescribed here. For example, although normal liver si-nusoids are characterized by a discontinuous pheno-type, in cirrhosis these delicate vascular structuresundergo what is commonly referred to as ‘‘capillariza-tion,’’ with more durable stellate cell coverage of moreclosely interconnected endothelial cells. These pheno-typic changes coincide with known functions of Ang1as a stabilizer of vessels.36 Therefore, our results mayalso help to explain how sinusoidal vasculature adoptsdistinct phenotypic changes in cirrhosis and use thisknowledge for designing future therapeutic interven-tions targeting this pathway. In total, this study under-scores the importance of considering both vasculatureand matrix as combined therapeutic targets of thera-pies aimed to ameliorate cirrhosis and itscomplications.
Acknowledgments: We thank Helen Hendricksonfor managerial support in the laboratory and TerriJohnson for secretarial assistance.
References1. Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher
CH, et al. Hepatic stellate cells secrete angiopoietin 1 that inducesangiogenesis in liver fibrosis. Gastroenterology 2008;135:1729-1738.
2. Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodelingin chronic liver disease: New targets for the treatment of portal hyper-tension? J Hepatol 2010;53:976-980.
3. Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derivedgrowth factor signaling through ephrin-b2 regulates hepatic vascularstructure and function. Gastroenterology 2008;135:671-679.
4. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al.Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-390.
5. Martinelli E, Troiani T, Morgillo F, Rodolico G, Vitagliano D, MorelliMP, et al. Synergistic antitumor activity of sorafenib in combinationwith epidermal growth factor receptor inhibitors in colorectal and lungcancer cells. Clin Cancer Res 2010;16:4990-5001.
6. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA,et al. Discovery and development of sorafenib: a multikinase inhibitorfor treating cancer. Nat Rev Drug Discov 2006;5:835-844.
7. Huebert RC, Jagavelu K, Liebl AF, Huang BQ, Splinter PL, LarussoNF, et al. Immortalized liver endothelial cells: a cell culture model forstudies of motility and angiogenesis. Lab Invest 2010;90:1770-1781.
584 THABUT, ROUTRAY, ET AL. HEPATOLOGY, August 2011

8. LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP,et al. Angiogenesis-independent endothelial protection of liver: role ofVEGFR-1. Science 2003;299:890-893.
9. McNeil E, Capaldo CT, Macara IG. Zonula occludens-1 function inthe assembly of tight junctions in Madin-Darby canine kidney epithe-lial cells. Mol Biol Cell 2006;17:1922-1932.
10. Das A, Fernandez-Zapico ME, Cao S, Yao J, Fiorucci S, Hebbel RP,et al. Disruption of an SP2/KLF6 repression complex by SHP isrequired for farnesoid X receptor-induced endothelial cell migration. JBiol Chem 2006;281:39105-39113.
11. Rockey DC, Chung JJ. Reduced nitric oxide production by endothelialcells in cirrhotic rat liver: endothelial dysfunction in portal hyperten-sion. Gastroenterology 1998;114:344-351.
12. Jorgensen SM, Demirkaya O, Ritman EL. Three-dimensional imagingof vasculature and parenchyma in intact rodent organs with X-raymicro-CT. Am J Physiol 1998;275:H1103-H1114.
13. Muthupillai R, Lomas DJ, Rossman PJ, Greenleaf JF, Manduca A,Ehman RL. Magnetic resonance elastography by direct visualization ofpropagating acoustic strain waves. Science 1995;269:1854-1857.
14. Manduca A, Oliphant TE, Dresner MA, Mahowald JL, Kruse SA,Amromin E, et al. Magnetic resonance elastography: non-invasive map-ping of tissue elasticity. Med Image Anal 2001;5:237-254.
15. Talwalkar JA, Yin M, Fidler JL, Sanderson SO, Kamath PS, EhmanRL. Magnetic resonance imaging of hepatic fibrosis: emerging clinicalapplications. HEPATOLOGY 2008;47:332-342.
16. Jagavelu K, Routray C, Shergill U, O’Hara SP, Faubion W, Shah VH.Endothelial cell toll-like receptor 4 regulates fibrosis-associated angio-genesis in the liver. HEPATOLOGY 2010;52:590-601.
17. Dechene A, Sowa JP, Gieseler RK, Jochum C, Bechmann LP, El FoulyA, et al. Acute liver failure is associated with elevated liver stiffness andhepatic stellate cell activation. HEPATOLOGY 2010;52:1008-1016.
18. Tugues S, Fernandez-Varo G, Munoz-Luque J, Ros J, Arroyo V, RodesJ, et al. Antiangiogenic treatment with sunitinib ameliorates inflamma-tory infiltrate, fibrosis, and portal pressure in cirrhotic rats. HEPATOLOGY
2007;46:1919-1926.19. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interac-
tions. Circ Res 2005;97:512-523.20. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated
cellular response to tissue injury. J Biol Chem 2000;275:2247-2250.21. Rippe RA, Almounajed G, Brenner DA. Sp1 binding activity increases
in activated Ito cells. HEPATOLOGY 1995;22:241-251.22. Ratziu V, Lalazar A, Wong L, Dang Q, Collins C, Shaulian E, et al.
Zf9, a Kruppel-like transcription factor up-regulated in vivo duringearly hepatic fibrosis. Proc Natl Acad Sci U S A 1998;95:9500-9505.
23. Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN,et al. Integration of flow-dependent endothelial phenotypes by Krup-pel-like factor 2. J Clin Invest 2006;116:49-58.
24. Jain RK. Molecular regulation of vessel maturation. Nat Med 2003;9:685-693.
25. Iyer R, Fetterly G, Lugade A, Thanavala Y. Sorafenib: a clinical andpharmacologic review. Expert Opin Pharmacother 2010;11:1943-1955.
26. Miele L, Beale G, Patman G, Nobili V, Leathart J, Grieco A, et al.The Kruppel-like factor 6 genotype is associated with fibrosis in nonal-coholic fatty liver disease. Gastroenterology 2008;135:282-291.
27. Narla G, DiFeo A, Yao S, Banno A, Hod E, Reeves HL, et al. Targetedinhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostatecancer cell growth and spread. Cancer Res 2005;65:5761-5768.
28. Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, UemuraM, et al. Increased stiffness of the rat liver precedes matrix deposition:implications for fibrosis. Am J Physiol Gastrointest Liver Physiol 2007;293:G1147-G1154.
29. Hennenberg M, Trebicka J, Stark C, Kohistani AZ, Heller J, Sauer-bruch T. Sorafenib targets dysregulated Rho kinase expression and por-tal hypertension in rats with secondary biliary cirrhosis. Br J Pharmacol2009;157:258-270.
30. Mejias M, Garcia-Pras E, Tiani C, Miquel R, Bosch J, Fernandez M.Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocol-lateral circulations in portal hypertensive and cirrhotic rats. HEPATOLOGY
2009;49:1245-1256.31. Patsenker E, Popov Y, Stickel F, Schneider V, Ledermann M, Sagesser
H, et al. Pharmacological inhibition of integrin alphavbeta3 aggravatesexperimental liver fibrosis and suppresses hepatic angiogenesis. HEPATO-
LOGY 2009;50:1501-1511.32. Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angio-
poietin-1 in vascular protection. Circ Res 2006;98:1014-1023.33. Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas
L, et al. Angiopoietin-1 is an antipermeability and anti-inflammatoryagent in vitro and targets cell junctions. Circ Res 2000;87:603-607.
34. Fukuhara S, Sako K, Noda K, Nagao K, Miura K, Mochizuki N. Tie2is tied at the cell-cell contacts and to extracellular matrix by angiopoie-tin-1. Exp Mol Med 2009;41:133-139.
35. Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, et al.Upregulation of proinflammatory and proangiogenic cytokines by lep-tin in human hepatic stellate cells. HEPATOLOGY 2005;42:1339-1348.
36. Wang YL, Hui YN, Guo B, Ma JX. Strengthening tight junctions ofretinal microvascular endothelial cells by pericytes under normoxia andhypoxia involving angiopoietin-1 signal way. Eye (Lond) 2007;21:1501-1510.
HEPATOLOGY, Vol. 54, No. 2, 2011 THABUT, ROUTRAY, ET AL. 585
Related Documents











