Comparative characterization of oligomeric precursors intended for injectable implants Alejandro López a *, Cecilia Persson a , Jöns Hilborn b and Ramiro Rojas b The use of injectable materials is a simple approach for drug delivery and tissue repair, in, e.g. minimally invasive surgery applications. If these materials are used past their glass transition temperature and have a low viscosity, they will be able to flow while delivered in situ. Whether these materials are to be used as low viscosity drug carriers or further crosslinked for tissue repair, there is a need for a better understanding of their handling properties. In this study, oligo(trimethylene carbonate) (oTMC) and oligo[D,L-lactide-co-(«-caprolactone)] (oDLLA-co-CL) of various molecular weights within a relevant injectability range were synthesized via ring-opening polymerization. The materials were comparatively characterized by 1 H NMR spectroscopy, differential scanning calorimetry, gel permeation chromatography, and rheological measurements. After comparing the viscosities and molecular weights of the materials, it was concluded that oDLLA-co-CLs were, gener- ally, better suited as an injectable in situ crosslinking network, whereas oTMCs were found to be better candidates as injectable drug carriers. This study provides useful data and guidelines on the use of these and other similar oligomers intended for injectable implants. Copyright © 2012 John Wiley & Sons, Ltd. Keywords: oligomer(s); polyester(s); polycarbonate(s); rheology; injectable implant(s) INTRODUCTION Injectable biomaterial systems are especially desirable in mini- mally invasive clinical treatments, due to their ease of handling and application, when restoration is required within a short period of time; e.g. vertebroplasty or kyphoplasty for vertebral augmentation, [1] or for filling defects with complex shapes. These types of materials can also be a vehicle for in situ delivery of drugs, growth factors, or cells; e.g. for intervertebral disc restoration. [2] Cyclic monomers such as trimethylene carbonate (TMC), D,L-lactide (DLLA), and e-caprolactone (e-CL), can be homo- and copolymerized via ring-opening polymerization, co-initiated by a diol, to form linear oligomers. These oligomers have a low glass transition temperature (T g ), low viscosity, and are able to flow at room temperature. They can readily be loaded with drugs to be used as delivery agents, or can further be functionalized with, e.g. acrylic or methacrylic end-groups to give them the ability to react in situ, via redox or UV radical initiation, to form covalently crosslinked networks. Degradable, in situ curing polymers of low molecular weight, have been thoroughly described in a series of US patents by Dunn et al. since the early 90s. [3] Since then, these types of materials have been potentially available for several medical applications requiring injectability, in situ curing chemistry, drug reservoir capabilities, tailorable mechanical properties, degradability, and biocompatibility. Poly(TMC), PTMC, is a degradable aliphatic polycarbonate that predominantly undergoes enzymatic degradation in vivo into non- acidic products. [4] Compression-molded high molecular weight PTMCs have been shown to have a Young’ s modulus of 3.0–6.8 MPa, a yield strength of 0.5–2.3 MPa, and a strain at break of 230– 1250%. [5] PTMC copolymerized with glycolide and p-dioxanone has been used as flexible suture materials (Maxon ™ and BioSyn ™ ), as well as tacks and screws for orthopedics (Acufex ™ ). [6] Additionally, it has been investigated as drug-releasing agent when copolymer- ized or blended with adipic anhydride. [7] PTMC has long been underestimated for medical device applications (i.e. load bearing) due to its relatively poor mechanical properties [8] and tackiness; however, the ongoing development of injectable biomaterials may find it advantageous for applications where the local pH must be maintained over a certain level during degradation. For instance, Amsden et al. [9] have shown the potential of low molecular weight PTMC for the localized delivery of the acid sensitive, vascular endo- thelial growth factor. Another important advantage of PTMC is that it undergoes surface degradation unlike polyesters, which undergo bulk degradation followed by swelling and collapse. [6] Furthermore, the surface degradation of PTMC is accelerated by the presence of certain endogenous enzymes, e.g. lipase. [4] On the other hand, the acidic products from the degradation of polyesters can auto-catalyze their hydrolysis, making the degradation of polyesters faster than that of the PTMC; this occurs in spite of the higher electrophilic character of the carbonyl carbon on the carbonate group, compared to that of the carbonyl carbon on the ester group. [10] On the other hand, the aliphatic polyesters poly(DLLA) (PDLLA), poly(e-CL) (PCL), and their copolymers are well-known degradable materials that have been used in several applications such as orthopedic implants, drug delivery, suture coatings, and den- tistry. [11,12] Mechanical properties of these polyesters reported in literature have pointed out the strong dependence of molecular weight and chemical structure; for instance, (i) PDLLA has Young’ s * Correspondence to: Alejandro López, Institutionen för teknikvetenskaper, Ångströmlaboratoriet, Lägerhyddsvägen 1, Box 538, 751 21 Uppsala, Sweden. E-mail: [email protected] a A. López, C. Persson Department of Engineering Sciences, Division of Applied Materials Science, The Ångström Laboratory, Uppsala University, Uppsala, Sweden b J. Hilborn, R. Rojas Department of Materials Chemistry, Division of Polymer Chemistry, The Ångström Laboratory, Uppsala University, Uppsala, Sweden Research article Received: 16 September 2011, Accepted: 8 April 2012, Published online in Wiley Online Library (wileyonlinelibrary.com) DOI: 10.1002/pat.3042 Polym. Adv. Technol. (2012) Copyright © 2012 John Wiley & Sons, Ltd.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Comparative characterization of oligomericprecursors intended for injectable implantsAlejandro Lópeza*, Cecilia Perssona, Jöns Hilbornb and Ramiro Rojasb
The use of injectable materials is a simple approach for drug delivery and tissue repair, in, e.g. minimally invasive surgeryapplications. If these materials are used past their glass transition temperature and have a low viscosity, they will be ableto flow while delivered in situ. Whether these materials are to be used as low viscosity drug carriers or further crosslinkedfor tissue repair, there is a need for a better understanding of their handling properties. In this study, oligo(trimethylenecarbonate) (oTMC) and oligo[D,L-lactide-co-(«-caprolactone)] (oDLLA-co-CL) of variousmolecular weightswithin a relevantinjectability range were synthesized via ring-opening polymerization. The materials were comparatively characterized by1H NMR spectroscopy, differential scanning calorimetry, gel permeation chromatography, and rheological measurements.After comparing the viscosities and molecular weights of the materials, it was concluded that oDLLA-co-CLs were, gener-ally, better suited as an injectable in situ crosslinking network, whereas oTMCs were found to be better candidates asinjectable drug carriers. This study provides useful data and guidelines on the use of these and other similar oligomersintended for injectable implants. Copyright © 2012 John Wiley & Sons, Ltd.
Keywords: oligomer(s); polyester(s); polycarbonate(s); rheology; injectable implant(s)
INTRODUCTION
Injectable biomaterial systems are especially desirable in mini-mally invasive clinical treatments, due to their ease of handlingand application, when restoration is required within a shortperiod of time; e.g. vertebroplasty or kyphoplasty for vertebralaugmentation,[1] or for filling defects with complex shapes.These types of materials can also be a vehicle for in situ deliveryof drugs, growth factors, or cells; e.g. for intervertebral discrestoration.[2] Cyclic monomers such as trimethylene carbonate(TMC), D,L-lactide (DLLA), and e-caprolactone (e-CL), can behomo- and copolymerized via ring-opening polymerization,co-initiated by a diol, to form linear oligomers. These oligomershave a low glass transition temperature (Tg), low viscosity, andare able to flow at room temperature. They can readily be loadedwith drugs to be used as delivery agents, or can further befunctionalized with, e.g. acrylic or methacrylic end-groups to givethem the ability to react in situ, via redox or UV radical initiation,to form covalently crosslinked networks. Degradable, in situcuring polymers of low molecular weight, have been thoroughlydescribed in a series of US patents by Dunn et al. since the early90s.[3] Since then, these types of materials have been potentiallyavailable for several medical applications requiring injectability,in situ curing chemistry, drug reservoir capabilities, tailorablemechanical properties, degradability, and biocompatibility.Poly(TMC), PTMC, is a degradable aliphatic polycarbonate that
predominantly undergoes enzymatic degradation in vivo into non-acidic products.[4] Compression-molded high molecular weightPTMCs have been shown to have a Young’s modulus of 3.0–6.8MPa, a yield strength of 0.5–2.3 MPa, and a strain at break of 230–1250%.[5] PTMC copolymerized with glycolide and p-dioxanonehas been used as flexible suture materials (Maxon™ and BioSyn™),as well as tacks and screws for orthopedics (Acufex™).[6] Additionally,it has been investigated as drug-releasing agent when copolymer-ized or blended with adipic anhydride.[7] PTMC has long been
underestimated for medical device applications (i.e. load bearing)due to its relatively poor mechanical properties[8] and tackiness;however, the ongoing development of injectable biomaterials mayfind it advantageous for applications where the local pH must bemaintained over a certain level during degradation. For instance,Amsden et al.[9] have shown the potential of low molecular weightPTMC for the localized delivery of the acid sensitive, vascular endo-thelial growth factor. Another important advantage of PTMC is thatit undergoes surface degradation unlike polyesters, which undergobulk degradation followed by swelling and collapse.[6] Furthermore,the surface degradation of PTMC is accelerated by the presence ofcertain endogenous enzymes, e.g. lipase.[4] On the other hand, theacidic products from the degradation of polyesters can auto-catalyzetheir hydrolysis, making the degradation of polyesters faster thanthat of the PTMC; this occurs in spite of the higher electrophiliccharacter of the carbonyl carbon on the carbonate group, comparedto that of the carbonyl carbon on the ester group.[10]
On the other hand, the aliphatic polyesters poly(DLLA) (PDLLA),poly(e-CL) (PCL), and their copolymers are well-known degradablematerials that have been used in several applications such asorthopedic implants, drug delivery, suture coatings, and den-tistry.[11,12] Mechanical properties of these polyesters reported inliterature have pointed out the strong dependence of molecularweight and chemical structure; for instance, (i) PDLLA has Young’s
* Correspondence to: Alejandro López, Institutionen för teknikvetenskaper,Ångströmlaboratoriet, Lägerhyddsvägen 1, Box 538, 751 21 Uppsala, Sweden.E-mail: [email protected]
a A. López, C. PerssonDepartment of Engineering Sciences, Division of Applied Materials Science, TheÅngström Laboratory, Uppsala University, Uppsala, Sweden
b J. Hilborn, R. RojasDepartment of Materials Chemistry, Division of Polymer Chemistry, TheÅngström Laboratory, Uppsala University, Uppsala, Sweden
Research article
Received: 16 September 2011, Accepted: 8 April 2012, Published online in Wiley Online Library
(wileyonlinelibrary.com) DOI: 10.1002/pat.3042
Polym. Adv. Technol. (2012) Copyright © 2012 John Wiley & Sons, Ltd.

modulus of 1.9–2.4 GPa and tensile strength of 29–35 MPa[8]; (ii)PCL has Young’s modulus of >400 MPa, tensile strength of 16–23MPa, and strain at break of >700%.[8,13] Due to the higher hydro-phobicity and the semicrystallinity of PCL, it has lower degradationrates compared to amorphous PDLLA. As a result, copolymers andblends of PDLLA and PCL are materials with a good compromisebetween mechanical properties and close-to-predictable degrada-tion rates. Therefore, they have been widely investigated as tissueengineering scaffolds and other medical device applications.[14,15]
Despite the extensive literature available for these kinds of mate-rials, in-depth characterization, including rheology of the oligomericdegradable precursors intended for injectable implants, is scarce.Most of the available literature revolves around the use of PTMCfor applications such as microstereolithography[16] or the synthesis,characterization and degradation behavior of PTMC[4,5,17] and PDLLAand PCL[14,18,19] -based networks as biomaterials.
To our knowledge, there are no studies that focus on fullycharacterizing oligo(TMC) (oTMC) and oligo[DLLA-co-(e-CL)](oDLLA-co-CL) precursors and assessing the effects of smallchanges in the molecular weight on their rheological behavior.For instance, Matsuda et al.[20] evaluated the effects of photocuringon the mechanical and degradation properties of oTMC cross-linked networks (oTMC with Mn� 650–3400 Da) without investi-gating the rheology of the oligomers. Amsden et al.[21] reportedthe properties of injectable oligo(e-CL) precursors (Mn� 560–1480Da) synthesized using different initiators; however, the rheologicalmeasurements were performed at 45�C, which is irrelevant for theintended in vivo applications. Timbart et al.[22] thoroughly evaluatedoTMC (Mn� 620–2400 Da) as an injectable biomaterial and con-cluded that, upon crosslinking, these polymers are well toleratedin vivo, using a subcutaneous rat model. The importance of compar-ative characterization is based on the fact that unlike their highmolecular weight counterparts, the properties of the oligomers,prior to crosslinking, can vary extensively with small changes inchemical structure and molecular weight. To control handling prop-erties of any injectable biomaterial based on these oligomers, theirphysicochemical properties need to be thoroughly investigated.
In this study, random co-oligomers of DLLA and e-CL, andoligomers of TMC were assessed within ranges of molecularweights (500–2000 Da) and temperatures (25 and 37�C) ofinterest for the fabrication of injectable implants. The aim wasto synthesize and compare the physicochemical and rheological
properties of oTMC and oDLLA-co-CL precursors intended to beused as injectable biomaterials. The oligomers were synthesizedand their physicochemical properties studied through 1H NMRspectroscopy, differential scanning calorimetry (DSC), and gelpermeation chromatography (GPC). The rheological properties,complex modulus (G*), and zero shear viscosity (�0) were alsoassessed at 25 and 37�C.
EXPERIMENTAL
Materials
The monomers DLLA (Purac, The Netherlands), TMC (BoehringerIngelheim, Germany), and the catalysts Sn(II)2-ethylhexanoate(Sigma-Aldrich, USA) and 4-dimethylaminopyridine (DMAP)(Acros Organics, Belgium) were used as received. The co-initiator1,4-butanediol (BD) (Sigma-Aldrich, USA) and the monomer e-CL(Sigma-Aldrich, USA) were distilled, whereas the N,N-diisopropy-lethylamine (DIPEA) (Sigma-Aldrich, USA) was dried over potassiumhydroxide before use.
Synthesis of hydroxy-terminated oligomers
Oligocarbonates with different molecular weights were synthe-sized by ring-opening polymerization of TMC (Fig. 1A). Themonomer-to-initiator ratio was varied between 1:3 and 1:17.The TMC and the co-initiator were heated from RT to 200�C,before the addition of the catalyst, (0.2% of the monomer) andmaintained there for 1.5 h, followed by 160�C for 3.5 h underAr atmosphere. The product was dissolved in dichloromethane(DCM) and triturated twice in cold hexane, followed by decanta-tion of the solvent mixture and rotoevaporation.Random co-oligoesters were synthesized by ring-opening po-
lymerization of DLLA and e-CL (Fig. 1B). The monomer-to-initiatorratio was varied between 1:4 and 1:10 for 1:2 DLLA/e-CL respec-tively. The DLLA, e-CL, and the co-initiator were heated fromRT to 170�C before the addition of the catalyst (0.2% of themonomer) and maintained there for 3 h followed by 120�Cfor 21 h under Ar atmosphere. The purification procedure wasthe same as for the oligocarbonates.
Figure 1. General reactions schemes: (A) Ring-opening polymerization of trimethylene carbonate; (B) Ring-opening polymerization of D,L-lactide ande-caprolactone; (C) Hydroxyl end-group acrylation with acryloyl chloride.
A. LÓPEZ ET AL.
wileyonlinelibrary.com/journal/pat Copyright © 2012 John Wiley & Sons, Ltd. Polym. Adv. Technol. (2012)
Synthesis of the oligomeric -a,v-acrylates
The hydroxy-terminated oligomers (1 eq) were acrylated usingacryloyl chloride (2 eq) in dry DCM at 0�C (Fig. 1C) in the presenceof DIPEA (2 eq) and DMAP (0.2 eq). The acryloyl chloride wasdissolved in dry DCM (0.1 %, v/v) and added dropwise using asyringe infusion pump at a speed of 10 mL/h. After rotoevapora-tion of excess solvent and un-reacted acryloyl chloride, bothoTMC-a,o-acrylate (oTMC-DA) and oDLLA-co-CL-a,o-acrylate(oDLLA-co-CL-DA) were purified by dissolution in tetrahydrofuran(THF) and triturated twice in 0.5M HCl followed by rotoevapora-tion after decantation of the aqueous phase. The products weredissolved in DCM, dried with magnesium sulfate and filtratedbefore removing the remaining solvent by rotoevaporationfollowed by complete drying under vacuum.
Materials characterization1H-NMR spectroscopy
The oligomers were characterized by 1H-NMR spectroscopy,and the molecular weights were determined via end-groupanalysis on a JEOL ECP-400 NMR (JEOL, Japan) using deuteratedchloroform (CDCl3) as the solvent.
Differential scanning calorimetry
DSC measurements were performed on a DSC Q1000 (TA Instru-ments, United States). All the samples were crimped betweenan aluminum pan and lid. A heat/cool/heat ramp experimentwas done from�80 to 200�C at a rate of 10�C/min. The Tg of eacholigomer was determined from the second heat ramp.
Gel permeation chromatography
GPC measurements were performed on a Viscotek TDA model301 equipped with a TSK-gel GMHXL column set. THF was usedas the eluent at 35�C and 1 mL/min. The number average molarmass (Mn) and the polydispersity index (PDi) were determinedrelative to polystyrene standards.
Rheometry and Viscometry
Inherent viscosity [�] measurements were performed on anUbbelohde suspended level dilution viscometer (Schott Instru-ments, Germany) at 25�C using chloroform as the solvent at aconcentration of 10 mg/mL.
A parallel plate AR 2000 rheometer (TA Instruments, UnitedStates) with a custom-made titanium geometry of 32 mm and agap of 500 mmwas used to further evaluate the rheological prop-erties. An oscillation procedure was first run in stress sweepmodeat a constant frequency of 1 Hz and a lower plate temperature of25�C, in order to determine the linear viscoelastic range (LVR). Afrequency sweep was then run at an oscillatory stress within thecorresponding LVR in order to determine the rheological para-meters: complex modulus (G*), storage modulus (G′), loss modu-lus (G00), and loss tangent (d). A flow procedure in a continuousrampmodewas run in order to determine the zero shear viscosity(�0) and assess injectability. The experiments were performed atboth 25 and 37�C, which are the possible working temperaturesassuming an initial working temperature of 25�C and reaching amaximum of 37�C in vivo before the onset of the curing reaction.
RESULTS AND DISCUSSION
Physicochemical properties
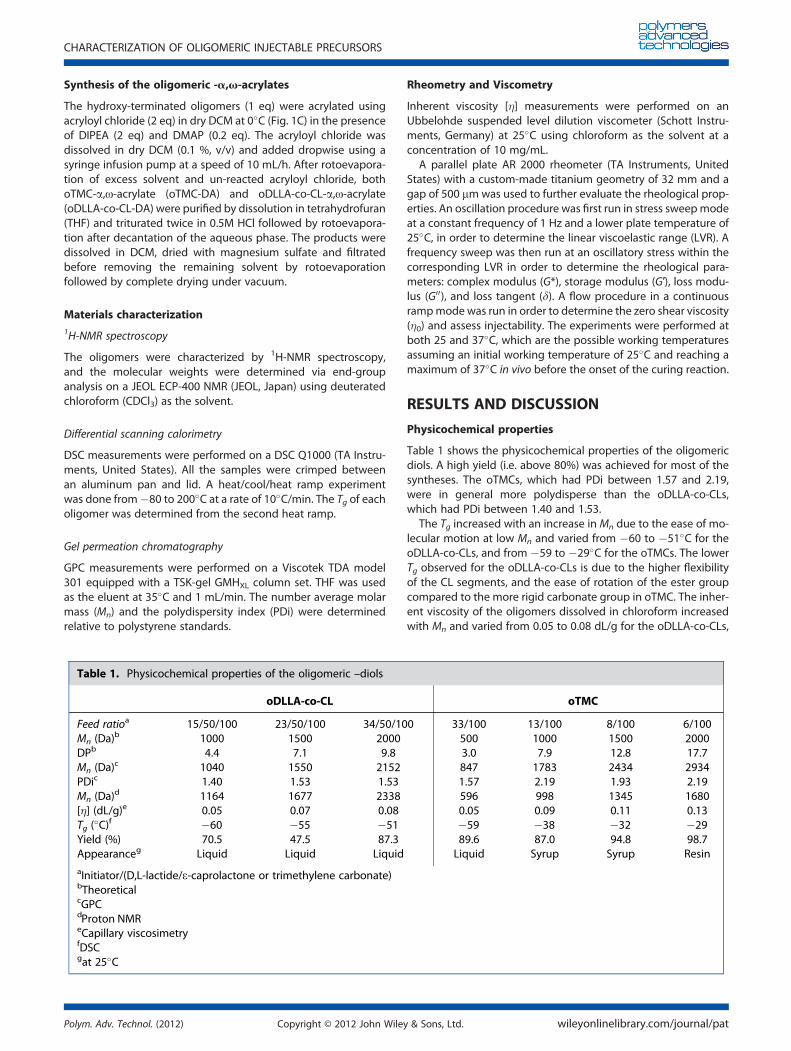
Table 1 shows the physicochemical properties of the oligomericdiols. A high yield (i.e. above 80%) was achieved for most of thesyntheses. The oTMCs, which had PDi between 1.57 and 2.19,were in general more polydisperse than the oDLLA-co-CLs,which had PDi between 1.40 and 1.53.
The Tg increased with an increase in Mn due to the ease of mo-lecular motion at low Mn and varied from �60 to �51�C for theoDLLA-co-CLs, and from �59 to �29�C for the oTMCs. The lowerTg observed for the oDLLA-co-CLs is due to the higher flexibilityof the CL segments, and the ease of rotation of the ester groupcompared to the more rigid carbonate group in oTMC. The inher-ent viscosity of the oligomers dissolved in chloroform increasedwith Mn and varied from 0.05 to 0.08 dL/g for the oDLLA-co-CLs,
CHARACTERIZATION OF OLIGOMERIC INJECTABLE PRECURSORS
Polym. Adv. Technol. (2012) Copyright © 2012 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/pat
and from 0.05 to 0.13 dL/g for the oTMCs at 25�C. The trend ofthe inherent viscosity values also correlated with the zero shearviscosity values obtained through rheological measurements.
1H-NMR spectroscopy
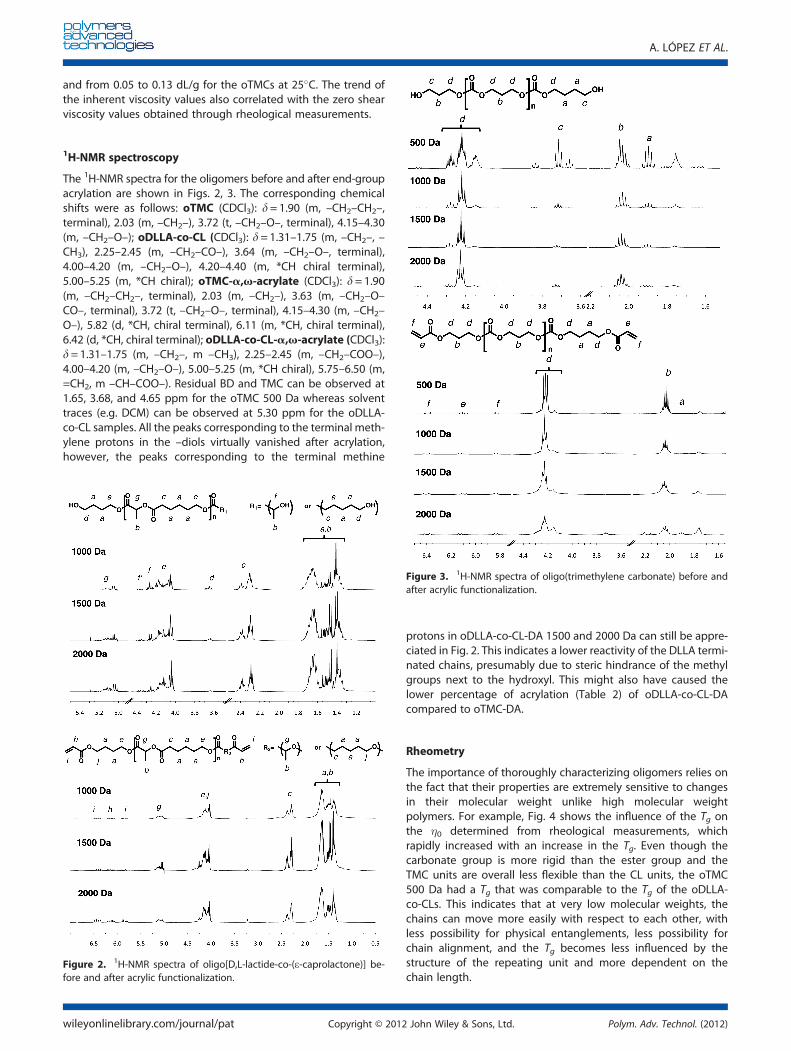
The 1H-NMR spectra for the oligomers before and after end-groupacrylation are shown in Figs. 2, 3. The corresponding chemicalshifts were as follows: oTMC (CDCl3): d= 1.90 (m, –CH2–CH2–,terminal), 2.03 (m, –CH2–), 3.72 (t, –CH2–O–, terminal), 4.15–4.30(m, –CH2–O–); oDLLA-co-CL (CDCl3): d=1.31–1.75 (m, –CH2–, –CH3), 2.25–2.45 (m, –CH2–CO–), 3.64 (m, –CH2–O–, terminal),4.00–4.20 (m, –CH2–O–), 4.20–4.40 (m, *CH chiral terminal),5.00–5.25 (m, *CH chiral); oTMC-a,v-acrylate (CDCl3): d=1.90(m, –CH2–CH2–, terminal), 2.03 (m, –CH2–), 3.63 (m, –CH2–O–CO–, terminal), 3.72 (t, –CH2–O–, terminal), 4.15–4.30 (m, –CH2–O–), 5.82 (d, *CH, chiral terminal), 6.11 (m, *CH, chiral terminal),6.42 (d, *CH, chiral terminal); oDLLA-co-CL-a,v-acrylate (CDCl3):d=1.31–1.75 (m, –CH2–, m –CH3), 2.25–2.45 (m, –CH2–COO–),4.00–4.20 (m, –CH2–O–), 5.00–5.25 (m, *CH chiral), 5.75–6.50 (m,=CH2, m –CH–COO–). Residual BD and TMC can be observed at1.65, 3.68, and 4.65 ppm for the oTMC 500 Da whereas solventtraces (e.g. DCM) can be observed at 5.30 ppm for the oDLLA-co-CL samples. All the peaks corresponding to the terminal meth-ylene protons in the –diols virtually vanished after acrylation,however, the peaks corresponding to the terminal methine
protons in oDLLA-co-CL-DA 1500 and 2000 Da can still be appre-ciated in Fig. 2. This indicates a lower reactivity of the DLLA termi-nated chains, presumably due to steric hindrance of the methylgroups next to the hydroxyl. This might also have caused thelower percentage of acrylation (Table 2) of oDLLA-co-CL-DAcompared to oTMC-DA.
Rheometry
The importance of thoroughly characterizing oligomers relies onthe fact that their properties are extremely sensitive to changesin their molecular weight unlike high molecular weightpolymers. For example, Fig. 4 shows the influence of the Tg onthe �0 determined from rheological measurements, whichrapidly increased with an increase in the Tg. Even though thecarbonate group is more rigid than the ester group and theTMC units are overall less flexible than the CL units, the oTMC500 Da had a Tg that was comparable to the Tg of the oDLLA-co-CLs. This indicates that at very low molecular weights, thechains can move more easily with respect to each other, withless possibility for physical entanglements, less possibility forchain alignment, and the Tg becomes less influenced by thestructure of the repeating unit and more dependent on thechain length.
Figure 2. 1H-NMR spectra of oligo[D,L-lactide-co-(e-caprolactone)] be-fore and after acrylic functionalization.
Figure 3. 1H-NMR spectra of oligo(trimethylene carbonate) before andafter acrylic functionalization.
A. LÓPEZ ET AL.
wileyonlinelibrary.com/journal/pat Copyright © 2012 John Wiley & Sons, Ltd. Polym. Adv. Technol. (2012)
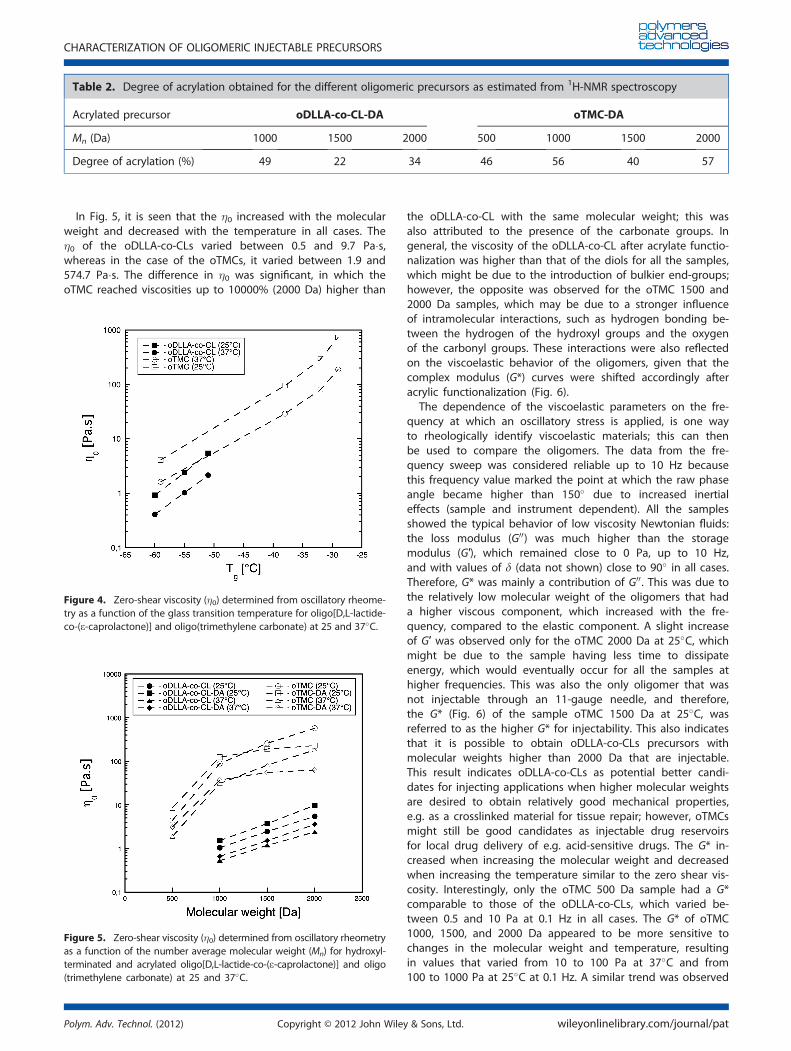
In Fig. 5, it is seen that the �0 increased with the molecularweight and decreased with the temperature in all cases. The�0 of the oDLLA-co-CLs varied between 0.5 and 9.7 Pa�s,whereas in the case of the oTMCs, it varied between 1.9 and574.7 Pa�s. The difference in �0 was significant, in which theoTMC reached viscosities up to 10000% (2000 Da) higher than
the oDLLA-co-CL with the same molecular weight; this wasalso attributed to the presence of the carbonate groups. Ingeneral, the viscosity of the oDLLA-co-CL after acrylate functio-nalization was higher than that of the diols for all the samples,which might be due to the introduction of bulkier end-groups;however, the opposite was observed for the oTMC 1500 and2000 Da samples, which may be due to a stronger influenceof intramolecular interactions, such as hydrogen bonding be-tween the hydrogen of the hydroxyl groups and the oxygenof the carbonyl groups. These interactions were also reflectedon the viscoelastic behavior of the oligomers, given that thecomplex modulus (G*) curves were shifted accordingly afteracrylic functionalization (Fig. 6).
The dependence of the viscoelastic parameters on the fre-quency at which an oscillatory stress is applied, is one wayto rheologically identify viscoelastic materials; this can thenbe used to compare the oligomers. The data from the fre-quency sweep was considered reliable up to 10 Hz becausethis frequency value marked the point at which the raw phaseangle became higher than 150� due to increased inertialeffects (sample and instrument dependent). All the samplesshowed the typical behavior of low viscosity Newtonian fluids:the loss modulus (G00) was much higher than the storagemodulus (G′), which remained close to 0 Pa, up to 10 Hz,and with values of d (data not shown) close to 90� in all cases.Therefore, G* was mainly a contribution of G00. This was due tothe relatively low molecular weight of the oligomers that hada higher viscous component, which increased with the fre-quency, compared to the elastic component. A slight increaseof G′ was observed only for the oTMC 2000 Da at 25�C, whichmight be due to the sample having less time to dissipateenergy, which would eventually occur for all the samples athigher frequencies. This was also the only oligomer that wasnot injectable through an 11-gauge needle, and therefore,the G* (Fig. 6) of the sample oTMC 1500 Da at 25�C, wasreferred to as the higher G* for injectability. This also indicatesthat it is possible to obtain oDLLA-co-CLs precursors withmolecular weights higher than 2000 Da that are injectable.This result indicates oDLLA-co-CLs as potential better candi-dates for injecting applications when higher molecular weightsare desired to obtain relatively good mechanical properties,e.g. as a crosslinked material for tissue repair; however, oTMCsmight still be good candidates as injectable drug reservoirsfor local drug delivery of e.g. acid-sensitive drugs. The G* in-creased when increasing the molecular weight and decreasedwhen increasing the temperature similar to the zero shear vis-cosity. Interestingly, only the oTMC 500 Da sample had a G*comparable to those of the oDLLA-co-CLs, which varied be-tween 0.5 and 10 Pa at 0.1 Hz in all cases. The G* of oTMC1000, 1500, and 2000 Da appeared to be more sensitive tochanges in the molecular weight and temperature, resultingin values that varied from 10 to 100 Pa at 37�C and from100 to 1000 Pa at 25�C at 0.1 Hz. A similar trend was observed
Table 2. Degree of acrylation obtained for the different oligomeric precursors as estimated from 1H-NMR spectroscopy
Acrylated precursor oDLLA-co-CL-DA oTMC-DA
Mn (Da) 1000 1500 2000 500 1000 1500 2000
Degree of acrylation (%) 49 22 34 46 56 40 57
Figure 4. Zero-shear viscosity (�0) determined from oscillatory rheome-try as a function of the glass transition temperature for oligo[D,L-lactide-co-(e-caprolactone)] and oligo(trimethylene carbonate) at 25 and 37�C.
Figure 5. Zero-shear viscosity (�0) determined from oscillatory rheometryas a function of the number average molecular weight (Mn) for hydroxyl-terminated and acrylated oligo[D,L-lactide-co-(e-caprolactone)] and oligo(trimethylene carbonate) at 25 and 37�C.
CHARACTERIZATION OF OLIGOMERIC INJECTABLE PRECURSORS
Polym. Adv. Technol. (2012) Copyright © 2012 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/pat
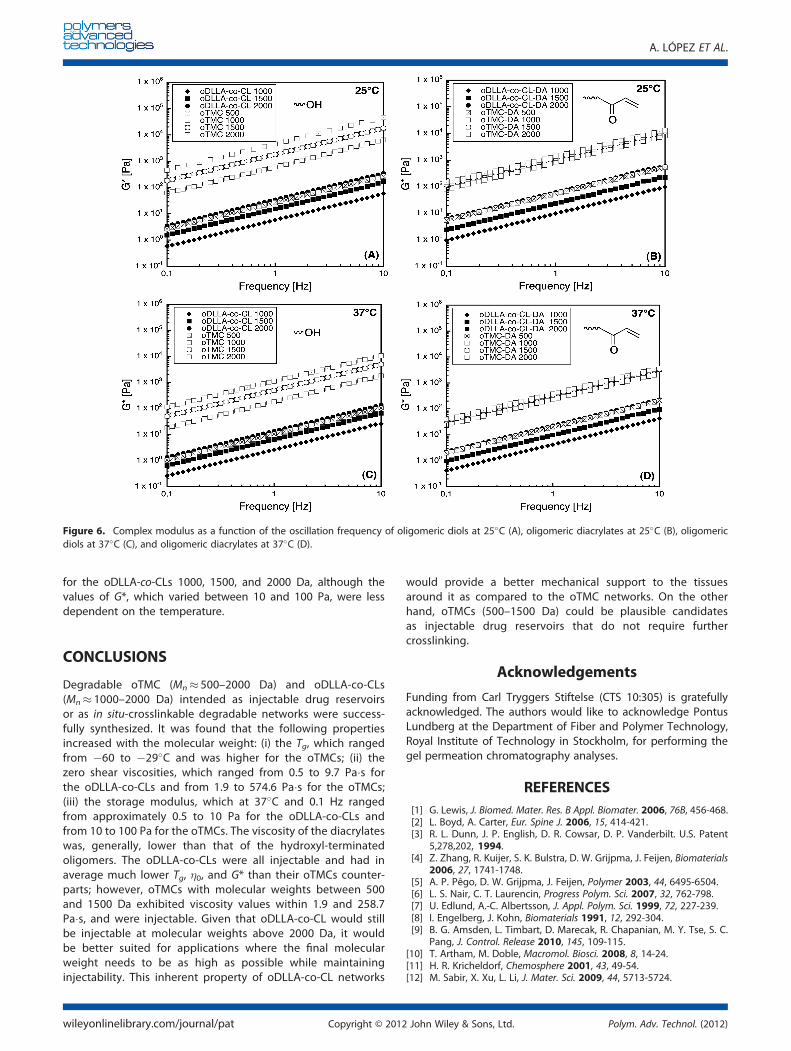
for the oDLLA-co-CLs 1000, 1500, and 2000 Da, although thevalues of G*, which varied between 10 and 100 Pa, were lessdependent on the temperature.
CONCLUSIONS
Degradable oTMC (Mn� 500–2000 Da) and oDLLA-co-CLs(Mn� 1000–2000 Da) intended as injectable drug reservoirsor as in situ-crosslinkable degradable networks were success-fully synthesized. It was found that the following propertiesincreased with the molecular weight: (i) the Tg, which rangedfrom �60 to �29�C and was higher for the oTMCs; (ii) thezero shear viscosities, which ranged from 0.5 to 9.7 Pa�s forthe oDLLA-co-CLs and from 1.9 to 574.6 Pa�s for the oTMCs;(iii) the storage modulus, which at 37�C and 0.1 Hz rangedfrom approximately 0.5 to 10 Pa for the oDLLA-co-CLs andfrom 10 to 100 Pa for the oTMCs. The viscosity of the diacrylateswas, generally, lower than that of the hydroxyl-terminatedoligomers. The oDLLA-co-CLs were all injectable and had inaverage much lower Tg, �0, and G* than their oTMCs counter-parts; however, oTMCs with molecular weights between 500and 1500 Da exhibited viscosity values within 1.9 and 258.7Pa�s, and were injectable. Given that oDLLA-co-CL would stillbe injectable at molecular weights above 2000 Da, it wouldbe better suited for applications where the final molecularweight needs to be as high as possible while maintaininginjectability. This inherent property of oDLLA-co-CL networks
would provide a better mechanical support to the tissuesaround it as compared to the oTMC networks. On the otherhand, oTMCs (500–1500 Da) could be plausible candidatesas injectable drug reservoirs that do not require furthercrosslinking.
Acknowledgements
Funding from Carl Tryggers Stiftelse (CTS 10:305) is gratefullyacknowledged. The authors would like to acknowledge PontusLundberg at the Department of Fiber and Polymer Technology,Royal Institute of Technology in Stockholm, for performing thegel permeation chromatography analyses.
REFERENCES[1] G. Lewis, J. Biomed. Mater. Res. B Appl. Biomater. 2006, 76B, 456-468.[2] L. Boyd, A. Carter, Eur. Spine J. 2006, 15, 414-421.[3] R. L. Dunn, J. P. English, D. R. Cowsar, D. P. Vanderbilt. U.S. Patent
5,278,202, 1994.[4] Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma, J. Feijen, Biomaterials
2006, 27, 1741-1748.[5] A. P. Pêgo, D. W. Grijpma, J. Feijen, Polymer 2003, 44, 6495-6504.[6] L. S. Nair, C. T. Laurencin, Progress Polym. Sci. 2007, 32, 762-798.[7] U. Edlund, A.-C. Albertsson, J. Appl. Polym. Sci. 1999, 72, 227-239.[8] I. Engelberg, J. Kohn, Biomaterials 1991, 12, 292-304.[9] B. G. Amsden, L. Timbart, D. Marecak, R. Chapanian, M. Y. Tse, S. C.
Pang, J. Control. Release 2010, 145, 109-115.[10] T. Artham, M. Doble, Macromol. Biosci. 2008, 8, 14-24.[11] H. R. Kricheldorf, Chemosphere 2001, 43, 49-54.[12] M. Sabir, X. Xu, L. Li, J. Mater. Sci. 2009, 44, 5713-5724.
Figure 6. Complex modulus as a function of the oscillation frequency of oligomeric diols at 25�C (A), oligomeric diacrylates at 25�C (B), oligomericdiols at 37�C (C), and oligomeric diacrylates at 37�C (D).
A. LÓPEZ ET AL.
wileyonlinelibrary.com/journal/pat Copyright © 2012 John Wiley & Sons, Ltd. Polym. Adv. Technol. (2012)

[13] P. Gunatillake, R. Mayadunne, R. Adhikari, In Biotechnology AnnualReview, Vol. 12 (Ed.: M. R. El-Gewely), Elsevier, 2006, 301-347.
[14] H. M. Younes, E. Bravo-Grimaldo, B. G. B. G. Amsden, Biomaterials2004, 25, 5261-5269.
[15] L. Calandrelli, A. Calarco, P. Laurienzo, M. Malinconico, O. Petillo, G.Peluso, Biomacromolecules 2008, 9, 1527-1534.
[16] G. t. R. P. Henry, A. Heise, D. Bottai, A. Formenti, A. Gorio, A. M. DiGiulio, C. E. Koning, Biomacromolecules 2008, 9, 867-878.
[17] C. Zhang, H. Subramanian, J. J. Grailer, A. Tiwari, S. Pilla, D. A.Steeber, S. Gong, Polym. Adv. Technol. 2009, 20, 742-747.
[18] A. O. Helminen, H. Korhonen, J. V. Seppälä, Macromol. Chem. Phys.2002, 203, 2630–2639.
[19] M.-H. Huang, S. Li, J. Coudane, M. Vert, Macromol. Chem. Phys. 2003,204, 1994-2001.
[20] T. Matsuda, I. K. Kwon, S. Kidoaki, Biomacromolecules 2003, 5,295-305.
[21] B. Amsden, A. Hatefi, D. Knight, E. Bravo-Grimaldo, Biomacromole-cules 2004, 5, 637-642.
[22] L. Timbart, M. Y. Tse, S. C. Pang, O. Babasola, B. G. Amsden,Macromol. Biosci. 2009, 9, 786-794.
CHARACTERIZATION OF OLIGOMERIC INJECTABLE PRECURSORS
Polym. Adv. Technol. (2012) Copyright © 2012 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/pat
Related Documents











