MOLECULAR AND CELLULAR NEXJROSCIENCES 3,433-445 (19%) Cochlear Disorder Associated with Melanocyte Anomaly in Mice with a Transgenic Insertional Mutation MASAYOSHI TACHIBANA,* YOSHINOBU HARA,?” DARSHAN VYAS,$ COLIN HODGKINSON,$ JORGEN FEX,* KENNETH GRUNDFAST,* AND HEINZ ARNHEITER$ *Laboratory of Molecular Biology, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, Maryland 20892; and fLaboratory of Neurochemistry and #Laboratory of Viral and Molecular Pathogenesis, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892 Received for publication January 27, 1992 We have generated eight lines of transgenic mice con- taining mouse vasopressin-&galactosidase fusion con- structs. One of these lines, VGA-g, harbors approximately 50 transgene copies at a single chromosomal site. When bred to transgene homozygosity, mice of this line showed a complete loss of skin pigmentation, microphthalmia, and cochlear abnormalities. The vascular stria of the co- chlea was thin and deficient in melanin pigment which is normally produced by presumably neural crest-derived melanocytes. The marginal cells of the stria were thin and lacked basal infoldings. Degeneration of outer hair cells was also observed in homozygous mice, but this al- teration may be secondary to the strial abnormalities. In contrast to homozygous VGA-9 mice, heterozygous VGA- 9 mice were pigmented and appeared to have no anatom- ical alterations in either eye or cochlea. Since the inte- grated transgene provides a marker for cloning an en- dogenous gene necessary for normal pigmentation and proper development of the inner ear, the transgenic line VGA-9 may become valuable for the study of the molec- ular genetics of inner ear disorders associated with pig- ment abnormalities in both mice and humans. o 1992 Academic Press, Inc. INTRODUCTION In laboratory mice, many neurological mutations in- volve lesions of the inner ear, either alone or in conjunc- tion with lesions in other parts of the nervous system or even other organ systems (1). However, few of these mu- tations have been characterized at the molecular level, chiefly because of difficulties in isolating the correspond- ing genes solely on the basis of their linkage with known genes in the genome. Transgenic insertional mutations affecting neural development provide the closely linked 1 Present address: Laboratory of Biochemical Genetics, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD 20892. genetic tags that may eventually allow the isolation of relevant endogenous genes. Here, we describe a line of transgenic mice, VGA-g, that harbor multiple copies of a chimeric construct composed of portions of the mouse vasopressin gene (2) fused with the Escherichia coli /3- galactosidase coding region. When bred to transgene ho- mozygosity, these mice display an insertional mutation that affects melanocytes and the development of the eyes and the stria vascularis of the cochlea. The interest in this particular insertional mutation is based on its phenotypic similarity to mice with mutations at the microphthalmia (mi) locus (with which the trans- genie insertion appears to be allelic) and to humans with Waardenburg syndrome. The mi locus of mice, located 46 map units from the centromer of mouse chromosome 6, is highly complex; that is, many different semidominant or dominant alleles are known that affect, to varying de- grees, melanocytes of the skin and of inner organs and bone marrow-derived cells such as mast cells and osteo- clasts (1, 3). To date, none of these mi alleles have been cloned. Waardenburg syndrome is a genetically hetero- geneous, autosomal dominant disorder with a highly pleiotropic phenotype that includes, as in mice with cer- tain mi alleles, pigment abnormalities of the skin and head and congenital unilateral or bilateral hearing im- pairment (4). Extensive phenotypic variations between families suggest the involvement of a multitude of differ- ent alleles of one locus, or more than one locus, and vari- ations within families imply the existence of modifier al- leles. By virtue of the presence or absence of dystopia canthorum (lateral displacement of the inner canthi), persons with Waardenburg syndrome are classified as Waardenburg type I (WS I) or Waardenburg type II (WS II) (5, 6). WS I is genetically linked to several loci on the long arm of human chromosome 2, including the placental alkaline phosphatase (ALPP) locus (7-9). This region is syntenic with a mouse chromosome 1 region to which another mouse mutation affecting pigmentation, Splotch (Sp) (1, 3), maps. Splotch mice have recently been rec- ognized to have a mutation in pax-3, a gene coding for a 433 1044.7431/92 $5.00 Copyright 0 1992 by Academic Press, Inc. All rights of reproduction in any form reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MOLECULAR AND CELLULAR NEXJROSCIENCES 3,433-445 (19%)
Cochlear Disorder Associated with Melanocyte Anomaly in Mice with a Transgenic Insertional Mutation
MASAYOSHI TACHIBANA,* YOSHINOBU HARA,?” DARSHAN VYAS,$ COLIN HODGKINSON,$ JORGEN FEX,* KENNETH GRUNDFAST,* AND HEINZ ARNHEITER$
*Laboratory of Molecular Biology, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, Maryland 20892; and fLaboratory of Neurochemistry and #Laboratory of Viral and Molecular Pathogenesis, National Institute of
Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892
Received for publication January 27, 1992
We have generated eight lines of transgenic mice con- taining mouse vasopressin-&galactosidase fusion con- structs. One of these lines, VGA-g, harbors approximately 50 transgene copies at a single chromosomal site. When bred to transgene homozygosity, mice of this line showed a complete loss of skin pigmentation, microphthalmia, and cochlear abnormalities. The vascular stria of the co- chlea was thin and deficient in melanin pigment which is normally produced by presumably neural crest-derived melanocytes. The marginal cells of the stria were thin and lacked basal infoldings. Degeneration of outer hair cells was also observed in homozygous mice, but this al- teration may be secondary to the strial abnormalities. In contrast to homozygous VGA-9 mice, heterozygous VGA- 9 mice were pigmented and appeared to have no anatom- ical alterations in either eye or cochlea. Since the inte- grated transgene provides a marker for cloning an en- dogenous gene necessary for normal pigmentation and proper development of the inner ear, the transgenic line VGA-9 may become valuable for the study of the molec- ular genetics of inner ear disorders associated with pig- ment abnormalities in both mice and humans. o 1992 Academic Press, Inc.
INTRODUCTION
In laboratory mice, many neurological mutations in- volve lesions of the inner ear, either alone or in conjunc- tion with lesions in other parts of the nervous system or even other organ systems (1). However, few of these mu- tations have been characterized at the molecular level, chiefly because of difficulties in isolating the correspond- ing genes solely on the basis of their linkage with known genes in the genome. Transgenic insertional mutations affecting neural development provide the closely linked
1 Present address: Laboratory of Biochemical Genetics, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD 20892.
genetic tags that may eventually allow the isolation of relevant endogenous genes. Here, we describe a line of transgenic mice, VGA-g, that harbor multiple copies of a chimeric construct composed of portions of the mouse vasopressin gene (2) fused with the Escherichia coli /3- galactosidase coding region. When bred to transgene ho- mozygosity, these mice display an insertional mutation that affects melanocytes and the development of the eyes and the stria vascularis of the cochlea.
The interest in this particular insertional mutation is based on its phenotypic similarity to mice with mutations at the microphthalmia (mi) locus (with which the trans- genie insertion appears to be allelic) and to humans with Waardenburg syndrome. The mi locus of mice, located 46 map units from the centromer of mouse chromosome 6, is highly complex; that is, many different semidominant or dominant alleles are known that affect, to varying de- grees, melanocytes of the skin and of inner organs and bone marrow-derived cells such as mast cells and osteo- clasts (1, 3). To date, none of these mi alleles have been cloned. Waardenburg syndrome is a genetically hetero- geneous, autosomal dominant disorder with a highly pleiotropic phenotype that includes, as in mice with cer- tain mi alleles, pigment abnormalities of the skin and head and congenital unilateral or bilateral hearing im- pairment (4). Extensive phenotypic variations between families suggest the involvement of a multitude of differ- ent alleles of one locus, or more than one locus, and vari- ations within families imply the existence of modifier al- leles. By virtue of the presence or absence of dystopia canthorum (lateral displacement of the inner canthi), persons with Waardenburg syndrome are classified as Waardenburg type I (WS I) or Waardenburg type II (WS II) (5, 6). WS I is genetically linked to several loci on the long arm of human chromosome 2, including the placental alkaline phosphatase (ALPP) locus (7-9). This region is syntenic with a mouse chromosome 1 region to which another mouse mutation affecting pigmentation, Splotch (Sp) (1, 3), maps. Splotch mice have recently been rec- ognized to have a mutation in pax-3, a gene coding for a
433 1044.7431/92 $5.00 Copyright 0 1992 by Academic Press, Inc.
All rights of reproduction in any form reserved.
434 TACHIBANA ET AL
DNA-binding protein (lo), and it has been demonstrated that members of families with ALPP-linked WS I have deletions or base substitutions in the coding region of HuP2, the human homologue of mouse Pax-3 (11, 12). However, other loci not linked to ALPP may be involved in different WS I families (13), and no linkage with genes mapping to human chromosome 2 has so far been found for WS II. Thus, it is likely that many individuals with Waardenburg syndrome have mutations in other genes. Some of these individuals may in fact have mutations in the human homologue of mouse mi, as the range of phe- notypes of the various mutant alleles of mi may reflect much of the variations observed among individuals with Waardenburg syndrome (14). The analysis of the trans- genie insertional mutation VGA-9 may thus become im- portant for the molecular analysis of a gene specifying pigmentation, and ear development and function, in both mice and humans.
As a first step toward the analysis of this mutation, we here provide a description of the structural abnormalities of the cochlea of VGA-9 homozygous transgenics. We pay particular attention to the vascular stria, since this struc- ture is commonly involved in mutations causing a com- bination of pigmentary and cochlear pathology (15-18).
MATERIALS AND METHODS
Transgene Construction
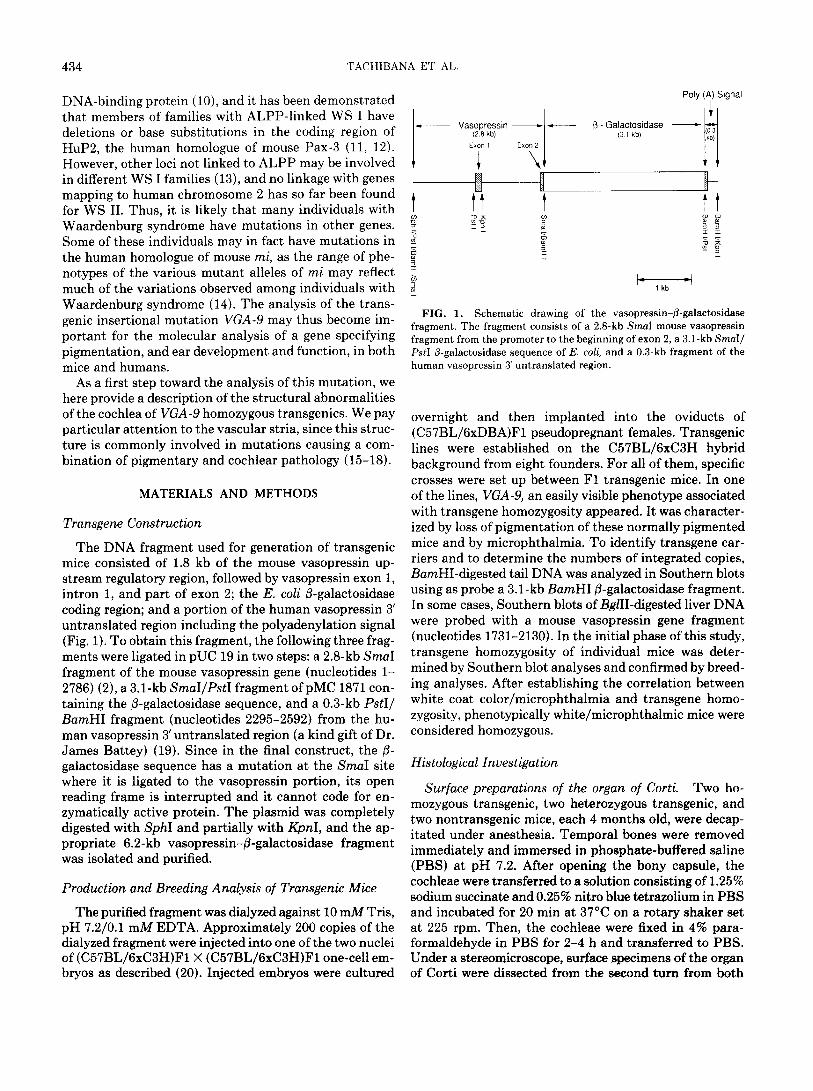
The DNA fragment used for generation of transgenic mice consisted of 1.8 kb of the mouse vasopressin up- stream regulatory region, followed by vasopressin exon 1, intron 1, and part of exon 2; the E. coli /3-galactosidase coding region; and a portion of the human vasopressin 3’ untranslated region including the polyadenylation signal (Fig. 1). To obtain this fragment, the following three frag- ments were ligated in pUC 19 in two steps: a 2.8-kb SmaI fragment of the mouse vasopressin gene (nucleotides l- 2786) (2), a 3.1-kb SmaI/PstI fragment of pMC 1871 con- taining the /3-galactosidase sequence, and a 0.3-kb PstI/ BamHI fragment (nucleotides 2295-2592) from the hu- man vasopressin 3’ untranslated region (a kind gift of Dr. James Battey) (19). Since in the final construct, the p- galactosidase sequence has a mutation at the SmaI site where it is ligated to the vasopressin portion, its open reading frame is interrupted and it cannot code for en- zymatically active protein. The plasmid was completely digested with SphI and partially with KpnI, and the ap- propriate 6.2-kb vasopressin-/3-galactosidase fragment was isolated and purified.
Production and Breeding Analysis of Transgenic Mice
The purified fragment was dialyzed against 10 mM Tris, pH 7.2/0.1 mM EDTA. Approximately 200 copies of the dialyzed fragment were injected into one of the two nuclei of (C57BL/6xC3H)Fl X (C57BL/6xC3H)Fl one-cell em- bryos as described (20). Injected embryos were cultured
- Vasopressin - (2.8 kb)
Exon 1 Exon 2
1
Poly (A) Signal I
_ - p Galactosidase - (3 1 kb)
5 2 I- -
FIG. 1. Schematic drawing of the vasopressin-@-galactosidase fragment. The fragment consists of a 2.8-kb SmaI mouse vasopressin fragment from the promoter to the beginning of exon 2, a 3.1-kb SmaI/ PstI (I-galactosidase sequence of E. coli, and a 0.3-kb fragment of the human vasopressin 3’ untranslated region.
overnight and then implanted into the oviducts of (C57BL/GxDBA)Fl pseudopregnant females. Transgenic lines were established on the C57BL/6xC3H hybrid background from eight founders. For all of them, specific crosses were set up between Fl transgenic mice. In one of the lines, VGA-g, an easily visible phenotype associated with transgene homozygosity appeared. It was character- ized by loss of pigmentation of these normally pigmented mice and by microphthalmia. To identify transgene car- riers and to determine the numbers of integrated copies, BarnHI-digested tail DNA was analyzed in Southern blots using as probe a 3.1-kb BamHI /I-galactosidase fragment. In some cases, Southern blots of BglII-digested liver DNA were probed with a mouse vasopressin gene fragment (nucleotides 1731-2130). In the initial phase of this study, transgene homozygosity of individual mice was deter- mined by Southern blot analyses and confirmed by breed- ing analyses. After establishing the correlation between white coat color/microphthalmia and transgene homo- zygosity, phenotypically white/microphthalmic mice were considered homozygous.
Histological Investigation
Surface preparations of the organ of Corti. Two ho- mozygous transgenic, two heterozygous transgenic, and two nontransgenic mice, each 4 months old, were decap- itated under anesthesia. Temporal bones were removed immediately and immersed in phosphate-buffered saline (PBS) at pH 7.2. After opening the bony capsule, the cochleae were transferred to a solution consisting of 1.25% sodium succinate and 0.25% nitro blue tetrazolium in PBS and incubated for 20 min at 37°C on a rotary shaker set at 225 rpm. Then, the cochleae were fixed in 4% para- formaldehyde in PBS for 2-4 h and transferred to PBS. Under a stereomicroscope, surface specimens of the organ of Corti were dissected from the second turn from both

TRANSGENIC MICE WITH COCHLEAR DISORDER 435
cochleae of an individual mouse. The specimens were mounted on glass slides in a solution of 50% glycerol/ 50% PBS and examined by light microscopy using a Reichert-Jung Polyvar microscope equipped for differ- ential interference contrast as well as epifluorescence mi- croscopy.
Surface preparations of the vascular stria. Cochleae were prepared from two 4-month-old mice belonging to one of the three groups of nontransgenics, heterozygous transgenics, and homozygous transgenics and from a lo- month-old homozygous transgenic and a lo-month-old nontransgenic mouse. The cochleae were immersed in PBS and the lateral walls of the cochlear duct (first and second turn) were dissected. The specimens were left un- fixed and mounted on glass slides. They were observed by differential interference contrast microscopy and an- alyzed for autofluorescence by epifluorescent illumination, using a 200-W mercury high-pressure bulb and a BP 450- 495 excitation filter combined with an LP 520 barrier filter. Some specimens were incubated in 15% hydrogen peroxide for 2 h at room temperature before being mounted.
Histological sections. Three to four mice belonging to each of the three groups of nontransgenic, heterozygous transgenic, and homozygous transgenic mice were used. They were between 1 and 4 months old and were perfused through the left ventricle with 2.7% glutaraldehyde in 0.1 M phosphate buffer (PB, pH 7.4) under anesthesia by Avertin (tribromoethanol in 2-methyl-2-butanol) applied intraperitoneally. The temporal bones were removed im- mediately following decapitation and immersed in fixative. The bony cochlea was opened to allow further infiltration of fixative, and the specimens were left overnight in fix- ative at 4°C. The cochleae were then transferred to PB and the second turn of the membraneous cochlea was dissected using a stereomicroscope. The dissected tissues were postfixed with 2% OsO.+, dehydrated by graded con- centrations of ethanol, and embedded in Spur-resin. Thin sections were stained with uranyl acetate and lead citrate and observed in a JEOL 100 CX II electron microscope. For light microscopy, the cochleae of a nontransgenic, a heterozygous, and two homozygous transgenic mice, each approximately 4 months old, were decalcified for 2 weeks in a solution of 3.8% EDTA and 3% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4). These cochlea were embedded in the Spur-resin without osmium fixation. Semithin sections of these preparations were stained with a solution of 0.5% methylene blue, 0.5% azur II, and 0.5% borax in distilled water and observed by light microscopy. Also, hematoxylin/eosin-stained paraffin sections of the eye of two animals of each group were examined by light microscopy.
The startle response to hand clapping was tested for mice of various ages and for all mice used for histological preparations.
RESULTS
Genetic and Southern Analysis
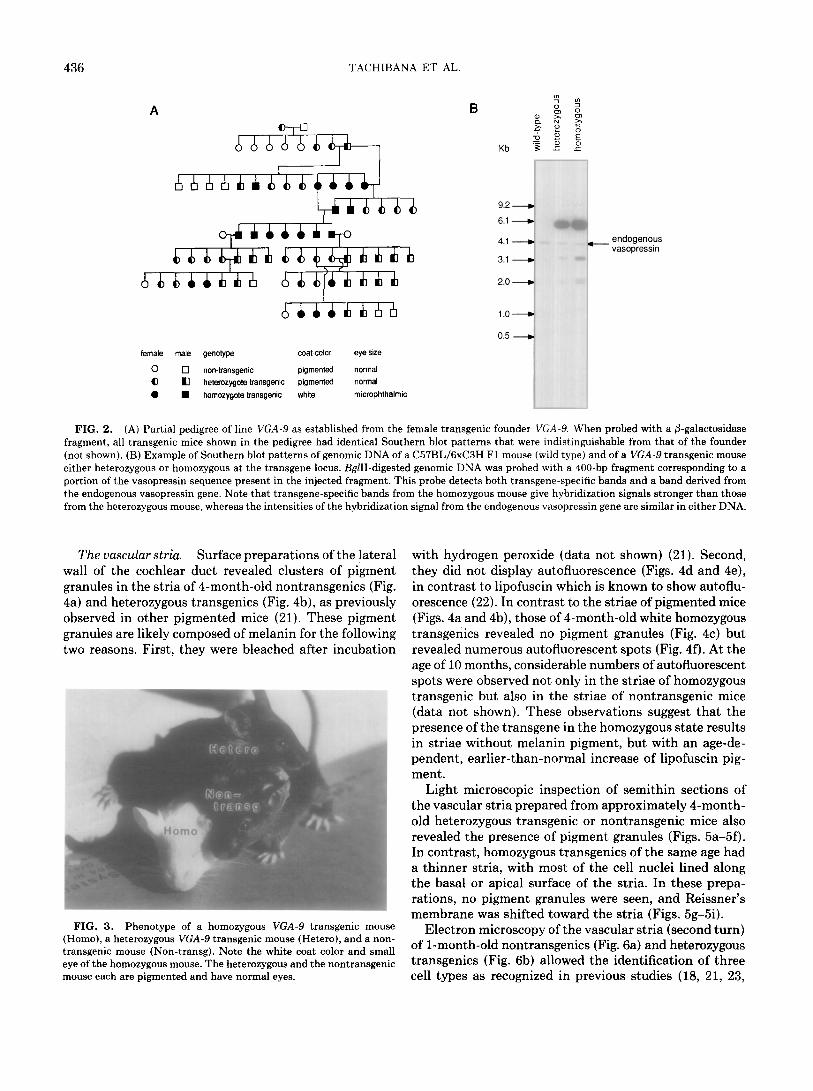
The VGA-9 founder animal contained approximately 50 copies of the chimeric transgene consisting of 1.8 kb of the mouse vasopressin upstream region, vasopressin exon 1 and intron 1, and part of exon 2 fused to the E. coli P-galactosidase coding region (Fig. 1). The founder mouse had a normally pigmented coat and eyes of normal size and gave birth to various litters of Fl transgenic and nontransgenic mice of normal appearance. However, in crosses between two Fl transgenics, 5 of 13 of the progeny were white and microphthalmic, whereas the remainder were pigmented and had normal eyes as indicated in the pedigree shown in Fig. 2A. Southern blot analysis revealed that all white/microphthalmic animals carried the trans- gene. When such white/microphthalmic mice were crossed with nontransgenic mice, all (16 of 16) progeny mice were pigmented, normophthalmic, and transgenic, indicating that the white/microphthalmic transgenics were homo- zygous at the transgene locus. Crosses between white/ microphthalmic mice produced only transgenic white/ microphthalmic progeny. The white/microphthalmic mice were consistently found to have transgene hybridization signals approximately twice as strong as those of pig- mented transgenics (for an example, see Fig. 2B), and this observation is consistent with the interpretation that white/microphthalmic mice are homozygous at the trans- gene locus. The restriction patterns of the genomic DNA of all transgenics were similar to that of the founder mouse, which suggests that the transgene array had in- tegrated at a single chromosomal site. Figure 3 shows the phenotypic appearance of a nontransgenic mouse and of a heterozygous and a homozygous transgenic mouse. Some of the pigmented heterozygous transgenics displayed a white belly spot or streak that was larger than those oc- casionally observed in nontransgenic mice (data not shown).
Histological Ihvestigation
It has been noted earlier that certain mutant mice with pigment abnormalities display hearing deficiencies. We noticed that the startle response of homozygous trans- genies to hand clapping disappeared at the age of about 1 month, whereas heterozygous and nontransgenic mice continued to show a startle response beyond this age. Therefore, we analyzed the cochleae of these animals his- tologically at both the light and the electron microscopic level.
The dissections of the inner ears revealed that the overall anatomy of the bone structure of the cochlea was not altered in homozygous or heterozygous transgenic mice when compared with that of nontransgenic mice. However, the histological analysis of the cochlea revealed a striking pathology confined to the homozygous trans- genies.
436 TACHIBANA ET AL.
female male genotype coat-color eye size
0 0 non-transgenic pigmented normal cl tl hetarozygota transgenic pigmented normal 0 H homozygote transgenic white microphthalmic
9.2 -
6.1 -
4.1 - Ima i_
3.1 - *I
2.0 - ‘“1
i.o-
0.5 ----,
- endogenous vasopressin
FIG. 2. (A) Partial pedigree of line VGA-9 as established from the female transgenic founder VGA-Y. When probed with a @-galactosidase fragment, all transgenic mice shown in the pedigree had identical Southern blot patterns that were indistinguishable from that of the founder (not shown). (B) Example of Southern blot patterns of genomic DNA of a C57BL/6xC3H Fl mouse (wild type) and of a VGA-9 transgenic mouse either heterozygous or homozygous at the transgene locus. BglII-digested genomic DNA was probed with a 400-bp fragment corresponding to a portion of the vasopressin sequence present in the injected fragment. This probe detects both transgene-specific bands and a band derived from the endogenous vasopressin gene. Note that transgene-specific bands from the homozygous mouse give hybridization signals stronger than those from the heterozygous mouse, whereas the intensities of the hybridization signal from the endogenous vasopressin gene are similar in either DNA.
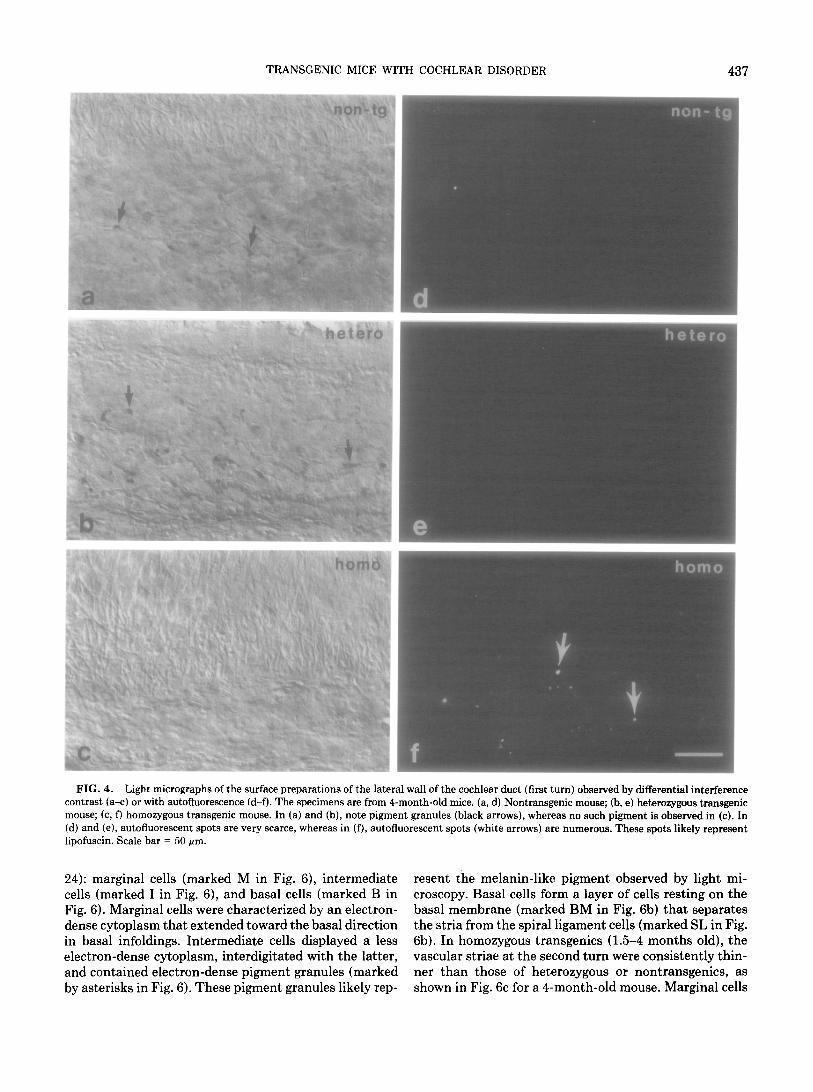
The vascular stria. Surface preparations of the lateral wall of the cochlear duct revealed clusters of pigment granules in the stria of 4-month-old nontransgenics (Fig. 4a) and heterozygous transgenics (Fig. 4b), as previously observed in other pigmented mice (21). These pigment granules are likely composed of melanin for the following two reasons. First, they were bleached after incubation
FIG. 3. Phenotype of a homozygous VGA-9 transgenic mouse (Homo), a heterozygous VGA-9 transgenic mouse (Hetero), and a non- transgenic mouse (Non-transg). Note the white coat color and small eye of the homozygous mouse. The heterozygous and the nontransgenic mouse each are pigmented and have normal eyes.
with hydrogen peroxide (data not shown) (21). Second, they did not display autofluorescence (Figs. 4d and 4e), in contrast to lipofuscin which is known to show autoflu- orescence (22). In contrast to the striae of pigmented mice (Figs. 4a and 4b), those of 4-month-old white homozygous transgenics revealed no pigment granules (Fig. 4c) but revealed numerous autofluorescent spots (Fig, 4f). At the age of 10 months, considerable numbers of autofluorescent spots were observed not only in the striae of homozygous transgenic but also in the striae of nontransgenic mice (data not shown). These observations suggest that the presence of the transgene in the homozygous state results in striae without melanin pigment, but with an age-de- pendent, earlier-than-normal increase of lipofuscin pig- ment.
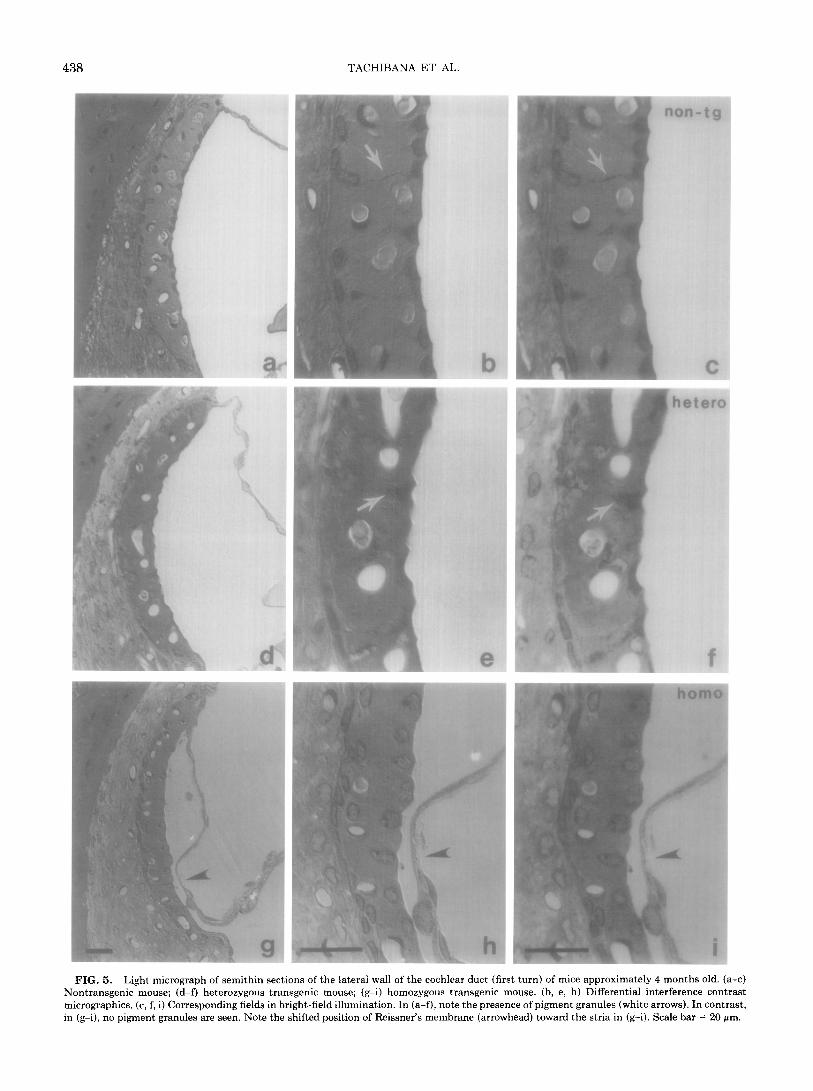
Light microscopic inspection of semithin sections of the vascular stria prepared from approximately 4-month- old heterozygous transgenic or nontransgenic mice also revealed the presence of pigment granules (Figs. 5a-5f). In contrast, homozygous transgenics of the same age had a thinner stria, with most of the cell nuclei lined along the basal or apical surface of the stria. In these prepa- rations, no pigment granules were seen, and Reissner’s membrane was shifted toward the stria (Figs. 5g-5).
Electron microscopy of the vascular stria (second turn) of l-month-old nontransgenics (Fig. 6a) and heterozygous transgenics (Fig. 6b) allowed the identification of three cell types as recognized in previous studies (18, 21, 23,
TRANSGENIC MICE WITH COCHLEAR DISORDER 437
FIG. 4. Light micrographs of the surface preparations of the lateral wall of the cochlear duct (first turn) observed by differential interference contrast (a-c) or with autofluorescence (d-f). The specimens am from 4-month-old mice. (a, d) Nontransgenic mouse; (b, e) heterozygous transgenic mouse; (c, f) homozygous transgenic mouse. In (a) and (b), note pigment granules (black arrows), whereas no such pigment is observed in (c). In (d) and (e), autofluorescent spots are very scarce, whereas in (f), autofluorescent spots (white arrows) are numerous. These spots likely represent lipofuscin. Scale bar = 50 pm.
24): marginal cells (marked M in Fig. 6), intermediate cells (marked I in Fig. 6), and basal cells (marked B in Fig. 6). Marginal cells were characterized by an electron- dense cytoplasm that extended toward the basal direction in basal infoldings. Intermediate cells displayed a less electron-dense cytoplasm, interdigitated with the latter, and contained electron-dense pigment granules (marked by asterisks in Fig. 6). These pigment granules likely rep-
resent the melanin-like pigment observed by light mi- croscopy. Basal cells form a layer of cells resting on the basal membrane (marked BM in Fig. 6b) that separates the stria from the spiral ligament cells (marked SL in Fig. 6b). In homozygous transgenics (1.5-4 months old), the vascular striae at the second turn were consistently thin- ner than those of heterozygous or nontransgenics, as shown in Fig. 6c for a 4-month-old mouse. Marginal cells
438 TACHIBANA ET AL
FIG. 5. Light micrograph of semithin sect ions of the lateral wall of the cochlear duct (fin rt turn) of mice approximately 4 months old. (a-c) Nontransgenic mouse; (d-f) heterozygous trar rsgenic mouse; (g-i) homozygous transgenic n noose. (b, e, h) Differential interference contrast micrographics. (c, f, i) Corresponding fields in b right-field illumination. In (a-f), note the preser ice of pigment granules (white arrows). In contrast, in (g-i), no pigment granules are seen. Note the shifted position of Reissner’s membrane (arrow head) toward the stria in (g-i). Scale bar = 20 pm.
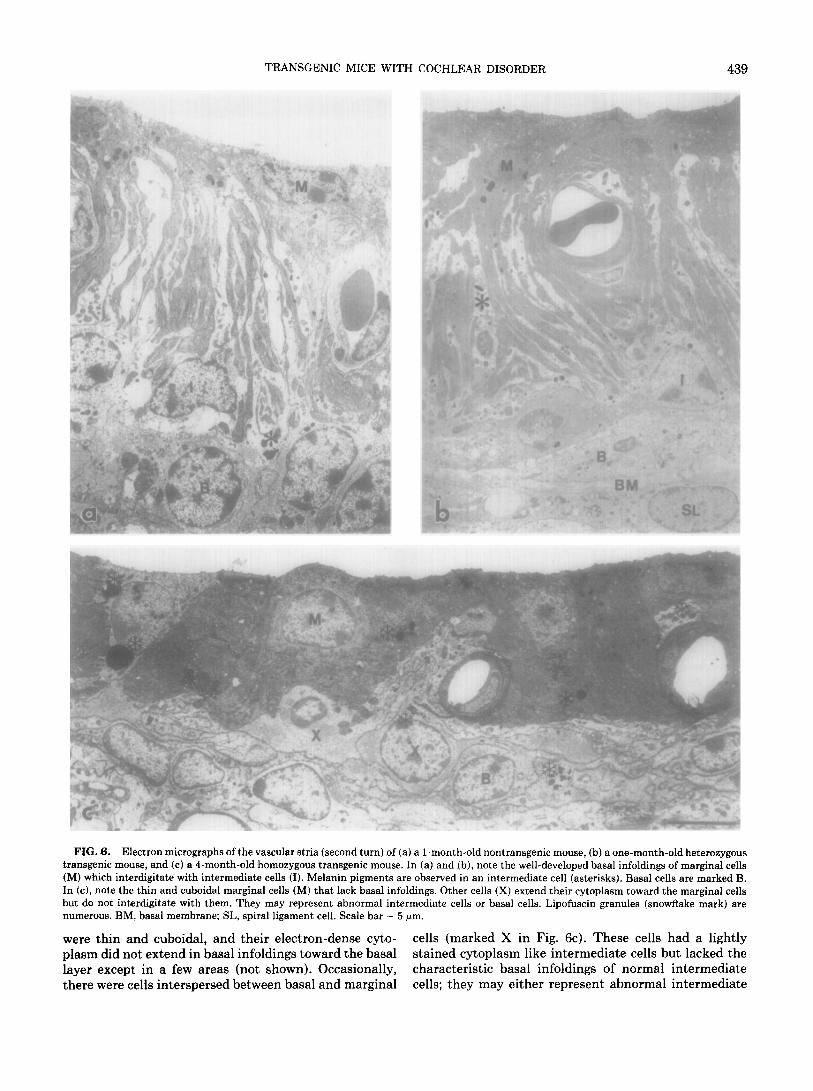
TRANSGENIC MICE WITH COCHLEAR DISORDER 439
FIG. 6. Electron micrographs of the vascular stria (second turn) of (a) a l-month-old nontransgenic mouse, (b) a one-month-old heterozygous transgenic mouse, and (c) a 4-month-old homozygous transgenic mouse. In (a) and (b), note the well-developed basal infoldings of marginal cells (M) which interdigitate with intermediate cells (I). Melanin pigments are observed in an intermediate cell (asterisks). Basal cells are marked B. In (c), note the thin and cuboidal marginal cells (M) that lack basal infoldings. Other cells (X) extend their cytoplasm toward the marginal cells but do not interdigitate with them. They may represent abnormal intermediate cells or basal cells. Lipofuscin granules (snowflake mark) are numerous. BM, basal membrane; SL, spira1 ligament cell. Scale bar = 5 pm.
were thin and cuboidal, and their electron-dense cyto- cells (marked X in Fig. 6~). These cells had a lightly plasm did not extend in basal infoldings toward the basal stained cytoplasm like intermediate cells but lacked the layer except in a few areas (not shown). Occasionally, characteristic basal infoldings of normal intermediate there were cells interspersed between basal and marginal cells; they may either represent abnormal intermediate

440 TACHIBANA ET AL
cells or abnormally positioned basal cells. There were abundant, heavily electron-dense granules visible by electron microscopy (snowflake marks in Fig. 6~). These granules most likely represent the lipofuscin granules identified by autofluorescence (see above).
The organ of Corti. Surface preparations of the organ of Corti (second turn) were observed by differential in- terference contrast microscopy. The organs of Corti of 4- month-old nontransgenics (Fig. 7a) and heterozygous transgenics (Fig. 7b) had three rows of outer and one row of inner hair cells. In contrast, in 4-month-old homozy- gous transgenics, outer hair cells were either absent or not properly arranged in three rows (Fig. 7~). However, the row of inner hair cells appeared largely intact (Fig. 7~). The stereocilia of both outer and inner hair cells were normal in nontransgenic (Figs. 8a and 8b) and hetero- zygous transgenic mice (Figs. 8c and 8d). In contrast, in homozygous transgenics, no stereocilia were seen in areas where outer hair cells were missing. In a small area where outer hair cells still could be identified by their shape and histochemical staining, stereocilia were mostly missing or, where present, were not arranged as seen in the pig- mented mice (Fig. 8e). The row of inner hair cells of the homozygous transgenics contained stereocilia. However, in some areas, inner hair cells occupied ectopic positions outside the regular single row of cells (Fig. Sf, arrow).
By light microscopy of semithin sections, the organ of Corti and the spiral ganglion of old heterozygous trans- genie mice seemed indistinguishable from those of non- transgenic control mice (for the first turn of the organ of Corti of approximately 4-month-old mice, see Figs. 9a- 9d). In contrast, in homozygous transgenics, the organ of Corti was disorganized. Outer hair cells were missing and their space was taken over by Deiters’ cells (Fig. 9e). However, the spiral ganglion and the cochlear nerve were normal (Fig. 9f).
By electron microscopy, the organs of Corti (at the sec- ond turn) of l-month-old nontransgenics (Fig. 10a) or heterozygous transgenics (Fig. lob) were normal. In con- trast, a 1.5-month-old, as well as a 4-month-old (Fig. lOc), homozygous transgenic showed degenerated outer hair cells (OHC) and swollen Deiters’ cells (D). The section depicted in Fig. 1Oc shows a Deiters’ cell with two nuclei, a phenomenon that has not been observed in the pig- mented mice. A 2-month-old homozygous transgenic had a normal appearance of the organ of Corti (not shown) even though the stria of the same animal showed a pa- thology similar to that described for all other homozygous transgenics. No pathological changes were seen in the inner hair cells of any of the mice analyzed, except for the occasional positioning of these cells in two rows in the homozygous transgenics.
DISCUSSION
VGA-9 mice, bred to homozygosity at the transgene locus, display a characteristic triad of symptoms: lack of
hetero
FIG. 7. Light micrographs of surface preparations of the organ of Corti (second turn) of 4-month-old mice, stained for succinic dehydro- genase and observed by differential interference contrast microscopy. (a) Nontransgenic mouse; (b) heterozygous transgenic mouse; (c) ho- mozygous transgenic mouse. Focus is at the level of the pillar cell body. In (a) and (b), note three rows of outer hair cells and one row of inner hair cells. In (c), there are no rows of outer hair cells visible, but a row of inner hair cells appears intact. Scale bar = 50 pm.
skin pigmentation, small red eyes, and a cochlear pa- thology consistently manifested in the vascular stria which was thin and free of melanin pigment. It is likely
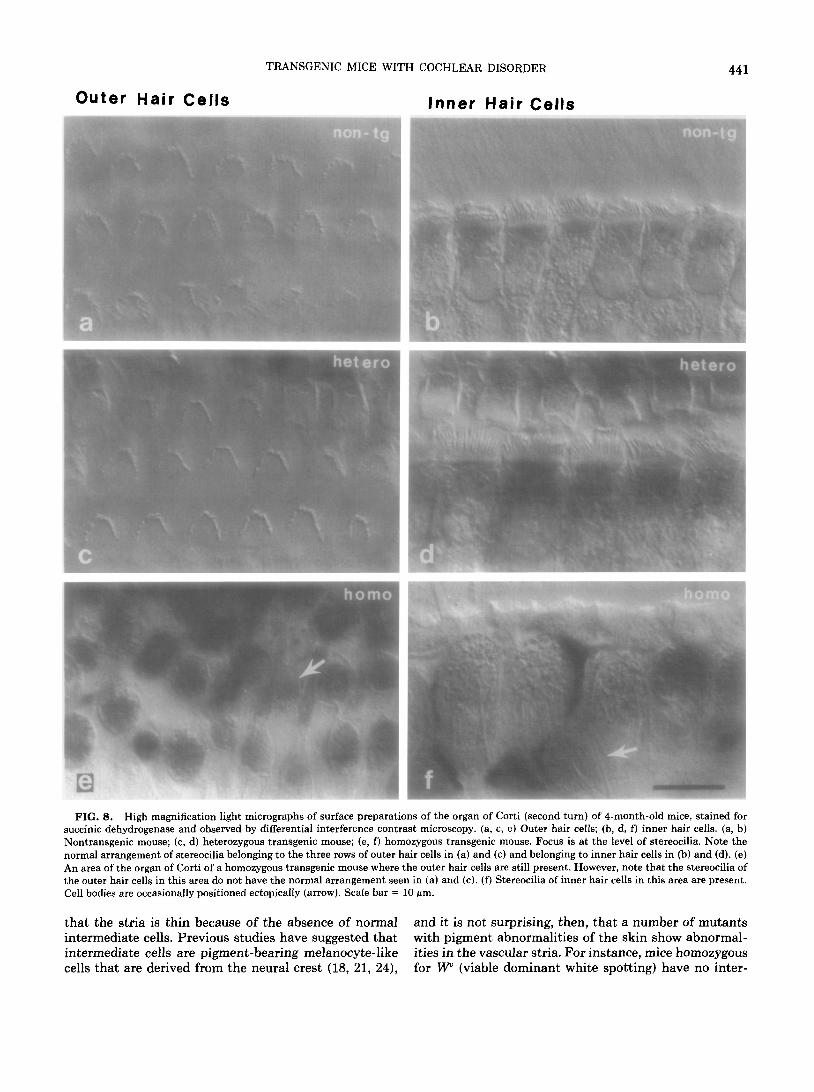
TRANSGENIC MICE WITH COCHLEAR DISORDER
Outer Hair Cells Inner Hair Cells
FIG. 8. High magnification light micrographs of surface preparations of the organ of Corti (second turn) of 4-month-old mice, stained for succinic dehydrogenase and observed by differential interference contrast microscopy. (a, c, e) Outer hair cells; (b, d, f) inner hair cells. (a, b) Nontransgenic mouse; (c, d) heterozygous transgenic mouse; (e, f) homozygous transgenic mouse. Focus is at the level of stereocilia. Note the normal arrangement of stereocilia belonging to the three rows of outer hair cells in (a) and (c) and belonging to inner hair cells in (b) and (d). (e) An area of the organ of Corti of a homozygous transgenic mouse where the outer hair cells are still present. However, note that the stereocilia of the outer hair cells in this area do not have the normal arrangement seen in (a) and (c). (f) Stereocilia of inner hair cells in this area are present. Cell bodies are occasionally positioned ectopically (arrow). Scale bar = IO am.
that the stria is thin because of the absence of normal and it is not surprising, then, that a number of mutants intermediate cells. Previous studies have suggested that with pigment abnormalities of the skin show abnormal- intermediate cells are pigment-bearing melanocyte-like ities in the vascular stria. For instance, mice homozygous cells that are derived from the neural crest (18, 21, 24), for W (viable dominant white spotting) have no inter-
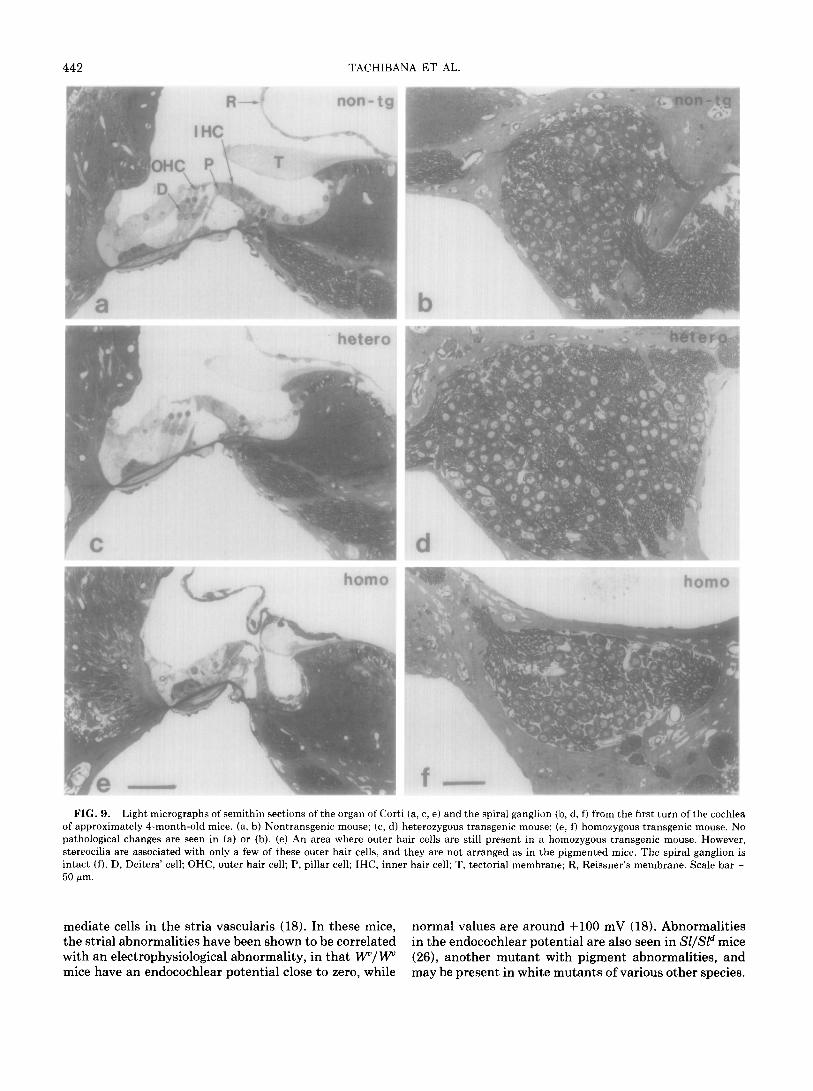
442 TACHIBANA ET AL.
FIG. 9. Light micrographs of semithin sections of the organ of Cort of approximately 4-month-old mice. (a, II) Nontransgenic mouse; (c, d) pathological changes are seen in (a) or (b). (e) An area where outer h stereocilia are associated with only a few of these outer hair cells, and intact (f). D, Deiters’ cell; OHC, outer hair cell; P, pillar cell; IHC, innc 50 pm.
mediate cells in the stria vascularis (18). In these mice, normal values are around +lOO mV (18). Abnormalities the strial abnormalities have been shown to be correlated in the endocochlear potential are also seen in Sl/Sld mice with an electrophysiological abnormality, in that W/W (26), another mutant with pigment abnormalities, and mice have an endocochlear potential close to zero, while may be present in white mutants of various other species.
i (a, c, e) and the spiral ganglion (b, d, f) from the first turn of the cochlea heterozygous transgenic mouse; (e, f) homozygous transgenic mouse. No
lair cells are still present in a homozygous transgenic mouse. However, they are not arranged as in the pigmented mice. The spiral ganglion is
!r hair cell; T, tectorial membrane; R, Reissner’s membrane. Scale bar =
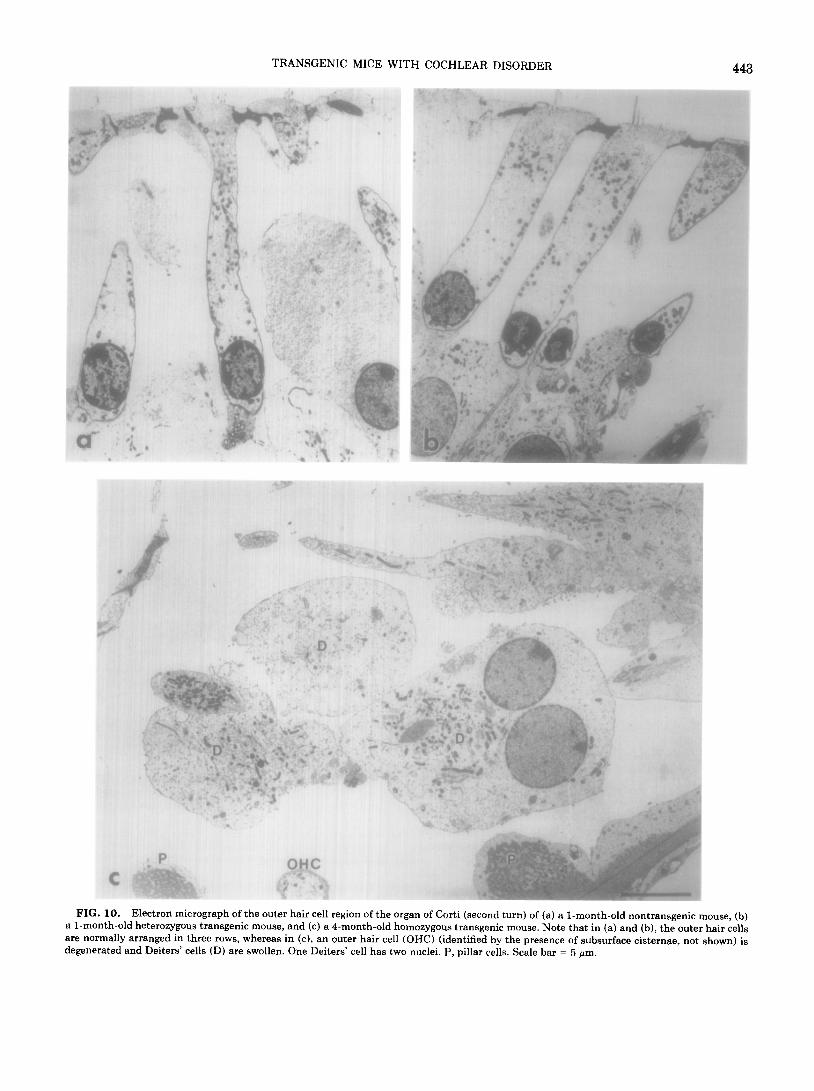
TRANSGENIC MICE WITH COCHLEAR DISORDER
FIG. 10. Electron micrograph of the outer hair cell region of the organ of Corti (second turn) of (a) a l-month-old nontransgenic mouse, (b) a l-month-old heterozygous transgenic mouse, and (c) a B-month-old homozygous transgenic mouse. Note that in (a) and (b), the outer hair cells are normally arranged in three rows, whereas in (c), an outer hair cell (OHC) (identified by the presence of subsurface cisternae, not shown) is degenerated and Deiters’ cells (D) are swollen. One Deiters’ cell has two nuclei. P, pillar cells. Scale bar = 5 Wm.

444 TACHIBANA ET AL.
In fact, Charles Darwin mentioned the relationship be- tween abnormalities of pigmentation and hearing im- pairment in white, blue-eyed cats already in 1859 (27).
In contrast to the consistent strial abnormalities found in homozygous transgenics, pathological changes in the organ of Corti were found to vary with the age of a mouse. The organ of Corti of a 2-month-old homozygous trans- genie was judged to be normal in the area examined by electron microscopy, even though its associated stria was abnormal. At 4 months of age, the organ of Corti contained large areas lacking outer hair cells, but these areas were occasionally adjacent to areas in which the outer hair cells were still present, even though their stereocilia lacked the characteristic positioning seen in nontransgenic or het- erozygous transgenic mice. These observations suggest that in homozygous transgenics, outer hair cells are first generated normally, but later degenerate. This interpre- tation is consistent with the observation that the startle response to sound was initially present, but was lost as the mice reached an age of about 1 month. Therefore, we would classify the pathology in the present mouse mutant as of the cochleosaccular and not the neuroepithelial type. Shift of Reissner’s membrane toward the stria (Figs. 5g- 5i) is one of the characteristic features of the cochleosac- cular abnormality (28).
While intermediate cells of the vascular stria are con- sidered to be derived from the neural crest, melanin-bear- ing cells in the pigment layer of the retina, the inner layer of the iris, and the ciliary body are derived from the optic cup and not the neural crest (25). Interestingly, homo- zygous transgenics show defects in pigmentation in both cochlea and the eye (data not shown). However, it is un- likely that the VGA-9 mutation is similar to the tyrosinase deficiency of the well-characterized oculocutaneous al- binism, in which amelanotic melanocytes are present, and which is not associated with strial abnormalities, hearing impairment, or microphthalmia (1, 3).
Of all the mouse mutants with pigment abnormalities, mice with mutations at the mi locus most closely resemble VGA-9 mice. Depending on the mi allele, and depending on whether a particular allele is present in the hetero- zygous or homozygous state or combined with another mi allele, such mice show varying degrees of white spotted- ness or complete loss of skin pigmentation, microphthal- mia, and hearing impairment (1, 3). In fact, preliminary breeding experiments suggest that the transgene insertion of VGA-9 mice is allelic with mi (Forrester, Bernstein and Arnheiter, unpublished observation). This is compatible with the interpretation that the transgene had integrated into, and disrupted, the mi locus on chromosome 6.
Experiments with mi/mi mast cells (29) or Miwh/+ me- lanocytes (reviewed in (3)) indicate that the phenotype of mice with mutations at mi is cell autonomous, that is, independent of a diffusible factor. Thus, it is likely that mi codes for an intracellular factor (and not a diffusible ligand such as a growth factor) and that with respect to the pathology of the inner ear, this intracellular factor is
necessary for the proper development of the presumably neural crest-derived intermediate cells of the vascular stria. Based on comparisons with pigment abnormalities in mice with mutations at two different loci, namely Sl, which codes for a growth factor-like ligand (30), and W, which codes for the corresponding tyrosin kinase receptor (31), it has been proposed that mi might be part of the Sl- W intracellular signaling pathway (29). However, the possibility remains that mi acts independently of this pathway. In any event, isolating the gene interrupted by the VGA-9 transgene insertion may lead to the identifi- cation of the mi gene, a gene which is associated with a fascinating set of mutations that affect pigmentation and hearing in mice and possibly humans.
ACKNOWLEDGMENTS
We thank Dr. Olivier Gout for help with the perfusions, and Drs. Mark Godec and Lynn Hudson for their critical reading of the manu- script. Thanks are also due to Ms. Marrianne Parakkal for her skillful technical assistance. During the course of this work, D.V. was a Howard Hughes Medical Institute-NIH Research Scholar.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
REFERENCES
Lyon, M. F., and A. G. Searle (1989). Genetic Variants and Strains of the Laboratory Mouse. Oxford Univ. Press, Oxford.
Hara, Y., J. Battey, and H. Gainer (1990). Structure of mouse vasopressin and oxytocin genes. Mol. Brain Res. 8: 319-324.
Silvers, W. K. (1979). The Coat Colors of Mice. Springer, New York.
Waardenburg, P. J. (1951). A new syndrome combining develop- mental anomalies of the eyelids, eyebrows and nose root with pig- mentary defects of the iris and head hair and with congenital deaf- ness. Am. J. Hum. &net. 3: 195-253.
Arias, S. (1971). Genetic heterogeneity in the Waardenburg syn- drome. Birth Defects Orig. Artic. Ser. 7(4): 87-101.
Hageman, M. J., and J. W. Delleman (1977). Heterogeneity in Waardenburg syndrome. Am. J. Hum. Genet. 29: 468-485.
Foy, C., V. Newton, D. Wellesley, R. Harris, and A. P. Read (1990). Assignment of the locus for Waardenburg syndrome type I to hu- man chromosome 2q37 and possible homology to the Splotch mouse. Am. J. Hum. Genet. 46: 1017-1023.
Ishikiriyama, S., H. Tonoki, Y. Shibuya, S. Chin, N. Harada, K. Abe, and N. Niikawa (1986). Waardenburg syndrome type I in a child with de novo inversion (2)(q35-q3’7.3). Am. J. Med. Genet. 33: 505-507.
Asher, Jr., J. H., R. Morell, and T. B. Friedman (1991). Waarden- burg syndrome (WS): The analysis of a single family with a WSI mutation showing linkage to RFLP markers on human chromosome 2q. Am. J. Hum. Genet. 48: 43-52.
Epstein, D. J., M. Vekemans, and G. Philippe (1991). splotch (Sp?, a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell 67: 767- 774.
Tassabehji, M., A. P. Read, V. E. Newton, R. Harris, R. Balling, P. Gruss, and T. Strachan (1992). Waardenburg’s syndrome pa- tients have mutations in the human homologue of thepan- paired box gene. Nature 355: 635-636.
Baldwin, C. T., C. F. Hoth, J. A. Amos, E. 0. da Silva, and A. Milunsky (1992). An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature 356: 637-638.

TRANSGENIC MICE WITH COCHLEAR DISORDER 445
13.
14.
15.
16.
17.
18.
19.
20.
21.
Farrer, L. A., K. M. Grundfast, J. Amos, K. S. Amos, J. H. Asher, P. Beighton, S. Diehl, J. Fex, C. Foy., T. B. Friedman, J. Greenberg, C. Hoth, M. Marazita, A. Milunsky, R. Morell, W. Nance, V. New- ton, R. Ramesar, T. B. San Augustin, J. Skare, C. A. Stevens, R. G. Wagner, E. R. Wilcox, I. Winship, and A. P. Read (1992). Waardenburg syndrome (WS) Type I is caused by defects at mul- tiple loci, one of which is near ALPP on chromosome 2-First report of the WS consortium. Am. J. Med. Genet. 50: 902-913. Asher, Jr., J. H., and T. B. Friedman (1990). Mouse and hamster mutants as models for Waardenburg syndromes in humans. J. Med. Genet. 27: 618-626. Deol, M. S. (1970). The relationship between abnormalities of pig- mentation and the inner ear. Proc. R. Sot. London A 175: 201- 217. Steel, K., C. Barkway, and G. R. Bock (1987). Strial dysfunction in mice with cochlea-saccular abnormalities. Hear. Res. 27: ll- 26. Schrott, A., and H. Spoendlin (1987). Pigment anomaly-associated inner ear deafness. Acta Oto Laryngol. (Stockholm) 103: 451-457. Steel, K., and C. Barkway (1989). Another role for melanocytes: Their importance for normal stria vascularis development in mammalian inner ear. Development 107: 453-463. Sausville, E., D. Carney, and J. Battey (1985). The human vaso- pressin gene is linked to the oxytocin gene and is selectively ex- pressed in a cultured lung cancer cell line. J. Biol. Chem. 260: 10832-10241. Hogan, B., F. Costantini, and E. Lacy (1986). Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Labo- ratory, Cold Spring Harbor, NY. Cable, J., and K. P. Steel (1991). Identification of two types of melanocyte within the stria vascularis of the mouse inner ear. Pigm. Cell Res. 4: 87-101.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
Harman, D. (1989). Lipofuscin and ceroid formation: The cellular recycling system. Adu. Exp. Med. Biol. 266: 3-15.
Rodriguez-Echandia, E. L., and M. H. Burgos (1965). The fine structure of the stria vascularis of the guinea pig inner ear. 2. Zelljorsch. 67: 600-619.
Hilding, D. A., and R. D. Ginzberg (1977). Pigmentation of the stria vascularis: The contribution of neural crest melanocytes. Acta Oto Laryngol. (Stockholm) 84: 24-37.
Le Douarin, N. (1982). The Neural Crest. Cambridge Univ. Press, London.
Schrott, A., I. Melichar, J. Popelir, and J. Syka (1990). Deterio- ration of hearing function in mice with neural crest defect. Hear. Res. 46: l-8.
Darwin, C. (1859). On the Origin of Species. Murray, London.
Steel, K. P., and G. R. Bock (1985). Genetic factors affecting hearing development. Acta Oto Laryngol. (Stockholm) Suppl. 421: 48-56.
Dubreuil, P., L. Forrester, R. Rottapel, M. Reedlik, J. Fujita, and J. Bernstein (1991). The c-fms gene complements the mitogenic defect in mast cells derived from mutant W mice but not mi (mi- crophthalmia) mice. Proc. Natl. Acad. Sci. USA 88: 2341-2345.
Zsebo, K. M., D. A. Williams, E. N. Geissler, V. C. Broudy, F. H. Martin, H. L. Atkins, R-Y. Hsu, N. C. Birkett, F. W. Jacobsen, K. E. Langley, K. A. Smith, T. Takeishi, B. M. Cattanach, S. J. Galli, and S. V. Suggs (1990). Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell 63: 213-224.
Geissler, E. N., M. A. Ryan, and D. E. Housman (1988). The dom- inant-white spotting ( W) locus of the mouse encodes the c-kitproto- oncogene. Cell 55: 185-192.
Related Documents











