Cofractionation of HMGB proteins with histone dimers Qinqin Zhuang a , Hugh Smallman a , Stanley J. Lambert a, , Sirirath S. Sodngam b , Colin D. Reynolds a , Katie Evans a , Mark J. Dickman c , John P. Baldwin a , Christopher M. Wood a,⇑ a School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool L3 3AF, UK b Natural Products Research Unit, Centre of Excellence for Innovation in Chemistry (PERCH-CIC), Department of Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand c ChELSI Institute, Department of Chemical and Biological Engineering, University of Sheffield, Sheffield S1 3JD, UK article info Article history: Received 2 August 2013 Received in revised form 31 October 2013 Accepted 1 November 2013 Available online 13 November 2013 Keywords: Histone octamer Histone dimer Histone tetramer HMGB interactions Protein interaction domains Pull-down assays abstract An effective and flexible method is presented that can be used to investigate cofractionation of groups of nuclear proteins. The method was used to analyze chromatin-related proteins, of which high-mobility group B (HMGB) proteins consistently cofractionated by cation-exchange chromatography with the his- tone dimer (H2A–H2B). This led to the hypothesis that the two form a complex, further suggested by gel filtration, in which the HMGBs with core histones eluted as a defined high-molecular-weight peak. A nec- essary requirement for further studying protein interactions is that the constituents are of the highest possible purity and the pure histone dimers and tetramers used in this study were derived from pure his- tone octamers with their native marks. There is a growing interest in protein–protein interactions and an increasing focus on protein-interaction domains: most frequently, pull-down assays are used to examine these. The technology presented here can provide an effective system that complements pull-down assays. Ó 2013 Elsevier Inc. All rights reserved. Introduction DNA in the eukaryotic cell nucleus is folded into several orders of structure. At the lowest order the nucleosome core particle con- tains 147 bp of DNA wound 1.65 times around a globular-protein structure known as the histone octamer. In addition, attached to the nucleosome core particle, there is an extra species-dependent length of DNA (20 to 50 bp) and a linker-histone protein H1 (in special cases H5 or H1 0 ). This combined structure is known as a nucleosome. Continuous sequences of nucleosomes fold into sev- eral levels of higher-order structure, tighter structures applying to inactive chromatin (heterochromatin) and looser structures to active chromatin (euchromatin). The structured part of the nucle- osome core particle and the octomeric core-histone structure are known to high resolution [1–4]. The DNA in the tight, unmodified structure of chromatin is not accessible to protein factors that are needed to carry out all the processes of the cell. To gain controlled access of factors to their target DNA sequence, a number of molecular pathways have evolved. These techniques are generically called nucleosome remodeling and consist of patterns of posttranscriptional modifications on the histones and related proteins, the replace- ment of canonical histones by their variants, and the application of ATP-based nucleosome remodeling [5]. The remodeling process is a continuous process—molecular fission–fusion dynamics [6]. The notion of dynamic nucleosomes implies that those nucleo- somes—or, more specifically, histone octamers—will be assembled and disassembled [7]. The histone octamer in vivo exists only as such when bound to DNA and linker histones. Assembly of the his- tone octamer is achieved by bringing together a tetramer config- ured as (H3–H4) 2 and two dimers, each of which is configured as H2A–H2B. The tetramer and dimer are accompanied to the assem- bly site by a specific set of molecules called histone chaperones. These molecules are quite structurally varied, but frequently have acidic C-terminal tails [8]. Firm evidence that chaperones such as Nap1, FACT, and Asf1 are also able to disassemble nucleosomes was, for a while, lacking. However, in vivo experiments using yeast have indicated that Asf1 not only assembles chromatin but also is a global chromatin disassembly factor [9]. Histone octamers in vitro and isolated from DNA can form un- der high-salt conditions [10]. However, at physiological pH the oct- amer exists as separate dimers and tetramers [10]. It is known from in vitro experiments that dimers and tetramers can succumb to nonspecific interactions with other proteins, DNA, and RNA [8,11]. Therefore, histone chaperones also serve to inhibit these nonspecific histone interactions. Salt-gradient experiments have shown that nucleosomes are assembled in a sequential fashion: 0003-2697/$ - see front matter Ó 2013 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.ab.2013.11.001 ⇑ Corresponding author. E-mail address: [email protected] (C.M. Wood). Deceased. Analytical Biochemistry 447 (2014) 98–106 Contents lists available at ScienceDirect Analytical Biochemistry journal homepage: www.elsevier.com/locate/yabio

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Analytical Biochemistry 447 (2014) 98–106
Contents lists available at ScienceDirect
Analytical Biochemistry
journal homepage: www.elsevier .com/locate /yabio
Cofractionation of HMGB proteins with histone dimers
0003-2697/$ - see front matter � 2013 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/j.ab.2013.11.001
⇑ Corresponding author.E-mail address: [email protected] (C.M. Wood).
� Deceased.
Qinqin Zhuang a, Hugh Smallman a, Stanley J. Lambert a,�, Sirirath S. Sodngam b, Colin D. Reynolds a,Katie Evans a, Mark J. Dickman c, John P. Baldwin a, Christopher M. Wood a,⇑a School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool L3 3AF, UKb Natural Products Research Unit, Centre of Excellence for Innovation in Chemistry (PERCH-CIC), Department of Chemistry, Faculty of Science, Khon Kaen University,Khon Kaen 40002, Thailandc ChELSI Institute, Department of Chemical and Biological Engineering, University of Sheffield, Sheffield S1 3JD, UK
a r t i c l e i n f o
Article history:Received 2 August 2013Received in revised form 31 October 2013Accepted 1 November 2013Available online 13 November 2013
Keywords:Histone octamerHistone dimerHistone tetramerHMGB interactionsProtein interaction domainsPull-down assays
a b s t r a c t
An effective and flexible method is presented that can be used to investigate cofractionation of groups ofnuclear proteins. The method was used to analyze chromatin-related proteins, of which high-mobilitygroup B (HMGB) proteins consistently cofractionated by cation-exchange chromatography with the his-tone dimer (H2A–H2B). This led to the hypothesis that the two form a complex, further suggested by gelfiltration, in which the HMGBs with core histones eluted as a defined high-molecular-weight peak. A nec-essary requirement for further studying protein interactions is that the constituents are of the highestpossible purity and the pure histone dimers and tetramers used in this study were derived from pure his-tone octamers with their native marks. There is a growing interest in protein–protein interactions and anincreasing focus on protein-interaction domains: most frequently, pull-down assays are used to examinethese. The technology presented here can provide an effective system that complements pull-downassays.
� 2013 Elsevier Inc. All rights reserved.
Introduction
DNA in the eukaryotic cell nucleus is folded into several ordersof structure. At the lowest order the nucleosome core particle con-tains 147 bp of DNA wound 1.65 times around a globular-proteinstructure known as the histone octamer. In addition, attached tothe nucleosome core particle, there is an extra species-dependentlength of DNA (20 to 50 bp) and a linker-histone protein H1 (inspecial cases H5 or H10). This combined structure is known as anucleosome. Continuous sequences of nucleosomes fold into sev-eral levels of higher-order structure, tighter structures applyingto inactive chromatin (heterochromatin) and looser structures toactive chromatin (euchromatin). The structured part of the nucle-osome core particle and the octomeric core-histone structure areknown to high resolution [1–4].
The DNA in the tight, unmodified structure of chromatin is notaccessible to protein factors that are needed to carry out all theprocesses of the cell. To gain controlled access of factors to theirtarget DNA sequence, a number of molecular pathways haveevolved. These techniques are generically called nucleosomeremodeling and consist of patterns of posttranscriptional
modifications on the histones and related proteins, the replace-ment of canonical histones by their variants, and the applicationof ATP-based nucleosome remodeling [5]. The remodeling processis a continuous process—molecular fission–fusion dynamics [6].
The notion of dynamic nucleosomes implies that those nucleo-somes—or, more specifically, histone octamers—will be assembledand disassembled [7]. The histone octamer in vivo exists only assuch when bound to DNA and linker histones. Assembly of the his-tone octamer is achieved by bringing together a tetramer config-ured as (H3–H4)2 and two dimers, each of which is configured asH2A–H2B. The tetramer and dimer are accompanied to the assem-bly site by a specific set of molecules called histone chaperones.These molecules are quite structurally varied, but frequently haveacidic C-terminal tails [8]. Firm evidence that chaperones such asNap1, FACT, and Asf1 are also able to disassemble nucleosomeswas, for a while, lacking. However, in vivo experiments using yeasthave indicated that Asf1 not only assembles chromatin but also is aglobal chromatin disassembly factor [9].
Histone octamers in vitro and isolated from DNA can form un-der high-salt conditions [10]. However, at physiological pH the oct-amer exists as separate dimers and tetramers [10]. It is knownfrom in vitro experiments that dimers and tetramers can succumbto nonspecific interactions with other proteins, DNA, and RNA[8,11]. Therefore, histone chaperones also serve to inhibit thesenonspecific histone interactions. Salt-gradient experiments haveshown that nucleosomes are assembled in a sequential fashion:
Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106 99
the histone tetramer is the first to be deposited onto DNA, followedby two dimers [8].
There are three classes of HMG (high-mobility group) proteins:HMGB, HMGN, and HMGA. The HMGA-group proteins containthree AT-hook domains that bind to the minor groove of A/T re-gions in B-form DNA. The HMGN group of proteins binds to nucle-osomes. Both these types have been reviewed in detail elsewhere[12]. In contrast to the core histones, HMGB amino acid residue se-quences are not conserved over all eukaryotes. However, in birdsand mammals conserved HMGB1, HMGB2, and HMGB3 proteinscontain two HMG boxes and one C-terminal acidic region [13–15]. Plants and insects have their own HMGB sequences [16,17].Single-celled yeast is an example of the lack of conservation inHMGBs, compared to multicellular eukaryotes. Although theHMGBs in yeast are not the same as in multicellular species, theydo have acidic C-terminal histone chaperones such as Chz1, ofknown partial structure in combination with the H2A.Z–H2B his-tone dimer [18]. Evidence is given in this paper that the HMGB pro-teins cofractionate preferentially with histone dimers, from whichan inference can be made that they may bind as histone dimerchaperones.
Chromatin complexes are fundamental to cellular processes andtherefore it is important to have a reliable technology that canexamine the components of those complexes, along with theirposttranslational modifications. For example, in transcription,chromatin complexes will form around the core (minimal pro-moter) of a gene and around regulatory promoters such as enhanc-ers and repressors. Should these chromatin assemblies bedisrupted in some way, then the correct spatiotemporal expressionof the gene can go awry [19].
The predominant technology for studying protein complexes ispull-down assays. In these procedures the proteins of a complexwill have an affinity tag attached, or less frequently antibodiescan be raised against key proteins. The tagged proteins can thenbe immunoprecipitated and analyzed using mass spectrometry[20]. This approach has a number of caveats. It is generally onlysmall scale and there can be problems with affinity tags or antibod-ies affecting or even disrupting the protein complex of interest. Theapproach demonstrated here does not use affinity tags or antibod-ies and there is no postprocessing using immunoprecipitation. Our
Table 1Formulation of solutions.
Solution Totalvol. Vt
(ml)
Vol. stockA (VA) (ml)
Vol. stockB (VB) (ml)
Vol. C (VC) (ml)
(1) PNE1 extraction buffer 500 20 0 480(2) Nuclei washing buffer 100 0.5 0 98(3) Nuclei lysing buffer 400 100 300 0(4) Octamer/PNE2
precipitation buffer200 200 0 0
(5) PNE2/octamer pptdissolving buffer
100 20 60 20
(6) Cation-exchangedialysis buffer
4000 200 0 3800
(7) Cation-exchangebinding buffer A
4000 200 0 3800
(8) Cation-exchange elutionbuffer B
1000 50 375 575
(9) Gel-exclusion buffer,fraction 5c
2000 100 450 1450
(10) PNE2/octamer pptdissolving and freezingbuffer
100 20 60 20 of glycerol
(11) Gel exclusion buffer,fraction 7b
1000 50 322.5 627.5
Stock solution A is 2 M KCl, 1 M K2HPO4, 1 M KH2PO4; stock solution B is 2.66 M KClhydrochloride. The pH value for all solutions used is 6.8.
method can be used for large-scale purification of chromatincomplexes, or individual proteins, that are compatible with down-stream crystallization studies and chemical crosslinking massspectrometry studies. For example, the procedures used hereinhave also allowed the production of pure native-histone dimers,tetramers, and octamers in considerable quantities.
Materials and methods
A simple method for fractionating all the native proteins in eukaryoticcell nuclei
A method has been developed that utilizes just two core stocksolutions from which are derived all necessary buffers. The twostock solutions are high-molarity forms of potassium chloride(KCl), dibasic potassium phosphate (K2HPO4), and monobasicpotassium phosphate (KH2PO4). They are used alone, or in combi-nation. Stock solution A is 2 M KCl, 1 M K2HPO4, 1 M KH2PO4; stocksolution B is 2.66 M KCl. In addition to the core stock solutions,ancillary solutions consist of C, deionized water; D, 1 M magne-sium chloride (MgCl2), and E, 250 mM benzamidine hydrochloride,which is used as a protease inhibitor. All the necessary buffer solu-tions were made by mixing the core stock solutions and ancillarysolutions in accordance with Table 1. The X and Y columns inTable 1 are the final molarities of the mixed solutions. Fig. 1summarizes the purification steps.
Earlier progenitors of the method have been used on chicken-erythrocyte cell nuclei to obtain highly pure histone octamers forcrystallization studies [3,4] and to analyze the posttranslationalmodifications of the linker histones from the same cell type [21].The flexibility of the method means it can be adapted to isolateand purify almost any nuclear protein. This capability has been uti-lized in earlier work by our group to produce high yields of theHMGB proteins and also the peptidyl-prolyl isomerase FKBP3 [22].
Release of proteins from intact nuclei
Chick-erythrocyte nuclei were initially prepared as described inearlier work [22]. Typically, the yield from this initial preparation,using 10 L of blood, was approximately 2 � 1012 nuclei. The nuclei
Vol. D(VD)(ml)
Vol. E(VE)(ml)
Equimolarphosphate (X)(mM)
KCl (Y)(mM)
Benzamidinehydrochloride(mM)
MgCl2
(mM)
1.75 5 80 80 2.5 3.50.35 1.0 10 10 2.5 3.50 4 500 2500 2.5 00 2 2000 2000 2.5 0
0 1 400 2000 2.5 0
0 40 100 100 2.5 0
0 0 100 100 0 0
0 0 100 1100 0 0
0 20 100 700 2.5 0
0 1 400 2000 2.5 0
0 0 100 960 0 0
. Other solutions: C, deionized water; D, 1 M MgCl2 and E, 250 mM benzamidine

Fig.1. Purification cycle showing how cePNE1, cePNE2, and cePNE3 are produced.
100 Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106
were washed three times in solution 1, centrifuging at 1500 rpmeach time. Centrifuging at these low speeds softly pelleted thenuclei, leaving a supernatant that contained large quantities ofsoluble proteins, which were designated as cePNE1 (chicken-erythrocyte protein nuclear extract 1). The three successive washeswere to extract the maximum possible yield of cePNE1 proteins(Fig. 1). The combined supernatants contained approximately 1 gof protein including high yields of HMG proteins, peptidyl-prolylisomerases, and histone deacetylases, but no histones. Otherproteins in this group included lamin A, elongation factor 1a, tauprotein, and endothelial differentiation factor 1. The proteins inthe cePNE1 sample were identified by mass spectrometry.
The cePNE1-depleted nuclei were dialyzed in solution 1, pre-pared in 20% glycerol instead of pure water, and frozen in approx-imately 45 50-ml Falcon tubes.
Lysis of cePNE1-depleted nuclei
With the nuclei depleted of many nonhistone proteins, thegroundwork had been laid for the production of chromatin-re-lated proteins with very little contamination. Lysis of nucleican in general be highly problematic, such that a large propor-tion of nuclei remains unlysed in a matrix of tightly gelledDNA, preventing penetration of the lysis buffer. This variabilityin lysis can drastically affect the amount of chromatin-related
proteins that can be retrieved. In this section, we show how tomaximize lysis of the nuclei to give a supernatant rich in solublechromatin-related proteins such as histones and HMGs. To initi-ate the process, one of the frozen Falcon tubes (about 40 ml) de-scribed at the end of the previous section was thawed in a coldroom, balanced with a Falcon tube containing a suitably denseliquid, and centrifuged at 1500 rpm for 15 min (Fig. 1).
After 15 min the solution was clearly separated into a super-natant and a pellet. The supernatant was discarded and the nu-clei pellet (13 ml of packed nuclei) was mixed with an equalvolume of solution 2 (Table 1). The mixture was dispersed, madeup to 80 ml by adding more of solution 2, and left stirring in abeaker. The intact cePNE1-depleted nuclei in suspension werethoroughly dispersed by transferring to a separate stirred beakerusing a 100-ll yellow pipette tip with a 50-ml syringe to furtherdisperse the suspension.
A small sample of the dispersed cePNE1-depleted nuclei wasused to determine the concentration of the nuclei using a hemocy-tometer. This worked out to be approximately 2.5 � 108 nuclei permilliliter (approximately 4 � 108/ml allowing for some clumpingin the hemocytometer). Therefore, the 80-ml mixture contained atotal of approximately 3 � 1010 nuclei.
In preparation for lysis, 52 ml of solution 3 (Table 1) was putinto each of six Ti45 centrifuge tubes. To each of the six tubeswas then added a 13-ml sample taken from the still-stirring nuclei
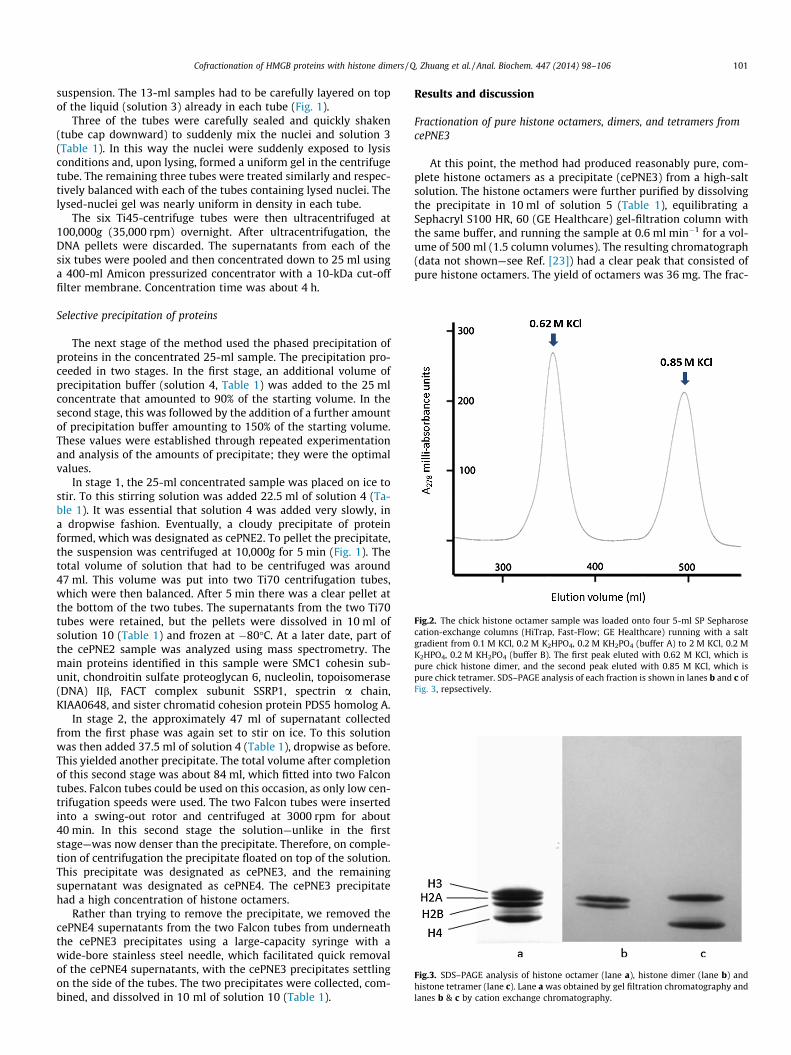
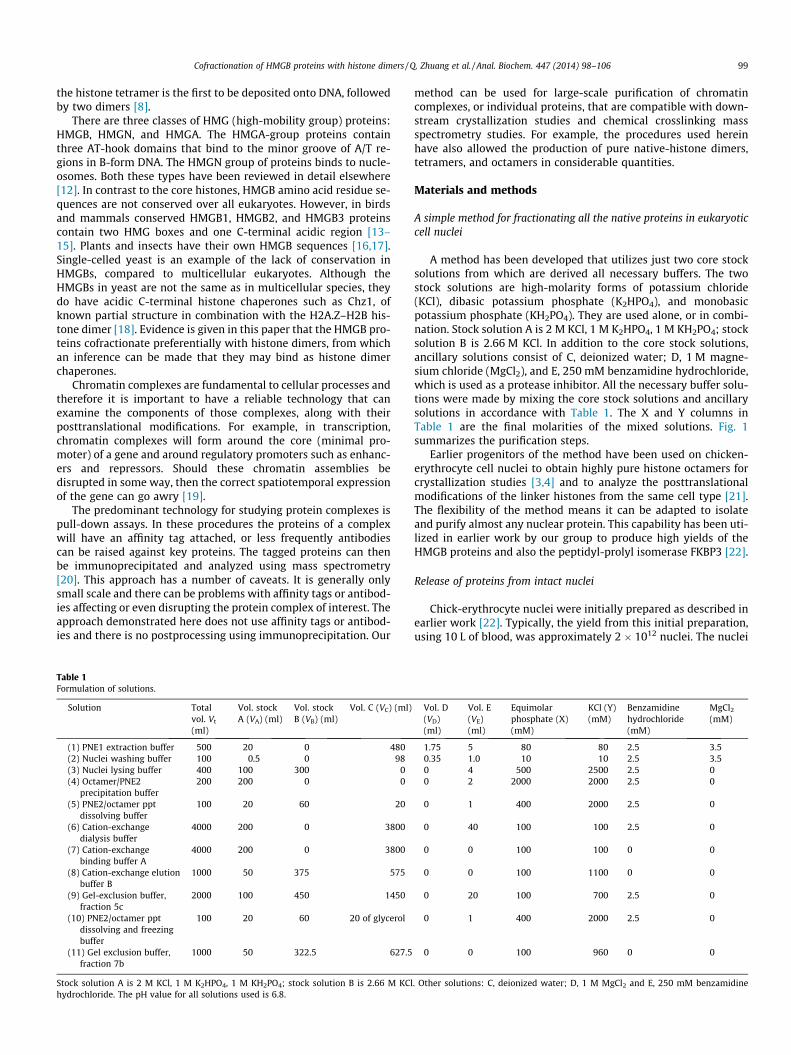
Fig.2. The chick histone octamer sample was loaded onto four 5-ml SP Sepharosecation-exchange columns (HiTrap, Fast-Flow; GE Healthcare) running with a saltgradient from 0.1 M KCl, 0.2 M K2HPO4, 0.2 M KH2PO4 (buffer A) to 2 M KCl, 0.2 MK2HPO4, 0.2 M KH2PO4 (buffer B). The first peak eluted with 0.62 M KCl, which ispure chick histone dimer, and the second peak eluted with 0.85 M KCl, which ispure chick tetramer. SDS–PAGE analysis of each fraction is shown in lanes b and c ofFig. 3, repsectively.
Fig.3. SDS–PAGE analysis of histone octamer (lane a), histone dimer (lane b) andhistone tetramer (lane c). Lane a was obtained by gel filtration chromatography andlanes b & c by cation exchange chromatography.
Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106 101
suspension. The 13-ml samples had to be carefully layered on topof the liquid (solution 3) already in each tube (Fig. 1).
Three of the tubes were carefully sealed and quickly shaken(tube cap downward) to suddenly mix the nuclei and solution 3(Table 1). In this way the nuclei were suddenly exposed to lysisconditions and, upon lysing, formed a uniform gel in the centrifugetube. The remaining three tubes were treated similarly and respec-tively balanced with each of the tubes containing lysed nuclei. Thelysed-nuclei gel was nearly uniform in density in each tube.
The six Ti45-centrifuge tubes were then ultracentrifuged at100,000g (35,000 rpm) overnight. After ultracentrifugation, theDNA pellets were discarded. The supernatants from each of thesix tubes were pooled and then concentrated down to 25 ml usinga 400-ml Amicon pressurized concentrator with a 10-kDa cut-offfilter membrane. Concentration time was about 4 h.
Selective precipitation of proteins
The next stage of the method used the phased precipitation ofproteins in the concentrated 25-ml sample. The precipitation pro-ceeded in two stages. In the first stage, an additional volume ofprecipitation buffer (solution 4, Table 1) was added to the 25 mlconcentrate that amounted to 90% of the starting volume. In thesecond stage, this was followed by the addition of a further amountof precipitation buffer amounting to 150% of the starting volume.These values were established through repeated experimentationand analysis of the amounts of precipitate; they were the optimalvalues.
In stage 1, the 25-ml concentrated sample was placed on ice tostir. To this stirring solution was added 22.5 ml of solution 4 (Ta-ble 1). It was essential that solution 4 was added very slowly, ina dropwise fashion. Eventually, a cloudy precipitate of proteinformed, which was designated as cePNE2. To pellet the precipitate,the suspension was centrifuged at 10,000g for 5 min (Fig. 1). Thetotal volume of solution that had to be centrifuged was around47 ml. This volume was put into two Ti70 centrifugation tubes,which were then balanced. After 5 min there was a clear pellet atthe bottom of the two tubes. The supernatants from the two Ti70tubes were retained, but the pellets were dissolved in 10 ml ofsolution 10 (Table 1) and frozen at �80�C. At a later date, part ofthe cePNE2 sample was analyzed using mass spectrometry. Themain proteins identified in this sample were SMC1 cohesin sub-unit, chondroitin sulfate proteoglycan 6, nucleolin, topoisomerase(DNA) IIb, FACT complex subunit SSRP1, spectrin a chain,KIAA0648, and sister chromatid cohesion protein PDS5 homolog A.
In stage 2, the approximately 47 ml of supernatant collectedfrom the first phase was again set to stir on ice. To this solutionwas then added 37.5 ml of solution 4 (Table 1), dropwise as before.This yielded another precipitate. The total volume after completionof this second stage was about 84 ml, which fitted into two Falcontubes. Falcon tubes could be used on this occasion, as only low cen-trifugation speeds were used. The two Falcon tubes were insertedinto a swing-out rotor and centrifuged at 3000 rpm for about40 min. In this second stage the solution—unlike in the firststage—was now denser than the precipitate. Therefore, on comple-tion of centrifugation the precipitate floated on top of the solution.This precipitate was designated as cePNE3, and the remainingsupernatant was designated as cePNE4. The cePNE3 precipitatehad a high concentration of histone octamers.
Rather than trying to remove the precipitate, we removed thecePNE4 supernatants from the two Falcon tubes from underneaththe cePNE3 precipitates using a large-capacity syringe with awide-bore stainless steel needle, which facilitated quick removalof the cePNE4 supernatants, with the cePNE3 precipitates settlingon the side of the tubes. The two precipitates were collected, com-bined, and dissolved in 10 ml of solution 10 (Table 1).
Results and discussion
Fractionation of pure histone octamers, dimers, and tetramers fromcePNE3
At this point, the method had produced reasonably pure, com-plete histone octamers as a precipitate (cePNE3) from a high-saltsolution. The histone octamers were further purified by dissolvingthe precipitate in 10 ml of solution 5 (Table 1), equilibrating aSephacryl S100 HR, 60 (GE Healthcare) gel-filtration column withthe same buffer, and running the sample at 0.6 ml min�1 for a vol-ume of 500 ml (1.5 column volumes). The resulting chromatograph(data not shown—see Ref. [23]) had a clear peak that consisted ofpure histone octamers. The yield of octamers was 36 mg. The frac-
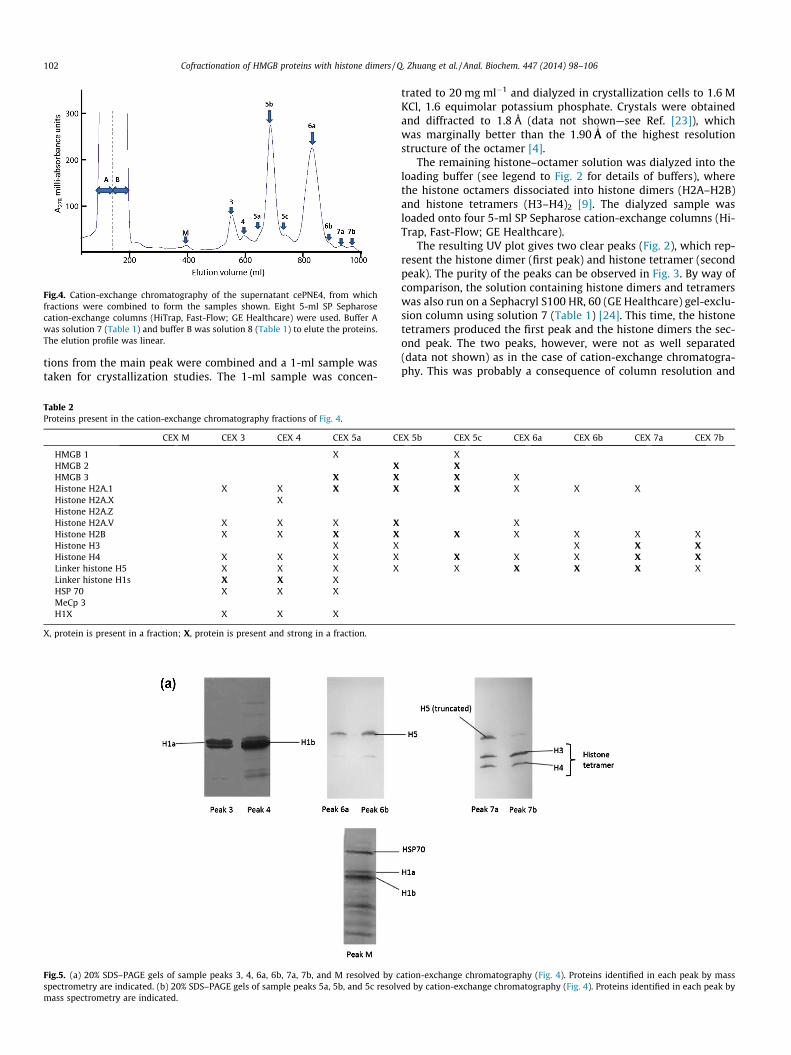
Fig.4. Cation-exchange chromatography of the supernatant cePNE4, from whichfractions were combined to form the samples shown. Eight 5-ml SP Sepharosecation-exchange columns (HiTrap, Fast-Flow; GE Healthcare) were used. Buffer Awas solution 7 (Table 1) and buffer B was solution 8 (Table 1) to elute the proteins.The elution profile was linear.
102 Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106
tions from the main peak were combined and a 1-ml sample wastaken for crystallization studies. The 1-ml sample was concen-
Table 2Proteins present in the cation-exchange chromatography fractions of Fig. 4.
CEX M CEX 3 CEX 4 CEX 5a C
HMGB 1 XHMGB 2 XHMGB 3 X XHistone H2A.1 X X X XHistone H2A.X XHistone H2A.ZHistone H2A.V X X X XHistone H2B X X X XHistone H3 X XHistone H4 X X X XLinker histone H5 X X X XLinker histone H1s X X XHSP 70 X X XMeCp 3H1X X X X
X, protein is present in a fraction; X, protein is present and strong in a fraction.
Fig.5. (a) 20% SDS–PAGE gels of sample peaks 3, 4, 6a, 6b, 7a, 7b, and M resolved by cspectrometry are indicated. (b) 20% SDS–PAGE gels of sample peaks 5a, 5b, and 5c resolvmass spectrometry are indicated.
trated to 20 mg ml�1 and dialyzed in crystallization cells to 1.6 MKCl, 1.6 equimolar potassium phosphate. Crystals were obtainedand diffracted to 1.8 Å (data not shown—see Ref. [23]), whichwas marginally better than the 1.90 ÅA
0
of the highest resolutionstructure of the octamer [4].
The remaining histone–octamer solution was dialyzed into theloading buffer (see legend to Fig. 2 for details of buffers), wherethe histone octamers dissociated into histone dimers (H2A–H2B)and histone tetramers (H3–H4)2 [9]. The dialyzed sample wasloaded onto four 5-ml SP Sepharose cation-exchange columns (Hi-Trap, Fast-Flow; GE Healthcare).
The resulting UV plot gives two clear peaks (Fig. 2), which rep-resent the histone dimer (first peak) and histone tetramer (secondpeak). The purity of the peaks can be observed in Fig. 3. By way ofcomparison, the solution containing histone dimers and tetramerswas also run on a Sephacryl S100 HR, 60 (GE Healthcare) gel-exclu-sion column using solution 7 (Table 1) [24]. This time, the histonetetramers produced the first peak and the histone dimers the sec-ond peak. The two peaks, however, were not as well separated(data not shown) as in the case of cation-exchange chromatogra-phy. This was probably a consequence of column resolution and
EX 5b CEX 5c CEX 6a CEX 6b CEX 7a CEX 7b
XXX XX X X X
XX X X X X
X X XX X X X XX X X X X
ation-exchange chromatography (Fig. 4). Proteins identified in each peak by massed by cation-exchange chromatography (Fig. 4). Proteins identified in each peak by
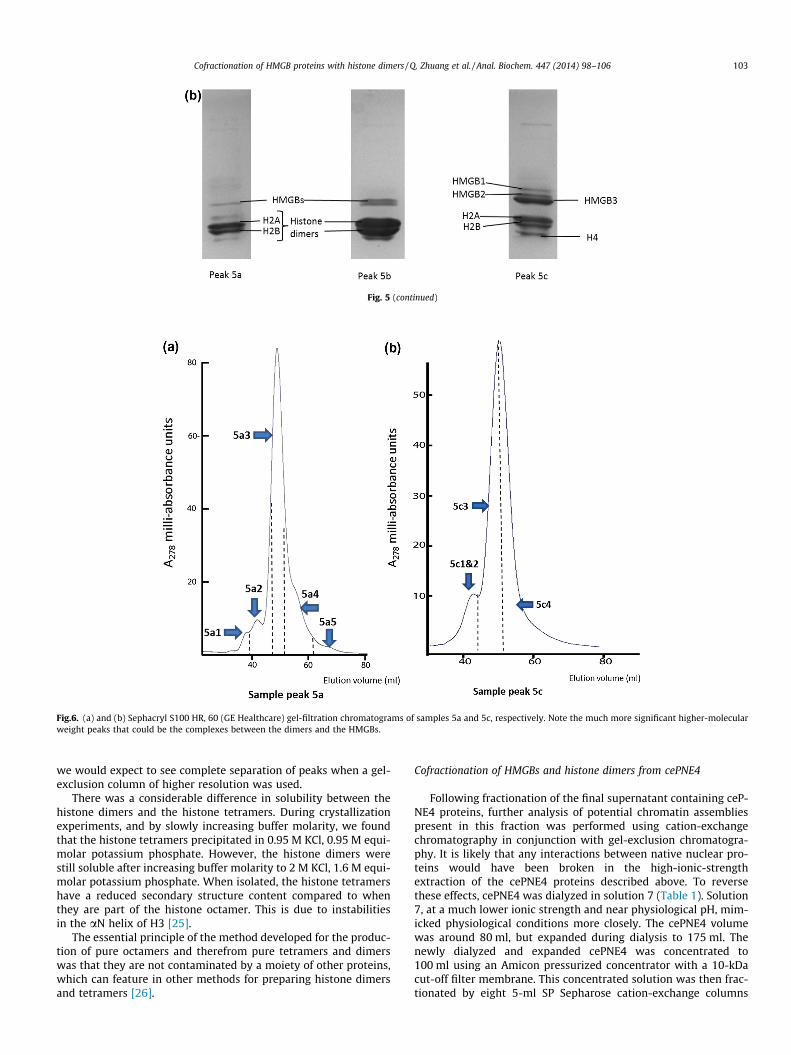
Fig.6. (a) and (b) Sephacryl S100 HR, 60 (GE Healthcare) gel-filtration chromatograms of samples 5a and 5c, respectively. Note the much more significant higher-molecularweight peaks that could be the complexes between the dimers and the HMGBs.
Fig. 5 (continued)
Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106 103
we would expect to see complete separation of peaks when a gel-exclusion column of higher resolution was used.
There was a considerable difference in solubility between thehistone dimers and the histone tetramers. During crystallizationexperiments, and by slowly increasing buffer molarity, we foundthat the histone tetramers precipitated in 0.95 M KCl, 0.95 M equi-molar potassium phosphate. However, the histone dimers werestill soluble after increasing buffer molarity to 2 M KCl, 1.6 M equi-molar potassium phosphate. When isolated, the histone tetramershave a reduced secondary structure content compared to whenthey are part of the histone octamer. This is due to instabilitiesin the aN helix of H3 [25].
The essential principle of the method developed for the produc-tion of pure octamers and therefrom pure tetramers and dimerswas that they are not contaminated by a moiety of other proteins,which can feature in other methods for preparing histone dimersand tetramers [26].
Cofractionation of HMGBs and histone dimers from cePNE4
Following fractionation of the final supernatant containing ceP-NE4 proteins, further analysis of potential chromatin assembliespresent in this fraction was performed using cation-exchangechromatography in conjunction with gel-exclusion chromatogra-phy. It is likely that any interactions between native nuclear pro-teins would have been broken in the high-ionic-strengthextraction of the cePNE4 proteins described above. To reversethese effects, cePNE4 was dialyzed in solution 7 (Table 1). Solution7, at a much lower ionic strength and near physiological pH, mim-icked physiological conditions more closely. The cePNE4 volumewas around 80 ml, but expanded during dialysis to 175 ml. Thenewly dialyzed and expanded cePNE4 was concentrated to100 ml using an Amicon pressurized concentrator with a 10-kDacut-off filter membrane. This concentrated solution was then frac-tionated by eight 5-ml SP Sepharose cation-exchange columns

Table 3Proteins present in gel-filtration chromatography fractions of Figs. 6a and 6b.
Peak 5c1 Peak 5c2 Peak 5c3 Peak 5c4
HMGB 1 XHMGB 2 XHMGB 3 XHistone H2A.1 X X X XHistone H2A.X X X XHistone H2A.ZHistone H2A.V X X X XHistone H2B X X X XHistone H3 X XHistone H4 X X X XLinker histone H5 X X XLinker histone H1s
X, protein is present in a fraction; X, protein is present and strong in a fraction.
104 Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106
(HiTrap, Fast-Flow; GE Healthcare). This number of columns wasused to greatly improve resolution—giving better separation be-tween peaks. The elution buffer used was solution 8 (Table 1).The elution was linear, run at 2 ml min�1, and 10-ml fractions werecollected. The resulting chromatogram plot can be seen in Fig. 4.The various peaks shown in Fig. 4 were analyzed using mass spec-trometry (Supplementary Information; Table 2) and SDS–PAGE(Fig. 5).
Peak 5b in Fig. 4 was assessed by UV spectrophotometry (datanot shown). The spectrum was that of pure protein (peak at278 nm) and with an optical density of 0.018 over a 1-mm pathlength. Using the Beer–Lambert Law, the optical absorbance is gi-ven by A = eCL, where e is the extinction coefficient, C is the con-centration, and L is the length, standardized at 1 cm. However,when 1-mm UV cells are used, the concentration has to be in-creased by a factor of 10. Thus, C = A/eL = 0.018/0.464 = 0.0387, giv-ing an actual concentration of 0.4 mg ml�1. There was 16 mg ofhistone proteins.
The above chromatography step was a subtle, but valuablechange from previous work [22], in which the nuclei were not de-pleted of the cePNE1 proteins and cation-exchange elution wasfrom a low pH (pH 5.2). At this pH, the HMGB proteins in the pre-vious work would have been mildly acid extracted, which wouldremove any interactions with histones. HMGBs in Ref. [21] elutedat a much lower ionic strength than they would have when usingphysiological pH levels.
In the present work HMGB proteins, at pH 6.8, were not elutedwith the H1 and H5 linker histones (Figs. 4 and 5a), but were elutedwith the H2A–H2B histone dimers in peaks 5a, 5b, and 5c (Fig. 5b).In peak 5c (Figs. 4 and 5b) all the HMGB proteins were present withthe histone dimer (and a lower level of histone H4). The assemblyformed a distinct, reasonably resolved protein peak. Core histonesH3 and H4 were also present in lesser quantities than the dimers(see Supplementary Information).
The isoelectric point (pI) for HMGB1, HMGB2, and HMGB3 inchick can be calculated as 5.62, 7.62, and 8.5, respectively, whereasthe pI of histones H2A and H2B is 10.9 and 10.3, respectively.Therefore, at pH 6.8 (the pH of the cation-exchange chromatogra-phy experiment), HMGB1 will be negatively charged and so shouldnot be binding at all, and yet it is present in peak 5c. Also, HMGB2and HMGB3 should be eluting a lot earlier than they have done.However, the three-dimensional structure of a protein can modu-late the theoretical value of its pI and therefore, while these figuresmay be interesting, they cannot be raised to the higher category ofevidence.
There was no sign of HMGB proteins in the H1 and H5 fractionsin Figs. 4 and 5a (peaks 3, 4, and 6) and if there were, the HMGBabsorbance should show up, because the molar-extinction
coefficients of HMGBs at 278 nm are at least five times those of lin-ker histones.
To see if the peak 5c proteins were interacting, the relevantfractions were combined, concentrated to 4 ml, and subjected togel filtration on a Sephacryl S100 HR, 60 (GE Healthcare) gel-filtra-tion column under the buffer conditions applying at the point oftheir elution in Fig. 4 (solution 9, Table 1). Fig. 6 shows the gel-fil-tration results for the concentrated sample from peak 5c: it showsa clear peak with a distinct shoulder to the left. Mass spectrometryanalysis of fractions in Fig. 6 (Supplementary Information) showedthat HMGB proteins were fractionated only in the first, smaller,peak with histone dimers (H2A–H2B) and some H4. The main peakin Fig. 6b contained only the dimers and H4. The fractions selectedfor identification by mass spectrometry are also shown in Fig. 6and the contents identified in each peak are shown in Table 3 (alsoSupplementary Information).
These results show the purification of a multimeric proteincomplex that contains HMGBs and the H2A–H2B histone dimer(with H3 and H4 as moieties) (Supplementary Information), sug-gesting that HMGBs are binding to H2A–H2B under the conditionsemployed in this study. Further work will be necessary to confirmthat HMGB proteins prefer to bind to histone dimers. It will beessential to improve the chromatography to extract peak 5c as afully resolved peak and to observe differences when the startingnuclei have not been depleted of HMGB proteins.
Conclusions
A highly efficient procedure has been presented that allows theselective extraction of four categories of native nuclear proteins.The biochemical procedures used in this work maintain proteinsin a state that is a close facsimile of their natural state—an essentialrequirement for successful studies of posttranslational modifica-tions. The procedure is flexible in that it can be easily adapted toproduce almost any group of proteins. The technology presentedherein was tailored to the purification of chromatin-related pro-teins for the purposes of identifying potential interactions. The fourgroups were defined as cePNE1, cePNE2, cePNE3, and cePNE4. ThecePNE1 sample contained proteins with a diverse range of func-tions; cePNE2 contained a range of proteins that were of interme-diate solubility.
The cePNE3 sample produced pure histone octamers when ana-lyzed using gel-exclusion chromatography. The resulting chro-matogram consisted of a well-defined, symmetric peak thatcontained octamers. The fractions that made up this peak were col-lected and dialyzed in a low-salt buffer and further analyzed usingcation-exchange chromatography. The resulting chromatographyenabled the separation and purification of histone dimers and his-tone tetramers. The low-salt buffer caused dissociation of the his-tone octamer. High-resolution crystals of the histone octamer wereproduced that diffract to 1.8 Å. The histone tetramers were precip-itated and crystallization studies with other proteins continue, butso far have not yielded any crystals. The histone dimers have beenshown to be extremely soluble and are still soluble in 2 M KCl,1.6 M equimolar phosphate.
The cePNE4 sample, which was the supernatant left after thesecond stage of precipitation, was analyzed by cation-exchangechromatography. The chromatography revealed the coelution ofHMGB proteins and histone dimers and a quantity of histoneH4. HMGB1 has a pI of 5.62 and was not expected to have beenbinding to a cation-exchange column at pH 6.8. Therefore,HMGB1 should, ordinarily, have been washed out as part ofthe nonbinding group of proteins. To resolve this conundrum,the cation-exchange fractions (peaks 5a and 5c) were subse-quently analyzed using gel-exclusion chromatography. The

Fig.7. (a) and (b) SDS–PAGE of the gel-filtration fractions of 5a and 5b, respectively. The gel in (a) shows that the HMGB proteins are present with core histones in the earlyfractions in the gel filtration, but only core histones are present in the later fractions. The gel in (b) shows more specific fractionation of HMGBs with all four core histones onlyin the earlier fractions. Histones H4 and H2A are more strongly represented with all three HMGB proteins. Histones H3 and H4 should not be present at all in Figs. 5 and 6unless they interact with other proteins, since Fig. 2 shows they would not be present in peak 5 of Fig. 4.
Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106 105
resulting chromatograms (Fig. 6a and b) showed two clear,slightly overlapping peaks. Mass spectrometry analysis of thegel-exclusion fractions identified the presence of both HMGBsand core histones.
The SDS–PAGE results for the gel-filtration fractions shown inFig. 7a and b correspond to Fig. 6a and b, respectively. The gel inFig. 7b shows more specific fractionation of HMGBs, with all fourcore histones only in the earlier fractions. Histones H4 and H2Aare more strongly represented with all three HMGB proteins. Notethat histones H3 and H4 should not be present in Figs. 6 and 7 un-less they interact with other proteins, since Fig. 2 shows theywould not be present in peak 5 of Fig. 4. Further, peak 5c inFig. 4 is a separate cation-exchange fraction.
These results demonstrate that HMGBs may be bound to corehistones and form a stable chromatin complex under bothcation-exchange and gel-filtration conditions used in these stud-ies. It is proposed that the HMGBs may act as histone chaperonesand stabilize histone octamers in nucleosomes with loosenedDNA.
When the cePNE4 sample was initially processed by cation-ex-change chromatography, several H2A variants were also detected.The variants found alongside H2A were H2A.V and H2A.X. TheH2A.V variant is conserved along its length across species andseems to be associated with heterochromatin [27].
The methods described herein allow the separation of groups ofnuclear proteins, dialyzing them in vitro into conditions near tothose in cell nuclei to study chromatin assemblies. In this studythere are indications that HMGB proteins interact with histone di-mers, although there is not yet a suggestion that there is prefer-ence for H2A variants in the dimers (Table 3 and SupplementaryInformation). Furthermore, the methods are easily scaled downand have been shown to work for nuclei from human cell culturesand human leukocytes [23].
Acknowledgments
We thank Patricia Burke and Glyn Hobbs from the School ofPharmacy and Biomolecular Sciences for their recent endeavorsin assessing doctoral theses from members of our group. Thechicken blood used in these experiments was kindly suppliedby Johnson and Swarbrick, Goosnargh Ltd, from their facility atSwainson House Farm, Goosnargh, Preston PR3 2JU, UK.
Appendix A. Supplementary Information
Supplementary information associated with this article can befound, in the online version, at http://dx.doi.org/10.1016/j.ab.2013.11.001.
References
[1] G. Arents, R.W. Burlingame, B.C. Wang, W.E. Love, E.N. Moudrianakis, Thenucleosomal core histone octamer at 3.1 Å resolution: a tripartite proteinassembly and a left-handed superhelix, Proc. Natl. Acad. Sci. USA 88 (1991)10148–10152.
[2] K. Luger, A.W. Mäder, R.K. Richmond, D.F. Sargent, T.J. Richmond, Crystalstructure of the nucleosome core particle at 2.8 Å resolution, Nature 38 (1997)251–260.
[3] L. Chantalat, J.M. Nicholson, S.J. Lambert, A.J. Reid, M.J. Donovan, C.D. Reynolds,C.M. Wood, J.P. Baldwin, Structure of the histone-core octamer in KCl/phosphate crystals at 2.15 A resolution, Acta Crystallogr. Sect. D: Biol.Crystallogr. 59 (2003) 1395–1407.
[4] C.M. Wood, J.M. Nicholson, S.J. Lambert, L. Chantalat, C.D. Reynolds, J.P.Baldwin, High-resolution structure of the native histone octamer, ActaCrystallogr. Sect. F: Struct. Biol. Cryst. Commun. 61 (2005) 541–545.
[5] S.J. Petesch, J.T. Lis, Overcoming the nucleosome barrier during transcriptelongation, Trends Genet. 28 (2012) 285–294.
[6] F. Aureli, C.M. Shaffner, C. Boesch, S.K. Bearder, J. Call, C.A. Chapman, R. Connor,A. Di Fiore, R.I.M. Dunbar, P. Henzi, K. Holekamp, A.H. Korstjens, R. Layton, P.Lee, J. Lehman, J.H. Manson, G. Ramos-Fernandez, K.B. Strier, C.P. van Shaik,Fission–fusion dynamics: new research frameworks, Curr. Anthropol. 49(2008) 627–654.
[7] F.X. Wilhelm, M.L. Wilhelm, M. Erard, M.P. Duane, Reconstitution ofchromatin: assembly of the nucleosome, Nucleic Acids Res. 5 (1978) 505–521.
[8] S.J. Elsässer, S. D’Arcy, Towards a mechanism for histone chaperones, Biochim.Biophys. Acta 2012 (1819) 211–221.
[9] M.W. Adkins, J.K. Tyler, The histone chaperone Asf1p mediates globalchromatin disassembly in vivo, J. Biol. Chem. 279 (2004) 52069–52074.
[10] T.H. Eickbush, E.N. Moudrianakis, The histone core complex: an octamerassembled by two sets of protein–protein interactions, Biochemistry 17 (1978)4955–4964.
[11] A.J. Andrews, X. Chen, A. Zevin, L.A. Stargell, K. Luger, The histone chaperoneNap1 promotes nucleosome assembly by eliminating nonnucleosomal histoneDNA interactions, Mol. Cell 37 (2010) 834–884.
[12] R. Reeves, Nuclear functions of the HMG proteins, Biochim. Biophys. Acta 1799(2010) 3–14.
[13] M. Stros, HMGB proteins: interactions with DNA and chromatin, Biochim.Biophys. Acta 1799 (2010) 101–113.
[14] K.T. Bauerle, E. Kamau, A. Grove, Interactions between N- and C-terminaldomains of the Saccharomyces cerevisiae high-mobility group proteinHMO1 are required for DNA bending, Biochemistry 45 (2006) 3635–3645.
[15] T. Ueda, H. Chou, T. Kawase, H. Shirakawa, M. Yoshida, Acidic C-tail of HMGB1is required for its target binding to nucleosome linker DNA and transcriptionstimulation, Biochemistry 43 (2004) 9901–9908.

106 Cofractionation of HMGB proteins with histone dimers / Q. Zhuang et al. / Anal. Biochem. 447 (2014) 98–106
[16] K.D. Grasser, D. Launholt, M. Grasser, High mobility group proteins of the plantHMGB family: dynamic chromatin modulators, Biochim. Biophys. Acta 1769(2007) 346–357.
[17] V. Aleporou-Marinou, C. Deli, Y. Ninios, B. Agelopoulou, H. Marinou, T.Patargias, A high mobility group protein from the Dipteran insects Ceratitiscapitata and Bactrocera oleae, Biochem. Genet. 41 (2003) 235–243.
[18] Z. Zhou, H. Feng, D.F. Hansen, H. Kato, E. Luk, D.I. Freedberg, L.E. Kay, C. Wu, Y.I.Bai, NMR structure of chaperone Chz1 complexed with histones H2A.Z-H2B,Nat. Struct. Mol. Biol. 15 (2008) 868–869.
[19] D.A. Kleinjan, V. van Heyningen, Long-range control of gene expression:emerging mechanisms and disruption in disease, Am. J. Hum. Genet. 76 (2005)8–32.
[20] M. Baker, The interaction map, Nature 484 (2012) 271–275.[21] A.P. Snijders, S. Pongdam, S.J. Lambert, C.M. Wood, J.P. Baldwin, M.J. Dickman,
Characterization of post-translational modifications of the linker histones H1and H5 from chicken erythrocytes using mass spectrometry, J. Proteome Res. 7(2008) 4326–4335.
[22] L.E. Foulger, C.G.T. Sin, Q.Q. Zhuang, H. Smallman, J.M. Nicholson, S.J. Lambert,C.D. Reynolds, M.J. Dickman, C.M. Wood, J.P. Baldwin, K. Evans, Efficientpurification of chromatin architectural proteins: histones, HMGB proteins andFKBP3 (FKBP25) immunophilin, RSC Adv. 2 (2012) 10598–10604.
[23] Q. Zhuang, Liverpool: Biochemical and structural studies of histone andassociated proteins from chick and human nuclei. John Moores University;2012. (Ph.D. thesis).
[24] S. Sodngam, Liverpool: Purification and structural studies of histone andhistone associated proteins. John Moores University; 2007. (Ph.D. thesis).
[25] A. Bowman, R. Ward, H. El-Mkami, T. Owen-Hughes, D.G. Norman, Probing the(H3–H4)(2) histone tetramer structure using pulsed EPR spectroscopycombined with site-directed spin labelling, Nucleic Acids Res. 38 (2010)695–707.
[26] G. Felsenfeld, R.H. Simon, A new procedure for purifying histone pairs H2A +H2B and H3 + H4 from chromatin using hydroxylapatite, Nucleic Acids Res. 6(1979) 689–696.
[27] S. Baldi, P.B. Becker, The variant histone H2A.V of Drosophila—three roles, twoguises, Chromasoma 4 (2013) 245–258.
Related Documents









![Histone Modification - fnkprddata.blob.core.windows.net · $ GTX117336 I H istone H 1 t a ntibody [N1C3] @ GTX21938 I Histone H1 antibody Acetylation $ GTX88006 I Histone H1 K25ac](https://static.cupdf.com/doc/110x72/5c66fbdf09d3f2e33b8ce2a6/histone-modification-gtx117336-i-h-istone-h-1-t-a-ntibody-n1c3-gtx21938.jpg)

